modelagem molecular aplicada ao planejamento … · de compostos bioativos: uma introdução....
TRANSCRIPT

MODELAGEM MOLECULAR APLICADA AO
PLANEJAMENTO DE COMPOSTOS
BIOATIVOS: PRINCÍPIOS E APLICAÇÕES
Dr. Carlos Mauricio R. Sant’Anna
GPAQ, Dequim, UFRuralRJ
LASSBio - UFRRJ

Bibliografia Indicada
C.M.R. Sant'Anna, 2014
2
Para quem está começando hoje:
SANT'ANNA, C. M. R., Métodos de Modelagem Molecular para Estudo e Planejamento
de Compostos Bioativos: Uma Introdução. Revista Virtual de Química, 1, 49 - 57, 2009.
SANT'ANNA, C. M. R., Glossário de Termos Usados no Planejamento de Fármacos
(Recomendações da IUPAC para 1997). Química Nova. , v.25, p.505 - 512, 2002.
Para quem quer saber mais:
Molecular Modelling: Principles and Applications ( A. Leach)
Introduction to Computational Chemistry (F. Jensen)
Computational Chemistry using the PC (D. W. Rogers)
Computational Chemistry (D. C. Young)
Introdução à Química Computacional (L. Alcácer)
Molecular Modeling: Basic Principles and Applications (H. D. Holtje)
Programas: http://www.click2drug.org/

O Que é Modelagem Molecular?
C.M.R. Sant'Anna, 2014
3
A Modelagem Molecular faz uso de diferentes teorias e programas de
computador para criar modelos da estrutura molecular e prever suas
propriedades (como sua energia).
Em Química Medicinal, a modelagem molecular é usada para se fazer o
Planejamento de Fármacos Auxiliado por Computadores (CADD, do
inglês Computer Assisted Drug Design).
As estratégias empregadas para o CADD são divididas em dois grandes
grupos, o Planejamento de Fármacos Baseado na Estrutura dos Ligantes
(LBDD, do inglês Ligand Based Drug Design) e o Planejamento de
Fármacos Baseado na Estrutura do Receptor (SBDD, do inglês Structure
Based Drug Design.
Muitas técnicas são aplicadas na construção dos modelos na Modelagem
Molecular e as mais importantes serão discutidas durante o Curso.

O Que a Modelagem Molecular pode
de fato fazer em Química Medicinal?
C.M.R. Sant'Anna, 2014
4
Isso? Ou isso?

Estratégias Gerais da Descoberta de fármacos
C.M.R. Sant'Anna, 2014
5
Protótipo
Otimização estrutural
“Screening”
Bibliotecas de produtos naturais (de plantas, animais marinhos,
processos fermentativos) ou de compostos sintéticos ou novas
estruturas obtidas com o uso de química combinatória
Protótipo
Otimização estrutural
Planejamento racional
Conhecimento parcial ou completo do mecanismo de
interação entre o composto bioativo (ligante)
e o alvo biológico
Modelagem
Molecular
Modelagem
Molecular
Modelagem
Molecular
Modelagem
Molecular

“Screening”: Estratégias reais X virtuais
C.M.R. Sant'Anna, 2014
6
Estágio de geração de protótipos
HTS
Protótipo (IC50 ~ 10 mM)
Planejamento da modificação
IC50 ~ 1-10 nM
“Screening” virtual
Biblioteca de estruturas reais
Biblioteca de estruturas virtuais
e/ou reais
Estágio de otimização
Modificação estrutural
experimental
computacional

Modelagem Molecular no Planejamento Racional de Compostos Bioativos
C.M.R. Sant'Anna, 2014
7
Modelagem Molecular: Geração de estruturas moleculares para...
Relações Qualitativas e Quantitativas
entre a Estrutura e a
Atividade
“Docking”
Planejamento “de novo”
Estudos de mecanismos de reação, etc...

Visualização em Modelagem Molecular: Gráficos Moleculares
C.M.R. Sant'Anna, 2014
8

Gráficos Moleculares
C.M.R. Sant'Anna, 2014
9
porina zirconoceno

Gráficos Moleculares
C.M.R. Sant'Anna, 2014
10
Complexo Proteína-RNA

Visualização de Modelos: Proteínas
C.M.R. Sant'Anna, 2014
11
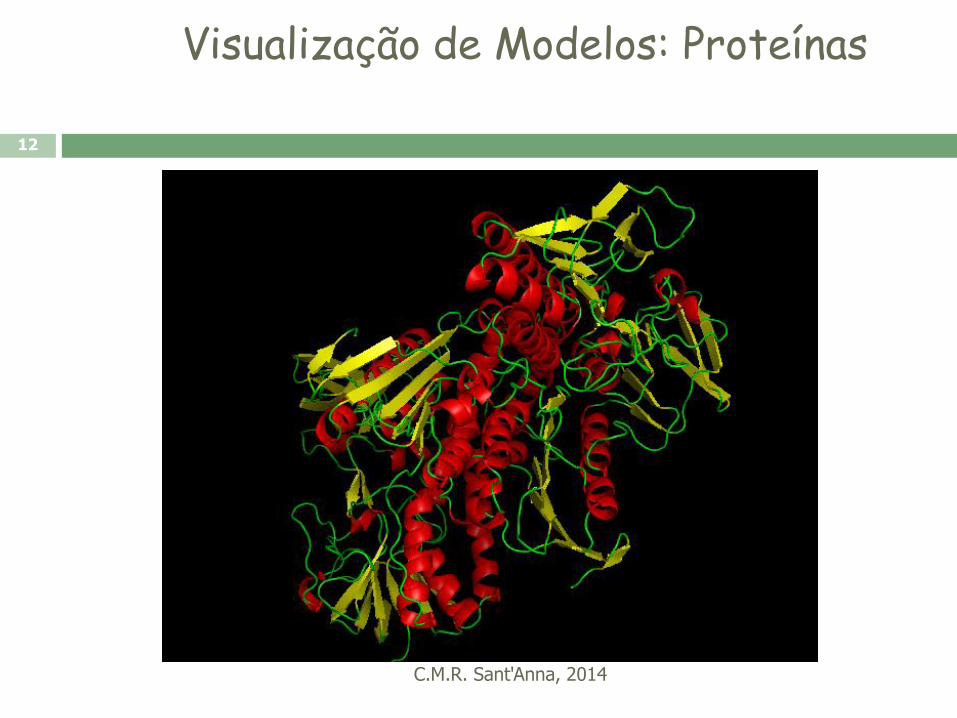
Visualização de Modelos: Proteínas
C.M.R. Sant'Anna, 2014
12

Visualização de Modelos: Superfícies
C.M.R. Sant'Anna, 2014
13

Visualização de Modelos: Superfícies
C.M.R. Sant'Anna, 2014
14
Potencial eletrostático mapeado sobre a superfície molecular (Bujnicki et al. BMC Bioinformatics 2001 2:2
doi:10.1186/1471-2105-2-2)

Definição de Geometria Molecular
C.M.R. Sant'Anna, 2014
15
Coordenadas cartesianas
Formato PDB

Definição de Geometria Molecular
C.M.R. Sant'Anna, 2014
16
http://www.rcsb.org/pdb/home/home.do

Modelagem Molecular:
Classificações dos Métodos
C.M.R. Sant'Anna, 2014
17
Quanto à Dinâmica do Método de Cálculo da Energia
•Métodos de otimização estrutural direta (busca do ponto estacionário mais próximo)
•Métodos de otimização múltipla (métodos estatísticos, análise sistemática...)
•Métodos dinâmicos: geração de trajetórias (dinâmica molecular...)
Quanto ao Tipo do Método de Cálculo da Energia
•Métodos de Campo de Força: mecânica molecular
•Métodos Quânticos: Semi-empíricos, ab initio HF, MP, DFT
•Métodos de Simulação: métodos de energia livre, métodos de perturbação termodinâmica, métodos de integração termodinâmica

Métodos Empíricos
C.M.R. Sant'Anna, 2014
18
ligações
Realidade
Ótimo

Métodos Empíricos
C.M.R. Sant'Anna, 2014
19
ângulos

Métodos Empíricos
C.M.R. Sant'Anna, 2014
20
Quanto maior o valor de k, maior a energia necessária para deformar uma distância ou um ângulo de ligação. Potenciais rasos são obtidos com valores de k entre 0,1 e 1.
Exemplos de parâmetros:
C sp3 – C sp3
– r0 = 1,526 Å
– kb = 310,0 kcal/(mol Å2)
− θ0 = 109,50°
– Kθ = 40,0 kcal/(mol rad2)
C sp2 – O sp2
– r0 = 1.250 Å
– kb = 656,0 kcal/(mol Å2)

Métodos Empíricos
C.M.R. Sant'Anna, 2014
21
diedros

Métodos Empíricos
C.M.R. Sant'Anna, 2014
22

Métodos Empíricos
C.M.R. Sant'Anna, 2014
23
termo de van der Waals termo eletrostático
regime de atração de van der Waals
regime de repulsão
energia ótima

l
lpot knkkllkE
22
0
2
0 cos1
0
'
000'
'
00' ''''
l
l
l l
ll llkkllllk
i ij ij
ij
ij
ij
r
B
r
Akk
612' '
'00' '''cos
i l
l
qil
li
k lk
lkkl
kl
lk
i ij ijq
ji
DDr
q
rDrD
coscoscos3cos23
Campo de força padrão para moléculas orgânicas
Campo de força compostos organometálicos
2
0llkE ll 2
0exp1 llDEl
2
0 kE nkE cos1
Classe I ou harmônicos:
Sem termos cruzados
Ex.: CHARMM, AMBER
Classe II:
Termos cruzados e séries
Ex.: MM4
C.M.R. Sant'Anna, 2014
24

Métodos Empíricos: Exemplos de Campos de Força
C.M.R. Sant'Anna, 2014
25
Campo de Força Sistemas Moleculares
EFF Alcanos MM3 Geral
CVFF Geral Tripos Geral MMFF Geral
Dreiding Geral Amber Proteínas, ácidos nucleicos, carboidratos CHARMM Proteínas
GROMOS Proteínas, ácidos nucleicos, carboidratos MOMEC Compostos de coordenação

A Superfície de Energia Potencial
C.M.R. Sant'Anna, 2014
26

Busca Direta de Mínimos de Energia
C.M.R. Sant'Anna, 2014
27
Ei
Emin
Ei
Emin

Análise Conformacional Sistemática:
Pesquisa de Grade
C.M.R. Sant'Anna, 2014
28
O número de conformações necessárias para varrer o espaço conformacional cresce rapidamente: quando o número de ligações com rotação livre é 6, por exemplo, o número de confôrmeros a ser avaliado é 46656 ( igual a 600).
O número de confôrmeros equivale a (3600/)n, onde é o incremento
usado no processo de varredura e n é o número de ligações avaliadas.

Dinâmica Molecular
C.M.R. Sant'Anna, 2014
29
O movimento molecular está presente em praticamente todos os processos químicos, como reações, interações intermoleculares, mudanças de fase, solubilizações ou simplesmente em vibrações moleculares (espectroscopia de IV).
As etapas em um processo de minimização de energia são dirigidas simplesmente para a busca do mínimo mais próximo. As etapas na dinâmica representam todas as mudanças nas posições atômicas no tempo (as velocidades).
Termodinâmica Quais estados são possíveis?
Cinética Como e com que velocidade os estados se interconvertem?

Dinâmica Molecular
C.M.R. Sant'Anna, 2014
30
Dê aos átomos posições iniciais
Calcule forças
Mova átomos:
Avance no tempo:
Repita tantas vezes quanto for necessário
defina Dt curto.
MD: Algoritmo simplificado

Dinâmica Molecular
C.M.R. Sant'Anna, 2014
31
Campo de força usado em DM deve ser apropriado para representar forças intermoleculares e vibrações longe do equilíbrio. Entre os campos mais usados estão o GROMOS e o OPLS.
Dinâmica de aquecimento
Dinâmica de equilibração
Dinâmica de produção
Análise das trajetórias

Método de Monte Carlo
C.M.R. Sant'Anna, 2014
32
http://www.cs.otago.ac.nz/cosc453/student_tutorials/monte_carlo.pdf
MC compara energias. Forças não são calculadas.
Gere a estrutura inicial R. Calcule V(R).
Modifique a estrutura para R’. Calcule V’(R’).
Repita para N etapas.
Algoritmo de Monte Carlo
No equilíbrio a T:

Métodos Quânticos: Introdução
C.M.R. Sant'Anna, 2014
33
E. Schrödinger
A mecânica quântica mostra que as partículas têm comportamento
ondulatório. Se é assim, como determinar sua energia?
é a chamada função de onda e
E é a energia.

Métodos Quânticos para Moléculas
C.M.R. Sant'Anna, 2014
34
•Nos métodos
quânticos, a energia da
estrutura molecular é
definida colocando-se
os núcleos em regiões
nas quais os elétrons
estão associados a
orbitais, regiões do
espaço definidas pela
resolução da equação
de Schrödinger.
Representação de orbitais moleculares
para a molécula de H2
HE
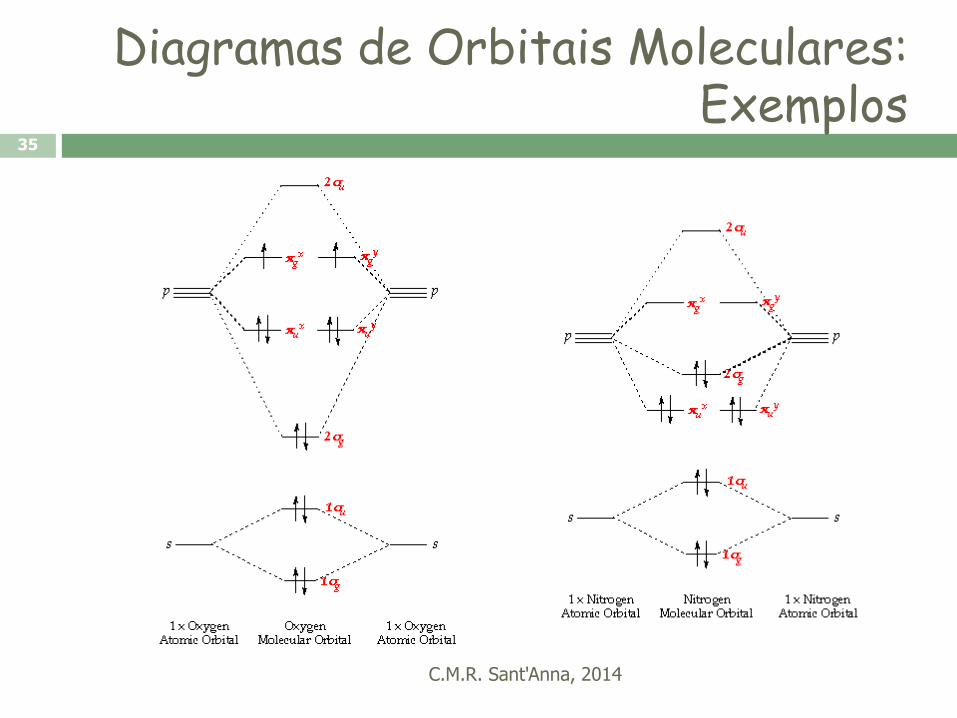
Diagramas de Orbitais Moleculares: Exemplos
C.M.R. Sant'Anna, 2014
35

Métodos Quânticos: Soluções Práticas
C.M.R. Sant'Anna, 2014
36
Métodos de orbital molecular Ab initio
Hartree-Fock, Perturbação Moeller-Plesset (MPn), Interação de configuração (CI), etc.
Teoria do funcional de densidade (DFT)
Métodos de orbital molecular semi-empíricos
Hückel, PPP, CNDO, INDO, MNDO, AM1
PM3, PM6, RM1…

Métodos Quânticos: Aproximações
C.M.R. Sant'Anna, 2014
37
•A resolução de (1.2) é chamada
dinâmica quântica e depende
de uma função de energia (a SEP) definida apenas pelas posições dos núcleos, E(R).
Métodos ab initio resolvem (1.3) diretamente.
Ex.: HF, MPx
Métodos semi-empíricos fazem aproximações que são compen-sadas com funções ajustadas empiricamente.
Aproximação de Born-Oppenheimer
•A descrição de moléculas é feita pela resolução da equação de Schrödinger na qual a função de onda é uma função das coordenadas dos núcleos (R) e dos elétrons (r):
H(R,r) = E(R,r) (1.1)
Como os núcleos são milhares de vezes mais pesados do que os elétrons, os movimentos nuclear e eletrônico podem ser considerados separadamente (Born-Oppenheimer):
H(R) = E (R) (1.2)
H(r;R) = E (r;R) (1.3)

Métodos Quânticos: Orbitais Atômicos
C.M.R. Sant'Anna, 2014
38

Conjuntos de Base
C.M.R. Sant'Anna, 2014
39
1. Mínimo
•O mais simples deste conjunto de bases ainda usado é o STO-3G (acrônimo para Orbitais do Tipo Slater simulados pela soma de 3 Gaussianas).
•O conjunto de bases mínimo tem apenas o número de bases necessário para acomodar os elétrons dos átomos e reter a simetria esférica. Assim, o conjunto STO-3G tem apenas uma função para o H (1s), cinco para o Li até o Ne (1s, 2s, 2px, 2py e 2pz) e 9 para os elementos do segundo período Na até o Ar (1s, 2s, 2px, 2py, 2pz, 3s, 3px, 3py e 3pz).
•É o conjunto de bases usados pelos hamiltonianos do programa Mopac.
•Problemas: estabilidade de anéis pequenos é superestimada

Conjuntos de Base
C.M.R. Sant'Anna, 2014
40
2. Conjunto de Bases de Valência Dividida Os orbitais são divididos em pelo menos duas partes: uma interna e compacta e outra externa, mais difusa. Os coeficientes das duas partes podem ser variados independentemente na combinação LCAO-MO. Assim, o tamanho do orbital pode ser variado dentro dos limites das funções interna e externa. Exemplos: 3-21G, 6-31G, 6-311G (notação de Pople)...
6-311G
Uma contração de 6 funções Gaussianas usadas para representar os orbitais de camada interna
Uma contração de 3 funções Gaussianas usadas para representar a parte interna dos orbitais de valência
1 função Gaussiana usada para representar a parte externa dos orbitais de valência
1 função Gaussiana usada para representar a parte intermediária dos orbitais de valência

Conjuntos de Base
C.M.R. Sant'Anna, 2014
41
3. Conjunto de Bases com Funções de Polarização •Melhoria nas funções de base foi obtida pela adição de funções do tipo d para todos os átomos “pesados” (diferentes de H). Por ser mais conveniente computacionalmente, são usadas 6 funções do tipo d (x2, y2, z2, xy, xz, yz), que equivalem a 5 funções d e a uma função s. •Essas funções permitem que o centro de um orbital se desloque para além do plano que contém o núcleo, levando à distribuições eletrônicas não-esféricas (anisotrópicas).
•A presença do conjunto de polarização é indicada por um asterisco. Ex.: 3-21G* •Funções de polarização também podem ser usadas para os átomos de H. Nesse caso, são usadas funções do tipo p. A presenças destas funções é indicada por um segundo asterisco. Ex.: 6-31G** •Um asterisco entre parênteses indica que as funções de polarização foram adicionadas apenas para os elementos do segundo período. •Uma maneira alternativa de indicar a presença de funções de polarização é colocar (d) ou (d,p) após o G.

Conjuntos de Base
C.M.R. Sant'Anna, 2014
42
4. Conjunto de Bases com Funções Difusas •Sistemas com elétrons mais fracamente ligados aos núcleos, como ânions ou mesmo átomos com elétrons não-ligantes, são melhor descritos pela inclusão de funções difusas. •Essas funções são conjuntos de orbitais s e p muito difusos, com expoentes entre 0,1 e 0,01. •A presença das funções difusas nos átomos “pesados” é indicada por um sinal “+” antes da letra G. Ex.: 6-31+G •A presença das funções difusas também nos átomos de H é indicada por um segundo sinal “+”. Ex.: 6-311++G •Processos que envolvem mudança no número pares de elétrons solitários, como reações de protonação, são melhor descritas com o uso das funções difusas. •A adição de funções difusas nos átomos de H tem, em geral, pouco efeito nos resultados, a não ser nos casos em que íons hidretos estejam envolvidos.

Método HF e Sistemas com Elétrons Desemparelhados
C.M.R. Sant'Anna, 2014
43
No método HF, os elétrons devem ocupar os orbitais em pares, ou seja, a mesma função de onda do orbital é usada para os elétrons nos 2 estados de spin ( e ); isso é chamado de método de Hartree-Fock restrito (RHF). Para sistemas com elétrons desemparelhados, como os radicais livres, uma alternativa é usar conjuntos separados de orbitais para os elétrons e ; isso é chamado de método de Hartree-Fock irrestrito (UHF). Por exemplo:
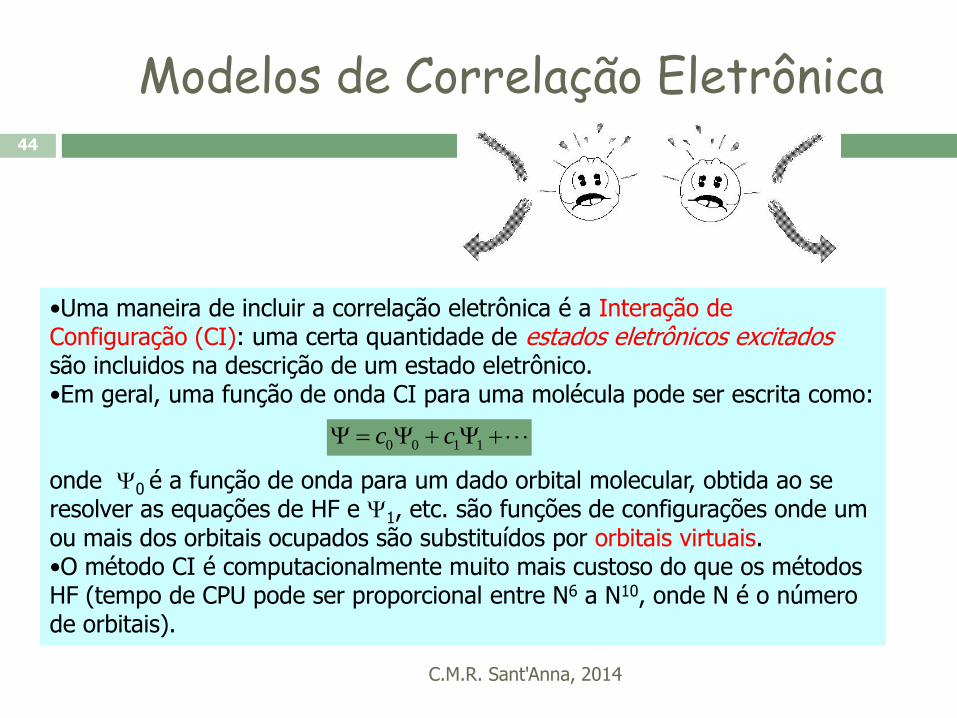
Modelos de Correlação Eletrônica
C.M.R. Sant'Anna, 2014
44
•Uma maneira de incluir a correlação eletrônica é a Interação de Configuração (CI): uma certa quantidade de estados eletrônicos excitados são incluidos na descrição de um estado eletrônico. •Em geral, uma função de onda CI para uma molécula pode ser escrita como:
onde 0 é a função de onda para um dado orbital molecular, obtida ao se resolver as equações de HF e 1, etc. são funções de configurações onde um ou mais dos orbitais ocupados são substituídos por orbitais virtuais. •O método CI é computacionalmente muito mais custoso do que os métodos HF (tempo de CPU pode ser proporcional entre N6 a N10, onde N é o número de orbitais).
1100 cc

Modelos de Correlação Eletrônica
C.M.R. Sant'Anna, 2014
45
•No Modelo de Möller-Plesset (MP), as funções de onda e as energias podem ser gradualmente melhoradas substituindo-se o hamiltoniano “verdadeiro” Hl por:
onde l é um parâmetro que varia entre 0 e 1, V é a “perturbação” e H0 é o
hamiltoniano de ordem zero. Assim, para as funções de onda e energias: onde Ei
(1) é a correção de primeira ordem da energia, Ei(2) é a de segunda
ordem, etc.: De acordo com a ordem da correção introduzida, temos os vários modelos de Möller-Plesset: MP2, MP3, MP4…
VHH ˆˆˆ0 ll
)2(2)1()0(
)2(2)1()0(
iiii
iiii
EEEE ll
ll

Métodos Semi-empíricos
C.M.R. Sant'Anna, 2014
46
Nos métodos semi-empíricos, a energia total da molécula é representada como a soma da energia eletrônica com a energia de repulsão entre os cernes. Um conjunto de treinamento de moléculas é selecionado, escolhido para cobrir o maior número de situações de ligação química possível. É aplicado um procedimento de mínimos quadrados não-linear com os valores dos parâmetros ajustáveis sendo as variáveis as propriedades do conjunto de treinamento como constantes a serem reproduzidas. Estas propriedades incluem calores de formação, variáveis geométricas, momentos de dipolo, energias de ionização, etc. Dependendo da escolha do conjunto de treinamento, do número de parâmetros ajustáveis e do modo de ajuste às propriedades experimentais, temos diferentes hamiltonianos; por exemplo: AM1 (M. J. S. Dewar et al.) PM3, PM5, PM6 e PM7 (J. J. P. Stewart) RM1 (G. B. Rocha et al.)

Métodos Semi-empíricos
C.M.R. Sant'Anna, 2014
47
RM1: a Reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I Gerd Bruno Rocha, Ricardo Oliveira Freire, Alfredo Mayall Simas, and James J. P. Stewart. Journal of Computational Chemistry 27(10), 1101-1111, 2006
1480 moléculas contendo H, C, N, O, P, S, F, Cl, Br e I.

C.M.R. Sant'Anna, 2014
48
J. J. P Stewart, J. Mol. Model. 2007, 13, 1173-1213. Parâmetros semi-empíricos para 70 elementos da tabela periódica.

C.M.R. Sant'Anna, 2014
49
J. J. P Stewart, J. Mol. Model. 2007, 13, 1173-1213.

Desempenho Relativo dos Métodos de Cálculo
de Modelagem Molecular
C.M.R. Sant'Anna, 2014
50
Objetivo
Mecânica
Molecular
Semi-empírico Ab Initio
HF MP2
Geometria
(grupo principal)
aceit.bom Bom Bom Bom
Geometria
(metais de transição)
- Bom Pobre ?
Geometria
(ests. de transição)
- Bom Bom Bom
Conformação Bom Pobre aceit.bom Bom
Termoquímica
(não isodêsmica)
- Pobre aceit.bom Bom
Termoquímica
(isodêsmica)
- aceitável Bom Bom
Custo muito baixo baixo alto muito alto

Um Modelo Quântico Alternativo: Teoria do Funcional de Densidade
C.M.R. Sant'Anna, 2014
51
Número de artigos com a palavra DFT no título a partir de 1980
Structure and Bonding, Vol. 150 Putz, Mihai V.; Mingos, D Michael P (Eds.) 2013, XII, 236 p.

Teoria do Funcional de Densidade
C.M.R. Sant'Anna, 2014
52
•Na Teoria do Funcional de Densidade (DFT), todas as propriedades do sistema são uma função de uma função (um funcional), a densidade eletrônica r (1o teorema de Hohenberg e Kohn). •Como obter a densidade eletrônica? A densidade eletrônica é expressa como uma combinação linear de funções de base matematicamente semelhantes aos orbitais HF, chamados orbitais de Khon-Sham. •Há várias formas de funcionais. Alguns foram desenvolvidos a partir da mecânica quântica fundamental (ab initio) e outros que foram parametrizados para melhor reproduzir dados experimentais (semi-empíricos).
r ii rr iii EK )(

Principais Funcionais de Densidade
C.M.R. Sant'Anna, 2014
53
Acrônimo Nome Tipo
X X alfa Troca
HFS Hartree, Fock e Slater HF com troca LDA
VWN Vosko, Wilks e Nusair LDA
BLYP Funcional de correlação de Becke com de troca de Lee, Yang, Parr
Gradiente-corrigido
B3LYP Termo 3 de Becke com de troca de Lee, Yang, Parr Híbrido
PW91 Perdue e Wang 1991 Gradiente-corrigido
G96 Gill 1996 Troca
P86 Perdew 1986 Gradiente-corrigido
B96 Becke 1996 Gradiente-corrigido
B3P86 Troca de Becke, correlação de Perdew Híbrido
B3PW91 Troca de Becke, correlação de Perdew e Wang Híbrido
Os funcionais mais simples tem apenas um termo de troca eletrônica (ex.: X); outros incluem termos de correlação eletrônica, como nos métodos da aproximação de densidade local (LDA) e de densidade de spin local (LSDA). Métodos mais complexos (e mais precisos) usam a densidade e o seu gradiente (gradiente-corrigido) e alguns misturam partes de diferentes funcionais (híbridos). Os mais completos têm uma qualidade equivalente a um cálculo MP2. Alguns exemplos:

Aplicações: Propriedades Eletrônicas
C.M.R. Sant'Anna, 2014
54
Cátion t-butil:
mapas de
potencial eletrostático
Formaldeído: LUMO Formaldeído: HOMO

Aplicações: Estudo da Reatividade Química
C.M.R. Sant'Anna, 2014
55
Representação do LUMO dos substratos acetaldeído e
triflúor-acetaldeído (método quântico semi-empírico AM1)
Representação superfície de potencial eletrostático dos substratos acetaldeído e
triflúor-acetaldeído (método quântico semi-empírico AM1)

Aplicações:
Estudo da Reatividade Química
C.M.R. Sant'Anna, 2014
56
SN2: representação do LUMO do substrato CH3Br obtido pelo método
quântico semi-empírico AM1 SN1: representação do LUMO do
intermediário cátion t-butil obtido pelo método quântico semi-empírico AM1

Qual o Melhor Modelo?
C.M.R. Sant'Anna, 2014
57
Os principais parâmetros para a escolha de um modelo teórico são o desempenho e o custo.
O sucesso de qualquer modelo depende primeiro da sua capacidade de reproduzir dados experimentais.
Mas um modelo precisa também ser prático, o que depende do tamanho do sistema em estudo e das facilidades computacionais disponíveis.
Um modelo prático provavelmente não vai ser o melhor tratamento possível para um determinado problema, mas aquele que produzirá as informações necessárias com uma precisão e um custo
razoáveis.

Métodos Híbridos
C.M.R. Sant'Anna, 2014
58
•Nos chamados métodos híbridos (ou combinados) QM/MM (do inglês Quantum Mechanics/Molecular Mechanics), os métodos clássico e quântico são usados ao mesmo tempo, mas em “camadas” diferentes deste sistema. •O cálculo quântico é aplicado em uma parte pequena da estrutura (chamada parte quântica), enquanto o restante da estrutura é tratado com um método clássico (a parte clássica). Vantagens: Como a região tratada quanticamente é pequena, os resultados podem ser alcançados com rapidez, mesmo que sejam usados conjuntos de base extensos. Problemas: definição da fronteira quântico/clássico (uso de hidrogênios para completar ligações cortadas? orbitais localizados congelados?) Exs.: integrated MO ‡MM (IMOMM); our own n-layered integrated MO and MM method (ONIOM); integrated MO MO method (IMOMO).
Warshel & Levitt, Journal of Molecular Biology, 103, 1976, 227–249

Modelagem de Proteínas
C.M.R. Sant'Anna, 2014
59
1. Modelagem Comparativa (por Homologia)
Esse método aproxima a estrutura tridimensional para sequências primárias de proteínas, usando como molde estruturas 3D conhecidas. É necessário ~30% de identidade entre as sequências da proteína a ser modelada (proteína-alvo) e do molde. As estruturas 3D são conhecidas através de dados de cristalografia de raios-X ou, mais raramente, dados de RMN. Muitas estão depositadas em bancos de acesso público, como o Protein Data Bank (PDB). Ex.: Swiss-Model, Modeller.
2. Modelagem ab initio (de novo)
Baseia-se em princípios físicos ao invés do uso de moldes. Há métodos que tentam mimetizar o processo de enovelamento de proteínas e outros que aplicam métodos estocásticos para pesquisar possíveis soluções estruturais. São métodos computacionalmente muito exigentes e foram aplicados com sucesso em proteínas pequenas. Ex.: I-TASSER, Rosetta@home

Modelagem de Proteínas
C.M.R. Sant'Anna, 2014
60
Etapas da Modelagem Comparativa (por Homologia) 1. Busca do molde a partir da sequência da proteína-alvo usando um algoritmo de similaridade. Ex.: Blast no Swiss-Model. (semelhança e resolução) 2. Alinhamento das sequências . Ex.: ClustalW e T-Coffee (problemas: inserções e deleções, loops). 3. O ‘backbone’ do alvo é construido sobre o do molde (C e ângulos e ). 4. Ajuste das cadeias laterais (bibliotecas de confôrmeros ajudam); eliminação de colisões (clashes) 5. Refinamento da estrutura por simples minimização de energia com mecânica molecular ou por dinâmica molecular.
inserção
deleções
Modelo estrutural
Alinhamento das sequências
Molde Estrutura 3D desconhecida
loop

Modelagem de Proteínas
C.M.R. Sant'Anna, 2014
61
>tr|Q4CQE2|Q4CQE2_TRYCR Ribose 5-phosphate isomerase, putative
OS=Trypanosoma cruzi GN=Tc00.1047053509199.24 PE=4 SV=1
MTRRVAIGTDHPAFAIHENLILYVKEAGDEFVPVYCGPKTAESVDYPDFASRVAEMVARK
EVEFGVLACGSGIGMSIAANKVPGVRAALCHDHYTAAMSRIHNDANIVCVGERTTGVEVI
REIIITFLQTPFSGEERHVRRIEKIRAIEASHAGKKGVQ
>[Template]|1nn4D|2.2|Structural Genomics RpiBAlsB
Length = 159
[Display Alignment in DeepView]
Score = 114 bits (286), Expect = 2e-26, Method: Composition-based stats.
Identities = 60/147 (40%), Positives = 93/147 (63%), Gaps = 3/147 (2%)
Query: 3 RRVAIGTDHPAFAIHENLILYVKEAGDEFVPVYCGPKTAESVDYPDFASRVAEMVARKEV 62
+++A G DH F + ++ ++ E G E + G ++E DYP +AS+VA VA EV
Sbjct: 14 KKIAFGCDHVGFILKHEIVAHLVERGVEVIDK--GTWSSERTDYPHYASQVALAVAGGEV 71
Query: 63 EFGVLACGSGIGMSIAANKVPGVRAALCHDHYTAAMSRIHNDANIVCVGERTTGVEVIRE 122
+ G+L CG+G+G+SIAANK G+RA +C + Y+A +SR HND N++ G R G+E+ +
Sbjct: 72 DGGILICGTGVGISIAANKFAGIRAVVCSEPYSAQLSRQHNDTNVLAFGSRVVGLELAKM 131
Query: 123 IIITFLQTPFSGEERHVRRIEKIRAIE 149
I+ +L + G RH +R+E I AIE
Sbjct: 132 IVDAWLGAQYEG-GRHQQRVEAITAIE 157
Arquivo FASTA (Swiss-Prot)
Alinhamento
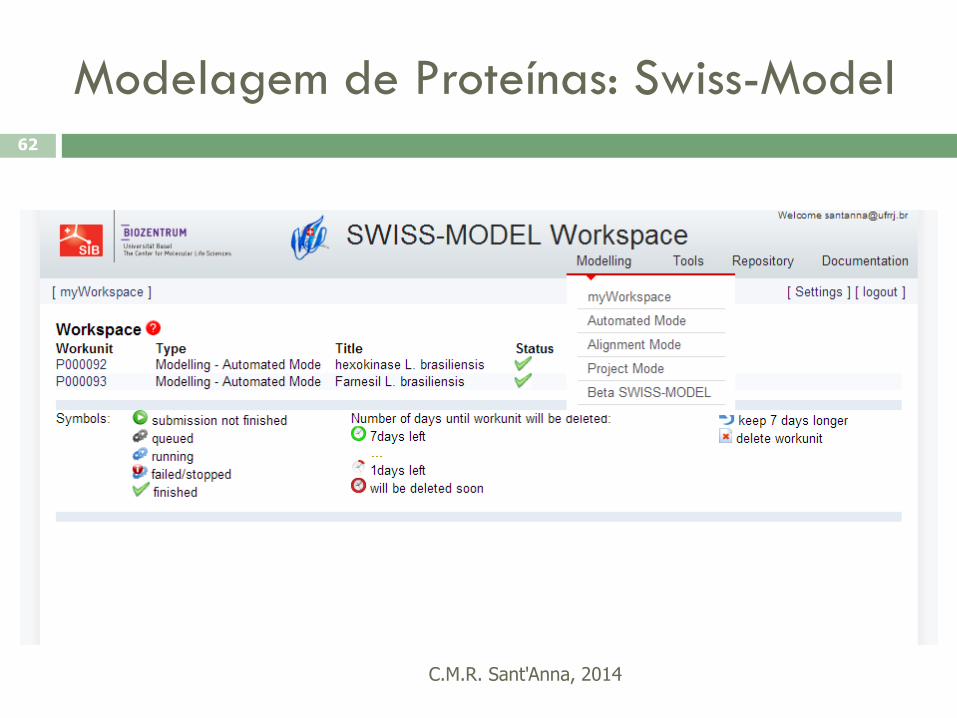
Modelagem de Proteínas: Swiss-Model
C.M.R. Sant'Anna, 2014
62

Modelagem de Proteínas: Swiss-Model
C.M.R. Sant'Anna, 2014
63

Modelagem de Proteínas
C.M.R. Sant'Anna, 2014
64
Molde Modelo
Modelo gerado a partir do servidor Swiss-Model (modo automático); otimização estrutural com campo de força GROMOS Ferramentas avaliam a qualidade do
modelo. Ex.: gráfico de Ramachandran (Swiss-Model), o servidor ANOLEA…

Modelagem de Proteínas
C.M.R. Sant'Anna, 2014
65
Há uma variedade de ferramentas desenvolvidas para avaliar a qualidade dos modelos, por exemplo: 1. Procheck (http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/): avalia a
qualidade estereoquímica da estrutura; 2. Molprobity (http://molprobity.biochem.duke.edu/): verifica os contatos entre
átomos do modelo e a presença de conformações não usuais da cadeia principal e das cadeias laterais dos aminoácidos;
3. ProSA (https://prosa.services.came.sbg.ac.at/prosa.php): usa potenciais de campos de força construídos a partir de uma análise estatística de estruturas 3D conhecidas para avaliar a qualidade do modelo.

Ancoramento Molecular (‘Docking’)
C.M.R. Sant'Anna, 2014
66
O ancoramento molecular é uma metodologia que tem como objetivo prever o modo de interação (orientação e conformação) do complexo formado entre uma molécula pequena (ligante) e um receptor, geralmente uma proteína. Há diferentes níveis de ancoramento: 1. Ancoramento rígido: apenas o ligante tem liberdade conformacional; 2. Ancoramento semi-rígido: cadeias laterais selecionadas e/ou moléculas de água são mantidas livres; 3. Ancoramento flexível: todas as espécie presentes no complexo são mantidas livres durante o procedimento.

Geração de poses: Estratégias
C.M.R. Sant'Anna, 2014
67
próxima geração
Criar população de cromossomos
Determinar o ajuste de cada
indivíduo
várias gerações
Selecionar próxima geração
Mostrar resultados
Realizar reprodução por cruzamento
Realizar mutação
AG: uma “máquina” para fazer evolução
Estratégias: Algoritmo genético Monte Carlo Construção incremental Métodos híbridos
Programa Estratégia
AutoDock AG/MC
FlexX CI
Glide Híbrido
GOLD AG
Evolução...
Charles Darwin 1809-1882

Algoritmo Genético mais Detalhado
C.M.R. Sant'Anna, 2014
68
1. Um conjunto de operadores de reprodução (“crossover”, mutação
etc) é escolhido. A cada operador é estipulado um “peso”.
2. Uma população inicial é randomicamente criada, e
a aptidão (“fitness”) de seus membros é determinada.
3. Um operador é escolhido utilizando seleções por
“roletas de cassino”, baseadas no peso de operadores.
4. Os “pais” requeridos pelos operadores são escolhidos utilizando
seleções por “roletas”, baseadas em aptidões escaladas.
5. O operador é aplicado e cromossomos “filhos”
são produzidos. Suas aptidões são avaliadas.
6. Se ainda não estão presentes na população, os cromossomos filhos
substituem os membros de menor aptidão (os piores ancorados).
7. Se N operações foram feitas, o cálculo é parado;
caso contrário, volta-se ao passo 3.
Um algoritmo genético (AG) é uma técnica de busca utilizada para achar soluções aproximadas em problemas de otimização e busca. Algoritmos genéticos são uma classe particular de algoritmos evolutivos que usam técnicas inspiradas pela biologia evolutiva como hereditariedade, mutação, seleção natural e recombinação.

Ancoramento Molecular (‘Docking’)
C.M.R. Sant'Anna, 2014
69
Representações Básicas da Proteína e do Ligante: a. Atômica: calcula-se a interação átomo-átomo; é
usada com funções de energia potencial. b. Representação em grade: o uso de grades é
feito para o cálculo de energia, acumulando informações sobre a contribuição na energia da proteína nos pontos em uma grade.
c. Superficial: utilizada para ancoramento entre duas proteínas (ancoramento proteína/proteína), através do alinhamento de pontos de cada superfície.
a
c
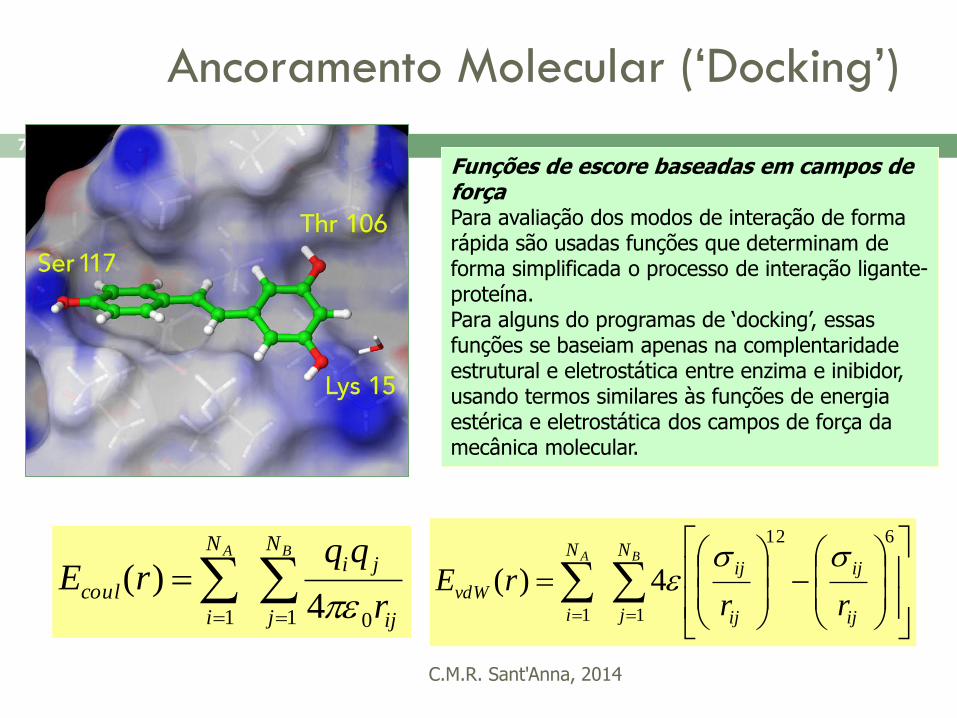
Ancoramento Molecular (‘Docking’)
C.M.R. Sant'Anna, 2014
70
Funções de escore baseadas em campos de força Para avaliação dos modos de interação de forma rápida são usadas funções que determinam de forma simplificada o processo de interação ligante-proteína. Para alguns do programas de ‘docking’, essas funções se baseiam apenas na complentaridade estrutural e eletrostática entre enzima e inibidor, usando termos similares às funções de energia estérica e eletrostática dos campos de força da mecânica molecular.
BA N
j ij
jiN
i
coulr
qqrE
1 01 4)(
BA N
j ij
ij
ij
ijN
i
vdWrr
rE1
612
1
4)(

Funções empíricas: Essas funções de escore reproduzem dados experimentais como
uma soma de várias funções de energia parametrizadas.
C.M.R. Sant'Anna, 2014
71
+ H3N
OH
Ângulos diedros com pequenas
barreiras em solução
Classificação das “poses”
DGint = SDGi
DGint = DGrot+ DGhb+ DGion+ DGaro + DGlipo + ...
H3N
OH O
NH
O
O
DGio
DGlipo
DGrot
DGaro
DGhb
Ângulos diedros
variados no docking
flexível O
NH
O
O
Sítio de interaçãona proteína
Ligante Receptor
“Poses” do complexo
ligante-receptor
Valores usados diretamente ou para gerar outros valores comparativos (escores)

Ancoramento Molecular (‘Docking’)
C.M.R. Sant'Anna, 2014
72
Algoritmos de Busca Aplicados a Ligantes Flexíveis a. Métodos sistemáticos: fragmentos moleculares são atracados no sítio, sendo
então ligados covalentemente um ao outro (construção incremental), podendo ser ainda divididos em regiões rígidas (“núcleos”) e flexíveis (cadeias laterais).
b. Métodos estocásticos: atuam promovendo mudanças aleatórias nas “poses” dos ligantes, de acordo com funções de probabilidade, implementadas principalmente por duas metodologias: “Monte Carlo” e “algoritmo genético” .
c. Métodos simulacionais: simula-se o movimento real do ligante no interior da proteína; a técnica de dinâmica molecular é a mais utilizada neste tipo de método.
Programa Estratégia
AutoDock AG/MC
FlexX CI
Glide Híbrido
GOLD AG

Ancoramento Molecular (‘Docking’)
C.M.R. Sant'Anna, 2014
73
Sobreposição das estruturas do FAD co-cristalizado (no padrão CPK de cores) e atracado (em ciano), após ancoramento com o programa GOLD 3.0 na enzima tripanotiona redutase. Átomos de H da proteína omitidos (Del Cistia, C.N., Tese de Doutorado, 2010)
“Redocking” é o processo no qual um ligante retirado da estrutura de um complexo é ancorado na forma ajustada do receptor. É um processo usado para verificar quais parâmetros para o processo de ancoramento são adequados e capazes de recuperar a estrutura do complexo e suas interações.
O “cross-docking” é feito com um conjunto de complexos, onde todos os ligantes são ancorados em todos os receptores. É útil para determinar a capacidade de separação dos ligantes pelo programa frente a um receptor.

Ajuste Induzido: Cadeias Laterais
C.M.R. Sant'Anna, 2014
74
74
Neuraminidase da influenza A complexada com o inibidor ácido 2-desoxi-2,3-desidro-N-acetilneuramínico (PDB 1NNB) e com o ácido 5-acetilamino-4-amino-6-(fenetilpropilcarbamoil)-5,6-diidro-4H-pirano-2-carboxílico (PDB 1BJI).
Glu276 Glu276
Arg224 Arg224

Ajuste Induzido: Cadeia Polipeptídica
C.M.R. Sant'Anna, 2014
75
75
Agonista 17β-estradiol (PDB 1ERE) no domínio de interação do ligante no receptor do estrogênio causa uma conformação diferente da hélice 12, quando comparada com conformação da hélice 12 com o ligante seletivo do receptor do estrogênio raloxifeno (PDB 1ERR).
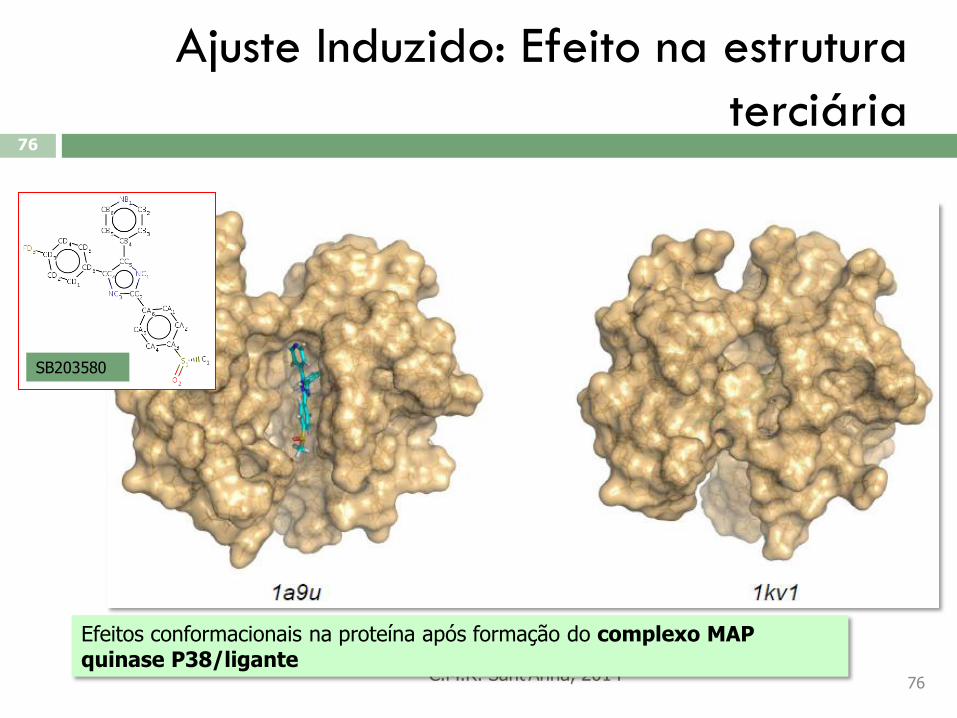
Ajuste Induzido: Efeito na estrutura
terciária
C.M.R. Sant'Anna, 2014
76
76
Efeitos conformacionais na proteína após formação do complexo MAP quinase P38/ligante
SB203580

Ajuste induzido: Soluções no “docking”
C.M.R. Sant'Anna, 2012
77
“Ensemble docking”
Sobreposição estrutural
Otimização estrutural
Huang e Zhou, Proteins, 2007, 6,399-421.
10 proteínas + 87 ligantes
“docking” comum
“ensemble docking”
75 % de acerto 93 % de acerto

Ajuste induzido: Soluções no “docking”
C.M.R. Sant'Anna, 2012
78
Escore da estrutura SB203580
SB203580

Triagem Virtual Baseada no “Docking”
C.M.R. Sant'Anna, 2014
79
Preparação da estrutura alvo
“docking”
Avaliação das poses (“scoring”)
Classificação das poses (“ranking”)
Pós-processamento
Preparação da biblioteca de compostos
Triagem virtual baseada na estrutura
(SBVS – Structure Based Virtual Screening)
Triagem virtual baseada no
“docking” (DBVS – Docking Based
Virtual Screening)

Seleção de Compostos para o DBVS
C.M.R. Sant'Anna, 2014
80
Bibliotecas: Próprias Públicas Comerciais
Comp. 2
Comp. 3
Comp. 2
Comp. 1 Enriquecimento de Bibliotecas (filtragem):
Filtros físico-químicos e/ou farmacológicos (ex.: solubilidade, Lipinski, ADME/Tox) Filtros de similaridade c/ compostos ativos conhecidos Filtros baseados no alvo

Aplicações no Estudo de Compostos
Bioativos
C.M.R. Sant'Anna, 2014
81

C.M.R. Sant'Anna, 2014 82
www.sciencemag.org SCIENCE 303
19 MARCH 2004
“Is there really a case where a drug that’s
on the market was designed by a
computer? When asked this, I invoke the
professorial mantra (“All questions are
good questions.”), while sensing that the
desired answer is “no”. Then, the
inquisitor could go back to the lab with the
reassurance that his or her choice to
avoid learning about computational
chemistry remains wise.”
“...the phrasing of the question suggests
misunderstanding and oversimplification
of the drug discovery process.”
“There is not going to be a “voila`” moment
at the computer terminal. Instead, there is
systematic use of wide ranging
computational tools to facilitate and
enhance the drug discovery process.”

Estratégias no Planejamento Racional
C.M.R. Sant'Anna, 2014
83
Métodos Dependentes do
Receptor/Enzima
Métodos Independentes do Receptor/Enzima
Quantidade
crescente de
informação
disponível
Modelagem por homologia
+
Ligantes conhecidos
(estrutura e
dados biológicos)
+
Seqüência do
receptor conhecida
Modelo do
Receptor
QSAR
(CoMFA, etc)
Interações
ligante-receptor
(docking, etc)
Novos
ligantes
+
Estrutura 3D do
receptor conhecida
Métodos Baseados no Mecanismo
Mecanismo de
ação conhecido
Dinâmica das
interações, reações

Exemplo 1: Inibidores da PDE4
C.M.R. Sant'Anna, 2014
84
O
OCH3
NOH
NN
N
O
Cl
N N
N CH3
O
Cl
NEtO
OH3C
H
N
N CH3
O
Cl
SEtO
OH3C
N
N
N CH3
OCH3
CH3
NCN
N CH3
O
NO
H3C
N
N
O
NO
H3C
CH3 N
N CH3
O
SH2N
OH3C
N
N CH3
O
S
NC
H3C
N
N
N
OCH3
CH3
NC
CH3
1 (rolipram) 2 (syntex 3) 3 4
5 6 7 8
9 10
Oliveira, Caffarena, Sant`Anna, Dardenne, Barreiro, 2006

Interação Ligante-Receptor: Aspectos Teóricos
C.M.R. Sant'Anna, 2014
85
+
[E(aq)] [I(aq)] [E•I(aq)]
bindbindbind STHG DDD
dabind KRTKRTG lnln D
Ki = Kd = [E(aq)] [I(aq)]
[E•I(aq)] _________
Trocas de energias envolvidas no processo de “binding”, como as energias de interação ligante-sítio e ligante-solvente.
Formas de distribuição das energias, classificadas como entropia translacional, rotacional, conformacional e vibracional.

Atividade Biológica: Construção de Modelos
Teóricos
C.M.R. Sant'Anna, 2014
86
Objetivo: proposição de um modelo teórico capaz de determinar a atividade biológica.
iKRTG lnD
543
2
21ln cNcHccGcKRT LRbindsolvi DD
ligsacompbind HHHH DDDDNLR: termo entrópico associado à perda de rotação em ligações após associação do ligante ao sítio ativo
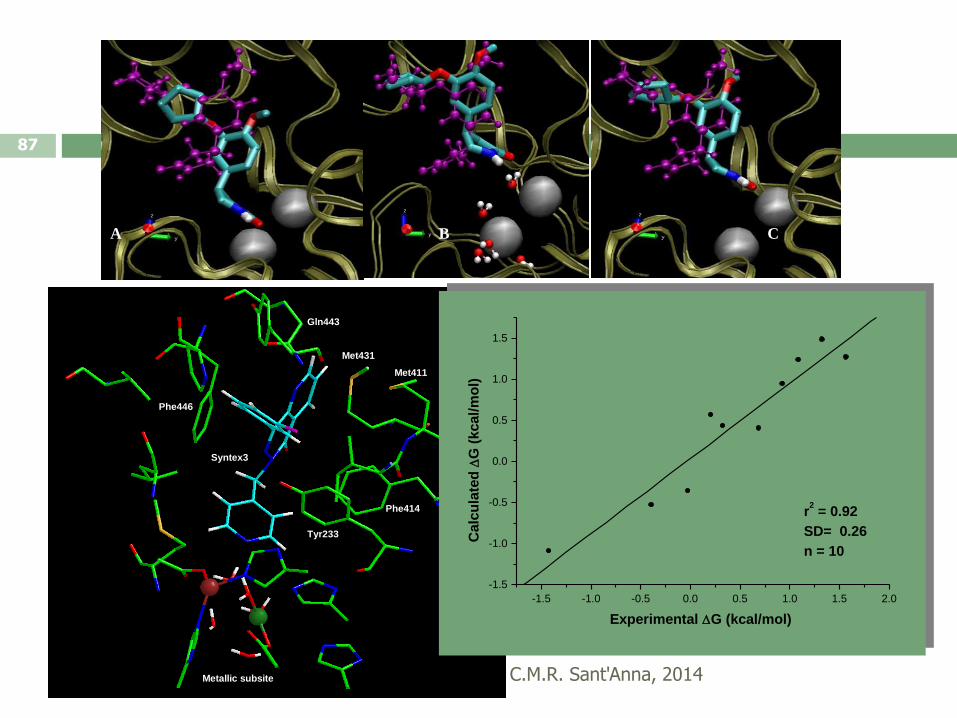
C.M.R. Sant'Anna, 2014
87
A B C
Syntex3
Phe446
Gln443
Met431
Met411
Phe414
Tyr233
Metallic subsite
-1.5 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
r2 = 0.92
SD= 0.26
n = 10
Calc
ula
ted
DG
(kcal/
mo
l)
Experimental DG (kcal/mol)

Exemplo 2: Modelo para a Inibição para a
Tripanotiona Redutase
C.M.R. Sant'Anna, 2014
88
Composto R1 R2 R3 R4
1 Cl (CH2)3NMe2 - H
2 COCH2CH3 (CH2)3NMe2 - H
3 COOH (CH2)3NMe2 - H
4 CONH2 (CH2)3NMe2 - H
5 H (CH2)2C(NH2)=NH - H
6 H (CH2)3N(CH2CH2)2NCH3 - H
7 H CO(CH2)2COOH - H
8 H COCH2Br - H
9 CF3 (CH2)2C(NH2)=NH - H
10 Cl (CH2)3NMe2 - COCH3
11 Cl (CH2)3NMe2 =O H
12 COCH3 COCH3 - H
13 COCH3 COCH3 - COCH3
14 Cl (CH2)3N+(Me)2CH2C6H5 - H
15 Cl (CH2)3N+(Me)2CH2C6H33,4-Cl - H
16 Cl (CH2)3N+(Me)2CH2C6H44-NO2 - H
17 Cl (CH2)3N+(Me)2CH2C6H33,4-CH3 - H
18 Cl (CH2)3N+(Me)2CH2C6H33,4-OCH3 - H
19 Cl (CH2)3N+(Me)2CH2C6F5 - H
20 Cl CH3 - H
TR de Trypanosoma cruzi*
*Bond et al., Structure Fold Des., 1999, 7, 81.
N
S
R1
R2
R4
R3
Khan et al., J. Med. Chem., 2000, 43, 3148.
Chan et al., J. Med. Chem., 1998, 41, 148.
“Docking” de fenotiazinas

C.M.R. Sant'Anna, 2014 89
“Docking” (GOLD) Melhores ajustes
(Goldscore)
Geração de descritores para os complexos (SILVER)
0 2 4 6 8
1
2
3
4
5
6
7
8
ln IC
50 c
alc
ln IC50
exp
Linear fit
R=0,904
SD=0,871
N=20
432150ln cDcDcDcIC AHHYDVDW Seleção de descritores e geração de correlação
DVDW é a contribuição em interações de van de Waals ligante/proteína, DHYD
o número de átomos hidrofóbicos do ligante acessíveis ao solvente, DAH o
número de aceptores de ligação hidrogênio do ligante e os coeficientes c1-c4
são obtidos ajustando-se a equação aos valores experimentais de ln (IC50)
Comp . l n (IC50) exp l n (IC50) calc
1 2,08 2,24
2 2,3 1,15
3 - 0,25 1,14
4 2,88 3,43
5 1,69 1,51
6 3,33 3,16
7 4,34 5,52
8 5,16 5,13
9 7,94 6,53
10 3,89 5,43
11 4,28 4,50
12 7,3 8,09
13 5,53 5,96
14 5,73 6,71
15 3,57 4,17
16 5,88 4,45
17 8,27 6,54
18 6,13 5,98
19 1,36 1,10
20 6,9 5,55
21 4,7 5,28
22 2,48 2,15
23 6,08 7,35

Exemplo 3: Mesoiônicos, Novos
Inibidores da TR
C.M.R. Sant'Anna, 2012
90
Espécie Inibição (%) da TR pelo
derivado p-NO2 (1 M)
L.
amazonensis
76
L. infantum 70
L. brasiliensis 69,5
T. cruzi 83
Inibição não competitiva
L. infantum
+
S
N N
NHR
R = H, m-OCH3, p-OCH
3, p-NO
2
Cl-
Rodrigues, R., et al., Bioorg. Med. Chem., 20, 1760 (2012)

C.M.R. Sant'Anna, 2012 91
+
S
N N
NHR
R = H, m-OCH3, p-OCH
3, p-NO
2
Cl-
TR L. infantum (2W0H)
TR L. amazonensis (modelo)
TR L. brasiliensis (modelo)
TR T. cruzi (1BZL)
Docking
Sítio da tripanotiona
Sítio do NADPH
Sítio do FAD
Rodrigues, R., et al., Bioorg. Med. Chem., 20, 1760 (2012)

C.M.R. Sant'Anna, 2012 92
+
S
N N
NHR
R = H, m-OCH3, p-OCH
3, p-NO
2
Cl-
Sítio do NADPH
Rodrigues, R., et al., Bioorg. Med. Chem., 20, 1760 (2012)

C.M.R. Sant'Anna, 2012 93
+
S
N N
NH
N+
O-
OCl-
Rodrigues, R., et al., Bioorg. Med. Chem., 20, 1760 (2012)

Exemplo 4: DBVS aplicado aos receptores A2A
C.M.R. Sant'Anna, 2013
94
Docking
545.000 compostos
Banco de dados inicial
Filtragem: 1.Propriedades adequadadas ao CNS; 2. Ausencia de furano ou xantina
Sequência primária do receptor A2A
Modelagem comparativa
MOE Modeller
Glyde 372 compostos
230 compostos
Disponibilidade comercial
Binding no receptor A2A
20 “hits” IC50 < 55 M
Otimização estrutural baseada nos resultados do “docking” de “hits” selecionados Langmead et al., J. Med. Chem. 2012, 55,
1904−1909
Adenosina
4 subtipos de receptores: A1, A2A, A2B, A3

C.M.R. Sant'Anna, 2013
95
Top 10 Hits
Otimização estrutural do composto 1
Langmead et al., J. Med. Chem. 2012, 55, 1904−1909
“Docking” do composto 15 (Biophysical Mapping)
Ki = 3,5 nM
Ki = 1,6 nM Ki = 0,1 nM

Referências
C.M.R. Sant'Anna, 2014
96 Harel, M., Kryger, G., Rosenberry, T. L., Mallender, W. D., Lewis, T., Fletcher,
R. J., Guss, J. M., Silman, I., Sussman, J. L. (2000) Protein Science 9, 1063.
Liverton, N. J.; Butcher, J. W.; Claiborne, C. F. et al. (1999) J Med Chem. 42, 2180.
Mutero, A., Pralavorio, M., Bride, J., Fournier, D. (1994) Prod. Natl. Acad. Sci. USA 91 5922.
Ott, K. et al., (1996) J. Mol Biol., 263, 329.
Sant’Anna, C. M. R. e Santos, A. C. S. (2002) J. Mol. Struc. Theochem, 585, 59.
Sant’Anna, C. M. R., Souza, V. P., Andrade, D. S. (2002) Int. J. Quantum Chem. 87, 311.
Sant’Anna, C. M. R., Viana A. S. Nascimento Junior, N. M. (2006) Bioorg. Chem. 34, 77.
Santos, V. M. R., Costa, J. B. N., Moya, G. E. M., Sant’Anna, C. M. R., (2004) Phosph. Sulf. Sil. Rel. Elem. 179, 173.
Santos, V. M. R., Costa, J. B. N., Moya, G. E. M., Cortes, W. S. e Sant’Anna, C. M. R., (2007) Bioorg. Chem. 35.

Referências
C.M.R. Sant'Anna, 2014
97
Silva, G. M. S., Sant’Anna, C. M. R., Barreiro, E. J. (2004) Bioorg Med Chem Lett. 2004, 12, 3159.
Stewart, J. J. P. J. Comp. Chem. 10 (1989) 209.
Walsh, S. B., Dolden, T. A., Moores, G. D., Kristensen, M., Lewis, T., Devonshire, A. L., Williamson, M. S. Biochem. J. 359 (2001) 175.
Stelmach, J. E.; Liu, L.; Patel, S. B.; et al. Bioorg Med Chem Lett. 2003, 13, 277.

Agradecimentos
Dr. João Batista N. Costa (PQ/UFRuralRJ)
Dr. Marco Edilson F. Lima
(PQ/UFRuralRJ)
Dra. Ana Cristina S. Santos
(PQ/UFRuralRJ)
Dr. Gonzalo E. Moya (PQ/PUFRuralRJ)
Dr. Wellington S. Cortes (PQ/UFRuralRJ
Dr. Eliezer Barreiro (PQ/LASSBio/UFRJ)
Dr. Anivaldo X. da Silva (PQ/UFRuralRJ)
Dra. Catarina del Cistia (PQ/UFRuralRJ)
Dra. Viviane Martins R. Santos (UFOP)
Dr. Arthur E. Kümmerle (PQ/UFRuralRJ
MSc. José Geraldo Rocha Jr. (PG/UFRRJ)
MSc. Saraí S. Souza (PG/UFRRJ)
MSc. Daniel Rosa da Silva (PG/UFRRJ)
MSc. Letícia Zampirolli (PG/UFRRJ)
MSc. Vinicius T. Gonçalves (PG/UFRRJ)
Sheisi Fonseca L. Silva (PG/UFRJ)
Larissa Henriques E. Castro (IC/UFRRJ)
Marcus Vinicius H. Silva (IC/UFRRJ)
Fernanda Guedes (PG/IQ e LASSBio/UFRJ)
98
C.M.R. Sant'Anna, UFRRJ, 2012
CNPq, Faperj,CAPES, INCT-INOFAR

C.M.R. Sant'Anna, UFRRJ, 2012 99