universidade de são paulo · aos meus queridos amigos do iq: roberto, pablo, andréa...
TRANSCRIPT

BIBLIOTECAINSTITUTO DE QUíMICAUniversidade de São Paulo
;X;gh~
Universidade de São PauloInstituto de Química
Estudo da Etapa de Quimiexcitação
do Sistema Peroxioxalato
(Tese de Doutorado)
Sandra Maria da Silva
Orientador: Josef Wilhelm Baader
São Paulo
04 de junho de 2004

DEDALUS-AceNo-CQ
111~JlIIII~111
Ficha CatalográficaElaborada pela Divisào de Biblioteca e
Documentaçào do Conjunto das Químicas da USP.
Silva. Sandra Maria daS586e Estudo da etapa de quimiexcitaçào do sistema
peroxioxalato I Sandra Maria da Silva. -- Sào Paulo,2004.
170p.
Tese (doutorado) Instituto de Química daUniversidade de Sào Paulo. Departamento de Quimicafundamental.
Orientador: Baader, Josef Wilhelm
I. Quimiluminescência: físico-química: Orgànica2. Mecanismo de reaçào : Fisico-química : Orgànica3. Cinética de reaçào: Físico-química: Orgânica 1. T.11. Baader, Josef Wílhelm, orientador.
547.135 CDD
.~.


Dedicatória
Dedico este trabalho,
Aos meus pais, Maria Izabel e Elias Martins por disseminar o carinho, respeito e
amizade entre nossa família e pela simplicidade com a qual conduzem a vida,
Aos meus queridos irmãos Vera Lúcia (sempre estressada), Antônio Sergio (sempre bem
humorado) e Elizabeth (nossa "irmãzinha"),
A minha queridissima sobrinha Yanka que, com seu sorriso, ilumina a nossa vida e ao
meu cunhado Roberto pela amizade e simpatia.

Agradecimentos
Agradecimentos
Em primeiro lugar gostaria de agradecer ao meu orientador, Prof. Or. Josef W.
Baader (Willi) , por ter me recebido em seu laboratório e me ensinado tudo o que sei
sobre química orgânica, pela amizade, simpatia e paciência. Obrigada!
Ao Cassius (agora, Professor Cassius) por ter me orientado na iniciação
científica, ajudando-me a dar os primeiros passos dentro de um laboratório.
Aos meus amigos de laboratório: Erick, Luiz Francisco ("Lolo"), Camila, e ao
Marcelo pela constante companhia e pelos divertidos comentários. Aos alunos que me
deram a oportunidade de orientá-los na iniciação científica: Fabiana, Juliano, Viviane e
LoIo. À Professora Paulete pela amizade e pelas agradáveis reuniões em sua casa. O
meu trabalho foi bastante agradável na companhia de vocês.
À Silvia (sempre simpática e sorridente) e ao Luiz (... onde está o Luiz?) pela
disponibilidade com que sempre me ajudaram.
Agradeço a todos que colaboraram com o desenvolvimento do meu trabalho, à
Professora Regina (Bloco 4) pelas medidas de viscosidade, ao Greg pelas medidas do
potencial de oxidação e a todos que em algum momento me cederam um reagente,
uma vidraria, etc ...
Agradeço também à Central Analítica, principalmente ao Fernando, Alessandra,
Mirian, Alfred e Márcio. Ao pessoal da seção de pós-graduação e da Biblioteca pela
prestação de serviço, disponibilidade e paciência com que sempre me auxiliaram.
Agradeço a Professora Oaisy pela ajuda na correção de alguns artigos e pelas
conversas agradáveis.
Especialmente agradeço a Fapesp (Fundação de Amparo à Pesquisa de São
Paulo), ao CNPq (Conselho Nacional de Pesquisa) e ao OAAD (Intercâmbio Acadêmico
Brasil-Alemanha) por financiarem o desenvolvimento deste trabalho.
Aos meus queridos amigos do IQ: Roberto, Pablo, Andréa "Carioca", Celize, João
Lago, Chang, Andréa Aguilar, Elaine, Geise, Fátima, Alessandra, Lilian, Perpétua,
Teresa, Marquinhos por me ajudar na correção do texto, etc... (Graças a Deus a lista é
grande!).
Especialmente, à Sarah, minha irmã espiritual e à Alba por estarem sempre por
perto e, também, à Ritinha, Abadia, Regina e Lurdinha.
Ao Sr. Antõnio, O.Regina, Priscila e a Oaiana pela agradável convivência e
amizade. Enfim, agradeço a todos que torceram por mim,
Obrigada.

Índice
BIBLIOTECAINSTITUTO DE QUíMICAUnIversidade de São Paulo
1 - Introdução 1
1.1 - Quimiluminescência 1
1.2 - Luminescência Induzida Quimicamente pela Troca de Elétron (CIEEL) 9
1.3 - O Sistema Peroxioxalato 12
1.4 - Aplicações Analíticas 21
1.5 - Referências 25
2 - Parte Experimental 30
2.1 - Aparelhagem 30
2.2 - Reagentes 31
2.3 - Solventes 32
2.4 - Determinação da concentração dos perácidos 5-7 e das soluções anidras de H20 2 33
2.5 - Estudo cinético dos derivados de O,O-hidrogênio monoperoxioxalato de arila (5-7) 33
2.6 - Estudo do sistema peroxioxalato variando a viscosidade 34
2.7 - Estudo do sistema peroxioxalato utilizando os ativadores 9-13 35
2.8 - Estudo do sistema peroxioxalato na ausência de H20 2 35
2.9 - Calibração da fotomultiplicadora 36
2.10 - Medida da viscosidade da mistura de tolueno com difenilmetano (C'3Hd 38
2.11 - Preparação dos cloretos de feniloxalila substituídos 38
2.11.1 - Tentativa de preparação do cloreto de p-nitrofeniloxalila (4) 41
2.11.2 - Preparação do bis(4-nitrofenil) oxalato (23) 42
2.12 - Síntese dos derivados de O.O-hidrogênio monoperoxioxalato de arila (5-8) 43
.2.12.1 - Tentativa de síntese do O,O-hidrogênio monoperoxioxalato de p-nitrofenila (8) a
partir do composto 4 45
2.12.2 - Segunda tentativa de síntese do O,O-hidrogênio monoperoxioxalato de p-
nitrofenila (8) a partir do composto 23 45
2.13 - Síntese do ácido p-metoxifeniloxálico (28) 46
2.14 - Síntese de derivados de oxazolonas 47
2.14.1 - Preparação do Mn02 em carvão ativo' 47
2.14.2 - 2[4-bromofenil]-5,5-dimetiloxazol-4-ona (14) 47
2.14.3 - 2-[4-benzilacetileno]-5,5-dimetiloxazol-4-ona (15) 49
2.14.4 - 2-[4-bromofenil]-5,5-dimetiloxazol-4-tiona (16) ' 51
2.14.5 - Androsta-1 ,4-dieno-3-tiona-17-ona (18) 52
2.14.6 - Androsta-1 ,4-dieno-3 hidrazona-17-ona (19) 53
2.14.7 - Androsta-1 ,4-dieno-3 diazo 17-ona (20)' 54
2.14.8 - 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilideno]-androsta-1 ,4-dien-17-ona (21)1 .. 55
2.15 -Referências _ 57

Índice
3 - Resultados 58
3.1 - Síntese e estudo dos derivados de O,O-hidrogênio monoperoxioxalato de arila (5-7) .. 58
3.1.1 - Síntese e caracterização 58
3.2 - Estudo das propriedades de quimiluminescência dos intermediários 5-7 69
3.3 - Estudo da eficiência de transferência de elétron na etapa de quimiexcitação 74
3.3.1 - Determinação do rendimento quântico singlete 74
3.3.2 - Rendimento quântico singlete na concentração infinita (cI>s"') 78
3.4 - Influência da viscosidade no rendimento quântico singlete (cI>s) 81
3.5 - Estudo do Sistema Peroxioxalato na ausência de H20 2 85
3.6 - Síntese de derivados de Oxazolonas (14-21) 89
3.6.1 - 2-[4-fenilacetileno]-5-dimetiloxazol-4-ona (15) 89
3.6.2 - 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilideno]androsta-1 ,4-dien-17-ona (21) 90
3.7 - Referências 93
4 - Discussão 95
4.1 - Síntese dos derivados do O,O-hidrogênio monoperoxioxalato (5-7) 95
4.2 - Estudo cinético dos perácidos 5 a 7 99
4.3 - Correlação de estrutura e reatividade 112
4.4 - Estudo da Eficiência de transferência de elétron na etapa de quimiexcitação 118
4.5 - Influência da viscosidade no rendimento quântico singlete 127
4.6 - Estudo do Sistema Peroxioxalato na ausência de H20 2 .....................•....................... 133
4.7 - Síntese de derivados de Oxazolonas (14-21) 135
4.7.1 - 2-[4-benzilacetileno]-5,5-dimetiloxazol-4-ona (15) 136
4.7.2 - 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilideno]-androsta-1 ,4-dien-17-ona (21) 137
4.8 - Referências 142
5 - Conclusão 147
6 - Anexos 148
7 - Curriculum Vitae 166

Índice
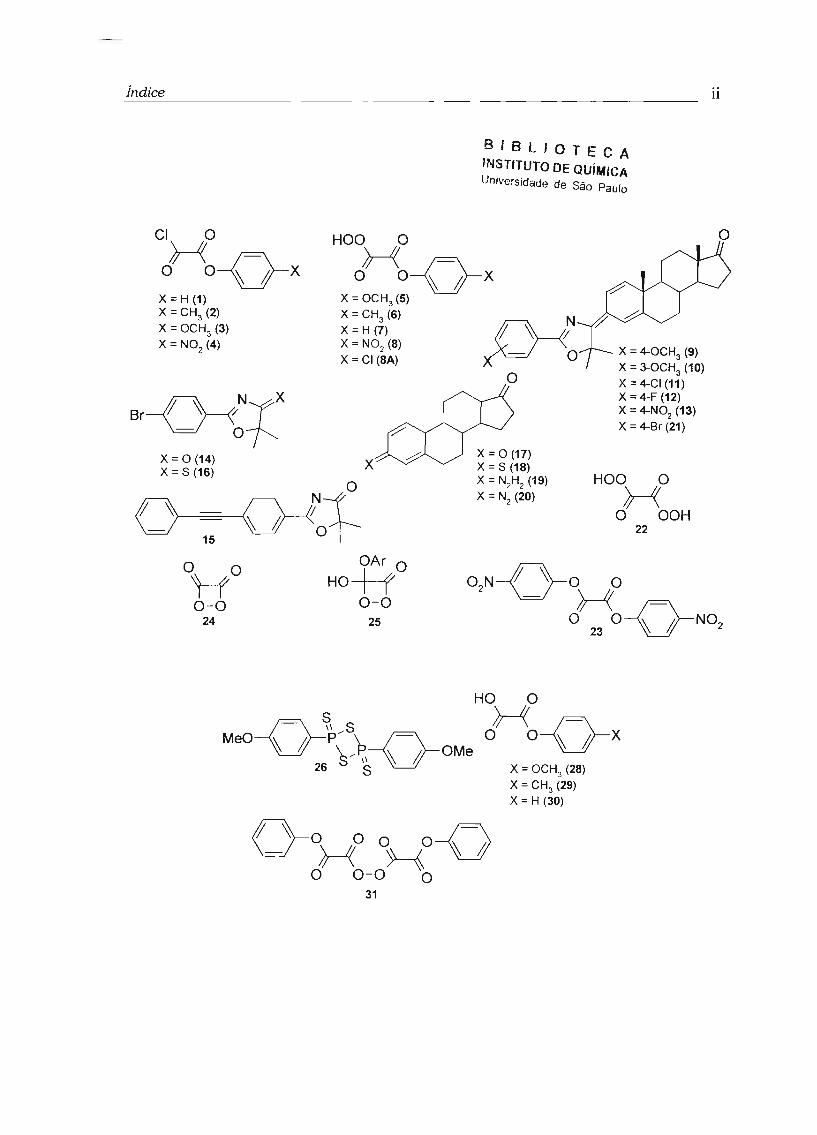
I - Lista de estruturas
Cloreto de feniloxalila (1)
Cloreto de p-metilfeniloxalila (2)
Cloreto de p-metoxifeniloxalila (3)
Cloreto de p-nitrofeniloxalila (4)
O,O-hidrogênio monoperoxioxalato de p-metoxiteniloxalila (5)
O,O-hidrogênio monoperoxioxalato de p-metilfeniloxalila (6)
O,O-hidrogênio monoperoxioxalato de fenila (7)
O,O-hidrogênio monoperoxioxalato de p-nitrofenila (8)
O,O-hidrogênio monoperoxioxalato de p-clorofenila (8A)
3-[2-(4-metoxifenil)-5, 5-dimetiloxazol-4-il idenojandrosta-1 ,4-dien-17-ona (9)
3-[2-(3-metoxifenil)-5, 5-dimetiloxazol]-4-ilideno]androsta-1 ,4-dien-17-ona (10)
3-[2-(4-clorofenil)-5,5-dimetiloxazol-4-ilidenojandrosta-1 ,4-dien-17-ona (11)
3-[2-(4-f1uorofenil)-5,5-dimetiloxazol-4-ilidenojandrosta-1,4-dien-17-ona (12)
3-[2-(4-nitrofenil)-5,5-dimetiloxazol-4-ilidenojandrosta-1 ,4-dien-17-ona (13)
2-[4-bromofenilj-5,5-dimetiloxazol-4-ona (14)
2-[4-fenilacetilenoj-5,5-dimetiloxazol-4-ona (15)
2-[4-bromofenil]-5,5-dimetiloxazol-4-tiona (16)
Androsta-1 ,4-dieno-3, 17-diona (ADD) (17)
Androsta-1 ,4-dieno-3-tiona-17-ona (18)
Androsta-1 ,4-dieno-3-hidrazona-17-ona (19)
Androsta-1 ,4-dieno-3-diazo-17-ona (20)
3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilidenoj-androsta-1 ,4-dien-17-ona (21)
Ácido peroxálico (22)
Bis(4-nitrofenil) oxalato (23)
1,2-dioxetanodiona (24)
1,2-dioxetanona (25)
Reagente de Lawesson (26)
Cloreto de bis(trifenilfosfina) paládio 11 [(Ph3P)zPdCb] (27)
ácido p-metoxifeniloxálico (28)
ácido p-metilfeniloxálico (29)
ácido feniloxálico (30)
anidrido do O,O-hidrogênio monoperoxioxalato de fenila (31)
Índice 11
BIBl.IOTECAINSTITUTO DE QUíMICAUniversidade de São Paulo
-o-~ O O
°2N
- ~/ ~NOÓ/\O~ 2
23
O
X = 4-0CH3 (9)
X = 3-0CH3 (10)X=4-CI(11)X = 4-F (12)X = 4-N0
2(13)
X = 4-Br (21)
HOO O
HO OOH
22
~O
X = 0(17)X = S (18)X = N
2H
2(19)
X = N2 (20)
HOO O
OHO-o-XX = OCH3 (5)X = CH
3(6)
X=H(J)X = N02 (8)
X = CI (8A)
OAr
HO-t-f°O-O
25
xO
~o+-o O
'HO-O
24
CI O
oHo-o-xx = H (1)X = CH3 (2)
X = OCH3
(3)X = N02 (4)
~N X
Br~_~~Tx =°(14)X=S(16)
HO O
-o~\ S H-o-MeO ~ /; p'" \~ O O ~ /; X
's/~\~OMe26 S X = OCH
3(28)
X =CH3 (29)
X = H (30)
o-~ O O O o-{)- H H
O o-o O31

Índice
II - Lista de siglas
• OPA - 9,1 O-difenilantraceno
• OBA - 9,1 O-dibromoantraceno
• CIEEL - Chemically Initiated electron exchange luminescence
(Luminescência induzida quimicamente pela troca de elétron)
• ACT - ativador
• IAE - intermediário de alta energia
• TCPO - bis(2,4,6-triclorofenil)antraceno
• TCP - 2,4,6-triclorofenol
• 001 - 1,1'-oxalyldiimidazole (di-imidazolida)
• IMI-H - imidazol
• 3-AFA - 3-aminofluoranteno
• ONPO - bis(3,5-dinitrofenil) oxalato
• PCPO - bis(pentaclorofenil) oxalato
• OMOA - 9,1 O-dimetoxiantraceno
• OCNA - 9,1 O-dicianoantraceno
• THF - tetrahidrofurano
• OFM - difenilmetano
• ACN - acetonitrila
• CCO - cromatografia em camada delgada
III

Estudo da etapa de quimiexcitação do sistema peroxioxalato IV
III - Resumo
o fenômeno de quimiluminescência é uma das mais fascinantes
demonstrações de conversão de energia química em energia de excitação. O
representante de maior eficiência dentro desta classe é a reação peroxioxalato,
pois possui rendimento de emissão de aproximadamente 30%, comparado
apenas a sistemas bioluminescentes, cujo rendimento é da ordem de 100%. O
sistema peroxioxalato consiste na reação de derivados fenólicos com
substituintes atraentes de elétrons do éster oxálico com peróxido de hidrogênio
catalisada por base na presença de um hidrocarboneto aromático
policondensado com baixo potencial de oxidação (Eox) e altos rendimentos de
fluorescência, denominado ativador. O mecanismo desta reação é bastante
complexo com várias etapas consecutivas e paralelas que antecedem a
emissão de luz.
O ponto chave do mecanismo é a etapa de quimiexcitação, onde um
intermediário de alta energia (IAE), formado em etapas anteriores, interage com
o ativador (ACT), resultando na formação de estados eletronicamente excitados
e subseqüentemente em emissão de luz. Para explicar esta interação, é
utilizado o mecanismo de Luminescência Induzida Quimicamente pela
Transferência de Elétron (UChemically Initiated Electron Exchange
Luminescence" - CIEEL), o qual envolve transferência e retro-transferência de
elétron. O ponto de controvérsia entre vários pesquisadores que estudam a
reação peroxioxalato está relacionado com a estrutura do intermediário de alta
energia (IAE), pois, de acordo, com os resultados destes trabalhos, diferentes
intermediários foram postulados (A-H).
OH o ArO O~N---L.J/ HN:d 6-6 O OOH
C D
° OJl . Jl.. ,o-oAr~N.:y°·o ArO Ã >=0
[[~)00o ArO o-io
N G O H
OH oAro-t-f
o-oB
0yo·o o
J--oVoAro NF~)
N
o oHo-o
A0yO-o
~N+oV:N:dHO íÍJJI- \1
E \ !J ~NN

Estudo da etapa de quimiexcitação do sistema peroxioxalato v
o enfoque geral deste trabalho foi estudar a natureza do IAE e a etapa
de quimiexcitação com o intuito de obter evidências sobre o mecanismo de
geração de estados excitados no sistema peroxioxalato. Para que isto fosse
possível, o trabalho foi abordado da seguinte maneira:
1- Síntese e caracterização de intermediários perácidos do tipo D
contendo OCH3, CH3, H e N02 substituídos no anel aromático. Do estudo
cinético da reação destes compostos, catalisada por imidazol, na presença de
9,10-difenilantraceno, foi possível propor um mecanismo completo e obter
informações a respeito da estrutura do IAE; através da correlação de Hammett
foram obtidas evidências para a ocorrência da 1,2-dioxetanodiona (A), como
IAE na reação peroxioxalato.
2- Estudo da eficiência de transferência de elétron na etapa de
quimiexcitação (mecanismo CIEEL) utilizando a reação do bis(2,4,6-triclorofenil)
oxalato (TCPO) com H20 2 catalisado por imidazol na presença de 3-(2-aril-5,5
dimetiloxazol-4-ilideno)androsta-1 ,4-dien-17-onas como ativadores. As
constantes de velocidade relativas da interação do ACT com o IAE mostram
correlação linear com o Eox do ACT e os rendimentos quânticos singlete em
concentrações infinitas do ACT (<!>s;XJ) correlacionam com as energias livres para
a formação do ativador no estado excitado ('ó'G*BET) através do processo de
retro-transferência de elétron. Desde modo, os resultados obtidos apresentam
evidências para a validade do mecanismo CIEEL utilizado para explicar a
formação de estados excitados na etapa de quimiexcitação da reação
peroxioxalato.
3- Com o objetivo de obter informações sobre a influência da viscosidade
no rendimento quântico singlete (<!>s) foram feitos estudos da reação do TCPO
catalisada por imidazol com H202 na presença de 9,1 O-difenilantraceno (OPA)
como ativador, utilizando-se misturas variadas de tolueno e difenilmetano. Foi
observado aumento de até 10 vezes no rendimento quântico singlete com o
aumento da viscosidade de 0,5 para 2,5 cP. Este comportamento é compatível

Estudo da etapa de quimiexcitação do sistema peroxioxalato VI
com o mecanismo CIEEL, indicando que a etapa de quimiexcitação ocorre em
uma seqüência de eventos, envolvendo intermediários íons radicais.
4- Síntese de derivados de oxazolonas esteroidais substituídos, com o
objetivo de obter compostos com novas propriedades foto-físicas e físico
químicas para aplicar no estudo da reação peroxioxalato, especificamente para
obter informações adicionais sobre a etapa de quimiexcitação. O outro interesse
nestes compostos era o de transformá-los em reagentes de derivatização para
futuramente empregá-los em trabalhos de química analítica que utilizam a
reação peroxioxalato. Foram preparados dois novos compostos fluorescentes,
além de vários intermediários sintéticos e estes derivados possuem potencial
para o uso como ACT em futuras aplicações analíticas.

Estudo da etapa de quimiexcitação do sistema peroxioxalato VIl
IV - Abstract
The phenomenon of chemiluminescence (CL) is one of the most
fascinating demonstrations of chemical energy conversion into excitation
energy. The most prominent example of this c1ass of reaction is the
peroxyoxalate system showing emission quantum yields of around 30 %,
comparable only to the 100 % efficiency bioluminescence systems. The
peroxyoxalate system consists in the base catalyzed reaction of activated
oxalate esters with hydrogen peroxide in the presence of polycondensed
aromatic hydrocarbons with low oxidation potentials (Eox) and high fluorescence
quantum yields, denominated activatars (ACT). The mechanism of this reaction
is quite complex with various subsequent and parallel reaction steps prior to light
emission.
The crucial mechanistic step is the chemiexcitation step, where a high
energy intermediate (HEI), formed in former reaction steps, interacts with the
ACT resulting in excited state formation and subsequent light emission. This
interaction can be understood on the basis of the Chemically Initiated Electron
Exchange (CIEEL) mechanism, which involves electron and back-electron
transfer steps. A controversial point between various authors who study
mechanistic aspects of the peroxyoxalate reaction is related to the nature of the
high-energy intermediate (HEI) and different proposal for its structure (A - I)
have been made.
OH o ArO O
r'N+-f HN=.! o-o O OOH
C O
O OJl _ Jl ,o-o
Ar~N>(O'o ArO J\ )=0[~>00o ArO O--{o
N G O H
OH O
ArO+-fO-O
B
01"°'0 °J... O J;-JZ OAro N
F~)N
o oHo-o
A
0yO·O
r'N+ot{N=.!HO íÍ ';)(.. l1
E~.J ~N
The main object of this work was to study the nature of the HEI and the
chemiexcitation step in arder to obtain evidence with respect to the

Estudo da etapa de quimiexcitação do sistema peroxioxalato Vlll
chemiexcitation mechanism in peroxyoxalate CL. For this purpose, the following
approach was used:
1 - Synthesis and characterization of peracid intermediates of type E,
containing different substituents (OCH3, CH3, H, N02). A kinetic study on the
imidazole catalyzed reaction of these compounds in the presence of 9,10
diphenylanthracene (OPA) allowed to propose a reaction mechanism and obtain
information with respect to the structure of the HEI; Hammett correlations clearly
indicate the occurrence of 1,2-dioxetanedione (A) as the HEI in the
peroxyoxalate reaction.
2 - The efficiency of electron transfer in the chemiexcitation step (CIEEL
mechanism) of the imidazole catalyzed reaction of bis(trichlorophenyl) oxalate
(TCPO) with H202 in the presence of 3-(2-aryl-5,5-dimethyloxazol-4
ylidene)androsta-1,4-dien-17-ones as activators was studied by measuring the
singlet quantum yieldso The relative rate constants for the interaction of the ACT
with the HEI show linear correlation with the activators oxidation potentials (Eox)
and the singlet quantum yields at infinite activator concentrations (<1>5") can be
correlated with the free energy change for the formation of the activators excited
singlet state (~G*BET) in the back-electron transfer processo Therefore, the
obtained results add additional evidence for the validity of the CIEEL scheme to
rationalize excited state formation in the peroxyoxalate CL.
3 - The imidazole catalyzed reaction of TCPO with H20 2 in the presence
of OPA as ACT was studied in mixtures of toluene and diphenymethane in order
to get information about the solvent viscosity influence on the singlet quantum
yields (<1>5)0 A change of the solvent viscosity from 0,5 to 2,5 cP results in an up
to ten fóld increase in <1>5, and this behavior is in agreement with the CIEEL
mechanism, indicating that the chemiexcitation step occurs in a sequence of
events involving discreet radical ion intermediates.
4 - Synthesis of steroid-substituted oxazolone derivatives with the aim to
obtain compounds with new photo physical and physical chemical properties for
applications in studies on the peroxyoxalate CL, specifically to obtain insights to

Estudo da etapa de quimiexcitação do sistema peroxioxalato 1X
the chemiexcitation step. An additional interest in these compounds is their use
as derivatization reagents for future utilization in analytical applications of the
peroxyoxalate system. In this context, two new f1uorescent derivatives have
been prepared, apart from several synthetic intermediates and these
compounds may have future utility as activators on peroxyoxalate system and
analytical applications.

Estudo da etapa de quimiexcitação do sistema peroxioxalato
v - Objetivo
x
o objetivo deste trabalho foi estudar a etapa de quimiexcitação da reação
peroxioxalato, através das seguintes abordagens:
(i)- Síntese de derivados do intermediário D com diferentes substituintes no anel
aromático, visando um estudo da correlação da estrutura e reatividade com o
intuito de obter informações sobre a estrutura do intermediário de alta energia
(IAE) que interage com o ativador (ACT) na etapa de quimiexcitação.
ArO O
HO OOH
D
(ii)- Estudo da etapa de transferência e retro-transferência de elétron que ocorre
na etapa de quimiexcitação através da reação do bis(2,4,6-triclofenil) oxalato
(TCPO) com HzOz catalisado por imidazol na presença de 3-(2-aril-5,5
dimetiloxazol-4-ilideno)androsta-1 ,4-dien-17-onas.
(iii)- Estudo da influência da viscosidade do solvente utilizando-se misturas de
tolueno e difenilmetano, sobre o rendimento quântico singlete (<Ds) da reação de
TCPO com HzOz catalisada por imidazol na presença de 9,1 O-difenilantraceno
(OPA) para obter evidências sobre a importância da gaiola de solvente no
processo de transferência e retro-transferência de elétron (etapa de
quimiexcitação).
(iv)- Síntese de derivados de oxazolonas esteroidais substituídos, com objetivo
de obter compostos com novas propriedades foto-físicas e físico-químicas para
aplicar no sistema peroxioxalato e em sistemas analíticos.

Introdução
1 - Introdução
1.1 Quimiluminescência
1
A emissão de luz pode ser resultado da incandescência ou luminescência.
No primeiro caso, a energia vibracional é convertida em energia radiante e
ocorre para todos os objetos na temperatura acima do zero absoluto
(-273,15 °C). No segundo caso, a emissão de luz é proveniente de um estado
eletrônico excitado. Deste modo, em fluorescência, a luz absorvida na banda de
absorção de comprimento de onda mais longo de uma molécula orgânica
produz o primeiro estado singlete excitado pela promoção de um elétron do
orbital ligante de mais alta energia para o orbital antiligante de mais baixa
energia. O processo reverso após aproximadamente 10-9 a 10-8 segundos gera
um fóton e a molécula volta para o estado fundamental. Naturalmente, quando
uma molécula orgânica absorve energia, outros processos podem ocorrer e não
apenas o de fluorescência. Uma molécula quando absorve energia pode sofrer
as seguintes transformações energéticas (Esquema 1):
Esquema 1
ts,~-
I::(So --- S1)1
-tt- So
1- promoção do elétron do estado singlete fundamental para o primeiro estado
singlete excitado, 2- retorno do elétron do estado singlete excitado para o
estado singlete fundamental, resultando em fluorescência, 3- conversão interna,

Introdução
BIBLIOTECAINSTITUTO DE QUíMICAUnIversidade de São Paulo
2
resultando no estado fundamental da molécula inicial ou um produto de reação
proveniente do estado singlete excitado, 4- conversão intersistemas (7):
promoção do elétron do estado singlete fundamental para o primeiro estado
triplete excitado. 5- fosforescência: emissão gerada pela transição entre
estados com multiplicidade de spins diferentes (neste caso singlete para
triplete) e 6 - conversão interna, resultando no estado fundamental da molécula
inicial ou um produto de reação proveniente do estado triplete excitado em um
produto de reação.
Na quimiluminescência o processo de emissão de luz é idêntico ao de
uma molécula foto-excitada, entretanto a fonte geradora da energia de
excitação é uma reação química. Para que ocorra uma reação que resulte em
emissão de luz são necessários alguns pré-requisitos fundamentais, os quais
envolvem a presença de um composto com capacidade de ser formado no
estado eletronicamente excitado, um emissor capaz de liberar esta energia de
excitação e um sistema de coordenada de reação que favoreça a formação de
um produto no estado excitado ao invés de formar o produto no estado
fundamental 1.
A + B ~-----...~ C* ... C + ilV
A pergunta que sempre surge diante desta definição é: Por que uma
reação química pode liberar sua energia em forma de luz e não em forma de
calor conforme ocorre na maioria das reações químicas?
Resumidamente, fatores energéticos e geométricos estão envolvidos na
formação de um estado excitado. Energeticamente, uma reação química na
qual a entalpia de ativação para a formação do produto no estado excitado é
menor que a entalpia para a formação do produto no estado fundamental,
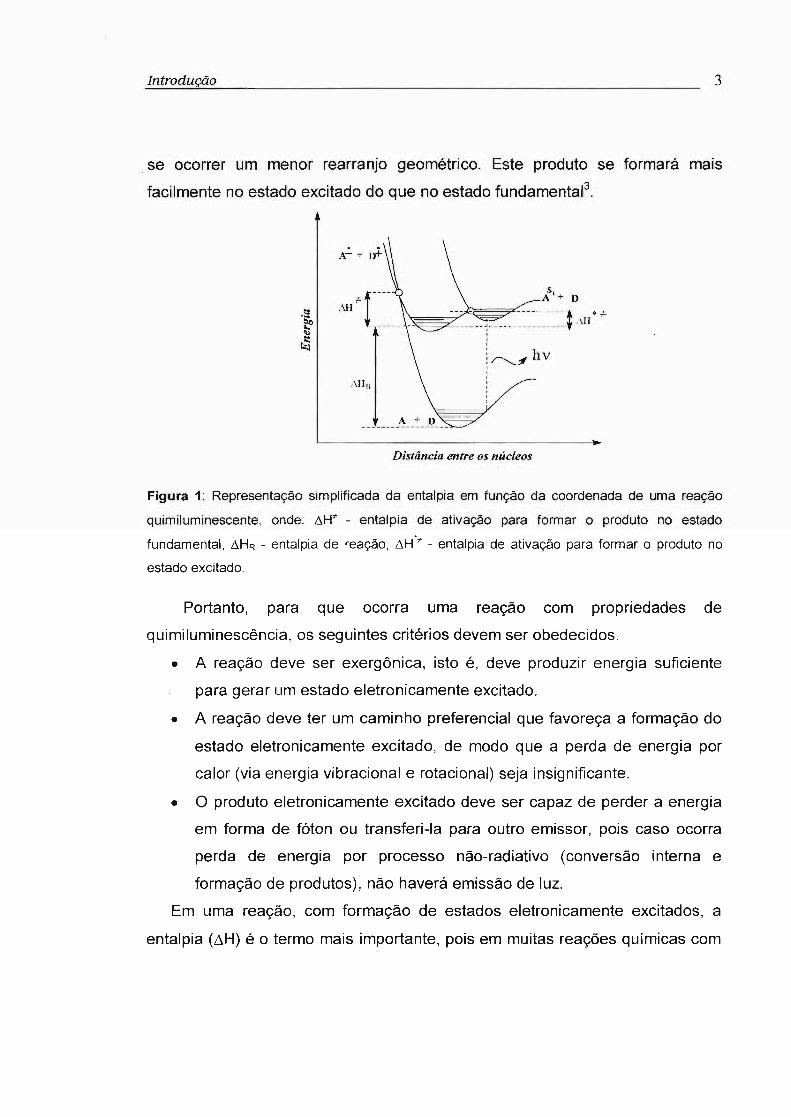
formará principalmente o produto no estado excitado (Figura 1). Por outro lado,
de acordo com o "Princípio de Frank Condon"2, uma transição eletrônica é tão
rápida que a geometria da molécula permanece inalterada (congelada) durante
a transição. Portanto, a formação do produto no estado excitado será favorecida
Introdução 3
se ocorrer um menor rearranjo geométrico. Este produto se formará mais
facilmente no estado excitado do que no estado fundamental3.
.:;~'I.l
~
A~ + rri-
~ s,---- A + D
MI1't \s=::d~~_~~_5~~~~~l.\1l *1'~hv
i\II R
A + D
Distância entre os núcleos
Figura 1: Representação simplificada da entalpia em função da coordenada de uma reação
quimiluminescente, onde: ~H'" - entalpia de ativação para formar o produto no estado
fundamental, ~HR - entalpia de reação, ~H·" - entalpia de ativação para formar o produto no
estado excitado.
Portanto, para que ocorra uma reação com propriedades de
quimiluminescência, os seguintes critérios devem ser obedecidos.
• A reação deve ser exergônica, isto é, deve produzir energia suficiente
para gerar um estado eletronicamente excitado.
• A reação deve ter um caminho preferencial que favoreça a formação do
estado eletronicamente excitado, de modo que a perda de energia por
calor (via energia vibracional e rotacional) seja insignificante.
• O produto eletronicamente excitado deve ser capaz de perder a energia
em forma de fóton ou transferi-Ia para outro emissor, pois caso ocorra
perda de energia por processo não-radiativo (conversão interna e
formação de produtos), não haverá emissão de luz.
Em uma reação, com formação de estados eletronicamente excitados, a
entalpia (i1H) é o termo mais importante, pois em muitas reações químicas com

Introdução 4
propriedades de quimiluminescência a mudança na entropia é pequena. Sendo
assim t..G e t..H são similares em magnitude. Porém esta afirmação é mais
adequada para reações de decomposição unimolecular, como por exemplo,
dioxetano ou reação de luminol, nos quais o fator entrópico não influência.
Para que uma reação produza um estado excitado o somatório da
entalpia de reação mais a entalpia de ativação que leva os produtos para o
estado fundamental deve ser maior que a energia para formar o produto no
estado excitado (t..HR + t..H# ~ t..Eex).
O fenômeno da emissão de luz através de reações químicas,
quimiluminescência, e seres vivos, bioluminescência, tem atraído o interesse de
inúmeros pesquisadores durante várias décadas. Embora a bioluminescência
seja descrita desde a Grécia antiga, a primeira reação quimiluminescente bem
caracterizada quimicamente, a oxidação do luminol (Esquema 2), só veio a ser
descrita por Albrecht em 19284. Um grande avanço no estudo mecanístico da
quimiluminescência foi alcançado com a primeira síntese de 1,2-dioxetano, um
peróxido cíclico de quatro átomos, por Kopecky em 19695,6, Contudo, o
mecanismo da maioria das reações orgânicas com propriedades de
quimiluminescência ainda não está completamente esclarecido,
especificamente no que diz respeito ao passo de excitação, ou seja, ao passo
elementar no qual a energia "química" da reação é transformada em energia de
excitação eletrônica.
Uma abordagem mecanística quantitativa considera que a eficiência de
uma reação quimiluminescente (<DcL) , pode ser determinada pela quantidade de
moléculas formadas no estado excitado singlete excitado (<Ds) multiplicado pelo
rendimento quântico de fluorescência do ativador (<DFI) , para o caso do produto
ser o próprio emissor.
cI>CL =cI>s X cI>FI Equação 1

Introdução 5
Se o produto formado transfere a sua energia para um outro emissor,
deve-se considerar o rendimento da eficiência de transferência de energia
(<1>te) 1.
<Del =<Ds X <DFI X <Dte Equação 2
Uma reação, cujo produto gerado no estado excitado é o próprio emissor,
é denominada de Quimiluminescência direta. Um exemplo clássico que
caracteriza este tipo de quimiluminescência é a reação de luminol, no qual o
produto da reação, 3-aminoftalato, é o responsável pela emissão de luz1
(Esquema 2).
Esquema 2
NH2 O
~NH H202/-0H
I I ~~ NH DMSO
O
po~O
O
Em reações classificadas como Quimiluminescência indireta, o emissor é
excitado através de uma transferência de energia de um produto formado no
estado excitado. A decomposição unimolecular de dioxetanos sintetizados por
Kopecky-Munford5,6 é um exemplo deste processo conforme mostrado no
esquema 3.

Introdução 6
Esquema 3
ySoO
-------;..~ DPASo +
hv (R2)
hv (R3)
DBASo + hv (R1)•
kF1 ySo--- +O
kF1(DBA)
IT-----sl
kF1(DPA)
Is-----sl
DBAS1
DPAS1
kTE
DPASo
I So 50YT1 +YO 51 O
DBA
..k1
ô++-O-O
DPA=
Br
DBA=~~
Br
Nesta decomposição pode ocorrer a formação de uma espécie no estado
fundamental, no estado triplete e no estado singlete (R2). A espécie formada no
estado singlete excitado pode decair diretamente para o estado singlete
fundamental (R2) ou ter a sua energia transferida para um aceptor de energia,
que neste exemplo é o OPA (R3). A emissão observada, neste caso, é
proveniente da excitação eletrônica do OPA.
Sabe-se que a emissão de fosforescência de espécies tripletes não é
observada normalmente em meio líquido, devido à presença de impurezas ou
de oxigênio, portanto para que isto seja possível geralmente é adicionado ao
meio um aceptor de energia triplete, como por exemplo, o 9,10
dibromoantraceno (R1). A transferência de energia triplete-singlete ocorre
através de um acoplamento spin-orbital causado pelo efeito de átomo pesado.
No caso da Quimiluminescência ativada, ocorre a reação bimolecular
entre um peróxido cíclico com anel tensionado, portanto, rico em energia e um
f1uoróforo, resultando na formação de estados excitados do aceptor. Neste
caso, o aceptor passa a ser chamado de ativador, pois o aumento da
concentração do aceptor acelera a decomposição do peróxido (Esquema 4).

Introdução 7
Esquema 4
"'" + hv0H-+o-o
Para explicar a geração de estados excitados em reações com
propriedades de quimiluminescência, Rauhut et aI. 7, 13 foram os primeiros a
propor um complexo de transferência de carga entre o peróxido rico em
energia, formado na reação, e o aceptor de energia. Além disso, baseado nos
resultados de rendimento quântico de quimiluminescência, foi observado que
quanto menor o potencial de oxidação, maior é a constante de velocidade de
formação de estados excitados. Da mesma forma, quanto menor a energia
singlete do ativador maior o rendimento quântico de quimiluminescência.
Posteriormente, Schuster8, estudando uma série de peróxidos cíclicos e
lineares isoláveis, constatou que tanto a velocidade de decomposição do
peróxido quanto a intensidade de emissão aumentavam na presença do
aceptor, sendo que a emissão observada era a fluorescência do mesmo e o
aceptor não sofria decomposição. Além disso, a velocidade dependia da
concentração do aceptor (ativador) e a constante de velocidade catalisada
dependia do potencial de oxidação do ativador e quanto menor o potencial de
oxidação maior era a formação de estados excitados do ativador.
Schuster8 também observou que, para todos os peróxidos cíclicos
estudados, o valor da constante de velocidade catalisada (kcat) correlacionava
linearmente com o potencial de oxidação do ativador, indicando que na etapa
limitante ocorria uma transferência de elétron (Esquema 5). Além disso, o
aumento da viscosidade de solventes com mesma constante dielétrica no
estudo do sistema difenoil-peróxido resultou no aumento do rendimento
quântico de quimiluminescência9.

Introdução
Esquema 5
kcatPeróxido .. hv
8
Jknsem emissão
v = kObS[Peróxido]
kobs =k1 + kcat[ACT]
km , ~ A.exp[:r(E= - E,," ;:)] Equaçio 3
Através destas observações Schuster e Ko09 postularam um mecanismo
que pudesse explicar a interação do peróxido rico em energia com o ativador, o
qual foi por eles denominado de mecanismo CIEEL ("Chemical/y Initiated
Electron Exchange Luminescence') , ou seja, Luminescência Induzida
Quimicamente pela Troca de Elétron8,9. Resumidamente, o valor de a presente
na equação de velocidade catalisada (Equação 3) está relacionado com a
extensão da ligação O-O no momento que ocorre a transferência de elétron do
ativador para o intermediário de alta energia (IAE). Este valor é semelhante ao
valor de a de Bronsted utilizado em catálise ácida geral, o qual mede a
transferência do próton no estado de transição. De acordo com a equação 3, o
valor de a é obtido da relação Inkcat vs. Eox.

Introdução 9
1.2 Luminescência Induzida Quimicamente pela Troca de
Elétron (CIEEL)
Esquema 6
O~ICiii)
~.
~O~O, O
O~ ",..~ ~ .. , I1
O-O~ \.$
(v)hv
.~s1.# oOH- +O-O
c~c
o r COCO IV) 2
2
A luminescência induzida quimicamente pela troca de elétron supõe as
seguintes etapas: (i)- o peróxido formaria um complexo de encontro com o
ativador dentro da gaiola de solvente, (ii)- o alongamento da ligação oxigênio
oxigênio diminuiria a energia do orbital anti-ligante permitindo a transferência de
elétron do ativador para o peróxido, formando um par de íons radicais dentro da
gaiola de solvente, (iii)- a c1ivagem do ânion radical da dioxetanona levaria à
formação do ânion radical do dióxido de carbono, uma espécie altamente
redutora, que (iv)- por uma retro-transferência de elétron levaria à formação do
ativador no estado eletronicamente excitado que (v)- pelo decaimento para o
estado fundamental emitiria luz (Esquema 6). Esta proposta foi recebida com
bastante entusiasmo pela comunidade acadêmica, pois podia ser utilizada para
explicar a geração de estados excitados na reação bioluminescente do vaga
lume10. A proposta mecanística feita por McElroy et a1. 11
, para a reação de
emissão de luz do vaga-lume envolvia a formação de uma dioxetanona, gerada
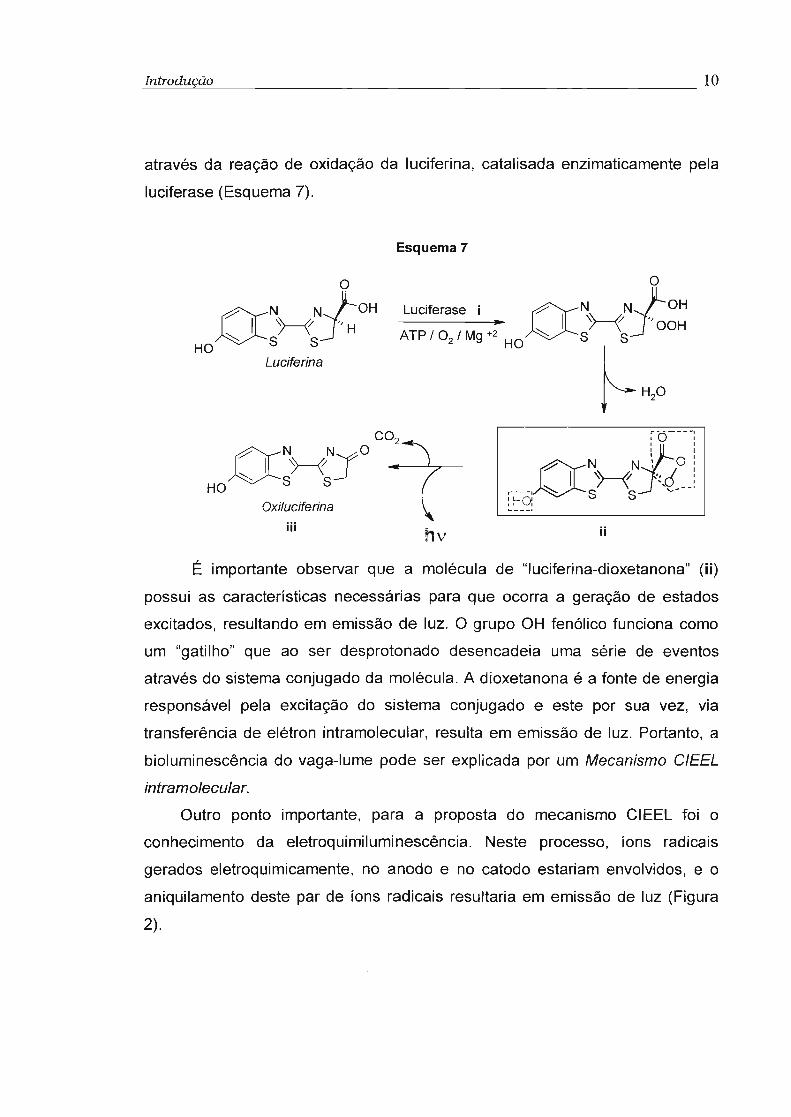
Introdução 10
através da reação de oxidação da luciferina, catalisada enzimaticamente pela
luciferase (Esquema 7).
Esquema 7
O NN'jOCOTI )----f~
HO S S
Oxil~~iferina
111 •nv
O
(yN->--/- f OH
HO~S SJ"H
Luciferina
o
Luciferase j ON N1a0H~ I \~ '/
ATP / O2
/ Mg +2 ~ S/ \ OOH
HO ~H'O
~~o---:
I I
-:? N N: O:I ~ '/ 'I :C---,Os~ "({--~
:Ho, S '-
jj
É importante observar que a molécula de "Iuciferina-dioxetanona" (ii)
possui as características necessárias para que ocorra a geração de estados
excitados, resultando em emissão de luz. O grupo OH fenólico funciona como
um "gatilho" que ao ser desprotonado desencadeia uma série de eventos
através do sistema conjugado da molécula. A dioxetanona é a fonte de energia
responsável pela excitação do sistema conjugado e este por sua vez, via
transferência de elétron intramolecular, resulta em emissão de luz. Portanto, a
bioluminescência do vaga-lume pode ser explicada por um Mecanismo CIEEL
intramolecular.
Outro ponto importante, para a proposta do mecanismo CIEEL foi o
conhecimento da eletroquimiluminescência. Neste processo, íons radicais
gerados eletroquimicamente, no anodo e no catodo estariam envolvidos, e o
aniquilamento deste par de íons radicais resultaria em emissão de luz (Figura
2).

Introdução 11
BA
--GhVBA*
_ + hv
t++~/ A B' ~
- -
- t+ ~+-B: "" -i
t-#
.A-
Lume 1-Homo t+
Figura 2: Representação do aniquilamento de íons radicais gerados eletroquimicamente
resultando em emissão de luz
Deste modo o mecanismo CIEEL é utilizado para explicar uma série de
reações com transferência de elétron que resultam em emissão de luz.
Entretanto, em relação à eficiência do sistema que gerou a proposta para a
luminescência induzida quimicamente pela troca de elétron (mecanismo CIEEL)
há uma certa controvérsia na literatura. Pois de acordo, com Catalani &
Wilson12, os rendimentos quânticos de quimiluminescência calculados para o
sistema difenoilperóxido/perileno observados por Schuster8 foram
superestimados em 1000 vezes. Catalani & Wilson 12 determinaram um
rendimento quântico de quimiluminescência de 10-4E mor1 contra 0,1 E mor1
observado por Schuster9. Em primeira análise, o que se pode concluir destes
trabalhos é que o sistema responsável pela formulação do Mecanismo CIEEL
possui uma eficiência extremamente baixa.

Introdução 12
1.3 O Sistema Peroxioxalato
o fenômeno de quimiluminescência é uma das mais fascinantes
demonstrações de conversão de energia química em excitação eletrônica.
Contudo, sistemas com propriedades de quimiluminescência geralmente
apresentam eficiência insatisfatória, tipicamente com valores próximos a 1%.
Entretanto, o sistema peroxioxalato é um excelente exemplo deste fenômeno,
pois sua eficiência é de aproximadamente 30%13. O sistema surgiu na década
de 60 quando Chandross14 misturou cloreto de oxalila com peróxido de
hidrogênio na presença de rubreno e observou emissão de luz. Atualmente o
sistema mais eficiente é formado pela reação de derivados de ésteres fenólicos
do ácido oxálico, com grupos aceptores de elétrons, com peróxido de
hidrogênio catalisado por uma base, na presença de hidrocarbonetos
aromáticos policondensados com baixo potencial de oxidação, denominado
ativador (ACT) (Esquema 8).
Esquema 8
° OArH + H2 0 2
ArO °ACT
-----;.... 2ArOH + 2C02 + hvbase
Rauhut et aI. 13, foram os primeiros a formular o mecanismo da reação
peroxioxalato e a propor a formação de um intermediário de alta energia (IAE)
que em um único passo deve liberar energia suficiente para provocar a
excitação do ativado r. Experimentalmente, o desenvolvimento da reação
peroxioxalato pode ser estudado pela emissão de luz, utilizando um fluorímetro
adequado ou pela liberação do fenol substituído através da sua absorção no
ultravioleta. As etapas nas quais estão envolvidas a formação e interação do
IAE com o ativador fazem parte da etapa rápida do processo, e portanto, não
podem ser observadas por cinética.
O mecanismo desta reação é complexo, com várias etapas consecutivas
e paralelas que antecedem a etapa de quimiexcitação. Do estudo cinético

BIBLIOTECA
Introdução INSTITUTO DE QUíMICAUniversidade de São Paulo 13
realizado em trabalhos anteriores, no mesmo grupo em que o projeto foi
desenvolvido, foi proposto um mecanismo simplificado para a reação principal
que leva à formação de estados excitados e atribuídas constantes de
velocidade para estas etapas (Esquema 9)15.16.
Esquema 9
Nt)N
/H
OH
2 C1--NC1
YCI
+
O OOHH +
N O
[~N
~ ) HzOz
etapa 2 rJ/
H
etapa 3Nt »NJ
/H
k3
Nt)N
/H
N
k1
O fJ), \L_l,--- T\\
k-1 Netapa 1 [~ O
N
N+ 2fJ
NH/
CI
CI ~' :Ho-P-C1
\={ OCI
CI . _) etapa 2Ak2 H O2 2
CI
+ °Ho-O-CIHOO OY~3'CI~CI
etapa3A yfN CI
~J O O
~ t-fO-O
OH
c'hc'yCI
Quimiexcitação
O O
t-f + CCCO-O
k4
etapa 4• ~) +hv
Estas informações são referentes às etapas lentas da reação que
ocorrem antes da etapa de quimiexcitação e podem ser resumidas da seguinte
maneira:
.:. A curva de formação do 2,4,6-triclorofenol foi monitorada por
espectrofotometria e analisada com a função f(t) = A [1-exp(-kobs1 t)].
.:. A curva de emissão de luz da reação peroxioxalato foi ajustada com
uma função do tipo: f(t) =A1exp(-kObs1 t)-A2 exp(-kobs2 t), onde: kobs1 é

Introdução 14
a constante observada para o decaimento da emissão de luz da
reação e kobs2 é a constante de velocidade observada para o
aumento da intensidade de luz, também chamada de constante de
subida.
•:. kobs1 depende da concentração de imidazol. Experimentos de
absorção (liberação do fenol) e emissão mostraram que esta é a
etapa mais lenta da reação. Em ambos os casos, a dependência com
a concentração de imidazol é complexa, sendo ajustada com uma
função binomial: kobs1 = k1(Bim)[IMI-H]+k1(Trim)[IMI-Ht Ademais, esta
etapa não depende da concentração de H20 2 em baixas
concentrações (Esquema 9 - etapa 1).
•:. Porém, kobs1 depende da [H202] para concentrações acima de 10
mM, pois nestas condições o TCPO sofre ataque nucleofílico direto
do H20 2, catalisado por imidazol (etapa 2A).
•:. A adição de 2,4,6-triclorofenol (TCP) nos experimentos de emissão
e absorção demonstrou que a reação inversa (k_1) não é importante
para as condições de reação empregadas (etapa 1), pois não
ocorreu variação no valor de kobs1 com o aumento da concentração
do 2,4,6-triclorofenol (TCP).
•:. kobs2 apresenta dependência linear com a concentração H20 2 e a
constante bimolecular obtida é atribuída ao ataque nucleofílico do
H20 2 ao 001. Esta etapa é catalisada por imidazol e apresenta
dependência de segunda ordem na concentração de imidazol.
.:. Em altas concentrações de H20 2 ocorre uma mudança do passo
limitante e esta etapa não depende mais da [H20 2]. Nestas
condições, o ataque do H20 2 ao 001 (di-imidazolida) não é mais a
etapa limitante, e sim a ciclização do perácido intermediário (etapa
3). Entretanto, esta etapa é catalisada por imidazol, no qual este atua
como catalisador básico.

Introdução 15
Os valores para as constantes de velocidade obtidas para cada etapa da
reação foram:
Tabela 1: Valores da constante de velocidade para cada etapa da reação do TCPO
com H20 2, catalisada por imidazol na presença de OPA16.
etapa Equação de velocidade Constante
1 kobs1 = k1(Bim)[IMI-H] + k1(Trim)[IMI-Hf k1(Bim) = 2,5 molLVk1(Trim) =900 moIL-2s-1
2,2A kobs2 = k2(Tetr)[H20 2][IMI-H]2 k2(Tetr) = 1,5 x 107moIL~~
3 kmáx =k3(Bim)[IMI-H] k3(Bim) = 320 moIL-'g'
O ponto culminante dessa reação é a etapa de quimiexcitação, na qual
ocorre a interação do intermediário de alta energia com o ativador (ACT), dentro
da gaiola de solvente, levando a emissão de luz.
Vários autores vêm contribuindo para que o sistema peroxioxalato possa
ser esclarecido. Cabe ressaltar que a maioria dos trabalhos apresentados na
literatura, com enfoque no estudo mecanístico, busca estabelecer qual é o
intermediário de alta energia que se forma na etapa de quimiexcitação.
Entretanto, há controvérsias entre estes grupos que, de acordo com a
interpretação dada aos resultados observados, propuseram diferentes
intermediários (A a H). (Figura 3).
OH O ArO O
~N-t-f HN:::::I o-o O OOH
C O
O °Jl _ Jl p-oAr:N)(O'O ArO Ã )=0
[ J000 ArO O~ON G O H
OH O
Aro-t-fo-o
B
0yO,o o
J---O~OAro NF~)
N
° oHo-o
A
0yO-o
r'N+oHN:::::I HO r.'J t. 11
E \\ !J ~NN
Figura 3: Intermediários de alta energia (IAE) propostos na reação peroxioxalato.

Introdução 16
Hohman et ai. 17, propuseram a estrutura D como intermediário de alta
energia (IAE), partindo da desprotonação do tri-isopropilsililperoxi-2,6
difluorofenil oxalato (I) e tri-isobutilperoxi-2,6-difluorofenil oxalato (J) com
fluoreto de tetrabutilamônio em ftalato de dimetila e em acetonitrila. Entretanto,
seus resultados não foram muito conclusivos em relação ao intermediário
proposto. Segundo seu trabalho, após a desproteção com fluoreto de
tetrabutilamônio, o íon percarboxilato formado interage rapidamente com o
ativador (ACT).
Q-F O OOS'R
f \~ 13
~ O~_ O
F I R=~sopropilaJ R=lsobutila
Hadd et ai. 18,19, em um estudo mais detalhado com respeito ao
mecanismo, discutiram a importância do papel catalítico do imidazol e a
formação da di-imidazolida como um intermediário importante para a eficiência
da reação. Nestes trabalhos, os autores defendem as estruturas C e F como
intermediários de alta energia20.
Stevani et a1. 21, reportaram a tentativa de capturar a 1,2-dioxetanodiona,
um possível intermediário de alta energia formado na reação peroxioxalato, pela
inserção de trifenilantimônio na ligação O-O deste peróxido cíclico. O produto
esperado nesta reação, 2,2,2-trifenil-2À5-1,3,2-dioxastilbolano-4,5-diona, foi
preparado por uma rota independente e caracterizado por RMN de 13C,
espectroscopia na região do infravermelho e espectrometria de massa.
Entretanto, não foi bem sucedida a detecção da 1,2-dioxetanodiona a partir da
reação do peroxioxalato com trifenilantimônio, provavelmente devido ao seu
tempo de vida, que não é o suficiente para reagir com o trifenilantimônio, ou
devido à decomposição catalítica do peróxido na presença de trifenilantimônio
(Esquema 10).

Introdução 17
Esquema 10
2N0N-H N~ OCI 3c L H-Q-~ O 0-q-CI O n
CI_ H - C1 O" C, ~N10 O ~ ~ CI~I r
c 2~ H,O,TCPO CI
CI
ROTA 1 I
(C6 H5)3Sb
O O O
HO o~ t-l HoH ~ p o-oOH .Sb /
HOCH,~'A~ / '\~ . ~ ~~~~
·',1
(C6H5hSb(OCH3)2
(C6H5)3SbBr2 [
NaOCH3~
NaBr~
Jonsson et aI. 22, estudaram a reação de TCPO com H20 2 catalisada por
uma série de derivados de imidazol, sendo que alguns deles são utilizados em
síntese de peptídeos, na presença de 3-aminofluroanteno (3-AFA) em
acetonitrila. O objetivo foi examinar as propriedades catalíticas destes derivados
na reação peroxioxalato. Os autores observaram eficiência catalítica com os
compostos 1,2,4-triazol, 1-metilimidazol, 2-metilimidazol, 4-metilimidazol e 4,5
tricloro-imidazol, enquanto que os compostos 2-mercapto-imidazol, 2-nitro
imidazol, 4-nitro-imidazol entre outros, não apresentaram emissão de luz
detectável nas condições investigadas. Além disso, a reação do sistema
peroxioxalato utilizando 1-metilimidazol (pKa = 7,06) possui uma intensidade 25
vezes menor do que com imidazol (pKa= 6,99) nas mesmas condições, embora
ambos os catalisadores tenham pKa's similares. Os autores atribuem este
comportamento ao fato, do intermediário gerado da reação entre TCPO e 1
metilimidazol, possuir carga positiva que o desestabíliza, fazendo com que o
equilíbrio desta reação seja desfavorável à formação dos produtos. Por outro

Introdução 18
lado, a mesma reação utilizando imidazol levaria a um intermediário mais
estável devido à transferência de próton do imidazol para outro imidazol
presente na solução. Assim, o equilíbrio da reação seria favorável à formação
dos produtos. Os autores concluíram que o catalisador tem um papel importante
para a eficiência da reação peroxioxalato, pois o intermediário formado deve ser
mais suscetível à reação com HzOz do que TCPO com HzOz.
Lee et ai. 23, estudaram a reação de DNPO com HzOz sem adição de
catalisador na presença de perileno em acetato de etila. Os autores observaram
que quando a reação era efetuada em baixas concentrações de HzOz produzia
um decaimento lento, enquanto que na presença de altas concentrações de
HzOz duas diferentes emissões eram observadas: um decaimento rápido
seguido de um decaimento lento. Para esta observação experimental, os
autores propuseram a formação de dois intermediários de alta energia (IAE), as
estruturas O e H (Figura 3). Estes autores também utilizaram TCPO e PCPO
em altas concentrações de HzOz e observaram que na reação com TCPO
ocorreu apenas um decaimento lento, enquanto que, com PCPO foi observado
um fraco decaimento rápido seguido de um decaimento lento. Com base nestes
resultados, os autores sugeriram que o IAE proveniente do decaimento rápido,
deveria ter em sua estrutura um forte grupo sacador de elétrons, pois a curva
de decaimento rápido depende da força do substituinte do anel aromático do
éster oxálico (DNP>PCP>TCP).
Em um outro trabalhoz4 publicado no mesmo ano, foi investigada a reação
de TCPO com HzOz catalisada por IMI-H na presença de perileno, com o
objetivo de elucidar as propriedades do possível IAE, mediada pela adição
variada de reagentes. Como fruto deste estudo, concluíram que as estruturas O
e H seriam os intermediários de alta energia (IAE). Além disso, também
observaram que a mesma reação sem adição de HzOz resultava em emissão
de luz. Assim, este IAE seria formado pela reação entre TCPO e IMI-H.
Entretanto, não foi proposta uma estrutura para este intermediário, sendo
designada pelos autores apenas como intermediário Z.

Introdução 19
Outros trabalhos relevantes são os de Shamsipur et ai.2~27, que
investigaram o sistema peroxioxalato na presença de derivados de cumarinas e
acriflavinas como ativadores. O interesse no estudo, destas espécies reside no
fato de possuírem propriedades antimicrobianas. No caso das acriflavinas, são
também, utilizadas como foto-sensitizadores para agentes anti-cancerigenos25.
Das estruturas citadas como intermediário de alta energia (A-H) foi
demonstrado que derivados da estrutura D não poderiam ser intermediários de
alta energia, visto que o O,O-hidrogênio monoperoxioxalato de p-c1orofenila, um
derivado análogo à estrutura D, não levava a emissão de luz em presença
apenas do ativador, necessitando de uma base para que tal evento fosse
observado7,28. Tal fato indica, que deveria ocorrer uma reação química, anterior
à emissão, provavelmente uma ciclização, originando, então, o intermediário de
alta energia. Neste trabalho foram também obtidas evidências experimentais
adicionais, ainda que indiretas, que sustentavam a hipótese da 1,2
dioxetanodiona (A) ser o intermediário reativo.
Bos R et aI. 29, reportaram pela primeira vez, o uso de cloreto de oxalila
isotopico C3C) com H202 na presença de 9,1 O-difenilantraceno (OPA) em THF
anidro para monitorar a formação do intermediário de alta energia por RMN de
13C, variando a temperatura entre -80 a +20 cC. Os autores observaram um
sinal de baixa intensidade em 8 154,5 que se tornava mais intenso a -70°C,
sendo este atribuído a 1,2-dioxetanodiona. Na temperatura de -60°C, o mesmo
sinal apresentou um alargamento. Além disso, foram observados outros sinais,
atribuídos a outros intermediários, como por exemplo, monocloreto de
peroxioxalila. No espectro obtido a +20 °c todos os sinais desapareciam, exceto
a ressonância em 8 160,4. Segundo os autores, este composto possuiria uma
estabilidade térmica relativa, porém, ainda, não foi possível identificá-lo.
Baseado nos resultados da RMN de 13C juntamente com os dados de cálculo
ab initio, propuseram a 1,2-dioxetanodiona como intermediário de alta energia
(IAE).

Introdução 20
De acordo com os vários trabalhos apresentados na literatura, pode-se
perceber que o mecanismo da reação e principalmente a estrutura do
intermediário de alta energia responsável pela emissão de luz, ainda, não estão
bem esclarecidos.
O interesse pela estrutura do IAE é grande, pois este é o responsável pela
emissão de luz que ocorre na etapa de quimiexcitação do sistema
peroxioxalato.
Rauhut et ai. 13,30,31 , foram os primeiros a propor a formação de um
intermediário meta-estável, provavelmente a 1,2-dioxetanodiona, capaz de
provocar a excitação do ativador, mediante excitação originada por um
complexo de transferência de carga. Dos experimentos de rendimento quântico
de quimiluminescência (<1>cd foi observado que quanto menor o potencial de
oxidação do ACT era maior era a velocidade de formação de estados excitados.
Além disso, quanto menor a energia singlete, maior o <1>CL.
Schuster et aI. 9, ao estudar uma série de peróxidos cíclicos e lineares
também observou que a velocidade de decomposição destes peróxidos era
acelerada em presença de ativadores com baixo potencial de oxidação e altos
rendimentos de fluorescência (<1>F) , além de ocorrer um aumento na intensidade
de emissão de luz. Com base nestas informações foi postulado o mecanismo
CIEEL. Na etapa de quimiexcitação da reação peroxioxalato estariam
envolvidas apenas duas espécies: O IAE e o ativador (ACT). Considerando que
o IAE é o mesmo para qualquer ativador utilizado, as informações obtidas desta
etapa seriam provenientes da mudança do <1>F, energia singlete e potencial de
oxidação dos ativadores.
Stevani et ai.32, estudaram a etapa de quimiexcitação da reação
peroxioxalato através da reação de TCPO com IMl-H e HzOz e da reação de
O,O-hidrogênio monoperoxioxalato de p-clorofenila catalisada por IMI-H na
presença de uma série de ativadores aromáticos policondensados, como por
exemplo: 9,1 O-difenilantraceno, rubreno, perileno entre outros. Para os dois
sistemas foram obtidas evidências experimentais que reforçaram a hipótese da

Introdução 21
transferência de elétron entre o ativador e o IAE, através da correlação linear
entre a constante de velocidade catalisada (kcat) e o potencial de oxidação;
conforme postulado pelo mecanismo CIEEL. Entretanto, para dois ativadores,
não comumente utilizados, 9,1 O-dimetóxi-antraceno (DMOA) e 9,10
dicianoantraceno (DCNA) foram observados desvios significativos. Até o
momento, não há uma explicação para tal comportamento. Isto mostra que
detalhes sobre este interessante mecanismo, ainda, precisam ser esclarecidos.
Silva et ai.33, reportaram a reação de TCPO catalisada por IMI-H com H202
na presença de vários derivados de oxazolinilidenos esteróides substituídos
como ativadores. Os resultados obtidos também se aplicam ao mecanismo
CIEEL, isto é, com transferência de elétron entre o IAE e o ACT. A informação
mais importante obtida neste trabalho foi mostrar que compostos com
esqueletos totalmente distintos das estruturas dos ativadores comumente
empregados no estudo do mecanismo CIEEL, também podem ser aplicados.
1.4 Aplicações Analíticas
De modo geral, a quimiluminescência tem sido largamente aplicada em
diversas áreas de biotecnologia, farmacologia, biologia molecular, clínica e
química, sendo que o sistema que mais se destaca é a reação peroxioxalato.
Portanto, torna-se importante conhecer o mecanismo desta reação para que
seja possível ampliar e melhorar a eficiência de sua aplicação. Do ponto de
vista analítico, esta reação é um das mais interessante reportada na literatura,
pois possui um alto rendimento quântico de quimiluminescência, comparado
apenas a sistemas biológicos, ou seja, é um sistema altamente sensível34,3S.
O sistema peroxioxalato possui uma série de aplicações, sendo que os
derivados de ésteres oxálicos mais utilizados são o bis(2,4,6-triclofenil) oxalato,
bis(2,4-dinitrofenil) oxalato e o bis[2-(3,6,9-trioxadeciloxicarbonil)-4-nitrofenil]
oxalat036,39. A primeira aplicação prática da reação peroxioxalato foi como "Iight
sticks", que são bastões de plástico contendo em seu interior os reagentes
necessários para que ocorra uma reação com propriedades de

Introdução 22
quimiluminescência. Estes bastões ao serem quebrados possibilitam a mistura
dos reagentes, o qual resulta na emissão de luz. Há uma série de aplicações
para estes bastões, sendo uma delas como sinalizadores em mergulho. Os
bastões são até hoje fabricados pela "American Cynamid Co.," que teve como
funcionário MM Rauhut, descobridor da reação peroxioxalato7.
Na literatura há um grande número de trabalhos, nos quais a
quimiluminescência da reação peroxioxalato é utilizada como ferramenta
analítica. Basicamente, qualquer sistema que forme peróxido de hidrogênio ou
o destrua, que forme uma substância com propriedades de fluorescência ou
contenha um composto que suprima a fluorescência, pode ser aplicado ao
sistema peroxioxalato. A seguir serão relatados alguns exemplos de aplicação
analítica da reação peroxioxalato com o intuito de ilustrar a sua importância.
Nozaki et aP7,38 e Ragab39 utilizaram a reação peroxioxalato,
especificamente o bis(2-(3,6-9-trioxadecaniloxicarbonil)-4-nitrofenil oxalato
(TDPO), para otimizar a produção de H20 2 produzido pela oxidação da
dopamina em meio básico. A dopamina é um neurotransmissor, geralmente,
presente em baixas quantidades em fluídos biologicos. Sob condições
fisiólogicas, pode gerar H20 2 enzimaticamente, resultando em dopamina
neurotóxica. O H20 2 produzido é detectado por HPLC pelo método de injeção
em fluxo. No intuito de otimizar o estudo, testou-se vários compostos cíclicos
nitrogenados, dentre eles imidazol como catalisadores da reação (Esquema
11 ).
~CHZCHZNHZ
HO :::::---. I + °Z
OH
Esquema 11
HI
N
~)•
pH alcalino J?CHZCH NHZ Z
-;:/
° I +
°HZOZ
Yamada et ai. 40, utilizaram a reação peroxioxalato para determinar a
presença de estradiol em plasma utilizando HPLC. O limite de detecção
observado foi de 15 fmol (4 pg). Estradiol é um dos mais importantes

BIBLIOTECAIntrodução INSTITUTO DE QUíMICA
Universidade ae ~ão Palllo 23
estrógenos secretado pelo ovário humano, sendo responsável pela manutenção
do sistema reprodutivo feminino. Além disso, derivados de estradiol têm sido
usados como agente anti-osteoporose. Como o estradiol não possui
propriedades de fluorescência, foi necessário derivatizá-Io com cloreto de
dansila (Esquema 12).
Esquema 12
OHOH aÇbS02C1
ao o::r Içól
'<::: +aÇb acetonalNaHC0 3 "&11L ""b ::r '- "" 110-- I N I o
"" I ""
/N, HO
Sun et al. 41, desenvolveram um método por HPLC para detectar a
presença de 4,4'-isopropylidenediphenol (bisfenol A, BPA) proveniente de
recipientes de policarbonato durante o processo de esterilização, como por
exemplo mamadeiras. Segundo os autores, pesquisadores tem mostrado que
BPA apresenta efeitos xeno-estrogênicos in vitro em concentrações de 6 ppb
(25 nM). Sendo assim, os autores julgaram importante desenvolver um método
altamente sensível para monitorar o nível de BPA (Esquema 13).
Esquema 13
7J~
o-- \ N~COCI + HO~--: H \==! I U
OH~ ~ DlB-C1 j BPA
4-(4,5-diphenyl-1 H-imidazol-2-yl)- TEA
benzoyl chloride
~# ~I~~O~/\O~N ~N~ "=I/~~N
~) H DIB-BPA H ~ ~
Pode ser observado que para a maioria das determinações analíticas é
necessário uma derivação da espécie a ser analisada, pois muitos compostos
não possuem propriedades de fluorescência. Além de cloreto de dansila, vários

Introdução 24
outros compostos são utilizados como reagentes de derivatização
(Esquema 14)42,
Esquema 14
9-metanol-antraceno
NH2
o ~ CHpH
~ ~H ~
0- "'o
LUMINARIN4
r i NHCOCH21
~
c:? ~ ,/ ~~
H5C2'N ~ o oI
C2H5 DelA
Estes exemplos correspondem a uma pequena demonstração das
possíveis aplicações envolvendo a reação peroxioxalato. Percebe-se que o
método mais utilizado é a cromatografia líquida de alta resolução (HPLC).
Porém, observa-se também o desenvolvimento da aplicação do sistema
peroxioxalato em eletroforese43,44, Em suma, o sistema peroxioxalato é aplicado
em uma série de sistemas, cujo objetivo é desenvolver um método simples, de
fácil aplicação, sensível e seletivo.

Introdução 25
1.5 Referências
1 Campbell AK, Chemiluminescence: Principies and Applications in Biology and Medicine.
Weinheim; Deerfield Beach, F1.: VCH; Chischester: Horwood, 1998.
2 Turro NJ, Modem Molecular Photochemistry. The Benjamin/Cummings Publishing Co., Inc.,
1978.
3 Hercules DM, "Chemiluminescence from Electron - Transfer Reactions", Aee. Chem. Res, 2,
301-7, 1969.
4 Albrecht HO, "Über die Chemilumineszenz des aminophthalsaurehydrazids",
Z. Physik. Chem. 136,321-30,1928.
5 Kopecky KR & Mumford C, "Abstraets, 51§! Annual Conferenee of the Chemieal Institute of
Canada", Vancouver BC, p. 41, 1968.
6 Kopecky KR & Mumford C, "Luminescence in the thermal decomposition of 3,3,4-trimethyl-1 ,2
dioxetane", Can. J. Chem., 47, 709-10, 1969.
7 Stevani CV, "Estudo Mecanístico do Sistema Peróxi-Oxalato", Tese de Doutorado,
Universidade de São Paulo, 1997.
8 Schuster GB, "Chemi-Iuminescence of organic peroxides-conversion of ground-state reactants
to excited-state products by the chemically-initiated electron-exchange luminescence
mechanism", Aee. Chem. Res., 12 (10),366-73, 1979.
9 Koo J-Y & Schuster GB, "Chemiluminescence of Diphenoyl Peroxide Chemically Initiated
Electron Exchange Luminescence. A New General Mechanism for Chemical Production of
Electronically Excited States", J. Am. Chem. Soe., 100,4496-4503, 1978.
10 Koo J-y, Schmidt SP & Schuster GB, "Bioluminescence of firefly-key steps in formation of
electronically excited-state for model systems", Prec. Nat/. Acad. Sei., USA, 75 (1),30-3, 1978.
11 McElroy WD, Seliger HH & White EH, "Mechanism of Bioluminescence, Chemiluminescence
and Enzyme function in the oxidation of firefly luciferin", Photochem. Photobiol., 10, 153-70,
1969.

Introdução 26
12 Catalani LH & Wilson T, "Electron Transfer and Chemiluminescence. Two Inefficient Systems:
1,4-Dimetoxy-9, 10-diphenyanthrace Peroxide and Diphenoyl Peroxide", J. Am. Chem. Soe., 111
(7),2633-9, 1989.
13 Rauhut MM, "Chemiluminescence from Concerted Peroxide Decomposition Reactions", Aee.
Chem. Res., 2, 80-7, 1969.
14 Chandross EA, "A New Chemiluminescent System", Tetrahedron Lett., 761-65, 1963.
15 Stevani CV, Lima DF, Toscano VG & Baader WJ, "Kinetic Studies on the Peroxyoxalate
Chemiluminescence Reaction: Imidazole as a Nucleophilic Catalyst", J. Chem. Soe. Perkin
Trans. 2, 989-95, 1996.
16 Silva SM, Casallanovo F, Oyamaguchi KH, Ciscato LFLM, Stevani CV & Baader WJ, "Kinetic
Studies on the Peroxyoxalate Chemiluminescence Reaction: determination of the cyclization
rate constant", Lumineseenee, 17,313-20, 2002.
17 Hohman JR, Givens RS, Carlson RG & Orosz G, "Synthesis and Chemiluminescence of a
Protected Peroxyoxalate", Tetrahedron Lett., 37(46),8273-6, 1996.
18 Hadd AG, Birks JW, "Kinetics and Mechanisms of the Nucleophilic Substitution Reaction of
Imidazole with Bis(2,4,6-trichlorophenyl) Oxalate and bis(2,4-dinitrophenyl) Oxalate", J. Org.
Chem., 61, 2657-63.
19 Hadd AG, Robinson AL, Rowten KL & Birks JW, "Stopped-Flow Kinetics Investigation of the
Imidazole-Catalyzed Peroxyoxalate Chemiluminescence Reaction", J. Org. Chem. 63, 3023-31,
1998.
20 Hadd AG, Seeber A, Birks JW, "Kinetics of two Pathways in Peroxyoxalate
Chemiluminescence" J. Org. Chem., 65, 2675-83, 2000.
21 Stevani C & Baader WJ, "Preparation and characterization of 2,2,2-triphenyl-2À.5-1,3,2
dioxastilbolane-4,5-dione as standard for an attempt to trap 1,2-dioxetanodione, a possible high
energy intermediate in peroxyoxalate chemiluminescence", J. Chem. Res. (S), 430-2, 2002.

Introdução 27
22 Jonsson T, Emteborg M & Irgum K, " Heterocyclic compounds as catalysts in the
peroxyoxalate chemiluminescence reaction of bis(2,4,6-trichlorophenyl) oxalate", Anal. Chim.
Acta, 361, 205-15, 1998.
23 Lee JH, Rock JC, Schlautman MA & Carraway ER, "Characteristics of key Intermediates
Generated in Uncatalyzed bis (2,4-dinitrophenyl) oxalate (DNPO) Chemiluminescence
Reactions", J. Chem. Soc., Perkin Trans. 2, 1653-57,2002.
24 Lee JH, Rock JC, Park SB, Schlautman MA & Carraway RE, " Study of the Characteristics of
Three High-Energy Intermediates in Peroxyoxalate Chemiluminescence (PO-CL) Reactions" J.
Chem. Soc., Perkin Trans. 2, 802-9, 2002.
25 Shamsipur M & Chaich MJ, "A Study of Chemiluminescence from Reactions of Peroxyoxalate
Esters, Hydrogen Peroxide and 7-amino 4-trifluorometylcumarin", Spectrosc. Letters, 34 (4),
459-68, 2001.
26 Shamsipur M & Chaich MJ, "Quenching effect of Triethylamine on Peroxyoxalate
Chemiluminescence in the Presence of 7-amino-4-trifluoromethylcumarin", Spectrochim. Acta
Part A, 57, 2355-58, 2001.
27 Shamsipur M, Chaich MJ & Karami AR, "A Study of Peroxyoxalate-Chemiluminescence of
Acriflavine" Spectrochim. Acta Part A, 59,511-7,2003.
28 Stevani CV, Campos IPAR & Baader WJ, "Synthesis and Characterisation of an Intermediate
in the Peroxyoxalate Chemiluminescence: 4-chlorophenyl O,O-hydrogen Monoperoxyoxalate",
J. Chem. Soc., Perkin Trans. 2, 1645-8, 1996.
29 Bos R, Barnett NW, Dyson GA, Um KF, Russel RA & Watson SP, "Studies on the mechanism
of the peroxyoxalate chemiluminescence reaction part 1. Confirmation of 1,2-dioxetanodione as
an intermediate using 13C nuclear magnetic resonance spectroscopy", Anal. Chim. Acta, 502,
141-147,2004.

Introdução 28
30 Rauhut MM, Bollyky LJ, Roberts BG, Loy M, Whitman RH, lannotta AV, Semsel AM & Clarke
RA, "Chemiluminescence from reactions of electronegatively substituted aryl oxalates with
hydrogen peroxide and f1uorescent compounds", J. Am. Chem. Soe., 89 (25), 1967.
31 Rauhut MM, Roberts BG & Sernsel AM, "A study of chemiluminescence from reactions of
oxalyl chloride, hydrogen peroxide, and fluorescent compounds", J. Am. Chem. Soe., 88 (15),
1966.
32 Stevani CV, Silva SM & Baader WJ, "Studies on the mechanism of the excitation step in
peroxyoxalate chemiluminescence", Eur. J. Org. Chem. 4037-46,2000.
33 Silva SM, Wagner K, Weiss D, Beckert R, Stevani CV & Baader WJ, "Studies on the
chemiexcitation step in peroxyoxalate chemiluminescence using steroid-substituted activators",
Lumineseenee, 17, 362-9, 2002.
34 Kuroda N, Hosoki N, Akiyama S & Givens RS, "Photografic detection of fluorescent-Iabelled
oligodeoxynucleotide in the blotting format by peroxyoxalate chemiluminescence." J. Biolumin.
Chemilumin. 13 (2), 101-5, 1998.
35 Ellingson A. & Karnes HT, "Investigation of far red dyes for use in peroxyoxalate
chemiluminescence detection and analysis of the CY5 derivative of amantadine hydrochloride in
human plasma." Biomedieal Chromatography, 12 (1),8-12,1998.
36 Nakamura MM, Saraiva SA & Coichev N, " Parameters Affecting the Peroxyoxalate
Chemiluminescence" Analytieal Letters, 32 (12), 2471-87, 1999.
37 Nozaki 0, Iwaeda T & Kato Y, "Amines for Detection of Dopamine by Generation of Hydrogen
Peroxide and Peroxyoxalate Chemiluminescence", J. Biolumin. Chemilumin., 11, 309-13,1996.
38 Nozaki O. "Detection of Substances with Alcoholic or Phenolic Hydroxyl Groups by Generation
of Hydrogen Peroxide with Imidazole and Peroxyoxalate Chemiluminescence", J. Biolumin.
Chemilumin. 10, 339-44, 1995.

Introdução 29
BIBLIOTECAINSTITUTO DE QUíMICAUniversidade de São Paulo
39 Ragab GH, Nohta H & Zaitsu K. "Chemiluminescence determination of catecholamines in
human blood plasma using 1,2-bis(3-chlorophenyl)ethylenediamine as pre-column derivatizing
reagent for liquid chromatography", Anal. Chim. Acta., 403, 155-60, 2000.
40 Yamada H, kuwara Y, Takasumo Y & Hayase T. "A new sensitive determination method of
estradiol in plasma using peroxyoxalate ester chemiluminescence combined with an HPLC
system.", Biomed. Chromatogr., 14, 333-7, 2000.
41 Sun Y, Wada M, AI-Dirbashi O, Kuroda N, Nakazawa H & Nakashima K. "High-performance
Iiquid chromatography with peroxyoxalate chemiluminescence detection of bisphenol A migrated
from polycarbonate baby bottles using 4-(4,5-diphenyl-1 H-imidazol-2-yl)benzoyl chloride as a
label", J. Chromatogr. B, 749, 49-56, 2000.
42 Ohba Y, Kuroda N & Nakashima K Anal. Chim. Acta, 465, 101-9, 2002.
43 Tsukagoshi K, Kameda T, Yamamoto M & Nakajima R. "Separation and determination of
phenolic compounds by capillary electrophoresis with chemiluminescence detection", J.
Chromatogr. A, 978,213-20,2002.
44 Appelblad P, Irgum K. "Separation and detection of neuroactive steroids from biological
matrices", J. Chromatogr. A, 955, 151-82,2002.

Parte Experimental 30
2 Parte Experimental
2.1 Aparelhagem
Para seguir a cinética de emissão proveniente das reações dos
perácidos e do sistema peroxioxalato, foi utilizado um espectrofluorímetro SPEX
FLUOROLOG 1681, com tensão de 750V na fotomultiplicadora, fenda de 1mm
e grade na condição de espelho. Com a grade do monocromador estando
posicionada na condição de espelho, a luz emitida pela reação é toda refletida
para a fotomultiplicadora sem a separação espectral. Além disso, foi utilizado
um outro espectrofluorímetro da marca Varian Cary Eclipse, tensão na
fotomultiplicadora de 750 V, fenda 20nm e grade também na condição de
espelho. Para registrar os espectros de absorção foi utilizado um
espectrofotômetro SHIMADZU Multispec1501. Nos experimentos efetuados na
ausência de H20 2 foi utilizado um contador de fóton equipado com um
refrigerador termoelétrico, modelo THORN EMI Gencom Inc 1kV. (Modelo
FACT 50 MK). A saída da fotomultiplicadora foi conectada a um amplificador
Princeton Applied Research (modelo 1121A).
Para medir a viscosidade das misturas de tolueno e difenilmetano foi
utilizado um reômetro Brookfield para baixa viscosidade, modelo LV DVIII cone
CP40 ângulo 0,8° volume = 0,5 mL e raio 2,4 em.
Os potenciais de oxidação dos ativadores 9 a 13 foram determinados em
um aparelho EG & G Princeton applied Research modelo 175, nas seguintes
condições: eletrodo de platina (trabalho), eletrodo Ago/Ag+ (referência), filtro 590
Hz, capacitor 15 nF, eletrólito: perclorato de tetrabutilamônio (0,1 M) em
acetonitrila.
Na sublimação do composto 4 foi utilizado um sublimador modelo: BÜCHI
Glass Oven B-580 e na cromatografia circular foi utilizado em um Chromatotron
79241.

Parte Experimental 31
. Para caracterizar os compostos, preparados neste trabalho, foram
utilizados os seguintes equipamentos: Bruker ORX 500, Bruker AC200, FT IR
Bomem MB100, Shimadzu CG MS-QP5050A, Elemental Analyzer 2400CHN
Perkin Elmer.
2.2 Reagentes
A piridina foi mantida em temperatura de refluxo por 4 h na presença de
NaOH e destilada a 111 oCo A trietilamina foi destilada de NaOH após refluxo de
3 horas. A solução de H20 2, utilizada na preparação dos compostos 5-8, foi
preparada a partir da extração de 20 mL de uma solução concentrada (60%) de
H20 2 com três porções de 15 mL de éter etílico, sendo a solução seca com
MgS04 a 5 oCo A concentração da solução resultante variava entre 5-8 M,
determinada por iodometria. O p-cresol foi recristalizado de hexano, o fenol de
éter de petróleo (40-60 °C), o p-metoxifenol de hexano e o p-nitrofenol em
tolueno/EtOAc 9:1. Todos foram secos à vácuo e armazenados sob P20 S. 9,10
difenilantraceno (OPA) (Aldrich), imidazol (Aldrich 99%), cloreto de p
bromofenilbenzoíla (Fluka), reagente de Lawesson (Fluka) (26), cloreto de
bis(trifenilfosfina) paládio 11 [(Ph3P)2PdCb] (27), fenilacetileno (Merck) e hidrazina
hidratada (Merck) foram utilizados sem purificação prévia.
Os compostos 9 a 13 foram preparados no laboratório do Prof. Rainer
Beckert1: 3-[2-(4-metoxifenil)-5,5-dimetiloxazol-4-i1ideno]androsta-1 ,4-dien-17
ona (9), 3-[2-(3-metoxifenil)-5,5-dimetiloxazol]-4-ilideno]androsta-1 ,4-dien-17-ona
(10), 3-[2-(4-c1orofenil)-5,5-dimetioxazol-4-ilideno]androsta-1 ,4-dien-17-ona (11),
3-[2-(4-f1uorofenil)-5,5-dimetiloxazol-4-ilideno]androsta-1 ,4-dien-17-ona (12) e 3
[2-(4-nitrofenil)-5,5-dimetiloxazol-4-ilideno]androsta-1 ,4-dien-17-ona (13).

Parte Experimental 32
2.3 Solventes
o acetato de etila foi seco sob CaCb por um dia, filtrado e submetido à
agitação mecânica por 30 minutos com 1 M de NaOH a O DC, sendo novamente
filtrado e destilado lentamente (p.e. = 77 DC) na presença de P20S.
O éter dietílico foi pré-seco com H2S04 (100 mL por litro), destilado e
aquecido à temperatura de refluxo por 4 h com Nao na presença de
benzofenona. Após esta etapa de purificação foi, então, redestilado na presença
de peneira molecular de 4 Á e armazenado também sob peneira molecular de 4
Á pré-ativada.
O tolueno e o THF utilizados na síntese dos derivados de oxazolona foram
tratados com sódio metálico e benzofenona, sendo destilados após a mistura
atingir a coloração azul característica de meio anidro.
O clorofórmio foi destilado de CaCI2 após 24 horas na presença do agente
secante.
O etanol e o metanol foram utilizados sem tratamento prévio. O tolueno
utilizado nos estudos de viscosidade foi destilado de P20 S e armazenado sob
peneira molecular 4A.
O difenilmetano (p.f. = 25,4 °C) também utilizado nos estudos da
viscosidade foi destilado à vácuo (90°C - 3 mmHg). As características físicas
destes solventes estão relacionadas na tabela 2.
Tabela 2: Características físico-químicas dos solventes utilizados no estudo da viscosidade.
solventeáTJ (mPas) E no
2U p.e (0C)
tolueno 0,560 2,379 1,4960 110
difenilmetano 1,945 2,54 1,5770 264
acetato de etila 0,423 6,08 1,372 76,5
aobtidos da literatura2

Parte Experimental 33
2.4 Determinação da concentração dos perácidos 5-7 e das
soluções anidras de H20 2
Na determinação da concentração de cada perácido foi empregada uma
cubeta de quartzo de caminho óptico 1,0 cm a temperatura de 25°C contendo
3,0 mL de uma solução 0,05 M de KI em tampão NaOAc/AcOH (pH = 3,8),
sendo a absorção determinada em 353 nm (E = 2,55.104 moi L-1 cm-1). Nesta
solução era adicionado 5 I-lL da solução do perácido (5-7), cuja diluição variava
de 5 a 20 vezes, pois dependia da concentração da solução estoque do
perácido a ser analisado. A concentração da solução estoque variou em torno
de aproximadamente 300mM.
Para determinar a concentração de H20 2 empregava-se a mesma
metodologia utilizada na determinação dos perácidos. Porém, era necessário
diluir 100 vezes, em água, a solução estoque de H20 2, cuja concentração era
de aproximadamente 2 M (1 mL de H20 2 60% em 10mL de acetato de etila
anidro) e adicionar 10 I-lL de solução de peroxidase HRP VI (/" = 403 nm,
E = 1,02.105 moi L-1 cm-1), cuja concentração final era 1.10-8 moi L-1
.
2.5 Estudo cinético dos derivados de O,O-hidrogênio
monoperoxioxalato de arila (5-7)
Em uma cubeta de quartzo de caminho óptico de 1,0 cm e volume de 3
mL foram adicionados 300 I-lL de uma solução estoque de 9,1 O-difenilantraceno
(10 mM), 5 I-lL a 2,5 mL de uma solução estoque de imidazol (300 mM) e 0,15
mL a 2,64 mL de acetato de etila. A reação foi iniciada com aproximadamente
30 I-lL do perácido (5-7) (50 mM), o volume do perácido variou de acordo com a
concentração da solução estoque, o qual foi determinada antes de cada
experimento. A concentração final para cada reagente era: [5], [6] e [7] =0,5, 0,1
e 0,5 mM respectivamente, [IMI-H]=0,5 mM a 250 mM e [DPA]=1 mM. Todas as

Parte Experimental 34
soluções foram preparadas em acetato de etila anidro, e os dados cinéticos
calculados a partir da intensidade de emissão de luz.
2.6 Estudo do sistema peroxioxalato variando a viscosidade
Em uma cubeta de quartzo, de caminho óptico de 1,0 cm a 25°C, cujo
volume máximo era 3,0 mL, foram adicionados 0,3 mL de solução de OPA (10
mM) em difenilmetano (OFM), 63,80 J.!L ou 31,40 J.!L de uma solução de
imidazol (23,50 mM) em tolueno e 3,5 J.!L de uma solução de H20 2, (2,10 mM)
em acetato de etila. O volume restante foi completado com tolueno e
difenilmetano (Tabela 3). A reação foi iniciada pela adição de 25 J.!L de uma
solução de TCPO (12 mM) em tolueno.
Tabela 3: Composição das misturas de solventes (tolueno e difenilmetano) utilizadas nos
experimentos.
DFM C13H12 tolueno
% (mL) (mL)
oa - 2,60
10b- 2,60
20b 0,3 2,31
40b 0,9 1,70
60b 1,5 1,10
SOb 2,1 0,51
90b 2,4 0,20
a solução estoque aeOPA em tolueno, Õ solução estoque de OPA em OFM.
As concentrações finais de cada reagente foram: [H202] = 2,5 mM,
[TCPO] = 0,1 mM, [IMI-H] = 1,0 mM ou [IMI-H] = 0,5 mM e [OPA] = 1,0 mM. A
solução de H20 2 foi preparada em acetato de etila, devido a sua baixa
solubilidade em tolueno/C13H1i No entanto, para reduzir a influência nas
características físico-químicas do sistema, preparou-se uma solução de H20 2
3M de modo que o volume da alíquota em acetato de etila fosse reduzido (2,5
J.!L), ou seja 0,08 %.

Parte Experimental 35
2.7 Estudo do sistema peroxioxalato utilizando os ativadores
9-13
Em uma cubeta de quartzo de caminho óptico de 1,0 cm a 25 aC, cujo
volume máximo era 3,0 mL, foram adicionados de 0,015 a 1,5 mL da solução
estoque do ativador (9-13), (solução estoque: 1,0 mM), 40 IlL de IMI-H,
(solução estoque de 75 mM), 30 IlL de HzOz (solução estoque de 1.OM) e 1,41
2,89 mL de acetato de etila, completando o volume para 2,95 mL. A reação foi
iniciada pela adição de 50 IlL da solução estoque do TCPO (6,1 mmol L-1). As
concentrações finais das soluções contidas na cubeta eram as seguintes:
[TCPO] = 0,10 mM, [IMI-H] = 1,0 mM, [HzOz] = 10 mM e [ACT]= 0,005-0,5 mM.
2.8 Estudo do sistema peroxioxalato na ausência de H20 2
Em uma cubeta de quartzo a 25 aC, cujo volume era de 6 mL foram
adicionados 106 IlL de IMI-H, 200 IlL de rubreno e 66 IlL de DNPO ou TCPO.
Os experimentos foram executados na presença (1,7 IlL) e ausência de
peróxido de hidrogênio. O restante do volume foi completado com acetato de
etila anidro para 4 mL. A concentração estoque e final corresponderam as
descritas na tabela 4:
Tabela 4: Concentrações dos reagentes utilizados no sistema peroxioxalatQ sem H20 2 .
Reagente [Reagente]sol.estoque [Reagente]final
(mmol L-1) (mmol L-1
)
IMI-H 75 2
TCPO 12 0,2
DNPO 12 0,2
Rubreno 1 0,05
H20 2 1,19 e-5 e 1,19 e-s 5 e-7 a 5 e-lO

Parte Experimental 36
2.9 Calibração da fotomultiplicadora
Como a intensidade de emissão de luz é medida em unidade arbitrária
(u.a.) ou em contagens por segundo (cs-1), unidades que dependem do
instrumento utilizado, é necessário fazer a calibração da fotomultiplicadora. A
calibração é necessária para que a intensidade de luz emitida possa ser
transformada em Einstein (E, unidade SI de quantidade de luz emitida). Este
procedimento foi efetuado utilizando-se a reação de luminol com H202,
catalisada por hemina, descrita na literatura3 e que foi estabelecida pelo nosso
grupo como reação padrão na determinação de rendimentos de
quimiluminescência4. Cabe ressaltar, que o rendimento quântico de
quimiluminescência da reação de luminol com H202 (<Dluminol = 0,0114 ± 0,0006
E mor1) é independente da concentração inicial do luminol e, também que, a
concentração do peróxido de hidrogênio deve ser 100 vezes maior que a
concentração do luminol. As soluções utilizadas, exceto a solução estoque do
luminol, devem ser preparadas, imediatamente, antes do experimento. Para
este procedimento são utilizadas as seguintes soluções: luminol (0,10 mM, em
solução tampão de fosfato pH 11,6, E = 7600 M-1cm-1), H20 2 (0,30 % em água
deionizada), e hemina bovina dissolvida em meio aquoso alcalino (duas
soluções com absorbância 0,6 e 0,2 medidas em 414 nm). Em uma cubeta
contendo 2,8 mL de solução de luminol posicionada no espectrofluorímetro sob
agitação são rapidamente adicionadas 100 /-lL de solução de H202, seguida da
adição de 100 /-lL da solução de hemina mais diluída (absorbância 0,2). A
adição da solução de hemina é acompanhada por um pulso de emissão de luz,
seguida de um decaimento relativamente rápido. Quando a intensidade atinge
aproximadamente 2% da intensidade máxima de luz, 100 /-lL da solução de
hemina mais concentrada (absorbância 0,6) são adicionados. Observa-se
novamente um pulso de luz, o qual retoma rapidamente à linha base. Este.procedimento é utilizado para garantir o consumo completo de luminol. Após a
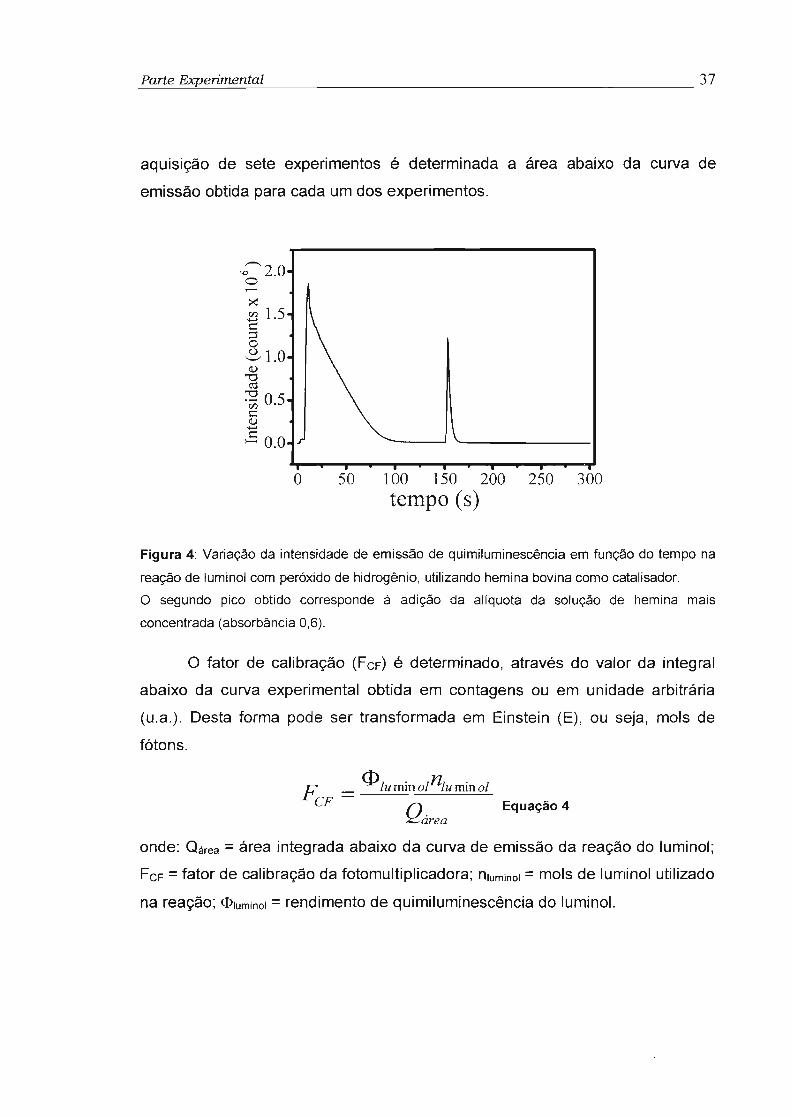
Parte Experimental 37
aqulslçao de sete experimentos é determinada a área abaixo da curva de
emissão obtida para cada um dos experimentos.
'0'-"'2.0o......~
~ 1.5e:::lo
2., 1.0-<1)
-g~ 0.5-e:<1).....
,.s O.OJ.r
O 50
- '''...... _100 150 200 250 300tempo (s)
Figura 4: Variação da intensidade de emissão de quimiluminescência em função do tempo na
reação de luminol com peróxido de hidrogênio, utilizando hemina bovina como catalisador.
O segundo pico obtido corresponde à adição da alíquota da solução de hemina mais
concentrada (absorbância 0,6).
o fator de calibração (FCF) é determinado, através do valor da integral
abaixo da curva experimental obtida em contagens ou em unidade arbitrária
(u.a.). Desta forma pode ser transformada em Einstein (E), ou seja, mols de
fótons.
F - cI>/umino/n/umino/CF -
QáreaEquação 4
onde: Qárea =área integrada abaixo da curva de emissão da reação do luminol;
FCF =fator de calibração da fotomultiplicadora; nluminol =mols de luminol utilizado
na reação; <1>luminol = rendimento de quimiluminescência do luminol.

Parte Experimental 38
2.10 Medida da viscosidade da mistura de tolueno com
difenilmetano (C13H12J.Para a determinação da viscosidade da mistura de tolueno com C13H12
em várias proporções foi utilizado um reâmetro Brookfield para baixa
viscosidade. As medidas foram executadas a 25°C.
Tabela 5: Viscosidade das misturas tolueno e difenilmetano em várias proporções.
DFM% 11 (cP)
O O,56a
10 0,703 ± 0,011
20 0,816 ± 0,035
40 1,050 ± 0,006
60 1,372±0,O12
80 2,166±0,021
90 2,573 ± 0,040
a obtido da Iiteratura2
2. 11 Preparação dos cloretos de feniloxalila substituídos
oHo-o-xCI O
..éter
O CI
H +CI O
A mesma metodologia sintética foi utilizada na preparação dos cloretos
de feniloxalila substituídos. Assim, será apresentado a seguir o procedimento
geral seguido da caracterização de cada composto.
OH
Qx
X =OCH3 (1), CH3 (2), H (3), N02 (4)
Procedimento geral:
Em um balão de 100 mL, contendo 106 mmol do fenol substituído
dissolvido em 60 mL de éter dietílico anidro, foram adicionados, gota a gota,
aproximadamente 106 mmol de cloreto de oxalila (9,2 mL, d = 1,455 g cm-3). A

Pnrlp FY['erimental
BIBLIOTECAINSTITUTO DE QUíMICAUniversidade de São Paulo
mistura foi mantida sob agitação à temperatura ambiente durante três dias.
Após este tempo, o solvente foi evaporado restando um sólido, exceto para o 2.
Este sólido foi destilado sob pressão reduzida, utilizando banho de silicone e
condensador de destilação aquecido [p.e/mmHg: 100°C /7 (1), 80°C / 1 (2),
145°C / < 1 (3)].
Rendimentos: 62 % (1),82 % (2) e 60 % (3).
Análise elementar
Composto 1 (%): observado 52,21 (C), 3,04 (H) e 19,28 (CI) / calculado 52,06
(C), 2,73 (H) e 19,21 (CI).
Composto 2 (%): observado 54,64 (C) e 3,70 (H) / calculado 54,43 (C) e 3,55
(H). Diferentemente dos outros compostos preparados, este composto é um
líquido viscoso, por isso não foi possível efetuar a determinação do cloro.
Composto 3 (%): observado 50,16 (C), 3,33 (H) e 16,35 (CI) / calculado 50,37
(C), 3,29 (H) e 16,52 (CI).
Infravermelho (KBr)
1 1776 e 1753 cm-1 (v, C=O), 689-761 cm-1 (õ, C-C/). 1173 cm-1
(õ, C-O éster) 1486 cm-1 (õ, C=C "anel aromático") e 3394 cm-1 (v, O-H)
(impureza do ácido correspondente).
2 1783 cm-1 (v, C=O), 712 cm-1 (õ, C-CI) 1249 cm-1 (õ, C-O éster), 1505 cm-1
(õ, C=C "anel aromático").
3 1737 e 1764 cm-1 (v, C=O), 1209 cm-1 (õ, C-O), 772 cm-1 (õ, C-CI),
1508 cm-1 (õ, C=C) e 3503 cm-1 (v, O-H) (impureza do ácido
correspondente) .

Parte Experimental 40
Espectrometria de Massa
1 (EI) Injeção direta, mlz (%): 184 (19,5) Me+, 156 (6,5), 121 (10,5), 93
(14,84) e 77 (100), além da presença do ácido correspondente, cujo pico
do íon molecular é 165 (3,07).
2 (CI) Ionização química, mlz (%) [198+Ht(O,13), 163 (0,13), [136+Ht
(9,89), 109 (100),91 (2,41).
3 (EI) Injeção direta m/z (%) 214/216 (5/2) Me+, 186 (1), 151 (1), 124 (100),
109 (86) e 81 (28). Há também a presença do ácido correspondente, cujo
pico do íon molecular é 196 (13).
RMN de 13C (ORX125 MHz, COCbl
3 4 5
a ti2 aM 3~ ~6 aMe
CI 1 a 7
(ú) 160,8 (C-1), 158,2 (C-6), '154,3 (C-2), 143,4 (C
3), 121,3 (C-4), 114,7(C-5), 55,6 (C-7) (Anexo 8)

Parte Experimental 41
2.11.1 Tentativa de preparação do cloreto de p-nitrofeniloxalila (4)
A tentativa de preparação do composto 4 foi feita de acordo com o
procedimento descrito na seção 2.11, contudo não foi obtido o produto. Cabe
dizer que, embora várias tentativas tenham sido feitas, não foi possível purificar
este composto por destilação à pressão reduzida. Embora, a mistura de reação
tenha sublimado dentro da aparelhagem de destilação, tentativas de purificação
por sublimação também não foram bem sucedidas, resultando em rendimentos
insatisfatórios. Apesar do teste com o produto sublimado, juntamente com
H20 2, imidazol e 9,1 O-difenilantraceno ter resultado em emissão de luz visível, o
resultado da análise elementar mostrou que este composto era o
p-nitrofenol. Por outro lado, o resultado do espectro na região do infravermelho
apresentou as seguintes bandas: 628-753 cm-1 (8, C-CI), 1202 cm-1 (8, C-O),
1757 cm-1 (v, C=O) banda fina e 3366 cm-1 (v, O-H) banda larga (proveniente
do fenol ou do ácido correspondente).

Parte Experimental 42
2.11.2 Preparação do bis(4-nitrofenil) oxalato (23)
6·N02
o CI
MCI o
éter
~N~..J
-02N-o-~O~__t-O--NO,_ O O
23
Em um balão de fundo redondo de 100 mL com três bocas foram
dissolvidos 10 g (72 mmol) de p-nitrofenol em 25 mL de éter dietílico anidro. A
solução foi resfriada a -10°C e 7,1 mL (50 mmol, d=0,72 g/cm3) de trietilamina
foram adicionados lentamente. Em seguida foram adicionados lentamente 2,4
mL (27,5 mmol, d=1,455 g/cm3) de cloreto de oxalila durante,
aproximadamente, 30 minutos. A reação ficou sob agitação à temperatura
ambiente durante 13 horas. O solvente foi evaporado à pressão reduzida,
restando um sólido marrom o qual foi lavado sob agitação magnética por uma
hora com éter de petróleo e, depois, mais uma hora com acetato de etila. O
produto foi recristalizado em acetato de etila. Rendimento: 47,8% (4,3 g)
Análise elementar:
(%): observado 50,60 (C), 2,41 (H) e 8,43 (N) I calculado 50,63 (C), 2,95 (H) e
8,40 (N).
Invravermelho (KBr)
1772 cm-1 (v, C=O), Õ (C-O) 1348 cm-\ (õ, C-N02) 886 cm-1, (õ, C=C) 1484
1616 cm-1.
Espectrometria de Massa (Impacto eletrônico)
m/z (%): 332 (1,33) Me+, 304 (29,49),166 (43,98),139 (35,24),122 (100,00).

Parte Experimental 43
2.12 Síntese dos derivados
monoperoxioxalato de arila (5-8)
de O,O-hidrogênio
Os perácidos utilizados neste trabalho foram preparados de acordo com
uma metodologia comum. Assim, a exemplo da preparação do cloreto de
feniloxalila substituídos, será apresentado um procedimento geral, dando
ênfase apenas nas diferenças entre cada preparação.
oHo-o-xCI ° + H1: X =H 2°2
2: X =CH3: X = OC~4: X =NO 3
2
o ~ oHo-o-xéter HOO ° 5: X =OCH3
6: X =CH 3
7: X= H8X =N02
Procedimento geral:
Em um balão contendo o cloreto de feniloxalila substituído (1-4) em 10
mL de éter dietílico anidro a -25 cC, foi adicionada, gota a gota durante 20
minutos, uma solução etérea de H202, contendo piridina, sob atmosfera de N2,
conforme mostra a tabela abaixo.
Após 30 minutos de agitação a -25 °C, a mistura foi lavada com uma
solução saturada e gelada de cloreto de amônio (3 x 20 mL) e a porção etérea
seca com sulfato de magnésio anidro por 10 minutos a O oCo Após este tempo o
solvente foi evaporado a -40 °C e substituído por 2-4 mL de acetato de etila
anidro. A reação foi acompanhada por cromatografia em camada delgada (CCO)
utilizando placa de Si02 com indicador de fluorescência, 60 F254, a -20 DC, e
solução de KI 10% como revelador. Foram observados os seguintes fatores de
retenção (Rf):
Composto 5: CH2CI2/EtOAc 9,5:0,5 (eluente) / Rf <;:;: 0,0 (H20 2), Rf =0,44 (5) e Rf
= 0,85 (p-metoxifenol).
Composto 6: CH2CI2/EtOAc 8,5:1,5 (eluente) / Rf = 0,63 (6) e Rf = 0,80 (p
metilfenol).
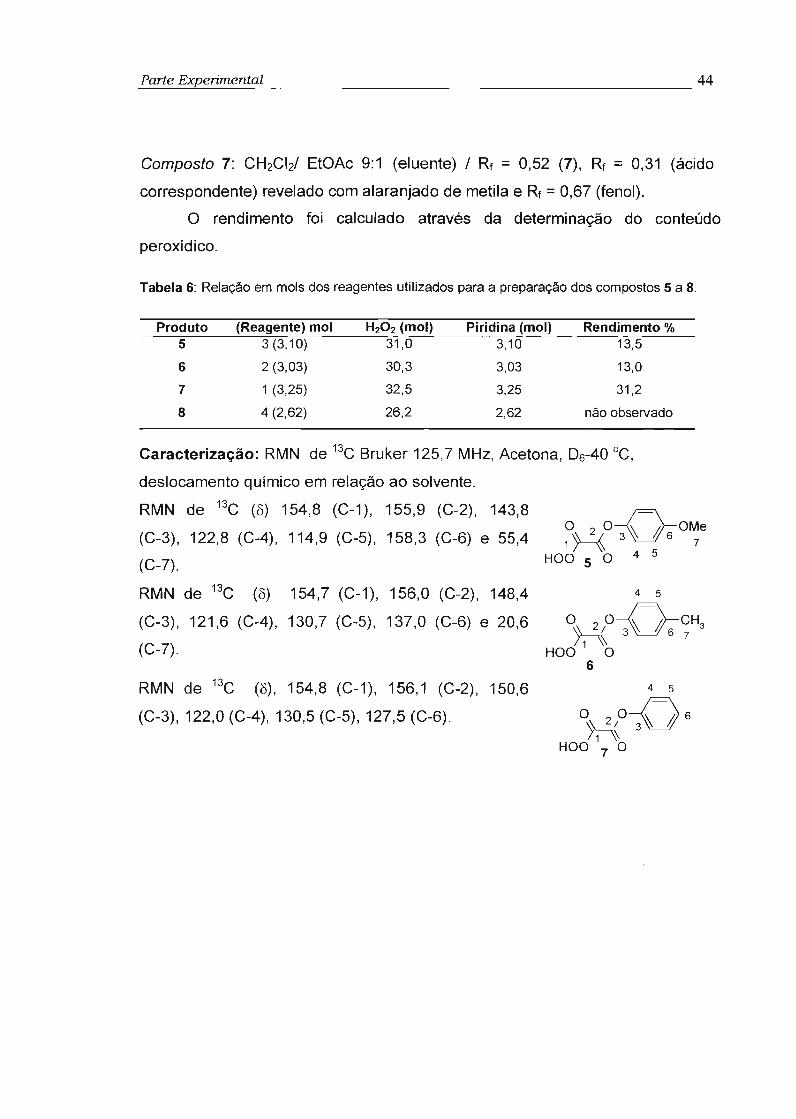
a -O2 a1M 3 ~ !J 6 aMe
Haa 5 a 4 5 7
4 5
a\~(a---D;-CH3Ha0'1\\ 6 7
6 a
Parte Experimental 44
Composto 7: CH2CI2/ EtOAc 9:1 (eluente) / Rf = 0,52 (7), Rf = 0,31 (ácido
correspondente) revelado com alaranjado de metila e Rf = 0,67 (fenol).
O rendimento foi calculado através da determinação do conteúdo
peroxídico.
Tabela 6: Relação em mais dos reagentes utilizados para a preparação dos compostos 5 a 8.
Produto (Reagente) moi HzOz (moi) Piridina (moi) Rendimento %5 3 (3,10) 31,0 3,10 13,5
6 2 (3,03) 30,3 3,03 13,0
7 1 (3,25) 32,5 3,25 31,2
8 4 (2,62) 26,2 2,62 não observado
Caracterização: RMN de 13C Bruker 125,7 MHz, Acetona, 06-40°C,
deslocamento químico em relação ao solvente.
RMN de 13C (8) 154,8 (C-1), 155,9 (C-2), 143,8
(C-3), 122,8 (C-4), 114,9 (C-5), 158,3 (C-6) e 55,4
(C-7).
RMN de 13C (8) 154,7 (C-1), 156,0 (C-2), 148,4
(C-3), 121,6 (C-4), 130,7 (C-5), 137,0 (C-6) e 20,6
(C-7).
RMN de 13C (8), 154,8 (C-1), 156,1 (C-2), 150,6
(C-3), 122,0 (C-4), 130,5 (C-5), 127,5 (C-6).
4 5
a\~ai )6Haa11\\7 a

BIBLIOTECA
Parte Experimental INSTITUTO DE QUíMICAURI"QrsidilQ'i! ~~ São Pilulo 45
2.12.1 Tentativa de síntese do O,O-hidrogênio monoperoxioxalato
de p-nitrofenila (8) a partir do composto 4
A primeira tentativa de preparação do composto 8 foi feita de acordo com
o procedimento descrito na seção 2.12. Entretanto, não foi observada a
presença de mancha peroxídica. eCD: CH2CI2/EtAc 9:1 (eluente) I Rf =0,05
(H20 2) e Rf=O,SO (p-nitrofenol).
2.12.2 Segunda tentativa de síntese do O,O-hidrogênio
monoperoxioxalato de p-nitrofenila (8) a partir do composto 23
° 0-o-NO H202
H~,1 2 -O-o2N-Q-o ° O THF- °HO ~ ,1 N0223 N HOO °8
Em um balão de 125 mL com três bocas foram dissolvidos 100 mg (0,3
mmol) de 23 em 40 mL de THF. Em seguida adicionaram-se 67,S ~L (0,15
mmol - 2,21 M) de H202 diluído em 5 mL de THF através de um funil de adição
e 1,2 ~L (0,015 mmol, d=1 ,05 g cm-3) de piridina em 5 mL através de outro funil
de adição. A reação foi conduzida a -40°C sob argônio e acompanhada por
CCD em placa de Si02 com indicador de fluorescência, a -20 DC, 60 F254,
eluídas em CH2Cb/HexlACN 5:2:3. As placas foram reveladas com uma
solução aquosa de KI 10%. Após 35 minutos de reação observou-se a presença
de uma substância com grupo funcional peroxídico (Rf = 0,51).
Seguiu-se o mesmo processo de lavagem utilizados na preparação dos
outros perácidos, entretanto não foi observado conteúdo peroxídico após a
lavagem com NH4CI.

Parte Experimental 46
2.13 Síntese do ácido p-metoxifeniloxálico (28)
°J--_t-Q-oMe H,O • 0:Y_t-Q-OM'CI 3 o O éter HO 28 O
N -40 DC
Em um balão de 50 mL foram dissolvidos 300 mg (1,4 mmol) de cloreto de
p-metoxifeniloxalila (3) em 10 mL de éter anidro. Durante 20 minutos foi
adicionada, gota a gota, uma solução etérea contendo água (0,40 mL / 22,22
mmol) e piridina (0,11 mU 1,4 mmol), sob atmosfera de Nz seco. Após 30
minutos de agitação a -40°C, a mistura reacional foi lavada com uma solução
saturada e previamente resfriada de cloreto de amônio (3 x 15 mL) e a porção
etérea seca com sulfato magnésio por 10 minutos a O oCo O solvente foi
evaporado a -10°C, obtendo-se um sólido branco. CCD -20°C', eluente:
CH2Cb/HEXlACN 15:3:2 e revelação com alaranjado de metila (Rf = 0,12).
RMN de 13C (Acetona-D6 , -40°C, 125 MHz)
(õ): 158,9 (C-1), 158,6 (C-2), 144,6 (C-3), 122,9 (C-4),
115,4 (C-5), 158,1 (C-6), 55,9 (C-7) (anexo 7).
4 5"
O ~-" \~?~OMe
H61\\ 6 7O

Parte Experimental 47
2. 14 Síntese de derivados de oxazolonas
2.14.1 Preparação do Mn02 em carvão ativo1•
Uma solução contendo 20 9 de KMn04 em 250 mL de água destilada foi
agitada com 10g de carvão ativo a temperatura ambiente por 48 horas. Após
este tempo a mistura foi filtrada em um funil de Büchner, lavada com água
destilada e seca à temperatura de 105 -110°C por 24 horas.
2.14.2 2[4-bromofenil]-5,5-dimetiloxazol-4-ona (14)
oQCI
I~~
Br
+ -JCNHO
1- CHCI3 , SnCl4..2- (C2Hs)3N, tolueno
3'
---!QA' N~OBr !j ~ /' 4'
4 - 1 o 5'3 2 l'
14
Em um balão de 125 mL de 3 bocas foram dissolvidos 8,7 9 (40 mmol)
de cloreto de p-bromofenil benzoíla em 40 mL de clorofórmio seco juntamente
com 3,4 9 (40 mmol) de cianidrina de acetona (d = 0,93 g/cm\ Em seguida,
utilizando um funil de adição, foram adicionados lentamente 10,4 9 (40 mmol)
de cloreto de estanho IV (d = 2,217 g/cm\ A reação permaneceu sob agitação
à temperatura ambiente sob argônio por 20 horas.
O sal formado foi filtrado em um funil de placa sinterizada, em seguida
adicionado a 150 mL de tolueno seco juntamente com 10 mL (71 mmol, d=0,72
g/cm3) de trietilamina e aquecido à temperatura de refluxo por 30 minutos. Após
este período o solvente da mistura reacional foi evaporado à pressão reduzida,
ao sólido restante foram adicionados mais 150 mL de tolueno, submetendo a
mistura novamente ao refluxo por 30 minutos. O solvente foi removido à
pressão reduzida e o produto recristalizado em etanol/tolueno.
Rendimento: 20% (2,14 g).
RMN de 1H (OMSO 0 6), Bruker 250MHz, (8): 7,8 (d, 2H, H-2, 3J=8,68 Hz), 8,1
(d, 2H, H-3, 3J=8,68 Hz) e 1,5 (s, 6H, CH3) (anexo 13).

Parte Experimental 48
RMN de 13C (8): 129,5 (C-1), 132,5 (C-2), 131,3 (C-3), 124,9 (C-4), 183,5 (C
2'), 193,6 (C-4'), 85,7 (C-5') e 22,7 (2 x CH3) (anexo 14).

Parte Experimental 49
2.14.3 2-[4-benzilacetileno]-5,5-dimetiloxazol-4-ona (15)
N O
BC-Q-fof14
(Ph3P)2PdC'!Cul
~~7 3'
tolueno/(C2H
S) N" 10 _ == - 2' ,,-N*O
D--==-~ 3 9 6 5 4 ~ /; 1 4'
_ 8 O_ _ H 15 3 2 l' 5'
Em um balão de 50 mL do tipo "Schlank" foram adicionados 0,5 9 (1,86
mmol) do composto 14 em 15 mL de tolueno seco juntamente com 20 mL (142
mmol, d=O,72 g/cm3) de trietilamina e 0,19 9 (1,86 mmol) de fenilacetileno (d =
0,93 g/cm\ Em seguida foram adicionados 0,0035 9 (0,0185 mmol) de Cul e
0,0130 9 (0,0186 mmol) de (Ph3P)2PdCb (11). A reação foi acompanhada por
cromatografia em camada delgada (CCO) em silica com uma mistura de
tolueno/EtOAc 9:1 como eluente. O produto desejado possuia um Rf = 0,39 e
fluorescência azul.
Rendimento: 13% (69 mg)
Análise elementar
(%): observado 76,03 (C), 5,44 (H), 4,57 (N) e 13,96 (O) I calculado 78,87 (C),
5,23 (H), 4,85 (N) e 11,06 (O).
Infravermelho (KBr)
2993 cm-1 (v, H-C), 2222 cm-1 (v, C-C), 1744 cm-1 (v, C=O), (v,C=N) 1604 cm-1.
Espectrometria de Massa (Impacto eletrônico)
m/z (%): 289 (93) M·+, 203 (100), 176 (10) e 43 (10)
RMN de 1H (COCI3, 300 MHz, TMS)
(õ) 7,7 (d, 2H, H-2, 3J=8,5Hz), 8,2 (d, 2H, H-3, 3J=8,5Hz,), 7,54 (m, 2H, H-8),
7,36 (m, 2H, H-9) e 1,61 (s, 6H, CH3).

Parte Experimental 50
RMN de 13C
(8) 130,3 (C-1), 129,9 (C-2), 132,4 (C-3), 125,3 (C-4), 88,5 (C-5), 94,1 (C-6),
122,4 (C-7), 131,8 (C-8), 128,5 (C-9), 129,1 (C-10), 184,2 (C-2'), 194,1 (C-4') e
85,7 (C-5').
UV "'máX 323 nm (Iage = 4,51)

Parte Experimental 51
2.14.4 2-[4-bromofenil]-5,5-dimetiloxazol-4-tiona (16)1
N O~//'r~Br~OI
14
3'
Reagente de Lawesson ~\\2'/N~S____________ Br I '\ '/ 4'
4 - 1 Otolueno 5'
3 2 l'
16
0,5 9 (1,86 mmol) do composto 14 foi adicionado em 15 mL de toluol e
em seguida 0,53 9 (1,21 mmol) do reagente de Lawesson (26), A mistura foi
aquecida à temperatura de refluxo por 10 horas, sendo que a formação do
produto foi acompanhada por CCO. O produto formado foi purificado por coluna
cromatográfica utilizando silica (Kiesegel Merck 0,040mm-O,063mm) como fase
estacionária e tolueno como fase móvel.
Rendimento: 47%. (0,25 g)
RMN de 1H (Bruker 250 MHz) COCb, TMS)
(8) 7,63 (d, 2H, H-2, J3=6,8HZ), 8,09 (d, 2H, H-3, J3=6,8Hz) e 1,63 (s, 6H, CH3).
RMN de 13C
(8) 124,5 (C-1), 132,6(C-2), 131,1(C-3), 124,5(C-4), 182,1(C-2'), 233,7(C-4'),
101 ,3(C-5') e 27,26 (2xCH3) (anexo15).

Parte Experimental 52
2.14.5 Androsta-1 ,4-dieno-3-tiona-17-ona (18)
o
o
Reagente de Lawesson•
THF
6
18
2g (7,04 mmol) de ADD (17) e 1,86g (4,22mmol) de reagente de
Lawesson foram adicionados em 50 mL de THF seco. Após 1 hora de agitação
à temperatura ambiente formou-se um produto azulo qual foi isolado em coluna
cromatográfica utilizando silica (Merck-Kiesegel 0,043mm-0,060mm) e
toluenofTHF 6:4. O produto purificado foi recristalizado de acetato de etila.
Rendimento: 88% (1,86 g).
Pf 162,3-163,6 DC (literatura 165-168 DC)5.
RMN de 1H (Varian 500 MHz, CDCb, TMS)
(5) 0,95 (s, 3H, CH3-18), 1,29 (s, 3H, CH3-19), 6,80 (d, 1H, J3 = 9,65 Hz, H-1),
6,86 (m, 1H, H-4) e 6,89 (dd, 1H, J3=9,64 Hz, J4=1 ,65 Hz, H-2).
RMN de 13C
(5) 133,5 (C-4), 135,6 (C-2), 147,4 (C-1), 160,9 (C-5), 218,9 (C-3) e 217,5 (C
17) (anexo16).
Invravermelho (KBr)
(v, C=O) 1737 cm-1, (5, C=S) 1134 cm-1
, (5, C=C) 1625 cm-1, (v, C-H) 2926 cm-1
Espectrometria de Massa (ionização química)
m/z (%): [300+CH5+r (M+) (100), [300+H+r (M+) (40), [268+H+] (11).

Parte Experimental 53
2.14.6 Androsta-1,4-dieno-3 hidrazona-17-ona (19)
H2NNH2 ·H20
s
THF, Qoe
..~
H2N-N
0,3 9 (1 mmol) do composto 18 foi adicionado em 30 mL de THF seco e a
temperatura desta solução foi diminuída para O oCo Em seguida foi adicionado
0,1 9 (2 mmol) de hidrazina hidratada. formou-se um produto azul-violeta que
permaneceu sob agitação por mais 10 minutos à temperatura ambiente. A
reação foi acompanhada por CCO (CHCb/MeOH 9:1), sendo o produto
purificado por coluna cromatográfica. Este composto foi caracterizado apenas
por infravermelho e comparado com a literatura5.
Rendimento: 91%.(0,27 g)
Infravermelho (KBr)
(v, N-H) 3386 cm-1, (v, N-H) 3221 cm-1
, (v, C=O) 1734 cm-1 e (5, C=N) 1658
cm-1.

Parte Experimental 54
2.14.7 Androsta-1,4-dieno-3 diazo 17-ona (20)1
o o
~
H2N-N
MnO/carvâo ativo
KOHaq. / Etp
3 g de Mn02/carvão ativo (1 :2) foram adicionados em 20 mL de éter
dietílico, sendo a mistura agitada no ultra-som durante 5 minutos. Em seguida
foi adicionado 0,27 g (0,91 mmol) do composto 19 juntamente com 5 gotas de
KOH aquoso. Em poucos minutos formou-se uma solução levemente azulada.
Após 1 hora de reação, a solução foi retirada e transferida para outro recipiente.
Ao resíduo foram adicionados mais 20 mL de éter dietílico e submetido à
agitação por mais 30 minutos à temperatura ambiente. É importante ressaltar
que, este produto é estável apenas por algumas horas, portanto é
recomendável executar a etapa seguinte, logo ao término desta reação.

Parte Experimental 55
2.14.8 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilideno]-androsta-1,4
dien-17-ona (21)1
N S
Br--0;fo+- +
N2
•3'
~1"N
Br~-2~3" r l'
18
0,108 9 (0,38 mmol) do composto 16 foi adicionado à solução do
composto 20, proveniente de 0,27g (O,91mmol) de 19, e a mistura da reação foi
submetida a 24 horas de repouso à temperatura ambiente, A reação
desprendeu N2 e precipitou enxofre. A formação do produto foi acompanhada
por CCO (tolueno/EtOAc 95:5). Ao final da reação observou-se a formação de
três substâncias amarelas fluorescentes: Rf = O, Rf = 0,38 e Rf = 0,9. As
substâncias foram separadas por cromatografia em coluna de sílica utilizando
tolueno como eluente. O produto corresponde ao Rf= 0,38.
Rendimento: 32% (E/Z 0,78/0,99)
Infravermelho (KBr)
(v, H-C) 2932 cm-1, (o, C=N) 1664 cm-1
, (v, C=O) 1737, (o, C-O) 1010 cm-1, (o,
C-Sr) 1068 cm-1.
RMN de 1H (Varian 300MHz, COCI3, TMS)
(o) 0,91 (s, 3H, CH3-18), 1,19 (s,3H, CH3-19), 1,66 (s, 6H, CH3-5'), 5,85 (d, 1H,
H-1E, J3 =10,1),5,87 (d, 1H, H-1Z, J3=10,1), 6,08 (s,1H, H-4E), 6,83 (s,1H, H
4Z), 7,02 (dd, 1H, H-2E, 3J=10,1 Hz, 4J=1,78 Hz), 6,25 (dd, 1H, H-2Z, J3=10,15,
J4=1,8), 7,51-7,61 (m, 2-H, 3"-aromático), 7,85-7,90 (m,2-H, 2 "-aromático).

Parte Experimental 56
BIBLIOTECAINSTITUTO DE QUíMICAUniversidade de São Paulo
RMN 13C (8 ppm)
(8) 13,83 (C-18), 14,17 (C-15), 21,78 (C-19), 21,00, 27,08 (2xCH2), 26,98 (C
CH3), 31,40, 32,35 (2xCH2), 35,62 (C-8), 35,71 (C-16), 41,44 (C-10), 47,77 (C
13), 50,95 (C-14), 52,99 (C-9), 87,30 (C-5'), 115,23 (C-4E), 125,87 (C-2Z),
117,15 (C-3), 117,30 (4-Z), 122,01 (C-4"), 126,98 (C-1"), 126,89 (C-2E),
125,87 (C-2Z), 129,58 (C-2"), 131,58 (C-3"), 134,24 (C-1E), 136,29 (C-1Z),
147,31 (C-5E), 145,61 (C-5Z), 149,57 (C-4'), 163,39 (C-2'), 219,81 (C-17).
Espectrometria de Massa (ionização química)
m/z (%): [520+H+r (M+) (81), 504 (17),442 (100), 426 (20).

Parte Experimental 57
2. 15Referências
I Wagner K, Weiss D & Beckert R, "New Luminescent Materiais Based on a Steroid Molecule",
Eur. J. Org. Chem., 3001-5, 2000.
2 Handbook Chemistry and physics 79 Ih, 1998-1999, CRC PRESS.
3 Lee J & Seliger HH "Absolute Spectral Sensitivity of Phototubes and the Application to the
Measurement of the Absolute Quantum Yields of Chemiluminescence and Bioluminescence"
Photochem. Photobiol, 4, 1015-48, 1965.
4 Stevani CV, Silva SM & Baader WJ, "Studies on the Mechanism of the excitation step in the
peroxyoxalate system. Eur. J. Org. Chem., 4037-46, 2000.
5 Weil3 D, Gaudig U & Beckert R, "Eine neue Methode zur Herstellung von Thioxosteroiden",
Synthesis, 751-2, 1992.

Resultados 58
3 Resultados
3.1 Síntese e estudo dos derivados de O,O-hidrogênio
monoperoxioxalato de arila (5-7)
3.1.1 Síntese e caracterização
A síntese dos intermediários (5-7) foi efetuada a partir dos cloretos de
feniloxalila substituídos (1-3) e, de uma maneira geral, obtidos com rendimentos
satisfatórios. Entretanto, devido à rápida hidrólise sofrida por estes compostos,
o ideal é armazená-los em ampolas condicionadas abaixo de -20 oCo A tabela 7
apresenta a relação em moi entre fenol e cloreto de oxalila utilizados na
preparação de cada derivado, juntamente com os rendimentos e pontos de
ebulição.
Tabela 7: Resultados obtidos na preparação dos compostos 1-4.
FenollCloreto de Oxalila Produto Destilação
mmoll mmol Rendimento (%) (oC I mmHg)
H (1) 1:1,5 62 100/7
CH3 (2) 1:1,5 82 80/1
OCH3 (3) 1:1,2 84 145/6
N02 (4) 1:1,4 6,33 sublimado
Exceto o composto 4, os demais (1-3) foram submetidos à destilação sob
pressão reduzida para separar o cloreto de fenila monosubstituído do derivado
dissubstituído, possivelmente formado pela reação com dois fenóis. Na
destilação do cloreto de feniloxalila (1) é importante mencionar que se a
destilação for feita sob pressão abaixo de 5 mmHg, o composto 1 é arrastado
para o "trap" de segurança. O cloreto de p-metilfeniloxalila (2) foi o único dos
três cloretos preparados que se apresentou como um líquido amarelado de
característica levemente oleosa, enquanto que os demais derivados formaram
um sólido branco, após o processo de destilação.

Resultados 59
A análise na região do infravermelho do composto 2 apresentou apenas
uma banda referente ao estiramento de carbonila em 1783 cm-1. Este fato
experimental nos levou a inferir que as freqüências das carbonilas, pertinentes
aos grupos éster e cloreto de acHa, estariam muito próximas, não sendo
possível identificá-Ias separadamente. Já os outros derivados (1 e 3)
apresentaram duas bandas, embora também muito próximas.
A reação de cada derivado do cloreto de feniloxalila com peróxido de
hidrogênio na presença de piridina em éter dietílico, resultou na formação dos
derivados do O,O-hidrogênio monoperoxioxalato de arila (5-7)
°H°-Q-XCI ° +1: X = H H
20
2
2: X = CH3
3: X = OCH4: X = NO 3
2
o . O~_t-Q-xéter HOO O 5: X = OCH3
6: X = CH3
7: X = H8 X = NOz
Cabe ressaltar, que os compostos (5-7) se apresentaram instáveis a
temperatura ambiente, decompondo-se rapidamente (no caso do composto 7, a
decomposição ocorreu em alguns minutos), sendo necessário armazená-los
a -80°C. A formação do produto foi acompanhada por cromatografia em
camada delgada (CCD), que ao ser revelada com solução aquosa de KI 10%
apresentou uma mancha escura característica de função peroxídica, devido à
oxidação do iodeto para iodo. Este comportamento foi observado para os três
derivados (5-7). Acompanhou-se, também, a formação do ácido correspondente
com alaranjado de metila (pH 3,2-4,4). Foi interessante observar que ao borrifar
a placa com alaranjado de metila, formava-se uma mancha branca na região do
perácido. Além da formação do ácido, também foi observada a presença do
fenol correspondente em algumas preparações.
Com o objetivo de separar o composto peroxídico das demais impurezas
presentes na amostra, tentou-se uma separação por cromatografia circular
dentro de uma câmara fria (7 DC), usando como eluente a mistura de

Resultados 60
CH2CI2/HexlACN 7:2: 1, obtendo-se a separação de duas substâncias não
peroxídicas, e uma terceira que ficou retida na placa, cuja revelação, com
solução de KI10%, também mostrou não ser peroxídica. O mesmo
comportamento foi observado na tentativa de purificação de um composto
análogo1, com o cloro como substituinte no anel aromático, por cromatografia
relâmpago em coluna, utilizando sílica, alumina e f10risil como fases
estacionárias, em várias misturas de CH2Cb/Hex a -40 oCo Em tal purificação,
não foi obtido sucesso, devido à decomposição do peróxido. Tal fato também foi
observado para alquil peroxioxalatos, os quais também sofreram decomposição
quando submetidos à cromatografia em coluna2.
Como a tentativa de purificação do composto 7 não foi bem sucedida,
abandonou-se a idéia de purificação, preocupando-se em analisar a influência
das impurezas na ocasião dos estudos cinéticos futuros.

Resultados 61
3.1.1.1 O,O-hidrogênio monoperoxioxalato de p-metoxifenila (5)
A espectroscopia na freqüência de RMN de 13C constituiu a técnica mais
adequada para caracterizar os compostos 5 a 7, uma vez que os compostos
preparados, geralmente continham como impurezas o derivado do ácido oxálico
e o fenol correspondente. Portanto, técnicas como espectroscopia na região de
infravermelho ou espectrometria de massas não seriam satisfatórias para
identificar cada composto presente na mistura. As carbonilas, tanto do perácido
como do ácido, possuem deslocamentos químicos muito próximos, que variam
entre 154 a 158 8, o que nos levou a preparar um dos compostos com alta
pureza para que este fosse submetido à análise espectroscópica detalhada. °composto 5 foi escolhido como composto modelo, pois, em relação aos outros,
foi o que apresentou maior facilidade de manipulação. A análise foi iniciada com
a preparação e caracterização por RMN de 13C (-40 De, Acetona 0 6 , Varian
ORX500) do ácido p-metoxifeniloxálico (28), resultando nos seguintes
deslocamentos químicos:
o 2 0 -/\ 7H~oMe
HO 1 o 4 5 6
(C-1 )
158,9
(C-2) (C-3) (C-4) (C-S) (C-6)
158,6 144,6 122,9 115,4 158,1
(C-7)
55,9
Na região de carbonilas de éster e ácido foram observados três sinais,
dois deles foram atribuídos aos carbonos carbonílicos 1 e 2 (8 158,9 e 158,6
respectivamente) e o deslocamento observado em 8 158,1 foi atribuído ao
carbono aromático 6. A atribuição dos deslocamentos químicos do anel
aromático foi feita em analogia aos dados relatados na literatura para o ácido p
c1orofeniloxálico3 (Anexo 1).
A análise da mistura contendo o perácido foi feita nas mesmas condições
do ácido p-metoxifeniloxálico (28). Assim, no espectro do O,O-hidrogênio
monoperoxioxalato de p-metoxifenila (5) foi observado a presença do composto
28 como impureza, mas não a presença do p-metoxifenol, devido à ausência de

OH493 I5 :::::-..
6OMe
Resultados 62
sinais em 8 149,9 e 8 151,2 provenientes dos deslocamentos
químicos dos carbonos 3 e 6 respectivamente. A atribuição dos
sinais do perácido foi feita com base no composto O,O-hidrogênio
monoperoxioxalato de p-c1orofenila (8A)3, através da contribuição
dada pelas carbonilas dos grupos éster e peróxido ao anel aromático. Desta
forma também, foi possível prever o deslocamento químico do anel aromático
para cada mudança de substituinte (Anexo 1).
Tabela 8: Deslocamentos químicos observados no O,O-hidrogênio monoperoxioxalato de
p-metoxifenila (5) e ácido p-metoxifeniloxálico (28), juntamente com as intensidades relativas de
cada. Acetona Os, -40 °C, Varian DRX500.
OH Obs. Cal. OOH Obs. Cal.
1 158,1 (1,18) 1S8,Sa 1 154,8 (0,75) 154,6a
2 157,9 (0,91) 157,8a 2 155,9 (0,92) 156,3a
O 2 O~ ~OMe 3 144,1 (1,00) 143,2 3 143,8 (1,00) 142,9H 3 6 7 4 122,8 (2,18) 123,2 4 122,7 (3,00) 123,1
X 1 O 4 55 114,8 (2,16) 115,7 5 114,9 (3,00) 115,8
6 158,4 (1,27) 158,0 6 158,3 (1,42) 158,3
7 55,49 (1,09) 54 7 55,47 (1,09) 54
a Deslocamentos químicos obtidos da literatura3
Os sinais observados na análise por RMN de 13C foram atribuídos ao
compostos 5 e 28 . Na região de carbonila de éster foram observados sinais em
8 154,8 e 155,9 (perácido) e 158,1 e 157,9 (ácido). Os sinais em 8 158,3 e
158,4 foram atribuídos ao carbono 6 do anel aromático do perácido e do ácido,
respectivamente (Anexo 4). Os sinais atribuídos aos carbonos 7 do perácido (8
55,47) e do ácido (8 55,49), por apresentarem sinais muitos próximos, foram
diferenciados com base em simulação feita pelo programa MESTRE C 2.3.
A adição de KI para reduzir o perácido, resultou no completo
desaparecimento dos sinais em 8 154,8 e 155,9 atribuídas as carbonilas 1 e 2
do perácido. Deste modo, foi possível distinguir os deslocamentos químicos das
carbonilas do perácido das carbonilas do ácido (Anexo 6). Em 8 162,5 foi
observado um sinal que pode ser referente à formação do ácido oxálico, pois na

Resultados 63
redução do perácido é possível também a formação deste tipo de composto,
cujo sinal pode ser observado em aproximadamente 6 16011. Ademais, os sinais
em 6 114,9; 6 122,8 e 6 143,8 (região do anel aromático) diminuíram de
intensidade em relação aos sinais (6 114,8; 6122,7 e 6144,1) dos carbonos do
anel aromático proveniente do ácido 28. Ainda, na região de aromáticos foram
observados sinais em 6 114,7 e 122,6 que, a princípio, poderiam ser atribuídos
à formação do p-metoxifenol. Contudo, não foram observados sinais em 6
149,9, 6 115,2 e 6 151,2, os quais seriam os sinais aproximados para o p
metoxifenol. Foram também observados sinais de baixa intensidade em 618,5; 6
69,4; 6 125,2 e 6 137,0. Porém, de acordo com os deslocamentos esperados
para os principais produtos da reação, não existe proposta para tais
observações. Isto também vale para o sinal observado em 6157,3. Finalmente,
os experimentos de adição de KI confirmam claramente a atribuição dos sinais
de RMN de 13C feita para o perácido 5, pois todos os sinais atribuídos a este
composto sofreram alterações consideráveis. Os sinais atribuídos as carbonilas
desapareceram completamente e os sinais atribuídos aos carbonos do anel
aromático diminuíram de intensidade em relação ao anel aromático do ácido 28
(Anexo 6).

Resultados 64
3.1.1.2 O,O-hidrogênio monoperoxioxalato de p-metilfenila (6)
De acordo com o resultado da análise em RMN de 13C, a amostra
analisada era composta basicamente do perácido 6 e do p-metilfenol. O
derivado ácido, geralmente encontrado em concentração expressiva, estava
presente na mistura em apenas 1%. Portanto, os sinais relacionados ao ácido
apresentaram fraca intensidade. Os deslocamentos químicos relacionados ao
composto 6 estão apresentados na tabela 9.
Tabela 9: Deslocamentos químicos observados no O,O-hidrogênio monoperoxioxalato de
p-metilfenila (6), juntamente com as intensidades relativas. Acetona D6, -40 DC, Varian DRX500.
OOH Obs. Cal.
1 154,7 (1,08) 154,63
2 156,0 (1,04) 156,33
O 20-Q;-CH3 3 148,4 (1,00) 147,8H 3 6 7 4 121,6 (2,80) 121,9
HOO 1 O 4 55 130,7(3,20) 130,9
6 137,0(1,05) 135,9
7 20,6 (1,09) 20
3 Deslocamentos químicos obtidos da literaturaJ
Além destes deslocamentos químicos, foram observados
também sinais relacionados ao p-metilfenol, cujos deslocamentos e
intensidades relativas foram: 8 155,7 (1,01), 115,4 (2,03), 130,4
(2,05), 128,3 (1,00), 20,3 (1,03), para os carbonos 3, 4, 5, 6 e 7
OH
Q'-'::: 4
I ~ 5
CH3
respectivamente.
Conforme fora mencionado, o ácido p-metilfeniloxálico apresentou
deslocamentos químicos de baixa intensidade (Anexo 9), porém não foi
possível identificar os deslocamentos químicos provenientes das carbonilas,
apenas os carbonos do anel aromático, cujos sinais foram 8 149,2, 136,7, 121,5
e 20,5. Ademais, foi observada a presença de éter dietílico (8 66,1 e 15,4)
proveniente da preparação de 6, uma vez que este fora usado como solvente, e

PO·""1tados
um deslocamento em õ 125,3 que não pode ser atribuído a nenhum dos
compostos presentes na mistura.
3.1.1.3 O,O-hidrogênio monoperoxioxalato de fenila (7)
Na análise do O,O-hidrogênio monoperoxioxalato de fenila (7) por RMN
de 13C foram observadas as presenças de ácido feniloxálico e fenol, além da
presença do composto 7.
Tabela 10: Deslocamentos químicos observados no O,O-hidrogênio monoperoxioxalato de
fenila (7) e ácido feniloxálico, juntamente com as intensidades relativas de cada. Acetona 0 6,
30 De, Varian DRX500.
OH Obs. Cal. OOH Obs. Cal.
1 158,8 (0,92) 158,Sa 1 154,8 (0,98) 1S4,6a
O O~ )6 2 157,9 (0,89) 157,8a 2 156,1 (0,53) 156,3a
1H2 3 150,9 (1,00) 151,1 3 150,6 (1,00) 150,8X O 4 5
4 122,0 (2,11) 122,1 4 121,9 (2,26) 122,0
5 130,4 (1,92) 130,1 5 130,5 (2,74) 130,2
6 127,2 (0,92) 126,5 6 127,5 (1,02) 126,8
a Deslocamentos químicos obtidos da literatura3
Os deslocamentos químicos observados em õ 150,6, 122,0, 130,5 e
127,5 foram atribuídos aos carbonos 3, 4, 5 e 6 do anel aromático de 7
respectivamente. No mesmo espectro de RMN de 13C também havia um
composto que não pode ser identificado, cuja quantidade na mistura era o
equivalente a 10,3%, segundo a integração dos sinais. Em primeira análise,
nossa proposta estrutural seria o peranidrido 31; muito embora, nem todos os
sinais relativos a este composto estivessem presentes. Os sinais excedentes
foram: õ 117,4,121,4,123,9,128,7, 153,Oe 157, 03 (Anexos 11 e 12).
4' 5'
6~02 o o oD6'\J3 n ~2~
5 4 o 10-0 l' o
31

Resultados 66
De fato, considerando que, a estrutura do peranidrido 31 é semelhante
à do composto 7, esperar-se-ia que os deslocamentos químicos para ambos
compostos fossem também semelhantes. Outro fato que corrobora este
argumento está relacionado com as intensidades relativas dos sinais de 31, que
deveriam ser 1:1: 1:2:2: 1. Entretanto, as intensidades relativas observadas
foram 1:3,5:3:3,6:3,2:0,36, respectivamente. Sendo assim não há uma proposta
estrutural consistente para os sinais observados.

O,O-hidrogênio
Resultados 67
3.1.1.4 Tentativa de preparação do
monoperoxioxalato de p-nitrofenila (8)
Na tentativa de preparação deste composto foram encontradas várias
dificuldades. Primeiramente, o cloreto de p-nitrofeniloxalila (4) não pôde ser
destilado, visto que sublimava, cristalizando ao longo da aparelhagem de
destilação. Por outro lado, a tentativa de purificação por sublimação apresentou
baixo rendimento, devido ao controle da temperatura, sempre abaixo de 110 aC,
para que não ocorresse degradação do composto 4. Ademais, as técnicas
utilizadas para a caracterização, principalmente a análise elementar, não
revelaram a presença de 4, embora o teste qualitativo de quimiluminescência
fosse positivo. Como os processos de purificação não foram eficientes, o cloreto
de p-nitrofeniloxalila (4) foi utilizado sem purificação prévia na tentativa de
preparação do perácido 8. A reação do composto 4 com H20 2 efetuada na
. presença de piridina foi acompanhada por CCO, sem, contudo, detectar a
presença de um composto peroxídico. Além disso, durante a extração, com
uma solução aquosa saturada de NH4CI, um processo necessário para retirar o
excesso de cloreto de piridínio e H202, observou-se uma coloração amarela.
Este fato indica que talvez tenha ocorrido a formação de p-nitrofenol, devido à
hidrólise do composto 8, eventualmente formado em pequenas quantidades, do
composto 4 ou proveniente da formação do ácido diperoxálico (22) (Esquema
15). Este último pode ser formado pela adição do peróxido de hidrogênio à
carbonila do éster. A formação de carga positiva no oxigênio ligado ao anel
aromático, devido ao efeito atraente de elétron do grupo nitro na posição para,
resultaria no aumento da reatividade da carbonila, ligada neste oxigênio, frente
ao ataque nucleofílico do peróxido de hidrogênio. Ademais o peróxido de
hidrogênio encontra-se em excesso estequiométrico de dez vezes.

Resultados 68
Esquema 15
OH
-O- O OOOH9O O ~ /; NOz N .. M +:::",. IM H,02 HOO O
CI 4 O 22 NOz
Tendo isto em vista, foi testada uma outra abordagem envolvendo a
reação de bis(4-nitrofenil) oxalato (23) com peróxido de hidrogênio na presença
de piridina visando a obtenção do composto 8 (Esquema 16). Este método, à
princípio, apresentava vantagens em relação ao método utilizado na obtenção
dos outros perácidos pois evitava a preparação do cloreto de p-nitrofeniloxalila
(5) bem como as dificuldades decorrentes de sua purificação. Neste caso, o
peróxido de hidrogênio foi usado como reagente limitante (relação molar
23:H20 2de 1:0,5) para evitar uma suposta formação de ácido diperoxálico (22).
Após 35 minutos de reação a -40°C sob argônio, detectou-se uma substância
peroxídica com Rf = 0,65 quando revelada com solução aquosa de KI 10%
(placa de Si02, CH2CI2/HexlACN 5:2:3, -20°C), que, à princípio, poderia ser o
perácido 8. Entretanto, após a lavagem com uma solução saturada e gelada de
cloreto de amônio não se observou mais a presença da substância peroxídica.
Na etapa de evaporação do solvente observou-se a formação de alguns cristais
brancos, que, de acordo com o teste de KI, não eram peroxídicos. Estes cristais
também não eram do ácido correspondente, cuja identificação foi feita com
alaranjado de meti Ia. O sobrenadante proveniente da formação dos cristais
também não continha substância peroxídica. Deste modo, cessou-se as
tentativas de preparar o composto 8.
Esquema 16
O 0-o-NOz O -OzN--O-~oMo .. o\\__l-o-NOz
_ 23 H,02 ~HOO O8

Resultados 69
3.2 Estudo das propriedades de quimiluminescência dos
intermediários 5-7
As propriedades de quimiluminescência da cada perácido foram
estudadas pela reação destes compostos (5-7), catalisados por imidazol na
presença de 9,1 O-difenilantraceno (DPA) em acetato de etila anidro a 25°C
(Esquema 17). A emissão de luz proveniente desta reação apresentou um
rápido aumento da intensidade de emissão seguida de um decaimento lento,
sendo que esta intensidade variou de acordo com a natureza do substituinte no
anel aromático.
Esquema 17
O OOH
x-o-oHox =OCH3 (5)
X =CH3 (6)X =H (7)
N~~N-H
•
{}o"l
O 1_"l ~-ú
~ ú
OH9 + 2CO, + hv
X
o aumento da concentração de imidazol resultou em um aumento da
constante de velocidade de decaimento (Figura 5). A curva experimental foi
ajustada com uma função de decaimento mono-exponencial de primeira ordem
(Equação 1), sendo a constante de velocidade observada (kobs) relacionada
com o consumo de cada perácido. É importante frisar que o mesmo
comportamento cinético foi observado para todos os perácidos 5-7.

Resultados 70
--[IMI-H]=O,3mM (1)--[IMI-H]=O,5mM (2)- [IMI-H]=1 ,OmM (3)--[IMI-H]=2,OmM (4)
2000 4000 6000Tempo (8)
6 --[IMI-H]=6mM (5)
--:,-.... -- [IMI-H]=3OmM (6)[IMI-H]=100mM (7) 00
<Jl UU -- [IMI-H]=250mM (8) '-"
'-" '"'" oo .......-. ......~
~
Q)Q)
-g -g"'Cl :g.~ 2 00
s:: 2Q)
~......s ......
O" , . , . ,
O 400 800 1200 O
Tempo (8)O
Figura 5: Emissão de luz proveniente da reação de O,O-hidrogênio monoperoxioxalato de
p-metoxifenila (5) em várias concentrações de imidazol na presença de 9,10-difenilantraceno
(OPA). Espectrofluorímetro nas seguintes condições: fenda 1mm, tensão na
fotomultiplicadora=750V e monocromador na condição de espelho.
f(t) =A *exp(-kobs.t) Equação 5
Assim, ao estudar como variava a intensidade de emissão de luz em
função da concentração de imidazol para os perácidos (5-7), observamos que
tal intensidade diminuía com o aumento da concentração de imidazol. Cabe
ressaltar que comportamento semelhante também foi observado na reação do
sistema peroxioxalat04 e no traoalho de Stevani ei at.
A seguir estão relacionadas tabelas com os valores das constantes de
velocidade observadas (kobs) , juntamente com os rendimentos quânticos
singlete (<1>5) obtidos no estudo de cada perácido. Observa-se que ocorre um
aumento na constante de velocidade (kobs) em função do aumento da
concentração de imidazol, para os três perácidos, de aproximadamente 60
vezes, no caso dos perácidos 5 e 6, e de aproximadamente 120 vezes no caso
do perácido 7. Por outro lado, observa-se também uma diminuição drástica do
rendimento quântico singlete (<1>5).
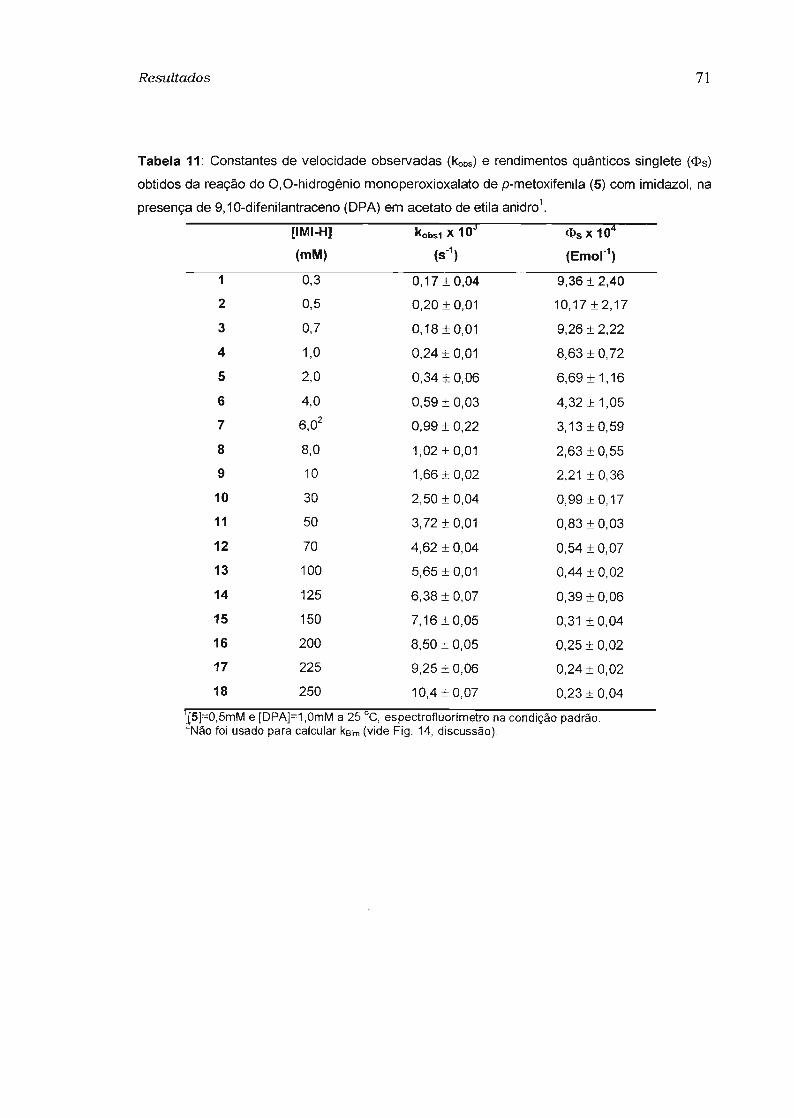
Resultados 71
Tabela 11: Constantes de velocidade observadas (kobs) e rendimentos quânticos singlete (<Ds)
obtidos da reação do O,O-hidrogênio monoperoxioxalato de p-metoxifenila (5) com imidazol, na
presença de 9,1 O-difenilantraceno (OPA) em acetato de etila anidro1.
[IMI-H] kobs1 X 103<Ds X 104
(mM) (S-1) (Emor1)
1 0,3 0,17 ± 0,04 9,36 ±2,40
2 0,5 0,20 ± 0,01 10,17 ± 2,17
3 0,7 0,18±0,01 9,26 ±2,22
4 1,0 0,24 ± 0,01 8,63 ± 0,72
5 2,0 0,34 ± 0,06 6,69 ± 1,16
6 4,0 0,59 ± 0,03 4,32 ± 1,05
7 6,020,99 ± 0,22 3,13±0,59
8 8,0 1,02 ± 0,01 2,63 ± 0,55
9 10 1,66 ± 0,02 2,21 ± 0,36
10 30 2,50 ± 0,04 0,99±O,17
11 50 3,72 ± 0,01 0,83 ± 0,03
12 70 4,62 ± 0,04 0,54 ± 0,07
13 100 5,65 ± 0,01 0,44 ± 0,02
14 125 6,38 ± 0,07 0,39 ± 0,06
15 150 7,16 ± 0,05 0,31 ± 0,04
16 200 8,50 ± 0,05 0,25 ± 0,02
17 225 9,25 ± 0,06 0,24 ± 0,02
18 250 10,4 ± 0,07 0,23 ± 0,04
)5]=o,5mM e [OP-A]=1,OmM a 2K"c, espectrofluorímetro na condição padrão.Não foi usado para calcular kBim (vide Fig. 14, discussão).

Resultados 72
Tabela 12: Constantes de velocidade observadas (kobS) e do rendimento quântico singlete (<1>s)
obtidos da reação do O,O-hidrogênio monoperoxioxalato de p-metilfenila (6) catalisada por
imidazol na presença de 9,1 O-difenilantraceno (OPA), utilizando acetato de etila anidro1.
[IMI-H]mM kobs X 103 5-' <1>s x 104 Emor1
1 0,5 0,19 ± 0,01 45,6 ±6,3
2 1,0 0,41 ± 0,12 27,4 ± 0,1
3 2,0 0,63 ± 0,10 17,2 ± 6,4
4 4,0 1,04 ± 0,06 10,8 ± 2,6
5 6,0 1,43 ± 0,11 7,71 ± 1,88
6 8,0 1,74±0,11 6,39 ± 1,81
7 10 1,92 ± 0,06 4,82 ± 0,71
B 20 2,86 ± 0,04 3,09 ± 0,68
9 40 4,05 ± 0,12 1,69 ± 0,06
10 60 5,11 ± 0,29 1,38 ± 0,02
11 80 6,13±0,83 0,91±0,13
12 100 6,14 ± 0,08 0,98 ± 0,21
13 120 7,04 ± 0,31 0,73 ± 0,04
14 130 7,37 ± 0,42 0,73 ± 0,06
15 150 8,31 ± 0,13 0,64 ± 0,04
16 170 8,95 ± 0,68 0,56 ± 0,01
17 200 10,20 ± 0,21 0,50 ± 0,02
18 230 11,80 ± 0,07 0,46 ± 0,01
19 250 12,7 ± 0,6 0,45 ±0,01
'[6]=0, 1mM, [OPA]=1,OmM a 25uC, espectrofluorímetro: fenda-8mm.
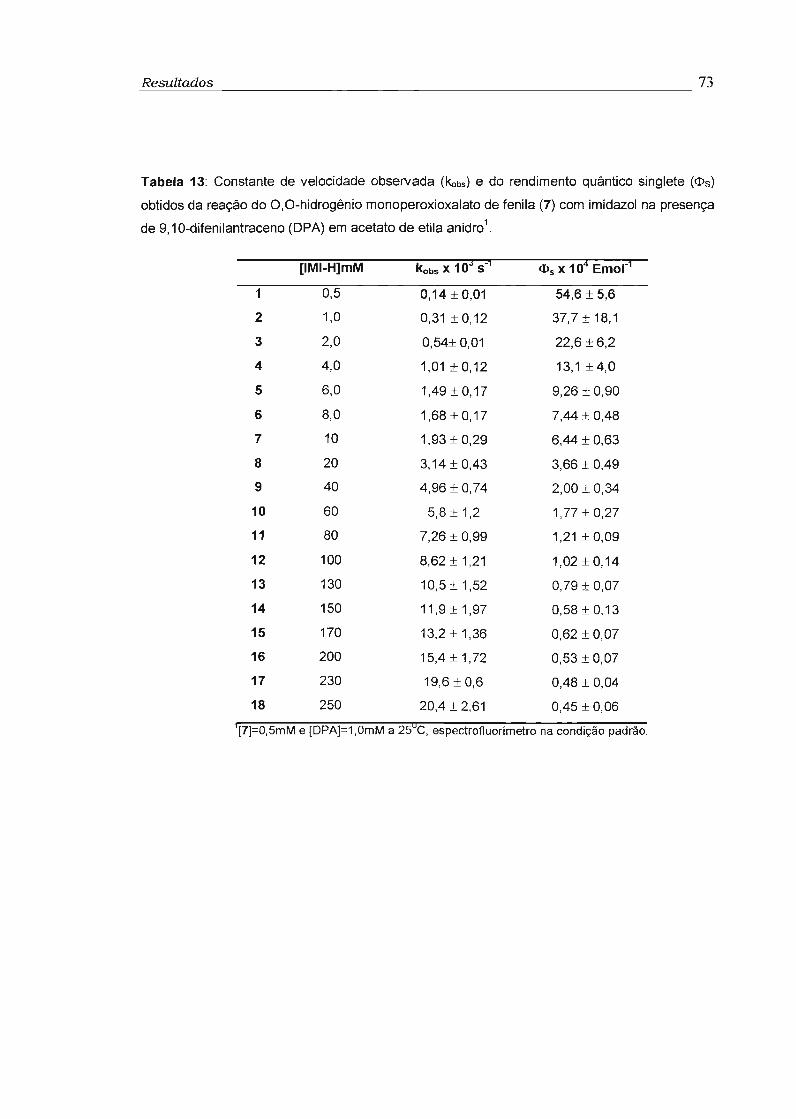
Resultados 73
Tabela 13: Constante de velocidade observada (kabS) e do rendimento quântico singlete (<I>s)
obtidos da reação do O,O-hidrogênio monoperoxioxalato de fenila (7) com imidazol na presença
de 9,1 O-difenilantraceno (OPA) em acetato de etila anidro1.
[IMI-H]mM kobs X 103 5-' <1>s x 104 Emor'
1 0,5 0,14 ± 0,01 54,6 ± 5,6
2 1,0 0,31 ± 0,12 37,7 ± 18,1
3 2,0 0,54± 0,01 22,6 ±6,2
4 4,0 1,01 ±0,12 13,1 ±4,O
5 6,0 1,49 ± 0,17 9,26 ± 0,90
6 8,0 1,68 ± 0,17 7,44 ± 0,48
7 10 1,93 ± 0,29 6,44 ±0,63
8 20 3,14 ± 0,43 3,66 ± 0,49
9 40 4,96 ± 0,74 2,00 ± 0,34
10 60 5,8 ± 1,2 1,77 ± 0,27
11 80 7,26 ± 0,99 1,21 ± 0,09
12 100 8,62 ± 1,21 1,02 ± 0,14
13 130 10,5±1,52 0,79 ± 0,07
14 150 11,9 ± 1,97 0,58±0,13
15 170 13,2±1,36 0,62 ± 0,07
16 200 15,4 ± 1,72 0,53 ± 0,07
17 230 19,6 ± 0,6 0,48 ± 0,04
18 250 20,4 ± 2,61 0,45 ± 0,06
'[7]=O,5mM e [DPA]-1,OmM a 25°C, espectrofluorímetro na condição padrão.

Resultados 74
3.3 Estudo da eficiência de transferência de elétron na etapa
de quimiexcitação.
3.3.1 Determinação do rendimento quântico singlete
Conforme foi apresentado na introdução deste trabalho, na etapa de
quimiexcitação ocorre interação do intermediário de alta energia (IAE) com o
ativador, resultando na excitação eletrônica deste último. Para que um
determinado composto possa atuar eficientemente como ativador é necessário
que este tenha um alto rendimento de fluorescência (<1>s), baixo potencial de
oxidação e que seja estável nas condições de reação.
Os resultados apresentados neste tópico estão relacionados com a
reação de bis(2,4,6-triclorofenil) oxalato (TCPO) com peróxido de hidrogênio
catalisado por· imidazol na presença de 3-(2-aril-5,5-dimetiloxazol-4
ilideno)androsta-1,4-dien-17-onas (9-13), denominados de ativadores (ACT),
conforme exemplificado no esquema 18.
Esquema 18
ACro
~hVX: 4-CI (11)X: 4-F (12)X: 4-N02 (13)
X: 4-0CH3 (9)X: 3-0CH3 (10)
X~No
I ACTI
CI
CI ~o~_l-h-cl~o or
CI TCPO
N~hdN-H \ H20 2
o Ot-f_/O-O
IAE
A reação apresentou um rápido aumento na intensidade de emissão de
luz seguida de um decaimento lento (Figura 6). A cinética foi monitorada por um
fluorímetro, registrando intensidade vs tempo e os experimentos realizados a
25°C. Da curva cinética foram obtidas duas constantes de velocidade,
denominadas de kobs1 e kobs2 através da função abaixo (Equação 6):

Resultados 75
f(/) = AI exp(-kobsl/) - A2 exp(-kobs2 /) Equação 6
,..-....-'~ 75
-v'-.-/o-
><: 50
~.....~ 25Cl)
~o
.~3
,~:PkObs1 (1) [ACT]=O,5mM
(2) [ACT]=O,1mM(3) [ACT]=O,025mM
o 100 200 300 400 500Tempo (s)
Figura 6: Emissão de luz proveniente da reação de bis(2,4,6-triclorofenil) oxalato (TCPO) com
peróxido de hidrogênio catalisado por imidazol na presença de 3-[2-(4-f1uorofenil)-5,5
dimetiloxazol-4-i1ideno]androsta-1,4-dien-17-ona (12) em acetato de etila anidro, nas seguintes
condições: [TCPO]=O, 1mM, [IMI-H]=1,OmM, [H202]=10mM e [12]=0,5mM, 0, 1mM e 0,025mM.
Com os resultados apresentados neste tópico foram obtidas informações
sobre o mecanismo CIEEL, utilizado para explicar a geração de estados
excitados em reações com propriedades de quimiluminescência. Determinou-se
o rendimento quântico singlete (<1>5)' de cada ativador, utilizando a seguinte
equação:
Área.fcF ·fsF<1> S = n.<1> F
Equação 7
onde: Área = área obtida abaixo da curva de emissão, fCF = fator de calibração da
fotomultiplícadora, fSF = fator de sensibilidade da fotomultiplicadora, n = número de mols do
TCPO e <I>F = rendimento quãntico de fluorescência do ativador.

Resultados 76
o fator de calibração da fotomultiplicadora foi obtido utilizando a reação
de luminol como padrão, para que fosse possível transformar a unidade de
contagens por segundo em Einstein por moi, uma unidade de fótons de luz. O
método é relativamente simples, sendo a calibração realizada a cada nova série
de experimentos e os valores obtidos tendo variações mínimas entre (5-6).10-17
E counts-1. Outro fator igualmente importante é o fator de sensibilidade da
fotomultiplicadora com o comprimento de onda (fs) (Tabela 14). Para obter este
fator para a emissão de quimiluminescência basta registrar um espectro de
fluorescência sem correção e outro espectro com correção (geralmente está
correção é feita pela solução de rodamina B presente nos aparelhos de
espectrofluorímetria) e dividir a área abaixo da curva do espectro corrigido pelo
espectro sem correção.
Tabela 14: Rendimento quântico de fluorescência (<1>5) e fator de sensibilidade da
fotomultiplicadora (fs) para os ativadores 9-13.
ACT <1>F! fs
9 4-0CH3 0,65 1,75
10 3-0CH3 0,78 1,65
11 4-CI 0,80 1,72
12 4-F 0,84 1,87
13 4-NOz 0,046 6,04
Além dos dois fatores mencionados, foi necessário determinar o
rendimento de fluorescência de cada ativador. Estas medidas foram feitas em
acetato de etila, utilizando 9-cianoantraceno como padrão secundário em
etanol6 , utilizando-se comprimento de onda de excitação em 330 nm e absorção
neste mesmo comprimento de onda de aproximadamente 0,05.

Resultados 77
Área(padrão)
Área(ACT)
_ <1> F(padrão) Abs(padrão) (n(EtOH) J2- X X Equação 8
<1> F(ACT) Abs(ACT) n(EtAc)
onde: Área(padrão) =área abaixo da curva de emissão do padrão, Área(ACn =área abaixo da curva
de emissão do ativador (ACT), neste caso os compostos 9 a 13, <1>F(padrão) =rendimento quântico
de fluorescência do padrão, <1>F(ACn =rendimento quântico de fluorescência do ACT, Abs(padrão) =absorção do padrão no Àex, Abs(Acn =absorção do ACT, n(EtOH) =índice de refração do solvente
utilizado no padrão; neste caso, etanol e n(EtAc) = índice de refração do acetato de etila utilizados
nos ativadores.

BIBLIOTEC-AResultados INSTITUTO DE auíMlc)L 78
UnIversidade de São Pau10
3.3.2 Rendimento quântico singlete na concentração infinita (<1>500)
o mecanismo CIEEL é iniciado pela transferência de elétron do ativador
para a 1,2-dioxetanodiona (kcat), a qual é a etapa limitante para a excitação
eletrônica em concorrência com a etapa que leva à decomposição escura (ko)
(Esquema 19).
Esquema 19
o OH +o-o
kcatACT
_ hv
1kD
sem emissão
A correlação entre o <I>s e a concentração do ativador pode ser deduzida
diretamente de um mecanismo simplificado (Esquema 20)
Esquema 20
2ArOH
kcat
o OIMI-H ~
.. .. .. I I +O-O
24
24 + ACT ... produtos + ACT*<I> co
5
ko24 .. produtos
kF1
ACT* ... ACT + hv<I>FI
TCPO + H20 2
Como os tempos de vida de 24 e do ACT* são muito curtos, é permitido
assumir a condição de aproximação do estado estacionário para estas duas
espécies, resultando na Equação 51.

Resultados 79
1 1 ( kD J 1-=-+ -<1>s <1>~ kCAT<1>~ (ACT] Equação 9
Com os valores do rendimento quântico singlete (<1>8), obtido em várias
concentrações de cada ativador (Anexo 2), foi possível construir um gráfico do
inverso do <1>s em função do inverso da concentração do ativador (1/<1>8 VS.
1/[ACT]), correlacionando o <1>8 e a concentração do ativador através da
equação 5, para a interação do ativador com o IAE (24).
4010 20 301/[ACT] (mM- I
)
R = 4-Oct-\ (Q), 3-OCH] (10).4-Cl (11), U (12), 4-N01 (13)
,~:~
O' , , . , , , ' , ,O
60
15
~
I
O 45E~'-'(/)30~-
Figura 6: Duplo recíproco do rendimento quãntico singlete (<1>s) em função da concentração dos
ativadores (9-13).
Do intercepto desta relação foi obtido o rendimento quântico na
concentração infinita (<1>800). Nesta condição admite-se que todo o intermediário
formado na etapa de quimiexcitação é interceptado pelo ativador, resultando na
eficiência máxima de emissão de luz. Além disso, pode-se obter da inclinação

Resultados 80
da reta o valor da relação kcat/ko, (1/cDoo8, onde 8 é o coeficiente angular da
relação 1/cDcL vs. 1/[ACT]) onde kcat é a constante de velocidade da interação do
ativador com o intermediário de alta energia (IAE), enquanto que o ko é a
constante de velocidade de decomposição do IAE e, portanto, independe do
ativador. Esta é uma medida indireta, pois kcat não pode ser medido diretamente
devido ao fato da etapa de interação do IAE com o ACT ser muito mais rápida
do que as etapas que levam a formação do IAE.
Os parâmetros de cDs, relação kcat/ko, potencial de oxidação de meio pico
(EOXp/2), assim como os rendimentos de fluorescência dos ativadores estão
relacionados na tabela 15.
Tabela 15: Parâmetros de quimiluminescência da reação de bis(2,4,6-triclofenil) oxalato com
peróxido de hidrogênio catalisado por imidazol na presença de 3-[2-(aril)-5,5-dimetiloxazol-4
ilideno]androsta-1 ,4-dien-17-onas 9-13.
EOX (a) cD; X 102kcAT/ko X 10-3pl2
Ativator <l>FL(mor1L)(VVS. SHE) (Emor1
)
9 4-0CH3 0,95 0,65 6,0 ± 0.2 13 ±4
10 3-0CH3 1,02 0,78 11 ± 2 8±1
11 4-CI 1,02 0,80 20 ±6 6±2
12 4-F 1,03 0,84 16 ± 1 5,0 ± 0,3
13 4-N02 1,09 0,046 75 ± 10 2,4 ± 0,4
a acetonitrila, SHE eletrodo padrão de hidrogênio
A variação no EOXp/2 é pequena com a mudança do substituinte;
entretanto, observa-se que a variação, do rendimento quântico singlete na
concentração infinita do ACT (cDsoo), é bastante pronunciada quando se compara
o derivado 4-N02 (13) com o 4-0CH3 (9). Além disso, observa-se também que
ocorre uma diminuição na relação kcat/ko em relação ao aumento do EOXp/2. É
importante mencionar que kobs1 e kobs2, obtidas das curvas cinéticas (Figura 6)
não dependem da natureza e concentração do ativador, indicando que a
interação do IAE com o ACT ocorre em etapas rápidas.

Resultados 81
3.4 Influência da viscosidade no rendimento quântico singlete
(rJJJ
De acordo com mecanismo postulado para a formação de estados
excitados na etapa de quimiexcitação do sistema peroxioxalato, as etapas de
transferência e retro-transferência de elétron entre o IAE e o ativador,
ocorreriam dentro de uma gaiola de solvente. Admitindo-se este pressuposto, já
que esta etapa depende da formação de um complexo de transferência de
carga, a polaridade e viscosidade do solvente influenciariam na eficiência de
formação de estados excitados. Supõe-se que em solventes de alta polaridade
ocorreria a solvatação das espécies, dificultando a aproximação necessária
para gerar o ativador no estado excitado. Por outro lado, em solventes com
baixa polaridade, não seria criada a condição de solvatação em volta das
espécies. Deste modo, também não haveria o processo de transferência e
retro-transferência de elétron.
Os experimentos apresentados neste tópico têm como objetivo verificar a
eficiência da suposta gaiola de solvente na troca de elétron que ocorre na etapa
de quimiexcitação. Para este propósito, foram utilizadas misturas de tolueno e
difenilmetano em várias proporções, resultando em um aumento da intensidade
de emissão com aumento da viscosidade (Figura 7). Da curva cinética foram
extraídas duas constantes de velocidade utilizando a equação 6.
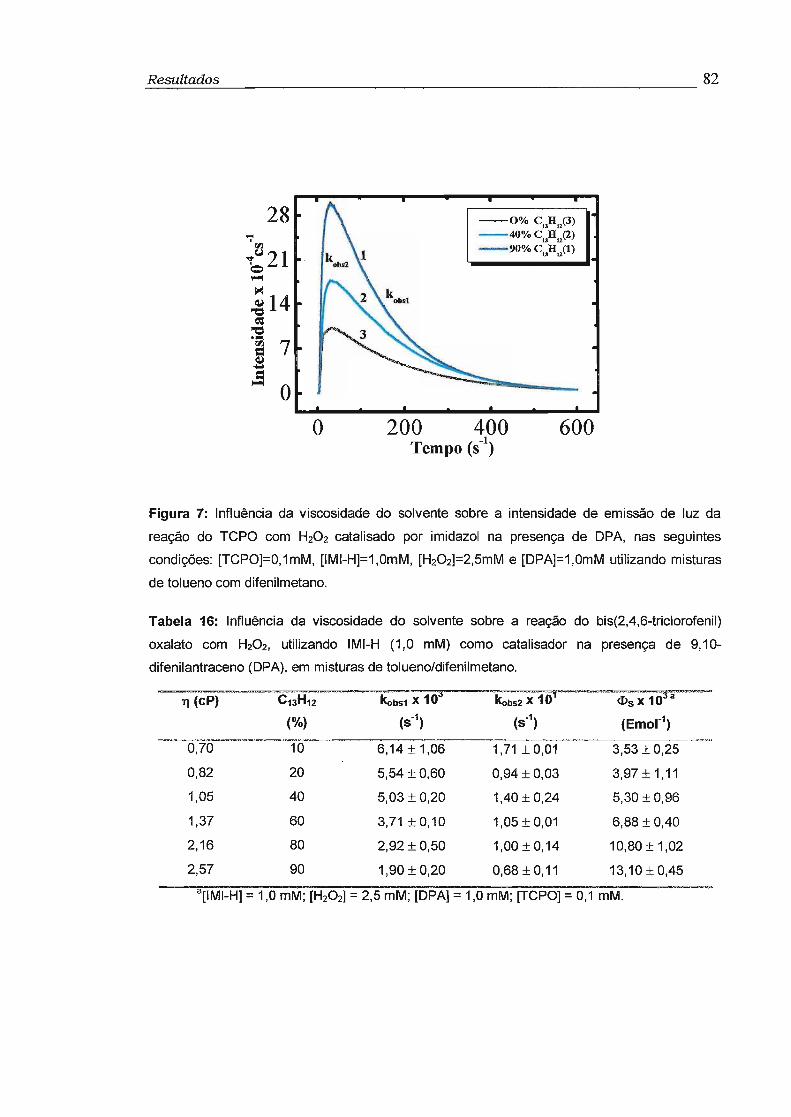
Resultados 82
--0% C13
H u(3)--40% C
13H
u(2)
--90% C13
Hu
(1)
600200 400Tempo (S-1)
o
28'";'
rLJ
~21-··~
~
~14~
"O....~~
~
Figura 7: Influência da viscosidade do solvente sobre a intensidade de emissão de luz da
reação do TCPO com H202 catalisado por imidazol na presença de OPA, nas seguintes
condições: [TCPO]=0,1mM, [IMI-H]=1,OmM, [H20 2]=2,5mM e [OPA]=1,OmM utilizando misturas
de tolueno com difenilmetano.
Tabela 16: Influência da viscosidade do solvente sobre a reação do bis(2,4,6-triclorofenil)
oxalato com H20 2, utilizando IMI-H (1,0 mM) como catalisador na presença de 9,10
difenilantraceno (OPA), em misturas de tolueno/difenilmetano.
TI (cP) C13H12 k~~1 i 103kobs2 X 10' •. <l>s x 103'"a
(%) (5-1) (5-
1) (Emor1)
0,70 10 6,14±1,06 1,71±0,01 3,53±0,25
0,82 20 5,54 ± 0,60 0,94 ± 0,03 3,97 ± 1,11
1,05 40 5,03 ± 0,20 1,40 ± 0,24 5,30 ± 0,96
1,37 60 3,71 ±0,10 1,05±0,01 6,88±0,40
2,16 80 2,92±O,50 1,OO±0,14 10,80±1,02
2,57 90 1,90±0,20 0,68±0,11 13,10±0,45
a[IMI_H] = 1,0 mM;[H20 2] = 2,5 mM; [ÕPAj = 1,0 mM; [TCPO] = 0,1 mM.
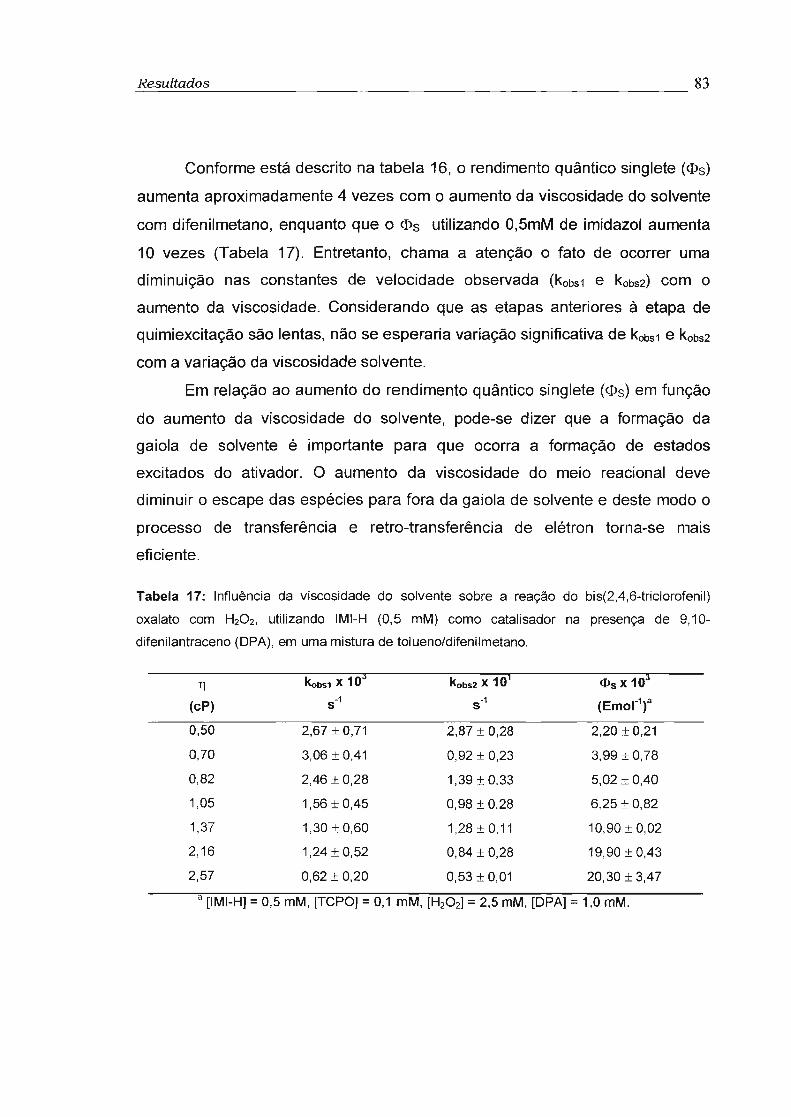
Resultados 83
Conforme está descrito na tabela 16, o rendimento quântico singlete (<1>s)
aumenta aproximadamente 4 vezes com o aumento da viscosidade do solvente
com difenilmetano, enquanto que o <1>s utilizando 0,5mM de imidazol aumenta
10 vezes (Tabela 17). Entretanto, chama a atenção o fato de ocorrer uma
diminuição nas constantes de velocidade observada (kobs1 e kobs2) com o
aumento da viscosidade. Considerando que as etapas anteriores à etapa de
quimiexcitação são lentas, não se esperaria variação significativa de kobs1 e kobs2
com a variação da viscosidade solvente.
Em relação ao aumento do rendimento quântico singlete (<1>s) em função
do aumento da viscosidade do solvente, pode-se dizer que a formação da
gaiola de solvente é importante para que ocorra a formação de estados
excitados do ativador. O aumento da viscosidade do meio reacional deve
diminuir o escape das espécies para fora da gaiola de solvente e deste modo o
processo de transferência e retro-transferência de elétron torna-se mais
eficiente.
Tabela 17: Influência da viscosidade do solvente sobre a reação do bis(2,4,6-triclorofenil)
oxalato com H20 2 , utilizando IMI-H (0,5 mM) como catalisador na presença de 9,10
difenilantraceno (OPA), em uma mistura de tolueno/difenilmetano.
TI kobs1 X 103
kobs2 X 10' <Ds X 103
(cP) S·1 S-1 (Emor1)a
0,50 2,67 ± 0,71 2,87 ± 0,28 2,20 ± 0,21
0,70 3,06 ± 0,41 0,92 ± 0,23 3,99 ± 0,78
0,82 2,46 ± 0,28 1,39 ± 0,33 5,02 ± 0,40
1,05 1,56 ± 0,45 0,98 ± 0,28 6,25 ± 0,82
1,37 1,30±O,60 1,28±O,11 10,90±0,02
2,16 1,24±0,52 0,84±0,28 19,90±0,43
2,57 0,62 ± 0,20 0,53 ± 0,01 20,30 ± 3,47
a [IMI-H] =0,5 mM, [TCPO] =0,1 mM, [H20 2] =2,5 mM, [OPA] =1,0 mM.

BIBLIOTECAResultados iNSTITUTO DE QUíMICA 84
Universidade de São Paulo
o fato do <1>s ser maior quando se utiliza 0,5 mM de imidazol em relação
à concentração de 1,OmM, não surpreende, pois já foi observado em trabalhos
anteriores do mesmo grupo em que o presente trabalho foi desenvolvido, que o
aumento da concentração de imidazol diminui o rendimento quântico singlete
(<1>s) 1.

Resultados 85
3.5 Estudo do Sistema Peroxioxalato na ausência de H20 2
Recentemente fora reportado na literatura um trabalho sobre a emissão de
luz do sistema peroxioxalato sem a adição de peróxido de hidrogênio? A reação
foi executada com TCPO ou ONPO catalisada por imidazol na presença de
perileno. Segundo estes resultados?, o intermediário de alta energia (IAE) é
proveniente de um derivado oxálico formado antes da 1,1'-di-imidazolida (001).
Vale lembrar que o 001 é formado pela substituição nucleofílica de dois
imidazóis no TCPO. Os autores não apresentaram qual seria a eficiência de
formação de estados excitados na reação sem a adição de peróxido de
hidrogênio, bem como as intensidades relativas na ausência e presença de
HzOz.
O estudo descrito no presente trabalho, relata a reação com TCPO nas
condições padrão, conforme descrito na legenda, cuja concentração de
peróxido de hidrogênio é 10mM (figura 9-curva A). A curva de emissão
apresentada na curva B é proveniente da reação com ONPO. Pode ser
observada que a intensidade de emissão do DNPO é dez vezes maior do que
com o TCPO nas mesmas condições. Por outro lado, diminuindo a
concentração do HzOz em 100 vezes, na reação de ONPO, observa-se que a
intensidade de emissão diminui em torno de 160000 vezes (curva C). Não foi
possível reduzir, ainda mais, a concentração de peróxido de hidrogênio, pois
abaixo de 0,5 IlM de HzOz a intensidade de emissão de luz era muito próxima
do limite de detecção do equipamento (aproximadamente 300 counts). Os
experimentos foram executados em um espectrofluorímetro do tipo Spex
Fluorolog 1681.
Como a emissão de luz abaixo de 0,5 IlM era pouco intensa, os
experimentos foram continuados em um "Photon Counter", aparelho mais
adequado para registrar sistemas de quimiluminescência com fraca intensidade
de emissão de luz.

Resultados 86
40
@)
•
~.,. 12 _
cn I e.
~ I· \8 8 .~b ~- ..~ 4;· \OI •..., ..- .5 ........ ..IE O •
o 20Tempo(s)
';;;' 0.16C::8 0.12~~
O-; 0.08<l.)
"âl:::! 0.04
5 l ·.5 0.00 •
600
1.2fi)
fi)-c=8 0.8~~
~
~
~ 0.4"O.;;=~ O.O~ • ~~
O 200 400Tempo (s)
o 17 34[H,O,j).l.M
51
Figura 8: Emissão de luz proveniente da reação do TCPO com HzOz catalisada por imidazol na
presença de perileno, nas seguintes condições: [IMI-H]=1,0 mM, [TCPO]=0,1 mM,
[perileno]=0,5mM e [H20 Z]=10mM (A), curva B nas mesmas condições de A, exceto o TCPO
que foi substituído por DNPO na mesma concentração e (C) variação da concentração de HzOz
em 100 vezes, nas seguintes condições de reação: [DNPO]=0,2mM, [IMI-H]=2,OmM,
[perileno]=0,05mM e [H202]=0,5f.1M a 50f.1M.
Após uma série de experimentos variando a concentração de peróxido de
hidrogênio entre 5.10-7 e 5.10-10 observou-se que a variação nas intensidades
era muito baixa. Além disso, foi atentado ao cuidado na manipulação dos
reagentes e da vidraria utilizada, pois foi observado que traços de impurezas
também geravam emissão de luz nas condições investigadas. As reações de
TCPO e DNPO sem adição de IMI-H e H20 2 apresentaram intensidade de
emissão de luz de aproximadamente 10000 counts S-1, considerando que a
intensidade da linha base era de 60 counts S-1, a intensidade observada na
reação sem catalisador e peróxido é significativa (Figura 9).

Resultados 87
"
la '''- i:-TCPO (1), l -ONPO(2)f ~
ç 8......-'1 2'-'
\";'o '\.
- 6 ~,-~
~"
V) ~..l.~--..§ 4 ~.~-""~~
oU
2l , :: ; ; ;' ; : .. -, ,
O 100 200 300 400 500 600tempo(s)
Figura 9: Emissão de luz proveniente da reação de bis(2,4,6-triclorofenil) oxalato (TCPO) e
bis(3,S-dinitrofenil) oxalato (DNPO) com perileno, na ausência de imidazol e H20 2, nas
seguintes condições: [TCPO] e [DNPO]=O,2mM, [IMI-H]=2mM,
Qualitativamente, as reações do TCPO e DNPO, com IMI-H e perileno,
sem adição e com adição de H20 2 (5,10-10 M) apresentaram intensidades de
emissão muito semelhantes (Figura 10). A diferença nas intensidades de
emissão é de aproximadamente quatro vezes no caso da reação do DNPO e
aproximadamente 3 vezes no caso da reação do TCPO. Supondo, que a
emissão de luz observada sem a adição de peróxido, seja causada pela
presença de H202 no solvente como impureza, esta concentração está abaixo
de 5.10-10 M.
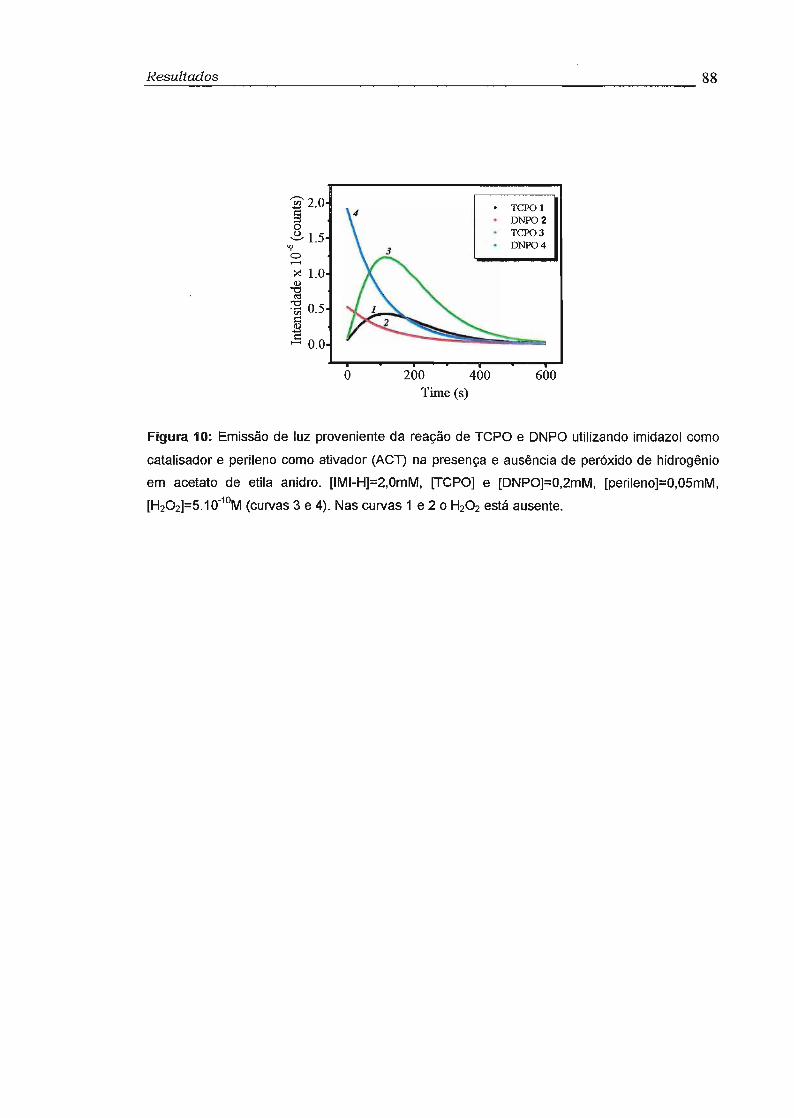
Resultados 88
S2.0g~ 1.5
'9o......~ 1.0
.g~ 0.5
~.s 0.0
O 200 400Time (8)
• TCPO 1• DNP02
TCP03• DNP04
600
Figura 10: Emissão de luz proveniente da reação de TCPO e DNPO utilizando imidazol como
catalisador e perileno como ativador (ACT) na presença e ausência de peróxido de hidrogênio
em acetato de etila anidro. [IMI-H]=2,OmM, (TCPO] e [DNPO]=O,2mM, [perileno]=O,OSmM,
[H202]=S.10·10M (curvas 3 e 4). Nas curvas 1 e 2 o H20 2 está ausente.

Resultados 89
3.6 Síntese de derivados de Oxazolonas (14-21)
o trabalho apresentado nesta seção teve como objetivo preparar
compostos com propriedades físico-químicas e foto-físicas distintas dos
compostos geralmente aplicados no sistema peroxioxalato, porém análogos aos
compostos 9 a 13. Além disso, um dos objetivos iniciais do projeto, era
converter estes compostos em reagentes de derivatização para aplicá-los em
sistemas biológicos e analíticos utilizando o sistema peroxioxalato. Esta parte
do trabalho foi desenvolvida durante três meses na universidade Friedrich
Schiller em Jena na Alemanha e financiado pelo DAAD (Intercâmbio acadêmico
Brasil-Alemanha).
3.6.1 2-[4-fenilacetileno]-5-dimetiloxazol-4-ona (15)
Esquema 21
N °Br---Q\ 1'1'-
14 OI(Ph3P)2PdCI/Cul
toluol/(C2HshN • <) <) ~N'1O
~H 15 0-r--
o composto 15 foi preparado pela reação de 14 com fenilacetileno
catalisado por bis(trifenilfosfina) paládio (11) em meio de tolueno e trietilamina,
de acordo com o descrito na literatura8. Entretanto, o rendimento da reação foi
insatisfatório devido à formação de um produto secundário. A reação foi
acompanhada por CCD, no qual, após 20 horas de agitação, à temperatura
ambiente sob argônio, não se observou a formação de produto. A mistura
reacional foi aquecida brandamente, obtendo-se três produtos após 2 horas.
Além do produto desejado, Rf = 0,39, foi isolado um produto secundário (Rf =0,92) que de acordo com a análise por espectrometria de massas sugeriu-se
ser o dímero do fenilacetileno, cujo sinal do íon molecular possui rnIz = 202.
Uma possível alternativa a fim de elevar o rendimento da oxazolona 15 seria
encontrar uma condição reacional que minimizasse a formação do dímero.

Resultados 90
3.6.2 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-i1ideno]androsta-1,4
d ien-17-ona (21)9
26
~
H2N-N
S
H2NNH2·H20 jTHF,OOC
KOH
THF
MnO,JAI20 3
•
°N °Br---ü-\ 'r~
14 °I26 J1010000
~~N SBr~IY~
16 °I
Esquema 22
°
Br--Q-f: ~ 21
26 = reagente de Lawesson
A preparação do composto 21 seguiu uma rota composta por cinco
etapas de reações. A reação do composto 14, com o reagente de Lawesson
(26), resultou no composto 16 cuja coloração era laranja intensa. O composto
foi caracterizado por RMN de 1H e RMN de 13C. A diferença marcante entre o
reagente de partida e o produto está no deslocamento da carbonila de 40 ppm
(RMN de 13C) ao ser convertida em tiocarbonila (Anexos 14 e 15). Da mesma
maneira, o composto 18 (anexo 16) foi preparado pela reação do composto 17
com 26, no qual também foi observada uma mudança brusca na coloração, na
medida que foi introduzido o grupo enxofre, formando um produto de coloração
roxa. A caracterização do composto 18 (anexo 16) foi feita por comparação com
o composto descrito na literatura10. O que chama a atenção é o fato de uma

Resultados 91
carbonila insaturada (C-3) ter preferência na substituição, frente a umaI
carbonila saturada (C-17), visto que carbonilas a,p-insaturadas são
estabilizadas por ressonância e, portanto deveriam ser menos reativas do que
as saturadas. Entretanto, observou-se que, se a reação for efetuada em THF à
temperatura ambiente durante uma hora, ocorre formação apenas do composto
18. Na caracterização foi dada maior atenção aos deslocamentos que
evidenciam a molécula quando esta sofre mudança do reagente para o produto.
No composto 17 (reagente de partida), as carbonilas possuem os seguintes
deslocamentos: C-3 (8 186) e C-17 (8 219,7). Ao ser convertido em produto, o
sinal referente à carbonila C-3 se desloca para 8217,5. Vale mencionar que, se
as duas carbonilas (C-3 e C-17) do composto 17 (reagente) sofressem
substituição, seria observado um deslocamento químico em 8 217,5 e em
aproximadamente 8 260, correspondente ao grupo tiocarbonila do carbono 17,
conforme previsão feita pelo programa de simulação de espectros NMR
Prediction11.
A conversão do composto 18 em 19 foi feita com hidrazina hidratada em
THF à OcC, e o produto caracterizado apenas por espectroscopia na região do
infravermelho e comparado com a Iiteratura9.
A preparação do composto diazo 20 via oxidação da hidrazona 19 requer
bastante cuidado para evitar a dimerização de 20. Esta reação foi feita em éter
com Mn02 em carvão ativo e submetida ao ultra-som por 5 minutos em meio
básico. Ao término da reação foi retirada uma solução fracamente azulada,
sendo o seu volume reduzido cuidadosamente à pressão reduzida, para que
não ocorresse dimerização. Acredita-se que esta dimerização ocorra pela
formação de um carbeno, devido à instabilidade do composto diazo 20, seguido
do acoplamento entre os carbenos formados na reação.
O rendimento de formação da olefina 21 foi de aproximadamente 30%,
entretanto, de acordo com a literatura, pode-se chegar a 40% dependendo do
substituinte. A reação ocorre sem necessidade de agitação, bastando adicionar
o sólido do composto 16 ao composto 20 e deixar em repouso durante 24

Resultados 92
horas. A reação ocorre via mecanismo de Barton Kellog (vide discussão), sendo
que, além da formação do produto desejado, foi detectada a formação do
dímero do composto 16, caracterizado por espectrometria de massa, cujo sinal
do íon molecular possui m1z =503.

Resultados 93
3.7 Referências
1 Stevani CV, "Estudo mecanístico do sistema peróxi-oxalato", Tese de Doutorado,
Universidade de São Paulo, 1997.
2 Bartlett PD & Pincock RE, Peresters. VI. Ethyl, Benzyl, p-nitrophenyl and p-methoxybenzoyl
t-butyl-peroxyoxalate, J. Am. Chem. Soc., 82 (7), 1769-73, 1960.
3 Stevani CV, Campos IPA & Baader WJ, "Synthesis and characterisation of an intermediate in
the peroxyoxalate chemiluminescence: 4-chlorophenyl O,O-hydrogen monoperoxyoxalate", J.
Chem. Soc., Perkin Trans. 2, 1645-8, 1996.
4 Silva SM, Casalianovo F, Oyamaguchi KH, Ciscato LFLM, Stevani CV & Baader WJ, "Kinetic
studies on the peroxyoxalate reaction: determination of the cyclization rate constant",
Luminescence, 313-20, 2002.
5 Silva SM, Wagner K, Weiss D, Beckert R & Baader WJ " Studies on the chemiexcitation step in
peroxyoxalate chemiluminescence using steroid-substituted activators" Luminescence, 17, 362
9,2002.
6 Melhuish WH, "Quantum efficiencies of f1uorescence of organic substances: effect of solvent
and concentration of the fluorescent solute" J. Phys. Chem., 65, 229-35, 1961.
7 Lee JH, Rock JC, Park SB, Schlautman MA & Carraway RE, " Study of the characteristics of
three high-energy intermediates in peroxyoxalate chemiluminescence (PO-CL) reactions" J.
Chem. Soc., Perkin Trans. 2, 802-9, 2002.
8 Sonogashira K, Tohda Y & Hagihara N, "Convenient synthesis of acetylenes-catalytic
substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines,"
Tetrahedron Lett., 50,4467-70, 1975.
9 Wagner K, Synthese und Eingenschaften neuer A-Ring substituierter Steroide-Ein Beitrag zur
modernen biochemischen Analytik, Dissertation, Universitat Jena, 1998.
10 Wei~ D, Gaudig U & Beckert R, "Eine neue Herstellung von gekreuzt-konjugierten 3
Thioxosteroiden", Synthesis, 751-2, 1992.

Resultados 94
11 Pretsch E, Bühlmann P, Affotter C & Badertscher M, Spektroskopische Daten zur
Struturaufklarung Organischer Verbingungen, Springer Velag, 2001.

Lnscussão 95
4 Discussão
4.1 Síntese dos derivados
monoperoxioxalato (5-7)
do O,O-hidrogênio
Os derivados 5 a 7 foram preparados através da reação dos derivados 1 a
3 com peróxido de hidrogênio na presença de piridina em éter anidro (Esquema
23). Inicialmente é importante destacar que para preparar os derivados do 0,0
hidrogênio monoperoxioxalato de arila (5-7) foram necessários alguns cuidados
para que não ocorresse degradação do produto, já que estes são instáveis à
temperatura ambiente. Por exemplo, foi necessária a rápida extração para isolar
o produto dos reagentes. Por isso, devido a sua instabilidade, tornou-se como
procedimento armazená-los a -80°C.
Esquema 23
°M°-o-X
CI ° + H1. 2°2
.X=H2: X = CH3: X =OC~3
o ° o----r>--X___N • \'-----1~• 1~
éter HOO 2 ° 4 5
5: X = OCH3
6: X = CH3
7: X= H
A formação dos compostos (5-7) foi acompanhada por cromatografia em
camada delgada (CCO) e a caracterização por RMN de 13C pelo. método
INVGATE (inverse gated coupling) (Tabela 18). O método permite a integração
dos sinais e com isso foi possível analisar a relação entre o ácido e o perácido
na amostra.
Os sinais dos compostos 5 a 7 apresentaram intensidades relativas
similares, cuja razão esperada entre os carbonos 1 a 6 era de 1:1:1:2:2: 1, muito
embora a integração referente aos sinais dos carbonos 4 e 5 de cada composto
tenha apresentado valores acima do esperado. Talvez isto esteja relacionado
com a determinação da área da integral, pois os sinais dos carbonos 4 e 5 do
perácidos são muito próximos dos sinais dos carbonos 4 e 5 dos ácidos
correspondentes, resultando na variação observada. Além disso, o efeito do

Lnscussão 96
substituinte sobre os deslocamentos químicos de cada perácido é pronunciado
nos carbonos 3 e 6.
Tabela 18: Deslocamentos químicos da RMN de 13C (I NVGATE) dos compostos 5 a 7
juntamente com as intensidades relativas, nas seguintes condições: Acetona 0 6 , 5 e 6 (-40 0c)
e 7 (-30°C), 125 MHz.
HOO O 4 5
~>r-{<o~xX =OCH3 (5)
X =CH3 (6)
X =H (7)
c1
2
3
4
5
6
7
5
154,8 (0,75)
155,9 (0,92)
143,8 (1,00)
122,8 (3,00)
114,8 (2,16)
158,3 (1,42)
55,47 (1,09)
6
154,7 (1,08)
156,0 (1,04)
148,4 (1,00)
121,6 (2,80)
130,7 (3,20)
137,0 (1,05)
20,6 (1,09)
7
154,8 (0,98)
156,1 (0,53)
150,6 (1,00)
122,0 (2,26)
130,5 (2,74)
127,5 (1,02)
A CCO, ao longo do trabalho, tornou-se um método bastante eficiente
para acompanhar a formação do produto. Geralmente, estes compostos (5-7)
continham, como impurezas, fenol e o ácido correspondente, que podiam ser
identificados com lâmpada UV, no caso do fenol, e com alaranjado de metila, no
caso do ácido. Foi interessante observar que o perácido, na presença de
alaranjado de metila, causava a formação de uma mancha branca na placa de
CCO, demonstrando a decomposição do alaranjado de metila na presença do
perácido. Tal comportamento tornou-se um método complementar para
identificação do perácido no produto bruto. Havia também a possibilidade de
formação do peranidrido (31) (Esquema 24), o qual apresentaria
comportamento similar ao perácido. Entretanto, segundo a literatura1, o excesso
de peróxido de hidrogênio impediria a formação de tal composto. Sendo assim,
a reação foi feita utilizando H20 2 em excesso de dez vezes.
Esquema 24
o CI
x-o-oHoH20 2O · o o_:-y.-rO -x
N x~oH o~ o 31
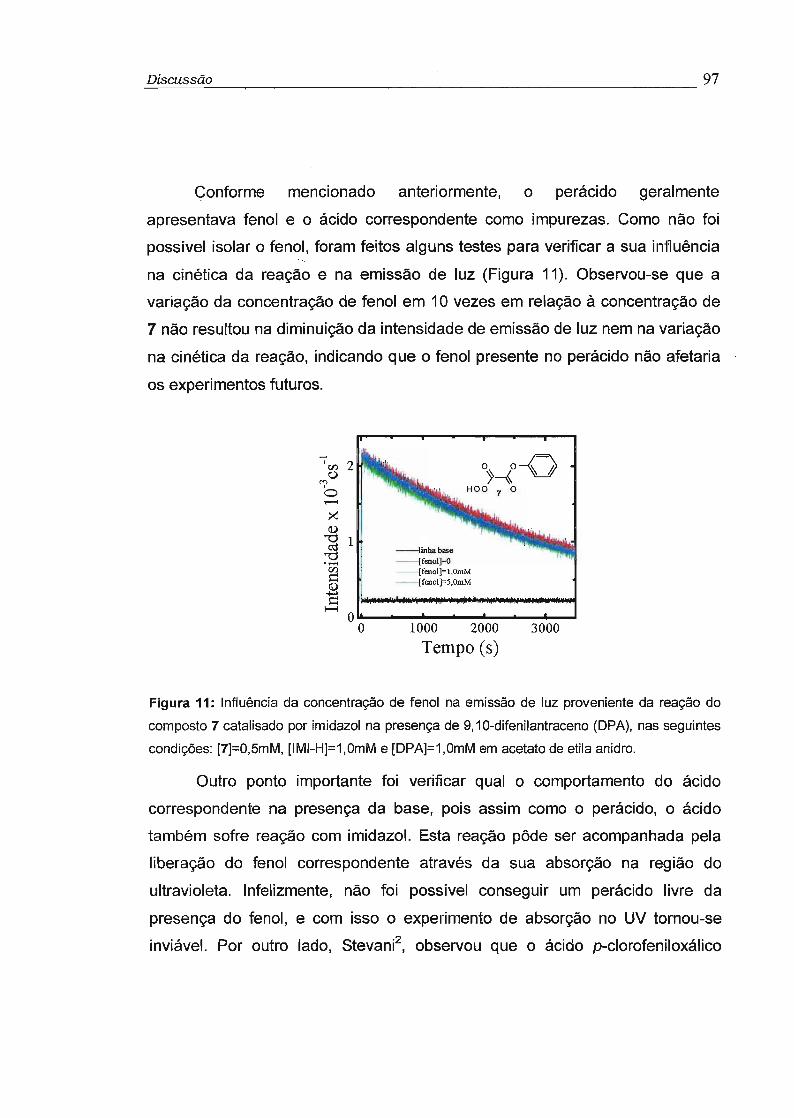
Lnscussão 97
Conforme mencionado anteriormente, o perácido geralmente
apresentava fenol e o ácido correspondente como impurezas. Como não foi
possível isolar o fenol, foram feitos alguns testes para verificar a sua influência
na cinética da reação e na emissão de luz (Figura 11). Observou-se que a
variação da concentração de fenol em 10 vezes em relação à concentração de
7 não resultou na diminuição da intensidade de emissão de luz nem na variação
na cinética da reação, indicando que o fenol presente no perácido não afetaria
os experimentos futuros.
-~ °-Yo__t-o'eI) 2U ,:
'" ', I HOO 7 oo /..,..-l
X(lJ
11~ -linha base-[fenol]=O
[fenol]~I,OmM
(lJ-[fenol]=5,OmM
~-oo 1000 2000 3000
Tempo (s)
Figura 11: Influência da concentração de fenol na emissão de luz proveniente da reação do
composto 7 catalisado por imidazol na presença de 9,10-difenilantraceno (DPA), nas seguintes
condições: [7]=O,5mM, [IMI-H]=1,OmM e [DPA]=1,OmM em acetato de etila anidro.
Outro ponto importante foi verificar qual o comportamento do ácido
correspondente na presença da base, pois assim como o perácido, o ácido
também sofre reação com imidazol. Esta reação pôde ser acompanhada pela
liberação do fenol correspondente através da sua absorção na região do
ultravioleta. Infelizmente, não foi possível conseguir um perácido livre da
presença do fenol, e com isso o experimento de absorção no UV tornou-se
inviável. Por outro lado, Stevani2 , observou que o ácido p-c1orofeniloxálico

.... 'w1..IVi. A
INSTITUTO DE QUIi,;iCAUnIversidade de São Paulo 98
Discussão
reagia 100 vezes mais lentamente do que o O,O-hidrogênio monoperoxioxalato
de p-c1orofenila (8A). Portanto, esta foi uma condição para admitir que
provavelmente os outros ácidos correspondentes (H, CH3 e OCH3) teriam
comportamento semelhante em relação aos perácidos.
O resultado mais importante da preparação dos compostos 5-7 foi
verificar que estes não reagem com o ACT na ausência de uma base. Fora
relatado na literatura que derivados dos compostos 5 a 7 seriam os
intermediários de alta energia (IAE) que na presença de um ativador (ACT),
resultariam na emissão de luz3. Entretanto, foi verificado que a emissão de luz
só é observada quando uma base adequada (pKa > 6,0) é utilizada. Este
resultado também foi observado por Stevani ao estudar O,O-hidrogênio
monoperoxioxalato de p-c1orofenila2.

Lnscussão 99
4.2 Estudo cinético dos perácidos 5 a 7
A reação dos compostos 5 a 7 foi catalisada por imidazol na presença de
9,10-difenilantraceno (OPA) em acetato de etila anidro, resultando em emissão
de luz, devido à desprotonação do mesmo por imidazol, saída do grupo de
partida e formação do IAE; que, por sua vez, interage com o OPA na etapa de
quimiexcitação provocando a sua excitação eletrônica. (Esquema 25).
Esquema 25
F N
-O- H-N~ -O-0Ho ~ li x _ 0Ho ~ li x
HOO ° -00 °JACT
Não emiteluz
J° °H .. 2C02o-o ~
ACT Acr
~hv
o uso de imidazol neste estudo merece alguns comentários, pois foi
observado que em estudos envolvendo o sistema peroxioxalato, os melhores
resultados foram observados com esta base, e isto inclui, também, trabalhos de
química analítica. Imidazol é uma molécula heterocíclica plana com seis
elétrons n. Em solução aquosa, um equilíbrio ácido-base é estabelecido com o
solvente.
H-N~ + H20+ 'L-N-H
k1
k_1
N~ + H30+ pKa =7,1'L-N-H
N~ + H20'L-N-H
k1
k.1
N~'L-N -
+ H30+ pKa = 14,2 - 14,6
As reações inversas são favoráveis, cujas constantes de velocidades são
controladas por difusão. O imidazol pode ser protonado no nitrogênio 3

BIBLIOTECADiscussão INSTITUTO DE QUíMICA 100
Unrversidade de São Paulo
formando o íon imidazolium (pKa=7, 1) ou pode ser desprotonado, em soluções
fortemente alcalinas, no nitrogênio 1 formando o íon imidazolida (pKa=14,2
14,6)4. O extenso interesse pelas propriedades catalíticas de imidazol começou
depois que Hartley e Massel observaram que éster metílico da benzoil
histidina catalisava hidrólise de acetato de p-nitrofenila. De lá para cá, uma
série de trabalhos relevantes podem ser encontrados na literatura; dentre eles
os trabalhos de Jencks et a/ 6.7
, Bruice et a/15., Fife8
, entre outros.
No estudo dos perácidos, apresentado neste trabalho, foi observado uma
dependência de primeira ordem em baixas concentrações de imidazol e uma
dependência de segunda ordem em altas concentrações, caracterizando uma
dependência complexa para os três casos (Figuras 12, 14 e 16).
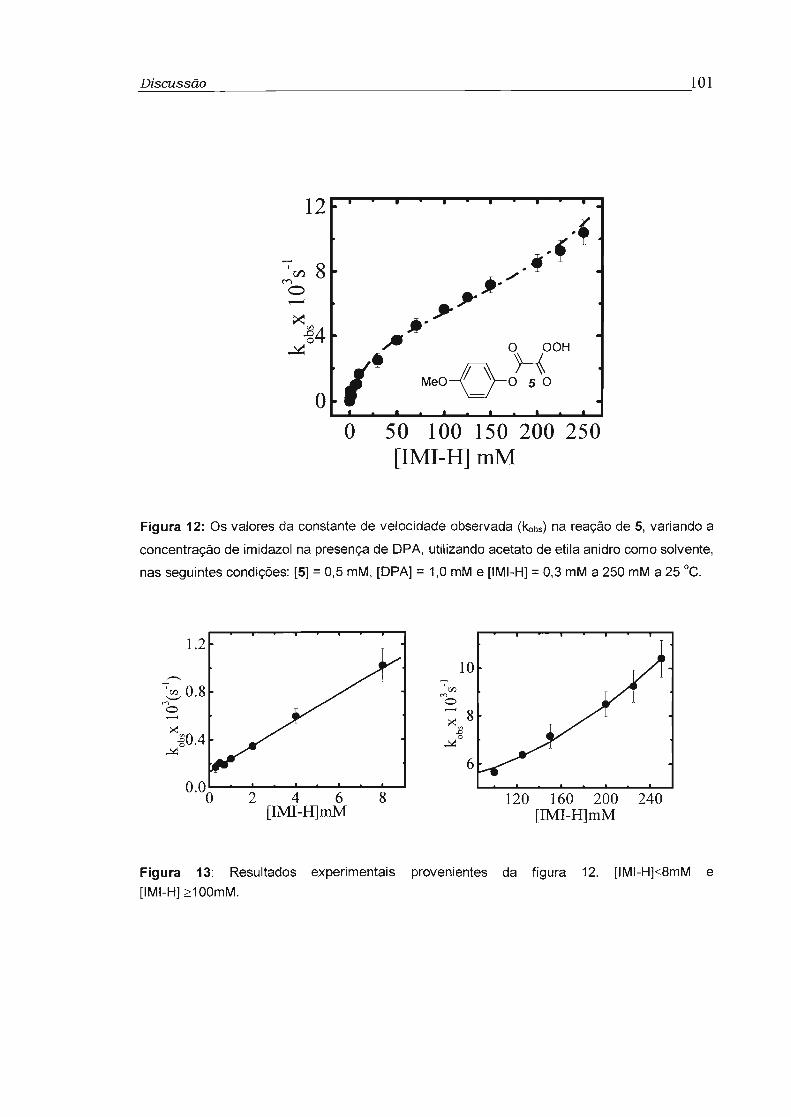
Lnscussão 101
12~~ ~ ~ ~ T •
.
.
....
o OOH
Me0-o-~ H_ o 5 O
81-
,~,f,f,,/ .
~'.,".,AI
O~,·
~
.g41~
tn("")
O....-..
I
o 50 100 150 200 250[IMI-H] mM
Figura 12: Os valores da constante de velocidade observada (kobS) na reação de 5, variando a
concentração de imidazol na presença de DPA, utilizando acetato de etila anidro como solvente,
nas seguintes condições: [5] = 0,5 mM, [DPA] = 1,0 mM e [IMI-H] = 0,3 mM a 250 mM a 25°C.
1.2
_-.",~0.8o........~
.g0.4...:.=:
0.00 246[IMI-H]mM
8
10
<'"IV)
o~ 8
E...:.=:0
6
120 160 200 240[IMI-H]mM
Figura 13: Resultados experimentais provenientes da figura 12. [IMI-H]:s:8mM e
[IMI-H] ~1 OOmM.
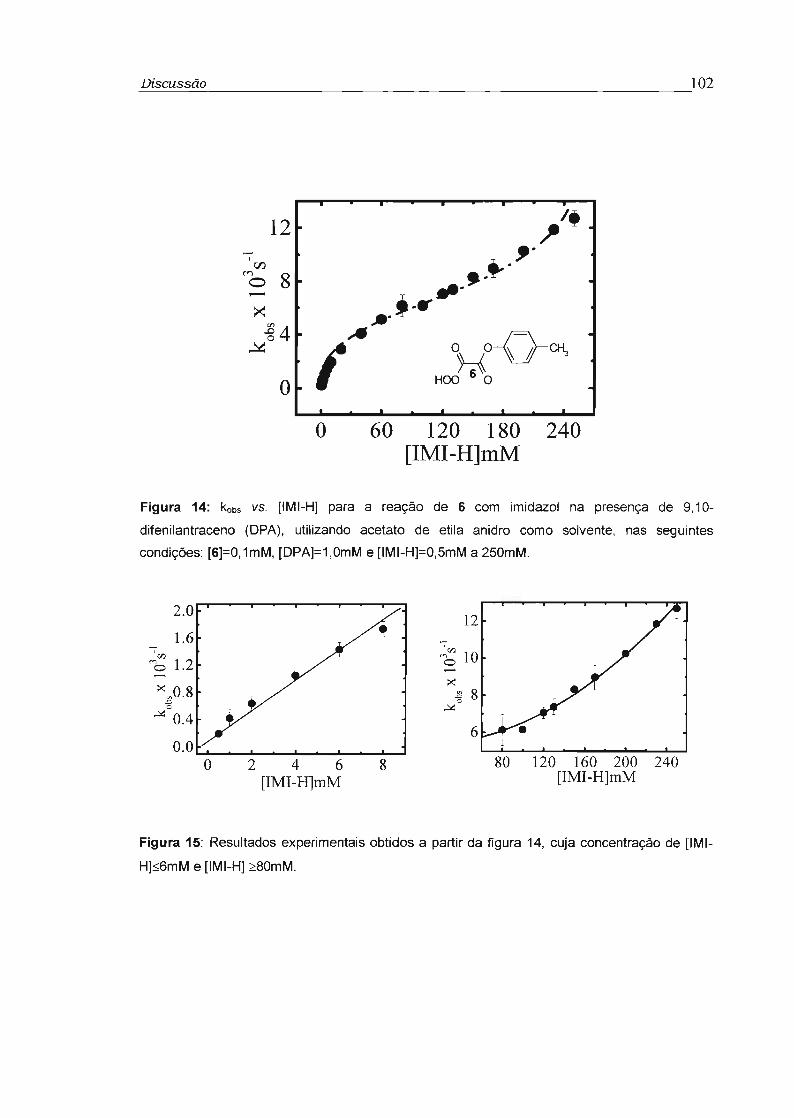
Lnscussão 102
.
~~
o~o--O-CH, .HOO 6 O
--.-
.
--.- ~
'tJ' .,......"...., ...(A
~
'"~41-
121-
OI-
~,U':l
r'"lo 81-.......
o 60 120 180 240[IMI-H]mM
Figura 14: kobs vs. [IM l-H] para a reação de 6 com imidazol na presença de 9,10
difenilantraceno (DPA), utilizando acetato de etila anidro como solvente, nas seguintes
condições: [6]=0,1mM, [DPA]=1,OmM e [IMI-H]=0,5mM a 250mM.
2.0
1.6"";"C/l
'b 1.2........
x~0.8.oo
~ 0.4
0.0_O 246
[IMI-H]mM8
12
'C/l
'b 10........X
1l 8~o
6
80 120 160 200 240[IMI-H]mM
Figura 15: Resultados experimentais obtidos a partir da figura 14, cuja concentração de [IMI
H]~6mM e [IMl-H] ~80mM.
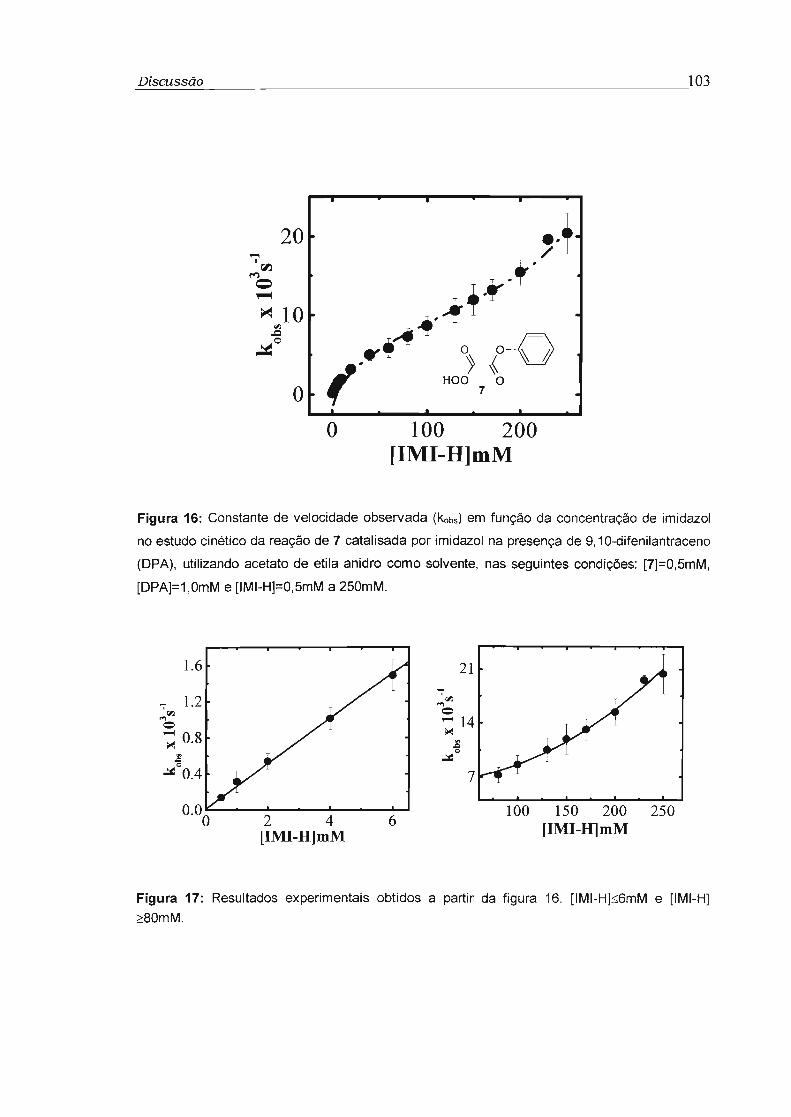
Discussão 103
,. ,.
20".....
I
~
1=~
~ 10"'"..c
~o
O"
,·f·...,-t 0
'"
T~" .(
'.... O0-0HooH
• 7 O .
O 100 200[IMI-H]mM
Figura 16: Constante de velocidade observada (kobs) em função da concentração de imidazol
no estudo cinético da reação de 7 catalisada por imidazol na presença de 9,10-difenilantraceno
(DPA), utilizando acetato de etila anidro como solvente, nas seguintes condições: [7]=O,5mM,
[DPA]=1 ,OmM e [IMI-H]=O,5mM a 250mM.
1.2
1.6
'7~
'O,...; 08~ ...
,Qo
~ 0.4
0.00 2 4[IMI-HJmM
6
21
'~..,o,...; 14~
1)~o
100 150 200 250[IMI-H]mM
Figura 17: Resultados experimentais obtidos a partir da figura 16. [IMI-H]:s;6mM e [IMI-H]
~80mM.

Lnscussão 104
o comportamento de kobs em função da concentração de imidazol pode
ser explicado pelo mecanismo proposto no esquema 26. Em baixas
concentrações de imidazol este agiria principalmente como catalisador básico
desprotonando o perácido, resultando em íon percarboxilato. Este por sua vez,
sofreria ciclização no passo lento, formando o intermediário de alta energia.
Neste passo ocorre a participação de apenas uma molécula de imidazol. Em
concentrações mais altas de imidazol, este agiria predominantemente como
catalisador nucleofílico (Esquema 26), com a participação de duas moléculas de
imidazol no passo lento7,a.
Esquema 26
catálise básica (A)
~l +NI
H
ArOH
°NOAr
HOO O
0N-HN~
knuC1
k1
k-1
o OArH
-~o
kcicl
+ H-N~I N-Hl:::::::::./ +
N0N - H~ + ArOH
o OOH
HN O
[~N
catálise nucleofílíca (B)
~
LN:J-H
[Ottj Z ~o,
De acordo com o mecanismo proposto, a contribuição do imidazol em
baixas concentrações pode ser deduzida a partir do seguinte esquema
mecanístico (Esquema 27).
Esquema 27
o OAr N k1 O OAr H,H + I.. "=l - - H + + N"=l
HOO O ~N-H k -00 O ~N-HPer IMI-H -1 Per- IMI-H
2+
°NOArkcicl
-00 O
O O• t-1
o-oIAE

Lnscussão 105
o aumento da concentração da base no meio reacional, favorece a
desprotonação do perácido, resultando na sua ciclização e na formação do
intermediário de alta energia (IAE).
Aplicando a aproximação do estado estacionário no perácido desprotonado
têm-se:
~[Per- ] = kl[PerUIMI - H]- k_
1[Per- J[IMI - H;]- kCiAPer-] = O
dt
[Per-]= kl[PerUIM! -H]k_1 [IMI - H 2 ]+ kCic/
A velocidade da reação é dada por: kobs = kcic/ [Per - ]
kCic/kl [IMI - H]kobs=~
k_1 LIMI - H 2 J+ kCic/
A equação não pôde ser mais simplificada porque não há os valores das
constantes de equilíbrio envolvidas em acetato de etila. Esta dedução está
relacionada apenas com a etapa de catálise básica específica. Para obter a
equação global correspondente aos dois mecanismos em questão, basta somar
uma equação de segunda ordem.
k = kCic/ k][IM~- H] + knuc/[IMI - Hfobs k_
1[IMI - H
2]+ kcic/
A maneira encontrada para se obter dados das curvas com dependência
complexa, foi analisá-Ias separadamente (Figuras 12, 14 e 16). Estas curvas
foram divididas em duas regiões, uma para baixas concentrações e outra para
altas concentrações de imidazol. Da região linear ([IMI-H] < 8,OmM), obteve-se
uma constante de velocidade bimolecular (kSim) utilizando a seguinte
equação:kobs=ksim[IMI-H] e da região de altas concentrações ([IMI-H] > 80mM) a
curva pôde ser ajustada com uma função de segunda ordem:
kobs=kTrim[IMI-H]2+C, obtendo-se, desta forma, a constante de velocidade
trimolecular (Tabela 19).

Lnscussão 106
As constantes de velocidade bimolecular (kbim) e trimolecular (ktrim) são
condizentes com o efeito eletrônico de seus substituintes, ou seja, diminuindo o
efeito atraente de elétrons dos substituintes observa-se uma diminuição na
constante de velocidade. Porém, no caso do derivado com o grupo metila 6, a
diferença não é muito acentuada quando se compara com o derivado não
substituído 7.
Tabela 19: Influência dos substituintes nas constantes de velocidade bimolecular (kBim) e
trimolecular (kTrim) dos derivados 5-7 e SA.
HOO o
OHO-D-R
R k Bim (M-'s") [IMI-H] ::; kTrim (M-2s") C [IMI-H] ~
(mM) (mM)
OCH3 (5) 0,108 ± 0,009 8 0,086 ± 0,003 0,0049- ± 0,0001 100
CH3 (6) 0,224 ± 0,010 6 0,122 ± 0,003 0,0053 ± 0,0001 80
H (7) 0,250 ± 0,006 6 0,234 ± 0,009 0,0062 ± 0,0003 80
"SA)8 0,96 ± 0,02 20 1,6 ± 0,2 0,040 ± 0,005 100
a Ref. 2
Em relação ao rendimento quântico singlete (<I>s) observa-se que
aumentando a concentração de imidazol o <l>s diminui (Tabela 20).
Correlacionando os diferentes perácidos, esta redução fica em torno de
aproximadamente 100 vezes, sendo que, a maior variação foi observada com o
perácido 7. Vale lembrar que a influência do imidazol sobre o <l>s também foi
observada com o bis(2,4,6-triclorofenil) oxalato. Neste caso foi verificado que o
imidazol, interage com o intermediário de alta energia (IAE), anulando a
interação deste com o ativador (ACT)9.
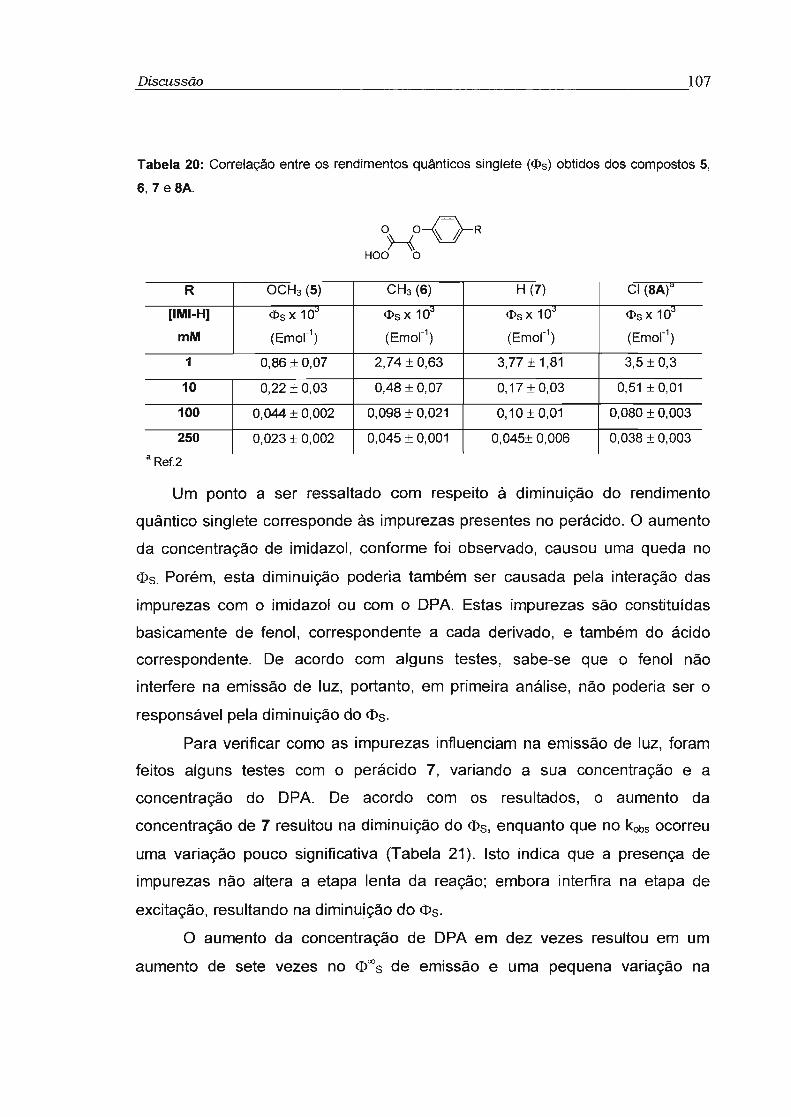
Lnscussão 107
Tabela 20: Correlação entre os rendimentos quânticos singlete (<I>s) obtidos dos compostos 5,
6,7 e 8A.
°'J_·l-o-R
HOO o
R OCH3 (5) CH3 (6) H (7) CI (8A)"
[IMI-H] <l>s x 10" <l>s x 10" <1>8 x 10" <1>8 x 10"
mM (Emor1) (Emor1
) (Emor1) (Emor1
)
1 0,86 ± 0,07 2,74 ± 0,63 3,77 ± 1,81 3,5 ± 0,3
10 0,22 ± 0,03 0,48 ± 0,07 0,17 ± 0,03 0,51 ± 0,01
100 0,044 ± 0,002 0,098 ± 0,021 0,10 ± 0,01 0,080 ± 0,003
250 0,023 ± 0,002 0,045 ± 0,001 0,045± 0,006 0,038 ± 0,003
a Ref.2
Um ponto a ser ressaltado com respeito à diminuição do rendimento
quântico singlete corresponde às impurezas presentes no perácido. O aumento
da concentração de imidazol, conforme foi observado, causou uma queda no
<!>s. Porém, esta diminuição poderia também ser causada pela interação das
impurezas com o imidazol ou com o OPA. Estas impurezas são constituídas
basicamente de fenol, correspondente a cada derivado, e também do ácido
correspondente. De acordo com alguns testes, sabe-se que o fenol não
interfere na emissão de luz, portanto, em primeira análise, não poderia ser o
responsável pela diminuição do <!>s.
Para verificar como as impurezas influenciam na emissão de luz, foram
feitos alguns testes com o perácido 7, variando a sua concentração e a
concentração do OPA. De acordo com os resultados, o aumento da
concentração de 7 resultou na diminuição do <!>s. enquanto que no kobs ocorreu
uma variação pouco significativa (Tabela 21). Isto indica que a presença de
impurezas não altera a etapa lenta da reação; embora interfira na etapa de
excitação, resultando na diminuição do <!>s.
O aumento da concentração de OPA em dez vezes resultou em um
aumento de sete vezes no <!>CXls de emissão e uma pequena variação na

Discussão 108
constante de velocidade observada (kobs). Fato também observado com o
perácido 8A2 e com o sistema peroxioxalat09. Ainda neste sentido,
experimentos sobre a influência de impurezas, contidas no perácido, na
fluorescência do 9,1 O-difenilantraceno (OPA) foram realizados. Ao registrar o
espectro de fluorescência do OPA na presença da solução do perácido 7,
verificou-se que estas supostas impurezas não seqüestram a fluorescência do
ACT (dados não mostrados).
Tabela 21: Influência da variação da concentração de 7 e da concentração de OPA na
constante de velocidade observada (kaoo) e na área de emissão de luz, nas seguintes
condições: [OPA]=1,OmM e [IMI-H]=1,OmM para os experimento com variação de 7 e [7]=0,5,
[IMI-H]=1 ,0mM para os experimentos com variação de OPA.
[7] kobs X 104
<l>s X 103 [DPA] kobs x 104
<l>s X 103
mM 5-1(Emor1
) mM 5.1 (Emor1)
0,25 3,66 4,36 0,5 1,82 2,11
1,0 2,97 1,86 1,0 2,49 2,60
1,5 3,07 1,76 2,5 2,46 6,55
5,0 1,65 15,8
A redução no rendimento quântico singlete (<1>s) , observado na reação
dos perácidos e também no sistema peroxioxalato, quando se aumenta a
concentração de imidazol pode ser, em uma primeira análise, associada ao
processo de supressão de energia, muito embora, na reação em questão não
ocorra transferência de energia. Na etapa de quimiexcitação do sistema
peroxioxalato ocorre a interação do IAE com o ativador, gerando o ativador no
estado excitado. Esta interação, a qual resulta em emissão de luz,
provavelmente ocorre em concorrência com outros caminhos que desativam a
formação de estados excitados.
Ao que tudo indica, o imidazol adicionado à reação, responsável pela
catálise nas etapas anteriores ao processo de quimiexcitação, contribui para
que ocorra um destes caminhos de desativação (Esquema 28).

Lnscussão 109
Esquema 28
OH° kcat .. ACT* J ... hv
T~~M~ ACT
ko /-::::-NH-N~
2C02 2C02sem emissão sem emissão
Com base nestas observações mecanísticas, de que maneira o imidazol
poderia atuar na supressão da emissão de luz? Em um estudo desenvolvido
anteriormente10, foi reportado que a variação na concentração de imidazol não
altera a fluorescência do 9,1 O-difenilantraceno (OPA) e, que, portanto, esta
interação não é fotofísica, isto é, atuação direta do imidazol sobre o estado
excitado do OPA. O caminho mais provável para que ocorra esta supressão é
através da interação do imidazol diretamente com o IAE por intermédio de uma
interação química. Considerando, que a concentração do ACT é inalterada, e
que a intensidade de emissão diminui com o aumento da concentração de
imidazol, podemos, à princípio, aplicar uma relação de Stern- Volmer11; já que o
imidazol, nesta reação, atua como um "seqüestrador' de emissão e, deste
modo, obter o rendimento singlete na concentração zero (<Das) de imidazol,
eliminando a sua interação com o ACT (Figura 18 e Tabela 22).
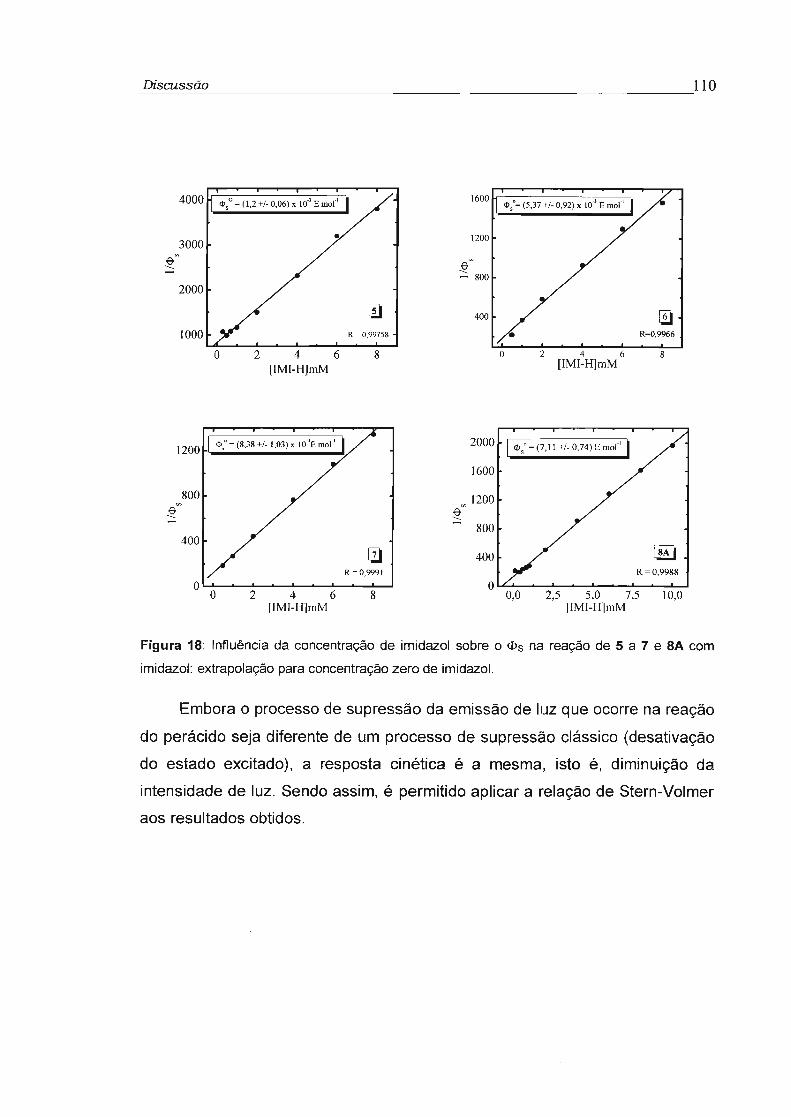
Lnscussão 110
1600L i i i i " Y ]
1200
12001-1 $:-(8,38 +/- 1.03) x 10·'E mor' l
800~'"
~~
- 800
400
2000
1600
1200~'"
o
..2 4 6
[IMI-H]mM
I <J)s" ~ (7,11 +/- 0,74) E mor' I
@}R~0,9%6
8
400
O' r, ! ! , i I
o 246[IMI-H]mM
oR =0,9991
8
800
400
0,0
[E}R ~0,9988
2,5 5,0 7,5 10,0[IMI-H]mM
Figura 18: Influência da concentração de imidazol sobre o <l>s na reação de 5 a 7 e 8A com
imidazol: extrapolação para concentração zero de imidazol.
Embora o processo de supressão da emissão de luz que ocorre na reação
do perácido seja diferente de um processo de supressão clássico (desativação
do estado excitado), a resposta cinética é a mesma, isto é, diminuição da
intensidade de luz. Sendo assim, é permitido aplicar a relação de Stern-Volmer
aos resultados obtidos.
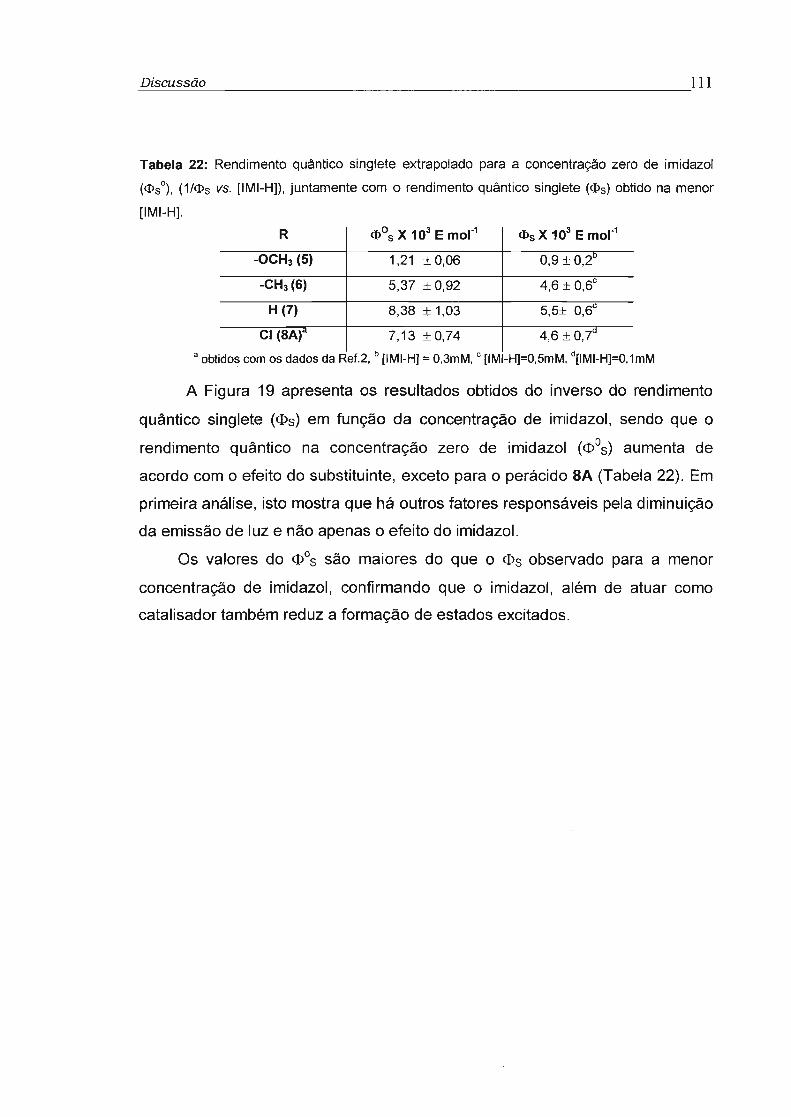
Lnscussão 111
Tabela 22: Rendimento quântico singlete extrapolado para a concentração zero de imidazol
(<1>so), (1/<1>s vs. [IMI-H)), juntamente com o rendimento quântico singlete (<1>s) obtido na menor
[IMI-H].
R <1>°s X 103 E mor1 <1>s X 103 E mor1
-OCH3 (5) 1,21 ± 0,06 0,9 ± 0,2D
-CH3 (6) 5,37 ± 0,92 4,6 ± 0,6c
H (7) 8,38 ± 1,03 5,5± O,6c
CI (8Ar 7,13 ± 0,74 4,6 ± 0,7°
a obtidos com os dados da Ref.2, b [IMI-H] = O,3mM, c [IMi-H]=O,5mM, d[IMI-H]=O,1mM
A Figura 19 apresenta os resultados obtidos do inverso do rendimento
quântico singlete (<1>5) em função da concentração de imidazol, sendo que o
rendimento quântico na concentração zero de imidazol (<1>°5) aumenta de
acordo com o efeito do substituinte, exceto para o perácido 8A (Tabela 22). Em
primeira análise, isto mostra que há outros fatores responsáveis pela diminuição
da emissão de luz e não apenas o efeito do imidazol.
Os valores do <1>°5 são maiores do que o <1>5 observado para a menor
concentração de imidazol, confirmando que o imidazol, além de atuar como
catalisador também reduz a formação de estados excitados.

Lnscussão 112
4.3 Correlação de estrutura e reatividade
Conforme fora mencionado na introdução, a estrutura do intermediário de
alta energia (IAE), que se forma na etapa de quimiexcitação, é um dos motivos
de controvérsia entre vários grupos de pesquisa. Para obter informações a
respeito deste IAE, foi feito um estudo de correlação de estrutura e reatividade,
calculando o valor de p, através dos gráficos do tipo logkbim e logk1rim VS cr
(Figura 19) com o intuito de obter evidências sobre a identidade estrutural do
intermediário de alta energia (Esquema 29). Os dois intermediários de alta
energia mais prováveis na reação peroxioxalato são a 1,2-dioxetanodiona (24) e
a 1,2-dioxetanona 25, os quais podem ser formados a partir de derivados de
perácidos12. A ciclização do perácido, após a desprotonação, pode ocorrer de
duas maneiras: (i) ataque intramolecular do íon percarboxilato, sem saída do
grupo de partida, resultando na formação da 1,2-dioxetanona (25), que, por sua
vez, pode agir diretamente como IAE ou formar 24 pela saída do grupo de
partida em uma etapa rápida; (ii) ataque intramolecular do íon percarboxílato,
acompanhado pela saída simultânea do grupo de partida fenolato, resultando
na 1,2-dioxetanodiona (24) (Esquema 29).
Esquema 29
rArO 0j* ~o~ - _0+{0 Ó~ hv8- (Y' O-O
8- 25O OArH lenta
-00 O
ii
rAro.~: O j*O~
'. O(Y'8-
O O
rttArO- 24
f- hv
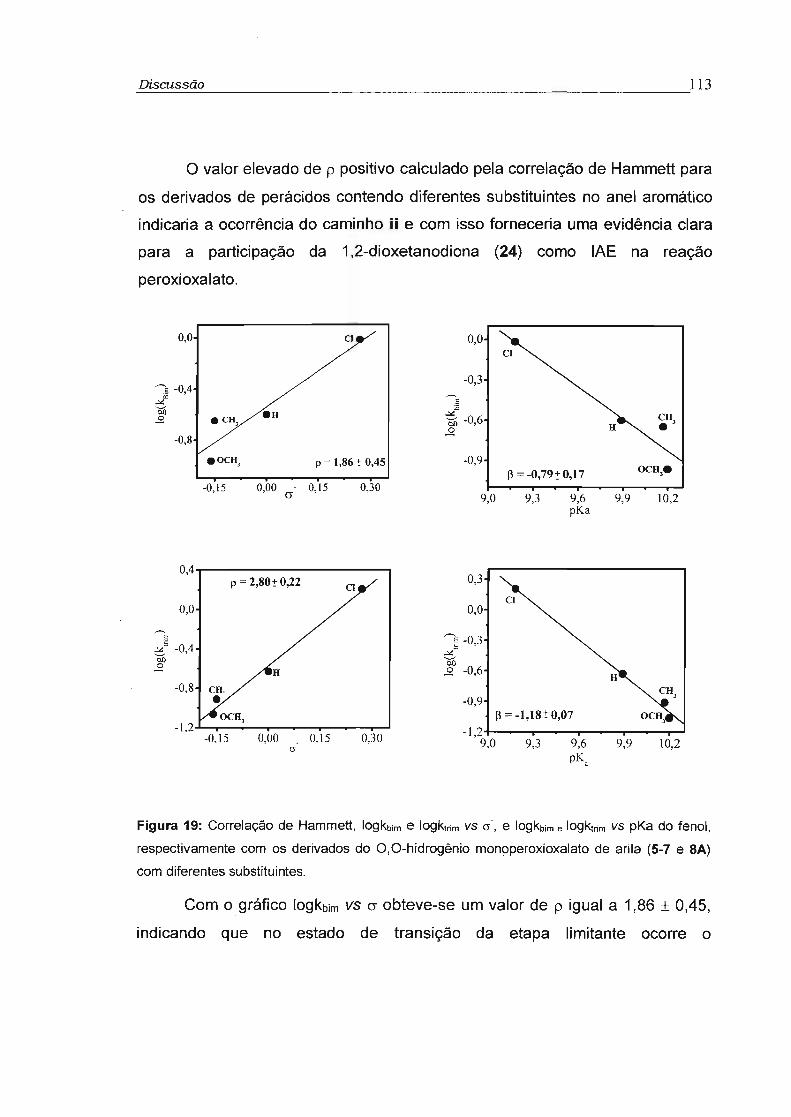
Lnscussão 113
o valor elevado de p positivo calculado pela correlação de Hammett para
os derivados de perácidos contendo diferentes substituintes no anel aromático
indicaria a ocorrência do caminho ii e com isso forneceria uma evidência clara
para a participação da 1,2-dioxetanodiona (24) como IAE na reação
peroxioxalato.
O,Oi CI""-- I 0,0
~CI
-0,3JJ -0,4
"-"'E'"..><..::It.".õ'õ1í eu bíí -0,6.9 e cu,.2
-0,8
e ocu, p = 1,86 "t 0,45 -O 9ocu,.
' ~ p = -0,79~ 0,170,30 i , i ,-0,15 0,00 (J 0,15
9,0 9,3 9,6 9,9 10,2pKa
0,4. •p = 2,80t 0,22 CI
0,3
0,0CI
0,0
~§..><" -O 4]i ,
-0,8u
-E -O 3"5 '
..><
]í -0,6
-0,9cu,
13=-1,18tO,07 OCU-1,2 i i , i i '
10,29,99,6pK
a
9,3-1,21 , i , i '
9,00,300,00 0,15cr
-0,15
Figura 19: Correlação de Hammett, logkbim e logktrim VS a", e logkbim e logktrim VS pKa do fenol,
respectivamente com os derivados do O,O-hidrogênio monoperoxioxalato de arila (5-7 e 8A)
com diferentes substituintes.
Com o gráfico logkbim VS cr obteve-se um valor de p igual a 1,86 ± 0,45,
indicando que no estado de transição da etapa limitante ocorre o

Lnscussão 114
desenvolvimento de carga negativa e, a partir do gráfico kbim VS pka obteve-se
PGP = -0,79 (inclinação Br0nsted).
De acordo com Jencks et af, correlações do tipo Hammett para reações
de acetato de arila substituídos com determinadas aminas exibem valores de p
aproximadamente igual a 2, indicando que estas reações apresentam grande
sensibilidade em relação à natureza do grupo de partida. Enquanto que,
nucleófilos básicos oxigenados exibem valores menores. A questão
fundamental neste tipo de reação está relacionada ao mecanismo operante, o
qual resulta na liberação do fenol substituído. Nestas reações, haveria a
formação de um intermediário tetraédrico na etapa limitante e saída do grupo de
partida em uma etapa posterior e rápida (Esquema 30A), ou a reação ocorreria
por um mecanismo concertado, sem a formação do intermediário (Esquema
30B)?
Esquema 30
GP =grupo de partidaNu =nucleófilo
lR-JJ: O· JcF . RJ(-- -GP NuNu o.
{ORrGPNu
lenta
A
B
oR~GpNu
_ O
rápida R~ Nu
Na hidrólise alcalina de tiobenzoatos de fenila substituídos, Um ei a/13.,
observaram um valor de p igual a 0,75 e P igual a -0,35. Com base nestes
resultados, os autores propuseram a formação de um intermediário na etapa
limitante da reação. Por outro lado, EI Seoud et al. 14, relataram valores de p
igual a 1,59 e P igual a -0,75, para a reação de hidrólise de 3,5-benzoatos de
fenila substituídos catalisada por imidazol. Os autores propuseram saída do
fenol na etapa limitante, ou seja, saída simultânea do fenol concomitante com o
ataque nucleofílico.

Lnscussão 115
A magnitude do valor de P indica a extensão da ligação do grupo de
partida no estado de transição da etapa limitante. Valores baixos, entre -0,2 e
-0,35 foram encontrados para reações de hidrólises de acetatos15 e cinamatos
de arila16, para aminólises de acetatos de arila6, e tioderivados17. Com base
nestes valores, foi proposta a formação de um intermediário tetraédrico na
etapa limitante. Por outro lado, também foram reportados trabalhos envolvendo
a saída do grupo de partida na etapa-limitante da velocidade, isto é, sem
formação de intermediário tetraédrico, cujos valores de Pforam acima de -0,75;
como por exemplo: reações de cinamatos de arila com íon N3- (PGP = -0,95),
aminólise de acetatos de arila, benzoatos com grupos pobres de partida (PGP =
1,40) e hidrólise de derivados de benzoatos de fenila substituídos catalisado por
imidazol (PGP= _0,75)14.
Diante do resultado obtido no presente trabalho (p =1,86 ± 0,45 e PGP =0,79), é possível propor que na reação de derivados de O,O-hidrogênio
monoperoxioxalato de fenila catalisada por imidazol, ocorra ataque
intramolecular do íon percarboxilato acompanhado pela saída simultânea do
grupo de partida, sendo portanto o caminho ii o mecanismo mais adequado e a
1,2-dioxetanodiona (24) o IAE mais provável (Esquema 29). Além disso, o valor
de PGP indica que no passo de ciclização, para a formação do IAE, mais da
metade da carga formal está localizada no fenolato.
As correlações obtidas com o logktrim VS cr e pKa indicam que ocorre a
formação de uma alta densidade de carga negativa, p = 2,80 ± 0,22 e
PGP = -1,18 ± 0,07, no estado de transição. Todavia, de acordo com o
mecanismo proposto (Esquema 26), esta é a etapa de adição nucleofílica do
imidazol à carbonila do éster, resultando na saída do fenolato. Nesta etapa não
ocorre a formação do IAE.
Observa-se que os valores de p e P são maiores na região de catálise
nucleofílica (p = 2,80 ± 0,22 e PGP = -1,18 ± 0,07) do que na região de catálise
básica (p = 1,86 ± 0,45 e PGP = -0,79). Na região de catálise nucleofílica (altas

Lnscussão 116
concentrações de imidazol), o ataque do imidazol à carbonila do éster e saída
do fenol, resulta em um intermediário (Esquema 31 B), diferente do
intermediário que seria esperado na catálise básica e, que, resultaria na saída
do fenol formando diretamente o intermediário de alta energia (IAE) (Esquema
31A). Portanto, o valor de p obtido nesta etapa não fornece nenhuma
informação sobre o IAE, pois este é formado em uma etapa posterior à saída do
fenolato.
Esquema 31
+ óOH
::::,...1
HOO O
oHN:)
~N
~O
O~-o
í'CN-H~HOO O _ ..
oHo-o l J
+1 U1J OHA~ O r O O
-a+t · H+ÓI\ O o-o::::,...o· cY
o· Intermediário dealta energia (lAE)
[
- 40 2
HOO O O oH - B HOOH,OPh
/0-0 · O 'N~ Y\0+] ·
. ~N-H N~N-HbJ
I I
N""'- "N""'- Intermediário de~N-H ~N-H reação
Deve-se destacar ainda, que os nucleófilos envolvidos nas etapas
correspondentes as constantes kbim e k trim são diferentes, ou seja, íon
percarboxilato e imidazol, respectivamente. Isto explica a diferença nos valores
de p e pobtidos.
Em resumo deve-se destacar que, o valor de p (log kBim VS cr) indica que
no estado de transição para a formação do IAE ocorre o desenvolvimento de
uma alta densidade de carga negativa. Este fato, estaria associado ao ataque
nucleofílico intramolecular do íon percarboxilato à carbonila do éster com
concomitante saída do grupo de partida, o que nos permite inferir que a 1,2
dioxetanodiona (24) seria o intermediário de alta energia que interage com o
ativador na etapa de quimiexcitação da reação peroxioxalato.
O gráfico de 10g(<D°s) VS. cr - (Figura 20) não apresentou correlação
significativa. De acordo, com a tabela 22, o <Dos é maior do que o <Ds obtido na
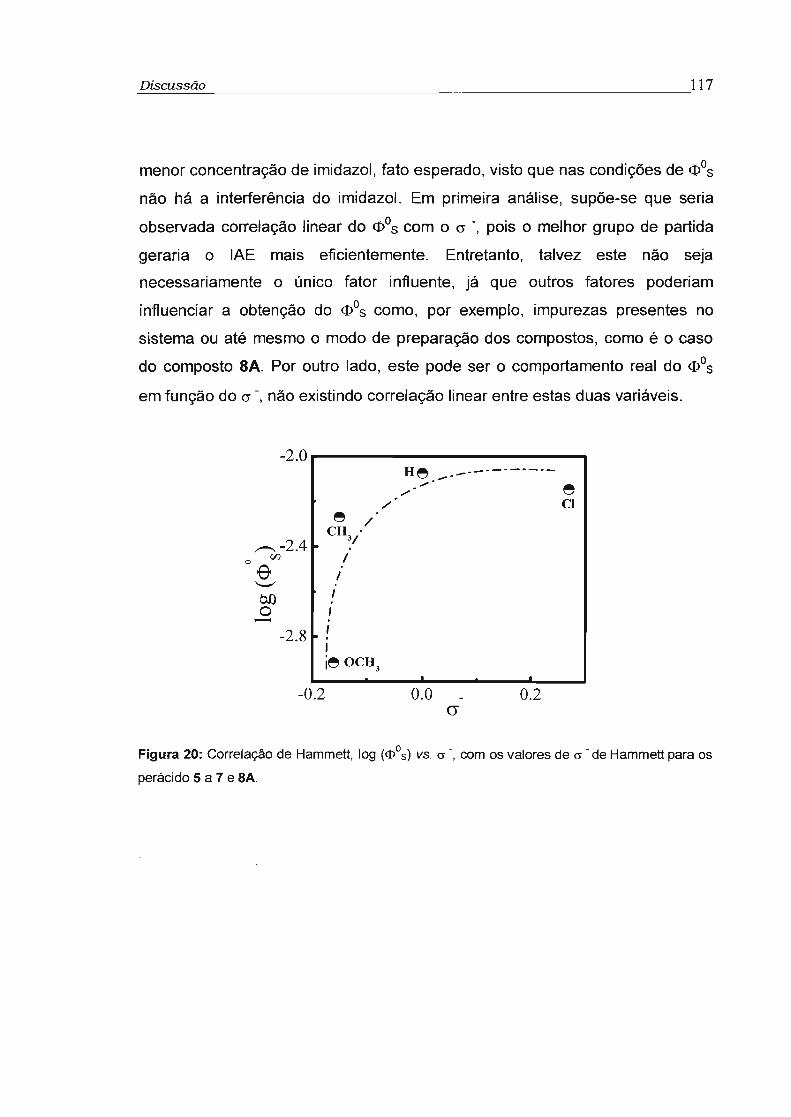
Lnscussão 117
menor concentração de imidazol, fato esperado, visto que nas condições de <1>°s
não há a interferência do imidazol. Em primeira análise, supõe-se que seria
observada correlação linear do <1>°s com o (J" -, pois o melhor grupo de partida
geraria o IAE mais eficientemente. Entretanto, talvez este não seja
necessariamente o único fator influente, já que outros fatores poderiam
influenciar a obtenção do <1>°s como, por exemplo, impurezas presentes no
sistema ou até mesmo o modo de preparação dos compostos, como é o caso
do composto 8A. Por outro lado, este pode ser o comportamento real do <1>°s
em função do (J" -, não existindo correlação linear entre estas duas variáveis.
-2.0. He _.---------.--...... e.....
/ CIe /
..-.--2.4CH3/.
C/) Io e I'.-'
b1) I
o ,......-2.8
,Iie OCH
3
• . • . •-0.2 0.0 - 0.2
cr
Figura 20: Correlação de Hammett, 109 (<1>0s) VS. cr -, com os valores de cr - de Hammett para os
perácido 5 a 7 e 8A.

Lnscussão 118
4.4 Estudo da Eficiência de transferência de elétron na etapa
de quimiexcitaçáo
Conforme fora dito anteriormente, a etapa de quimiexcitação ocorre em
uma etapa rápida da reação, e, portanto não pode ser avaliada por cinética.
Esta é a etapa a qual envolve a interação do IAE com o ativador dentro da
gaiola de solvente mediada por uma transferência de elétron. Considerando que
nesta etapa há apenas o envolvimento de duas substâncias, a mudança na
estrutura de uma delas poderia trazer informações sobre o processo no qual
elas estão inseridas. Alteração na estrutura do ativador mudaria principalmente
o rendimento de fluorescência (<DF) e o potencial de oxidação. O mecanismo
utilizado para explicar a geração de estados excitados que ocorre na etapa de
quimiexcitação é o mecanismo CIEEL (vide introdução) (Esquema 32). Os
resultados apresentados nesta parte do trabalho, têm como objetivo principal
obter informações sobre o mecanismo CIEEL do sistema peroxioxalato
proposto neste trabalho18. O intermediário de alta energia (IAE) postulado nesta
reação é a 1,2-dioxetanodiona, conforme evidência indireta reportada na
literatura19, e nos resultados obtidos neste trabalho.
Esquema 32
C02~~:i)
fI", +
co '" I'"2... '"
/
o O
'M'o-o~+~_(I)-
hv
ô- "'11'1111'"111 B+
(v)
f1
'>. "'I",
"'I
.. oco (iv)
2
o OH_
o. O ~

Lnscussão 119
De acordo com o mecanismo CIEEL proposto, ocorre a transferência de
elétron do ACT para o IAE no passo limitante da reação, acompanhada pela
c1ivagem da ligação O-O do peróxido. A c1ivagem da ligação carbono-carbono
do peróxido leva à formação do ânion radical do C02, ainda associado ao cátion
radical do ACT dentro da gaiola de solvente (Esquema 32). Em seguida, o par
de íons radical sofre retro-transferência (BET), levando a formação de CO2 e do
ativador no estado singlete excitado (kBET*) ou no estado fundamental (kBET) , ou
ainda, através do escape das espécies da gaiola de solvente, formando os
produtos no estado fundamental. Considerando cada caminho que leva a
formação do estado excitado, juntamente com os que o desativam, o
rendimento para cada etapa pode ser obtido de acordo com o esquema 33.
Esquema 33
o O lO O J~ ij ~ ijc-c kcAT c-c .
TCPO ~. I I -----=--ACT I I ACT+etapa 0-0 O O
Um;,","' ko O j"=k co? + ACT~ -
~ [ CcY2 + ACT+ ] -----------.. CO2
+ ACT*k UET*
jk'= j~"
<1> = constanteR
2C02
<1>CA:;'
ACT + 2C02
kcAT[ACnkcAT[ACT] + ko
CO2 + ACT
kcuv~uv= kcuv + kESC
hv + ACT
kuET*
~;* kUET + kSET*+ k'ESC
Dos gráficos do inverso do rendimento quântico singlete (1/<t>s) em
relação ao inverso da concentração do ACT (1/[ACT]) dos ativadores 9 a 13
(Figura 6-resultados) foram obtidos os parâmetros de quimiluminescência kcat/ko
e <t>oos (Tabela 15-resultados). Os valores de In(kcat/ko) apresentam correlação
linear com o potencial de oxidação de meio-pico (EOX1/2) dos ativadores (Figura
21), conforme seria esperado pelo mecanismo. A transferência de elétron do
ativador para o intermediário de alta energia (24) é proposta como a etapa
limitante do mecanismo, sendo que neste par o potencial de redução
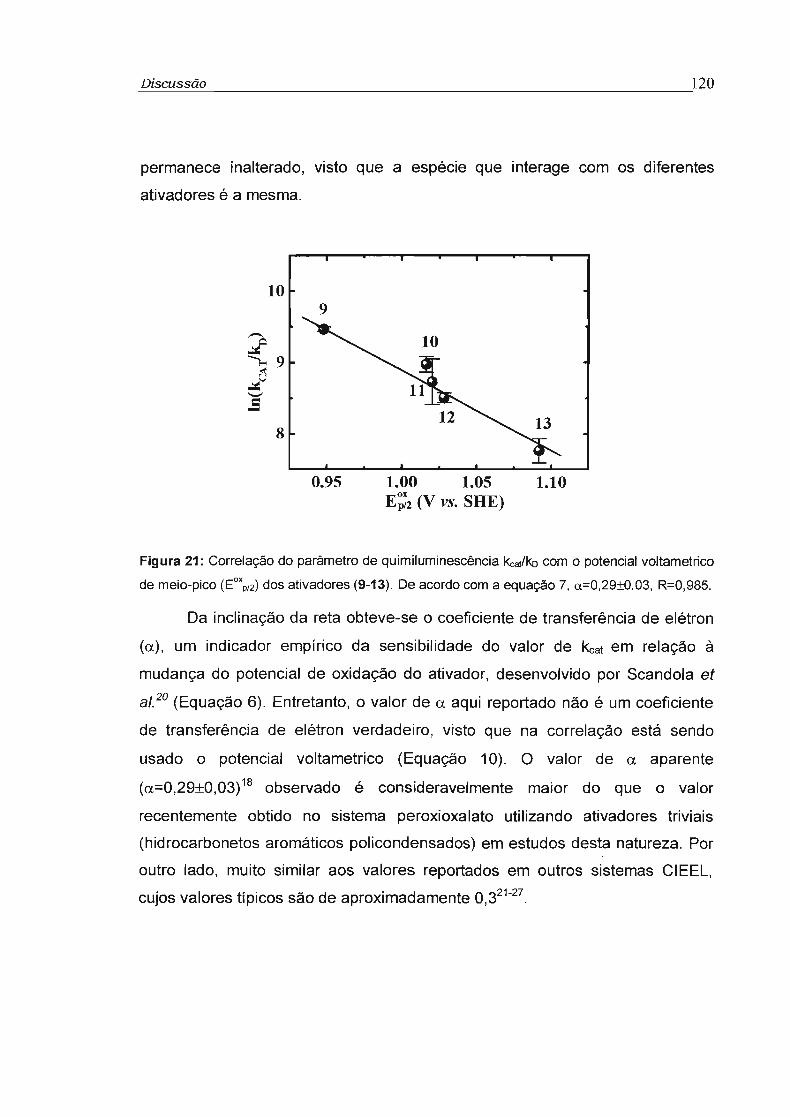
J)i"'rllssão
permanece inalterado, visto que a espécie que interage com os diferentes
ativadores é a mesma.
109
~'f... 9
6~'-'
=-8
0.95 1.00 1.05E;:2 (V vs. SHE)
1.10
Figura 21: Correlação do parâmetro de quimiluminescência kcat/ko com o potencial voltametrico
de meio-pico (Eoxp/2) dos ativadores (9-13). De acordo com a equação 7, a=O,29±O,03, R=O,985.
Da inclinação da reta obteve-se o coeficiente de transferência de elétron
(a), um indicador empírico da sensibilidade do valor de !<cat em relação à
mudança do potencial de oxidação do ativador, desenvolvido por Scandola et
al. 2o (Equação 6). Entretanto, o valor de a aqui reportado não é um coeficiente
de transferência de elétron verdadeiro, visto que na correlação está sendo
usado o potencial voltametrico (Equação 10). O valor de a aparente
(a=0,29±0,03)18 observado é consideravelmente maior do que o valor
recentemente obtido no sistema peroxioxalato utilizando ativadores triviais
(hidrocarbonetos aromáticos policondensados) em estudos desta natureza. Por
outro lado, muito similar aos valores reportados em outros sistemas CIEEL,
cujos valores típicos são de aproximadamente 0,321-27
.

Discussão 121
kCAT = AeXp[-a(Eox -Ered~J]RT Ro&
ln(kCAT )= lnA +aB-(;T )Eox
e2 Eonde:B= +~
Ro&RT RT
Equação 10
onde: A e B são constantes, a é O coeficiente de transferência de elétron, R é a constante geral
dos gases (8,613 e-15 eV K"\ T é a temperatura em Kelvin (295±2K), Eox é o potencial de
oxidação do ACT, Ered é o potencial de redução do IAE, que no caso é constante, pois a
espécie é a mesma para todos os ativadores, e é a carga do elétron, Ro é a distância entre os
íons no complexo de transferência de carga no ponto de transição e E é a constante dielétrica
do solvente.
Ao contrário do comportamento observado para os valores de kcat/ko, não
há correlação entre o In(<Dsoo) e o EO\/2 dos ativadores (Figura 22). Esta
correlação não é esperada pelo mecanismo CIEEL, pois <Dsoo é obtido pela
extrapolação do gráfico duplo-recíproco na concentração infinita do ativador,
portanto não depende da constante de velocidade de interação do IAE com o
ACT (kcat) e conseqüentemente é independente do potencial de oxidação dos
ativadores (Esquema 33). Porém, o 13, com maior potencial de oxidação,
resultou no rendimento de excitação mais alto (vide resultados), enquanto que
os ativadores mais facilmente oxidáveis (9-10) resultaram em rendimentos mais
baixos. A correlação empírica entre a intensidade de quimiluminescência e o
potencial de oxidação dos ativadores tem sido reportada na literatura em vários
estudos envolvendo o mecanismo CIEEL, nos quais os maiores rendimentos
quântico foram obtidos com ativadores com baixo potencial de oxidaçã022-26
.
Entretanto, a intensidade de emissão em certas concentrações de ativadores
depende de ambos, <Ds00 e kcat. Os resultados da literatura são, em uma primeira
análise, contrários aos obtidos no presente trabalho. Entretanto, analisando
estas observações mais detalhadamente é necessário verificar dois pontos
importantes que poderiam esclarecer esta aparente contradição: (i) Na maioria
dos trabalhos reportados na literatura, como por exemplo, Catheral e
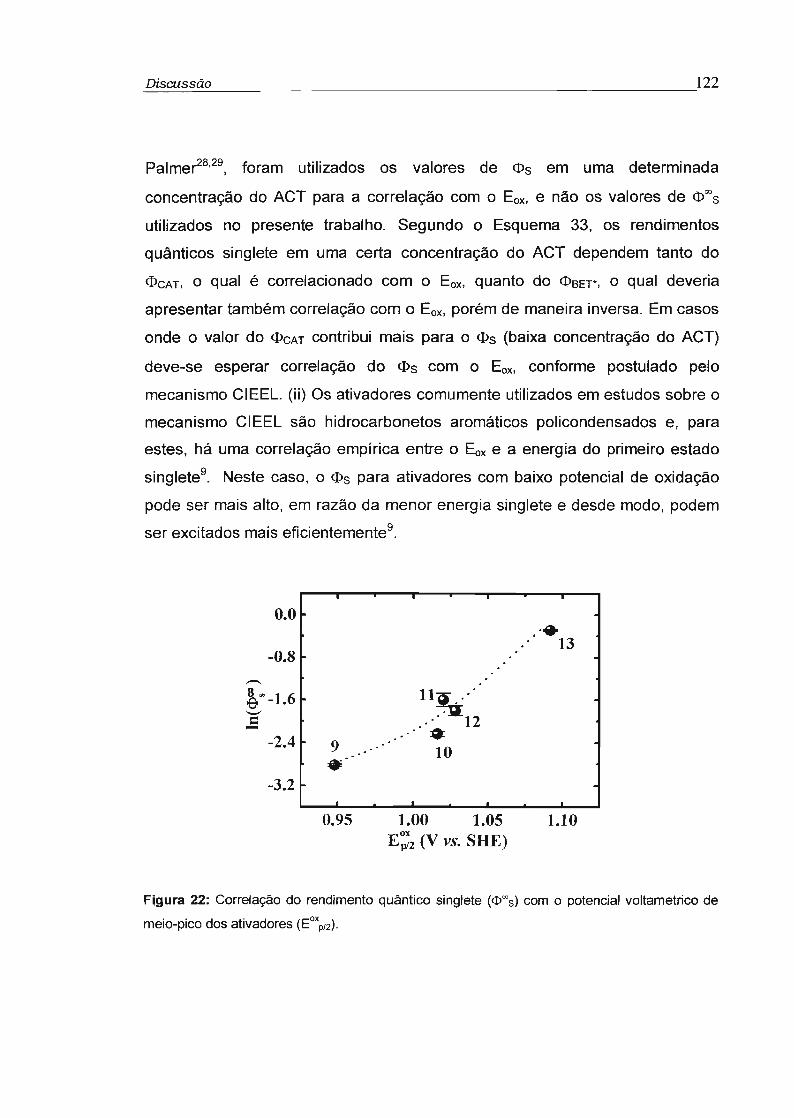
Lnscussão 122
Palme,-28,29, foram utilizados os valores de <1>s em uma determinada
concentração do ACT para a correlação com o Eox, e não os valores de <1>CXJs
utilizados no presente trabalho. Segundo o Esquema 33, os rendimentos
quânticos singlete em uma certa concentração do ACT dependem tanto do
<1>CAT, o qual é correlacionado com o Eox, quanto do <1>BET*, o qual deveria
apresentar também correlação com o Eox, porém de maneira inversa. Em casos
onde o valor do <1>CAT contribui mais para o <1>s (baixa concentração do ACT)
deve-se esperar correlação do <1>s com o Eox, conforme postulado pelo
mecanismo CIEEL. (ii) Os ativadores comumente utilizados em estudos sobre o
mecanismo CIEEL são hidrocarbonetos aromáticos policondensados e, para
estes, há uma correlação empírica entre o Eox e a energia do primeiro estado
singlete9. Neste caso, o <1>s para ativadores com baixo potencial de oxidação
pode ser mais alto, em razão da menor energia singlete e desde modo, podem
ser excitados mais eficientemente9.
• . • . • .O.O~
e'.e 13
-0.8 .. .
---t--1•6t 11~,e -
e··· 12-2.4 ' :O:
9 10.,-3.2 ~
• . •0.95 1.00 1.05 1.10
E;2 (V vs. SHE)
Figura 22: Correlação do rendimento quãntico singlete (<1>"'5) com o potencial voltametrico de
meio-pico dos ativadores (Eoxp/2).
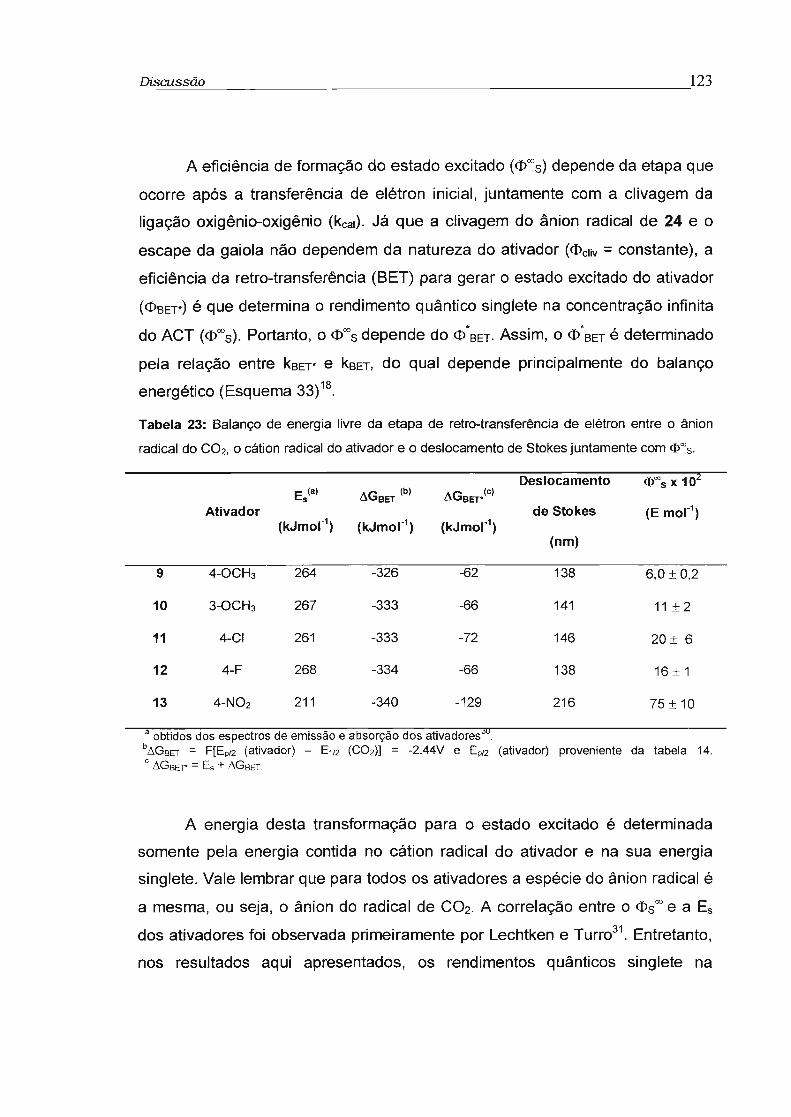
Lnscussão 123
A eficiência de formação do estado excitado (<1>oos) depende da etapa que
ocorre após a transferência de elétron inicial, juntamente com a c1ivagem da
ligação oxigênio-oxigênio (kcat). Já que a c1ivagem do ânion radical de 24 e o
escape da gaiola não dependem da natureza do ativador (<1>cliv =constante), a
eficiência da retro-transferência (BET) para gerar o estado excitado do ativador
(<1>SET*) é que determina o rendimento quântico singlete na concentração infinita
do ACT (<1>oos). Portanto, o <1>oos depende do <1>*SET. Assim, o <1>*SET é determinado
pela relação entre kSET* e kSET, do qual depende principalmente do balanço
energético (Esquema 33)18.
Tabela 23: Balanço de energia livre da etapa de retro-transferência de elétron entre o ânion
radical do COz, o cátion radical do ativador e o deslocamento de Stokes juntamente com <1>"'s.
DeslocamentoEs(a) ~GBET
(b)~GBET'
(e)
Ativador de Stokes(kJmor1
) (kJmor1) (kJmor1
)
(nm)
9 4-0CH3 264 -326 -62 138
10 3-0CH3 267 -333 -66 141
11 4-CI 261 -333 -72 146
12 4-F 268 -334 -66 138
13 4-NOz 211 -340 -129 216
<1>"'5 X10'
(E mor1)
6,0 ± 0,2
11 ± 2
20 ± 6
16 ± 1
75 ± 10
a -obtidos-dos espectros de emissão e absorção dos ativadores'30.b.6.GBET = F[Ep/2 (ativador) - E1/2 (C02)] = -2.44V e Ep/2 (ativador) proveniente da tabela 14.c .6.GBET" =Es + .6.GBET
A energia desta transformação para o estado excitado é determinada
somente pela energia contida no cátion radical do ativador e na sua energia
singlete. Vale lembrar que para todos os ativadores a espécie do ânion radical é
a mesma, ou seja, o ânion do radical de COzo A correlação entre o <1>soo e a Es
dos ativadores foi observada primeiramente por Lechtken e Turro31. Entretanto,
nos resultados aqui apresentados, os rendimentos quânticos singlete na

Lnscussão 124
concentração infinita (<1>500) de vários ativadores podem ser correlacionados com
o balanço energético do processo de retro-transferência de elétron (BET)18. A
mudança de energia livre associada com o processo de BET do ânion radical
de C02 para o cátion radical dos ativadores, o qual resulta em produtos no
estado fundamental (~GBET), pode ser estimado do potencial de meio-pico dos
ativadores (Tabela 23) e do potencial de redução do C02 em acetonitrila,
E1/2=-2,44 V vs.SHE32.
Conforme o esperado, esta mudança de energia livre, ~GBET, é maior
para ativadores com maior potencial de oxidação (Tabela 23), muito embora
haja uma diferença relativamente pequena entre os valores. O balanço de
energia livre proveniente do processo de retro-transferência (BET) para obter o
ativador no estado singlete excitado (S1) é obtido considerando a energia
singlete (Es ) dos ativadores e a ~GBET (Figura 23) O valor de ~GBET· é muito
similar para os ativadores 9 a 12, porém um valor significativamente maior é
obtido para o ativador 13, principalmente devido a sua energia singlete mais
baixa. Uma correlação razoável, porém não linear, foi obtida entre o ~GBET·
calculado e o <1>500 determinado experimentalmente para os ativadores 9 a 13
(Figura 24).

Lnscussão 125
Figura 23: Potencial de energia livre para o processo de retro-transferência de elétron (BET-)
entre o anion radical do CO2 e o cátion radical do ativador.
i • i . i . i . i . i
O~ ........-...... . 13. -
~-1[...
~ 12 ......
:s l1~.l ....-2 /
10.-0:I
-3'- *9i , i • i • i • i • i
60 75 90 105 120 135L\GBET.(kJmor
1)
Figura 24: Correlação do parâmetro de quimiluminescência <1>500 com a mudança na energia
livre na etapa de retro-transferência para a formação do estado excitado do ativador (~G8ET-).

Lnscussão 126
o ativador que possui o ~GBET* mais favorável também possui o
rendimento quântico singlete na concentração infinita (<DsOO) mais alto, e os
ativadores 9 a 12 para os quais os valores de ~GBET* são próximos, também
possuem rendimentos de excitação (<DsOO) similares. Além do balanço de energia
mais favorável, o <Dsoo obtido com o ativador 13 deve estar relacionado com
fatores geométricos, visto que este ativador possui um deslocamento de Stokes
de 216 nm contra aproximadamente 140 nm observado nos ativadores 9 a 12.
Este fato indica que o primeiro estado singlete excitado do ativador 13 é
estruturalmente diferente do estado fundamental e isto deve ser uma razão
adicional para a maior eficiência para a formação do estado excitado na etapa
de retro-transferência do ânion radical do C02 para o cátion radical do ativador
13, presumindo que este radical deva ser estruturalmente similar ao estado
singlete excitado.
Até o inicio deste estudo, nenhum composto com estrutura diferente de
hidrocarboneto aromático policondensado havia sido utilizado no estudo do
mecanismo CIEEL. Sendo assim, este trabalho mostra que compostos, como,
por exemplo, derivados de esteróides, com estruturas e propriedades foto
físicas diferentes dos ativadores comumente utilizados podem ser aplicados
neste tipo de estudo e os resultados obtidos demonstram claramente a validade
do mecanismo CIEEL para o passo de quimiexcitação da reação peroxioxalato.

Lnscussão 127
4.5 Influência da viscosidade no rendimento quântico singlete
A mudança de viscosidade do meio não afeta ou, em alguns casos,
interfere muito pouco na eficiência de transferência radiativa. Entretanto, pode
influenciar substancialmente a polarização da emissão de luz e a eficiência de
certos processos não-radiativos. Geralmente, estes parâmetros são relevantes
em processos de transferência de energia dependentes da formação de
complexos de carga, os quais são estabilizados pelo aumento da viscosidade33.
Em outras palavras, a mudança do solvente poderia ou não aumentar o
rendimento de emissão de luz em processos fotofísicos.
No caso de processos quimiluminescentes, com formação de complexo de
carga e transferência de elétron, a viscosidade desempenha um papel
fundamental. Como por exemplo, utilizando-se o sistema difenoil-peróxido, o
rendimento de emissão mais baixo foi observado em solventes polares, não
viscosos como acetonitrila e diclorometano. Por outro lado, utilizando ftalato de
dimetila, o qual tem constante dielétrica semelhante ao dicloro metano, porém
mais viscoso, o rendimento de emissão foi aumentado em três vezes. Koo e
Schuste~2 observaram que o rendimento de emissão mais alto foi observado
com éter dietílico (baixa constante dielétrica e viscosidade), enquanto que,
utilizando benzeno como solvente, menos polar e mais viscoso do que éter
dietílico, o rendimento de emissão foi cinco vezes menor. Isto mostra que a
constante dielétrica ou a viscosidade não são os únicos parâmetros que podem
influenciar a transferência de elétron em reações com propriedades de
quimiluminescência.
Adam et a/. 34, estudaram o efeito do aumento da viscosidade, utilizando
benzeno e difenilmetano, sobre a eficiência de transferência de elétron de um
dioxetano-spiroadamantil substituído. Neste trabalho, o autor mostra que para
a escolha do solvente deve-se levar em consideração a teoria de transferência
de elétron de Marcus35.

Lnscussão 128
Segundo esta teoria, qualquer alteração na constante dielétrica do
solvente pode afetar a velocidade de transferência de elétron, pois esta é
controlada pela energia de reorganização que engloba reorganização das
ligações (estiramento, deformação angular e movimento torsional) e
reorganização do solvente (mudança eletrostática em volta dos reagentes).
Uma transferência de elétron ocorre de acordo com o principio de Franck
Condon, ou seja, a transferência é muito mais rápida que o movimento nuclear.
Desse modo, a velocidade de transferência de elétron é controlada apenas pela
energia de reorganização36.
e2 (1 1 1XII)110; =4 2r + 2r ---;- n2 - & Equação 11
D A AD
onde:
e =carga do elétron
ra e rD = são os raios moleculares do aceptor de elétron e do doador de elétron,
respectivamente.
rDA =representa o diâmetro de colisão do doador de elétron e do aceptor de elétron.
s =constante dielétrica do solvente
n =índice de refração do solvente.
De acordo com a equação acima, a utilização de solventes com
polaridades diferentes, deveria afetar o processo de transferência de elétron,
principalmente quando o fator ~2 - X) difere significativamente de um
solvente para o outro. Na prática, esta teoria é utilizada apenas como uma
ferramenta para a escolha da mistura de solventes mais adequada, de modo
que o efeito observado seja proveniente apenas da mudança da viscosidade.
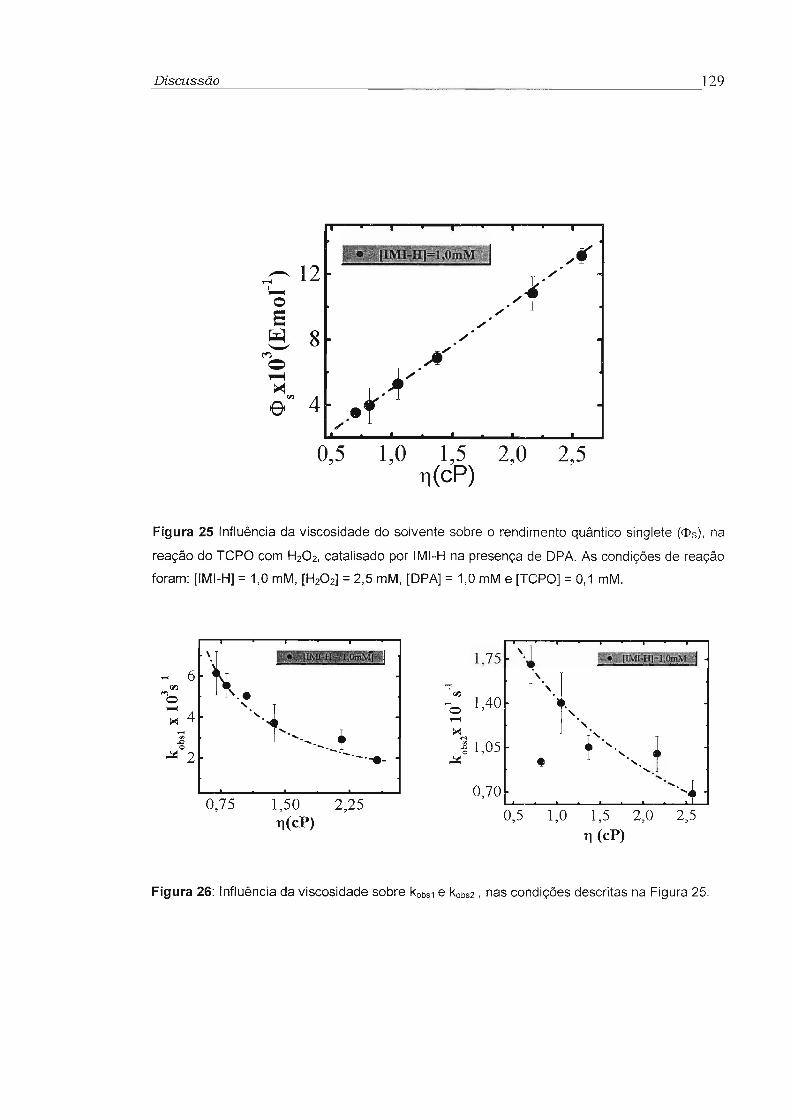
Discussão 129
/f'~ 12 4'/-I-o ,/ 18 /
/
~ 8 /'-" ,A/~
o~ .•f"-+/~
'"e/ . ,
0,5 1,0 1,5 2,0 2,511 (cP)
Figura 25 Influência da viscosidade do solvente sobre o rendimento quântico singlete (<Ds) , na
reação do TCPO com HzOz, catalisado por IMI-H na presença de DPA. As condições de reação
foram: [IM l-H] =1,0 mM, [HzOz] =2,5 mM, [DPA] =1,0 mM e [TCPO] =0,1 mM.
0,75 1,50 2,2511(cP)
-~o
~ 2
2,52,01,0
•
\'1\. li\Bt:
,·t,.~,.,. t1 ' .
'-'-...........~
1,75
0,70, , , , , ,I ,
0,5
~N
~ 1,05~
-171
-o 1,40-\>.,J~!$2!*~~~ ~~r_:i~ti>~
6 ~ '.~ ~
Tl.~ 1". ""'.-.J
.-.-....-
";'",171
o-~ 4
Figura 26: Influência da viscosidade sobre kobs1 e kobsZ , nas condições descritas na Figura 25.
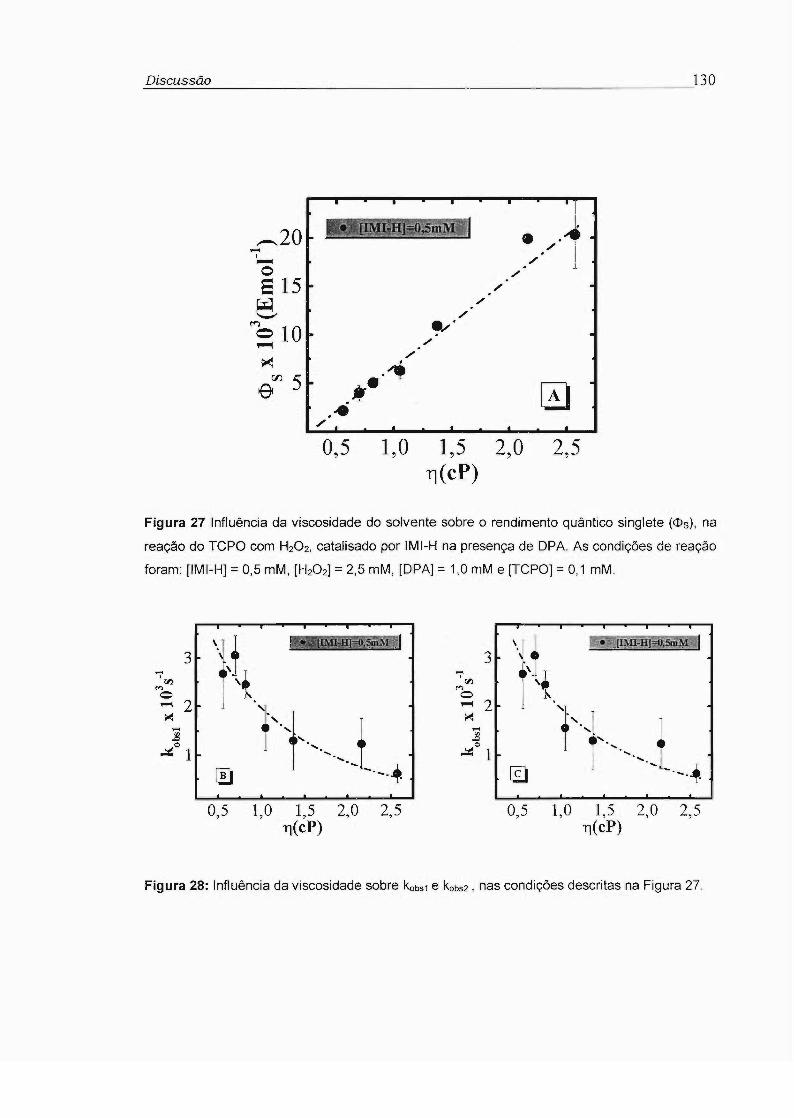
Discussão 130
,/.,/.
,/
e.....,/.
,/
.~. '-
0ff.4
'/, , , .0,5 1,0 1,5 2,0 2,5
11 (cP)
,/
_,-20I-oe 15~eQ 10,.....(
~
erJi
5
K Ú '0 f(· i:r~ ,4i"<'t: . õ:Y."~':)l ....~~~~-!tt! II ~ \ -/"1
Figura 27 Influência da viscosidade do solvente sobre o rendimento quântico singlete (<I>s) , na
reação do TCPO com H20 2, catalisado por IMI-H na presença de DPA. As condições de reação
foram: [IM l-H] = 0,5 mM, [H20 2] = 2,5 mM, [DPA] = 1,0 mM e [TCPO] = 0,1 mM.
~'::. '~~5)Pl1tlJ,~ 'Mlml~Sin"1W.:·'1 1.,~.
~
1'-1 lT 1 ..... ·..... ·.....·-1 T-.-..,@}
3
-:E~Ol
~
'~...,o...-( 2
'1 1'1~
"1'"1'"'"'-,-1,_,[!J
3'~...,~ 2~
~o
~ 1
0,5 1,0 1,5 2,0 2,5l1(CP)
0,5 1,0 1,5 2,0 2,5l1(CP)
Figura 28: Influência da viscosidade sobre kobs1 e kobs2 , nas condições descritas na Figura 27.

Lnscussão 131
No estudo apresentado neste trabalho utilizando tolueno e difenilmetano,
esta condição foi respeitada. A reação do bis(2,4,6-triclorofenil) oxalato com
peróxido de hidrogênio, utilizando imidazol como catalisador na presença de
9,10-difenilantraceno (OPA) em misturas variadas de tolueno/difenilmetano
apresentou um aumento no rendimento quântico singlete (<1>8) à medida que
aumentou-se a viscosidade do solvente com difenilmetano. Sabe-se que na
reação peroxioxalato há várias etapas consecutivas e paralelas que levam a
formação do intermediário de alta energia (IAE) e que são regidas pela
reatividade. Após a formação do intermediário, em etapas anteriores, ocorre a
interação deste com o OPA, via transferência de elétron dentro da gaiola de
solvente (Esquema 32).
O aumento no rendimento quântico singlete (<1>8) em função do aumento
da viscosidade (vide resultados), indica um comportamento compatível com o
mecanismo CIEEL (Esquema 32), isto é, a etapa de quimiexcitação ocorre
dentro de uma gaiola de solvente e o aumento da viscosidade do solvente,
diminuiu a difusão das espécies responsáveis pela excitação do ativador, para
fora da gaiola, resultando no aumento da emissão de luz.
Por outro lado, as constantes de velocidade observadas (kobs1 e kobs2)
apresentaram uma variação nos seus valores, indicando que a reação torna-se
mais lenta (Figuras 26 e 28). Entretanto, isto não seria um comportamento
esperado, visto que a etapa de formação do intermediário de alta energia (IAE)
não é regida somente pela difusão, mas também pela reatividade. Portanto, o
rendimento da reação, <1>R, deveria manter-se constante na reação (Esquema
33), na prática as constantes de velocidade (kobs1 e kobs2) não deveriam sofrer
variação. O fato é que até o momento não há uma proposta para tal
observação. Provavelmente, trabalhos futuros poderão responder tal questão.
De acordo com os resultados obtidos, o rendimento quântico singlete
(<1>8) aumenta até 10 vezes com o aumento da viscosidade de 0,5 para 2,5 cP.
Utilizando uma mistura de solventes semelhantes, benzeno e difenilmetano,
Adam et al. 34, observaram um aumento de 2,5 vezes no <1>8 do sistema

Discussão 132
spiroadamantil. Com base nestes resultados, propuseram que a eficiência da
quimiexcitação depende da formação da gaiola de solvente, uma vez que, a
etapa de retro-transferência de elétron exerce papel importante no mecanismo,
constituindo um mecanismo CIEEL intermolecular.
O que se pode concluir dos nossos resultados é que o mecanismo, o
qual resulta na excitação do ativador, ocorre em vários passos consecutivos, e
não em um processo "concertado" envolvendo transferência de carga em um
único passo conforme foi sugerido por Wilson37, como uma possível alternativa
ao mecanismo CIEEL.

Discussão 133
4.6 Estudo do Sistema Peroxioxalato na ausência de H20 2
A emissão de luz observada no sistema peroxioxalato sem adição de
peróxido de hidrogênio apresentou intensidade de emissão muito baixa38. Lee
et aI. 38, propuseram que o intermediário formado antes da 1,1'-di-imidazolida
(001) (Esquema 34), denominado de "Z", seria o responsável pela emissão de
luz. Os autores relataram que o intermediário "Z", muito instável, interage com o
ativador resultando em emissão de luz. Porém não foi proposto, qual seria este
intermediário.
k4
Esquema 34
o:JArOrt~ ~N
k., o Ni'J k2 o N
fJi f· Hk"l N ArO oF~H ~ArO- j
H-N~ k5
pNwN~
~N Zo N-.!J r~\'----i ACT ACr
não emite .. ACT ~i- "'o Y
0'J_tAr+ <;N)
ArO o ,N hH
hv
No presente trabalho, a intensidade de luz observada na reação de
TCPO e ONPO sem adição de H20 2 , apresentou intensidade de emissão de
aproximadamente 3 vezes menor do que a intensidade observada com a
concentração de H20 2 igual a 5.10-1oM (vide resultados). Esta concentração é
tão baixa que poderia ser proveniente de outras fontes de contaminação,
inclusive de traços de H20 2 presentes no solvente. Após vários experimentos
verificou-se que utilizando uma concentração tão baixa, qualquer traço de
impureza que gerasse uma fraca emissão de luz, poderia ser detectado pelo
aparelho sem ser necessariamente proveniente de um intermediário não
peroxídico formado na reação peroxioxalato. Deste modo, é necessário ter
cautela ao inferir novas informações a respeito do mecanismo, visto que se

Lnscussão 134
trata de um sistema bastante complexo. Por fim, independentemente da
estrutura do intermediário de alta energia formado na etapa de quimiexcitação,
a sua contribuição é insignificante para a eficiência do processo global que leva
a emissão de luz nas condições geralmente utilizadas na reação. A diferença
na intensidade de emissão de luz entre a reação de DNPO com adição de
peróxido de hidrogênio ([H202 = 5.1 0-5M]) , observado no espectrofluorímetro, e
sem adição de H20 2 observado no contador de fótons é de aproximadamente
160000 vezes. Sendo assim, podemos concluir que na reação peroxioxalato
não há um caminho de reação sem o envolvimento de peróxido de hidrogênio
que leve a formação eficiente de estados eletronicamente excitados.

Discussão 135
4.7 Síntese de derivados de Oxazolonas (14-21)
Conforme foi mencionado na introdução, a reação peroxioxalato é um
dos sistemas mais sensíveis em determinações de compostos fluorescentes
reportado na literatura. Por isso, têm sido largamente utilizados em aplicações
de química analítica, incluindo determinação de ácidos graxos, esteróides, etc...
Entretanto, em alguns compostos, a reação de derivatização, antes da análise,
é necessária para introduzir propriedades fluorescentes dentro da molécula, a
ser analisada, para aumentar a sua sensibilidade. Para este propósito, vários
tipos de reagentes de derivatização são comumente utilizados.
A quimiluminescência tem sido largamente aplicada em diversas áreas
de biotecnologia, farmacologia, biologia molecular, clínica e química, sendo que
o sistema que mais se destaca é a reação peroxioxalato. Portanto, torna-se
importante conhecer o mecanismo deste sistema para que seja possível ampliar
e melhorar a eficiência de sua aplicação. Do ponto de vista analítico, este
sistema é um dos mais interessante reportado na literatura, pois possui um alto
rendimento quântico de quimiluminescência, comparado apenas a sistemas
biológicos, ou seja, para um químico analítico é considerado altamente sensível.
Neste contexto, o objetivo deste trabalho foi desenvolver algumas
metodologias para a síntese de novos derivados de esteróides para o estudo
mecanístico do sistema peroxioxalato e para futuras aplicações analíticas.
Para preparar estes compostos, foram utilizadas as reações de Barton
Kellog39 e Sonogashira4o, os quais resultaram nos compostos 15 e 21. A reação
de Barton-Kellog é um caminho alternativo para a preparação de olefinas
trissubstituidas e tetrassubstituidas (Esquema 35).
Esquema 35R1 X R3 R1 R3
X X .. >=< +x+yR2 y R4 R2 R4
A reação de Sonogashira possibilita que um hidrogênio acetilênico possa
ser facilmente substituído por iodoarenos, bromoalcenos ou bromopiridinas na

Lhscussão 136
presença de uma quantidade catalítica de dicloreto de bis(trifenilfosfina)paládio
iodeto cuproso em dietilamina sob condições brandas.41,42 (Esquema 36).
Esquema 36
1-0 + H== H (Ph3P)PdClz
Cul EtzNH•~
A reação é iniciada pela adição do haleto ao Pdo gerado in situ a partir do
Pd(II). O intermediário arilpaládio forma um complexo com o alceno resultando
na formação da ligação carbono-carbono e regeneração do Pdo, sendo que o
Cul tem a função de co-catalisador40.
4.7.1 2-[4-benzilacetileno]-5,5-dimetiloxazol-4-ona (15)
Na preparação do composto 15 foi observada a formação de um produto
secundário em grande quantidade, o dímero do fenilacetileno. De acordo, com o
mecanismo proposto por Sonogashira para a formação destes compostos, a
reação seria iniciada pela inserção de duas moléculas de fenilacetileno ao
complexo de paládio, seguida da eliminação do dímero para gerar o Pdo
(Esquema 37). Vale lembrar que o complexo de paládio é usado em
quantidades catalíticas; portanto, bastaria uma quantidade também catalítica de
fenilacetileno para gerar o Pdo no primeiro passo. Após a formação da espécie
reduzida, este entra no ciclo da reação, no qual seria regenerado sem a
necessidade de consumir mais fenilacetileno no primeiro passo. Entretanto, o
rendimento da reação foi baixo, devido a fonnação do dímero do fenilacetileno.
Segundo o mecanismo proposto, o fenilacetileno tem duas funções na reação:
contribuir para a redução do paládio, e neste caso ocorre a formação do dímero
e agir como reagente de acoplamento. Talvez uma maneira de melhorar o
rendimento da reação, seria adicionar, aos poucos, uma quantidade maior de
fenilacetileno durante a reação.

Lnscussão 137
Esquema 37
CuI + N(C,H,)3
(Ph3PhPdClz _ .. (Ph3P)zPd(-C=C-Ar)z ). (Ph3PhPdO
2O-=-HAr Ar
(Ph,P.Pd'"",.~::(
~14~:rO15 ~
P1Ph0\3Nf=0Br-Pd !j ~ ,-
I - OPPh3
PPh3 ~ + N(C H,)
~~d~NrO/~:~I ~-b~ ~
PPh3
4.7.2 3-[2-(4-bromofenil)-5,5-dimetiloxazol-4-ilideno]-androsta-1,4
dien-17-ona (21)
A síntese do composto 21 foi iniciada pela preparação do derivado de
oxazolona 14. Na literatura43 foi descrito um método para síntese do sal de
oxazolônio, partindo de a-hidróxinitrilas e cloreto de ácido na presença de ácido
de Lewis, conforme demonstrado na reação de cianidrina de acetona com
cloreto de p-bromobenzoíla e tetracloreto de estanho (Esquema 38). O sal é
convertido em derivado de oxazolona quando tratado com trietilamina utilizando
tolueno como solvente.
De acordo com a proposta para o mecanismo, a reação seria iniciada
pela adição nucleofílica do grupo CN à carbonila do cloreto de benzoíla
formando uma amida com carga. A etapa seguinte foi o ataque nucleofílico do
grupo OH à mesma carbonila formando um anel cíclico que, após alguns
equilíbrios, resulta na formação o sal (M). O tratamento de M com trietilamina
resulta na oxazolona 14.
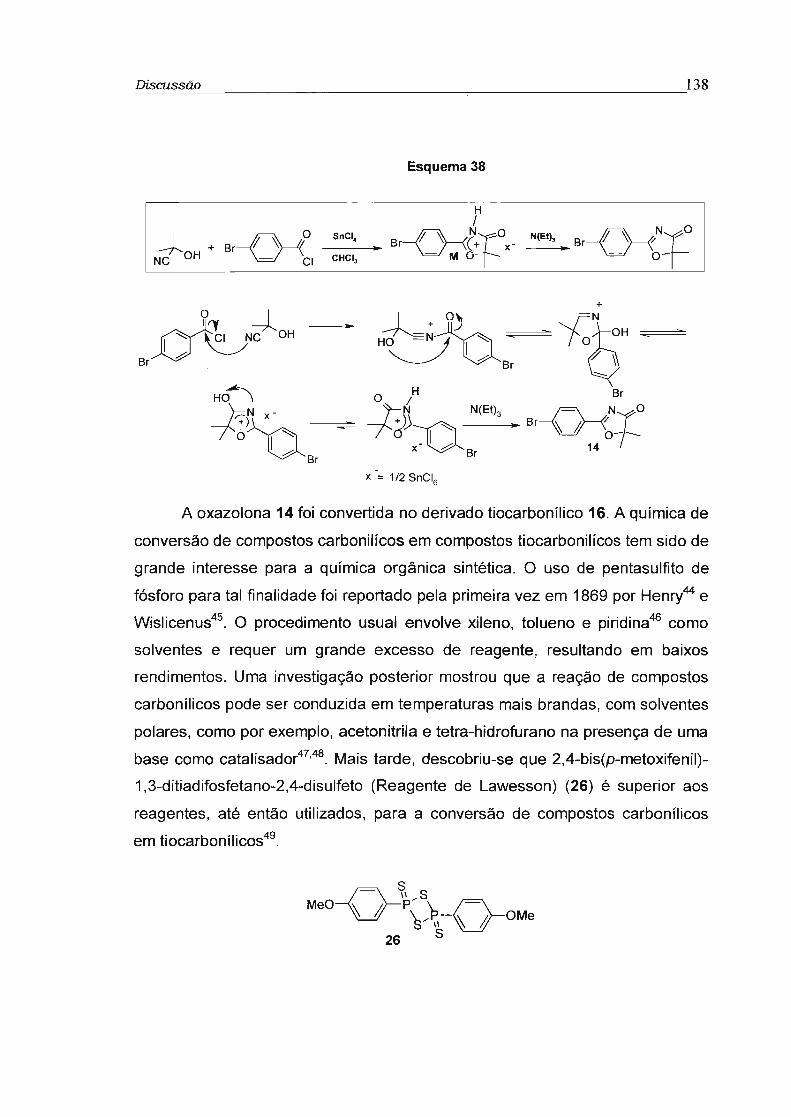
Lnscussão 138
Esquema 38
H/
I --cH\\o 5nCI. ~(N1=0--+-- + Br \ • Br~; + x-NC OH - CI CHCI, - M o·
N(Et), Br~- ijNyO
- \J\0-r
+
'foqN OH
7' \~
BrNyO
- Br-Q--fo;--14
I + O)---f-....=:~NfQ ~HO I
h- Br
HO I N(Et)3
-M~:-
..
HO~
~~llABr
~Ory ~
I rCI NC OH
Br h- "---/
x = 1/2 SnCls
A oxazolona 14 foi convertida no derivado tiocarbonílico 16. A química de
conversão de compostos carbonilícos em compostos tiocarbonilícos tem sido de
grande interesse para a química orgânica sintética. O uso de pentasulfito de
fósforo para tal finalidade foi reportado pela primeira vez em 1869 por Henry44 e
Wislicenus45. O procedimento usual envolve xileno, tolueno e piridina46 como
solventes e requer um grande excesso de reagente, resultando em baixos
rendimentos. Uma investigação posterior mostrou que a reação de compostos
carbonílicos pode ser conduzida em temperaturas mais brandas, com solventes
polares, como por exemplo, acetonitrila e tetra-hidrofurano na presença de uma
base como catalisador47,48. Mais tarde, descobriu-se que 2,4-bis(p-metoxifenil)
1,3-ditiadifosfetano-2,4-disulfeto (Reagente de Lawesson) (26) é superior aos
reagentes, até então utilizados, para a conversão de compostos carbonílicos
em tiocarbonílicos49.
S-o\\SV-p/ OMeMeO ~ t1 's/~ ~ t1S26

Discussão 139
Conforme relatado por Cava e Levinson49, este reagente não é estável
em temperaturas acima de 110°C, sofrendo polimerização lenta e outras
decomposições. Há uma série de especulações a cerca do mecanismo de
reação de 26 com compostos carbonílicos. Entretanto, Lawesson sugeriu que
uma espécie dipolar é formada em baixas concentrações, semelhante ao ilídeo
de Wittig. O ataque nucleofílico desta espécie à carbonila e "colapso" da nova
espécie dipolar formada, resulta em um anel heterocíclico de quatro membros,
cuja clivagem gera o composto tiocarbonílico 16 e uma nova espécie dipolar de
fósforo (Esquema 39).
Esquema 39
ÚOMe
o-~' ~s I s
"'::::: P, ~'p:::"'" - > - 11I s " 2 Me0-O--1.P-S-MeO ~ S II +
-o-~ ~ -~y9!MeO _ ~-s Br~o~.-
o
B'~:+s-~-O--OMe
s
~Ni=~~Br-~~T-s V o:-
~/,N+S -0--+Br~ + MeO ~ ~ ~-S-
o o16
A etapa seguinte foi a formação do derivado diazo 20, partindo-se
inicialmente do composto 17. Conforme a literatura50, não é possível a reação
direta de 17 com hidrazina hidratada devido à formação de uma mistura de
produtos, pois a reação não é seletiva. A alternativa encontrada foi preparar um
derivado 3-tiocarbonílico 18. A conversão de 17 em 18 ocorreu quase que
quantitativamente na presença de 26 (Esquema 40). Sendo a tiocarbonila da
posição 3 mais reativa do que a carbonila da posição 17, esperar-se-ia que a
formação do composto 19, a partir do 18, fosse mais favorecida. A formação da
hidrazona (19) é semelhante à reação de redução de cetonas e aldeídos de
Wollf-Kishner. Entretanto, na redução Wollf-Kishner a hidrazona não é

Lnscussão 140
isolada5o. A formação do composto diazo 20 ocorreu via reação de oxidação,
em meio básico, com dióxido de manganês em carvão ativo.
Esquema 40
A
o
H2NNH2'xH,o~ "--........,,,- 7'THF,OOC 19
/. .&H
2N-Nd9!
0
26
s-7'.& 18o~26 =Reagente de LawessonlTHFA =MnO!carvão ativo/KOH
A etapa final para a formação do composto 21 foi o acoplamento dos
compostos 16 e 20 via reação de 8arton- Kellog. Geralmente, a formação de
olefinas tetrassubstituidas é seriamente afetada pelo impedimento estérico, por
isso seus rendimentos são, na maioria das vezes, baixos39. A reação de Wittig
tem sido largamente utilizada na preparação de olefinas dissubstituídas, mas o
rendimento de formação diminui à medida que se aumenta a substituição, como
no caso de olefinas trissubstituídas e tetrassubstituídas. A olefinação de 8arton
Kellog51 consiste em um caminho alternativo para a formação de olefinas com
impedimento estérico; como foi o caso da formação do composto 21. A adição
do sólido do composto 16 ao composto 20 gerou o composto 21 e mais dois
compostos, de acordo com a CCO. Para um destes compostos foi sugerida a
dimerização do composto 16, de acordo com a análise de espectrometria de
massa.
Esquema 41
N S
"'--Q:ot- + #
N2
N2+ ,..
N
Br~
Esta parte do trabalho tinha como objetivo desenvolver algumas
metodologias para a síntese de novos derivados de esteróides para o estudo
mecanístico do sistema peroxioxalato e futuras aplicações analíticas.

DiscussãoB , B L , O T E c A 141
INSTITUTO DE (,}UiIll.1C["Universidade de São Paulo
o composto 21 é o material de partida para que outros compostos
possam surgir, como por exemplo, substituição de bromo, via reação de
Sonogashira, por derivados que possam aumentar a conjugação do sistema.
Isto poderia promover o deslocamento da emissão de luz para comprimentos de
onda mais longos, e talvez aumentar o rendimento de fluorescência.
Esta etapa do trabalho fazia parte de um projeto que deveria ser
desenvolvido em três meses e cujo objetivo final seria preparar uma série de
compostos com propriedades fluorescentes para que fossem aplicados na
reação peroxioxalato e em sistemas analíticos. Sendo assim, foram preparados
dois compostos: 16 e 21. Além disso, foram preparados outros compostos (14,
15, 18, 19 e 20), os quais foram utilizados como reagentes intermediários de
reação.
O ponto importante desta etapa do trabalho, foi ter a oportunidade de
conciliar o trabalho de estudo cinético da reação peroxioxalato com a síntese de
compostos com propriedades fluorescentes que poderiam ser aplicados nesta
reação.

Lnscussão 142
4.8 Referências
1 Stevani CV & Baader WJ. "Kinetics studies on the chemiluminescence decomposition of and
isolation intermediate in the peroxyoxalate reaction. J.Phys.Org.Chem., 10, 593-9. 1997.
2 Stevani CV, "Estudo mecanístico do sistema peróxi-Oxalato", Tese de Doutorado,
Universidade de São Paulo, 1997.
3 Hohman JR, Givens RS, Carlson RG & Orosz G, "Synthesis and chemiluminescence of a
protected peroxyoxalate" Tetrahedron Lett., 37(46),8273-6, 1996.
4 Yagil G, "The proton dissociation constant of pyrrole, indole and related compounds",
Teifahedron, 23, 2855-61, 1967.
5 Hartley BS and Massey V, Annu. Rep. Prog. Chem., 51, 311, 1954, apud Hõfle G, Steglich W
& Vorbrüggen H, "4-Dialkylaminopyridines as highly active acylation catalysts", Angew.Chem.
Int. Ed. Engl., 17, 569-83, 1978.
6 Jencks PW & Gilchrist M, "Nonlinear structure-reactivity correlations. The reactivity of
nucleofilic reagents toward esters", J. Am. Chem. Soc., 2622-37, 1968.
7 Kirsch JF & Jencks W, "Base catalysis of imidazole catalysis of ester hydrolysis" J. Am. Chem.
Soc., 86, 833-7, 1964.
8 Fife TH, "Kinetic and mechanistic effects of ease of C-N bond breaking in amide hydrolisis. The
mechanisms of hydrolysis of N-acylimidazoles and N-acylbenzimidazoles" Acc. Chem. Res. 26,
.325-31, 1993.
9 Stevani CV, Silva SM & Baader WJ, "Studies on the mechanism of the excitation step in the
peroxyoxalate system. Eur. J. Org. Chem., 4037-46, 2000.
10 Dados apresentados em relatório interno.
11Turro NJ, Modem Molecular Photochemistry, The BenjaminlCummings Publishing Company,
Inc., 1978.
12 Stevani CV & Baader WJ, "O sistema quimiluminescente peróxi-Oxalato" Química Nova,
22(6), 715-23, 1999.

Lnscussão 143
13 Um l-H, Lee J-Y, Kim HT & Bae SK, "Curved hammett plot and hydrolysis of O-aryl
thionobenzoates: change in rate-determining step versus ground-state stabílization", J. Org.
Chem., 69,2436-41,2004.
14 EI Seoud OA, Ruasse M-F & Rodrigues WA, "Kinetics and mechanism of phosphate
catalysed hydrolysis of benzoate esters: comparison with nucleophilic catalysis by imidazole and
o-iodosobenzoate", J. Chem. Soc., Perkin Trans. 2, 1053-8, 2002.
15 Bruice TC, Fife TH, Bruno JJ & Brandon NE, "Hydroxyl group catalysis. 11. The reactivity of the
hydroxyl group of serine. The nucleophilicity of alcohols and the ease of hydrolysis of their acetyl
esters as related to their pKa", Biochemistry, 1, 7-12, 1962.
16 Suh J & Lee BH, "Structure-reactivity relationships in nucleophilic reactions on cinnamoyl
azide and phenyl cinnamates. Kinetic stability of the acyl azide and reiative leaving ability of
nitrogen and oxygen", J. Org. Chem., 45, 3103-7,1980.
17 Castro EA, Leandro L, Quesieh N & Santos JG, "Kinetics and mechanisms of the reactions of
3-methoxyphenyl, 3-ehlorophenyl and 4-eyanophenyl 4-nitrophenyl thionocarbonatos with
alicyclic amines", J. Org. Chem, 66, 6130-5, 2001.
18 Silva SM, Wagner K, Weiss D, Beckert R & Baader WJ " Studies on the chemiexcitation step
in peroxyoxalate chemiluminescence using steroid-substituted activators" Luminescence, 17,
362-9, 2002.
19 Stevani CV, Lima DF Toscano VG & Baader WJ, "Kinetic studies on the peroxyoxalate
chemiluminescent reaction: imidazole as a nucleophilic catalyst. J. Chem. Soc. Perkin Trans. 2,
989-95, 1996.
20 Scandola F, Balzani V & Schuster GB. "Free-energy relationships for reversible and
irreversible electron-transfer processes." J. Am. Chem. Soc., 103, 2519-23, 1981.
21 Schuster GB, "Chemiluminescence of organic peroxides. Conversion of ground-state
reactants to excited-state products by the chemically initiated electron-exchange luminescence
mechanism." Acc.Chem. Res. 12, 366-373, 1979.

Lnscussão 144
22 Koo J-Y, Schuster GB, "Chemiluminescence of diphenoyl chemically initiated electron
exchange luminescence. A new general mechanism for chemical production of electronically
excited states." J. Am. Chem. Soc., 100, 4496-503, 1978.
23 Schmidt SP, Schuster GB, "Chemiluminescence of dimethyldioxetanone. Unimolecular
generation of excited singlet and tríplet acetone. Chemically initiated electron-exchange
luminescence, the primary Iight generating reaction." J. Am. Chem. Soe., 102, 306-14, 1980.
24 Dixon BG, Schuster GB, "Investigation of the mechanism of the unimolecular and the electron
donor-catalyzed thermal fragmentation of secondary peroxy esters. Chemiluminescence of 1
phenylethyl peroxyacetate by the chemically initiated electron-exchange luminescence
mechanism." J. Am. Chem. Soe; 101, 3116-8, 1979.
25 Smith JP, Schrock AK &, Schuster GB, "Chemiluminescence of organic peroxides. Thermal
generation of an o-xylylene peroxide." J. Am. Chem. Soc., 104, 1041-47, 1982.
26 Darmon MJ, Schuster GB. Thermal chemistry of cyciopropyl-substituted malonyl peroxides. A
new chemiluminescent reaction. J. Org. Chem., 47, 4658-64, 1982.
27 Adam W & Cueto O. "Fluorescer-enhanced chemiluminescence of a-peroxylactones via
electron exchange." J. Am. Chem. Soc., 101,6511-15,1979.
28 Catheral CLR & Palmer F. "Chemiluminescence from reactions of bis(pentachlorophenyl)
oxalate, hydrogen peroxide and f1uorescent compounds - Kinetics and mechanism", J. Chem.
Soe., Faraday Trans 2, ao, 823-34, 1984.
29 Catheral CLR & Palmer F. "Chemiluminescence from reactions of bis(pentachlorophenyl)
oxalate, hydrogen peroxide and fluorescent compounds - Role of the fluor and nature of
chemieletronic process (es), J. Chem. Soc., Faraday Trans 2, ao, 837-49, 1984.
30 Melhuish WH, "Quantum efficiencies of f1uorescence of organic substances: effect of solvent
and concentration of the fluorescent solute." J. Phys. Chem, 65, 229-35, 1961.
31 Lechtken P & Turro NJ. "Peroxyoxalate chemiluminescence. Chemiexcitation of high-energy
excited states in acceptor molecules." MoI. Photoehem., 6,95-9, 1974.

niCrJlssão
32 Chang M-M, Saji T & Bard AJ, "Electrogenerated chemiluminescence. 30. Electrochemical
oxidation of oxalate ion in the presence of luminescers in acetonitrile solutions." J. Am. Chem.
SOC., 99, 5399-403, 1977.
33 Campbell AK, Chemiluminescence -Principies and applications in biology and medicine,
Weinheim; Deerfield Beach, FI.: VCH; Chichester: Horwood, 1988.
34 Adam W, Bronstein I, Trofimov AV & Vasil' ev RF. "Solvent-cage effect (viscosity dependence)
as diagnostic probe for the mechanism of the intramolecular chemically initiated electron-
exchange luminescence (CIEEL) triggered from a spiroadamantyl-substituted dioxetane." J. Am.
Chem. Soc., 121, 958-961,1999.
35 Marcus RA. "On the theory of oxidation-reduction reactions involving electron transfer" Chem.
Phys., 24 (5),966-78, 1956.
36 Eberson, L "Electron transfer reactions in organic chemistry" Springer-Velarg, Berlim, 1987.
37 Frimer A, Singlete Oxygen, vol.2, CRC Press. Boca Raton, 1985.
38 Lee JH, Rock JC, Park SB, Schlautman MA & Carraway ER, "Study of the characteristics of
three high-energy intermediates generated in peroxyoxalate chemiluminescence (PO-CL)
reactions", J. Chem. Soc., Perkin Trans. 2, 2002, 802-9.
39 Barton DHR & Willis BJ, "Olefin synthesis by two-fold extrusion processes. Part I. Preliminary
experiments." J. Chem. Soco Perkin', 305-10, 1972.
40 Sonogashira K, Tohda Y & Hagihara N, "Convenient synthesis of acetylenes-catalytic
substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines."
Tetrahedron Lett., 4467-70, 1975.
41 Hudson RHE, Li G & Tse J, "The use of sonogashira coupling for the synthesis of modified
uracil peptide nucleic acid." Tetrahedron Lett., 43, 1381-6,2002.
42 Albrecht M Blau O & Rõttele H, "An ethylene-linked catechol/8-hydroquinoline derivative and
its dinuclear gallium (111) complex." New J. Chem., 24, 619-22, 2000.

Lnscussão 146
43 Dorofeenko GN, Karpenko VD & Ryabukhin Yu I, "Synthesis of 4(5H)-oxazolonium salts and
4(5H)-oxazolones from a-hidroxy nitriles", J. Org. Chem. USSR (Engl. Transl.) 14, 1767-71,
1978.
44 Henry L, "Über eine neue Bildguns-und Darstellungs-weise der Nitrile", Ann.Chem.Pharm.,
152, 148-52, 1869.
45 Wislicenus J, Z.Chem., 324, 1869, apud [49].
46 Campaigne E, "Thiones & thials", Chem. Rev., 1,39,1946.
47 Scheeren JW, Ooms PHJ & Nivard RJF, "General procedure for conversion of a carbonyl
group into a thione group with tetraphosphorus decasulfide", Synthesis 149-51, 1973.
48 Dash B, Dora EK & Panda CS, 'Triethylamine solubilized phosphorus pentasulfide as thiation
reagent a novel route to totally thiated heterocycles", Heterocycles 19 (11), 2093, 1982.
49 Cava MP, Levinson MI, "Thionation reactions of Lawesson's reagents", Tetrahedron, 41,
5061-87, 1999.
50 Wagner K, Weiss D & Beckert R, "New nitrogen derivatives of steroids by regioselective
reactions of 3-thioxoandrosta-1 ,4-dien-17-one." Synthesis, 1245-7, 1995.
51 Wagner K, Weiss D & Beckert R, "New luminescent materiais based on a steroid molecule",
Eur. J. Org. Chem., 3001-5, 2000.

Conclusão 147
5 Conclusão
Os resultados obtidos no estudo da etapa de quimiexcitação da reação
peroxioxalato foram bastante satisfatórios, contribuindo para o esclarecimento
desta etapa fundamental para a eficiência de geração de estados excitados na
reação peroxioxalato, sendo que, a etapa de quimiexcitação ocorre conforme o
mecanismo de Luminescência Induzida Quimicamente pela Transferência de
Elétron (CIEEL). De acordo com os dados, o intermediário de alta energia (IAE)
que interage com o ativador na etapa de quimiexcitação é a 1,2-dioxetanodiona.
Isto quer dizer que, este IAE se forma devido ao ataque nucleofílico
intramolecular do íon percaboxilato à carbonila do éster concomitantemente
com à saída do grupo fenolato, via um mecanismo concertado, portanto exclui a
outra proposta para o IAE que seria a 1,2-dioxetanona 25.
Em relação ao processo de transferência e retro-transferência de elétron
que ocorre entre o intermediário e o ativador, ficou provado que esta interação
depende do potencial de oxidação do ativador, devido à correlação da
constante de velocidade relativa com o potencial de oxidação, bem como os
rendimentos quânticos singlete, também, correlacionam com as energias livres
para gerar o ativador no estado excitado. Nos estudos do efeito da viscosidade
sobre o rendimento quântico singlete foi comprovado que, a gaiola de solvente
exerce um papel fundamental para a eficiência do processo de transferência e
retro-transferência de elétron, indicando que estas ocorrem em vários passos
envolvendo íons radicais, conforme a hipótese CIEEL
Os compostos preparados, 15 e 21, serão aplicados como precursores
de reagentes de derivatização em estudos analíticos, envolvendo o sistema
peroxioxalato.

AnAXOS
6 Anexos
o2~OOH
~o1 o4 '9' I I :> OAyOOH + CI O5~
+
6 --l o XCI 128,5ppmy
õ (pprn)a = Y + x'tS + 128,5
3 149,3 = Y + -1,5 + 128,5 Y =+22,3
4 123,8 = Y + 1,8 + 128,5 Y = -6,5
5 130,4 = Y + 0,2 + 128,5 Y =+1,7
6 132,4 = Y + 5,6 + 128,5 Y =-1,7
• Ret. 3 - Discussãõ,""l' Ewing DE, Org. Magn. Reson., 12,499, 1979.
Anexo 1: Determinação do valor da contribuição do grupo Y para o deslocamento químico dos
carbonos aromáticos dos compostos 5 a 7. O mesmo raciocínio foi utilizado para calcular os
deslocamentos dos ácidos correspondentes.

Anexos 149
Anexo 2: Rendimento quãntico singlete (<1>s) obtido na reaçâo do bis(2,4,6-triclorofenil) oxalato
(TCPO) com H20 2 catalisado por IMI-H na presença dos compostos 9-13 (ACT), nas seguintes
condições: (TCPO) =0,1 mM, [H20 2) =10mM, [IMI-H) =1,OmM e [ACT) =1,0 mM.
~)Jxk/ "o x =4-OCH, (9)
X = 3-OCH, (10)X =4-C1 (11)X =4-F (12)X =4-N02 (13)
o
[9] <1>s x 103 [10] <1>s x 103 [11] <1>s x 103
mmol L-1Emor1 mmol L-1
E mor1 mmol L-1E mor1
0,005 3,6 ± 0,2 0,01 8,7 ± 0,4 0,005 9±1
0,025 14 ± 1 0,05 36±3 0,01 15 ±2
0,05 24±2 0,1 55±2 0,025 29±4
0,1 35±2 0,25 68± 17 0,05 45±6
0,25 46±2 0,5 93±7 0,1 86 ± 10
0,5 48±3 0,25 135 ± 21
0,5 117 ± 28
[12] l1>s x 103 [13] l1>s x 103
mmol L-1E mor1 mmol L-1
E mor1
0,005 4,5 ± 0,1 0,005 18 ± 1
0,025 21 ± 1 0,01 32±3
0,05 38±4 0,025 56±9
0,1 63±4 0,05 103 ± 16
0,25 97±4 0,1 197±5
0,5 117 ±6 0,25 366 ±30
0,5 700 ± 34
I-.
._-_
._-_
.-----------
..•"
----
----
_...
_..--
_._.
1-2
98
03
/02
12
00
4
e Q Q
.... .., '" UJ O ru
Sd
nd
rd5
M4
tA
coco
lOru
1'1
("'1
lO('
"}__
_O
C\,
lOO
CT
lOlC
Tl
'Q'C
Tl
.....
O'l
OlC
DO
CD
(D
CD
CD
r---l.
llq
"",C
Tl
~~~~~~~~
~~v
('l
)'I
r--
.....
ln0
'1r-
--r-
-<
Dr-
--
C\,
lf'.
.l'Q
''Q
'
~~:::::
\lY
CTl
1Il
l"l
r---
C'\J
ç~
oo
ln
l.1
10
cn
oq
'q..
..O
l(D
r---
{Y
lC\
,l
l.l1
oO
lOlC
Jl
cn
cn
mC
"'lC
'\JC
'\JC
'\JC
\JC
\J
HO
OO
7
1~
--.J
>-O
Me
O0
-3
'\J6
4I
P
Cu
rrcn
tO
ata
Para
.ule
rsNA
ME
San
dra
13C
EXPN
O3
PAOC
NOI
f2-
Aeq
ulsH
lon
Par
alil
lete
rsD
ate_
2004
030-
4riu
10
.45
lNS
fAU
Hsp
ect
PR
OB
rD5
IrII'll
l>.Ja
I13
CI
P!A.
PRO
G.g
lg3
0TO
6553
6SO
LVEN
TA
eeto
neNS
610
OS•
SWH
3267
g.73
8It
.rI
ORE
S0
.'9
86
53
It.
AO1.
0027
508
see
RG2
0'8
0OW
15.3
00us
ecDE
6.0
0us
eeIE
30
0.0
Kai
30.0
0000
000
see
diI
0.03
0000
00se
e
::r:. ~ CJl
11
·_··
····
13C
8.0
0u
Stc
3.0
0d8
125.
7716
22-4
~l
......
....
CII",.
,...nllU
CIPI P
Usr
OI
••••
••••
CH"N
NEL
f2.
CPO
PRG
2••It"
6N
JC2
IHPC
P02
tOa.
oous
eePL
2-3
.00
48PL
I215
.00
d8sr
02
500.
1320
005
MHz
1
'11HO
O
l~~o~e
Ipf
OO~
4I
A
IT
I"pp
m20
0,
-r--
'--r
175
I lôO
"-"
-I
12
j',"
",--
, 100
I 75I
-",-
-,--
--r'
"T
-r
I_
-,-,
-A,
r-',
.......50
25o
F2-
Pro
eess
lng
para
llel
ers
SI
3276
8sr
125
7577
378
>ti
.
IoIlw
E"
SSB
OLB
200
It.
GBO
PC1
.40
laNH
Ap
lot
para
ll'le
lerS
CX2
3.0
0eM
CY6
00
,00
el
F1P
23
0.0
00
ppll
FI28
924.
28Hz
F"2P
-10
.00
0p
ptl
r2-1
257.
58It
.PP
HC
H10
...3
..7
8PP
M/e
lllH2
CHt3
t2.
25..6
4H
z/e
ll
An
exo
3:E
spec
tro
deR
MN
de13
e(a
ceto
na-O
s,-4
0De
,12
5M
Hz)
doa
,a-h
idro
gê
nio
mon
oper
oxio
xala
tode
p-m
etox
ifeni
la(5
)(P
),co
nten
doco
mo
impu
reza
oác
ido
p-m
etox
ifeni
loxá
lico
(28)
(A)
......
VI o

~
~J~Int!Qral
CD)(
o~
ã:;:;,(D
~l 3n>oCI>lJ)
~~ -o(Dn-O!.....o.w,-oü,~ o
CD___
.....1.3946 (D-i31.7107..... (D
1.3236 COãi;~~ O
co
a.o!J.oooe
n>,lJ)
&lI .....
'" O-O:;;1 ::J
iil~
iü,J -.o'
;:;,J '"cn
-1g.;-I.t---- '"1.1392
,-----;:;, Ul-
Ul
i
~~
~-I~J
~;;;=====~-;-
~s::;====~-:>
5::===~-~;...,;
ppm
--15B,4IB
--15B.30B
--156,126
--157.902
--155,903
--154.B33
:C' ::>~,Q.I!
Ullzl ~I;\
!
soxauv

~II.
pp~
-14).IU3
___114.858
-114.794
___122.773
-122.714
:b
-=:3~=:;:::==::::::;:~~=====~-t:-:....-_~-t::-
~lI3."ii9ã5-
J~'"j
~t..j~!
"'-I0.0i'-1
~
!lU
:>.-\ c,~I
II
"1'j<.
s~.
~J:>.UI
~j~-vLN
~I
::1J
"1I
"~nm- ~J~i
I
~
~1
1
.g-JnteOt'il..
...,CI>
<On;;oO<llQ):::l~Q)
o3~~o
lÇIsoxouv

--137.003
»"O::J"O
ilntgralPP'
CD"~ )COIcnã:CO3li>ococn
"'OcoSlcn--l(3
o
W0.69S2~"'-u
~ISB.622
li>=________IS8.296
"'O~a"--IS8.097O-~157.899cn0.2S03::=li>
-I~~~~;
~157.349
Dla.
~J '&Ou>,a.:t>
_,UgC'
CO-,nn?
V-e-
ZS~1~
::o1COj
<OI~J
iiiii O
a.
"'CO
~-,O-O;1 ::J
,jli>
! COIli>::J~li>~Ui ..,.......11.~.6
O...-~~ll')iO
3U>~:b---'1"'1
~VI
Õ·1.0000==:I::.
_144.071
~~-143.820
soxauv

~ ~ C/)
t,.
13C
1.2
0u
sec
1.0
008
t2'5
.77
16
22
4H
Hl
20
02
12
13
15
.46
spect
5I.
Du
al
tJC
I19
PQ
3065
536
CO
CI3
25
77 •
32
67
9.7
38
Hl
O..
f98
65
3H
lI.
0027
508
sec
3276
815
.)0
0u
sec
6.0
0u
sec
30
0.0
K2
.00
00
00
00
uc
0.03
0000
00se
cO
.00
00
20
00
sec
Da
te_
Ti.
eIN
STRU
HPl
lOB
tilPU
LPRO
GTO SO
LvEN
TltS OS SW
HF1
lJAE5
.0 RG OW OE TE O, 011
012
....
....
....
CH
Atf
El
NUCI
PI PLl
SFO
I
f2-
AC
Qu
ull
lon
Pll
"all
eter
s
••••
••••
CH
AttN
fl'2
..
CPI
lPR
G2
walt
l16
NU
Ca
IHP
CP
02tO
a.0
0U
Sl!C
Pl2
0.0
0dB
?l1
21
1.0
0d
8P
U)
t7.0
0d
eS
F02
500
13
20
00
5H
Hz
f2-
Pro
cess
inQ
paru
ete
rsS
I32
768
SFJ2
5.7
5768
03H
Hl
NO
"EM
55
8o
lB1
.00
til
08o
PC1.
-40
Cur
renl
Dal
4P
arl
l.ete
rsNA
HEsa
nd
raEx
PNa
..PR
OCN
OI
10N
HA
plo
tpa
raC
lele
rsCX
23
.00
c.CY
too.
OO
c.
FIP
2]0
.00
0P
P.
FI
2892
4.27
Hz
F2P
-10
.00
0P
P.
F2-1
25
7.5
8H
zPP
HCH
10
.04
78
pp
./c:
.H
204
1312
.2'5
415
Hz/c
.o
2550
U'l
U'l
OlO
U"ll
D...
..(9
1(I
JC
J)(
'\JO
lJ"')
OU
")U
"lO
O'q
IC
lO
10
1tT
l"Q
'"alM
C1
l1"lC
'\J('
\j...
..O
CT
IC
Or--r--c.Q
'q'(
'f1
..
..
..
..
..
..
Ln
OO
OO
OO
lC
Jlcn
(J)O
'lO
'l(7
lL
'l(T
')C
"1
I"l(
\'"l(
'l")n
Jru
(\J
ru
C\J
ru
C\l
7510
0
~ ::;
13
/12
/20
02
~ CD '" '"
125
'"01 U"l :;San
dra
SM-1
9
150
"'~'"
"''''
C
D"'_
CDCD
<Xl
U"l
U"l
U"l
I~
175
200
....'"
"<
Xl
-0
1
<D
U"l
<:>
<:>
'"'"
E g
rpp
m
\1)
~(
II
II~\
))JJJJ
HO
O
,~
-os7
LtO
203~
h,O
Me
C4
5.:
J
-;
3
1.Z. ,
,,
,I
I,
,f--r--r-'-
,-,
r--,--,
II
,I
"'-
-',
II
II
II
iI
,•
An
exo
7:E
spec
tro
deR
MN
de13
C(a
ceto
na-0
6.-4
0°C
,12
5M
Hz)
doá
cid
op-
met
oxife
nilo
xálic
o(2
8).
.... VI
+:o.

F2-
"'CQUlsll~on
Par
8l1
eter
s
Cur
TeM
Oat
aP
ar-a
-ete
rsN
AH
fS
anel
raEK
fJtiO
2PA
OCN
OI
San
dra
SM-1
81
3/1
2/2
00
2
Mq
'".n
<O<
D'"
<D
qm
'"<D
mM
qIn
mq
"ru
ruM
M...
..ru
m...
..<O
go
<Xl
q'"
-q
.....
<O
lD.n
lD.n
Inq
'"-
"....
......
In
\11I
II
\V4
15
CI
O7
l~O-
-.l)
OM
eO3~
45
Dat
!_TI..
INSJ
ALt
4PA
OB
tIlPU
lPR
OG
TO SOLv
ENr
NS OS SOH
FIO
OES
AO RG 00 DE TE OI dll
dl2
2002
1213
12
.03
SlJ
ect
5••
Oua
lIJ
C/
llilP
OJO
65
53
6C
OC
I] 99,
32
67
9.7
38
Hz
O.•
98
65
3"1
,.0
02
75
08
sec
32
76
81
5.3
00
use
c6
.00
uu
e3
00
.0te
2.00
0000
00se
c0
.03
00
00
00
Sll!:C
O.0
0002
000
sec
::t:. ;::s ~ ~
Ó.2
3
-f
....
....
.CH
AIfE
"LfI·
..•
..•..•
•NU
CIIl
CPt
1.2
0\l
llc
P\.I
1.0
0d
8S
fOI
t25
.17
1622
04"H
z
....
....
...
CH
AN
NEl
f.2..
CPO
PRG
21l
f,,)
tzI6
t«.JC
2IH
PC
P02
10
0,0
0us
eeP
L20
.00
dBP
U:!
17.0
0d8
Pll
31
7.0
0dB
Sf0
25
00
.13
20
00
5M
Hz
f2-
Pro
ce5s
lnQ
par
e_el
ers
SI32
768
5f
125.
7577
9042
HH
z)(
)W(H
SSB
OLB
1.0
0H
zG8
OPC
t."O
ppm
200
-i
..,...-,--,---r--I-----r--r--r--I-----r-r---.-.,--~-.
I'
Ir
Ii
175
150
125
100
7550
25
10f'lH
Ap
lot
jJa
ralle
ters
()(
23
.00
c.CY
15
.00
c.
FIP
2]0
.00
0p
p.
FI
2139
24.2
9H
zF2
P-ta
.00
0p
p.
F2
-12
57
.58
Hz
PPH
CHta
.434
78P
Pll
/c.
HZC
N13
12.2
552!
5H
z/c.
An
exo
8:E
spe
ctro
de
RM
Nd
e13
C(C
DC
I 3.
12
5M
Hz)
do
clo
reto
de
p-m
eto
xife
nilo
xalil
a(3
).-VI V
I
r--------
,~ ;::! ~ o C/
)
11••
••••
••,3
C8
.00
use
c3
.00
d81
25
.17
16
22
4N
Hz
f2-
Acq
uU
ltlo
nP
aram
eter
sOa
te200.0~05
TI.
e10
.22
tNST
RUIo
4sp
ect
PRO
BtIJ
5••
Oua
1lJ
e!
PlA.
.PRO
GZ
9193
0TO
6553
6SO
LV
ENf
CO
CI]
NS50
6OS
OSH
H3
26
79
.73
8HZ
FlO
RE
S0
.49
86
53
Hz
AOI
.00
27
50
8se
cRG
24
57
6
OHIS
.30
0u
sec
DE6
.00
usee
Te:
30
0.0
KO
IJO
.00
00
00
00
!le
ccl
ll0
.03
00
00
00
sec
Cu
rren
tO
ata
Par
alle
ters
NANE
Sa
nd
raB
CEX
PNO
!PA
UCNO
I
••••
••••
CHI~fL
NUCI
PI Pu
SfO
\
~
tOo
oo
oo
01
cn
mcn
cn
cn
oo
olC
lr.D
(T1M(T')(T')(T')C'\JC\J(\JruC\J(\JruC\JC\J~
"'l
rnrn
'<::I'
l!l
.......
('1
lOC
\Jcn
"'"o
QJ
(J'I
om
l!ll!1
01
0"'l
l!l0
l0
lo
;rC
D(Y
lrrlr---Q
1C
\J
orr
lC\J
....
....
....
OO
lC
Dr-..
.lO
'V(T
llD
l!l('\
J'V
.....
Sa
nd
raSM
51T
"23
3K
05
/04
/20
04
15h
ora
sI
lOç
(\)
'"Q
)r--
-...
..--
-(\
.1ru
lDl!
l0
1'1
lfl
l:O
.......
C\J
"'l
O(T
1.....
..(T
l"'1
cn
cn
CD
(T')
.....
mr---ru
O"""''''''''C
'\JV
Or--l'
lD(T
lC'\
J(T
1I''-L
!1
'q"'Ç
lDl!
1'Q
"Q
)(D
r-..
..lD
OO
OC
Dln
....
....
....
...
lfl
~~~~~~~~~~~~~~~:::
~\\/~\V~
HO
OO
7
~~O-
-D&CH3
•S
P
m '" " o '"
E o.
o.
~ >-
QOH
49
'
5~
~F
CH
3
1F1P
....
....
.O
1ANN
ELf2
-..
CPO
PRG
2Illa
llZ
l6NU
C2IH
PCP0
21
20
.00
use
cPL
2-]
.00
aBP
Ll2
t7.0
0da
5f02
500.
1]20
005
HHz
',II
,. ,L,~ :~ ; :-T
pprn
"I-1 20
0I
175
1 150
~I
.,-
·,-·
-r--
T-1
-.-
--;---
.----
r-r
-r'
l~
100
~
"'-
r-r
50
....,-]
'"m
'"C
D
'"'"
'"..
..O
N
T-.
.,..
..--
,.·.
...-
r--r
··-r
-25
O
F2-
Pro
cess
lng
par
alle
ters
SI32
768
Sf
125.
7577
292
HH
zlfO
HEM
SSB
oLB
2.0
0H
zG
8o
PC1
...0
10NI
+Rp
lot
par
alle
ters
cx23
.00
CI
CY1
5.0
0e.
FIP
230.
000
pp
.fi
2B92
<4.2
BH
lF2
P-1
0.0
00
pp
.f2
-t2
57
.58
Hz
PPM
CH10
..043
..78
pp
./c.
HZC
"13
12.2
5464
Hz/
cl
An
exo
9:E
spec
tro
deR
MN
de13
e(a
ceto
na-0
6,-4
0De
.125
MH
z)do
O,O
-hid
rogê
nio
mon
oper
oxio
xala
tod
ep-
met
ilfen
ila(6
)(P
),co
nte
nd
oco
mo
impu
reza
p-m
etilf
enol
(F).
..... VI
0'1

>~ai)(
o..çã: (1)
3Q)oro(J)
"O(1)
U..,o.<0
"Oo..,(1).
3ãl
<Q.Q).o0ro@..,CTo~.iil(1)
D>:::l~Q)..,o3~
S·
~~Tiniêõrêi'--"-'--·-·----..,.---·---·1-········...............T.-.----..----r..---..--r-....-....·._-'"['ppm
~p...
~\)
__156.016~
~W-155.724
üi-'>-.---TI
<n"~--154.742
-~ -..-,.,,
<n-o-0.0037<::--149.204
-~G-;--148.439U">- ..
-O'" ..::>~:I:
C>
-O~
-O-O'"-
00:0z
'"I ,b•••-'"-
;,I
'9~
I ,I "..:::,...1 I
o....~-0...o-
~
A-w:I:"-'" og
-,u;
0,2721'::::=1="...,....__137.O17
::>.6\
o
..:.,-O
0:0-0-136731:;:
i-1,.,,--'
w"I..._::I.
!J'l:..-,..-
-CnL130.741 ,~-o-~130.692
....-~
--------130.392 o--
j1.0000::=C'"--128.288
T1
0.0536>~- ;;;..:j--125.335l.J1-.:
,
j-..J:-...-/121.719 ,
0.7230==-.;:)
"i~121.594
121.475
-1,::32.0347==--115.428<n
-O..J::-
]'"T1
~;
j
soxauv
.Ane
xo11
:E
spec
trode
RM
Nde
13
C(a
ceto
na-D
6,-3
0D
C,
125
MH
z)do
O,O
-hid
rogê
nio
mon
oper
oxio
xala
tode
feni
la(7
)(P
),co
nten
doco
mo
impu
reza
so
ácid
op-
met
oxife
nilo
xálic
o(3
0)(A
)e
feno
l(F)
.
San
dra
SM
50T
=2
43
K3
0/0
3/2
00
4d
I=
30
se
[15
ho
rasl
"'"
'"o
";;;
'"'"
"_
Cl
.....C
urr
ent
Dat
aP
ara-
eter
lll
(Tl
'"(T
l'"
cr>
"(T
l
'"C
l<=
>co
NAI4E
San
dr.
13
CE
coC
lo
-(T
l.....
oo
(Tl
colO
'"Q
co.....
.....'"
"'"
"(T
lo
<=
>ai
EXPN
O7
Q
'"'"
'"'"
"''"
"''"
'""'
'""
PAOC
NO1
II
II
II
II
III
IF2
•A
cqu
\Slu
on
Pir
uete
rsD
ate_
2004
0329
Tll
e1
1..
8IN
STAU
Nsp
ect
PR
e6t()
5H
Oua
lIX
I
H?>
r---
!t_
Pll.P
RO
G'9
19
30
lO65
536
SCLv
ENT
Ace
tone
O2
0-0
6NS
565
OSO
OH
•-
6S
Iti
3267
9.73
8H
z
·0A
FlO
RES
0.•
9965
3HZ
'01
.002
7508
sec
RO24
576
5::
"..
HO
OO
O.
IS.3
00
usec
DE6
.00
useI
:
•1H
-oTE
30
0.0
teF
O2
O3~
116
OI
]O.0
0000
000
se<
011
O.0
3000
000
s.e
p•
5••
••••
••O
1A1f
<EL
fi••
••••
••NU
CIIJ
CP
I1
.00
UI'C
PLI
'.0
0oa
SfO
I12
5.71
1622
....
,
iR3
A••
••••
••O
1.lf.
f4El
'2••
••••
••
2A
el'O
PRG
2w
altZ
l6HU
C2IH
PCP0
212
0.0
0us
ecPL
2-3
.00
06
iPP
U2
17.0
0dS
SF02
500
.132
OD
D5
MHl
1?
rF2
-pr
OC
l!SSl
ngp
ara
.ete
rs
oGp
SI
3276
6
3PSF
125.
7577
236
MH
l
f3F
H!J
IjE
.55
11o
IJI
L62
.00
Hz
)1li
J/
/1G6
o
)~
1~/I
IJv
PC1
.40
i
!~II
10N~
p)a
tpar~let!rs
ex2
].0
0C
I
i.;;;
o.....
""'
(Tl
'"'"
ey4
0.0
0C
I
'""
"''"
<=>o
.....
ILo
.....(T
l
"o
Cl
cr>
Z;;FI
P16
2.00
0pp
o!~
oC
l(T
l-
'"(T
l<=>
i~-
<=>o
oo
<=>-
<=>F
I20
372.
75H
'F2
I'I4
B.O
OO
pp
Ir
,--~~.-
.,._
.-,-
--r-
--....
-1
··
--r-
I.,.~-
-,I
'1--
....
.--r
---
T--·T-~j
,i
,F2
1861
2.14
Hz
pprn
160
15
81
56
15
41
52
15
0PP
HCH
0.6
08
70
PP
l/ca
H2CH
76,5
4818
Hz/
CI
.__.
---
-----
§=z
OJ-
cn~
-f
...,-
OJ
~.
-f
a.c
,-II
I-f
g-o
a.o
OCl
lm
cno
-I
lll'
Co
_,m
~!n
c:o
õ»
~ ;:::l ~ ~ .,.
.- ~:' ".

::t. ;:s ~ ~
I1••
••••
••1
](
8.0
0us
ec]
.00
081
25
.17
16
22
4N
Hz
••••
••••
OtA
JfifL
NUCI
PI Pu
5FO
I
F2-
Pro
cess
lng
par
afll
eter
,S
I]2
76
8S
f1
25
.75
17
2]6
HHz
MON
EM5~
Ola
2.0
0tu
G8
OPC
1."
0
F2-
Acq
ul51
110n
P"ra
lete
rsD
ale
200<
4032
9H
.et1
.48
lNST
RU
Msp
ect
PRO
BttJ
5••
Oua
IIl
el
Pll.P
RO
Gzg
lgJO
106
55
]6S
OlV
ENT
Ace
ton
eNS
585
OSO
SWH
32
67
9,7
38
Hl
FIO
RES
0.4
98
65
3H
lAO
1.0
02
75
08
see
RG2<
4516
Dlt
15
.30
0us
ecOE
6.0
0U
SK
TE]0
0.0
KO
IlO
,OO
DO
Oaa
ose
e01
1O
.030
0000
0se
e
....
....
.O
1ANN
ELf2
..
CPQ
PRG
2lia
Itz
16N
UC2
IHPC
P02
12
0.0
0us
ecP
l2-3
.00
el8
PLI2
11
.00
de
5F02
50
0.1
32
00
05
HH
l
Cur
rent
Dat
aP
ar..
ete
rsN
ANE
Sa
nd
ra13
CEX
PNO
7PR
OCHO
I
laN~
Plo
tp
ara.
ele,
.sex
2]
.00
c.CY
40
.00
c.F
IP1
32
.00
0PP
•F
It6
60
0.0
2H
zF2
P1
16
.00
0P
P.
F214
587.
90H
zPP
HCH
0.6
95
65
PP
Il/C
.I'tZ
CH8
7.4
83
63
Hz/
c.
-r---'-
-·-
T..
-I
.~
118
I-r
--r-
122
120
1--
·'--r--~-.--
124
r-""
""""
"---
.---
r-...
r-
123
126
San
dra
SM50
j'2
43
K3
0/0
3/2
00
4d
l'3
0se
c15
ho
ras
I
.....
<.0
...,
.....
LI1
lDC
Ccn
"'"
-'"
,.,t!
1lT
'1lO
U(D
(T
'JúJ
U"l
CD
Oll""-rrJ
(\J"Q
'oo
'""
"-
"C
Dru
ru....
.tl
lfT
lo
"Q'
(T')
cc(D
(\J
0'1
,.....
lO'"
C\J
"'"
~(Y
"lo
cn
r---
lC)
vo
"o
ocn
cn
alC
Da
Je
D"
",.,
,.,,.,
C\J
("\J
....
....
....
....
....
.C
D"
~~~~
~~~~
C\J
C\J
~~
C\J~~~~~~
:::
\1\/
\\1/
II\I
I\\
\IIi
E g ! !-
~
511
D
I
bASF
??
6Pbf
=? .
I){
I~
--'
\..J
A;.
J~U
J1
1)
+o
Isl!~(~
~~!~I
'U"l
C\J
o'C
D'"
o.
olC
lo
.C\J
o~
~-_
.,.
.•
-I
I,-
--,-
130
PP
"
An
exo
12:
Idem
aoes
pect
ro11
,po
rém
regi
ãod
ean
elar
omát
ico.
.... VI
\O

::t:. ;::s ~ ~S
andr
aSM
9
2003
0605
B.2
1sp
ect
5M
I8
80
88
19
3032
768
OHSO 16 2
51
75
.98
3H
z0.
1579
5BH
z3
.16
54
38
9se
c.5
6.\
9660
0us
ec/.
00
U,"
100.
0K
1.00
0000
00'"
tOff4
Ap
tot
pa
ram
ete
rsex
20.0
0C~
ev0.
00C
II
FIP
B.5
29p
p.
FI21
31.3
2H
zF2
P0.
0477
pplll
F211
9.36
HzPP
HCH
0.4
02
58
ppm
/cm
HZCH
100.
6981
9HZ/c~
..~
••••
•C
HAN
Nf'L
fI
..
MJC
IIH
Pt
12.4
0us
ecP
u6.
00dB
SFO
I25
0.13
1890
1H
11
F2
-A
CQ
uis
it10
0P
ara
flle
ters
F2
-P
roce
ssIO
gp
ara
llete
f"s
SI32
76B
SF25
0.1
3000
00H
'lzO
IlHEM
SSB
OLB
0.]
0H
zGB
OPC
16.0
0
Cu
rre
ot
Da
taP
ara
llete
rsNA
HEJu
n04-
2<l0
3EX
PNO
t60
PROC
NOI
Dat
eTI
l11e
INST
IUl
PRO
B>Il
PlLP
RO
GTO SO
LVEN
!NS OS SN
HFl
OR
ES
AO RG OH DE TE OI
/
~ll1"""01(TlOCD""'M""'C'\JOO1l.Clcn
r--'V-"r--.lt1l!1<DO(T)~(Tl""'(Y1'V'V
aJ-'C
tt.D
Oll
DC
IlO
tDm
,...
.(T
l'V
tDaJ
('\J
C'\
l...
..O
O1
01
rrlL
CiU
)C
\Jr--.l
l10
1U
)fT
lL!lL!lt.l1ll1"Q'"r---l.Cl~r""lr""lC'\lruooo
.,
,,.....
,
~\~\[J~
/.J x
~~IC
JI
IS3
'
.-gl ~
,..
II
3·
""-t;I<
::tI
1.-11
,1.JI~
..~
I~(
1!(I~!
ru
CD
ru
ow
oo
0"1
r--.
<D
CD
oru
mC
\j(T
lIIlm
mr--.
01O
Ola
J<
...C
Ll1
.....
aJ
OlC
D<
DC
DC
IllJ
"lo
:t
.111
.
n2.,
I!~
tCO~(TltO
C'1
lDíO
tDll
l_
ML
I1I.
O(l)
01
I:COr--L!l~C"'1
g~~~~~
~~3
o c
J
---
1r-r-r~~'1......-rT
rr'
1'I
"l
I1
"1'1
I1
II-'-r-r~'-"''''''''-I
''''''
-1-1
-1'1
rr-l-
j'I
Irr-r'-l-"'~
ppm
87
65
43
2I
An
exo
13:
Esp
ectr
ode
RM
Nde
'H(D
MS
O,
250
MH
z)do
2-[4
-bro
mof
enil]
-5,5
-dim
etilo
xazo
l-4-
ona
(14)
...... 0
\ o
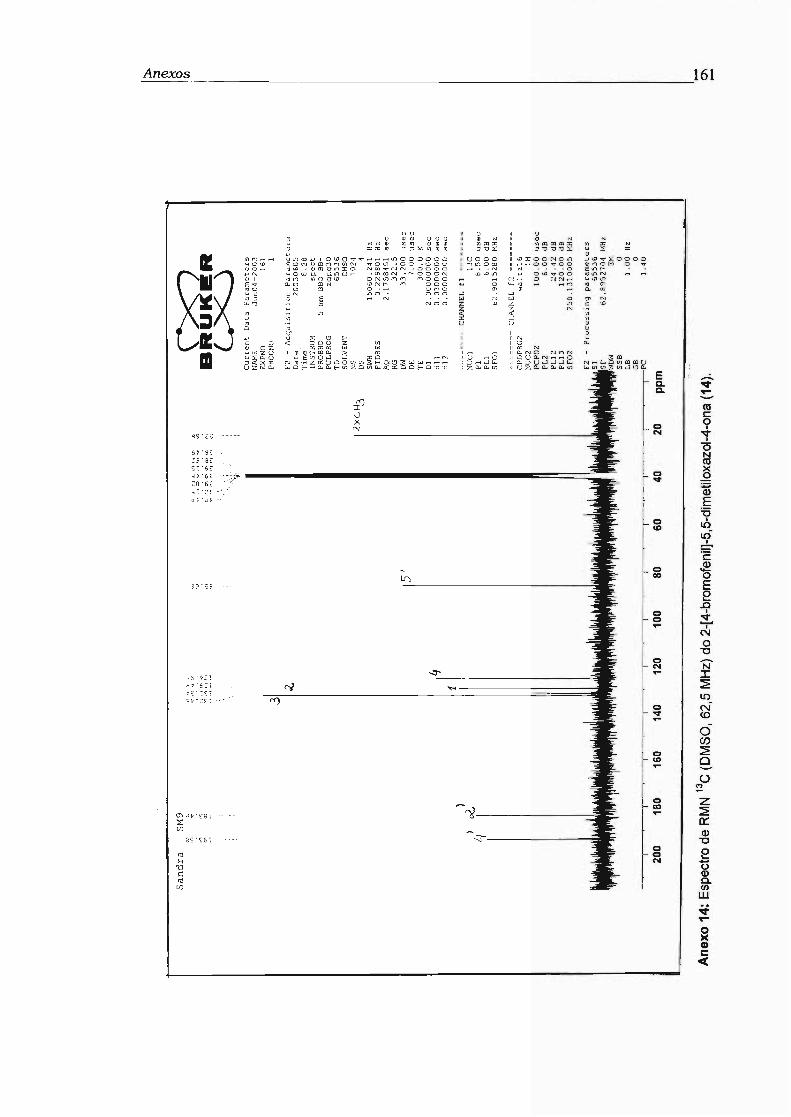
San
dra
SM9 ,., m
,'j
.1
,.,,.,
3
w '" "'',"..ll
"li
Lú
l(·,
loC
....
mv
.......
'JJ
V
';1
0'1
0\0
\00
«
""
''',
..1
1')
""
,'1
'~i
;:; " "B~R
L>
<-J
Cu
rren
tD
ata
Pa
ram
eter
sN
AM
EJu
n0
4-2
00
3E
XP
NO
16
]P
RO
CN
O]
).
;:l ~ c til
F2-
Acq
uis
iti
on
Pa
ra
mete
rs
Da
te2
00
30
60
5l'
ime-
8.2
8IN
ST
RU
Msp
ect
PR
OB
HD
5m
mB
BO
BB
-P
ULP
RO
Gzg
pg
30
TO
65
53
6S
OL
VE
NT
DM
SO
NS
10
24
DS
4SW
}J1
50
60
,24
1fi
zF
IDR
ES
0.2
29
80
1H
zI\
Q2
.17
58
45
1se
eR
G3
22
.5OW
33
.20
0Il
:-ie
C
DE7
,00
use
eT
E3
00
.0K
Dl
2.0
00
00
00
0se
ed
ll0
.03
00
00
00
se
ed
]Z0
,00
00
20
00
se
e
)~)
LI
~ 14
,51
2xC
H3
NU
C]
Pl
PL
lS
FO
l
CH
I\N
NE
I.f1
••••••••
13
C!:
j.SD
usec
6.0
0d
B6~,
90
15
28
0M
Hz
CP
DP
RG
2N
UC
2P
CP
02
PL
2P
Ll2
PL
l3S
F0
2
CII
I\N
NE
Lf2
.w
alt
z1
6 IH1
00
.00
usec
6.0
0d
B2
4.4
2d
B1
20
.00
dB
25
0.1
31
00
05
MH
z
Pruc~::;~ing
pa
ram
eter
::;
65
53
66
2,8
95
27
04
MH
zE
M O1
.00
fiz
O1
.40
ppm
2040
II
l'~-í-"""
II
II
II
I
200
180
160
140
120
100
8060
Ane
xo14
:Es
pect
rode
RM
N13
C(D
MS
O,
62,5
MH
z)do
2-[4
-bro
mof
enil]
-5,5
-dim
etilo
xazo
l-4-o
na(1
4).
......
0'1 ......

San
dra
S~
li
~ ;:::J ~ C CIl
"•••
,....
.CHA~l
f.2..
...,.
••••
:ao..:a
:":"
.CH
AN
NEl
fI."
,.••
••:
r2-
Acq
uisi
tlon
Par
am!t
ers
NU
CIt)
CP
\e
.50
usec
PU
6.0
0CI
BS
fOI
62
.90
15
28
0/'
til
wal
lZ1
6 IHtO
a00
use
e6
00
ae.2.
4.4
2d6
1.20
00d
a.l!
5013
1000
5I+
lz
2(0
)06
16
15.•
1sp
ett
5m
lllB
80B
B
zgpg
3065
536
Co
ei)
10
2. •
10
03
2.3
93
Hz
0.2
87
36
0H
z1
.74
00
30
8se
c2
03
.226
550
usec
700
use
c3
00
.0K
200
0000
00se
c0,
0300
0000
'ec
0.00
0020
00se
c
CPQ
PRG
2N
UC.
2P
CP
02
PC
'PL
l.2P
U3
SF0
2
Curr~nt
Dat
aP
araf
llet
t:rs
NA
IEJu
nl6
-,2
00
3E
XPf
()61
PROC
NOI
Oa
tefim
eII\
STA
lt4PR
OB
Ifl
PLLP
ilQG
TO SOLV
ENT
NS OS SHH
FlO
RES
AO RG O.
DE TE Dl
dll
dl2
g., )( ('> ,;
~.,
'.(
Ii
"'
!"
··.:;·i/
r-,
~~~
::~
~~~
~A;~f;
tt~~~
." 0:1 '"
02<:\l/
I'.
;~"T
.,",
1'.::
0;.1
:")
:.:
:,.:
..,';
{'.i
"".
Ir:
0''':
~::.:
"-~.
,r'
~gõ,;
~::;
é;'.:;
':i
:Il
'U m !
3' N
S-A
2'4*,
Br~O
5'3
2l'
~-
:~l
n;
t,;'
r.:':.
II
II
.----
--.--
-1-.-
----
'T
J-r-
'-----
.----.
------
--r-'
II
ppm
225
200
175
150
125
100
7550
25
An
exo
15:
Esp
ectr
od
eR
MN
13C
(CO
CI 3
,62,
SM
Hz)
do2-
[4-b
rom
ofen
il]-5
,5-d
imet
iloxa
zol-
4-tio
na(1
6),
~)
.2,)
14
5)
F2-
Pro
cess
lng
para
mele
rs
SI65
536
Sf
62
695.
2390
I+lz
HO
HEM
sse
oL6
I00
Hz
GSo
PC1
..40
10~
plo
lp
ara
llle
lers
CJ(
.20.
00CI
'ICY
OO
Oel
'lfl
P2.
4376
0pp
mn
1533
2.61
Hz
F2P
la.2
06pp
mF
26
.41
.93
HZPP
MCM
116
76
69
ppm
/cl'l
l
HZC
H7
34
.53
41
2H
Z/C
m
-0"1 N
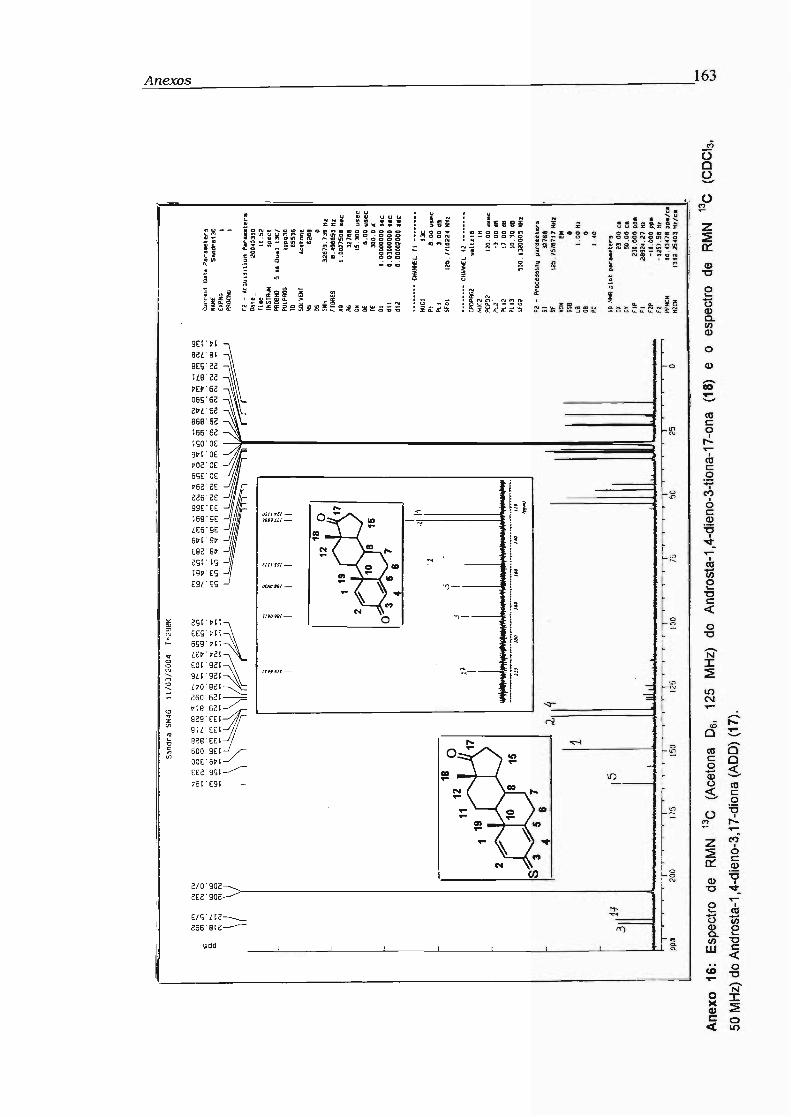
2••
4
).
;:l ~ CIl
CU
"''''.l
ltD
lla
Par'
Rte
rsNA
NES
an
dr.
t.JC
EIlP
,.oI
PAO
CtIJ
I
F2-
AtQ
u\s
ltio
np
.,...d
ers
Ott
t_20
041:
1310
fiM
1t.
52
U~SfFUt
sp
ect
PA08
tf)
5..
DlH
1ll
el
PU
lPA
OG
19P
930
TD65
536
SOL
VfJ
rtfA
teto
nl!
16
6261
DSo
SWH
32
61
9.1
31
Hz
flll
llt:
SO
.098
l!S
3H
zAO
I.0
02
75
0'
":C
Illõ]2
76
.OM
15
.30
0u
se
cDE
1.0
0u
.ec
TE3
00
.0I(
OI
1.0
00
00
00
0se
c11
110
.03
00
00
00
tec
1'2
0.00
0a20
00u
c
••••
••••
CH
.lIK
L"
••••••••
NUCI
IXP
Ia.
DO
U$I
!CP
U:J
.OO
dl!t
SfO
IIl
S.1
11
52
24
MH
z
F2-
Pro
cl!S
sinQ
p.r
alll
!ter
s51
3276
1SF
125
7578
7()
MH
lMO
MEM
SSB
ol8
1.0
0H
lGB
oPC
1.•
0
••••
••••
Ctu
ffol
fL'2
.rP
OPA
G2
••1
tlll
NU
C2
'HPC
P02
12
0.0
0u
se
cPL
2'·]
.00
dll
PL12
17
.00
d8P
lI3
10
.30
.1S
f02
500
.13
2000
511
Hz
~~~~ll
1 1
..
~fT'l_m~1.l1~(TlNNOOOOmmmmmNNC%]~
~~~~'q'(Tl(T'l~MfT'lfTlfT'lMfT'lNNNNruNN
__
~~ru(Tlmr---~lOru""lmqW~_mruo~_m~w
~w~mq~~l.ONm~Oq~mmqmfT'l~~N~
,.....~
.....
ru...
..m
mM
mru
(T
'lN
......omm~~~~~~_
~ ~ I .5J
II
~..
.-_
,"'.
.,..
....
....
..ro
-•.
'"
IHU
IJ11
1l
'-'
1
o18
San
dra
5H46
11
/03
/20
04
r-2
98
K
"'1l"
"'l
OC
TIC
O[D
(J)"
"lr
u....
...t.
CC
"1
r---
Olf
TlC
'U0
l1
T1
00
ru
-ru
-C
J'I
qr---0
f1
1Ii'l
fT
'l1
.l1
(\,I
(Tlo
(Z)r
---U
J<
D0
0""'''''''
ql.O
tn
-.
..
.fT
lI.O
O'l
l.O
C"'l
C"1
Mm
cn
mw
l.O
'q'"
Qv
q-
~~~~~c;::~~~~~~~~~:::
1\\~\lIp~
rufT
lN
ru
CJ'I
r---
fTl,
.....
0l1
Sl
Na
e"
..
a.C%
]r--
-c.o
lOa.
_~
00
NN
NN
\/ 'i
I-r
-rI
iI
.-r-~--r--r-
II'
i
200
175
150
125
pp~
3··H
5
r-I
i
100
,J
',
i•
II
..---
T---.
------
-.I
~~
~O
10~NR
,,'o
tp
,"aa
l!te
rsex
n.oo
t.CY
50.0
0ta
rlP
23
0.0
00
PP
.FI
28
92
4.2
1tu
F2P
-lO
.OD
O91
'.F2
-12
57
.58
"zPP
HCH
10
.43
47
8tl
pa
/c..
t12C
H1
31
2.2
5.4
03
Hl/
C.
An
exo
16
:E
spe
ctro
deR
MN
13C
(Ace
tona
06
•12
5M
Hz)
doA
nd
rost
a-1
,4-d
ien
o-3
-tio
na
-17
-on
a(1
8)e
oes
pect
rode
RM
N13
C(C
OC
I3•
50M
Hz)
doA
nd
rost
a-1
,4-d
ien
o-3
,17
-dio
na(A
OO
)(1
7).
......
0'1
W

/
~r;'o
~~~.
10~~;-~1'o
,-
32
l'
Cur
rent
Cat
aP
aral
lele
rsNA
Mt:
San
dra
EXPN
O3
PRO
CNO
1
f'2-
Acq
ul5
1tl
on
Par
alll
eter
sO
ole
_2
00
.05
13
Tim
e15
.20
INS
[RUM
spe
et
PRO
Bf(J
5m
.O
ual
13C
IPU
LPRO
Gzg
30TO
6553
6SO
LVEN
TCO
C13
NS32
OSO
SWH
3255
.20B
Hz
FlO
RE
S0
.0.9
67
1H
z~O
10.0
6637
95se
eRG
406
.•OW
15
3.6
00
usee
DE6
.00
usee
TE3
00
.0K
OI
I.0
0000
000
see
::t:. ~ ~ O (J)
I1••
••••
••
IH9
.50
usee
3.0
0dB
13
1.0
.5'*'Z
....
,••
•CH
ANNE
L
NU
CI
PI
PU
SfO
I30
0
'Q!"'-
q...
...C
\JO
O...
..0
0lD
ru
v"'T
0'l0
'll"
"l
CD
oLt
1...
...0
CO
OlL
IlN
<D
oO
1V
.....
O'lr-
--L
f1C
T1
....
oO
OlD
l11
ru
ru
ru
oo
ruru
......
..--
1..
....
....
....
....
..o
o
\(~
\/1\/
San
dra
SM53
13/0
5/20
04
~oru~ruNooMw~~omMrn~Mom~
OO~Mooo~mo~oom~ru~oo~~N~~
ovm~~m~~mOMm~m~~oow~w
N~mrnoo~w~~~w~vmoooo~~~w
('\J
ru<
r'I
....
..lO
t.DlO
lDL!
1Lt
1LI
1ll
11.
!1(T
)l"
'1M
M'r')
('1
l\.l
't.!l~~~p;;;5
e c. c.
F'2-
Pro
cess
ing
para
lnet
ers
SI
3276
BSF
30
0.1
30
00
40
MHz
wOw
E~
SSB
OLB
0.2
0H
zGB
OPC
1.0
0
I- c o ~ c
jí L1
JJ~1~
(~~~~
,;
._"._
_._...
..
.--.-J~
~~15
1 .... 1!1
(T1
1!1 o
J__
laro
Ml
plo
tpa
rall
eter
sCX
23
.00
emCY
12
.50
e.
FIP
10
.00
0pp
oF
I3
00
1.3
0H
zF2
P-0
.50
0pp
oF2
-15
0.0
6H
zPPHC~
O.•
5652
ppm
/eo
HZCM
137.
015B
5H
zlcm
An
exo
17:
Esp
ectr
ode
RM
Nde
1H(C
DC
b,30
0M
Hz)
do2-
[4-f
enila
cetil
eno]
-5,5
-dim
etilo
xazo
l-4-
ona
(15)
.
ppm
I' 9I 6
r-"
5-,
--r-
'4
I1-
·-y-'
--'-
-r--
,-r-
l-.r-
-r--
"2
Io
......~

E o. o.
..... '"C\J...
...O
IQ
J
II
~cinlJra
SM~]
1)/
05
/20
04
"~
~
""
Uo~
I~
~-
..
VI
rI~
'J
.lIX.
San
dra
SM53
13/0
5/20
04
Wt"1
rr1
O'llD
'QO
l,{"lm
t!1
(..,
lOo
V..
....
...
,.....
oo
'"o
O'l
....
"'1,...
....,.,.
....,O
........
..o
)N
UJ""''''''lfliJ'1
NlD
oo
rr1
OlC
DM
('I"
')O
'l...
..li
1(T
)(T
1..
..v
,...
,...
,.C
\JO
UJ
.....M
C\J
o('
\J..
....
....
-O
CT
lO'lC
Dl.
Il"
-J
"'f
CO
Ln
,...
..,.
,.,,
....
.WO
IM
'"o
(ri
(T)
C"'l
MM
C"\J
(\J
ruC
\JC
\JQ
laJ(D
",,
....
.,r--,,
....
.C
\JC
\J-
I..
....
....
....
....
....
....
....
....
....
....
....
...
~V~
I\I~
II
I
r
Cur
rent
Dat
aP
aru
ete
rsN
ANE
Sa
nd
raEX
PI<O
4PR
OCN
OI
F2-ÀCQu~51tlon
Pa
rafl
lete
rs0
0«
_20
0405
13fi
lie
15
.59
IN5T
RUH
,pec
tPR
OB
tll5
...
Qua
IIX
/PU
LPRO
G.g
pg
30
106
55
36
SOLV
ENT
CO
C13
NS40
96OS
OSN
H1
66
32
.39
3H
.fl
OR
ES
0.2
67
36
0H
.AO
1.7
"00
30
8se
cRG
3276
6OH
26
.55
0u
sec
DEf:i
,00
usec
TE3
00
.0K
013.
0000
0000
sec
ti11
0.03
0000
00se
cd
l20
.00
00
20
00
soe
). ~
I1••
••••
••
l3C
8.04
0us
ec6
.00
de
75
.47
60
50
5H
H.
....
....
CHA
NN
El
NU
CIPI P
US
fOI
••••
••••
CHA
NN
ELT2
••••
••••
CPO
PAG
2••
1t.
16
I<oC
2IH
PCP0
29
0.0
0u
sec
PL2
3.0
0.6
PL12
21
.66
.aP
U3
12
0.0
0d
aS
f02
30
0.1
31
20
05
HH
.
..
I 1/.
" ..,J.-,-_.
__.~_,_,..
__1.
I •<':i
I l,>ti
....-_.
.........
_--...~_.~-'.~
I(] "L,.
I no
...)..,
,...1..
L,
-.-
r ll.?
An
exo
18:
Esp
ectro
deR
MN
de13
C(C
OC
I 3•75
MH
z)do
2-[4-fenilacetileno]-5,5:dimetiloxazol-4~ona(15).·
'11
,(,'
1·
-T·r
-·-
,-r-
··"-
-I-r
--,
rpp
m20
017
5I
'r"-,.
._,o
-"--
-r15
012
5
4K
C.H
3
51
6.5
I
T-'\
-'"
T--'-
-'r
--r-r'
T"M
T-,-
r-1
--"'-
r--r
--l
Ii
I"'-
r-r--
\00
7550
25
O
f2-
Pro
cess
lng
par
amel
ers
SI
3276
65
f7
5.4
67
747
0H
H.
NON
EMss
eO
La
1.0
0H
.G
6o
PC1.
•0
10~~
plo
tpa
ram
eter
sCX
23
.00
CII
Cy
t2.5
0C
II
F\P
23
0.0
00
PP
'F
I\7
35
7.5
6N
.F2
P-1
0.0
00
pp
,F2
'75
4.6
6H
.PP
HCH
10
.43
47
8P
Pll
/c.
HlC
N7a
7.•
6950
H7
/C'
-0'1 Vl