funÇÕes nucleares de fukui e a representaÇÃo de …livros01.livrosgratis.com.br/cp145610.pdf ·...
TRANSCRIPT

INSTITUTO MILITAR DE ENGENHARIA
1◦ TEN TATIANA FERNANDES DE MORAES
FUNÇÕES NUCLEARES DE FUKUI E A REPRESENTAÇÃODE ÁTOMOS DEFORMADOS EM MOLÉCULAS DA
DENSIDADE ELETRÔNICA: APLICAÇÕES À MOLÉCULADE RDX
Dissertação de Mestrado apresentada ao Curso deMestrado em Química do Instituto Militar de Engenha-ria, como requisito parcial para obtenção do título deMestre em Química.
Orientador: Itamar Borges Jr., D.Sc.
Rio de Janeiro
2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

c2010
INSTITUTO MILITAR DE ENGENHARIAPraça General Tibúrcio, 80-Praia VermelhaRio de Janeiro-RJ CEP 22290-270
Este exemplar é de propriedade do Instituto Militar de Engenharia, que poderá incluí-loem base de dados, armazenar em computador, microfilmar ou adotar qualquer forma dearquivamento.
É permitida a menção, reprodução parcial ou integral e a transmissão entre bibliote-cas deste trabalho, sem modificação de seu texto, em qualquer meio que esteja ou venhaa ser fixado, para pesquisa acadêmica, comentários e citações, desde que sem finalidadecomercial e que seja feita a referência bibliográfica completa.
Os conceitos expressos neste trabalho são de responsabilidade do(s) autor(es) e do(s)orientador(es).
M827 Moraes, T.F.Funções Nucleares de Fukui e a Representação
de Átomos Deformados em Moléculas da DensidadeEletrônica: aplicações à molécula de RDX, Tatiana Fer-nandes de Moraes. – Rio de Janeiro: Instituto Militarde Engenharia, 2010.
71 p.: il, graf., tab.
Dissertação: (mestrado) – Instituto Militar de Enge-nharia, Rio de Janeiro, 2010.
1. Molécula de RDX. 2. Funções Nucleares de Fukui.3. Decomposição da densidade eletrônica.
CDD 539.12
2

INSTITUTO MILITAR DE ENGENHARIA
1◦ TEN TATIANA FERNANDES DE MORAES
FUNÇÕES NUCLEARES DE FUKUI E A REPRESENTAÇÃO DEÁTOMOS DEFORMADOS EM MOLÉCULAS DA DENSIDADE
ELETRÔNICA: APLICAÇÕES À MOLÉCULA DE RDX
Dissertação de Mestrado apresentada ao Curso de Mestrado em Química do InstitutoMilitar de Engenharia, como requisito parcial para obtenção do título de Mestre emQuímica.
Orientador: Itamar Borges Jr., D.Sc.
Aprovada em 05 de Fevereiro de 2010 pela seguinte Banca Examinadora:
Itamar Borges Jr., D.Sc. do IME - Presidente
Pierre Mothé Esteves, D.Sc. da UFRJ
Ardson dos Santos Vianna Jr., D.Sc. do IME
Antônio dos Santos Lima, D.Sc. do IME
Rio de Janeiro2010
3

Ao meu eterno namorado, amigo e cúmplice, meumarido Robson. A meus pais, Celso e Dulce, minhasfontes de incentivo e determinação. E a meu irmão,Celso Júnior, por tornar possível o impossível emminha vida.
4

AGRADECIMENTOS
Em primeiro lugar, a Deus, esta Força eterna, por permitir-nos a vida em toda a sua
plenitude.
Ao meu marido Robson, agradeço toda a compreensão e todo o amor, sempre pre-
sentes neste período de estudos. Seu apoio, seu carinho e sua presença foram determi-
nantes para a conclusão desta etapa de meus estudos.
A meus pais, por todo seu esforço em me fornecer uma sólida base de educação e
valores que me permitiram as condições de chegar a este momento de minha vida.
À Wendy, por me lembrar que a vida é muito mais do que apenas uma tela de
computador.
Ao meu orientador, Dr. Itamar Borges Jr., pela disponibilidade e prontidão a atender
e esclarecer minhas dúvidas, pelo conhecimento transmitido ao longo destes dois anos,
seja em aulas ou apenas em conversas de trabalho.
Por fim, a todos que de alguma forma contribuíram com esta jornada que aqui se
encerra.
5

"A coragem é a primeira qualidade humana, poisgarante todas as outras."
Aristóteles
6

SUMÁRIO
LISTA DE ILUSTRAÇÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
LISTA DE TABELAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
LISTA DE ABREVIATURAS E SÍMBOLOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1 INTRODUÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
1.1 Conceitos Fundamentais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
1.2 Classificação dos explosivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
1.3 O RDX . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
1.4 Estudos Teóricos da Decomposição do RDX . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2 FUNDAMENTOS TEÓRICOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.1 Aproximação de Born-Oppenheimer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.2 Método Hartree-Fock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.3 Conjuntos de Funções de Bases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
2.4 Correlação Eletrônica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
2.5 Teoria do Funcional da Densidade (DFT) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
2.6 Otimização de Geometrias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
3 METODOLOGIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
4 RESULTADOS E DISCUSSÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
5 CONCLUSÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
6 REFERÊNCIAS BIBLIOGRÁFICAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
7 APÊNDICES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
7.1 Trabalho baseado nesta dissertação aceito para publicação no Interna-
tional Journal of Quantum Chemistry em 2010 . . . . . . . . . . . . . . . . . . . . . . . . . . 60
7

LISTA DE ILUSTRAÇÕES
FIG.1.1 Modelo de Deflagração de um Propelente (Onde Vg é a velocidade
de recuo, e Vd é a velocidade direta). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
FIG.1.2 Trem de Ignição. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
FIG.1.3 Fluxograma de Classificação de Substâncias Explosivas (Adaptado
de (MEYER, 2002)). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
FIG.1.4 Síntese do RDX e HMX.(Adaptado de: Revista
eletrônica do Departamento de Química - UFSC -
http://www.qmc.ufsc.br/qmcweb/artigos/c4.html - acessado
em 18 de dezembro de 2009.) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
FIG.1.5 CL-20. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
FIG.1.6 Mecanismos de decomposição do RDX (Adaptado de (SWADLEY,
2007)). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
FIG.3.1 Superfície de deformação atômica na molécula da água. (a)
Vermelho: regiões de acúmulo de elétrons. (b) Azul: regiões
de depleção de elétrons. (c) Composição de (a) e (b). Va-
lores de contorno: ±0.05au (elétron/bohr3).(Adaptado de:
http://web.uam.es/departamentos/ciencias/qfa/DAM/definition.html
- acessado em 12 de janeiro de 2010.) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
FIG.4.1 Escala de energias relativas para os confôrmeros do RDX represen-
tados em vista frontal. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
FIG.4.2 Confôrmero AAA do RDX: (a) Superfície de deformação atômica
na molécula. Azul: regiões de depleção de elétrons. Vermelho:
regiões de acúmulo de elétrons. Valores de contorno: +0.020 e
−0.020 (elétron/bohr3). (b) Vetores de dureza nuclear (verde) e
índice de reatividade (rosa). (c) vista frontal. . . . . . . . . . . . . . . . . . . . . . . 43
FIG.4.3 Confôrmero AAE do RDX: (a) Superfície de deformação atômica
na molécula. Azul: regiões de depleção de elétrons. Vermelho:
regiões de acúmulo de elétrons. Valores de contorno: +0.020 e
−0.020 (elétron/bohr3). (b) Vetores de dureza nuclear (verde) e
índice de reatividade (rosa). (c) vista frontal. . . . . . . . . . . . . . . . . . . . . . . 44
FIG.4.4 Confôrmero AEE do RDX: (a) Superfície de deformação atômica
8

na molécula. Azul: regiões de depleção de elétrons. Vermelho:
regiões de acúmulo de elétrons. Valores de contorno: +0.020 e
−0.020 (elétron/bohr3). (b) Vetores de dureza nuclear (verde) e
índice de reatividade (rosa). (c) vista frontal. . . . . . . . . . . . . . . . . . . . . . . 45
FIG.4.5 Confôrmero EEE do RDX: (a) Superfície de deformação atômica
na molécula. Azul: regiões de depleção de elétrons. Vermelho:
regiões de acúmulo de elétrons. Valores de contorno: +0.020 e
−0.020 (elétron/bohr3). (b) Vetores de dureza nuclear (verde) e
índice de reatividade (rosa). (c) vista frontal. . . . . . . . . . . . . . . . . . . . . . . 46
9

LISTA DE TABELAS
TAB.4.1 Magnitudes dos vetores de dureza e índice de reatividade nuclear
para cada átomo do confôrmero AAA do RDX. . . . . . . . . . . . . . . . . . . . . . 48
TAB.4.2 Magnitudes dos vetores de dureza e índice de reatividade nuclear
para cada átomo do confôrmero AAE do RDX. . . . . . . . . . . . . . . . . . . . . . 49
TAB.4.3 Magnitudes dos vetores de dureza e índice de reatividade nuclear
para cada átomo do confôrmero AEE do RDX. . . . . . . . . . . . . . . . . . . . . . 50
TAB.4.4 Magnitudes dos vetores de dureza e índice de reatividade nuclear
para cada átomo do confôrmero EEE do RDX. . . . . . . . . . . . . . . . . . . . . . 51
10

LISTA DE ABREVIATURAS E SÍMBOLOS
ABREVIATURAS
A - Axial
AIM - Atoms in Molecules (Átomos de Bader em moléculas)
Al - Alumínio
BD - Base dupla
BS - Base simples
C - Carbono
C4 - Mistura explosiva composta por 91% de RDX e 9% de aditivos plas-
tificantes
Cl - Cloro
DAM - Deformed Atoms in Molecules (Átomos Deformados em Moléculas)
DFT - Density Functional Theory (Teoria do Funcional de Densidade)
DMA - Distributed Multipole Analysis (Análise de Multipolos Distribuídos)
E - Equatorial
G - Gaussianas
H - Hidrogênio
HDS - Hidrodessulfurização
HF - Método Hartree-Fock
HMX - Her Majesty’s eXplosive
IME - Instituto Militar de Engenharia
INDO - Intermediate-Neglect of Differential Overlap
IRMPD - InfraRed MultiPhoton Dissociation (Dissociação MultiFotônica no
Infravermelho)
M - Número de Massa
NC - NitroCelulose
NG - NitroGlicerina
OST - Oxi-S-Triazina
PABX - Polímero explosivo insensível contendo RDX
11

PETN - Nitropenta
PN - Pólvora Negra
RDX - Royal Demolition eXplosive
S - Enxofre
STO - Orbitais Tipo Slater em inglês
TAZ - 1,3,5 triazina
TNT - Trinitrotolueno
12

SÍMBOLOS
∆Hf - variação da entalpia de formação
∆Gf - variação da entropia de formação
H - operador Hamiltoniano
Ψ - função de onda
r - coordenadas eletrônicas
R - coordenadas nucleares
E - energia do sistema
ǫi - energia do i-ésimo orbital
Jij - integral de Coulomb
Kij - integral de troca
VNN - energia potencial de interação núcleo-núcleo
cik - coeficientes da combinação linear de orbitais atômicos
χk - orbitais atômicos na representação dos orbitais moleculares
Frs - operador de Fock
csi - coeficientes da equação com o operador de Fock
ǫi - energias da equação com o operador de Fock
Frs− - aproximação dos coeficientes para o operador de Fock
Eexata - energia exata não-relativística do sistema
Ψ(n)0 - correção de n-ésima ordem para a função de onda do estado funda-
mental
λ - parâmetro aplicado na aproximação da função de onda
ET - energia cinética
EV - inclui a contribuição da atração elétron-núcleo e da repulsão núcleo-
núcleo
EJ - repulsão elétron-elétron
EXC - termo de troca e correlação
N - número de elétrons no sistema
υ(r) - potencial externo definido pelas cargas nucleares
Fi - força eletrostática total em um núcleo i em uma molécula
ρ(r) - densidade de elétrons em r
13

ri - vetor separação entre o núcleo i e a coordenada r
Rij - vetor separação entre os núcleos i e j
Zi - carga do i-ésimo núcleo
χ - eletronegatividade
Qi - distorção do núcleo da molécula
Φi - reatividade nuclear
Gi - dureza nuclear
F+i e F−
i - forças de Hellmann-Feynmann
14

RESUMO
A estrutura eletrônica e o início do processo de decomposição dos quatro confôrmerosda molécula de RDX (1,3,5-trinitro-s-triazina, ou ciclotrimetilenotrinitramina) foram es-tudados com a Teoria do Funcional de Densidade (DFT), empregando o funcional B3LYPe a base gaussiana 6-311 + G (2d, p). A densidade eletrônica calculada em nível DFTfoi decomposta em contribuições atômicas usando o método de átomos deformados emmoléculas (DAM), que permitiu identificar as regiões de acúmulo e depleção de elétronsem cada confôrmero. As funções nucleares Fukui de dureza e de índice de reatividadenuclear foram calculadas. Ambas as funções são vetores atômicos, propriedades intrínse-cas da molécula, que fornecem informações sobre o início do processo de fragmentação,e foram ainda discutidas através da análise da densidade eletrônica. A decomposiçãoda estrutura eletrônica indica que os grupos NO2 equatoriais(E) são menos volumososque os grupos axiais (A), com o primeiro contribuindo mais para o aumento de elétronsdeslocalizados no anel. No que diz respeito à decomposição, a ruptura de uma ligaçãoNN é o processo mais provável para os confôrmeros AAA, AAE e EEE, enquanto quepara a confôrmero AEE eliminação HONO é favorecida, concordando, parcialmente, comestudos anteriores.
15

ABSTRACT
The electronic structure and the onset of decomposition processes of the four con-formers of the RDX molecule were studied with the Density Functional Theory (DFT),the B3LYP functional and the 6-311+G(2d,p) Gaussian basis set. The computed DFTelectron density was decomposed into atomic contributions using the Deformed Atoms inMolecules (DAM) method, which allowed identifying the regions of electron accumulationand electron depletion of each conformer. The nuclear Fukui functions nuclear stiffnessand the nuclear reactivity index were then calculated. Both functions are atomic vectors,intrinsic molecular properties, which provide information for the onset of the fragmenta-tion process, and were further discussed through the analysis of the DAM electron density.The computed decomposed electronic structure indicate that equatorial (E) groups areless bulky than axial (A) groups, with the former contributing to the increase of thering delocalized electrons. Concerning the decomposition, the RDX N-N bond cleavageis the most probable process for the AAA, AAE and EEE conformers, while for the AEEconformer HONO elimination is favored, partially agreeing with previous studies.
16

1 INTRODUÇÃO
A nova Estratégia Nacional de Defesa (BRASIL, 2008) fundamenta-se na busca de
solução pacífica das controvérsias e no fortalecimento da paz e da segurança interna-
cionais. Desta forma, assume uma postura dissuasória, baseando-se na capacidade de
defesa de suas Forças Armadas. Deste modo, torna-se imprescindível o desenvolvimento
científico e tecnológico do país. Para realizar este trabalho é necessário formar pessoal
capacitado, e a melhor formação para este fim é por meio da pesquisa básica.
Neste quadro, insere-se a pesquisa básica de ponta. Em particular, ferramentas
da química teórica e computacional podem ser aplicadas a problemas relevantes para a
Defesa, o que é o objetivo geral desta proposta de dissertação de mestrado. Em particular,
o uso de tais ferramentas permite a concepção, avaliação e caracterização de explosivos e
propelentes através da modelagem molecular aplicada ao estudo de materiais energéticos,
como o RDX (1,3,5-trinitro-s-triazina, ou ciclotrimetilenotrinitramina) de largo uso pelo
Exército Brasileiro, dentre outros. Além disto, com a modelagem molecular torna-se
possível investigar novas moléculas, reduzindo a custosa etapa de síntese, com potencial
para constituírem novos explosivos e propelentes mais eficientes e seguros.
A formação de pessoal capacitado em desenvolver pesquisa básica na área de ma-
teriais energéticos segue os interesses do Exército Brasileiro, ao demonstrar capacidade
científica em concepção, avaliação e no desenvolvimento de novos materiais explosivos e
propelentes. Além disso, um trabalho de mestrado para investigar propriedades físicas e
químicas de materiais energéticos, como o supracitado RDX, permitirá adquirir conhe-
cimentos para a concepção de novos materiais com propriedades melhoradas e de maior
eficiência.
Desta forma, nesta dissertação estudou-se as possíveis formas de decomposição para
os quatro possíveis confôrmeros do RDX. Em uma primeira etapa, otimizou-se a ge-
ometria do estado fundamental de cada conformação com o método DFT, o funcional
B3LYP e a base gaussiana 6-311+G(2d,p). Fazendo-se uso da geometria otimizada do
estado fundamental, calculou-se, então, com o mesmo nível de cálculo, o single-point para
o cátion e ânion de cada confôrmero. Complementando o trabalho, na segunda etapa,
fez uso do programa DAMQT (LOPEZ, 2009) para decompor a densidade eletrônica e,
posteriormente, calcular as forças de Helmann-Feynman necessárias para a obtenção das
17

funções nucleares de Fukui.
O trabalho desta dissertação foi apoiado por verbas de vários órgãos de fomento
(FAPERJ, CNPq, CAPES) e a partir de 2010 será financiado também pelo Plano Básico
de Ciência e Tecnologia do Exército Brasileiro.
1.1 CONCEITOS FUNDAMENTAIS
Alguns conceitos podem ser apresentados de forma a contextualizar esta dissertação.
Faremos esta exposição baseada em (CORNER, 1950). Assim:
Materiais Energéticos são substâncias químicas ou misturas de substâncias quími-
cas capazes de se transformarem rapidamente, liberando uma grande quantidade de calor
(reações exotérmicas), em gases, ondas de choque ou fragmentação.
Combustão ou Queima é uma reação química exotérmica de oxi-redução. Normal-
mente o termo combustão é utilizado para definir uma reação (queima) de uma substância
combustível em presença de uma oxidante. A reação de combustão tem lugar na super-
fície da substância combustível e depois de iniciada é auto-sustentada pela transferência
de calor liberado na geração dos produtos da combustão.
Deflagração é uma combustão muito rápida, ocorre com velocidade de chama de 1
a 100 m/s, sendo o que acontece em maior frequência nas indústrias. O aumento na ve-
locidade de reação é obtido por um contato mais íntimo entre as substâncias combustível
e oxidante (fornecedoras de oxigênio), ou pela existência de elementos combustíveis (C,
H, S, Al, Cl) e oxigênio na mesma substância, como ocorre nas pólvoras. O mecanismo
da deflagração é o mesmo da combustão, porém com maior velocidade de reação. Os
fatores determinantes da velocidade de reação são a taxa de transferência de calor dos
produtos da reação para a superfície da massa do material e a taxa de decomposição da
substância. A deflagração não produz ondas de choque.
Detonação é um tipo específico de reação exotérmica, que está sempre associada a
uma onda de choque sobre o material. A velocidade de detonação é função da onda de
choque e normalmente varia de 1500 a 9000 m/s.
Explosão é um processo caracterizado por súbito aumento de volume e grande libe-
ração de energia, geralmente acompanhado por altas temperaturas e produção de gases.
Uma explosão provoca ondas de pressão ao redor do local onde ocorre. Explosões podem
ser originadas em fenômenos físicos ou químicos. Note-se que, dependendo do tipo de
explosivo, este pode se decompor por detonação ou deflagração.
Energia de Ativação é a energia necessária para iniciar uma reação química, no
18

FIG. 1.1: Modelo de Deflagração de um Propelente (Onde Vg é a velocidade de recuo, eVd é a velocidade direta).
caso, a reação de decomposição de um material. Teoricamente, é aceito que o início da
reação é causado pela elevação da temperatura em uma pequena porção do material,
conhecida como hot spot. Não é necessário que o estímulo seja executado ao longo do
mesmo. Quaisquer estímulos que possam provocar a elevação da temperatura, tais como:
calor, choque, centelha elétrica etc podem gerar a energia de ativação necessária para a
iniciação de um material explosivo.
A pólvora negra, considerada o primeiro material energético, e a azida de chumbo
são iniciadas por chama, as espoletas das munições das armas de fogo por choque e os
outros explosivos, como as dinamites, por forte pressão, como a produzida por outros
explosivos.
Trem de Ignição é o dispositivo utilizado para ignitar as cargas de propelentes.
FIG. 1.2: Trem de Ignição.
Explosivos são substâncias capazes de, com muita rapidez, se transformarem em
gases, produzindo calor intenso e pressões elevadas.
19

1.2 CLASSIFICAÇÃO DOS EXPLOSIVOS
Classificações de materiais energéticos foram empreendidas por muitos cientistas du-
rante o século XX(URBÁNSKY, 1964):
• Quanto à natureza química:
a) Compostos nitro; b) Ésteres nítricos; c) Nitraminas; d) Derivados dos ácidos
clórico e perclórico; e) Azidas; f) Vários compostos capazes de produzir uma ex-
plosão, por exemplo, fulminatos, acetilidas, compostos enriquecidos de nitrogênio,
por exemplo, tetrazeno, peróxidos e ozonidas, etc.
• Quanto à velocidade de decomposição:
Altos Explosivos - detonam, ou seja, possuem velocidade acima da velocidade do
som no meio. A decomposição é praticamente instantânea. Consequentemente sua
ação é rápida e violenta.
Baixos Explosivos ou Propelentes - deflagram, ou seja, possuem velocidade abaixo
da velocidade do som no meio. Queimam instantaneamente durante um certo
período de tempo.(PONT, 1975)
• Quanto à constituição
Explosivos Químicos - substâncias puras (TNT - Trinitrotolueno, NG - Nitroglice-
rina, etc).
Explosivos Mecânicos - Misturas de substâncias não explosivas quando isoladas (PN
- Pólvora Negra).
Explosivos Mistos - Misturas de dois ou mais explosivos ou destes com substâncias
não explosivas (dinamites).
• Quanto à fase
Explosivos sólidos (TNT), líquidos (NG), gasosos (mistura de Acetileno e Oxigênio).
• Quanto à finalidade ou emprego
a) Explosivos Iniciadores (trem de explosão) - Aqueles que são empregados, em
mistos, para a iniciação ou excitação das cargas explosivas. São muito sensíveis ao
atrito, calor e choque. Quando sob os efeitos do fogo explodem sem incendiar-se.
Os principais são: azida de chumbo, estifinato de chumbo (ou trinitrorresorcinato
de chumbo), fulminato de mercúrio e tetraceno.
20

b) Explosivos Reforçadores - Servem como intermediários entre o iniciador e a carga
explosiva propriamente dita. Podem ser iniciados pelo calor, atrito ou choque.
Podem detonar quando queimados em grande quantidade. Incluem-se nesse tipo
de explosivos: Ciclonita (RDX), Nitropenta (PETN) e Tetril.
c) Explosivos de Ruptura - Constituem os alto-explosivos propriamente ditos. São
quase todos tóxicos. Os principais são: Ácido pícrico, Amatol, Composição à base
de RDX, Pentolite, Picrato de amônio (Explosivo D), Picratol, Tetritol, Torpex,
Tritonal, Trotil, Haleita e Ednatol.
d) Pólvoras - São utilizadas para propulsão ou projeção, e classificam-se em: pólvo-
ras químicas e mecânicas. Sua estabilidade é afetada pela umidade e pela tempe-
ratura elevada.
d.1) Pólvora Química- Queimam rapidamente e, sob condições especiais de inicia-
ção, podem detonar com procedimento igual ao de qualquer outro explosivo. As
poeiras das pólvoras químicas são sensíveis ao atrito, chama e centelhas. São usadas
na propulsão de projéteis. As pólvoras químicas recebem denominações conforme
o número de suas bases ativas1: base simples (BS), base dupla (BD), existindo
pólvoras de base tripla (NG, NC, nitroguanidina).
d.2) Pólvora mecânica (pólvora negra, marrom e de mina) - Contém nitrato de
potássio ou de sódio, enxofre e carvão, em mistura íntima. Muito sensível ao atrito,
chama, calor e choque, propriedade que a torna um dos mais perigosos explosivos
para manuseio. É higroscópica. Usada em granadas de exercícios, petardos, cordéis,
espoletas, tiros de salvas e minas terrestres.
e) Artifícios - São destinados a produzir efeitos visuais ou auditivos, ou provocar
inflamação ou detonação de explosivos.
1Base ativa é a substância responsável pelo potencial energético da pólvora.
21

e.1) Iniciadores - Destinados à inflamação ou detonação (mechas, cordéis deto-
nantes, estopins, acionadores e acendedores).
e.2) Pirotécnicos - São os que produzem efeitos luminosos, fumígenos, incendiários
etc.
• Classificação geral
A classificação geral das substâncias explosivas pode ser vista na Figura 1.3.
FIG. 1.3: Fluxograma de Classificação de Substâncias Explosivas (Adaptado de(MEYER, 2002)).
1.3 O RDX
O RDX foi preparado pela primeira vez em 1899 pelo alemão Henning para uso
medicinal. Seu valor como explosivo foi reconhecido em 1920 por Hertz, que o desenvolveu
a partir da nitração direta da hexamina. Uma exemplificação pode ser visualizada na
Figura 1.4. Entretanto, nesta época, o processo já era caro e o rendimento da reação
era pequeno, não se caracterizando um bom produto para produção em larga escala
(AKHAVAN, 2004).
Em 1925, Hale do Arsenal de Picatinny em Nova Jersey (EUA) desenvolveu um pro-
cesso de produção de RDX com rendimento de 68%. Contudo, uma melhora substancial
22

só ocorreu em 1940, quando Meissner desenvolveu um método contínuo de produção do
RDX . Ross e Schiessler, ambos do Canadá, desenvolveram um processo que não requeria
a hexamina como reagente. No mesmo tempo, Bachmann desenvolveu um processo de
produção do RDX a partir da hexamina com elevado rendimento.
O produto de Bachmann ficou conhecido como B RDX e possuía um grau de impureza
de 8% a 12%. As propriedades explosivas desta impureza mais tarde foram utilizadas e
o explosivo HMX, também conhecido como "Octogen", foi desenvolvido. O processo de
Bachmann foi adotado pelo Canadá durante a Segunda Guerra Mundial e, mais tarde,
pela Companhia Tennessee-Eastman. Este processo era mais econômico e, também, levou
a descoberta de muitos novos explosivos. A rota de síntese do RDX puro, ou seja, sem
impurezas, foi desenvolvida por Brockman e o produto ficou conhecido como A RDX.
Na Grã-Bretanha o Departamento de Pesquisa de Armamento em Woolwich começou
a desenvolver uma rota de fabricação de RDX após a publicação da patente de Herz em
1920. Uma planta-piloto em pequena escala que produzia 75 quilos de RDX por dia foi
instalado em 1933 e operou até 1939. Outra planta foi instalada em 1939 em Waltham
Abbey e uma usina em larga escala foi erguida em 1941 perto de Bridgewater. O RDX não
foi usado como o preenchimento principal de reservatórios e bombas britânicas durante
a Segunda Guerra Mundial, mas foi adicionado ao TNT para aumentar o poder das
composições explosivas na Alemanha, França, Itália, Japão, Rússia, E.U.A., Espanha e
Suécia.
A pesquisa continuou durante a Segunda Guerra Mundial para desenvolver novos e
mais potentes explosivos e composições explosivas. Torpex (TNT / RDX / alumínio)
e ciclotetrametilenotetranitramina, conhecido como Octogen (HMX), tornaram-se
disponíveis ao final da II Guerra Mundial. Em 1952, uma composição explosiva chamada
"Octol" foi desenvolvida, que continha 75% HMX e 25% TNT. Explosivos plásticos
moldáveis também foram desenvolvidos durante a Segunda Guerra Mundial, estes muitas
vezes contínham vaselina ou nitrocompostos líquidos gelatinizados para dar uma con-
sistência de plástico.
Em seu estado puro e sintetizado o RDX, também conhecido como Hexogen, Ciclonita
e ciclotrimetiltrinitramina, é um sólido cristalino branco com elevado ponto de fusão de
204◦C, tornando difícil a utilização em fundição. O RDX puro é muito sensível a iniciação
por impacto ou fricção e é dessensibilizado por revestimento dos cristais com cera, óleo
ou graxa. Este pode também ser misturado com geléia de minerais ou similares para
formar explosivos plásticos. Uma composição explosiva insensível contendo RDX pode
23

ser obtida através da incorporação de cristais de RDX em uma matriz de polímero. Este
tipo de composição é conhecido como um polímero explosivo (PABX) e é menos sensível à
iniciação acidental. O RDX tem uma alta estabilidade química e grande poder explosivo
comparado com TNT e ácido pícrico. É estável em estocagem e é considerado um dos
mais poderosos e rompedores dentre os alto-explosivos militares. É difícil para dissolver
o RDX em líquidos orgânicos, mas ele pode ser recristalizado em acetona.
É importante salientar que a sigla é uma apologia ao seu poder: Royal Demolition
eXplosive, RDX.
FIG. 1.4: Síntese do RDX e HMX.(Adaptado de: Revista eletrônica do Departamentode Química - UFSC - http://www.qmc.ufsc.br/qmcweb/artigos/c4.html - acessado em
18 de dezembro de 2009.)
Por ser extremamente potente, esta nova classe de explosivos possui qualidades su-
periores aos demais: estabilidade, maleabilidade e resistência ao calor. Estes explosivos
podem ser moldados e aquecidos sem perigo de uma detonação indesejada. Entretanto,
basta um pulso elétrico para que o processo se inicie: o RDX é capaz de derrubar pare-
des de concreto ou mesmo de aço. A chamada dinamite militar é uma mistura de 75%
RDX, 15% TNT e 10% de aditivos estabilizantes e plastificantes. A mistura explosiva
conhecida como C4 é composta por 91% RDX e 9% de aditivos plastificantes. O poder
de detonação do C4 é suficiente, por exemplo, para gerar ondas de compressão capazes de
iniciar a fissão nuclear de uma bomba de urânio-235, sendo de uso exclusivo das Forças
Armadas e policiais. Exemplos de outros explosivos militares à base de RDX:
• Composição A (revestido de cera, explosivo granular consistindo de RDX e cera
plastificante)
• Composição A5 (mista com 1.5% ácido esteárico)
24

• Composição B (misturas fundidas de RDX e TNT)
• Composição C (um explosivo plástico de demolição consistindo de RDX, outros
explosivos, e plastificantes)
• Composição D, HBX (misturas fundidas de RDX, TNT, alumínio pulverizado, e
cera D-2 com cloreto de cálcio)
Em 1987, o laboratório do US Naval Air Warfare Center Weapons Division sintetizou
uma outra nitroamina cíclica: a hexanitrohexaazaisowurtzitana, designada como CL-20,
que pode ser visto na Figura 1.5. Assim como RDX, o CL-20 é estável e maleável, mas
cerca de 20% mais poderoso. Em um teste do exército americano, um projetil de 30mm
foi detonada em um cartucho carregado com CL-20, e foi capaz de penetrar em 7 placas
de 1 polegada de aço inox.
FIG. 1.5: CL-20.
Como é evidente a importância deste explosivo não somente no âmbito do Exército
Brasileiro, os possíveis mecanismos de decomposição em fase gasosa sugeridos, até então,
por pesquisadores podem ser vistos na Figura 1.6 (SWADLEY, 2007).
1.4 ESTUDOS TEÓRICOS DA DECOMPOSIÇÃO DO RDX
Uma vez que materiais energéticos (explosivos e propelentes) necessitam de requisitos
muito peculiares como alta densidade de energia, insensibilidade a choques mecânicos,
resistência à decomposição química, síntese barata com reagentes facilmente disponibi-
lizados no mercado e habilidade de formulação com outros materiais para fabricação de
dispositivos práticos, ainda hoje existe uma grande escassez de materiais que atendam
adequadamente a todos estes requisitos (MURRAY, 1998; RICE, 2006). A obtenção
25

FIG. 1.6: Mecanismos de decomposição do RDX (Adaptado de (SWADLEY, 2007)).
de novos materiais energéticos com melhoradas propriedades físico-químicas constitui-se
num grande auxiliar no que diz respeito à necessidades da Defesa Nacional. Para isto,
é fundamental um estudo melhor da físico-química dos materiais energéticos. Mesmo os
mais conhecidos explosivos militares necessitam de uma análise mais acurada de suas
propriedades em nível molecular.
Além disso, o desenvolvimento, a fabricação, o teste e a utilização de um novo ma-
26

terial energético despende muito tempo e recurso. A eliminação prévia sem grande custo
de qualquer candidato devido a problemas de sensibilidade ou de performance é um
grande facilitador (ZHANG, 2005; BORGES JR, 2008). Assim, a capacidade de previsão
e avaliação de cada estágio do desenvolvimento é altamente desejável. Portanto, deve-se
desenvolver a capacidade de prever as mais diversas propriedades dos materiais que são
associadas com performance e sensibilidade antes de despender recursos em sua síntese
(MURRAY, 1998; POLITZER, 2001; RICE, 2002, 2006; BORGES JR, 2008).
Dentro desta idéia, alguns países como Estados Unidos da América, Taiwan e China
vêm realizando pesquisa sobre materiais energéticos. Em especial sobre o RDX, que, como
dito anteriormente, é um importante ingrediente na constituição de vários propelentes e
explosivos.
Em uma breve pesquisa das últimas décadas, alguns trabalhos se destacam. Por
exemplo, em 1997, na Califórnia, (WU, 1997) foi feito um estudo sobre o mecanismo da
decomposição do RDX. A investigação foi feita usando a teoria do funcional de densi-
dade, utilizando-se, dentre outros, os funcionais B-PW91, B3-PW91, B-LYP, e B3-LYP.
Os cálculos foram realizados com as bases "double zeta": D95V, D95V+ (função difusa)
e cc-pVDZ. Calculando-se, assim, a energia potencial para dois caminhos de reação pos-
síveis. Desta forma, foi assegurado que sua conclusão final não dependia de métodos
computacionais. O primeiro, a ruptura da ligação N − NO2 e o segundo, a fissão das
ligações simétricas formando três moléculas de metil nitro amina (mecanismos a e b na
Figura 1.6).
Os autores encontraram uma barreira de ativação para a fissão das ligações simétricas
18 Kcal/mol maior que da ligação N − NO2, diferença esta que é bastante significativa.
Além disto, os cálculos, também realizados por estes cientistas, da teoria do estado
de transição e cinética indicaram que os dois mecanismos diferem muito pouco, não se
constituindo num diferencial de taxas de reação. Sugeriu-se, então, que a ruptura da
ligação N − NO2 é mais provável de acontecer.
Já em 2000, também na Califórnia, foi estudado o Mecanismo de Decomposição
Unimolecular do RDX, agora com outro foco (CHAKRABORTY, 2000).
Existem dois mecanismos de reação: decomposição da ligação simétrica para formar
três moléculas de CH2NNO2(M=74, onde M é o número de massa) e clivagem ho-
molíticada ligação NN para formar NO2(M=46) mais o RDR(M=176) com subseqüente
decomposição de vários produtos. Estudos experimentais de dissociação multifotônica
no infravermelho (IRMPD)(ZHAO, 1988) do RDX concluíram que o caminho primário
27

dominante de decomposição do RDX é a fissão simétrica tripla. Já o estudo de fotólise
ultravioleta (BOTCHER, 1994, 1993; PACE, 1991; CHOI, 1995) observou como meca-
nismo principal a homólise da ligação N-NO2. A utilização conjunta de termogravimetria
(BEHRENS, 1990b,a, 1991, 1992)com espectrometria de massa sugeriu a formação de oxi-
s-triazina (OST) via eliminação de uma molécula de HNO e duas de HONO. Por outro
lado, cálculos teóricos, como em (WU, 1997), sugerem que a quebra homolítica requer
menos energia.
Assim sendo, eles estudaram, além destes, um terceiro mecanismo: eliminação suces-
siva de HONO para formar 3 HONO(M=47) mais a estável 1,3,5 triazina (TAZ)(M=81),
com o método DFT/B3LYP e com a base gaussiana 6-31G(d), incluindo as barreiras de
todos os produtos.
Seus cálculos concluíram que o caminho energeticamente favorável é a eliminação de
HONO para formar TAZ e três HONO (com adicional decomposição do TAZ a 3HCN). A
clivagem da ligação NN para formar RDR é bastante favorecida também, mas a energia
necessária para a decomposição subseqüente o prejudica. Esta eliminação de HONO se
constitui também no caminho de decomposição primária mais exotérmica e pode explicar
a energia liberada observada na decomposição do RDX. Desta forma, está evidente que
a energia liberada na fase gasosa do RDX vem de reações secundárias.
Em 2007, foram feitas pesquisas sobre Mecanismos de Reação do RDX descobertos
pela Teoria do Funcional de Densidade, usando o método B3-LYP 6-311G(2d,p)++ e
6-21G(d,p) (SWADLEY, 2007).
Em trabalhos anteriores diversos mecanismos para a decomposição do RDX foram
propostos, mas nenhum argumento que apresente um caminho definitivo. Assim sendo,
Swadley focou sua pesquisa na Teoria do Funcional de Densidade usando, em particular
a função nuclear de Fukui, neste caso, funções nucleofílica e eletrofílica, para avaliar
os efeitos de aumentar e diminuir a pertubação eletrônica dos confôrmeros do estado
gasoso e da estrutura do cristal de RDX. Uma vez que esta função é a medida do estresse
físico que a carga da população de elétrons impõe sobre o núcleo, obtém-se, então, uma
informação importante do papel de cada átomo na decomposição.
Diferentemente dos demais cientistas, então, estes otimizaram as estruturas das con-
formações AAA, AAE, AEE, EEE do RDX usando Gaussian03 (versão que também
foi usada nesta dissertação), o método DFT B3-LYP e o conjunto de funções de base
6-311G++(2d,p).
Desta forma, na fase vapor, o RDX apresentou ambos os mecanismos de homólise da
28

ligação NN e o de eliminação de HONO como favoráveis. O cálculo da função de Fukui
para os conformêros AAA e AAE apresentou como melhor caminho a homólise NN, já
para os conformêros AEE e EEE, a eliminação de HONO mostrou-se mais viável. O
a-RDX sólido mostrou-se mais favorável ao mecanismo de homólise da ligação NN.
Também em 2007, Liu e Zeg (LIU, 2007) realizaram um estudo computacional do me-
canismo de decomposição do explosivo RDX baseado na energia de ligação calculada pelo
método semi-empírico "intermediate-neglect of differential overlap" (INDO). Elaborado
pelo próprio autor, esse ajuda a determinar o perfil da decomposição com a determinação
do ∆Hf e ∆Gf .
Deste modo, concluiu-se que a eliminação cis-HONO possui menor energia de ativação
que a eliminação trans, sendo o mecanismo de reação mais favorável.
Em recente contribuição a esta área, em (BORGES JR, 2008), foram avaliadas a
conformação e a distribuição de carga dos diazociclopropanos. Apesar de não se tratar
do RDX, foco deste trabalho, a metodologia usada é semelhante a que foi usada aqui.
Trata-se do método de análise de multipolos distribuídos (DMA) baseado na idéia de que
o sistema molecular é dividido em regiões. Este considera a densidade eletrônica como a
soma de produtos de funções de base do átomo central.
A concepção química das moléculas como a de átomos ligeiramente distorcidos não
tem uma tradução unívoca em termos de densidade de elétrons. A partir deste fato,
segue-se que qualquer partição da densidade molecular em contribuições atômicas é in-
trinsecamente arbitrária e deve ser justificada em termos de sua praticidade e utilidade
conceitual. Assim, do ponto de vista prático, a utilidade de uma partição pode ser consi-
derada em termos de sua capacidade de facilitar o cálculo dos vários tipos de densidade de
elétrons que tenham interesse em aplicações químicas quânticas (potenciais eletrostáticos
moleculares, forças, momentos multipolares, etc.). No que diz respeito ao desenvolvi-
mento conceitual, o ponto importante é o seu significado químico e, em particular, a
criação de um vínculo da Química Quântica com as noções básicas da química empírica.
Dado o evidente interesse do tema, diversas partições da densidade molecular em con-
tribuições atômicas têm sido propostas, e os pesquisadores em Química Quântica podem
escolher, entre as descrições complementares, o que que consideram mais adequada para
o problema em estudo (RICO, 2004).
O estudo da densidade e do papel desempenhado pela sua representação atômica é
proposto como um caminho para a racionalização do comportamento químico. Como
este comportamento tem sido muito apresentado em termos dos conceitos básicos da
29

química empírica estrutural, uma ligação direta entre ambas as abordagens é procurada,
usando a representação exata da densidade fornecida por átomos deformados em molécu-
las. Observando-se que os termos esféricos dos pseudoátomos não podem ser os principais
responsáveis pelo comportamento químico, alguns pesquisadores estudaram pequenas de-
formações não-esféricas para relacioná-las com os conceitos básicos da química empírica
estrutural. Pares isolados, individuais, ligações duplas, triplas, de diferentes classes de
átomos, grupos funcionais, e assim por diante, são acompanhadas pelas deformações da
densidade.
A densidade eletrônica apresenta um papel cada vez mais central para o desenvolvi-
mento conceitual e prático da química teórica. Na aproximação de Born-Oppenheimer (o
paradigma no estudo da estrutura molecular), a energia eletrônica incluindo a repulsão
nuclear é a energia potencial do movimento dos núcleos e, como conseqüência, as compo-
nentes da força que agem sobre um núcleo são determinadas pelas suas derivadas, com
relação a suas coordenadas. O teorema eletrostático de Hellmann-Feynman afirma que as
derivadas são iguais aos componentes da força eletrostática gerada pela nuvem eletrônica
mais os núcleos restantes. Assim, as forças podem ser obtidas de duas maneiras: a partir
da energia eletrônica através de suas derivadas ou a partir da densidade de eletrônica
usando a eletrostática clássica. A primeira forma é cara computacionalmente e não pro-
porciona uma visão química. A segunda despende um menor tempo computacional para
aplicar e fornece uma grande percepção química.
No entanto, embora o teorema eletrostático seja conhecido há mais de 60 anos, as
suas possibilidades têm sido pouco exploradas. Houve duas razões principais para isso. A
primeira é que o cumprimento do teorema requer densidades calculadas com alta quali-
dade. Em particular, conduz a resultados desastrosos se utilizadas densidades calculadas
com conjuntos de base pequenos. A segunda razão vem do fato de que, para extrair
informações químicas do teorema, precisa-se de uma boa representação da densidade
calculada. (RICO, 2005)
Como se percebe, existem muitos estudiosos em diversos países investigando as pos-
síveis formas de decomposição do RDX utilizando vários métodos diferentes. Entretanto,
no Brasil não se tem conhecimento de resultados neste assunto. Portanto, esta dissertação
realizou um estudo deste processo, porém fazendo uso da técnica de átomos deformados
em moléculas. Além disto, escolheu-se a fase gasosa devido a sua importância na previsão
do comportamento de materiais energéticos (BORGES JR, 2008).
30

2 FUNDAMENTOS TEÓRICOS
Os fenômenos relacionados ao universo atômico dependem das descrições prove-
nientes da chamada mecânica quântica. A diferença fundamental desta última em relação
à mecânica newtoniana está no fato de que toda informação ou toda modelagem de qual-
quer sistema microscópico está assentada em um caráter intrínsecamente probabilístico.
A Química Quântica é um ramo especializado tanto da Química como da Física que,
usando as ferramentas da Mecânica Quântica, visa explicar e prever o comportamento de
sistemas físico-químicos microscópicos, tais como átomos, moléculas, íons e redes cristali-
nas.
Com o excepcional progresso das técnicas computacionais, pode-se estudar sis-
temas cada vez mais complexos, devido a grande quantidade de métodos computacionais
disponíveis. E, assim sendo, esta dissertação está fundamentada na Química Quântica
para descrever o comportamento Físico-Químico Molecular.
2.1 APROXIMAÇÃO DE BORN-OPPENHEIMER
Um sistema físico é descrito pela seguinte equação dinâmica, a equação de
Schrödinger, que para um estado estacionário (ATKINS, 1997) é dada por:
HΨ (R, r) = EΨ (R, r) (2.1)
onde H é o operador Hamiltoniano, Ψ é a função de onda, dependente das coordenadas
eletrônicas (r) e nucleares (R), e E é a energia do sistema. Então, para resolver esta
equação deve-se encontrar as funções de onda que a satisfazem e os seus autovalores E
de energia.
Haja vista que os elétrons se movimentam com velocidades bastante superiores
aos dos núcleos, podemos tratar estes como fixos. Esta é a aproximação de Born-
Oppenheimer.
Para o átomo de hidrogênio, os únicos termos que compõem o operador Hamiltoniano
são a energia cinética do elétron (assumindo o núcleo fixo) e a energia potencial interação
elétron-núcleo. Assim, para este sistema pode-se empregar o sistema de separação de
variáveis, no qual a eq. de Schrödinger é dividida em três equações mais simples que
podem ser resolvidas de forma exata.
31

2.2 MÉTODO HARTREE-FOCK
Quando o sistema possui vários elétrons, não se pode fazer uso do método de sepa-
ração de variáveis, fazendo-se necessário considerar a interação repulsiva elétron-elétron.
É necessário, então, se buscar soluções numéricas ou analíticas aproximadas. Um exemplo
destes tipos de soluções é o método Hartree-Fock (FORESMAN, 1996).
O início da busca por soluções numéricas ou aproximadas consiste em escrever a
função de onda em termos de funções mais simples ϕi (ri), onde cada uma depende
das coordenadas apenas do elétron i. Funções deste tipo são denominadas de orbitais
moleculares ou atômicos, dependendo do sistema em estudo.
Desta forma, Douglas Hartree conseguiu encontrar soluções numéricas usando o pro-
duto das funções ϕi (ri), chamadas de orbitais-spin:
Ψ (r1, r2, · · · , rn) = ϕ1 (r1) ϕ2 (r2) · · ·ϕn (rn) (2.2)
Entretanto este processo não era condizente com o Princípio da Exclusão de Pauli,
que matematicamente requer que o sinal da função de onda seja invertido quando se troca
as coordenadas de dois elétrons. Isso não ocorre na Eq. 2.2, pois ϕ1 (r1) ϕ2 (r2) · · ·ϕn (rn)
é o mesmo que ϕ2 (r2) ϕ1 (r1) · · ·ϕn (rn), por exemplo.
Então, Vladimir Fock, para incluir a antissimetria da função de onda, trabalhou com
a função em forma de determinante:
Ψ (r1, r2, · · · , rn) =1√n!
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
ϕ1 (r1) ϕ1 (r2) · · · ϕ1 (rn)
ϕ̄1 (r1) ϕ̄1 (r2) · · · ϕ̄1 (rn)...
.... . .
...
ϕk (r1) ϕk (r2) · · · ϕk (rn)
ϕ̄k (r1) ϕ̄k (r2) · · · ϕ̄k (rn)
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
∣
(2.3)
onde 1/√
n! é uma constante de normalização e a barra sobre as funções serve para
identificar o spin eletrônico. Esta função ficou conhecida como determinante de Slater.
Fock, usando o método variacional e funções de onda tipo determinante de Slater,
deduziu a seguinte equação para a energia molecular:
EHF = 2
n/2∑
i=1
ǫi −n/2∑
i=1
n/2∑
j=1
(2Jij − Kij) + VNN (2.4)
onde ǫi é a energia do orbital i, Jij é a "integral de Coulomb" e diz respeito à repulsão
elétron-elétron; Kij é a "integral de troca", que surge da antissimetria da função de
32

onda (Princípio de Pauli); VNN é a energia potencial de interação núcleo-núcleo. Este
procedimento utilizado é chamado de "Método Hartree-Fock" (HF).
Entretanto, apesar da aproximação de Hartree-Fock para a função de onda estar
baseada na redução de equações íntegro-diferenciais para as formas ótimas dos orbitais-
spin pelo uso do método variacional, ainda faz-se necessário conhecer os orbitais molecu-
lares.
Rothaan propôs representar os orbitais moleculares como combinações lineares de
orbitais atômicos:
ϕi (ri) =∑
k
cikχk = ci1χ1 + ci2χ2 + · · · + cinχn (2.5)
sendo cik os coeficientes e χk funções atômicos. As funções χk utilizadas para a
construção de cada orbital molecular constituem um "conjunto de funções de base",
sendo as mais comuns as gaussianas, ou seja, funções do tipo e−αx2
.
Deve-se, então, determinar o conjunto ótimo de valores para os coeficientes da expan-
são da Eq. 1.5. Assim sendo, a substituição da Eq. 2.5 na Eq. 2.5 , seguida de rearranjos
fornece:
n∑
s=1
csi (Frs − ǫiSrs) = 0 (2.6)
Onde Frs é o "operador de Fock". Nessa equação, deve-se obter os coeficientes csi.
As energias Ei da equação anterior são os autovalores do operador de Fock G:
Fϕi = ǫiϕi (2.7)
Entretanto, antes, faz-se necessário conhecer os orbitais moleculares, que dependem
dos coeficientes cik. Mas para isto, precisa-se do operador F que depende dos coeficientes.
Este tipo de problema deve ser resolvido iterativamente.
Parte-se, então, de um conjunto inicial de coeficientes para obter os orbitais mole-
culares e seus autovalores iniciais. Por sua vez, os coeficientes Frs são utilizados para
encontrar um conjunto de valores melhorados para os cik, obtendo-se um novo sistema
de equações lineares que gera um conjunto melhorado de coeficientes dos orbitais. Este
procedimento de cálculos sucessivos é repetido até que a diferença entre duas iterações
consecutivas encontre-se dentro de um limite pré-estabelecido, atingindo a convergência.
A densidade eletrônica determina a distribuição de carga elétrica da molécula ou do
orbital e, esta, gera um campo elétrico. Se este campo elétrico, antes desconhecido, for
33

utilizado em um novo cálculo e o resultado for o próprio campo, diz-se que o campo é
"auto-consistente". Deste modo, o método HF é conhecido também como método do
"campo auto-consistente".
2.3 CONJUNTOS DE FUNÇÕES DE BASES
Os orbitais atômicos são construídos a partir das funções atômicas e, desta forma,
torna-se primordial a escolha correta do conjunto de funções de base para o resultado
final.
Inicialmente, em função de restrições computacionais, escolhia-se o chamado conjunto
mínimo, composto apenas dos orbitais atômicos do átomo livre. Por exemplo, para o
átomo de hidrogênio, os orbitais do tipo Slater (STO) são ótimas aproximações para as
soluções exatas da equação de Schrödinger. Porém tais funções dificultam os cálculos
devido a sua forma matemática.
Neste contexto, as funções gaussianas possuem propriedades que facilitam a mani-
pulação e a substituição dos STO. Entretanto, uma gaussiana não consegue reproduzir
o comportamento de um STO próximo à origem, fazendo-se necessário combinar várias
gaussianas para aproximar o comportamente de um STO.
Assim sendo, um dos primeiros conjuntos elaborados foi o STO-3G, que possui três
gaussianas combinadas para aproximar para um orbital do tipo Slater. Tais gaussianas
são denominadas de primitivas. Contudo do ponto de vista químico apresenta pouca
adaptabilidade para descrever orbitais de valência por não incluir funções de polarização.
Para solucionar o problema de demanda computacional, desenvolveram-se conjuntos
de base com valência dividida. Desta forma, ao invés de combinar três gaussianas para
descrever um orbital de valência numa única contração, pode-se dividir em dois grupos,
um com duas Gaussianas e um outro com uma única Gaussiana. A otimização destes
grupos pode ser realizada separadamente, produzindo orbitais que melhor se adaptam a
cada ambiente químico.
A base 3-21G representa os orbitais internos por uma combinação de três Gaussianas
primitivas, e cada orbital de valência é representado por dois grupos de funções, um
formado por duas Gaussianas primitivas e o outro por apenas uma.
Pode-se melhorar a base, aumentando-se o número de gaussianas. Por exemplo, a
base 6-31G utiliza 6 gaussianas primitivas para representar os orbitais internos e divide
cada orbital de valência em dois grupos um de 3 e um de 1 gaussiana primitiva. Os
resultados das bases com valência dividida são bastante superiores aos dos conjuntos
34

mínimos, mas dispende maior tempo de cálculo.
Apesar da divisão da camada de valência melhore a adequabilidade das bases facili-
tando que os orbitais atômicos se expandam ou se contraiam, mas não permite polariza-
ção, que é fundamental para a descrição acurada das ligações químicas, logo, de todas
as propriedades moleculares. Seu uso é indicado por uma letra entre parêntesis, ou por
asteriscos: 6-31G(d,p) indica o uso da base 6-31G com a inclusão de funções do tipo p
no hidrogênio e funções do tipo d nos átomos pesados; também se pode usar 6-31G**.
Ânions e átomos com pares isolados exigem que as regiões distantes dos seus núcleos
sejam melhor descritas, portanto adiciona-se mais uma Gaussiana de expoente pequeno
ao conjunto de bases, fazendo com que a função adicionada decaia "lentamente" com
o raio atômico e possua valores significativos regiões afastadas dos núcleos. Estas são
chamadas de funções difusas e são representadas pelo símbolo "+". Por exemplo, 6-
31G+(d,p). Ao adicionar função difusa ao átomo de hidrogênio tem-se, por exemplo,
6-31G++(d,p).
2.4 CORRELAÇÃO ELETRÔNICA
O método Hartree-Fock representa cada elétron com uma função de onda dependente
apenas das coordenadas do próprio elétron. Desta forma, a probabilidade de se encontrar
um elétron próximo ao núcleo depende apenas de sua posição em relação ao próprio
núcleo, mas não em relação aos demais elétrons, ou seja, não considera as interações
instantâneas entre esses elétrons (FORESMAN, 1996). Assim, os movimentos não estão
correlacionados.
Este tratamento, dado por Hartree e Fock, faz com que parte da energia seja negli-
genciada. A esta energia dá-se o nome de energia de correlação eletrônica:
Ecorr = Eexata − EHF (2.8)
onde Eexata é a energia exata não-relativística do sistema.
Assim sendo, foram desenvolvidos alguns métodos para incluir correlação eletrônica,
como Teoria de Pertubação de Moller-Plesset e a Teoria do Funcional de Densidade (TFD,
em inglês DFT).
35

2.5 TEORIA DO FUNCIONAL DA DENSIDADE (DFT)
Thomas, Fermi e Dirac, em 1920, expressaram a energia de um sistema como uma
função de sua densidade eletrônica, E = E [ρ (r)], que por sua vez é uma função das
coordenadas espaciais, r. Assim sendo, trata-se a energia como um funcional de densidade
eletrônica.
Assim, é importante salientar que a estrutura atual da DFT deve-se aos trabalho
de Hohenberg, Kohn e Shan, em 1964 e 1965, propuseram que a densidade eletrônica
determina todas as propriedades do estado fundamental de um sistema e desenvolveram
a seguinte expressão para a energia molecular:
E = ET + EV + EJ + EXC (2.9)
Onde ET é a energia cinética, EV inclui a contribuição da atração elétron- núcleo e
da repulsão núcleo-núcleo, EJ é a repulsão elétron-elétron e EXC é o termo de troca e cor-
relação. A energia de troca resulta da antisimetria das funções de onda para elétrons, já a
energia de correlação da influência instantânea que um elétron exerce sobre o movimento
dos demais.
2.6 OTIMIZAÇÃO DE GEOMETRIAS
Descreveu-se vários métodos, entretanto não discutiu-se a respeito de qual geometria
deve ser utilizada em tais cálculos. Por exemplo, ao alterar-se o comprimento de uma
ligação, altera-se também a energia da molécula. A busca é, sempre, pela estrutura
que fornece a menor energia. Dentro desta idéia, pode-se recorrer a dados experimentais
como difração de raio-X ou microondas. Porém, para muitas moléculas tais dados não são
encontrados na literatura. Este processo de determinar qual o valor mínimo da energia
do sistema é denominado de otimização de geometria (FORESMAN, 1996). Não se pode
deixar de observar que ao aumentar a complexidade do sistema, aumenta-se também o
número de parâmetros a serem otimizados (comprimentos de ligações e ângulos).
Este problema é resolvido ao se determinar as derivadas da energia relativas a cada
parâmetro. Assim, os algoritmos utilizados hoje são construídos de modo a procurar por
mínimos de energia. As derivadas de energia possuem expressões analíticas conhecidas
para a maioria dos métodos teóricos.
Resumindo, parte-se de uma estrutura inicial, com parâmetros estruturais aproxima-
dos, calcula-se a energia da molécula e determina-se o gradiente da energia em relação
36

aos parâmetros. O processo se repete até que o gradiente esteja abaixo de um valor
considerado razoável dizendo-se, então, que a geometria está otimizada ou convergida.
37

3 METODOLOGIA
Há um número infinito de maneiras possíveis para se decompor uma densidade
eletrônica molecular. Os métodos de partição de densidade de elétrons podem ser se-
parados em dois tipos principais: espaço e partições no átomo central (LOPEZ, 2009).
A primeira abordagem divide a densidade em regiões distintas separadas, cada uma
correspondendo a um domínio atômico. Um método bem conhecido deste tipo é o Áto-
mos em Moléculas de Bader (AIM)(BADER, 1994). A segunda abordagem atribui uma
distribuição de carga centrada em um determinado átomo, e a cada dois centros de dis-
tribuição é dividido entre os dois átomos envolvidos.
A diferença entre os vários métodos de partição centrados em átomos está relacionada
em como a distribuição de dois centros estão divididas. Dois exemplos de métodos de
partição de átomos são a Análise de Multipolos Distribuídos de Stone (DMA) (STONE,
1997) e Átomos Deformados em moléculas(DAM) (RICO, 2005). Em trabalhos anteri-
ores, explorou-se o DMA para estudar os efeitos de microondas em processos catalíticos
de hidrodessulfuração (HDS) (BORGES JR, 2007) e propriedades de moléculas com po-
tencial para ser novos materiais energéticos (BORGES JR, 2008).
Esta análise consiste em descrever uma distribuição de cargas moleculares em termos
de momentos de multipolo distribuídos em um número de posições dentro da molécula,
podendo ser derivados da função de onda sem necessidade de ajuste. Desta forma, são
sensíveis a qualidade da função de onda e dos conjuntos de base.
Neste trabalho, usou-se outra decomposição possível da densidade eletrônica, o
método de átomos deformados em moléculas (DAM)(LOPEZ, 2009; RICO, 2005) para
decompor o cálculo da densidade eletrônica molecular B3LYP e calcular as funções nu-
cleares Fukui.
As moléculas podem ser consideradas como núcleos em meio a uma nuvem de carga
dos seus elétrons. As características dessa nuvem são determinadas pela densidade
eletrônica, dada pela probabilidade de encontrar um életron por unidade de volume (den-
sidade de probabilidade). A densidade eletrônica determina as forças químicas e, por sua
vez, o comportamento químico de qualquer sistema molecular.
No método DAM são atribuídas a cada átomo distribuições de carga centradas em
seu núcleo mais a influência dos átomos mais próximos, levando, desta forma, a uma den-
38

sidade molecular dividida em densidades pseudoatômicas deformadas. Estas podem ser
representadas com acurácia em termos do produto dos harmônicos regulares pelos fatores
radiais e, assim, a soma das densidades atômicas reproduzem exatamente a densidade da
molécula inteira (RICO, 1999, 2002). O resultado é um retrato detalhado da densidade
eletrônica molecular com as regiões de acúmulo e de depleção de elétrons reproduzindo
as esperadas intuições químicas.
Um exemplo pode ser visto na Figura 3.1, onde estão representadas, para a molécula
de água, as superfícies de deformação atômica positivas (regiões de acúmulo de elétrons) e
negativas (regiões de depleção de elétrons), além da composição das duas, usando valores
de contorno de ±0.05au(elétron/bohr3). Para este caso em particular, as regiões foram
desenhadas separadamente para uma maior clareza do resultado.
(a) (b) (c)
FIG. 3.1: Superfície de deformação atômica na molécula da água. (a) Vermelho: regiõesde acúmulo de elétrons. (b) Azul: regiões de depleção de elétrons. (c) Composição de
(a) e (b). Valores de contorno: ±0.05au (elétron/bohr3).(Adaptado de:http://web.uam.es/departamentos/ciencias/qfa/DAM/definition.html - acessado em 12
de janeiro de 2010.)
Este método de decompor a densidade eletrônica das moléculas permite calcular
muitas propriedades moleculares, tais como potencial eletrostático molecular e forças
eletrostáticas. Assim, o DAM mapeia as regiões de depleção e acúmulo de elétrons na
molécula, e a partir destas densidades foram calculadas duas funções vetoriais para cada
átomo nos confôrmeros de RDX. A função Gi que representa a rigidez nuclear é definida
pela variação da dureza molecular após deformação molecular e a função Φi que repre-
senta o índice de reatividade nuclear sendo definida pela variação da eletronegatividade
após a deformação molecular. Ambas as funções são vetores tridimensionais.
39

A idéia de funções nucleares de Fukui decorre do teorema eletrostático de Hellmann-
Feynmann (LEVINE, 1991). Segundo este teorema a força eletrostática total Fi em um
núcleo i em uma molécula, devido aos elétrons e aos núcleos remanescentes, é dada por
Fi =
[
Zi
∫
ρ (r)ri
r3i
d3r −∑
i6=j
ZjRij
R3ij
]
(3.1)
onde ρ (r) é a densidade de elétrons em r, ri é o vetor separação entre o núcleo i e a
coordenada r, e Rij é o vetor separação entre os núcleos i e j, com cargas Zi e Zj, respec-
tivamente. Baseado nas forças de Hellmann-Feynmann, Ordon e Komorowski (ORDON,
1998; KOMOROWSKI, 2003) definiram duas funções resposta nucleares de Fukui. Uma
delas é a derivada da eletronegatividade (χ) com respeito à distorção (dos núcleos) da
molécula Qi, chamada de reatividade, representada pelo vetor nuclear Φi:
Φi =∂χ
∂Qi
∼=1
2
(
F+i − F−
i
)
(3.2)
O outro é a derivada da dureza (η) com respeito à distorção (dos núcleos) da molécula
Qi, a dureza, representada pelo vetor nuclear Gi:
Gi =∂η
∂Qi
∼= −1
2
(
F+i + F−
i
)
(3.3)
As forças de Hellmann-Feynmann F+i e F−
i são calculadas a partir da densidade DAM
das espécies catiônicas e aniônicas, de cálculos single-point na geometria convergida do
estado fundamental da molécula neutra de cada confôrmero. Swadley e Li estudaram a
decomposição dos quatro confôrmeros do RDX, mas usaram diferentes funções nucleares
de Fukui, e as forças nucleofílica e eletrofílica, respectivamente, definidas como Φ+i∼= F+
i
e Φ−i∼= −F+
i (SWADLEY, 2007).
Os vetores Gi e Φi são propriedades intrínsecas da molécula. Eles foram original-
mente definidos para fornecer informações sobre o início de uma fragmentação molecular
(LUTY, 2002). Em um trabalho recente, foi mostrado para uma molécula diferente, a his-
tamina, utilizando a decomposição DAM da densidade eletrônica, que ambas as funções
nucleares de Fukui Gi e Φi possuem uma interpretação simples e clara como se segue
(BORGES JR, 2009). Uma magnitude grande para o vetor Gi em um sítio atômico
indica uma considerável região de depleção de elétrons, ou seja, um sítio ácido de Lewis
particularmente propenso a ganhar elétrons, a ataques nucleofílicos e onde a dureza tem
a maior variação. Por outro lado, uma magnitude grande para o vetor Φi representa
uma região de acúmulo de elétrons particularmente grande, portanto, um sítio de base
40

de Lewis suscetível a ionização, a ataques eletrofílicos e onde a eletronegatividade varia
mais. Esta interpretação é confirmada neste trabalho para os quatro confôrmeros do
RDX, como mostrado no capítulo de resultados.
A vantagem desta proposta com respeito às anteriores é a combinação dos cálculos
de funções de Fukui com a visualização da densidade eletrônica via DAM, o que leva à
interpretações químicas mais claras.
Inserido neste assunto, faz-se necessário mencionar a Teoria de Pearson (COSTA,
2005), que classifica os ácidos e bases em duros e moles. Nas bases de Lewis moles, o
átomo que contém o par de elétrons livre é pouco eletronegativo, fazendo com que tal
base possua uma nuvem eletrônica facilmente polarizável. Na situação oposta, tem-se
as bases duras, cujo átomo que contém o par de elétrons não compartilhado de ele-
vada eletronegatividade, sendo pouco polarizáveis. Os ácidos de Lewis moles são sis-
temas pouco eletronegativos, ou seja, facilmente polarizáveis e os ácidos duros, têm alta
eletronegatividade, são pouco polarizáveis. Tal conceito é de grande importância na
compreensão da reatividade e da estabilidade das moléculas.
A geometria do estado fundamental de cada uma das quatro conformações possíveis
do RDX foi otimizada com o método DFT e o funcional B3LYP (KOCH, 2001). Cálculos
de frequências confirmaram o caráter de mínimo de cada estrutura (RICE, 2002, 2006;
VLADIMIROFF, 2002). Comparações com os cálculos anteriores ratificaram a geometria
convergida. A base gaussiana utilizada foi 6-311 + G (2d, p). Cálculos de single-point
para o cátion e ânion de cada confôrmero, usando a geometria otimizada do estado
fundamental, foram realizados com o mesmo nível de cálculo.
Usou-se o programa DAMQT (LOPEZ, 2009) para a decomposição da densidade
B3LYP em densidades deformadas e, a partir deles, para calcular as forças de Helmann-
Feynman necessárias para as funções nucleares de Fukui.
O DAM permitiu mapear as regiões moleculares da depleção e acúmulo de elétrons,
e a partir desta densidade foram calculadas duas funções vetor para cada átomo i na
molécula: a função Gi - rigidez nuclear - é a variação da dureza com a deformação
molecular e a função Φi - o índice de reatividade nuclear - é a variação da eletronegativi-
dade com a deformação molecular. Ao combinar a visualização da densidade eletrônica
DAM com os vetores Gi e Φi calculados, estabeleceu-se, uma interpretação clara dos dois
vetores, apresentada um pouco mais acima. Ambas as propriedades fornecem informações
sobre o início da fragmentação molecular.
41

4 RESULTADOS E DISCUSSÃO
A Figura 1.6 representa os mecanismos possíveis de decomposição do RDX
(SWADLEY, 2007). As Figuras 4.2, 4.3, 4.4 e 4.5 mostram a densidade eletrônica
decomposta segundo o método DAM (a), e ambas as funções nucleares Fukui (b), para
os confôrmeros AAA, AAE, AEE e EEE, respectivamente, com índice "A" representando
um grupo NO2 em posição axial e "E" em posição equatorial. As Tabelas 4.1, 4.2, 4.2
e 4.2 apresentam as funções nucleares Fukui calculadas para cada confôrmero.
FIG. 4.1: Escala de energias relativas para os confôrmeros do RDX representados emvista frontal.
O confôrmero AAE tem a mais baixa energia, com as outras estruturas seguintes
na seguinte ordenação energética (energias relativas em kJ/mol entre parênteses): AAA
(3,11), AEE (3,47), EEE (18,72), conforme Figura 4.1. Portanto, a fase gasosa do RDX
provavelmente tem três confôrmeros de menor energia - AAA, AAE e AEE - interconver-
síveis, ou seja, coexistindo. Energias similares foram previamente obtidas (SWADLEY,
2007).
As superfícies DAM obtidas mostram as regiões de densidade eletrônica molecular
42

(a)
(b)
(c)
FIG. 4.2: Confôrmero AAA do RDX: (a) Superfície de deformação atômica namolécula. Azul: regiões de depleção de elétrons. Vermelho: regiões de acúmulo de
elétrons. Valores de contorno: +0.020 e −0.020 (elétron/bohr3). (b) Vetores de durezanuclear (verde) e índice de reatividade (rosa). (c) vista frontal.
com acúmulo e depleção de elétrons, no que diz respeito à distribuição atômica esférica.
Como esperado, a superfície de densidade deformada muda de um confôrmero para outro.
Em particular, quanto maior o número de grupos NO2 em posição E (equatorial), mais
43
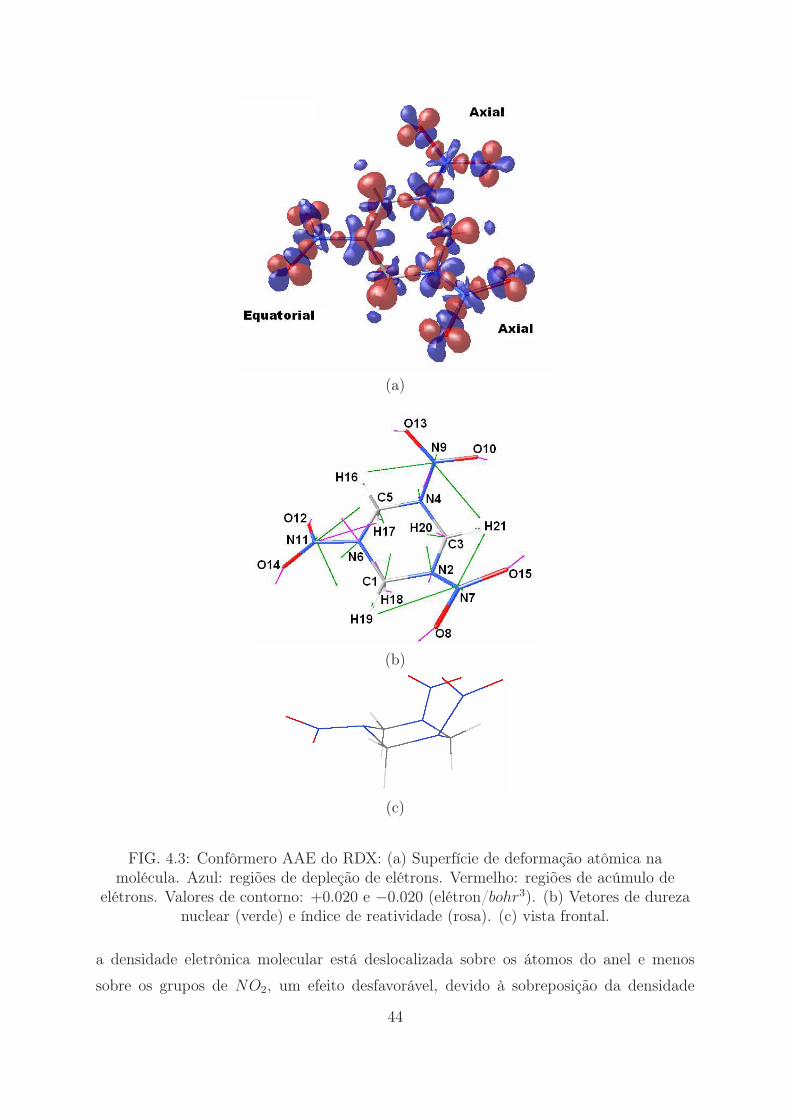
(a)
(b)
(c)
FIG. 4.3: Confôrmero AAE do RDX: (a) Superfície de deformação atômica namolécula. Azul: regiões de depleção de elétrons. Vermelho: regiões de acúmulo de
elétrons. Valores de contorno: +0.020 e −0.020 (elétron/bohr3). (b) Vetores de durezanuclear (verde) e índice de reatividade (rosa). (c) vista frontal.
a densidade eletrônica molecular está deslocalizada sobre os átomos do anel e menos
sobre os grupos de NO2, um efeito desfavorável, devido à sobreposição da densidade
44

(a)
(b)
(c)
FIG. 4.4: Confôrmero AEE do RDX: (a) Superfície de deformação atômica na molécula.Azul: regiões de depleção de elétrons. Vermelho: regiões de acúmulo de elétrons.
Valores de contorno: +0.020 e −0.020 (elétron/bohr3). (b) Vetores de dureza nuclear(verde) e índice de reatividade (rosa). (c) vista frontal.
eletrônica fora do plano (elétrons pi) dos átomos de carbono do anel e dos átomos de
oxigênio mais próximos dos grupos NO2 vizinhos. Isto é visto mais claramente quando
a densidade de elétrons DAM do confôrmero AAA (Figura 4.2) é comparada com a do
45
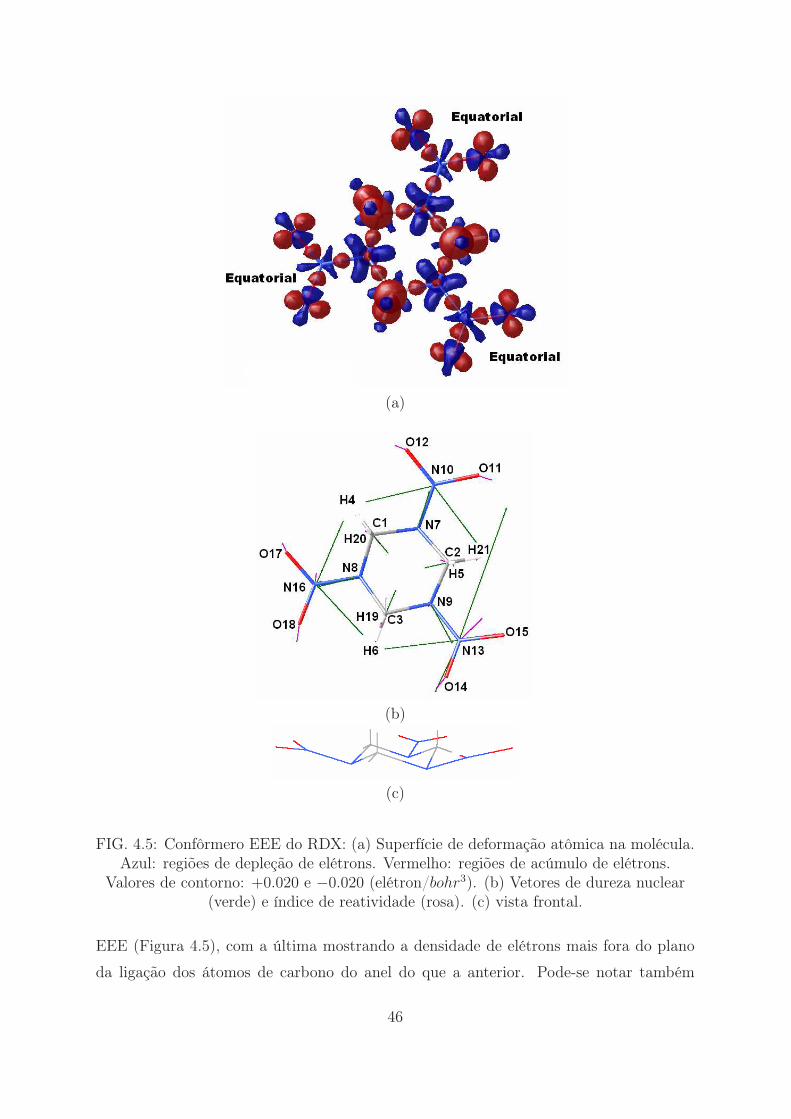
(a)
(b)
(c)
FIG. 4.5: Confôrmero EEE do RDX: (a) Superfície de deformação atômica na molécula.Azul: regiões de depleção de elétrons. Vermelho: regiões de acúmulo de elétrons.
Valores de contorno: +0.020 e −0.020 (elétron/bohr3). (b) Vetores de dureza nuclear(verde) e índice de reatividade (rosa). (c) vista frontal.
EEE (Figura 4.5), com a última mostrando a densidade de elétrons mais fora do plano
da ligação dos átomos de carbono do anel do que a anterior. Pode-se notar também
46

que a densidade eletrônica dos grupos nitro no confôrmero AAA é mais volumosa do
que no EEE. Esse comportamento das densidades eletrônicas pode explicar a ordem de
estabilidade encontrada, com os confôrmeros que possuem dois ou três grupos nitro na
posição equatorial (E), sendo menos estáveis.
Os pares isolados do átomo de oxigênio dos grupos NO2 são claramente vistos com
as densidades DAM calculadas, com a sua extensão espacial sendo maior para os grupos
axiais do que para os grupos equatoriais, pois, como discutido no último parágrafo, um
grupo equatorial contribui para uma maior deslocalização de elétrons dos átomos de
carbono do anel. As ligações dos átomos de nitrogênio possuem três regiões de depleção
de elétrons, apontando para os vértices de um triângulo imaginário, devido aos vizinhos
oxigênios serem mais eletronegativos.
Em geral, as regiões de acúmulo e depleção de elétrons seguem claramente a diferença
de eletronegatividade dos átomos vizinhos, com acúmulo de elétrons em átomos mais
eletronegativos e vice-versa, como seria de esperar da intuição química. Os átomos de
hidrogênio e os átomos de carbono do anel (ao longo das ligações com os átomos de
N do anel) são as regiões de depleção de elétrons, porque são, respectivamente, ligados
ao carbono mais eletronegativo e a átomos de nitrogênio. Inversamente, os átomos de
oxigênio e nitrogênio, este último ao longo das ligações do anel, são regiões de acúmulo
de elétrons pela mesma razão.
Pode-se observar para os quatro confôrmeros do RDX (Figuras 4.2b, 4.3b, 4.4b e
4.5b) que, de acordo com a definição das funções nucleares Fukui, um vetor de dureza
nuclear Gi em um sítio atômico indica uma região de depleção de elétrons, assim, um sítio
de ácido de Lewis suscetível ao ganho de elétrons, a ataques nucleofílicos e onde a dureza
varia especialmente. Por outro lado, uma magnitude grande do vetor que representa o
índice de reatividade representa uma região de acúmulo de elétrons, portanto um sítio
de base de Lewis onde a eletronegatividade varia bastante. Foi verificado em trabalho
anterior uma conexão direta entre os vetores e e as regiões de depleção e acúmulo de
elétrons, respectivamente, obtidas a partir da decomposição da densidade eletrônica pelo
método DAM para a molécula da histamina (BORGES JR, 2009).
Uma vez que os vetores calculados fornecem informações do início da fragmentação
molecular, estes resultados proporcionam algumas indicações sobre isso. Para o con-
fôrmero AAA (Figura 4.2), os vetores de dureza nuclear Gi têm os maiores valores sobre
os átomos O dos grupos NO2, perto de 0,35 u.a., com os átomos N do anel com valores
cerca de 43% menores, assim, esses sítios são propensos ao ganho de elétrons, ataques
47

TAB. 4.1: Magnitudes dos vetores de dureza e índice de reatividade nuclear para cada átomo do confôrmero AAA do RDX.
ÁtomoCoordenadas Componentes Vetoriais Gi Magnitude Componentes Vetoriais Φi Magnitude
x y z x y z Gi x y z Φi
N1 -1,21346 -0,70059 0,79471 0,04142 0,02430 0,14153 0,14946 -0,00805 -0,00426 0,00059 0,00913
C2 -1,23842 0,71500 1,15801 0,06867 0,03526 0,04165 0,08771 0,02011 -0,00722 -0,01915 0,02869
N3 0,00000 1,40118 0,79471 0,00000 0,03322 0,14181 0,14565 0,00000 0,02391 0,00031 0,02391
C4 1,23842 0,71500 1,15801 0,06867 0,03526 0,04165 0,08771 -0,02011 -0,00722 -0,01915 0,02869
N5 1,21346 -0,70059 0,79471 0,04142 0,02430 0,14153 0,14946 0,00805 -0,00426 0,00059 0,00913
C6 0,00000 -1,43000 1,15801 0,00000 0,06869 0,02782 0,07411 0,00000 0,01261 -0,00532 0,01369
N7 0,00000 2,20404 -0,37318 0,00000 0,00940 0,00344 0,01001 0,00000 -0,02724 0,03662 0,04564
O8 1,09429 2,54505 -0,80196 0,30697 0,10654 0,11739 0,34549 0,01080 0,00055 -0,00523 0,01201
N9 1,90876 -1,10202 -0,37318 0,00589 0,00102 0,02019 0,02106 -0,00956 0,00790 0,01987 0,02342
O10 1,65693 -2,22021 -0,80196 0,05900 0,31907 0,11794 0,34525 -0,00715 -0,00967 -0,00469 0,01291
N11 -1,90876 -1,10202 -0,37318 0,00589 0,00102 0,02019 0,02106 0,00956 0,00790 0,01987 0,02342
O12 -2,75122 -0,32484 -0,80196 0,24537 0,21291 0,12002 0,34633 -0,00626 0,00875 -0,00260 0,01107
O13 -1,09429 2,54505 -0,80196 0,30697 0,10654 0,11739 0,34549 -0,01080 0,00055 -0,00523 0,01201
O14 2,75122 -0,32484 -0,80196 0,24537 0,21291 0,12002 0,34633 0,00626 0,00875 -0,00260 0,01107
O15 -1,65693 -2,22021 -0,80196 0,05900 0,31907 0,11794 0,34525 0,00715 -0,00967 -0,00469 0,01291
H16 2,08533 1,20397 0,68544 0,01391 0,00850 0,00839 0,01833 -0,00115 -0,00113 -0,00231 0,00282
H17 1,33321 0,76973 2,24713 0,00116 0,00128 0,03507 0,03511 -0,00032 -0,00079 -0,00058 0,00103
H18 0,00000 -1,53946 2,24713 0,00000 0,00191 0,03603 0,03608 0,00000 0,00094 -0,00155 0,00181
H19 0,00000 -2,40793 0,68544 0,00000 0,01629 0,00793 0,01812 0,00000 0,00154 -0,00186 0,00241
H20 -1,33321 0,76973 2,24713 0,00116 0,00128 0,03507 0,03511 0,00032 -0,00079 -0,00058 0,00103
H21 -2,08533 1,20397 0,68544 0,01391 0,00850 0,00839 0,01833 0,00115 -0,00113 -0,00231 0,00282
48

TAB. 4.2: Magnitudes dos vetores de dureza e índice de reatividade nuclear para cada átomo do confôrmero AAE do RDX.
ÁtomoCoordenadas Componentes Vetoriais Gi Magnitude Componentes Vetoriais Φi Magnitude
x y z x y z Gi x y z Φi
C1 0,65586 -1,33132 0,65083 -0,02697 0,07658 -0,02862 0,08609 0,00807 0,01574 -0,00076 0,01770
N2 -0,79198 -1,15104 0,73897 -0,01328 0,03499 -0,14431 0,14908 0,00238 -0,00683 -0,00044 0,00725
C3 -1,24259 0,13542 1,26669 0,05253 -0,00583 -0,05606 0,07705 0,00553 -0,00099 -0,01444 0,01549
N4 -0,53854 1,26116 0,65551 -0,02216 -0,03705 -0,13998 0,14649 0,00162 0,00548 0,00151 0,00591
C5 0,91451 1,13051 0,56565 -0,02894 -0,06423 -0,02452 0,07459 0,01541 -0,01800 0,00488 0,02419
N6 1,32164 -0,17601 0,05206 0,09834 -0,02131 0,07743 0,12697 -0,00679 0,00030 -0,04148 0,04203
N7 -1,60482 -1,79852 -0,22467 -0,01107 -0,00801 -0,01231 0,01839 0,01337 0,01060 0,01727 0,02428
O8 -1,09054 -2,72151 -0,84208 -0,13487 0,27035 0,17422 0,34876 0,00936 -0,00755 -0,00367 0,01257
N9 -1,20617 1,99585 -0,35595 -0,00983 0,01549 -0,01506 0,02374 0,01129 -0,01295 0,01260 0,02131
O10 -2,42199 1,86830 -0,41185 0,34045 0,02532 0,01484 0,34171 -0,00703 -0,00354 0,00036 0,00788
N11 2,70701 -0,16782 -0,24665 -0,00334 0,01661 0,00514 0,01771 -0,05192 0,00935 -0,00176 0,05278
O12 3,37130 0,72314 0,26570 -0,21297 -0,24999 -0,14775 0,36011 0,00801 0,01159 0,01303 0,01919
O13 -0,51594 2,74756 -1,03130 -0,18393 -0,22209 0,18992 0,34529 0,00897 0,00080 -0,00220 0,00927
O14 3,12477 -1,09490 -0,92753 -0,12945 0,26343 0,19292 0,35124 0,00524 -0,01152 0,00096 0,01269
O15 -2,76805 -1,42548 -0,29788 0,33275 -0,09343 0,01731 0,34605 -0,01104 0,00731 -0,00152 0,01333
H16 1,31922 1,91831 -0,06303 -0,01131 -0,01530 0,00026 0,01903 -0,00150 -0,00397 0,00419 0,00596
H17 1,31256 1,21920 1,58145 -0,01401 -0,00702 -0,03017 0,03400 -0,00354 0,00205 0,00368 0,00550
H18 1,03411 -1,43106 1,67315 -0,01409 0,00524 -0,03224 0,03557 -0,00509 -0,00097 0,00530 0,00741
H19 0,88369 -2,22707 0,08039 -0,00877 0,01793 0,00035 0,01996 -0,00125 0,00272 0,00417 0,00513
H20 -1,00964 0,14795 2,33615 -0,01120 -0,00045 -0,03014 0,03216 -0,00140 0,00050 0,00048 0,00156
H21 -2,31304 0,24272 1,11742 0,01210 -0,00121 -0,01120 0,01653 0,00028 -0,00007 -0,00221 0,00223
49
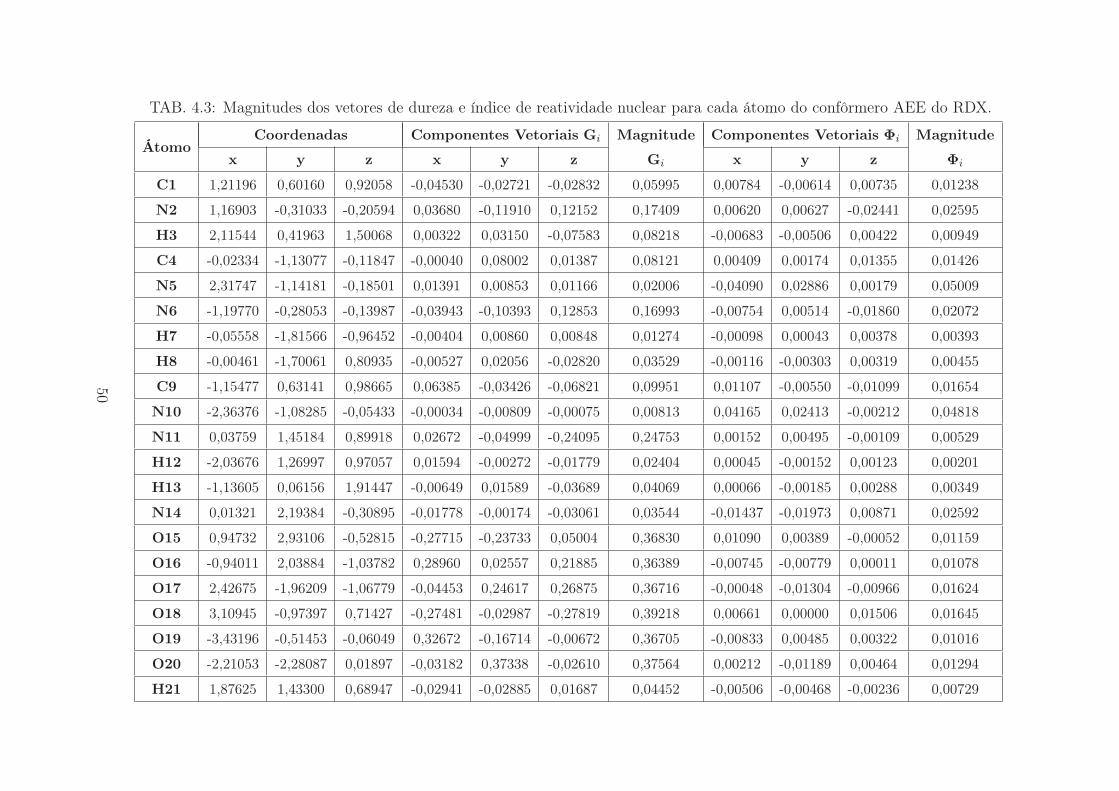
TAB. 4.3: Magnitudes dos vetores de dureza e índice de reatividade nuclear para cada átomo do confôrmero AEE do RDX.
ÁtomoCoordenadas Componentes Vetoriais Gi Magnitude Componentes Vetoriais Φi Magnitude
x y z x y z Gi x y z Φi
C1 1,21196 0,60160 0,92058 -0,04530 -0,02721 -0,02832 0,05995 0,00784 -0,00614 0,00735 0,01238
N2 1,16903 -0,31033 -0,20594 0,03680 -0,11910 0,12152 0,17409 0,00620 0,00627 -0,02441 0,02595
H3 2,11544 0,41963 1,50068 0,00322 0,03150 -0,07583 0,08218 -0,00683 -0,00506 0,00422 0,00949
C4 -0,02334 -1,13077 -0,11847 -0,00040 0,08002 0,01387 0,08121 0,00409 0,00174 0,01355 0,01426
N5 2,31747 -1,14181 -0,18501 0,01391 0,00853 0,01166 0,02006 -0,04090 0,02886 0,00179 0,05009
N6 -1,19770 -0,28053 -0,13987 -0,03943 -0,10393 0,12853 0,16993 -0,00754 0,00514 -0,01860 0,02072
H7 -0,05558 -1,81566 -0,96452 -0,00404 0,00860 0,00848 0,01274 -0,00098 0,00043 0,00378 0,00393
H8 -0,00461 -1,70061 0,80935 -0,00527 0,02056 -0,02820 0,03529 -0,00116 -0,00303 0,00319 0,00455
C9 -1,15477 0,63141 0,98665 0,06385 -0,03426 -0,06821 0,09951 0,01107 -0,00550 -0,01099 0,01654
N10 -2,36376 -1,08285 -0,05433 -0,00034 -0,00809 -0,00075 0,00813 0,04165 0,02413 -0,00212 0,04818
N11 0,03759 1,45184 0,89918 0,02672 -0,04999 -0,24095 0,24753 0,00152 0,00495 -0,00109 0,00529
H12 -2,03676 1,26997 0,97057 0,01594 -0,00272 -0,01779 0,02404 0,00045 -0,00152 0,00123 0,00201
H13 -1,13605 0,06156 1,91447 -0,00649 0,01589 -0,03689 0,04069 0,00066 -0,00185 0,00288 0,00349
N14 0,01321 2,19384 -0,30895 -0,01778 -0,00174 -0,03061 0,03544 -0,01437 -0,01973 0,00871 0,02592
O15 0,94732 2,93106 -0,52815 -0,27715 -0,23733 0,05004 0,36830 0,01090 0,00389 -0,00052 0,01159
O16 -0,94011 2,03884 -1,03782 0,28960 0,02557 0,21885 0,36389 -0,00745 -0,00779 0,00011 0,01078
O17 2,42675 -1,96209 -1,06779 -0,04453 0,24617 0,26875 0,36716 -0,00048 -0,01304 -0,00966 0,01624
O18 3,10945 -0,97397 0,71427 -0,27481 -0,02987 -0,27819 0,39218 0,00661 0,00000 0,01506 0,01645
O19 -3,43196 -0,51453 -0,06049 0,32672 -0,16714 -0,00672 0,36705 -0,00833 0,00485 0,00322 0,01016
O20 -2,21053 -2,28087 0,01897 -0,03182 0,37338 -0,02610 0,37564 0,00212 -0,01189 0,00464 0,01294
H21 1,87625 1,43300 0,68947 -0,02941 -0,02885 0,01687 0,04452 -0,00506 -0,00468 -0,00236 0,00729
50
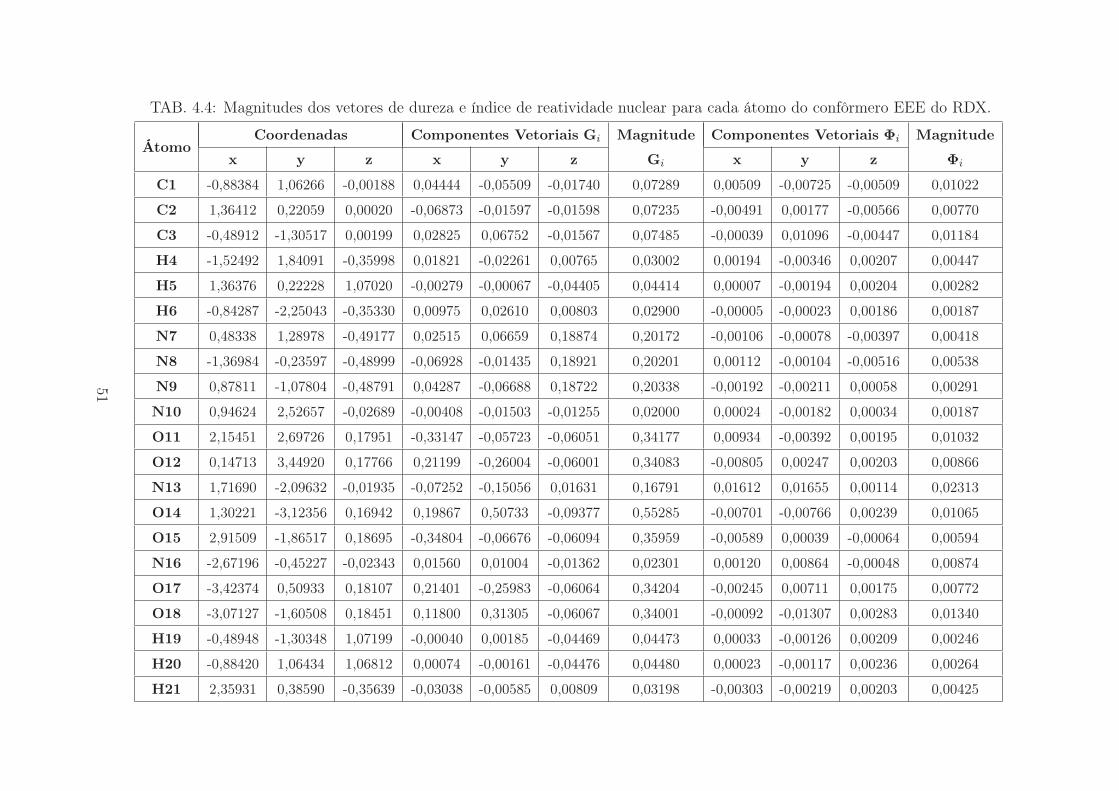
TAB. 4.4: Magnitudes dos vetores de dureza e índice de reatividade nuclear para cada átomo do confôrmero EEE do RDX.
ÁtomoCoordenadas Componentes Vetoriais Gi Magnitude Componentes Vetoriais Φi Magnitude
x y z x y z Gi x y z Φi
C1 -0,88384 1,06266 -0,00188 0,04444 -0,05509 -0,01740 0,07289 0,00509 -0,00725 -0,00509 0,01022
C2 1,36412 0,22059 0,00020 -0,06873 -0,01597 -0,01598 0,07235 -0,00491 0,00177 -0,00566 0,00770
C3 -0,48912 -1,30517 0,00199 0,02825 0,06752 -0,01567 0,07485 -0,00039 0,01096 -0,00447 0,01184
H4 -1,52492 1,84091 -0,35998 0,01821 -0,02261 0,00765 0,03002 0,00194 -0,00346 0,00207 0,00447
H5 1,36376 0,22228 1,07020 -0,00279 -0,00067 -0,04405 0,04414 0,00007 -0,00194 0,00204 0,00282
H6 -0,84287 -2,25043 -0,35330 0,00975 0,02610 0,00803 0,02900 -0,00005 -0,00023 0,00186 0,00187
N7 0,48338 1,28978 -0,49177 0,02515 0,06659 0,18874 0,20172 -0,00106 -0,00078 -0,00397 0,00418
N8 -1,36984 -0,23597 -0,48999 -0,06928 -0,01435 0,18921 0,20201 0,00112 -0,00104 -0,00516 0,00538
N9 0,87811 -1,07804 -0,48791 0,04287 -0,06688 0,18722 0,20338 -0,00192 -0,00211 0,00058 0,00291
N10 0,94624 2,52657 -0,02689 -0,00408 -0,01503 -0,01255 0,02000 0,00024 -0,00182 0,00034 0,00187
O11 2,15451 2,69726 0,17951 -0,33147 -0,05723 -0,06051 0,34177 0,00934 -0,00392 0,00195 0,01032
O12 0,14713 3,44920 0,17766 0,21199 -0,26004 -0,06001 0,34083 -0,00805 0,00247 0,00203 0,00866
N13 1,71690 -2,09632 -0,01935 -0,07252 -0,15056 0,01631 0,16791 0,01612 0,01655 0,00114 0,02313
O14 1,30221 -3,12356 0,16942 0,19867 0,50733 -0,09377 0,55285 -0,00701 -0,00766 0,00239 0,01065
O15 2,91509 -1,86517 0,18695 -0,34804 -0,06676 -0,06094 0,35959 -0,00589 0,00039 -0,00064 0,00594
N16 -2,67196 -0,45227 -0,02343 0,01560 0,01004 -0,01362 0,02301 0,00120 0,00864 -0,00048 0,00874
O17 -3,42374 0,50933 0,18107 0,21401 -0,25983 -0,06064 0,34204 -0,00245 0,00711 0,00175 0,00772
O18 -3,07127 -1,60508 0,18451 0,11800 0,31305 -0,06067 0,34001 -0,00092 -0,01307 0,00283 0,01340
H19 -0,48948 -1,30348 1,07199 -0,00040 0,00185 -0,04469 0,04473 0,00033 -0,00126 0,00209 0,00246
H20 -0,88420 1,06434 1,06812 0,00074 -0,00161 -0,04476 0,04480 0,00023 -0,00117 0,00236 0,00264
H21 2,35931 0,38590 -0,35639 -0,03038 -0,00585 0,00809 0,03198 -0,00303 -0,00219 0,00203 0,00425
51

nucleofílicos e onde a dureza varia mais. Os valores restantes são cerca de uma ordem de
magnitude menor. A magnitude do vetor que representa o índice de reatividade é uma
ordem menor que o vetor de dureza, por isso, a eletronegatividade varia menos que a
dureza, e a ionização é mais provável neste sítio. O maior valor para o vetor é encontrado
no átomo de nitrogênio de um dos grupos nitro (0,046 u.a., N7), 37% maior do que o
valor dos átomos adjacentes de C do anel e 49% maior do que o vetor sobre os átomos
de N dos outros grupos nitro. Esta assimetria inesperada dos átomos de N dos grupos
nitro AAA foi anteriormente observada por Luty e colaboradores e Swandley Li (LUTY,
2002; SWADLEY, 2007) e, sendo uma indicação da resposta diferente dos átomos de
nitrogênio com simetria equivalente. Estes resultados para ambas as funções nucleares
Fukui também indicam que uma ligação NN de um dos grupos nitro serão mais afetadas
em um processo de decomposição, levando a sua liberação. Portanto, um mecanismo de
ruptura concertada dos grupos nitro para confôrmero AAA seria improvável, o que está
de acordo com trabalhos anteriores, apesar de ambos os vetores nos átomos de carbono do
anel serem apreciáveis e não se pode desconsiderar a possibilidade de quebra simultânea
de diferentes partes do anel.
Para o confôrmero AAE (Figura 4.3), os vetores de dureza nuclear de todos os átomos
de oxigênio possuem valores na faixa de 0,35-0,36 u.a., com o maior valor em um átomo
de oxigênio equatorial (O12). O maior vetor índice de reatividade (0,05278 u.a.) está no
átomo de nitrogênio (N11) do grupo equatorial, e no átomo de N do anel ligado a ele (N6,
0,04203 u.a.) - ver Figura 4.3. Este último também tem um valor apreciável do vetor de
dureza (0,12697 u.a.). Esta situação aponta para uma liberação do grupo nitro equatorial
como mais provável, o que está em acordo com Swandley e Li (SWADLEY, 2007). A
eliminação HONO aqui parece improvável porque os valores dos vetores de índice de
reatividade dos átomos de hidrogênios, incluindo os que estão próximos dos átomos de
O equatoriais, à semelhança dos confôrmeros AAA e AAE, são muito pequenos quando
comparados com o confôrmero AEE que favorecem este mecanismo- ver discussão no
próximo parágrafo.
No confôrmero AEE (Figura 4.4), os vetores de dureza nuclear Gi têm os maiores
valores sobre os átomos de oxigênio dos grupos nitro. Um dos átomos de oxigênio em um
grupo NO2 equatorial (O18), mais perto do grupo A, tem o valor mais elevado (0,3922
u.a.), o átomo equivalente no outro grupo de nitro (O19) tem 0,3671 u.a., enquanto os
átomos de O restantes tem valores na faixa de intervalo 0,3639-0,3756 u.a. Estes valores
são bastante próximos, o que favoreceria diferentes formas de eliminação HONO. Em
52

particular, a ligação de O18 a um átomo de hidrogênio para formar HONO seria o mais
favorecido com um dos átomos de hidrogênio mais próximos (H3), pois o valor do seu
vetor Gi é mais do que duas vezes os valores para os valores de outros átomos H. Os
outros vetores relevantes Gi estão nos átomos de N do anel, com o átomo de N ligado ao
grupo axial tendo 0,2475 u.a. enquanto os outros dois têm cerca de 0,17 u.a. O vetor de
índice de reatividade nuclear em um átomo de N de um dos grupos nitro equatorial (N10),
com magnitude de 0,04818 u.a., e o átomo do anel preso a ele (N6), com 0,02072 u.a.,
favorecem uma única liberação deste grupo NO2 ou; a formação de uma ligação deste
grupo nitro com átomos de hidrogênio vizinho, este último pode ser liberado a partir do
átomo de carbono C9 ao lado do grupo axial porque tem um vetor índice de reatividade
maior do que no átomo C4. Este quadro global é ainda apoiado ao se considerar o
acúmulo de elétrons nos átomos de oxigênio perto de uma região de depleção de elétrons
dos átomos de hidrogênio vizinhos (Figura 4.4a).
Finalmente, para o confôrmero EEE (Figura 4.5), como na estrutura AAA, obteve-
se um vetor Gi assimétrico em um dos átomos de oxigênio de um grupo nitro (O14),
com sua magnitude sendo cerca de 38% maior do que para os outros átomos de oxigênio
de simetria equivalente - isso também é visto claramente na Figura 4.5. O átomo de
nitrogênio do mesmo grupo nitro (N13) tem um vetor Gi quase 10 vezes maior do que
os valores dos átomos de nitrogênio equivalentes (N10 e N16). A magnitude do vetor
índice de reatividade no mesmo átomo (N13) é quase o dobro do segundo maior vetor.
Os átomos de N do anel, ligado a cada um dos grupos nitro têm magnitudes do vetor
Gi de 0,20 u.a.. Estes resultados, juntamente com a pequena magnitude de ambos os
vetores nos átomos de carbono e hidrogênio, indicam um mecanismo preferido de saída
de um dos grupos nitro EEE (composto pelos O14, O15 e átomos N13). Por outro lado,
os cálculos de Swandley e ponto Li sugerem a HONO eliminação para este confôrmero,
um processo que não está de acordo com nossos resultados.
53

5 CONCLUSÕES
O trabalho desta dissertação corresponde ao tema IME/25 da pós-graduação iniciada
em 2008, solicitado pelo CTEx e pelo IME, cuja área de conhecimento é a modelagem
molecular baseada em métodos de química quântica computacional para estudos de ma-
teriais energéticos, e com as seguintes aplicações: (1) caracterização em nível molecular
de explosivos e propelentes e, (2) desenvolvimento de novos materiais de explosivos e
propelentes. Este tópico se encontra inserido no Plano Básico de Ciência e Tecnologia,
nos grupos finalísticos GAM e GMF, e esta oficial está nomeada para o CTEx onde os
conhecimentos adquiridos serão aplicados em projetos de interesse da Força.
Neste contexto, faz-se necessário salientar que materiais energéticos (explosivos e
propelentes) necessitam obedecer a requisitos específicos tais como: alta densidade de
energia; insensibilidade a choques mecânicos; resistência à decomposição química; síntese
barata a partir de reagentes prontamente disponíveis; e, habilidade para serem formulados
com outros materiais para a fabricação de dispositivos práticos2. Assim, para obter-se
novos materiais que possuam tais requisitos é fundamental compreender melhor a física
e a química dos materiais energéticos. Até mesmo explosivos mais empregados, como o
RDX (1,3,5-trinitro-s-triazina, ou ciclotrimetilenotrinitramina), carecem de conhecimento
acurado das suas propriedades em nível molecular.
Desta forma, a pesquisa básica em química teórica e computacional é aplicada a
problemas de Defesa, como no caso deste trabalho, que estudou as possíveis formas de
decomposição para os quatro confôrmeros do RDX. Molécula escolhida por apresentar
diversas aplicações militares e civis.
O trabalho foi desenvolvido em algumas etapas. Primeiro, otimizou-se a geometria
do estado fundamental de cada uma das quatro conformações possíveis do RDX com o
método DFT, o funcional B3LYP e a base gaussiana 6-311 + G (2d, p). Desta forma,
os cálculos de frequências confirmaram o caráter mínimo de cada estrutura, que quando
comparadas com os cálculos anteriores de outros pesquisadores ratificaram a geometria
convergida (SWADLEY, 2007). Calculou-se com o mesmo nível o single-point para o
cátion e ânion de cada confôrmero, usando a geometria otimizada do estado fundamental.
Numa segunda etapa, usou-se o programa DAMQT (LOPEZ, 2009) para a decom-
2adaptado de http://www.llnl.gov/str/Simpson.html
54

posição da densidade eletrônica, calculada com o funcional B3LYP, em densidades defor-
madas e, a partir deles, para calcular as forças de Helmann-Feynman necessárias para as
funções nucleares de Fukui, que foram utilizadas.
Quando a distribuição da densidade eletrônica dos átomos da molécula é examinada,
noções da química clássica podem ser observadas, tais como pares isolados, ligações quími-
cas simples, duplas e triplas, aromaticidade e grupos funcionais. Além disto, os conceitos
de dureza e índice de reatividade nuclear, em combinação com as densidades eletrôni-
cas, forneceram subsídios para a avaliação de mecanismos de reação. Como esperado, a
superfície de densidade deformada muda de um confôrmero para outro.
Na análise realizada, a partir dos parâmetros acima, pode-se concluir que a primeira
etapa mais provável da decomposição é a ruptura da ligação N-N para os confôrmeros
AAA e AAE, e a eliminação de HONO para o confôrmero AEE. Ratificando o resultado
obtido anteriormente em outros trabalhos (SWADLEY, 2007), que utilizaram parâme-
tros distintos para a avaliação: funções nucleares de Fukui para ataques nucleofílicos e
eletrofílicos.
Já na análise do confôrmero EEE concluiu-se que a primeira etapa mais provável da
decomposição é também a ruptura da ligação N-N. Este resultado, difere da pesquisa
citada, mostrando ser interessante o desenvolvimento de um trabalho futuro com a fi-
nalidade de ratificar os resultados encontrados na literatura utilizando-se os mesmos
parâmetros de pesquisa.
Além desta pesquisa, esta oficial e seu orientador chegaram a iniciar trabalho se-
melhante para as moléculas de nitramida e dimetilnitramida. Entretanto, o mesmo não
foi concluído a tempo de ser exposto e discutido nesta dissertação. Este trabalho pode
ser continuado através da investigação de outras moléculas de materiais energéticos, per-
mitindo uma maior capacidade científica na área em foco.
Este trabalho permitiu a participação desta oficial no Simpósio Brasileiro de Química
Teórica no ano de 2009 e a confecção de um artigo aceito. Além, evidentemente, de sua
conclusão ter atingido uma estratégia de caráter dissuasório outrora mencionada. E,
ainda, a formação em química quântica permite o trabalho em projetos que envolvam
a necessidade do entendimento de processos físico-químicos em nível molecular e em
problemas de inovação tecnológica.
55

6 REFERÊNCIAS BIBLIOGRÁFICAS
AKHAVAN, J. The Chemistry of Explosives. RSC Paperbacks, UK, 2 edição, 2004.
ATKINS, P. W. e FRIEDMAN, R. S. Molecular Quantum Mechanics. OxfordUniversity Press, Oxford, 3 edição, 1997.
BADER, R. F. W. Atoms in Molecules: A Quantum Theory. Oxford UniversityPress, Oxford, 1994.
BEHRENS, R. J. Thermal decomposition of energetic materials: tempo-ral behaviors of the rates of formation of the gaseous pyrolysis prod-ucts from condensed-phase decomposition of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine. J. Phys. Chem., 94(17):6706–6718, Agosto 1990a.
BEHRENS, R. J. Thermal decomposition of hmx and rdx: Decomposition processes andmechanisms based on stmbms and tof velocity-spectra measurements. Em BULUSU,
S. N., editor, Chemistry and Physics of Energetic Materials, 1990b.
BEHRENS, R. J. e BULUSU, S. Thermal decomposition of energetic materials.2. deuterium isotope effects and isotopic scrambling in condensed-phasedecomposition of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine. J. Phys.Chem., 95(15):5838–5845, Julho 1991.
BEHRENS, R. J. e BULUSU, S. Thermal decomposition of energetic materials.3. temporal behaviors of the rates of formation of the gaseous pyrolysisproducts from condensed-phase decomposition of 1,3,5-trinitrohexahydro-s-triazine (rdx). J. Phys. Chem., 96(22):8877–8891, Outubro 1992.
BORGES JR, I., SILVA, A., AGUIAR, A., BORGES, L., SANTOS, J. e DIAS, M. Den-sity functional theory molecular simulation of thiophene adsorption on mos2including microwave effects. J. Molec. Structure (THEOCHEM), 822(1-3):80–88,2007.
BORGES JR, I. Conformations and charge distributions of diazocyclopropanes.International Journal of Quantum Chemistry, 108(13):2615–2622, 2008.
BORGES JR, I. e LIMA, A. L. S. AN PINTO, A. C. submetido, 2009.
BOTCHER, T. R. e WIGHT, C. A. Transient thin film laser pyrolysis of rdx. J.Phys. Chem., 97(36):9149, Setembro 1993.
BOTCHER, T. R. e WIGHT, C. A. Explosive thermal decomposition mechanismof rdx. J. Phys. Chem., 98(21):5441, Maio 1994.
BRASIL. Decreto no 6703/2008 - Estratégia Nacional de Defesa. Poder Execu-tivo, 18 de dezembro, 2008.
56

CHAKRABORTY, D., MULLER, R. P., DASGUPTA, S. e GODDARD, W. A. Themechanism for unimolecular decomposition of rdx (1,3,5-trinitro-1,3,5-triazine), an ab initio study. J. Phys. Chem. A, 104(11):2261–2272, 2000.
CHOI, M., KIM, H. e CHUNG, C. Ft-ir spectra of photochemical reaction productsof crystalline rdx. J. Phys. Chem., 99(43):15785–15789, Outubro 1995.
CORNER, J. The Theory of the Interior Ballistics of Guns. John Wiley & Sons,N.Y., 1950.
COSTA, P., FERREIRA, V., ESTEVES, P. e VASCONCELLOS, M. Ácidos e Basesem Química Orgânica. Bookman, Porto Alegre, 2005.
FORESMAN, J. B. e FRISCH, A. Exploring Chemistry with Electronic StructureMethods. Gaussian, Inc., Pittsburgh, PA, 2 edição, 1996.
KOCH, W. e HOLTHAUSEN, M. C. A Chemist’s Guide to Density FunctionalTheory. Wiley-VCH Verlag GmbH, Weinheim, 2001.
KOMOROWSKI, L. e ORDON, P. Fluctuations in electronegativity and globalhardness induced by molecular vibrations. J. Molec. Structure (THEOCHEM),630, 2003.
LEVINE, I. N. Quantum Chemistry. Prentice Hall, Englewood Cliffs(NJ), 1991.
LIU, M. H. e ZHENG, G. F. Computacional study of unimolecular decompositionmechanism of rdx explosive. J. Chem. Theory Comput., 6(2):341–351, 2007.
LOPEZ, R., RICO, J. F., RAMIREZ, G., EMA, I. e ZORRILLA, D. Damqt: A pack-age for the analysis of electron density in molecules. J. of Computer PhysicsCommunications (COMPHY), 180, 2009.
LUTY, T., ORDON, P. e ECKHARDT, C. J. A model for mechanochemical trans-formations: Applications to molecular hardness, instabilities, and shockinitiation of reaction. Journal of Chemical Physics, 117(4):1775–1785, Julho 2002.
MEYER, R., KOHLER, J. e HOMBURG, A. Explosives. Wiley-VCH, Weinheim, 5edição, 2002.
MURRAY, J. S., LANE, P. e POLITZER, P. Effects of strongly electron-attractingcomponents on molecular surface eletronic potencials: Application to pre-dicting impact sensitivities of energetic molecules. Molecular Physics, 93(2):187–194, 1998.
ORDON, P. e KOMOROWSKI, L. Nuclear reactivity and nuclear stiffness indensity functional theory. Chemical Physics Letters, 292(1-2):22–27, Julho 1998.
PACE, M. D. Epr spectra of photochemical nitrogen dioxide formation in mono-cyclic nitramines and hexanitrohexaazaisowurtzitane. J. Phys. Chem. B, 95(15):5858, Julho 1991.
57

POLITZER, P., MURRAY, J. S., SEMINARIO, J. M., LANE, P., GRICE, M. E. eCONCHA, M. C. Computational characterization of energetic materials. J.Molec. Structure (THEOCHEM), 573(1):1–10, 2001.
PONT, D. Manual Du Pont para o uso de explosivos. Du Pont do Brasil SA., SãoPaulo, 1975.
RICE, B. M. e BYRD, E. F. C. Theoretical chemical characterization of energeticmaterials. Materials Research Society, 21(10):2444–2452, 2006.
RICE, B. M. e HARE, J. J. A quantum mechanical investigation of the rela-tion between impact sensitivity and the charge distribution in energeticmolecules. J. Phys. Chem. A, 106(9):1770–1783, 2002.
RICO, J. F., LÓPEZ, R., EMA, I. e RAMÍREZ, G. Analysis of the molecular density:Sto densities. Journal of Chemical Physics, 117(2):533–540, 2002.
RICO, J. F., LÓPEZ, R., EMA, I. e RAMÍREZ, G. Electrostatic potentials andfields from density expansions of deformed atoms in molecules. J Comput.Chem., 25, 2004.
RICO, J. F., LÓPEZ, R., EMA, I. e RAMÍREZ, G. Deformed atoms in molecules:analytical representation of atomic densities for gaussian type orbitals. J.Molec. Structure (THEOCHEM), 727, 2005.
RICO, J. F., LÓPEZ, R. e RAMÍREZ, G. Analysis of the molecular density. Journalof Chemical Physics, 110(9):4213–4220, 1999.
STONE, A. J. The Theory of Intermolecular Forces. Oxford University Press,Oxford, 1997.
SWADLEY, M. J. e LI, T. Reaction mechanism of 1,3,5-trinitro-s-triazine(rdx)deciphered by density functional theory. J. Chem. Theory Comput., 3(2):505–513, 2007.
URBÁNSKY, T. Chemistry and Technology of Explosives. The Macmillan Com-pany, 1964.
VLADIMIROFF, T. e RICE, B. M. Reinvestigation of the gas-phase structureof rdx using density functional theory predictions of electron-scatteringintensities. J. Phys. Chem. A, 106(43):10437–10443, Outubro 2002.
WU, C. J. e FRIED, L. E. Ab initio study of rdx decomposition mechanisms. J.Phys. Chem. A, 101(46):8675–8679, 1997.
ZHANG, C., SHU, Y., HUANG, Y., ZHAO, X. e DONG, H. Investigation of corre-lation between impact sensitivities and nitro group in nitro compounds. J.Phys. Chem. B, 109(18):8978–8982, 2005.
ZHAO, X., HINSTA, E. J. e LEE, Y. T. Infrared multiphoton dissociation of rdxin a molecular beam. J. Chem. Phys., 88(2):801, Janeiro 1988.
58

7 APÊNDICES
59

7.1 TRABALHO BASEADO NESTA DISSERTAÇÃO ACEITO PARA PUBLI-
CAÇÃO NO INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY EM
2010
60

61

62

63

64

65

66

67

68

69

70

71

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo