microestrutura e propriedades mecÂnicas de … · 3 instituto militar de engenharia maria cecilia...
TRANSCRIPT

3
INSTITUTO MILITAR DE ENGENHARIA
MARIA CECILIA CORRÊA DE SÁ E BENEVIDES DE MORAES
MICROESTRUTURA E PROPRIEDADES MECÂNICAS DE
COMPÓSITOS ALUMINA-ZIRCÔNIA PARA PRÓTESES DENTÁRIAS
Proposta de Tese de Doutorado apresentada ao Curso de Doutorado em Ciência dos Materiais do Instituto Militar de Engenharia.
Orientadores: Carlos Nelson Elias – D. Sc. Jamil Duailibi Filho – D. Sc.
Aprovada em 20 de setembro de 2004 pela seguinte Banca Examinadora:
_________________________________________________________________
Carlos Nelson Elias – D. Sc. - IME - Presidente
_________________________________________________________________
Jamil Duailibi Filho – D. Sc. - INT
_________________________________________________________________
Luis Henrique Leme Louro – Ph. D. - IME
_______________________________________________________________
Maria Cecília de Souza Nóbrega – D. Sc. - UFRJ
_________________________________________________________________
Cecília Zavaglia – D. Sc. – Unicamp
_______________________________________________________________
José Carlos da Rocha– D. Sc. - INT
_______________________________________________________________
Antônio José do Nascimento Dias – D. Sc. – INT
Rio de Janeiro
2004

4
AGRADECIMENTOS
Ao Prof. Carlos Nelson Elias, orientador desta tese, pelo interesse, otimismo e
empenho na realização deste trabalho, assim como pela amizade, compreensão e
incentivo constantes.
Ao Dr. Jamil Duailibi Filho, pelo apoio fundamental para a realização desta tese.
À todos os professores, funcionários e técnicos do Departamento de Engenharia
Mecânica e de Materiais DE-4 do IME e do Departamento de Química, que direta ou
indiretamente contribuíram para a execução deste trabalho.
Um agradecimento especial aos engenheiros Carlos Roberto e Carlos Gomes, e
aos técnicos: Leonardo, Joel, Cristóvão, Miguel, Anderson pela grande colaboração.
À minha família, pelo grande apoio e compreensão durante este período.
Ao IME, pela oportunidade de realização do Doutorado.
A DPCM-INT (Divisão de Processamento e Caracterização de Materiais), ao
DEMP (Divisão de Ensaios de Materiais e Produtos), aos funcionários e técnicos dos
laboratórios (Laboratório de Processamento de Pós, LATEP; Laboratório de
Metalografia e Dureza, LAMED; e Laboratório de Análises Inorgânicas, LABAI), pelo
acolhimento e pela grande colaboração na realização de ensaios experimentais
desta tese.
Aos colegas e bolsistas do INT, Leandra, Antonio José, Cássio, Brant, Carla,
Marcelo, André pela grande colaboração.
Ao Departamento de Ciência dos Materiais e Metalurgia da PUC-RJ (Lab.de
Difração de Raios-x (LDRX) pela realização de medidas experimentais para este
trabalho.
Aos amigos e colegas da pós-graduação do IME, Itamar, Vivian, Elaine, Marcos,
Mário Henrique, Solange, Sheila, Leonardo, Felipe, Souza Lima, Cardoso, Ricardo,
pela cooperação e convívio durante estes três anos e meio.
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-CAPES, pelo
apoio financeiro.
À Conexão Sistemas de Prótese pelo fornecimento de material específico
necessário ao desenvolvimento da tese.

5
SUMÁRIO
LISTA DE ILUSTRAÇÕES .................................................................................. 9
LISTA DE TABELAS ......................................................................................... 15
LISTA DE ABREVIATURAS E SÍMBOLOS ....................................................... 17
LISTA DE SIGLAS ............................................................................................ 18
1 INTRODUÇÃO ..................................................................................... 21
2 OBJETIVO ............................................................................................ 25
3 REVISÃO BIBLIOGRÁFICA................................................................ 26
3.1 Cerâmicas como biomateriais .............................................................. 26
3.1.1 Biocerâmicas........................................................................................ 26
3.1.2 Alumina como biomaterial .................................................................... 28
3.1.3 Alumina em próteses ortopédicas ........................................................ 30
3.1.4 Alumina em implantes dentários .......................................................... 32
3.1.5 Processos industriais para obtenção de pós de zircônia...................... 35
3.1.6 Zircônia em prótese ortopédicas .......................................................... 36
3.1.7 Radioatividade de biomateriais cerâmicos ........................................... 38
3.1.8 Pilares cerâmicos ................................................................................. 38
3.2 Cerâmicas a base de alumina .............................................................. 44
3.2.1 Estrutura cristalina da alumina ............................................................. 44
3.3 Cerâmicas a base de zircônia .............................................................. 45
3.3.1 Estrutura cristalina da zircônia ............................................................. 45
3.3.1.1 Zircônia monoclínica............................................................................. 46
3.3.1.2 Zircônia tetragonal................................................................................ 47
3.3.1.3 Zircônia cúbica ..................................................................................... 48
3.3.2 Equilíbrio da fase binária ...................................................................... 49
3.3.2.1 Zircônia pura ........................................................................................ 49
3.3.2.2 Zircônia parcialmente estabilizada (PSZ) ............................................. 51

6
3.3.2.3 Zircônia tetragonal policristalina (TZP) ................................................. 51
3.3.2.4 Zircônia totalmente estabilizada ........................................................... 51
3.3.3 Transformação martensítica (T- M) ...................................................... 52
3.3.4 Mecanismos de tenacificação .............................................................. 53
3.3.4.1 Aumento de tenacidade induzido por tensão........................................54
3.3.4.2 Microtrincamento.................................................................................. 56
3.3.4.3 Tensões superficiais compressivas...................................................... 57
3.4 Compósitos a base de alumina e zircônia ............................................ 57
3.5 Processamento .................................................................................... 59
3.5.1 Aditivos de processamento .................................................................. 59
3.5.2 Moagem ............................................................................................... 60
3.5.3 Dispersão dos pós................................................................................ 62
3.5.4 Conformação por prensagem uniaxial.................................................. 69
3.5.4.1 Característica dos pós para prensagem............................................... 70
3.5.4.2 Mecânica da compactação................................................................... 73
3.5.5 Secagem e evaporação de ligante ....................................................... 77
3.5.6 Sinterização das cerâmicas à base de óxidos...................................... 79
3.5.6.1 Aspectos teóricos......... ........................................................................ 79
3.6 Identificação das fases da zircônia e transformação martensítica ....... 84
3.7 Propriedades mecânicas ...................................................................... 85
3.7.1 Dureza.................................................................................................. 85
3.7.2 Tenacidade à fratura ............................................................................ 87
3.7.3 Resistência mecânica........................................................................... 98
3.7.4 Degradação das propriedades mecânicas da zircônia ........................107
3.7.5 Resistência ao desgaste .....................................................................113
4 MATERIAIS E MÉTODOS ...................................................................114
4.1 Caracterização dos pós........................................................................114
4.1.1 Espectrometria de fluorescência de raios-X .........................................114
4.1.2 Difração de raios X...............................................................................115
4.1.3 Área específica de superfície ..............................................................115
4.1.4 Análise de tamanho de partículas ........................................................117
4.1.5 Microscopia eletrônica de varredura (MEV)................................. ........118

7
4.2 Materiais utilizados...............................................................................118
4.2.1 Pós cerâmicos......................................................................................118
4.2.1.1 Alumina ................................................................................................118
4.2.1.2 Zircônia.................................................................................................118
4.2.2 Aditivos.................................................................................................119
4.3 Processamento ....................................................................................121
4.3.1 Metodologia de Análise ........................................................................121
4.3.2 Preparação das composições ..............................................................121
4.3.3 Moagem ...............................................................................................128
4.3.4 Secagem dos pós.................................................................................128
4.3.5 Desaglomeração ..................................................................................128
4.3.6 Conformação dos corpos de prova ......................................................128
4.3.7 Cálculo da densidade a verde ..............................................................129
4.3.8 Retirada do ligante ou plastificante .....................................................130
4.3.9 Sinterização..........................................................................................131
4.4 Caracterização da microestrutura e propriedades do material sinterizado
.....................................................................................................132
4.4.1 Avaliação das propriedades físicas ......................................................133
4.4.1.1 Densidade e porosidade.......................................................................133
4.4.1.2 Avaliação das fases cristalinas e transformação martensítica .............135
4.4.2 Avaliação das características microestuturais......................................138
4.4.3 Avaliação das propriedades mecânicas ...............................................140
4.4.3.1 Determinação dos valores de dureza...................................................140
4.4.3.2 Determinação do fator de intensidade de tensão crítica – KIC..............143
4.4.3.3 Determinação da resistência mecânica à flexão ..................................145
4.5 Tratamento estatístico dos dados ........................................................148
5 RESULTADOS E DISCUSSÃO ............................................................153
5.1 Caracterização dos pós........................................................................153
5.1.1 Área específica de superfície ..............................................................153
5.1.2 Tamanho de partículas.........................................................................153
5.1.3 Forma da partícula ...............................................................................156
5.1.4 Caracterização das fases presentes ....................................................156

8
5.2 Caracterização e determinação das propriedades do material sinterizado
.....................................................................................................161
5.2.1 Composições analisadas......................................................................161
5.2.2 Densidade e dependência da microestrutura .......................................162
5.2.3 Difração de raios-X: fases cristalinas e transformação martensítica ...168
5.2.4 Tamanho de grâo .................................................................................178
5.2.5 Propriedades mecânicas ......................................................................185
5.2.5.1 Módulo de elasticidade.........................................................................185
5.2.5.2 Carga de indentação e dureza Vickers.................................................186
5.2.5.3 Valores de resistência mecânica..........................................................189
5.2.5.4 Valores de tenacidade à fratura ...........................................................197
5.2.5.5 Sistema de trinca..................................................................................204
5.2.5.6 Uso de equações para determinação do KIC pelo método de indentação
...................................................................................................206
5.2.5.7 Análise das superfícies de fratura ........................................................208
5.2.5.8 Relação entre geometria da trinca e tenacidade à fratura....................210
5.2.5.9 Resistência ao desgaste ......................................................................214
5.2.5.10 Microscopia eletrônica de transmissão................................................216
6 CONCLUSÕES......................................................................................220
6.1 Conclusões.............................................................................................220
6.2 Sugestões ..............................................................................................222
7 REFERÊNCIAS BIBLIOGRÁFICAS.......................................................222
8 ANEXOS.................................................................................................243
8.1 Anexo 1 ..................................................................................................244
8.2 Anexo 2 ..................................................................................................252

9
LISTA DE ILUSTRAÇÕES
FIG. 1.1 Implante osseointegrado e restauração protética. ..................................24
FIG. 3.1 Prótese de quadril em alumina. ..............................................................30
FIG. 3.2 Implante de Tübingen. ............................................................................33
FIG. 3.3 Cilindros híbridos para testes em animais. .............................................33
FIG. 3.4 Protótipos implantados em animais. .......................................................34
FIG. 3.5 Implante híbrido(metal-cerâmica). ..........................................................34
FIG. 3.6 Preparação de zircônia parcialmente estabilizada com ítria a partir de
oxicloreto de zircônio. .............................................................................36
FIG. 3.7 Implante dentário substituindo um elemento dentário. ..........................39
FIG. 3.8 Pilares metálicos em titânio. ...................................................................40
FIG. 3.9 Retração gengival e exposição do metal. ...............................................40
FIG. 3.10 Pilar CerAdapt em alumina e seu parafuso de fixação. ..........................42
FIG. 3.11 Pilar Cerabase em alumina e sua base de titânio...................................43
FIG. 3.12 Pilar pré-fabricado Zi-Real em zircôna. ..................................................43
FIG. 3.13 Sequência de utilização do pilar: (a) Implante osseointegrado de titânio
instalado; (b) Pilar cerâmico fixado ao impante; (c) Coroas cerâmicas
cimentadas. ............................................................................................44
FIG. 3.14 Estrutura cristalina da alumina. ..............................................................45
FIG. 3.15 Estrutura cristalina da zircônia monoclínica............................................47
FIG. 3.16 Estrutura cristalina da zircônia tetragonal...............................................47
FIG. 3.17 Estrutura cristalina da zircônia cúbica. ...................................................48
FIG. 3.18 Diferentes estruturas cristalinas da ZrO2 cúbica, tetragonal e
monoclínica, respectivamente. ...............................................................48
FIG. 3.19 Diagrama de fases dos sistema ZrO2/Y2O3. ...........................................50
FIG. 3.20 Representação esquemática das tensões geradas em uma matriz que
possui uma região que envolve uma trinca, quando esta região é
submetida a uma transformação de fase expansiva. As tensões
residuais induzidas na matriz vizinha à região agem no sentido de fechar
a trinca....................................................................................................54
FIG. 3.21 Mecanismo de aumento de tenacidade por transformação da zircônia..55

10
FIG. 3.22 Moinho atritor..........................................................................................60
FIG. 3.23 Formação de aglomerados na ausência de forças repulsivas................63
FIG. 3.24 Partículas dispersas e defloculadas. ......................................................63
FIG. 3.25 Formação de uma dupla camada de cargas elétricas ao redor de uma
partícula carregada negativamente em meio aquoso.............................65
FIG. 3.26 Estrutura molecular do ácido cítrico(o símbolo Cit representa o grupo
citrato). ...................................................................................................66
FIG. 3.27 Micrografias eletrônicas de varredura após polimento e ataque térmico da
amostra Al2O3 - 15% 3Y-TZP sem a utilização do dispersante: (a)
elétrons retroespalhados e (b) elétrons secundários em diferentes
magnitudes. ............................................................................................69
FIG. 3.28 Estágios da compactação: (a) Enchimento; (b) Fechamento; (c)
Prensagem; (d) Ejeção. ..........................................................................70
FIG. 3.29 Compactação de esferas finas no interstício de partículas grosseiras. ..71
FIG. 3.30 Distribuição de tamanho de partículas cerâmicas. .................................72
FIG. 3.31 Estrutura de poros em um arranjo de partículas anisométricas. (a)
arranjo randômico, (b) arranjo ordenado. ...............................................72
FIG. 3.32 Moléculas de ligante reduzem a densidade de empacotamento sob
baixas tensões efetivas, A – partículas, B- Moléculas de polímeros. ....73
FIG. 3.33 Forças normais e tangenciais nos contatos entre partículas. .................74
FIG. 3.34 Alterações microestruturais durante a compactação. .............................75
FIG. 3.35 Defeitos nos compactos prensados a seco. ...........................................76
FIG. 3.36 Perfil de distribuição da pressão na prensagem uniaxial. .......................77
FIG. 3.37 Fluxograma geral dos processos de sinterização...................................80
FIG. 3.38 Mecanismos de transporte de massa. ....................................................82
FIG. 3.39 Configuração da ponta da trinca e base da mecânica da fratura linear
elástica. ..................................................................................................87
FIG. 3.40 Modos básicos de carregamento envolvendo diferentes deformações da
superfície das trincas..............................................................................88
FIG. 3.41 Modelo de Johnson da cavidade esférica em expansão. .......................90
FIG. 3.42 Sequência da iniciação e propagação de trincas na indentação de um
material duro...........................................................................................91
FIG. 3.43 Comparação dos sistemas de trincas ao redor de impressão Vickers. ..94

11
FIG. 3.44 Sistemas de trincas (a): Palmquist; (b): “half-penny”. .............................94
FIG. 3.45 Desenho esquemático do sistema IF produzido por um indentador
Vickers ...................................................................................................96
FIG. 3.46 Diferença entre sistema radial/mediano e Palmqvist após polimento da
superfície. ...............................................................................................97
FIG. 3.47 (a) defeito semi-elíptico em barra submetida a flexão em quatro pontos.
em (b), a profundidade do defeito, a, é menor do que sua semilargura, c,
e em (c) a é maior do que c..................................................................101
FIG. 3.48 Zona frontal do processo, zona da marola e zona de ponteamento da
trinca em propagação. ..........................................................................103
FIG. 3.49 Processos de absorção de tensões (a) deflexão (“deflection”) da trinca
por decoesão (“debonding”) na interface; (b) microtrincamento na zona
frontal do processo (“branching”); (c) escoamento plástico na zona
frontal do processo; (d) transformação de fase com expansão de volume
e/ou microtrincamento (“microcracking”)na zona da marola; (e)
ponteamento da trinca por fibras ou whiskers; (f) ponteamento
(“bridging”) da trinca por grãos e travamento (g) ponteamento
viscoelástico. ........................................................................................104
FIG. 3.50 Gráfico esquemático da curva-R crescente..........................................105
FIG. 3.51 Campos de tensões em um viga prismática carregada em três pontos,
franjas isocromáticas - campo escuro. .................................................107
FIG. 4.1 Metodologia de análise.........................................................................122
FIG. 4.2 Massas e volumes utilizados para a preparação das misturas de
diferentes composições. .......................................................................125
FIG. 4.3 Razão de aspecto do corpo verde para prensagem uniaxial. ...............129
FIG. 4.4 Variação da temperatura do forno durante a evaporação do ligante. ...130
FIG. 4.5 Variação da temperatura do forno durante a sinterização das
composições a 1600°C.........................................................................132
FIG. 4.6 Variação da temperatura do forno durante a sinterização das
composições a 1500°C.........................................................................132
FIG. 4.7 Difratograma da Y-ZrO2 Tosoh com 2θ na faixa de 27° a 33°, mostrando
a ocorrência do do pico tetragonal a= (101)T, e dos picos monoclínicos
b = ( 1 11) M e c = (111)M. .....................................................................137

12
FIG. 4.8 Distância entre as indentações Vickers conforme norma ASTM C -
1327 - 99. ............................................................................................141
FIG. 4.9 Indentações Vickers aceitáveis de acordo com a norma ASTM C - 1327 -
99............. ............................................................................................141
FIG. 4.10 Indentações Vickers não aceitáveis conforme norma ASTM C - 1327 -
99 . .......................................................................................................142
FIG. 4.11 Esquema do ensaio de flexão em 4 pontos... .......................................145
FIG. 5.1 Distribuição de tamanho de partículas para todas as amostras. ..........154
FIG. 5.2 Distribuição de tamanho de partículas para amostras ZT e alumina. ...155
FIG. 5.3 Distribuição de tamanho de partículas para amostras ZM e alumina. ..155
FIG. 5.4 Micrografia de solução bem diluída dos pós: (a)100 ZT , (b) 100 A , (c)
100 ZM. ................................................................................................156
FIG. 5.5 Difratograma do pó da amostra 100 ZT................................................157
FIG. 5.6 Difratograma do pó para amostra 80 ZT...............................................157
FIG. 5.7 Difratograma do pó para amostra 21 ZT...............................................158
FIG. 5.8 Difratograma do pó para amostra 15 ZT...............................................158
FIG. 5.9 Difratograma do pó para amostra 10 ZT...............................................158
FIG. 5.10 Difratograma do pó para amostra 5 ZT.................................................159
FIG. 5.11 Difratograma do pó para amostra A. ....................................................159
FIG. 5.12 Difratograma do pó para amostra 100 ZM............................................160
FIG. 5.13 Difratograma do pó para amostra 80 ZM..............................................160
FIG. 5.14 Difratograma do pó para amostra 15 ZM..............................................160
FIG. 5.15 Micrografias eletrônicas de varredura utilizando elétrons
retroespalhados, onde as partículas escuras e claras são alumina e
zircônia, respectivamente, para as amostras: (a) 100 ZT, (b) 80 ZT, (c)
21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j) 15
ZM. .......................................................................................................167
FIG. 5.16 Difratograma da amostra 100 ZT após sinterização. ............................168
FIG. 5.17 Difratograma da amostra 80 ZT após sinterização. ..............................169
FIG. 5.18 Difratograma da amostra 21 ZT após sinterização. ..............................169
FIG. 5.19 Difratograma da amostra 15 ZT após sinterização. ..............................169
FIG. 5.20 Difratograma da amostra 10 ZT após sinterização. ..............................169
FIG. 5.21 Difratograma da amostra 5 ZT após sinterização.................................170

13
FIG. 5.22 Difratograma da amostra 100 ZM após sinterização. ...........................170
FIG. 5.23 Difratograma da amostra 80 ZM após sinterização. ............................170
FIG. 5.24 Difratograma da amostra 15 ZM após sinterização. ............................171
FIG. 5.25 Difratograma da amostra 100 ZT após fratura.....................................171
FIG. 5.26 Difratograma da amostra 80 ZT após fratura.......................................171
FIG. 5.27 Difratograma da amostra 21 ZT após fratura.......................................172
FIG. 5.28 Difratograma da amostra 15 ZT após fratura.......................................172
FIG. 5.29 Difratograma da amostra 10 ZT após fratura.......................................172
FIG. 5.30 Difratograma da amostra 5 ZT após fratura.........................................173
FIG. 5.31 Difratograma da amostra 100 ZM após fratura....................................173
FIG. 5.32 Difratograma da amostra 80 ZM após fratura......................................173
FIG. 5.33 Difratograma da amostra 15 ZM após fratura......................................174
FIG. 5.34 Tamanho médio de grão versus ZrO2 Tosoh (% peso). ......................180
FIG. 5.35 Tamanho médio de grão versus ZrO2 Mel Chemicals (% peso). .........181
FIG. 5.36 Micrografias eletrônicas de varredura das amostras com superfícies
polidas e termicamente atacadas (1500°C 10min): (a) 100 ZT, (b) 80 ZT,
(c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j)
15 ZM. ..................................................................................................184
FIG. 5.37 Diagrama de Weibull para amostra 100 ZT. .........................................194
FIG. 5.38 Diagrama de Weibull para amostra 80 ZT. ...........................................194
FIG. 5.39 Diagrama de Weibull para amostra 21 ZT. ...........................................195
FIG. 5.40 Diagrama de Weibull para amostra 10 ZT. ..........................................195
FIG. 5.41 Diagrama de Weibull para amostra 5 ZT. ............................................195
FIG. 5.42 Diagrama de Weibull para amostra A. ..................................................196
FIG. 5.43 Diagrama de Weibull para amostra 100 ZM. ........................................196
FIG. 5.44 Diagrama de Weibull para amostra 80 ZM. ..........................................196
FIG. 5.45 Diagrama de Weibull para amostra 15 ZM. ..........................................197
FIG. 5.46 Dureza e tenacidade à fratura versus composição (zircônia Tosoh)
utilizando equação de Anstis, G. R. et al., 1981. .................................198
FIG. 5.47 Dureza e tenacidade à fratura versus composição (zircônia Mel
Chemicals) utilizando equação de Anstis, G. R. et al., 1981. ..............198
FIG. 5.48 Micrografias da amostra 100 ZT antes e depois do polimento com pasta
de diamante de 15 μm..........................................................................205

14
FIG. 5.49 Micrografias eletrônicas de varredura das superfícies de fratura (a) 100
ZT, (b) 80 ZT, (c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM,
(i) 80 ZM, (j) 15 ZM...............................................................................209
FIG. 5.50 Micrografias eletrônicas de varredura das trincas induzidas por
indentação nas superfícies das amostras: (a) 100 ZT, (b) 80 ZT, (c) 21
ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j) 15
ZM. .......................................................................................................212
FIG. 5.51 Exemplo dos caminhos de trinca típicos para os materiais
examinados
..............................................................................................................214
FIG. 5.52 Imagem em campo claro(MET) de (a) ZrO2-Al2O3(80 ZT) onde alumina é
o grão escuro........................................................................................217
FIG. 5.53 Espectro de EDS para o grão de alumina. ...........................................218
FIG. 5.54 Espectro de EDS para o grão de zircônia.............................................219

15
LISTA DE TABELAS
TAB. 3.1 Propriedades de biomateriais comerciais de alumina e o padrão da ISO-
6474..........................................................................................................29
TAB. 3.2 Propriedades de biomateriais comerciais de zircônia e o padrão da ISO-
13356........................................................................................................37
TAB. 3.3 Pilares cerâmicos comerciais. ..................................................................42
TAB. 3.4 Características dos polimorfos da zircônia ...............................................46
TAB. 3.5 Raios iônicos de alguns elementos estabilizadores das formas
polimórficas de altas temperaturas da zircônia e a razão (R) entre raio
iônico do elemento estabilizante e raio do zircônio...................................49
TAB. 3.6 Características dos três estágios da sinterização.....................................81
TAB. 3.7 Mecanismos de transporte de massa. ......................................................83
TAB. 4.1 Teor de óxidos presente no pó de alumina – A – 1000SG - Alcoa .........118
TAB. 4.2 Teor dos óxidos presentes no pó de zircônia TZ - 3YSB – Tosoh..........119
TAB. 4.3 Teor de óxidos presente no pó de zircônia XZO708/14 - Mel Chemicals.119
TAB. 4.4 Especificação do ácido cítrico monohidratado P. A. – Vetec..................120
TAB. 4.5 Especificação do álcool etílico P. A. - Vetec...........................................120
TAB. 4.6 Especificação do polyetileno glycol 400 – (ATPEG 400). .......................121
TAB. 4.7 Massas e volumes utilizados para a preparação das misturas de
diferentes composições. .........................................................................125
TAB. 4.8 Equações propostas na literatura e utilizadas para determinação de
KIC...........................................................................................................144
TAB. 4.9 Dados típicos para um experimento de um fator. ...................................149
TAB. 4.10 ANOVA para experimento de um fator. .................................................151
TAB. 5.1 Área específica de superfície dos pós (m2/g). ........................................153
TAB. 5.2 Tamanho médio de partícula . ................................................................155
TAB. 5.3 Densidade teórica da alumina SG-1000- Alcoa (g/cm3). ........................161
TAB. 5.4 Densidade teórica da zircônia Mel Chemicals (g/cm3)............................161
TAB. 5.5 Densidade teórica da zircônia Tosoh (g/cm3). ........................................161

16
TAB. 5.6 Composição das amostras (%peso) e densidade teórica das amostras
(g/cm3). ...................................................................................................161
TAB. 5.7 Densidade e porosidade das amostras. ..............................................162
TAB. 5.8 Análise de variância (ANOVA) para as médias de densidade. .............163
TAB. 5.9 Teste LSD para a variável densidade das amostras de 1 a 5. .............164
TAB. 5.10 Teste LSD para a variável densidade das amostras de 6 a 10. ...........164
TAB. 5.11 Retração linear das amostras. ..............................................................166
TAB. 5.12 Análise quantitativa das fases presentes obtida pelo método de
Rietveld.................................................................................................174
TAB. 5.13 Fração volumétrica de zircônia monoclínica presente nos pós, após
sinterização e após fratura, obtida pelo método interno. ......................175
TAB. 5.14 Extensão da transformação induzida por tensão a partir de dados obtidos
pelo método interno..............................................................................176
TAB. 5.15 Extensão da transformação induzida por tensão a partir de dados obtidos
pelo método de Rietveld. ......................................................................177
TAB. 5.16 Tamanho médio de grão........................................................................179
TAB. 5.17 Módulo de elasticidade das amostras calculado pela regra da mistura.185
TAB. 5.18 Medidas de dureza Vickers (HV). ..........................................................186
TAB. 5.19 Análise de variância(ANOVA) para as médias de dureza. ...................188
TAB. 5.20 Teste LSD para a variável dureza das amostras de 1 a 5. ...................188
TAB. 5.21 Teste LSD para a variável dureza das amostras de 6 a 10. .................189
TAB. 5.22 Resultados do ensaio de flexão em 4 pontos. ......................................190
TAB. 5.23 Análise de variância(ANOVA) para as médias de resistência à flexão.191
TAB. 5.24 Teste LSD para a variável resitência à flexão das amostras de 1 a
5. ..........................................................................................................191
TAB. 5.25 Teste LSD para a variável resistência à flexão das amostras de 6 a
10. ........................................................................................................192
TAB. 5.26 Medidas de densidade e tenacidade à fratura (KIC)..............................197
TAB. 5.27 Análise de variância (ANOVA) para médias de KIC. .............................202
TAB. 5.28 Teste LSD para a variável KIC das amostras de 1 a 5. .........................202
TAB. 5.29 Teste LSD para a variável KIC das amostras de 6 a 10. .......................203
TAB. 5.30 Medidas do comprimento médio de trinca e razão c/a. ........................205
TAB. 5.31 Valores de densidade, dureza, KIC e resistência ao desgaste. .............215

17
LISTA DE ABREVIATURAS E SÍMBOLOS
ABREVIATURAS
PSZ - zircônia parcialmente estabilizada
Y-PSZ - zircônia parcialmente estabilizada com ítria
TZP - zircônia tetragonal policristalina
Y-TZP - zircônia tetragonal policristalina estabilizada com ítria
YSZ - zircônia totalmente estabilizada com ítria
ATZ - zircônia tenacificada por alumina
ZTA - alumina tenacificada por zircônia
PI - ponto isoelétrico
PCZ - ponto de carga zero
Mg-PSZ - zircônia parcialmente estabilizada com óxido de magnésio
SÍMBOLOS
μm - micrometro
mm - milímetro
cm - centímetro
g - grama
kg - kilograma
MPa - mega Pascal
GPa - giga Pascal
HV - dureza Vickers
N - newton
kgf - kilograma-força
m - metro
°K - kelvin
°C - celsius
W - watts

18
LIISTA DE SIGLAS
MEV Microscópio Eletrônico de Varredura
MET Microscópio Eletrônico de Transmissão
INT Instituto Nacional de Tecnologia
IME Instituto Militar de Engenharia
ISO International Standards Organization
ASTM American Standard for Testing Materials
ABNT Associação Brasileira de Normas Técnicas

19
RESUMO
Materiais cerâmicos apresentam muitas qualidades incluindo estabilidade
química, elevada dureza, resistência ao desgaste. Por outro lado, devido a sua fragilidade, podem sofrer falha sob níveis de tensões relativamente baixos. A alumina é um material cerâmico biocompatível, apresenta boa resistência ao desgaste, mas exibe moderada resistência à flexão e tenacidade. A zircônia é também inerte em meio fisiológico, apresenta menor módulo de elasticidade, maior resistência à flexão e tenacidade à fratura. O aumento de tenacidade verificado para a zircônia está relacionado com a transformação induzida por tensão das partículas de zirconia tetragonal em monoclínica e o microtrincamento associado a esta transformação. Assim, compósitos a base destes materiais são promissores candidatos a utilização como material de implantes. No presente trabalho, foram analisadas dez diferentes composições onde o teor de zircônia estabilizada com ítria (Y-TZP) adicionada à alumina variou de 5 % a 80 % em peso. A difração de raios-X e o método de Rietveld foram utilizados para a quantificação das fases presentes e a avaliação da extensão da transformação de fase da zircônia. Observações da microestrutura e determinação de propriedades mecânicas dos materiais sinterizados foram realizadas. Mecanismos de fratura presentes nestes compósitos são discutidos para melhor entendimento das falhas. Os materiais investigados apresentaram densidade na faixa de 99,13 % a 99,86 % da densidade teórica, que aumentou com o teor de zircônia no compósito. Estes sistemas podem atingir resistência à flexão 93 % e tenacidade à fratura 29 % superiores, quando comparados com a alumina pura. O modo de fratura dos materiais investigados foi predominantemente intergranular, e a fratura transgranular foi observada para alumina pura e compósitos com 5 % de zircônia. O aumento de tenacidade à fratura está relacionado principalmente com a maior densidade, homogeneidade da microestrutura e foi proporcional a quantidade de zircônia tetragonal disponível para transformação, indicando que o mecanismo de tenacificação por transformação e o microtrincamento associado a esta transformação tem importante papel nestes compósitos. A resistência à flexão aumentou com o refinamento da microestrutura, sendo mais sensível ao tamanho de grão do que a tenacidade à fratura.

20
ABSTRACT
Ceramic materials have many qualities including chemical stability, high
hardness and good wear resistance. However, due to their fragility they can present catastrofic failure under relatively low stress. The alumina is a ceramic material biocompatible, has good wear resistance but exhibits moderate flexural strength and fracture toughness. Zirconia is inert in physiological environment, has lower elasticity modulus, higher flexural strength and fracture toughness. The increase in toughness verified for zirconia is related to the stress induced tranformation of zirconia tetragonal into monoclinic and the microcracking associated with this transformation. Thus, composites based on these materials are promising candidate for utilization as implant material. In the present work, ten different compositions were yttria stabilised zirconia (Y-TZP) content added to alumina varied from 5 to 80 wt. %. The X-ray diffraction and the Rietveld method were used for the quantification of the present phases and to compute the extent of zirconia phase transformation. Microstructure observations and determination of mechanical properties from sintered materials were performed. Fracture mechanisms present in these composites are discussed to better understanding the failure. The materials investigated showed densities in the range of 99,13 % to 99,86 % of theoretical density , which increased with zirconia content. These systems can achieve a flexural strength 93 % and fracture toughness 29 % superior when compared to the pure alumina ceramics. The fracture mode of the investigated materials were predominantly intergranular and the transgranular mode were observed for pure alumina and composites with 5 wt. %. The increase in fracture toughness were mainly related to a higher density, homogeneous microstructure and were proportional to the tetragonal zirconia available for transformation, indicating that the transformation toughness and the microcracking associated to this transformation have an important role in these composites. The flexural strength can be increased with the microstructure refinement, been more sensitive to grain size than fracture toughness.

21
1 INTRODUÇÃO
O desenvolvimento de tecnologias para a produção de novos materiais tem sido
motivada pela demanda de materiais que executem novas funções ou
desempenhem antigas funções de forma mais adequada. A síntese e o
processamento de materiais cerâmicos avançados teve um desenvolvimento
acelerado na década de 70 e têm alcançado resultados promissores, verificando-se
inclusive, uma contínua evolução no desenvolvimento e uso destes materiais
voltados para aplicações estruturais. Cerâmicas estruturais devem possuir elevada
resistência mecânica até altas temperaturas, elevada dureza, estabilidade
dimensional, boa resistência ao ataque químico e ao desgaste. As principais
aplicações são: componentes de motores, ferramentas de corte, materiais sujeitos a
abrasão e ao ataque químico e peças para implantes ósseos e dentários (Rezende,
D. T., 1997).
Uma grande evolução nas técnicas de restauração dentária vem sendo
estabelecida pelo uso de materiais cerâmicos. Esses materiais apresentam
vantagens relativas, devidas ao ótimo desempenho das suas propriedades
funcionais, principalmente estética, biocompatibilidade e resistência química. A
tendência das técnicas de cerâmica dental vem sendo a substituição da subestrutura
metálica das restaurações, inclusive das restaurações sobre implantes, visando uma
melhor estética e utilizando para isso cerâmicas de maior tenacidade à fratura,
minimizando sua fragilidade.
A implantodontia é a especialidade da odontologia que visa a reposição de
elementos dentários perdidos ou removidos. Após 20 anos de reabilitação protética
sobre implantes, constatou-se a importância da estética. Os pilares metálicos em
algumas situações comprometiam a estética. Com base nisso, os sistemas de
implantes desenvolveram os componentes protéticos cerâmicos. Porém, eles
apresentam dificuldades mecânicas, devido a fragilidade. O segundo problema é
que os pilares cerâmicos a base de alumina e Y-TZP, comercialmente disponíveis,
são importados e de elevado custo, não havendo tecnologia nacional para produção
de pilares cerâmicos.
A utilização de cerâmicas a base de alumina e zircônia de altas densidades vem
sendo propostas como material para implantes. A alumina demonstra

22
biocompatibilidade e resistência ao desgaste, contudo, oferece moderada resistência
à flexão e baixa tenacidade. A zircônia é também inerte em meio fisiológico porém,
apresenta vantagem sobre a alumina pois tem maior resistência à flexão, maior
tenacidade à fratura e menor módulo de elasticidade. Porém, as propriedades
mecânicas ainda não são satisfatórias. Além disso, a zircônia se torna
esteticamente bastante interessante quando polida.
As técnicas utilizadas no processamento cerâmico podem ser o método
convencional de escultura da massa cerâmica ou por alguns procedimentos
avançados já existentes, que utilizam os mais variados métodos de conformação.
Métodos como cerâmica infiltrada com vidro, ou vitro-cerâmica centrifugada a partir
de vidro fundido, usinagem de um bloco cerâmico, e modelagem de cerâmica sob
pressão a quente, têm sido propostos (Phillips, R., 1996). Entretanto, por utilizar
equipamentos especiais de processamento, apresentam um alto custo de aquisição,
o que resulta no custo elevado da restauração cerâmica.
A alumina mantem-se na liderança da indústria de cerâmicas avançadas, devido
ao seu custo moderado, sua versatilidade e comprovado desempenho. As
composições cerâmicas a base de zircônia, entretanto, apresentam maior
crescimento, visto que novas variedades com maior resistência estão sendo
desenvolvidas (Oliveira, A. P. A. , 1997).
Diante das evidências do papel da zircônia como agente tenacificador de
cerâmicas utilizadas para fins estruturais, vários sistemas cerâmicos a base de
zircônia têm sido estudados. Tal reforço é conseqüência da transformação de fase
tetragonal para monoclínica das partículas de zircônia finamente dispersas na matriz
e ocorre por meio de dois mecanismos: a) formação de microtrincas devido a
expansão volumétrica que acompanha a transformação t → m, onde é gerado um
estado de compressão e ocorre a formação de microtrincas ao redor das partículas.
Estas microtrincas irão absorver energia durante o processo de desenvolvimento das
trincas, aumentando a tenacidade da cerâmica; b) transformação induzida por
tensão que ocorre quando a trinca encontra partículas de zircônia tetragonal que se
transformam em zircônia monoclínica. Estas partículas de zircônia tetragonal podem
ser obtidas pela adição de óxidos de terras raras que estabilizam a fase t-ZrO2, ou
pela simples compressão exercida pelos outros grãos. A transformação absorve

23
parte da energia necessária para a propagação da trinca, ocorrendo um aumento da
tenacidade à fratura (Rezende, D. T., 1997).
A adição de zircônia na alumina como aditivo de sinterização vem sendo
praticada a longo tempo com objetivo de densificação de cerâmicas a base de
alumina. Todavia, o conceito de tenacificação de cerâmicas de alumina por
dispersão de partículas de zircônia em uma matriz somente foi reconhecida nos
últimos 20 anos. A introdução de pequena quantidade de zircônia em alumina como
aditivo de sinterização leva a formação de solução sólida a qual promove o processo
de densificação pela introdução de defeitos (Wang, J. & Stevens, R., 1989). Por
outro lado, a microestrutura de compósitos a base de alumina e zircônia é
caracterizada pela presença de duas fases distintas, ao invés de uma solução sólida
(Wang, J. & Stevens, R., 1989). É conhecido que a adição de uma segunda fase
resulta em uma melhoria de propriedades como resistência à flexão e tenacidade à
fratura (Claussen, N.,1976; Lange, F.F., 1982c; Hori, S. et al., 1986; Casellas, D.,
1999, Evans, A. G., 1990). Por esta razão, estes materiais são promissores em
diversas aplicações que requerem elevada dureza, alta resistência ao desgaste e
relativa tenacidade à fratura.
Compósitos de alumina e zircônia são conhecidos como ZTA (“zirconia
toughened alumina” ou matriz de alumina reforçada com partículas de zircônia), e
ATZ (“alumina toughened zirconia” ou matriz de zircônia estabilizada reforçada com
partículas de alumina) (De Aza, A. H. et al., 2002). Com ambos materiais bifásicos
(ZTA e ATZ), é esperada a obtenção de maiores valores de tenacidade à fratura
quando comparado com os materiais cerâmicos monofásicos(Fantozi G. & Orange
G., 1986; Karihaloo, B.L., 1991; Becher, P.F. et al., 1993; Rühle, M. et al., 1984;
Gregori, G. et al., 1999).
De acordo com Gregori, G.et al., 1999, os compósitos a base de alumina e
zircônia apresentam uma associação com ganho de tenacidade mantendo as
propriedades peculiares da alumina como excelente resistência ao desgaste e
estabilidade química. Todas estas características qualificaram o ZTA para diversas
aplicações e o tornam um promissor candidato a utilização como material de
implantes, uma vez que a comunidade de biomateriais já está familiarizada com a
alumina e a zircônia separadamente. Apesar disso, muito pouco foi publicado na

24
literatura sobre estes compósitos como biomateriais (Piconi, C. & Maccauro, G.,
1999).
Desta forma, o presente trabalho introduz a utilização de compósitos cerâmicos
alumina-zircônia como biomateriais a serem utilizados em pilares cerâmicos para
restaurações sobre implantes osseointegrados de titânio. Através da avaliação das
propriedades mecânicas e observação da microestrutura dos compósitos obtidos, o
presente trabalho visa determinar a faixa de composição mais promissora para sua
utilização nesta aplicação odontológica específica. (FIG. 1.1).
FIG. 1.1 Implante osseointegrado e restauração protética.
Pilar Coroa
Implante

25
2 OBJETIVO
As cerâmicas como a alumina e a zircônia são utilizadas como biomateriais na
medicina e odontologia em diversas aplicações. Os mecanismos de tenacificação
da zircônia e ainda os compósitos de alumina e zircônia parecem ser promissores
neste campo.
Os pilares para restaurações sobre implantes utilizados atualmente são
metálicos, o que compromete a estética. Alguns pilares importados em alumina e
zircônia Y-TZP estão disponíveis, porém com elevado custo não havendo tecnologia
nacional de pilares cerâmicos.
Assim, 10 composições foram sinterizadas a partir de parâmetros de
processamento otimizados obtidos na literatura (dispersante, ligante, tempo de
moagem, elementos de moagem, temperatura de sinterização, etc.) e através da
avaliação das fases presentes e sua quantificação, das propriedades mecânicas e
observação da microestrutura dos compósitos obtidos, o presente trabalho visa
determinar a faixa de composição mais promissora para sua utilização nesta
aplicação odontológica específica, ou seja, em pilares cerâmicos para restaurações
sobre implantes osseointegrados de titânio.

26
3 REVISÃO BIBLIOGRÁFICA 3.1 CERÂMICAS COMO BIOMATERIAIS
A finalidade deste capítulo é apresentar as informações de interesse disponíveis
na literatura, de modo a compreender melhor as biocerâmicas, suas aplicações,
processamento e suas propriedades.
3.1.1 BIOCERÂMICAS
As cerâmicas são materiais inorgânicos, não metálicos. Consistem de
elementos metálicos e não-metálicos que apresentam ligações covalentes e/ou
iônicas. (Smith, 1998).
Biocerâmicas são cerâmicas especiais usadas na Medicina e Odontologia para a
substituição ou reconstrução de partes afetadas ou destruídas do sistema
esquelético. Podem ser classificadas em reabsorvíveis (fosfato tri-cálcio), bioativas
(biovidros, biovitro-cerâmicas e hidroxiapatita) e bioinertes (carbono, alumina
sinterizada e zircônia estabilizada com ítria) (Kunes et al., 2000).
As biocerâmicas inertes são usadas principalmente na substituição de ossos,
próteses de quadril e implantes dentários. As mais utilizadas em implantes
cirúrgicos são a alumina e a zircônia. Estas cerâmicas atendem a esta demanda em
função de suas propriedades mecânicas e de corrosão, pureza química e
biocompatibilidade. Elas apresentam pequena ou nenhuma alteração química
durante longo tempo de exposição ao ambiente fisiológico. Mesmo nos casos em
que estas biocerâmicas apresentam degradação química ou mecânica com o tempo,
a concentração de produtos de degradação em tecidos adjacentes é facilmente
controlada por mecanismos reguladores naturais do corpo humano. A resposta dos
tecidos envolve a formação de uma membrana fibrosa muito fina, micrométrica, ao
redor do material do implante (Hulbert, S.F., 1993).
O potencial dos cerâmicos como biomateriais está na compatibilidade com o
ambiente fisiológico. Biocerâmicas são compatíveis porque são compostos de íons
comumente encontrados no ambiente fisiológico (ex: cálcio, potássio, magnésio e

27
sódio, etc...) e de íons que apresentam baixa toxidade aos tecidos do corpo (ex:
zircônio, titânio, etc...).
Inicialmente, os materiais bioinertes mais frequentemente usados eram os
cerâmicos à base de alumina sinterizada (α -Al2O3, corundum), que pode exibir
resistência à flexão em torno de 380 MPa ou superior.
Dentre as principais vantagens dos materiais com zircônia estão seus elevados
valores de resistência mecânica e tenacidade à fratura, os quais são obtidos devido
a transformação de fase da zircônia, de tetragonal para fase monoclínica. Todavia,
a resistência mecânica de um material, depende fortemente da seleção dos pós
iniciais e da microestrutura final do corpo, a qual é resultado do processamento.
Os pós utilizados para preparação de biocerâmicas (ex: implantes), devem ser
caracterizados por um pequeno tamanho de partícula inicial, uma estreita
distribuição de tamanho de partícula, forma de partícula isométrica e elevada pureza
química, especialmente a ausência de sódio, ferro e silício.
Ossos e dentes, tecidos duros do corpo humano, apresentam um componente
inorgânico, que é um análogo sintético dos tecidos calcificados dos vertebrados
(Kong, Y. M. et al., 2004). Este componente inorgânico consiste de hidroxiapatita
(Ca10(PO4)6(OH)2, HA). Na maioria dos casos, eles também apresentam um
componente orgânico, freqüentemente o colágeno. Esmalte, a camada mais externa
do dente é o material mais duro do corpo humano e consiste de aproximadamente
92% de hidroxiapatita.
O dente em função, é submetido a um dos mais inóspitos ambientes do corpo
humano. Eles estão sujeitos a grandes variações de temperatura, variando do frio
do gelo (O°C) até cafés e sopas quentes. O PH varia na faixa de 0,5 a 8. Somado a
isto, as tensões associadas à mastigação onde tensões cíclicas podem variar de 20
a 100 MPa. Além de se manter estáveis neste ambiente e aptos a suportar as
cargas de mastigação, materiais dentários precisam satisfazer outro critério, que é a
estética. Qualquer material dentário que seja utilizado em regiões de maior
solicitação estética, devem ter cor e translucidez mais próximo possível do dente
natural. (www.azom.com).
As cerâmicas dentárias foram inicialmente usadas a 225 anos atrás. A primeira
aplicação foi a dentadura de porcelana, material largamente utilizado em dentística
nos dias de hoje.

28
As principais aplicações de cerâmicos na odontologia incluem: restaurações,
coroas e “veneers” (Anderson, M. & Óden, A., 1993, Graber, G. & Besimo, C., 1995,
Hegenbarth, E. A., 1996, Marinello, C. P. et al., 1997, Razzog, M. E., et al., 1997,
Óden, A. et al., 1998, Luthard, R. G. et al., 1999, Cando, D. C., 2001, Ardlin, B. I.,
2002, Velasco, R. V., et al., 2003, Bona, A. D. et al., 2003), pilares endodônticos
(Fischer, H. et. al., 2002), brackets ortodônticos (Keith, O. et al., 1994), pilares
cerâmicos para implantes (Prespitino, V. & Ingber, A., 1996, Anderson, B. et al.,
1999, Nishioka, R. S., et al., 2003), todas visando a saúde e a estética.
As restaurações tradicionais eram de amálgama utilizando prata, estanho e
mercúrio. Rapidamente restaurações de resina ganharam popularidade (Kelly, J. R.
et al., 1996). Apesar das vantagens das restaurações de resina, estes materiais
podem sofrer desgaste quando aplicados a superfícies de mastigação. Por esta
razão, coroas totalmente cerâmicas também vem ganhando popularidade.
Sistemas CAD_CAM permitem aos dentistas usinar a cerâmica individualmente
para cada caso e cimentar a coroa numa mesma consulta. Os materiais adequados
a esta aplicação são: porcelana feldspática reforçada com leucita, alumina com
poros interconectados infiltrados com vidro após usinagem para fornecer
translucidez, espinel poroso infiltrado com vidro, zirconia porosa infiltrada com vidro.
Cerca de 75% das coroas no mercado consumidor de aproximadamente 30 milhões
de coroas somente nos Estados Unidos, são feitas com porcelana feldspática em
base metálica. A porcelana é também usada em veneers para recobrir dentes
anteriores danificados. Núcleos cerâmicos também são utilizados em algumas
coroas. Coroas totalmente cerâmicas são normalmente recobertas com porcelana
de forma que a cor e translucidez desejadas possam ser obtidas.
Implantes dentários são utilizados como uma alternativa a pontes onde o dente
foi perdido ou removido (Adell, R. et al., 1981). Os elementos dentários são também
feitos de porcelana fundida com metal, onde um implante metálico biocompatível
como o titânio é ancorado dentro da mandíbula e a coroa de porcelana é fixada ao
implante. O implante metálico pode ser recoberto por hidroxiapatita para facilitar a
ligação com o osso e acelerar a osseointegração (www.azom.com).
3.1.2 ALUMINA COMO BIOMATERIAL

29
Resistência mecânica à flexão, resistência à fadiga e tenacidade à fratura de
policristais α -Al2O3 são uma função do tamanho de grão e da porcentagem de
aditivos de sinterização, isto é, da pureza. Estas e outras propriedades estão
resumidas na TAB. 3.1 para um material cerâmico comercial para implante, bem
como os padrões da International Standards Organization ISO-6474 para implantes
de alta pureza a base de alumina. Extensivos testes têm mostrado que implantes
cerâmicos de alumina que atingem ou excedem os padrões da ISO apresentam
excelente resistência à fadiga estática e dinâmica e resistem ao crescimento
subcrítico de trinca e falha por impacto.
TAB. 3.1 – Propriedades de biomateriais comerciais de alumina e o padrão da ISO-6474.
Propriedades
Unidades
Implantes cerâmicos de
alumina comerciais
ISO - 6474
Al2O3 % > 99,7 > 99,5 SiO + Na2O (Outros óxidos) % < 0,02 < 0,1
Densidade g/cm3 ≥ 3,97 ≥ 3,94 Porosidade % < 0,1 -
Tamanho médio de grão (μm) 3,6 ≤ 4,5 Resistência à flexão (MPa) MPa > 500 > 450 Resistência à compressão MPa 4100 - Acabamento superficial Ra μm 0,02 -
Módulo de elasticidade GPa 380 - Tenacidade à fratura K1c MPa.m1/2 4-6 - Coeficiente de expansão
térmica
x 10-6 / °C 8
-
Condutividade térmica W/m°K 30 - Dureza GPa 22 -
Cerâmicas de alumina de alta densidade e elevada pureza (>99,5%) são usadas
em próteses de quadril e implantes dentários devido a combinação de excelente
resistência à corrosão, boa biocompatibilidade, alta resistência ao desgaste e
moderada resistência mecânica. Apesar de alguns implantes dentários serem
constituídos por monocristais de safira, a maioria dos dispositivos de cerâmicas de
alumina são policristais de grãos muito finos (α-Al2O3) produzidos por prensagem e
sinterização em temperaturas na faixa de 1400°C-1800°C dependendo das
propriedades do material puro. Uma pequena quantidade de óxido de magnésio -
MgO (< 0,5%) é usada como inibidor de crescimento de grão e é essencial para que
se obtenha um corpo sinterizado de alta densidade com uma microestrutura de

30
grãos finos. É ainda muito importante que a quantidade de SiO2 e óxidos alcalinos
seja inferior a 0,1%, porque eles impedem a densificação e promovem o crescimento
de grão. É ainda essencial que a quantidade de CaO seja inferior a 0,1% uma vez
que sua presença leva a uma redução da resistência à fadiga estática.
3.1.3 ALUMINA EM PRÓTESES ORTOPÉDICAS
A alumina tem sido usada como material para confecção de próteses de quadril
e joelho por ser uma cerâmica bioinerte e com alta resistência ao desgaste. Cerca
de meio milhão de próteses de quadril foram implantadas até 1993 com articulação
de rótula de alumina(esfera em alumina) em substituição do componente cabeça de
fêmur e que este número vem aumentando em pelo menos 100.000 por ano. Na
Fig. 3.1 é apresentada uma prótese de quadril em alumina.
FIG. 3.1 Prótese de quadril em alumina.
O principal problema das próteses totais de quadril é o desgaste. Numerosos
estudos clínicos indicam que a utilização de esferas de alumina como cabeças de
fêmur em atrito com a carcaça de rolamento da esfera também em alumina, reduz o
desgaste em até 10 vezes e em 2 vezes ou mais quando a carcaça de rolamento de
esfera é confeccionada em polietileno (UHMWPE).

31
A primeira utilização clínica de uma prótese total de quadril foi reportada por
Boutin em 1971.
Em 1981, o mesmo autor apresentou 1.330 casos, onde foram citadas 6
fraturas de esferas, 4 fraturas de conexões e 7 casos com severo desgaste. As
taxas de desgaste observadas por Boutin, J. M. & Blanquaert, D., 1981 estavam na
faixa de 5-9 μm por ano.
Dorlot, J. M. et al., 1986 reportaram taxa de desgaste inferiores a 1 μm por ano.
Plitz, W. & Hoss, H. V., 1982 apresentaram taxas de desgaste consideradas
significantes em próteses totais de quadril toda em alumina.
Sedel, L. et al.,1988, apresentaram resultados de próteses totais de quadril em
alumina de 116 pacientes com menos de 50 anos realizadas entre abril de 1977 e
agosto de 1989. A análise mostrou 98,5% de probabilidade de conservação das
próteses por mais de 10 anos. Em comparação, Harris, W. H. & Mulroy, R. D. J.,
1990, apresentaram resultados mostrando que 42% de próteses totais de polietileno
(UHMWPE) e metálicas necessitavam de revisão dos componentes em menos de 10
anos.
Sedel, L. et al., 1991, apresentaram um estudo clínico de 10 anos com 187
próteses totais de quadril em alumina. A principal causa de falha foi perda de um
componente por falta de assepsia. As fraturas foram observadas em 5 pacientes.
Todas as falhas mecânicas ocorreram em componentes fabricados antes de 1979.
O desenvolvimento de alumina que atinge ou excede os padrões da ISO, reduziram
o número de falhas mecânicas e no caso de Sedel, L. 1991 et al., as falhas foram
completamente eliminadas.
Estudos clínicos tem comparado taxas de desgaste dos componentes da
conexão de polietileno (UHMWPE) contra bolas de alumina versus bolas metálicas.
Existe uma considerável variação de dados mas em cada caso, a taxa de desgaste
para sistemas com bolas metálicas é muito maior do que com bolas de alumina.
Um estudo apresentado por Oonishi, H. et al., 1988, envolvendo 956 próteses
totais de quadril com bolas ou cabeças de alumina e conexão UHMWPE realizadas
entre 1977 e 1988 e 117 próteses com bolas ou cabeças metálicas e conexão
UHMWPE realizadas entre 1975 e 1981, utilizou técnicas de difração de raios-x para
avaliação do desgaste. A taxa de desgaste para a combinação alumina- UHMWPE
foi 0,098 mm ao ano e 0,245 mm ao ano para a combinação metal- UHMWPE.

32
Dörre, E., 1991, apresentou resultados de taxas de desgaste anuais de 16 anos
de sua experiência clínica.
Para a combinação liga Co-Cr-Mo/UHMWPE, a taxa obtida foi de 200 μm / ano;
Para alumina/UHMWPE, a taxa ficou na faixa de 20 -130 μm / ano; E para
alumina/alumina, foi de 2 μm /ano.
As propriedades de desgaste da alumina estão relacionadas com sua pequena
rugosidade superficial e alta energia de superficie, resultando na forte e rápida
adsorção de moléculas biológicas. Estas camadas de moléculas adsorvidas
fornecem um recobrimento líquido que limita a direção de contato das superfícies
sólidas das articulações. A alta energia de superfície da alumina é demonstrada
pelas medidas do ângulo de contato. Com uma estrutura de alumina monofásica de
grãos finos, a rugosidade superficial é menor do que em ligas bifásicas Co-Cr-Mo.
Próteses totais de joelho consistindo de um componente fêmur em alumina com
um componente tíbia em UHMWPE, foram testadas clínicamente. Outras próteses
em cerâmica de ossos dos dedos, cotovelos, tornozelos, ombros e pulsos também
foram testadas e tiveram sucesso igual ou maior do que outros sistemas
confeccionados com outros materiais.
3.1.4 ALUMINA EM IMPLANTES DENTÁRIOS
Em 1964, Sandhaus solicitou a patente para um implante dentário cerâmico em
alumina densa (Hulbert, S. F., 1993).
Em 1975, Schulte e Heimke introduziram um implante dentário cerámico
confeccionado em alumina densa, conhecido como implante de Tübingen,
apresentado na FIG. 3.2(Schulte, W., 1976; Schulte, W. & Heimke, G.1976; Schulte,
W., 1984, Schulte, W. et al., 1992;).
Uma avaliação de resultados com 610 implantes de Tübingen após 10 anos,
mostrou uma taxa de sucesso de 84,5%. Outro estudo mais recente com
aproximadamente 1300 implantes, apresentou uma taxa de sucesso de 92,5%.
Porém, a taxa de fratura dos implantes foi considerada alta, sendo posteriormente
substituído por implantes de titânio.

33
FIG. 3.2 Implante de Tübingen.
Foram confeccionados cilindros híbridos(metal-cerâmica) para testes em
animais, apresentados na FIG. 3.3.
FIG. 3.3 Cilindros híbridos para testes em animais.
Estes protótipos foram implantados em animais para avaliação. A FIG. 3.4
mostra os protótipos instalados em mandíbula de animais.

34
FIG. 3.4 Protótipos implantados em animais.
Como próximo passo, foi desenvolvido um implante híbrido (metal-cerâmica),
apresentado na FIG. 3.5. Este implante foi idealizado em metal com a região de
pescoço em cerâmica. A região de pescoço ficaria em contato com o tecido
gengival, devido ao melhor acabamento superficial da cerâmica, promovendo a
manutenção do tecido gengival.
FIG. 3.5 Implante híbrido(metal-cerâmica).

35
3.1.5 PROCESSOS INDUSTRIAIS PARA OBTENÇÃO DE PÓS DE ZIRCÔNIA
As duas maiores fontes de zircônio são a zirconita (ZrO2-SiO2, ZrSiO4) e a
badeleyita (ZrO2), ambas contendo aproximadamente 2% em peso de háfnia (HfO2)
em relação à zircônia. Quimicamente e estruturalmente, a háfnia e a zircônia são
tão semelhantes que , na maioria dos casos, a presença da háfnia pode ser
ignorada.
O mineral contendo zircônio mais comum e mais amplamente distribuído é a
zirconita. Grandes depósitos secundários de zirconita formaram areias de praia
como resultado do intemperismo das rochas ígneas e da concentração natural
devido à sua alta densidade. Depósitos de zirconita são encontrados na Austrália,
Estados Unidos, Índia, Malásia, Brasil, China, África do Sul, Sri Lanka, Tailândia e na
antiga União Soviética (Oliveira, A. P. A. , 1997).
A badeleyita é uma ocorrência natural da zircônia. Sua maior fonte é a mina de
Palabora na África do Sul. Este mineral pode ser usado como um precursor de
zircônia de alta pureza, mas é mais freqüentemente usado como fonte direta de
zircônia comercial.
A maior parte da zircônia é produzida a partir da zirconita e pode ser classificada
convenientemente pela técnica utilizada na decomposição do minério, isto é, por
processamento químico ou térmico. A zircônia obtida através da rota térmica tende
a ser mais pura que a badeleyita, entretanto possui maiores níveis de impurezas do
que os produtos derivados da rota química. A decomposição química da zirconita
requer elevadas temperaturas e o uso de agentes agressivos, sendo que as rotas
mais comuns empregadas são a carbo-cloração e fusão alcalina. Todas as rotas
químicas passam por uma etapa onde as espécies zircônio estão em solução.
Através do controle criterioso dos processos de cristalização e precipitação, os
produtos intermediários e, conseqüentemente, a zircônia podem ser produzidos com
tamanho e forma das partículas controlados. O processamento químico permitiu aos
produtores de zircônia agrupar produtos não só por suas aplicações, mas também
para atingir necessidades específicas do consumidor.
A decomposição química é utilizada em processos comerciais adotados pela
Tosoh Corporation, no Japão, na produção de zircônia e zircônia parcialmente
estabilizada com ítria (Y-PSZ). Tais processos envolvem a precipitação aquosa de

36
hidróxidos de zircônio e ítria a partir do oxicloreto de zircônio e do cloreto de ítrio,
respectivamente (FIG. 3.6). Estes precipitados são submetidos a uma operação de
secagem para se obter Y-PSZ que é sinterizada a 1400°C (Oliveira, A. P. A., 1997).
FIG. 3.6 Preparação de zircônia parcialmente estabilizada com ítria a partir de oxicloreto de zircônio.
3.1.6 ZIRCÔNIA EM PRÓTESES ORTOPÉDICAS
A alumina demonstrou sua biocompatibilidade e resistência ao desgaste,
contudo, exibiu moderada resistência à flexão e tenacidade. Por esta razão, o
diámetro de cabeças de fémur ficou limitado a 32 mm. A zircônia é também inerte
em meio fisiológico porém, apresenta uma vantagem sobre a alumina pois tem maior
resistencia à flexão, maior tenacidade à fratura e menor módulo de elasticidade.
A zircônia sugerida para implantes cirúrgicos se apresenta em dois tipos
básicos: zircônia tetragonal estabilizada com ítria (Y-TZP) e zircônia parcialmente
estabilizada com óxido de magnesio (Mg-PSZ). As propriedades de materiais
cerâmicos a base de zircônia de elevada pureza utilizado em implantes comerciais,
juntamente com o padrão da “International Standard Organization” - ISO 13356 são
apresentadas na TAB. 3.2.

37
TAB. 3.2 Propriedades de biomateriais comerciais de zircônia e o padrão da ISO-13356.
Propriedades
Unidade
Implantes cerâmicos de
zircônia comerciais
Y-TZP
Implantes cerâmicos de
zircônia comerciais
Mg-PSZ
ISO - 13356
Composição
ZrO2 + 3% mol
Y2O3
ZrO2 + 8%-10% mol
MgO
ZrO2 + HfO2 > 93,6% HfO2 < 5% Y2O3 = 5,1 ± 0,25 % Al2O3 < 0,5% Outros óxidos < 0,5% U, TH óxidos < 20 ppm
Densidade g/cm3 > 6,00 5,74-6,00 > 6,00 Porosidade % < 0,1 - -
Tamanho médio de grão (μm) 0,2-0,4 12-30 < 0,6 Resistência à flexão (MPa) MPa 900-1200 450-700 > 900 Resistência à compressão MPa 2000 2000 -
Módulo de elasticidade GPa 210 210 - Tenacidade à fratura K1c MPa.m1/2 7-10 7-15 - Coeficiente de expansão
térmica x10-6/°C 11 7-10 -
Condutividade Térmica W/m°K 2 2 - Dureza (HV) GPa 12 12 -
A zircônia é adequada para superfícies de apoio em próteses totais de quadril.
Todavía, existem três principais controvérsias a respeito da zircônia. A primeira é a
redução da resistência com o tempo em meio fisiológico. A segunda diz respeito as
suas propriedades de desgaste e a terceira é o potencial radioativo deste material.
A transformação martensítica de fase tetragonal para monoclínica prejudicial que
ocorre em zircônia dopada com ítria devido ao envelhecimento em água bem como
a consequente redução da tenacidade que a acompanha, têm sido documentada.
Todavía, testes de simulação em fluidos humanos e em animais têm mostrado
somente pequenos decrécimos na resistencia à fratura e tenacidade. A resistência
observada após 2 anos é ainda muito maior que a da alumina testada sob as
mesmas condições.
A zircônia foi testada como componente de articulação em próteses totais de
quadril contra UHMWPE e apresentou resultados de desgaste inferiores ao sistema
alumina/ UHMWPE.

38
3.1.7 RADIOATIVIDADE DE BIOMATERIAIS CERÂMICOS
A zircônia é freqüentemente acompanhada por elementos radioativos de vida
média longa, tais como o tório e o urânio. A separação destes elementos é difícil e
cara. Dois tipos de radiação dizem respeito a zircônia: gama e alfa. A radioatividade
gama de próteses de cabeça de fêmur de alumina, zircônia e ligas Co-Cr foram
medidas. A alumina apresentou a menor radioatividade e a zircônia e as ligas Co-
Cr apresentaram valores semelhantes. Os dados sugerem que o nível de radiação
gama não é o principal problema em biocerâmicas de zircônia. Todavia, quantidade
significante de radiação alfa tem sido observada em cerâmicas de zircônia com fins
a implantes cirúrgicos. As partículas alfa devido a sua alta capacidade de ionização,
destroem células de tecidos moles e duros. De acordo com Hulbert, S. F., 1993, a
emissão alfa observada em próteses de cabeça de fêmur em zircônia é pequena
porém, questões sobre os efeitos de longo tempo da emissão de radiação alfa em
cerâmicas de zircônia devem ser investigados.
A zircônia é refinada a partir de minério natural, zircon e usualmente contém
traços de outros elementos dependendo da fonte do minério original. Em particular,
zircônia típica contém traços de elementos da série dos actinídeos tais como 226Ra e 228Th. A radioatividade da zircônia é desprezível. Por exemplo, a radiação emitida
pela zircônia estabilizada com ítria (3% mol) é da mesma ordem de grandeza que o
da alumina, os quais são ambas diversas ordens de grandeza menores que a
tipicamente medida para a água, leite, vegetais e carne na Europa. A radioatividade
de cabeças de fêmur em cerâmica Y-TZP mostrou ser similar as de alumina e das
ligas de cromo-cobalto aceitas para implantações em humanos. A dose de radiação
de cada material é bem inferior aos limites de radiação aceitáveis especificados na
Europa para exposição externa do corpo humano e interna de órgãos e tecidos, e
além disso, não era maior do que a radiação ambiente. Resultado semelhante foi
obtido em estudos de diversos pós de zircônia comercialmente disponíveis medidos
pelo laboratório de radiação australiano (Standard, O., 1995).
3.1.8 PILARES CERÂMICOS
A implantodontia é a especialidade da odontolgia que visa a reposição de
elementos dentários perdidos ou removidos por meio de implantes dentários. Na

39
FIG. 3.7 pode ser visto a reposição de um elemento dentário perdido por um
implante de titânio inserido na mandíbula e a restauração protética ancorada ao
implante.
FIG. 3.7 Implante dentário substituindo um elemento dentário.
Como foi visto, alguns materiais foram utilizados no passado para a confecção
de implantes dentários, dentre eles, a alumina. Estes implantes apresentaram
excelente biocompatibilidade, porém, por problemas de fratura, foram abandonados.
Após alguns estudos em ortopedia, o médico Brånemark, P.I., (1987), verificou
que o titânio era um excelente material para utilização em implantes dentários e
introduziu o conceito da osseointegração. De acordo com Brånemark, existe uma
correlação direta entre o osso estruturado vivo e a superfície do implante quando
este está submetido às cargas mastigatórias. A partir daí, os implantes
osseointegrados dominaram o mercado e se tornaram uma rotina para os
profissionais da área.
A restauração protética sobre implantes envolve uma conexão entre o implante
metálico e a coroa, conhecida como pilar e a coroa cerâmica propriamente dita.
Este pilar é metálico, de titânio comercialmente puro ou liga de titânio. O pilar
metálico é acoplado ao implante e fixado a ele por meio de um parafuso de titânio. A
FIG. 3.8 apresenta dois pilares metálicos em titânio sendo um reto e outro angulado.

40
FIG. 3.8 Pilares metálicos em titânio.
Por razões estéticas, alguns pilares começaram a ser fabricados com materiais
cerâmicos, buscando minimizar problemas decorrentes de retração gengival e
conseqüente exposição do metal. A FIG. 3.9 ilustra a exposição do metal,
comprometendo a estética.
FIG. 3.9 Retração gengival e exposição do metal.
Atualmente, o sistema totalmente cerâmico está disponível em cinco tipos de
processos distintos: (La Croix, S. P., 1998).
a) Processo convencional por escultura da porcelana feldspática.
b) Processo de cerâmica usinada (Cerec), que consiste do torneamento de
uma estrutura utilizando um dispositivo que copia a superfície de um

41
padrão (metal, cerâmica ou polímero), e transfere estes dados para uma
unidade fresadora onde um bloco cerâmico é usinado. Processo de
cerâmica moldada sob pressão (IPS Empress), que consiste no
aquecimento e compressão da cerâmica em um molde sob pressão
(Anusavice, K. J., 1998).
c) Processo de cerâmica infiltrada (In-Ceram), onde um núcleo de Al2O3 ou
MgAl2O4 (Spinell), parcialmente sinterizado possuindo uma matriz com
espaços que são preenchidos com vidro fundido por atração capilar.
d) Processo de cerâmica injetada por centrifugação (Dicor) (Rosenblum, M.
et al., 1997).
Com exceção do processo convencional, as técnicas citadas utilizam cerâmicas
de alta temperatura de sinterização. Devido aos avanços significativos dessas
cerâmicas de nova geração, a partir de 1990, tornou-se possível a execução de
próteses dentais totalmente cerâmicas. Há uma tendência natural de substituição da
subestrutura metálica por cerâmicas de maior resistência mecânica (Phillips, R.,
1996; Rosenblum, M. et al., 1997).
Materiais como a alumina, e mais recentemente a zircônia tetragonal
policristalina estabilizada com ítria (Y-TZP), vem sendo utilizados na confecção de
pilares totalmente cerâmicos para implantes osseointegrados.
Os pilares cerâmicos importados comercialmente disponíveis em alumina e
zircônia foram desenvolvidos para sistemas de implante específicos. Não existe
tecnologia nacional de pilares cerâmicos. Os pilares importados, apresentam um
custo elevado, o que inviabiliza a sua utilização em larga escala. A TAB. 3.3
apresenta os pilares comerciais disponíveis. Observando a tabela é possível
perceber que alguns pilares são pré-fabricados, o que exige que sejam usinados
pelos profissionais de odontologia na forma adequada para em seguida serem
utilizados. São eles o Cerabase, CerAdapt e o Zi-Real. O pilar denominado
Procera é um tipo de pilar personalizado, ou seja, ele é confeccionado a partir de
um molde, e já vem pronto para imediata utilização pelos profissionais de
odontologia.

42
TAB. 3.3 Pilares cerâmicos comerciais.
SISTEMA NOME
COMERCIAL
TIPO MATERIAL
Friadent
(Alemanha)
Cerabase
Pré-fabricado
Al2O3
(com encaixe em titânio)
Nobel Biocare
(Suécia)
Procera
CerAdapt
Personalizado
Pré-fabricado
Al2O3
Al2O3
3i
(EUA)
Zi- Real
Pré-fabricado
Y-TZP
(com cilindro na base em titânio)
A FIG. 3.10 apresenta um pilar cerâmico pré-fabricado comercialmente
conhecido como CerAdapt, o qual é totalmente confeccionado em alumina de alta
densidade, bem como seu parafuso de fixação.
FIG. 3.10 Pilar CerAdapt em alumina e seu parafuso de fixação.
A FIG. 3.11 apresenta o pilar pré-fabricado Cerabase. O Cerabase é constituído
de um corpo confeccionado em alumina, e o encaixe deste corpo com o implante é
feito através de uma base em titânio, diferentemente do pilar CerAdapt, que como
visto anteriormente, é totalmente cerâmico. Este pilar também é fixado ao implante
por meio de um parafuso.

43
É possível notar ainda na FIG. 3.11 que o pilar se apresenta com quatro
diâmetros diferentes, sendo cada um para diâmetro de implante diferente.
FIG. 3.11 Pilar Cerabase em alumina e sua base de titânio.
A FIG. 3.12 apresenta um pilar pré-fabricado conhecido como Zi-Real, o qual é
confeccionado em zircônia tetragonal policristalina estabilizada com ítria.
O pilar Zi-Real apresenta um corpo cerâmico o qual envolve um cilindro de
titânio presente na base do pilar. A fixação com o implante é feita com um parafuso
de titânio.
Na FIG. 3.12, são apresentados ainda dois diâmetros diferentes para este pilar.
FIG. 3.12 Pilar pré-fabricado Zi-Real em zircôna.

44
Pilares personalizados Procera são fabricados em alumina de alta densidade e
pelo sistema CAD-CAM, ou seja, o material é fresável através da utilização de
desenho (forma desejada) e processamento computadorizado.
Para um melhor entendimento da seqüência de utilização de um pilar, a
FIG.3.13(a) apresenta a primeira fase que consiste na instalação do implante
osseointegrado de titânio.
(a) (b) (c)
FIG. 3.13 Sequência de utilização do pilar: (a) Implante osseointegrado de titânio instalado; (b) Pilar cerâmico fixado ao impante; (c) Coroas cerâmicas cimentadas.
Após esta etapa, o pilar cerâmico já usinado na forma adequada é acoplado ao
implante e fixado a ele por meio de um parafuso, o que pode ser visto na FIG. 3.13b.
Em seguida, as coroas metalocerâmicas são cimentadas nos pilares cerâmicos,
chegando desta forma ao resultado final do tratamento, conforme FIG. 3.13c.
3.2 CERÂMICAS A BASE DE ALUMINA
A finalidade deste item é apresentar as informações de interesse disponíveis na
literatura, de modo a compreender melhor a estrutura cristalina da alumina.
3.2.1 ESTRUTURA CRISTALINA DA ALUMINA
A estrutura cristalina da α-alumina conhecida como corundum é a forma
termodinamicamente estável da alumina em todas as temperaturas. Ela apresenta
uma estrutura cristalina romboédrica compreendendo uma rede hexagonal de
empacotamento onde os íons Al 3+ estão ordenados simetricamente em dois terços
dos interstícios octaedrais (FIG. 3.14). Na estrutura corundum os íons Al 3+ estão
coordenados a seis íons de oxigênio (Feighery, A. J. & Irvine, J. T. S., 1999).

45
FIG. 3.14 Estrutura cristalina da alumina.
3.3 CERÂMICAS A BASE DE ZIRCÔNIA
A finalidade deste item é apresentar as informações de interesse disponíveis na
literatura, de modo a compreender melhor a estrutura cristalina dos polimorfos da
zircônia, o equilíbrio das fases, a transformação de fase tetragonal para monoclínica,
bem como os mecanismos de tenacificação por ela produzidos.
3.3.1 ESTRUTURA CRISTALINA DA ZIRCÔNIA
A zircônia pura é polimórfica. Ela apresenta três estruturas cristalinas:
monoclínica (estável até 1173 ºC), tetragonal (até 2370 ºC) e cúbica com sua
estabilidade garantida até a temperatura de fusão de 2680 ºC. A zircônia cúbica é
baseada na estrutura cristalina da fluorita , onde os átomos de zircônio ocupam a
2/3 dos Interstícios octaédricos ocupados por Al3+.

46
posição na rede cúbica de face centrada (CFC) (0,0,0) e o oxigênio a posição
(1/4,1/4,1/4). As estruturas tetragonal e monoclínica são consideradas distorções da
estrutura anterior (Stevens, R., 1986).
Dados cristalográficos dos polimorfos da zircônia são apresentados na TAB. 3.4.
TAB. 3.4 Características dos polimorfos da zircônia
Estrutura
cristalina Cúbica Tetragonal Monoclínica
a = 5,124 Å a = 5,094 Å a = 5,156 Å
b = 5,191 Å
Parâmetro
de rede c = 5,177 Å c = 5,304 Å *O parâmetro de rede varia com o ânion e sua concentração.
3.3.1.1 ZIRCÔNIA MONOCLÍNICA
A forma natural da zircônia, a badeleita (“baddeleyite”), apresentada na FIG.
3.15 contém aproximadamente 2% de HfO2 (óxido de háfnio), o qual é tão similar a
zircônia em estrutura e propriedades químicas, que tem pequeno efeito (Stevens,
R.,1986).
Os íons Zr4+ tem número de coordenação sete com os íons de oxigênio
ocupando os interstícios tetraedrais, sendo que a distância média entre o íon de
zircônia e três dos sete íons de oxigênio é de 2,07 Å . Já a distância média entre o
íon de zircônia e os outros quatro íons de oxigênio é de 2,21 Å. Assim, um dos
ângulos (134,3°) na estrutura difere significantemente do valor tetraedral (109,5°).
Desta forma, a estrutura dos íons oxigênio não é planar e uma curvatura ocorre no
plano dos quatro oxigênios e o plano dos três oxigênios é completamente irregular.
Este fato ode ser útil na explicação do comportamento de maclagem da
badeleita, onde é pouco comum cristais não maclados. O plano de maclação é
composto de íons oxigênio os quais se movem de sua posição de equilíbrio
resultando em uma distorção mínima (Stevens, R., 1986).

47
FIG. 3.15 Estrutura cristalina da zircônia monoclínica.
3.3.1.2 ZIRCÔNIA TETRAGONAL
Na sua forma tetragonal (FIG. 3.16), os íons Zr4+ apresentam número de
coordenação oito onde novamente surge uma distorção devido ao fato de quatro
íons de oxigênio estarem a uma distância de 2,065 Å na forma de um tetraedro
plano e os outros quatro a uma distância de 2,455 Å em um tetraedro alongado e
com rotação de 90°.
Por simplicidade, os parâmetros da rede para a forma tetragonal têm sido
freqüentemente descritos em termos da simetria tetragonal cúbica de face centrada,
ao invés de corpo centrado, com o objetivo de relacionar com a estrutura de face
centrada fluorita (CaF2) (Stevens, R., 1986).
FIG. 3.16 Estrutura cristalina da zircônia tetragonal.

48
3.3.1.3 ZIRCÔNIA CÚBICA
A estrutura cúbica da zircônia (fluorita) pode ser vista na FIG. 3.17 como uma
rede cúbica simples com oito íons de oxigênio, os quais são rodeados por um
arranjo cúbico de cátions, isto é, os oxigênios ocupam os interstícios tetraedrais de
uma rede cúbica (CFC) de empacotamento de cátions (Feighery & Irvine, 1999).
FIG. 3.17 Estrutura cristalina da zircônia cúbica.
Na FIG. 3.18 são apresentadas as três estruturas cristalinas da zircônia.
FIG. 3.18 Diferentes estruturas cristalinas da ZrO2 cúbica, tetragonal e monoclínica , respectivamente.

49
3.3.2 EQUILÍBRIO DA FASE BINÁRIA
3.3.2.1 ZIRCÔNIA PURA
A zircônia pura tem um alto ponto de fusão (2680 ºC) e uma baixa
condutividade térmica. O polimorfismo da zircônia restringe o seu uso na indústria
cerâmica. Durante o aquecimento, a zircônia passará por um processo de
transformação de fase. A mudança em volume associada a essas transformações
podem provocar tensões externas e até mesmo fratura do material, tornando
impossível o uso da zircônia pura em muitas aplicações.
Adições de certas quantidades de óxidos estabilizantes, sendo os mais
comuns óxidos de terras raras ou compostos semelhantes como CaO, CeO2, MgO e
Y2O3 estabilizam a forma cúbica evitando transformações de fase durante o
aquecimento e resfriamento. Os estabilizantes mais adequados são óxidos cujos
cátions possuam estrutura cristalina cúbica e razão da diferença entre raios dos
cátions em relação ao raio do zircônio inferior a 40 % (Oliveira, A. P. A., 1997). A
TAB. 3.5 apresenta alguns óxidos estabilizantes e suas respectivas razões
percentuais.
TAB. 3.5 Raios iônicos de alguns elementos estabilizadores das formas polimórficas de altas temperaturas da zircônia e a razão (R) entre raio iônico do elemento estabilizante e raio do
zircônio.
Elemento Raio iônico (Å) R(%)
Zr4+ 0,84 -
Ba2+ 1,42 +69
Ca2+ 1,12 +33
Ce4+ 0,97 +15
Hf4+ 0,83 -1
Mg2+ 0,89 +6
Sc3+ 0,87 +3,6
Sr2+ 1,26 +50
Y3+ 1,019 +21
Yb3+ 1,125 +36
Os sistemas de óxidos estabilizantes podem ser classificados como
precipitados e de soluções sólidas.

50
Os sistemas precipitados são aqueles em que o estabilizante possui baixa
solubilidade sólida na rede da zircônia em temperatura onde a migração dos cátions
ainda é ativa (T>1400 °K). Materiais produzidos a partir de tais elementos
estabilizantes (ex: Ca e Mg) em geral são parcialmente estabilizados, ou seja,
possuem as estruturas tetragonal e monoclínica.
Sistemas de soluções sólidas ocorrem quando a solubilidade do estabilizante
(ex: Y2O3 e CeO2) é tal que cessa a mobilidade do cátion e este é retido em solução
sólida em temperaturas relativamente baixas. Estes sistemas podem formar
policristais de zircônia tetragonal, dependendo do teor de estabilizante.
A FIG. 3.19 apresenta o diagrama de fases do sistema ZrO2 / Y2O3. No
diagrama de fase do sistema ZrO2 / Y2O3 , é possível observar que a região de
estabilidade da solução sólida tetragonal é bastante ampla e que a temperatura da
transformação t → m é bastante baixa. O óxido de ítrio ou ítria é extraído do
mineral xenotímio e esta é utilizada como aditivo de sinterização em sialons e nitreto
de silício. É usada também para a estabilização da zircônia e para melhorar à
tenacidade à fratura (Kumar, A., S., et al., 2004b).
FIG. 3.19 Diagrama de fases dos sistema ZrO2/Y2O3.

51
Cerâmicas de zircônia são geralmente produzidas na faixa de 2,5 % - 3,5 %
de Y2O3 pelo aquecimento a 1400-1500°C seguido de resfriamento rápido, gerando
assim policristais de ZrO2 tetragonal. Materiais de zircônia parcialmente estabilizada
também podem ser produzidos para teores de Y2O3 entre 3 e 6 % em mol.
3.3.2.2 ZIRCÔNIA PARCIALMENTE ESTABILIZADA(PSZ)
A zircônia parcialmente estabilizada (PSZ) é uma mistura de polimorfos da
zircônia fase cúbica (Css) e fase tetragonal metaestável (Tss) ou monoclínica (Mss).
Usualmente, a zircônia parcialmente estabilizada (PSZ) consiste de quantidades
maiores que 8% mol (2,77 % peso) de MgO; 8 % mol (3,81 % peso) de CaO ou
ainda 3 % - 4 % mol (5,4 % - 7,1 % peso) de Y2O3.
A zircônia PSZ típica apresenta uma microestrutura de grãos cúbicos cujo
tamanho está na faixa de 40 μm - 60 μm com precipitados submicrométricos
tetragonais e monoclínicos, finamente dispersos (Heuer, A. H. et al., 1988).
3.3.2.3 ZIRCÔNIA TETRAGONAL POLICRISTALINA (TZP)
Uma pequena quantidade de estabilizante adicionada a zircônia pura levará a
sua estrutura a uma fase tetragonal em temperaturas superiores a 1000°C e uma
mistura de fase cúbica e monoclínica ou fase tetragonal em temperaturas menores.
Portanto, a zircônia parcialmente estabilizada é também conhecida como zircônia
tetragonal policristalina (TZP).
A zircônia tetragonal policristalina (TZP) consiste de uma quantidade de
estabilizante menor do que a PSZ, por exemplo, 4 % - 5 % peso de Y2O3.
A zircônia TZP típica apresenta uma microestrutura de grãos de zircônia
predominantemente tetragonais na faixa de 1 μm a 5 μm (Heuer, A. H. et al., 1988).
3.3.2.4 ZIRCÔNIA TOTALMENTE ESTABILIZADA
É necessário que seja adicionada uma quantidade maior que 16 % mol (7,9%
peso) de CaO, ou 16 % mol (5,86 % peso) de MgO, ou ainda 8 % mol (13,75 % peso
de Y2O3, para que se possa obter uma estrutura de zircônia totalmente estabilizada.

52
Sua estrutura se torna uma solução sólida cúbica, a qual não apresenta
transformação de fase da temperatura ambiente até 2500°C
(www.standfordmaterial.com).
3.3.3 TRANSFORMAÇÃO MARTENSÍTICA (T-M)
A transformação das partículas de zircônia de fase tetragonal para fase
monoclínica é do tipo martensítica, sendo acompanhada por um aumento de
volume de 3 a 5 %, o que é suficiente para exceder o limite elástico mesmo em
pequenos grãos de zircônia monoclínica e esta expansão de volume somente pode
ser acomodada pela formação de trincas, o que inviabiliza a utilização da zircônia
pura. Porém, com a adição de alguns óxidos (MgO, CaO, CeO2, Y2O3 e terras
raras), as fases cúbica e tetragonal podem ser parcialmente ou totalmente
estabilizadas, possibilitando a obtenção de propriedades mecânicas necessárias
para sua utilização (Stevens, R., 1986; Wang, J. & Stevens, R.,1989).
A transformação ocorre por processo sem difusão, não é termicamente ativada,
é uma transformação militar, onde a quantidade transformada não depende do
tempo, para uma dada temperatura, uma fração da fase original se transforma
rápido, cessando a transformação, a fração transformada é função da temperatura, a
velocidade da transformação é alta, não há mudança de composição do produto
formado (Porter, D. A & Easterling, K. E., 1992). A transformação é
termodinamicamente reversível a T= 1174 °C (Stevens, R. 1986), a mudança de
posição atômica é feita bruscamente, possuindo uma curva de histerese térmica
entre ciclos de resfriamento e aquecimento. A transformação inicia-se no ciclo de
resfriamento à temperatura de transformação martensítica (Ms) que, para
monocristais de ZrO2 ou corpos densos de ZrO2 policristalina, situa-se entre 950 °C
e 850 ºC. O valor de Ms é influenciado por diversos parâmetros, tamanho, formato e
localização (inter ou intragranular) das partículas de ZrO2, quantidade de óxidos
estabilizadores, diferença de coeficiente de dilatação térmica, e outros (Bressiani, J.
C., & Bressiani, A. H. A., 1988).
A característica mais importante do sistema ZrO2-Y2O3 é o decréscimo da
temperatura de transformação tetragonal-monoclínica com o aumento da quantidade
de ítria (eutetóide a 4,6 % mol). Com isso, partículas maiores de zircônia

53
estabilizada podem ficar retidas na forma tetragonal metaestável (Stevens, R.,
1986). Este fato também ocorre no sistema ZrO2-CeO2, porém não ocorre nos
sistemas ZrO2-MgO e ZrO2-CaO.
Adicionando ítria à zircônia, inibimos a transformação martensítica. O
decréscimo da temperatura de transformação com adições de ítria produz uma
menor mudança na temperatura e menos deformação térmica (Heuer, H. A. 1987).
A estabilização da fase tetragonal é afetada pelo tamanho de partícula e módulo
de elasticidade do compósito, assim como pelo estabilizante e sua concentração
(Shulman, H.S., et al., 1998).
Caso o grão de zircônia esteja isolado, a transformação t → m ocorre uma vez
que as condições de nucleação sejam atingidas. No entanto, quando este grão está
imerso em uma matriz, a variação de volume (3 - 5 %) e a deformação associada a
esta transformação não podem ser aliviadas por mecanismos difusionais, mas
devem se acomodar por deformação plástica ou elástica na região próxima ao grão.
Conseqüentemente surge uma energia de deformação associada ao grão e à matriz
na vizinhança do grão. A produção desta energia de deformação, que resulta na
compressão da partícula, adiciona um termo extra à energia livre do sistema,
tornando a transformação menos provável. Para que esta transformação prossiga, o
sistema deve ser superresfriado a fim de aumentar a força motriz química para a
transformação. Para que a fase tetragonal seja retida, é necessário o controle de
fatores microestruturais(tamanho de grão) e químicos(teor de óxido estabilizante).
3.3.4 MECANISMOS DE TENACIFICAÇÃO
É conhecido na literatura o potencial da zircônia por aumentar tanto a resistência
como a tenacidade de cerâmicos por utilização da transformação de partículas
tetragonais metaestáveis induzida pela presença de um campo de tensões na ponta
da trinca (Stevens, R.,1986, etc...). A mudança de volume e deformação cisalhante
desenvolvida na reação martensítica foram reconhecidas por agir em oposição a
abertura de uma trinca e ainda por aumentar a resistência do cerâmico à
propagação da trinca. É reconhecido que além da deflexão da trinca, a qual pode
ocorrer em cerâmicos bifásicos, a transformação de fase tetragonal para
monoclínica pode desenvolver significante melhorIa na tenacidade dos cerâmicos

54
através de três diferentes mecanismos que serão descritos a seguir: Aumento de
tenacidade induzido por tensão, microtrincamento e tensões superficiais
compressivas (Wang, J. & Stevens, R., 1989; Cannon, W. R., 1989; Justo, L. F. et
al., 1990).
3.3.4.1 AUMENTO DE TENACIDADE INDUZIDO POR TENSÃO
O campo de tensão da trinca pode iniciar a transformação martensítica, desde
que as partículas transformadas se expandam contra uma matriz, formando um
campo de tensões compressivas superficiais próxima à ponta da trinca, que está
diretamente envolvida com a absorção de energia e inibição de propagação da trinca
(Cannon, W. R., 1989), como pode ser visto na FIG. 3.20.
FIG. 3.20 Representação esquemática das tensões geradas em uma matriz que possui uma região que envolve uma trinca, quando esta região é submetida a uma transformação de fase
expansiva. As tensões residuais induzidas na matriz vizinha à região agem no sentido de fechar a trinca.
A mudança de fase tetragonal para monoclínica ocorre associada ao campo de
tensão de uma trinca em propagação. Grandes tensões trativas são geradas ao
redor de uma trinca, principalmente na ponta da trinca. Estas tensões relaxam a
restrição elástica imposta pela matriz às partículas de zircônia tetragonal e, se elas
são suficientemente grandes, a tensão trativa atuando sobre as partículas de ZrO2
irá favorecer a transformação das mesmas para a fase monoclínica. A expansão
volumétrica (3% a 5 %) e a deformação cisalhante (1% a 7 %) desenvolvida durante
a transformação resultam em uma deformação compressiva na matriz. Tais tensões
fecham a trinca e agem como uma barreira energética para o crescimento da trinca.
Como esses fenômenos ocorrem associados à trinca em propagação, um trabalho
extra é requerido para propagar a trinca através da microestrutura cerâmica, o que

55
se traduz em um aumento da tenacidade e da resistência mecânica (Stevens, R.,
1986; Hannink, R. H. J., 2000; Wang, J. & Stevens, R., 1989). Este mecanismo de
tenacificação está ilustrado na Figura 3.21.
FIG. 3.21 Mecanismo de aumento de tenacidade por transformação da zircônia.
Existe uma faixa de tamanho crítico da zircônia dentro da qual as partículas
tetragonais podem ser transformadas por tensão. Se as partículas são menores que
o tamanho crítico, elas não irão transformar, se maiores, irão se transformar
espontaneamente. Este intervalo crítico depende da matriz utilizada, da quantidade
de óxidos estabilizadores, e da fração volumétrica da zircônia (Bressiani, J. C. &
Bressiani, A. H. A.,1988).
A medida em que a quantidade de óxido estabilizante é aumentada, ocorre o
decréscimo da energia livre associada à transformação e desta forma, partículas
maiores são induzidas a permanecer na forma tetragonal metaestável (Stevens,
1986).
Duas aproximações semi-quantitativas da transformação foram apresentadas. O
modelo inicial proposto por Evans (Ruf, H. & Evans, A. G.,1983; McMeeking, R. M. &
Evans, A. G., 1982), baseou-se na mudança da energia total e explica o campo de
tensão na ponta da trinca como uma zona do processo. A aproximação de Lange
(Lange, F. F., 1982a, Lange, F. F. 1982b) é associada à um modelo termodinâmico,
considerando as condições para reter a zircônia tetragonal metaestável na matriz
com o objetivo de aumentar a tenacidade. Lange demonstrou que a mudança da

56
energia livre ΔG, associada com a transformação pode ser alterada com a
temperatura e a composição usada, e a maximização da tensão que induz a
transformação pode ser obtida pelos seguintes métodos:
a) maximizando a fração volumétrica das partículas de zircônia
tetragonal retidas à temperatura ambiente;
b) aumentando o módulo de elasticidade do compósito pela adição
de uma segunda fase quimicamente compatível e com um módulo
elástico maior. No caso a Al2O3 tem o módulo elástico
aproximadamente duas vezes maior do que o da ZrO2 (380 e 210
GPa respectivamente);
c) diminuindo a variação da energia livre associada com a
transformação martensítica. Para esta transformação, a energia
livre diminui com o aumento da temperatura e do teor de
estabilizante (Y2O3). (Justo, L.F. et al., 1990).
3.3.4.2 MICROTRINCAMENTO
O microtrincamento pode ser induzido pela incorporação de partículas de ZrO2
em uma matriz cerâmica como a zircônia cúbica ou alumina por exemplo.
No resfriamento, na temperatura de transformação (T T - M), a expansão de
volume de 3% a 5% ocorre nas partículas de zircônia. Tensões tangenciais são
geradas ao redor das partículas transformadas, as quais induzem a nucleação de
microtrincas na matriz.
O microtrincamento é responsável pelo aumento da energia de absorção durante
a propagação de uma trinca, aumento conseqüentemente a tenacidade do cerâmico.
A condição ótima é atingida quando as partículas são grandes o suficiente para
sofrerem transformação, mas pequenas o suficiente para promover um
microtrincamento limitado.
Objetivando obter tenacidade máxima, a fração volumétrica de inclusões de
zircônia deve estar em um nível ótimo. A tenacidade atinge um valor máximo e a
partir daí, as microtrincas geradas pelas partículas de zircônia irão interagir umas
com as outras resultando em um decréscimo da resistência (Clausen, N. J., 1976).

57
3.3.4.3 TENSÕES SUPERFICIAIS COMPRESSIVAS
O desenvolvimento de camadas superficiais compressivas desenvolvidas na
zircônia a partir de transformação é um fenômeno bem conhecido (Reed, J. S. &
Lejus, A. M., 1977; Lange, F. F. & Evans, A. G., 1979). Estas tensões se
desenvolvem como resultado da transformação de partículas de zircônia de
tetragonal para monoclínica (t → m) na superfície ou em sua vizinhança, devido a
ausência de restrição próxima a superfície livre. Um considerável aumento na
resistência à fratura do cerâmico pode ser obtido.
A usinagem foi reconhecida como o mais eficiente método de indução da
transformação de partículas tetragonais, já que tensões compressivas podem ser
geradas a uma profundidade de 10 μm a 100 μm.
O efeito do aumento de resistência é conhecido ser dependente da severidade
da usinagem (Pascoe, R. T. & Garvie, R. C., 1977). A quantidade de fase
monoclínica presente na superfície decresce rapidamente quando a superfície é
cuidadosamente polida em camadas. O benefício máximo é alcançado se a
espessura da camada de tensão compressiva for maior que o tamanho crítico da
falha, porém pequena quando comparada com a seção transversal do cerâmico.
O desenvolvimento de uma camada superficial compressiva no cerâmico quando
usinado é provavelmente o mais significante fenômeno associado ao aumento de
tenacidade por transformação em cerâmicos.
3.4 COMPÓSITOS A BASE DE ALUMINA E ZIRCÔNIA
A adição de zircônia na alumina como aditivo de sinterização vem sendo
praticada a longo tempo com objetivo de densificação de cerâmicas a base de
alumina. Todavia, o conceito de tenacificação de cerâmicas de alumina por
dispersão de partículas de zircônia em uma matriz somente foi reconhecida nos
últimos 20 anos. A introdução de pequena quantidade de zircônia em alumina como
aditivo de sinterização leva a formação de solução sólida a qual promove o processo
de densificação pela introdução de defeitos.(Wang, J. & Stevens, R., 1989).
A microestrutura de compósitos a base de alumina e zircônia é caracterizada
pela presença de duas fases distintas, ao invés de uma solução sólida. (Wang, J. &
Stevens, R., 1989).

58
Compósitos de alumina e zircônia são conhecidos como ZTA(“zirconia
toughened alumina” ou alumina tenacificada por zircônia), e ATZ(“alumina
toughened zirconia” ou zircônia tenacificada por alumina). (De Aza, A. H. et al.,
2002).
O compósito conhecido como ZTA consiste de uma matriz de alumina reforçada
com partículas de zircônia. Estas partículas podem estar estabilizadas ou não. O
compósito conhecido como ATZ consiste de uma matriz de zircônia estabilizada
reforçada com partículas de alumina.(De Aza, A. H. et al., 2002).
A adição de uma segunda fase resulta em uma melhoria de propriedades como
resistência à flexão e tenacidade à fratura (Claussen, N.,1976; Lange, F.F., 1982c;
Hori, S. et al., 1986; Casellas, D., 1999, Evans, A. G., 1990). Por esta razão, estes
materiais são promissores em diversas aplicações que requerem elevada dureza,
alta resistência ao desgaste e relativa tenacidade à fratura.
Com ambos materiais bifásicos (ZTA e ATZ), é esperada a obtenção de maiores
valores de tenacidade à fratura quando comparado com os materiais cerâmicos
monofásicos, porém, maiores valores são esperados para os compósitos ZTA
(Fantozi G. & Orange G., 1986; Karihaloo, B.L., 1991; Becher, P.F. et al., 1993;
Rühle, M. et al., 1984; Gregori, G. et al., 1999).
Adicionalmente, no caso da matriz de zircônia estabilizada (ATZ), problemas
relacionados com a estabilidade hidrotérmica permanecem devido à necessidade da
adição de ítria para estabilização da zircônia (Green, D. J. et al.,1989). Por outro
lado, com uma matriz de alumina esta adição pode ser evitada uma vez que esta
age restringindo as partículas de zircônia, retendo a zircônia tetragonal no estado
metaestável, tenacificando o material (Green, D. J. et al.,1989; Claussen, N., 1976).
Além disso, a dureza do compósito com matriz de alumina (ZTA) seria maior,
porque a alumina apresenta maior dureza que a zircônia, tendo portanto maior
resistência ao desgaste (Heros, R. & Willmann, G.,1998).
De acordo com Gregori, G., (1999), o compósito ZTA apresenta uma associação
de elevada tenacidade com propriedades peculiares da alumina como excelente
resistência ao desgaste e estabilidade química. Todas estas características
qualificaram o ZTA para diversas aplicações e o tornam um promissor candidato a
utilização como material de implantes, uma vez que a comunidade de biomateriais já
está familiarizada com a alumina e a zircônia separadamente. Apesar disso, muito

59
pouco foi publicado na literatura sobre o ZTA como biomaterial (Piconi, C. &
Maccauro, G., 1999).
3.5 PROCESSAMENTO
A finalidade deste item é apresentar as principais etapas do processamento
cerâmico que estão relacionadas com este trabalho.
3.5.1 ADITIVOS DE PROCESSAMENTO
Além dos aditivos de sinterização como por exemplo a utilização de MgO na
alumina para controlar o crescimento de grão, os principais aditivos normalmente
empregados num processamento cerâmico são os ligantes, plastificantes,
dispersantes e lubrificantes.
A adição dos ligantes visa a obtenção de compactos a verde com maior
resistência mecânica ao manuseio. O plastificante modifica o ligante para torná-lo
mais flexível. O dispersante é usado para melhorar a dispersão dos pós e evitar a
formação de aglomerados. Algumas vezes lubrificantes são requeridos para
reduzir o atrito entre partículas e na parede do molde, diminuindo o desgaste da
matriz e a pressão de ejeção. (Reed, J., 1987; Mesquita, C. E. M, 2001).
A presença dos aditivos permite que as partículas do pó cerâmico deslizem para
um novo arranjo mais empacotado, promove a distribuição de pressão de forma
equivalente durante o processo de prensagem (Richerson, D., 1992; Reed, J., 1988),
minimiza o surgimento de defeitos como delaminações ou trincas e além disso,
promove uma distribuição homogênea na densidade do corpo a verde, fato este que
é fundamental durante o processo de sinterização e para as propriedades do produto
final (Duailibi, J. F. & Rocha, J. C., 1988).
O estado de aglomeração do pó cerâmico influencia o processo de conformação
dos mesmos. Os aglomerados dependem da distribuição de tamanho, forma e
carga superficial das partículas. Os ligantes, plastificantes e dispersantes tem o
papel de alterar estas características (Duailibi, J. F. & Rocha, J. C., 1988).
Na conformação por prensagem a seco ou semi-seco, a adição de 1% a 3%
peso de ligantes e/ou plastificantes é recomendada. Embora sejam adicionadas em
pequenas quantidades e além disso sejam eliminadas posteriormente, não

60
aparecendo no produto final, são essenciais na obtenção de um produto cerâmico
final com um menor número de defeitos (La Croix, S. P., 1998, Oliveira, L. G., 2002).
Na conformação por extrusão e injeção, o teor de plastificantes deve ser superior,
em torno de 20% a 30% peso (Duailibi, J. F. & Rocha, J. C., 1988; Rocha, J. C. &
Duailibi, J. F., 1991).
3.5.2 MOAGEM
A moagem é um processo físico que utiliza forças mecânicas para reduzir os
tamanhos de partículas que constituem um pó cerâmico. Este preocesso tem sido
utilizado para a produção em escala de laboratório e industrialmente de vários pós
cerâmicos.
Os equipamentos mais utilizados para a moagem são os moinhos de bolas ou
cilindros. A redução do tamanho de partícula ocorre devido ao impacto das bolas ou
cilindros, também conhecidos como corpos moedores, contra o material sob
moagem e pelo atrito entre as partículas e as paredes do moinho. Os meios
moedores, corpos moedores ou elementos de moagem são geralmente constituídos
de materiais como alumina, zircônia e outros.
As variáveis que controlam a eficiência de moagem em um moinho são a
velocidade do moinho e os tamanhos das partículas. A tensão de cisalhamento e a
freqüência de rotação devem garantir a total estabilização da suspensão, ou seja,
um valor constante da viscosidade para uma determinada amostra em função do
tempo.
Em moinhos atritores (FIG. 3.22), existem limites bem definidos com relação à
sua capacidade de receber e transferir energia para os elementos de moedores e
conseqüentemente às partículas.
FIG. 3.22 Moinho atritor.

61
Um aumento da velocidade de rotação do moinho aumenta a potência
transferida, porém existe um limite, pois eventualmente a carga centrifugará e a
potência transferida irá diminuir. Nestes moinhos a velocidade é crítica e varia
inversamente com a raiz quadrada do diâmetro do mesmo (Costa, B.J., 1988). É
possível aumentar a potência através do aumento do diâmetro do moinho, mas após
um certo tamanho crítico (da ordem de 6 m de diâmetro), a eficiência da cominuição
diminuirá drasticamente (Tavares, L. M. M., 2001; Conley, R. F., 1992).
A utilização da moagem ultrafina na preparação de pós cerâmicos vem sendo
estudada atualmente. A moagem ultrafina permite a obtenção de pós cerâmicos com
alta área específica e a produção de peças com alta densidade e boa sinterabilidade
(Tavares, L. M. M., 2001).
O número excessivo de parâmetros que influenciam o processo de moagem,
geram uma otimização complexa. Gao, M. W. & Forsseberg, E. (1992), estudaram
quatro parâmetros importantes simultaneamente em moinho de bolas rotativo, como
o tamanho dos meios moedores, carga de sólidos na polpa de moagem, tamanho e
taxa da alimentação de topo. A partir dos resultados obtidos no trabalho, concluíram
que os corpos moedores na faixa de 0,8 mm a 1 mm eram mais eficientes, por
produzirem maior área superficial específica com menor consumo de energia.
A carga dos meios moedores deve ocupar entre 50 e 80 % do volume total do
moinho (Tavares, L. M. M., 2001). Se considerarmos constante o número de
colisões entre os meios moedores para um dado volume de carga e a velocidade de
rotação do moinho, então a única maneira de se aumentar o número de colisões
entre os meios moedores seria aumentando o seu número, ou seja, reduzindo o
diâmetro dos meios moedores, dentro dos limites baseados na razão entre o
tamanho inicial das partículas e o tamanho dos corpos moedores. Podem ainda ser
utilizados meios moedores de diferentes tamanhos para aumentar o número de
colisões, devido ao maior empacotamento dos interstícios (Tavares, L. M. M., 2001).
A porcentagem de sólidos na polpa de moagem contendo corpos moedores
deve ser a maior possível a fim de se reduzir o desgaste dos meios moedores e
aumentar a taxa de cominuição do material, sem que haja uma resistência excessiva
ao escoamento, mantendo baixa a viscosidade do fluido (Tavares, L. M. M., 2001).
A moagem a úmido geralmente é mais eficiente que a seco, devido à
possibilidade do meio líquido transportar o material através dos meios moedores. O

62
transporte do material no moinho depende das propriedades físicas da polpa, tais
como a fluidez, estado de dispersão, densidade dos sólidos e dos meios moedores.
(Tavares, L. M. M., 2001).
Oliveira, L. G., 2002, estudou a influência do tempo de moagem e diâmetro de
corpos moedores para pós de zircônia em moinho atritor na distribuição final de
tamanho de partícula e área de superfície específica. Foram avaliados corpos
moedores de diferentes diâmetros. Para o diâmetro de 5,3 mm o tempo de moagem
ideal foi de 5,5 horas de moagem. Para menores diâmetros (0,7 - 1,2 mm), o tempo
de moagem para se obter um tamanho de partícula relativamente baixo era de no
máximo 2h.
3.5.3 DISPERSÃO DOS PÓS
De acordo com Onoda, G.Y., 1977, aglomerado é um termo que descreve uma
pequena quantidade de massa possuidora de poros interconectados (FIG. 3.23). O
aglomerado compreende as partículas primárias ligadas ou unidas por forças de
superfície (atração eletrostática e van der Walls) e por pontes sólidas resultantes da
sinterização (fusão, reação química e ação de um ligante).
A dispersão de pós (FIG. 3.24) se faz necessária para a obtenção de uma alta
densidade a verde e uma microestrutura homogênea, sem a presença de
aglomerados, melhorando assim as propriedades mecânicas dos corpos sinterizados
(Oliveira, I. R. et al., 2000; Wei, W. J. et al., 1999; Schaw, D. J., 1975).
Quando partículas sólidas são suspensas em um líquido polar, elas ficam
carregadas superficialmente e se movimentam aleatoriamente no meio dispersor.
Devido a esse movimento, vão ocorrer colisões entre as partículas e a estabilidade
da solução vai depender das forças de interação entre as partículas. A estabilidade
da suspensão depende do sinal e da magnitude da energia total de interação entre
as partículas.
Para se obter suspensões estáveis, forças repulsivas (eletrostáticas) devem
superar as forças atrativas (London van der Walls) entre as partículas.

63
FIG. 3.23 Formação de aglomerados na ausência de forças repulsivas.
FIG. 3.24 Partículas dispersas e defloculadas.
A estabilização de suspensões ou interação repulsiva é gerada por três
mecanismos diferentes:
a) estabilização eletrostática: é o resultado da dispersão em um líquido
polar, que produz forças repulsivas que diminuem quando há um
aumento da distância entre as partículas (forças coulombianas);
b) estabilização estérica: onde a superfície das partículas adsorvem
macromoléculas presentes na suspensão(polímeros de cadeias
longas) formando barreiras repulsivas, dificultando a aproximação das
partículas por impedimento estérico ou;
c) estabilização eletroestérica: adsorção específica de moléculas com
grupos ionizáveis ou polieletrólitos na superfície das partículas, no
qual os íons provenientes da dissociação desses grupos ionizáveis

64
adicionam uma barreira eletrostática ao efeito estérico.(Oliveira, I. R.
et al., 2000).
Existem duas formas de se evitar a ocorrência de aglomeração de pós muito
finos:
a) o aumento das cargas elétricas das partículas (a superfície de qualquer
sólido tem uma carga superficial resultante, existe falta ou excesso de
elétrons em relação ao número necessário para balancear os prótons dos
átomos da superfície) ou;
b) pela ação de dispersantes sintéticos (poliméricos ou polieletrólitos-
polímero carregado – se adsorvem fortemente sobre as partículas em
suspensão e formam barreiras repulsivas – eletrostáticas e/ou estéricas –
que superam as forças de atração entre as partículas).
Como em suspensões estáveis, as forças repulsivas superam as forças
atrativas, para conservar a neutralidade, as partículas carregadas atraem íons de
carga oposta (contra-íons) que vão ser adsorvidos à superfície, formando uma
primeira camada conhecida como camada de Stern, sendo mais interna e de menor
espessura.
Uma segunda camada conhecida como camada de Gouy ou camada difusa,
mais externa e de maior espessura onde estão distribuídos de maneira difusa os
íons de cargas opostas (contra-íons) e os íons de mesma carga (co-íons) (Hunter, R.
J., 1986; Oliveira, L. G., 2002).
Isto produz um gradiente, resultando em uma camada carregada de líquido, a
qual se estende a uma certa distância a partir da superfície da partícula. Quando
ocorre a aproximação entre as partículas, há uma interação entre as camadas
carregadas do líquido, gerando uma força de repulsão entre elas (Pereira, A. P. D.,
2001).
A FIG. 3.25 mostra a formação de uma dupla camada de cargas elétricas ao
redor da partícula carregada negativamente em meio líquido.

65
Contra-íon positivo
Co-íon negativo
Partícula negativa
Camada de Stern
Camada difusa
Íons em equilíbrio com a solução
FIG. 3.25 Formação de uma dupla camada de cargas elétricas ao redor de uma partícula carregada negativamente em meio aquoso.
Para se medir a estabilidade de uma suspensão, ou seja, a magnitude da
repulsão ou atração entre partículas, é utilizado o potencial zeta (ζ). O potencial
zeta é o potencial elétrico que existe através da interface de todos os sólidos e
líquidos e representa a diferença de voltagem entre a superfície da chamada
camada “difusa” (de íons neutralizantes) e o restante do volume no qual a partícula
encontra-se dispersa. A suspensão mais estável é aquela que apresenta maior
potencial zeta (ζ) (Wei, W. J. et al., 1999; Shaw, D. J., 1975; Oliveira, L.G. et al.,
2002). Esta técnica se aplica para avaliar a dispersão em suspensões com baixa
concentração de sólidos.
É difícil obter uma solução homogênea, bem estabilizada, dispersa com alta
concentração de sólidos (óxidos) e baixa viscosidade sem a utilização de
polieletrólitos tais como os dispersantes ou defloculantes (Fengqiu, T. et al., 2000;
Wang, J. & Gao, L., 2000).
Existe uma estreita correlação entre o estado de dispersão das partículas de um
pó em suspensão, o pH e a viscosidade. Variações no pH provocam mudanças na
carga superficial das partículas, nas forças de interação entre as mesmas, no
potencial zeta, acarretando um aumento ou diminuição do seu estado de dispersão,

66
e conseqüentemente uma variação na viscosidade (Pereira, A. P. D., 2001; Pileggi,
R. G. et al., 2000; Ortega, F. S. et al, 1997a; Ortega, F. S. et al, 1997b; Ortega, F. S.
et al, 1997c; Kamiya, H. et al., 1999; Bergtröm, L. et al., 1999).
O valor de pH no qual a neutralidade é alcançada, igualando-se o número de
cargas negativas da molécula adsorvente com o número de cargas positivas, é
denominado ponto isoelétrico (PI) do sistema partícula / adsorvente.
O ponto de carga zero (PCZ), conhecido como o ponto no qual o potencial zeta
é igual a zero, o número de sítios de cargas positivas produzidas pelos pós de
zircônia contendo os íons H+ adsorvidos é igual aos sítios de cargas negativas pela
adsorção de íons OH-, então a carga superficial é nula. No PCZ as partículas em
suspensão podem ser facilmente aglomeradas e floculadas. Perto do PCZ, a força
repulsiva da dupla camada elétrica é pequena e a força de atração de van der Waals
gera uma aglomeração e o potencial zeta distante do PCZ separa as partículas
umas das outras favorecendo a dispersão. Assim, o pH correspondente ao PCZ é o
que se deve evitar quando se deseja obter uma eficiente dispersão de pós (Oliveira,
L. G., 2002)
Superfícies em contato com o meio aquoso se apresentam mais freqüentemente
com carga negativa, pois os cátions estão mais hidratados apresentando uma maior
tendência a permanecer no meio aquoso, enquanto os ânions hidratados e mais
polarizados apresentam maior tendência a serem adsorvidos especificamente.
O Ácido Cítrico HOC(CH2CO2H)2CO2H é um polieletrólito aniônico usado no
processamento cerâmico devido a forte liberação de íons H3O+ em solução aquosa,
e sua estrutura molecular, está indicada na FIG. 3.26.
C
CH2
COO- H+
COO- H+
H2C
H+ COO-
HO
H3Cit
FIG. 3.26 Estrutura molecular do ácido cítrico(o símbolo Cit representa o grupo citrato).
A dissociação dos grupos carboxílicos em água pode originar íons de citrato de
valência -1, -2 ou -3, de acordo com as seguintes equações: (Oliveira, I. R., 2000;
Oliveira, L. G., 2002).

67
H3Cit H+ + H2Cit - (3.1)
H2Cit - H+ + HCit 2- (3.2)
HCit 2- H+ + Cit 3- (3.3)
A valência dos íons de citrato é maior com o aumento do pH. Em virtude do
tamanho reduzido da molécula de ácido cítrico, os íons de citrato originados a partir
da dissociação deste dispersante não apresentam variação conformacional
observada no caso dos polieletrólitos (Oliveira, L. G., 2002).
O Calgon (metahexafosfato de sódio - Na4P2O7), também é um polieletrólito
aniônico usado no processamento cerâmico devido à presença de grupos (P2O7)2-.
Kunes, K. et al. (2000), avaliaram a estabilização de suspensões de
biocerâmicas contendo alumina e zircônia e concluíram que a quantidade ótima de
defloculante está relacionada com a mínima viscosidade aparente. Para todas as
suspensões analisadas, os autores concluíram que a menor viscosidade aparente
era obtida com adição de 0,6 % em peso de defloculante.
Oliveira, L. G. et al., 2002 analisaram a carga superficial e o grau de dispersão
de suspensões de zircônia e de misturas de alumina e zircônia estabilizada com
cálcia e ítria em suspensões aquosas. Foram avaliados três diferentes
dispersantes: o Calgon (metahexafosfato de sódio – Na4P2O7), o Tamol (sal sódico
de um ácido policarboxílico) e o ácido cítrico. O estudo foi realizado por meio de
medidas do potencial zeta e da reologia da suspensões. O ajuste de PH foi
realizado com ácido clorídrico (HCl) e hidróxido de potássio (KOH). Os autores
verificaram que para as suspensões aquosas de zircônia sem dispersante, o ponto
de carga zero (PCZ) era aproximadamente 7, deslocando-se para regiões mais
ácidas utilizando dispersantes aniônicos. Foi visto ainda que em condição
fortemente ácida (PH<3) ou fortemente básica (PH>12), a força iônica diminui,
sendo necessária maior quantidade de HCl ou KOH para ajuste de PH resultando no
decréscimo do potencial zeta (explicado pela teoria da dupla camada). O PH ideal
para uma boa defloculaçâo destes pós foi verificado estar nas faixas 3 - 5 e 9 - 11.
Com uso do dispersante de Calgon (0,1 % p/v), o PCZ se localiza em PH < 2, as
partículas podem ser bem dispersas em suspensão em PH na faixa de 6 - 12 e
parcialmente aglomeradas em PH < 4. Com o ácido cítrico (0,005 % p/v), em PH < 8

68
ocorre uma leve aglomeração e em PH > 8 as partículas estão no estado
monodisperso.
As curvas de potencial zeta em função do PH para baixas concentrações dos
dispersantes Calgon (0,1% p/v) e para o ácido cítrico (0,005 % p/v), ambas em
PH=10, mostraram os maiores valores de potencial zeta de ζ = -57,6 mV e -51,6 mV,
respectivamente. Os autores concluíram que tanto o Calgon quanto o ácido cítrico
são bons dispersantes para pós de zircônia e misturas de alumina e zircõnia, uma
vez que apresentaram valores de potencial zeta bem próximos, porém, o ácido
cítrico foi mais efetivo que o Calgon, pois a densidade teórica relativa de amostras
contendo ácido cítrico alcançou o valor de 98,20 % da densidade teórica, sendo
superior a densidade obtida utilizando o Calgon (96,88 % da densidade teórica) para
as mesmas condições de moagem. O trabalho mostrou ainda que para pós de
alumina zircônia estabilizada com ítria, moagem por 1,5 h em álcool, utilizando 0,6 %
em peso de ácido cítrico, sem ajuste de PH, forneceu melhores resultados de
resistência mecânica.
O estudo da reologia para a suspensão da mistura de alumina e zircônia
estabilizada com ítria, contendo 50% peso de sólidos na solução e PH=11(ajustado
com hidróxido de amônio - NH4OH), onde foi analisada a variação da viscosidade
em função do teor de dispersante utilizado, mostrou que a concentração de 0,6 %
em peso de ácido cítrico foi a que apresentou a menor viscosidade (η = 4,512
mPa.s). Na ausência do dispersante, foram encontrados aglomerados de zircônia
na amostra contendo alumina e 15% em peso de zircônia estabilizada com ítria (3%
mol), como pode ser visto na FIG. 3.27.
Kelso, J. F. & Ferrazolli, T. A. (1989), mostraram que diferenças na química
superficial de dois tipos de alumina provocavam efeitos significativos na mobilidade
eletroforética das partículas, sendo necessárias diferentes quantidades de
dispersantes para estabilizar as suspensões. Foi verificado que na ausência de
dispersante ambos os pós não dispersavam bem em água para valores de pH entre
8 e 9, pois nesta faixa de pH as mobilidades eletroforéticas dos pós são próximas de
zero, levando a uma baixa repulsão eletrostática.

69
(a)
(b)
FIG. 3.27 Micrografias eletrônicas de varredura após polimento e ataque térmico da amostra Al2O3 - 15% 3Y-TZP sem a utilização do dispersante: (a) elétrons retroespalhados e (b) elétrons
secundários em diferentes magnitudes.
A presença do dispersante promoveu um aumento, em módulo, das mobilidades
eletroforéticas das partículas, propiciando um aumento nas forças repulsivas,
evitando a floculação dos pós.
3.5.4 CONFORMAÇÃO POR PRENSAGEM UNIAXIAL A prensagem é uma etapa do processamento em que se efetua
simultaneamente, a conformação e a compactação do pó cerâmico. A mecânica da
conformação depende da consistência e das propriedades reológicas do material.
Materiais de granulometria grosseira e pós granulados são normalmente
conformados por compactação uniaxial em uma matriz rígida de pós secos (Reed,
1987).
O processo de conformação de materiais cerâmicos por prensagem uniaxial a
seco é um dos mais simples e mais utilizado. O pó cerâmico com pequeno teor de
umidade ou contendo um ligante preenche um molde metálico ao qual é aplicada
uma determinada pressão, através de um punção, para a formação do compacto a
verde. È um processo relativamente de baixo custo e pode ser utilizado para a
obtenção de cerâmicas de formas variadas em alta escala de produção. São três os
estágios de compactação: A prensagem consiste de três etapas (FIG. 3.28):
Preenchimento da matriz, compactação e conformação, e por fim a ejeção (Reed, J.,
1987).

70
FIG. 3.28 Estágios da compactação: (a) Enchimento; (b) Fechamento; (c) Prensagem; (d) Ejeção.
A matriz e os punções devem possuir elevada resistência à abrasão e ajuste
deslizante para evitar que partículas do pó prensado penetrem no interstício entre a
parede da matriz e a superfície externa lateral do punção durante a prensagem,
provocando um desgaste prematuro do conjunto. O tempo de prensagem varia
desde fração de segundos para peças pequenas até alguns minutos para peças
maiores.
3.5.4.1 CARACTERÍSTICA DOS PÓS PARA PRENSAGEM
Os resultados finais atingidos durante a sinterização são fortemente
influenciados pelas características do pó: tamanho e distribuição de tamanho de
partícula, forma da partícula, estrutura e condição de superfície além do
processamento cerâmico.
Um bom escoamento dos pós é importante para se obter uma densidade
uniforme de preenchimento e permitir maiores velocidades de compactação. Pós
que apresentam boa densidade de enchimento, na forma de grânulos
aproximadamente esféricos com superfície lisa, são os mais indicados para a
operação de prensagem devido a sua elevada fluidez.
Pós aglomerados na forma de grânulos preenchem mais facilmente a matriz,
pois podem sofrer deformações que resultam em melhor empacotamento e
conseqüentemente maior densidade a verde. O material granulado, geralmente
obtido por “spray drying”, contém defloculante, ligante, plastificante e algumas vezes
um lubrificante, um umectante e antiespumante (Reed, J., 1987; Mesquita, C. E. M.,
2001).

71
A presença de mais de 5 % de finos pode prejudicar o escoamento, pois podem
penetrar na folga existente entre o punção e a matriz aumentando o atrito e
dificultando o escapamento do ar aprisionado. Os problemas de compactação são
minimizados quando a densidade de enchimento da matriz for elevada. Neste caso,
a quantidade de ar entre as partículas do pó fica diminuída. Pós que não
apresentam esta característica provocam heterogeneidades na densidade de
enchimento e geram compactos com defeitos que são repassados para o produto
sinterizado (Reed, J., 1987).
Uma distribuição bimodal, constituída por partículas pequenas distribuídas nos
interstícios das partículas maiores, reduz a porosidade e o tamanho dos poros. A
mistura de tamanho de partículas para atingir uma elevada densidade de
compactação é uma prática comum no processamento de cerâmicas com alta
densidade final. Na FIG. 3.29, pode ser visto que a densidade de compactação
aumenta com o aumento da razão entre os tamanhos das partículas (Reed, J.,
1987).
FIG. 3.29 Compactação de esferas finas no interstício de partículas grosseiras.
A maioria dos materiais cerâmicos é produzida a partir de pós que apresentam
uma distribuição bimodal de tamanho de partícula. A distribuição de tamanho dos
pós granulados produzidos por “spray drying” ou pela moagem fina de minerais
geralmente se aproxima de uma distribuição log-normal, como pode ser vista na
FIG.3.30, onde fN e fM são a fração numérica e a de massa respectivamente (Reed,
J., 1987).

72
FIG. 3.30 Distribuição de tamanho de partículas cerâmicas.
As forças de adesão entre partículas e a floculação retardam o movimento das
partículas e dificultam a compactação. A configuração randômica de partículas
anisométricas apresenta geralmente uma porosidade maior e uma maior variedade
de tamanho de poros quando comparada com um arranjo ordenado de partículas
anisométricas (FIG. 3.31).
FIG. 3.31 Estrutura de poros em um arranjo de partículas anisométricas. (a) arranjo randômico, (b) arranjo ordenado.
A adsorção de moléculas de ligante e de partículas coloidais pode dificultar o
movimento das partículas, reduzindo a densidade de compactação dos pós
cerâmicos, se este impedimento ou barreira não for suplantado pela tensão aplicada.
No impedimento estérico, o mesmo é tanto maior quanto maior o peso molecular do
ligante e quanto maior forem as forças de ligação entre as moléculas (FIG. 3.32).

73
FIG. 3.32 Moléculas de ligante reduzem a densidade de empacotamento sob baixas tensões efetivas, A – partículas, B- Moléculas de polímeros.
A presença de partículas finas nos interstícios de partículas maiores, em mistura
com o ligante, podem recobrir as partículas maiores, reduzindo a densidade.
Quanto menor for o tamanho de partícula e maior a concentração do ligante, mais
difícil se torna a obtenção de uma compactação homogênea (Reed, J., 1987).
Desta forma, as principais características do pó cerâmico para prensagem são:
um fácil escoamento para o enchimento da cavidade da matriz com eficiência, uma
alta densidade de volume (distribuição bi-modal das partículas), ser estável nas
condições ambientes, causar desgaste mínimo da matriz, não aderir ao punção,
para que o preeenchimento do molde seja eficiente e a densidade a verde a maior
possível.
3.5.4.2 MECÂNICA DA COMPACTAÇÃO
Em um sistema de partículas sob compressão, a pressão aplicada é suportada
pelas tensões mecânicas de contato entre as partículas e pela pressão do fluido
contido nos poros. A diferença entre a pressão aplicada (Pa), e a pressão do fluido
(u), é denominada tensão efetiva(σef) (Reed, J., 1987).
σef = Pa – u ( 3.4 )
A tensão efetiva é também chamada de tensão interparticular, mas não deve ser
confundida com a tensão local de contato entre as partículas. A tensão local é
função da tensão efetiva, da pressão dos poros e da pressão da rede devido às

74
interações atrativas e repulsivas (R-A). A tensão de contato (σc) pode ser expressa
como:
σc = [ σef – (R – A)] (AT/Ac) + u ( 3.5 )
Onde Ac é a área média dos contatos entre partículas na área de seção
transversal AT.
Em um sistema insaturado, a deformação elástica, a deformação permanente e
a tensão de escoamento do corpo dependem da tensão efetiva. A tensão efetiva
pode variar com o tempo e a temperatura quando a pressão dos poros varia com o
tempo e a temperatura (Reed, J., 1987).
Um sistema de partículas no interior de uma matriz rígida submetido a
compressão uniaxial é apresentado na FIG. 3. 33.
FIG. 3.33 Forças normais e tangenciais nos contatos entre partículas.
As forças nos pontos de contato entre as partículas podem ser decompostas em
normal (N) e tangencial (T). As partículas são comprimidas de forma não uniforme,
de tal forma que as partículas na forma de fibras ou placas podem fletir, sofrer
rotação e translação. Estes movimentos, levam a uma nova distribuição de tensões,
causando redução ou algumas vezes o aumento de volume e distorção na forma,
provocando uma deformação plástica e também fratura do corpo cerâmico.
O deslizamento entre as partículas é dificultado devido a adesão química e ao
atrito. A força que resiste ao cisalhamento (TR), pode ser calculada como TR = f.N,
onde f é o coeficiente de atrito e N é a força normal. Quando a tensão tangencial (T)

75
for maior que a de resistência ao cisalhamento (T > TR ) em todos os pontos de
contato entre as partículas.
Durante a compactação a tensão aplicada é transmitida através dos pontos de
contato entre as partículas, provocando deformação do granulado por deslizamento
e rearranjo das partículas. Essas deformações reduzem a porosidade e aumentam
o número de contatos intergranulares. A compactação ocorre pela fratura e
deformação do granulado, o que reduz o volume dos interstícios e elimina os poros
no meio do granulado deformado, promovendo uma maior condensação das
partículas cerâmicas (Richerson, D., 1992; Reed, J., 1987).
Na compactação dos pós cerâmicos são considerados três estágios (Reed,
1987). São eles:
1) Densificação superior a partir da densidade de enchimento devido ao
escorregamento e rearranjo dos grânulos;
2) Deformação e fratura dos grânulos reduzindo o volume dos interstícios
intergranulares;
3) Este estágio inicia quando a maioria dos grandes poros (intergranularea)
desaparecem. A pressão aplicada que neste estágio é mais elevada
promove o deslizamento e o rearranjo das partículas ou fragmentos de
fratura no interior dos grânulos (intragranulares), levando a uma melhor
densificação do corpo cerâmico. As mudanças na forma dos grânulos de
esféricos para grânulos deformados e na distribuição bimodal de tamanho de
poros durante a compactação pode ser vista na FIG. 3. 34.
FIG. 3.34 Alterações microestruturais durante a compactação.

76
Durante a prensagem, a compressão elástica dos grânulos começa no estágio 2
e aumenta no estágio 3. A energia elástica armazenada produz um aumento nas
dimensões da peça prensada durante a ejeção, conhecido como efeito mola. Este
efeito pode causar defeitos no corpo cerâmico durante a sua ejeção da matriz. A
força de ejeção depende do efeito mola do compactado, da velocidade de ejeção, da
lubrificação das paredes da matriz, e das condições superficiais da matriz. A
lubrificação pode diminuir a pressão de ejeção e reduzir o desgaste da matriz.
Ligantes e plastificantes podem melhorar a adesão de partículas, promovendo
maior resistência ao manuseio do corpo verde. Este, deve ser capaz de resistir a
solicitação mecânica durante a ejeção e a manipulação sem apresentar falhas e
deve ter uma microestrutura uniforme. Os principais defeitos em compactos
prensados a seco são a laminação e os defeitos de extremidade (FIG. 3.35). Estes
defeitos são decorrentes de tensões produzidas pelo efeito mola diferencial durante
a ejeção. O efeito mola diferencial surge devido a existência de gradientes de
pressão interna gerados pelo atrito com as paredes da matriz, gradientes de energia
elástica armazenados devido a grânulos não uniformes e quando e o efeito mola do
compactado for maior que o da matriz.
FIG. 3.35 Defeitos nos compactos prensados a seco.
O método de conformação por prensagem uniaxial apresenta uma limitação. As
forças de fricção do pó contra a parede do molde afetam a velocidade de
prensagem, provocam o surgimento de gradientes de pressão e como
conseqüência, à gradientes de densidade no compacto formado. Este efeito é tanto
maior quanto menor for a densidade aparente do pó cerâmico, pois maior será o

77
percurso do punção no molde, resultando em perdas de pressão durante a
prensagem. Estudos sobre o movimento dos pós durante a compactação indicam
que o deslocamento axial dos pós é maior no centro do que próximo as paredes da
matriz e que o deslocamento diminui a medida que se afasta do punção. A pressão
máxima está localizada próxima aos vértices do topo e a pressão mínima está
próxima aos vértices do fundo, como pode ser visto na FIG. 3.36.
FIG. 3.36 Perfil de distribuição da pressão na prensagem uniaxial.
3.5.5 SECAGEM E EVAPORAÇÃO DE LIGANTE
A secagem é uma importante operação antes da sinterização. Consiste na
remoção de líquido do interior de um material poroso, por meio de transporte e
evaporação. Deve ser cuidadosamente controlada , pois tensões produzidas pelo
diferencial de contração podem causar defeitos no produto final.
O transporte de energia térmica, dá origem aos mecanismos de secagem. O
líquido é transportado através dos vazios para os meniscos por forças dirigentes
motivadas pela evaporação e através dos poros para a superfície pelo vapor. As
forças dirigentes são originadas pela diferença de pressão que existe no equilíbrio
entre a superfície curva e o interior do material cerâmico. A migração do líquido
para esta superfície ocorre por fluxo capilar, difusão química e difusão térmica
(Reed, J., 1988)
As forças de capilaridade estão presentes na fase inicial da secagem,
provocando o movimento da umidade no sentido da evaporação superficial,
aumentando a densificação do corpo pelo contato direto entre as partículas. A
P1 > P2 > P3 > P4

78
difusão ocorre quando as partículas estão totalmente em contato e a contração não
é mais possível.
A retirada de umidade do interior do corpo ocorre por razões energéticas, pois a
evaporação do líquido dos poros faz com que a fase sólida fique exposta, logo, uma
interface mais energética, a interface sólido/vapor, substitui a interface sólido/líquido.
Então, para evitar este aumento de energia do sistema, o líquido tende a se expandir
do interior do corpo para encobrir esta parte sólida, reduzindo a energia do sistema
(Pereira, A. P. D., 2001).
Com a remoção do líquido e conseqüente redução da distância inter-partículas
ocorre a primeira contração do corpo cerâmico.
Existem alguns métodos de retirada de ligantes, sendo o mais utilizado o
aquecimento lento das amostras desde a temperatura ambiente até um valor
máximo de aproximadamente 600°C.
A etapa da remoção de ligantes do corpo cerâmico é crítica por três razões:
a) Defeitos podem ser originados por exemplo, bolhas e trincas, durante
uma retirada rápida do ligante. Em taxas muito lentas, além do custo,
poros estáveis podem ser formados, pois os gases produzidos
permanecem muito tempo no interior do poro durante a expansão
térmica.
b) Tempos de retirada de ligante podem ser comercialmente inaceitáveis em
função do tamanho da peça.
c) Presença de resíduos resultantes de remoção incompleta do ligante, são
prejudiciais pois durante a sinterização há formação de bolhas de ar e
poros.
Dentre os principais fatores que afetam a remoção do ligantes estão: a taxa de
aquecimento, o tipo de atmosfera, a geometria da peça, características do pó e do
ligante e a porosidade a verde.
A queima afeta os ligantes de formas diferentes. Ligantes de baixo peso
molecular são removidos por evaporação sem que ocorra a quebra de sua estrutura

79
química. Os ligantes de alto peso molecular sofrem degradação térmica com a
quebra de suas cadeias. A presença do oxigênio acelera a degradação do ligante
em função do processo de oxidação. Assim, o sucesso da remoção de ligantes
depende do equilíbrio entre as taxas de difusão e evaporação ou degradação.
A retirada de ligantes pode introduzir defeitos de duas maneiras. Na primeira o
ligante sai e deixa um espaço interparticular, gerando poros que podem aumentar de
tamanho durante a etapa de sinterização. Na segunda, a pressão de capilaridade
dos poros causa o rearranjo das partículas possibilitando o aparecimento de trincas.
Por outro lado, a redistribuição do ligante e a contração decorrentes da retirada do
ligante, resultam em redução do volume total da porosidade aberta.
As características dos pós podem afetar a retirada de ligantes. O tamanho, a
forma e área superficial das partículas, a homogeneidade são aspectos importantes.
Corpos cerâmicos produzidos a partir de pós finos, que adsorvem o ligante mais
facilmente, apresentam maior dificuldade para a remoção do ligante sem que haja a
introdução de defeitos.
3.5.6 SINTERIZAÇÃO DAS CERÂMICAS À BASE DE ÓXIDOS
3.5.6.1 ASPECTOS TEÓRICOS
Sinterização é um processo no qual pequenas partículas são ligadas por difusão
no estado sólido. Neste processo, o tratamento térmico imposto resulta na
transformação de um compacto poroso em um produto cerâmico coerente e denso.
O processo de sinterização é controlado por:
a) pelas propriedades características do compacto (densidade a verde,
estrutura de poros, tamanho e distribuição de partículas);
b) atmosfera de sinterização (oxidante, redutora ou inerte);
c) pressão
d) temperatura (incluindo as taxas de aquecimento e resfriamento)
e) tempo de permanência na temperatura de sinterização(patamar)

80
Os processos de sinterização podem ser classificados de acordo com o
fluxograma geral apresentado na FIG. 3.37 (Marchi, J., 1999). O presente trabalho
envolve o processo de sinterização sem pressão no estado sólido monofásico.
PROCESSO DE SINTERIZACÃO
sem pressão com aplicação de pressão externa
Estado sólido Via Fase líquida
Multifásico
MonofásicoLíquido
transiente
baixa pressão
Líquido persistente escoamento
plástico
alta pressão
fluxo viscoso ou escoamento por
fluênciamultifásico
ou supersólidus
reativo ou solução sólida
FIG. 3.37 Fluxograma geral dos processos de sinterização.
A sinterização do estado sólido é definida como sendo um processo de remoção
de poros localizados entre partículas agrupadas e acompanhada por contração
(“shrinkage”) do componente combinado com o crescimento e formação de ligações
fortes entre partículas adjacentes.
A força motriz da sinterização do estado sólido é a redução da área de superfície
(energia de superfície) na qual ocorre uma substituição de superfícies mais
energéticas sólido-vapor, existente nos poros, por superfícies de menor energia
sólido-sólido dos contornos de grão. Portanto, quanto mais fino for o pó do
compacto verde, maior será a força motriz para sinterização, pois maior será a
energia de superfície a ser reduzida.
O estágio inicial da sinterização compreende o rearranjo das partículas do pó e
a formação de pescoços (microestrutura com grande gradiente de curvatura) no
contato entre elas. Devido ao melhor empacotamento das partículas, pode ser
obtida uma densificação de 50 a 60 % da densidade teórica neste estágio.

81
O estágio intermediário caracteriza-se pelo crescimento do tamanho do pescoço.
A quantidade de porosidade é consideravelmente reduzida e isto faz com que as os
centros das partículas se aproximem, causando contração no componente. A
densidade chega a aproximadamente 90 % da teórica. Neste estágio, são formados
os contornos de grão, que se movimentam de modo que alguns grãos cresçam a
custa dos outros. Este estágio perdura enquanto existirem canais interconectando os
poros (porosidade aberta), e termina quando os poros se tornarem isolados
(porosidade fechada), aprisionando o gás do ambiente de sinterização.
O estágio final é caracterizado pela eliminação lenta dos poros fechados por
difusão de vacâncias ao longo dos contornos de grão. Devem ser criadas condições
para que os poros fiquem atrelados ao contorno de grão por:
a) serem mais facilmente eliminados por difusão pelo contorno de grão, que
age como sumidouro de vacâncias, desta forma promovendo um ganho
de densidade.
b) Inibirem o crescimento de grão pelo travamento dos contornos de grão ou
seja, redução de sua mobilidade. Esta inibição também pode ocorrer por
inclusões de 2ª fase nos contornos de grão (Kingery, 1960).
Assim, não é vantagem a eliminação dos poros no início da sinterização, pois a
capacidade de travamento dos contornos fica diminuída sem a presença dos poros.
Se houver um crescimento excessivo de grão, há risco de se deixar poros isolados,
no interior dos grãos, os quais serão difíceis de serem eliminados, pois a difusão
pela rede cristalina é lenta. A TAB. 3.6 relaciona o processo de densificação, a
perda de área específica, a densificação e o coalescimento nos diferentes estágios
da sinterização (Marchi, J., 1999).
TAB. 3.6 Características dos três estágios da sinterização
Estágio Processo de densificação
Perda de área específica
Densificação Coalescimento
Inicial Crescimento do pescoço
Significante
> 50 %
Pequena Mínimo
Intermediário Arredondamento dos poros e
Perda da porosidade aberta
Significante Aumento do tamanho de grão e

82
alongamento poros
Final Fechamento de poros e
densificação final
Desprezível Lenta e relativamente
mínima
Crescimento extensivo de grãos
e poros
Em resposta a força motriz para a sinterização(redução da área de superfície),
surgem fluxos de massa atuando por meio de vários mecanismos de transporte.
Estes podem ser classificados em transporte pela superfície e transporte pelo
volume.
A FIG. 3.38 apresenta de forma esquemática, os mecanismos de difusão. A área
de contacto entre as duas partículas é um dos meios pelo qual a difusão irá se
processar (Rocha, J. C., 1981).
FIG. 3.38 Mecanismos de transporte de massa.
O transporte pela superfície envolve o crescimento do pescoço sem mudar a
distância entre os centros das partículas, portanto, sem densificar. O fluxo de massa
origina-se e termina na superfície da partícula. Os mecanismos de transporte por
superfície incluem: a difusão pela superfície (1), difusão pelo volume (2), e a
evaporação e condensação (3). Os mecanismos 1 e 3 são os dois principais
mecanismos de transporte, que conduzem a um excessivo tamanho de grão.
O transporte pelo volume conduz à contração, por aproximação dos centros das
partículas, portanto, conduz à densificação; quando a matéria provêm do interior de
duas partículas e o depósito de matéria se dá na região do pescoço. Os
mecanismos de transporte por volume incluem: difusão pelo volume (4), difusão pelo
3
2
1
6 4 5

83
contorno de grão (6), e fluxo termoplástico ou fluxo viscoso (5), sendo este último de
pouca importância para cerâmicos cristalinos. A TAB. 3.7 apresenta os mecanismos
de transporte de massa e suas características.
TAB. 3.7 Mecanismos de transporte de massa.
Número do mecanismo
Caminho do transporte
Fonte de massa
Destino
1 Difusão pela superfície superfície 2 Difusão pelo volume superfície 3 Evaporação e condensação superfície 4 Difusão pelo volume Contorno de grão 5 Difusão pelo volume Discordâncias 6 Difusão pelo contorno de grão Contorno de grão
P E S C O Ç O
O transporte por difusão de superfície (1) ocorre pelo movimento ao longo das
superfícies das partículas, que são rugosas e apresentam defeitos, tais como
vacâncias e degraus. Os átomos se movimentam entre essas regiões de defeitos
por meio de um processo termicamente ativado.
A difusão por volume (2), envolve o movimento de vacâncias através da
estrutura cristalina, sendo a taxa de difusão função da temperatura, curvatura e
composição das partículas.
O mecanismo de evaporação e condensação (3) é caracterizado por um
aumento da pressão de vapor com a temperatura e o fluxo de massa é maior via
fase vapor. A evaporação ocorre a partir de superfícies planas ou convexas(raio de
curvatura positiva), enquanto que a condensação ocorre em regiões de formato
côncavo(pequeno raio de curvatura negativa), os pescoços(menor pressão), devidos
aos diferenciais de pressão de vapor. É um processo importante para pós com alta
área de superfície específica.
A difusão pelo volume ou pela rede (4) ocorre devido ao gradiente de
concentração de vacâncias, da maior para a menor concentração, ou seja, da
superfície côncava para a convexa, com fluxo de massa para o pescoço e
conseqüente retração. A difusividade é função do número e mobilidade de
vacâncias.

84
O transporte por fluxo termoplástico ou viscoso (5) ocorre por movimento de
discordâncias sob tensão aplicada. Durante o aquecimento, há geração de
discordâncias que interagem com as vacâncias durante a sinterização.
A difusão pelo contorno de grão (6), envolve a remoção de matéria ao longo do
contorno e deposição na interseção dos contornos com a superfície do pescoço. Há
um transporte de poros via contornos de grão com o prosseguimento da
sinterização. Este mecanismo é dominante para a densificação na maior parte dos
cerâmicos, como por exemplo, a alumina. Já o mecanismo de difusão pelo volume
geralmente fica restrito a estrutura de defeitos nos cerâmicos.
3.6 IDENTIFICAÇÃO DAS FASES DA ZIRCÔNIA E TRANSFORMAÇÃO MARTENSÍTICA
Diversos pesquisadores têm se utilizado da difração de raios-X para identificar
as fases cristalinas (monoclínica, tetragonal e cúbica) da zircônia obtida por
precipitação ou co-precipitação (Oliveira, A. P. A., 1997). Esta identificação é
extremamente importante para a zircônia, pois para que o material produzido possa
ser utilizado em cerâmicas avançadas torna-se necessária à estabilização das fases
tetragonal ou cúbica à temperatura ambiente. Os difratogramas de raios-X da
zircônia são de difícil análise, pois a maioria dos picos associados à fase cúbica e
tetragonal coincidem.
A seleção da região de 2θutilizada para a identificação das fases tetragonal e
cúbica da zircônia é bastante complexa. Srinivasan, R. et al., 1991, prepararam três
amostras, uma monoclínica, uma tetragonal e uma cúbica. Apesar da diferença
entre o padrão de raios-X das estruturas cúbica e tetragonal em relação ao padrão
da estrutura monoclínica ter sido evidente, as reflexões referentes ao plano (111)
das fases tetragonal e cúbica foram coincidentes.
A faixa de 2θ entre 71° e 76° contendo os planos 004 e 400 para amostras
tetragonais e cúbicas, vem sendo estudada por pesquisadores para distinguir as
estruturas tetragonal e cúbica da zircônia. A curva obtida para a amostra tetragonal
mostrou que os picos referentes aos planos (004) e (400) da fase tetragonal podiam
ser observados claramente. Na curva para a amostra cúbica, foi verificado que o
pico cúbico (400) era bastante acentuado.

85
3.7 PROPRIEDADES MECÂNICAS
3.7.1 DUREZA
A dureza não é uma propriedade fundamental de um material, seu valor varia
com o método de teste utilizado. A dureza de um material é usualmente medida
pela resistência à indentação ou penetração por algum corpo duro. Ela pode ser
ainda descrita como a resistência a abrasão e ao desgaste, corte, usinagem, choque
e ainda resistência ao risco (Askeland, D. R.,1990).
A dureza é definida pela pressão de indentação, a qual pode ser obtida pela
razão entre a carga de indentação aplicada e a área da impressão superficial
residual (Tanaka, K. et al., 1999). Desta forma, para um estudo dimensional
elementar, a dureza (H) pode ser calculada usando a equação 3.1, a seguir, quando
são conhecidos a semi-diagonal de indentação(a) e a carga de indentação (P).
22aPH = ( 3.6 )
As técnicas de impressão elástica ou técnicas de indentação são as mais usuais
para medir a dureza de um material, por sua simplicidade e por poder ser realizada
em pequenas amostras (Anton, R. J. & Subhash, G., 2000; Jiang, L. Z. & Sun, C. T.,
2001). O indentador de diamante (em forma esférica, cone ou pirâmide) é forçado
contra a superfície da amostra por um intervalo de tempo determinado e mede-se o
tamanho da impressão plástica residual na superfície do material após a remoção do
penetrador.
No processo de impressão Vickers, o trabalho externo aplicado pelo indentador
é consumido na deformação e no processo de fratura do material. O trabalho é
convertido em um componente de energia de deformação, proporcional ao volume
da pirâmide Vickers, e um componente de energia superficial, proporcional à área de
contato da pirâmide (Marchi, J., 1999).

86
Investigações confirmam que a dureza de um dado cerâmico é função da carga
de teste aplicada, onde medidas experimentais de dureza aumentam com o
decréscimo da carga de teste (Gong, J. et al., 1999).
A medida em que a carga de teste aumenta, as indentações tornam-se maiores,
e a energia é absorvida via deformação volumétrica e propagação da trinca. Isto
significa que um maior volume de material é deformado e gera-se um aumento na
área de impressão da trinca, e a dureza diminui até que se atinja uma transição para
a dureza constante. As explicações para a diminuição da dureza com o aumento da
carga baseiam-se em erros devido às medidas experimentais e/ou erros
relacionados com fatores estruturais intrínsicos das amostras, tais como: a
recuperação elástica da impressão quando a carga é removida, a qual é
proporcionalmente mais acentuada para pequenas impressões; e a presença de
microtrincas causadas pela indentação. O processo dominante é o da deformação
quando cargas baixas são utilizadas. Para altas cargas aplicadas, o processo de
fratura é dominante (Marchi, J., 1999).
É difícil estabelecer uma carga universal para a realização dos testes de dureza,
pois a manifestação da trinca é diferente em cada material cerâmico. Comparações
entre dureza obtida por vários penetradores também não devem ser feitas devido às
diferenças entre as sensibilidades quanto à carga, geometria do indentador e
propensões às trincas.
Os parâmetros que influenciam no teste de dureza são:
a) O tamanho e morfologia de grão: quanto menor o grão, e mais alongado,
maior a dureza;
b) a carga aplicada: a taxa de aplicação da carga não deve produzir
componentes laterais no movimento do penetrador, e todo movimento
deve ser completamente vertical;
c) a temperatura: o aumento da temperatura gera um amolecimento do
material;
d) a pureza: impurezas podem causar o endurecimento do material ou uma
fase secundária vítrea pode ocasionar a diminuição da dureza;
e) a superfície: deve estar polida e plana;

87
f) a porosidade: quanto maior, menor a dureza, pois diminui o volume
resistente à solicitação mecânica, sendo portanto a densidade um fator
primordial (Marchi, J., 1999).
3.7.2 TENACIDADE À FRATURA
A mecânica da fratura é o estudo das tensões e deformações causadas por
trincas estacionárias ou dinâmicas. Seu objetivo básico é prever a velocidade de
propagação das trincas e a variação da resistência mecânica com o tempo. Para
isso, é necessário o conhecimento da natureza dos processos que atuam na ponta
da trinca, região dominante – K (FIG. 3.39) de forma a bloquear o seu avanço.
FIG. 3.39 Configuração da ponta da trinca e base da mecânica da fratura linear elástica.
A concentração de tensões na ponta da trinca é caracterizada em termos do
fator de intensidade de tensões, K. Com relação a este fator, existem vários modos
(FIG.3.40 ) de abertura da trinca que podem ser aplicados (modo I, II e III).
Modo I Modo IIModo III
Zona do
processo
Região dominante - K
trinca

88
FIG. 3.40 Modos básicos de carregamento envolvendo diferentes deformações da superfície das trincas.
O modo I, representado por KI, refere-se a tensão trativa aplicada na direção y
normal às faces da trinca. Esse é o modo usual para o teste de tenacidade à fratura.
O modo II, modo de cisalhamento frontal, refere-se a uma tensão de cisalhamento
aplicado no plano da trinca normal à aresta frontal da trinca. O modo III, modo de
cisalhamento paralelo, é relacionado a tensões cisalhantes aplicadas paralelamente
à aresta frontal da trinca.
A propagação do defeito em forma de trinca ocorrerá quando o fator de
intensidade de tensões, na ponta da trinca, alcançar o valor crítico (Kc), levando a
fratura do material.
O processo de fratura é constituído de duas partes: início da trinca e propagação
da trinca. A fratura pode ser classificada como fratura dúctil, quando acompanhada
previamente de deformação plástica apreciável e como fratura frágil, quando a
deformação não é mensurável macroscopicamente, sendo esta o modo comum de
resposta final às solicitações mecânicas dos materiais cerâmicos à temperatura
ambiente.
A tenacidade à fratura é uma propriedade que representa a resistência à
propagação de uma trinca num determinado material, sendo assim, crítica para
materiais com aplicações estruturais. Por ser uma propriedade importante
principalmente para materiais cerâmicos estruturais, várias técnicas de avaliação e
medida de tenacidade foram desenvolvidas, utilizando macroentalhes e microfalhas.
A literatura apresenta uma grande variação nos valores de tenacidade à fratura e
parâmetros de crescimento de trinca para materiais similares quando a geometria da
amostra e/ou o método de teste são diferentes. O espalhamento destes resultados
pode ser em parte atribuído a diferenças na composição e microestrutura dos
materiais testados e também a diferenças reais no comportamento de fratura
(trincamento rápido, crescimento lento de trinca, curva-R crescente, etc...)
Os materiais cerâmicos, devido à combinação de ligações covalentes e iônicas,
tem intrinsecamente baixa tenacidade. Nos últimos anos, tem sido realizado grande
esforço de investigação para melhorar a tenacidade dos materiais cerâmicos.
O método de fratura por indentação (IF - “indentation fracture”) é uma das
técnicas mais simples para determinação da tenacidade à fratura de materiais

89
frágeis como as cerâmicas, sendo assim, comumente utilizado (Anstis G. R. et al.,
1981; Chiang S. S. et al., 1982; Tsai, Y. M., 1984; Cook, R. F. & Pharr, G. M., 1990;
Pajares, A. et al., 1996; Tanaka, K. et al., 1999; Anton, R. J. & Subhash, G., 2000;
Jiang, L. Z. & Sun, C. T., 2001). Esta técnica permite a avaliação da tenacidade em
pequenas amostras, de materiais amorfos ou policristalinos, podendo ser útil no
estudo para o desenvolvimento de materiais. Em comparação com outros métodos
convencionais para obtenção do valor de tenacidade à fratura, o método IF oferece
simplicidade e economia nos procedimentos de teste, porém, com uma pequena
margem de erro. Um erro de 10 % pode ser obtido quando o módulo de Young (E) é
conhecido, ou 30 % quando E não é conhecido.
A principal desvantagem da técnica IF é a dispersão (30 – 40 %) nos valores
obtidos. Isto se deve a campos de tensões residuais associados com a produção da
trinca. Além disso, a técnica apresenta grande dificuldade na avaliação do
comprimento de trinca e dificuldades na seleção de uma fórmula aceitável entre as
várias equações já propostas para cálculo de KIC. A dificuldade na medição do
comprimento da trinca pode ser parcialmente superada pela adição de uma camada
de ouro de 20 nm sobre a superfície antes da indentação. A técnica de IF também
requer minimização das tensões compressivas residuais. As tensões residuais
combinadas com a tensão aplicada produzem uma baixa resistência à fratura e
resultam em um valor de KIC menor. A eliminação da tensão residual pode ser feita
através da remoção de material ou recozimento. A remoção de material é o método
preferido, uma vez que o recozimento pode conduzir a cura ou cegamento de
defeitos, e pode, em alguns materiais, levar a uma indesejável mudança
microestrutural. O polimento espelhado da superfície a ser identada também diminui
a tensão residual (Marchi, J., 1999; Anstis, G. R. et al., 1981; Basu, D. & Sarkar, B.
K., 1996; Muchtar, A. & Lim, L. C., 1998; Piorino, F.N. et al., 1990; Oliveira, L. G.,
2002). As amostras devem ser polidas na superfície a ser identada com pasta de
diamante de 15 a 0,25 μm, sucessivamente, para eliminação de porosidade
superficial e remoção de tensões compressivas superficiais (Oliveira, L. G., 2002).
O método da fratura por indentação, proposto há 20 anos atrás, é um método de
medida de tenacidade à fratura que envolve a medida das trincas que surgem a
partir dos vértices de uma indentação Vickers como função direta da carga de
indentação. A base teórica para o método por impressão Vickers está estabelecida

90
em termos da mecânica da fratura para impressão em função de uma deformação
elástica/plástica (FIG.3.41).
FIG. 3.41 Modelo de Johnson da cavidade esférica em expansão.
A resposta do material sujeito à penetração por pirâmides rígidas é complexa do
ponto de vista da análise do campo de tensões. Competem nesse caso processos
de deformação induzida por tensões de corte e processos de densificação ou
compactação de zonas porosas.
O modelo da cavidade esférica em expansão é aplicado a matérias frágeis
sujeitos a indentação. Apesar da resposta frágil à maioria das solicitações
mecânicas, os cerâmicos, tais como os metais, exibem deformação permanente
quando sujeitos à ação superficial de indentadores rígidos e agudos. A deformação
permanente de um material é determinada por diversos processos em escala
atômica: movimento das discordâncias, difusão de defeitos, deslizamento dos
contornos de grão e maclagem.
Um núcleo esférico imediatamanete abaixo do indentador é sujeito a um campo
hidrostático de pressão igual a pressão de contato, seguindo-se uma região
deformada plasticamente, de deformação radial decrescente até a fronteira com o
domínio puramente elástico. A fronteira elasto-plástica, na qual o estado de tensão
é puramente de tração, é uma região potencialmente geradora de trinca nos
cerâmicos. Desta forma, os ensaios de dureza por indentação, em particular os de
dureza Vickers, são utilizados não só na medida da resistência à deformação
permanente, mas também como uma medida da resistência à propagação de
trincas, permitindo a determinação da tenacidade à fratura por indentação. A

91
seqüência da iniciação e propagação de trincas decorrentes da indentação de um
material duro é apresentada na FIG. 3.42 (Rezende, D. T., 1997).
FIG. 3.42 Sequência da iniciação e propagação de trincas na indentação de um material duro.
(a) o indentador produz uma zona inelástica de deformação;
(b) para um valor crítico de carga, desenvolve-se uma trinca induzida pela
deformação, a trinca mediana, no eixo da carga a partir da zona
deformada inelasticamente. Podem ter início ainda as trincas
superficiais perpendiculares à direção de aplicação da carga, conhecidas
como trincas radiais;
(c) o incremento da carga intensifica o crescimento estável das trincas,
resultando num sistema composto radial/mediano;
(d) com a retirada do indentador, a trinca mediana fecha, porém não
desaparece;
(e) prosseguindo com o descarregamento, a tensão de indentação evolui
para valores residuais. Ocorre a nucleação de novas trincas na fronteira

92
elasto-plástica com propagação paralela à superfície, as trincas laterais.
As tensões residuais contribuem, também, para a propagação das
trincas radiais;
(f) após a completa remoção da carga, as trincas laterais se desenvolvem
até a superfície podendo causar fragmentação paralela à superfície; a
indentação Vickers em materiais frágeis, após a remoção da carga,
apresenta tipicamente quatro trincas radiais e eventualmente,
fragmentação lateral.
A pirâmide de diamante Vickers é adotada como indentador padrão. A
geometria Vickers produz um sistema de duas trincas, com as trincas dispostas em
ângulos retos. Esta modalidade apresenta maior facilidade na reprodução de traços
de trincas radiais bem definidas sobre superfícies cerâmicas. Entretanto
indentadores Knoop também podem ser usados. A Indentação Knoop cria uma trinca
simples (única), prontamente tratável para a aplicação da mecânica da fratura de
uma falha na superfície. As trincas produzidas com indentador knoop são muito
mais rasas (superficiais) que as produzidas com o indentador Vickers com a mesma
carga, além disso, apresentam alguma recuperação da deformação elástica após a
remoção do indentador.
Uma característica específica do padrão (impressão) produzido por um
indentador agudo (“sharp”) é a formação de uma zona plástica central e suas
associadas tensões residuais imediatamente abaixo da área de contato. A
preocupação principal das técnicas de microtrincamento induzido por indentação
para a determinação da tenacidade à fratura tem sido justamente como tratar o
campo de tensão residual complexo produzido pela indentação.
Os modelos de tenacidade, baseados no desenvolvimento de trincas
governadas pelo campo de tensão elástico-plástico produzido pela aplicação de uma
carga, são usualmente classificados de acordo com a forma geométrica da trinca
abaixo da indentação, em dois sistemas principais: (Song, Y. K. & Varin, R. A.,
1997).
a) Sistema radial mediano ou sistema mediano (”half-penny”): Trincas que
se formam como resultado de indentação Vickers, chegam a superfície
como radial mediana. Neste sistema, a força motriz para o campo de

93
tensão da trinca é formada por uma componente elástica, responsável
pelo crescimento da trinca durante o carregamento, e uma componente
plástica, responsável pela extensão da trinca superficial durante o
descarregamento. A uma dada carga crítica de indentação, uma trinca
inicia-se abaixo do ponto de contato devido à geração de uma
componente de tensão trativa. A trinca que se forma abaixo da superfície
é comumente referida como trinca mediana. Para o indentador piramidal
Vickers, a trinca mediana aparece na forma de duas trincas circulares
mutuamente perpendiculares e na direção paralela às diagonais da
indentação. Quando a carga é retirada e o indentador é removido, as
trincas medianas de subsuperfície tornam-se instáveis e propagam-se na
direção da superfície identada dando assim origem às trincas radiais.
b) Sistema Palmqvist: trincas radiais que se formam durante o
carregamento localizado causado por deformação plástica associada ao
contato. Em alguns materiais frágeis, em corpos de prova com superfície
polida e com tenacidade relativamente alta, trincas de superfície radiais
rasas, conhecidas como trincas de Palmqvist podem se formar a cargas
de indentação relativamente baixas antes da trinca mediana de
subsuperfície.
A FIG. 3.43 apresenta um esquema de comparação entre as geometrias da
trinca (Palmquist e “half-penny”) ao redor de impressão Vickers, vistas em seção
transversal. A FIG. 3.44 representa os mesmos sistemas, porém de uma vista
tridimensional. (Marchi, J., 1999).

94
FIG. 3.43 Comparação dos sistemas de trincas ao redor de impressão Vickers.
FIG. 3.44 Sistemas de trincas (a): Palmquist; (b): “half-penny”.
A técnica IF requer uma medição direta do tamanho da trinca “halfpenny” ou
Palmqvist obtida por indentação Vickers. Uma vasta literatura existe sobre as
relações quantitativas entre o comprimento da trinca superficial introduzida por
indentação e a tenacidade à fratura. Essas análises usualmente envolvem
diferentes suposições sobre a geometria da trinca e zona de deformação, algumas
das quais não podem ser aplicáveis para um dado material.
Para identações Vickers, considerando-se que o comprimento da trinca é muito
superior a semi-diagonal da impressão e a geometria é centrada em relação ao
ponto de aplicação da carga na zona de deformação, temos que a expressão básica
que avalia a tenacidade à fratura do material é dada pela equação 3.7 (Anstis, G. R.
et al., 1981, Wang, J.et al., 2002),
23cPKc β= ( 3.7 )

95
onde P é a carga de indentação, β é uma constante para um dado material e
uma dada geometria do indentador. Considerações detalhadas sobre o modo com
que o volume de impressão plástica é acomodado pela matriz elástica nos arredores
mostram que esta constante depende da razão entre o módulo de elasticidade (E) e
a dureza (H).
Os valores de KIC podem variar em função da taxa de teste, por causa dos
efeitos de temperatura ou ambiente. Cargas aplicadas por longos períodos de
tempo podem causar extensão de trinca, levando a obtenção de valores de KIc
menores que aqueles medidos em outros métodos. Este fenômeno de dependência
do tempo é conhecido como crescimento lento de trinca (Slow Crack Growth - SCG).
SCG pode ser significante até mesmo para tempos relativamente curtos, podendo
ser minimizado pelo controle preciso da temperatura, e pela mudança no ambiente,
como por exemplo, o uso de ambiente inerte.
A FIG. 3.45 mostra um desenho esquemático do padrão de indentação
fratura/deformação para geometrica Vickers: a e c são as dimensões características
da impressão plástica e trinca radial/mediana, respectivamente. Em uma análise
dimensional pode ser demonstrado que esses parâmetros estão relacionados
diretamente com a dureza (H) e a tenacidade à fratura (KIC) do material identado.
Indentação Vickers
Trincas radiais na superfície c
a
Trinca lateral sob a superfície

96
FIG. 3.45 Desenho esquemático do sistema IF produzido por um indentador Vickers.
Desde a observação, por Palmqvist em 1957, da extensão de trincas radiais
superficiais que ocorrem quando materiais não dúcteis são indentados, têm-se
aperfeiçoado cada vez mais os modelos, estudando-se as relações entre os campos
de tensão existentes no material e a deformação (Evans, A. G. & Charles, E. A.,
1976; Lawn, B. R. & Marshall, D. B., 1979).
Novas equações propostas são utilizadas para se obter valores mais próximos
do verdadeiro valor de tenacidade à fratura do material, conhecendo o comprimento
da trinca (c) e metade da diagonal de indentação (a), a tenacidade à fratura pode ser
calculada usando a expressão universal de Liang, K. M. et al., 1990. nas equações
3.8 e 3.9 e a validade dessas equações deverá ser verificada.
( ) 51,118/4,0
5,0
−
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
Φ⎟⎠⎞
⎜⎝⎛ Φ ac
Ic
ac
EH
HaK α ( 3.8 )
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛
+−
−=4
10548114
ννα ( 3.9 )
Onde α é uma constante adimensional relacionada a razão de Poisson (ν), como
pode ser verificada na equação acima. Φ é um fator de limitação (estabelecido por
Liang como ≈3), a é a semi-diagonal da impressão Vickers, c é o comprimento da
trinca somado com a semi-diagonal da impressão (c= l + a). E é o módulo de
Young, H é a dureza da amostra testada.
Uma maneira simples de diferenciar as trincas “half-penny” ou radial/mediana da
trinca Palmqvist é o polimento de camadas superficiais. As trincas “half-penny”
permanecem conectadas aos vértices da diagonal enquanto que as trincas do tipo
Palmqvist se separam da diagonal, como pode ser visto na FIG.3.46 (Song, Y. K. &
Varin, R. A., 1998).
Sistema de trinca radial/mediano

97
FIG. 3.46 Diferença entre sistema radial/mediano e Palmqvist após polimento da superfície.
Uma outra maneira de distinguir o sistema de trinca é pela razão entre o
comprimento da trinca e a carga de indentação (Song, Y. K. & Varin, R. A., 1998).
Alguns procedimentos permitem identificar o tipo de sistema envolvido: sistemas
do tipo Palmqvist, P/l é independente da carga, para sistemas “half-penny”, P/c3/2 é
independente da carga. Para os dois sistemas, ln P versus ln l deve ser linear
(Song, Y. K. & Varin, R. A., 1998). A tangente observada no sistema Palmqvist deve
ser entre 0,5 e 1 e, no sistema “half-penny”, deve ser entre 1 e 2; as trincas são
Palmquist se a razão c/a for menor do que três, e “half-penny” se for maior ou igual a
três (Marchi, J., 1999).
De acordo com Wang, J. et al. (2002), que se baseou no estudo de Niihara et al.,
(1982), a trinca Palmqvist pode ser observada quando c/a for menor que 2,5 e a
trinca “half-penny” quando a razão c/a for maior que 2,5.
Ao longo dos anos, diversos resultados de tenacidade à fratura e modelos de
indentação de materiais resistentes ao desgaste abrasivo (Berns, H. & Franco, S. D.,
1997), compósitos de alumina e zircônia (Lange, F. F., 1982c; Casellas, D. et al.,
1997; Zhou, D. & Low, I. M., 1996; Nawa, M. et al., 1998; Celli, A. et al., 2003),
cerâmicas contendo zircônia (Dauskardt, R. H. et al., 1987; Gogotsi, G. A. et al.,
1995; Matsuzawa, M. et al., 1999, Tanaka, K. et al., 1999) e compósitos Al2O3-ZrO2-
SiC (Niihara, K. et al., 1994), foram reportados na literatura.
Sistema de trinca Palmqvist
Não polida Polida
Não polida Polida

98
Bucko, M. M. & Haberko, K.,(1999), estudaram a anisotropia mecânica de
cerâmicas de alumina tenacificada por zircônia. Inclusões de zircônia (distribuída na
matriz de alumina) formam partículas alongadas com uma orientação cristalográfica
devido à compactação uniaxial. Este material apresentou diferentes valores de
Módulo de Young, comprimento de trinca, KIC nas direções paralela e perpendicular
à força de compactação. Pandolfelli, V. C. et al.,(1991), verificaram a correlação
existente entre a técnica de medida da tenacidade e o grau de transformabilidade de
zircônias tetragonais.
3.7.3 RESISTÊNCIA MECÂNICA
Cerâmicos apresentam muitas qualidades atrativas incluindo estabilidade
química, elevada dureza, resistência ao desgaste e elevado ponto de fusão. Estas
propriedades são essenciais para o uso de cerâmicos em componentes que são
aplicados em condições ambientais extremas. Devido a sua fragilidade, falha
catastrófica sob níveis de tensões relativamente baixos é um problema crítico no
design de estruturas utilizando cerâmicas. A natureza basicamente frágil dos
cerâmicos resulta de suas ligações iônicas e covalentes as quais fornecem um
limitado número de sistemas de deslizamento independentes, os quais são
necessários para se atingir uma deformação plástica homogênea comumente
observada nos materiais metálicos. Devido à ausência de mecanismos de
deformação plástica, a ponta de uma trinca em um material frágil é considerada
como sendo atomicamente aguda e é usualmente considerado que ela se extende
através de um processo de ruptura de ligações atômicas.
A resistência mecânica dos materiais é devida às forças coesivas entre os
átomos, e o seu valor teórico varia de E/5 a E/10, conforme o modelo empregado
para o seu cálculo, onde E é o módulo de elasticidade do material. Os resultados
experimentais mostram, entretanto, que os valores da resistência mecânica são de
100 a 1000 vezes menores que os valores da resistência teórica esperada.
A primeira explicação da discrepância observada entre a resistência medida e
a resistência teórica em materiais frágeis foi proposta por Griffith (Griffith, A. A.,
1920). A teoria de Griffith, na sua forma original, é aplicável somente a um material
perfeitamente frágil, como o vidro. Griffith propôs que um material frágil contém uma

99
população de pequenas trincas, nas extremidades das quais existe uma
concentração de tensões de magnitude tal, que o valor da resistência coesiva teórica
é alcançado nestas regiões do material, provocando a fratura do mesmo, ainda que
a tensão nominal aplicada esteja bem abaixo do valor teórico da resistência.
Quando uma trinca se propaga, produz um aumento na área de suas superfícies
laterais. Isso requer uma certa quantidade de energia para vencer a força coesiva
dos átomos, ou ainda, exige um aumento na energia de superfície. A fonte de
energia para suprir a energia extra para o aumento da superfície é a energia de
deformação elástica, a qual é liberada com a extensão da trinca. De acordo com
Griffith, uma trinca irá se propagar quando o decréscimo na energia de deformação
elástica for pelo menos igual a energia necessária para a criação de uma nova
superfície de trinca.
Griffith considerou as trincas como tendo forma elíptica e obteve uma relação
entre o valor da resistência mecânica σ , o módulo de elasticidade E, a energia
superficial 0γ , e o tamanho do defeito crítico, ac. Para o estado de tensão plana, ou
seja, placas finas, temos:
21
0
.2
⎟⎟⎠
⎞⎜⎜⎝
⎛=
CaE
πγ
σ ( 3.10 )
e para o estado de deformação plana, ou placa espessa em relação ao
tamanho da trinca, temos:
( )21
20
.12
⎟⎟⎠
⎞⎜⎜⎝
⎛
−=
CaE
πυγ
σ ( 3.11 )
Mais tarde foi reconhecido que os valores de 0γ são muito pequenos para
explicar a resistência mecânica conforme a teoria de Griffith. Esta observação levou
a substituição de 0γ por iγ , energia específica efetiva para iniciação da fratura, que
explica melhor os resultados experimentais. Outra modificação foi a introdução de
uma constante Y, que é função da geometria do corpo de prova e do tipo de ensaio
mecânico. Essas modificações devidas a Davidge, R. W. & Evans, A. G., (1970),
resultaram na chamada equação básica da resistência mecânica:
21
21⎟⎟⎠
⎞⎜⎜⎝
⎛=
C
i
aE
Yγσ ( 3.12 )

100
Para defeitos superficiais tem-se que Y ≅ 2,0 e para defeitos abaixo da
superfície Y ≅ 1,8. Esta equação mostra que a resistência mecânica de um material
frágil está relacionada à energia de fratura iγ , ao módulo de elasticidade E e ao
tamanho do defeito mais crítico Ca , os quais dependem da microestrutura do
material. É possível observar a partir desta equação que para obtermos um aumento
da resistência do material, deve ser reduzido o tamanho do defeito crítico, o que
pode ser feito através de rigoroso controle do processamento cerâmico, ou ainda,
aumentar iγ , o que poderia ser feito por exemplo, com adições de zircônia na
alumina, o que aumentaria as tensões compressivas as quais deveriam ser
vencidas, promovendo assim um aumento da resistência do material.
Evans, e Tappin (Evans, A. G. & Tappin, G., 1972), modificaram a equação
3.12 pela introdução de um novo parâmetro Z, que leva em consideração a forma
do defeito, resultando na expressão mais geral:
2
12
⎟⎟⎠
⎞⎜⎜⎝
⎛=
c
i
aE
YZ γ
σ ( 3.13 )
O termo Z é conhecido como parâmetro de forma de trinca, e o seu valor para
o caso de trincas elípticas pode ser calculado a partir das seguintes expressões:
Para a ≤ c:
∫ ⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛−−=
2
0
21
22
2
sen11
π
θcaZ ( 3.14 )
e para a ≥ c:
21
2
0
22
2
sen11∫ ⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛−−=
π
θac
caZ ( 3.15 )
onde a é a profundidade da trinca e c sua semilargura, isto é, a e c são os semi-
eixos da trinca elíptica, conforme mostrado na FIG. 3.47.

101
FIG. 3.47 (a) defeito semi-elíptico em barra submetida a flexão em quatro pontos. Em (b), a profundidade do defeito, a, é menor do que sua semilargura, c, e em (c) a é maior do que c.
Irwin, G. R., 1957, estabeleceu a relação entre o fator de intensidade de
tensão crítico da mecânica da fratura e parâmetros do material, como o módulo de
elasticidade e a energia de fratura, através da expressão:
( )21
2 EK iIC γ= ( 3.16 )
Pode ser observado que KIC é uma constante do material uma vez que é
função dos parâmetros E e iγ , que por sua vez são constantes do material.
Devido a essa expressão, a equação 3 pode ser escrita da seguinte maneira:
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛=
21
1
C
IC
a
KY
σ ( 3.17 )
ou ainda: iIC aYK σ= ( 3.18 )
De acordo com esta equação, quando a tensão aplicada e o tamanho do
defeito são tais que atinja o valor crítico KIC, a fratura do material ocorrerá.
Desta forma, a teoria de Griffith trouxe uma contribuição fundamental ao
estudo de materiais frágeis, ficando estabelecido que a resistência dos materiais não
é uma característica própria do material, mas um parâmetro que depende fortemente

102
do tamanho dos defeitos, falhas e trincas do material. A presença de uma trinca
decresce a resistência da estrutura a qual decresce progressivamente com o
aumento do tamanho da trinca.
Além disso, é reconhecido que o espalhamento da resistência mecânica
observada nos materiais frágeis como os cerâmicos é proveniente de uma
distribuição de defeitos ou falhas. Este fato levou ao tratamento da probabilidade de
fratura e ao desenvolvimento de teorias como a estatística de Weibull. Todavia, foi
uma evolução no campo da engenharia conhecida como mecânica da fratura que
formalizou estes conceitos tendo como base os conceitos básicos propostos por
Griffith. A resistência de um material é determinada por dois fatores, a tenacidade à
fratura ou resistência a propagação da trinca e o maior ou mais severo defeito. O
principal problema dos materiais cerâmicos é a tenacidade à fratura, a qual é
inerentemente muito baixa, de tal forma que estes materiais são muito sensíveis a
falhas e defeitos muito pequenos, mesmo em baixos níveis de tensões.
O desenvolvimento de tecnologias para a produção de novos materiais tem
sido motivada pela demanda de materiais que executem novas funções ou
desempenhem antigas funções de forma mais adequada. A elaboração de materiais
cerâmicos avançados teve início na década de 70 e têm alcançado resultados
promissores, verificando-se inclusive, uma contínua evolução no desenvolvimento e
uso destes materiais voltados para aplicações estruturais. Tais melhorias foram
realizadas através da disponibilidade de pós finos de elevada pureza bem como a
incorporação de mecanismos de aumento da tenacidade tais como a transformação
de fase do tipo martensítica induzida por tensão, microtrincamento, reforços por
fibras, whiskers, e etc...
A resistência dos materiais cerâmicos está diretamente relacionada ao tamanho
do defeito mais crítico existente no material testado. O defeito mais crítico é aquele
que, devido ao seu tamanho, orientação e localização no volume testado do
material, apresenta a maior probabilidade de fratura, ou seja, aquele defeito cuja
extremidade se encontra o maior fator de intensidade de tensão. Como para todos
os materiais uma amostra nunca é exatamente igual a outra, o tamanho, a
orientação e a localização do defeito mais crítico não serão os mesmos de amostra
para amostra, e de acordo com a equação básica da resistência mecânica, a
resistência mecânica será diferente para cada amostra. Por isso, em um material

103
cerâmico os valores de resistência mecânica medidos apresentam um certo
espalhamento devido a grande variedade de defeitos presentes. Este fato, é um dos
grandes inconvenientes no emprego de materiais cerâmicos. Para caracterizar a
resistência mecânica de uma maneira bem definida há necessidade de se utilizar
tratamentos estatísticos. Esses tratamentos foram desenvolvidos pela primeira vez
por Weibull, que relacionou a resistência mecânica σ de um material com a
probabilidade de fratura F, que é a probabilidade de se encontrar num volume
testado V sob a tensão σ um defeito de características tais que o material frature
sob aquela tensão aplicada. A essa correlação entre probabilidade de fratura e
resistência mecânica dá-se o nome de distribuição de Weibull.
Numerosos processos de microfratura e mecanismos os quais ocorrem não
somente na zona frontal do processo (na frente de uma trinca aguda), mas também
na zona deixada para trás no avanço da ponta da trinca em propagação (zona da
marola – “following crack wake”) e zona de ponteamento de trinca (“crack bridging
region”) como na FIG. 3.48, podem defender a ponta da trinca de tensões aplicadas
longe desta região e assim tenacificam o material.
FIG. 3.48 Zona frontal do processo , zona da marola e zona de ponteamento da trinca em propagação.
A FIG. 3.49 apresenta vários processos de absorção de tensões por materiais
frágeis.
Ponteamento da trinca
Zona da marola
Zona frontal do processo

104
FIG. 3.49 Processos de absorção de tensões (a) deflexão (“deflection”) da trinca por decoesão (“debonding”) na interface; (b) microtrincamento na zona frontal do processo (“branching”);
(c) escoamento plástico na zona frontal do processo; (d) transformação de fase com expansão de volume e/ou microtrincamento (“microcracking”)na zona da marola; (e) ponteamento da
trinca por fibras ou whiskers; (f) ponteamento (“bridging”) da trinca por grãos e travamento (g) ponteamento viscoelástico.
Nos cerâmicos, são os processos que ocorrem na região atrás da trinca em
propagação ou zona de marola é que são substanciais no efeito tenacificador. Os
fenômenos que ocorrem nesta região, tais como microtrincamento e ramificações,
formação de pontes de fratura, transformação de fases e atrito entre grãos, resultam
em uma curva-R crescente, isto é, a resistência à propagação da trinca KR, aumenta
com o crescimento ou extensão da trinca( Δ a), levando a tenacificação cumulativa.
Na FIG. 3.50 é apresentado um gráfico esquemático de uma curva-R crescente
e as contribuições ao processo de microfratura da zona frontal do processo Δ KFP
(a0) e da zona da marola Δ KW ( Δ a) ao aumento da tenacidade. As conseqüências
destes efeitos são significativos e criam dificuldades na determinação experimental
da tenacidade à fratura de materiais frágeis. A medida da tenacidade sofre um
espalhamento em função do método de medida utilizado, da geometria e dimensões
da amostra.

105
FIG. 3.50 Gráfico esquemático da curva-R crescente.
Os materiais cerâmicos, em função principalmente do tipo de ligação química
apresentam altos valores de resistência teórica. A resistência à compressão dos
cerâmicos é alta porque os defeitos inerentes ao material (defeitos superficiais,
poros, inclusões, grãos grandes gerados durante o processamento), permanecem
fechados durante a solicitação em compressão, sendo necessário então outros
mecanismos que irão consumir mais energia para fraturar o material.
Por outro lado, quando o material cerâmico é solicitado em tração, ele apresenta
resistência bem menor porque permite a abertura e crescimento dos defeitos
internos, os quais atingem um valor crítico que leva à fratura do material. O ensaio
de tração submete um volume maior, de maneira uniforme, a carga aplicada.
Porém, a confecção do corpo de prova é difícil e de alto custo e susceptível a erros
experimentais (Anstis, G. R. et al.,1981).
Os ensaios de flexão tem sido utilizados na avaliação da resistência dos
cerâmicos por ser de baixo custo, por gerar dados precisos, pela facilidade de
preparação de corpos de prova, e pela simplicidade na execução do ensaio
propriamente dito.
Carvalho, E. A. & Duarte, E. T. V., (2000), apresentaram os fatores geométricos
e físicos que controlam o estado de tensões em ensaios de flexão a 3 ou 4 pontos,
muito empregados na determinação de propriedades mecânicas em materiais
Extensão da trinca ( Δ a) →
Res
istê
ncia
ao
cres
cim
ento
da
trin
ca K
R

106
frágeis, como os cerâmicos. O efeito das tensões de contato, desalinhamento no
carregamento, atrito e concentração de cargas tem efeito dominante no resultado
obtido em ensaios de flexão, porém as formulações tradicionalmente empregadas
para a determinação de estado de tensões não levam em conta estes efeitos,
causando um mascaramento de resultados, usualmente atribuído a natural alta
dispersão dos valores de resistência mecânica obtidos quando do estudo de
materiais frágeis em geral e cerâmicos em particular. O efeito de cada fator foi
analisado isoladamente e em conjunto com outros, resultando em formulações
matemáticas que levam em conta os mesmos na conversão de cargas para tensões
de ruptura. Métodos experimentais e numéricos tais como a fotoelasticidade e o
método de elementos finitos foram empregados para corroborar as hipóteses
apresentadas.
Os ensaios tradicionais de flexão admitem apenas como existentes ou
significativas, as tensões originárias da flexão da região estudada. Como
conseqüência a formulação empregada para converter uma carga aplicada em
tensão reflete apenas este componente do estado de tensões reinantes. Uma
análise cuidadosa de ensaios de tensão revela a existência de outros componentes
de tensão, originários das imperfeições geométricas do corpo de prova bem como
dos desalinhamentos do dispositivo de ensaio empregado, como tensão de flexão,
de contato, devido a curvatura do corpo de prova, de torsão, tensões cisalhantes
transversais e de atrito (Carvalho, E. A. & Duarte, E. T. V., 2000).
A formulação para ensaio de flexão em três pontos que leva em conta somente
os efeitos de flexão sobre o estado geral de tensões tem para uma viga de seção
retangular, está descrita na equação 3.19, onde P é a carga, L a distância entre
cutelos, b a largura do corpo de prova e h sua altura (Carvalho, E. A. & Duarte, E. T.
V., 2000; Davidge, R. W. & Evans, A. G., 1970; Piorino, F. N. et al., 1992).
2P3
Flex bh2PL3
=σ ( 3.19)
Frocht apresenta a equação 3.20, chamada de Wilson-Stokes que leva em conta
o efeito da carga concentrada, onde: x varia de zero (abaixo do cutelo superior) até a
altura total (h), na base do corpo de prova (Carvalho, E. A. & Duarte, E. T. V., 2000)

107
bhP
2hx
2h
4L
bhP123
P3stokes ππ
σ +⎟⎠⎞
⎜⎝⎛ −⎟
⎠⎞
⎜⎝⎛ −= ( 3.20 )
Na FIG. 3.51 é apresentado o campo de tensões cisalhantes máximas (linhas
pretas). Este resultado obtido pela técnica da fotoleasticidade apresenta o complexo
campo total de tensões que atua em uma viga prismática carregada a três pontos,
indicando que a expressão (3.19) é precária ao tentar descrever o campo de tensões
atuantes neste corpo.
FIG. 3.51 Campos de tensões em um viga prismática carregada em três pontos, franjas isocromáticas - campo escuro.
Carvalho, E. A. & Duarte, E. T. V., (2000) compararam resultados de flexão em
três pontos obtidos utilizando as duas equações e concluíram que existe uma
diferença de 10,6 % entre os mesmos. Isto significa dizer que se a propriedade for
medida no ponto oposto ao cutelo superior que aplica a carga, e avaliada pela
equação 3.19, esta será 10,6 % superior ao valor obtido com a equação de Wilson-
stokes, com abordagem mais realística.
Ensaios de flexão em três pontos e quatro pontos (Yang, C. C. T. & Wei, W. C.
J., 2000; Tuan, W. H. et al, 2002) vem sendo largamente utilizados para avaliação
da resistência mecânica de materiais cerâmicos. O ensaio de flexão em quatro
pontos apresenta resultados mais representativos, embora inferiores aos obtidos em
ensaios de três pontos, uma vez que envolve um volume ensaiado maior e ainda um
maior número de defeitos presentes que podem levar à fratura do material.
3.7.4 DEGRADAÇÃO DAS PROPRIEDADES MECÂNICAS DA ZIRCÔNIA

108
De acordo com Piconi, C. & Macauro, G. (1999), a zircônia pode apresentar
degradação da sua resistência mecânica em meio aquoso ou vapor d`água na faixa
de temperatura crítica de 200-300°C, de acordo com as seguintes características:
a) Os efeitos do envelhecimento são: redução da resistência, tenacidade
e densidade, e um aumento da quantidade de fase monoclínica;
b) A degradação das propriedades mecânicas ocorre devido a
transformação de fase T → M com micro e macrotrincas no material;
c) A transformação de fase T → M começa na superfície do material e
progride para o seu interior;
d) A redução do tamanho de grão ou aumento da concentração de óxido
estabilizante , reduz a taxa de transformação.
Utilizando técnicas atuais e trabalhando em nanoescala, pesquisadores tem
inibido o processo de degradação pela adição de pequenos traços de materiais tais
como a alumina à zircônia, sem comprometer sua tenacidade. A adição destes
materiais tem como alvo a prevenção do processo de degradação na zircônia a partir
de sua superfície.(www.azom.com).
A zircônia estabilizada com ítria (Y-TZP) tem sido considerada material de
escolha para aplicações médicas, tais como cabeça de fêmur em reconstruções
totais de quadril, implantes dentários devido a excelente resistência e tenacidade em
temperatura ambiente. Todavia, a desintegração da estabilidade de fase e
propriedades mecânicas após longo uso em temperaturas entre 100 e 400°C em ar
ou água limita as aplicações clínicas de cerâmicas biocompatíveis a base de zircônia
quando a esterilização do material é realizada em autoclave (vapor d`´agua e
temperatura), ao invés de esterilização com plasma.
A tentativa de minimizar a degradação em baixas temperaturas (“Low Thermal
Degradation”) da Y-TZP inclui a redução do tamanho de grão, um aumento no
conteúdo de estabilizante (Y2O3, CeO2) ou ainda a formação de compósitos com
Al2O3. A adição de partículas de alumina impede a relaxação da rede de zircônia
tetragonal, sob tensão, durante o processo de envelhecimento uma vez que a
relaxação é responsável pela degradação e é governada pela difusão de vacâncias
de oxigênio quando recozida em baixas temperaturas (Lee, D. Y. et al., 2000).

109
No trabalho de Shimizu, K. et al., (1993), com o objetivo de estimar a
degradação das propriedades mecânicas da zircônia com o tempo, peças de
zircônia foram colocadas em solução salina a 95°C por 3 anos e suas propriedades
foram examinadas. Não foi observado nenhum decréscimo relevante na resistência
à flexão sob tais condições. Os autores concluíram que a zircônia pode ser usada
clinicamente por reter uma resistência à flexão superior a 700 MPa.
A transformação de fase fica favorecida com o recozimento a baixas
temperaturas e/ou exposição a vapor d`água. Assim, o envelhecimento de
cerâmicas de zircônia em condições fisiológicas tem sido questionado. Resultados
confusos foram reportados em anos anteriores tornando difícil o entendimento por
parte dos não ceramistas. É importante notar que o envelhecimento é uma
característica de todos os biocerâmicos, como alumina e zircônia, sendo dependente
da microestrutura e composição química do material (Cales, B., 1994). O
envelhecimento de materiais para implante a base de alumina foi observado nas
condições in-vitro e in-vivo. O decréscimo da resistência à fratura foi observado
para a alumina com relativamente baixa porosidade ou com contaminação de Ca
após poucas semanas in-vitro e in-vivo. Todavia, é bem conhecido que a alumina de
elevada pureza e densidade é totalmente estável no interior do corpo. A situação é
similar para a zircônia, exceto que o envelhecimento foi observado em vapor e em
altas temperaturas (150 °C – 400 °C) e não investigado em condições fisiológicas.
O vapor a altas temperaturas não é representativo do ambiente fisiológico. Desta
forma, a extrapolação de resultados a partir de altas temperaturas não é correto uma
vez que a taxa de transformação poderia ser diferente para altas temperaturas e a
temperatura do corpo humano. Ainda, o envelhecimento deve ser analisado para
cada material sob condições representativas para cada caso e qualquer
generalização deve ser evitada. Para adição de 3% mol de ítria (5,1 ± 0,3% em
peso), é esperado um tamanho de grão médio inferior a 0,8 μm. Porém, tamanhos
muito pequenos (inferiores a 0,3 μm) devem ser evitados devido ao baixo valor
obtido para as propriedades mecânicas. De acordo com Chevalier et al., 1999, o
crescimento lento de trinca depende diretamente do tamanho médio de grão e
maiores valores de tenacidade à fratura são obtidos para maiores tamanhos de grão.
A faixa de tamanho entre 0,4 - 0,8 μm poderia ser considerada uma faixa de ótimo.
De acordo com Calès, B., (1994), estudos realizados in-vivo (condições humanas) e

110
in-vitro (solução de Ringer) por 2 anos foram feitos com biocerâmicos de zircônia.
Na condição in-vivo, a resistência á fratura foi verificada antes e após a implantação
em pacientes. Os autores concluíram que não houveram diferenças significantes
nos valores de resistência mecânica, tanto in-vivo quanto in-vitro com nível de
significância de 99%.
No trabalho de Guo, X. & He, J., (2003), a transformação espontânea de fase
tetragonal para monoclínica sofrida pela zircônia, em presença de água ou vapor
d`água quando recozida em temperaturas relativamente baixas (65 °C – 400 °C),
mostrou ser devida a aniquilação de vacâncias de oxigênio. De acordo com os
autores, muito esforço tem sido feito para explicar a degradação. Alguns
mecanismos típicos podem ser resumidos a seguir: Lange, F. F. et al., 1986,
observou a presença de cristalitos de Y(OH)3 em cerâmicas de zircônia dopada com
ítria após degradação, sugerindo que o vapor d`água reagiu com Y2O3 formando os
cristalitos, os quais extraíram localmente a ítria dos grãos tetragonais, transformando
os grãos tetragonais em monoclínicos. Por outro lado, Sato, T. & Shimada, M.,
(1985), propuseram que o vapor d`água reagiria com ligações Zr-O-Zr e ligações
Zr-OH eram formadas. Todavia, outro trabalho mostra que a dissociação de H2O na
superfície da zircônia e a formação de ligações Zr-OH e Y-OH, resulta na
deformação da rede na superfície e promove a transformação tetragonal →
monoclínica. Foi também proposto que a reaçâo dos grupos hidroxila OH – nos
pontos mais ativos (Y2O3) forma um oxi-hidróxido da espécie YO(OH), responsável
chave para a degradação de Y-ZrO2. Todavia, nenhum dos mecanismos acima
pode explicar o efeito do tamanho de grão. Alguns autores sugerem que as
vacâncias de oxigênio desempenham um importante papel na estabilização da
zircônia. Através da criação e aniquilação de vacâncias de oxigênio na zircônia,
alguns autores provaram que existe um mínimo e um máximo críticos de
concentração de vacâncias de oxigênio para cada uma das fases: cúbica, tetragonal
e monoclínica. Uma variação suficiente na concentração de vacâncias de oxigênio
resulta na transformação de fase, ou seja, quando a concentração de vacâncias de
oxigênio é reduzida de tal forma que a fase tetragonal não é mais estável, a
transformação ocorre. Se a quantidade transformada é suficientemente grande, há
uma expansão de volume associada à transformação gerando trincas nas camadas

111
superficiais e estas geram novas superfícies para reagirem com as moléculas de
água levando a seguir a transformação espontânea.
A melhoria da estabilidade da Y-TZP com a adição de alumina foi reportada na
literatura (Feighery, A. J. & Irvine, J. T. S., 1999, Oblak, C. et al., 2004) e atribuída
ao decréscimo no tamanho de grão e ao maior módulo de elasticidade, o qual
aumenta a restrição elástica da matriz Y-TZP, permitindo a retenção da zircônia
tetragonal. De acordo com os autores acima citados, não foi verificado a
degradação mecânica para a zircônia Tosoh com 3% mol de Y2O3 e adição de 20%
de Al2O3.
De acordo com Li, J. et al. (2001), dentre os óxidos como SiO2, Fe2O3, Al2O3,
etc., somente a alumina pode efetivamente suprimir a degradação da zircônia. De
acordo com os autores, o fenômeno da degradação pode ser resumido assim: 1) a
transformação é controlada pela quebra de ligações Zr-O-Zr durante a reação com a
água, a qual resulta em alívio da restrição agindo nos grãos tetragonais e
promovendo a transformação de fase. 2) A nucleação da fase monoclínica é
causada pela acumulação de deformação devido a difusão de OH – pela formação
de ligações Zr-OH e Y-OH e a expansão da rede. 3) A desestabilização da fase
tetragonal é causada pelo decréscimo da quantidade de ítria nos grãos tetragonais
devido a formação de hidróxido de ítria. Ainda de acordo com os autores, a adição
de alumina não altera o mecanismo de degradação de Y-TZP, porém, ela pode
prevenir o prosseguimento do processo de degradação quando o volume
transformado atinge um certo valor.
Cordingley, R. et al., 2003 afirmam que a degradação em baixas temperaturas
da zircônia tetragonal policristalina ocorre como resultado da transformação
espontânea de fase da zircônia tetragonal para fase monoclínica durante
envelhecimento a 130 °C - 300°C, possivelmente em ambiente aquoso, levando a
um decréscimo da resistência devido a formação de microtrincas que acompanham
a transformação. Os autores relatam ainda que novas cerâmicas livres de
degradação estão em estudo no Japão, tais como os seguintes compósitos: 80% Y-
TZP (sendo 90 %mol de ZrO2, 6 %mol de Y2O3, 4 %mol de Nb2O5) e 20 % alumina e
o outro material é composto de 70 %vol. TZP (estabilizada com 10% de CeO2) e 30
% vol.de alumina e 0,05% mol. de TiO2.

112
Watanable, M. et al., 1984, identificou um tamanho de grão crítico abaixo do qual
nenhuma transformação ocorre durante 1000 h de envelhecimento a 300° C. Por
outro lado, Li, J. et al., 2001, reportaram que a transformação de fase em baixa
temperatura se torna mais difícil com o aumento do tamanho de grão. O efeito do
tamanho de grão na transformação é complicado por diversos fatores, resultando em
uma relação do tipo “U” entre a facilidade de transformação e o tamanho de grão. É
importante lembrar que a taxa de degradação da resistência não é a mesma para
todas as cerâmicas Y-TZP. Existe evidência experimental de que a Y-TZP está apta
a manter boas propriedades mecânicas em diversos ambientes por longos períodos
porém, conclusões sobre a sua estabilidade devem ser evitadas pois este
comportamento é peculiar para cada material e técnica de fabricação. Swarb, J. J.,
1991, estudou sete cerâmicas Y-TZP de diferentes fabricantes e todos eles se
comportaram de forma bem diferente. A variabilidade do comportamento ao
envelhecimento está relacionada com as diferenças de parâmetros microestruturais,
tais como: concentração e distribuição do estabilizante, tamanho de grão,
distribuição e população de falha, tensões residuais, densidade, etc. Desta forma, é
necessário conduzir experimentos de envelhecimento em diferentes cerâmicos
individualmente para se obter informação sobre o seu comportamento ao
envelhecimento. Apenas limitadas informações estão disponíveis sobre a
degradação de cerâmicas expostas a água em temperatura ambiente,
especialmente em estudos in-vivo. Resultados conflitantes foram reportados por
Drummond, J. L., 1989 e Thomson, I. & Rawlings, R. D., 1990. Drummond verificou
redução de 20% na resistência em amostras TZP após envelhecimento por 730 dias
em solução salina a 37°C. Thomson, I. & Rawlings, R. D., 1990 não observaram
redução da resistência em amostras envelhecidas com 10% de aumento de fase
monoclínica após 18 meses a 37°C em solução de Ringer, quando comparadas com
amostras iniciais. Os autores concluíram que a resistência nas amostras testadas
era controlada por defeitos mais do que por transições de fase.
No trabalho de Ardlin, B., 2002, duas cerâmicas Y-TZP utilizadas em
reconstruções dentárias foram testadas com objetivo de avaliar a estabilidade
química, e o efeito do envelhecimento (4% ácido acético a 80°C por 168h) na
resistência à flexão, estruturas cristalina e superficial. Os resultados mostraram que
as duas cerâmicas apresentaram alta resistência, a qual não foi afetada pelo

113
envelhecimento e alta estabilidade química. Todavia, os autores verificaram que as
estruturas cristalinas e superficiais foram afetadas. Ocorreu a transformação de fase
tetragonal para monoclínica e pequenas elevações foram observadas na superfície
após o envelhecimento. A solubilidade química ficou bem abaixo dos valores limite
para cerâmicas dentárias (ISO 6872:1995).
3.7.5 RESISTÊNCIA AO DESGASTE
A resistência ao desgaste dos materiais cerâmicos é uma função que depende
não apenas da dureza (H), mas também da tenacidade do material (KIC).
Evans, A. G. & Marshaw, T. R. (1976), desenvolveram a equação 3.21,
relacionando o volume de material removido (V) pelo sistema de indentação em
operação de desbaste ou em desgaste por abrasão:
××××
α 4/52/14/3
IC
PNHK
1V ( 3.21 )
onde “N” é o número de partículas abrasivas, sendo “P” a força normal e “ ”a
distância percorrida. A resistência ao desgaste (1/V) comporta-se então como uma
função linear de KIC e H conforme a equação :
2/14/3IC HK
V1
×α ( 3.22 )
A importância de se buscar sistemas cerâmicos com maior tenacidade à fratura,
está na relação (KIC)3/4. (H)1/2, ou seja, a resistência ao desgaste, é muito mais
sensível à variação na tenacidade em função do maior expoente ¾ (Duailibi, J. F., et
al., 1999).

114
4 MATERIAIS E MÉTODOS
A metodologia empregada neste trabalho teve por objetivo:
• A produção e a caracterização de compósitos cerâmicos a base de
alumina e zircônia;
• Caracterização microestrutural e determinação das propriedades
mecânicas dos materiais produzidos em função da composição química.
4.1 CARACTERIZAÇÃO DOS PÓS
Os pós iniciais utilizados foram a alumina da Alcoa (A-1000-SG) e a zircônia
estabilizada com ítria de dois diferentes fornecedores: Tosoh Corporation, Japão
(TZ-3YSB ) e Mel Chemicals, Inglaterra (XZO708/14).
Os pós iniciais bem como as composições após moagem em moinho de bolas e
desaglomeração, foram caracterizados pela realização dos seguintes ensaios:
a) Determinação das impurezas (óxidos) presentes nos pós por análise
de espectrometria de fluorescência de raios-X (INT).
b) Determinação das fases presentes nos pós por análise por difração
de raios-X em um difratômetro Philips (INT), empregando a radiação
Kα de um tubo de cobre de λ=1,5405 Å.
c) Determinação da área específica de superfície dos pós, medidas
através do método BET (Brunauer-Emmett-Teller), de adsorção de
gases, com o equipamento Asap 2000- Micromeritcs (IME).
d) Determinação do diâmetro médio das partículas por espalhamento de
laser (granulômetro a laser), com o equipamento Cilas 1064 (INT).
e) Determinação da morfologia e tamanho das partículas através de
observação em microscopia eletrônica de varredura (MEV) no
microscópio JEOL JSM - 5800LV (IME).
4.1.1 ESPECTROMETRIA DE FLUORESCÊNCIA DE RAIOS-X
A determinação dos óxidos presentes nos pós utilizados neste trabalho foi feita
através da espectrometria de fluorescência de raios-X. Nesta técnica, raios–X são
empregados para estimular a radiação característica secundária para análises

115
qualitativas dos principais elementos presentes. Os raios-X característicos são
difratados por um cristal de análise e o ângulo de difração juntamente com a
intensidade difratada fornecem informação sobre o elemento desejado bem como
sobre sua concentração.
4.1.2 DIFRAÇÃO DE RAIOS X
Os pós cerâmicos obtidos após moagem e desaglomeração, foram submetidos
ao ensaio de difração de raios-x para identificação das fases presentes.
Este meio de análise baseia-se no princípio de espalhamento que um feixe
monocromático de raios-x sofre quando incide sobre um material. Alguns destes
feixes espalhados se cancelam, entretanto quando o raio-x atinge certos planos
cristalográficos em ângulos específicos ocorre uma interferência construtiva e tal
fenômeno é chamado de difração de raios-x a qual obedece a lei de Bragg,
conforme a equação abaixo:
hkl2d
nλsenθ = ( 4.1 )
onde: θ é a metade do ângulo entre o feixe difratado e a direção do feixe original;
λ é o comprimento de onda do raios-x; n é um múltiplo inteiro do comprimento de
onda, e dhkl é a distância interplanar dos planos que difratam.
A análise por difração dos pós foi realizada no difratômetro PW3710 BASED da
Phillips, utilizando a radiação Cu Kα (1,54056 Å), voltagem 35KV e corrente de
20mA, com passos de 0.02° para o ângulo de detecção. A varredura foi realizada
para o ângulo 2θ na faixa de 5° a 80°.
4.1.3 ÁREA ESPECÍFICA DE SUPERFÍCIE
As medidas da área específica de superfície dos pós foram determinadas
através do método de BET (Brunauer-Emmett-Teller), o qual utiliza a adsorsão física
de um gás por superfícies sólidas, utilizando o equipamento Mj – micromeritcs ASAP
2000(IME).

116
Usualmente o nitrogênio é empregado na temperatura criogênica (77,35K) e em
baixa pressão para mantê-lo gasoso (1atm de pressão).
O cálculo da área específica de superfície pode ser realizado através de
isotermas de adsorção. De uma maneira simplificada, isto é feito medindo-se a
quantidade molar n de um gás adsorvido (ou volume adsorvido Vo) a uma
temperatura T constante, por uma superfície sólida, em função da pressão do gás P.
A isoterma é geralmente construída ponto a ponto pela entrada e saída de
quantidades conhecidas de gás, durante um determinado tempo para permitir o
equilíbrio em cada ponto.
No método BET, para determinação da área específica de superfície obtida a
partir dos dados da isoterma de adsorção física do gás, é utilizada a seguinte
equação
[ ] 0mmoa PP
CV1C
CV1
PPVP
⋅−
+=−
( 4.2 )
onde:
Va é o volume do gás adsorvido pela amostra;
Vm é o volume de gás adsorvido, quando uma superfíce está recoberta por uma
camada monomolecular;
P é a pressão absoluta de um gás sobre a amostra;
P0 é a pressão de saturação do gás adsorvido;
X é o peso do adsorvido a uma dada pressão relativa P/P0; Xm é o peso do
adsorvido requerido para cobrir a superfície da amostra com uma camada molecular
e C é uma constante que é função da energia de interação adsorvido/adsorvente.
De acordo com a equação de BET, uma relação linear é dada se um gráfico de
[ ]PPVPoa −
em função de 0P
P é construído, na região de 0,05 <
0PP
< 0,35,
cuja inclinação a e o intercepto i são dados por:
CV1Ca
m
−= e
CV1im
= ( 4.3 )

117
A área específica de superfície do material S, pode então ser calculada a partir
do volume do gás adsorvido, Vm pela equação:
o
am
mVσNVS = ( 4.4 )
onde:
σ = área superficial ocupada por uma molécula do gás;
m = massa da amostra;
Na = número de Avogrado;
Vo = volume molar do gás.
Antes da determinação das isotermas é necessário remover os gases
adsorvidos no material. Para isso, as amostras foram desgaseificadas a
temperaturas de 150°C em estufa por, no mínimo 2horas (Chinelatto, A. S. A.,
2002).
4.1.4 ANÁLISE DE TAMANHO DE PARTÍCULAS
A distribuição granulométrica foi determinada por espalhamento de luz, através
da utilização do granulômetro a laser, Cilas 1064 (Compagnie Industrielle Des
Lasers). Este equipamento utiliza uma pequena alíquota do pó em estudo (que é
dispersa em água destilada e deionizada e em seguida é bombeada continuamente
através de uma célula com paredes de vidro sofrendo uma mistura ultra-sônica por
alguns segundos. Então a dispersão passa por um laser e retorna a câmara de
agitação, formando assim um ciclo continuo. A média do sinal de luz difratado em
alguns segundos é registrada. O número de ciclos como também o índice de
refração do líquido e da amostra devem ser adicionados como dados de entrada,
para que o programa possa estabelecer a curva para distribuição granulométrica.
As análises foram realizadas com tempo de ultra-som de 720 s e o meio
dispersante foi a água com ajuste de pH (10) com hidróxido de amônio
(aproximadamnente 5 gotas de NH4OH) para fugir do PZC da zircônia

118
(aproximadamente pH=7) e adição de hexametafosfato de sódio (0,1%p/v de
Calgon, Na4P2O7) como dispersante.
4.1.5 MICROSCOPIA ELETRÔNICA DE VARREDURA (MEV)
A morfologia e tamanho das partículas foram observadas por microscopia
eletrônica de varredura (MEV) no microscópio JEOL JSM - 5800LV. O pó foi
disperso em ethanol (0,01g do pó em 60ml de ethanoll) e depositado sobre um
suporte de latão. Após a evaporação do etanol o pó foi recoberto com ouro antes de
ser analisado.
As demais propriedades foram obtidas do fabricante ou de referências
consultadas.
4.2 MATERIAIS UTILIZADOS
4.2.1 PÓS CERÂMICOS
4.2.1.1 ALUMINA
A alumina utilizada foi a A-1000 SG da Alcoa, EUA. O tamanho médio de
partícula (D50) de 0,44 μm (Granulômetro a laser). De acordo com o fabricante o
tamanho médio de partícula era de 0,41μm (Sedígrafo-Namp). A TAB. 4.1
apresenta os teores dos óxidos presentes no pó, de acordo com o fornecedor Alcoa.
TAB. 4.1 Teor de óxidos presente no pó de alumina – A – 1000SG - Alcoa
Óxido Quantidade (%) Al2O3 99,83
SiO2 0,0153
Fe2O3 0,0243
Na2O 0,0663
CaO 0,0267
MgO 0,0397
B2O3 0,00233
4.2.1.2 ZIRCÔNIA

119
Neste trabalho, foi utilizada a zircônia de dois fornecedores diferentes:
a) Zircônia tetragonal policristalina estabilizada com 3% mol ou 5,1%
peso de ítria (Y2O3), YTZ-3YSB (LOTE S306276B) da Tosoh
Corporation, Japão. Apresentava tamanho médio de partícula de
0,35 μm (dado fornecido pelo fabricante). A TAB. 4.2 apresenta a
quantidade de impurezas (óxidos) presentes na zircônia parcialmente
estabilizada com Y2O3 da Tosoh, fornecida pelo fabricante.
TAB. 4.2 Teor dos óxidos presentes no pó de zircônia TZ - 3YSB – Tosoh.
Óxido Quantidade (%) Zr(Hf)O2 94,82
Y2O3 5,16
SiO2 0,003
Fe2O3 0,004
Na2O 0,004
Al2O3 < 0,005
b) Zircônia tetragonal policristalina estabilizada com 3% mol ou 5,1%
peso de ítria (Y2O3), 3Y-YZO-708/14 da Mel Chemicals, Inglaterra.
Apresentava tamanho médio de partícula de 0,19 μm (Granulômetro
a laser). A TAB. 4.3 apresenta a quantidade de impurezas(óxidos)
presentes na zircônia parcialmente estabilizada com Y2O3 da Mel
Chemicals, obtida através da análise química de espectrometria de
fluorescência de raios-X.
TAB. 4.3 Teor de óxidos presente no pó de zircônia XZO708/14 - Mel Chemicals.
Óxido Quantidade (%)ZrO2 90,466 HfO2 2,834 Y2O3 5,859 Fe2O3 0,030 SO3 0,270
Al2O3 0,511 Ta2O5 0,030
4.2.2 ADITIVOS
Os aditivos utilizados neste trabalho foram o dispersante, o solvente e o ligante:

120
a) O dispersante utilizado foi o ácido cítrico monohidratado P. A. da
Vetec Química Fina Ltda., lote 012193, de fórmula molecular
C6H8O7.H2O A especificação do produto de acordo com o
fabricante é apresentada na TAB. 4.4.
TAB. 4.4 Especificação do ácido cítrico monohidratado P. A. – Vetec.
Testes Limites Resultados Cor Incolor a branco Incolor
Aspecto Cristais a pó cristalino
Pó cristalino
Teor Min. 99,5% 100% Insolúveis Máx. 0,005% 0,005%
Resíduo após ignição Máx. 0,02% 0,02% Cloreto Máx. 0,001% 0,001% Sulfato Máx. 0,002% 0,002%
Metais pesados (como Pb) Máx. 0,0002% 0,0002%
Ferro Máx. 0,0003% 0,0003% Oxalato (C2O4)
(Limite aprox. 0,05%) Máx. 0,05% 0,05
Subst. Carbonizáveis pelo H2SO4 quente
(tartaratos, etc...)
Passa no teste Passa no teste
b) O solvente utilizado foi o álcool etílico da Vetec Química Fina
Ltda., lote 013471, de fórmula molecular C2H6O. A especificação
do produto de acordo com o fabricante é apresentada na TAB.4.5.
TAB. 4.5 Especificação do álcool etílico P. A. - Vetec
Testes Limites Resultados Cor Apha 10 < Apha 10
Aspecto Líquido claro Líquido claro Dosagem (por volume) Min. 99,5% 99,85%
Água Máx. 0,2% 0,14% Resíduo após evaporação Máx. 0,001% 0,00063%
Metanol Máx. 0,1% 0,1% Acetona e álcool isopropílico Passa no teste Passa no teste
Substâncias que reduzem o KMnO4 Passa no teste Passa no teste Substâncias que escurecem pelo H2SO4 Passa no teste Passa no teste
Base titulável Máx. 0,0002 meq/g* < 0,0002 meq/g Ácido titulável Máx. 0,0005 meq/g* < 0,0005 meq/g
Solubilidade em água Passa no teste Passa no teste Óleo fusel Passa no teste Passa no teste
* Conforme especificação ACS.
c) O ligante ou plastificante utilizado neste trabalho foi o polyetileno
glycol 400 – P1. 023 Synth (Carbowax 400) (ATPEG 400). A

121
especificação do produto de acordo com o fabricante é
apresentada na TAB. 3.6.
TAB. 4.6 Especificação do polyetileno glycol 400 – (ATPEG 400).
Testes Resultados Densidade / 25° 1,11 – 1,14
PH sol. 5% 4,5 – 7,5 Máx. de impurezas -
Ponto congelamento 4° - 8° Viscosidade 6,8 – 8,0
4.3 PROCESSAMENTO
4.3.1 METODOLOGIA DE ANÁLISE
As principais etapas do processamento experimental estão representadas no
fluxograma da FIG. 4.1 indicando os parâmetros utilizados no processamento e a
metodologia de análise.
4.3.2 PREPARAÇÃO DAS COMPOSIÇÕES
Deste ponto em diante será utilizada a seguinte nomenclatura para os óxidos
puros e compósitos: “ZT” indica zircônia Tosoh, “ZM” indica zircônia Mel Chemicals e
“A” indica alumina. O índice XX representa o teor de zircônia (% peso) presente nos
compósitos. Por exemplo, 80ZT significa compósito com zircônia da Tosoh
contendo 80 % em peso de zircõnia.
Neste trabalho, foram preparadas dez diferentes amostras onde foi variado o
teor de zircônia, descritas a seguir:
1- Amostra 100 ZT: Amostra contendo 100 % peso de ZrO2 Tosoh. Não foram
utilizados o solvente, o dispersante nem o ligante. Este material já contém ligante
depositado no pó por “spray drier” e já vem pronto para prensagem, assim, não foi
necessário realizar a moagem em moinho.

122
FIG. 4.1 Metodologia de análise
2- Amostra 80 ZT: Amostra contendo 80 % peso de ZrO2 Tosoh e 20 % peso de
Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm), por 1,5h
ZrO2 (3Y-TZP- Tosoh) (6.21 m2/g; d50 = 0.35μm)
Al2O3 – SG-1000 - Alcoa (10.47m2/g; d50 = 0.44μm)
ZrO2 (3Y-TZP- MelChemicals) (11.10 m2/g; d50 = 0.19μm)
Material: óxidos puros + compósitos com zircônia variando teor de ZrO2 de 5 % a 80 % peso.
Moinho atritor – 50 % peso de sólidos – 1.5 h – 350 rpm, Meios moedores de zircônia ( Ø = 2 mm).
Secagem com evaporador rotatório e estufa 60°C /24h; Desaglomeração (65 mesh Tyler – 212 μm).
Prensagem uniaxial ( 5 x 6 x 80 mm). P = 55
Evaporação do ligante – 6° C/min a 600°C/1,5h.
Densidade à verde (razão peso/vol.).
Densidade sinterizada (Método de Arquimedes)
Sinterização em ar, T = 1500°C – 1600°C/ 2 h, conforme a
Caracterização da microestrutura
Propriedades mecânicas
DRX após sinterização, MEV, EDS, e MET.
1. Dureza Vickers – Hv - (98.1 N / 10s). 2. Tenacidade à fratura - KIC (Método de indentação). 3. Resistência à flexão em 4 pontos –σ4 p .
Aditivos: álcool etílico; 2 % peso PEG; 0.6% peso ácido cítrico.
Análise geral dos resultados
DRX dos pós
DRX após fratura

123
em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos. Como
dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Não foi
utilizado o polyetilieno glycol 400.
3- Amostra 21 ZT: Amostra contendo 21 % peso de ZrO2 Tosoh e 79 % peso de
Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm), por 1,5h
em álcool etílico P.A., rotação de 350 rpm, e 50 % em peso de sólidos. Como
dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos 15
min finais da moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
4- Amostra 15 ZT: Amostra contendo 15 % peso de ZrO2 Tosoh e 85 % peso de
Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm), por 1,5h
em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos. Como
dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos 15
min finais da moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
5- Amostra 10 ZT: Amostra contendo 10 % peso de ZrO2 Tosoh e 90 % peso de
Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm), por 1,5h
em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos. Como
dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos 15
min finais da moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
6- Amostra 5 ZT: Amostra contendo 5 % peso de ZrO2 Tosoh e 95 % peso de
Al2O3. . Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm), por
1,5h em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos. Como
dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos 15
min finais da moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
7- Amostra A: Amostra contendo 100 % peso de Al2O3. Foi realizada moagem
com corpos moedores de zircônia (φ = 2 mm), por 1,5h em álcool etílico P.A.,
rotação

124
de 350 rpm e 50 % em peso de sólidos. Como dispersante foi utilizado o ácido
cítrico monohidratado P.A. (0,6 % peso). Nos 15 min finais da moagem, foi
adicionado o polyetileno glycol 400 (2 % peso).
8- Amostra 100 ZM: Amostra contendo 100 % peso de ZrO2 Mel Chemicals. Foi
realizada moagem com corpos moedores de zircônia (φ = 2 mm), por 1,5h em álcool
etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos. Como dispersante foi
utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos 15 min finais da
moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
9- Amostra 80 ZM: Amostra contendo 80 % peso de ZrO2 Mel Chemicals e 20 %
peso de Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2 mm),
por 1,5h em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de sólidos.
Como dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 % peso). Nos
15 min finais da moagem, foi adicionado o polyetileno glycol 400 (2 % peso).
10- Amostra 15 ZM: Amostra contendo 15 % peso de ZrO2 Mel Chemicals e 85
% peso de Al2O3. Foi realizada moagem com corpos moedores de zircônia (φ = 2
mm), por 1,5 h em álcool etílico P.A., rotação de 350 rpm e 50 % em peso de
sólidos. Como dispersante foi utilizado o ácido cítrico monohidratado P.A. (0,6 %
peso). Nos 15 min finais da moagem, foi adicionado o polyetileno glycol 400 (2 %
peso).
As composição das amostras, as respectivas massas gastas de pós cerâmicos e
de dispersante, bem como o volume gasto de ligante e de solvente utilizados para a
sua preparação, são apresentadas na TAB. 4.2.
A pesagem das massas utilizadas para a preparação das composições foi
realizada em balança digital (Mettler P 1200 – INT).
A seguir são apresentados os cálculos realizados para a determinação das
massas e volumes gastos para a obtenção das amostras apresentadas na TAB. 4.2.
O solvente utilizado foi o álcool etílico P.A.

125
Exemplo de cálculo do volume de álcool para 50% peso de sólidos na polpa de
moagem, para a amostra 21 ZT (21% ZrO2 + 79% Al2O3):
207 g de sólidos => 21% ZrO2 = 43,5 g + 79% Al2O3 = 163,5 g.
207 g de solvente => álcool etílico ( ρ = 0,9 g/cm3)
Temos que:
vm
=ρ onde: ρ = densidade; m = massa e v = volume;
Então:
mlvvv
m 2302079,0 =⇒=⇒=ρ .
TAB. 4.7 Massas e volumes utilizados para a preparação das misturas de diferentes composições.
Massa (g)
Massa (g)
Vol. (ml)
Vol. (ml)
Amostra
Composição ZircôniaTosoh
Zircônia
Mel Chemicals
Alumina
Ácido cítrico
PEG
ÁlcoolEtílico
100 ZT 100 % ZrO2 150,0 - - - - - 80 ZT 80 % ZrO2 +
20 % Al2O3
165,6 - 41,4 2,48 - 230
21 ZT 21 % ZrO2 + 79 % Al2O3
43,5 - 163,5 2,48 3,7 230
15 ZT 15 % ZrO2 + 85 % Al2O3
31,0 - 176,0 2,48 3,7 230
10 ZT 10 % ZrO2 + 90 % Al2O3
20,7 - 186,30 2,48 3,7 230
5 ZT 5 % ZrO2 + 95 % Al2O3
10,4 - 196,7 2,48 3,7 230
A 100 % Al2O3 - - 207,0 2,48 3,7 230 100 ZM 100 % ZrO2 - 150,0 - 1,90 2,7 167 80 ZM 80 % ZrO2 +
20 % Al2O3 - 165,6 41,4 2,48 3,7 230
15 ZM 15 % ZrO2 + 85 % Al2O3
- 31,0 176,0 2,48 3,7 230
Exemplo de cálculo do volume de álcool para 50 % peso de sólidos na polpa de
moagem, para a amostra 100 ZM (100 % ZrO2):

126
ml167vv
15090vm
=⇒=⇒=ρ , .
O dispersante utilizado neste trabalho foi o ácido cítrico monohidratado P. A.,
com concentração de 0,6 % peso para 100 g de solução.
Exemplo do cálculo da massa de ácido cítrico para a amostra 21 ZT (21 %
ZrO2 + 79 % Al2O3):
0,6 g => 100 g (óxidos + álcool)
x => 414 g (207 g de óxidos + 207 g de álcool)
x = 2,48 g.
Exemplo do cálculo da massa de ácido cítrico para a amostra 100 ZM
(100% ZrO2):
0,6 g => 100 g (óxidos + álcool)
x => 317 g (150 g de óxidos + 167 g de álcool)
x = 1,90 g.
Com o objetivo de conferir ao material certa resistência mecânica a verde após a
etapa de compactação e assim facilitar o manuseio das amostras, foi utilizado como
ligante ou plastificante o polyetileno glycol 400 (PEG 400 – 2 % peso), o qual foi
adicionado somente nos 15 min finais da moagem.
Exemplo do cálculo da massa e volume de PEG 400 para a amostra 21 ZT (21%
ZrO2 + 79 % Al2O3):
2 g => 100 g de óxidos
x => 207 g de óxidos
x = 4,14 g => Por pesagem correspondeu a 3,7 ml.
Exemplo do cálculo do volume de PEG 400 para a amostra 100 ZM (100 %
ZrO2 ):
207 g => 3,7 ml
150 g => x
x = 2,7 ml.

127
A carga de peso de sólidos na polpa de moagem pode ser calculada:
207 g óxidos => x
207 g (óxidos) + 2,48 g (ácido cítrico)+ 207 g (álcool) => 100 %
x = 50 % de peso de sólidos na polpa de moagem.
A relação entre óxidos e corpos moedores é normalmente de 1 para 4 e pode
ser calculada (Chinelatto, A. S. A., 2002):
Densidade corpos moedores de zircônia = 6,02 g / cm2.
P/ 600 g de corpos moedores (φ=2 mm), temos v= 150 ml (100 ml serão de
corpos moedores, os restantes 50ml serão descontados p/ obtermos vazios entre os
corpos moedores).
600 g corpos moedores => 100 ml
150 g óxidos (100 % ZrO2) => x
x = 25 ml
Assim, os óxidos ocupam ¼ do volume ocupado pelos corpos moedores, sendo
a relação entre óxidos e corpos moedores é portanto de 1 para 4.
Para uma boa eficiência de moagem, o preenchimento do moinho deve estar
na faixa de 60 a 80 %.
O cálculo do preenchimento da moagem para amostra 15 ZT, 15 % ZrO2 + 85 %
alumina (207g óxidos) c/ densidade da composição pela regra das misturas = 4,27
g/cm3, por exemplo, é:
550 ml => 100 % de preenchimento
428,48 ml (230 ml álcool + 150 ml corpos moedores + 49,64 ml óxidos) => x
x = 78,11 %
O cálculo do preenchimento da moagem para a amostra 100 ZM (100 % ZrO2)
(150 g óxidos) c/ densidade = 5,99 g / cm3, por exemplo, é:
550 ml => 100% de preenchimento
342,04 ml (167 ml álcool +150 ml corpos moedores + 25,04 ml óxidos ) => x
x = 62,19 %

128
4.3.3 MOAGEM
As condições utilizadas na moagem e desaglomeração foram obtidas da
literatura.
As moagens foram realizadas em moinho atritor vertical Szegvari Attritor System
– Union Process – INT. Foi utilizado jarro de zircônia (550ml), corpos moedores de
zircônia da Netsch (φ= 2 mm) e velocidade de 350 rpm. O tempo total de moagem
foi de 1,5h.
Os materiais eram colocados dentro do jarro do moinho atritor já em movimento,
na seguinte ordem: Inicialmente era colocado o ácido cítrico monohidratado (0,6 %
peso) já dissolvido no álcool etílico P. A. Em seguida, eram colocados os corpos
moedores de zircônia. Após esta etapa, eram adicionados os pós: primeiro o pó de
alumina e depois o pó de zircônia. Após o tempo de 1:15 h, ou seja, nos 15 minutos
finais do tempo total de moagem, era adicionado o polietilenoglycol (2 % peso) e a
moagem continuava portanto por mais 15 min.
4.3.4 SECAGEM DOS PÓS
Após cada uma das moagens, as composições foram colocadas por 1h no
rotoevaporador – Fisatom série 781743 Mod. 550 – INT, com o objetivo de evaporar
o solvente, e reaproveitar o álcool etílico. Em seguida, a secagem das composições
foi realizada em estufa G. Siebert – INT por aproximadamente 24h na temperatura
de 60°C.
4.3.5 DESAGLOMERAÇÃO
Após a secagem, os pós foram desaglomerados em gral e pistilo de ágata. O
peneiramento das composições foi Foram utilizadas peneiras da série Tyler(65
mesh), abertura de 212 μm /ABNT/ASTM 70). Em seguida as peneiras foram
colocadas no vibrador Bertel – INT, por 60 minutos. O restante do pó foi novamente
desaglomerado e peneirado.
4.3.6 CONFORMAÇÃO DOS CORPOS DE PROVA

129
A técnica de conformação dos corpos de prova utilizada foi a prensagem uniaxial
por ser de baixo custo, rápida e de fácil realização. As desvantagens de sua
utilização são: distribuição não uniforme da porosidade, defeitos de compactação e a
limitação de ser aplicada para corpos verdes com razão de aspecto H/D ≤ 1,
conforme FIG. 4.3.
FIG. 4.2 Razão de aspecto do corpo verde para prensagem uniaxial.
A prensagem uniaxial compreende a compactação simultânea com a
conformação de um pó confinado em uma matriz rígida de aço endurecido. Neste
trabalho, utilizou-se a prensa Gabbrrielli Pressa L4 – INT, usando o método dos dois
punções móveis e camisa flutuante, com a finalidade de uma distribuição mais
homogênea da pressão no material compactado.
A carga de prensagem foi de 26,53 ton. = 70 kg / cm2. Após a prensagem as
dimensões típicas dos corpos de prova foram 5 x 6 x 80 mm.
A partir da força de prensagem, o valor da pressão aplicada foi de:
F = 26,53 ton
A = 480 mm2 = 4,8 cm2. Para as 10 cavidades, A = 48 cm2.
P = F/A = 26,53 ton /48 cm2 = 0,55 ton/cm2 = 550 kgf / cm2 = 5500 N / cm2 =
5500 x 104 N / m2 = 55 MPa.
P= 55 MPa.
4.3.7 CÁLCULO DA DENSIDADE A VERDE
Para o cálculo da densidade a verde das amostras compactadas, foram
realizadas três medidas de cada uma de suas dimensões utilizando um paquímetro
com precisão de ± 5,0 x 10-3 mm, para o cálculo do volume das mesmas. Em
seguida as amostras foram pesadas em uma balança Mettler P-1200, com
aproximação de 1,0 x 10-4 . A densidade a verde foi obtida pela razão massa/volume
g/cm3) e expressa como percentual da densidade teórica.
D H
1≤DH

130
4.3.8 RETIRADA DO LIGANTE OU PLASTIFICANTE
O ligante ou plastificante é um composto molecular que deve ser eliminado antes
da sinterização porque em altas temperaturas ele se transforma em carbono fixo
formando inclusões que promoveriam a diminuição da resistência mecânica e além
disso, os ligantes gerariam gases e produziriam defeitos durante a sinterização.
Neste trabalho, a retirada do ligante foi utilizada a queima lenta, que consiste na
queima do ligante no forno, a uma taxa de aquecimento controlada e lenta até que
se atinja a temperatura de 600 °C, onde a amostra permaneceu durante algumas
horas, para garantir a saída eficiente do ligante. A retirada do ligante das amostras
deste trabalho foi realizada em forno tipo mufla Maitec FE 1600 – INT, com taxa de
aquecimento de 6°C/min e um patamar de 180 minutos a 600°C. A taxa de
resfriamento foi de 10°C/min.
A FIG. 4.4 apresenta a variação de temperatura do forno durante a retirada do
ligante.
FIG. 4.3 Variação da temperatura do forno durante a evaporação do ligante.
Uma taxa de aquecimento muito lenta é além de comercialmente inviável,
responsável pelo aparecimento de defeitos definitivos no corpo devido aos gases
formados durante a queima, os quais permanecem muito tempo no interior dos
poros, expandindo-os. Já uma taxa de aquecimento muito rápida pode provocar a
formação de trincas devido à rápida contração do corpo durante a retirada do

131
material orgânico. Tais defeitos normalmente não são eleiminados na sinterização e
comprometem as propriedades mecânicas do corpo sinterizado. Logo, a taxa de
aquecimento deve ser cuidadosamente selecionada e controlada para evitar o
crescimento excessivo, pressões internas e minimizar danos na amostra (Pereira, A.
P. D., 2001).
4.3.9 SINTERIZAÇÃO
A sinterização é uma das etapas fundamentais do processamento cerâmico, pois
nesta etapa ocorre a consolidação do corpo cerâmico. As sinterizações das
amostras do presente trabalho foram realizadas em forno Maitec FE 1600 – INT.
Para as composições 3, 4, 5, 6, 7 e 10, a temperatura de sinterização foi de
1600°C (Casellas, D. et al., 1999; Oliveira, L. G., 2002; Tuan, W. H. et al., 2002;
Celli, A. et al., 2003; Casellas, D. et al., 2003). A taxa de aquecimento foi de 6
°C/min até 1100°C, 3°C/min até 1400°C e 5°C/min até 1600°C. O tempo de patamar
em 1600°C foi de 2 horas (Casellas, D. et al., 1999; Oliveira, L. G., 2002; Lin, J. D. &
Duh, J. G., 2002 ). A taxa de resfriamento foi de 5°C/min até 1400°C e de 3°C/min
até 1100°C, como pode ser visto na FIG. 4.5 indicado pela linha vermelha. A partir
de 1100 °C, o resfriamento se fez automaticamente pelo forno, como pode ser visto
na FIG. 4.5, indicado pela linha azul.
Para as composições 1, 2, 8 e 9, a temperatura de sinterização foi de 1500°C
(Stevens, R., 1986; Oliveira, L. G., 2002; Lin, J. D. & Duh, J. G., 2002; Celli, A. et al.,
2003), pois há um crescimento excessivo dos grãos de zircônia a 1600°C (Stevens,
R., 1986; Oliveira, L. G., 2002). A taxa de aquecimento foi de 6° C/min até 1100 °C,
3°C/min até 1400°C e 5°C/min até 1500°C. O tempo de patamar em 1500°C foi de 2
horas (Casellas, D. et al., 1999; Oliveira, L. G., 2002; Lin, J. D. & Duh, J. G., 2002 ).
A taxa de resfriamento foi de 5°C/min até 1400°C e de 3°C/min até 1100°C, como
pode ser visto na FIG. 4.6 indicado pela linha vermelha. A partir de 1100 °C, o
resfriamento se fez automaticamente pelo forno, como pode ser visto na FIG. 4.6,
indicado pela linha azul.
Durante a sinterização, foram utilizados contra pesos de alumina tenacificada
por zircônia para evitar o empenamento. As taxas de aquecimento e resfriamento
utilizadas foram baixas visando garantir a integridade do dispositivo de mufla, feito
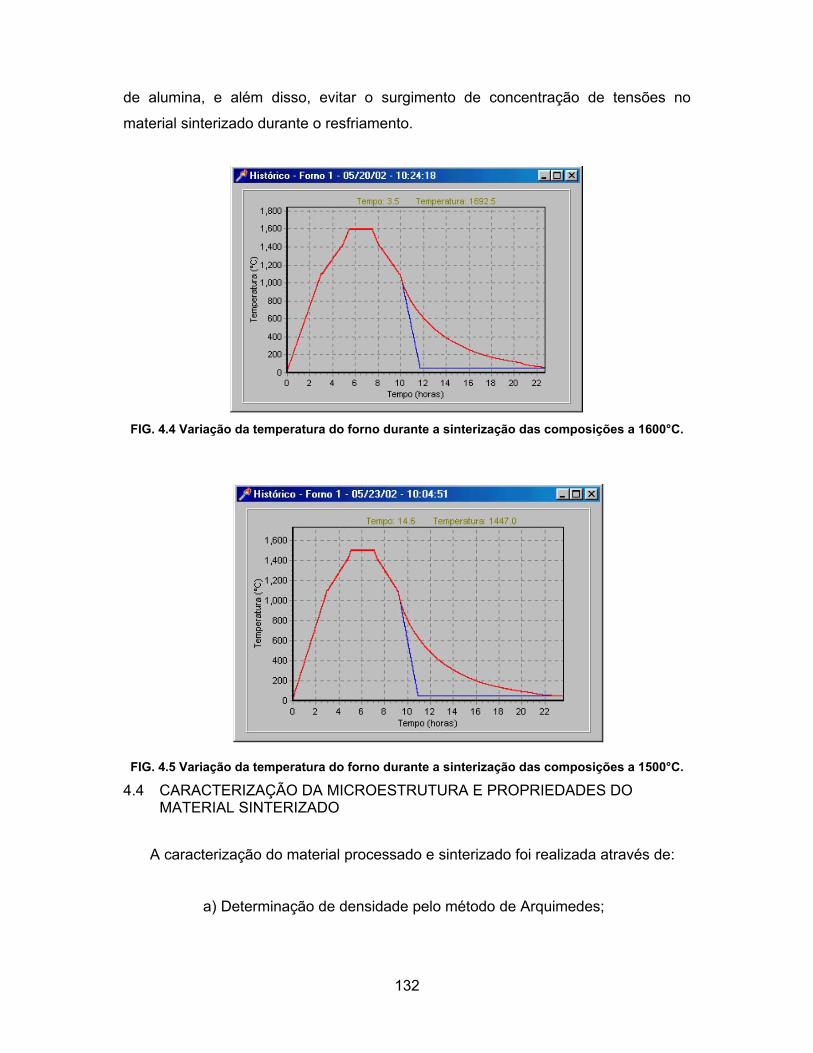
132
de alumina, e além disso, evitar o surgimento de concentração de tensões no
material sinterizado durante o resfriamento.
FIG. 4.4 Variação da temperatura do forno durante a sinterização das composições a 1600°C.
FIG. 4.5 Variação da temperatura do forno durante a sinterização das composições a 1500°C.
4.4 CARACTERIZAÇÃO DA MICROESTRUTURA E PROPRIEDADES DO MATERIAL SINTERIZADO
A caracterização do material processado e sinterizado foi realizada através de:
a) Determinação de densidade pelo método de Arquimedes;

133
b) Identificação das fases cristalinas presentes por difração de raios-X
das amostras após sinterização e após a fratura;
c) Observação da microestrutura por microscopia eletrônica de
varredura (MEV) e microscopia eletrônica de transmissão (MET) e dos
elementos químicos presentes por espectroscopia de energia
dispersiva (EDS), medida de tamanho médio de grãos.
d) Comportamento relativo às propriedades mecânicas: dureza Vickers,
tenacidade à fratura, resistência à flexão em quatro pontos,
resistência ao desgaste e investigação dos mecanismos de fratura
presentes nas amostras.
4.4.1 AVALIAÇÃO DAS PROPRIEDADES FÍSICAS
4.4.1.1 DENSIDADE E POROSIDADE
A metodologia utilizada para os ensaios é uma adaptação da NBR 6220 da
ABNT proposta por Rocha, J. C. et al., 1990, para ser utilizada em materiais
cerâmicos de alto desempenho e da norma ASTM C 373-88.
Seguindo este método, as amostras foram colocadas sobre uma tela metálica,
imersas em um recipiente com água destilada, sem entrar em contato com o fundo
do recipiente, fervidas durante duas horas para a eliminação do ar dos poros e
resfriadas à temperatura ambiente. O processo de resfriamento pode ser acelerado
com água destilada corrente mantendo a amostra imersa o tempo todo.
A massa imersa (Mi) foi determinada com aproximação de 1,0 x 10-4 g. A
pesagem foi feita com a amostra apoiada em um suporte e mergulhada em um
volume de 150ml de água destilada, à temperatura ambiente. A temperatura da água
foi medida para determinação de sua densidade.
Após a leitura da massa imersa, a amostra foi retirada rapidamente da água com
uma pinça e envolvida em um pano úmido, para a remoção das partículas de água
que se encontravam na superfície. O CP foi pesado com aproximação de 1,0 x 10-4 g
para a determinação da massa saturada ou úmida (Mu). O tempo de leitura deve ser
o mínimo possível, para que não haja evaporação da água residual da amostra.

134
Os corpos de prova foram secos em estufa à 125oC, durante 50 minutos. As
amostras foram resfriadas por um período de 40 minutos em dessecador e pesadas
para a determinação da massa seca (Ms). A partir dos dados obtidos, foram
calculados:
Volume aparente (Va)
O volume aparente representa o volume do material sólido mais o volume dos
poros abertos e fechados (cm3). Foi calculado pela expressão:
Va = (Mu - Mi) / ρH2O(T) ( 4.5 )
Onde ρH2O(T) é a densidade da água na temperatura T.
Massa específica aparente ou densidade aparente(Mea)
A Massa específica aparente é o quociente da massa de cada corpo de prova
seco pelo volume aparente (g/cm3). Foi calculada pela expressão:
Mea = Ms / Va ( 4.6 )
A porosidade aparente (Pa) é o quociente do volume dos poros abertos de cada
corpo de prova pelo volume aparente do mesmo e foi calculada pela seguinte
equação:
Pa = (Mu – Ms) / (Va/ρH2O(T)) ( 4.7 )
O valor da densidade teórica para as diferentes composições desenvolvidas
neste trabalho, foi calculada pela regra das misturas, utilizando-se a equação 4.8,
onde ρT é a densidade teórica da composição (g/cm3), wi é a porcentagem em peso
e ρi a densidade de cada óxido.
∑=
i
iT w
ρ
ρ 1 ( 4.8 )

135
A porosidade percentual, P, foi determinada indiretamente através da medida da
densidade, ρ, utilizando-se o princípio de Archimedes, que trata do empuxo sofrido
por um corpo imerso em um líquido, como a água, utiliazndo-se a seguinte equação:
P = [(ρt -ρ)/ρ] x 100 ( 4.9 )
Onde ρt é a densidade teórica e ρ é dado pela seguinte equação:
ρ = (Mar . ρ H2O – MH2O . ρar) / (Mar – MH2O) ( 4.10 )
Onde: Mar é a massa da amostra em ar, MH2O a massa da amostra em água, ρar
a densidade do ar e ρH2O a densidade da água a temperatura T. A porosidade
determinada por este método é bastante precisa para valores de P < que 5 %, isto é,
para porosidade fechada.
4.4.1.2 AVALIAÇÃO DAS FASES CRISTALINAS E TRANSFORMAÇÃO MARTENSÍTICA
A transformação martensítica pode ser avaliada pela fração volumétrica da
zircônia monoclínica em relação ao total de zircônia utilizando a difração de raios-X.
(Gregori, G. et al., 1999). No presente trabalho, medidas de difração de raios-X
foram realizadas nas amostras tendo como objetivo a determinação e quantificação
das fases resultantes do processo de sinterização. A quantificação da fase
monoclínica presente nas amostras após a sinterização e após a fratura foi realizada
para a avaliação da extensão da transformação de fase induzida por tensão que
ocorre em cerâmicas contendo zircônia.
As medidas foram realizadas utilizando difratômetro Philips XPERT Pro, tubo de
cobre, radiação CuKα (λ=1,54056 Å), tempo de 2s, passo de 0,05°, voltagem de
30kV e corrente de 40 mA. A quantificação das fases por Rietveld foi realizada
utilizando o programa TOPAS Bruker versão 2.1.
A partir dos trabalhos prévios realizados por Toraya, H., 1984; Gutzov, S. et al.,
1994; Kim, B. K. et al., 1997; e Oliveira, A P. A., 1997, a quantificação da fração
volumétrica da fase monoclínica (FM) pelo método interno foi calculada a partir das

136
intensidades integradas dos picos monoclínicos ( 1 11)M e (111)M e do pico tetragonal
(101)T, os quais são apresentados na FIG. 4.7, usando as seguintes equações:
M
MM X31101
X3111F.
.+
= ( 4.11 )
onde,
( ) ( )( ) ( ) ( )TMM
MMM 101111111
111111X++
+= ( 4.12 )
onde:
( 1 11)M, (111)M e (101)T, representam a intensidade integrada dos picos
difratados nos planos monoclínicos ( 1 11)M e (111)M e no plano tetragonal (101)T.
O método de Rietveld também foi utilizado para a determinação quantitativa das
fases presentes nas amostras investigadas neste trabalho.(Young, R. A., 1995; Hill,
R. J.,1991; Foschini, C. R., 1994; Hill, R. J. & Howard, C. J., 1987).
O método de Rietveld é uma poderosa ferramenta para quantificação de fases e
o ajuste de resultados experimentais é dado pela seguinte equação (Young, R. A.,
1995):
( ) bikki2
kK
kci yAP22FLsy +θ−θφ= ∑ ( 4.13 )

137
27 28 29 30 31 32 33
2700
3600
4500
cb
a
Inte
nsid
ade
u.a.
2θ
FIG. 4.6 Difratograma da Y-ZrO2 Tosoh com 2θ na faixa de 27° a 33°, mostrando a ocorrência do do pico tetragonal a= (101)T, e dos picos monoclínicos b = ( 1 11) M e c = (111)M.
Onde:
s é o fator de escala; K representa os índices de Miller h, k,l para as reflexões
de Bragg; Lk contém os fatores de Lorentz-polarização e de multiplicidade; φ é a
função do perfil de reflexão; Pk é a função de orientação preferencial; A é o fator de
absorção; Fk é o fator de estrutura para o Kth reflexão de Bragg e ybi é a intensidade
do ruído de fundo para o ith passo.
O fator yci calculado é então utilizado o método de mínimos quadrados com
objetivo de minimizar a diferença entre os perfis observado e calculado, de acordo
com a seguinte equação:
( )2
ciii
iy yywS −= ∑ (4.14 )

138
onde: wi = 1/yi; yi é a intensidade observada no ith passo e yci é a intensidade
calculada no ith passo. Sy é a quantidade residual do processo de minimização e
indica a qualidade do ajuste.
Observando a equação 4.13, é possível verificar que os cálculos incluem fatores
cristalográficos como fator cristalino, orientação preferencial, posição atômica na
estrutura cristalina e tamanho cristalino. É necessário também incluir nos cálculos
fatores associados com a óptica, tal como comprimento de onda da fonte de raios-X,
aberturas, monocromadores e distâncias do goniômetro. O cálculo de Rietveld leva
em conta fatores como o equipamento, a amostra (estrutura cristalina) e por fim o
próprio fenômeno da difração, os quais estão representados na equação 4.13
descrita acima.
4.4.2 AVALIAÇÃO DAS CARACTERÍSTICAS MICROESTUTURAIS
Observações da microestrutura das amostras sinterizadas foram realizadas em
um microscópio eletrônico de varredura, da marca JEOL- JSM-5800LV.
O microscópio eletrônico de varredura (MEV) utiliza um feixe focado de elétrons
de alta energia que sistematicamente varre a superfície da amostra. A interação do
feixe com a amostra produz um grande número de respostas que podem ser
convertidas em um sinal eletrônico. Este sinal eletrônico é reproduzido sobre um
tubo de raio catódico. A varredura do feixe é sincronizada com a varredura do tubo
de raio catódico, produzindo uma relação de 1:1 entre os pontos da amostra e os de
tubos de raio catódico.
Para a avaliação da morfologia das partículas, utiliza-se sinais emitidos pelos
elétrons secundários (energia inferior a 50 eV), resultantes da ejeção de elétrons
fracamente ligados. Estes elétrons não são muito afetados pelo número atômico do
elemento constituinte da amostra, mas são bastante sensíveis ao relevo e aos
campos magnéticos ou elétricos presentes na superfície da amostra ou acima dela.
O preparo das amostras para observação da microestrutura por microscopia
eletrônica de varredura (MEV) foi realizado através do corte com disco de borda
diamantada e embutimento das amostras em baquelite (a quente). Cada baquelite
apresentava quatro pequenas amostras de uma mesma composição.

139
Depois do embutimento em baquelite das amostras, foi realizado o desbaste
manual ou automático em lixas d’água de 180 até 1000 mesh, para a remoção total
do material de embutimento e obtenção de uma superfície plana.
Após o desbaste manual, as amostras foram polidas com pastas de diamante na
seqüência de 15, 9, 6, 3,1 e 0,25 μm. Durante esta etapa, as amostras foram
observadas em microscópio OLYMPUS BME-3 de platina invertida, com o auxílio de
um analisador de imagem IA-3001 da LECO, para a avaliação do processo de
polimento, comportamento quanto aos riscos, poros e arrancamentos. O tempo de
polimento não era fixo, era função da evolução do processo, isto é, só se trocava de
pasta quando o defeito médio na superfície polida, o qual era quantificado através do
microscópio óptico, estivesse na ordem de grandeza que o tamanho do abrasivo da
pasta.
Ao final do polimento as amostras foram retiradas do embutimento, atacadas
termicamente a 1500 ºC por aproximadamente 6 a 10 minutos ao ar no forno Netzch
(IME) buscando garantir uma boa revelação dos contornos de grão. A seguir, as
amostras foram recobertas com um filme de ouro de 80 nm (Balzers 07-120B-IME),
que possibilitou a condução dos elétrons e permitiu melhor contraste no exame ao
microscópio. Foram utilizadas imagens obtidas com elétrons secundários (SE) para
determinação do tamanho de grão e imagens com elétrons retro-espalhados (BSE)
para avaliação da homogeneidade da microestrutura. A voltagem utilizada foi de
20kV.
O tamanho de grão foi determinado pelo método das interseções lineares
metalografia quantitativa, de acordo com a norma ASTM E-112/96, utilizando a
seguinte equação:
TG = ( L / i ) x ( 1 / M ) ( 4.11 )
Onde:
TG = tamanho de grão;
L = comprimento das linhas usadas pelo método quantitativo;
i = o número de interceptos ao longo da L;
M = Magnitude do aumento utilizado.

140
O preparo das amostras para observação das superfícies de fratura por
microscopia eletrônica de varredura (MEV), foi realizada após a fratura das mesmas
em ensaio de flexão em quatro pontos. As amostras foram cortadas para se obter a
altura de 1cm e em seguida foi feita a deposição de um filme de ouro (80nm) para
permitir a condução dos elétrons. Para a observação das superfícies de fratura
foram utilizados os elétrons secundários (SE).
As amostras que apresentaram melhores propriedades mecânicas foram
preparadas para observação em microscopia eletrônica de transmissão (MET) no
microscópio JEOL EM – 2010. Para isto, as amostras foram cortadas com disco
diamantado de 5 μm, sendo obtidas amostras em fatias com 150 a 200 μm de
espessura. Em seguida, as amostras foram cortadas com ultrassom para obtenção
do diâmetro de 3mm (Disc cutter - Gatan- modelo 601) sendo utilizado carbeto de
silício em pó grit 120 da Buehler. As amostras foram lixadas com lixas de SiC para
obtenção da espessura de 70 a 100 μm. Em seguida, para obtenção da calota foi
utilizado o dimple grinder - Gatan – modelo 656, com pasta de 15 μm, carga de 30 g
e velocidade 2. Após esta etapa, as amostras foram polidas no Precision íon
polishing system-Gatan – modelo 691, utilizando ângulo de 4°, energia do feixe de 5
KeV. O tempo total de polimento necessário para obtenção do padrão no MET foi
de 21h. Foram realizadas imagens das amostras e figuras de difração foram obtidas
bem como foram realizadas análises de EDS (Espectroscopia de energia
dispersiva). A observação no MET foi realizada utilizando voltagem de 200 kV.
4.4.3 AVALIAÇÃO DAS PROPRIEDADES MECÂNICAS
4.4.3.1 DETERMINAÇÃO DOS VALORES DE DUREZA
A metodologia utilizada para a determinação dos valores de dureza das
amostras, obedeceu à norma ASTM C 1327-99, a qual fornece o método de teste
padrão para a obtenção da dureza Vickers de cerâmicas avançadas.
Foram realizadas aproximadamente 30 impressões Vickers nas superfícies de
cada uma das amostras, as quais já estavam embutidas e polidas, conforme já
descrito no item 4.4.2.

141
A carga aplicada no ensaio de dureza (durômetro Zwich) foi de 10 Kgf (98N)
durante dez segundos.
As impressões foram realizadas de tal forma que a distância entre os centros
das impressões era de pelo menos cinco vezes a medida da semi-diagonal de
indentação mais o tamanho da trinca(5c) ou quatro vezes a diagonal de
indentação(4d), conforme FIG. 4.8.
FIG. 4.7 Distância entre as indentações Vickers conforme norma ASTM C-1327-99.
Somente as diagonais de impressão consideradas aceitáveis dentro dos padrões
da norma ASTM C-1327, foram medidas com o auxílio do analisador de imagens IA
3001 da LECO acoplado a um microscópio Olympus BME 3 de platina invertida
(INT), com aumento de 100X.
FIG. 4.8 Indentações Vickers aceitáveis de acordo com a norma ASTM C-1327-99

142
FIG. 4.9 Indentações Vickers não aceitáveis conforme norma ASTM C-1327-99.
De acordo com a norma ASTM C-1327-99 e após a medição das diagonais de
impressão, foram calculadas a dureza Vickers (GPa), e a dureza Vickers (HV10) a
partir das equações 4.12 e 4.13, respectivamente:
( )2dP0,0018544 HV = ( 4.12 )
Onde:
HV = Dureza Vickers (GPa)
P = carga aplicada (N)
d = média aritmética do comprimento das duas diagonais (mm).
( )2dP0,1891488 HV = ( 4.13 )
Onde:
HV = Dureza Vickers (HV10)
P = carga aplicada (N)
d = média aritmética do comprimento das duas diagonais (mm).

143
4.4.3.2 DETERMINAÇÃO DO FATOR DE INTENSIDADE DE TENSÃO CRÍTICA – KIC
A metodologia utilizada para a determinação dos valores de tenacidade à fratura
das amostras obedeceu a norma ASTM C 1421-99, a qual fornece o método de
teste padrão para a obtenção da tenacidade à fratura de cerâmicas avançadas em
temperatura ambiente.
Conforme descrito no item anterior, foram realizadas 30 impressões Vickers nas
superfícies de cada uma das amostras, as quais já estavam embutidas e polidas.
Cada impressão apresentava dois pares de trincas radiais, o que gerou um total de
60 pares de trincas.
Nos cálculos, foram utilizadas para cada amostra pelo menos 40 pares de
trincas perfeitas, ou seja, aquelas que não apresentavam interações com
imperfeições de polimento e desvios da trajetória da trinca nucleada.
A medida do comprimento das trincas e a observação de sua geometria foram
realizadas logo após o ensaio de dureza, buscando evitar o crescimento lento de
trinca após a impressão, iniciado pelo campo de tensão que atua após o
carregamento (Marchi, J., 1999). As trincas foram medidas e sua geometria
observada com o auxílio do analisador de imagens IA 3001 da LECO acoplado a um
microscópio Olympus BME 3 de platina invertida (INT), com aumento de 100X.
As equações propostas na literatura e utilizadas para o cálculo dos valores de
tenacidade são apresentadas na TAB. 4.7. Três equações foram escolhidas dentre
as diversas existentes e os valores obtidos a partir destas equações foram
comparados.
Os valores de tenacidade à fratura encontrados neste trabalho, foram corrigidos
levando-se em conta a densidade do material, uma vez que o aumento da
porosidade reduz a tenacidade por diminuição da área resistente e pelo efeito de
concentração de tensão nos poros. A correção foi realizada a partir da seguinte
equação (Duailibi, J. F., 1994):
bp
ICdICp .eKK −= ( 4.17 )

144
Onde:
KICp = Tenacidade à fratura do material corrigida, considerando a porosidade
(MPa.m1/2);
KICd = Tenacidade à fratura do material suposto denso (MPa.m1/2);
b = 5,3 ± 0,4 (Nos cálculos, adotou-se b = 5,3);
p = porosidade aparente do material.
TAB. 4.8 Equações propostas na literatura e utilizadas para determinação de KIC.
Referência Bibliográfica
Equação proposta de tenacidade à fratura KIC
Anstis, G. R. et. al., 1981
23
21
016,0c
PHEK IC ⎟
⎠⎞
⎜⎝⎛= ( 4.14 )
Casellas, D. et al, 1997 23
21
024,0cP
HEKIC ⎟
⎠⎞
⎜⎝⎛= ( 4.15 )
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛=
−
φφ 5,05
223
129,0 HaHE
acK IC ( 4.16a )
para 5,2≥ac
Niihara, K., 1983 ⎟⎟
⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛=
−
φαφ 5,05
221
035,0 HaHE
alK IC ( 4.16b )
para 5,225,0 ≤≤al
Onde:
KIC = tenacidade à fratura do material (MPa.m1/2);
P = carga aplicada (N);
E = módulo de elasticidade do material, calculado pela regra das misturas(GPa);
a = semi-diagonal da impressão Vickers(m);
l = comprimento da trinca (m);
c = l+a (m);
H = dureza do material (GPa) calculada conforme equação 4.12 .

145
Com a finalidade de se determinar o se o tipo de trinca presente no sistema
estudado neste trabalho era Palmqvist ou ““half-penny””, as superfícies polidas das
amostras contendo as impressões Vickers foram ligeiramente desbastadas com
pasta de diamante de 15 μm. Além disso, a razão c/a foi avaliada com o mesmo
objetivo, conforme o estudo de Niihara et al., 1982, para o qual a trinca Palmqvist
pode ser observada quando c/a for menor que 2,5 e a trinca “half-penny” quando a
razão c/a for maior que 2,5.
4.4.3.3 DETERMINAÇÃO DA RESISTÊNCIA MECÂNICA À FLEXÃO
A metodologia utilizada para a determinação dos valores de resistência à flexão
das amostras, obedeceu a norma ASTM C 1161-90, a qual fornece o método de
teste padrão para a obtenção da resistência à flexão de cerâmicas avançadas em
temperatura ambiente.
A resistência mecânica foi avaliada por testes de flexão em quatro pontos em
uma máquina de ensaios universal EMIC modelo DL-10000(IME). A velocidade do
travessão foi de 0,5 mm/min e a célula de carga de 100kg foi utilizada para a
amostra de alumina. Para todas as outras amostras, a célula de carga utilizada foi
de 500 kg.
Um esquema do aparato utilizado no ensaio de flexão em quatro pontos é
apresentado na FIG. 4.11. Neste trabalho, a distância entre os roletes inferiores(L)
era de 30mm, e a distância entre os roletes superiores era de 15mm (L/2).
FIG. 4.10 Esquema do ensaio de flexão em 4 pontos.
Roletes inferiores
Corpo de prova Roletes
superiores

146
A fórmula padrão utilizada para a determinação da resistência de uma viga por
flexão em quatro pontos, a qual consta na norma ASTM C 1161-90, foi utilizada
neste trabalho e está descrita a seguir:
24PtsFLEX 4bd
3PLσ = ( 4.18 )
Após os ensaios de flexão, os fragmentos de fratura foram retidos e preservados
para posterior análise fratográfica. A fratura original foi observada em MEV segundo
a norma ASTM C1322-96a.
A dispersão da resistência uniaxial é bem modelada pela estatística de Weibull
(Absi, J. & Gandus, J. C. 2002). Esta análise é conhecida como o modelo do elo
mais fraco para uma medida da distribuição da resistência, conforme a equação
4.19.
∫ ⎟⎟⎠
⎞⎜⎜⎝
⎛ −−−=
V
m
o
u dVσσσexp1F ( 4.19 )
Onde:
F = probabilidade de falha para a amostra;
V = volume da amostra;
σ = máxima resistência à tensão para um dado ponto;
σu = tensão limite (abaixo do qual nenhuma falha irá ocorrer);
σo = resistência característica; e
m =módulo de Weibull.
A equação 4.19 é conhecida como distribuição de Weibull de três parâmetros,
porque três parâmetros definem a forma da distribuição. O módulo de Weibull
caracteriza o espalhamento nas resistências e a resistência característica que define

147
o quanto alta ou baixa são as resistências. Altos valores de σu, σo e m são
desejados.
A tensão limite σu, é a tensão abaixo da qual a falha não irá ocorrer e
corresponde a idéia ou conceito de um tamanho máximo de falha. Na prática, σu é
usualmente considerado igual a zero. Esta consideração resulta em uma
superestimativa da probabilidade de falha, o que é desejável em projetos de cálculo
de engenharia (Sullivan, J. D. & Lauzon, P. H., 1986). Todavia, existem casos na
literatura em que ele não foi considerado igual a zero. São casos raros e requerem
extensivos testes para verificação (Sullivan, J. D. & Lauzon, P. H., 1986; Lewis, D.,
1979).
Para o caso de σu ser considerada nula, a equação 4.19 pode ser reescrita na
equação 4.20, conhecida como distribuição de Weibull de dois parâmetros, método
mais comumente utilizado para análises de rotina.
∫ ⎟⎟⎠
⎞⎜⎜⎝
⎛−−=
V
m
o
dVσσexp1F ( 4.20 )
Os diagramas de Weibull são uma maneira conveniente de se apresentar os
dados de resistência. O diagrama apresenta eixos especialmente escolhidos para
linearizar os dados. Se os dados se ajustam a uma distribuição de três parâmetros,
então:
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛ −−−=
m
o
u
σσσVexp1F ( 4.21 )
( )m
o
VexpF-1 ⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛σσ
−= ( 4.22 )

148
m
0σσV
F11lnF)ln(1 ⎟⎟
⎠
⎞⎜⎜⎝
⎛=
−=−− ( 4.23 )
mlnσmlnσlnVF1
1lnln 0 +−=⎥⎦
⎤⎢⎣
⎡⎟⎠⎞
⎜⎝⎛
− ( 4.24 )
Os valores de resistência ou módulo de ruptura (σ) foram calculados usando a
equação 4.18.
O diagrama de Weibull foi plotado tendo como eixo das ordenadas os valores de
ln (ln (1/(1-F))), e como eixo das abscissas os valores de ln σ. O módulo de Weibull
(m) é o coeficiente angular ou inclinação da reta.
As resistências foram colocadas em ordem crescente e para cada resistência foi
associado uma probabilidade.
A resistência mecânica foi avaliada para uma probabilidade de fratura de 50 %
(σ50%) usando a estimativa da seguinte equação:
n0,5jF −
= ( 4.25 )
onde:
n = número total de amostras; e
j = enésimo resultado de uma população de n amostras.
A estimativa mostrou ser menos tendenciosa, promovendo o menor
espalhamento para análises de resistência.(Trusttrum, K. & Jayatilaka, A. De S.,
1979; Absi, J. & Gandus, J. C. 2002).
4.5 TRATAMENTO ESTATÍSTICO DOS DADOS
Neste trabalho, foram realizados um grande número de ensaios para cada
composição. Como em qualquer experimento, os resultados bem como as

149
conclusões obtidas a partir dos mesmos, dependem em grande parte da maneira
como estes dados foram coletados. Métodos estatísticos devem ser utilizados para
analisar os dados de tal forma que os resultados e conclusões sejam válidas e
objetivas.
Com o objetivo de se avaliar a significância do efeito da adição da zircônia na
alumina, foi realizado o tratamento estatístico de todos os resultados obtidos neste
trabalho.
A fim de se obter conclusões válidas e objetivas ao se comparar as médias que
foram obtidas a partir dos resultados dos ensaios realizados para determinação da
densidade, dureza, tenacidade e resistência à flexão, foi utilizada a análise de
variância. A análise de variância (ANOVA) é a técnica mais utilizada no campo da
inferência estatística, sendo considerada como procedimento apropriado para o teste
de igualdade de diversas médias (Montgomery, D. C., 1997).
Consideremos a tratamentos ou diferentes níveis (ex: 5 %, 10 %, 15 %, 80 % ,
etc.) (diferentes composições) de um único fator (ex: teor de zircônia presente na
amostra) e que desejamos compará-los. A resposta observada (ex: resistência
mecânica) a partir de cada um dos a tratamentos é uma variável aleatória. Cada um
dos valores yij representa a j-ésima observação para o tratamento i. Existem n
observações ou replicatas para o i-ésimo tratamento. O número total de
observações ou replicatas do experimento é definido como N, sendo igual ao
produto a x n.
A TAB. 4.9 apresenta os dados típicos para um experimento de um fator.
TAB. 4.9 Dados típicos para um experimento de um fator.
TRATAMENTO
(NÍVEL)
OBSERVAÇÕES
TOTAIS MÉDIAS
1 y11 y12 y1. 1.y
−
2 y21 y22 y2.
2.y−
...
...
...
...
...
a ya1 yan ya. a.y
−
y..
..y−

150
As observações podem ser descritas por um modelo estatístico linear:
ijiijy ετμ ++= , {i = 1,2......a} ( 4.26 )
{j = 1,2.......n}
Onde:
ijy = (ij)-ésima observação
μ = média global
iτ = efeito do i-ésimo tratamento
ijε = componente do erro aleatório
Usualmente, os cálculos para análise de variância podem ser realizados em um
computador utilizando um software com capacidade de analisar dados de
experimentos. Neste trabalho, a análise de variância (ANOVA) foi obtida com o
programa Statistica 6.0 (StatSoft).
O nome análise de variância é derivado de um particionamento da variabilidade
total ou soma dos quadrados total (SST) (equação 4.27) em suas duas partes
componentes, que são conhecidas como: a soma dos quadrados das diferenças
entre as médias dos tratamentos e a média global (SStrat)(equação 4.28) mais a
soma dos quadrados das diferenças das observações dentro dos tratamentos a
partir da média do tratamento (SSE)(equação 4.29).
A soma dos quadrados total é dada por:
∑∑= =
−=a
1i
n
1jTSS
Ny
yij
2..2 (4.27 )
e a soma dos quadrados dos tratamentos é dada por:
Nyy
N1SS
2..
a
1i
2i.Trat −= ∑
=
( 4.28 )
A soma dos quadrados do erro é obtida por subtração, assim:

151
SSE = SST - SSTrat. ( 4.29 )
Temos ainda que o grau de liberdade (GL) para o tratamento é dado por a -1,
para o erro é igual a N -a e o grau de liberdade total é igual a N –1. O grau de
liberdade total é a soma dos graus de liberdade do tratamento e do erro.
A TAB.4.10 Apresenta a tabela da ANOVA para o experimento de um fator
TAB. 4.10 ANOVA para experimento de um fator.
Fonte de
variação
Soma dos
quadrados(SS)
Graus de
liberdade(GL)
Média dos
quadrados(MS)
F0
P-
valor Entre
tratamentos SSTrat. a -1
1aSSMS Trat.
Trat. −=
Erro (dentro dos
tratamentos)
SSE N -a
aNSSMS E
E −=
Total SST N -1 -
E
Trat.0 MS
MS F =
< 0,01
A partir da tabela da ANOVA, duas hipóteses serão testadas com o objetivo de
se avaliar a igualdade das médias. A primeira hipótese apresentada na equação
4.30 é H0, para a qual as médias de a tratamentos são iguais.
aμμμH 210 === …: ( 4.30 )
A segunda hipótese que está representada na equação 4.31 é H1, para a qual
pelo menos um par de médias difere significantemente.
ji1 μμ:H ≠ ⇒ para pelo menos um par (i,j) ( 4.31 )
Devemos rejeitar H0 e concluir que existem diferenças entre as médias ou que o
fator analisado(ex:percentual de zircônia) afeta significantemente a variável
analisada(ex: resistência mecânica), quando:
F0 > F (α, a-1, N-a)
Onde: α = nível de significância (ex: 0,01, 0,05). O valor de F(α, a-1, N-a) é tabelado
e o valor de F0 pode ser obtido da tabela da ANOVA, dividindo-se a média dos
quadrados dos tratamentos (MSTrat.) pela média dos quadrados do erro (MSE).

152
Outra forma de verificar a rejeição da hipótese nula (H0) é através do P-valor
<0,01.
A partir do momento em que se rejeita a hipótese nula e conclui-se que as
médias diferem significantemente, testes devem ser realizados para comparar todos
os possíveis pares de médias.
Alguns testes como o método de Fisher LSD (mínimas diferenças significantes),
teste de Duncan, teste de Newman-Keuls, teste de Tukey podem ser utilizados para
este fim. Neste trabalho o teste utilizado para comparação entre os pares de médias
foi o teste Fisher LSD, por ser considerado um poderoso teste se aplicado após o
teste F na análise de variância com significância de 5 % (α = 0,05).
Como deseja-se comparar pares de médias, então é necessário testar a
seguinte hipótese: H0 : μi = μj para todo i ≠ j.
Este teste estatístico Fisher LSD pode ser resumido nas equações 4.32 a 4.33.
O valor de LSD pode ser obtido a partir da equação 4.32 é chamado de mínima
diferença significante.
⎟⎟⎠
⎞⎜⎜⎝
⎛+= −
jiEaNα/2, n
1n1MStLSD ( 4.32 )
No caso de experimentos balanceados, ou seja, quando o número de
observações ou réplicas é igual para todos os tratamentos, temos que:
n
2MStLSD EaNα/2, −= ( 4.33 )
Para realizar o teste LSD, simplesmente são comparadas as diferenças
observadas entre cada par de médias em módulo com o valor de LSD obtido a partir
das equações 4.32 ou 4.33. Desta forma, se ji yy − > LSD, pode ser concluído
que a população de médias μi e μj diferem significantemente.

153
5 RESULTADOS E DISCUSSÃO 5.1 CARACTERIZAÇÃO DOS PÓS
5.1.1 ÁREA ESPECÍFICA DE SUPERFÍCIE
Na TAB. 5.1 são apresentados os valores da área específica de superfície dos
pós (óxidos puros e compósitos) após desaglomeração, obtidos por BET.
TAB. 5.1 Área específica de superfície dos pós (m2/g).
Amostra Área específica de superfície (m2/g)
100 ZT 6,21 80 ZT 8,92 21 ZT 8,32 15 ZT 8,29 10 ZT 10,08 5 ZT 9,83
A 10,48 100 ZM 11,10 80 ZM 10,51 15 ZM 8,27
A partir dos resultados da Tab. 5.1 é possível verificar que 100 ZT e 80 ZT
apresentaram menor área de superfície específica quando comparadas com as
amostras 100 ZM e 80 ZM, respectivamente. A amostra 100 ZT não foi submetida
ao processo de moagem e desaglomeração tendo assim apresentado o menor valor
de área específica de superfície.
5.1.2 TAMANHO DE PARTÍCULAS
As curvas de distribuição de tamanho de partículas para os pós (óxidos puros e
compósitos, após desaglomeração e secagem, analisados neste trabalho são
apresentadas nas FIG. 5.1, FIG. 5.2 e FIG. 5.3. Os valores utilizados para a
confecção das curvas de distribuição de tamanho de partículas foram obtidos a partir
do gralunômetro a laser (espalhamento a laser).

154
Tamanhos característicos às proporções em que 10, 50 e 90 % do material é
passante foram determinados e representados respectivamente por D10, D50 e
D90. Enquanto o D50 representa o tamanho mediano do material, a razão D90/D10
oferece uma indicação da homogeneidade do material. Os resultados estão
apresentados na TAB. 5.2.
0,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
80,00
90,00
100,00
0,010,1110100Tamanho de partícula (µm)
Pass
ante
acu
mul
ado
(%)
100 ZT
80 ZT
21 ZT
15 ZT
10 ZT
5 ZT
100 A
100 ZM
80 ZM
15 ZM
FIG. 5.1 Distribuição de tamanho de partículas para todas as amostras.

155
0,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
80,00
90,00
100,00
0,010,1110100
Tamanho de partícula (µm)
Pass
ante
acu
mul
ado
(%)
100 ZT
80 TZ
21TZ
15 TZ
10 TZ
5 TZ
A
FIG. 5.2 Distribuição de tamanho de partículas para amostras ZT e alumina.
0,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
80,00
90,00
100,00
0,010,1110100
Tamanho de partícula (µm)
Pass
ante
acu
mul
ado
(%)
100 ZM
80 ZM
15 ZM
A
FIG. 5.3 Distribuição de tamanho de partículas para amostras ZM e alumina.
TAB. 5.2 Tamanho médio de partícula .

156
Tamanho médio de partículas (μm)
Amostr
a D 10 D 50 D 90
100 ZT 0,36 0,97 3,84 80 ZT 0,19 0,80 3,65 21 ZT 0,42 1,07 4,18 15 ZT 0,29 0,79 2,49 10 ZT 0,20 0,72 2,05 5 ZT 0,41 1,02 3,58
A 0,11 0,44 2,10 100 ZM 0,06 0,19 0,45 80 ZM 0,21 0,74 2,35 15 ZM 0,32 0,92 3,05
5.1.3 FORMA DA PARTÍCULA
A caracterização da forma das partículas dos pós iniciais foi realizada a partir de
solução bem diluída dos pós e com o auxílio do MEV. As micrografias são
apresentadas na FIG. É possível verificar a presença de partículas submicrométricas
e com forma arredondada e de aglomerados formados por pequenas partículas.
0.5 μm
0.5 μm
0.5 μm
(a) (b) (c)
FIG. 5.4 Micrografia de solução bem diluída dos pós: (a)100 ZT , (b) 100 A , (c) 100 ZM.
5.1.4 CARACTERIZAÇÃO DAS FASES PRESENTES
Os difratogramas dos pós são apresentados nas FIG. 5.5. a FIG. 5.14 (linha
azul). A linha vermelha é a linha do ajuste realizado pelo programador para
determinação e quantificação das fases presentes. Pode ser observado que as
fases presentes nos óxidos puros e compósitos foram α−alumina, zircônia
monoclínica (“baddeleyite”) e zircônia tetragonal.

157
FIG. 5.5 Difratograma do pó da amostra 100 ZT.
FIG. 5.6 Difratograma do pó para amostra 80 ZT.
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 65,94 Baddeleyite 34,36
Inte
nsid
ade
u. a
.
2 θ
Corundum 20,64 ZrO2 - T 48,77 Baddeleyite 30,59
Corundum 77,44 ZrO2 - T 13,95 Baddeleyite 8,61

158
FIG. 5.7 Difratograma do pó para amostra 21 ZT.
FIG. 5.8 Difratograma do pó para amostra 15 ZT.
FIG. 5.9 Difratograma do pó para amostra 10 ZT.
Inte
nsid
ade
u. a
.
2 θ
In
tens
idad
e u.
a.
2 θ
Corundum 83,76 ZrO2 - T 9,98 Baddeleyite 6,26
Inte
nsid
ade
u. a
.
2 θ
Corundum 88,79 ZrO2 - T 6,79 Baddeleyite 4,42
Corundum 94,29 ZrO2 - T 3,39 Baddeleyite 2,37

159
FIG. 5.10 Difratograma do pó para amostra 5 ZT.
0 10 20 30 40 50 60 70 80 90
0
500
1000
1500
2000
2500
3000
25.48
35.0643.26
52.42
57.38
68.12
Alumina
Inte
nsid
ade
u. a
.
2θ
FIG. 5.11 Difratograma do pó para amostra A.
Inte
nsid
ade
u. a
.
2 θ

160
80787674727068666462605856545250484644424038363432302826242220181614121086
1,600
1,500
1,400
1,300
1,200
1,100
1,000
900
800
700
600
500
400
300
200
100
0
ZrO2T 70.80 %Baddeleyite 29.20 %
FIG. 5.12 Difratograma do pó para amostra 100 ZM.
FIG. 5.13 Difratograma do pó para amostra 80 ZM.
FIG. 5.14 Difratograma do pó para amostra 15 ZM.
Inte
nsid
ade
u. a
.
2 θ
Corundum 20,35 ZrO2 - T 64,44 Baddeleyite 15,21
Inte
nsid
ade
u. a
.
2 θ
Corundum 83,80 ZrO2 - T 12,67 Baddeleyite 3,47
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 70,80 Baddeleyite 29,20

161
5.2 CARACTERIZAÇÃO E DETERMINAÇÃO DAS PROPRIEDADES DO MATERIAL SINTERIZADO
5.2.1 COMPOSIÇÕES ANALISADAS
Neste trabalho, foram analisadas dez diferentes composições onde foi variado o
percentual de zircônia presente na amostra. Os valores das densidades teóricas dos
pós iniciaIs de alumina, zircônia Tosoh e zircônia Mel Chemicals, foram utilizados no
cálculo das densidades teóricas das dez amostras estudadas neste trabalho e estão
apresentadas nas TAB. 5.3, TAB. 5.4 e TAB. 5.5, respectivamente. A composição e
a densidade teórica das amostras analisadas são apresentados na TAB. 5.6.
TAB. 5.3 Densidade teórica da alumina SG-1000- Alcoa (g/cm3).
Óxidos % peso Wi=%peso/100Densidade Di
(g/cm³) Wi/Di SOMA Wi/Di 1/SOMA= D.Teórica
Al2O3 99,22 0,9922 3,97 0,2502 0,2524 3,96 Fe2O3 0,52 0,0052 5,24 0,0010 SiO2 0,11 0,0011 2,65 0,0004 SO3 0,15 0,0015 1,97 0,0008
TAB. 5.4 Densidade teórica da zircônia Mel Chemicals (g/cm3).
Óxidos % peso Wi=%peso/100Densidade Di
(g/cm³) Wi/Di SOMA Wi/Di 1/SOMA= D.Teórica
Y2O3 5,86 0,0586 5,01 0,0117 0,1669 5,99 ZrO2 90,46 0,9047 6,05 0,1495 Al2O3 0,51 0,0051 3,97 0,0013 SO3 0,27 0,0027 1,97 0,0014
Fe2O3 0,03 0,0003 5,24 0,0001 HfO2 2,83 0,0283 9,68 0,0029 Ta2O5 0,03 0,0003 8,20 0,0001
TAB. 5.5 Densidade teórica da zircônia Tosoh (g/cm3).
Óxidos % peso Wi=%peso/100Densidade Di
(g/cm³) Wi/Di SOMA Wi/Di 1/SOMA= D.Teórica
Al2O3 0,01 0,0001 3,97 0,0000 0,1670 5,99 Y2O3 5,16 0,0516 5,01 0,0103 Fe2O3 0,01 0,0001 5,24 0,0000 ZrO2 94,82 0,9482 6,05 0,1567 SiO2 0,01 0,0001 2,65 0,0000 Na2O 0,01 0,0001 2,27 0,0000
TAB. 5.6 Composição das amostras (%peso) e densidade teórica das amostras (g/cm3).

162
Amostra
Al2O3 (% peso)
ZrO2 Tosoh(% peso)
ZrO2 MCH
(% peso)
ρT (g/cm3)
100 ZT - 100 - 5,99 80 ZT 20 80 - 5,43 21 ZT 79 21 - 4,27 15 ZT 85 15 - 4,17 10 ZT 90 10 - 4,10 5 ZT 95 5 - 4,03
A 100 - - 3,96 100 ZM - - 100 5,99 80 ZM 20 - 80 5,43 15 ZM 85 - 15 4,17
5.2.2 DENSIDADE E DEPENDÊNCIA DA MICROESTRUTURA
As sinterizações das amostras 100 ZT, 80 ZT e 100 ZM e 80 ZM foram
realizadas em forno na temperatura de 1500 °C por 2h, pois há um crescimento
excessivo dos grãos de zircônia a 1600 °C. Todos os outros materiais foram
sinterizados na temperatura de 1600 °C por 2h. Os valores obtidos para a
densidade a verde, densidade aparente e porosidade aparente são apresentados na
TAB. 5.7.
TAB. 5.7 Densidade e porosidade das amostras.
Amostra ρverde (% Teórica)
ρaparente (% Teórica)
Paparente (%)
100 ZT 45,80 ± 0,27 99,46 ± 0,08 0,63 ± 0,40 80 ZT 48,64 ± 1,04 99,60 ± 0,46 0,62 ± 0,12 21 ZT 54,91 ± 0,43 99,55 ± 0,04 0,40 ± 0,20 15 ZT 55,53 ± 0,96 99,52 ± 0,35 0,54 ± 0,10 10 ZT 52,20 ± 1,87 99,35 ± 0,20 0,50 ± 0,14 5 ZT 54,29 ± 1,21 99,29 ± 0,10 0,44 ± 0,21
A 55,92 ± 1,69 99,13 ± 0,13 0,43 ± 0,13 100 ZM 43,88 ± 0,68 99,35 ± 0,92 0,60 ± 0,31 80 ZM 43,81 ± 1,73 99,86 ± 0,06 0,70 ± 0,23 15 ZM 54,14 ± 0,44 99,41 ± 0,38 0,75 ± 0,12
A partir dos dados da TAB. 5.7 é possível verificar que os óxidos puros e
compósitos apresentaram densidade na faixa de 99,13 % a 99,86 % da densidade
teórica.
A porosidade total menor que 1vol.% é requerida para cerâmicas de alumina
(ASTM F-603-00) e zircônia (ASTM F-1873-98), para uso em implantações
cirúrgicas.

163
Resultados da literatura mostram que a densidade influencia a dureza,
resistência à flexão e tenacidade à fratura (Lin, J. D. & Duh, J. G., 2002; Kunes, K.
et al., 2000; Heijman, M. J. G. W., 2002). Geralmente, amostras cerâmicas
sinterizadas com maior densidade relativa exibem melhores propriedades
mecânicas. Os poros funcionam como concentradores de tensões e além disso, há
diminuição da área resistente. Todavia, existe sempre a influência da uniformidade
da microestrutura sobre as propriedades mecânicas. Para materiais com elevada
tenacidade é esperado um espalhamento na medida de tenacidade à fratura como
observado nas medidas de resistência à fratura se a distribuição da porosidade não
for homogênea.
Os maiores valores de densidade foram obtidos para 80 ZT e 80 ZM (TAB. 5.7).
Partindo da alumina pura (A) até 21 peso% de zircônia Tosoh (21 ZT), foi possível
observar que a densidade aumentou com o aumento do teor de zircônia, indicando
que a presença de partículas de zircônia contribuiu para a densificação da matriz de
alumina. A zircônia deve ter um tamanho médio de partícula reduzido para ocupar as
posições intergranulares da alumina após a sinterização, bem dispersas na
microestrutura, controlando o crescimento dos grãos de alumina, evitando a
presença de agregados que prejudicariam as propriedades mecânicas do material
sinterizado. Da mesma forma, partindo da zircônia pura (100 ZT) para 80 ZT, a
adição de alumina melhorou a densificação do compósito.
Com o objetivo de verificar o efeito do fator ou tratamento composição na
variável densidade, a análise de variância (ANOVA) foi realizada com auxílio do
programa Statistica 6.0 e está apresentada na TAB. 5.8, como descrito no item 4.2.
TAB. 5.8 Análise de variância (ANOVA) para as médias de densidade.
STAT. TABELA DA ANOVA
Resumo de todos os efeitos; projeto: (densidade.sta) 1 – COMPOSIÇÃO
Efeito
Grau de liberdade
do tratamento
Media dos quadrados
do tratamento
Grau de liberdade do erro
Média dos quadrados do erro
F
p-valor
1 9 .1179996 20 .040635 2.903819 .022566
A partir dos dados apresentados na TAB. 5.8, temos que o valor de Fcalculado
(2.903819) é maior que o valor de Ftabelado (2,39), então a hipótese Ho de médias
iguais deve ser rejeitada, ou seja, é possível afirmar que existem diferenças

164
significantes com 95 % de confiança entre pelo menos duas médias de densidade
encontradas.
Para determinar as diferenças significantes entre as médias, foi utilizado o teste
LSD, conforme já descrito no item 4.5.
As TAB. 5.9 e TAB. 5.10 apresentam o resultado do teste LSD para as amostras
de 1 a 5 e 6 a 10, respectivamente. As diferenças significantes entre as médias são
apresentadas nas tabelas TAB. 5.9 e TAB. 5.10 e são identificadas com um símbolo
(*).
TAB. 5.9 Teste LSD para a variável densidade das amostras de 1 a 5.
STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (densidade.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{1} 99.46426
{2} 99.59698
{3} 99.55141
{4} 99.51863
{5} 99.35323
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.429518 .602332 .744614 .507654 .305194 .055229 .495516 .026008* .745055
.429518
.784677
.639183
.154200
.077850 .010070* .149080 .125721 .269361
.602332
.784677
.844162
.242634
.129388 .018459* .235295 .075481 .400447
.744614
.639183
.844162
.326953
.182024 .028183* .317816 .051198 .516788
.507654
.154200
.242634
.326953
.709605
.188574
.984522 .005920* .733771
TAB. 5.10 Teste LSD para a variável densidade das amostras de 6 a 10.
STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (densidade.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{6} 99.29106
{7} 99.12919
{8} 99.35000
{9} 99.86000
{10} 99.41000
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.305194
.077850
.129388
.182024
.709605
.337103
.724025 .002493* .478269
.055229 .010070* .018459* .028183* .188574 .337103
.194758 .000251* .103462
.495516
.149080
.235295
.317816
.984522
.724025
.194758
.005663* .719279
.026008* .125721
.075481 .051198 .005920* .002493* .000251* .005663*
.012786*
.745055
.269361
.400447
.516788
.733771
.478269
.103462
.719279 .012786*
De acordo com o teste é possível notar a partir dos dados das TAB. 5.9 e TAB.
5.10 que a densidade da amostra 80 ZM é significantemente diferente das

165
densidades das amostras 100 ZT, 10 ZT, 5ZT, A, 100 ZM e 15 ZM e é equivalente
as médias das amostras 80 ZT, 21 ZT, 15 ZT. As amostras 100 ZT e 100 ZM só
apresentam diferenças significantes com a amostra 80 ZM. A alumina apresentou
diferenças significantes com as amostras 80 ZT, 21 ZT, 15 ZT e 80 ZM.
Uma pequena e estreita distribuição de tamanho de partícula inicial bem como
uma distribuição bimodal, permite um preenchimento mais eficiente das cavidades
da matriz para prensagem uniaxial, promovendo melhor densificação dos pós.
Nos sistemas investigados neste trabalho, as suspensões apresentavam carga
de 50% peso de sólidos na polpa de moagem e microestrutura uniforme, não sendo
encontrados aglomerados. Os aglomerados nos corpos verdes são fonte de falhas
durante a sinterização, comprometendo as propriedades do material. A carga de
sólidos presente na polpa de moagem é discutida na literatura como fator importante
na obtenção de maiores densidades. No presente trabalho, a carga de sólidos na
polpa de moagem foi de 50 % peso, tendo sido este percentual retirado da literatura.
Resultados recentes confirmam a adequação deste percentual. Liu, X. et al., 2002
encontraram menores densidades relativas durante a sinterização nos compósitos
com carga de sólidos de 40% vol. A medida que a carga de sólidos aumentou, as
densidades aumentaram. Acima de 50 % vol. de sólidos, foram detectados
aglomerados que permaneceram nos corpos verdes e agiram como fonte de falhas
nos compactos sinterizados.
Neste trabalho, o ligante utilizado foi o polietileno glicol, tendo sido utilizado 2 %
em peso, conforme descrito na literatura. O solvente para este ligante é o álcool. A
utilização do ligante tem como objetivo dar maior resistência a verde possibilitando o
manuseio das amostras. O dispersante utilizando nas moagens foi o ácido cítrico
(0,6 % peso), com o objetivo de evitar a formação de aglomerados. No trabalho de
Kunes, K. et al., 1999, o percentual de 0,6 % em peso de dispersante nas
suspensões de alumina-zircônia investigadas foi o que promoveu a menor
viscosidade aparente para os três tipos de dispersantes testados, sendo efetivo para
prevenir aglomerados os quais são fontes de imperfeições durante a sinterização.
Mukherjee, A. et al., 2001, investigou a função de dispersantes e tamanho de
partículas do pó na reologia das suspensões e os efeitos correspondentes na
densidade a verde bem como na densidade sinterizada de zircônia estabilizada com
ítria. Os autores concluíram que a melhor dispersão foi obtida com o uso de

166
dispersante e com pós mais finos e que a reologia das suspensões têm um profundo
efeito na densidade a verde e na densidade sinterizada das amostras.
Oliveira, L. G., 2002, observou em seu trabalho que os pós de zircônia
parcialmente estabilizada com ítria na ausência de dispersante e falta de ajuste do
pH na moagem, formam um material sinterizado com uma microestrutura
heterogênea, contendo agregados duros de zircônia, que geram valores de KIC
inferiores ao valor obtido para a alumina pura, indicando a falta de reforço. No
entanto, foi observado que a adição de ácido cítrico (0,6 % peso em álcool) na
moagem proporcionou a obtenção de compósitos com valores de resistência à
flexão cerca de 17 % superior ao obtido para a alumina pura, bem como um
aumento do parâmetro “m” da distribuição de Weibull, indicando uma melhora da
confiabilidade do material em termos de dispersão dos resultados de resistência
mecânica.
As microestruturas homogêneas mostram uma segunda fase bem dispersa nos
compósitos e pode ser vista na FIG. 5.15.
As micrografias das seções transversais polidas foram obtidas no MEV com a
utilização de elétrons retroespalhados. A diferença em contraste está principalmente
associada com o número atômico das fases. Regiões claras correspondem a
números atômicos altos (Zr) e regiões escuras correspondem a baixo número
atômico (Al).
Os índices de retração referente as 3 dimensões lineares (altura, largura e
comprimento) das amostras obtidas após sinterização é apresentado na TAB. 5.11.
TAB. 5.11 Retração linear das amostras.
Retração linear (%) Amostra
Comprimento Largura Espessura 100 ZT 23,47 ± 0,11 22,61 ± 0,67 22,53 ± 0,70 80 ZT 22,04 ± 0,21 21,70 ± 0,58 21,70 ± 0,52 21 ZT 18,90 ± 0,18 18,88 ± 0,47 18,90 ± 0,92 15 ZT 18,42 ± 0,36 17,97 ± 0,37 18,36 ± 1,08 10 ZT 18,94 ± 0,78 18,75 ± 0,60 18,89 ± 0,74 5 ZT 18,91 ± 0,46 18,89 ± 1,02 18,71 ± 0,35
A 18,05 ± 0,74 17,41 ± 0,45 17,29 ± 1,23 100 ZM 23,99 ± 0,30 22,91 ± 2,41 22,11 ± 1,60 80 ZM 24,91 ± 0,07 23,81 ± 0,43 22,85 ± 2,64 15 ZM 19,03 ± 0,09 18,30 ± 0,08 18,17 ± 0,29

167
(a) (b) (c)
(d) (e) (f)
(g) (h) (i)
(j)
FIG. 5.15 Micrografias eletrônicas de varredura utilizando elétrons retroespalhados, onde as partículas escuras e claras são alumina e zircônia, respectivamente para as amostras: (a) 100
ZT, (b) 80 ZT, (c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j) 15 ZM.

168
5.2.3 DIFRAÇÃO DE RAIOS-X: FASES CRISTALINAS E TRANSFORMAÇÃO MARTENSÍTICA
Um dos problemas encontrados em materiais que apresentam várias fases
polimórficas,é a quantificação das fases presentes. Neste trabalho, foi utilizada a
difração de raios-X e dois métodos de quantificação das fases: o método interno, e o
método de Rietveld.
Os difratogramas das amostras após a sinterização são apresentados nas
FIG.5.16. a FIG. 5.24 (linha experimental em azul). Os difratogramas das amostras
após a fratura são apresentados nas FIG. 5.25 a FIG. 5.33 (linha experimental em
azul). A linha vermelha é a linha do ajuste realizado pelo programador para
determinação e quantificação das fases presentes. Pode ser observado que as fases
presentes nos óxidos puros e nos compósitos foram α−alumina, zircônia monoclínica
(baddeleyite) e zircônia tetragonal.
FIG. 5.16 Difratograma da amostra 100 ZT após sinterização.
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 90,29 Baddeleyite 9,71
Inte
nsid
ade
u. a
.
2 θ
Corundum 27,02 ZrO2 - T 68,06 Baddeleyite 4,92

169
FIG. 5.17 Difratograma da amostra 80 ZT após sinterização.
FIG. 5.18 Difratograma da amostra 21 ZT após sinterização.
FIG. 5.19 Difratograma da amostra 15 ZT após sinterização.
FIG. 5.20 Difratograma da amostra 10 ZT após sinterização.
Inte
nsid
ade
u. a
.
2 θ
Corundum 81,65 ZrO2 - T 18,13 Baddeleyite 0,22
Inte
nsid
ade
u. a
.
2 θ
Corundum 85,74 ZrO2 - T 14,02 Baddeleyite 0,24
Inte
nsid
ade
u. a
.
2 θ
Corundum 90,72 ZrO2 - T 8,94 Baddeleyite 0,34

170
FIG. 5.21 Difratograma da amostra 5 ZT após sinterização.
FIG. 5.22 Difratograma da amostra 100 ZM após sinterização.
FIG. 5.23 Difratograma da amostra 80 ZM após sinterização.
Inte
nsid
ade
u. a
.
2 θ
Corundum 95,44 ZrO2 - T 4,56
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 95,96 Baddeleyite 4,04
Inte
nsid
ade
u. a
.
2 θ
Corundum 19,45 ZrO2 - T 73,36 Baddeleyite 7,19

171
FIG. 5.24 Difratograma da amostra 15 ZM após sinterização.
FIG. 5.25 Difratograma da amostra 100 ZT após fratura.
FIG. 5.26 Difratograma da amostra 80 ZT após fratura.
Inte
nsid
ade
u. a
.
2 θ
Corundum 85,09 ZrO2 - T 14,91
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 89,65 Baddeleyite 10,35
Inte
nsid
ade
u. a
.
2 θ
Corundum 26,73 ZrO2 - T 66,77 Baddeleyite 6,80

172
FIG. 5.27 Difratograma da amostra 21 ZT após fratura.
FIG. 5.28 Difratograma da amostra 15 ZT após fratura.
FIG. 5.29 Difratograma da amostra 10 ZT após fratura.
Inte
nsid
ade
u. a
.
2 θ
Corundum 77,58 ZrO2 - T 20,58 Baddeleyite 1,84
Inte
nsid
ade
u. a
.
2 θ
Corundum 86,62 ZrO2 - T 13,14 Baddeleyite 0,24
Inte
nsid
ade
u. a
.
2 θ
Corundum 91,09 ZrO2 - T 8,71 Baddeleyite 0,20

173
FIG. 5.30 Difratograma da amostra 5 ZT após fratura.
FIG. 5.31 Difratograma da amostra 100 ZM após fratura.
FIG. 5.32 Difratograma da amostra 80 ZM após fratura.
Inte
nsid
ade
u. a
.
2 θ
Corundum 94,92 ZrO2 - T 4,54 Baddeleyite 0,54
Inte
nsid
ade
u. a
.
2 θ
Corundum 21,82 ZrO2 - T 70,51 Baddeleyite 7,67
Inte
nsid
ade
u. a
.
2 θ
ZrO2 - T 89,90 Baddeleyite 10,10

174
FIG. 5.33 Difratograma da amostra 15 ZM após fratura.
A TAB. 5.12. apresenta a quantificação das fases presentes, zircônia tetragonal
(ZrO2- t), zircônia monoclínica (ZrO2- m) e α-alumina (α- Al2O3), utilizando o método
de Rietveld, para os pós, para as amostras após sinterização e para as amostras
após a fratura.
TAB. 5.12 Análise quantitativa das fases presentes obtida pelo método de Rietveld.
Pó (%)
Após sinterização (%)
Após fratura (%)
Amostra
ZrO2 - t ZrO2 - m α-Al2O3 ZrO2 - t
ZrO2 - m
α-Al2O3
ZrO2 - t
ZrO2 - m α-Al2O3
100 ZT 65,94 34,36 - 90,29 9,71 - 89,65 10,35 - 80 ZT 48,77 30,59 20,64 68,06 4,92 27,02 66,77 6,80 26,73 21 ZT 13,95 8,61 77,44 18,13 0,22 81,65 20,58 1,84 77,58 15 ZT 9,98 6,26 83,76 14,02 0,24 85,74 13,14 0,24 86,62 10 ZT 6,79 4,42 88,79 8,94 0,34 90,72 8,71 0,20 91,09 5 ZT 3,39 2,37 94,29 4,56 - 95,44 4,54 0,54 94,92
A - - 100 - - 100 - - 100 100 ZM 70,80 29,20 - 95,96 4,04 - 89,90 10,10 - 80 ZM 64,44 15,21 20,35 73,36 7,19 19,45 70,51 7,67 21,82 15 ZM 12,67 3,47 83,80 14,91 - 85,09 15,89 1,18 82,93
Na TAB. 5.12, é possível verificar a partir da fração de zircônia tetragonal e
monoclínica presentes nas amostras, que a fração de zircônia tetragonal retida após
a sinterização das amostras foi superior a 90% em relação ao total de zircônia
presente para todas as amostras investigadas e que esta fração aumentou com o
aumento da quantidade de zircônia no compósito. Por exemplo, a soma da fração
tetragonal (68,06 %) e monoclínica (4,92 %) para a amostra 80 ZT após a
sinterização é de 72,98 %. A fração tetragonal para esta amostra é de 68,06 %, o
Inte
nsid
ade
u. a
.
2 θ
Corundum 82,93 ZrO2 - T 15,89 Baddeleyite 1,18

175
que corresponde a 93,26 % do total de zircônia presente nesta amostra (72,98 %)
após a sinterização.
Pode ser observado também na TAB. 5.12 que a fração de zircônia tetragonal
presente na amostra 21 ZT e na amostra 15 ZM após a fratura aumentou quando
comparada com a fração de zircõnia tetragonal para estas amostras após
sinterização. Este fato pode ser explicado pela não homogeneidade da
transformação das partículas tetragonais em monoclínicas.
Para realização do ajuste pelo método de Rietveld, não foi considerada
nenhuma orientação preferencial das fases em estudo, assim como o tamanho do
cristalito. Na TAB. 5.12 foram observadas diferenças para o percentual de alumina
presente nas condições após sinterização e após fratura em relação a composição
das amostras. A existência de uma orientação preferencial pode alterar a
concentração de uma fase presente resultando na diminuição da concentração da
mesma. Ainda, a redução do tamanho do cristalito acarreta um aumento da largura
do pico a meia altura, promovendo um aumento da área integrada sob o mesmo e
assim, um aumento da concentração da fase.
Os cálculos das frações volumétricas de zircônia apresentados na TAB. 5.13, foi
realizado a partir de métodos tradicionais, como o método interno, descritos na
literatura. Nestes métodos, é feita a deconvolução de picos isolados, utilizando a
intensidade integrada de cada pico para o cálculo das frações volumétricas. Já o
método de Rietveld utiliza o padrão de difração total, integrando a área sob os picos
com uma função matemática que considera o caráter gaussiano e lorentziano das
curvas. Assim, os resultados obtidos desta forma são considerados mais precisos
(Frâncio, E. & Lima, B. N., 2002).
A partir dos dados apresentados nas TAB. 5.12 e 5.13 foram observadas
diferenças entre os valores obtidos para as frações de zircônia monoclínica presente
nos pós, pelo método interno e pelo método de Rietveld para as amostras ZT. As
diferenças encontradas para estas amostras foram de no máximo 1,5 pontos
percentuais. Para as amostras ZM estas diferenças foram maiores, sendo a maior
diferença de 6,15 pontos percentuais obtida para a amostra 100 ZM. Para a
amostra 80 ZM a diferença foi de 3,14 pontos percentuais.
TAB. 5.13 Fração volumétrica de zircônia monoclínica presente nos pós, após sinterização e após fratura, obtida pelo método interno.

176
ZrO2 - Monoclínica (%)
Amostra Pó Após
sinterização Após fratura
100 ZT 35,79 9,41 10,87 80 ZT 29,11 4,30 6,29 21 ZT 8,77 - - 15 ZT 6,01 - - 10 ZT 4,27 - - 5 ZT 2,35 - -
A - - - 100 ZM 35,35 5,31 8,89 80 ZM 18,35 6,43 7,49 15 ZM - - -
A partir dos resultados de fração de zircônia monoclínica presente nas amostras
após sinterização e após fratura, apresentados nas TAB. 5.12 pelo método interno e
na TAB. 5.13 pelo método de Rietveld, é possível verificar que a quantidade de
zircônia monoclínica aumentou após a fratura, indicando que o mecanismo de
transformação induzida por tensão é um importante mecanismo que opera durante a
fratura destes materiais.
A difração de raios-X não permite detectar a presença de pequenas quantidades
de uma segunda fase. De acordo com o trabalho de Sallé, C. et al., 2003, para
compósitos alumina-zircônia com menos de 10 % peso, a difração de raios X não
permite detecção da zircônia tetragonal.
No presente trabalho, não foi possível detectar zircônia monoclínica apenas para
a amostra 15 ZM após sinterização conforme apresentado na TAB. 5.12. Porém,
para todas as amostras nas três diferentes condições investigadas (pó, após
sinterização e após fratura), foi possível obter a fração de zircônia tetragonal
presente nas mesmas, pelo método de Rietveld.
Nas TAB. 5.14 e TAB. 5.15 é apresentada a quantidade de transformação
induzida por tensão verificada para as diferentes amostras analisadas, a partir dos
dados de fração volumétrica de zircônia monoclínica obtidos pelo método interno e
pelo método de Rietveld, respectivamente.
TAB. 5.14 Extensão da transformação induzida por tensão a partir de dados obtidos pelo método interno.

177
ZrO2 - Monoclínica (%) Amostra Pó Após
sinterização Após
fratura
Transformação induzida por tensão (%)
100 ZT 35,79 9,41 10,87 1,46 80 ZT 29,11 4,30 6,29 1,99 21 ZT 8,77 - - - 15 ZT 6,01 - - - 10 ZT 4,27 - - - 5 ZT 2,35 - - -
A - - - - 100 ZM 35,35 5,31 8,89 3,58 80 ZM 18,35 6,43 7,49 1,06 15 ZM - - - -
TAB. 5.15 Extensão da transformação induzida por tensão a partir de dados obtidos pelo método de Rietveld.
ZrO2 - Monoclínica (%) Amostra Pó Após
sinterização Após
fratura
Transformação induzida por tensão (%)
100 ZT 34,36 9,71 10,35 0,64 80 ZT 30,59 4,92 6,80 1,88 21 ZT 8,61 0,22 1,84 1,62 15 ZT 6,26 0,24 0,24 - 10 ZT 4,42 0,34 0,20 - 5 ZT 2,37 - 0,54 -
A - - - - 100 ZM 29,20 4,04 10,10 6,06 80 ZM 15,21 7,19 7,67 0,48 15 ZM - - 1,18 -
Para avaliação da extensão da transformação induzida por tensão apenas
frações superiores a 0,5 % foram consideradas confiáveis. Desta forma não foram
calculados os percentuais de zircônia monoclínica transformada para as amostras 15
ZT, 10 ZT, 5 ZT e 15 ZM, uma vez que para estas amostras foram encontradas
frações de zircônia monoclínica após sinterização ou após fratura muito pequenas,
estando dentro da faixa de erro das medidas realizadas. A partir dos resultados de transformação induzida por tensão apresentados nas
TAB. 5.14 pelo método interno e TAB. 5.15 pelo método de Rietveld, é possível
verificar que a quantidade de zircônia monoclínica transformada pode ser detectada
para as amostras que apresentaram maiores frações de zircônia tetragonal retida
após a sinterização, tais como 100 ZT, 80 ZT, 21 ZT, 100 ZM e 80 ZM (TAB. 5.12).
A fração transformada para os compósitos (80 ZT e 21 ZT) foi maior do que para a

178
zircônia pura (100 ZT) e que esta fração aumentou com o aumento de zircônia nos
compósitos, tendo seu maior valor para o compósito 80 ZT (TAB. 5.14 e TAB. 5.15).
A fração de zircônia monoclínica transformada a partir da sinterização até a
fratura, medida pelo método interno (TAB. 5.14), variou na faixa de 1,46 % a 1,99 %
para amostras ZT e 1,06 % a 3,58 % para amostras ZM. A fração transformada
medida pelo método interno (TAB. 5.15) variou na faixa de 0,64 % a 1,88 % para as
amostras ZT e 0,48 % a 6,06 % para as amostras ZM.
No trabalho de Casellas, D. et al., 1999 foram investigados compósitos de
alumina-zircônia com 5 vol.%, 15 vol.% e 30 vol.% tendo sido verificado que
somente 5% da zircônia tetragonal disponível se transformou em monoclínica
durante a fratura. Já nos materiais submetidos a tratamento térmico de crescimento
de grão, esta porcentagem subiu para 25%. O tratamento térmico aumenta o
tamanho de grão, conhecido parâmetro de controle da transformação induzida por
tensão.
A partir da análise da quantidade de fase monoclínica, a transformação induzida
por tensão parece ser segundo os autores, o principal mecanismo de aumento de
tenacidade no ZTA (Casellas, D., 1999). O segundo mecanismo que age nestes
compósitos, o microtrincamento foi também discutido. O microtrincamento ao redor
de partículas transformadas de zircônia causam dispersão de energia e tenacificam
o material. Quanto maior a transformação de partículas de zircônia, maior o
aumento da tenacidade por microtrincamento. Segundo os autores, este mecanismo
opera no mesmo sentido da transformação de fase e é mais relevante nos
compósitos com maior teor de zircônia.
5.2.4 TAMANHO DE GRÂO
A TAB. 5.16 apresenta os tamanhos médios de grãos de zircônia e alumina para
as amostras analisadas neste trabalho.
Para a obtenção das medidas através do método das interseções lineares, foram
utilizadas três imagens de microscopia eletrônica de varredura com mesmo aumento
para cada amostra polida e termicamente atacada (1500 °C /10min.), como as
apresentadas na FIG. 5.36. As áreas foram selecionadas aleatoriamente. Os
valores médios foram obtidos com base na análise de pelo menos 150 grãos.

179
Os valores obtidos para os tamanhos médios de grãos de zircônia e alumina
presentes nas amostras contendo zircônia Tosoh e zircônia Mel Chemicals são
apresentados nas FIG. 5.34 e FIG. 5.35, onde é possível observar que o principal
efeito da adição de zircônia na alumina é a redução do tamanho de grão da matriz
alumina e conseqüentemente a redução do tamanho da população de falha,
melhorando as propriedades mecânicas do compósito. É possível verificar ainda
que com o aumento do teor de zircônia ZT (FIG. 5.34) ou ZM (FIG. 5.35) no
compósito, ocorre o aumento do tamanho de grão de zircônia. A adição de 20 % de
alumina a zircônia também promoveu a redução do tamanho de grão da zircônia em
relação a zircônia pura, de acordo com Sharif, A. A. & Mecartney, M. L., (2004).
TAB. 5.16 Tamanho médio de grão
Tamanho médio de grão Amostra
Al2O3 (μm) ZrO2 (μm)
Ratio Al2O3/ ZrO2
100 ZT - 0,71 ± 0,07 - 80 ZT 0,90 ± 0,04 0,69 ± 0,05 1,30 21 ZT 1,14 ± 0,14 0,57 ± 0,06 2,00 15 ZT 1,43 ± 0,12 0,50 ± 0,02 2,86 10 ZT 1,57 ± 0,09 0,43 ± 0,03 3,65 5 ZT 1,86 ± 0,08 0,36 ± 0,13 5,17
A 3,75 ± 013 - - 100 ZM - 0,75 ± 0,24 - 80 ZM 0,86 ± 0,10 0,65 ± 0,15 1,32 15 ZM 1,57 ± 0,07 0,55 ± 0,18 2,85
As partículas de zircônia são distribuídas uniformemente nos compósitos ZTA e
estão localizados nos contornos de grão da alumina e desta forma a alumina é
refinada por travamento dos contornos de grão da alumina (Calderon-Moreno, J. M.
& Schehl, M., 2004, Sharif, A. A. et al., 2004). De acordo com Casellas, D. et al.,
1997, a adição de zircônia tem um grande efeito no aumento da resistência à fratura,
porém, sua influência na tenacidade à fratura é muito menor. A razão para este
comportamento pode ser a ausência de dois dos principais mecanismos de aumento
de tenacidade do ZTA: ponteamento de grão e a tenacificação por transformação. O
ponteamento de grão é desprezível devido ao pequeno tamanho de grão da alumina
(somente pequenas pontes foram encontradas em alumina pura). O mecanismo de
aumento de tenacidade por transformação é também praticamente ausente devido a

180
somente 5% de zircônia tetragonal se transformar nas superfícies de fratura. Uma
explicação para isto seria o fato do tamanho médio das partículas de zircônia ser tão
pequeno que não permite ativar a transformação de fase. O tamanho de partícula
da zircônia é um conhecido parâmetro de controle da transformação de fase
induzida por tensão. De fato, os autores verificaram o aumento da tenacidade à
fratura quando o tamanho de grão da matriz de alumina e das partículas de zircônia
foram aumentados com tratamentos térmicos de crescimento de grão (Casellas, D.
et al, 2003). Tal aumento é explicado pela influência que o tamanho das partículas
de zircônia exerce no aumento de tenacidade por transformação. De acordo com os
autores, a extensão do tratamento térmico reduz a dureza, fato esperado uma vez
que a dureza está inversamente relacionada com a maior facilidade de
transformação.
0 20 40 60 80 1000.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
Tam
anho
méd
io d
e gr
ão (μ
m)
ZrO2 Tosoh (% peso)
alumina zircônia
FIG. 5.34 Tamanho médio de grão versus ZrO2 Tosoh (% peso).

181
0 20 40 60 80 100
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
Tam
anho
méd
io d
e gr
ão (μ
m)
ZrO2 Mel Chemicals (% peso)
alumina zircônia
FIG. 5.35 Tamanho médio de grão versus ZrO2 Mel Chemicals (% peso).
Casellas, D. et al., 1999, afirma que a microestrutura grosseira em materiais
bifásicos é fortemente dependente da fração volumétrica da segunda fase. Em
materiais contendo uma pequena quantidade de segunda fase, uma força que
funciona como um obstáculo é introduzida, travando os contornos de grão e
restringindo o crescimento de grãos da matriz, como encontrado pelos autores para
o ZTA 5 vol.%. Quando o conteúdo desta segunda fase aumenta para 10 %, as
partículas tendem a formar núcleos (“clusters”), os quais são mais efetivos em travar
os contornos. Foi observado para o ZTA 15 vol. %, a tendência para a formação de
núcleos e maior grau de controle do crescimento dos grãos da matriz do que para o
ZTA5 vol.%. Com adições de segunda fase maiores que 30 vol.%, os grãos de
alumina cresceram com dificuldade devido ao alto conteúdo de zircônia envolvido. A
maior quantidade de segunda fase restringiu o contato entre os grãos da matriz e a
microestrutura grosseira foi limitado devido a fatores como: limitada solubilidade
sólida, relativamente grande comprimento do caminho de difusão e restrições físicas
de ambas as fases. O sistema alumina-zircônia exibe solubilidade limitada (Betz, U.
et al., 2000), então, o engrossamento das partículas de zircônia se dá por difusão
intergranular entre núcleos vizinhos ou por coalescência de partículas dentro dos

182
núcleos (“clusters”). No ZTA 5 % as partículas de zircônia crescem lentamente
devido ao relativamente grande caminho para difusão. No ZTA15 % e 30 %, as
partículas de zircônia crescem mais facilmente, devido ao menor caminho de difusão
para a zircônia e o maior número de núcleos. Todavia, estes núcleos não
coalescem totalmente, o que pode estar associado com o baixo raio de curvatura da
interface da zircônia e seu alto ponto de fusão (2660°C), levando a um decréscimo
da força motriz de crescimento de grão (Casellas, D. et al., 2003).
O tamanho da partícula tetragonal e o conteúdo de estabilizante tem grande
influência na tensão requerida para nuclear a transformação de partículas
tetragonais (Gupta. T. K.. et al., 1978). Um tamanho de grão pode ser definido para
o qual a transformação induzida por tensão em temperatura ambiente seja ativada.
Com tamanhos de grão maiores a transformação ocorre espontaneamente no
resfriamento a partir da temperatura de tratamento térmico sem qualquer efeito
benéfico para as propriedades mecânicas. Então, um tamanho de grão crítico para
a transformação de fase no resfriamento pode também ser definido. Ambos os
tamanhos de grão crítico aumentam com o módulo de elasticidade do compósito, o
que está relacionado com as restrições impostas pela matriz. Uma vez que o
módulo de elasticidade decresce com a adição de zircônia, de acordo com a regra
das misturas, ambos os tamanhos de grão críticos serão menores em compósitos
com maior teor de zircônia. É sabido que a retenção da fase tetragonal é
criticamente governada pelo tamanho de grão. Isto é, redução do tamanho de grão é
prevista para aumentar a tensão crítica que induz a transformação de fase tetragonal
para monoclínica.(Gupta, T. K. et al., 1978).
O equilíbrio entre as fases tetragonal e monoclínica pode ser descrito em termos
do tamanho de grão crítico: em uma determinada temperatura, para cada
composição, existe um tamanho de grão acima do qual o material é estável na fase
monoclínica e abaixo do qual ele é estável na fase tetragonal (Bravo-Leon, A et al.,
2002)
No trabalho de Tuan, W. H., et al., 2002, o tamanho dos grãos de alumina nos
compósitos contendo t-ZrO2 foi menor do que nos compósitos contendo m-ZrO2,
indicando que o agente estabilizante Y2O3, pode impedir o crescimento dos grãos de
alumina. Apesar da carga iônica do ítrio ser a mesma do alumínio, o íon ítrio é muito
maior que o íon alumínio (0,89 Å versus 0,53 Å). Íons de ítrio tendem a segregar nos

183
contornos de grão da alumina, reduzindo a energia de deformação elástica. Apesar
da solubilidade da ítria em alumina ser extremamente baixa (< 10 ppm), íons de ítrio
podem bloquear a difusão de íons ao longo dos contornos de grão, levando a
redução da densificação e medida de tamanho de grão. Segundo os autores,
apesar do baixo teor de ítria nos compósitos, a quantidade é suficiente para suprimir
o crescimento excessivo dos grãos da matriz de alumina.
Observações das amostras em MEV (JSM-5200LV, Joel, Japan), após
sinterizadas e recobertas por uma fina camada de ouro de 80nm (Balzers 07-120B)
revelaram microestruturas homogêneas, livre de aglomerados, poros ou crescimento
excessivo dos grãos de alumina, como pode ser visto na FIG. 5.36.
A prevenção do crescimento excessivo dos grãos de alumina é sempre de
grande importância durante a sinterização de cerâmicos de elevada resistência
(Yang, H.et al., 2001). No presente estudo, o crescimento excessivo dos grãos de
alumina não foi observado na temperatura de sinterização de 1600° C. Foi
encontrado que amostras contendo mais de 5% em peso de zircônia, efetivamente
inibiram o crescimento do grão de alumina e a ocorrência de crescimento anormal ou
exagerado.
Lange, F. F. & Hirlinger, M. M., 1984 reportaram que o crescimento exagerado
da alumina foi prevenido por inclusões de zircônia em frações volumétricas
superiores a 5%. O crescimento anormal de grãos em materiais com α-alumina foi
reportado na literatura em presença de uma fase líquida induzida pela presença de
impurezas tais como Ca e Na(Poulon-Quintin, A et al., 2004). O crescimento
exagerado aumenta as tensões nos contornos e leva a formação de microtrincas,
prejudiciais às propriedades mecânicas.
A razão entre os tamanhos de grão Al2O3/ZrO2 (TAB. 5.16) decresceu com o
aumento da quantidade de zircõnia. Este comportamento é consistente com
análises de inclusões de segunda fase travando os contornos de grão da matriz,
ficando o tamanho de grão da alumina dependente da dispersão, tamanho e
crescimento da menor fase (zircônia).
O efeito de travamento da zircônia no crescimento do grão de alumina é distinto.
Ou seja, com o aumento do teor de zircônia, dois fenômenos são óbvios. Um é a
mudança na morfologia dos grãos de alumina. Ela muda gradualmente da forma de
placas para forma equiaxial (FIG. 5.36). O efeito supressivo da zircônia na formação

184
(a) (b) (c)
(d) (e) (f)
(g) (h) (i)
(j)
FIG. 5.36 Micrografias eletrônicas de varredura das amostras com superfícies polidas e termicamente atacadas (1500°C 10min): (a) 100 ZT, (b) 80 ZT, (c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5
ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j) 15 ZM.

185
de grãos de alumina na forma de placas é grande. O outro fenômeno é o
decréscimo do tamanho de grão da alumina com o aumento do teor de zircônia
(TAB. 5.16). Para os compósitos com pequenas percentagens de zircônia, as
partículas de zircônia (grãos claros) podem ser ineficientes no travamento dos
contornos de grão da alumina, de tal forma que o crescimento exagerado ou anormal
pode ocorrer. Algumas partículas de zircônia podem ficar aprisionadas dentro dos
grãos de alumina, como pode ser visto para a amostra 5 ZT na FIG. 5.36(f).
5.2.5 PROPRIEDADES MECÂNICAS
5.2.5.1 MÓDULO DE ELASTICIDADE
Os valores de módulo de elasticidade das amostras analisadas estão
apresentados na TAB. 5.17 e foram calculados pela regra da mistura partindo-se de
E = 210 GPa para a zircônia (Picconni & Macauro,1999, Bravo-Leon, A., et al., 2002)
e E = 380 GPa para a alumina. (Picconni, C. & Maccauro, G., 1999), a partir da
seguinte expressão:
⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎟⎠
⎞⎜⎜⎝
⎛=
322 OAl
32
ZrO
2
Compósito
EO%pesoAl
E%pesoZrO
1E ( 5.1 )
TAB. 5.17 Módulo de elasticidade das amostras calculado pela regra da mistura.
Amostra
100 ZT 80 ZT 21 ZT 15 ZT 10 ZT 5 ZT A 100 ZM 80 ZM 15 ZM
Módulo de
elasticidade
(GPa)
210,00 230,64 324,79
338,85
351,54 365,22 380,00 210,00 230,64 338,85
O módulo de elasticidade da zircônia, 210 GPa, é menor que o da alumina,
então o módulo de elasticidade dos compósitos decresce com o aumento do teor de
zircônia (TAB.5.17).

186
É importante lembrar que a medida experimental do módulo de elasticidade
pode fornecer informações a partir do interior dos compósitos. A presença de
porosidade e microtrincas podem reduzir o valor do módulo de elasticidade do
compósito (Tuan, W. H. et al., 2003).
A onda de ultrasom penetra através das amostras, diferentemente da análise de
raios-X por exemplo, a qual detecta somente a região próxima à superfície.
Tuan, W. H. et al., 2003 mediu o modulo de elasticidade com onda de ultrasom e
por comparação plotou a curva (linear) prevista pela regra das misturas. Somente
foram observados diferenças muito pequenas no valor do módulo de elasticidade
para os compósitos analisados quando comparados com os previstos pela regra da
mistura. Este mesmo comportamento foi observado no trabalho de Choi, S. R. &
Basal, N. P., 2004.
Podemos justificar a utilização deste método para determinação do módulo de
elasticidade dos compósitos por termos amostras com elevadas densidades e um
pequeno número de defeitos.
5.2.5.2 CARGA DE INDENTAÇÃO E DUREZA VICKERS
Na TAB. 5.18 estão apresentados os valores de dureza Vickers obtidos com
aplicação de carga de 10Kgf ou 98N, por 10s para as diferentes composições
analisadas.
TAB. 5.18 Medidas de dureza Vickers (HV).
Amostra Dureza (GPa)
100 ZT 13,20±0,48 80 ZT 15,44±0,46 21 ZT 17,14±0,38 15 ZT 17,38±0,23 10 ZT 17,41±0,21 5 ZT 17,48±0,76 A 17,53±0,52
100 ZM 12,78±0,34 80 ZM 14,32±0,77 15 ZM 16,98±0,38
A seleção da carga de indentação é um aspecto crítico na medida de dureza e
tenacidade à fratura usando o método de indentação Vickers, uma vez que a

187
quantidade de carga poderia afetar o tamanho da indentação e o comprimento da
trinca, sendo dependente da dimensão da amostra e da microestrutura do material.
De acordo com Krell, A., 1995, muitos cerâmicos que exibem um decréscimo da
dureza com o aumento de carga podem ter este problema solucionado se a medida
da dureza Vickers for realizada em cargas mais elevadas, preferencialmente acima
de 50N, onde a influência da carga na medida da dureza decresce. Tanto a dureza
quanto o KIC variam com a carga aplicada.
De acordo com o trabalho apresentado por Lin, J. D. & Duh, J. G., 2002, a
dureza das amostras de zircônias tetragonais dopadas com ítria permanece dentro
da faixa de dureza constante para cargas de 49 a 490N. Os autores observaram
ainda que para cargas inferiores a 49N houve um espalhamento dos dados
provavelmente devido a um menor comprimento do indentador.
Pode ser observado na TAB. 5.18 que com o aumento do percentual de
zircônia presente no compósito, ou ainda, com a redução do percentual de alumina,
a dureza dos compósitos diminui. Os valores de dureza das amostras obedecem a
uma regra das misturas linear (Lange, F. F., 1982c , Chen, Z., et al., 2004, Choi, S.
R. & Bansal, N. P., 2004, Casellas et al., 2004). Este fato deve considerar a
diferença de dureza entre a alumina e a zircônia, considerando as faixas de valores
de dureza respectivas, isto é, 17 - 18 para alumina e 12-13 GPa para a zircônia
tetragonal, como encontrado neste trabalho e por Casellas, D. et al., 2003.
Elevadas durezas implicam em boa resistência ao risco e ao desgaste, porém,
acarretam maior dificuldade de usinagem. Materiais com menor dureza seriam mais
indicados para serem utilizados na confecção de pilares cerâmicos uma vez que
após sinterização dos pilares, os mesmos necessitam ser usinados pelos
profissionais de prótese dentária para a obtenção da forma final adequada para
adaptação do pilar ao elemento dentário a ser reabilitado.
Com o objetivo de verificar o efeito do fator ou tratamento composição na
variável dureza, a análise de variância (ANOVA) foi realizada com auxílio do
programa Statistica 6.0 e está apresentada na TAB. 5.19, como descrito no item 4.2.
A partir dos dados apresentados na TAB. 5.19, temos que o valor de Fcalculado
(249.3360) é maior que o valor de Ftabelado (1.88), então a hipótese Ho de médias
iguais deve ser rejeitada, ou seja, é possível afirmar que existem diferenças

188
significantes com nível de confiança de 95% entre pelo menos duas médias de
dureza encontradas.
TAB. 5.19 Análise de variância(ANOVA) para as médias de dureza. STAT. TABELA DA ANOVA
Resumo de todos os efeitos; projeto: (dureza.sta) 1 – COMPOSIÇÃO
Efeito
Grau de liberdade
do tratamento
Media dos quadrados
do tratamento
Grau de liberdade do erro
Média dos quadrados do erro
F
p-valor
1 9 82.28285 222 .330008 249.3360 0.00
Para determinar as diferenças significantes entre as médias, foi utilizado o teste
LSD, conforme já descrito no item 4.5. As TAB. 5.20 e TAB. 5.21 apresentam o
resultado do teste LSD para as amostras de 1 a 5 e 6 a 10, respectivamente. As
diferenças significantes entre as médias são apresentadas nas tabelas TAB. 5.20 e
TAB. 5.21 e são identificadas com um símbolo (*).
De acordo com o teste é possível notar a partir dos dados das TAB. 5.20 e 5.21
que os valores de dureza para cada uma das amostras 100 ZT, 80 ZT, 100 ZM e 80
ZM, apresentaram diferenças significantes com relação aos valores de dureza
obtidos para todas as outras amostras e inclusive entre elas. Por exemplo, podemos
afirmar com 95 % de confiança que a amostra 100 ZT apresentou dureza inferior a
amostra 80 ZT, por exemplo. A alumina (A) apresentou dureza significantemente
diferente das durezas obtidas para as amostras 100 ZT, 80 ZT, 21 ZT, 100 ZM, 80
ZM e 15 ZM.
TAB. 5.20 Teste LSD para a variável dureza das amostras de 1 a 5. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (dureza.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{1} 13.20182
{2} 15.44069
{3} 17.14346
{4} 17.38198
{5} 17.41457
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.000000* 0.000000* 0.000000* 0.000000* 0.000000* 0.000000* .013952* .000000* 0.000000*
.000000*
.000000* .000000* .000000* .000000* .000000* 0.000000* .000000* .000000*
0.000000* .000000*
.160515 .110925 .046919* .026437* 0.000000* 0.000000* .333869
0.000000* .000000* .160515
.847621 .555717 .391289 0.000000* 0.000000* .017618*
0.000000* .000000* .110925 .847621
.691211 .502916 0.000000* 0.000000* .010354*

189
TAB. 5.21 Teste LSD para a variável dureza das amostras de 6 a 10. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (dureza.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{6} 17.48195
{7} 17.53092
{8} 12.78202
{9} 14.32283
{10} 16.98112
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
0.000000* .000000* .046919* .555717 .691211
.777887 0.000000* 0.000000* .003126*
0.000000* .000000* .026437* .391289 .502916 .777887
0.000000* 0.000000* .001560*
.013952* 0.000000* 0.000000* 0.000000* 0.000000* 0.000000* 0.000000*
.000000* 0.000000*
.000000* .000000* 0.000000* 0.000000* 0.000000* 0.000000* 0.000000* .000000*
0.000000*
0.000000* .000000* .333869 .017618* .010354* .003126* .001560* 0.000000* 0.000000*
5.2.5.3 VALORES DE RESISTÊNCIA MECÂNICA
Os valores de resistência mecânica de materiais frágeis como os cerâmicos,
usualmente exibem um grande espalhamento (superior a 100 %) mesmo para
cerâmicos de alta performance (Absi, J. & Glandus, J. C., 2002). Este conhecido
fenômeno é proveniente de uma distribuição de defeitos ou falhas. Este fato levou
ao tratamento da probabilidade de fratura e ao desenvolvimento de teorias como a
estatística de Weibull.
O primeiro e mais importante passo para a melhoria da resistência e
confiabilidade dos cerâmicos é reduzir o tamanho de trincas e defeitos. Isto pode
ser alcançado a partir de uma microestrutura refinada, de elevada densidade. O
refinamento da microestrutura pode ser obtido através do uso de pós com as
seguintes características: elevada pureza, pequeno tamanho de partícula inicial e
uma estreita distribuição de tamanho de partículas e ainda com um controle rigoroso
das etapas de processamento.
A TAB. 5.22 apresenta os valores de resistência mecânica dos materiais
investigados. São apresentados valores de tensão máxima (σmáx), tensão para 50 %
de probabilidade de fratura (σ 0,5), tensão média (σméd), parâmetro de Weibull “m” e o
coeficiente de correlação R para as amostras ensaiadas.
A partir dos dados apresentados na TAB. 5.22 é possível verificar uma tendência
das tensões máximas aumentarem com o aumento de zircônia no compósito e

190
também com a densidade dos mesmos, quando comparadas com a tensão máxima
obtida para a alumina pura.
TAB. 5.22 Resultados do ensaio de flexão em 4 pontos.
Amostra
Densidade aparente
(%Teórica) σmáx (MPa) σ 0,5 (MPa) σméd (MPa) “m” R2
100 ZT 99,46±0,08 736,55 354,53 385,08 ± 192,88 6,3099
2,2371
0,9805
0,921
80 ZT 99,60±0,46 755,35 479,23 447,62 ± 159,94 12,889
3,1807
0,9695
0,9806
21 ZT 99,55±0,04 510,79 307,41 315,67 ± 100,19 5,3727 0,9418
15 ZT 99,52±0,35 491,14 315,41 323,77 ± 73,50 5,6968 0,9587
10 ZT 99,35±0,20 473,13 329,63 342,91 ± 75,73 5,6529 0,9208
5 ZT 99,29±0,10 441,40 340,16 324,67 ± 67,13 5,8284 0,9827
A 99,13±0,13 396,71 334,75 329,38 ± 40,59 9,6677 0,9906
100 ZM 99,35±0,92 635,02 232,06 315,11 ± 192,92 9,2031
3,8641
0,9383
0,9538
80 ZM 99,86±0,06 763,70 328,21 344,43 ± 148,29 11,802
4,4339
0,9565
0,9474
15 ZM 99,41±0,38 471,38 284,09 288,67 ± 72,12 4,9407 0,9846
Os maiores valores de resistência foram obtidos para as amostras 80 ZT e 80
ZM, as quais apresentaram também as maiores densidades. Estes compósitos
apresentaram resistência à flexão cerca de 90 % a 93 % superiores aos valores
encontrados para a alumina, respectivamente. Estes resultados mostraram que a
adição da segunda fase promoveu um compósito com microestrutura refinada. De
acordo com Spriggs, R. M. & Vasilos, T., 1963, a resistência dos cerâmicos é
inversamente proporcional a raiz quadrada do tamanho de grão. Como pode ser
visto na TAB. 5.16, dentre as amostras ZT, a amostra 80 ZT apresentou o menor
tamanho de grão para a alumina e dentre as amostras ZM, a amostra 80 ZM
também foi a que apresentou o menor tamanho de grão para a alumina. Além disso,
as maiores densidades (TAB. 5.7) e a maior extensão da transformação induzida por
tensão das partículas de zircônia tetragonal (TAB. 5.14 e TAB. 5.15) foram também
obtidas para estas amostras, promovendo o aumento de resistência mecânica
observado.

191
Com o objetivo de verificar o efeito do fator ou tratamento composição na
variável resistência à flexão (σméd), a análise de variância (ANOVA) foi realizada com
auxílio do programa Statistica 6.0 e está apresentada na TAB. 5.23, como descrito
no item 4.2.
TAB. 5.23 Análise de variância(ANOVA) para as médias de resistência à flexão. STAT. TABELA DA ANOVA
Resumo de todos os efeitos; projeto: (flexão.sta) 1 – COMPOSIÇÃO
Efeito
Grau de liberdade
do tratamento
Media dos quadrados
do tratamento
Grau de liberdade do erro
Média dos quadrados do erro
F
p-valor
1 9 64761.06 322 12920.33 5.012339 .000002
A partir dos dados apresentados na TAB. 5.23, temos que o valor de Fcalculado
(5.012339) é maior que o valor de Ftabelado (1.88), então a hipótese Ho de médias
iguais deve ser rejeitada, ou seja, é possível afirmar que existem diferenças
significantes com nível de confiança de 95 % entre pelo menos duas médias de
resistência à flexão encontradas.
Para determinar as diferenças significantes entre as médias, foi utilizado o teste
LSD, conforme já descrito no item 4.5. As TAB. 5.24 e TAB. 5.25 apresentam o
resultado do teste LSD para as amostras de 1 a 5 e 6 a 10, respectivamente. As
diferenças significantes entre as médias são apresentadas nas tabelas TAB. 5.24 e
TAB. 5.25 e são identificadas com um símbolo (*).
TAB. 5.24 Teste LSD para a variável resitência à flexão das amostras de 1 a 5. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (flexão.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{1} 385.0844
{2} 447.6249
{3} 315.6747
{4} 323.7657
{5} 342.9098
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.031119* .014646* .029817* .139521 .033424* .051281 .017700* .160270 .000796*
.031119*
.000003*
.000010*
.000217*
.000013*
.000031*
.000006*
.000328*
.000000*
.014646* .000003*
.764473 .320463 .740753 .616894 .984186 .301780 .324499
.029817* .000010* .764473
.481774 .973217 .836491 .758350 .454868 .197546
.139521 .000217* .320463 .481774
.505690 .623933 .329677 .956755 .049972*

192
TAB. 5.25 Teste LSD para a variável resistência à flexão das amostras de 6 a 10. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (flexão.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{6} 324.6723
{7} 329.3804
{8} 315.1137
{9} 344.4291
{10} 288.6661
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.033424* .000013* .740753 .973217 .505690
.863538 .735594 .477822 .189279
.051281 .000031* .616894 .836491 .623933 .863538
.616665 .591272 .140695
.017700* .000006* .984186 .758350 .329677 .735594 .616665
.310943 .353648
.160270 .000328* .301780 .454868 .956755 .477822 .591272 .310943
.047234*
.000796* .000000* .324499 .197546 .049972* .189279 .140695 .353648 .047234*
De acordo com o teste é possível notar a partir dos dados apresentados nas
TAB. 5.24 e 5.25 que o valor obtido para a tensão média (σméd) da amostra 80 ZT
apresentou diferenças significantes com relação aos valores de resistência obtidos
para todas as outras amostras. A tensão média (σméd) para a amostra 80 ZM não
apresentou diferença significante da amostra 100 ZM, mas foi superior ao valor
encontrado para a 15 ZM. A tensão média (σméd) encontrada para a alumina (A) só
apresentou diferença significante com o valor obtido para amostra 80 ZT, indicando
que a alumina apresentou uma boa resistência à flexão.
As tensões médias (σ méd) e as tensões para 50 % de probabilidade de fratura
(σ 0,5) apresentadas na TAB. 5.22, mostraram seus maiores valores para as
amostras com 80 % de zircônia, tanto para as que contém zircônia ZT como para as
que contém zircônia ZM.
Durante a retífica das amostras, foi observado uma série de problemas no
equipamento, a retificadora mostrou um desempenho cada vez pior, comprovando
que a retificadora passou por um processo de deterioração. No princípio suspeitou-
se também do material que estava sendo estudado até que se constatou a pane da
máquina. Apesar do equipamento apresentar esse problema foram obtidos
resultados válidos que serviram para uma comparação entre as diferentes
composições, uma vez que o problema da retificadora se manteve o mesmo durante
todo o processo.
O número de defeitos tende a ser maior próximo à superfície das amostras já
que a contaminação pode ser facilmente introduzida durante vários estágios do

193
processamento. A resistência mecânica do material com superfície retificada é maior
do que a do material de superfície sinterizada. A causa do aumento da resistência
mecânica pela retificação é a remoção dos severos defeitos presentes na superfície
sinterizada, que são substituídos entretanto por novos defeitos menos severos
introduzidos pela própria retificação. O processo de retífica pode alterar o tamanho
do defeito crítico. O tamanho do defeito introduzido pela retificação é função da
interação entre rebolo e amostra, isto é, depende da severidade da retificação e de
parâmetros do material, como dureza e resistência ao desgaste.
A retificação com a capacidade de promover a transformação das partículas de
zircônia na região da superfície pode ser usada para introduzir tensões superficiais
compressivas que aumentam a resistência à flexão das cerâmicas tenacificadas com
zircõnia consideravelmente (Lino, U. R. A., 1982; Borsa, C. E., 1991; Tuan, W. H. &
Kuo, J. C., 1999). Por outro lado, se altas tensões são desenvolvidas durante o
processo de retificação, ou seja, se a retífica é severa, existe a possibilidade de se
introduzir defeitos superficiais profundos que agem como concentradores de tensões
e podem influenciar de maneira negativa na resistência mecânica e no módulo de
Weibull se o seu maior comprimento exceder a espessura da camada superficial
compressiva introduzida pela retificação das amostras. Alguns trabalhos também
mostram que o efeito do endurecimento superficial é desprezível devido ao
desenvolvimento de temperaturas localmente durante o processo de retificação
(Oblak, C. et al., 2004). Valores elevados para o coeficiente de Weibull “m” são
desejáveis uma vez que está relacionado com a confiabilidade do cerâmico. De
acordo com o trabalho de Ban, S. & Anusavice, K. J. 1990 e Oblak, C. et al., 2004,
valores para “m” na faixa de 5 a 15 são aceitáveis para cerâmicas dentárias.
É possível verificar que as tensões (σ 0,5 ) e (σ méd) para as amostras ZM foram
inferiores as obtidas para as amostras ZT com mesma composição. As amostras
ZM apresentam menor dureza que as amostras ZT como pode ser visto na TAB.
5.18. Além disso, estes resultados podem indicar falha no reforço, presença de
defeitos introduzidos durante o seu processamento e o mais significativo em função
da deteorização do equipamento utilizado, seria a geração de defeitos superficiais
críticos no processo de usinagem para a retificação de algumas amostras, como
descrito anteriormente.

194
Os diagramas de Weibull estão apresentados nas FIG. 5.37 a FIG. 5.45. O
diagrama de Weibull apresentou duas regiões bem definidas para as amostras
sinterizadas 100 ZT, 80 ZT, 100 ZM, 80 ZM e 15 ZM, o que sugere a existência de
duas populações de defeito crítico controlando a resistência mecânica (defeitos
oriundos do processamento e defeitos oriundos da retificação como discutido
anteriormente).
Para as outras amostras, o diagrama de Weibull sugere a atuação de um
mesmo tipo de defeito controlador da resistência.
y = 2,2371x - 13,694R2 = 0,921
y = 6,3099x - 34,856R2 = 0,9805
-5-4-3-2-1012
4,7 4,9 5,1 5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.37 Diagrama de Weibull para amostra 100 ZT.
y = 12,889x - 69,634R2 = 0,9695
y = 3,1807x - 19,808R2 = 0,9806
-5-4-3-2-1012
4,7 4,9 5,1 5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.38 Diagrama de Weibull para amostra 80 ZT.

195
y = 5,3727x - 30,995R2 = 0,9418
-5-4-3-2-1012
4,7 4,9 5,1 5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.39 Diagrama de Weibull para amostra 21 ZT.
y = 5,6529x - 33,403R2 = 0,9208
-5-4-3-2-1012
4,7 4,9 5,1 5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.40 Diagrama de Weibull para amostra 10 ZT.
y = 5,8284x - 34,124R2 = 0,9827
-5-4-3-2-1012
4,7 4,9 5,1 5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.41 Diagrama de Weibull para amostra 5 ZT.
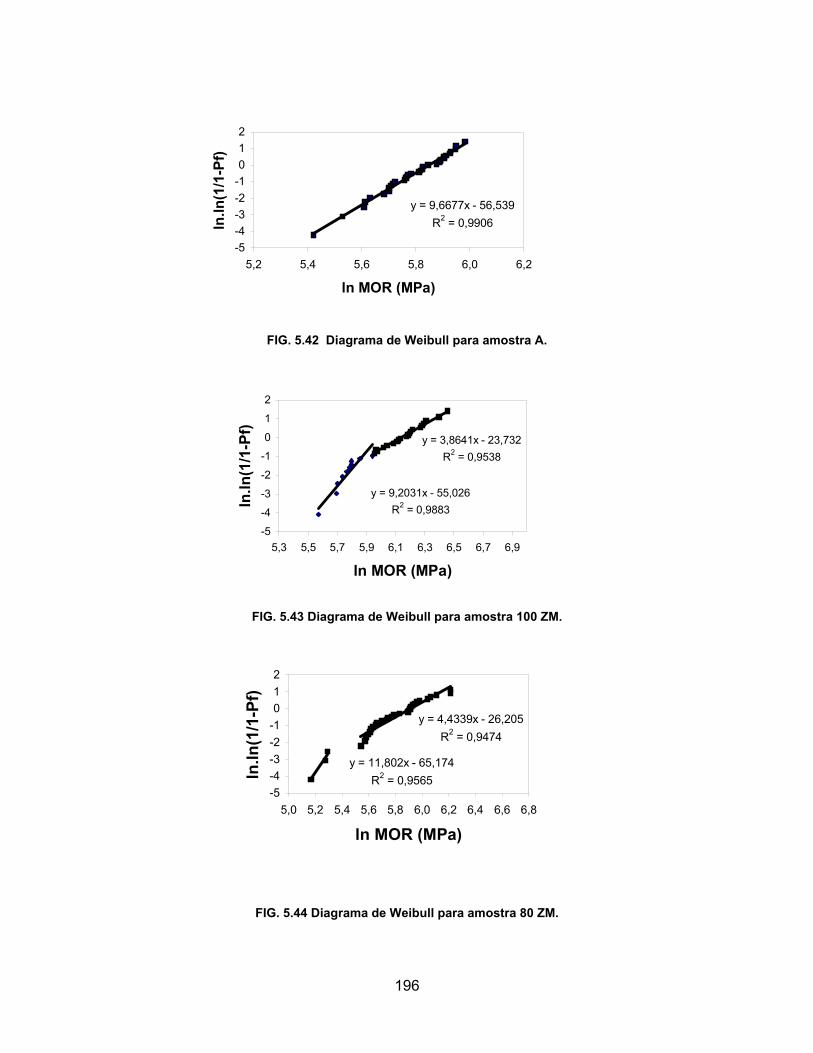
196
y = 9,6677x - 56,539R2 = 0,9906
-5-4-3-2-1012
5,2 5,4 5,6 5,8 6,0 6,2
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.42 Diagrama de Weibull para amostra A.
y = 3,8641x - 23,732R2 = 0,9538
y = 9,2031x - 55,026R2 = 0,9883
-5
-4
-3
-2
-1
0
1
2
5,3 5,5 5,7 5,9 6,1 6,3 6,5 6,7 6,9
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.43 Diagrama de Weibull para amostra 100 ZM.
y = 11,802x - 65,174R2 = 0,9565
y = 4,4339x - 26,205R2 = 0,9474
-5-4-3-2-1012
5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.44 Diagrama de Weibull para amostra 80 ZM.

197
y = 4,9407x - 29,101R2 = 0,9846
-5
-4
-3
-2
-1
0
1
2
5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4
ln MOR (MPa)
ln.ln
(1/1
-Pf)
FIG. 5.45 Diagrama de Weibull para amostra 15 ZM.
5.2.5.4 VALORES DE TENACIDADE À FRATURA
A TAB. 5.26 apresenta os valores de tenacidade à fratura obtidos para os
materiais analisados, utilizando o método de indentação e três diferentes equações
propostas na literatura. Os valores apresentados na TAB. 5.26 foram corrigidos de
acordo com a porosidade como descrito no item 4.4.3.2.
TAB. 5.26 Medidas de densidade e tenacidade à fratura (KIC).
K IC
(MPa . m1/2) K IC
(MPa . m1/2) K IC
(MPa . m1/2) Amostra Densidade
relativa (% teórica) Anstis, G. R. et al.,
1981 Casellas, D. et al.,
1997 Niihara, K.,
1983 100 ZT 99,46 ± 0,08 4,77 ± 0,24 7,16 ± 0,36 6,82 ± 0,19 80 ZT 99,60 ± 0,46 5,00 ± 0,32 7,49 ± 0,48 7,15 ± 0,27 21 ZT 99,55 ± 0,04 4,57 ± 0,69 6,85 ± 1,04 6,54 ± 0,96 15 ZT 99,52 ± 0,35 4,43 ± 0,54 6,65 ± 0,81 6,34 ± 0,74 10 ZT 99,35 ± 0,20 4,29 ± 0,38 6,43 ± 0,57 6,14 ± 0,53 5 ZT 99,29 ± 0,10 4,25 ± 0,60 6,38 ±0,89 6,09 ± 0,89
A 99,13 ± 0,13 4,06 ± 0,36 6,10 ± 0,54 5,81 ± 0,54 100 ZM 99,35 ± 0,92 5,08 ± 0,43 7,61 ± 0,65 7,26 ± 0,35 80 ZM 99,86 ± 0,06 5,22 ± 0,40 7,82 ± 0,65 7,46 ± 0,29 15 ZM 99,41 ± 0,38 4,63 ± 0,59 6,94 ± 0,88 6,62 ± 0,81
Os valores de dureza obtidos na TAB. 5.18 e os valores de KIC obtidos utilizando
a equação de Anstis (TAB. 5.26), em função do percentual em peso de zircônia,
para as amostras com zircônia Tosoh e zircônia Mel Chemicals são apresentados na
FIG. 5.46 e FIG. 5.47, respectivamente.

198
0 20 40 60 80 100
456789
1011
12131415161718
Pro
prie
dade
s M
ecân
icas
ZrO2 Tosoh (% peso)
HV (GPa) KIC (MPa.m1/2 )
FIG. 5.46 Dureza e tenacidade à fratura versus composição (zircônia Tosoh) utilizando equação de Anstis, G. R. et al., 1981.
0 20 40 60 80 100
456789
101112131415161718
Pro
prie
dade
s m
ecân
icas
ZrO2 Mel Chemicals (% peso)
HV (GPa) KIC (MPa.m1/2 )
FIG. 5.47 Dureza e tenacidade à fratura versus composição (zircônia Mel Chemicals) utilizando equação de Anstis, G. R. et al., 1981.
A observação destes gráficos sugerem que a dureza obedece uma regra das
misturas linear. A partir das FIG. 5.46 e FIG. 5.47 pode ser observado que existe

199
uma relação inversa entre os valores de dureza e tenacidade à fratura nas amostras
investigadas. Os valores de dureza diminuem enquanto os de tenacidade
aumentam gradualmente com o teor de zircônia, independentemente da equação
utilizada no cálculo de KIC. Uma vez que o grau de deformação pode ser
considerado como o da transformação, ou ainda, a dureza está inversamente
correlacionada com a facilidade de transformação, uma relação inversa entre dureza
e tenacidade à fratura pode ser esperada, resultado também verificado por Lin, J-D
& Duh, J-G., 2002 e Casellas, D. et al., 2003.
A adição de zircônia alterou o comportamento do compósito. Provavelmente, a
transformação de fase ocorre, e esta, tem uma função importante nos compósitos
alumina-zircônia.
Para a análise comparativa dos valores de KIC entre as diferentes amostras,
serão utilizados os valores obtidos com a utilização da equação proposta por Antis.
Pode ser verificado na TAB. 5.26 que a partir da zircônia pura que a adição de 20 %
de alumina na matriz de zircônia, promoveu os maiores valores de KIC, como para as
amostras 80 ZT (5,00 ± 0,32 MPa.m1/2) e 80 ZM (5,22 ± 0,40 MPa.m1/2), sendo estes
23% e 29% superiores ao valor de KIC obtido para a alumina pura (4,06 ± 0,36
MPa.m1/2), respectivamente. É possível observar ainda que estas amostras
apresentaram maior densidade.
Celli, A. et al., 2003 concluiram que os maiores valores de KIC para os
compósitos alumina-zircônia investigados foi obtido com as composições de 60 % e
80% ZrO2, sendo o valor de KIC para 80 % ZrO2 igual a 5,0 ± 0,2, utilizando a
equação de Anstis, semelhante aos encontrados neste trabalho.
A partir da análise do percentual de transformação induzida por tensão, ou
seja, a diferença entre as frações monoclínicas presentes nas amostras após
sinterização e após a fratura (TAB. 5.14 e TAB. 5.15), pode ser verificado que a
amostra 80 ZT apresentou uma percentagem maior de tranformação quando
comparada com a zirconia pura (100 ZT), e que as maiores percentagens foram
encontradas para as amostras contendo maior teor de zircônia. A transformação
induzida por tensão parece ser um importante mecanismo de aumento de
tenacidade no ATZ. O segundo mecanismo que age nestes compósitos, o
microtrincamento foi também investigado. O microtrincamento ao redor de
partículas transformadas de zircônia causam dispersão de energia e tenacificam o

200
material (Kumar, A. S. et al., 2004). Quanto maior a transformação de partículas de
zircônia, maior o aumento da tenacidade por microrincamento. Este mecanismo
opera no mesmo sentido da transformação de fase e é mais relevante nos
compósitos com maior teor de zircônia.
Além de maior retenção da fase tetragonal nos compósitos 80 ZT e 80 ZM,
outros mecanismos que promovem o aumento de tenacidade tais como o
ponteamento de trinca por grãos e a deflexão de trinca podem ser ativados quando
alumina é adicionada a Y-TZP como uma segunda fase dispersa, tal como nas
amostras 80 ZT e 80 ZM (compósitos ATZs).
É bem conhecido que a adição de alumina a zircônia tetragonal policristalina
estabilizada com ítria pode ser justificada pela vantagem na obtenção de um material
com maior módulo de elasticidade e microestrutura refinada, geralmente produzindo
cerâmicos com maior tenacidade. Glass, S. J. & Green, D.J., 1996 explicaram o
aumento de KIC a partir da zircônia pura para compósitos contendo 4 % vol. Al2O3,
como um processo de tenacificação dos contornos de grão da zircônia, devido a
presença de impurezas de alumina e conseqüentemente um aumento no modo de
fratura transgranular. Por outro lado, Lange, F. F., 1982a, observou que a adição de
alumina a zircônia causa um aumento no KIC, porém não tão alto quanto esperado
pela teoria. Este fato foi explicado pela retenção incompleta da fase tetragonal da
zircônia, devido a menores tamanhos de grão ou a diferenças de composição.
Lange enfatizou que o KIC destes materiais era fortemente influenciado pela
extensão da transformação da zircônia.
As amostras com 100 % de zircônia, apresentaram valores de KIC igual a 4,77 ±
0,24 MPa.m1/2 para 100 ZT e 5,08 ± 0,43 MPa.m1/2 para 100 ZM, sendo cerca de
18% e 25 % superiores ao encontrado para a alumina, respectivamente. O valor de
KIC encontrado para 100 ZT foi 11 % superior ao encontrado por Celli, A. et al., 2003
para zircônia pura (4,3 ± 0,1 MPa.m1/2). Valores superiores para zircônia Mel
Chemicals podem ser explicados pela melhor densificação do material (99,86%
teórica), menor tamanho inicial de partícula, estreita distribuição de tamanho de
partícula e maior área de superfície específica quando comparada com a zircônia
Tosoh.

201
A alumina pura neste trabalho apresentou KIC igual a 4,06 ± 0,39 MPa.m1/2,
sendo este valor semelhante ao encontrado por Celli, A. et al., 2003 (KIC = 4,2 ± 0,4
MPa.m1/2), utilizando a equação de Anstis.
Nos compósitos com matriz de alumina ou mesmo com um maior percentual de
alumina (ZTA), o aumento na quantidade de zircônia tetragonal, geralmente envolve
um considerável aumento do KIC. (Celli, A. et al., 2003). Para Tomaszewski, H.,
1988, o aumento de KIC é devido a transformação de fase da zircônia. Hori, S. et al.,
1985 justifica o aumento com a hipótese de outros mecanismos tais como a
formação de microtrincas ou a ramificação de trincas. No trabalho de Srdic, V. &
Radonjic, L., 1993, foram comparados compósitos obtidos a partir de diferentes
ciclos de sinterização. Os autores concluíram que o incremento na tenacidade à
fratura é devida principalmente a presença de uma microestrutura homogênea,
maior fração de zircônia tetragonal (t-ZrO2) e maior densidade. Lange, F. F., 1982c
observou aumento de KIC em compósitos de alumina-zircônia quando da adição de
zircônia tetragonal (dopada com 2 % mol de ítria). Por outro lado, a adição de fase
cúbica (zircônia com 7,5 % de ítria) reduziu a tenacidade à fratura destes
compósitos, o que foi explicado como resultado de tensões residuais associadas
com o diferencial de expansão térmica. O autor verificou também que a tenacidade
decresceu com o aumento do teor de fase monoclínica. Sendo a fase monoclínica
da zircônia a de maior volume, agiria como centro dissipador de energia elástica
transformando-a em energia de superfície de novas microtrincas, acomodando
assim esforços externos aplicados no material.
Segundo Celli, A. et al., 2003, em função das discrepâncias entre dados
experimentais e as diferentes interpretações do mesmo fenômeno, um entendimento
dos mecanismos de fratura atuantes nos compósitos requerem uma análise mais
detalhada.
Neste trabalho, foi possível observar a partir da alumina pura até 21 ZT ou
15ZM, que quanto maior o teor de zircônia adicionado na alumina, maior foi o valor
de KIC. Os valores de KIC para as amostras 21 ZT (4,57 ± 0,69), 15 ZT (4,43 ± 0,54)
e 15 ZM (4,63 ± 0,59) foram cerca de 13 %, 9 % e 14 % superiores ao valor
encontrado para a alumina pura.
Com o objetivo de verificar o efeito do fator ou tratamento composição na
variável tenacidade à fratura (KIC), a análise de variância (ANOVA) foi realizada com

202
auxílio do programa Statistica 6.0 e está apresentada na TAB. 5.27, como descrito
no item 4.2.
TAB. 5.27 Análise de variância (ANOVA) para médias de KIC. STAT. TABELA DA ANOVA
Resumo de todos os efeitos; projeto: (kIC.sta) 1 – COMPOSIÇÃO
Efeito
Grau de liberdade
do tratamento
Media dos quadrados
do tratamento
Grau de liberdade do erro
Média dos quadrados do erro
F
p-valor
1 9 5.148852 306 .277698 18.54116 .000000
A partir dos dados apresentados na TAB. 5.27, temos que o valor de Fcalculado
(18.54116) é maior que o valor de Ftabelado (1.88), então a hipótese Ho de médias
iguais deve ser rejeitada, ou seja, é possível afirmar que existem diferenças
significantes com nível de confiança de 95% entre pelo menos duas médias de KIC
encontradas.
Para determinar as diferenças significantes entre as médias, foi utilizado o teste
LSD, conforme já descrito no item 4.5. As TAB. 5.28 e TAB. 5.29 apresentam o
resultado do teste LSD para as amostras de 1 a 5 e 6 a 10, respectivamente. As
diferenças significantes entre as médias são apresentadas nas tabelas TAB. 5.28 e
TAB. 5.29 e são identificadas com um símbolo (*).
TAB. 5.28 Teste LSD para a variável KIC das amostras de 1 a 5. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (kic.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{1} 4.770004
{2} 5.004733
{3} 4.566418
{4} 4.433585
{5} 4.291161
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.083001 .129294 .012537* .000298* .000001* .000000* .023373* .000766* .412359
.083001
.001293* .000025* .000000* .000000* .000000* .602935 .103748 .023095*
.129294 .001293*
.345362 .036250* .001193* .000239* .000175* .000001* .646391
.012537* .000025* .345362
.230601 .019420* .004127* .000002* .000000* .209607
.000298* .000000* .036250* .230601
.293870 .087185 .000000* .000000* .026808*

203
TAB. 5.29 Teste LSD para a variável KIC das amostras de 6 a 10. STAT. TABELA DA ANOVA
Teste LSD; variável DENSIDADE (Kic.sta) Diferenças significantes após o teste de hipótese nula. PRINCIPAL EFEITO: COMPOSIÇÃO
COMPOSIÇÃO
{6} 4.165843
{7} 4.064655
{8} 5.075010
{9} 5.224964
{10} 4.639454
100 ZT {1} 80 ZT {2} 21 ZT {3} 15 ZT {4} 10 ZT {5} 5 ZT {6} A {7} 100 ZM {8} 80 ZM {9} 15 ZM {10}
.000001* .000000* .001193* .019420* .293870
.413864 .000000* .000000* .001699*
.000000* .000000* .000239* .004127*
.087185 .413864
.000000* .000000* .000381*
.023373* .602935 .000175* .000002* .000000* .000000* .000000*
.263461 .006530*
.000766* .103748 .000001* .000000* .000000* .000000* .000000* .263461
.000274*
.412359 .023095* .646391 .209607 .026808* .001699* .000381* .006530* .000274*
De acordo com o teste é possível notar a partir dos dados apresentados nas
TAB. 5.28 e TAB. 5.29 que o valor de KIC encontrado para a amostra 80 ZT foi
semelhante, ou seja, não apresentou diferenças significantes com os valores
encontrados para as amostras 100 ZT, 100 ZM e 80 ZM, porém foi superior ao KIC
encontrado para todas as outras amostras. Da mesma maneira, o valor de KIC para
a amostra 80 ZM foi semelhante apenas ao encontrado para as amostras 100 ZM e
80 ZT. A alumina (A) apresentou KIC semelhante ao KIC das amostras 10 ZT e 5 ZT,
porém foi inferior e apresentou diferenças significantes com todas as outras
amostras, mostrando que a adição de zircônia nos compósitos efetivamente
aumentou a tenacidade à fratura dos mesmos.
A formação da fase tetragonal tem dois efeitos benéficos: Um dos fatores é a
não formação de microtrincas no material devido a expansão volumétrica decorrente
da transformação do tipo martensítica. A formação em excesso de microtrincas
pode degradar sua resistência mecânica. Outro efeito é que quando ocorre a
transformação de fase T → M, a expansão resultante cria zonas de compressão que
dificultam a propagação de trincas a partir dos defeitos do material.
No trabalho de Casellas, D. et al., 1997, o aumento na tenacidade por adição de
zircônia na alumina foi considerado pequeno, uma vez que no máximo 5% de ZrO2
se transformou nas superfícies de fratura. De acordo com os autores, a razão para
este fato pode ser o pequeno tamanho médio das partículas de ZrO2, que não
permite a transformação de fase. Os autores encontraram maiores valores de
tenacidade à fratura quando o tamanho de grão das partículas de zircônia foram

204
aumentados com tratamentos térmicos de crescimento de grão. Casellas, D. et al.,
2003 verificou ainda que o percentual de transformação de fase aumentou de 5%
para mais de 9% após tratamento térmico a 1600 °C por 15 horas e para 11% após
tratamento térmico a 1600 °C por 30 horas nos compósitos ZTA.
De acordo com Casellas, D. et al., 2003, do ponto de vista da resistência ao
crescimento de trinca, a tenacidade à fratura do compósito ZTA pode ser vista como
a soma da tenacidade intrínseca da matriz de alumina e o incremento de tenacidade
devido a alguns mecanismos introduzidos pelas partículas de zircônia tais como: (1)
tenacidade por transformação, (2) microtrincamento relacionado à transformação, (3)
efeitos de tensões internas, como resultado das diferenças de expansão térmica
entre as fases que induz tensões residuais compressivas na matriz de alumina.
5.2.5.5 SISTEMA DE TRINCA
O indentador Vickers produz pelo menos dois tipos de sistemas de trincas, isto
é, um sistema de trinca radial/mediano ou um sistema de trinca Palmqvist. De
maneira geral, o material com baixa tenacidade apresenta sistema radial/mediano
enquanto que o material com alta tenacidade apresenta o sistema Palmqvist. De
fato, a maioria dos materiais irá exibir ambos sistemas de trincas dependendo da
carga aplicada. È possível verificar a dependência do comprimento da trinca com a
carga aplicada. Para a trinca Palmqvist temos que P/l é independente da carga, ou
seja, o comprimento da trinca varia linearmente com a carga e para sistemas de
trinca radial/mediana o comprimento de trinca varia com P2/3.
O trabalho de Bravo-Leon, A. et al., (2002) mostra a presença do sistema
Palmqvist para cerâmicas de zircônia tetragonal dopada com ítria (Y-TZP), utilizando
uma carga de 198 N. Outros autores também encontraram o sistema Palmqvist para
este mesmo material em cargas de indentação inferiores a 625 N, o que indicaria a
prevalência do sistema Palmqvist em cerâmicas Y-TZP.
Com o objetivo de verificar o sistema de trinca presente no material, foi utilizado
um procedimento simples, que é o polimento da camada superficial. No sistema
radial/mediano, a trinca irá permanecer sempre conectada ao vértice da indentação
e a trinca do sistema Palmqvist irá se destacar do vértice da indentação.
A FIG. 5.48 apresenta as micrografias obtidas por meio de microscópio ótico da
indentação Vickers com carga de 10 Kgf ou 98 N da amostra 100 ZT antes e depois

205
do polimento, onde é possível notar que as trincas se destacam do vértice da
indentação após o polimento.
(a) (b)
FIG. 5.48 Micrografias da amostra 100 ZT antes e depois do polimento com pasta de diamante de 15 μm.
Outra maneira de se verificar o sistema de trinca presente no material é através
da razão c/a. Se a razão for menor que 2,5 é do tipo Palmqvist. Se maior que 2,5 é
do tipo radial/mediano (Niihara, K. et al., 1983, Niihara, K., 1983; Wang, J. et al.,
2002).
A TAB. 5.30 apresenta os valores obtidos para a razão c/a e o valor médio dos
comprimentos de trinca (c) medidos no microscópio ótico com auxílio do analisador
de imagens para cada amostra.
TAB. 5.30 Medidas do comprimento médio de trinca e razão c/a.
Material Densidade aparente
(% teórica) c (μm) c/a
100 ZT 99,46 ± 0,08 117,32 ± 3,80 2,00 ± 0,07 80 ZT 99,60 ± 0,46 112,22 ± 4,92 2,06 ± 0,09 21 ZT 99,55 ± 0,04 130,25 ± 10,78 2,53 ± 0,21 15 ZT 99,52 ± 0,35 137,30 ± 10,82 2,68 ± 0,21 10 ZT 99,35 ± 0,20 137,88 ± 8,09 2,69 ± 0,15 5 ZT 99,29 ± 0,10 140,11 ± 13,47 2,75 ± 0,27
A 99,13 ± 0,13 146,31 ± 8,86 2,90 ± 0,17 100 ZM 99,35 ± 0,92 117,18 ± 6,30 1,97 ± 0,11 80 ZM 99,86 ± 0,06 110,98 ± 6,81 2,01 ± 0,08 15 ZM 99,41 ± 0,38 134,10 ± 10,89 2,58 ± 0,21
100 μm100 μm

206
As amostras 100ZT, 80ZT, 100ZM e 80ZM apresentam trincas do tipo Palmqvist
para a carga de indentação de 98 N. Todas as outras amostras apresentaram trincas
do sistema “half-penny”. Basu, D. & Sakar, B. K., 1996, determinaram a tenacidade
de alumina e ZTA (5 %, 10 %, 15 % e 20 % vol) pelo método de indentação e
também encontraram trincas do tipo “half-penny” para estes compósitos..
Para cada amostra, o valor de a (semi-diagonal da indentação) é
aproximadamente constante e desta forma, a razão c/a aumenta com o aumento do
comprimento da trinca (Wang, J. et al, 2002).
Este fato também pode ser observado na TAB. 5.30. Em testes de medida de
tenacidade por indentação, quanto maior a razão c/a, maior é o comprimento da
trinca e menor é a tenacidade à fratura da amostra analisada.
É possível observar que para trincas de menor comprimento, as quais foram
obtidas para as amostras 80 ZT e 80 ZM (TAB. 5.30), maiores são os valores de KIC
(TAB. 5.26), independentemente da equação utilizada para o seu cálculo, o que
permite concluir que a tendência observada para os valores de KIC para as amostras
deste trabalho não está relacionada com efeitos de curva-R, onde a resistência à
propagação da trinca aumentaria com o aumento do comprimento da trinca ou a
tenacidade aumentaria com o aumento do comprimento da trinca. Este resultado
também foi observado no trabalho de Bravo-Leon, A. et al., 2002 quando avaliaram
amostras nanocristalinas de zircônia tetragonal com baixo teor de ítria (1% e 1,5%).
5.2.5.6 USO DE EQUAÇÕES PARA DETERMINAÇÃO DO KIC PELO MÉTODO DE INDENTAÇÃO
A literatura mostra que dentre todos os métodos para medição da tenacidade em
cerâmicos, o método de indentação quando comparado com outros métodos,
apresenta os menores valores de KIC (Madruga, T. P. et al., 1986; Casellas, D. et al.,
1999).
De maneira geral, o emprego de diferentes métodos para medida da tenacidade
podem fornecer diferentes valores possivelmente devido a diferentes mecanismos
de trincamento. Por outro lado, o grande argumento para este fato pode ser a
utilização de diferentes equações. Desta forma, os valores de tenacidade dependem

207
significantemente do método utilizado e no caso do método de indentação da
equação utilizada para a obtenção do valor de KIC.
Na TAB. 5.26 é possível observar que existem diferenças entre os valores de KIC
obtidos pelo método de indentação em função da equação utilizada.
A partir da TAB. 5.26, foi possível verificar que para a alumina pura, a equação
de Anstis é a que fornece valor de KIC mais próximo dos valores encontrados na
literatura para este material. A equação de Casellas D. et al., 1997 e a equação de
Niihara, K., 1983 forneceram valores de KIC que são 50 % e 43 % superiores aos
valores obtidos para os mesmos materiais utilizando a equação de Anstis, G. R. et
al, 1981.
É importante observar ainda que para a zircônia pura e compósitos alumina-
zircônia, a equação proposta por Anstis apresenta valores inferiores ao encontrados
na literatura. Este fato confirma a necessidade de se indicar a equação utilizada
para determinação da tenacidade à fratura por indentação devido as discrepâncias
entre os valores encontrados.
No trabalho de Casellas et al., 1999, foi avaliada a tenacidade à fratura de
compósitos alumina-zircônia (ZTA) com 5 %, 15 % e 30 % de adição de zircônia em
alumina, utilizando cinco diferentes métodos: SENB, SEVNB, SCF, ISB e IM. Os
resultados mostraram que as medidas realizadas a partir do método IM foram
inferiores as encontradas para os outros métodos independentemente da
composição do ZTA. Uma das explicações dadas pelos autores para este fato foram
os parâmetros de ajuste nas correspondentes equações. O baixo KIC obtido em
amostras com tensões residuais produzidas por indentação, podem ser atribuídos ao
fato de que a equação proposta por Anstis et al., 1981, com os parâmetros A=0,016
± 0,004 e n=0,5, subestima o campo de tensões residuais.
Apesar da equação de Anstis ser largamente aplicada para estimativas da
tenacidade à fratura por indentação (Casellas, D. et al., 2003, Chen, Z. et al., 2004),
seus resultados são somente números médios calculados para ajustar os resultados
para uma ampla faixa de materiais. Desta forma, desvios podem ser esperados
para materiais específicos.
No caso da alumina, estes valores são utilizados e apresentam bons resultados.
Por outro lado, em cerâmicas a base de zircônia, estes parâmetros não levam a
valores atuais de tenacidade à fratura de materiais que apresentam transformação

208
de fase induzida por tensão, como no trabalho apresentado por Kaliszewski, M. S.,
1994.
Para cerâmicas a base de zircônia, o valor de A deve ser aumentado em pelo
menos 20 % para levar em conta a força motriz adicional da trinca devido a
transformação induzida por tensão. Desta forma, é proposto que o valor de A para o
ZTA seja maior do que os propostos por Anstis et al., 1981. Os autores propuseram
uma correção para o ZTA, onde A = 0,025, valor similar ao usado por
Ramachandran, N. & Shetty, D. K., 1991 em alumina e carbeto de silício
tenacificados com wiskers, no qual os autores encontraram A = 0,023.
Casellas, D. et al, 1997 estudaram três diferentes composições de ZTA e
utilizaram em seus cálculos de tenacidade à fratura os valores de 0,024 e 0,5 para
os parâmetros A e n, respectivamente.
De acordo com Sakai, M. & Bradt R. C., 1993 equações empíricas como a
equação de Anstis G. R. et al, 1981, só devem ser aplicadas a sistemas que
apresentem trincas induzidas por indentação do tipo medial/radiano ou “half-penny”.
Como neste trabalho o objetivo é avaliar a variação da tenacidade à fratura com
o teor de zircônia adicionado à alumina, a equação de Anstis parece ser mais
adequada para se obter uma comparação entre a alumina pura e os compósitos
formados a partir de adições de zircônia à alumina, uma vez que o valor de KIC para
a alumina obtido por esta equação foi o mais próximo dos valores encontrados na
literatura.
5.2.5.7 ANÁLISE DAS SUPERFÍCIES DE FRATURA
A FIG. 5.49 apresenta as superfíces de fratura das amostras analisadas neste
trabalho após ensaio de flexão em quatro pontos.
Todas as fraturas tiveram início na superfície sob tração e se localizou entre os
roletes inferiores. As amostras testadas falharam devido a presença de micro
defeitos e trincas superficiais produzidas no polimento. Não foram encontrados
defeitos grosseiros, tais como aglomerados ou grandes poros. Imagens da
superfície de fratura da alumina contendo poros esféricos, e os baixos valores de
resistência à flexão encontrados para esta amostra, confirmaram que os poros agem
como defeitos que limitam a resistência, como pode ser visto na FIG. 5.49(g).

209
(a) (b) (c)
(d) (e) (f)
(g) (h) (i)
(j)
FIG. 5.49 Micrografias eletrônicas de varredura das superfícies de fratura (a) 100 ZT, (b) 80 ZT, (c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h) 100 ZM, (i) 80 ZM, (j) 15 ZM.
poros

210
As amostras 100 ZT,100 ZM, 80 ZT e 80 ZM apresentaram uma superfície de
fratura plana, todavia, para as outras amostras investigadas, foi observado uma
superfície de fratura um pouco mais rugosa. Este fato pode ser atribuído a trincas
caminhando em diferentes planos, tanto de forma intergranular como transgranular,
devido a deflexão e ponteamento da trinca pela presença das partículas equiaxiais
de ZrO2. Assim, as trincas nestes compósitos estariam mais sujeitas a estes
mecanismos (deflexão e ponteamento), os quais estariam contribuindo para o
aumento da resistência à fratura.
As observação das superfícies de fratura mostrou que o modo de fratura
predominante nas amostras foi o modo intergranular. A fratura transgranular foi
facilmente detectada para as amostras A, 5 ZT, as quais apresentam os maiores
tamanhos de grão de alumina e os menores valores de resistência à flexão,
sugerindo que a resistência é dependente do tamanho de grão da alumina.
No trabalho de Choi, S. R. & Bansal, N. P., 2004, o efeito das tensões residuais
e/ou microtrincamento devido a diferença entre os coeficientes de expansão térmica
da alumina e da zircõnia foram examinados por ciclagem térmica em compósitos
contendo zircônia totalmente estabilizada com 10 % mol de ítria (10-YSZ) e
reforçada com 0 - 30% mol de alumina. Os autores concluíram que este efeito foi
desprezível uma vez que não afetou a resistência à flexão dos compósitos
investigados.
O microtrincamento provavelmente associado a uma maior fração de partículas
de zircõnia na fase tetragonal transformadas em monoclínica sob a influência de
uma tensão aplicada, foi observado nas superfíces de fratura (FIG. 5.49) para as
amostras com maior teor de zircônia (100 ZT, 80 ZT, 21 ZT, 100 ZM, 80 ZM) e está
indicado com setas brancas, mostrando que este mecanismo pode aumentar a
tenacidade nestes compósitos.
5.2.5.8 RELAÇÃO ENTRE GEOMETRIA DA TRINCA E TENACIDADE À FRATURA
O modo de fratura intergranular e/ou transgranular foi investigado para cada
amostra a partir das imagens obtidas no MEV das trincas introduzidas em suas
superfícies polidas e termicamente atacadas (FIG 5.50). Os resultados mostraram

211
que o modo de fratura predominante nas amostras analisadas neste trabalho foi o
modo intergranular.
Como visto anteriormente, o valor de KIC das amostras analisadas neste trabalho
aumentou com o aumento do teor de zircônia no compósito, indicando que o
mecanismo de tranformação por tensão das partículas de zircônia tem importante
papel no comportamento dos compósitos alumina-zircônia.
Para cada amostra analisada, o esquema de um segmento típico de trinca
emergindo dos vértices da indentação é apresentado na FIG. 5.51. É possível
verificar que a forma da trinca é dependente da amostra e que a não linearidade das
trincas é comum a todas as amostras. As trincas se apresentam bem diferentes
umas das outras, especialmente as amostras com um maior teor de alumina, sendo
necessário uma análise mais detalhada para evidenciar diferenças em suas
características geométricas.
As amostras com maior percentual de alumina, quando analisadas de A até 21
ZT e de A até 15 ZM, apresentaram um decréscimo no tamanho médio de grão de
alumina com o aumento do teor de zircônia (TAB. 5.16). Para estas amostras, a
tortuosidade (ângulos de desvio) do caminho da trinca diminuiu com o decréscimo
do tamanho de grão da alumina na amostra. Desta forma, as trincas tendem a
seguir os contornos de grão da alumina. Na FIG. 5.51 é possvel verificar que o
comprimento médio dos segmentos lineares diminui e que o número de desvios
aumenta com o decréscimo do tamanho de grão da alumina (por ex: de A até 21
ZT). Este fato confirma que o mecanismo de deflexão da trinca predomina. É
razoável supor que a deflexão da trinca e ponteamento em grãos de zircônia seja
uma das razões para o aumento da tenacidade à fratura (Miyazaki, H. et al., 2004,
Casellas, D. et al., 2003). Como pode ser visto na TAB. 5.26, a tenacidade à
fratura aumenta com o decréscimo do tamanho de grão da alumina ou com o
aumento do teor de zircônia no compósito. Assim, para as amostras com maior teor
de alumina, temos que para um mesmo comprimento total de trinca, o caminho total
da trinca (energia dissipada na formação de novas superfícies) aumenta com o
aumento do teor de zircônia no compósito. Para a amostra de alumina pura (A) as
trincas são caracterizadas por um caminho tortuoso, com pequeno número de
desvios e com maiores ângulos, denotando a eficiência do mecanismo de deflexão
de trinca.

212
(a) (b) (c)
(d) (e) (f)
(g) (h) (i)
(j)
FIG. 5.50 Micrografias eletrônicas de varredura das trincas induzidas por indentação nas superfícies das amostras: (a) 100 ZT, (b) 80 ZT, (c) 21 ZT, (d) 15 ZT, (e) 10 ZT, (f) 5 ZT, (g) A, (h)
100 ZM, (i) 80 ZM, (j) 15 ZM.

213
Analisando agora as amostras com maior teor de zircônia, 100 ZT, 80 ZT, 100
ZM e 80 ZM, são caracterizadas por um perfil de trincas aproximadamente linear,
onde pode ser visto que a tortuosidade das trincas não muda muito apesar da
variação da composição e da microestrutura (FIG. 5.51). Este fato sugere que a
fratura intergranular pode não ser o principal mecanismo de fratura para estas
amostras. Na literatura, a fratura intergranular tem sido reportada como o principal
modo de fratura para a zircônia tetragonal, como observado neste trabalho. De
acordo com o trabalho de Kaliszewski, M. S. et al., 1994, a transformação
martensítica não tem um papel muito significante no aumento da tenacidade em
amostras de zircônia. Marshall, D. B. et al., 1994, propuseram que a morfologia de
fratura mista para Y-TZP seria resultado da fratura intergranular de grãos tetragonais
e da fratura transgranular de grãos cúbicos.
Para as amostras 80 ZT e 80 ZM é possível assumir um mecanismo de fratura
que cause elevados valores de KIC sem introduzir grandes variações na geometria
da trinca. Glass, S. J. & Green, D. J., 1996, encontraram que pequenas adições de
alumina (4%) na zircônia tenacificam os contornos de grão da zircônia, induzindo
fratura transgranular e assim, um aumento no kIC. A fratura transgranular pode
ocorrer, por exemplo, e a deflexão de trinca é provavelmente um processo
secundário (somente uma pequena diminuição do comprimento médio dos
segmentos lineares e um pequeno aumento do número médio de desvios é
detectado com a redução do tamanho de grão da zircônia na amostra 80% ZT
quando comparada com a 100 ZT, por exemplo). Isto indica que outros importantes
mecanismo de fratura, tal como a transformação martensítica e o microtrincamento
têm uma função significante no aumento da tenacidade para os compósitos com
maior teor de zircônia. O microtrincamento que está associado a transformação das
partículas de zircônia tetragonais causa dispersão de energia e aumenta a
tenacidade do material. Quanto maior a transformação das partículas de zircônia,
maior o aumento da tenacidade por microtrincamento. O microtrincamento opera no
mesmo sentido da transformação de fase e parece ser mais relevante nos
compósitos com maior teor de zircônia, o que foi verificado com a análise da
extensão da transformação de fase induzida por tensão.

214
100 ZT
80 ZT
21 ZT
15 ZT
10 ZT
5 ZT
A
100 ZM
80 ZM
15 ZM
FIG. 5.51 Exemplo dos caminhos de trinca típicos para os materiais examinados.
5.2.5.9 RESISTÊNCIA AO DESGASTE
A TAB. 5.31 apresenta os valores de resistência ao desgaste aferidos para as
amostras analisadas com base na literatura e utilizando a equação proposta por
Evans, A. G. & Marshall, T. R., (1976), já descrita anteriormente no item 3.85. Esta
equação foi utilizada por outros autores como no trabalho de Baldon, J. G., (1985) e
Duailibi, J. F., (1999), para avaliar a resistência ao desgaste de materiais cerâmicos.
De acordo com os resultados apresentados na TAB. 5.31, a resistência ao
desgaste partindo da amostra A até 80 ZT, aumentou com o aumento do teor de
zircônia no compósito e com o aumento da tenacidade à fratura. O maior valor de
resistência ao desgaste foi obtido para as amostras 80 ZT e 80 ZM, as quais

215
apresentam a maior densidade e menor porosidade dentre as amostras
investigadas.
TAB. 5.31 Valores de densidade, dureza, KIC e resistência ao desgaste.
K IC (MPa. m1/2) Amostra
Densidade relativa
(% teórica)
HV (GPa) Anstis, G. R. et al.,
1981
Resistência ao desgaste
(KIC) 3/4 . (HV) 1/2
100 ZT 99,46 ± 0,08 13,20 ± 0,48 4,77 ± 0,24 11,73 80 ZT 99,60 ± 0,46 15,44 ± 0,46 5,00 ± 0,32 13,14 21 ZT 99,55 ± 0,04 17,14 ± 0,38 4,57 ± 0,69 12,94 15 ZT 99,52 ± 0,35 17,38 ± 0,23 4,43 ± 0,54 12,73 10 ZT 99,35 ± 0,20 17,41 ± 0,21 4,29 ± 0,38 12,44 5 ZT 99,29 ± 0,10 17,48 ± 0,76 4,25 ± 0,60 12,38
A 99,13 ± 0,13 17,53 ± 0,52 4,06 ± 0,36 11,98 100 ZM 99,35 ± 0,92 12,78 ± 0,34 5,08 ± 0,43 12,01 80 ZM 99,86 ± 0,06 14,32 ± 0,77 5,22 ± 0,40 13,07 15 ZM 99,41 ± 0,38 16,98 ± 0,38 4,63 ± 0,59 13,01
O desgaste de cerâmicos é uma função complexa da microestrutura, tamanho
de grão e forma, os quais influenciam na dureza e tenacidade (Hsu, S. M. & Shen,
M., 2004). De acordo com os resultados de Baldon, J. G., 1985, quanto maior os
valores de dureza e a tenacidade à fratura, maior a resistência ao desgaste do
material.
Na TAB. 5.31 é possível verificar ainda que a alumina pura (A) e a zircônia pura
(100 ZT) apresentaram boa resistência ao desgaste apesar da superior tenacidade à
fratura e resistência à flexão encontradas para a amostra 100 ZT, resultado também
encontrado por Medvedovski, E., 2001.
O comportamento ao desgaste do ZTA foi avaliado experimentalmente por K.
Cherif, K. et. Al., 1996. De acordo com os autores, o principal mecanismo de
desgaste para a alumina e o ZTA foi identificado como abrasão e arrancamento por
trincamento inter e transgranular. Foi observado ainda que a resistência ao
desgaste aumentou com o teor de zircônia adicionado até um valor ótimo. Acima de
um valor ótimo, a transformação espontânea da zircônia criou microtrincas na matriz,
reduzindo a restência ao desgaste. Uma vez que a presença de zircônia tetragonal

216
efetivamente aumenta a resistência do material, a microestrutura deve ser
controlada durante a fabricação no sentido de evitar a transformação espontânea (t
→ m).
A resistência ao desgaste é dependente da tenacidade à fratura (Affatato, S., et
al., 1999a , Affatato, S., et al., 1999b, Affatato, S., et al., 2001a, Affatato, S., et al.,
2001b, Xu, C. et al., 2001), então compósitos alumina-zircônia com maior teor de
zircônia e maior tenacidade à fratura como para as amostras 80 ZT and 80ZM,
apresentam maior resistência ao desgaste do que a alumina ou zircônia puras
(TAB.5.31). Comportamento semelhante foi observado por outros autores quando
estudaram a influência das propriedades do material no desgaste da zircônia, como
nos trabalhos de Serra, E. et al., 2002; Yang, C.C. T. & Wei, W. C. J., 2000. Os
autores observaram que a maior tenacidade à fratura e menor tamanho de grão
podiam compensar a baixa dureza para este material, sendo a maior resistência ao
desgaste atribuída à maior tenacidade à fratura quando comparada à alumina.
Affatato, S. et al., 2001a, investigaram o desgaste de dois compósitos alumina-
zircônia e compararam os resultados com alumina e zircônia puras. Os autores
verificaram que o compósito contendo 60 % de zircônia apresentou melhor
resistência aos desgaste do que a zircônia pura.
A rugosidade superficial e a porosidade obtidas a partir de diferentes processos
de acabamento podem podem levar a diferentes taxas de desgaste (Piconi, C. &
Maccauro, G., 1999).
Diferentes componentes cerâmicos (válvulas e outros), bem como as próteses e
cerâmicas dentárias estão sujeitos a fricção contínua de vários materiais, muitas
vezes em ambientes fortemente corrosivos. Cerãmicos usados para proteção ao
desgaste são densas e de baixa porosidade, podendo ser a alumina, a zircônia e
outros óxidos.
5.2.5.10 MICROSCOPIA ELETRÔNICA DE TRANSMISSÃO
Observações em microscopia eletrônica de transmissão (MET) indicaram um
tamanho de grão similar ao obtido com a utilização de imagens do MEV e o método
das interseções lineares, indicando que não existem subgrãos como pode ser visto

217
na FIG.5.52 (a). As microtrincas causadas pela transformação de fase da zircônia
tetragonal em monoclínica não foram facilmente observadas com MET.
As observações em MET indicam que o contorno de fase Al203-ZrO2 é
caracterizado por contato direto dos cristais, como pode ser visto na FIG. 5.52(a).
(a) (b)
(c) (d)
FIG. 5.52 Imagem em campo claro(MET) de (a) ZrO2-Al2O3(80 ZT) onde alumina é o grão escuro
e a zircônia é o claro; Figura de difração de : (b) Al2O3, eixo de zona ⎥⎦⎤
⎢⎣⎡ −
112 ; (c) Y-ZrO2-t , eixo de
zona ⎥⎦⎤
⎢⎣⎡ −
100 ; (d) Y-ZrO2-m, eixo de zona [ ]001 .
220
200
214
202

218
Padrões de difração por elétrons não apresentaram qualquer anel difuso
(FIG.5.52b, FIG.5.52c e FIG.5.52d), indicando a ausência de fase vítrea entre grãos
ou em pontos triplos, estando este resultado de acordo com as observações de
Ross, I. M., et al., 2001.
Espectroscopia dispersiva de raios-X (EDS) foi realizada no interior dos grãos de
alumina (FIG. 5.53) e zircônia (FIG. 5.54).
FIG. 5.53 Espectro de EDS para o grão de alumina.

219
FIG. 5.54 Espectro de EDS para o grão de zircônia.
Não foram observadas diferenças entre os espectros dos interiores e dos
respectivos contornos de grão. Investigações dos contornos de grãos e pontos
triplos foram realizadas e não foi detectada a presença de ítria em quantidades
notáveis, de acordo com o trabalho de Acalá, J. & Anglada, M., 1997. Ross, I. M., et
al., 2001, observaram a partir de análises com EDS um ligeiro aumento na
concentração de ítria nos contornos de grãos adjacentes. Os autores também
encontraram traços de alumínio nos contornos de grão, mas não no interior dos
grãos.

220
6 CONCLUSÕES 6.1 CONCLUSÕES
1) A análise dos compósitos alumina-zircônia mostrou que a adição de
zircônia produziu compósitos com maiores densidades, maior resistência
à flexão e maior tenacidade à fratura. Os compósitos alumina-zircônia
podem atingir resistência à flexão 93% e tenacidade à fratura 29%
superiores quando comparados com a alumina pura. Estes resultados
mostram que sob certas condições, componentes cerâmicos resistentes
ao desgaste podem ser preparados. Foi verificado ainda que existe uma
dependência inversa entre os valores de KIC e os valores de dureza.
2) Os compósitos alumina-zircônia com maior teor de zircônia investigados
neste trabalho, contendo 80 % peso de zircônia e 20 % de alumina,
demonstraram ser um material mais adequado para a utilização em
pilares cerâmicos para implantes dentários do que os óxidos puros
atualmente em uso, uma vez que apresentaram maior resistência à flexão
e tenacidade à fratura. Ainda, a dureza obtida para estes compósitos
pode ser adequada para a obtenção da forma final por usinagem dos
pilares cerâmicos.

221
3) A zircônia pura e os compósitos com 80 % peso de zircônia apresentaram
sistema de trinca do tipo Palmqvist sob carga de indentação de 98,1 N. A
alumina e os compósitos com menor teor de zircônia apresentaram
trincas do sistema radial/mediano ou “half-penny”.
4) O uso de diferentes métodos e as diferentes equações para determinação
dos valores de tenacidade à fratura por indentação mostraram diferenças
de até 50 % nos valores de KIC obtidos para uma mesma amostra.
Assim, a identificação do método e da equação utilizada para a
determinação da tenacidade dos cerâmicos é fundamental evitando
comparações que podem levar a conclusões errôneas.
5) O aumento de tenacidade está relacionado com a presença de uma
microestrutura homogênea, maior densidade e maior fração de zircônia
tetragonal disponível para transformação, indicando que este aumento é
principalmente devido ao mecanismo de tenacificação por transformação.
Como segundo mecanismo que age nestes compósitos, o
microtrincamento também foi discutido e foi observado nas superfícies de
fratura da zircõnia e compósitos com maior teor de zircônia. A resistência
à flexão aumentou com o refinamento da microestrutura, sendo mais
sensível a medidas de tamanho de grão do que a tenacidade.
6) A análise das superfícies de fratura e dos caminhos das trincas mostrou
que o modo de fratuta intergranular foi predominante. O modo
transgranular foi observado para a alumina pura e compósitos com 5 de
zircônia.
7) A análise de raios-X mostrou que as fases cristalinas presentes nos
óxidos puros e nos compósitos foram a α-alumina, zircônia tetragonal e
zircônia monoclínica. A fração de zircônia tetragonal retida após a
sinterização foi de aproximadamente 90% para todas as amostras e
aumentou com o aumento de zircônia. A quantidade de zircônia
monoclínica presente nas amostras após a fratura aumentou, mostrando
que a transformação induzida por tensão é um importante mecanismo de
aumento de tenacidade nestes compósitos.

222
6.2 SUGESTÕES
1) Os compósitos alumina-zircônia com maior teor de zircônia investigados
neste trabalho, demonstraram ser um material adequado para a utilização
em pilares cerâmicos para implantes osseointegrados. Todavia, outros
trabalhos incluindo testes clínicos com estes compósitos devem ser
realizados a fim de se obter resultados mais confiáveis de sua
performance nesta aplicação odontológica específica.
2) As amostras foram conformadas na forma de barras retangulares por
prensagem uniaxial. Como sugestão, pode se estudar a obtenção de
moldes flexíveis e a prensagem isostática para obtenção da forma final
dos pilares para implantes.
7 REFERÊNCIAS BIBLIOGRÁFICAS ABSI J. & GLANDUS J.C., Numerical separation of bi-modal strength
distributions, Journal of the European Ceramic Society 22, p. 593-601, 2002.
ADELL, R., LEKHOLM, U., BRÅNEMARK, P. I. A 15 year study of
osseointegrated implants in the treatment of edentulous jaws. Int. Journal
Oral Surgery, v.10, p. 387-416, 1981.
AFFATATO, S., FERNADES, B., TUCCI, A., ESPOSITO, L., TONI, A Isolation and
morphological characterisation of UHMWPE wear debris generated in vitro.
Biomaterials, 22, p. 2325-2331, 2001a.
AFFATATO, S., GOLDONI, M., TESTONI, M., TONI, A. Mixed-oxides prosthetic
ceramic ball heads. Part 3: effect of the ZrO2 fraction on the wear of
ceramic on ceramic hip joint prostheses. A long term in vitro wear study.
Biomaterials. 22, p. 717-723, 2001b.
AFFATATO, S., TESTONI, M., CACCIARI, G.L.et al .Mixed oxides prosthetic
ceramic ball heads. Part 1: effect of the ZrO2 fraction on the wear of
ceramic on polyethylene joints. Biomaterials, 20, p.971-975, 1999a.
AFFATATO, S., TESTONI, M., CACCIARI, G.L.et al. Mixed-oxides prosthetic
ceramic ball heads. Part 2: effect of the ZrO2 fraction on the wear of

223
ceramic on ceramic joints. Biomaterials. 20, p. 1925-1929, 1999b.
ALCALÁ, J. & ANGLADA, M. High-temperature crack growth in Y-TZP. Materials
Science and Engineering A, 232 (1997) 103-109.
ALCALÁ, J. & ANGLADA, M. High-temperature crack growth in Y-TZP. Materials
Science and Engineering A, 232, 103-109, 1997.
ANDERSON, B., SCHÄRER, P., SIMION, M. et al. Ceramic implant abutments
used for short-span fixed partial dentures: A prospective 2-year
multicenter study. The International Journal of Prosthodontics, v. 12, p. 318-
324, 1999.
ANDERSSON, M. & ODÉN, A. A new all-ceramic crown. Acta Odontol. Scand., v.
51, p. 60-64, 1993.
ANSTIS, G. R., CHANTIKUL, P., LAWN, B. R. MARSHALL, D. B. A critical
evaluation of indentation techniques for measuring fracture toughness: I
direct crack measurements. J. of American Ceramic Society. V. 64 (9), p. 533-
538, 1981.
ANTON, R. J. & SUBHASH, G. Dynamic vickers indentation of brittle materials.
Wear, v. 239. p. 27-35, 2000.
ANUSAVICE, K.J. Materiais Dentários. Editora Guanabara Koogan 10ª Edição,
cap. 26, 1998.
ARDLIN, B.I. Transformation-toughened zirconia for dental inlays, crowns and
bridges: chemical stability and effect of low-temperature aging on flexural
strength and surface structure, Dental Materials 18 (8), p. 590-595, 2002.
ASKELAND, D. r. The science and engineering of materials. Chapman & Hall, 2ª
edição, London, 1992.
ASTM C1161-90, Standard Test Method for Flexural Strength of Advanced
Ceramics at Ambient Temperature, p. 329-335, 1990.
ASTM C1322-96A Standard Practice for Fractography and Characterization of
Fracture Origins in Advanced Ceramics. p. 1-42, 1996.
ASTM C1327-99. Standard test method for vickers indentation hardness of
advanced ceramics. p.1-8, 1999.
ASTM C-1421-99. Standard test method for determination of fracture toughness
of advanced ceramics at ambient temperature. p. 1-32, 1999.

224
ASTM C373-88, Standard Test Method for water absorption, bulk density,
apparent porosity and apparent specific gravity of fired whiteware
products. P. 115-116, 1988.
ASTM E-112-96. Standard test methods for determining average grain size. p.
297-322, 1996.
ASTM F-1873-98. Standard specification for high-purity dense yttria tetragonal
zirconium oxide polycrystal(Y-TZP) for surgical implant applications.
p.1349-1351, 1998.
ASTM F-603,00, Standard specification for high-purity aluminium oxide for
medical application. p. 103-105, 2000.
BALDON, J. G., Cutting tool materials: mechanical properties-wear resistance
relationships, Am. So. Lubricat. Eng. Trans., 29, p. 347-352, 1985.
BAM, S. ANUSAVICE, K. J. Influence of test method on failure stress of brittle
dental materials. J. Dent. Res., 69, p. 1791-1799, 1990.
BASU, D., SARKAR, B. K., Toughness determination of zirconia toughened
alumina ceramics from growth of indentation-induced cracks, J. Mater.
Res., v. 11, n. 12, p. 3057-3062, 1996.
BECHER, P. F., ALEXANDER, K. B., WARMICK, W. Influence of ZrO2 grain size
and content on the transformation response in the Al2O3- ZrO2(12% mol
CeO2) system. J. AM. CERAM. SOC. V. 76 P. 657-663, 1993.
BERGTRÖM, L., BLOMBERG, E., PEDERSEN, G., Interparticle forces and
rheological properties of ceramic suspensions, key Engineering Materials,
v. 159-160, pp. 119-126, 1999.
BERNS, H., FRANCO, S. D., Caracterização in situ de propriedades mecânicas
de materiais resistentes ao desgaste abrasivo usando o método da
indentação, Cerâmica, v. 43, n. 281-282, pp. 128-132, 1997.
BETZ U., STURM A., LÖFFLER W., WIEDENMANN A., HAHN H. Microstructural
development during final-stage sintering of nanostructured zirconia based
ceramics, Materials Science and Engineering A , 281 68-74, 2000.
BONA A. D., ANUSAVICE K. J., DEHOFF P.H. Weibull analysis and flexural
strength of hot-pressed core and veneered ceramic structures. Dental
Materials 19, p. 662–669, 2003.

225
BORSA, C. E. Efeito da adições de TiO2 e fase vítrea no compósito Al2O3-ZrO2.
TESE DE MESTRADO, IME-RJ, 1991.
BOUTIN, J.M. & BLANQUAERT, D. Rev. Chir. Orthopaedic. V.67 P.279-287, 1981.
BRÅNEMARK, P.I., ZARB, G.A., ALBREKTSSON, T. Tissue-integrated
prostheses: osseointegration in clinical dentistry. QUINTESSENCE
VERLAGS – GMBH – BERLIM, 1987.
BRAVO-LEON, A. , MORIKAWA, Y., KAWAHARA, M., MAYO, M. J. Fracture
toughness of nanocrystalline tetragonal zirconia with low yttria content.
Acta Materialia, 50, p. 4555-4562, 2002.
BRESSIANI, J.C. & BRESSIANI, A.H.A. Cerâmicas á base de zircônia.
INFORMATIVO INT, V.20, N.41, P. 24-27, 1988.
BROOKS C. R. AND MCGILL B. L.. Microscopy to Fractography. Materials
Characterization, 33, p. 195-243, 1994.
BUCKO, M. M., HABERKO, K., Mechanical anisotropy in the zirconia toughened
alumina, J. Mat. Sci., v. 34, pp. 6157-6163, 1999.
BURGER,G., & SERGO, W. V. Piezo-spectroscopic analysis of the residual
stress in zirconia-toughened alumina ceramics: the influence of the
tetragonal-to-monoclinic transformation. Materials Science and Engineering
A 271 (1999) 401-406.
CALDERON-MORENO, J. M. & SCHEHL, M. Microstructure after superplastic
creep of alumina–zirconia composites prepared by powder alcoxide
mixtures. Journal of the European Ceramic Society , 24, p. 393-397, 2004.
CALÉS, B. Zirconia ceramic for improved hip prostheses. A review. 6th
Biomaterials Symposium “Ceramic implant materials in orthopedic surgery”,
Göttingen Germany, 1994
CANDO, D. C. , Zirconia: una alternativa fiable. Labor Dental, 4 (2) p. 1-7, 2001.
CANON, W.R. Transformation toughened ceramics for structural applications.
In: Torti, M.L., Treatise on Materials Science and Technology, 1 ed. V. 29,
chapter 5, New Jersey, USA, Academic Press Inc., p. 195-225, 1989.
CARVALHO, E. A., DUARTE, E. T. V., Ensaios de Flexão: Uma revisão dos
modelos matemáticos empregados na sua análise, In: Anais do 14º
Congresso Brasileiro de Cerâmica, p. 1-21, 2000.

226
CASELLAS, D., NAGL, M. M. LLANES, L., ANGLADA, M., ET AL. Fracture
toughness of alumina and ZTA ceramics: microstructural coarsening
effects. J. of Materials Processing and Technology, 143-144, p. 148-152, 2003.
CASELLAS, D., NAGL, M. M., ET AL., Growth of Surface Indentation Cracks in
Alumina and Zirconia Toughened Alumina, key Engineering Materials., v. 127-
131, pp. 895-902, 1997.
CASELLAS, D., RÀFOLS, I., LLANES, L., ANGLADA, M. Fracture toughness of
zirconia-alumina composites. Int. J. of Refractory Metals & Hard Materials.
V.17 p. 11-20, 1999.
CELLI A., TUCCI A., ESPOSITO L. ET AL. Fractal analysis of cracks in alumina-
zirconia composites. J. Eur. Ceram. Soc. 23, p. 469-479, 2003.
CHEN, Z., CHAWLA, K. K., KOOPMAN, M. Microstructure and mechanical
properties of in situ synthesized alumina/Ba-β-alumina/zirconia
composites. Materials Science and Engineering A, 367, p. 24-32, 2004.
CHERIF, K., GUEROULT, B., RIGAUD, M. Wear behaviour of alumina toughened
zirconia materials. Wear, 199, p. 112-121, 1996.
CHEVALIER, J., OLAGNON, G., FANTOZI, G. Crack propagation and fatigue in
zirconia-based composites. Composites: Part A, 30, p. 525-530, 1999.
CHIANG, S. S., MARSHALL, D. B., EVANS, A. G. The response of solids to
elastic/plastic indentation I: Stresses and residual stresses. J. of Applied
Physics. V. 53 p. 312-317, 1982.
CHINELATTO, A. S. A., Evolução microestrutural durante a sinterização de
pós finos e de alta pureza de alumina. Tese de Doutorado, UFSCar, São
Carlos, SP. 2002.
CHOI, S. R. & BANSAL, N. P. Mechanical behavior of zirconia/alumina
composites. Ceramics International, Article in press, 2004.
CLAUSSEN, N.J. Fracture toughness of Al2O3 with an unstable ZrO2 dispersed
second phase. J. AMERICAN CERAM. SOC., 61 P. 49-51, 1976.
CONLEY, R. F., Atrrition Milling of Industrial Minerals, Ultrafine Grinding and
Separation of Industrial Minerals, CHAPTER 4, PP. 37-48, 1992.
COOK, R. F. & PHARR, G. M. Direct observation of indentation cracking in
glass and ceramics. J. of American Ceramic Society. V. 73 n. 4 p. 787-817,

227
1990.
CORDINGLEY, R., KOHAN, L., BEN-NISSAN, B., PEZZOTTI, G. Zirconia and
partially stabilised zirconia as an orthopaedic biomaterial – Characteristics,
properties, performance and applications. J. of the Australasian Ceramic
Society, 39, p. 20-28, 2003.
COSTA, B. J. Processamento de materiais cerâmicos. Boletim técnico Instituto
de Tecnologia do Paraná., 1988.
DAUSKARDT, R. H., YU, W., RITCHIE, R. O., Fatigue Crack Propagation in
Transformation-Toughened Zirconia Ceramic, J. Am. Ceram. Soc., v. 70, n.
10, pp. C-248-252, 1987.
DAVIDGE, R. W. & EVANS, A. G. THE STRENGTH OF CERAMICS. MAT. SCI.
ENG., 6, P. 281-298, 1970.
De AZA, A.H., CHEVALIER, J., FANTOZZI, G. ET AL. Crack grouth resistance of
alumina, zirconia and zirconia toughened alumina ceramics for joint
prostheses. Biomaterials, 23, p. 937-945, 2002.
DORLOT, J.M., CHRISTEL, P., SEDEL,J. Biological and biomechanical
performances of biomaterials. Eds. Christel, P., Meunier, A., Lee, A.J.C.
Elsevier, Amsterdam, p. 495-500. 1986.
DÖRRE, E. Problems concerning the industrial production of alumina ceramic
components for hip joint prosthesis. In: Bioceramics and the human body.
Eds.Ravaglioli, A., Krajewski, A. Elsevier Applied Science. London and New
York. p.454-460. 1991.
DRUMMOND, J. L. In-vitro aging of yttria-stabilised zirconia. J. Amer. Ceram.
Soc., 72(4), p. 675-676, 1989.
DUAILIBI, J F., DIAS, A. J. N., VILARDO, C. A., Aplicação de Cerâmicas de alta
resistência ao desgaste em moldes de extrusão utilizados na indústria de
cerâmica vermelha; In: Anais do 45º Congresso Brasileiro de Cerâmica, Maio
1999.
DUAILIBI, J. F. & ROCHA, J. C., Cerâmicas de alta alumina; Informativo INT, v.
20, n. 41, pp. 19-23, 1988.
DUAILIBI, J. F., DIAS, A. J. N., VILARDO, C.A. Aplicação de cerâmicas de alta
resistência ao desgaste em moldes de extrusão utilizados na indústria de

228
cerâmica vermelha. In: Anais do 45º Congresso Brasileiro de Cerâmica, maio
de 1999.
DUAILIBI, J. F., Efeito da adição do Ferro e do Silício na Densificação,
Microestrutura e Propriedades Mecânicas do Nitreto de Silício. Tese de
D.Sc., IPEN/USP, São Paulo, SP, Brasil, 1994.
EVANS, A G. & TAPPIN, G. Effects of microstructure in the stress to propagate
inherent flaws. Proc. Brit. Ceram. Soc., 20, p. 275-297, 1972.
EVANS, A. G. Perspective on the development of high-toughness ceramics. J.
Am. Ceram. Soc. 73 p. 187-206, 1990.
EVANS, A. G., CHARLES, E. A., Fracture Tougness Determination by
Indentation, J. Am. Ceram. Soc., v. 59, n. 7-8, pp. 371-372, Jul/Aug. 1976.
EVANS, A. G., MARSHALL, T. R., Quasi-static solid particle damage in brittle
solids - I. Observations, analysis and implications, Acta Metal. v. 24, p. 939-
956, 1976.
FANTOZZI, G. & ORANGE, G. Thermomechanical properties of zirconia
toughened alumina materials. In: eds. Moya, J.S., De Aza, S. editors.
Processing of advanced ceramics. Soc. Esp. Ceram. Vidr. Arganda Del Rey
Madrid, Spain, p. 187-215, 1986.
FEIGHERY, A.J. & IRVINE, J.T.S. Effect of alumina additions upon electrical
properties of 8% mol yttria-stabilised zirconia. Solid State Ionics, v.121
p.209-216, 1999.
FENGQIU, T., XIAOXIAN, H., ET AL., Effect of dispersants on surface chemical
properties of nano-zirconia suspensions, Ceramic International, v. 26, pp.
93-95, 2000.
FISCHER, H., & MARX, R. Fracture toughness of dental ceramics: comparision
of bending and indentation method. Dental Materials, 18(1) (2002) 12-19.
FISCHER, H., RENTZSCH, W., MARX, R. Elimination of low-quality ceramic
posts by proof testing. Dental Materials, 18(8), p. 570-575, 2002.
FOSCHINI, C. R., SOUZA, D.M.P.F., PAULIN Fº, P.I. Análise quantitativa e
qualitativa de fases em zircônia através do método de Rietveld. Anais do
11º CBCIMat, p. 837-840, 1994.
FRÂNCIO, E. & LIMA, B. N. Comparação dos resultados obtidos para análise

229
quantitativa de fases dos polimorfos da zircônia por difração de raios X
com aplicação do método de Rietvel e métodos tradicionais. Capturado em:
http:// www.nit.ufscar.br/cbecimat/resumos/ceramica/TC101-034.htm EM
04/10/02.
GAO, M. W., FORSSEBERG, E., Increasing the Specific Surface Area of
Dolomite by Stirred Ball Milling, In: Kawatra, S. K., Comminution – Theory and
Practice, S. Komar Kawatra, 2 ed., chapter 12, Society for Mining, Metallurgy and
Exploration Inc., pp. 153-170, 1992.
GARVIE, R. C., Critical Size Effects in Alumina-Zirconia Alloys, In: Advances in
Ceramics - Science and Technology of Zirconia III, v. 24, Tokyo, Japan, The
American Ceramic Society, p. 55-69, 1988.
GLASS S. J. & GREEN D. J. Mechanical properties of infiltrated alumina-Y-TZP
composites. J. Amer. Ceram. Soc., 79, p. 2227-2239, 1996.
GOGOTSI, G. A., DUB, S. N., LOMONOVA, E. E. ET AL. Vickers and knoop
indentation behaviour of cubic and partially stabilized zirconia crystals. J.
of European Society, v. 15. p.405-413, 1995.
GONG, J., WU, J., GUAN, Z. Analysis of the indentation size effect on apparent
hardness for ceramics. Materials Letters. V. 38. p. 197-201, 1999.
GRABER, G. & BESIMO, C. The DCS high-performance ceramic-system: A new
means for computer-assisted fabrication of metal-free zirconium oxide
crowns and fixed partial dentures. QDT, p. 195-199, 1995.
GREEN, D. J., HANNINK, R. H. J., SWAIN, M. V. Transformation toughening of
ceramics. Boca Raton, FL.: CRS Press, Inc. p. 232, 1989.
GREGORI G., BURGER W., SERGO V. Piezo-spectroscopic analysis of the
residual stress in zirconia-toughened alumina ceramics: the influence of
the tetragonal-to-monoclinic transformation. Materials Science and
Engineering A 271, p. 401-406, 1999.
GRIFFITH, A. A. The phenomena of rupture and flow in solids. Phil. Trans. Roy.
Soc., A221, p. 163-198, 1920.
GUO, X. & HE, J. Hydrothermal degradation of cubic zirconia. Acta Materialia,
2003. Article in press.
GUPTA, T. K., LANGE, F. F., BECHTOLD, J. H. Effect of stress-induced phase

230
transformation on properties of polycrystalline zirconia containing
metastable tetragonal phase. J. Mater. Sci., 13, p. 1464-1470, 1978.
GUTZOV, S.; PONAHLO, J.; LENGAVER, C. L.; BERAN, A. Phase
characterization of precipitated zirconia. J. Am. Ceram. Soc., 77(6), p. 649-
652, 1994
HANNINK, R. H. J. Transformation toughening in zirconia- containing
ceramics. J. Am. Ceram. Soc., v. 83, n.3, p. 461-487, 2000.
HARRIS, W.H. & MULROY, R.D.JR., Improved cementing techniques: Effect on
femoral and socket fixation at 11 years. Transactions of the society for
biomaterials, v. 13, p. 147, 1990.
HEGENBARTH, E. A. Procera aluminium oxide ceramics: A new way to
achieve stability, precision, and esthetics in all-ceramic restorations. QDT,
p. 21-34, 1996.
HEIJMAN M.J.G.W., BENES N.E., TEM ELSHOF J.E., VERWEIJ H. Quantitative
analysis of the microstructural homogeneity of zirconia-alumina
composites. Materials Research Bulletin, 1904, p. 1-10, 2002.
HEROS, R. WILLMANN, G. Ceramics in total hip arthroplasty: history,
mechanical properties, clinical results and current manufacturing state of
the art. Seminars Arthroplasty. V.9 p. 114-122, 1998.
HEUER, A. H. Transformation toughening in ZrO2-containing ceramics. Journal
Am. Ceram. Soc., v. 70, n. 10, p. 689-698, 1987.
HEUER, A.H., CHAIM, R., LANTERI, V. Review: Phase transformations and
microestructural characterization of alloys in the system ZrO2-Y2O3.
Advances in Ceramics. V.24 Science and Technology of Zirconia III, p. 3-20,
1988.
HILL, R. J. & HOWARD, C. J. Quantitative phase analysis from neutron powder
diffraction data using the Rietveld method. J. Appl. Cryst. V. 20, p. 467-474,
1987.
HILL, R. J. Expanded use of the Rietveld method in studies of phase abundance
in multiphase mixtures. Power Diffraction, v. 6, n.2, p. 74-77, 1991.
HOME PAGE: www.azom.com. (capturado em 20/03/2004).
HOME PAGE: www.geocities.com. (capturado em 05/06/2002).

231
HOME PAGE: www.standformaterials.com. (capturado em 05/06/2002).
HORI S., YOSHIMURA M., SOMIYA S. Strength-toughness relation in sintered
and isostatically hot-pressed ZrO2- toughened Al2O3. J. Am. Ceram. Soc., 69
p. 169-172, 1986.
HSU, S. M. & SHEN, . Wear prediction of ceramics. Wear, 256, p. 867–878,
2004.
HULBERT, S.F. The use of alumina and zircônia in surgical implants. In:
Hench, L.L. & Wilson, J. Advanced series in ceramics. An introduction to
Bioceramics. World Cientific, v.1, cap.2, p. 25-36. 1993.
HUNTER, ROBERT J., Zeta Potencial in Colloid Science, Principles and
Applications, 2 ed., London, Academic Press Inc., pp.1-31, 69-74, 98-117,
258-295, 1986.
INTERNATIONAL STANDARD ISO – 13356 Specifications for implants for
surgery ceramic materials based on zirconia.
INTERNATIONAL STANDARD ISO – 6474 Specifications for implants for
surgery ceramic materials based on alumina.
INTERNATIONAL STANDARD ISO 6872:1995 Dental ceramics. International
Organization for Standardization, Genève, Switzzerland.
IRWIN, G. R. Analysis of stress and strain near to the end of a crack traversing
a plate. J. Appl. Mech., 24, p. 361-364, 1957.
JIANG, L. Z. & SUN, C. T. Analysis of indentation cracking in piezoceramics.
Int. J. of Solids and Structures. V.38, p. 1903-1918, 2001.
JUSTO, L.F, MELLO, F.C.L., DEVEZAS, T. Conjugados cerâmicos nos sistemas
Al2O3- ZrO2 e ZrO2-CeO2. In: Anais do 9º CBECIMAT, p. 439-441, São Paulo,
Dez, 1990.
KALISZEWSKI M.S., BEHRENS G., HEUER A.H. ET AL. Indentation studies on
Y2O3 – stabilised ZrO2: I. Development of indentation-induced cracks. J.
Am. Ceram. Soc. 77 (1994) 1185-1193.
KAMIYA, H., FUKUDA, Y., ET AL., Effect of Polymer Dispersant Structure on
Eletrosteric Interation and Dense Alumina Suspension Behavior, J. Am.
Ceram. Soc., 82, (12) , p. 3407-3412, 1999.
KARIHALOO B. L. Contributions of t-m phase transformation to the toughening

232
of ZTA. J. Am. Ceram. Soc., 74, p.1703-1706, 1991.
KEITH, O., KUSY, R. P., WHITEY, J. Q., Zirconia brackets: an evaluation of
morphology and coefficients of friction. Am. J. Orthod. Dentofacial
Orthop.,106 (6) (1994) 605-614.
KELLY, J. R., NISHIMURA, I., CAMPBELL, D. Ceramics in dentistry: Historical
roots and current perspectives. The Journal of Prosthetic Dentistry, v.78, n.1,
p. 18-32, 1996.
KELSO, J. F. & FERRAZZOLLI, T. A. Effect of powder surface chemistry on the
stability of concentrated aqueous dispersions of alumina. J. Am. Ceram.
Soc., 72(4), p. 625-627, 1989.
KIM, B. K; HAHN, J. W; HAN, K. R. Quantitative phase analysis in tetragonal
monoclinic two phase zirconia by Raman spectroscopy. J. Mat. Sci. Letters,
16, p. 669-671, 1984.
KINGERY, W. D. Introduction to ceramics. Ed. John Willey & Sons, Inc, New
York, 2ª edição, 1960.
KONG, Y. M., BAE, C. J., LEE, S. H., KIM, H. W., KIM, H. E. Improvement in
biocompatibility of ZrO2-Al2O3 nano-composite by addition of HA.
Biomaterials, Article in press, 2004.
KRELL, A. Load dependence of hardness in sintered submicrometer Al2O3 e
ZrO2. J. Am. Ceram. Soc.. 78(5) (1995) 1417-1419.
KUMAR, A.S., DURAI, A.R., SORNAKUMAR, T. Development of alumina–ceria
ceramic composite cutting tool. International Journal of Refractory Metals &
Hard Materials, 22, p. 17–20, 2004.
KUMAR, A.S., DURAI, A.R., SORNAKUMAR, T. Yttria ceramics: cutting tool
application. Materials Letters, 58, p. 1808– 1810, 2004.
KUNES, K., HAVRDA, J., HRONÍKOVÁ, K. et al. Stabilization of bioceramic
suspensions prepared from alumina-containing zirconia powders. Ceramics-
Silikáty, .44 (1) p. 1-8, 2000.
LA CROIX, S. P. Prensagem uniaxial de cerâmica dental. Dissertação de
mestrado, COPPE-UFRJ, 1998.
LANGE F. F. & HIRLINGER M. M. Hindrance of grain growth in Al2O3 by ZrO2
inclusions. J. Amer. Ceram. Soc., 67(3), p.164-168, 1984.

233
LANGE, F. F. Transformation toughening: Part 1 – Size effects associated with
the thermodynamics of constrained transformations. J. Mat. Sci., v.17, p.
225-234, 1982a.
LANGE, F. F. Transformation toughening: Part 2 – Contribution to fracture
toughness. J. Mat. Sci., v.17, p. 235-239, 1982b.
LANGE, F. F. Transformation toughening: Part 4 – Fabrication fracture
toughness and strength of Al2O3- ZrO2 composites. J. Mat. Sci., 17, p. 247-
254,1982c.
LANGE, F. F. & EVANS, A. G. Erosive damage depth in ceramics: A study on
metastable tetragonal zirconia. J. Am. Ceram. Soc., v. 62, p. 62-65, 1979.
LANGE, F. F., DUNLOP, G. L., DAVIS, B. I. Degradation during aging of
transformation-toughed ZrO2-Y2O3 materials at 250°C. J. Am. Ceram. Soc.,
69, p. 237-240, 1986.
LAWN, B. R., MARSHALL, D. B., Hardness, Toughness and Brittleness: An
Indentation Analysis, J. Am. Ceram. Soc., v. 62, n. 7-8, pp. 347-350, Jul/Aug.
1979.
LEE, D.Y., KIM, D-J., SONG, Y-S. Chromaticity, hydrothermal stability, and
mechanical properties of t-ZrO2/Al2O3 composites doped with yttrium,
niobium, and ferric oxides. Material Science and Engineering, A 289, p. 1-7,
2000.
LEWIS, D. Effects of surface treatment on the strength and thermal shock
behaviour of a commercial glass ceramic. Ceramic Bull., (6), v. 58, p. 599-
605, 1979.
LI, J.; ZHANG, L., SHEN, Q., HASHIDA, T. Degradation of yttria stabilizaed
zirconia at 370 K under a low applied stress. Materials Science and
Engineering, A 297, p. 26-30, 2001.
LIANG, K. M., ORANGE, G., FANTOZZI, G., Evaluation by indentation of fracture
toughness of ceramic materials, J. Mater. Sci., v. 25, pp. 207-214, 1990.
LIN J.D. AND DUH J.G. Fracture toughness and hardness of ceria- and yttria-
doped tetragonal zirconia ceramics. Materials Chemistry and Physics. 78, p.
253-261, 2002.
LINO, U. R. A., Efeito do acabamento superficial e tratamentos térmicos sobre a

234
resistência mecânica de alumina sinterizada., Tese de mestrado, IME-RJ,
1982.
LIU, X., HUANG, Y., YANG, J. Effect of reological properties of the suspension
on the mechanical strength of Al2O3- ZrO2 composites prepared by
gelcasting. Ceramics International, 28, p. 159-164, 2002.
LUTHARD, R.G., SANDKUHL, O., REITZ, B. Zirconia-TZP and alumina-advanced
technologies for the manufacturing of single crowns. The European Journal
of Prosthodontics and Restorative Dentistry, v. 7, p. 113-119, 1999.
MADRUGA, T. P., COSTA, R. C., ACCHAR, W. Estudo da influência da pré-
trinca no valor de tenacidade à fratura de alumina sinterizada de alta
pureza. Cerâmica, 32, p.105-108, 1986.
MARCHI, J. Estudo de sinterização de cerâmicas à base de nitreto de silício
utilizando-se como aditivos óxidos de cério e alumínio. Tese de mestrado,
IPEN, São Paulo, SP, Brasil, 1999.
MARINELLO, C.P., MEYENBERG, K.H., ZITZMANN, N. et al. Single-tooth
replacement: Some clinical aspects. Journal of Esthetic Dentistry, v. 9, p.
169-178, 1997.
MARSHALL D. B., DRANSMANN G.W., STEINBRECH R. W., PAJARES A.,
GUIBERTEAU F., CUMBRERA F. L, DOMINGUEZ-RODRIGUEZ A.
Indentation studies on Y2O3-stabilized ZrO2: I Development of indentation
induced cracks. J. Amer. Ceram. Soc., 77 (5), p. 1185-1193, 1994.
MATSUZAWA, M., YAJIMA, N., HORIBE, S., Damage accumulation caused by
cyclic indentation in zirconia ceramics, J. Mat. Sci., v. 34, pp. 5199-5204,
1999.
MCMEEKING, R. M. & EVANS, A. G. MECHANICS OF TRANSFORMATION-
TOUGHENING IN BRITTLE MATERIALS. J. AM. CERAM. SOC. V. 65, N. 5, P.
242-246, 1982.
MEDVEDOVSKI, E. Wear-resistant engineering ceramics. Wear, 249, p. 821-
828, 2001.
MESQUITA, C. E. M. Densificação quase-isostática do compósito Al2O3-TiC
obtido por SHS. Tese de doutorado, IME – RJ, 2001.
MILLEDING, P., CARLÉN, A., WENNERBERG, A. et al. Protein characterisation

235
of salivary and plasma biofilms formed in vitro on non-corroded and
corroded dental ceramics materials. Biomaterials, v.22, p. 2545-2555, 2001.
MIYAZAKI, H., YOSHIZAWA, Y., HIRAO, K. Preparation and mechanical
properties of 10 vol.% zirconia/alumina composite with fine-scale fibrous
microstructure by co-extrusion process. Materials Letters, 58, p. 1410– 1414,
2004.
MONTGOMERY, D. C., Design and Analysis of experiments. 4 ed., chapter 4,
USA, John Wiley & Sons, Inc., p. 126-145, 1997.
MUCHTAR, A., LIM, L. C., Indetation fracture toughness of high purity
submicron alumina, Acta Mater., v. 46, n. 5, pp. 1683-1690, 1998.
MUKHERJEE, A., MAITI, B., DAS SHARMA, A., ET AL. Correlation between
slurry rheology, green density and sintered density of tape cast yttria
stabilised zirconia. Ceramics International 27, p. 731-739, 2001.
NAWA, M., BAMBA, N., SEKINO, T. ET AL. The effect of TiO2 addition on
strengthening and toughening in intragranular type of 12Ce-TZP/Al2O3
nanocomposites. J. of European Society, 18. p. 209-219, 1998.
NIIHARA, K. Fracture mechanics analysis of identation-induced Palmqvist
crack in ceramics. J. Mat. Sci. Let., 2, p. 221-223, 1983a.
NIIHARA, K., MORENA, R., HASSELMAN D.P.H. In: Bradt R.C., Evans, A.G.,
Hasselman D.P.H, Lange, F.F., editors: Fracture mechanics of ceramics, New
York, Plenum: 5, p. 97, 1983b.
NIIHARA, K., ÜNAL, N., NAKAHIRA, A., Mechanical properties of (Y-TZP)-
alumina-silicon carbide nanocomposites and the phase stability of Y-TZP
particles in it, J. Mat. Sci., v. 29, pp. 164-168, 1994.
NISHIOKA, R. S., BOTTINO, M. A., SOUZA, F. A., LOPES, A. G. Carga imediata
e restauração protética definitiva com pilares protéticos personalizados. Rer. Bras. de Implantodontia e Prótese sobre Implantes. 10, n. 38, p. 98-102,
2003.
OBLAK, C., JEVNIKAR, P., KOSMAC, T. ET AL. Fracture resistance and
reliability of new zirconia posts. J Prosthet Dent, 91, p. 342,348, 2004.
ÓDEN, A., ANDERSSON, M., ONDRACEK, I.K. et al. Five-year clinical evaluation
of Procera AllCeram crowns. The Journal of Prosthetic Dentistry, v.80, n.4, p.

236
450-456, 1998.
OLIVEIRA, A. P. A. Influência de fatores físico-químicos na produção de pós de
zircônia. Tese de doutorado. PUC-RJ, 1997.
OLIVEIRA, I. R., PANDOLFELLI, V. C., ET AL., Dispersão e Empacotamento de
Partículas, 1 ed., São Paulo, SP, Fazendo arte editorial, pp. 25-91, 2000.
OLIVEIRA, L. G. Efeito do processamento nas propriedades mecânicas da
alumina tenacificada por zircônia parcialmente estabilizada com cálcia ou
ítria. Dissertação de mestrado, COPPE-UFRJ. 2002.
OLIVEIRA, L.G., DUAILIBI, Fh, J., NÓBREGA, M. C. S. Efeito do tipo de eletrólito
em suspensões de zircônia e misturas de alumina com zircônia
parcialmente estabilizada com cálcia e ítria. Proceedings of the 46th Annual
Meeting of the Brazilian Ceramic Society, São Paulo, Brasil, 2002.
ONODA, G. Y. Ceramic processing before firing. John Willey & Sons, 1977.
OONISH, H., IGAKI, H., TAKAYAMA, Y. Comparision of wear of uhmpwe
polyethylene sliding against metal and alumina in total hip prostheses. In:
Proceedings of 1st International Symposium on Ceramics in Medicine. Eds.
Oonishi, H., Aoki, H., Sawai, K. Ishiyaku EuroAmérica, Inc. p. 272-277, 1988.
ORTEGA, F. S., PANDOLFELLI, V. C., ET AL., Aspectos da reologia e da
estabilização de suspensões cerâmicas. Parte I: Fundamentos, Cerâmica,
43 (279) , p.5-10, Jan/Fev 1997a.
ORTEGA, F. S., PANDOLFELLI, V. C., ET AL., Aspectos da reologia e da
estabilização de suspensões cerâmicas. Parte II: Mecanismos de
Estabilidade Eletrostática e Estérica. Cerâmica, 43(280), p.77-83, Mar/Abr
1997b.
ORTEGA, F. S., PANDOLFELLI, V. C., ET AL., Aspectos da reologia e da
estabilização de suspensões cerâmicas. Parte III: Mecanismos de
Estabilização Eletroestérica de Suspensões com Alumina, Cerâmica, 43(281). p.113-119, 1997c.
PAJARES, A., GUIBERTEAU, F., CUMBRERA, F. L. ET AL. Analysis of kidney-
shaped indentation cracks in 4Y-PSZ. Acta Mater. V. 44 p. 4387-4394, 1996.
PANDOLFELLI, V.C., RODRIGUES, J. A., ET AL., Correlação entre a técnica de
medida da tenacidade e o grau de transformabilidade de zircônias

237
tetragonais; In: Anais do XXXV Congresso Brasileiro de Cerâmica e III
Iberoamericano de cerâmica, vidro e refratário, pp. 640-646, Maio 1991.
PASCOE, R.T. & GARVIE, R.C. Ceramic microstructures. Ed. Fulrath, R.M. &
Pask, J.A.. Westview Press. Boulder. P. 774-785, 1977.
PEREIRA, A. P. D. Comportamento mecânico de laminados cerâmicos de
Al2O3/ ZrO2 produzidos via gelcasting. Tese de doutorado. IME-RJ, 2001.
PHILLIPS, R. Science of dental materials. W.B.Saunders Co. Tenth edition,
Philadelphia. USA, 1996.
PICONI, C. & MACCAURO, G. Zirconia as a ceramic biomaterial. Biomaterials,
20, p. 1-25, 1999.
PILEGGI, R. G., STUDART, A. R., PANDOLFELLI, V. C., Um modelo para
previsão da viscosidade mínima de suspensões cerâmicas, Cerâmica, v.
46, n. 299, pp.160-165, 2000.
PIORINO, F. N., FURLAM, J. P., ET AL., Estudo Comparativo para ensaios de
tenacidade à fratura em cerâmicos de alto desempenho, Cerâmica., v. 36, n.
245, pp. 105-108, Set/Out. 1990.
PIORINO, F. N.; SILVA, C. R. M.; TAVARES, N. S.; Técnicas experimentais para
caracterização de Cerâmicos Estruturais – Propriedades Mecânicas, In:
Anais do 36º Congresso Brasileiro de Cerâmica, Caxambu, p. 765-772, Jun.
1992.
PLITZ, W. & HOSS, H.U. In: Biomaterials. Eds. Winter, G.D., Gibbons, D.F.,
Plenck, H. John Willey , New York, p. 187-196, 1982.
PORTER, D. A. & EASTERLING, K. E. Phase transformations in metals and
alloys. Chapman & Hall, 2º Edition, 1992.
POULON-QUINTIN, A., BERGER, M.H., BUNSELL, A.R. Mechanical and
microstructural characterization of Nextel 650 alumina–zirconia fibres.
Journal of the European Ceramic Society, 24, p. 2769–2783, 2004.
PRESTIPINO, V. & INGBER, A. All-ceramic implant abutments: Esthetic
indications. Journal of Esthetic Dentistry, 8, p. 255-262, 1996.
RAMACHANDRAN, N., SHETTY, D. K. Rising crack-growth-resistance (R-curve)
behaviour of toughened alumina and silicon carbide. J. Am. Ceram. Soc.,
74, p. 2634-2641, 1991.

238
RANGERT, B., JEMT, T., JORNEUS, L. Forces and moment on Brånemark
implants. Int. Journal Oral Maxillofacial Implants. v.4, p.241-247, 1989.
RAZZOOG, M.E., LANG, L.A., McANDREW, K.S. All-Ceram crowns for single
replacement implant abutments. The Journal of Prosthetic Dentistry, v.78 p.
486-489, 1997.
REED, J. Introduction to the principles of ceramic processing. John Willey &
Sons. New York – USA, 1987.
REED, J. S. & LEJUS, A. M. Effect of grinding and polishing on near surface
phase transformations in zirconia. Mat. Res. Bull. V. 12, p. 949-954, 1977.
REZENDE, D. T., Influência da adição de zircônia na tenacidade à fratura do
nitreto de silício obtido via sinterização normal. Dissertação de mestrado,
PUC-RJ, 1997.
RICHERSON, D. Modern ceramic engineering. Marcel Dekker Inc. New York –
USA, 1992.
ROCHA, J. C. Produção de alumina sinterizada a partir do pó e otimização dos
parâmetros de sinterização para máxima resistência mecânica. Tese de
mestrado, Rio de Janeiro, RJ, Brasil, 1981.
ROCHA, J. C., DUAILIBI, J. F., Avaliação do efeito do sistema ligante PVA/PEG
na microestrutura do compacto a verde, sinterabilidade e resistência
mecânica da alumina, In: Anais do XXXV Congresso Brasileiro de Cerâmica e
III Iberoamericano de cerâmica, vidro e refratário, pp. 515-521, Maio 1991.
ROCHA, J. C., MAIA, R. A., GOMES, M., Metodologia de ensaio para avaliação
de propriedades físicas em materiais cerâmicos, Informativo INT, 1990.
ROSENBLUM, M. ET AL. A review of all-ceramic restorations. Journal of
American Dental Association. ADA Publishing Co, Inc, USA, march, 1997.
ROSS, I. M., RAINFORTH, W. M., MCCOMB, D.W., SCOTT, A.J., BRYDSON, R.,
The role of trace additions of alumina to yttria-tetragonal zirconia
polycrystals (Y-TZP), Scripta Materialia 45, 653-660, 2001.
RUF, H. & EVANS, A. G. Toughening by monoclinic zirconia. J. Am. Ceram.
Soc. V. 66, n. 5, p. 328-332, 1983.
RÜHLE, M., STECKER, A., WAIDELICH, D., KRAUS, B. In situ observations of
stress-induced phase transformation in ZrO2 containing ceramics. In: eds.

239
Claussen, N. Rülle, M., Heuer, A. Advances in ceramics. Science and
Technology of Zirconia II. Columbus: American Ceramic Society, 12, p. 256-
274, 1984.
SAKAI, M. & BRADT, R. C., Fracture toughness testing of brittle materials.
International Materials Reviews, v. 38 n.2, p. 52-78, 1993
SALLÉ, C; GROSSEAU, P.; GUILHOT, B. ET AL. Detecção de zircônia tetragonal
em alumina-zirconia powders by thermoluminescence. Journal of European
CERAMIC SOCIETY V.23, P. 667-673, 2003.
SATO, T. & SHIMADA, M. Transformation of yttria-doped tetragonal ZrO2
polycrystals by annealing in water. J. Am. Ceram. Soc., 68, p. 356-369, 1985.
SCHULTE, W. Das Tübinger sofortimplantat, Monatliche Zeitschrift für
zahnärte, Quintessenz, Verlag, 27, p. 1-7, 1976.
SCHULTE, W. The intraosseous Al2O3 (Frialit) Tübingen implant.
Developmental status after eight years. Quintessence Int., 15, p. 9-26, 1984.
SCHULTE, W., D`HOEDT, B., AXMANN, D., GOMEZ, G. The first 15 years of the
Tübingen implant and its further development to the Frialit-2 system.
Zeitschr f Zahnärztl Impl., 8, p. 3-22, 1992.
SCHULTE, W., HEIMKE, G. The Tübingen immediate implant. Quintessenz, 27,
p. 17-23, 1976.
SEDEL, L., CHRISTEL, P., MEUNIER, A., BOUTIN, P. Alumina/bone interface.
Experimental and clinical data. In: Proceedings of 1st International Symposium
on Ceramics in Medicine. Ed. Ooninishi, H., Aoki, H., Sawai, K. Ishiyaku
EuroAmérica, Inc. p. 262-271. 1988.
SEDEL, L., MEUNIER, A., NIZARD, R.S., WITVOET, J. Ten year survivorship of
cemented ceramic-ceramic total hip replacement. In: Bioceramics, v.4 Eds.
Bonfield, W., Hastings, G.W., Tanner, K.E. Butterworth-Heinemann, Ltda.,
London, UK, p.27-37. 1991.
SERRA, E., TUCCI, A, ESPÓSITO, L. PICONI, C. Volumetric determination of the
wear of ceramics for hip joints. Biomaterials, 23, p. 1131-1137, 2002.
SHARIF, A. A., MECARTNEY, M. L. Superplasticity in cubic yttria stabilized
zirconia with 10 wt.% alumina. Journal of the European Ceramic Society, 24,
p. 2041–2047, 2004.

240
SHAW, D. J., Introdução à química dos colóides e de superfícies, 1 ed., São
Paulo, SP, Tradução: Juergen Heinrich Maar, Edgard Blücher, pp. 31-36,102-
152, 1975.
SHIMIZU, K., OKA, M., KUMAR, P., KOTOURA, Y., YAMAMURO, T., MAKINOUCHI,
K., NAKAMURA, T. Time-dependent changes in the mechanical properties
of zirconia ceramic. J. of Biomedical Materials Research, 27, p. 729-734,
1993.
SHULMAN, H. S., RYAN M..J., BAXTER N.I., BROWN I.W.M. Zrconia
Toughened Alumina (ZTA) – Optimisation of the Toughening Mechanism.
Journal of the Australasian Ceramic Society, Vol. 34, no. 2, p. 127-31 ,1998.
SMITH, W.F. Princípios de ciência e engenharia dos materiais. Mc Graw-Hill de
Portugal Ltda., 3ª Edição, 1998.
SONG, Y. K. & VARIN, R. A. Indentation microcraking and toughness of newly
discovered ternary intermetallic phases in the Ni-Si-Mg system. Intermetallics, v.6 p. 379-393, 1998.
SPRIGGS, R. M. & VASILOS, T. Effect of grain size on transverse bend strength
of alumina and magnesia. J. Am. Ceram . Soc., 46 (1963) 224-228.
SRDIC, V. & RADONJIC, L. Processing of the alumina-zircônia composite and
its mechanical properties. In: Third Euro-Ceramics, v. 3, ed. P. urán and J. F.
Fernández, p. 701-706, 1993.
SRINIVASAN, R., RAMAYI, V. M., SUJIT, K. R., PADMANBHA, K. K. N.
Dehydration and crystallization kinetics of zirconia-yttria gels. J. Am.
Ceram. Soc., 78(2), p. 429-432, 1995.
STANDARD, O. Application of Transformation-Toughened Zirconia Ceramics
as Bioceramics. Ph.D Thesis. University of New South Wales, Australia, 1995.
STEVENS, R. Zirconia and zirconia ceramics. 2 ed. Magnesium elektron ltda. P.
5-51, 1986.
SULLIVAN, J. D., LAUZON, P. H., Experimental propability estimators for
Weibull plots. J. Mater. Sci. Letters, v. 5, pp. 1245-1247, 1986.
SWARB, J. J. Low temperature degradation of Y-TZP materials. Journal of
Materials Science, 26, p. 6706-6714, 1991.
TANAKA, K., KANARI, M., MATSUI, N. A continuum dislocation model of vickers

241
indentation on a zirconia. Acta Mater. V. 47, n. 7. p. 2243-2257, 1999.
TAVARES, L. M. M., Moagem ultrafina, COPPE-UFRJ-COT 760, 2001.
THOMSON, I. & RAWLINGS, R. D. Mechanical behaviour of zirconia and
zirconia-toughened alumina in a simulated body environment. Biomaterials,
11, p. 505-508, 1990.
TOMASZEWSKI, H., Toughening effects in Al2O3- ZrO2 system. Ceram. Intern.,
14, p. 117-125, 1988.
TORAYA, H; YOSHIMURA, M; SOMIYA, S. Calibration curve for quantitative
analysis of the monoclinic-tetragonal ZrO2 system by X-ray diffraction.
Communications of the American Ceramic Society, C-119, 1984.
TRUSTTRUM, K. & JAYATILAKA, A. DE S. On estimating the Weibull modulus
for a brittle material. J. Mater. Sci, v.14 p. 1080-1084, 1979.
TSAI, Y. M. Indentation of a penny-shaped crack by an oblate spheroidal rigid
inclusion in a transversely isotropic medium. J. of Aplied Mechanics. V.51
p.811-815, 1984.
TUAN W. H, CHEN R.Z., WANG C.H., CHENG P.S., KUO P. S. Mechanical
properties of Al2O3//ZrO2 composites. J. European Ceram Soc. 22, p. 2827-
2833, 2002.
TUAN, W. H. & KUO, J. C. Contribution of residual stress to the strength of
abrasive ground alumina. J. Eur. Ceram. Soc. 19 p. 1593–1597, 1999.
VELASCO, R. V. C., SEKITO, T. J., ANDRADE, H. F. ET AL, Desempenho clínico
das restaurações do tipo inlay/onlay de cerâmica. Revista Brasileira de
Odontologia, v. 60, n. 6, p. 380-383, 2003.
WANG J. AND STEVENS R. Review zirconia-toughened alumina(ZTA) ceramics.
J. Mat. Science., 24, p. 3421-3440, 1989.
WANG, J., GAO, L., Surface and electrokinetic properties of Y-TZP suspensions
stabilized by polyelectrolytes, Ceramic International, v. 26, pp. 187-191, 2000
WANG, J., GONG, J., GUAN, Z. Variation in the identation toughness of silicon
nitride. Materials letters. Article in press, 2002.
WATANABE, M., LIO, S., FUKUURA, I. Advances in ceramics, v. 12, Science and
Technology of zirconia II. Edited by N. Claussen, M. Ruhle and A. H. Heuer.
American Ceramic Society, Columbus, OH, p. 391, 1984.

242
WEI, WEN-CHENG J., WANG, SHENG-CHANG, HO, FANG-YUAN, Eletrokinetic
Properties of Colloidal Zirconia Powders in Aqueous Suspension”, J. Am.
Ceram. Soc., v. 82, n. 12, pp. 3385-3392, 1999.
WILLMANN, G. Ceramics for total hip replacement – what a surgeon should
know. Orthopedics, v. 21, p. 173-177, 1998.
XU, C., AI, X., HUANG, C., Fabrication of na advanced ceramic tool material.
Wear, 249, 503-508, 2001.
YANG H., GAO L., SHAO G., XU R., HUANG P. Grain boundary glassy phase of
silicon nitride ceramics, Ceramics International, 27, 603-605, 2001.
YANG, C-C T., & WEI, W-C. J. Effects of material properties and testing
parameters on wear properties of fine-grain zirconia(TZP). Wear, 242, p. 97-
104, 2000.
YOUNG, R. A. The Rietveld method., Oxford Science. p.4, 1995.
ZHOU, D., LOW, I. M., Influence of residual stresses on indentation crack
patterns in zirconia toughened alumina and ZrO2 multilayer composites, J.
Mat. Sci. Letters, v. 15, pp. 695-698, 1996.

243
8 ANEXOS

244
8.1 ANEXO 1

245

246

247

248

249

250

251

252
8.2 ANEXO 2

253

254

255

256

257

258