helena brentani [email protected] professora ... · 2 -0.10 0.49 0.24 0.06 0.46 ... ... o...
TRANSCRIPT

Análise de expressão gênica
Helena Brentani
Professora Departamento de Psiquiatria da FMUSP

DNA
RNA
Proteínas
Funçõescelulares

Como estudar o transcriptoma
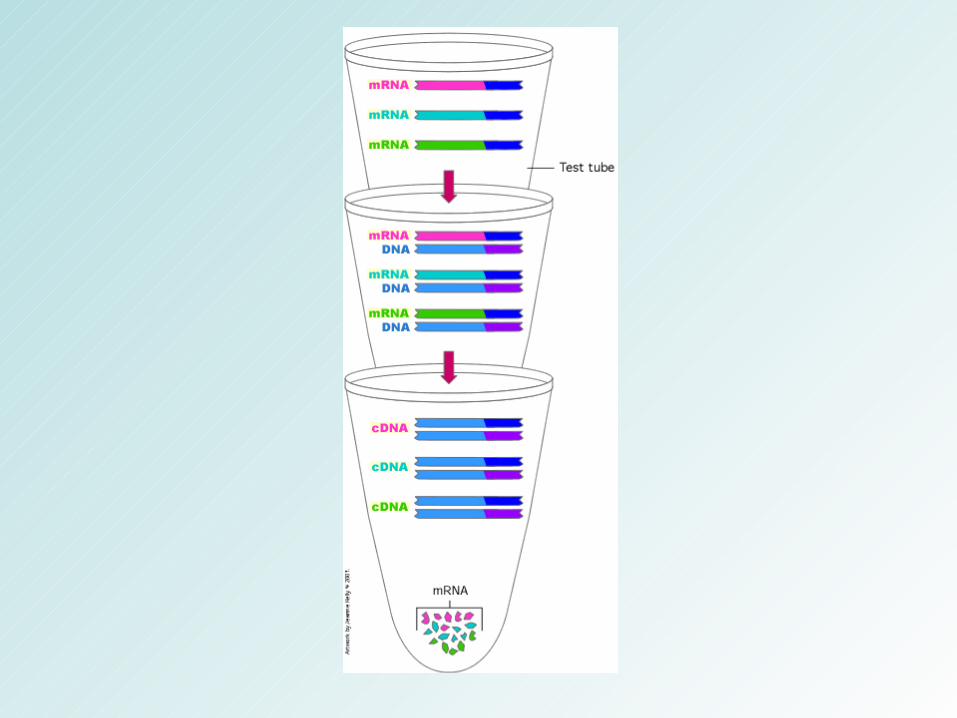

Análises em larga-escala
ESTs; SAGE; Microarray; RNA-seq
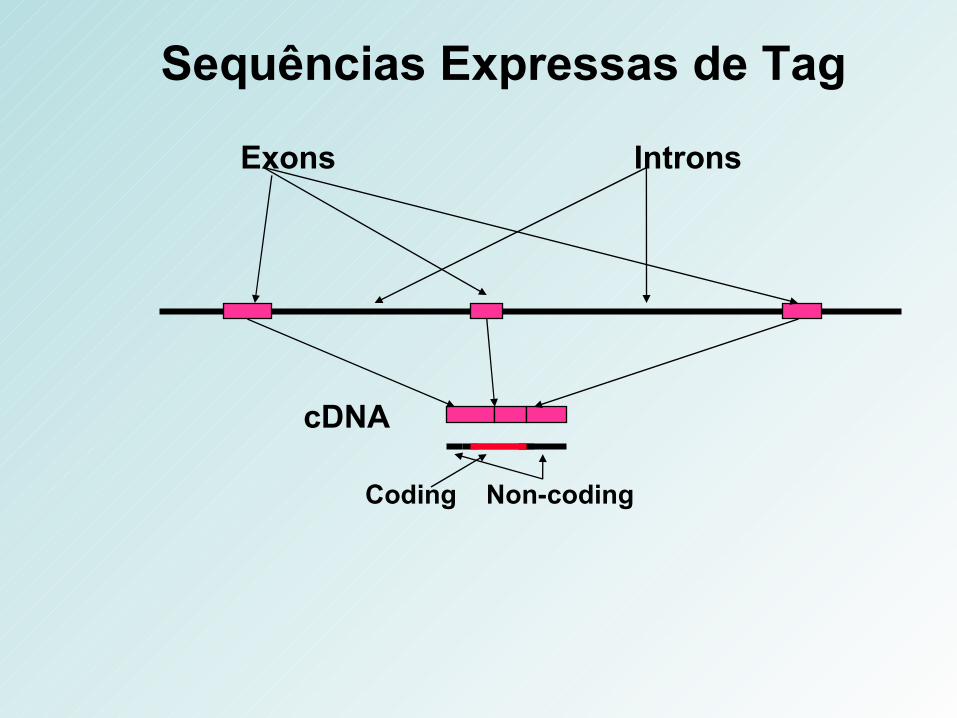
Exons Introns
cDNA
Coding Non-coding
Sequências Expressas de Tag
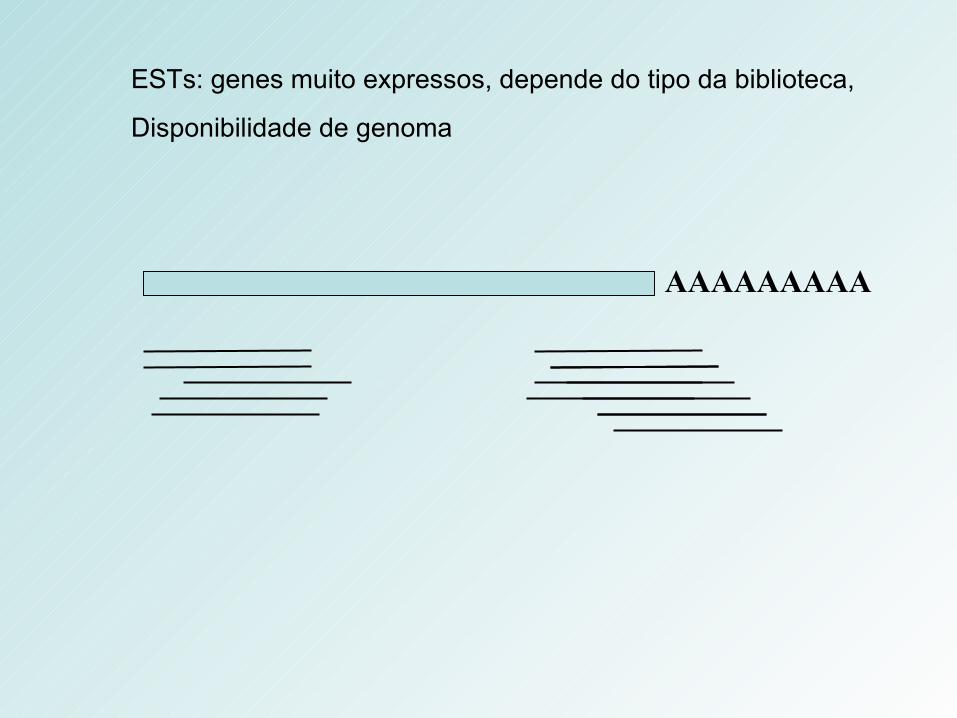
AAAAAAAAA
ESTs: genes muito expressos, depende do tipo da biblioteca,
Disponibilidade de genoma
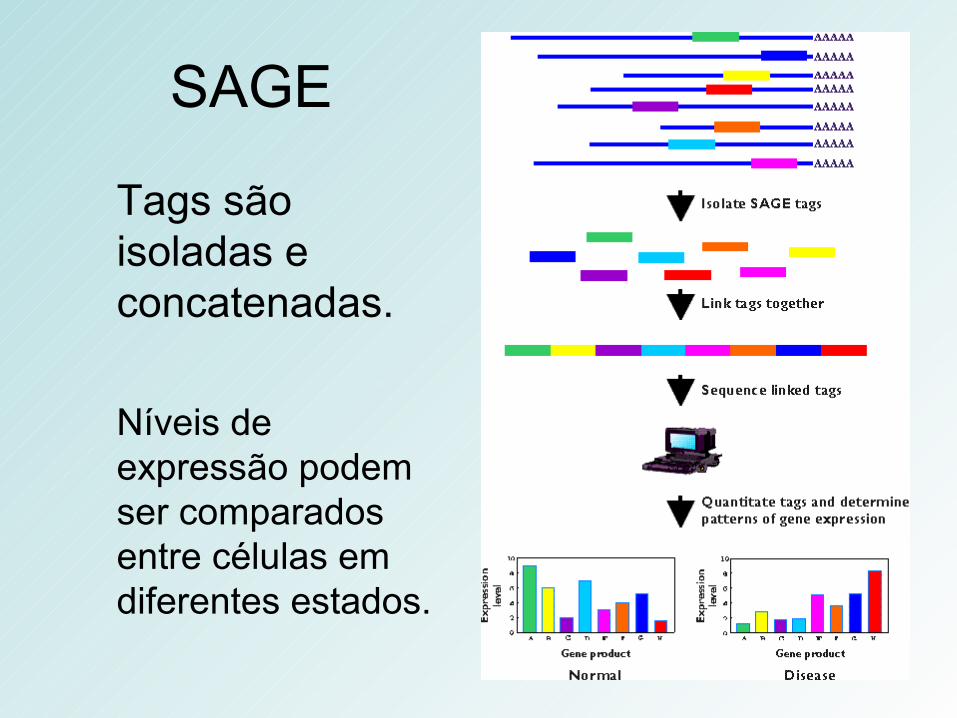
SAGE
Tags são isoladas e concatenadas.
Níveis de expressão podem ser comparados entre células em diferentes estados.

SAGE – pros e contras
Vantagens
•Não há hibridização nem referência – as medidas são relativas somente ao total de tags da biblioteca;
•Teoricamente, todos os mRNAs são medidos através das tags – não é preciso fixá-los;
•Maior sensitividade
Shui Qing YE, Tera LAVOIE, David C USHER, Li Q. ZHANG - Cell Research 2002; 12(2):105-115
Dificuldades
•Relacionar tag com transcrito;
•Custo elevado;

MicroArray

Microarray – prós e contras
Vantagens:
•Custo relativamente baixo
•Muitos transcritos (600-25k)
•Relação cDNA->gene quase inequívoca
Desvantagens
•Baixa reproducibilidade
•Two-colors: hibridização competitiva, diferença de marcação de um mesmo cDNA
•One-color: falta de parâmetro para comparar lâminas
•Dificuldade de mesclar experimentos numa mesma análise

As cinco etapas da análise de expressão gênica
Preparação das amostras
Reação Bioquímica
Spot identification
Análise dos dados
Questão Biológica/desenho
experimental

Verificação biológicae interpretação
Experimento de Microarray
Desenho experimental
Análise de imagem
Normalização
Questão biológica(baseada em hipóteses ou
exploratória)
TesteEstimaçã
oDiscriminaç
ão
AnáliseClusterizaçã
o
Medida daqualidade
Falhou
Passou
Pré-processamento

Desenho experimental
• Número de indivíduos para cada classe– Estimar a variabilidade biológica entre os indivíduos
da mesma classe
• Número de replicatas– Estimar a variabilidade experimental
• Tipo de desenho experimental– Reference design
– Balanced Block design
– Loop design
• Número limitado de bibliotecas, arrays...

Preparação das Amostras
– Extração do RNA total– Qualidade do RNA– Amplificação– Controle da amplificação– Tipos de protocolos

Identificação de Pontos
Histograma de pixel
Intensidade de um único ponto
• O sinal do ponto é quantificado.
„Donuts“
Média / Média / MedianaMediana / / ModoModo // 75% quantil 75% quantil

Dados brutos não sãoconcentrações de mRNA
• Contaminação do tecido
• Degradação do RNA
• Eficiência na amplificação
• Eficiência da transcrição reversa
• Eficiência na Hybridização e especificidade
• Identificação de clones e mapeamento
• Redimento da PCR e contaminação
• Eficiência dos pontos
• Suporte de ligação ao DNA
• Questões relacionadas a
outros fabricantes de
arranjos
• Segmentação da imagem
• Quantificação do sinal
• Correção do fundo
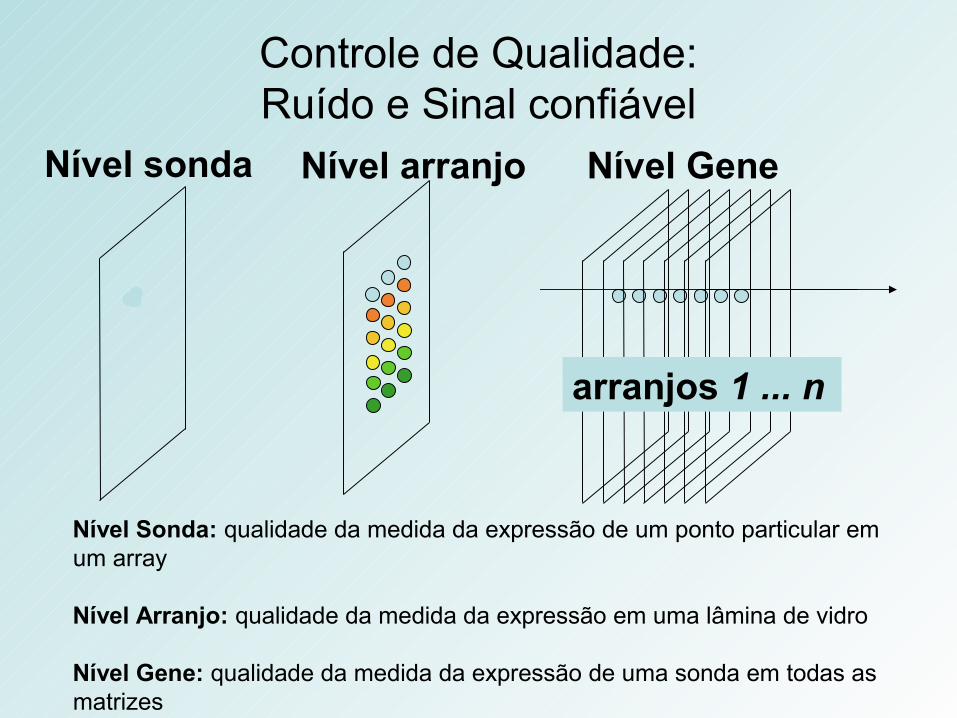
Controle de Qualidade:Ruído e Sinal confiável
arranjos 1 ... n
Nível arranjo Nível GeneNível sonda
Nível Sonda: qualidade da medida da expressão de um ponto particular em um array
Nível Arranjo: qualidade da medida da expressão em uma lâmina de vidro
Nível Gene: qualidade da medida da expressão de uma sonda em todas as matrizes

Controle de Qualidade do Nível da Sonda
• Pontos individuais impressos no slide• Fontes:
– impressão defeituosa, distribuição desigual, contaminação por detritos, magnitude do sinal em relação ao ruído, medição deficiente dos pontos;
• Inspeção visual:– cabelos, pó, arranhões, bolhas de ar, as regiões escuras, regiões com
neblina;• Qualidade do Ponto:
– Brilho: primeiro plano em relação a razão fundo– Uniformidade: variação da intensidades em pixel e a razão da intensidade
de um ponto– Morfologia: área, perímetro, circularidade.– Tamanho do Ponto: número de pixels no primeiro plano
• Ação:– medidas definidas para NA (valores não presentes).– local procedimentos de normalização, que representam idiossincrasias
regionais.– use weights for measurements to indicate reliability in later analysis.

Controle de Qualidadodo Nível do Arranjo
• Problemas:– Defeito de fabricação do arranjo– Problema com a extração de RNA– Falha na reação de rotulagem– Más condições de hibridização– Leitor avariado
• Medidas de Qualidade:– Porcentagem de pontos em sinal (~30% pontos excluídos) – Calibre da intensidade– (Av. Primeiro plano)/(Av. Fundo) > 3 nos dois canais– Distribuição do sinal da área dos pontos– Valor do ajuste necessário: sinais que alteram substancialmente
para comparar slides.

Controle de Qualidadedo Nível do Gene
Gene g• Hibridização ruim no canal de
referencia. Pode introduzir viés na mudança do fold.
• Algumas probes pode não hibridizar
bem com o RNA.
• Problemas de impressão: como
todos os pontos de um bom relatório,
podem ter baixa qualidade.
• Baixa qualidade – Contaminação
• Genes com baixo sinal de consistência no canal de referência são suspeitos

Gene
Amostras mRNA
Nível de expressão do gene ou razão por gene i na amostra de RNA j.
M =Log2 (intensidade vermelho/ intensidade verde)
Função (PM, MM) do MAS, dchip ou RMA
amostra1 amostra2 amostra3 amostra4 amostra5 …
1 0.46 0.30 0.80 1.51 0.90 ...2 -0.10 0.49 0.24 0.06 0.46 ...3 0.15 0.74 0.04 0.10 0.20 ...4 -0.45 -1.03 -0.79 -0.56 -0.32 ...5 -0.06 1.06 1.35 1.09 -1.09 ...
A =Média: log2(intensidade vermelho ), log2(intensidade verde)
Função (PM, MM) do MAS, dchip ou RMA
Dados de Expressão Gênica
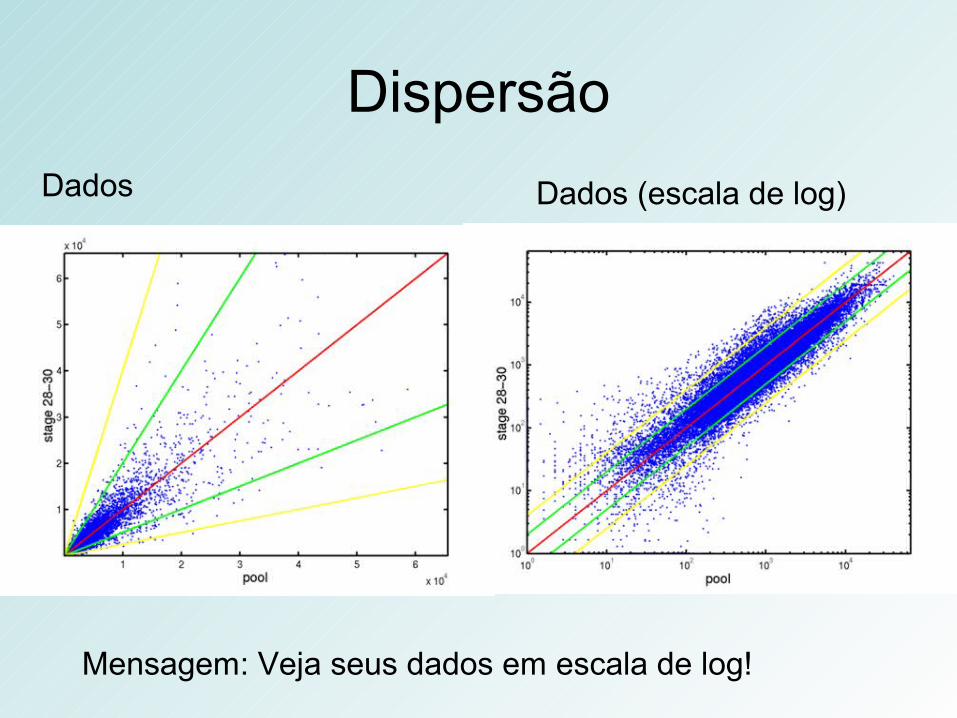
Dados Dados (escala de log)
Dispersão
Mensagem: Veja seus dados em escala de log!

MA Plot
A = 1/2 log2(RG)
M =
log
2(R
/G)

Centralização da Mediana
Log S
inal,
centr
aliz
ado n
o 0
Uma das estratégias mais simples é a de trazer todos os centros "da matriz de dados ao mesmo nível.
Assunção: a maioria dos genes permanecem inalterados entre as condições.
A mediana é mais robusta a outliers do que a média.
Divide todas as medições de expressão de cada matriz pela mediana.

Problemas de Centralização da Mediana
Verde Log
Ver
mel
ho L
og
Dispersão do sinal logado depois da Centralização da Mediana
A = (Verde log+ Vermelho log) / 2
M =
Ver
mel
ho L
og –
Ver
de L
og
M-A Plot dos mesmos dados
Centralizar a Mediana é um método global. Não ajusta para efeitos locais, efeitos dependente da intensidade, efeitos de impressão, etc

Normalização por Lowess
A = (Verde Log + Vermelho Log) / 2
M =
Ver
mel
ho L
og –
Ver
de L
og
Local estimado Use a estimativa para
dobrar a banana em linha reta

Partindo de Dados Conhecidos
Gene
Amostras mRNA
amostra1 amostra2 amostra3 amostra4 amostra5 …
1 0.46 0.30 0.80 1.51 0.90 ...2 -0.10 0.49 0.24 0.06 0.46 ...3 0.15 0.74 0.04 0.10 0.20 ...4 -0.45 -1.03 -0.79 -0.56 -0.32 ...5 -0.06 1.06 1.35 1.09 -1.09 ...
Ok, agora nós temos certeza que nosso dado é de alta qualidade e sistemático, efeitos não biológicos foram removidos.O resultado é uma matriz de expressão gênica
Já um resultado? Não! São apenas dados, não conhecimento.Precisamos usar estes dados para responder a uma questão científica.

Desenho do Experimento
• Tipo I: (n = 2)– Como é que este gene expresso no alvo 1 em
relação ao alvo 2?
– Que genes mostram alta/baixa regulação entre os dois alvos?
• Tipo II: (n > 2)– Como é que a expressão do gene A varia ao longo
do tempo, tecidos ou tratamentos?– Será que algum dos perfis de expressão exibem
padrões similares de expressão?

Tipos de experimentose a ferramenta adequada
Três tipos básicos de perguntas que os experimentos de microarray se propõe a resolver:
Genes diferencialmente expressos
•Encontrar genes que se comportem de maneira diferente em duas classes, com evidência estatística
•Teste T, Teste exato de Fischer, Chi quadrado, Fold change, BER
Padrões de expressão
•Encontrar listas de genes que tenham comportamento semelhante
•Clustering hierárquico
Classificação de amostras
•Encontrar uma lista reduzida de genes (1 para cada ~20 amostras) cujo comportamento permita predizer alguma informação sobre a amostra, e então construir um classificador
•Encontrar os genes (data mining): SVM-FS, busca exaustiva
•Construir o classificador: SVM, plano de Fischer, CART
Networks
•Encontrar redes de relação
entre genes
•Correlação, TOM, entropia

Métodos estatísticos usuais de análise de expressão diferencial
• Média/desvio padrão de classes• Fold change• SAM• Teste t student• wilcoxon• Bayes Error Rate• ANOVA• Correção multi-testes (Bonferroni, pFDR)

Padrões de Expressão Gênica
• Dados “Eisen”ized (dendrogramas)
• Mapas Auto-Organizados
• Análises dos Componentes Principais
• Agrupamento de K-médias

Classificação por SVM
• O SVM tenta encontrar um hiperplano separador ótimo entre os membros de duas classificações iniciais.
Separating hyperplane

Máquinas de Vetores de Suporte (SVM, Support Vector Machines)
Hiperplano separadorde margem maximal
Pontos mais próximosa hiperplano separador = vetores de suporte

O quão bem nós fizemos?
O classificador geralmente irá agir pior que antes:
Erro de teste > Erro de treinamento
Mesmo classificador (= linha)
Novos dados das mesmas classes
Erro de treinamento: O quão bem nós fazemos no dados nos quais treinamos o classificador?
Mas quão bem nós faremos no futuro, em novos dados?
Erro de teste: O quão bem o classificador generaliza?

EASE(Expression Analysis Systematic Explorer)
A análise do EASE identifica temas biológicos prevalentes em clusters de genes.
A significância de cada tema identificado é determinada por sua prevalência no cluster e na população de genes da qual o cluster foi criado.

Sistema de arquivos EASE

-Quais são alguns dos temas biológicos predominantes representados em um cluster e como deveria ser atribuída significância a um tema biológico descoberto?
Considere certo cluster…

Exemplo:
Tamanho da População: 40 genesTamanho do Cluster: 12 genes
10 genes, mostrados em verde, têm um tema biológico comum e 8 ocorrem no cluster.

A frequência do tema na população é 10/40 = 25%
A frequência do tema no cluster é 8/12 = 67%
40
12
10
8
* 80% dos genes relacionados ao tema na populaçãoterminaram em um cluster relativamente pequeno.
E
Considere o resultado

Atribuindo Significância às Descobertas
Teste Exato de Fisher, distribuição Hipergeométrica, Cálculo das probabilidades.
8 2
4 26
in out
in
out
Cluster
Tema p ≈ .0002
( 2x2 matriz de contingência)

Conjunto de genes cujos perfis de expressão são preditivos de algum outro.
Genes com baixa entropia(variabilidade mínima nos experimentos)são excluídos da análise.
H = -Σp(x)log2(p(x))x=1
10
Pode ser usado para identificar correlações negativas entre genes.
Redes de Relevância

Redes de Relevância
Coeficientes de correlação fora dos limites definidos pelos limiares máximo e mínimo são eliminados.
A
D
E B
C
.28
.75
.15.37
.40
.02
.51
.11
.63
.92A
D
E B
C
Tmin = 0.50O padrão de expressão da cada gene comparado aos de todos os outros.
À capacidade de cada gene de predizer a expressão de cada um dos outros genes é atribuído um coeficiente de correlação
Tmax = 0.90
As relações entre genes que restam definem as subredes

Microarray
• 1990s tecnologia escolhida para estudos em large-scale expressão gênica
• Habilidade em verificar simultaneamente milhares de transcritos, levou importantes avanços para:
– Identificação de genes diferencialmente expressos
– Farmacogenômica
– Evolução da regulação gênica
• Limitações– Níveis de hibridização do Background (sonda que ocorre independentemente do
nível de expressão correspondente do transcrito)
– Precisão da medida de expressão, particularmente por transcritos presentes em baixa abundância

Sequenciadores Segunda Geração
• Roche/454 and AB SOLiD– Construção in vitro adaptadores (adaptadores de amarelo e
verde)
– Multi-template PCR, com um único par de primers.
– Emulsão em água e óleo.
– Adição de Beads
Morozova, O., Hirst, M., & Marra, M. A. (2009). Applications of New Sequencing Technologies for Transcriptome Analysis. Annual Review of Genomics and Human Genetics. doi: 10.1146/annurev-genom-082908-145957.

Sequenciadores Segunda Geração
• Illumina– Construção in vitro adaptadores (adaptadores de amarelo e
verde)
– Amplificação por bridge PCR (ponte)
– Os dois primers podem são fixados
Morozova, O., Hirst, M., & Marra, M. A. (2009). Applications of New Sequencing Technologies for Transcriptome Analysis. Annual Review of Genomics and Human Genetics. doi: 10.1146/annurev-genom-082908-145957.

RNA-SeqNext-Generation Sequencing
(NGS)

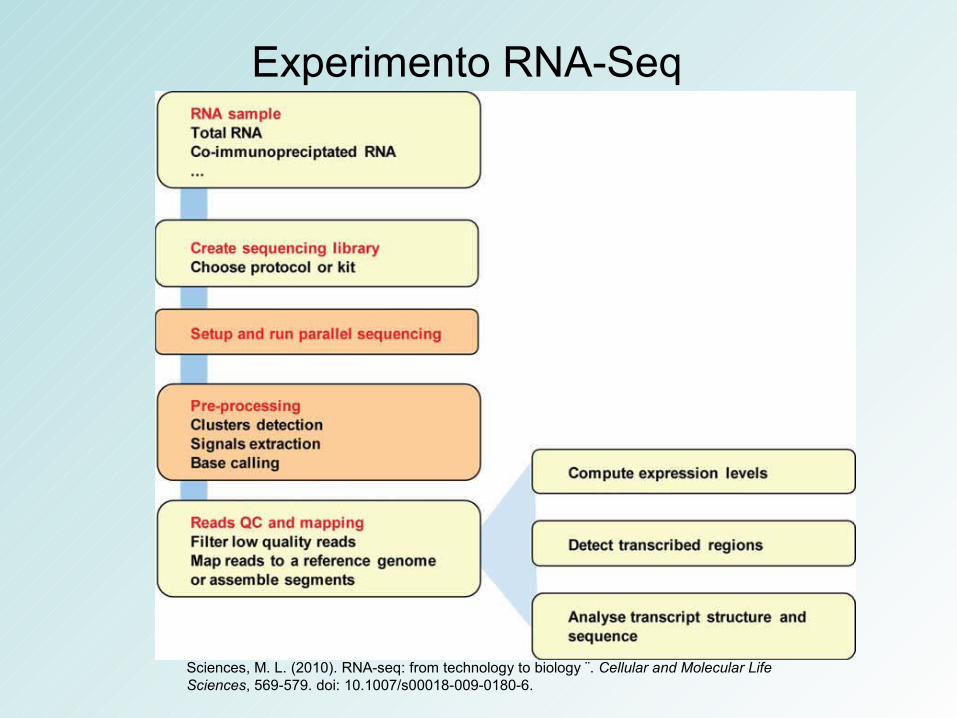
Experimento RNA-Seq
Sciences, M. L. (2010). RNA-seq: from technology to biology ¨. Cellular and Molecular Life Sciences, 569-579. doi: 10.1007/s00018-009-0180-6.

Qualidade•
Diferentes plataformas NGS, diferentes formas de avaliar e filtrar a qualidade da sequência.
Exemplo: Sequência 454
>FPO586001EP0DK rank=0421118 x=1819.0 y=1494.0 length=495
TACCTCTCCGCGTAGGCGCTCGTTGGTCCAGCAGAGGCGGCCGCCCTTGCGCGAGCAGAA
TGGCGGTAGGGGGTCTAGCTGCGTCTCGTCCGGGGGGTCTGCGTCCACGGTAAAGACCCC
GGGCAGCAGGCGCGCGTCGAAGTAGTCTATCTTGCATCCTTGCAAGTCTAGCGCCTGCTG
CCATGCGCGGGCGGCAAGCGCGCGCTCGTATGGGTTGAGTGGGGGACCCCATGGCATGGG
GTGGGTGAGCGCGGAGGCGTACATGCCGCAAATGTCGTAAACGTAGAGGGGCTCTCTGAG
TATTCCAAGATATGTAGGGTAGCATCTTCCACCGCGGATGCTGGCGCGCACGTAATCGTA
TAGTTCGTGCGAGGGAGCGAGGAGGTCGGGACCGAGGTTGCTACGGGCGGGCTGCTCTGC
TCGGAAGACTATCTGCCTGAAGATGGCATGTGAGTTGGATGATATGGTTGACGCTGGAAG
ACGTTGAAGCTGGCG
>FPO586001EP0DK rank=0421118 x=1819.0 y=1494.0 length=495
40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 34 34 21 21 21 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 33 33 33
40 40 40 40 39 39 39 40 26 26 26 26 26 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 22 22 22 22 22 22 40 40 40 40 40 40 40 40 40
26 26 26 25 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40
40 40 40 40 40 40 40 40 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 40 40 39 39 40 28 30 30 30 30 40
34 40 37 37 37 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 34 24 19 17 17 28 36 36 40 36 28 17 17 19 30 40 40 40 40
40 35 34 34 39 39 40 40 40 40 40 40 40 40 39 35 34 34 34 40 40 34 39 39 40 39 39 39 39 39 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40
40 40 35 34 34 40 40 40 40 40 40 40 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 28 28 28 36 34 34 28 32 28 28 22 22 19 32 30 30 28 27 27
Default Phred paraQualidade = 20

RNA-Seq mapeamento
• Contagem confiável no mapeamento contra o genoma de referência ou de novo assembly.
• Desafios para obter mapeamento com eficiência e confiabilidade– Recursos computacionais para mapear grande número de reads
– Taxa de erro relativamente alta. (o número de alinhamentos, não perfeitos, deve ser considerado).
– Taxa de erro é particularmente relevante para detecção de Single Nucleotide Polymorphism (SNP)
– Distinguir erros de sequenciamento de SNPs
– Viés e artefatos da preparação das bibliotecas e procedimentos de sequenciamento.

RNA-Seq Quantificação
Quantificar o número de genes que foi representado
• O número de reads por gene
• Tamanho do gene representado
– Genes grandes
• Protocolos foram utilizados na construção da biblioteca
– Diferentes plataformas e protocolos
C = Número de reads mapeados
N = Número total de reads mapeados
L = Tamanho total dos éxons
Mortazavi et al., 2008

RNA-Seq
• Contagem confiável do mapeamento gerado contro o genoma de referência.
• Possibilidade de encontrar rearranjos, modificações pós-transcricionais que não foram mapeados diretamente no genoma de referência
• de novo assembly de dados de RNA-Seq
• Grande cobertura
• Alta sensibilidade que permite a detecção de mais transcritos, comparado com o que é detectado com Microarray.
Microarray RNA-Seq
O scaner retorna a intensidade do sinal para cada probe do experimento de array
O número de reads mapeado para qualquer região do genoma representa o sinal

RNA-Seq x Microarray(expressão gênica)
• Illumina
• Affymetrix U133 Plus 2 arrays
• Procurar genes diferencialmente expressos entre amostras de:
– Rim
– Fígado

RNA-Seq x Microarray(expressão gênica)
• Para analisar a diferença de achados, utilizaram a técnica Quantitative PCR – (qPCR) para testar a diferenças de expressão entre as amostras de Rim e Fígado.
• 5 genes considerados diferencialmente expressos apenas nos dados de sequenciamento foram escolhidos (MMP25, SLC5A1, MDK, ZNF570, GPR64).
• 6 genes considerados diferencialmente expressos nos dados de array foram escolhidos (C16orf68, CD38, LSM7, S100P, PEX11A, GLOD5)
.
RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Research, 1509-1517. doi: 10.1101/gr.079558.108.

• Dados do sequenciamento– Dos 5 genes, 4 foram confirmados por qPCR. O gene (ZNF570)
como falso positivo.
• Dados de array– Dos 6 genes, apenas 2 foram confirmados (CD38 e GLOD5).
RNA-Seq x Microarray(expressão gênica)

Aderbal R. T. Silva, Paulo J. S. Silva, Cecília Feio, Luis P. Camargo, Lea T. Grinberg, Renata E. L. Ferreti, Renata Leite, José M. Farfel, Cesar H. Torres, Dirce M. Carraro, Diogo Patrão, and
Brazilian Aging Brain Study Group
Marcadores biológicos de diagnóstico precoce e prognóstico em AlzheimerUsando dados de expressão gênica

O PROBLEMA

Cadastro inicial do paciente e
amostras
Nro do VO, Nome do paciente, Questionários (opcionais)Qtd tubos por região do cérebroQtd tubos de sangue, timo, rim, etc
Geração do código de barras
Para cada amostra cadastrada, o sistema gera e imprime um código de barras único
Armazenamento das amostras
Freezer específico para cada tipo de amostra, controle de posição por código de barras.
Retirada das amostras
Em qual cuba de formaldeído está o hemisfério esquerdo?Em que freezer está o tubo de sangue?
Transformação das amostras
Hemisfério em cuba de FA => K7K7 => Lâmina HELâmina HE => Lâmina coradaTecido/sangue => RNA e/ou DNA
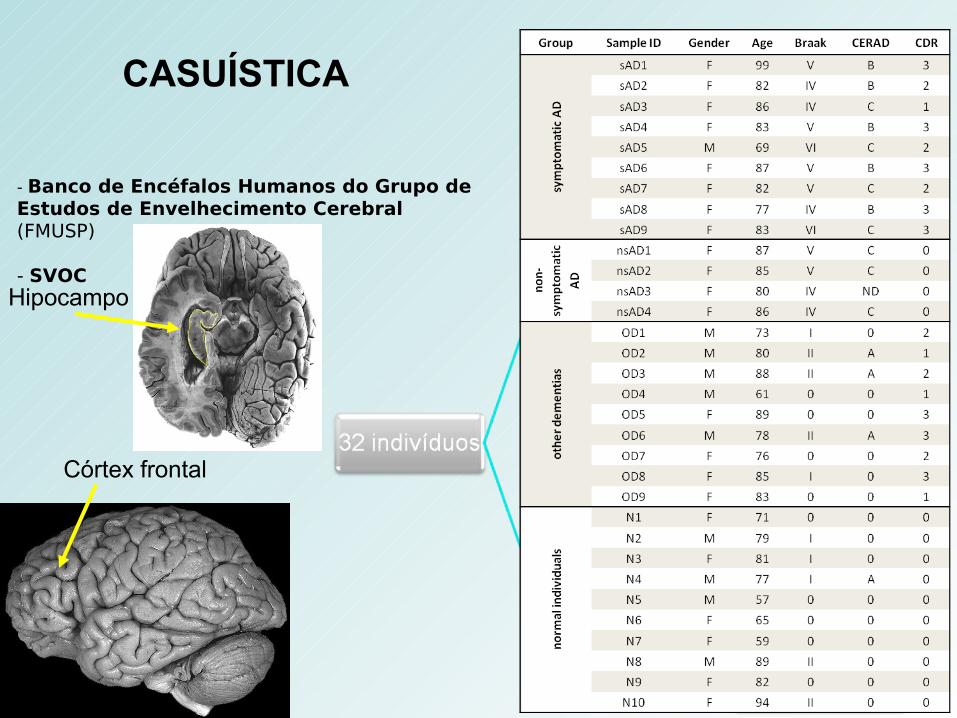
- Banco de Encéfalos Humanos do Grupo de Estudos de Envelhecimento Cerebral (FMUSP)
- SVOC
Córtex frontal
Hipocampo
CASUÍSTICA

Tecido ExperimentalsAD/nsAD/OD/N
Tecido de ReferênciaApanhado de 15 linhagens celulares
Isolação Total de RNAMini Kit RNeasy - QIAGEN
Amplificação de mRNA2 ciclos
abordagem com polimerase T7
Trascrição reversa,rotulada com fluorescência
Cy3 (verde) e Cy5 (vermelho)
Plataforma de cDNA personalizada: - 4.800 cDNA sequências- 4.608 genes humanos
- 192 controles positivos e negativos
Captura da Intensidadedo Sinal (Scanner)
Análise estatística
ANOVA (p < 0.05)
Processos biológicos (GO) (p < 0.05) - WebGestalt
classificadores lineares, com 3 genes
MATERIAL E MÉTODOS

A pergunta?
+ -
+DA definida
(sintomática)Outras
Demências
- DA assintomática
Indivíduos Normais
Apresentação Clínica de Demência
Neuropatologia da Doença de Alzheimer (DA)
Reserva Cognitiva
Capacidade de tolerar alterações relacionadas a idade/doença no cérebro sem desenvolver sintomas ou sinais claros

Resultados
• Genes diferencialmente expressos
DAa → DAd

Resultados• Processos Biológicos (GO)

Resultados• Rede dos processos biológicos
Module 1 Module 2
Up-reguladosDown-regulados

Resultados
• Vias de Sinalização (KEGG)

Resultados
• Clusterização Hierárquica (genes fold ≥│1,8│)
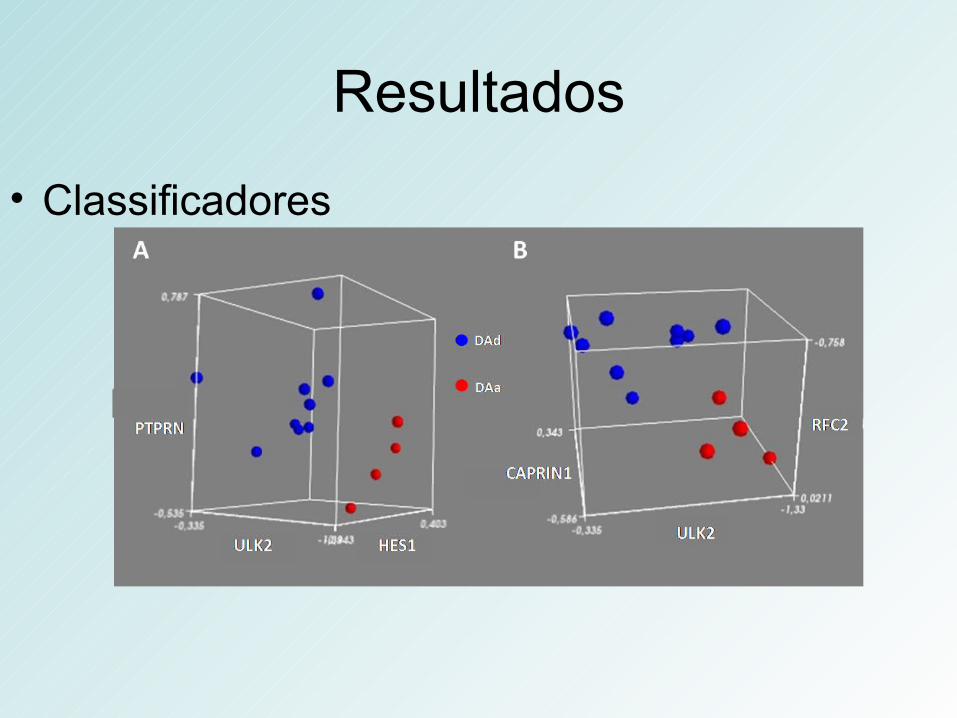
Resultados
• Classificadores

Conclusões• Genes que distinguem DAa de DAd estão
envolvidos, principalmente, com ciclo celular e plasticidade sináptica.
• HIPÓTESE
Neurônios erroneamente convertem sinais que seriam utilizados para plasticidade sináptica na ativação do ciclo celular, o que, subsequentemente, leva-os à morte.
DA assintomática
Neurônios diferenciados, após se retirarem do ciclo celular, são capazes de usar alternativamente mecanismo, essencialmente desenvolvido para controlar proliferação, para controlar plasticidade sináptica.
DA ‘sintomática’
Arendt T, Bruckner NK. Biochim Biophys Acta. 2007; 1772:413-421.Frank CL, Tsai LH. Neuron. 2009; 62:312-326.

Análise global de transcrição por sequenciamento paralelo para avaliar ativação gênica mediada por
super expressão de ERBB2

• HER2/neu (também conhecido como ErbB-2, ERBB2)
• Significa: Human Epidermal growth factor Receptor 2, é um proteína que confere maior agressividade em câncer de mama. É membro da família ErbB, mas conhecida como a família do receptor epidérmico do fator de crescimento.
• 20 – 30 % de câncer de mama super expressa HER2 – Herceptin
ERBB2

Experimento• Método para comparação de Whole Transcriptome com múltiplas
amostras
• Avaliação da arquitectura e as alterações quantitativas da transcrição.
– DpnII-restriction
– 4nt-tagging Barcoding
– Roche-454 platform
• Hb4a and C5.2 (super expressa ERBB2) (Harris et al., Int J Cancer. 1999 Jan 29;80(3):477-84)

Figure 1. Schematic representation of cDNA Figure 1. Schematic representation of cDNA libraries. libraries.
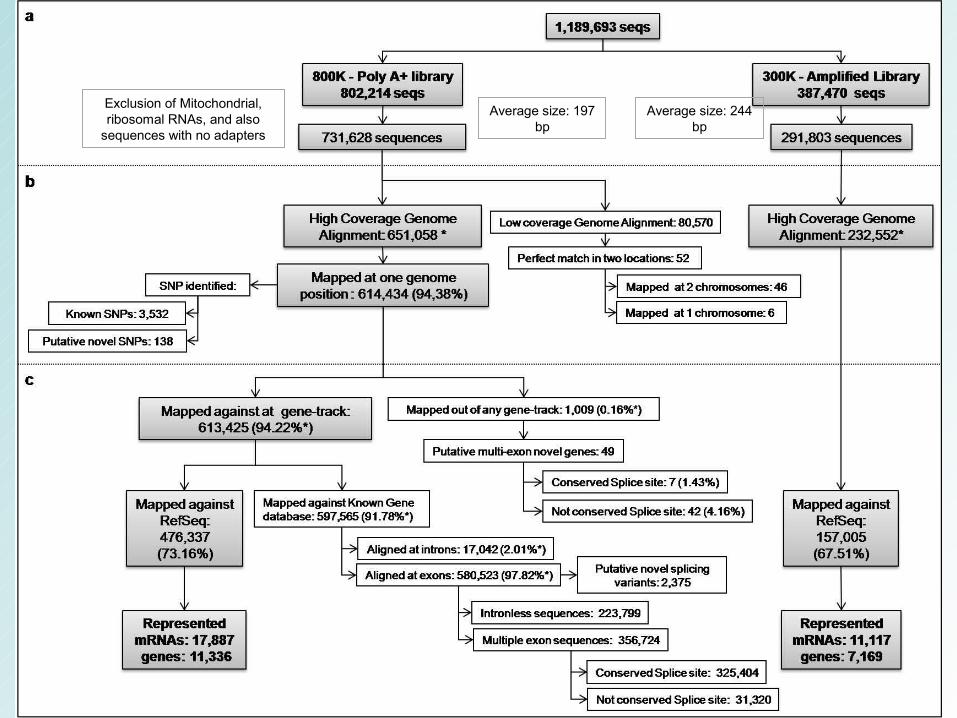
Exclusion of Mitochondrial, ribosomal RNAs, and also
sequences with no adapters
Average size: 197 bp
Average size: 244 bp

A – A frequência dos reads :distribuição dos reads ao longo da posição dos transcritos B – A posição relativa da sequence da Poly A+ biblioteca em relação ao tamanho do transcrito. A espessura das barras corresponde à freqüência de seqüências em cada grupo.
C, D – Dados da biblioteca amplificada.
Frequência da posição relativa de transcritos no RefSeq

Números
• 800K dataset
– 17,887 RefSeq (38.93% - 45,946 entradas do RefSeq - build 36).
– 11,366 human genes (40.74% - 27,827 genes - 7,562 and 3,804 genes were represented by one or 2 to 20 transcripts, respectively).
– Par de bases 23.48% - 14,208,089 nt of the potential 60,500,115 nt were covered
• 300K dataset – 11,117 RefSeq (24.20% - 45,946 entradas do RefSeq - build 36). – 7,1691 human genes (25.76% - 27,827 genes).
– base-pair representation was of 13.32% - 5,661,345 nt of the potential 60,500,115 nt were covered
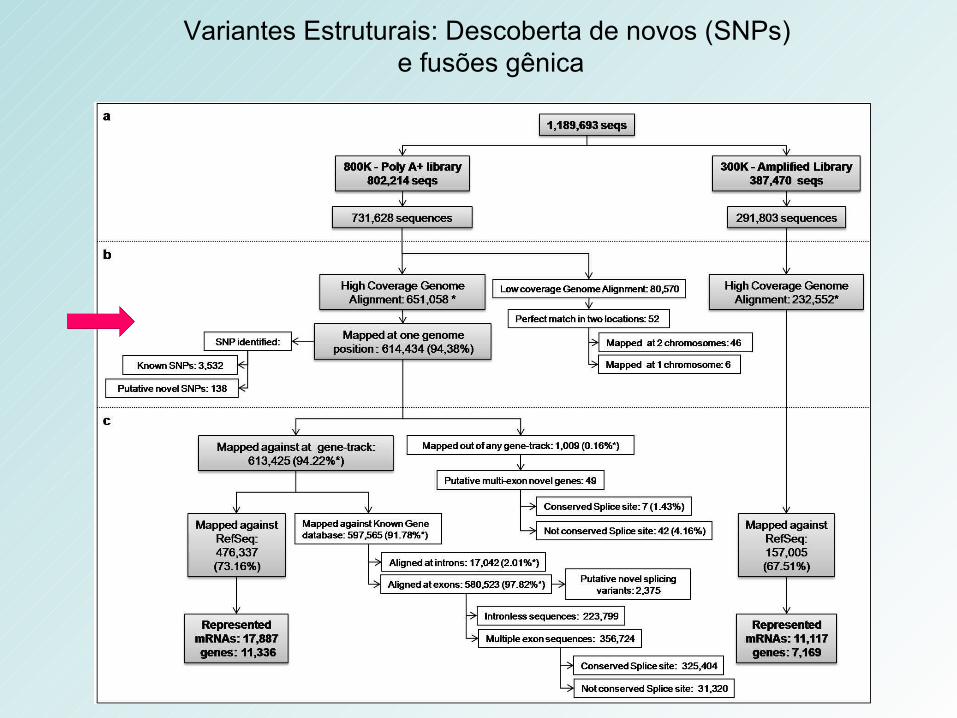
Variantes Estruturais: Descoberta de novos (SNPs) e fusões gênica

Validation of novel SNPs. The eletropherogram represents the validation of the SNPs for each gene. The SNPs from the HB4a and C5.2 cell lines are shown separately and classified as homo or heterozygosis.
18 SNPs mapping to exonic regions were selected to validation -genomic DNA of the two cell lines
High validation rate (89%) - 16 out of 18 SNPs
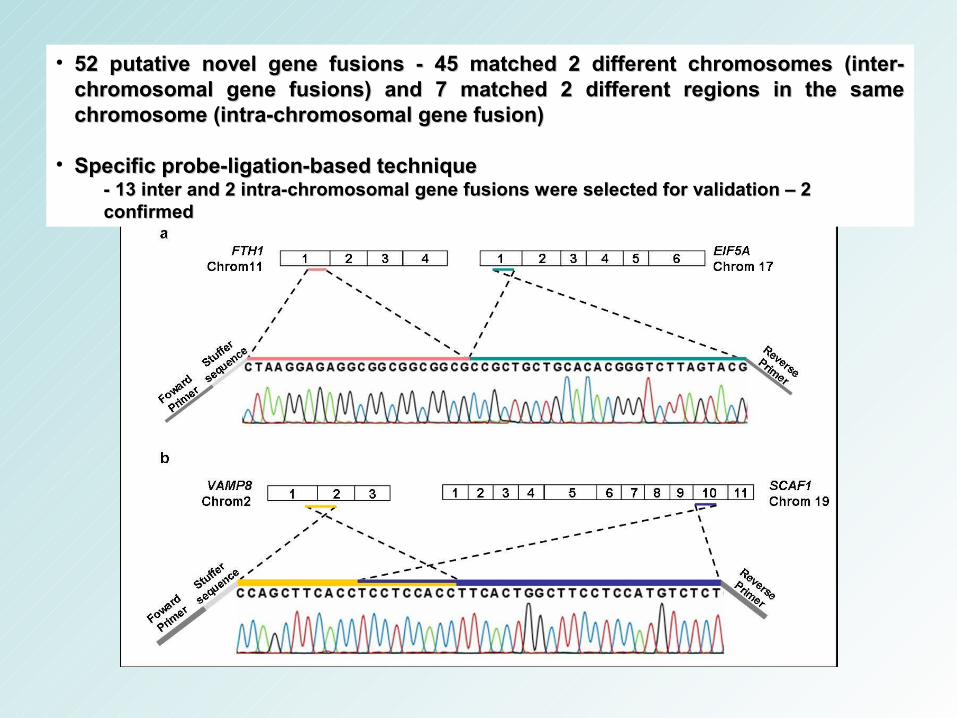
• 52 putative novel gene fusions - 45 matched 2 different chromosomes (inter-52 putative novel gene fusions - 45 matched 2 different chromosomes (inter-chromosomal gene fusions) and 7 matched 2 different regions in the same chromosomal gene fusions) and 7 matched 2 different regions in the same chromosome (intra-chromosomal gene fusion) chromosome (intra-chromosomal gene fusion)
• Specific probe-ligation-based techniqueSpecific probe-ligation-based technique- 13 inter and 2 intra-chromosomal gene fusions were selected for validation – 2 - 13 inter and 2 intra-chromosomal gene fusions were selected for validation – 2 confirmedconfirmed
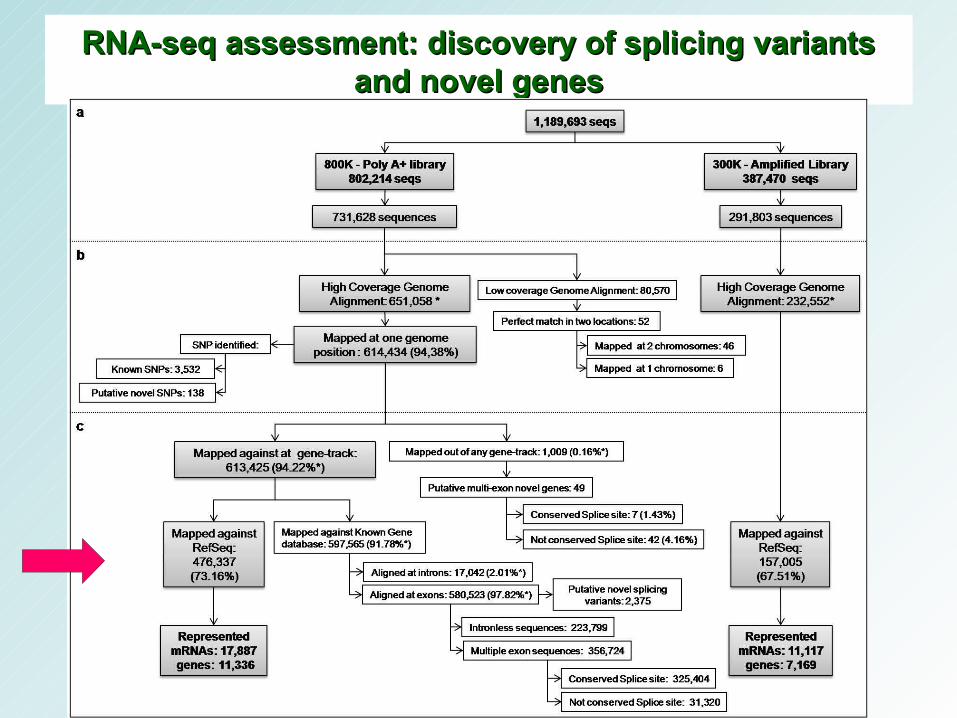
RNA-seq assessment: discovery of splicing variants RNA-seq assessment: discovery of splicing variants and novel genesand novel genes
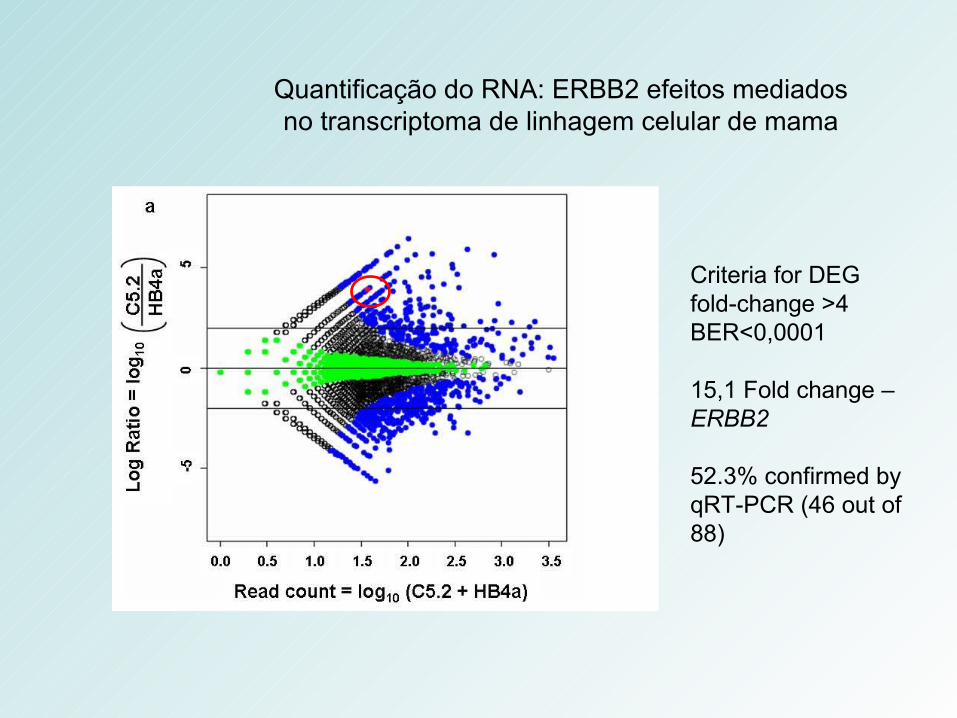
Quantificação do RNA: ERBB2 efeitos mediados no transcriptoma de linhagem celular de mama
Criteria for DEG fold-change >4 BER<0,0001
15,1 Fold change – ERBB2
52.3% confirmed by qRT-PCR (46 out of 88)

Conclusão NGS• NGS revolucionando pesquisas genômicas em um contexto rápido
e rentável comparado com tradicional Sanger.
• Custo reduzido se comparado ao número de reads gerado por corrida.
• RNA-Seq permite quantificar transcriptoma independente do tamanho do transcrito.
• Identificar possíveis rearranjos cromossômicos, SNPs, splice alternativo.





Agradecimentosao time do LBHC
Renato Puga
Leandro Lima
Barbara Mello
Aderbal Silva
Cecília Feio
César Torres
Hospital A.C. Camargo
Laboratório de Genômica
Dra. Dirce Maria Carraro
Elisa Napolitano
Gustavo Molina