medida de atividade total e específica (após) diálise ... experimental/2017... · determinação...
TRANSCRIPT

Diálise
Eletroforese(SDS-PAGE)
SequenciamentoeIdentificação deproteínas
Sayuri Miyamoto 2017
Cromatografia de troca-iônica
Levedura - Lise
Medida de atividade total e específica (após)
Medida de atividade total e específica (antes)

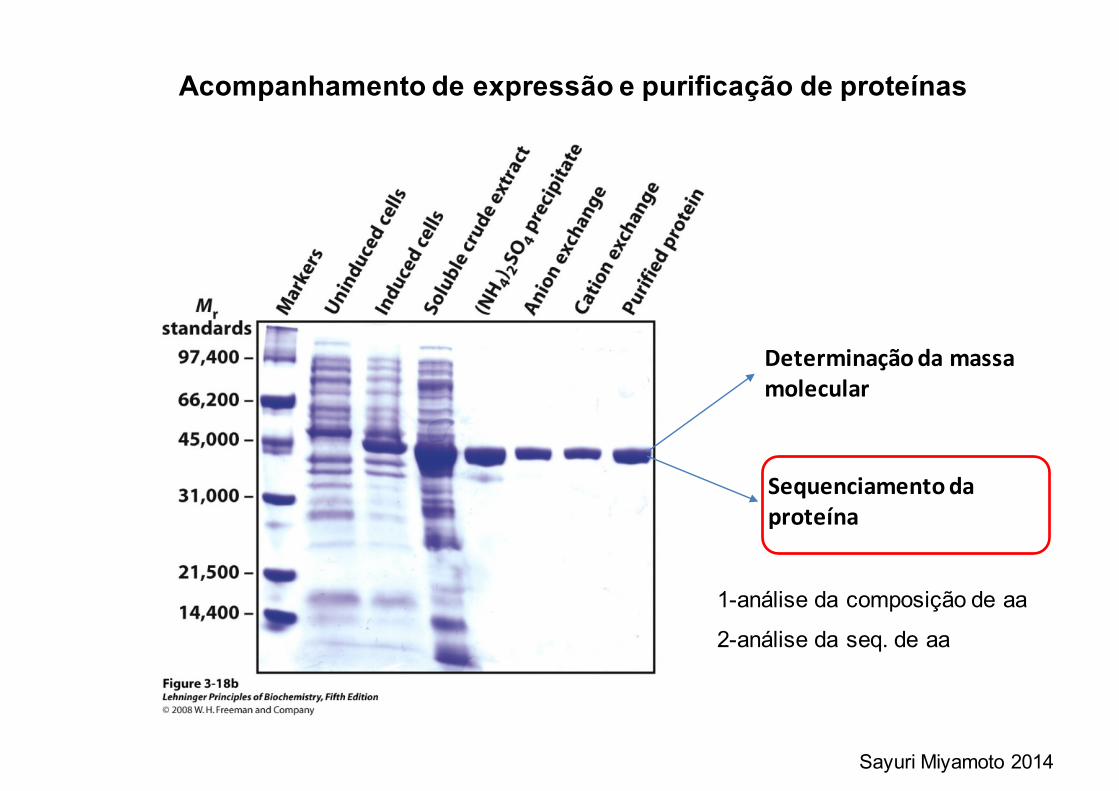
Acompanhamento de expressão e purificação de proteínas
Determinaçãodamassamolecular
Sequenciamentodaproteína

DeterminaçãodeMassaMoleculardaProteína
SDS-PAGECromatografia deexclusãoEspectrometriademassas
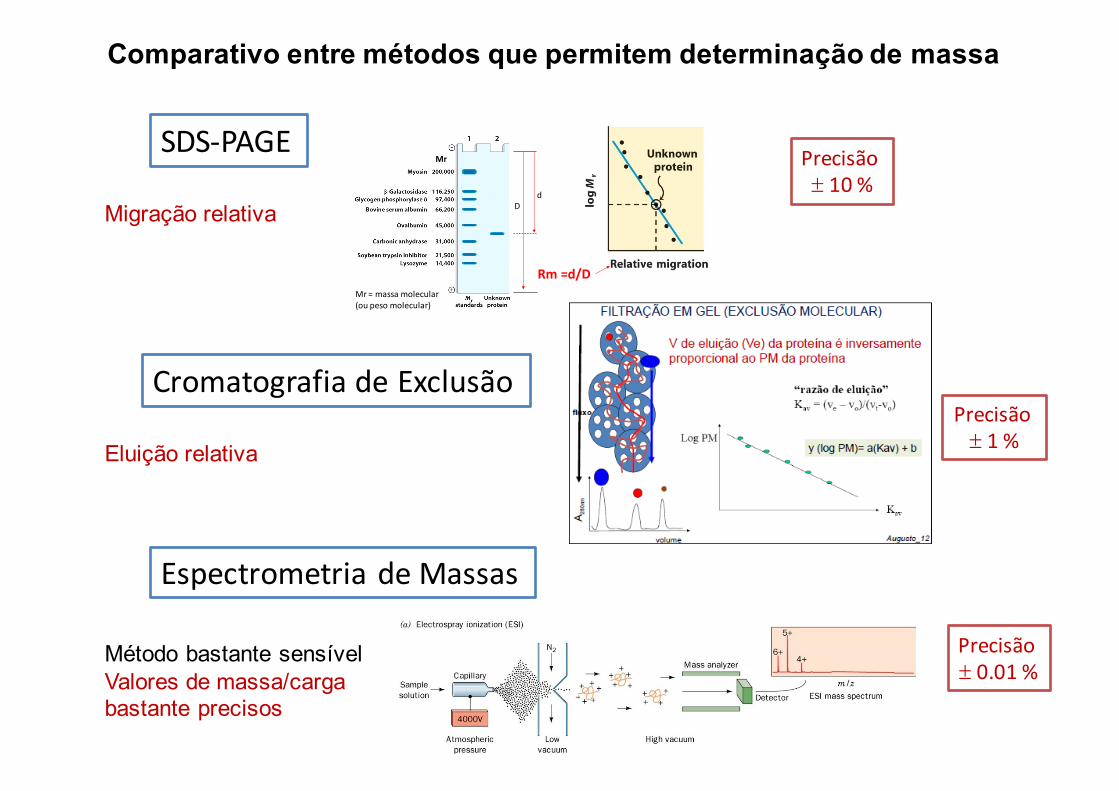
dD
Rm =d/D
Mr
Mr=massamolecular(oupesomolecular)
SDS-PAGE
Migração relativa
CromatografiadeExclusão
Eluição relativa
Precisão± 10%
Precisão± 1%
Método bastante sensívelValores de massa/carga bastante precisos
EspectrometriadeMassas
Precisão± 0.01%
Comparativo entre métodos que permitem determinação de massa

EspectrometriadeMassasComo funciona a técnica?
Espectrometria de massas (MS, Mass Spectrometry) é uma técnica analíticaextremamente sensível que permite determinar a massa molecular.Ela também fornece informações acerca da estrutura da molécula.
O equipamento que faz a medida da massa é o Espectrômetro de Massas.
MALDI-TOF Q-TOF
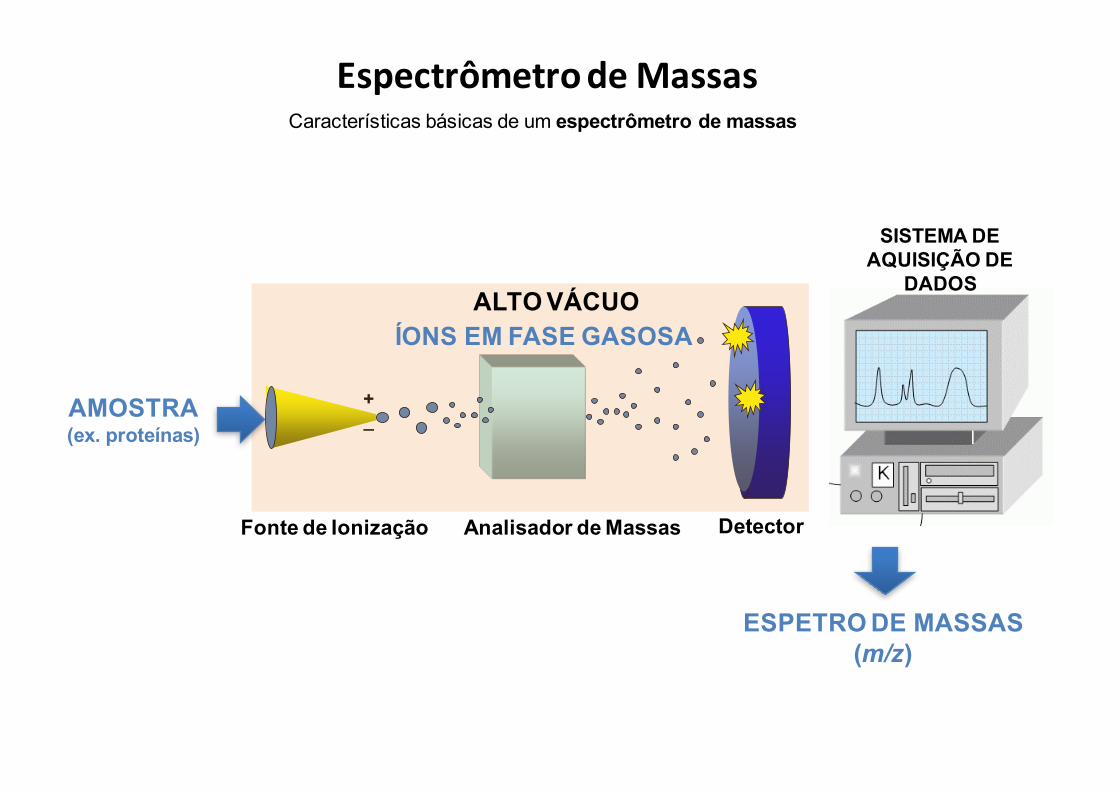
Características básicas de um espectrômetro de massas
EspectrômetrodeMassas
Fonte de Ionização
SampleInlet
+_
Analisador de Massas Detector
ESPETRO DE MASSAS(m/z)
ÍONS EM FASE GASOSA
AMOSTRA(ex. proteínas)
SISTEMA DE AQUISIÇÃO DE
DADOSALTO VÁCUO
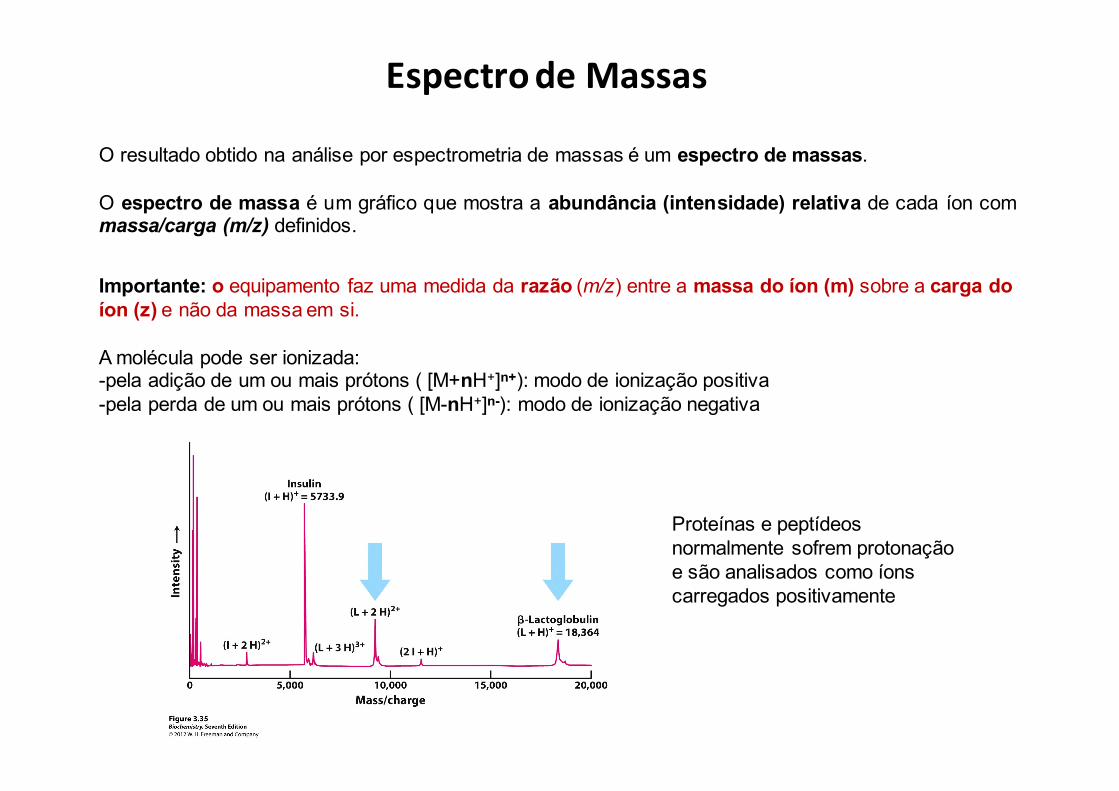
O resultado obtido na análise por espectrometria de massas é um espectro de massas.
O espectro de massa é um gráfico que mostra a abundância (intensidade) relativa de cada íon commassa/carga (m/z) definidos.
EspectrodeMassas
Importante: o equipamento faz uma medida da razão (m/z) entre a massa do íon (m) sobre a carga do íon (z) e não da massa em si.
A molécula pode ser ionizada:-pela adição de um ou mais prótons ( [M+nH+]n+): modo de ionização positiva-pela perda de um ou mais prótons ( [M-nH+]n-): modo de ionização negativa
Proteínas e peptídeosnormalmente sofrem protonaçãoe são analisados como íonscarregados positivamente

Ionização
IONIZAÇÃO (formação de íons com carga positiva ou negativa) é o primeiro passo
fundamental para que a molécula possa ser introduzida no analisador de massas
Duas principais técnicas de ionização ”BRANDAS (Soft)” empregadas para análise de
biomoléculas:
ELECTROSPRAY e o MALDI (Matrix-Assisted Laser Desorption/Ionization).
NobelPrize inChemistry,2002
John B. Fenn is the chemist who invented the ELECTROSPRAY method (1988). Professor of Chemistry at Yale University, USA, and at Virginia Commonwealth University, USA
Koichi Tanaka research engineer who developed the MALDI technique (1987).Shimadzu Corporation, Kyoto, Japan.
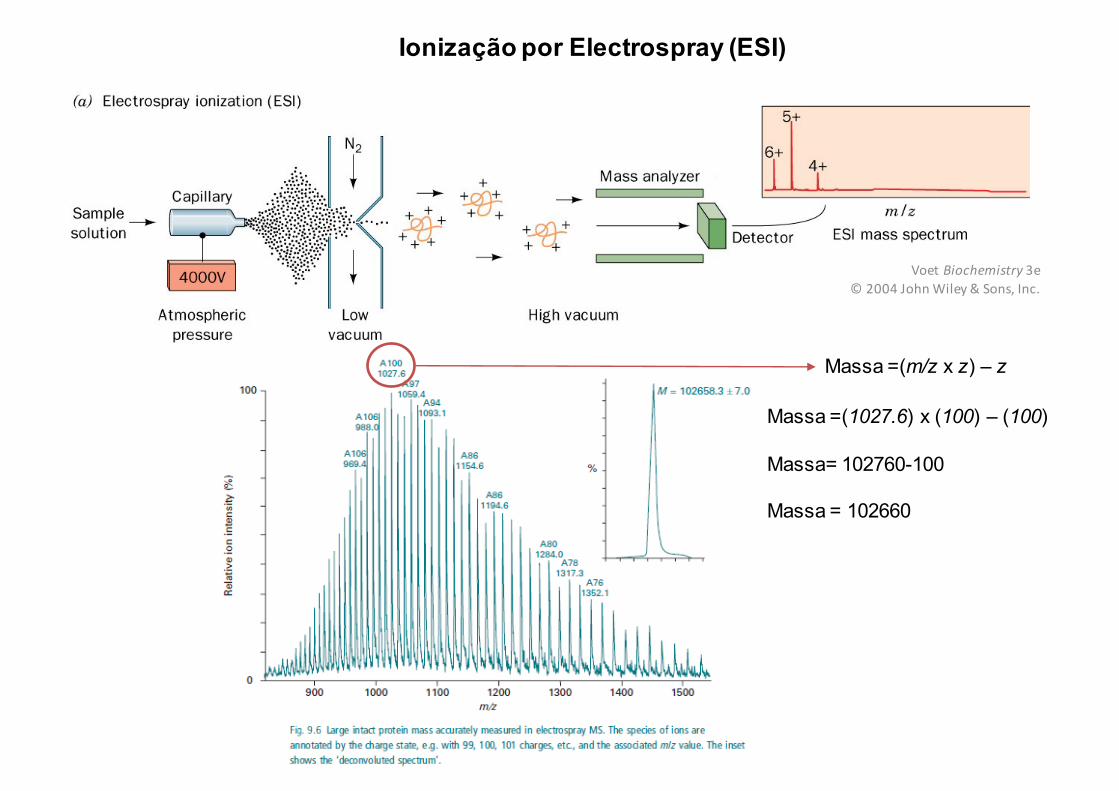
Voet Biochemistry 3e©2004JohnWiley&Sons,Inc.
Ionização por Electrospray (ESI)
Massa =(m/z x z) – z
Massa =(1027.6) x (100) – (100)
Massa= 102760-100
Massa = 102660
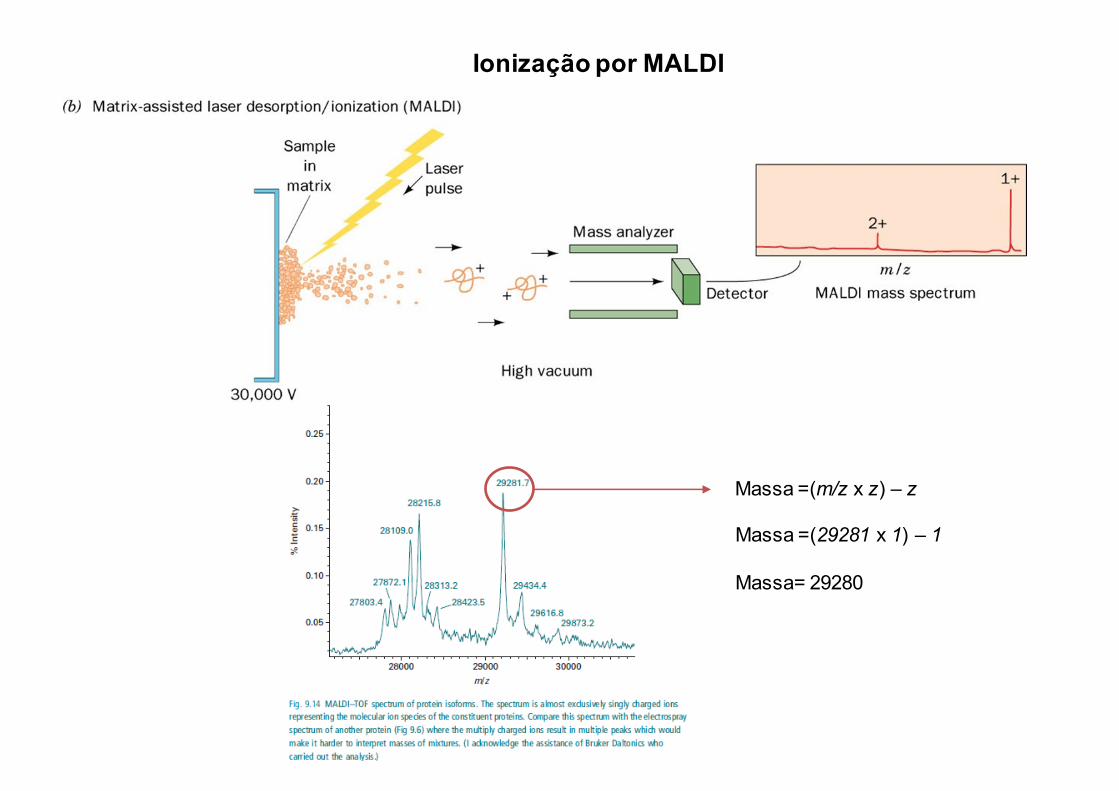
Ionização por MALDI
Massa =(m/z x z) – z
Massa =(29281 x 1) – 1
Massa= 29280

Espectros de Massa de Proteína Intacta
ESI MALDI
Sayuri Miyamoto 2014
Acompanhamento de expressão e purificação de proteínas
Determinaçãodamassamolecular
Sequenciamentodaproteína
1-análise da composição de aa
2-análise da seq. de aa
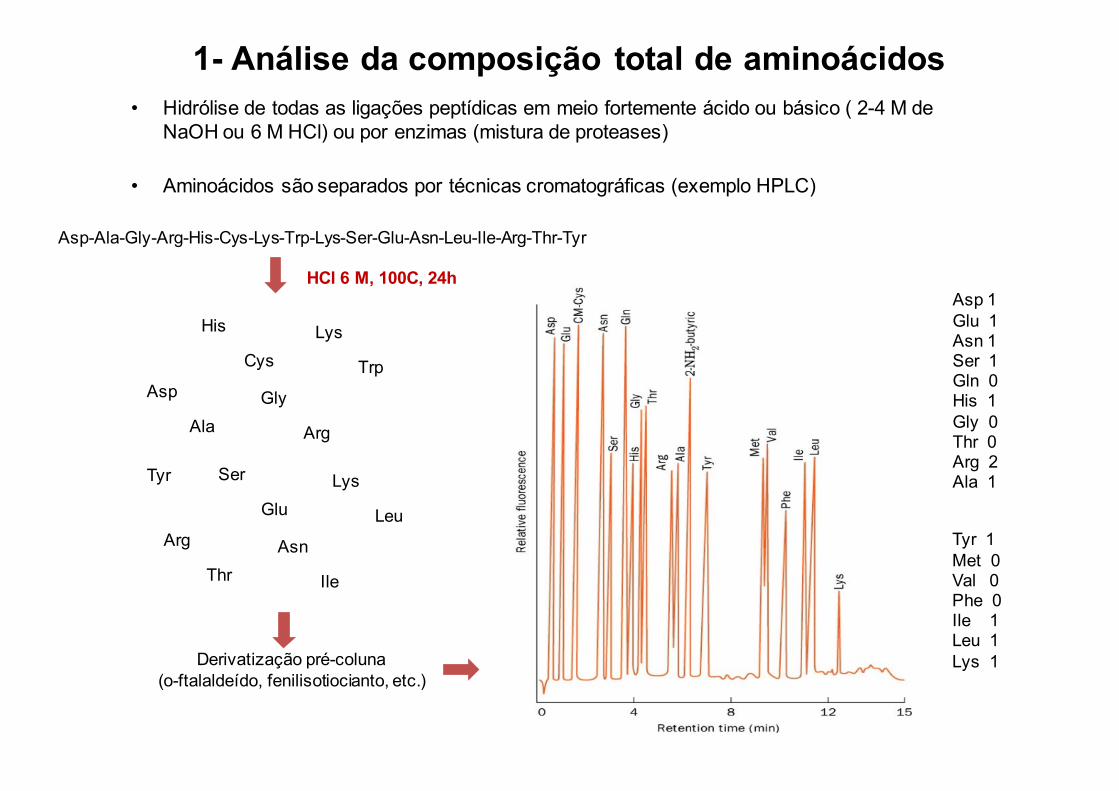
1- Análise da composição total de aminoácidos• Hidrólise de todas as ligações peptídicas em meio fortemente ácido ou básico ( 2-4 M de
NaOH ou 6 M HCl) ou por enzimas (mistura de proteases)
• Aminoácidos são separados por técnicas cromatográficas (exemplo HPLC)
Asp-Ala-Gly-Arg-His-Cys-Lys-Trp-Lys-Ser-Glu-Asn-Leu-Ile-Arg-Thr-Tyr
Asp 1Glu 1Asn 1Ser 1Gln 0His 1Gly 0Thr 0Arg 2Ala 1
Tyr 1Met 0Val 0Phe 0Ile 1Leu 1Lys 1
Asp
AlaGly
Arg
His
CysLys
Trp
Arg
ThrAsn
Ile
Ser
GluLys
Leu
Tyr
HCl 6 M, 100C, 24h
Derivatização pré-coluna(o-ftalaldeído, fenilisotiocianto, etc.)

Tripsina (Cliva após resíduos básicos Lys e Arg)
Fragmentos Peptídicos
+H3N- -COO-
II. Clivagem da cadeia polipeptídica usando métodos de clivagem específicos. (ex. Proteases como a Tripsina).
2-SequenciamentodeProteínas
I. Redução de pontes de disulfeto usando agentes redutores como o 2-mercaptoetanol.É importante alquilar as cisteínas para evitar que as pontes de disulfetos se formem
novamente.
* O uso combinado de mais de um método de clivagem ajuda no ordenamento das sequência de peptídeos, principalmente no método de Sanger

2-SequenciamentodeProteínas
Degradação de Edman(Sequenciamento a partir do N-terminal)
Sequenciamento através de análisepor MS e/ou MS/MS
MALDI-TOF Q-TOF
Limitado a oligopeptídeos de até 50 aa
III. Sequenciamento dos fragmentos peptídicos
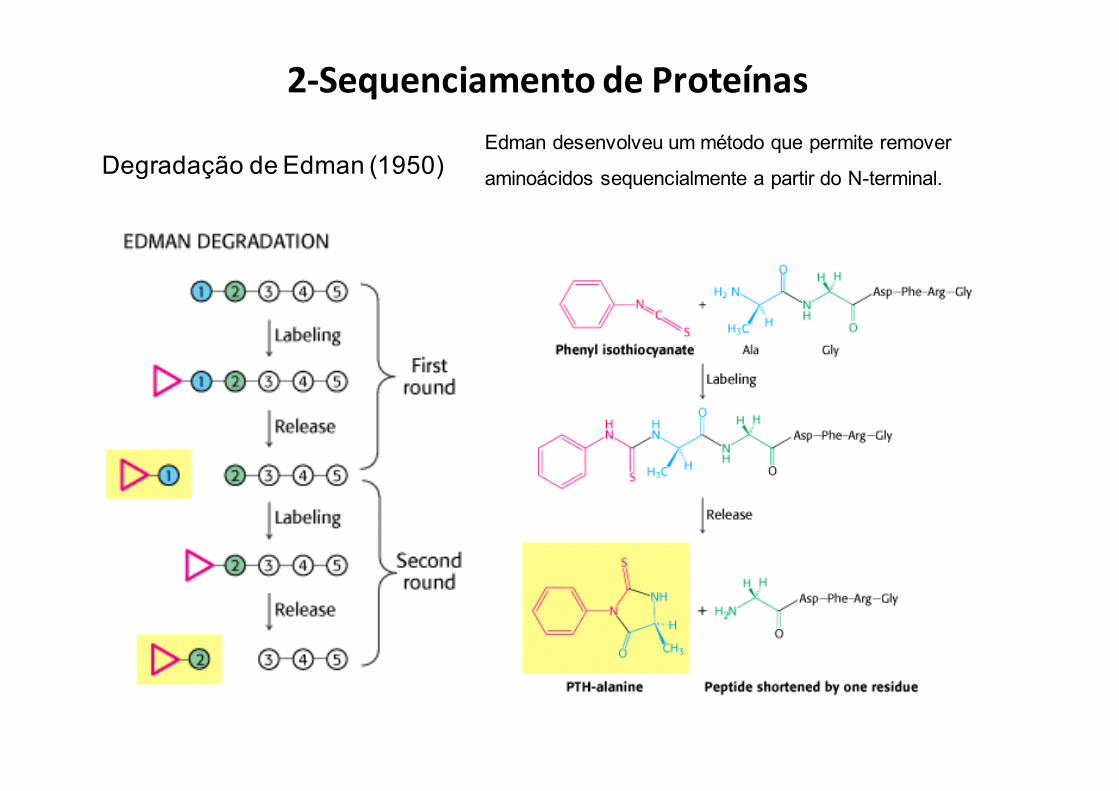
2-SequenciamentodeProteínas
Degradação de Edman (1950)Edman desenvolveu um método que permite remover
aminoácidos sequencialmente a partir do N-terminal.

Usar mais de um método de clivagem da cadeia polippetídica....
Asp-Ala-Gly-Arg-His-Cys-Lys-Trp Lys-Ser-Glu-Asn-Leu-Ile-Arg-Thr-Tyr
Exemplo: Quimotripsina (Cliva após resíduos aromáticos)
Tripsina (Cliva após resíduos básicos Lys e Arg)
Como ordenar a sequência de peptídeos??
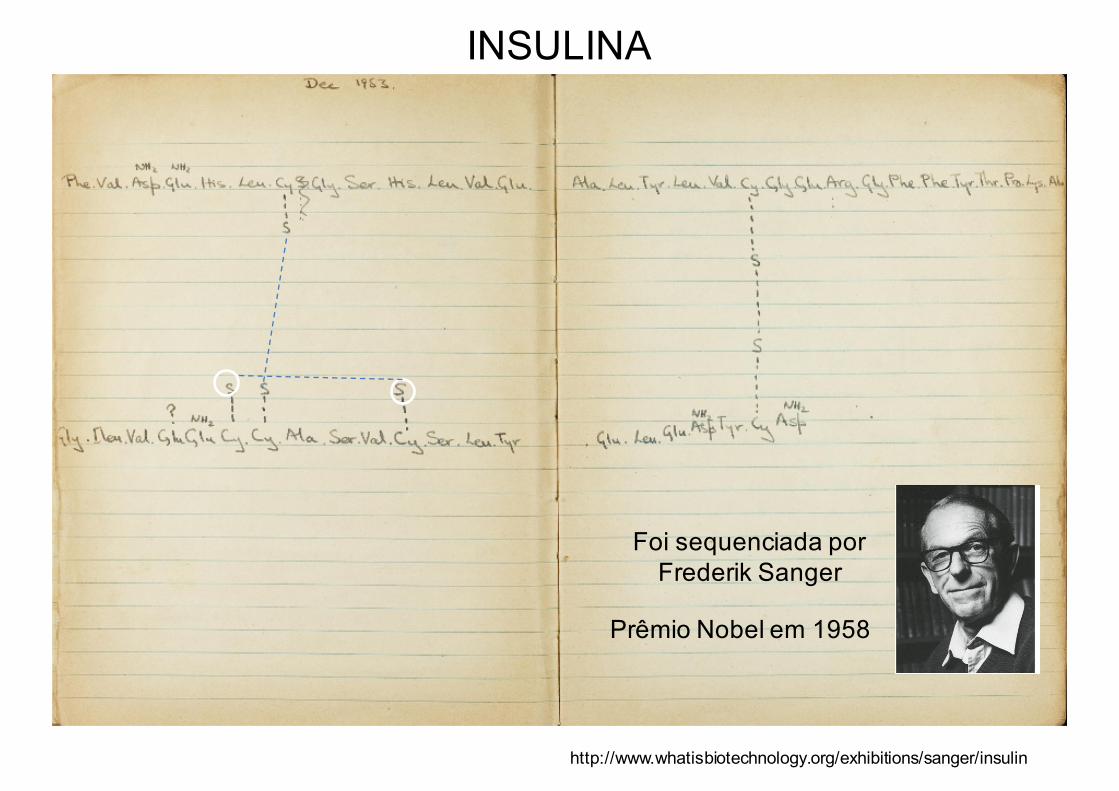
Foi sequenciada por Frederik Sanger
Prêmio Nobel em 1958
INSULINA
http://www.whatisbiotechnology.org/exhibitions/sanger/insulin

Limitações:Tempo de análise longo!Grande quantidade de amostra!Muito lento para os padrões de hoje!!!
Hoje em dia o sequenciamento é feito por espectrometria de massas. (+rápido, requer pouquíssima amostra)
Sequenciadores Automáticos
Degradação de Edman

EspectrometriadeMassas
AnálisedemassadeProteínasIntactas(MS)AnálisedemassadePeptídeos(MS)
Sequenciamento

Wenk, M.R. 2010 Cell 143, 188.
Espectrometria de massas propiciou grandes avanços nas áreas de Proteômica, Glicômica e Lipidômica

Sequenciamentodeproteínasporespectrometriademassas
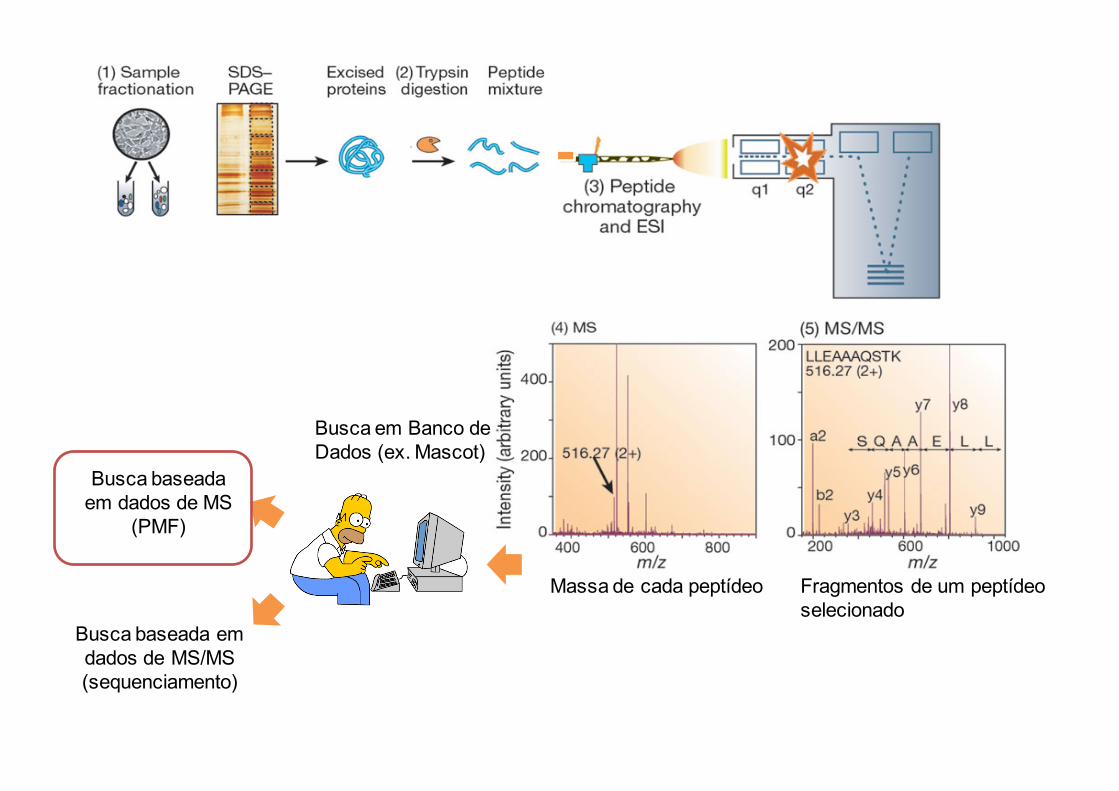
Massa de cada peptídeo Fragmentos de um peptídeo selecionado
Busca baseada em dados de MS/MS(sequenciamento)
Busca em Banco de Dados (ex. Mascot)
Busca baseada em dados de MS
(PMF)

Exemplo de análise de glicosidase por
SDS-PAGE e espectrometria de massas
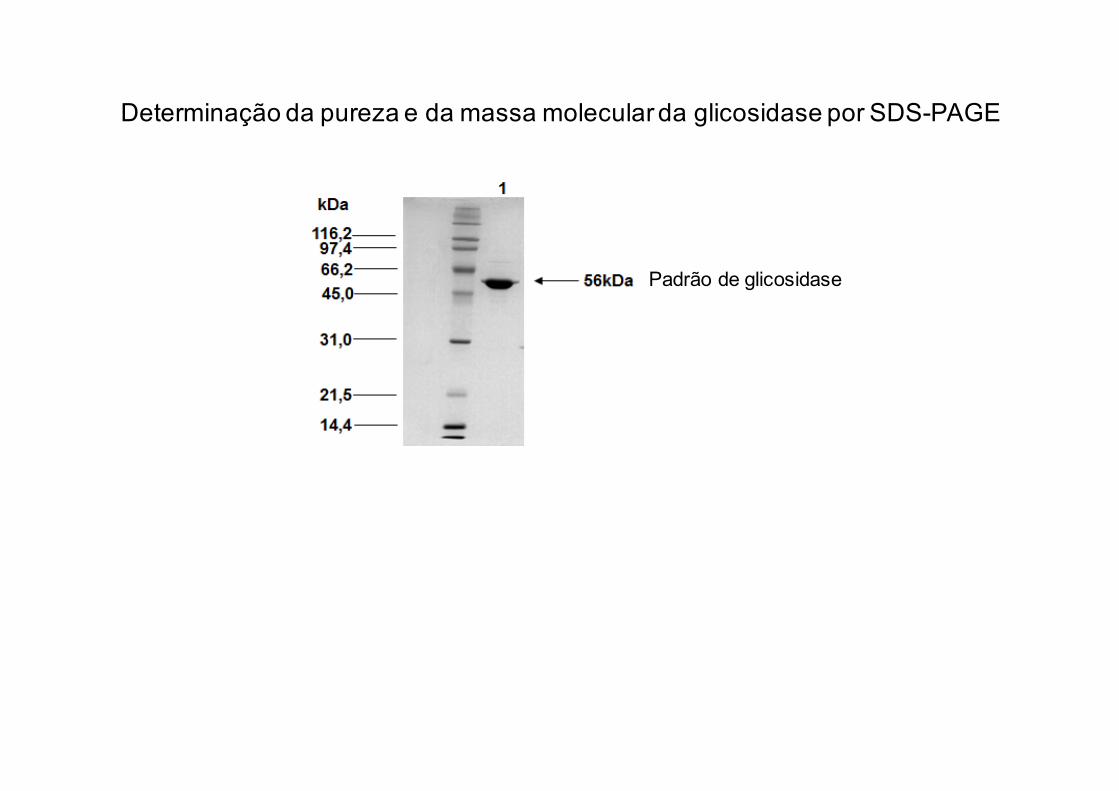
Padrão de glicosidase
Determinação da pureza e da massa molecular da glicosidase por SDS-PAGE
57288.292
0
1000
2000
3000
Inte
ns. [
a.u.
]
40000 45000 50000 55000 60000 65000 70000 75000m /z
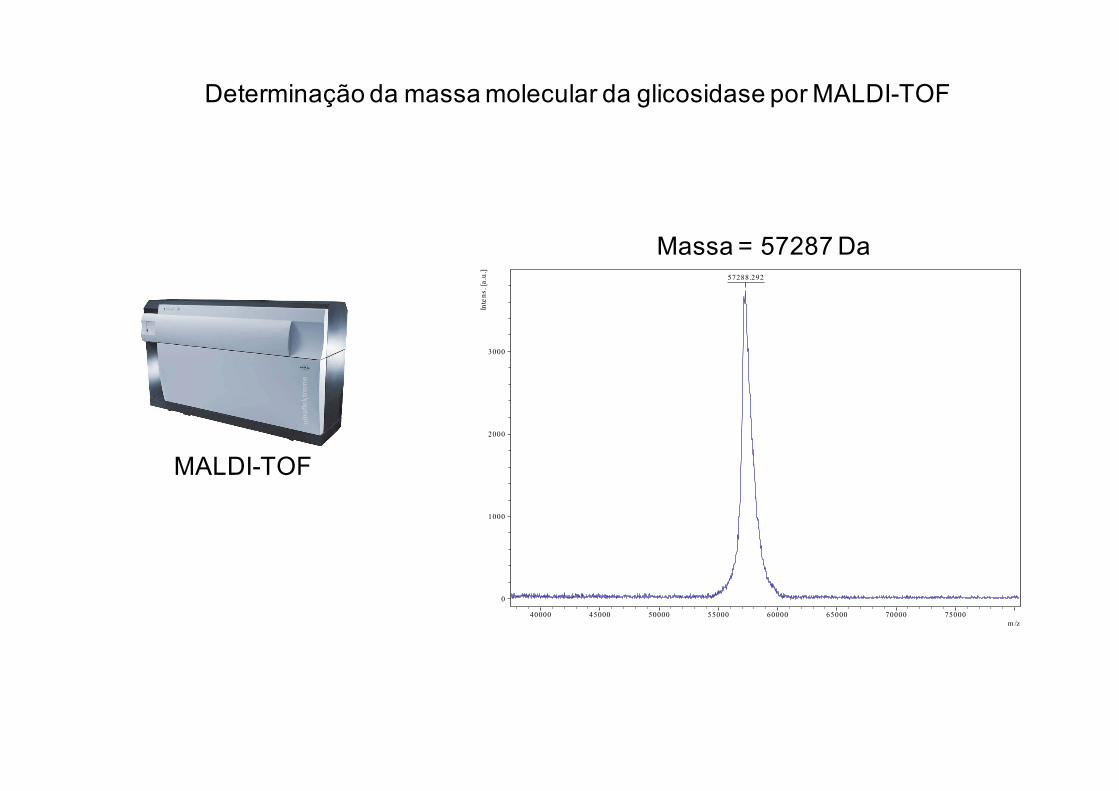
Massa = 57287 Da
Determinação da massa molecular da glicosidase por MALDI-TOF
MALDI-TOF
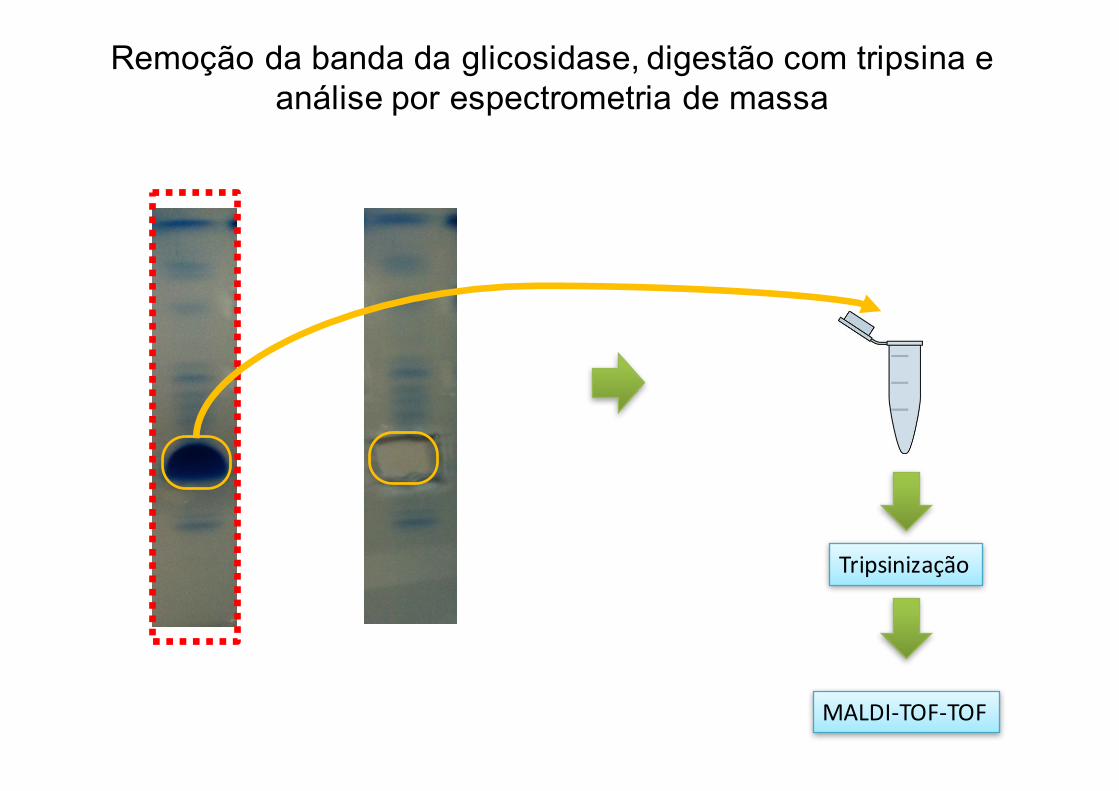
Tripsinização
MALDI-TOF-TOF
Remoção da banda da glicosidase, digestão com tripsina e análise por espectrometria de massa
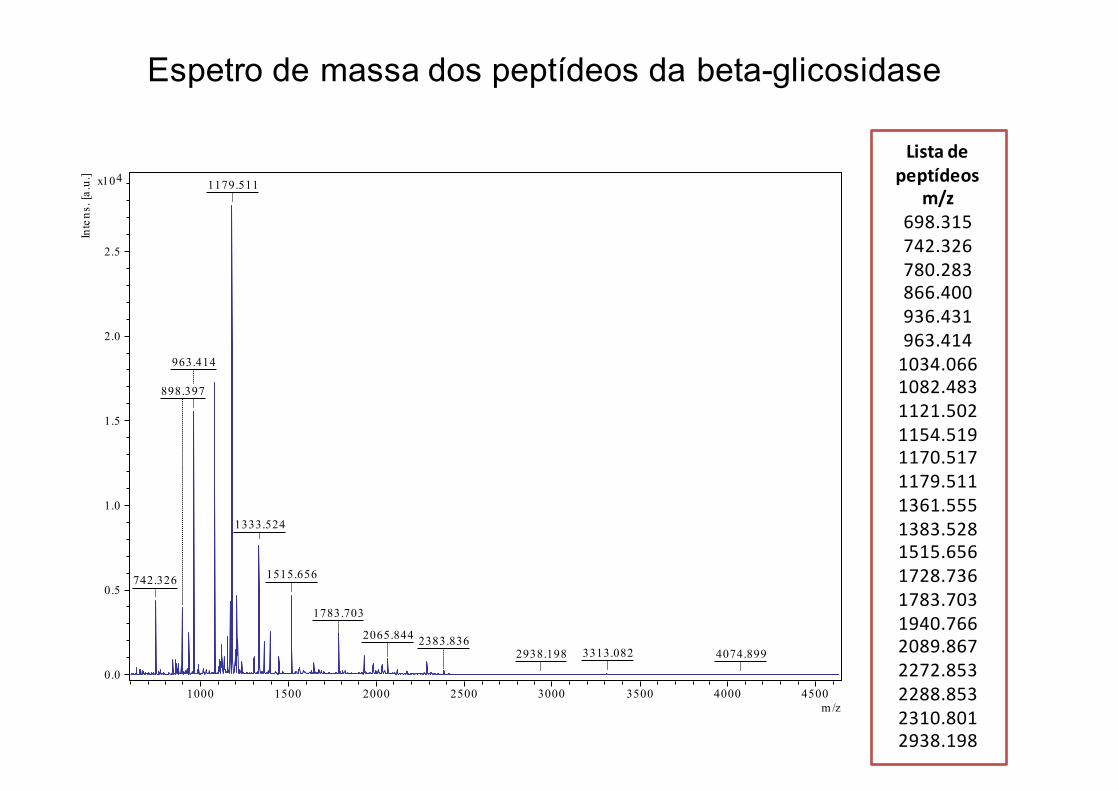
1179.511
963.414
1333.524
1515.656742.326
898.397
1783.703
2065.844 2383.8363313.0822938.198 4074.899
0.0
0.5
1.0
1.5
2.0
2.5
4x10
Inte
ns. [
a.u.
]
1000 1500 2000 2500 3000 3500 4000 4500m /z
Espetro de massa dos peptídeos da beta-glicosidase
Listadepeptídeos
m/z698.315742.326780.283866.400936.431963.4141034.0661082.4831121.5021154.5191170.5171179.5111361.5551383.5281515.6561728.7361783.7031940.7662089.8672272.8532288.8532310.8012938.198

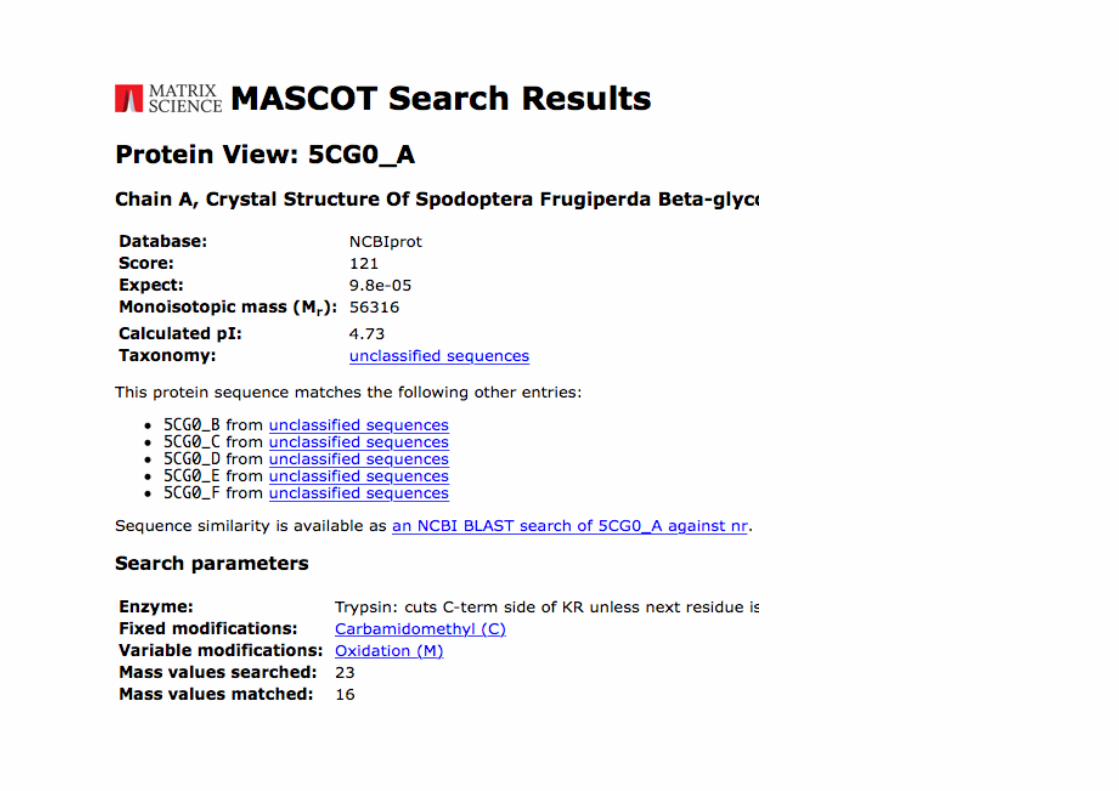
Busca no Mascot utilizando as massas dos peptídeos detectados
Listadepeptídeos
m/z698.315742.326780.283866.400936.431963.4141034.0661082.4831121.5021154.5191170.5171179.5111361.5551383.5281515.6561728.7361783.7031940.7662089.8672272.8532288.8532310.8012938.198

Swiss-ProtSe a proteína é de um organismo bem caracterizado comohumanos, camundongos, levedura, ou arabdopsis, o Swiss Prot é o recomendado
NCBIprotSe a proteína é de bactéria ou planta, usar o NCBIprot, pois é um banco de dados mais amplo que o Swiss-Prot
Banco de Dados (Database)

Modificações fixasExemplo mais comum é a alquilação de cisteínas.O alquilante mais comum é a iodoacetamida (carbamidomethyl) ouácido iodoacético (carboxymethyl).
Modificações
Modificações variáveisExemplo:- Oxidações que ocorrem durante o processamento de amostra(oxidação de metionina)- Modificações pós-traducionais (fosforilação)

Scoresacimade93:significantes (p<0.05)
Oscore obtidonabuscafoide121.


Massa de cada peptídeo Fragmentos de um peptídeo selecionado
Busca baseada em dados de MS/MS(sequenciamento)
Busca em Banco de Dados (ex. Mascot)
Busca baseada em dados de MS
(PMF)
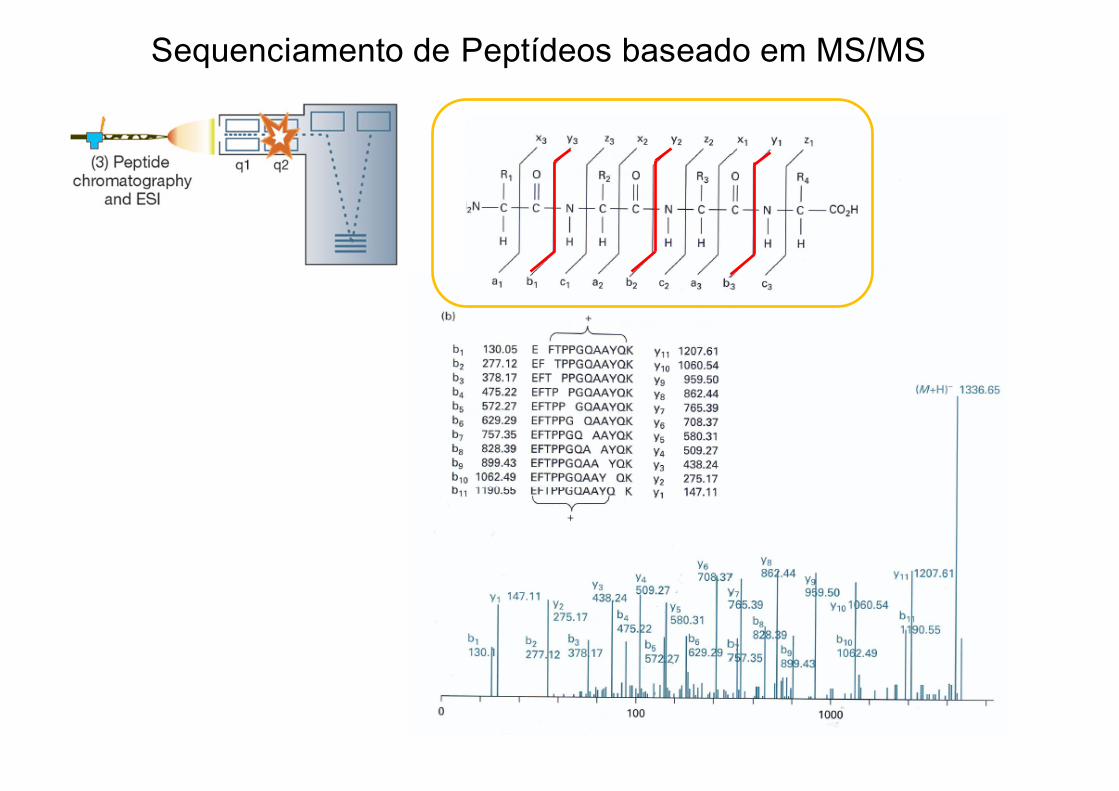
Sequenciamento de Peptídeos baseado em MS/MS
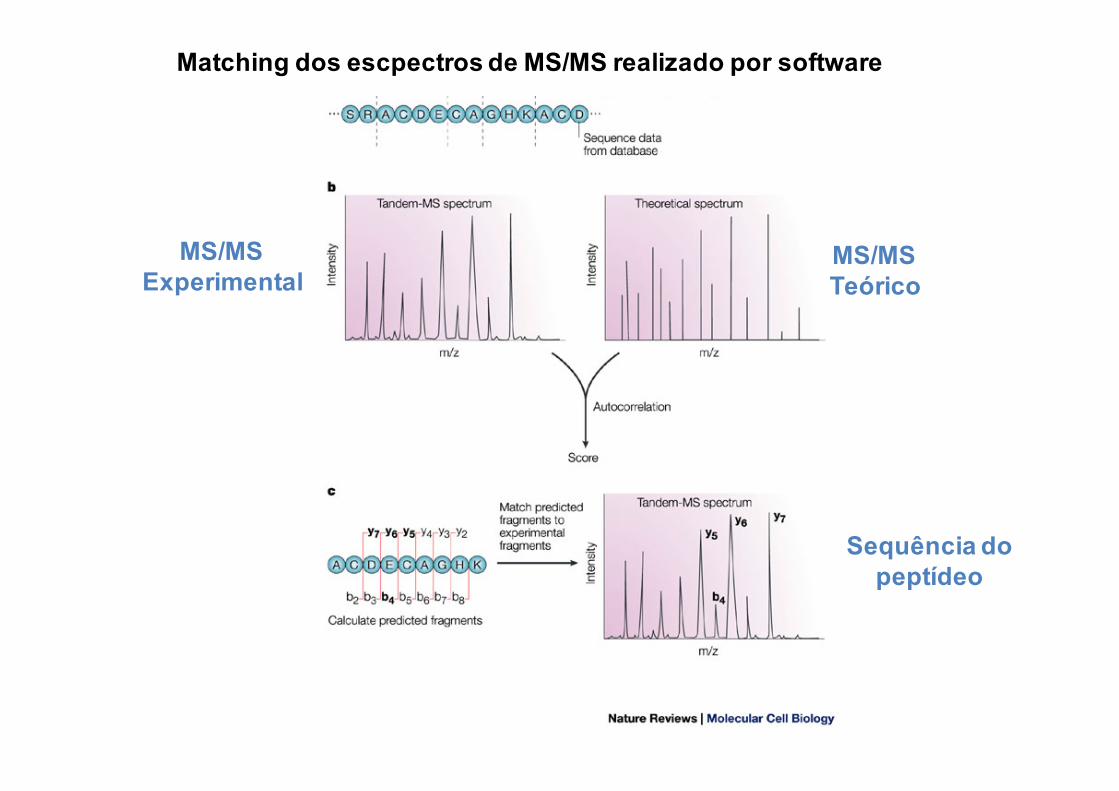
Matching dos escpectros de MS/MS realizado por software
MS/MSExperimental
MS/MSTeórico
Sequência do peptídeo

Resumo: Identificação de Proteínas/Enzimas
1. Determinação da massa molecular • SDS-PAGE• cromatografia de exclusão molecular• espectrometria de massas (MALDI-TOF, Q-TOF)
2. Determinação da sequência de aminoácidos• Degradação de Edman• Espectrometria de massas
Em ambos os casos proteínas precisam ser clivadas especificamente (ex. Proteases) para gerar peptídeos menores.
A espectrometria de massas é a técnica mais precisa e mais utilizadaatualmente.

Eletroforese em gel bidimensional das proteínas de E. coli
- mais de 2,000 proteínas podem ser visualizadas
Lenhinger
Utilizada para comparar níveis de expressão de proteínas
Proteínas de interesse podem ser identificadas utilizando-se anticorpos
Proteínas são identificadas por espectrometria de massas
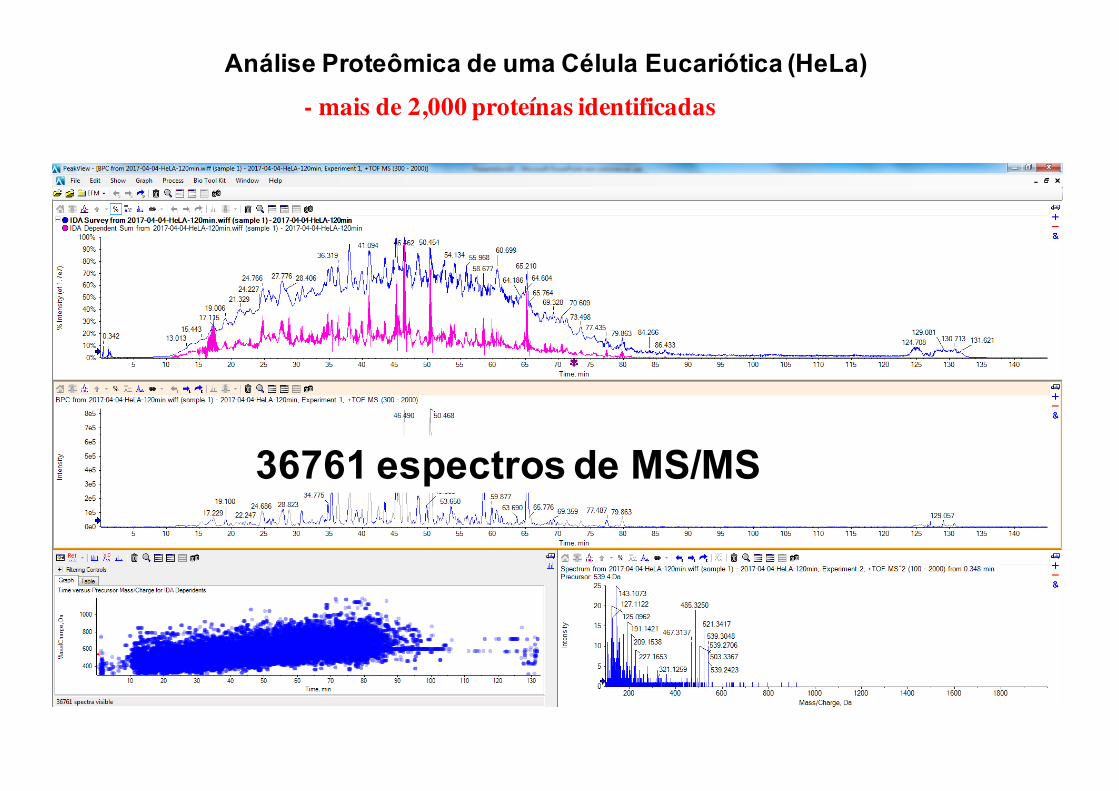
36761 espectros de MS/MS
Análise Proteômica de uma Célula Eucariótica (HeLa)- mais de 2,000 proteínas identificadas

Avisos:
Gabaritos e materiais da aula disponíveis na página do Sandro
Entregar exercício 1
Resolver exercício 2 em casa
Próxima aula (20/Junho) teremos plantão de dúvidas na sala 4
Prova 2 será no dia 27/Junho no Anfiteatro Vermelho