seminario para periodistas “mitos y realidades de … · ‐ medicamentos genéricos marco legal...
TRANSCRIPT

1
SEMINARIO PARA PERIODISTAS
“Mitos y realidades de los medicamentos genéricos”

2
Índice
1.‐Visión General del Sistema Nacional de Salud. Estructura reguladora
Estructura reguladora
El Sistema de Precios de Referencia
‐ Aplicación del precio de referencia en la financiación pública de los medicamentos
Papel de las Comunidades Autónomas en la gestión de la prestación farmacéutica
Medidas de contención del gasto farmacéutico
Causas del incremento del gasto sanitario
Qué es un incremento del gasto sostenible
2.‐ Medicamentos de marca
Contribución del medicamento en la erradicación de enfermedades y en el aumento de la esperanza de vida de la
sociedad
El valor de los medicamentos de marca
‐ I+D+i: ciclo de vida de un medicamento
‐ Calidad, eficacia, seguridad y accesibilidad
Marco nacional y europeo del medicamento de marca: Normativa sobre la patente
3.‐ Medicamentos genéricos
Marco legal
Autorización y registro de medicamentos genéricos
4.‐Prescripción y dispensación de medicamentos
5.‐ Mitos y realidades del medicamento genérico y de marca
6.‐ Glosario de términos
7.‐ Bibliografía

3
1. Visión General del Sistema Nacional de Salud. Estructura reguladora
Estructura reguladora en España
La producción, introducción en el mercado y utilización racional de los medicamentos es
probablemente una de las actividades más intensamente reguladas en las relaciones comerciales
actuales.
En España el Decreto que regula las Ordenanzas de Farmacia de 1927 es considerado como la
primera disposición que reguló el registro, producción y dispensación de los medicamentos de
producción industrial. A este reglamento le seguiría casi cuarenta años más tarde el Decreto
2464/1963 que reguló los laboratorios de especialidades farmacéuticas, su distribución, publicidad y
seguimiento después de su comercialización. Desde ahí se sucedieron una serie de normativas que
actualizaban a la existente y que perseguían la armonización con las directivas europeas y que
culminaron con la Ley 25/1990 de 20 de Diciembre del Medicamento. Dado el marco europeo en el
que se opera, la Ley del Medicamento ha sufrido sucesivas modificaciones y desarrollos para
adaptarla a la evolución de las directivas europeas, y de obligada trasposición a la normativa de los
Estados Miembros, hasta derivar en la actual Ley 29/2006 de 26 de julio, de garantías y uso racional
de los medicamentos y productos sanitarios, auténtico marco legislativo para el entorno de la
industria farmacéutica.
Dentro de los organismos que integran la estructura reguladora en España mencionaremos los
siguientes:
1. Ministerio de Sanidad y Política Social.‐ Le corresponde la política del Gobierno en materia de
salud, de planificación y asistencia sanitaria y de consumo, así como el ejercicio de las
competencias de la Administración General del Estado para asegurar a los ciudadanos el

4
derecho a la protección de la salud. La estructura orgánica del Ministerio de Sanidad y Política
Social sufrió una profunda remodelación como consecuencia de la conclusión del proceso
transferencial a las Comunidades Autónomas a finales del año 2001. Entre sus funciones ha
adquirido una especial importancia la labor de coordinación de las diferentes CCAA para
garantizar una homogeneidad asistencial.
2. Dirección General de Farmacia y Productos Sanitarios.‐ Es el órgano al que le corresponde la
dirección, desarrollo y ejecución de la política farmacéutica del departamento, así como el
ejercicio de las funciones que competen al Estado en materia de financiación pública y fijación
del precio de los medicamentos así como de los productos sanitarios dispensados a través de
receta oficial y, determinación de las condiciones especiales de su prescripción y dispensación
en el Sistema Nacional de Salud, en particular el establecimiento de visados previos a la
dispensación.
3. Agencia Española del Medicamento.‐ Se crea por la Ley 66/1997 de 30 de Diciembre de
Medidas Fiscales, Administrativas y de Orden Social. Nace como reflejo de las Agencias Europeas
de autorización de medicamentos y productos sanitarios, e inspiradas a su vez en el modelo de
la Food and Drug Administration (FDA). El objetivo principal de la Agencia Española es garantizar
que los medicamentos de uso humano y veterinarios que sean aprobados y autorizados en
nuestro territorio respondan a los criterios fundamentales de eficacia, seguridad y calidad.
El Sistema de Precios de Referencia
Ley 29/2006 de 26 de Julio, de Garantías y Uso Racional de los medicamentos y productos sanitarios ,
en su artículo 93, introduce una profunda reforma del sistema de precios de referencia y establece
que la financiación pública de medicamentos estará sometida al sistema de precios de referencia.
El precio de referencia será la cuantía con la que se financiarán las presentaciones de medicamentos
incluidas en cada uno de los conjuntos que se determinen, siempre que se prescriban y dispensen a
través de receta médica oficial del Sistema Nacional de Salud.
Se entiende por conjunto la totalidad de las presentaciones de medicamentos financiadas que
tengan el mismo principio activo e idéntica vía de administración entre las que existirá, al menos, una
presentación de medicamento genérico. Las presentaciones indicadas para tratamientos en pediatría
constituirán conjuntos independientes.
El Ministro de Sanidad y Consumo, previo acuerdo de la Comisión Delegada del Gobierno para
Asuntos Económicos e informe del Consejo Interterritorial del Sistema Nacional de Salud,
determinará, con la periodicidad que reglamentariamente se fije, dichos conjuntos, así como sus
precios de referencia y podrá fijar umbrales mínimos para estos precios, en ningún caso inferiores a
dos euros. El precio de referencia será, para cada conjunto, la media aritmética de los tres
costes/tratamiento/día menores de las presentaciones de medicamentos en él agrupadas por cada
vía de administración, calculados según la dosis diaria definida.

5
Reglamentariamente se podrán prever los supuestos, requisitos y procedimientos en los que
determinadas innovaciones galénicas que se consideren de interés por añadir mejoras en la utilidad
terapéutica, puedan quedar excluidas del sistema de precios de referencia durante cinco años.
Transcurridos los cinco años, la innovación galénica se integrará en el conjunto de referencia.
Los medicamentos genéricos no podrán superar el precio de referencia del conjunto
correspondiente. Asimismo, no podrán superar el precio de referencia las presentaciones de
medicamentos que no dispongan de iguales presentaciones de medicamentos genéricos a efectos de
la sustitución que establece el apartado siguiente, en tanto se mantenga la situación de no
disponibilidad.
Si bien el modelo elegido por el Estado garantiza la obtención del medicamento al precio más bajo
posible, puede decirse / deducirse que esto mismo beneficia a aquellas empresas que comercializan
sus productos a estrictos costes directos con mínimo valor añadido, fabricación, garantía de calidad,
I+D+i, abocando a aquellas operadoras que soportan importantes costes indirectos a cambiar
completamente su modelo de negocio o a desaparecer, de ahí, la importancia de introducir en la ley
el párrafo de garantía de abastecimiento.
‐Aplicación del precio de referencia en la financiación pública de los medicamentos.
Tal y como está establecido, el precio de referencia es la cuantía máxima que el Sistema Nacional de
Salud financiará para una especialidad farmacéutica financiable con fondos públicos pertenecientes a
sanidad. La Ley de Garantías establece que las especialidades genéricas no podrán superar el precio
de referencia.
Este principio no afecta a las marcas, que podrán mantener su precio superior al de referencia, pero
en este caso, no serían financiadas con fondos públicos. En el momento de la dispensación en la
oficina de farmacia, cuando se prescriba un medicamento de marca y si la misma supera el precio de
referencia, el farmacéutico deberá sustituirla por la especialidad farmacéutica genérica a precio de
referencia, no obstante, si el medicamento de marca no supera el precio de referencia, no deberá
aplicarse la sustitución por parte del farmacéutico.
El aspecto negativo que han asumido las compañías farmacéuticas, en algunos casos, es que debido a
que determinadas formas farmacéuticas de una sustancia no son atractivas para el
operador/laboratorio de especialidades genéricas, sólo la compañía farmacéutica innovadora
abastece el mercado. Por tanto, puede verse como injusto o desequilibrado que se deba penalizar a
una compañía farmacéutica por cubrir y satisfacer necesidades médicas para la sociedad.
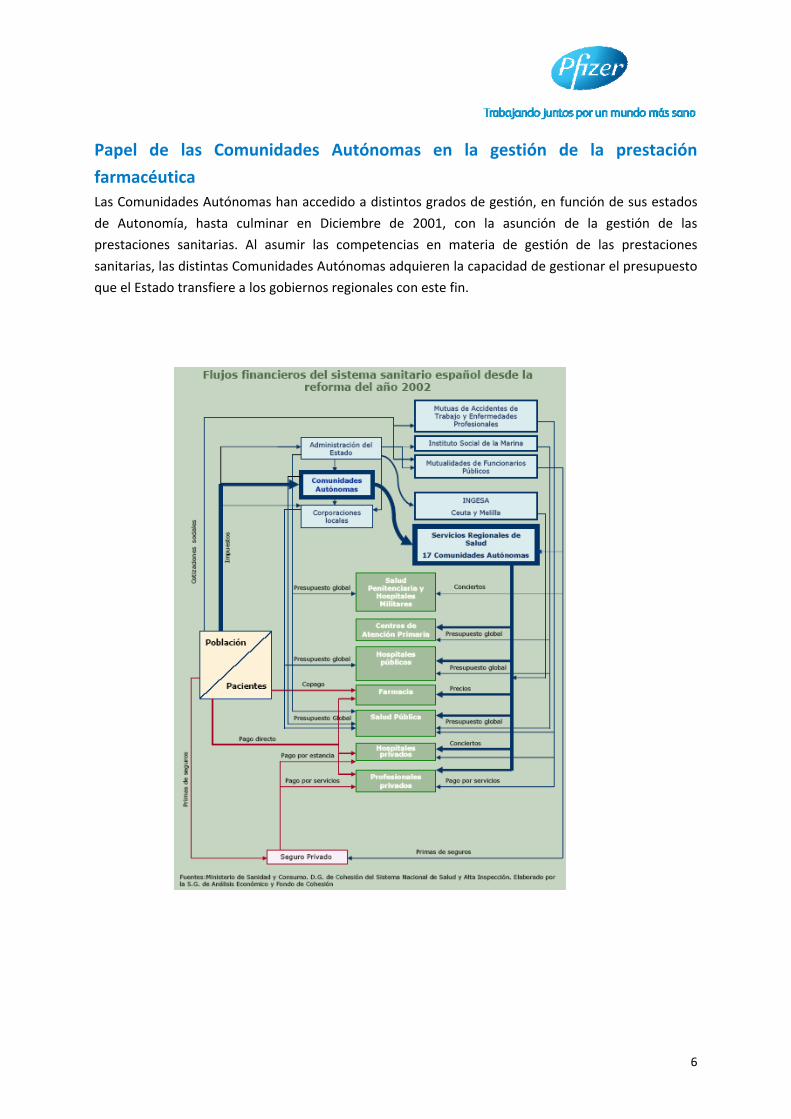
6
Papel de las Comunidades Autónomas en la gestión de la prestación
farmacéutica
Las Comunidades Autónomas han accedido a distintos grados de gestión, en función de sus estados
de Autonomía, hasta culminar en Diciembre de 2001, con la asunción de la gestión de las
prestaciones sanitarias. Al asumir las competencias en materia de gestión de las prestaciones
sanitarias, las distintas Comunidades Autónomas adquieren la capacidad de gestionar el presupuesto
que el Estado transfiere a los gobiernos regionales con este fin.

7
Competencias de las Comunidades Autónomas:
1. Gestión de la prestación farmacéutica del SNS. En concreto:
a. Preparación y difusión de guías fármaco‐terapéuticas para conseguir un uso
adecuado de los medicamentos.
b. Instauración de medidas para la contención del gasto farmacéutico. Por su parte el
Estado establecerá el precio industrial máximo y determinará las condiciones de
financiación por el SNS.
c. Disposiciones en materia de establecimiento de oficinas de farmacia, sus medios y
normas de funcionamiento, así como de los almacenes de distribución farmacéutica
y de los Servicios de Farmacia Hospitalaria.
2. Labores de inspección en laboratorios farmacéuticos
3. Autorización de ensayos clínicos post‐autorización y la normativa de funcionamiento de los
Comités Éticos. Deben ser también informadas de los ensayos clínicos que se realicen en su
territorio.
4. Autorización y seguimiento de la publicidad dirigida a profesionales sanitarios.
5. Establecimiento y mantenimiento de un sistema de farmacovigilancia
De acuerdo con el White Paper de IMS Health, Nuevos modelos de negocio para una sanidad
transferida, la consecuencia directa de la descentralización de las competencias sanitarias ha sido la
alteración de las relaciones de las compañías farmacéuticas con los decisores del SNS con la creación
de 17 sistemas sanitarios, en los que cada comunidad apuesta cada vez más por adoptar medidas en
el control efectivo del gasto o por lograr el tratamiento más efectivo para cada paciente.
En este nuevo escenario se ha producido además un cambio en el poder de influencia de los agentes
de mercado y han emergido los denominados nuevos “stakeholders”, muchos de ellos ubicados en el
ámbito regional. Estos nuevos agentes influyen cada vez más en la toma de decisiones relativas a
establecimiento de precios, reembolso, acceso al mercado y protocolos de tratamiento.
Actualmente las compañías que operan en España deben hacer frente, como decíamos, a la
descentralización del poder de decisión. Los pagadores del SNS tienen dos vías para hacer frente a la
creciente presión económica: las decisiones de establecimiento de precio y reembolso, y el acceso a
las medicinas. Si bien las decisiones de precio y reembolso son tomadas a través del Ministerio de
Sanidad, son las CC.AA. quienes deciden en lo que se refiere al acceso a diferentes opciones de
tratamiento.

8
Causas del incremento del gasto sanitario
Podemos identificar las siguientes razones:
1. Envejecimiento de la población
2. Mejora de las técnicas diagnósticas que incrementa el número de tratamientos aplicados, así
para enfermedades con Alzheimer u osteoporosis (densitometrías, DEXA, etc).
3. Aparición de nuevas patologías crónicas. Ej. Sida.
4. Aparición de medicamentos innovadores para el tratamiento de enfermedades con un mal
pronóstico. Ej: cáncer, enfermedades cardiovasculares.
5. Aparición de técnicas sanitarias de alta tecnología, quirúrgicas, diagnósticas, etc.
Dado que para la Administración puede resultar complicado intervenir directamente sobre estos
parámetros, debido a que los ciudadanos necesitan disponer de una Sanidad eficiente y equipada
con las últimas técnicas médicas para ofrecer un servicio de calidad a los pacientes, los Estados
suelen aplicar medidas de racionalización del gasto farmacéutico dirigido a moderar el consumo de
medicamentos.
Qué es un incremento de gasto sostenible
Todos los estados admiten que un incremento periódico del gasto farmacéutico es lógico y
razonable, al ser un indicador de que la sociedad está aplicando adecuadamente las nuevas técnicas
disponibles, compatibilizándolas con un incremento gradual de la población protegida.
Hasta ahora se había consolidado la opinión que un aumento porcentual del mercado equivalente al
PIB+ 1,8 unidades porcentuales es razonable; de hecho, las medidas que tienden a aplicarse han
perseguido este objetivo. Así:
1. Reducción generalizada de precios. En determinados países de la UE, por ejemplo, esta
medida está siendo asumida por la Industria Farmacéutica con crecientes dificultades al abrir
una brecha importante en el diferencial de precios frente al resto de estados europeos.
2. Colaboración del beneficiario en el coste del medicamento.
3. Financiación selectiva
4. Precios de referencia
5. Guías de prescripción
6. Imposición de límites de prescripción
7. Imposición de medidas restrictivas a la prescripción (visados)
8. Fomento de la prescripción de genéricos
La introducción del genérico en los mercados ha servido, tanto para su establecimiento de
base para el cálculo del precio de referencia, como para que los SNS fomenten su
prescripción, como una medida más de la racionalización del gasto farmacéutico.
9. Dispensación en unidosis
10. Aplicación de descuentos o aportaciones de los operadores al SNS

9
Recientemente, y en el marco actual de crisis a nivel global al que asistimos, Farmaindustria ha
firmado con el gobierno español, el denominado Pacto Sectorial para la sostenibilidad del sistema
sanitario. A través de este pacto el sector de la Industria Farmacéutica de nuestro país se ha
comprometido al mantenimiento del volumen total de empleo del sector durante los próximos tres
años, así como a mejorar la cualificación y la calidad del mismo.
2. Medicamentos de marca
Contribución del medicamento en la erradicación de enfermedades y en el
aumento de la esperanza de vida de la sociedad
La esperanza de vida de la sociedad ha aumentado exponencialmente en las últimas décadas,
permitiendo reducir las tasas de mortalidad y vivir con una mayor calidad de vida. Varios son los
factores que han contribuido a esta situación, como la mejora en los hábitos de vida, la exposición al
riesgo, la educación, las condiciones materiales de las personas, y muy especialmente las
innovaciones científicas y los medicamentos.
Desde una perspectiva histórica el papel del medicamento ha sido determinante para la erradicación
de muchas enfermedades. El valor del medicamento puede ser evaluado desde perspectivas como su
contribución económica, su contribución social, pero queremos destacar su beneficio terapéutico y
su aportación a aliviar el sufrimiento de las personas.
Si hacemos un balance son muchas las evidencias y estudios que ponen de manifiesto el impacto del
medicamento sobre la calidad de vida de las personas. Mencionamos sólo unos ejemplos:
Enfermedades Cardiovasculares. Estudios documentados en el New England Journal of Medicine
(NEJM) muestran cómo el desarrollo de las antihipertensivos ha permitido disminuir los accidentes
isquémicos cardiovasculares y cerebrovasculares.
Cáncer. La lucha contra el cáncer tiene una de sus principales esperanzas puestas en el desarrollo de
nuevos medicamentos. La tasa de supervivencia a los cinco años ha aumentado desde un 10% en
1960 a un 50% en 1990. La mejora observada en estas cifras tiene una relación directa con la
implantación de un diagnóstico precoz de las neoplasias más comunes, así como con el efecto
progresivo y significativo de los tratamientos. Esto es especialmente cierto para ciertos tumores
como el de mama, un tipo de tumor donde más han caído las tasas de mortalidad.
VIH/Sida. Gracias al desarrollo de los antirretrovirales se ha contribuido de forma muy notable a
reducir la mortalidad en pacientes infectados por el VIH y a retrasar el desarrollo de la enfermedad,
hasta el punto de que hoy el Sida está considerado como una enfermedad crónica.

10
El valor de un medicamento de marca
Detrás de una marca siempre hay un esfuerzo investigador, una aplicación de conocimientos
adquiridos a lo largo de los años, que culminan en un medicamento y en una solución para los
pacientes. De hecho suele establecerse un período de entre 8 y 10 años para poner un medicamento
a disposición de pacientes y profesionales sanitarios. Este esfuerzo innovador es fundamental para
seguir cubriendo las necesidades de los pacientes.
El desarrollo de medicamentos innovadores o de marca supone un gran esfuerzo por parte de las
compañías biomédicas, ya que su existencia depende de procesos largos, complejos y costosos y la
tasa de éxito, que un medicamento salga al mercado, no se corresponde en la mayoría de las veces
con el esfuerzo puesto en su desarrollo.
Los medicamentos de marca son desarrollados principalmente por la Industria Farmacéutica a través
de Centros de Investigación independientes y acuerdos de colaboración con Hospitales.
Los nuevos fármacos nacen de la necesidad de tratar las enfermedades o los síntomas para los que no existe
un tratamiento eficaz o de mejorar los ya existentes.
‐ I +D+ i: ciclo de vida de un medicamento
La industria biomédica proporciona a la sociedad, a través de la I+D+i, uno de los bienes más
preciados y que más contribuyen a prolongar la vida de la población. Uno de los grandes avances
terapéuticos de la medicina moderna: el medicamento.
Así como en los seres humanos, en la vida de un medicamento podemos distinguir diferentes
momentos: su gestación, nacimiento, crecimiento – madurez, vejez y muerte.
“El valor de un comprimido no responde a lo que cuesta hacerlo, sino al
conocimiento acumulado de investigación básica y clínica que ha permitido su
desarrollo” Dr. Juan Álvarez, director médico de Pfizer.

11
A pesar de que las fases de investigación son cada vez más complicadas y más caras, Pfizer es una de
las compañías que más invierte en I+D + i de cualquier sector. Contamos con unos 12.000
investigadores en el mundo y un presupuesto consolidado de Pfizer de unos 11.000 millones de
dólares.
‐ Calidad, eficacia, seguridad y accesibilidad
Para las grandes compañías biomédicas, que sus medicamentos sean seguros y efectivos para los pacientes
es uno de sus objetivos fundamentales. Esta misma idea se traslada a la investigación y a los estudios clínicos
que realizan.
Es por este motivo, que un medicamento de marca ofrece a los pacientes un riguroso control de calidad y
seguridad, y de la misma forma intenta cada día mantenerse cercano a los pacientes, tanto directamente
como a través de las asociaciones, facilitándoles el acceso a las terapias más innovadoras y a la información
más completa sobre todos los aspectos relativos a la salud y a la enfermedad.
En el caso de Pfizer, el principal compromiso con los pacientes es poner a su disposición soluciones
terapéuticas que contribuyan a salvar vidas, mejorar su salud y conseguir un mundo más sano. Esta tarea se
desarrolla bajo tres premisas: Seguridad, Transparencia y Colaboración.
Marco nacional y europeo del medicamento de marca: Normativa sobre la
patente
La investigación y desarrollo de medicamentos necesita un marco estable para su crecimiento,
principalmente porque es el sector del mundo en el que más se investiga. En los países desarrollados,
aproximadamente un 15% de los recursos que la industria privada destina a investigar proceden del
sector farmacéutico.
Por ello, es necesario que los agentes que más invierten en la actividad de I+D puedan seguir
desarrollándola, generando más valor y garantizando unos elevados requisitos de calidad, lo cual
lleva implícito una inversión acorde al panorama del sector y a las necesidades del mismo.
La actividad investigadora y productiva del sector farmacéutico tiene como meta el desarrollo de
medicamentos de calidad que mejoren la calidad de vida de los ciudadanos y favorezcan el desarrollo
de nuevas terapias que cubran las necesidades médicas no satisfechas.
El lanzamiento de un nuevo medicamento es un proceso largo y costoso. Poner un medicamento a
disposición de los pacientes y de los profesionales sanitarios requiere investigar en paralelo una
media de 10.000 moléculas, de las que sólo una, tras 12 ó 14 años de investigación, se podrá
convertir en un medicamento eficaz.
Ese proceso implica una inversión que ronda los 800 o 1.000 millones de euros además del trabajo de
un amplio equipo multidisciplinar y la participación de miles de pacientes. Asimismo, exige una
exhaustiva batería de controles y exámenes por parte de los organismos reguladores y agencias
evaluadoras de medicamentos más exigentes del mundo.

12
Sin embargo, a pesar de este esfuerzo investigador, el sector farmacéutico es el menos rentable en
cuanto a patentes registradas se refiere. La limitación del tiempo de patentes de los medicamentos
conlleva el riesgo de que las empresas biomédicas no cuenten con los recursos necesarios para
garantizar un correcto proceso de investigación.
Dada la estrecha relación del medicamento con el ámbito social y sanitario, ningún otro sector tiene
un grado de intervención y de control tan elevado como el farmacéutico. Se regula toda la actividad
investigadora, productiva y comercial, además de la autorización de la puesta en el mercado y precio
de los productos, afectando también al sistema de patentes.
El Acuerdo sobre los ADPIC (Aspectos de los Derechos de Propiedad Intelectual relacionados con el
Comercio), desarrollado por la Organización Mundial del Comercio (OMC), dispone que todas las
invenciones deben poder ser protegidas por una patente durante veinte años, bien se trate de un
producto (como un medicamento) o de un método (método de producción de un ingrediente que
entra en la composición de un medicamento).
Sin embargo, las diferencias en la aplicación de las leyes comunitarias en materia de patentes han
provocado que en la actualidad algunos medicamentos gocen de protección de patente en la UE y no
en España, pudiendo aparecer, por tanto, su genérico en nuestro país, pero no en Europa.
Esta situación implica efectos negativos directos para las compañías ubicadas en España como son la
desaparición anticipada del mercado de los productos innovadores.
La industria biomédica española considera clave para el sector la aplicación nacional de los acuerdos
ADPIC al encontrarse en una situación de desequilibrio respecto a sus socios europeos en cuanto a la
protección de la propiedad industrial de los medicamentos.
Las patentes representan el modo de garantizar y asegurar las inversiones en innovación e
investigación, que no se impulsarían sin una protección de los derechos de la industria.
En la actualidad, los medicamentos pueden ser patentados a través de dos vías principales (patente
de procedimiento o de proceso y patente de producto), en ambos casos su protección tiene validez
de 20 años.
Este es el plazo previsto para que las compañías biomédicas puedan amortizar la inversión y obtener
los beneficios correspondientes a su actividad de investigación y empresarial.
En este sentido, la armonización de los plazos de las patentes y el apoyo a la investigación y
desarrollo por parte de los gobiernos europeos, contribuirán a garantizar la capacidad innovadora
del sector farmacéutico.

13
3. Medicamentos genéricos
Según la Ley de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios de julio de
2006, un genérico es “todo medicamento que tenga la misma composición cualitativa y cuantitativa
en principios activos y la misma forma farmacéutica, y cuya bioequivalencia1 con el medicamento de
referencia haya sido demostrada por estudios adecuados de biodisponibilidad”2.
La bioequivalencia en los genéricos significa que han demostrado previamente a la solicitud y
autorización de comercialización su bioequivalencia con el medicamento de referencia mediante los
estudios clínicos adecuados. Un estudio de bioequivalencia es un ensayo clínico comparativo entre
dos formulaciones donde se comprueba que ambas formulaciones proporcionan niveles del fármaco
en la circulación sistémica comparables. Por tanto, sus niveles en el lugar de acción serán semejantes
y su perfil de eficacia y seguridad han de ser equivalentes.
Debe ofrecer la misma seguridad, calidad y eficacia que cualquier otro medicamento. Es el
Ministerio de Sanidad, a través de la Agencia Española del Medicamento y Productos Sanitarios
(AEMPS) el que autoriza y aprueba un medicamento genérico, de igual modo que cualquier otro
medicamento. Todos los medicamentos aprobados por el Ministerio de Sanidad, han de pasar por los
mismos controles de calidad, seguridad y eficacia.
Por definición, los medicamentos genéricos deberían suponer un menor costo frente al
medicamento de marca debido a una menor inversión en investigación, desarrollo y promoción. Sin
embargo, en España, este diferencial no es tan acusado debido a la implantación del sistema de
precios de referencia que permite tener en una banda muy estrecha de precios medicamentos de
marca y medicamentos genéricos dentro de un mismo conjunto.
Las primeras especialidades genéricas aparecieron en el mercado español en mayo de 1997.
Actualmente, el mercado de genéricos en España integra 139 principios activos. El número de
especialidades farmacéuticas genéricas (EFG) aprobadas a finales de enero de 2007 por la Agencia
Española del Medicamento asciende a 3.313.
1 La bioequivalencia es la igualdad de efectos biológicos de dos medicamentos, hasta el punto de poder ser intercambiados sin cambios significativos de sus efectos terapéuticos y adversos. 2 Fracción administrada de la dosis de un medicamento que alcanza su diana terapéutica, o lo que es lo mismo, que llega hasta el tejido sobre el que realiza su actividad.

14
Marco legal
‐ Ley 29/2006 de 26 de Julio, de Garantías y Uso Racional de los medicamentos y productos
sanitarios
‐ Real Decreto 1345/2007, de 11 de octubre por el que se regula el procedimiento de
autorización, registros y condiciones de dispensaciones de dispensación de los
medicamentos de uso humano fabricados industrialmente.
‐ Orden SCO/2007, de 11 de octubre, de autorización
‐ Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con
medicamentos.
De acuerdo con la normativa existente se establece lo siguiente para los medicamentos genéricos:
o Todo medicamento que tenga la misma composición cualitativa y cuantitativa en principios
activos y la misma forma farmacéutica, y cuya bioequivalencia con el medicamento de
referencia haya sido demostrada por estudios adecuados de biodisponibilidad. Las diferentes
sales, ésteres, éteres, isómeros, mezclas de isómeros, complejos o derivados de un principio
activo se considerarán un mismo principio activo, a menos que tengan propiedades
considerablemente diferentes en cuanto a seguridad y/o eficacia, en cuyo caso el solicitante
deberá facilitar datos suplementarios para demostrar la seguridad y/o eficacia de la
diversidad de sales, ésteres derivados de un principio activo autorizado.
o El solicitante podrá estar exento de presentar los estudios de biodisponibilidad si puede
demostrar que el medicamento genérico satisface los criterios pertinentes definidos en las
correspondientes directrices detalladas.
o Los medicamentos genéricos de un medicamento de referencia, no se comercializarán hasta
transcurridos diez años desde la fecha de la autorización inicial del medicamento de
referencia.
o Este periodo de diez años se ampliará hasta un máximo de once si, durante los primeros
ocho años, el titular de la autorización de comercialización del medicamento de referencia
obtiene una autorización para una o varias indicaciones terapéuticas nuevas y, durante la
evaluación científica previa a su autorización, se establece que dichas indicaciones aportarán
un beneficio clínico significativo en comparación con las terapias existentes.
o Cuando el medicamento de referencia no esté autorizado en España, el solicitante deberá
indicar en la solicitud el nombre del Estado miembro en que esté o haya sido autorizado y la
fecha de autorización.

15
Las Autoridades Sanitarias autorizan genéricos una vez ha transcurrido un plazo determinados por
los Estados Miembros para las autorizaciones nacionales o por la Comisión Europea para las
autorizaciones por procedimiento centralizado desde la primera autorización del correspondiente
medicamento innovador en algún país de la Unión Europea. Este periodo se denomina período de
exclusividad.
La directiva 2004/27/CE establece un periodo de exclusividad de datos único, armonizando en toda
Europa este período y fijándolo en diez años. Es muy importante tener en cuenta estos aspectos de
la normativa, dado que puede haber un medicamento genérico autorizado pero que infrinja una o
varias patentes.
Los medicamentos genéricos deberán designarse con la denominación oficial española de principio
activo y, en su defecto, con la denominación común internacional, o bien, si ésta no existiese, con la
denominación común usual o científica de dicha sustancia, acompañada, en su caso, del nombre o
marca del titular o fabricante. Se identificarán, además, con las siglas EFG (Equivalente Farmacéutico
Genérico). (Artículo 14 de la Ley 29/2006 de 26 de julio, de garantías y uso racional de los
medicamentos y productos sanitarios)
Autorización y registro de medicamentos genéricos
El procedimiento de autorización tiene por objeto comprobar que el medicamento:
Alcanza los requisitos de calidad establecidos.
Es seguro, no produciendo en condiciones normales de utilización efectos tóxicos o
indeseables desproporcionados al beneficio que procura.
Es eficaz en las indicaciones terapéuticas aprobadas.
Está correctamente identificado y va acompañado de la información precisa para su
utilización.
A los efectos del registro, se entiende una solicitud genérica como la que se refiere a una
solicitud esencialmente similar a otra innovadora. Además se sustenta con un dossier
abreviado y se presenta a registro después de transcurrido un periodo de tiempo
determinado desde la autorización de la innovación en la Unión Europea.

16
4. La prescripción y la dispensación de medicamentos
Según se desprende de la Declaración de la Comisión Central de Deontología sobre la Libertad de
Prescripción, el médico está, por principio, “obligado a prescribir a sus pacientes aquellas terapias o
medicamentos que mejor vayan a solventar la enfermedad, siguiendo criterios de racionalidad y buen
sentido económico”, No obstante, las decisiones tomadas por el médico a la hora de prescribir se ven
sometidas a distintas influencias.
Las variables que influyen en la toma de decisiones de prescripción son de tipo científico, económico,
deontológico y social, eminentemente, y están relacionadas entre sí.
Dentro de las variables económicas destaca la implicación para el Sistema Nacional de Salud y, en último
término, para el Gobierno, en cuanto al control del gasto sanitario se refiere. No obstante, es importante
señalar que no siempre la mejor terapia es la de menor coste.
Desde el punto de vista científico, es fundamental que la terapia establecida sea eficaz, y que conlleve el
menor número de riesgos o efectos adversos posibles para el paciente. Pero además, actualmente los
médicos cuentan con una extensísima cantidad de información en cada área de actuación, lo que puede
dificultar la elección de un medicamento u otro.
En cuanto a los aspectos sociales, hay que destacar la importancia de que el paciente esté informado de la
terapia que va a seguir, los objetivos de la misma, tiempo estimado de duración del tratamiento e incluso
alternativas en caso de que el medicamento prescrito no resulte eficaz.
Este aspecto tiene especial relevancia en cuanto que el paciente está cada vez más informado y
concienciado sobre su patología y sobre posibles terapias; probablemente, incluso haya consultado con
otros médicos antes de comenzar un tratamiento (sobre todo en el caso de patologías graves).
En cuanto a la dispensación de los medicamentos en las oficinas de farmacia, de acuerdo con la Ley
29/2006 de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, los farmacéuticos,
como responsables de la dispensación de medicamentos a los ciudadanos, velarán por el cumplimiento de
las pautas establecidas por el médico responsable del paciente en la prescripción.
La dispensación de medicamentos de acuerdo con el sistema de precios de referencia se realizará conforme
a los siguientes criterios:
1. Cuando se prescriba un medicamento que forme parte de un conjunto y que tenga un precio igual o
inferior al de referencia no procederá la sustitución.
2. Cuando se prescriba un medicamento que forme parte de un conjunto y que tenga un precio superior al
de referencia, el farmacéutico deberá sustituirlo por el de menor precio e idéntica composición cualitativa y

17
cuantitativa en principios activo, forma farmacéutica, vía de administración, dosificación y presentación que
el medicamento prescrito y, en caso de igualdad de precio, por el medicamento genérico.
Dado que los genéricos vienen a establecer la cuantía máxima que el estado financie por un
medicamento con cuenta a los fondos públicos, esto mismo ha propiciado que los medicamentos de
marca establezcan sus precios al de referencia, contribuyendo de está forma también a la
sostenibilidad del sistema sanitario.
3. Cuando la prescripción del médico se efectúe por principio activo sometido a precio de referencia,
el farmacéutico dispensará el medicamento de menor precio y, en caso de igualdad de precio, un
genérico, si lo hubiere.
Para comprobar que el medicamento genérico dispensado es equivalente a una prescripción
anterior, los consumidores pueden comprobar en la etiqueta el nombre genérico de su principio
activo.
Algunas voces apuntan que las diferencias de color, tamaño o forma de los medicamentos genéricos
entre ellos y con respecto al medicamento de referencia pueden contribuir a la disminución de la
adherencia terapéutica de los pacientes. Este aspecto cobra una especial relevancia en el caso de
pacientes ancianos, enfermos crónicos y/o polimedicados.
En este sentido, la Organización Mundial de la Salud (OMS) afirma en su publicación Drugs for the
elderly que es necesario que los médicos y farmacéuticos se aseguren de que los pacientes sigan
recibiendo comprimidos del mismo tamaño, forma y color que los administrados previamente. Este
documento afirma que los pacientes sufrirán también confusión si el nombre del medicamento que
reciben pasa de ser del registrado al genérico.
Además, hay que tener en cuenta que en la prescripción por principio activo, al paciente no se le
dispensa solamente este principio activo, sino un medicamento que contiene otros componentes,
llamados excipientes, que generalmente son inocuos, pero que en ocasiones pueden modificar la
actividad farmacológica del principio activo llegando a producir efectos adversos.
Por tanto, la prescripción de un medicamento por parte del médico ha de ser responsable y
contemplar, además de criterios estrictamente económicos, aspectos tan importantes como el
cumplimiento terapéutico del paciente o la respuesta del enfermo ante el tratamiento actual entre
otras consideraciones.
18
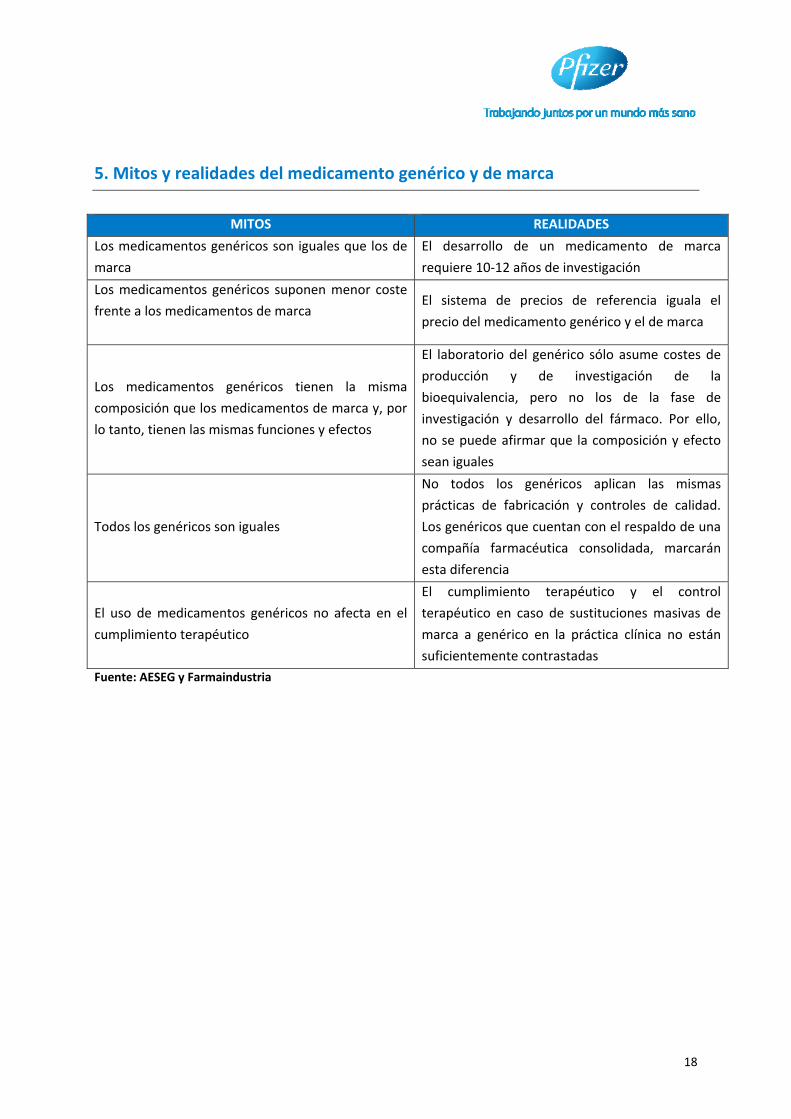
5. Mitos y realidades del medicamento genérico y de marca
MITOS REALIDADES
Los medicamentos genéricos son iguales que los de
marca
El desarrollo de un medicamento de marca
requiere 10‐12 años de investigación
Los medicamentos genéricos suponen menor coste
frente a los medicamentos de marca
El sistema de precios de referencia iguala el
precio del medicamento genérico y el de marca
Los medicamentos genéricos tienen la misma
composición que los medicamentos de marca y, por
lo tanto, tienen las mismas funciones y efectos
El laboratorio del genérico sólo asume costes de
producción y de investigación de la
bioequivalencia, pero no los de la fase de
investigación y desarrollo del fármaco. Por ello,
no se puede afirmar que la composición y efecto
sean iguales
Todos los genéricos son iguales
No todos los genéricos aplican las mismas
prácticas de fabricación y controles de calidad.
Los genéricos que cuentan con el respaldo de una
compañía farmacéutica consolidada, marcarán
esta diferencia
El uso de medicamentos genéricos no afecta en el
cumplimiento terapéutico
El cumplimiento terapéutico y el control
terapéutico en caso de sustituciones masivas de
marca a genérico en la práctica clínica no están
suficientemente contrastadas
Fuente: AESEG y Farmaindustria

19
6. Glosario de Términos
EFG
Equivalente Farmacéutico Genérico. Los medicamentos genéricos deberán designarse con una
denominación oficial española de principio activo (DOE) y, en su defecto, con la denominación
común internacional (DCI) o bien, si ésta no existiese, con la denominación común usual o científica
de dicha sustancia, acompañada, en su caso, del nombre o marca del titular o fabricante. Se
identificarán, además, con las siglas EFG.
Medicamento genérico de marca
Son medicamentos de la misma calidad que el medicamento de marca, cuya patente ya venció, pero
que no han presentado aún los estudios que permitan a la Secretaría de Salud catalogarlos como
Genéricos Intercambiables. Estos medicamentos se comercializan con un nombre comercial.
Bioequivalencia
La bioequivalencia demuestra la intercambiabilidad entre el medicamento genérico y el
medicamento innovador desde el punto de vista de la calidad, seguridad y eficacia. Los estudios de
bioequivalencia se realizan para demostrar que el medicamento genérico es equivalente e
intercambiable con el medicamento innovador en términos de eficacia terapéutica.
Conjunto
Es la totalidad de las presentaciones de medicamentos financiadas que tengan el mismo principio
activo, y entre las que exista, al menos, un medicamento genérico, quedando excluidas las “formas
farmacéuticas innovadoras”.
El precio de referencia para cada conjunto se calcula haciendo la media aritmética de los tres costes
tratamiento/día menores las presentaciones de medicamentos de dicho conjunto, para cada vía de
administración según la dosis diaria definida. Las tres presentaciones farmacéuticas seleccionadas
para fijar el precio de referencia deberán pertenecer a tres grupos empresariales diferentes.
Sustitución por el farmacéutico
Cuando se prescriba un medicamento que forme parte de un conjunto y que tenga un precio
superior al de referencia, el farmacéutico deberá sustituirlo por el de menor precio e idéntica
composición cualitativa y cuantitativa en principios activos, forma farmacéutica, vía de
administración, dosificación y presentación que el medicamento prescrito y, en caso de igualdad de
precio, por el medicamento genérico. En el caso de que la prescripción se efectúe por principio activo
sometido a precio de referencia, el farmacéutico dispensará el medicamento de menor precio y, en
caso de igualdad de precio, el medicamento genérico.

20
7. Bibliografía
AESEG. ‘Impacto económico de un cambio con carácter retroactivo en la regulación de patentes
farmacéuticas’.
Disponible en:
http://www.aeseg.es/Estudo‐Impacto_economico.pdf
AESEG. ‘La investigación del sector farmacéutico europeo. El caso de España’.
Disponible en: http://www.aeseg.es/restringido2/informeDEF2.pdf
Asociación Española de Derecho Farmacéutico (ASEDEF). Edición 2008.
Comisión Central de Deontología de la Organización Médica Colegial Española.
Disponible en: http://www.unav.es/cdb/ccdomc00b.html
Díez M.V., Errecalde M.F. ‘Aclaraciones al concepto de genérico’. Información Terapéutica del
Sistema Nacional de Salud. Vol.22‐Nº3‐1998.
Disponible en: http://www.msc.es/biblioPublic/publicaciones/docs/generico.pdf
Doctor Jose Luis Alloza: Artículo sobre Farmacología social publicado en la revista Drug Information
Journal.
Disponible en: http://www.essentialdrugs.org/efarmacos/archive/200411/msg00025.php
El ciclo vital del medicamento y Metodologías de la investigación: Dr. Federico Tobar.
Disponible en: http://www.federicotobar.com.ar/metodologia.php
Kristof Roox, Julia Pike, Andrew Brown, Stefan Becker et al. ‘Barreras de entrada a los medicamentos
genéricos en la Unión Europea: Un análisis de las debilidades del actual sistema europeo de patentes
y su impacto en el acceso en el mercado de los medicamentos genéricos’. Aeseg. 2008.
“La industria y el mercado del genérico en España”, Aeseg, 14 octubre 2009.
Disponible en: http://www.aeseg.es/industria_gen_Esp.pdf
Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios
Álvarez Cedrón , Benéitez Palomeque, Cabrera García, Mingote Muñiz, Tejero Tojo. Marketing
Farmacéutico.
Organización Médica Colegial Española, Facultad de Medicina, Ciencias y Farmacia.
Disponible en: http://www.unav.es/cdb/ccdomc03a.html
Organización Mundial del Comercio. Los ADPIC y las patentes de productos farmacéuticos.
Disponible en: http://www.wto.org/spanish/tratop_s/trips_s/factsheet_pharm00_s.htm

21
Portal de la Unión Europea. Síntesis de la legislación de la UE: Productos farmacéuticos y cosméticos.
Disponible en:
http://europa.eu/legislation_summaries/internal_market/single_market_for_goods/pharmaceutical
_and_cosmetic_products/l21168_es.htm
Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización,
registro y condiciones de dispensación de los medicamentos de uso humano fabricados
industrialmente.