universidade de sÃo paulo faculdade de medicina de ... · evaluation of their transfusional...
TRANSCRIPT

UNIVERSIDADE DE SÃO PAULO
FACULDADE DE MEDICINA DE RIBEIRÃO PRETO
PROGRAMA DE MESTRADO EM HEMOTERAPIA E BIOTECNOLOGIA
Jamile Souza Nicanor
Desenvolvimento e aplicação de um programa computacional para armazenamento de
dados e seguimento de pacientes em hemoterapia
Ribeirão Preto
2017

Jamile Souza Nicanor
Desenvolvimento e aplicação de um programa computacional para armazenamento de
dados e seguimento de pacientes em hemoterapia
Dissertação apresentada ao Programa de
Mestrado em Hemoterapia da Faculdade de
Medicina de Ribeirão Preto, da Universidade
de São Paulo, como requisito para a
obtenção do título de Mestre em Ciências.
Orientadora: Profa. Dra. Ana Cristina da Silva Pinto
Versão corrigida. A versão original encontra-se disponível tanto na Biblioteca da Unidade que
aloja o Programa, quanto na Biblioteca Digital de Teses e Dissertações da USP (BDTD).
Ribeirão Preto
2017

AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE
TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA
FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE.
Ficha catalográfica
Serviço de documentação
Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo
Nicanor, Jamile Souza
Desenvolvimento e aplicação de um programa computacional para
armazenamento de dados e seguimento de paciente em hemoterapia.
Ribeirão Preto, 2017.
90 p.
Dissertação apresentada à Faculdade de Medicina de Ribeirão
Preto/USP. Área de concentração: Hemoterapia e Medicina
Transfusional. Orientadora: Ana Cristina Silva Pinto
1.Sistemas de informação em saúde. 2. Anemia Falciforme. 3.
Transfusão sanguínea.

FOLHA DE APROVAÇÃO
Nome: Nicanor, Jamile Souza
Título: Desenvolvimento e aplicação de um programa computacional para armazenamento de
dados e seguimento de pacientes em hemoterapia.
Dissertação apresentada à Faculdade de
Medicina de Ribeirão Preto da
Universidade de São Paulo para a obtenção
do título de Mestre em Ciências.
Área de concentração: Hemoterapia e
Medicina Transfusional
Aprovado em:
Banca examinadora
Prof. Dr.: Instituição:
Assinatura:
Prof. Dr.: Instituição:
Assinatura:
Prof. Dr.: Instituição:
Assinatura:

“É preciso que eu suporte 2 ou 3 larvas se quiser conhecer as borboletas.”
Antoine de Saint Exupéry
Dedico este trabalho aos meus pais
que me ensinaram a acreditar em
mim, seguir meus sonhos e
agradecer sempre por tudo que me
é ofertado.

AGRADECIMENTOS
A Deus pela iluminação, força e esperança, ajudando-me a não desistir diante dos obstáculos.
As minhas mães de coração Mary, Glória, Nilsa e Itana pela atenção, carinho e amparo.
À minha irmã Luana e amigas Maíra, Lívia e Pâmela, por toda ajuda e incentivo.
A Edmilson por acreditar em mim e me incentivar mesmo diante das dificuldades.
Ao programador, Dmeval da Costa Chaves Neto, por confiar em mim e por todo seu empenho
para o desenvolvimento do Software.
Aos meus familiares, amigos e colegas de trabalho, principalmente da HEMOBA, pelo apoio e
acolhimento.
Aos meus pequenos pacientes por cada gesto de carinho, mesmo quando precisam de cuidado.
A todos aqueles que estiveram ao meu lado e acreditaram que completaria esta etapa de minha
vida.

RESUMO
NICANOR, J.N. Desenvolvimento e aplicação de um programa computacional para
armazenamento de dados e seguimento de pacientes em hemoterapia, 90 f. Dissertação.
Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, 2017.
Os sistemas de informação em saúde permitem melhorias no processo de gestão e
assistência ao paciente. A fundação HEMOBA é responsável pela assistência médica e
transfusional de grande parcela da população baiana, pelo SUS, em seu ambulatório
transfusional e de atendimento à pacientes com doenças hematológicas benignas. A doença
falciforme (DF) é uma das doenças hereditárias mais prevalentes no mundo, sendo a Bahia o
estado brasileiro onde é encontrada sua maior prevalência. A transfusão sanguínea é uma das
bases para o seu tratamento. Não existe sistema informatizado no atendimento desses
pacientes nem banco de dados sobre essa população na instituição. OBJETIVO: Desenvolver
um Software para seguimento transfusional em hemoterapia, com ênfase no acompanhamento
de pacientes falciformes, possibilitando descrever e analisar aspectos transfusionais dessa
população e dos doadores. MATERIAIS E MÉTODOS: Foi desenvolvido um Software,
denominado BDTrans, utilizando a metodologia de Ciclo de Vida de Desenvolvimento de
Sistemas, que possibilitou um estudo analítico, transversal, com abordagem qualitativa e
quantitativa, dos dados extraídos do sistema. Foram acompanhadas crianças falciformes, de 0
a 18 anos, em regime transfusional, atendidas no ambulatório da Fundação HEMOBA.
RESULTADOS: Foram incluídos 108 pacientes, destes 58% apresentavam DTC alterado e
38% apresentavam passado de AVC. As crianças residentes em Salvador correspondiam a
34%. A maioria das transfusões foi iniciada na faixa etária entre 2-8 anos (61%) e 9-12 anos
(24,7%). 28% dos pacientes iniciou o regime transfusional em 2011, seguido dos anos de 2012
com 16%. A prevalência de aloimunização eritrocitária foi de 26%, sendo os principais
aloanticorpos contra antígenos do sistema Rh e Kell. CONCLUSÃO: A utilização do
Software possibilitou tornar os dados mais acessíveis para análise clínica e epidemiológica,
auxiliando na gestão e assistência à saúde. O sistema poderá ser utilizado como base para o
desenvolvimento de um programa de fidelização de doadores fenotipados em um futuro
próximo.
Palavras-chaves: Sistemas de informação em saúde. Anemia Falciforme. Transfusão
sanguínea.

ABSTRACT
NICANOR, J. N. Development and application of a computer program for data storage
and follow-up of patients in hemotherapy, 90 f. Dissertation. Faculty of Medicine of Ribeirão
Preto, University of São Paulo, Ribeirão Preto, 2017.
Softwares built for data collection from Health Centers are important tools for patient
care and for public health decisions. The HEMOBA foundation is responsible for health
assistance of a great proportion of the population from the State of Bahia, including medical
care and transfusional assistance to patients with benign hematological disorders. Sickle cell
disease is one of the most prevalent hereditary disorders in the world, and the State of Bahia
has the highest prevalence of the S mutation in Brazil. Blood transfusion is one of the
treatment modalities for sickle cell patients, and there is no software for data collection of the
clinical and transfusional program. OBJECTIVE: to develop a software to help follow up the
transfusional program of the HEMOBA foundation, including the sickle cell patients,
collecting transfusional data and blood donor information. MATERIALS AND METHODS:
we developed a software called “BDTrans”, using the methodology of the “Cicle of Life
System Development”, that allowed an analytical, transversal study with both qualitative and
quantitative approach of the data extracted from the program. Sickle cell children from 0 to
18 years who were on transfusion at The HEMOBA foundation were the subject of our study.
RESULTS: 108 sickle cell patients were included, 58% had brain vasculopathy and 38% had
a history of stroke. 34% of the children lived in Salvador. The age range of the children when
the transfusion program started was between 2-8 years (61%) and 9-12 years (24.7%). 28%
of the patients started the transfusional program in 2011, and 16% started in 2012. The
prevalence of red cell alloimmunization was 26%, most of the antibodies were against Rh and
Kell blood groups. CONCLUSION: The utilization of the BDTrans Software promoted the
collection of clinical and transfusional data from sickle cell patients, possibilitating the
evaluation of their transfusional program, and providing important information to the public
health system. The software can also be used to develop a fidelization program of phenotyped
blood donors in the near future.
Key words: Health information systems. Sickle cell anemia. Blood transfusion.

LISTA DE ILUSTRAÇÕES
Quadro 1 – Etapas para o modelo da prototipação............................................................................45
Quadro 2 – Fluxograma de dados......................................................................................................48
Quadro 3 – Desenho do Estudo.........................................................................................................60
Figura 1 – Relações do banco de dados.............................................................................................51
Figura 2 – Tela de acesso ao programa.............................................................................................52
Figura 3 – Guia inicial.......................................................................................................................53
Figura 4 – Guia de dados do paciente (1ª parte)................................................................................54
Figura 5 – Grupo de dados do paciente (2ª parte).............................................................................54
Figura 6 – Lista de pacientes cadastrados.........................................................................................55
Figura 7 – Guia para solicitação da transfusão.................................................................................56
Figura 8 – Guia de dados da transfusão............................................................................................56
Figura 9 – Guia de dados da transfusão (rastreamento de transfusões solicitadas)..........................57
Figura 10 – Guia de relatórios..........................................................................................................57
Figura 11 – Dados administrativos...................................................................................................58

LISTA DE TABELAS
Tabela 1 – Características epidemiológicas e clínicas dos pacientes.......................................61
Tabela 2 – Ano do início do esquema transfusional crônico.....................................................62
Tabela 3 – Fenótipo eritrocitário dos pacientes.........................................................................63
Tabela 4 – Fenótipo eritrocitário dos doadores.........................................................................65
Tabela 5 - Comparação entre fenótipos eritrocitários de pacientes e doadores........................67
Tabela 6– Dados da transfusão..................................................................................................68
Tabela 7 – Comparação entre diferentes fenótipos eritrocitários......................................72
Gráfico 1 – Número de anticorpos por paciente alo ou autoimunizados..................................66
Gráfico 2 – Anticorpos identificados........................................................................................66

LISTA DE ABREVIATURAS:
AF Anemia Falciforme
AIH Autorizações de Internação Hospitalar
AIT Ataques Isquêmicos Transitórios
Anvisa Agência Nacional de Vigilância Sanitária
APAE Associação de Parentes e Amigos do Excepcional
AVC Acidente Vascular Cerebral
BDTrans Banco de Dados Transfusional
cGMP Monofosfato Ciclíco de Guanosina
CH Concentrado de Hemácias
CONITEC Comissão Nacional de Incorporação de Tecnologia
do SUS
CVO Crise vasoclusiva
DCV Doença Cerebrovascular
DF Doença Falciforme
DTC Doppler Transcraniano
FDA Food and Drug Administration
Glu Ácido Glutâmico
GTP Guanosina Trifosfato
HAS Hospital Santo Antônio
Hb Hemoglobina
HbF Hemoglobina F
HbS Hemoglobina S
HbSBeta tal Hemoglobina SBeta talassemia
HbSC Hemoglobina SC
HbSD Hemoglobina SD

HD Hard Disk
HEMOBA Fundação de Hematologia e Hemoterapia da Bahia
HEMOCAD Sistema Nacional para Cadastro de Unidades de
Hemoterapia
HEMOPROD Sistema de Informação de Produção Hemoterápica
HEMOVIDA Sistema de Gerenciamento de Serviços de
Hemoterapia
HLA Antígeno de Histocompatibilidade Principal
HU Hidroxiuréia
HUPES Hospital Universitário Professor Edgar Santos
IIS Infartos Isquêmicos Silenciosos
ICAM 1 Intracellular Adhesion Molecule 1
NO Óxido Nítrico
NOTIVISA Sistema Nacional de Notificações para a Vigilância
Sanitária
PNIIS Política Nacional de Informação e Informática em
Saúde
PAI Pesquisa de Anticorpos Irregulares
RNM Ressonância Nuclear Magnética
SCNES Sistema do Cadastro Nacional de Estabelecimentos
de Saúde
SE Sequestro Esplênico
SIA/SUS Sistema de Informação Ambulatorial do SUS
SIH/SUS Sistema de Informação Hospitalar do SUS
SIS Sistemas de Informação em Saúde
SNIS Sistema Nacional de Informação em Saúde
SIT Controlled Trial of Transfusion for Silent Cerebral
Infarcts in Sickle Cell Anemia
STA Síndrome Torácica Aguda

STOP Stroke Prevention Study in Silckle Cell Desease
SUS Sistema Único de Saúde
SWiCH Stroke with Transfusions Changing to Hydroxyurea
TWiCH Transcranial Doppler Ultrasounds with
Transfusions Changing to Hydroxyurea
Val Valina
VCAM 1 Vascular Cell Adhesion

SUMARIO
1. INTRODUÇÃO 13
1.1. Motivação do estudo 13
1.2. Sistema de Informação e Saúde 14
1.3. A Doença Falciforme 20
1.3.1. Conceitos 20
1.3.2. Manifestações Clínicas 22
1.3.3. Tratamento 26
1.3.4. Transfusão Sanguínea na Doença Falciforme 28
2. JUSTIFICATIVA 42
3. HIPÓTESE 42
4. OBJETIVOS 43
4.1. Objetivos Gerais 43
4.2. Objetivos Específicos 43
5. MATERIAIS E MÉTODOS 44
5.1. Tipo da Pesquisa 44
5.2. Local da Pesquisa 46
5.3. População 46
5.3.1. Critérios de Inclusão 47
5.3.2. Critérios de Exclusão 47
5.4. Desenvolvimento do Sistema 47
5.4.1. Levantamento de Necessidades 47
5.4.2. Projeto Inicial 49
5.4.3. Protótipo: Construção, Avaliação e Refinamento 50
5.4.4. Desenvolvimento 50
5.5. Aspectos éticos 58
5.6. Análise estatística 59
6. RESULTADOS 60
7. DISCUSSÃO 69
8. CONCLUSÃO 76
REFERÊNCIAS BIBLIOGRÁFICAS
ANEXOS

13
1. INTRODUÇÃO
1.1. Motivação do Estudo
A informatização ainda não é a realidade de grande parcela das instituições públicas de
saúde, como os ambulatórios espalhados por todo o país, que assistem pacientes com diversas
patologias.
A Fundação HEMOBA é o único Banco de Sangue público do Estado da Bahia e
responsável pelo suporte hemoterápico na maioria das instituições públicas de saúde do Estado
e algumas unidades particulares. Utiliza sistemas de informação para controle e gestão de todo
processo de triagem, coleta, produção de hemocomponentes, testagens sorológicas e
imunohematológicas e liberação dos hemocomponentes para transfusão. Como software
principal adota o Sistema de Gerenciamento de Serviços de Hemoterapia (HEMOVIDA),
disponibilizado pelo Ministério da Saúde para os serviços hemoterápicos vinculados ao Sistema
Único de Saúde (SUS). Também emprega os outros sistemas de informação disponibilizados
pela Agência Nacional de Vigilância Sanitária (Anvisa): Sistema de Informação de Produção
Hemoterápica (HEMOPROD), Sistema Nacional de Notificações para a Vigilância Sanitária
(NOTIVISA).
A instituição promove a assistência hemoterápica e hematologia a grande parcela da
população baiana, através de seu ambulatório transfusional e de assistência aos pacientes com
doenças hematológicas benignas. Porém os dados ambulatoriais não são informatizados, apesar
de serem registrados de forma manual em papel e armazenados conforme legislação. A ausência
de informatização e, por consequência, de um banco de dados, dificulta o pronto conhecimento
dos dados clínicos e transfusionais da população assistida.
Entre os pacientes atendidos na fundação, a população falciforme se destaca por sua
forte representação numérica, aspectos clínicos e principalmente por suas particularidades
transfusionais. A Bahia é o Estado brasileiro com maior população de pacientes falciformes,
em sua maioria acompanhados no ambulatório da HEMOBA. Muitas crianças falciformes
necessitam manter regimes transfusionais crônicos por diversas alterações, como a doença
cerebrovascular. A assistência transfusional desses pacientes é dificultada pelas altas taxas de
aloimunização eritrocitária encontradas nessa população levando muitas vezes à necessidade

14
de transfusão de sangue fenotipado. A disponibilização de concentrado de hemácias fenótipo
compatível em tempo hábil é um grande problema para a instituição apesar de todos os esforços
empregados e na fenotipagem de grande número de doadores.
As informações coletadas e documentadas manualmente ficam restritas ao setor onde
foram geradas: prontuário com os dados clínicos, prescrições e registros multiprofissionais são
restritos ao ambulatório; os dados laboratoriais de compatibilidade e liberação dos
hemocomponentes para transfusão ficam armazenados nos laboratórios relacionados e no
sistema específico.
A criação de um banco de dados que possibilite unificação dessas informações,
armazenamento de dados, emissão de relatórios auxiliará sobremaneira ao conhecimento da
população analisada, possibilitando melhoria das políticas públicas.
O presente trabalho enfoca o desenvolvimento de uma ferramenta informacional para
gestão de dados em hemoterapia com ênfase nas informações clínicas e transfusionais referentes
às crianças falciformes. A população foi escolhida por apresentar grande representatividade
entre os pacientes transfundidos na instituição e pelas suas particularidades transfusionais e
dificuldades técnicas para manter o esquema transfusional adequado para essa população, como
disponibilidade de sangue fenotipado em tempo hábil. Portanto, o desenvolvimento do sistema
visa um acompanhamento clínico mais detalhado do esquema transfusional, bem como oferece
a base computacional para uma futura implantação de um programa de fidelização de doadores
fenotipados para esses pacientes.
1.2. Sistema de Informação e Saúde
A tecnologia é uma grande marca dos tempos modernos. A sociedade atual é
considerada uma sociedade do conhecimento e da tecnologia. (LORENZETTI et al., 2012) A
informação é base do conhecimento, um meio ou material necessário para extrair e construir o
conhecimento, alicerce dessa sociedade. Embora estivesse sempre presente em outros períodos
históricos, a informação não tinha a importância que passou a ter na sociedade pós-capitalista.
(MIRANDA, 2004)

15
A gestão de serviços de saúde constitui uma prática administrativa que tem a finalidade
de otimizar o funcionamento das organizações de forma a obter o máximo de eficiência, eficácia
e efetividade, garantindo a melhor utilização dos recursos disponíveis para atingir um objetivo.
Neste processo o gestor utiliza conhecimentos, técnicas e procedimentos que lhe permitem
conduzir o funcionamento dos serviços na direção dos objetivos definidos.(TANAKA;
TAMAKI, 2012) A informação constitui uma ferramenta na produção de conhecimentos
válidos que podem orientar a tomada de decisão dos gestores e organizações. (PINHEIRO et
al., 2016)
Na medicina moderna, conhecimentos médicos e informações sobre os pacientes
apresentam constante crescimento, não sendo mais possível gerenciá-los através de métodos
tradicionais baseados em papel. (PEREZ; ZWICKER, 2010) O conhecimento acumulado
através de estudos, pesquisas e avaliações passadas necessitam ser acessados pelo gestor, em
tempo hábil, para que se estabeleça o processo de tomada de decisão. (TANAKA; TAMAKI,
2012)
Os sistemas de informação em saúde (SIS) permitem coletar, processar, armazenar e
distribuir a informação para apoiar o processo de tomada de decisão e auxiliar no controle das
organizações de saúde. Possibilitam a realização de pesquisas, fornecendo evidências e
auxiliando no processo de ensino.(MARIN, 2010) Para seu adequado funcionamento as
informações devem ser confiáveis, dotadas de relevância e com significado e pertinência no
processo decisório, estando prontamente disponíveis quando requeridas. (MINISTÉRIO DA
SAÚDE, 2013a)
O’Brien e Marakas (2008), definem um sistema de informação “ como um conjunto
integrado de recursos, que é composto por pessoas, dados, software, hardware e redes de
comunicação. Quando exposto de forma organizada, é capaz de receber os dados coletados e
transformá-los e organizá-los em informações úteis para a sociedade.” (apud PEREZ;
ZWICKER, 2010)
O atendimento em saúde pressupõe o envolvimento e a participação de múltiplos
profissionais, atuando em distintas áreas em uma mesma instituição ou locais diferentes. As
informações obtidas desses profissionais apresentam grande heterogeneidade de dados,
portanto precisam ser integradas para garantir a continuidade do processo do cuidado. (MARIN,
2010) Desta forma, os sistemas de informação em saúde devem satisfazer aos diversos usuários

16
envolvidos e lidar com mudanças de processos e métodos de trabalho profundamente arraigados
nesse ambiente profissional.(PEREZ; ZWICKER, 2010)
As informações relacionadas com o setor da saúde podem ser processadas de diversas
formas, como registros eletrônicos de saúde, inscrição informatizada de pedidos de
fornecedores, suporte de decisão, relatórios eletrônicos de resultados, prescrição eletrônica,
telemedicina, entre outros. (CHAUDHRY et al., 2006) Assim como em diferentes ambientes
de atenção à saúde, tais como laboratórios, ambulatórios, hospitais, unidades administrativas,
unidades de assistência da enfermagem, da nutrição e esferas maiores como secretarias e órgãos
gestores da saúde. (PANTANOWITZ; HENRICKS; BECKWITH, 2007) (MARIN, 2010) A
utilização funcional desses sistemas pode envolver a documentação clínica (informação / dados
de saúde), gestão de resultados, apoio à decisão, comunicação eletrônica e conectividade, apoio
ao doente, processos administrativos, relatórios de saúde da população, por conseguinte. Desta
forma a utilização dos SIS vem apresentando papel cada vez mais relevante, envolvendo uma
ampla gama de utilidades.(CHAUDHRY et al., 2006)
O desenvolvimento de ferramentas de informática capazes de coordenar e integrar todas
as informações em saúde, oriundas de diferentes setores e com múltiplas finalidades é
fundamental para garantir a qualidade do atendimento.(MARIN, 2010)
Uma revisão sistemática evidenciou a importância da utilização da tecnologia de
informação em saúde no aumento da aderência a protocolos e “guidelines”, aumento da
capacidade de atendimento e vigilância aos agravos, redução das taxas de erro e otimização
variável da utilização do tempo.(CHAUDHRY et al., 2006)
O desenvolvimento de ferramentas computacionais está fortemente relacionado ao
processo de inovação. A inovação envolve a busca e descoberta, experimentação,
desenvolvimento, imitação e adoção de novos produtos, novos processos de produção e novas
formas organizacionais. Não necessariamente inovação refere-se a algo novo ou inédito, pode
estar relacionada a submissão de uma ideia já existente a uma nova forma de realiza-la ou a
uma nova situação, desta forma caracterizando algo novo para o usuário ou entidade onde foi
adotada. (PEREZ; ZWICKER, 2010)
A inovação em saúde pode apresentar dois aspectos distintos. A primeira refere-se a seu
contexto típico, relacionada à tecnologia de alta sofisticação, como equipamentos. A segunda
envolve os sistemas de informação, com introdução ou não de elementos computacionais e

17
intervenção nos processos de trabalho, muitas vezes não bem aceitas pelo usuário. (PEREZ;
ZWICKER, 2010)
Pinheiro (2016) em seu estudo entrevistando gestores municipais de saúde evidenciou
muitas dificuldades para a implantação dos sistemas de informação em saúde no Brasil.
Observou haver a necessidade de fortalecimento de uma cultura informacional nas secretarias
municipais que consolide o uso da informação para a construção do conhecimento e o seu
compartilhamento para subsidiar o processo decisório, implicando no aperfeiçoamento do SUS.
(PINHEIRO et al., 2016)
No âmbito do SUS, todo esse desenvolvimento tecnológico e de conhecimento, atrelado
ao crescente uso dos sistemas de informação desenvolvidos nas mais diversas formas e
instituições, motivou a criação da Política Nacional de Informação e Informática em Saúde
(PNIIS). “O propósito de sua formulação foi de promover o uso inovador, criativo e
transformador da tecnologia da informação a fim de melhorar os processos de trabalho em
saúde e, assim, resultar em um Sistema Nacional de Informação em Saúde (SNIS) articulado e
que produza informações para os cidadãos, a gestão, a prática profissional, a geração de
conhecimento e o controle social, garantindo ganhos de eficiência e qualidade mensuráveis
através da ampliação de acesso, equidade, integralidade e humanização dos serviços de saúde,
contribuindo, dessa forma, para a melhoria da situação de saúde da
população.”(MINISTÉRIO DA SAÚDE, 2016a)
A PNIIS trabalha com as informações em saúde como insumos potenciais para usuários
e profissionais de saúde, bem como subsídio para a gestão, vigilância e atenção à saúde.
(MINISTÉRIO DA SAÚDE, 2016a) Portanto, a informação em saúde deve ser entendida como
um instrumento de apoio decisório nos vários níveis que constituem o SUS, para que haja o
conhecimento da realidade socioeconômica, demográfica e epidemiológica na rotina diária dos
serviços de saúde, no planejamento, na gestão, na organização e na avaliação das
ações.(MINISTÉRIO DA SAÚDE, 2013a)
Os sistemas de informação, no âmbito do SUS, podem ser empregados em nível local
(unidades básicas de saúde, unidades de pronto atendimento, hospitais, serviços de
hemoterapia, laboratórios, etc.) possibilitando a análise da situação de saúde com base nas
diferentes microrregiões e nas condições de vida da população na determinação do processo
saúde-doença. “Nesse processo, a responsabilidade do nível local é de grande importância

18
para o SUS, não apenas na alimentação dos sistemas de informação em saúde, mas também
na sua organização e gestão.”(MINISTÉRIO DA SAÚDE, 2013a)
Os serviços de hemoterapia consistem em serviços de saúde diretamente relacionados
com a prática hemoterápica, por consequência com o ciclo do sangue. No Brasil, são
estruturados de acordo com complexidade técnica, capacidade de produção, demanda
assistencial e distribuição geográfica e populacional, desta forma podem consistir desde
unidades de coleta, agencias transfusionais, bancos de sangue a hemocentros. Em todos os
níveis de assistência, os serviços de hemoterapia produzem grande quantidade de informação,
portanto requerem métodos adequados para coleta, processamento e armazenamento dos dados,
aumentando a segurança transfusional, proporcionando melhoria contínua do processo de
gestão e da atenção ao paciente. (BATISTA et al., 2015)
A portaria nº 158 de 04 de fevereiro de 2016, que regulamenta as atividades
hemoterápicas no Brasil, estabelece que “os serviços de hemoterapia devem ter sistemas de
registro apropriado que permitam a rastreabilidade da unidade de sangue ou componente,
desde a sua obtenção até o seu destino final, incluindo-se os resultados dos exames de
laboratório referentes ao produto”. Sugere que os registros referentes à doação e à transfusão
sejam, preferencialmente, informatizados e garantam a confidencialidade das informações.
(MINISTÉRIO DA SAÚDE, 2016b)(MINISTÉRIO DA SAÚDE, 2013a)
Nos últimos anos, vêm sendo implementados vários SIS no Brasil, envolvendo os
serviços de hemoterapia assim como outros setores da saúde.
O Sistema do Cadastro Nacional de Estabelecimentos de Saúde (SCNES) é a base para
cadastro hospitalar e ambulatorial do SUS, permitindo a visualização da realidade da rede
assistencial existente e suas potencialidades, visando auxiliar no planejamento em saúde, em
todos os níveis de governo.(MINISTÉRIO DA SAÚDE, 2013a)
O Sistema de Informação Ambulatorial do SUS (SIA/SUS) é utilizado em unidades
ambulatoriais credenciadas ao SUS para garantir o registro dos quantitativos e valores a serem
pagos aos prestadores de serviços, produzindo informações locais que são consolidadas
nacionalmente. (MINISTÉRIO DA SAÚDE, 2013a)
O Sistema de Informação Hospitalar do SUS (SIH/SUS) processa informações dos
serviços hospitalares prestados por meio das Autorizações de Internação Hospitalar (AIH),

19
informações referentes às transfusões hospitalares, tabelas de Procedimentos, Medicamentos,
Órteses, Próteses e Materiais Especiais, permitindo ao SUS efetuar os pagamentos pelos
serviços prestados.(MINISTÉRIO DA SAÚDE, 2013a)
Alguns SIS envolvem diretamente os serviços de hemoterapia, abordando quatro
aspectos principais: o cadastro de serviços de hemoterapia, a produção hemoterápica nacional,
a notificação de eventos adversos relacionados ao uso de sangue e hemocomponentes e o
gerenciamento do ciclo do sangue.
O Sistema Nacional para Cadastro de Unidades de Hemoterapia, denominado
HEMOCAD é gerido pela Anvisa e alimentado pelas vigilâncias sanitárias locais. Envolve um
amplo número de variáveis de dados específicos para o registro das atividades executadas por
cada serviço de hemoterapia, as relações entre os serviços, suas distribuições por unidade
federada e região geográfica. (MINISTÉRIO DA SAÚDE, 2013a)
Com a finalidade de melhorar o gerenciamento do Programa Nacional do Sangue, foi
desenvolvido o HEMOPROD, também gerido pela Anvisa. Consiste em um sistema não
informatizado no qual todos os dados de identificação do serviço, triagem, coleta, exames
realizados, produção de hemocomponentes e reações transfusionais são inseridos em planilhas
do ExcelR e encaminhados para às Vigilâncias Sanitárias Estaduais e Municipais.
(MINISTÉRIO DA SAÚDE, 2013a)
O NOTIVISA, consiste em um sistema informatizado na plataforma web para receber
as notificações de eventos adversos e as queixas técnicas relacionadas com os produtos sob
vigilância sanitária. Desta forma, os eventos adversos relacionados ao uso de
hemocomponentes deverão ser registrados no sistema, possibilitando adotar medidas sanitárias
pertinentes. (MINISTÉRIO DA SAÚDE, 2013a)
Para o gerenciamento das atividades hemoterápicas, com foco na segurança dos
processos, foi desenvolvido o HEMOVIDA. Ele consiste em um programa disponibilizado para
os serviços públicos de hemoterapia do país, permitindo a informatização do processo produtivo
e desta forma agregando maior controle e segurança ao processo hemoterápico. Possibilita o
registro informatizado das ações de captação de doadores, triagem clínica, coleta,
processamento, triagem sorológica e imunohematológica no sangue do doador/receptor e
transfusão. (MINISTÉRIO DA SAÚDE, 2013a)

20
Vários outros sistemas de informação existem no pais, sejam de empresas comerciais
ou desenvolvidos no próprio serviço de hemoterapia. Permitem, em sua maioria, o
gerenciamento modular das etapas de produção de hemocomponentes e transfusão.
(MINISTÉRIO DA SAÚDE, 2013a)
Os sistemas desenvolvidos internamente possibilitam maior adequação a realidade
local. São geralmente motivados por necessidades próprias da instituição onde estão sendo
implementados. Portanto, assumem papel inovador para atender a uma demanda do serviço de
hemoterapia contribuindo para a melhoria da assistência e da gestão, inclusive com melhor
alocação de recursos e insumos para atender os objetivos almejados.
1.3. A Doença Falciforme
1.3.1. Conceitos
A doença falciforme (DF) é uma das doenças hereditárias mais prevalentes no mundo e
a mais frequente mutação da cadeia da hemoglobina. Apresenta uma incidência mundial
estimada de mais de 300.000 novos casos por ano, principalmente na África Subsaariana,
Península Arábica e Índia. Estima-se que anualmente 500.000 indivíduos nasçam portando pelo
menos um alelo s no mundo. (PIEL et al., 2013)
A DF foi primeiro descrita por Herrick em um relato de caso publicado em 1910.
(HERRICK, 1910) Foi a primeira doença a ter suas bases moleculares descritas na história, em
1949, por Linus Pauling, configurando um importante marco para a ciência. (PAULING et al.,
1949)
Atualmente, sabe-se que é uma doença genética, de transmissão mendeliana,
caracterizada por uma mutação GAG→GTG no códon 6 do gene da beta globina, que leva a
produção de uma valina (Val) no lugar de um ácido glutâmico (Glu).(STEINBERG, 2008)
Desta forma, há substituição de um aminoácido hidrofílico (Glu) por um hidrofóbico (Val),
levando a formação da hemoglobina S (HbS), que polimeriza quando desoxigenada, conferindo
a característica rígida e em forma de “meia lua” à hemácia falciforme.(MEIER; MILLER, 2012)

21
O termo DF refere-se a toda hemoglobinopatia caracterizada pela presença da HbS que
apesar da diferente nomeação, apresentam características fisiopatológicas e clínicas
semelhantes, com variação na gravidade. Quando o alelo beta S (S) está em homozigose, o
fenótipo apresentado é HbSS, caracterizando Anemia Falciforme (AF). Quando em
heterozigose e associado a outras alterações da hemoglobina como hemoglobina C, D ou Beta,
chama-se hemoglobinopatia SC (HbSC), SD (HbSD), Sbeta talassemia (HbStal),
respectivamente. (BALLAS et al., 2010)
O gene S é amplamente distribuído na África, Mediterrâneo e partes da Índia.(TERESA
et al., 2008) Três quartos dos nascidos no mundo com DF estão na África Subsaariana.
(WANG et al., 2013) Essa distribuição genética tem relação com a origem africana da HbS e
sua associação com a proteção contra malária. (PAGNIER et al., 1984) Indivíduos com traço
falciforme apresentam menor parasitemia pelo plasmódium e, quando infectados, manifestam
malária de forma mais leve. (ALLISON, 1954)
Ao longo dos anos, o gene S vem sendo distribuído pelo mundo devido às
imigrações.(PIEL et al., 2014) Com a descoberta das Américas e sua colonização houve a
disseminação do alelo S no continente, através do tráfico de escravos africanos. Desta forma,
os países que receberam maior contingente desses escravos, como o Brasil, países caribenhos
e partes dos Estados Unidos, apresentam maior prevalência do gene mutado. (HUTTLE et al.,
2015)
No Brasil, o gene S é encontrado em 7% da população de origem africana, com a
prevalência estimada da HbS de 1:1000 no país, sendo 1:600 no Estado da Bahia, onde houve
a maior colonização africana, seguido de 1:1300 em Minas Gerais e 1:1335 no Rio de Janeiro.
(HUTTLE et al., 2015) (SERJEANT, 2013)
No Estado da Bahia, o teste de triagem neonatal vem confirmando a alta prevalência da
DF e sua distribuição predominante na região litorânea, Recôncavo Baiano e alto e médio São
Francisco, locais de intensa movimentação de escravos africanos. (AMORIM, 2010)

22
1.3.2. Manifestações Clínicas
A DF é caracterizada por uma anemia hemolítica, com manifestações clínicas variáveis a
depender de fatores genéticos e individuais.
A HbS apresenta afinidade pelo oxigênio reduzida em relação à variante normal e, quando
desoxigenada, tende a polimerização e consequente rigidez eritrocitária, menor
deformabilidade, perda de partes da membrana da célula, adquirindo a forma falcizada.
(ODER et al., 2016) (IUGHETTI; BIGI; VENTURELLI, 2016) Com a reoxigenação, a
hemácia falciforme chega a adquirir novamente sua forma arredondada. Porém, os repetidos
episódios de falcização e perda de membrana, tornam-na irreversivelmente deformada, sendo,
por conseguinte hemolizada. (MANWANI et al., 2013)
A hemólise na DF, é bastante expressiva, chegando a diminuir a sobrevida das hemácias
de habitualmente 120 dias para apenas 2 a 21 dias. Com a destruição intravascular dos
eritrócitos são liberados na circulação hemoglobina livre e heme, ambos potencialmente
reativos. (KATO; GLADWIN; STEINBERG, 2007) A hemoglobina livre consome o óxido
nítrico (NO), convertendo-o em metahemoglobina e nitrato. O NO é responsável pelo
relaxamento da musculatura lisa e consequente vasodilatação, via sua ligação a guanilato
ciclase, possibilitando a conversão do GTP (guanosina trifosfato) em cGMP (monofosfato
cíclico de guanosina). Desta forma, a depleção do NO promove vasoconstricção, ativação e
agregação plaquetária, maior expressão de moléculas de adesão como VCAM-1, ICAM-1, P-
selectina e E-selectina. A biodisponibilidade do oxido nítrico também é diminuída pelo heme,
através da produção de espécies reativas de oxigênio e da arginase, liberada pelos eritrócitos
hemolisados, que destrói a L-arginina, um substrato para a produção de NO. (KATO;
GLADWIN; STEINBERG, 2007)
As características físicas das hemácias falciformes, associadas à sua maior adesão ao
endotélio, plaquetas e leucócitos, levam à lentificação e diminuição do fluxo sanguíneo, micro
obstrução, chegando a causar isquemia e consequente processo inflamatório, dor e lesão de
órgãos- alvo. (ODER et al., 2016)
As complicações decorrentes da DF podem ocorrer em qualquer órgão ou tecido. Estão
relacionadas a tríade fisiopatológica de hemólise crônica, vaso oclusão e intensa inflamação,
associada ou não a fatores desencadeantes, tais como estresse, infecção ou desidratação. O
paciente falciforme pode apresentar manifestações como crise dolorosa, priaprismo, síndrome

23
torácica aguda (STA), acidente vascular cerebral (AVC), dentre outras.(MEIER; MILLER,
2012) A maioria dos óbitos desses pacientes ocorre até a terceira década de vida,
principalmente por STA, sequestro esplênico e crise álgica prolongada. (KARACAOGLU et
al., 2016) Na infância, a mortalidade é maior nos 2 primeiros anos de vida, principalmente por
infecção e sequestro esplênico. (FERNANDES et al., 2010)
A dor é uma das manifestações mais características e mais debilitantes da DF. Estima-se
que 60% desses pacientes apresentem pelo menos um episódio de dor intensa por ano, sendo
considerada a principal causa de hospitalização nessa população. (IUGHETTI; BIGI;
VENTURELLI, 2016) A crise vasoclusiva (CVO) pode ser aguda, crônica, até incapacitante.
(BALLAS, 2015) Alguns fatores desencadeantes foram descritos, tais como: estresse,
aumento da viscosidade sanguínea, diminuição de fluxo sanguíneo, hemólise ou uma
combinação de outros fatores menores.(MANWANI et al., 2013)
Uma manifestação álgica comum no paciente com DF é o priapismo. Consiste em uma
ereção peniana persistente e dolorosa sem estimulação sexual, que acomete 11% dos doentes
falciformes. (SHIGEHARA; NAMIKI, 2016) Nos pacientes com HbSS a prevalência chega
a ser de 5,6% enquanto que, naqueles com HbSC é de 1,1%.(FURTADO et al., 2012) O
indivíduo pode evoluir com perda da função erétil pelo priapismo prologado ou recorrente,
desta forma, o objetivo primário do tratamento é conseguir a detumescência da ereção peniana,
preservando sua função. (SHIGEHARA; NAMIKI, 2016) O tratamento deve ser instituído o
mais rápido possível e compreende desde hidratação, intervenção urológica à transfusão
sanguínea. (YAWN et al., 2014)
A Síndrome torácica aguda é outra manifestação comum no paciente falciforme. É
caracterizada por febre, desconforto respiratório agudo e novo infiltrado pulmonar a
radiografia de tórax.(DEBAUN; STRUNK, 2016) Acredita-se que 30% dos pacientes
falciformes irão apresentar STA pelo menos uma vez na vida.(IUGHETTI; BIGI;
VENTURELLI, 2016) Não apresenta etiologia bem definida, porém foram relatados fatores
de risco como episódio prévio, valor alto de hemoglobina ou leucócitos, baixos níveis de
hemoglobina fetal (HbF) e associação com Asma. (DEBAUN; STRUNK, 2016) (LAMARRE
et al., 2012) Para tratamento dos pacientes com STA, a transfusão de hemácias tem papel bem
estabelecido. Pode ser realizada transfusão simples ou eritrocitoaférese, com benefício
similar, sendo a última forma mais efetiva segundo alguns estudos.(SAYLORS et al., 1996)

24
Outra complicação frequente no paciente falciforme é o sequestro esplênico (SE),
principalmente na criança. Ocorre em 30% dos pacientes menores de 6 anos. (MEIER;
MILLER, 2012) É caracterizado por aumento súbito do volume do baço desencadeado pela
apreensão dos eritrócitos falcizados e outras células sanguíneas no interior do órgão,
diminuição dos valores de hemoglobina, reticulocitose e plaquetopenia. (BALLAS et al.,
2010) Pode ser classificado em sequestro esplênico maior ou menor, conforme sua
característica clínica. No primeiro, ocorre rápido aumento do baço e colapso circulatório,
potencialmente fatal. No segundo, também ocorre aumento rápido do baço, mas a redução dos
níveis de hemoglobina é menor (valores absolutos acima de 6 g/dL). (MEIER; MILLER,
2012) Uma indicação frequente para tratamento inicial de SE é a transfusão de eritrócitos para
controlar o choque e aliviar os sintomas da anemia. As opções de tratamento, a longo prazo,
consistem na realização de esplenectomia, programa de transfusão crônica ou a observação
cuidadosa para os sinais iniciais de SE até que o órgão se torne gradualmente não funcional
(atrofia esplênica) a depender da gravidade do episódio inicial e da idade do indivíduo.
(OWUSU-OFORI S, 2015)
O ambiente inflamatório associado a oclusão microvascular contribuem para o
desenvolvimento da doença cerebrovascular (DCV), outra importante manifestação da DF.
(ADAMS, 2007) As alterações neurológicas associadas à DF compreendem desde infartos
isquêmicos silenciosos (IIS), aos ataques isquêmicos transitórios (AIT) e AVC clinicamente
manifesto.
O IIS é a complicação neurológica mais comum da DF. Consiste em alteração no exame
de ressonância nucelar magnética (RNM) do cérebro, porém o paciente mantém exame
neurológico normal e não apresenta história compatível com AVC prévio. (DEBAUN et al.,
2012) Pelo menos 27% dos pacientes com AF terão um IIS antes dos 6 anos de idade.
(KWIATKOWSKI et al., 2009) Já o AIT consiste em outra complicação neurológica da DF e
um fator de risco importante para AVC (risco relativo 56.0). (BROUSSE; KOSSOROTOFF;
DE MONTALEMBERT, 2015) Tem por definição um evento que resultou em déficits
neurológicos focais com duração mínima de 24 horas, sem anormalidades nas imagens
ponderadas, T2 ou FLAIR, na RNM de cérebro indicativas de um infarto agudo e sem outra
causa explicável. (DEBAUN, 2014) A presença de AIT ou IIS, principalmente antes de 6 anos
de idade, configuram risco elevado de AVC.(MIRRE et al., 2010)

25
A alteração cerebrovascular mais catastrófica é o AVC. Em 75% dos casos decorrem de
lesões isquêmicas, principalmente infartos territoriais com falha de perfusão ou possivelmente
embolia arterial, envolvendo geralmente as grandes artérias do círculo de Willis, com dano
neurológico irreversível.(ADAMS, 2007) Apenas 25% dos AVCs ocorrem por hemorragia.
(ADAMS et al., 1998, apud STOCKMAN, 1972) A prevalência do AVC chega a 11% na
população falciforme, com maior incidência na primeira década de vida. (TALAHMA;
STRBIAN; SUNDARARAJAN, 2014 apud, HENE-FREMPONG, 1998)
Alguns fatores de risco estão associados ao AVC na população falciforme: baixa
disponibilidade de oxigênio devido à baixa saturação de oxigênio ou diminuição abrupta da
hemoglobina; presença de vasculopatia cerebral comprometendo o fluxo sanguíneo cerebral;
infecção aguda com febre, aumentando a demanda metabólica cerebral; fatores de risco
cardiovascular (hipertensão, diabetes melitus, dislipidemia, fibrilação atrial e doença renal);
infarto cerebral prévio, principalmente nos 2-3 primeiros anos após evento; aumento rápido
da hemoglobina >12g/dl, tanto após sequestros esplênicos ou hepáticos ou regimes
transfusionais. (DEBAUN; KIRKHAM, 2016,)
Estudos comprovam alta recorrência de AVC ou alterações neurológicas em pacientes
falciformes após o primeiro episódio. (SCOTHORN et al., 2002) (PEGELOW et al., 1995)
O doppler transcraniano (DTC) tornou-se um importante aliado para a detecção de
crianças falciformes que desenvolvem a vasculopatia cerebral e apresentam risco de AVC,
possibilitando intervenção precoce e eficaz. O DTC vem sendo utilizado como método não
invasivo para avaliar a velocidade do fluxo sanguíneo nas grandes artérias do círculo de Willis
desde 1982.(AASLID; MARKWALDER; NORNES, 1982) Porém, depois do estudo STOP,
vários centros no mundo inteiro adotaram o DTC como exame de triagem de doença
cerebrovascular obrigatório para crianças entre 2 e 16 anos. A população falciforme apresenta
velocidade de fluxo geralmente aumentada quando comparada a população geral, pela anemia
grave, porém, torna-se mais elevada quando algum grau de estenose reduz o diâmetro da luz
arterial. (ADAMS, 2007) A velocidade elevada, acima de 170cm/s, principalmente acima de
200cm/s, na artéria cerebral média ou carótida interna, consiste em um poderoso indicador de
DCV. (ADAMS et al., 1992)
No Brasil, a Portaria Ministerial 473, de 06 de abril de 2013, fornece as diretrizes para a
realização do DTC na população falciforme. Inclui os pacientes de 2 a 16 anos, com qualquer

26
fenótipo, dando-se preferência para o HbSS e Hb S/beta talassemia. A velocidade normal
considerada é de 70 – 170 cm/s, sendo o intervalo para repetição do exame dependente da
velocidade de fluxo. Desta forma, pacientes com DTC normal deverão repetir o exame
anualmente, caso velocidade entre 171-184 cm/s deve-se repetir o exame após 3 meses, porém
caso entre 185-199 cm/s repete-se com 1 mês, se 2 exames alterados, deve-se considerar o
risco de AVC. Indivíduos com velocidades <70 cm/s ou >200cm/s devem repetir o exame
com um mês, porém caso esteja >220 cm/s, preconiza-se considerar o risco elevado de AVC
e instituir terapêutica imediata, como transfusão de hemácias. (MINISTÉRIO DA SAÚDE,
2013b)
Outras manifestaçãoes clínicas comuns nos pacientes com DF são alterações retinianas,
ulceras crônicas, necrose asséptica de cabeça de fêmur dentre outras. O acompanhamento
clínico do paciente falciforme visa justamente a qualidade de vida, prevenção de lesão de
órgão alvo e controle de complicações já estabelecidas, minimizando suas sequelas.
1.3.3. Tratamento
Muitos avanços no tratamento da DF foram alcançados nos últimos anos desde o uso da
penicilina profilática, vacinação contra germes encapsulados, uso de Hidroxiuréia (HU) e dos
quelantes de ferro, e sobretudo relacionados às transfusões sanguíneas, com grande benefício
principalmente para a população pediátrica.
Pacientes com DF são especialmente propensos a infecções do trato respiratório e da
corrente sanguínea (sepse), principalmente causadas por germes encapsulados tais como
Haemophilus influenzae, Neiserria meningitidis, Staphylococcus aureus, sobretudo o
Pneumococo. As infecções ocorrem, em parte, devido ao baço não funcionante decorrente dos
múltiplos infartos, assim como por alterações no complemento, imunoglobulinas e leucócitos.
(HIRST; OWUSU-OFORI, 2002) Desde a década de 80 vem sendo documentado o benefício
da profilaxia com penicilina para os pacientes falciformes com 3-5 anos de idade, chegando a
84% de diminuição de infecções nessa população. (GASTON et al., 1986) A
antibioticoprofilaxia deve ser instituída precocemente, por volta dos 4 meses de vida, com
benefício documentado até os 5 anos de idade, quando associada a vacinação

27
antipneumocócica.(HIRST; OWUSU-OFORI, 2002) (FALLETTA et al., 1995) Nos pacientes
submetidos a esplenectomia, a penicilina profilática deve ser mantida por pelo menos 3 anos
após a realização do procedimento, associada a prévia vacinação contra germes encapsulados,
apresentando comprovada eficácia em diminuir o risco de infecções graves.(WRIGHT;
HAMBLETON; THOMAS, 1999)
A única medicação modificadora da DF, com impacto direto sobre a doença e aprovada
pela Food and Drug Administration (FDA), nos Estados Unidos, e pela Anvisa, no Brasil, é a
Hidroxiuréia (HU), também conhecida como hidroxicarbamina. (QUARMYNE et al., 2016)
A HU atua como inibidor da ribonucleotido redutase, reduzindo a proliferação celular, e assim
causando um estresse na medula óssea, que responde a esse estímulo com aumento do número
de células F, produtoras de hemoglobina fetal. Nas décadas de 80 e 90 muitos estudos já
relatavam seu papel na DF, principalmente no aumento da hemoglobina fetal.(RODGERS et
al., 1990) (CHARACHE et al., 1987) Atualmente, sabe-se que também promove o aumento
do tamanho do eritrócito, aumenta sua deformabilidade, além de diminuir o número de
leucócitos, reticulócitos e moléculas de adesão, reduzindo a oclusão vascular.(IUGHETTI;
BIGI; VENTURELLI, 2016)
O estudo Baby HUG, avaliou 193 crianças em uso de Hidroxiuréia e evidenciou a
importância da medicação em reduzir episódios dolorosos, STA e necessidade transfusional,
sem aumento do risco de toxicidade na população pediátrica. (THORNBURG et al., 2012) Os
benefícios associados à Hidroxiuréia (diminuição de crises de dor e hospitalizações),
possibilitam inclusive uma redução de custo no tratamento desses
pacientes.(STALLWORTH; JERRELL; TRIPATHI, 2010)
No Brasil, o Ministério da Saúde através da Comissão Nacional de Incorporação de
Tecnologia do SUS (CONITEC) preconiza o uso de Hidroxiuréia para os pacientes
falciformes acima de 2 anos (podendo ser utilizado após os 9 meses de vida, em situações
especiais), mediante a presença de algumas manifestações clínicas da DF, na dose inicial de
15mg/kg/dia, com aumento de 5mg/kg/dia a cada 4 semanas, até dose máxima de 35mg/kg/dia
ou dose menor mediante toxicidade. (MINISTÉRIO DA SAÚDE, 2016c)
Nos últimos anos, outros avanços foram desenvolvidos no tratamento do paciente
falciforme, visando sobretudo a cura da doença. O transplante de medula óssea é a única
terapêutica curativa da doença em uso clínico até o momento. (ARNOLD et al., 2016) No

28
Brasil, o transplante de medula óssea aparentado HLA idêntico foi aprovado como opção
terapêutica oficial no âmbito do SUS apenas recentemente (Portaria do MS n. 30, de 30 de
junho de 2015).
Outra modalidade terapêutica que promete a cura da doença falciforme é a terapia gênica.
Mas esta modalidade ainda está em estudos pré-clínicos, porém com grande promessa para
um futuro próximo. (RAI; MALIK, 2016)
1.3.4. A transfusão Sanguínea na Doença Falciforme
A transfusão sanguínea sempre constituiu um forte pilar no tratamento dos pacientes
falciformes. Artigos da década de 50 já mostravam benefícios aos pacientes com as
transfusões de hemácias de indivíduos saudáveis. (WAHL; QUIROLO, 2009 apudi MADEL,
1958) Inicialmente visava-se o incremento dos valores da hemoglobina, atualmente os
objetivos transfusionais incluem o alívio dos sintomas de anemia, redução relativa na
percentagem de eritrócitos em circulação contendo HbS e melhoria na capacidade de
transporte de oxigênio, minimizando complicações da doença. (STEINBERG, 2008). A
indicação transfusional para o paciente falciforme tem relação com as sintomatologias agudas
ou crônicas da doença e podem consistir em transfusões eventuais (agudas) ou regimes
transfusionais regulares (crônicos). (KOEHL et al., 2016)
As transfusões eventuais, geralmente, priorizam a melhoria no transporte de oxigênio em
relação a diminuição da concentração de HbS. (WAYNE; KEVY; NATHAN, 1993) O nível
de hemoglobina basal do paciente deve ser valorizado, visto que os indivíduos se ajustam
fisiologicamente a certo grau de anemia, portanto a transfusão normalmente é desnecessária
quando objetiva apenas o incremento desse valor. Estados que causem exacerbação da anemia
e seus sintomas devem orientar a decisão quanto ao valor de hemoglobina pós-transfusão
desejada e como será realizada a transfusão.(CHOU; LIEM; THOMPSON, 2012) (WAHL;
QUIROLO, 2009)
Para as transfusões agudas, são normalmente utilizados 10-20 ml de CH para crianças e
1-3 unidades de CH para adultos, com o cuidado em manter os níveis de hemoglobina pós

29
transfusionais abaixo de 11g/dl, desta forma evitando sobrecarga volêmica, aumento de
viscosidade sanguínea e suas consequências, como edema pulmonar e AVC. (JOHNSON,
2005) (TELEN, 2001) (CHOU; FASANO, 2016) Em situações como sequestro esplênico a
transfusão de alíquotas pequenas de CH, como 5ml/Kg, monitorando o tamanho do baço, nível
de Hb e status cardiovascular, mostram-se mais seguras, diminuindo o risco de
hiperviscosidade pós resolução do sequestro. As transfusões agudas normalmente são
utilizadas nos casos de anemia sintomática, sequestros, crises hemolíticas, crises aplásticas,
perda sanguínea. (WAYNE; KEVY; NATHAN, 1993)
Uma indicação de transfusão aguda frequente é a realização de cirurgia. O estudo TAPS1
avaliou pacientes falciformes com mais de 1 ano de idade, submetidas a cirurgias de baixo e
médio riscos em 22 cidades de alguns países, randomizando-os em um grupo com transfusão
pré-operatória e outro sem transfusão. No grupo transfundido profilaticamente, caso o
paciente apresentasse Hb <9 era feita uma transfusão simples, porém com Hb > 9 era feita
uma transfusão de troca objetivando HbS <60%, até 10 dias antes da cirurgia. No estudo foi
observada maior incidência de complicações no grupo sem transfusão pré-operatória do que
nos submetidos a transfusão profilática. (HOWARD et al., 2013) Contudo, uma revisão da
Cochrane de 2016, mostrou a existência de poucos estudos randomizados capazes de
comparar os protocolos transfusionais pré-operatórios mais agressivos em relação aos mais
conservadores na redução de complicações relacionadas a cirurgia e/ou á DF. (ESTCOURT
et al., 2016)
Nos regimes transfusionais regulares (crônicos) as transfusões são realizadas, geralmente,
a cada 3 a 5 semanas. (WARE; KWIATKOWSKI, 2013) Objetiva certo grau de elevação do
valor da hemoglobina, suprimindo desta forma a eritropoiese do próprio paciente e como
consequência diminuindo a produção de hemácias falcizadas, além do aumento de hemácias
circulantes normais provenientes da transfusão. (WAYNE; KEVY; NATHAN, 1993)
As principais indicações para os regimes transfusionais crônicos são a doença
cerebrovascular, complicações pulmonares e cardíacas, sequestro esplênico
recorrente.(WAYNE; KEVY; NATHAN, 1993) Algumas indicações são pouco embasadas
pela literatura, tais como ulceras crônicas e priaprismo.(MIAN et al., 2015) O valor desejado
1 Transfusion Alternatives Preoperatively in Sickle Cell Disease

30
de manutenção das hemácias do próprio paciente, ou seja, da HbS, é de 30% a 50%, a depender
da indicação transfusional. A HbS < 30% mostrou-se eficaz na prevenção primária e
secundária do AVC, com o cuidado de evitar o aumento excessivo do valor final da
hemoglobina. (WAHL; QUIROLO, 2009) Muitas vezes esses objetivos não são alcançados
devido a vários fatores como quantidade de sangue administrado, método utilizado, intervalo
entre as transfusões. (WARE, 2007)
As transfusões nos pacientes falciformes podem ser realizadas de forma simples, por troca
manual ou por eritrocitoaférese (troca automatizada). No primeiro caso o concentrado de
hemácias é transfundido de forma simples, com gotejamento direto na veia do paciente,
geralmente utilizadas por pacientes com valores de Hb mais baixos (Hb <8,5g/dL). No
segundo caso, transfusão de troca manual, uma quantidade de sangue do paciente é retirada
manualmente antes da transfusão do concentrado de hemácias, sendo preferível para pacientes
com Hb mais altas, acima de 8,5 – 9,0 g/dl. Já na eritrocitoaférese, uma máquina de aférese
permite, através de centrifugação, a retirada das hemácias falciformes do indivíduo e troca de
parte do sangue do paciente por hemácias do doador. (WARE, 2007) Quando possível, as
transfusões de troca devem ser preferidas às transfusões simples, uma vez que substituem
parte das hemácias falciformes do indivíduo por células normais, além de reduzir a
viscosidade do sangue e limitar a sobrecarga de ferro. (KOEHL et al., 2016)
Existem vários protocolos para transfusão de troca manual. Aloni et al, adotando o
protocolo com remoção de 30-40 ml/kg de sangue, transfusão de 15-25 ml/kg de CH e infusão
de 15-25 ml/kg de solução salina a cada 3 – 5 semanas com objetivo de manter HbS< 30% e
Hb entre 9 -10,5g/dL verificou boa tolerância por parte dos pacientes, com baixa incidência
de reações adversas e melhora na sobrecarga de ferro transfusional, limitando a necessidade
de terapia quelante. (ALONI et al., 2015)
Mian et al, comparou 2 protocolos para troca manual, um baseado por unidade e outro
baseado por peso, em pacientes falciformes com idade >16 anos. No primeiro, utilizado no
Canadá, foi preconizada a realização de uma flebotomia de 500ml seguida de infusão de
500ml de soro fisiológico e posterior transfusão de 02 unidades de CH. O segundo protocolo
foi realizado em Londres. Nele a transfusão de troca é baseada no peso do paciente, desta
forma utiliza-se flebotomia de 15 ml/kg com reposição 1:2, ou seja, mesma quantidade de
salina para o dobro de CH, porém, antes da troca, pode ser necessária realização de flebotomia
conforme o valor de Hb pré-transfusional: se a Hb> 7,0 g/dL retirar 5ml/Kg de sangue e

31
infusão de 5ml/Kg de solução salina; se a Hb estiver entre 6,0 e 7,0 g / dL não realizar
flebotomia previamente à troca; se Hb for <6,0 g/dL deve-se transfundir 10ml/kg de CH antes
da troca. No estudo foi verificado que o protocolo baseado em unidade foi mais eficaz em
atingir o alvo de HbS, sem necessitar de volume maior de CH. (MIAN et al., 2015)
A eritrocitoaférese é empregada mais frequentemente em pacientes em regime
transfusional crônico, como nos casos de pacientes após AVC ou com DTC alterado. Também
é utilizada em algumas situações agudas, menos frequentemente, como em pacientes com
STA. Apresenta maior eficiência e rapidez em diminuir HbS para um valor alvo, mantendo o
hematócrito/hemoglobina dentro do limite esperado, do que a transfusão de troca
manual.(QUIROLO et al., 2015)(MICHOT et al., 2014)
Para a realização da transfusão com troca automatizada, pode ser necessário grande
volume de hemácias, em média de 15 a 25ml/kg, a cada 5 a 6 semanas, chegando a utilizar
quantidade 20 a 50% maiores de CH do que as transfusões simples ou de troca manual,
expondo o indivíduo a um número grande de doadores e, consequentemente, a mais antígenos
eritrocitários. (MICHOT et al., 2014)
A eritrocitoaférese é um procedimento dispendioso e invasivo. (MIAN et al., 2015) Para
ser realizada é importante que o paciente tenha acesso venoso calibroso, preferencialmente 2
acessos venosos, quando não possível torna-se necessária a utilização de cateter venoso
central.(WARE, 2007) É um procedimento mais caro, que necessita de equipamento adequado
e equipe capacitada o que não está disponível em muitos centros. (WARE, 2007) (KIM, 2014)
O volume extracorpóreo no procedimento não deve ultrapassar 15% do volume sanguíneo
total do indivíduo. Em situações que excedam esse valor, como em crianças, deve ser feito
um primer de hemácias antecedendo o procedimento. (KIM, 2014) Atualmente, alguns
equipamentos apresentam menor volume extracorpóreo, sendo mais seguros para utilização
em crianças menores de 20Kg. (PERSEGHIN et al., 2013) (CHOU; FASANO, 2016)

32
Nas últimas 3 décadas, estudos com a prevenção primária ou secundária de acidente
vascular encefálico (AVC) como STOP2, TWiCH3 e SWiCH4 evidenciaram a importância dos
regimes transfusionais crônicos nos pacientes com DF e risco de AVC. (BROUSSE;
KOSSOROTOFF; DE MONTALEMBERT, 2015)
O estudo STOP avaliou se a transfusão sanguínea seria capaz de diminuir a HbS para
valores abaixo de 30%, diminuindo também o risco do primeiro AVC ou IIS na população
falciforme com alteração no DTC. Foram avaliadas 2000 crianças falciformes, destas 130
apresentaram DTC alterado sendo incluídas no estudo. Este Trial trouxe importantes repostas
para a ciência, consistindo em um marco no tratamento dos pacientes falciformes.(ADAMS
et al., 1998)
O estudo STOP II avaliou o risco de AVC ou DTC alterado em pacientes falciformes com
alteração prévia do DTC após normalização das velocidades de fluxo e suspensão das
transfusões. Teve que ser descontinuado por evidente risco de AVC quando interrompidas as
transfusões. (ADAMS; BRAMBILLA, 2005)
Mantendo a linha de investigação sobre doença cerebrovascular foi realizado o estudo
SWiTCH que analisou os pacientes falciformes em regime transfusional crônico por AVC, os
separando em dois grupos: Hidroxiuréia e flebotomia; transfusão e quelação de ferro. O
objetivo primário foi avaliar o papel das diferentes condutas analisadas na prevenção
secundária de AVC e sobrecarga de ferro, uma inevitável complicação transfusional. Concluiu
haver benefício com a prática de transfusão e quelação na população avaliada. (WARE;
HELMS, 2012)
Visando minimizar os riscos dos regimes transfusionais crônicos foi realizado o estudo
TWiTCH5, avaliando 121 pacientes com AF com DTC alterado sem AVC prévio. Concluiu
haver segurança na substituição das transfusões sanguíneas crônicas por HU, em pacientes
2 Stroke Prevention Study in Silckle Cell Desease
3 Transcranial Doppler Ultrasounds with Transfusions Changing to Hydroxyurea
4 Stroke with Transfusions Changing to Hydroxyurea
5 TCD With Transfusions Changing to Hydroxyurea

33
com DTC alterado, após pelo menos 1 ano de transfusão crônica e com DTC normalizado.
(WARE et al., 2016). Outros estudos vêm demostrando resultados também satisfatórios com
a troca das transfusões por HU após normalização das velocidades do DTC. (BERNAUDIN
et al., 2016)
Na infância, os regimes transfusionais crônicos são utilizados na DF para outras situações
além da doença cerebrovascular, como na prevenção da recorrência de sequestros esplênicos,
naquelas pacientes sob risco, até que completem seu calendário vacinal básico e sejam
submetidos a esplenectomia. Outras indicações consistem em síndromes torácicas agudas
recorrentes, além de algumas com menos evidências, como priapismo ou dor
recorrente.(O’SUOJI et al., 2013)
No estudo SIT6, apesar do objetivo primário ser testar a hipótese da menor recorrência de
infarto cerebral silencioso entre as crianças submetidas à terapia com transfusão sanguínea
regular do que entre aquelas que receberam tratamento padrão, verificou-se que a incidência
CVO, STA, priaprismo e nova necrose avascular sintomática do quadril foram
significativamente menores no grupo em regime transfusional crônico quando comparado
com o grupo controle. (DEBAUN, 2014)
As transfusões sanguíneas, apesar de cada dia mais seguras, guardam em si riscos tais
como reações transfusionais agudas e tardias, transmissão de patógenos, sobrecarga de ferro
e formação de anticorpos. (CHOU; LIEM; THOMPSON, 2012) Diante da maior utilização
das transfusões nos pacientes falciformes alguns problemas adquiriram maior relevância como
a alo e autoimunização eritrocitária e, uma das suas consequências, as reações transfusionais
hemolíticas tardias (RTHT), (CHOU et al., 2013) Nos pacientes aloimunizados o objetivo
transfusional de diminuir a hemoglobina S e aumentar o aporte de oxigênio pode ser
prejudicado devido a hemólise e menor sobrevida das hemácias, além de haver dificuldade de
encontrar sangue compatível, pela presença do aloanticorpo. (PINTO; BRAGA; SANTOS,
2011)
Os mecanismos relacionados com aloimunização eritrocitária não estão totalmente
elucidados. O ponto de partida para a formação de aloanticorpos são as diferenças antigênicas
6 Silent Cerebral Infarct Multi-Center Clinical Trial

34
entre as hemácias do doador/feto e do paciente/gestante. Neste caso, as hemácias
transfundidas ou provenientes do feto expressam antígenos em sua superfície geneticamente
ausentes no receptor. Esses antígenos têm epítopos reconhecidos pelas moléculas HLA de
classe II do receptor que os apresentam aos linfócitos T CD4 desencadeando a resposta imune,
com consequente ativação dos linfócitos B e formação de anticorpos. (HIGGINS; SLOAN,
2008) (YAZDANBAKHSH; WARE; NOIZAT-PIRENNE, 2012) (KÖRMÖCZI; MAYR,
2014) Pacientes falciformes aloimunizados apresentam maior porcentagem de células T CD4
de memória do que os não aloimunizados (57% x 49%), sem alteração significativa de outras
células do sistema imune, mostrando relação com as altas taxas de aloimunização encontradas
nesta população. (NICKEL et al., 2015)
Na população geral que recebeu transfusão é estimada uma prevalência de 4% de
aloimunização.(HIGGINS; SLOAN, 2008) As taxas de aloimunização mudam na literatura
conforme a doença apresentada pelo paciente, sendo 1% no caso de leucemia linfoide aguda,
3% em leucemia mieloide aguda, 5-45% nas talassemias. (HENDRICKSON; TORMEY,
2016) Na população falciforme a incidência de aloimunização eritrocitária é considerada
maior, porém é bastante variável. Nos Estados Unidos foi evidenciada incidência de
aloimunização eritrocitária em DF de 7% a 47%. Porém, na Uganda e na Jamaica essa
incidência atinge somente 6,1% a 2,6%, respectivamente. (CHOU, 2013) (OLUJOHUNGBE
et al., 2001) (NATUKUNDA et al., 2010) A diferença entre esses dados pode estar relacionada
com a origem étnica dos doadores de sangue e pacientes transfundidos. Nos Estados Unidos
a maioria dos doadores é branca e os pacientes com DF são afrodescendentes, já na Jamaica
e Uganda essa diferença entre doadores e pacientes torna-se menor. (NATUKUNDA et al.,
2010) (OLUJOHUNGBE et al., 2001) (YAZDANBAKHSH; WARE; NOIZAT-PIRENNE,
2012)
No Brasil, a incidência de aloimunização na população falciforme apresenta variação
entre os diferentes estudos e protocolos de compatibilidade antigênica utilizados. Murao e
Viana evidenciaram frequência de aloimunização de 9,9% nos pacientes falciformes. Em
contrapartida, Zanette et al, constatou uma prevalência de 49% de pacientes falciformes
adultos aloimunizados.(ZANETTE et al., 2010)(MURAO; VIANA, 2005)
Atualmente são descritos 339 antígenos eritrocitários, sendo 297 alocados em 33 sistemas
e os demais em 6 coleções, a série 700 de baixa frequência populacional e a série 901 de alta
frequência populacional.(STORRY et al., 2014) A prevalência de alguns antígenos é mais

35
elevada em determinados grupos étnicos, como os antígenos C e E do grupo sanguíneo (RH),
K1 do Kell (KEL), Fya do Duffy (FY), Jkb do Kidd (JK) e S do grupo sanguíneo MNS que são
mais frequentemente encontrados em indivíduos brancos do que em pessoas de ascendência
africana. Desta forma, os anticorpos contra esses antígenos são mais frequentes em pacientes
com DF. (VICHINNSKY et al., 1990)
Alguns antígenos eritrocitários são considerados mais imunogênicos do que outros. A
imunogenicidade desses antígenos é tipicamente expressa pela fração de indivíduos negativos
para determinando antígeno que desenvolvem anticorpo contra o antígeno quando exposto a
01 unidade de concentrado de hemácias com positividade para o antígeno em questão.
(STACK; TORMEY, 2016, apudi GILBLETT, 1961) Utilizando um modelo de cálculo
proposto por Gilblett, tendo como parâmetro a imunogenicidade do antígeno K, Stack e
Tormey, estabeleceram quais o antígenos eritrocitários são considerados mais imunogênicos,
sendo eles: K>Jka>Lua >E>P1>c>M>Leb >C>Lea >Fya >S. (STACK; TORMEY, 2016)
Diversos protocolos para compatibilização de hemácias baseadas em uma seleção
específica de antígenos são encontrados na literatura, visando diminuir as taxas de
aloimunização na população falciforme. Castro et. al., avaliou 5 protocolos distintos de
compatibilização fenotípica eritrocitária: protocolo 1 -compatibilidade C, c, E, e; protocolo 2
- C, c, E, e, K; protocolo 3 - C, c, E, e, K, S; protocolo 4 - C, c, E, e, K, S, Fya; protocolo 5 -
C, c, E, e, K, S, Fya, Jkb. Atingiu taxas de prevenção de aloimunização de 52,9% com o
protocolo 1, pouca variação com os protocolos 2 e 3, já com os protocolos 4 e 5 atingiu as
taxas de 68,2% e 70,8%, respectivamente. Por outro lado, o fenótipo D-, C-, c+, E-, e+, K-,
S-, Fy (a-), Jk (b-), requerido no protocolo 5 (com menor incidência de aloimunização
eritrocitária) ocorre em apenas 0,6% dos doadores brancos, sendo esses antígenos negativos
22,7 vezes menos prevalentes do que aqueles requeridos para a correspondência fenotípica no
protocolo 2, talvez este último seja o protocolo mais factível de ser implementado. (CASTRO
et al., 2002)
Atualmente há consenso na literatura sobre a necessidade de transfusão de hemácia pelo
menos com compatibilidade com os antígenos ABO, C, E e K, visando diminuir a
aloimunização eritrocitária e as reações hemolíticas tardias, para aqueles pacientes em risco.
(Yawn, et al., 2014). A extensão da compatibilidade para outros antígenos como Duffy, Kidd
ou MNS, diminui a aloimunização, porém traz dificuldades para a prática clínica pela sua
menor disponibilidade na população. (CHOU et al., 2013) A legislação Brasileira, através da

36
portaria nº 158, de 04 de fevereiro de 2016, sugere a extensão da compatibilidade fenotípica
para os sistemas mais imunogênicos (Rh, Kell, Duffy, Kidd e MNS) no caso de pacientes que
poderão entrar em esquema de transfusão crônica, como ocorre com as pacientes falciformes.
(MINISTÉRIO DA SAÚDE, 2016d)
Apesar das transfusões fenótipo compatíveis, alguns pacientes falciformes transfundidos
ainda adquirem aloanticorpos. Pessoas de origem africana possuem variantes de alguns
antígenos eritrocitários, principalmente do grupo sanguíneo Rh. Esses antígenos foram
descritos como dois tipos "parciais" ou "fracos". São codificadas por mutações nos genes
relacionados com os grupos sanguíneos, como mutações pontuais, mutações missense
múltiplas ou alelos híbridos de RHD e RHCE, do grupo sanguíneo Rh. Os antígenos parciais,
não apresentam sua expressão completa na superfície da hemácia, portanto, caso o indivíduo
receba transfusão contendo o antígeno “completo”, poderá desenvolver aloanticorpo. Os
pacientes com antígenos fracos apresentam diminuição da quantidade de antígenos expressos,
porém não perdem partes dos mesmos, desta forma, não são frequentemente aloimunizados.
(NOIZAT-PIRENNEA; TOURNAMILLE, 2011)
Devido ao aumento da aloimunização, mesmo com o uso de protocolos para
compatibilidade das transfusões de acordo com a fenotipagem eritrocitária, o estudo molecular
dos grupos sanguíneos, através da genotipagem, vem sendo empregada visando minimizar os
riscos transfusionais. A tipagem eritrocitária baseada no DNA proporciona maior precisão e
maior informação sobre os antígenos eritrocitários quando comparados aos métodos de
hemaglutinação. (CASAS et al., 2015) A utilização da genotipagem possibilita identificar
indivíduos com variantes de RhD e RhCE melhorando a prática transfusional, especialmente
para pacientes com DF.(GASPARDI et al., 2016) A utilização de concentrado de hemácias
com compatibilidade genotípica pode ser uma estratégia para os pacientes sem fenotipagem
eritrocitária e recentemente transfundidos, além de diminuir a taxa de aloimunização e as
RTHT, no entanto apresenta a dificuldade como a necessidade de genotipagem de grande
número de doadores e seu alto custo. (GASPARDI et al., 2016) (FICHOU et al., 2016)
Apesar de a diferença antigênica ser o fator mais importante para aloimunização
eritrocitária, vários outros fatores de risco também foram relacionados. Alguns estudos
associam o aumento no risco de formação de aloanticorpo diretamente ao número de
transfusões, independentemente da idade. Também estão associados, o estado inflamatório do
indivíduo, a idade avançada e o sexo feminino.(VICHINNSKY et al., 1990) (MURAO;

37
VIANA, 2005) (HENDRICKSON et al., 2006) Transfusões esporádicas, por complicação
aguda, apresentam maior risco de aloimunização quando comparada ao paciente em regime
transfusional crônico. (SINS et al., 2016)
O status inflamatório do paciente guarda relação direta com o risco de desenvolver
aloanticorpos. Pacientes falciformes transfundidos em vigência de eventos inflamatórios,
principalmente STA e CVO, apresentam alto risco de aloimunização. (FASANO et al., 2014)
Yazdanbakhsh, descreve sua hipótese sobre a associação entre estado inflamatório e
aloimunização. Aborda o papel da hemioxigenase (HO-1) em lizar o heme em compostos
como ferro, bilirrubina e monóxido de carbono, diminuindo seu potencial oxidativo e
inflamatório. Diante dos baixos níveis de HO-1 expresso pelos monócitos dos pacientes
falciformes, maior é o status inflamatório desses indivíduos. O ambiente pró-inflamatório,
desregula a resposta imune das células T aos eritrócitos transfundidos, levando a uma resposta
patogênica e consequente aloimunização. (YAZDANBAKHSH, 2015)
Michot et al, verificou a prevalência de aloimunização eritrocitária de 33% em pacientes
falciformes submetidos a eritrocitoaférese e 22% nos pacientes submetido a transfusão
convencional (transfusão simples com ou sem troca manual). Quando ajustada esta
prevalência pela quantidade de CH utilizado, verifica-se menor taxa de aloimunização nos
pacientes submetidos a eritrocitoaférese, assim como menores taxas de reação transfusional
hemolítica tardia.(MICHOT et al., 2014) O procedimento de aférese leva a retirada de plasma
do paciente e talvez com isto ocorra diminuição de citocinas e mediadores inflamatórios,
diminuindo a resposta imune com formação de anticorpos.(UHLMANN; SHENOY;
GOODNOUGH, 2014)
Alguns pacientes aparentam desenvolver aloanticorpos com frequência maior que a
esperada, são os chamados “respondedores”. Eles, apesar da mesma exposição aos fatores de
risco para aloimunização que a população geral, “não respondedora”, apresentam maior
formação de aloanticorpos. (HIGGINS; SLOAN, 2008) Vários mecanismos vêm sendo
relacionados a maior frequência de aloimunização nestes indivíduos respondedores tais como,
polimorfismos do HLA (Antígeno de Histocompatibilidade Principal), genes relacionados
com produção de citocinas e com o estado de inflamação. (SIPPERT et al., 2016)
(HENDRICKSON et al., 2006)

38
Os alelos HLA medeiam a resposta imune a diversos antígenos. Estudos mostram forte
associação entre o locus HLA DRB1 com o desenvolvimento de anticorpos contra antígenos
eritrocitários, principalmente o HLA DRB1*15 com formação de múltiplos aloanticorpos.
(SCHONEWILLE et al., 2014) (HOPPE et al., 2009) Em contrapartida, o alelo HLA DQ
parece estar relacionado com a menor incidência de aloimunização eritrocitária, constituindo
um fator aparentemente protetor em pacientes com DF. (TATARI-CALDERONE et al., 2016)
A presença de alguns alelos HLA associam-se com maior risco de formação de anticorpo
contra determinado antígeno eritrocitário específico como ocorre com o HLA DRB1*04 e
HLA DRB1*1501 com formação de anti-Fya, HLA-DRB1*0701 com anti-Dia, HLA
DRB1*11 e HLA DRB1*13 com anti-K. (CHIARONI et al., 2009) (BALEOTTI et al., 2014)
(CHIARONI et al., 2006)
A aloimunização traz consigo complicações clínicas importantes. Telen et. al, descrevem
a associação entre aloimunização eritrocitária na DF e dor crônica, risco de falência de órgãos
e menor sobrevida do paciente. (TELEN et al., 2015) Pacientes aloimunizados podem
apresentar anticorpos clinicamente significantes, capazes de desencadear hemólise, desta
forma, devem ser transfundidos com hemácias negativas para o antígeno para o qual apresenta
o aloanticorpo, dificultando a localização de bolsa fenótipo compatível para a transfusão.
(NOIZAT-PIRENNE, 2013)
A prática transfusional apresenta risco de reações transfusionais agudas ou tardias. As
reações agudas mais frequentes na população geral são a Reação Transfusional Febril não
Hemolítica (1-3% por unidade transfundida) seguida da Reação Alérgica. (SANDERS et al.,
2005) (DELANEY et al., 2016) No primeiro caso, o paciente apresenta febre associada ou não
a calafrios e/ou dor causadas por citocinas pró-inflamatórias ou anticorpos no receptor contra
antígenos presentes no hemocomponente. A segunda reação transfusional é mediada por
histamina e caracterizada por manifestações alérgicas como placas urticariformes, prurido,
edema, tosse até 4h após uso do hemocomponente. (DELANEY et al., 2016) (SANITÁRIA,
2015)
Uma reação transfusional diretamente relacionada a aloimunização eritrocitrária e mais
frequente em pacientes falciformes é a Reação Transfusional Hemolítica Tardia (RTHT),
muitas vezes associadas à Síndrome de Hiperemólise, potencialmente fatal. A RTHT pode ser
definida por sinais de hemólise acelerada (incluindo hemoglobinúria, icterícia, palidez,
aumento da desidrogenase láctica e/ou da bilirrubina e/ou anemia mais acentuada do que

39
detecta antes da transfusão), sem razão óbvia ou perda sanguínea, iniciando pelo menos 3 dias
após a transfusão, mais frequentemente entre o 5° e o 15° dias, podendo ocorrer intensificação
dos sintomas da doença como CVO ou STA. (DESSAP et al., 2016)
Vários artigos relacionam a aloimunização eritrocitária com RTHT. Normalmente os
pacientes com RTHT apresentam testes de triagem pré-transfusionais normais, porém
possuem histórico de aloanticorpos prévios, os quais devem estar em títulos bastante
diminuídos (evanescentes) por isso não detectados no momento da nova transfusão. A nova
exposição ao antígeno leva ao recrudescimento dos títulos do aloanticorpo, principalmente
IgG, com mudança de sua subclasse e aumento do seu potencial citotóxico. Grande parte das
hemácias transfundidas são então destruídas por macrófagos, no fígado e no baço, que
expressão em sua membrana receptores contra porção Fc das imunoglobulinas. A opsonização
do complemento pode sinergizar a hemólise mediada por IgG através de um receptor C3b em
macrófagos.(NOIZAT-PIRENNE, 2012) Outro grupo de pacientes pode possuir anticorpos
recém diagnosticados e classificados como não hemolíticos, porém, sua patogenicidade pode
ser mediada por células efetoras como macrófagos e células NK ativadas pela DF, desta forma
tornam-se capazes de desencadear hemólise. (DESSAP et al., 2016) (HABIBI et al., 2016)
A Hiperemólise é caracterizada pela queda acentuada da hemoglobina, marcada por sinais
de exacerbação da hemólise e destruição de hemácias autólogas, não apenas as transfundidas.
Os mecanismos evolvidos não estão bem elucidados, porem podem não guardar relação com
aloanticorpos.(BALLAS et al., 2010)
A sobrecarga de ferro é uma reação transfusional tardia muito frequente no paciente
falciforme, principalmente, naqueles em regime transfusional crônico. O ferro deve estar
sempre ligado a proteínas, sendo a transferrina a sua proteína ligante plasmática. Quando em
excesso, além da capacidade de ligação com a transferrina, fica livre, sendo capaz de causar
danos oxidativos nas células. O coração, fígado e órgãos endócrinos são mais susceptíveis à
oxidação pelo mineral.(WARE; KWIATKOWSKI, 2013) (WOOD et al., 2016) A terapia de
quelação de ferro visa evitar a intoxicação pelo ferro transfusional e remover o mineral
acumulado. A quelação pode ser iniciada em crianças com mais de 2 anos, após 1 ou 2 anos
de transfusão crônica e níveis de ferritina >1000 ng/ml obtidas em 2 dosagens diferentes sem
evento agudo. O objetivo é reduzir os estoques de ferro, mantendo ferritina entre 500-1500,
ferro hepático entre 2-7mg/g ou T2* cardíaco > 20ms.(WOOD et al., 2016)(WARE;
KWIATKOWSKI, 2013)

40
Existem três quelantes de ferro disponíveis: Defesoxamina, Deferasirox e Deferiprone.
(MEERPOHL et al., 2014) (WARE; KWIATKOWSKI, 2013) A Deferoxamina foi o primeiro
quelante de ferro a ser utilizado, ainda na década de 70, com grande eficácia apesar da
dificuldade de adesão ao tratamento por não apresentar formulação oral, sendo utilizada
apenas por via subcutânea ou venosa. Em seguida foi desenvolvido o Deferiprone, com
apresentação oral, porém com alguns efeitos colaterais e baixa eficácia a longo
prazo.(RAGHUPATHY; MANWANI; LITTLE, 2010) Por último foi desenvolvido o
Deferasirox, com apresentação oral e comodidade posológica de uma única administração por
dia. (RAGHUPATHY; MANWANI; LITTLE, 2010)
Alguns estudos foram realizados comparando os diferentes quelantes de ferro na
população falciforme. Foi evidenciada equivalência entre Deferasirox e Deferoxamina na
eficácia e tolerabilidade, porém com menor adesão ao tratamento com o último.
(VICHINSKY et al., 2007) (VICHINSKY et al., 2013)(JORDAN et al., 2012)
Em algumas situações torna-se necessária a combinação de diferentes quelantes, devendo,
tal medida, ser considerada caso o paciente não apresenta redução do ferro ou mostre sinais
de toxicidade da medicação ou dificuldade em tomá-la. (WARE; KWIATKOWSKI, 2013)
O Ministério da Saúde, através da Portaria Ministerial 1324, de 25 de novembro de 2013,
normatiza a utilização dos quelantes de ferro pelo SUS. Preconiza que sejam utilizados nos
pacientes falciformes, mediante comprovação de sobrecarga transfusional de ferro, nas doses
preconizadas: Deferasirox, 20mg/kg/dia, via oral, 1 vez ao dia, com dose máxima de
40mg/dia; ou Deferoxamina, 20-60mg/kg/dia, subcutâneo, por infusão durante 8 à
24h.(MINISTÉRIO DA SAÚDE, 2013c)
Os pacientes falciformes formam uma das populações mais beneficiadas pela prática
transfusional, porém também em maior risco de suas complicações. A adoção de medidas
hemoterápicas direcionadas para essa população tais como fenotipagem eritrocitária para pelo
menos Rh e Kell e se possível para Duffy, MNS e Kidd, leucodepleção, transfusão de
hemocomponentes isentos de HbS, aumentam a segurança transfusional. O conhecimento do
histórico transfusional desses pacientes torna-se fundamental para facilitar a adoção de
condutas em situações de emergência, evitar RTHT no caso de pacientes com aloanticorpos
evanescentes e facilitar a localização de bolsas fenótipo compatíveis para esses indivíduos.
(HARM et al., 2014)

41
Grande número de pacientes com hemoglobinopatias, na Bahia, são acompanhados
ambulatoriamente na HEMOBA (Hemocentro Coordenador da Bahia), que possui atualmente
cerca de 2000 pacientes com doença falciforme e idade entre 0 e 18 anos cadastrados. No
Estado existem poucas instituições públicas que fazem o seguimento desses pacientes, sendo
a maioria na capital baiana. A Bahia possui aproximadamente 14 milhões de habitantes, destes
apenas 2,6 milhões residem na capital, onde localiza-se o hemocentro. Os demais chegam a
viajar mais de 800km para manterem acompanhamento hematológico. No hemocentro elas
são acompanhadas pelo hematologista pediátrico, equipe multiprofissional composta por
enfermagem, psicologia, odontólogo, fisioterapeuta, são realizas coletas de alguns exames e
transfusões sanguíneas, em nível ambulatorial. No protocolo institucional preconiza-se
realização de transfusões com compatibilidade fenotípica ABO, Rh, Kell, Duffy, MNS e Kidd
para a população falciforme, exceto algumas exceções. Porém a transfusão de sangue
fenótipo-selecionado aumenta custos e pode dificultar encontrar doador compatível com o
fenótipo extendido. (Le, et al., 2014) Não existe sistemas informatizados no atendimento
desses pacientes nem banco de dados sobre essa população, desta forma não é possível
conhecer e acompanhar a realidade desses pacientes, bem como suas características
transfusionais.

42
2. JUSTIFICATIVA DO PROJETO
A Bahia é o Estado brasileiro com maior população de portadores de doença falciforme.
A transfusão sanguínea constitui um importante pilar no tratamento desses pacientes. A
HEMOBA é o único banco de sangue público do estado sendo responsável pelo
acompanhamento clínico e transfusional de grande parcela dessa população. Atualmente, no
cadastro da HEMOBA, constam aproximadamente 2080 pacientes com doença falciforme em
idade entre 0-18 anos. Inexiste sistema informatizado unificando os dados clínicos e
transfusionais da população falciforme assistida no ambulatório da instituição. Existe uma
carência de dados sobre os pacientes falciformes, desta forma um sistema informatizado para
seguimento desses pacientes, com ênfase em seus aspectos transfusionais, pode contribuir
sobremaneira para a organização e armazenamento dos dados, conhecimento dessa população,
possibilitando melhoria no seu atendimento e orientação das políticas de saúde não só no
Estado da Bahia, assim como em todo o país.
3. HIPÓTESE
A construção de um software para seguimento transfusional das crianças com doença
falciforme no HEMOBA auxiliará no conhecimento desta população e no seguimento clínico
da terapia transfusional, possibilitando adoção de medidas tanto para melhorar a assistência
médica junto a esses pacientes, quanto para orientar políticas de saúde pública para essa
população.

43
4. OBJETIVOS
4.1. Objetivos Gerais
- Desenvolver um Software para seguimento transfusional em hemoterapia, com ênfase
no acompanhamento de pacientes falciformes.
- Descrever e analisar aspectos transfusionais das crianças com idade entre 0 e 18 anos
com doença falciforme, acompanhados no ambulatório da HEMOBA, em Salvador.
4.2. Objetivos Específicos
- Desenvolver e testar um Software para coleta de dados demográficos e transfusionais de
crianças falciformes seguidas no ambulatório da HEMOBA.
- Identificar o número total de crianças entre 0 e 18 anos com doença falciforme em
acompanhamento na HEMOBA em regime transfusional crônico, e coletar dados
demográficos como sexo, idade, tipo de hemoglobina, cidade de origem, assim como coletar
dados clínicos, como as complicações mais frequentes dos pacientes (AVC, STA, Priapismo,
SE, DTC alterado e outras) e uso de medicamentos, como hidroxiuréia e quelantes de ferro.
- Identificar dados transfusionais como: indicação transfusional, número de transfusões
ambulatoriais, presença de aloimunização eritrocitária, especificidade do anticorpo, tipo de
hemocomponente transfundido, período entre as transfusões, valor de hemoglobina pré-
transfusional e as reações transfusionais mais frequentes.
- Identificar os principais fenótipos eritrocitários da população de doadores e dos
pacientes, e correlacionar os antígenos eritrocitários dos doadores com os dos pacientes
falciformes.

44
5. MATERIAS E MÉTODOS
O estudo foi realizado em duas etapas. Na primeira etapa foi desenvolvido um Software
para coleta e armazenamento de dados, possibilitando interface adequada com o usuário e
permitindo emissão de relatórios. O Software foi alimentado pela autora com os dados clínicos
e transfusionais estabelecidos, os quais foram retirados das planilhas transfusionais e
prontuários do ambulatório, laboratório de testagem imunohematológica e programa existente
para liberação de hemocomponentes. Na segunda etapa os dados armazenados no banco de
dados do programa foram avaliados.
5.1. Tipo da Pesquisa:
O estudo foi composto por duas metodologias distintas. A primeira envolveu o
desenvolvimento do Software e a segunda a análise dos dados obtidos pelo programa.
O Software foi desenvolvido utilizando a metodologia de Ciclo de Vida de
Desenvolvimento de Sistemas (SDLC – Systems Development Life Cycle) que possibilita
definir as diferentes estratégias para criação, projeto, desenvolvimento e implementação de
sistemas, permitindo pontos de verificação e controle. Esta etapa foi realizada pela
pesquisadora em conjunto com um programador.
Para a SDLC são adotados modelos de execução como Clássico, Prototipação, Espiral e
Rápido.
Para o presente trabalho foi escolhido o modelo de Prototipação, desta forma permitindo
o sequenciamento das informações e melhor interface com o usuário. O projeto foi desenhado
conforme Quadro 1.

45
A partir do banco de dados do Software foi realizado um estudo analítico, transversal,
com abordagem qualitativa e quantitativa dos dados coletados nesta segunda etapa da
pesquisa.
Quadro 1: Etapas para o modelo da prototipação

46
5.2. Local da Pesquisa:
Todas as etapas do estudo ocorreram na HEMOBA.
A instituição conta com um ambulatório de assistência a pacientes com doenças
hematológicas benignas e uma sala de transfusão ambulatorial. No local trabalham médicos,
enfermeiros, técnicos de enfermagem, recepcionistas, além de psicólogo, fisioterapeuta e
dentista.
O processo de transfusão no ambulatório tem inicio com a solicitação do
hemocomponente realizada pelo médico assistente do paciente ou plantonista do ambulatório,
em impresso apropriado. Em seguida, o paciente é encaminhado para a sala de transfusão onde
é assistido pela enfermagem, responsável pela programação da transfusão conforme
solicitação médica e encaminhamento do paciente para coleta de amostras sanguíneas para os
testes necessários, conforme data da programada para a transfusão.
Para dar suporte às transfusões existe um laboratório no qual os hemocomponentes
solicitados para os pacientes do ambulatório são preparados seguindo a legislação vigente e
protocolos internos, sendo posteriormente liberados para transfusão.
Os pacientes recebem a transfusão do hemocomponente na sala de transfusão aos
cuidados da equipe de enfermagem e médico plantonista responsável pelo setor.
5.3. População:
O Software foi desenvolvido para uso em qualquer ambulatório transfusional, com ênfase
na população de interesse deste estudo. A população de análise foi restrita às crianças com
doença falciformes devido às suas peculiaridades transfusionais e carência de dados clínicos
e transfusionais dessa população. Desta forma, foram avaliadas todas as crianças com doença
falciformes que receberam transfusão ambulatorial e foram acompanhados pela equipe médica
do ambulatório do HEMOBA, no período de 1 ano (de janeiro de 2015 a janeiro de 2016).

47
5.3.1. Critérios de inclusão:
- Crianças entre 0 e 18 anos com doença falciforme acompanhadas no ambulatório do
HEMOBA que realizaram mais do que 1 transfusão no ambulatório transfusional da
instituição.
5.3.2. Critérios de exclusão:
- Pacientes que não sejam acompanhados por hematologista pediatra no ambulatório da
HEMOBA.
- Pacientes que não tenham comparecido a pelo menos 1 consulta com o médico
hematologista pediatra no período do estudo.
5.4. Desenvolvimento do Sistema
Baseado no modelo da Prototipação o sistema foi desenvolvido em etapas bem
estabelecidas, porém não hierárquicas. Desta forma, foi possível retornar às etapas já
realizadas, quando requerido pelo programador ou usuário, para realização dos ajustes
necessários.
5.4.1. Levantamento de Necessidades:
Foi inicialmente feita uma revisão de literatura sobre doença falciforme para embasar os
pontos de relevância concernentes à doença e seus aspectos transfusionais. Portanto, foi visto
a necessidade da inclusão de informações referentes à antígenos eritrocitários dos pacientes e

48
doadores dos quais receberam transfusão, histórico de aloimunização eritrocitária, reações
transfusionais, indicação das transfusões, medicações de uso habitual do paciente falciforme.
Após avaliação de pontos críticos de interesse foi verificada a rotina da instituição e
possibilidade de acesso aos dados. Foram analisadas quais informações eram utilizadas
rotineiramente no ambulatório da HEMOBA para a transfusão dos pacientes e quais poderiam
ser acessadas com frequência através do prontuário dos pacientes, livro de registro de
enfermagem da sala de transfusão e registros do laboratório de imunohematologia do doador
e receptor, por serem fontes mais acessíveis para coleta de dados.
Com base em todas essas informações foi desenvolvido um fluxograma de informações
de relevância e com factibilidade para que se iniciasse o desenvolvimento do projeto,
conforme apresentado no Quadro 2.
Quadro 2: Fluxograma de dados

49
5.4.2. Projeto Inicial:
Um projeto inicial foi construído com base nos dados de interesse. Foi estabelecida a
necessidade de desenvolvimento de um Software denominado Banco de Dados Transfusional
(BDTrans). Para sua realização foram utilizadas as ferramentas:
• Visual Studio 2015
• Linguagem de Programação C#
• Banco de Dados do Microsoft AccessR
Foi optado por usar a linguagem de programação C# por ser uma linguagem bastante
difundida entre programadores de sistemas e frequentemente utilizada no âmbito profissional
facilitando, desta forma, a manutenção do programa nos mais diferentes locais de sua
instalação.
Com relação ao banco de dados foi escolhido o Microsoft AccessR, pelas características
do programa e a forma como foi pensado. O AccessR possibilita o armazenamento de dados em
Desktop e permite migração futura dos dados para um programa com maior capacidade de
armazenamento como MySQL ou SQL Server.
Um dos requisitos para o desenvolvimento do sistema foi a utilização de uma boa
interface com o usuário. O programa foi desenhado objetivando a facilidade de instalação e
operação, além da sua capacidade de ser conectado a uma fonte de dados. Desta forma, não
precisa de instalação prévia, requer apenas a pasta do arquivo de dados do Microsoft AccessR,
com armazenamento das informações diretamente em Hard Disk (HD), facilmente operado por
um usuário comum.
O programa conta com o módulo de conexão a dados permitindo conexões múltiplas,
bem como o módulo de login dando maior segurança aos dados.

50
5.4.3. Protótipo: Construção, Avaliação e Refinamento.
Foi desenvolvido inicialmente um banco de dados utilizando o programa Microsoft
AccessR. Os dados foram estruturados em 3 tabelas básicas: Identificação, Doador,
Transfusões. (ANEXOS A, B, C)
No banco de dados foram inseridos os dados iniciais da pesquisa baseados nas
necessidades levantadas. Esses dados coletados foram acompanhados possibilitando a
correção de falhas, como dados sem relevância ou impossíveis de serem acessados.
5.4.4. Desenvolvimento:
O Software BDtrans foi desenvolvido utilizando as informações contidas no banco de
dados do Microsoft AccessR confeccionado na primeira etapa deste trabalho, após seu
aprimoramento.
Foi necessário modificar as tabelas iniciais do banco de dados estabelecendo 4 tabelas:
Identificação, Pedido de Transfusão, Transfusão e Usuário. Após correções das tabelas foram
criadas as relações. (Figura 1)
O BDTrans permite o interfaceamento dos dados transfusionais dos pacientes falciformes
com o usuário e, desta forma, facilita a visualização dos dados incluídos, impressão de
relatórios e cruzamento de informações de pacientes e doadores para localização de fenótipos
eritrocitários, além do histórico transfusional dos pacientes.

51
Figura 1: Relações do banco de dados
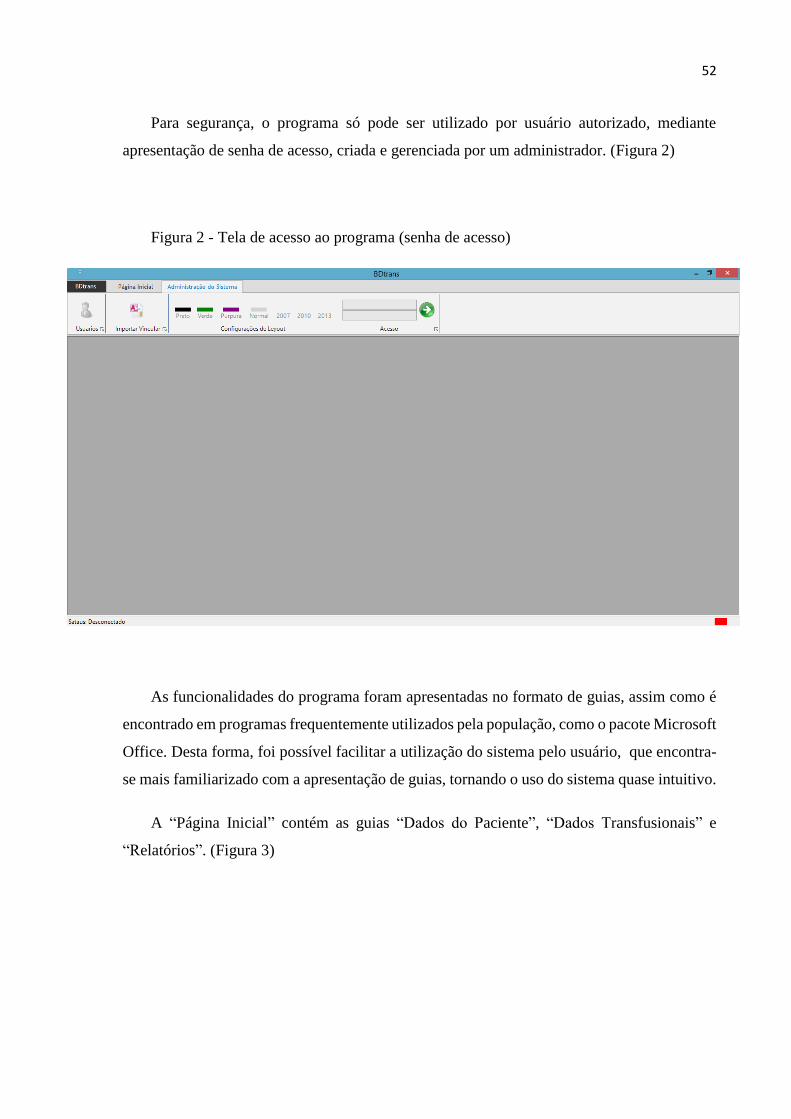
52
Para segurança, o programa só pode ser utilizado por usuário autorizado, mediante
apresentação de senha de acesso, criada e gerenciada por um administrador. (Figura 2)
Figura 2 - Tela de acesso ao programa (senha de acesso)
As funcionalidades do programa foram apresentadas no formato de guias, assim como é
encontrado em programas frequentemente utilizados pela população, como o pacote Microsoft
Office. Desta forma, foi possível facilitar a utilização do sistema pelo usuário, que encontra-
se mais familiarizado com a apresentação de guias, tornando o uso do sistema quase intuitivo.
A “Página Inicial” contém as guias “Dados do Paciente”, “Dados Transfusionais” e
“Relatórios”. (Figura 3)

53
Figura 3 - Guia inicial
A guia de “Dados” é subdividida em 3 campos. No primeiro, constam os dados da
fenotipagem eritrocitária do paciente para os principais antígenos eritrocitários: ABO, Rh,
Kell, MNS, Duffy, Kidd. No segundo, são inseridos os dados de identificação do paciente
como número do registro, nome do paciente, sexo, data de nascimento, endereço, nome da
mãe, CPF, número do cartão do SUS, além do diagnóstico, médico assistente, detecção de
anticorpo contra antígenos eritrocitários. No último campo constam os dados clínicos
relevantes como uso de Hidroxiuréia, quelantes de ferro, ácido fólico, Pen V Oral, outras
medicações, além da dosagem de ferritina e observações clínicas relevantes. (Figuras 4 e 5)
Em seguida à guia “Dados” está a guia “Pacientes” que possibilita visualizar todos os
pacientes cadastrados e buscar pelo paciente específico. (Figura 6)
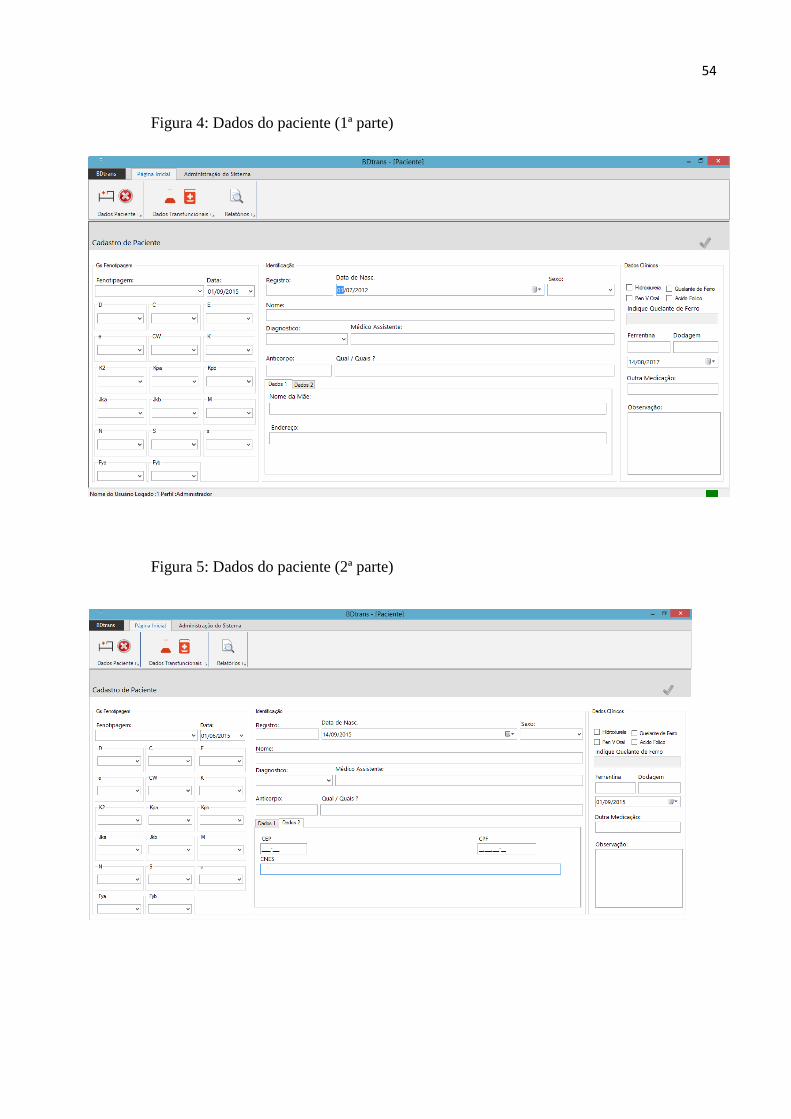
54
Figura 4: Dados do paciente (1ª parte)
Figura 5: Dados do paciente (2ª parte)

55
Figura 6 – Lista de pacientes cadastrados
Para inserir dados em qualquer outro campo, primeiramente faz-se necessário preencher
a guia de cadastro do paciente.
A guia “Dados Transfusionais” está subdividida em duas guias. A primeira é a guia
“Solicitação da Transfusão” contendo dados relevantes para solicitar a transfusão como: tipo
de transfusão (programada, urgente, não urgente), data da solicitação da transfusão, data
programada para a transfusão, tipo de hemocomponente solicitado, modificações dos
hemocomponentes (lavado, filtrado, irradiado) além da opção de hemocomponente
fenotipados, por fim a indicação da transfusão (Figura 7). A segunda guia, denominada
“Dados da Transfusão” possibilita a inserção dos dados relacionados com a liberação do
hemocomponente para uso transfusional como: número da bolsa do hemocomponente, data
da transfusão, PAI, compatibilidade, Hb pré transfusional, tipo de hemocomponente
transfundido com local para inserção caso fenotipado, filtrado, irradiado, lavado, além da
fenotipagem do doador para os grupos sanguíneos ABO, Rh, Kell, Duffy, Kidd e se realizou
transfusão de troca ou não. É possível rastrear as transfusões já solicitadas para o paciente,
visualizando inclusive àquelas que ainda não foram realizadas. (Figuras 8,9)
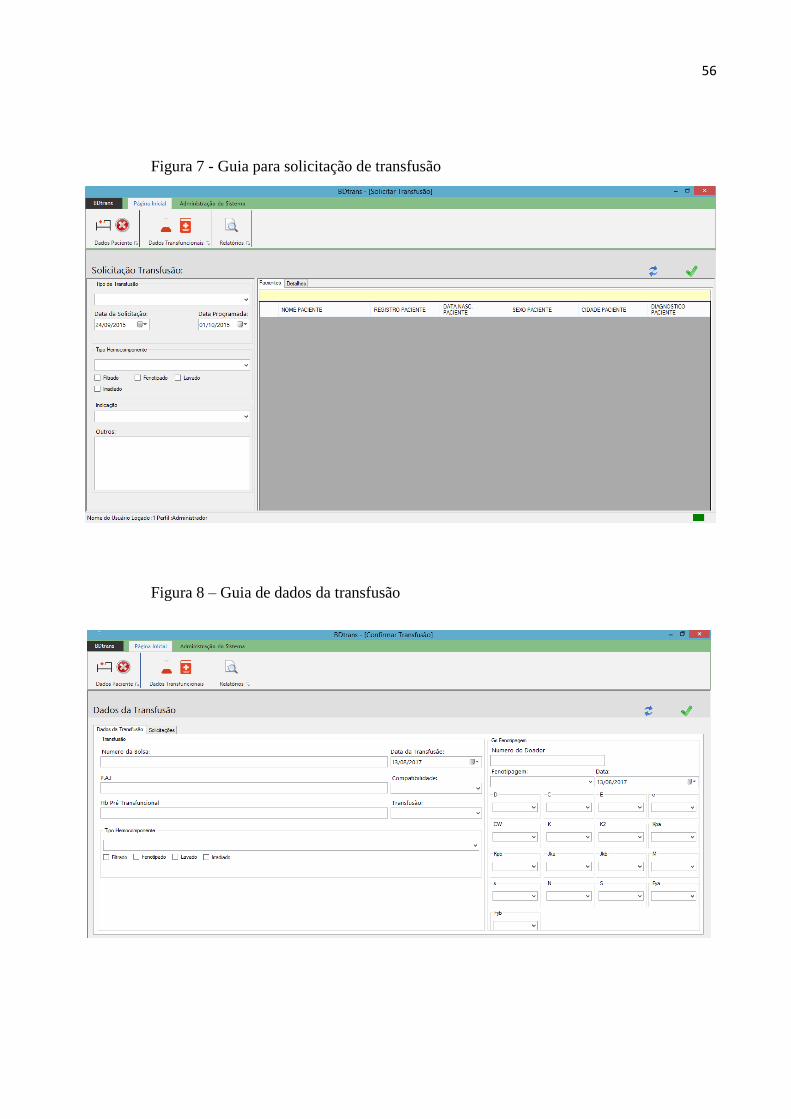
56
Figura 7 - Guia para solicitação de transfusão
Figura 8 – Guia de dados da transfusão

57
Figura 9 – Guia de dados da transfusão (rastreamento de transfusões solicitadas)
Na guia “Relatório” é possível emitir relatório dos dados, cruzar alguns dados e os
imprimir. Neste módulo é possível localizar o paciente por nome, localizar o doador por grupo
sanguíneo/fenotipagem, localizar o paciente por grupo sanguíneo/fenotipagem emitir relatório
contendo todas as transfusões de cada paciente. (Figura 10)
Figura 10: Guia de relatórios

58
No módulo “Administração do Sistema” constam os campos: importar (importa arquivos
para serem incorporados ao programa como o banco de dados do AccessR), usuário (para
inclusão de usuários mediante o cadastramento de senha). (Figura 11)
Figura 11: Dados administrativos
5.5. Aspectos Éticos:
O projeto foi aprovado pela diretoria do HEMOBA, inscrito na plataforma Brasil, sendo
autorizado pelo Comitê de Ética em 10 de maio de 2015.
Por tratar-se de pesquisa com dados de prontuário não foi necessário termo de
consentimento livre e esclarecido para cada indivíduo envolvido com a pesquisa.
Os dados que possam identificar os pacientes do estudo não serão divulgados.

59
5.6 Análise estatística:
Para a análise descritiva, as variáveis quantitativas foram representadas por suas médias
e desvios-padrão quando suas distribuições eram normais e por medianas e intervalos
interquartis quando não normais com outliers importantes. A definição de normalidade foi feita
através de análise gráfica e teste de Shapiro-Wilk. As variáveis categóricas foram representadas
através de frequências e porcentagens.
Foi aplicada tabela de contingência e Teste Exato de Fisher para comparar as diferenças
entre fenótipo de pacientes e doadores.
Análise em caráter descritivo, em virtude da amostra de conveniência e objetivos do
estudo. As análises foram conduzidas com o software IBM Statistical Package for the Social
Sciences (SPSS ®, Chicago, IL, EUA) 20.0.

60
6. RESULTADOS: ANÁLISE DE DADOS TRANSFUSIONAIS DE CRIANÇAS
FALCIFORMES EM REGUIME TRANSFUSIONAL CRÔNICO
No período entre janeiro de 2015 e janeiro de 2016, foram realizadas no ambulatório da
HEMOBA 3101 transfusões, sendo 1588 em pacientes falciformes pediátricos, o que
corresponde a 51,2% do total de transfusões.
Durante o estudo, 250 crianças entre 0 e 18 anos receberam transfusão de CH na
HEMOBA. Destas, 108 foram incluídas no estudo, baseado nos critérios de inclusão e
exclusão. Dos pacientes excluídos, 76 faziam acompanhamento em outras instituições, 20
pacientes não estavam em regime transfusional crônico e 46 não tiveram os prontuários
localizados, conforme quadro 3. (Quadro 3)
Durante o período do estudo, esses 108 pacientes receberam um total de 864 transfusões.
Esses pacientes tiveram suas transfusões acompanhadas, com o registro dos doadores dos
quais receberam os hemocomponentes, bem como os fenótipos eritrocitários.
Quadro 3: Desenho do Estudo de Dados Transfusionais
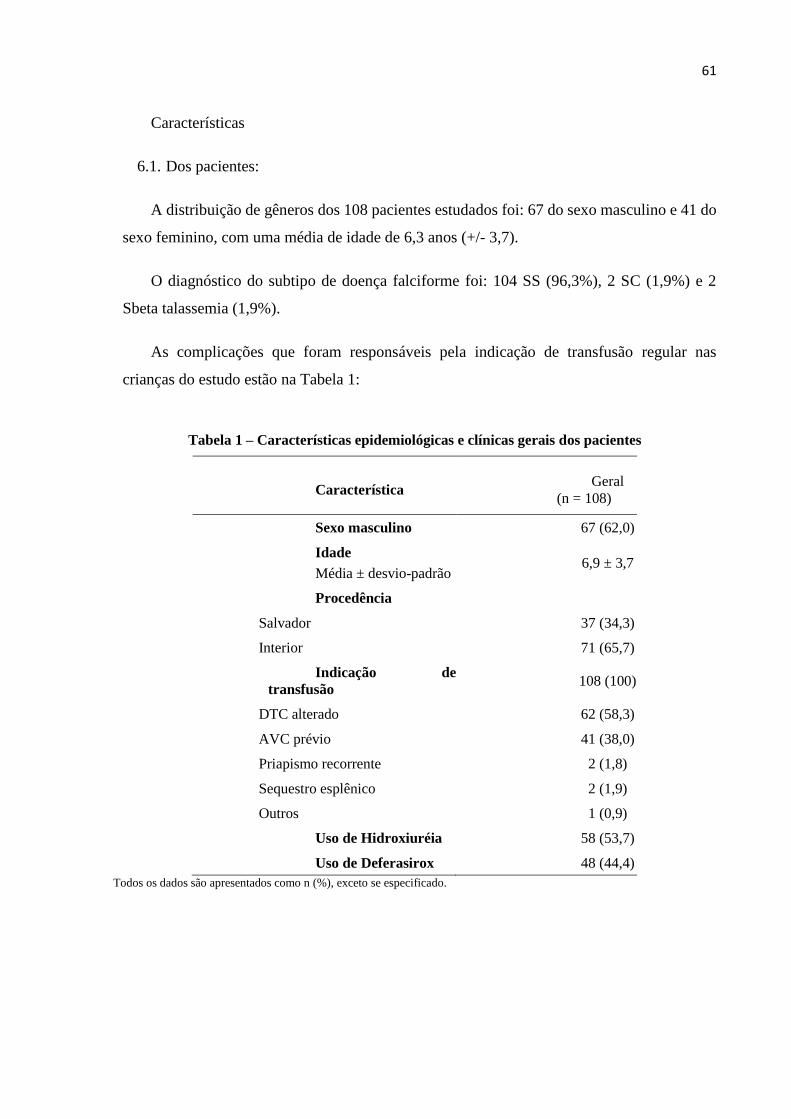
61
Características
6.1. Dos pacientes:
A distribuição de gêneros dos 108 pacientes estudados foi: 67 do sexo masculino e 41 do
sexo feminino, com uma média de idade de 6,3 anos (+/- 3,7).
O diagnóstico do subtipo de doença falciforme foi: 104 SS (96,3%), 2 SC (1,9%) e 2
Sbeta talassemia (1,9%).
As complicações que foram responsáveis pela indicação de transfusão regular nas
crianças do estudo estão na Tabela 1:
Tabela 1 – Características epidemiológicas e clínicas gerais dos pacientes
Característica Geral
(n = 108)
Sexo masculino 67 (62,0)
Idade
Média ± desvio-padrão 6,9 ± 3,7
Procedência
Salvador 37 (34,3)
Interior 71 (65,7)
Indicação de
transfusão 108 (100)
DTC alterado 62 (58,3)
AVC prévio 41 (38,0)
Priapismo recorrente 2 (1,8)
Sequestro esplênico 2 (1,9)
Outros 1 (0,9)
Uso de Hidroxiuréia 58 (53,7)
Uso de Deferasirox 48 (44,4)
Todos os dados são apresentados como n (%), exceto se especificado.

62
Os pacientes residentes em Salvador representaram 34%, com os demais distribuídos
entre 62 cidades.
O início do esquema transfusional variou de 2004 a 2015, conforme tabela 2.
Tabela 2. Ano do início do esquema transfusional
Ano de início da
transfusão
Número de
pacientes
% do total de 108
2004 1 0,92
2005 3 2,77
2006 1 0,92
2007 3 2,77
2008 12 11,11
2009 14 12,96
2010 5 4,62
2011 32 29,62
2012 17 15,74
2013 8 7,40
2014 5 4,62
2015 7 6,48
Todos os dados são apresentados como n (%).

63
Além da transfusão dos concentrados de hemácias, alguns pacientes recebiam outros
medicamentos, como Hidroxiuréia e quelante de ferro (Deferasirox) como consta na Tabela
1.
Os fenótipos eritrocitários dos pacientes estão listados na Tabela 3. Além do sistema ABO
e RH, os sistemas Kell, Kid, Duffy, Lewis e MNS foram estudados.
Como mostra a Tabela 4, o grupo sanguíneo mais prevalente entre os pacientes foi o grupo
O (47,2%), seguido pelo A (33,3%), os grupos B (16,7%) e AB (2,8%) foram mais raros.
Tabela 3 – Fenótipos eritrocitários dos pacientes (N = 108)
Antígenos Positivo Negativo Não realizado
Tipagem
ABO
A 36 (33,3) - -
B 18 (16,7) - -
AB 3 (2,8) - -
O 51 (47,2) - -
D 98 (90,7) 0 (9,3) 0 (0,0)
C 45 (41,7) 2 (48,1) 11 (10,2)
E 13 (12,0) 4 (77,8) 11 (10,2)
e_ 97 (89,8) 0 (0,0) 11 (10,2)
C 89 (82,4) 7 (6,5) 12 (11,1)
CW 1 (0,9) 91 (84,3) 16 (14,8)
K 3 (2,8) 91 (84,3) 14 (13,0)
Jka 46 (42,6) 3 (2,8) 59 (54,6)
Jkb 26 (24,1) 23 (21,3) 59 (54,6)
M 29 (26,9) 15 (13,9) 64 (59,3)
N 33 (30,6) 11 (10,2) 64 (59,3)
S 21 (19,4) 8 (25,9) 59 (54,6)
s_ 48 (44,4) 0 (0,0) 60 (55,6)
Fya 18 (16,7) 30 (27,8) 60 (55,6)
FYb 20 (18,5) 7 (25,0) 61 (56,5)
Todos os dados são apresentados como n (%).

64
6.2. Características dos doadores:
A maioria dos doadores apresentou fenotipagem estendida para os sistemas Kell, Kidd,
Duffy, Lewis e MNS, como mostra a Tabela 4.
Durante o período do estudo, foram analisadas 864 transfusões de 837 doadores.
Do total de doadores, 811 eram doadores de primeira vez (96,9%), 25 de segunda vez
(3%) e apenas 1 doador com 3 ou mais doações (0,1%).
Como podemos observar, a maioria dos doadores pertence ao grupo sanguíneo O
(71,4%), seguidos pelo A (20,7%), B (7,5%) e AB (0,4%).
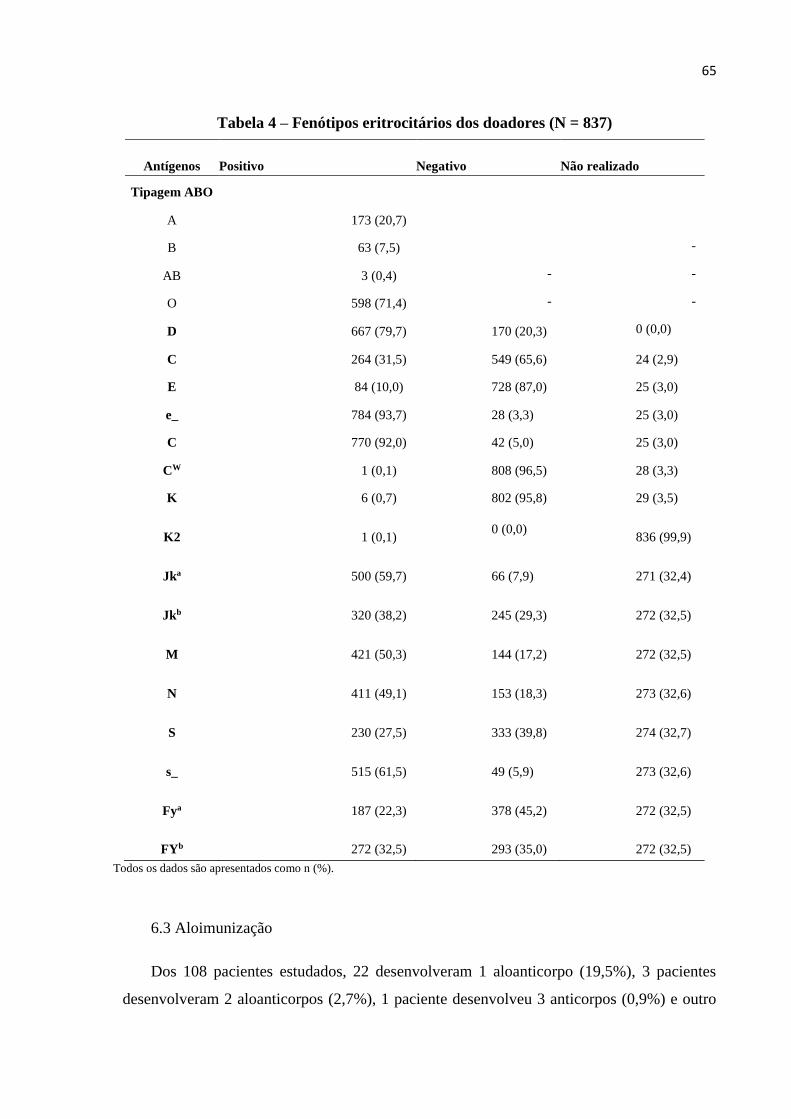
65
Tabela 4 – Fenótipos eritrocitários dos doadores (N = 837)
Antígenos Positivo Negativo Não realizado
Tipagem ABO
A 173 (20,7)
B 63 (7,5) -
AB 3 (0,4) - -
O 598 (71,4) - -
D 667 (79,7) 170 (20,3) 0 (0,0)
C 264 (31,5) 549 (65,6) 24 (2,9)
E 84 (10,0) 728 (87,0) 25 (3,0)
e_ 784 (93,7) 28 (3,3) 25 (3,0)
C 770 (92,0) 42 (5,0) 25 (3,0)
CW 1 (0,1) 808 (96,5) 28 (3,3)
K 6 (0,7) 802 (95,8) 29 (3,5)
K2 1 (0,1) 0 (0,0)
836 (99,9)
Jka 500 (59,7) 66 (7,9) 271 (32,4)
Jkb 320 (38,2) 245 (29,3) 272 (32,5)
M 421 (50,3) 144 (17,2) 272 (32,5)
N 411 (49,1) 153 (18,3) 273 (32,6)
S 230 (27,5) 333 (39,8) 274 (32,7)
s_ 515 (61,5) 49 (5,9) 273 (32,6)
Fya 187 (22,3) 378 (45,2) 272 (32,5)
FYb 272 (32,5) 293 (35,0) 272 (32,5)
Todos os dados são apresentados como n (%).
6.3 Aloimunização
Dos 108 pacientes estudados, 22 desenvolveram 1 aloanticorpo (19,5%), 3 pacientes
desenvolveram 2 aloanticorpos (2,7%), 1 paciente desenvolveu 3 anticorpos (0,9%) e outro

66
paciente com 4 aloanticorpos (0,9%), como observamos no gráfico 1. Além disso, 5 pacientes
desenvolveram apenas autoanticorpos.
Gráfico 1: Número de anticorpos por pacientes alo ou autoimunizados
No gráfico 2, abaixo, encontra-se a identificação dos aloanticorpos desenvolvidos pelos
pacientes.
Gráfico 2: Anticorpos identificados
Aloimunização
sem ac
1 ac
2 ac
3 ac
4 ac
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
1900ral
Aloanticorpos
Aloanticorpos

67
Na tabela 5 são comparados os fenótipos eritrocitários dos pacientes e dos doadores, em
porcentagem de antígeno positivo para os grupos sanguíneos mais imunogênicos.
Tabela 5– Comparação entre frequências de fenótipos de pacientes e doadores
Antígenos
presentes
Paciente
(n= 108)
Doador
(n= 811)
Valor de P
(Exato de Fischer)
A 36 (33,3) 173 (20,7) P<0.0001
B 18 (16,7) 63 (7,5) 0,0031
AB 3 (2,8) 3 (0,4) 0,0224
O 51 (47,2) 598 (71,4) P<0.0001
D 98 (90,7) 667 (79,7) 0,0058
C 45 (41,7) 264 (31,5) 0,0385
E 13 (12,0) 84 (10,0) 0,5016
e_ 97 (89,8) 784 (93,7) 0,1516
C 89 (82,4) 770 (92,0) 0,0037
K 3 (2,8) 6 (0,7) 0,0730
Jka 46 (42,6)
500 (59,7) 0,0009
Jkb 26 (24,1)
320 (38,2) 0,0040
M 29 (26,9)
421 (50,3) P<0.0001
N 33 (30,6)
411 (49,1) 0,0003
S 21 (19,4)
230 (27,5) 0,0826
s_ 48 (44,4)
515 (61,5) 0,0008
Fya 18 (16,7)
187 (22,3) 0,2145
FYb 20 (18,5)
272 (32,5) 0,0027
Foi aplicada tabela de contingência e Teste Exato de Fisher
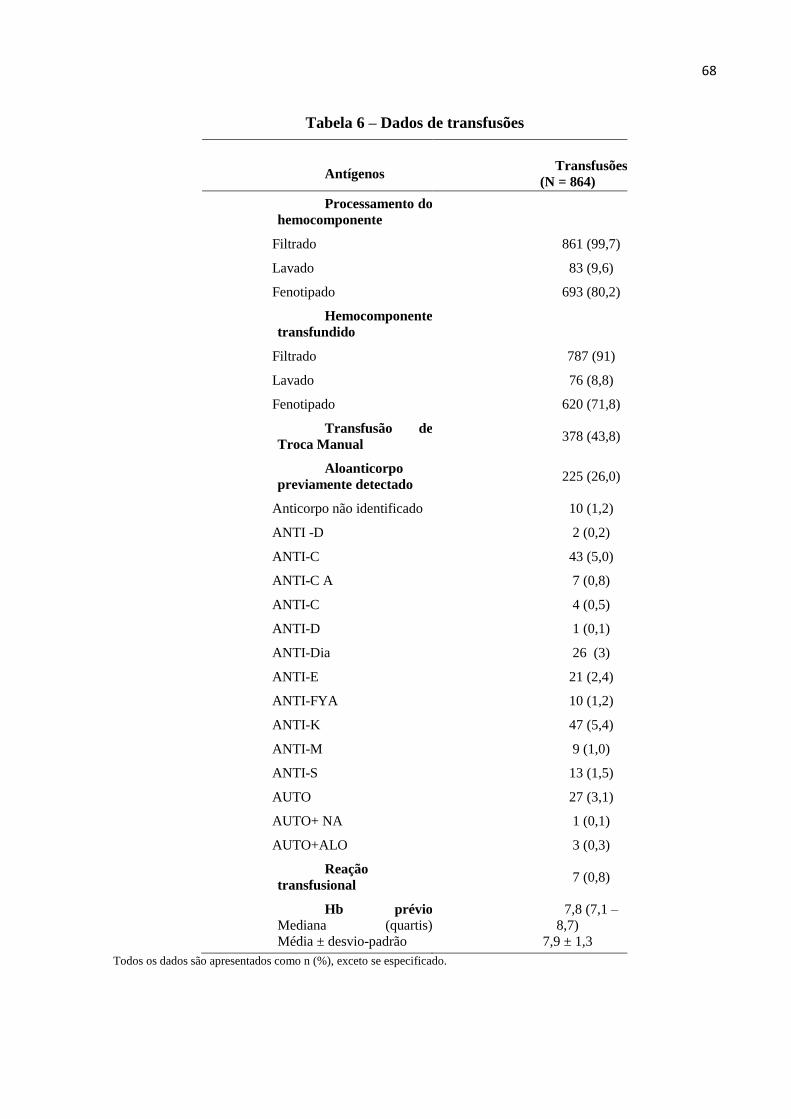
68
Tabela 6 – Dados de transfusões
Antígenos Transfusões
(N = 864)
Processamento do
hemocomponente
Filtrado 861 (99,7)
Lavado 83 (9,6)
Fenotipado 693 (80,2)
Hemocomponente
transfundido
Filtrado 787 (91)
Lavado 76 (8,8)
Fenotipado 620 (71,8)
Transfusão de
Troca Manual 378 (43,8)
Aloanticorpo
previamente detectado 225 (26,0)
Anticorpo não identificado 10 (1,2)
ANTI -D 2 (0,2)
ANTI-C 43 (5,0)
ANTI-C A 7 (0,8)
ANTI-C 4 (0,5)
ANTI-D 1 (0,1)
ANTI-Dia 26 (3)
ANTI-E 21 (2,4)
ANTI-FYA 10 (1,2)
ANTI-K 47 (5,4)
ANTI-M 9 (1,0)
ANTI-S 13 (1,5)
AUTO 27 (3,1)
AUTO+ NA 1 (0,1)
AUTO+ALO 3 (0,3)
Reação
transfusional 7 (0,8)
Hb prévio
Mediana (quartis)
Média ± desvio-padrão
7,8 (7,1 –
8,7)
7,9 ± 1,3
Todos os dados são apresentados como n (%), exceto se especificado.

69
7. DISCUSSÃO
A Doença Falciforme apresenta alta prevalência no Brasil e principalmente na Bahia.
(HUTTLE et al., 2015) É uma doença sistêmica cujas complicações podem acometer diversos
órgãos e tecidos e levam a diminuição da qualidade de vida e da sobrevida do
paciente.(BADAWY et al., 2016)
Várias políticas públicas estão sendo desenvolvidas para a atenção ao paciente
falciforme. No Brasil, o Ministério da Saúde, desde meados da década de 90, vem
implementando ações públicas voltadas para a doença, culminando com a portaria nº 1.391, de
16 de agosto de 2005, que instituiu a Política Nacional de Atenção Integral às Pessoas com
Doença Falciforme e outras Hemoglobinopatias, no âmbito do SUS. (DINIZ; GUEDES, 2005)
Porém, a falta de sistemas informatizados, tanto em nível local como nacional, dificulta o
conhecimento epidemiológico, demográfico e clínico desses pacientes e a melhorias nas
políticas de atenção à saúde dos portadores dessa doença tão prevalente no país.
Atualmente, no Estado da Bahia, o acompanhamento dos pacientes falciformes é
realizado, através do Sistema Único de Saúde (SUS), em alguns ambulatórios, principalmente
em Salvador e região metropolitana. Para a assistência hematológica das crianças falciformes,
estão disponíveis na capital baiana os ambulatórios da Fundação HEMOBA, Associação de
Pais e Amigos dos Excepcionais (APAE), Hospital Universitário Professor Edgar Santos
(HUPES), Hospital Santo Antônio (HSA), Hospital Martagão Gesteira e unidades de saúde da
prefeitura. Todas estas unidades, com exceção do Hospital Martagão Gesteira, utilizam o
ambulatório de transfusão da HEMOBA para suporte transfusional.
O desenvolvimento e a aplicação do sistema de informação chamado “BDTrans”
possibilita a coleta informatizada de dados demográficos, clínicos e transfusionais dos pacientes
assistidos na instituição possibilitando melhorias na assistência os pacientes.
O presente estudo foi realizado na HEMOBA, onde foram avaliados 174 pacientes
transfundidos e acompanhados no ambulatório da instituição, com inclusão total de 108
crianças falciformes que receberam transfusão ambulatorial. Foram excluídos 20 pacientes por
terem feito transfusão esporádica e 46 por falha na coleta de dados. A falta de informatização

70
do arquivo e prontuários do ambulatório dificultaram a localização e coleta de dados clínicos
por isso o grande número de pacientes excluídos.
De acordo com dados da triagem neonatal nos anos de 2009 - 2010, no Estado da Bahia,
os locais com maior incidência de novos casos de Doença Falciforme foram: Salvador, com
23,2% dos casos, seguidos por Feira de Santana (4,6%), Lauro de Freitas (2,2%) e Camaçari
com 18 casos (1,9%). Os demais casos estavam distribuídos entre outros 413 municípios.
(AMORIM, 2010) No estudo aqui descrito muitos dos pacientes avaliados eram residentes em
cidades do interior do estado, 67%.
A causa mais comum para manutenção de regime transfusional crônico em pacientes
com DF são as complicações cerebrovasculares. Estima-se que 8-10% das crianças com AF
terão um AVC até a idade adulta, com 50% a 90% de chance de apresentar um segundo evento.
As transfusões regulares diminuem o risco do segundo AVC para 10-20%. Desta forma, uma
das principais indicações de transfusão crônica é a prevenção primária ou secundária do AVC.
(WARE, 2007)
O estudo STOP avaliou a velocidade de fluxo nas artérias cerebrais médias de crianças
com HbSS ou HbSb0Tal evidenciando 9,3% de alteração nos exames. A velocidade de fluxo ao
DTC > 200cm/s, repetida e confirmada, confere risco de 46% da criança evoluir com AVC nos
próximos 39 meses, caso não inicie esquema de transfusões.(ADAMS et al., 1998) Segundo
Adams, o grupo etário onde o DTC apresentou maior alteração foi entre 2-8 anos (10,7%),
seguido de 9-12 anos (9,4%). (ADAMS, 2000, 347) A transfusão regular para manter a HbS
<30% deve ser oferecida como tratamento inicial dessas crianças, com idades entre 2-16 anos,
consideradas de alto risco para um primeiro AVC com base no DTC. (DAVIS et al., 2016)
Das crianças falciformes incluídas no presente estudo, 96% possuíam fenótipo SS, com
mediana de idade de 11 anos. As indicações para as transfusões foram: DTC alterado, em 58%
dos casos, enquanto AVC correspondeu a 38%, portanto a doença cerebrovascular consistiu
mais de 90% das indicações para as transfusões. Apenas 2 crianças (1,8%), fizeram transfusões
por sequestro esplênico, que também consiste em uma indicação frequente de transfusão na DF.
O sequestro esplênico acomete principalmente crianças de 3 meses a 6 anos, com 75% dos
casos ocorrendo até 26 meses de vida e taxas de recorrência de 67%, com mortalidade de 0,53%.
(REZENDE et al., 2009) (BROUSSE et al., 2012) A baixa prevalência de outras causas para
transfusão além da DCV talvez guarde relação com o fato de muitas crianças menores de 5

71
anos, faixa etária de maior prevalência de sequestro esplênico, serem acompanhadas em outros
ambulatórios, como APAE, e transferidas para a HEMOBA apenas após esta idade.
A maioria das transfusões foi iniciada na faixa etária entre 2-8 anos (61%) e 9-12 anos
(24%), nas mesmas faixas etárias onde foi verificada maior alteração de DTC no estudo STOP.
(ADAMS et al., 1998) Vinte e nove porcento, iniciaram o regime transfusional em 2011,
seguido dos anos de 2012 com 15% dos casos e 2009 com 13%. O DTC começou a ser realizado
na instituição do estudo em 2009 e talvez, por esse motivo, ocorreu o aumento significativo de
crianças falciformes em regime transfusional crônico a partir daquele ano.
A crescente indicação dos regimes transfusionais crônicos em pacientes com DF traz
consigo os riscos inerentes a transfusão, tais como RTHT, aloimunização, reações
transfusionais agudas, transmissão de patógenos e sobrecarga de ferro induzida por transfusão.
(CHOU; LIEM; THOMPSON, 2012)
A aloimunização eritrocitária apresenta prevalência variável na literatura. Estudos
mostram variação na prevalência de 6 – 60% a depender das variáveis associadas,
principalmente semelhança fenotípica eritrocitária entre doadores e
pacientes.(OLUJOHUNGBE et al., 2001)(NATUKUNDA et al., 2010)(MURAO; VIANA,
2005)(MILLER et al., 2013) No estudo houve uma prevalência de aloimunização de 23%. Em
análise previa realizada na HEMOBA, foi verificada aloimunização em mais de 60% dos
pacientes falciformes transfundidos com idade acima de 18 anos e com variadas indicações de
transfusão, utilizando apenas a fenotipagem Rh e Kell, o que torna as variáveis bastante
diferentes da presente análise. (ZANETTE et al., 2010) Provavelmente, a utilização rotineira
de hemácias fenótipo compatíveis seja responsável pela diminuição das taxas de
aloimunização.(DESSAP et al., 2016)
Desde 2010 a Fundação HEMOBA realiza fenotipagem estendida para os pacientes
falciformes, com coleta do exame preferencialmente quando ainda na infância e antes da
primeira transfusão. Dos pacientes avaliados, 87% apresentavam pelo menos a fenotipagem Rh
e Kell e aproximadamente 40% apresentavam fenotipagem para Kidd, Duffy e MNS,
provavelmente porque foram fenotipados antes de 2010.
No presente estudo, os grupos sanguíneos mais prevalentes nos pacientes foram: O
(47,2%), seguido pelo A (33,3%), B (16,7%) e AB (2,8%). Com relação a fenotipagem
estendida, 84,3% dos pacientes eram negativos para K, 77,8% para E e 48,1% para C, 21% para

72
JKb, 25,9% para S e 27,8% para FYa. Na literatura verifica-se prevalências variáveis O (61,1%),
seguido por A (20,4%), B (13,0%) e AB (5,6%), demais antígenos descritos em Tabela 6.
Talvez a diferença fenotípica entre os estudos envolva diferenças étnicas entre as populações
avaliadas. O Brasil possui a população mais heterogênea do mundo, decorrente de cinco séculos
de relações inter étnicas de povos dos três continentes: colonizadores europeus, escravos
africanos e os ameríndios. (PARRA et al., 2003)
Tabela 7: Comparação entre fenótipos eritrocitários de pacientes falciformes em diferentes estudos
% Antígeno
Negativo
Estudo Atual
(N:108 pacientes)
Karafin et al, 2015
(N: 64 pacientes)
Pinto et al, 2011
(N: 102 pacientes)
K 91 94 92
C 52 70 46
E 84 74 71
Jkb 23 37 49
Fya 30 82 40
O desenvolvimento de aloanticorpos parece ser uma resposta multifatorial, envolve
aspectos do paciente e do doador. (CHOU; LIEM; THOMPSON, 2012) (YAZDANBAKHSH;
WARE; NOIZAT-PIRENNE, 2012) Aspectos genéticos, idade, sexo, número de transfusões e
estado inflamatório do indivíduo guardam relação com a formação de aloanticorpos.(MURAO;
VIANA, 2005) (SINS et al., 2016) . (YAZDANBAKHSH, 2015) (FASANO et al., 2014)
Estudos mostram prevalência de 45% de aloimunização eritrocitária nos pacientes falciformes
em regime transfusional crônico e 12% naqueles fazendo uso de transfusões eventuais, com
predominância de anticorpos contra antígenos do sistema Rh. (CHOU et al., 2013) No presente
estudo foram avaliadas pacientes com mais de 1 transfusão anual, na faixa etária pediátrica,
com predominância do sexo masculino (67%).
A DF, pela sua origem africana, é mais prevalente em afrodescentes. Países com menor
diferença étnica entre sua população, consequentemente menor diferença entre os fenótipos
eritrocitários, apresenta baixa incidência de aloimunização.(OLUJOHUNGBE et al.,
2001)(NATUKUNDA et al., 2010)(CHOU; LIEM; THOMPSON, 2012)(YAZDANBAKHSH;

73
WARE; NOIZAT-PIRENNE, 2012) Avaliando os doadores foi possível observar que a maioria
era do grupo sanguíneo O (71,4%), seguidos pelos grupos A (20,7%), B (7,5%) e AB (0,4%).
A instituição preconiza a fenotipagem Rh e Kell de indivíduos do grupo O, caso o doador seja
C e E negativos a fenotipagem é estendida. Desta forma, 95% dos doadores eram negativos
para K, 87% para E e 42% para C, 29% para JKb, 39% para S e 45% Fya. Os dados foram
parecidos com os encontrados na literatura O (51,3%), seguida de A (25,6%), B (19,2%) e AB
(3,9%), exceto pela maior frequência de doadores O e menor de doadores B. (KARAFIN et al.,
2015) Talvez esses dados tenham relação com a preferência em realizar a fenotipagem de
indivíduos do grupo O.
Karafin et.al, avaliou 6066 doadores de sangue afrodescendentes nos Estados Unidos
verificado que a maioria (56,9%) doou apenas uma vez em 3 anos, 20,4% doaram duas vezes.
A mediana de doações ao longo de 3 anos foi de uma doação, com média de doações de 2,1.
(KARAFIN et al., 2015, 1401). No presente estudo, avaliando as doações durante um ano, 811
eram doadores de primeira vez (96,9%), 25 de segunda vez (3%) e apenas 1 doador com 3 ou
mais doações (0,1%). A baixa prevalência de doadores de repetição dificulta a construção de
um programa de fidelização de doadores para cada paciente falciforme em regime transfusional
crônico.
A literatura mostra maior prevalência dos aloanticorpo anti-C, anti-E, anti-K em ordem
decrescente, na população com DF. (VICHINNSKY et al., 1990) (ZANETTE et al., 2010)
(KARAFIN et al., 2015) (MILLER et al., 2013) No presente estudo foram encontrados 38
aloanticorpos. A maioria dos pacientes apresentavam apenas um aloanticorpo (19,5%), porém
houve pacientes com 2 (2,7%), 3(0,9%) e 4(0,9%) aloanticorpos. Além disso, 5 pacientes
desenvolveram apenas autoanticorpos.
Os aloanticorpos mais prevalentes foram anti-C (23,6%), anti-E (15,8%), anti-K (13,2%)
e anti-D (10,5%). Chama a atenção a prevalência de anti-D, que pode estar relacionada com
variantes nos genes RHD e RHCE, frequentes em afrodescendentes.(FLEGEL, 2011) Em um
estudo realizado do Hemocentro de Campinas, foi visto que pacientes com variantes RHD e
RHCE podem desenvolver alo e autoanticorpo, inclusive anti-D, mediante D
fraco.(GASPARDI et al., 2016) Sippert et al, demonstrou que 65% dos pacientes com DF
aloimunizados para antígenos do sistema Rh eram portadores de alelos variantes de RH e que
42% dos anticorpos encontrados tiveram significância clínica.(SIPPERT et al., 2015, 72)

74
Embora a tipagem de antígenos antes da transfusão de pessoas com DF e a transfusão de
concentrado de hemácias fenótipo compatível, a taxa de aloimunização permanece bastante
elevada nos pacientes com DF e pode dever-se em grande parte a transfusões recebidas em
instituições que não proporcionam hemocomponente fenótipo compatível. (MILLER et al.,
2013)
Uma das complicações dos regimes transfusionais crônicos é a sobrecarga de ferro. A
quelação de ferro com Deferasirox estava sendo utilizada por 44% dos pacientes no presente
estudo. No final do seguimento, no estudo STOP, 41% dos pacientes em regime transfusional
crônico faziam uso de Deferasirox.(ADAMS et al., 1998) No estudo TWiTCH, após 4 anos de
transfusões regulares 95% dos pacientes estavam fazendo uso de quelantes de ferro. (WOOD
et al., 2016)
As transfusões em pacientes falciformes podem ser realizadas de forma simples, por troca
manual ou por eritrocitoaférese. A transfusão de troca está mais indicada para os regimes
transfusionais crônicos por possibilitar maior diminuição da HbS pela retirada de parte dos
eritrócitos autólogos, diminuir a sobrecarga de ferro a longo prazo, e evitar o aumento da
viscosidade sanguínea. (SWERDLOW, 2006) A decisão de utilizar transfusão simples ou de
troca depende da situação clínica específica, dos recursos disponíveis e da capacidade de
estabelecer um acesso venoso adequado. A troca manual é um procedimento seguro, eficaz,
principalmente na redução da sobrecarga de ferro, e de menor custo em relação a
eritrocitoaférese para os pacientes falciformes. (KOEHL et al., 2016)(WARE, 2007) Na
instituição onde ocorreu o estudo não é realizada eritrocitoaférese, sendo preconizada a
realização de transfusão de troca manual com base no valor da hemoglobina pré-transfusional.
Foi verificado no estudo que, das 864 transfusões, 378 foram transfusão de troca manual, com
média de Hb 9,0 g/dl. No grupo de transfusão simples, a média de Hb foi 7,0 g/dl.
Com a maior utilização das transfusões aumenta o risco de reações transfusionais.
Segundo a Anvisa, em 2014 foram notificadas no Brasil mais de 11000 reações transfusionais,
com Reação Febril não Hemolítica responsável por 47,8% das notificações, seguida de reação
alérgica com 40,2%. A RTHT correspondeu apenas a 0,1% das notificações.(SANITARIA,
2015) Estima-se que a incidência de RTHT na população falciforme seja bem maior do que as
relatadas visto a dificuldade do diagnóstico. A RTHT simula complicações da própria doença
falciforme, como crise de dor e hemólise, e, muitas vezes, o paciente apresenta testes
diagnósticos negativos. (DE MONTALEMBERT et al., 2011) Com relação às reações

75
transfusionais agudas, foram 2 reações alérgicas e 1 hipotensão, esta última em um paciente
com Hb10,7, submetido a transfusão de troca manual. Dos hemocomponentes utilizados 8
pacientes utilizaram concentrado de hemácias lavados (total de 77 componentes transfundidos),
620 CH filtrados e fenotipados e somente 167, filtrados sem fenotipagem. A leucodepleção,
através da filtragem do hemocomponente, apresenta eficácia na prevenção de reação febril não
hemolítica, principalmente quando realizada pré estocagem (BIANCHI et al., 2016)
(DELANEY et al., 2016)
Assim como as transfusões, a Hidroxiuréia possui papel importante no controle das
complicações da DF e na melhoria da qualidade de vida desses pacientes.(THORNBURG;
CALATRONI; PANEPINTO, 2011) Na prevenção de recorrência de AVC, não mostrou
superioridade em relação a transfusão, que continua sendo a terapêutica mais indicada. (WARE;
HELMS, 2012) Já na prevenção primária do AVC, o estudo TWiTCH comprovou a eficácia da
troca do regime transfusional crônico por Hidroxiuréia nos pacientes já cursando com DTC
normal.(WARE et al., 2016). No presente estudo a Hidroxiuréia estava sendo utilizada por
53,7% das crianças analisadas, na sua maioria cursando com doença cerebrovascular (DTC
alterado ou AVC), porém o motivo do uso da medicação não pode ser relatado.

76
8. CONCLUSÃO
O desenvolvimento e a aplicação do sistema de informação denominado “BDTrans” nos
permitiu um maior conhecimento acerca da terapia transfusional das crianças falciformes
assistidas na HEMOBA e possibilitou o reconhecimento de problemas nesse processo para
orientação de futuras políticas de saúde pública voltadas para essa população.
O presente estudo permitiu conhecer as principais indicações para regimes transfusionais
em pacientes falciformes pediátricos, com destaque para a doença cerebrovascular e
principalmente sua profilaxia primária.
Foi possível traçar um perfil epidemiológico desses pacientes e analisar dados clínicos
como o uso de hidroxiuréia e quelantes de ferro, porém não foi possível relatar dados
laboratoriais como eletroforese de hemoglobina pré transfusional e dosagem de ferritina visto
que, apesar de sua importância para o acompanhamento transfusional desses pacientes, esses
exames não são realizados na instituição.
O acesso informatizado aos dados transfusionais permitiu correlacionar o fenótipo
eritrocitário da população falciforme e da doadora facilitando para que, em um futuro
próximo, seja desenvolvido um programa de fidelização de doadores fenotipados para cada
paciente, diminuindo assim a exposição de pacientes a doadores e reduzindo a chance de
aloimunização.
Houve diferença estatística entre os fenótipos dos doadores de sangue e os pacientes
falciformes o que pode explicar em parte a alta taxa de aloimunização nesses pacientes.
O sistema poderá ser utilizado para acompanhamento transfusional de outras populações
de pacientes, além das crianças falciformes. Com ele poderão ser realizados estudos adicionais
permitindo seguimento a longo prazo de pacientes que requeiram suporte transfusional,
possibilitando seu melhor conhecimento.

77
REFERÊNCIAS BIBLIOGRÁFICAS
AASLID, R.; MARKWALDER, T. M.; NORNES, H. Noninvasive
transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries.
Journal of neurosurgery, v. 57, n. 6, p. 769–774, 1982.
ADAMS, R. et al. The use of transcranial ultrasonography to predict stroke
in sickle cell disease. N Engl J Med, v. 326, n. 9, p. 605–610., 1992.
ADAMS, R. J. et al. Stroke prevention trial in sickle cell anemia.
Controlled clinical trials, v. 19, n. 1, p. 110–129, 1998.
ADAMS, R. J. Lessons From the Stroke Prevention Trial in Sickle Cell
Anemia (STOP) Study. J Child Neurol 2000, v. 15, p. 344–349, 2000.
ADAMS, R. J. Big strokes in small persons. Archives of neurology, v. 64,
n. 11, p. 1567–74, 2007.
ADAMS, R. J.; BRAMBILLA, D. Discontinuing prophylactic transfusions
used to prevent stroke in sickle cell disease. N Engl J Med, v. 353, n. 26, p. 2769–
78, 2005.
ALLISON, A. C. Protection afforded by sickle-cell trait against subtertian
malarial infection. British Medical Journal, v. 1, p. 290–294, 1954.
ALONI, M. N. et al. A pilot study of manual chronic partial exchange
transfusion in children with sickle disease. Hematology (Amsterdam,
Netherlands), v. 20, n. 5, p. 284–8, 2015.
AMORIM, T. Avaliação Do Programa De Triagem Neonatal da Bahia entre
2007 e 2009 - As Lições da Doença Falciforme. Gaz. méd. Bahia, v. 3, p. 10–13,
2010.
ARNOLD, S. D. et al. Haematopoietic stem cell transplantation for sickle
cell disease ??? current practice and new approaches. British Journal of
Haematology, v. 174, n. 4, p. 515–525, 2016.
BADAWY, S. M. et al. Health-related quality of life and adherence to
hydroxyurea in adolescents and young adults with sickle cell disease. Pediatric
blood & cancer, n. October, p. 1–8, 2016.
BALEOTTI, W. et al. HLA-DRB1*07:01 allele is primarily associated with
the Diego a alloimmunization in a Brazilian population. Transfusion, v. 54, n. 10,
p. 2468–2476, 2014.
BALLAS, S. K. et al. Definitions of the phenotypic manifestations of sickle
cell disease. American journal of hematology, v. 85, n. 1, p. 6–13, jan. 2010.
BALLAS, S. K. Pathophysiology and principles of management of the

78
many faces of the acute vaso-occlusive crisis in patients with sickle cell disease.
European journal of haematology, v. 95, n. 2, p. 113–23, 2015.
BATISTA, J. et al. Regulação de sangue no Brasil : contextualização para o
aperfeiçoamento. Rev Panam Salud Publica, v. 38, n. 4, p. 333–338, 2015.
BERNAUDIN, F. et al. Long-term treatment follow-up of children with
sickle cell disease monitored with abnormal transcranial Doppler velocities. Blood,
v. 127, n. 14, p. 1814–1823, 2016.
BIANCHI, M. et al. Leucoreduction of blood components: an effective way
to increase blood safety? Blood Transfusion, v. 14, n. 2, p. 214–27, 2016.
BROUSSE, V. et al. Acute splenic sequestration crisis in sickle cell disease:
cohort study of 190 paediatric patients. British journal of haematology, v. 156, n.
5, p. 643–8, mar. 2012.
BROUSSE, V.; KOSSOROTOFF, M.; DE MONTALEMBERT, M. How I
manage cerebral vasculopathy in children with sickle cell disease. British Journal
of Haematology, v. 170, p. 615–625, 2015.
CASAS, J. et al. Changing practice: Red blood cell typing by molecular
methods for patients with sickle cell disease. Transfusion, v. 55, n. 6, p. 1388–
1393, 2015.
CASTRO, O. et al. Predicting the effect of transfusing only phenotype-
matched RBCs to patients with sickle cell disease: theoretical and practical
implications. Transfusion, v. 42, p. 684–690, 2002.
CHARACHE, S. et al. Hydroxyurea-induced augmentation of fetal
hemoglobin production in patients with sickle cell anemia. Blood., v. 69, n. 1, p.
109–116., 1987.
CHAUDHRY, B. et al. Improving Patient Care Systematic Review : Impact
of Health Information Technology on. Ann Intern Med., v. 144, p. 742–752, 2006.
CHIARONI, J. et al. HLA-DRB1 polymorphism is associated with Kell
immunisation. British Journal of Haematology, v. 132, n. 3, p. 374–378, 2006.
CHIARONI, J. et al. Positive association of DRB1*04 and DRB1*15 alleles
with Fya immunization in a Southern European population. Transfusion, v. 49, n.
11, p. 2412–2417, 2009.
CHOU, S. T. et al. High prevalence of red blood cell alloimmunization in
sickle cell disease despite transfusion from Rh-matched minority donors. Blood, v.
122, n. 6, p. 1062–1071, 2013.
CHOU, S. T. Transfusion therapy for sickle cell disease: a balancing act.
Hematology / the Education Program of the American Society of Hematology.
American Society of Hematology. Education Program, v. 2013, p. 439–46, jan.
2013.

79
CHOU, S. T.; FASANO, R. M. Management of Patients with Sickle Cell
Disease Using Transfusion Therapy: Guidelines and Complications.
Hematology/Oncology Clinics of North America, v. 30, n. 3, p. 591–608, 2016.
CHOU, S. T.; LIEM, R. I.; THOMPSON, A. A. Challenges of
alloimmunization in patients with haemoglobinopathies. British Journal of
Haematology, v. 159, n. 4, p. 394–404, 2012.
DAVIS, B. A. et al. Guidelines on red cell transfusion in sickle cell disease.
Part I: Principles and laboratory aspects. British Journal of Haematology, p. 3–
18, 2016.
DE MONTALEMBERT, M. et al. Delayed hemolytic transfusion reaction
in children with sickle cell disease. Haematologica, v. 96, n. 6, p. 801–7, jun. 2011.
DEBAUN, G. Controled trial of transfusions for silent cerebral infarcts in
sickle cell anemia. N Engl J Med, v. 8, n. 371, p. 699–710, 2014.
DEBAUN, M. R. et al. Silent cerebral infarcts : a review on a prevalent and
progressive cause of neurologic injury in sickle cell anemia Review article Silent
cerebral infarcts : a review on a prevalent and progressive cause of neurologic injury
in sickle cell anemia. Blood, v. 119, p. 4587–4596, 2012.
DEBAUN, M. R.; STRUNK, R. C. The intersection between asthma and
acute chest syndrome in children with sickle-cell anaemia. The Lancet, v. 387, n.
10037, p. 2545–2553, 2016.
DELANEY, M. et al. Transfusion reactions: Prevention, diagnosis, and
treatment. The Lancet, v. 388, n. 10061, p. 2825–2836, 2016.
DESSAP, M. et al. A diagnostic nomogram for delayed hemolytic
transfusion reaction in sickle cell disease. American Journal of Hematology, v.
91, n. 12, p. 1181–1184, 2016.
DINIZ, D.; GUEDES, C. Confidencialidade, aconselhamento genético e
saúde pública: um estudo de caso sobre o traço falciforme. Cad Saúde Pública, v.
21, n. 3, p. 747–755, 2005.
ESTCOURT, L. J. et al. Preoperative blood transfusions for sickle cell
disease. Cochrane Database of Systematic Reviews, n. 4, p. 1–69, 2016.
FALLETTA, J. M. et al. Discontinuing penicillin prophylaxis in children
with sickle cell anemia. Prophylactic Penicillin Study II. The Journal of
pediatrics, v. 127, n. 5, p. 685–690, 1995.
FASANO, R. M. et al. Red blood cell alloimmunization is influenced by
recipient inflammatory state at time of transfusion in patients with sickle cell
diseasE.pdf. Blood Journal, p. 1–10, 2014.
FERNANDES, A. P. P. C. et al. Mortality of children with sickle cell
disease: a population study. Jornal de Pediatria, v. 0, n. 0, p. 279–284, 2010.

80
FICHOU, Y. et al. The experience of extended blood group genotyping by
next-generation sequencing (NGS): investigation of patients with sickle-cell
disease. Vox Sanguinis, v. 111, n. 4, p. 418–424, 2016.
FLEGEL, W. A. Molecular genetics and clinical applications for RH.
Transfusion and Apheresis Science, v. 44, n. 1, p. 81–91, 2011.
FURTADO, P. S. et al. The prevalence of priapism in children and
adolescents with sickle cell disease in Brazil. International Journal of
Hematology, v. 95, n. 6, p. 648–651, 2012.
GASPARDI, A. C. et al. Clinically relevant RHD-CE genotypes in patients
with sickle cell disease and in African Brazilian donors. Blood transfusion, v. 14,
p. 449–454, 2016.
GASTON, M. H. et al. Prophylaxis with oral penicillin in children with
sickle cell anemia. A randomized trial. The New England journal of medicine, v.
314, n. 25, p. 1593–1599, 1986.
HABIBI, A. et al. Delayed hemolytic transfusion reaction in adult sickle-
cell disease: presentations, outcomes, and treatments of 99 referral center episodes.
American Journal of Hematology, v. 91, n. 10, p. 989–994, 2016.
HARM, S. K. et al. A centralized recipient database enhances the serologic
safety of RBC transfusions for patients with sickle cell disease. American Journal
of Clinical Pathology, v. 141, n. 2, p. 256–261, 2014.
HENDRICKSON, J. E. et al. Recipient inflammation affects the frequency
and magnitude of immunization to transfused red blood cells. Transfusion, v. 46,
n. 9, p. 1526–1536, 2006.
HENDRICKSON, J. E.; TORMEY, C. A. Red Blood Cell Antibodies in
Hematology/Oncology Patients: Interpretation of Immunohematologic Tests and
Clinical Significance of Detected Antibodies. Hematology/Oncology Clinics of
North America, v. 30, n. 3, p. 635–651, 2016.
HERRICK, J. B. Peculiar elongated and sickle-shaped red blood corpuscles
in a case of severe anemia. 1910. The Yale journal of biology and medicine, v. 5,
p. 179–184, 1910.
HIGGINS, J.; SLOAN, S. Stochastic modeling of human rbc
alloimmunization: evidence for a distinct population of immunological responders.
Blood, v. 112, n. 6, p. 2546–2553, 2008.
HIRST, C.; OWUSU-OFORI, S. Prophylactic antibiotics for preventing
pneumococcal infection in children with sickle cell disease. Cochrane Database
of Systematic Reviews, n. 3, p. CD003427, 2002.
HOPPE, C. et al. HLA type and risk of alloimmunization in sickle cell
disease. American Journal of Hematology, v. 84, n. 7, p. 462–464, 2009.
HOWARD, J. et al. The Transfusion Alternatives Preoperatively in Sickle

81
Cell Disease (TAPS) study: A randomised, controlled, multicentre clinical trial.
The Lancet, v. 381, p. 930–938, 2013.
HUTTLE, A. et al. Sickle Cell in Sickle Cell Disease in Latin America and
the United States. Pediatr Blood Cancer, v. 62, p. 1131–1136, 2015.
IUGHETTI, L.; BIGI, E.; VENTURELLI, D. Novel insights in the
management of sickle cell disease in childhood. World journal of clinical
pediatrics, v. 5, n. 1, p. 25–34, 2016.
JOHNSON, C. S. Arterial blood pressure and hyperviscosity in sickle cell
disease. Hematology/Oncology Clinics of North America, v. 19, n. 5, p. 827–837,
2005.
JORDAN, L. B. et al. Persistence and compliance of deferoxamine versus
deferasirox in Medicaid patients with sickle-cell disease. Journal of Clinical
Pharmacy and Therapeutics, v. 37, n. 2, p. 173–181, 2012.
KARACAOGLU, P. K. et al. East Mediterranean region sickle cell disease
mortality trial: retrospective multicenter cohort analysis of 735 patients. Annals of
Hematology, v. 95, n. 6, p. 993–1000, 2016.
KARAFIN, M. S. et al. Barriers to using molecularly typed minority red
blood cell donors in support of chronically transfused adult patients with sickle cell
disease. Transfusion, v. 55, n. 6, p. 1399–1406, 2015.
KATO, G. J.; GLADWIN, M. T.; STEINBERG, M. H. Deconstructing
sickle cell disease: Reappraisal of the role of hemolysis in the development of
clinical subphenotypes. Blood Reviews, v. 21, n. 1, p. 37–47, 2007.
KIM, H. C. Red cell exchange: special focus on sickle cell disease.
Hematology / the Education Program of the American Society of Hematology.
American Society of Hematology. Education Program, v. 2014, n. 1, p. 450–
456, 2014.
KOEHL, B. et al. Comparison of automated erythrocytapheresis versus
manual exchange transfusion to treat cerebral macrovasculopathy in sickle cell
anemia. Transfusion, v. 56, n. 5, p. 1121–1128, 2016.
KÖRMÖCZI, G. F.; MAYR, W. R. Responder individuality in red blood
cell alloimmunization. Transfusion Medicine and Hemotherapy, v. 41, n. 6, p.
446–451, 2014.
KWIATKOWSKI, J. L. et al. Silent Infarcts in Young Children with Sickle
Cell Disease Janet. Br J Haematol., v. 146, n. 3, p. 300–305, 2009.
LAMARRE, Y. et al. Hemorheological risk factors of acute chest syndrome
and painful vaso-occlusive crisis in children with sickle cell disease.
Haematologica, v. 97, n. 11, p. 1641–1647, 2012.
LORENZETTI, J. et al. Technology, technological innovation and health: a
necessary reflection. Text Context Nursing, v. 21, n. 2, p. 432–439, 2012.

82
MANWANI, D. et al. Vaso-occlusion in sickle cell disease :
pathophysiology and novel targeted therapies. American Society of Hematology,
v. 122, p. 362–369, 2013.
MARIN, H. DE F. Sistemas de informação em saúde: considerações gerais.
Journal of Health Informatics, v. 2, n. 1, p. 20–24, 2010.
MEERPOHL, J. J. et al. Deferasirox for managing iron overload in people
with myelodysplastic syndrome. The Cochrane database of systematic reviews,
v. 10, n. 5, p. CD007461, 2014.
MEIER, E. R.; MILLER, J. L. Sickle cell disease in children. Drugs, v. 72,
n. 7, p. 895–906, 2012.
MIAN, H. S. et al. Optimal Manual Exchange Transfusion Protocol for
Sickle Cell Disease: A Retrospective Comparison of Two Comprehensive Care
Centers in the United Kingdom and Canada. Hemoglobin, v. 269, n. April 2017, p.
1–6, 2015.
MICHOT, J. et al. Immunohematologic tolerance of chronic transfusion
exchanges with erythrocytapheresis in sickle cell disease. Transfusion, v. 55, n. 2,
p. 357–363, 2014.
MILLER, S. T. et al. Red blood cell alloimmunization in sickle cell disease:
prevalence in 2010. Transfusion, v. 53, n. 4, p. 704–709, 2013.
MINISTÉRIO DA SAÚDE, B. Manual Técnico em Hemoterapia.
Brasilia -DF: [s.n.].
MINISTÉRIO DA SAÚDE, B. PORTARIA No 473, DE 26 DE ABRIL DE
2013. 2013b.
MINISTÉRIO DA SAÚDE, B. Protocolo Clínico e Diretrizes Teraputicas
da Sobrecarga de Ferro. Diário Oficial da União, v. 7042, p. 48–52, 2013c.
MINISTÉRIO DA SAÚDE, B. POLÍTICA NACIONAL DE
INFORMAÇÃO E INFORMÁTICA EM SAÚDE. 1a. ed. Brasilia - DF: [s.n.].
MINISTÉRIO DA SAÚDE, B. PORTARIA No 158, DE 04 DE
FEVEREIRO DE 2016. 2016b.
MINISTÉRIO DA SAÚDE, B. Protocolo Clínico E Diretrizes Terapêuticas
Da Doença Falciforme. 2016c.
MINISTÉRIO DA SAÚDE, B. Portaria no 158, de 04 de fevereiro de
2016, 2016d.
MIRANDA, S. V. Identificando competências informacionais. Ciência da
Informação, v. 33, n. 2, p. 112–122, 2004.
MIRRE, E. et al. Feasibility and efficacy of chronic transfusion for stroke
prevention in children with sickle cell disease. European journal of haematology,

83
v. 84, n. 3, p. 259–265, mar. 2010.
MURAO, M.; VIANA, M. B. Risk factors for alloimmunization by patients
with sickle cell disease. Brazilian journal of medical and biological research =
Revista brasileira de pesquisas médicas e biológicas / Sociedade Brasileira de
Biofísica ... [et al.], v. 38, n. 5, p. 675–82, maio 2005.
NATUKUNDA, B. et al. Red blood cell alloimmunization in sickle cell
disease patients in Uganda. Transfusion, v. 50, n. 1, p. 20–25, 2010.
NICKEL, R. S. et al. Immunophenotypic parameters and RBC
alloimmunization in children with sickle cell disease on chronic transfusion.
American Journal of Hematology, v. 90, n. 12, p. 1135–1141, 2015.
NOIZAT-PIRENNE, F. Relevance of alloimmunization in haemolytic
transfusion reaction in sickle cell disease. Transfusion Clinique et Biologique, v.
19, n. 3, p. 132–138, 2012.
NOIZAT-PIRENNE, F. Relevance of blood groups in transfusion of sickle
cell disease patients. C. R. Biologies, v. 336, n. 3, p. 152–158, 2013.
NOIZAT-PIRENNEA, F.; TOURNAMILLE, C. Relevance of RH variants
in transfusion of sickle cell patients. Transfusion clinique et biologique, v. 18, n.
5–6, p. 527–535, 2011.
O’SUOJI, C. et al. Alloimmunization in Sickle Cell Anemia in the Era of
Extended Red Cell Typing. Pediatr Blood Cancer, v. 60, p. 1487–1491, 2013.
ODER, E. et al. New developments in anti-sickling agents: can drugs
directly prevent the polymerization of sickle haemoglobin in vivo ? British Journal
of Haematology, v. 175, n. 1, p. 24–30, 2016.
OLUJOHUNGBE, A et al. Red cell antibodies in patients with homozygous
sickle cell disease: a comparison of patients in Jamaica and the United Kingdom.
British journal of haematology, v. 113, n. 3, p. 661–665, 2001.
OWUSU-OFORI S, R. T. Splenectomy versus conservative management
for acute sequestration crises in people with sickle cell disease (review). Cochrane
Database of Systematic Reviews, v. 2015, n. 9, 2015.
PAGNIER, J. et al. Evidence for the multicentric origin of the sickle cell
hemoglobin gene in Africa. Proceedings of the National Academy of Sciences of
the United States of America, v. 81, n. 6, p. 1771–3, mar. 1984.
PANTANOWITZ, L.; HENRICKS, W. H.; BECKWITH, B. A. Medical
Laboratory Informatics. Clinics in Laboratory Medicine, v. 27, n. 4, p. 823–843,
2007.
PARRA, F. C. et al. Color and genomic ancestry in Brazilians. Proc Natl
Acad Sci U S A, v. 100, n. 1, p. 177–182, 2003.
PAULING, L. et al. Sickle cell anemia, a molecular disease. Science, v. 110,

84
n. 2835, p. 443–548, 1949.
PEGELOW, C. H. et al. Risk of recurrent stroke in patients with sickle cell
disease treated with erythrocyte transfusions. The Journal of pediatrics, v. 126, n.
6, p. 896–899, 1995.
PEREZ, G.; ZWICKER, R. Fatores determinantes da adoção de sistemas de
informação na área de saúde: um estudo sobre o prontuário médico eletrônico.
Revita de Administração Mackenzie, v. 11, n. 1, p. 174–200, 2010.
PERSEGHIN, P. et al. Erythrocyte-Exchange With the OPTIATM Cell
Separator in Patients With Sickle-Cell Disease Paolo. Journal of clinical …, v. 28,
p. 411–415, 2013.
PIEL, F. B. et al. Global epidemiology of Sickle haemoglobin in neonates:
A contemporary geostatistical model-based map and population estimates. The
Lancet, v. 381, n. 9861, p. 142–151, 2013.
PIEL, F. B. et al. Global migration and the changing distribution of sickle
haemoglobin: A quantitative study of temporal trends between 1960 and 2000. The
Lancet Global Health, v. 2, n. 2, p. e80-89, 2014.
PINHEIRO, A. L. S. et al. Gestão da saúde: o uso dos sistemas de
informação e o compartilhamento de conhecimento para a tomada de decisão.
Texto & Contexto - Enfermagem, v. 25, n. 3, p. 1–9, 2016.
PINTO, P. C. A.; BRAGA, J. A. P.; SANTOS, A. M. N. DOS. Risk factors
for alloimmunization in patients with sickle cell anemia. Revista da Associação
Médica Brasileira (1992), v. 57, n. 6, p. 668–73, 2011.
QUARMYNE, M.-O. et al. Hydroxyurea effectiveness in children and
adolescents with sickle cell anemia: A large retrospective, population-based cohort.
American Journal of Hematology, v. 0, n. 0, p. 1–5, 2016.
QUIROLO, K. et al. The evaluation of a new apheresis device for automated
red blood cell exchange procedures in patients with sickle cell disease.
Transfusion, v. 55, n. 4, p. 775–781, 2015.
RAGHUPATHY, R.; MANWANI, D.; LITTLE, J. A. Iron overload in
sickle cell disease. Adv Hematol, v. 2010, p. 272940, 2010.
RAI, P.; MALIK, P. Gene therapy for hemoglobin disorders - a mini-review
Parul. J Rare Dis Res Treat., v. 1, n. 2, p. 25–31, 2016.
REZENDE, P. V et al. Acute splenic sequestration in a cohort of children
with sickle cell anemia. Jornal de pediatria, v. 85, n. 2, p. 163–169, 2009.
RODGERS, G. et al. Hematologic responses of patients with sickle cell
disease to treatment with hydroxyurea. N Engl J Med., v. 322, n. 15, p. 1037–
1045., 1990.
SANDERS, R. P. et al. Premedication with acetaminophen or

85
diphenhydramine for transfusion with leucoreduced blood products in children.
British Journal of Haematology, v. 130, n. 5, p. 781–787, 2005.
SANITARIA, A. N. DE V. Boletim de Hemovigilância no 7Brasilia - DF,
2015.
SANITÁRIA, A. N. DE V. Marco Conceitual e Operacional de
Hemovigilância : Guia para a Hemovigilância no BrasilBrasilia - DFAnvisa, ,
2015.
SAYLORS, R. L. et al. Comparison of Automated Red Cell Exchange
Transfusion and Simple Transfusion for the Treatment of Children With Sickle Cell
Disease Acute Chest Syndrome. Journal of Clinical Oncology, v. 14, n. 5, p.
1526–1531, 1996.
SCHONEWILLE, H. et al. HLA-DRB1 associations in individuals with
single and multiple clinically relevant red blood cell antibodies. Transfusion, v.
54, n. 8, p. 1971–1980, 2014.
SCOTHORN, D. J. et al. Risk of recurrent stroke in children with sickle cell
disease receiving blood transfusion therapy for at least five years after initial stroke.
The Journal of pediatrics, v. 140, n. 3, p. 348–354, 2002.
SERJEANT, G. R. The natural history of sickle cell disease. CSH
perspectives, v. 3, n. 10, p. a011783, out. 2013.
SHIGEHARA, K.; NAMIKI, M. Clinical Management of Priapism: A
Review. The world journal of men’s health, v. 34, n. 1, p. 1–8, 2016.
SINS, J. W. R. et al. Early occurrence of red blood cell alloimmunization in
patients with sickle cell disease. American Journal of Hematology, v. 91, n. 8, p.
763–769, 2016.
SIPPERT, E. et al. Variant RH alleles and Rh immunisation in patients with
sickle cell disease. Blood Transfusion, v. 13, n. 1, p. 72–77, 2015.
SIPPERT, E. Â. et al. Red blood cell alloimmunization in patients with
sickle cell disease: correlation with HLA and cytokine gene polymorphisms.
Transfusion, v. 0, n. October, p. 1–11, 2016.
STACK, G.; TORMEY, C. A. Estimating the immunogenicity of blood
group antigens: a modified calculation that corrects for transfusion exposures.
British Journal of Haematology, v. 175, n. 1, p. 154–160, 2016.
STALLWORTH, J. R.; JERRELL, J. M.; TRIPATHI, A. Cost-
effectiveness of hydroxyurea in reducing the frequency of pain episodes and
hospitalization in pediatric sickle cell disease. [s.l: s.n.]. v. 85
STEINBERG, M. H. Sickle cell anemia, the first molecular disease:
overview of molecular etiology, pathophysiology, and therapeutic approaches.
TheScientificWorldJournal, v. 8, p. 1295–324, jan. 2008.

86
STORRY, J. R. et al. International Society of Blood Transfusion Working
Party on red cell immunogenetics and blood group terminology: Cancun report
(2012). Vox Sanguinis, v. 107, n. 1, p. 90–96, 2014.
SWERDLOW, P. S. Red Cell Exchange in Sickle Cell Disease.
Hematology, v. 2006, n. 1, p. 48–53, 2006.
TALAHMA, M.; STRBIAN, D.; SUNDARARAJAN, S. Sickle Cell
Disease and Stroke. Stroke, v. 45, p. e98-100, 2014.
TANAKA, O. Y.; TAMAKI, E. M. O papel da avaliação para a tomada de
decisão na gestão de serviços de saúde The role of evaluation in decision-making
in the management of health services. Ciência & Saúde Coletiva, v. 17, n. 4, p.
821–828, 2012.
TATARI-CALDERONE, Z. et al. Protective effect of HLA-DQB1 alleles
against alloimmunization in patients with sickle cell disease. Human
Immunology, v. 77, n. 1, p. 35–40, 2016.
TELEN, M. J. Principles and problems of transfusion in sickle cell disease.
Seminars in Hematology, v. 38, n. 4, p. 315–323, 2001.
TELEN, M. J. et al. Antibody Specificites And Association With Chronic
Pain And Decreased Survival. Transfusion, v. 55, p. 1378–1387, 2015.
TERESA, M. et al. Frequency and Origins of Hemoglobin S Mutation in
African-Derived Brazilian Populations Frequency and Origins of Hemoglobin S
Mutation in. Hum Biol, v. 79, n. 6, p. 667–677, 2008.
THORNBURG, C. D. et al. Impact of hydroxyurea on clinical events in the
BABY HUG trial. Blood, v. 120, n. 22, p. 4304–4310, 2012.
THORNBURG, C. D.; CALATRONI, A.; PANEPINTO, J. A. Differences
in Health-Related Quality of Life in Children With Sickle Cell Disease Receiving
Hydroxyurea. Journal of Pediatric Hematology/Oncology, v. 33, n. 4, p. 251–
254, 2011.
UHLMANN, E. J.; SHENOY, S.; GOODNOUGH, L. T. Successful
treatment of recurrent hyperhemolysis syndrome with immunosuppression and
plasma-to-red blood cell exchange transfusion Erik. Transfusion, v. 54, n.
February, p. 384–388, 2014.
VICHINNSKY, E. P. et al. Alloummunization in Sickle Cell Anemia and
Transfusion of Racially Unmatched Blood. The New England journal of
medicine, v. 322, n. 23, p. 1617–1621, 1990.
VICHINSKY, E. et al. A randomised comparison of deferasirox versus
deferoxamine for the treatment of transfusional iron overload in sickle cell disease.
British Journal of Haematology, v. 136, n. 3, p. 501–508, 2007.
VICHINSKY, E. et al. Efficacy and safety of deferasirox compared with
deferoxamine in sickle cell disease: Two-year results including pharmacokinetics

87
and concomitant hydroxyurea. American Journal of Hematology, v. 88, n. 12, p.
1068–1073, 2013.
WAHL, S.; QUIROLO, K. C. Current issues in blood transfusion for sickle
cell disease. Curr Opin Pediatr, v. 21, n. 1, p. 15–21, 2009.
WANG, Y. et al. Sickle cell disease incidence among newborns in New
York State by maternal race/ethnicity and nativity. Genetics in Medicine, v. 15, n.
3, p. 222–8, 2013.
WARE, H. M.; KWIATKOWSKI, J. L. Evaluation and treatment of
transfusional iron overload in children. Pediatric Clinics of North America, v. 60,
n. 6, p. 1393–1406, 2013.
WARE, R. E. Principles and Indications of Chronic Transfusion Therapy
for Children With Sickle Cell Disease. Clinical Advances in Hematology &
Oncology, v. 5, n. 9, p. 686–688, 2007.
WARE, R. E. et al. Hydroxycarbamide versus chronic transfusion for
maintenance of transcranial doppler flow velocities in children with sickle cell
anaemia - TCD with Transfusions Changing to Hydroxyurea (TWiTCH): A
multicentre, open-label, phase 3, non-inferiority trial. The Lancet, v. 387, n. 10019,
p. 661–670, 2016.
WARE, R. E.; HELMS, R. W. Stroke With Transfusions Changing to
Hydroxyurea (SWiTCH). Blood, v. 119, n. 17, p. 3925–3932, 2012.
WAYNE, A. S.; KEVY, S. V.; NATHAN, D. G. Transfusion Management
of Sickle Cell Disease. Journal of the American Statistical Association, v. 81, p.
1109–1123, 1993.
WOOD, J. C. et al. Organ iron accumulation in chronically transfused
children with sickle cell anaemia: Baseline results from the TWiTCH trial. British
Journal of Haematology, v. 172, n. 1, p. 122–130, 2016.
WRIGHT, J.; HAMBLETON, I.; THOMAS, P. Postsplenectomy disease
course in homozygous sickle cell disease. J Pediatr, v. 134, p. 304–309, 1999.
YAWN, B. et al. Management of Sickle Cell Disease. JAMA, v. 312, n. 10,
p. 1033–1048, 2014.
YAZDANBAKHSH, K. Mechanisms of sickle cell alloimmunization.
Transfusion Clinique et Biologique, v. 22, n. 3, p. 178–181, 2015.
YAZDANBAKHSH, K.; WARE, R. E.; NOIZAT-PIRENNE, F. Red blood
cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and
transfusion management. Blood, v. 120, n. 3, p. 528–537, 19 jul. 2012.
ZANETTE, A. M. D. et al. Alloimmunization and Clinical Profile off Sickle
Cell Disease Patientes From Salvador - Brazil. Ethnicity & Disease, v. 20, p. 136–
141, 2010.

88
APÊNDICES
ANEXO A – Formulário de identificação do paciente

89
ANEXO B - Formulário de dados das transfusões

90
ANEXO C – Formulário de dados do doador