física atômica e molecular
TRANSCRIPT

.
Aulas em Fısica para pos-graduacao
Fısica Atomica e Molecular
Ph.W. CourteilleUniversidade de Sao Paulo
Instituto de Fısica de Sao Carlos17 de novembro de 2017

2

Sumario
0 Preface 1
0.1 Organizacao do curso . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
0.2 Literatura recomendada . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1 Revisao da fısica moderna e da mecanica quantica 3
1.1 Constantes em fısica atomica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.1.1 Unidades atomicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2 Modelos do atomo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.1 Modelo do Democritos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.2 Modelo de Thomson e experimento do Rutherford . . . . . . . . . . . . . 6
1.2.3 Emissao de radiacao no modelo planetario . . . . . . . . . . . . . . . . . . 9
1.2.4 Efeito Zeeman no modelo planetario . . . . . . . . . . . . . . . . . . . . . 10
1.2.5 Teoria de Bohr e suas limitacoes . . . . . . . . . . . . . . . . . . . . . . . 12
1.3 Formalismo da mecanica quantica . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
1.3.1 Equacao de Schrodinger . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.3.2 Caracterizacao de sistemas por operadores . . . . . . . . . . . . . . . . . . 14
1.4 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.4.1 Modelos do atomo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.4.2 Formalismo da mecanica quantica . . . . . . . . . . . . . . . . . . . . . . 16
2 Rotacoes / Potenciais centrais 17
2.1 Partıcula num potencial central . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.1.1 Hamiltoniano em coordenadas esfericas . . . . . . . . . . . . . . . . . . . 17
2.1.2 Separacao do movimento radial . . . . . . . . . . . . . . . . . . . . . . . . 19
2.2 Tratamento quantico do hidrogenio . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.2.1 O modelo de Bohr . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.2.2 O teorema virial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.3 Momento angular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.3.1 Operador do momento angular orbital . . . . . . . . . . . . . . . . . . . . 25
2.3.2 Algebra SU(2) do momento angular e spin . . . . . . . . . . . . . . . . . . 26
2.3.3 O spin do eletron . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
2.4 Acoplamento de momentos angulares . . . . . . . . . . . . . . . . . . . . . . . . . 28
2.4.1 Sistema de dois eletrons . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
2.4.2 Estados singleto e tripleto . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.4.3 Bases desacopladas e acopladas . . . . . . . . . . . . . . . . . . . . . . . . 31
2.4.4 Coeficientes de Clebsch-Gordan . . . . . . . . . . . . . . . . . . . . . . . . 33
2.5 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
2.5.1 Partıcula num potencial central . . . . . . . . . . . . . . . . . . . . . . . . 35
2.5.2 Tratamento quantico do hidrogenio . . . . . . . . . . . . . . . . . . . . . . 36
2.5.3 Momento angular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
2.5.4 Acoplamento de momentos angulares . . . . . . . . . . . . . . . . . . . . . 38
3

4 SUMARIO
3 Metodos de aproximacao 41
3.1 Perturbacoes estacionarias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.1.1 Metodo de perturbacao independente do tempo . . . . . . . . . . . . . . . 41
3.1.2 TPIT com estados degenerados . . . . . . . . . . . . . . . . . . . . . . . . 43
3.2 Metodo variacional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.2.1 A fracao de Rayleigh . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.2.2 Metodo de Rayleigh-Ritz . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.3 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.3.1 Perturbacoes estacionarias . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.3.2 Metodo variacional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
4 Subestrutura de atomos hidrogenoides 53
4.1 Estrutura fina e equacao de Dirac . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
4.1.1 Correcao para velocidades relativısticas . . . . . . . . . . . . . . . . . . . 54
4.1.2 Correcao pelo acoplamento spin-orbita . . . . . . . . . . . . . . . . . . . . 55
4.1.3 Interacao eletron-nucleo nao-local . . . . . . . . . . . . . . . . . . . . . . . 57
4.1.4 Resumo das correcoes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.1.5 Deslocamento de Lamb . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.2 Estrutura hiperfina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.2.1 Acoplamento ao spin do nucleo . . . . . . . . . . . . . . . . . . . . . . . . 60
4.2.2 Interacao quadrupolar eletrica . . . . . . . . . . . . . . . . . . . . . . . . 62
4.3 Atomos exoticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
4.3.1 Positronio e muonio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
4.3.2 Atomos hadronicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
4.3.3 Hidrogenio muonico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
4.3.4 Atomos de Rydberg . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
4.4 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
4.4.1 Estrutura fina e equacao de Dirac . . . . . . . . . . . . . . . . . . . . . . 67
4.4.2 Estrutura hiperfina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
4.4.3 Atomos exoticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
5 Atomos com spin em campos externos 69
5.1 Partıculas carregada em campo eletromagneticos . . . . . . . . . . . . . . . . . . 69
5.1.1 Lagrangiano e hamiltoniano de partıculas carregadas . . . . . . . . . . . . 69
5.1.2 Acoplamento mınimo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.2 Interacao com campos magneticos . . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.2.1 Efeito Zeeman normal da estrutura fina . . . . . . . . . . . . . . . . . . . 70
5.2.2 Efeito Zeeman anomalo . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.2.3 Efeito Paschen-Back e campos magneticos intermediarios . . . . . . . . . 72
5.2.4 Efeito Zeeman da estrutura hiperfina . . . . . . . . . . . . . . . . . . . . . 72
5.2.5 Efeito Paschen-Back da estrutura hiperfina . . . . . . . . . . . . . . . . . 74
5.2.6 Estrutura hiperfina em regime de campos intermediarios . . . . . . . . . . 74
5.3 Interacao com campos eletricos . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.3.1 Efeito Stark . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.4 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.4.1 Partıculas carregada em campo eletromagneticos . . . . . . . . . . . . . . 77
5.4.2 Interacao com campos magneticos . . . . . . . . . . . . . . . . . . . . . . 77
5.4.3 Interacao com campos eletricos . . . . . . . . . . . . . . . . . . . . . . . . 79

SUMARIO 5
6 Interacao de luz com atomos monoeletronicos 81
6.1 Transicoes entre estados atomicos . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
6.1.1 Perturbacao dependente do tempo por uma onda plana . . . . . . . . . . 81
6.1.2 Absorcao e emissao estimulada . . . . . . . . . . . . . . . . . . . . . . . . 82
6.1.3 Emissao espontanea . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
6.2 Transicoes dipolares . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
6.2.1 Aproximacao dipolar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
6.2.2 Regras de selecao e transicoes eletronicas . . . . . . . . . . . . . . . . . . 87
6.2.3 Resumo das regras de selecao inclusive estrutura fina . . . . . . . . . . . . 89
6.3 Linhas espectrais e tempos de vida . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.3.1 Largura natural de uma transicao . . . . . . . . . . . . . . . . . . . . . . 90
6.3.2 Alargamento de linha homogeneo . . . . . . . . . . . . . . . . . . . . . . . 91
6.3.3 Alargamento de linha inomogeneo . . . . . . . . . . . . . . . . . . . . . . 91
6.3.4 Equacoes de Bloch opticas . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
6.4 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
6.4.1 Transicoes entre estados atomicos . . . . . . . . . . . . . . . . . . . . . . . 92
6.4.2 Transicoes dipolares . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
6.4.3 Linhas espectrais e tempos de vida . . . . . . . . . . . . . . . . . . . . . . 92
6.4.4 Equacoes de Bloch opticas . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
7 Atomos de multiplos eletrons 95
7.1 Simetrizacao de bosons e fermions . . . . . . . . . . . . . . . . . . . . . . . . . . 95
7.1.1 O princıpio de Pauli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
7.1.2 Consequencias para estatıstica quantica . . . . . . . . . . . . . . . . . . . 98
7.2 Helio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
7.2.1 O estado fundamental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
7.2.2 Estados excitados . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
7.3 Estrutura da casca eletronica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
7.3.1 Modelo de Thomas-Fermi . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
7.3.2 Metodo de Hartree . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
7.3.3 Metodo de Hartree Fock . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
7.4 O sistema periodico dos elementos . . . . . . . . . . . . . . . . . . . . . . . . . . 112
7.4.1 Modelo de camadas eletronicas . . . . . . . . . . . . . . . . . . . . . . . . 113
7.4.2 Alcalinos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
7.4.3 Acoplamento LS e jj . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
7.4.4 Resumo dos graus de liberdade de um atomo . . . . . . . . . . . . . . . . 118
7.5 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
7.5.1 Simetrizacao de bosons e fermions . . . . . . . . . . . . . . . . . . . . . . 119
7.5.2 Helio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
7.5.3 Estrutura da casca eletronica . . . . . . . . . . . . . . . . . . . . . . . . . 120
7.5.4 O sistema periodico dos elementos . . . . . . . . . . . . . . . . . . . . . . 120
8 Moleculas dimericas 123
8.1 A ligacao molecular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
8.1.1 Ligacao ionica e covalente . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
8.1.2 Aproximacao de Born-Oppenheimer e a molecula H+2 . . . . . . . . . . . . 125
8.1.3 Combinacao linear de orbitais atomicos e a molecula H2 . . . . . . . . . . 127
8.1.4 Teoria dos orbitais moleculares . . . . . . . . . . . . . . . . . . . . . . . . 128

6 SUMARIO
8.1.5 Ligacao de valencia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1308.2 Estrutura rovibracional dos potenciais moleculares . . . . . . . . . . . . . . . . . 134
8.2.1 As equacoes radial e angular . . . . . . . . . . . . . . . . . . . . . . . . . 1348.2.2 Estados moleculares vibracionais . . . . . . . . . . . . . . . . . . . . . . . 1358.2.3 O princıpio de Franck-Condon . . . . . . . . . . . . . . . . . . . . . . . . 1388.2.4 Progressao rotacional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1408.2.5 Computacao dos estados vibracionais . . . . . . . . . . . . . . . . . . . . . 142
8.3 Forcas de van der Waals e acoplamento ao spin . . . . . . . . . . . . . . . . . . . 1458.3.1 Modelos analıticos para potenciais de curto e longo alcance . . . . . . . . 1468.3.2 Acoplamento de spins em dımeres, numeros quanticos moleculares . . . . 1478.3.3 Os casos de acoplamento de Hund . . . . . . . . . . . . . . . . . . . . . . 148
8.4 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1498.4.1 Ligacao molecular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1498.4.2 Estrutura rovibracional dos potenciais moleculares . . . . . . . . . . . . . 1508.4.3 Forcas de van der Waals e acoplamento ao spin . . . . . . . . . . . . . . . 151
9 Colisoes 1539.1 Teoria de espalhamento . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
9.1.1 Equacao de Lippmann-Schwinger . . . . . . . . . . . . . . . . . . . . . . . 1539.1.2 Pacotes de onda . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1559.1.3 Aproximacao de Born . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1579.1.4 Potenciais esfericos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1579.1.5 Fase e comprimento de espalhamento . . . . . . . . . . . . . . . . . . . . . 1599.1.6 Teorema optico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
9.2 Colisoes de atomos frios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1629.2.1 Estados ligados e ressonancias em colisoes frias . . . . . . . . . . . . . . . 1639.2.2 Colisoes entre partıculas identicas . . . . . . . . . . . . . . . . . . . . . . 1649.2.3 Colisoes de atomos quentes . . . . . . . . . . . . . . . . . . . . . . . . . . 166
9.3 Exercıcios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1669.3.1 Teoria de espalhamento . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1669.3.2 Colisoes de atomos frios . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167

0 SUMARIO

Capıtulo 0
Preface
A fısica dos atomos e tem uma historia de quase 2500 anos, comecando com o postulado daexistencia do atomo por Democrito ate sua descricao pela teoria da mecanica quantica. De fato,a necessidade de entender o atomo e sua interacao com a luz foi a primeira motivacao parao desenvolvimento da mecanica quantica, tal que os cursos em fısica atomica e em mecanicaquantica geralmente tem uma grande sobreposicao tematica.
O aluno ouvinte deste curso desejando aprofundar seu conhecimento da materia e encorajadotambem consultar, alem de outros livros, a apostila do curso de Mecanica quantica aplicada domesmo autor.
1. Conceitos fundamentais. O atomo classico. Metodos aproximativos em mecanica quantica.2. Atomos hidrogenoides especiais. Positronio. Muonio. Atomos de Rydberg. Estrutura finae hiperfina de atomos hidrogenoides. Estrutura eletronica de atomos alcalinos. 3. Interacaode atomos de um eletron com radiacao. Hamiltoniano basico e transicoes eletronicas. Regrasde selecao. Formas de linhas de absorcao. Modelo de dois nıveis: equacoes opticas de Bloch.4. Atomos de multiplos eletrons. Atomo de helio. Modelo de Thomas-Fermi pata atomosmulti-eletronicos. Metodo de Hartree-Fock. 5. Moleculas. Aproximacao de Born-Oppenheimer.Nıveis rotacionais e vibracionais. Espectro de atomos diatomicos. 6. Colisoes atomicas. Po-tencial de espalhamento e metodos de solucao. Colisao eletron-atomo. Colisao atomo-atomo.7. Aplicacoes de fısica atomica. Metrologia. Laser e maser. Confinamento de atomos e efeitoscoletivos. Confinamento de ıons e observacao de pulos quanticos. Astrofısica.
0.1 Organizacao do curso
A apostila foi desenvolvida para o curso Fısica Atomica e Molecular (SFI5814) oferecido pelo Ins-tituto de Fısica de Sao Carlos (IFSC) da Universidade de Sao Paulo (USP). O curso e destinadoa estudantes em Fısica de pos-graduacao. A apostila e uma versao preliminar continuamentesujeita a correcoes e modificacoes. Notificacoes de erros e sugestoes de melhoramento sempre saobem-vindas. A apostila incorpora exercıcios as solucoes das quais podem ser obtidas do autor.
Informacoes e anuncios a respeito do curso serao publicados na pagina web:http://www.ifsc.usp.br/ strontium/ − > Teaching − > SFI5814
A avaliacao do estudante sera feita baseado em provas escritas e um seminario sobre umtopico especıfico. No seminario o estudante apresentara um topico em 15 minutos. Ele tambementregara um trabalho cientıfico de 4 paginas em forma digital. Topicos possıveis sao:- Condensacao de Bose-Einstein,- O metodo de Hartree-Fock,- A aproximacao WKB,- O metodo de simulacao de Monte Carlo da funcao de onda,- A radiacao do corpo negro e sua influencia sobre os estados dos atomos,
1

2 CAPITULO 0. PREFACE
- O efeito Zeno quantico,- Evolucao temporal de uma partıcula livre descrita por um pacote de onda gaussiano,- As equacoes de Bloch: derivacao e interpretacao,- Atomos exoticos: o muonio,- O salto quantico. A sua historia e observacao,- O atomo de helio,- O efeito Stark quadratico e dinamico,- Calculo de efeito fotoeletrico a partir da regra de ouro de Fermi,- O metodo de combinacao de orbitais atomicos (LCAO),- Moleculas ultrafrias.
0.2 Literatura recomendada
P.W. Atkins e R.S. Friedman, Molecular Quantum Mechanics (Oxford University 1997, 2001)I.N. Levine, Quantum Chemistry, (Boston, Allyn and Bacon, 1983)H.A. Bethe, R. Jackiw, Intermediate Quantum Mechanics, 2nd ed. (W.A. Benkamin, Inc)J.I. Stienfeld, Molecules and Radiation, (The MIT Press)A. Corney, Atomic and Laser Spectroscopy, (Clarendon Press - Oxford)B.H. Bransden, C.J. Joachain, Physics of Atoms and Molecules, (John Wiley & Sons)C. Cohen-Tannoudji, B. Diu, F. Laloe, Quantum mechanics, vol. 1, (Wiley Interscience)

Capıtulo 1
Revisao da fısica moderna e damecanica quantica
1.1 Constantes em fısica atomica
Constantes fısicas:Velocidade da luz c = 299792458 m/s
Constante de Planck ~ = 1.05457266× 10−34 Js
Unidade de massa atomica u = 1.6605402× 10−27 kg
Constante de Boltzmann kB = 1.380658× 10−23 J/K
Constante de Faraday F = 96485.309 C/mol
Permeabilidade µ0 = 10−7 Vs/Am
Constante de gravitacao γ = 6.67259× 10−11 m3kg−1s−2
Massa do eletron me = 9.1096× 10−12 kg
Carga do eletron e = 1.60217733× 10−19 C
Fator g do eletron g = 2.002319304386
Constantes derivadas:Constante de estrutura fina α = e2/4πε0~c ≈ 1/137
Constante de Avogadro NA = 1/uA × 1g/mol = 6.0221367× 1023 1/mol
Constante do gas R = NAkB = 8.314510 J/mol K
Permitividade ε0 = 1/µ0c2 = 8.8542× 10−12 As/Vm
Raio de Bohr aB = 4πε0~2/mee2 = 0.529× 10−10 m
Magneton de Bohr µB = e~/2me = 9.27× 10−24 J/T
Raio de classico do eletron re = α2aBConstante de Rydberg R∞ = mecα
2/2h = 13.7 eV
Comprimento de onda de Compton λC = h/mec
Secao de choque de Thomson σe = (8π/3)r2e
Massa do muon mµ = 105.658389 MeV
Massa do proton mp = 938.27231 MeV
Massa do neutron mp = 939.56563 MeV
Massa do deuteron md = 1875.61339 MeV
3

4 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
1.1.1 Unidades atomicas
Um sistema de unidade comumente usado em fısica atomica sao as unidades atomicas. Estesistema se basea no sistema de unidades gaussianas (CGS) 1 definido por,
ecgs = e/√
4πε0 , aB = 1/α× ~/mec = ~2/mee2cgs , ~ = 1 . (1.1)
Com isso damos a energia em termos de e2cgs/aB, o vetor de onda em termos de 1/aB, a distancia
em termos de aB e a massa em termos de me, tal que,
E = E/(e2cgs/aB) , (1.2)
k = kaB ,
R = R/aB ,
µ = µ/me .
Esta notacao simplifica as formulas. Por exemplo:
k =√
2µ~2 (E − V ) fica k =
√2µ(E − V ) , (1.3)
V = C6e2cgsa
5B
R6 fica V = C6
R6.
1.2 Modelos do atomo
1.2.1 Modelo do Democritos
”Os princıpios de toda a realidade sao os atomos e o vazio enquanto as outras coisas sao merasopinioes.”Esta e uma citacao do filosofo grego Democrito 400 ante Cristo e ante Socrates. Juntocom seu professor Leucipo, ele formou a primeira ideia de partıculas indivisıveis: os atomos.
Figura 1.1: Democrito e poeira num raio de sol.
A obra de Democrito sobreviveu apenas na forma de relatos de segunda mao a maioria sendoescritas por Aristoteles que tambem, defendendo a ideia do contınuo, era o maior crıtico da teoriado Democrito. Aristoteles disse que o raciocınio que guiou Democrito para afirmar a existenciados atomos foi o seguinte. Para que um corpo possa mudar a sua forma, e necessario que as suaspartes podem se mover. Isso pressupoe o vazio no qual a materia se desloca. Mas, se a materiase dividisse em partes sempre menores infinitamente no vazio, ela nao teria consistencia. Nadapoderia se formar porque nada poderia surgir da diluicao sempre cada vez mais infinitamente
1Consulte a apostila Eletrodinamica do mesmo autor.

1.2. MODELOS DO ATOMO 5
profunda da materia no vazio. Daı concluiu que, a divisao da materia nao pode ser infinita, istoe, ha um limite indivisıvel, o atomo. ”Ha apenas atomos e vazio”, disse ele.
Observando partıculas de poeira num movimento de turbilhao dentro de um raio de sol,Democrito foi levado a ideia de que os atomos se comportariam da mesma maneira, colidindoaleatoriamente, alguns se aglomerando, outros se dispersando, outros ainda nunca se juntandocom outro atomo.
A consistencia dos aglomerados de atomos, que faz com que algo pareca solido, lıquido,gasoso ou anımico (que e o estado de espırito) seria entao determinada pela forma dos atomosenvolvidos e seu arranjo espacial. Desse modo, os atomos de agua sao lisos e escorregadios; osatomos de aco possuem um formato com bordas afiadas, que os prendem solidamente entre si;os atomos de sal, como demonstra o seu gosto, sao asperos e pontudos; os atomos de ar saopequenos e pouco ligados, penetrando todos os outros materiais; e os atomos da alma e do fogosao esfericos e muito delicados.
Figura 1.2: Atomos numa gota de agua, atomos de aco e de ar, atomos anımicos e o modelo doatomo de Bohr.
Sabemos hoje em dia que a primeira teoria da estrutura da materia do Democrito estavabem perto da verdade: Realmente existem partıculas indivisıveis chamado de atomos compostospor um nucleo e uma camada eletronica, e o espaco entre os nucleos atomicos e bastante vazio.
A hipotese atomica viria a renascer na idade moderna com Boyle, Clausius, Maxwell e Boltz-mann devido ao sucesso das explicacoes das propriedades de um gas por meio da chamada teoriacinetica, onde assumiam o gas constituıdo de moleculas identicas que colidiam elasticamente en-tre si e com as paredes do recipiente que as contem. A descoberta do atomo atraves das leisdas proporcoes em quımica e o estabelecimento do numero de Avogadro fortaleceram consi-deravelmente a hipotese atomica da materia que foi definitivamente consagrada com os variosexperimentos classicos que estabeleceram a carga do eletron e a relacao da massa entre eletronse protons.
No inıcio do seculo 19 a natureza atomica da materia tinha definitivamente sido estabele-cida, e tambem ja era relativamente bem conhecida a composicao basica dos atomos. Sabia-se,atraves de experimentos, que eletrons podiam ser removidos de atomos neutros criando ıons po-sitivamente carregados, e que dependendo do tipo de atomo, somente um determinado numerode eletrons podia ser removido de cada um. Este numero provou ser dependente de cada especiee denominado de numero atomico Z. Estas informacoes foram fundamentais para o estabele-cimento da composicao basica dos atomos. A questao que surgia nesta altura dizia respeito asdimensoes e configuracoes do sistema atomico. Como as cargas e massas estariam distribuıdasnesta entidade?

6 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
1.2.2 Modelo de Thomson e experimento do Rutherford
A estrutura interna de um corpo pode ser estudada jogando feixes de pequenas partıculas contrao corpo. A deteccao da distribuicao angular do espalhamento permite medir o fator estruturado corpo. Na cristalografia jogamos raios X para moleculas supercomplicadas para aprender aarquitetura p.ex. de proteınas. E na medicina os raios X revelam a estrutura interna do corpohumano. Obviamente, a tecnica do espalhamento e uma ferramenta extremamente poderosa,utilizada em muitas areas da fısica moderna.
Em um serie de experimentos feitos antes de 1911 o Ernest Rutherford analisou a estruturainterna de atomos de ouro usando partıculas α, isto e, atomos He2+. O experimento realizadopor Geiger, Marsden e Rutherford consistia em observar a deflexao de partıculas α provenientede um feixe colimado quando espalhado por uma fina folha metalica (ouro de espessura ∼ 1 µm)cuidadosamente obtidas por eletrodeposicao [vide Fig. 1.3(c-d)].
Figura 1.3: Comparacao do espalhamento de Rutherford por eletrons livres o eletrons fortementeligados a nucleos pequenos. (a) Atomo tipo ”pudim de passas”de Thomson; (b) atomo planetariode Rutherford. (c) O espalhamento de Rutherford por um atomo pudim de passas e (d) poratomo planetario.
O modelo do atomo proposto por Joseph John Thomson propoe uma estrutura tipo de pudimde passas: os eletrons seriam distribuıdos de forma homogenea dentro de um nucleo extenso (detamanho 0.1 nm) de carga positiva assim compensando a carga. As partıculas α penetrariamo nucleo de ouro, percebido como quase homogeneo, mas sofreriam multiplas deflexoes devidoas colisoes com os eletrons desordenados dentro do nucleo. Como os eletrons sao muito leves,o angulo de deflexao θ seria pequeno, mesmo apos muitas colisoes. Para este modelo espera-seuma dependencia gaussiana do angulo de deflexao das partıculas α dada pela secao transversalde espalhamento [vide Fig. 1.3(a-b)],
dσ
dΩ∝ e−θ2/θ2
0 , (1.4)
onde θ0 e um angulo pequeno.No entanto, as medidas realizadas neste espalhamento de Rutherford mostraram resultados
diferentes:
• Para um angulo de espalhamento θ fixo a quantidade de partıculas espalhadas dentro deum elemento de angulo solido dΩ e proporcional a espessura da folha metalica.

1.2. MODELOS DO ATOMO 7
• Para um determinado angulo fixo e uma dada folha metalica a quantidade de partıculas es-palhadas em dΩ varia inversamente com E2
kin, onde Ekin e a energia cinetica das partıculasα.
• Para uma determinada energia e uma dada folha metalica, o numero de partıculas espa-lhadas em dΩ e proporcional a (sin θ
2)−4.
• Para uma determinada energia e espessura da folha o numero de partıculas espalhadasem dΩ numa determinada direcao e proporcional a Z2
tg, onde Ztg e o numero atomico doelemento que constitui a folha.
A deflexao extremamente rara de partıculas α e a sua distribuicao angular pode ser entendidapela suposicao, que a carga positiva esta localizada em um volume muito pequeno (∼ 1 fm ouseja 10000 vezes menor do que do proprio atomo). Este volume e chamado de nucleo atomico,dai a denominacao de modelo nucleado. Uma vez que a maioria das partıculas passam atraves dafolha de ouro sem impedimento, deve haver uma grande folga entre os nucleos. Os eletrons, quese movem em relacao ao diametro do nucleo grande espaco vazio (vacuo) em torno do nucleo,protegem a carga nuclear positiva, de modo que o atomo aparecer exteriormente neutro.
Figura 1.4: (Esquerda:) Trajetoria da partıcula α. (Direita:) Ilustracao da secao de choque.
Derivamos agora a formula de espalhamento de Rutherford a partir da hipotese de um nucleopuntiforme. Devido a acao repulsiva da forca de Coulomb,
F =ZαZtge
2
4πε0r2, (1.5)
temos para a trajetoria da partıcula α (Zα = 2) uma hiperbole [vide Fig. 1.4(a)]. O grandesemi-eixo da hiperbole pode ser determinado a partir do seguinte ansatz,
Ekin =ZαZtge
2
4πε0
1
2a, (1.6)
onde 2a e a distancia mınima da partıcula α, quando ela colide com o nucleo numa colisaocentral 2. A distancia a depende da energia cinetica e pode ser usada tambem para colisoesnao centrais. O parametro de colisao b e a distancia mınima da partıcula α do nucleo, se elacontinuasse voar numa linha reta. De fato a partıcula α sera defletida por um angulo θ. Dageometria da hiperbole, como 2φ+ θ = 180, obtemos a seguinte equacao:
tanφ = ba = tan
(90 − θ
2
)= cot
(θ2
), (1.7)
2Numa colisao central, quando a partıcula α chega a distancia mınima 2a, a sua energia cinetica inicial Ekin etotalmente convertida em energia potencial.

8 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
e portanto
cot θ2 =b
a=
8πε0Ekin
ZαZtge2b , (1.8)
substituindo a pela formula (1.6). Derivando esta ultima formula obtemos uma relacao entre alargura db do cone oco e a largura pertinente dθ do angulo de deflexao θ.
− 1
2 sin2 θ2
dθ =8πε0Ekin
ZαZtge2db . (1.9)
Seja ntg =NtgV a densidade das partıculas do alvo (Ntg atomos por volume V ) e x a espessura
da pelıcula. Entao σ = ANtg
= V/xNtg
= 1ntgx
e a secao transversal media por atomo sentida pelapartıcula α na transicao atraves da pelıcula. σ se chama corte transversal. A probabilidadeP (θ)dθ para a partıcula α de ficar num anel numa distancia b do nucleo (cuja area e 2πbdb)sendo espalhado para o angulo θ entao e dada por,
P (θ)dθ =2πbdb
σ= ntgx2πbdb . (1.10)
Estas partıculas, isto e, o numero dN das N partıculas sao defletidas para o cone oco com aprobabilidade,
dN
N= P (θ)dθ = ntgx2π
ZαZtge2
8πε0Ekincot
θ
2· ZαZtge
2
8πε0Ekin· 1
2 sin2 θ2
dθ = ntgxZ2αZ
2tge
4
64πε20E
2kin
· cos θ2sin3 θ
2
dθ ,
(1.11)onde substituımos os parametros b e db pelas expressoes (1.8) e (1.9). O angulo solido do conepode ser exprimido por,
dΩ = 2π sin θdθ = 4π sin θ2 cos θ2dθ . (1.12)
Assim, o numero dN de partıculas espalhadas para o angulo solido dΩ fica,
dN
N= ntgx
Z2αZ
2tge
4
256π2ε20E
2kin
· 1
sin4 θ2
dΩ . (1.13)
Isso e a formula de espalhamento de Rutherford. Frequentemente, a formula e exprimida com asecao eficaz diferencial dσ
dΩ . Temos,
dN
N=dσ
σ= ntgxdσ , (1.14)
e portanto
dσ
dΩ=
(ZαZtge
2
4πε0 · 4Ekin
)21
sin4 θ2
, (1.15)
comdN
dΩ= Nntgx
dσ
dΩ. (1.16)
E preciso fazer alguns comentarios:
• O angulo θ = 0 nao e definido, pois existe um angulo mınimo de deflexao θmin. Este eadotado, quando a partıcula α se move na distancia b = bmax do atomo, isto e, na borda

1.2. MODELOS DO ATOMO 9
da area circular da secao transversal. Para um parametro de colisao b maior, a partıcula αfica no campo do proximo atomo vizinho, e o angulo de deflexao aumento de novo. Temos:
σ =A
Ntg= πb2max e θmin
2 ' tan θmin2 =
ZαZtge2
8πε0Ekin · bmax, (1.17)
simplesmente invertendo a formula (1.8). Para parametros de impacto muito grandes, istoe, a partıcula α passa o atomo fora da camada eletronica, os eletrons do atomo protegema carga do nucleo, um efeito chamado screening.
• Para energias muito elevadas, a distribuicao da carga nuclear sobre um volume finitoinfluencia o espalhamento, implicando a necessidade de correcoes na formula de Rutherford.Alem disso, em curtas distancias internucleares surgem as forcas nucleares em cima dainteracao eletromagnetica.
• A integral sobre a distribuicao de probabilidade P (θ)dθ e normalizada,
π∫
θmin
P (θ)dθ = 1 . (1.18)
Similarmente temos para as integrais de superfıcie∫
θ>θmin
dσ
dΩdΩ = σ . (1.19)
0 50 100 150
10-30
10-25
φ
N
Figura 1.5: Dependencia angular da secao transversal correspondendo aos modelos de Thomson(verde) e Rutherford (vermelho).
Rutherford derivou a formula (1.15) descrevendo o espalhamento das partıculas α dentro dafısica classica. Uma derivacao a partir das leis governando a mecanica quantica usando a apro-ximacao de Born mostra, que a formula de Rutherford descreve o espalhamento corretamenteem primeira ordem, e que efeitos puramente quanticos apresentam apenas pequenas correcoes.Revisaremos o espalhamento de Rutherford nos Excs. 1.4.1.1 e 1.4.1.2 e discutiremos o efeito descreening no Exc. 1.4.1.3.
1.2.3 Emissao de radiacao no modelo planetario
No modelo planetario proposto por Rutherford imagina-se os eletrons girando em torno de umnucleo positivamente carregado em orbitas circulares. Este movimento dos eletrons deveria obe-decer as leis da teoria eletrodinamica de Maxwell. Vamos agora calcular algumas consequenciasdesta imagem.

10 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
Tratamos agora o atomo como um rotor onde a partıcula negativa, o eletron, orbita apartıcula positiva. O momento dipolar e,
p0 = −eR . (1.20)
Calculamos no Exc. 1.4.1.4 a potencia emitida pela aceleracao a = ω2R do eletron em suatrajetoria circular,
P =µ0ω
4p20
12πc. (1.21)
A energia inicial do eletron girando em torno do nucleo (para um atomo de hidrogenio Z = 1),
E =p2
2me− e2
4πε0r=meω
2r2
2− e2
4πε0r, (1.22)
e dissipada por radiacao da potencia (1.21), isto e,
−P =dE
dt= meω
2rdr
dt+
e2
4πε0r2
dr
dt= 2meω
2rdr
dt. (1.23)
A ultima equacao supoe o equilıbrio entre a forca centrifuga e a forca de Coulomb,
meω2r =
e2
4πε0r2, (1.24)
permitindo relacionar a frequencia de revolucao ω ao raio instantaneo da orbita r(t). Resolvendoa Eq. (1.23) por r e substituindo a potencia pela relacao (1.21) e a frequencia ω pela relacao(1.24), obtemos
dr
dt= − P
2meω2r= − µ0ω
2e2
24πmecr = − e4
96π2ε20m2ec
3
1
r2. (1.25)
Integracao desta equacao da,
t− t0 = −32π2ε20m2ec
3
e4[r3 − r3(t0)] . (1.26)
Agora inserindo t0 = 0 e supondo r(t0) = aB, o tempo τ dentro do qual a perda de energiadevido a emissao de radiacao diminuı o raio da orbita do eletron ate r = 0 e,
t = τ =32π2ε20m
2ec
3a3B
e4. (1.27)
Insercao dos valores da o tempo de decaimento τ ∼ 10−10 s. Este e o efeito chamado de catastrofede radiacao do modelo classico do atomo.
1.2.4 Efeito Zeeman no modelo planetario
O movimento orbital do eletron corresponde a um anel de corrente I = e/T = eω/2π que produzum momento magnetico orbital que, como mostrado no Exc. 1.4.1.5, pode ser calculado seguinteas leis do electromagnetismo,
~µ` = IAn =eω
2ππr2n , (1.28)
onde A = πr2 e a area da trajetoria. Introduzindo o momento angular L = meωr2n obtemos
em notacao vetorial,
~µ` =e
2meL . (1.29)

1.2. MODELOS DO ATOMO 11
Imaginamos agora este atomo na presenca de um campo magnetico B na direcao que cha-maremos de z. Isso resulta numa precessao do momento magnetico ao redor do campo (como nocaso da precessao de um piao na presenca de uma campo gravitacional) governada pela equacao,
dL
dt= ~µ` ×B =
e
2meL×B = −ΩL × L ,
com ΩL = e2me
B representando a frequencia de precessao e sendo chamado de frequencia de
Larmor. E evidente, que a presenca do campo magnetico altera consideravelmente o estado doatomo, chegando mesmo a produzir profundas modificacoes na frequencia da orbita do eletronω0 e portanto no estado energetico do atomo. Esta alteracao e denominada de efeito Zeeman.
O efeito Zeeman pode ser calculado imaginando que o campo tem uma direcao qualquer comrelacao a L. Neste caso, a equacao descrevendo o movimento eletronico pelo equilıbrio entre aforca centrıfuga e a forca de Coulomb, sera influenciado pela forca de Lorentz,
mer +meω20r = FL = −ev ×B . (1.30)
onde mr e a forca centrıfuga devida ao movimento circular do eletron e meω20r a forca centrıpeta
devida a forca atrativa de Coulomb exercida pelo nucleo. Assumindo a direcao de B = Bez comB = 2meΩL/e, as equacoes de movimento podem ser decompostas em
x+ ω20x+ 2ΩLy = 0 (1.31)
y + ω20y − 2ΩLx = 0
z + ω20z = 0 .
A direcao z nao e alterada. Com o ansatz x = aeiωt e y = beiωt obtemos o sistema de equacoes,
a(ω20 − ω2) + 2iΩLωb = 0 (1.32)
b(ω20 − ω2)− 2iΩLωa = 0 ,
que apresenta solucao nao trivial para a e b quando a determinante dos coeficientes de a e bzera:
0 =
∣∣∣∣ω2
0 − ω2 2iΩLω−2iΩLω ω2
0 − ω2
∣∣∣∣ = ω4 − (2ω20 + 4Ω2
L)ω2 + ω40 . (1.33)
Obtemos
ω = ω1,2 =
√ω2
0 + 2Ω2L ± 2ΩL
√ω2
0 + Ω2L = ω0 ± ΩL +
1
2
Ω2L
ω0+ ... , (1.34)
ou, como ΩL ω, obtemos ω1,2 = ω0 ∓ ΩL. O resultado e um desdobramento dos nıveis deenergia proporcional ao campo magnetico,
∆E = 2~ΩL =~eme
B = 2µBB , (1.35)
onde a abreviacao µB = e~/2me ' 9.27 · 10−24 JT−1 se chama magneton de Bohr.
Apesar do calculo classico mostrar desvios em comparacao com observacoes experimentais,ele e bastante importante na ilustracao de varios aspectos em que encontraremos equivalenciaem mecanica quantica.

12 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
Exemplo 1 (Experimento de Stern-Gerlach): Entre varios experimentos historicos re-alizados para aumentar o conhecimento da estrutura atomica, um dos mais importantes eo experimento realizado por Otto Stern e Walter Gerlach em 1922, parta medir o momentomagnetico de atomos. Os resultados deste experimento demonstraram mais uma vez a neces-sidade de novos conceitos para explicar as observacoes. Se utilizarmos a regra de quantizacaode Bohr, L = n~, dentro da formula (1.29) obtemos,
~µ = −µBL
~.
Na presenca de um campo magnetico o dipolo sofre uma interacao W = −~µ ·B, e portantouma forca,
F = −~µ · ∇B .
Submetendo feixes de atomos a gradientes de campos magneticos e detectando esta forca,
Stern e Gerlach conseguiram medir o momento magnetico produzido pela rotacao dos eletrons
em torno dos nucleos atomicos.
1.2.5 Teoria de Bohr e suas limitacoes
O modelo classico do atomo planetario fornece uma ilustracao mecanica do mundo microscopico,mas falha em explicar quantitativamente observacoes experimentais, como a natureza discretados espectros atomicos.
A radiacao emitida por atomos de hidrogenio so acontece em linhas discretas espectralmentemuito finas. As linhas observadas sao agrupadas em series chamadas de Lyman, Ballmer eoutras,
1
λ= RH
µ
me
(1
m2− 1
n2
), (1.36)
onde m e n sao numeros inteiros. RH = (1/4πε)2(mee4/4π~3c) e a constante de Rydberg e
µ = memat/(me +mat) a massa reduzida.A natureza discreta das linhas espectrais e o problema da catastrofe de radiacao levaram
Niels Bohr para formular os seguintes postulados de Bohr:
1. Existem orbitas estacionarias especıficas, onde os eletrons nao emitem energia.
2. Cada emissao ou absorcao de energia de radiacao por eletrons vem com uma transicaoentre orbitas estacionarias. A radiacao emitida durante essa transicao e homogenea.
3. As leis da mecanica descrevem o equilıbrio dinamico de eletrons em estados estacionarios,mas nao descrevem a transicao do eletron entre orbitas estacionarias.
Assim, o modelo de Bohr preve a quantizacao dos nıveis de energia, conhecida como a primeiraquantizacao da mecanica quantica. Os raios das orbitas possıveis podem ser calculados postu-lando, que o momento angular orbital seja quantizado em unidades de ~, ou seja, os eletronsformam ondas estacionarias de Broglie ao longo das orbitas 3. Discutimos o modelo de Bohr nosExcs. 1.4.1.6 e 1.4.1.7.
No retrato proposto por Bohr, o decaimento radiativo e corresponde a uma transicao abruptade um eletron entre uma orbita exterior (mais energetica) e uma orbita interior (menos energetica).
3Uma generalizacao da teoria de Bohr foi fornecido por Arnold Sommerfeld. Assumindo orbitas elıpticaspara os eletrons que poderia explicar algumas caracterısticas da estrutura fina, se a massa do eletron foi tratadorelativisticamente. As premissas basicas foram 1. orbitas estaveis para a atracao de Coulomb e equilibrada poruma forca centrıfuga, 2. quantizacao da fase de espaco
∫rqdq = nq~, e 3. momento angular
∫Ldθ = nθ~.

1.3. FORMALISMO DA MECANICA QUANTICA 13
Como as energias das orbitas estacionarias sao definidas com muita precisao, a radiacao emitidae monoenergetica, isto e, o espectro consiste em linhas caracterısticas.
Notamos aqui, que a imagem da transicao abrupta do eletron entre estados discretos, cha-mada de salto quantico, nao teve a bencao do Schrodinger. Ele imaginava para os eletrons,dentro da sua teoria da mecanica quantica ondulatoria, orbitais em forma de ondas em vez detrajetorias planetarias, assim evitando o problema de radiacao por desaceleracao de cargas e oconceito do salto quantico. Seguinte ele, a energia dos orbitais e gradualmente transformada emradiacao 4.
1.3 Formalismo da mecanica quantica
Nesta secao faremos uma revisao muito rapida dos fundamentos da mecanica quantica. Asdemais nocoes que emprestaremos da mecanica quantica repetiremos, quando precisamos 5.
Na mecanica quantica desenvolvida por Erwin Schrodinger em 1925, chamada de mecanicadas ondas, o estado de um sistema e caracterizado por uma funcao ψ (chamada de funcao deonda) de uma variavel representando um grau de liberdade do sistema. Em fısica atomica oumolecular, quando a funcao de onda descreve uma partıcula, ela frequentemente depende desua posicao no espaco, ψ(r), da sua velocidade ψ(v), ou da sua energia ψ(E). A interpretacaofısica da autofuncao e aquela de uma densidade de probabilidade de encontrar o sistema numdado valor do grau de liberdade, p.ex. |ψ(r)|2 e a distribuicao de probabilidade de encontraruma partıcula na posicao r. As grandezas fısicas mensuraveis, como a posicao ou a velocidadede uma partıcula, sao representadas por operadores denotados por um chapel, p.ex. L para omomento angular de uma partıcula. Estes operadores, chamados de observaveis, sao lineares eagem sobre as funcoes de onda. Exemplos sao os operadores diferenciais representando a energia,o momento linear e o momento angular,
H = i~d
dte p = −i~∇ e L = −i~ d
dφ. (1.37)
No entanto, existem outras formulacoes da mecanica quantica. Na mecanica das matrizesinventada simultaneamente por Werner Heisenberg, os estados sao representadas por vetores,uma vez que temos definido uma base ortonormal, p.ex.,
〈1| =(1 0
)e |1〉 =
(10
), (1.38)
adotando a notacao Bra-Ket introduzida por Paul Adrien Maurice Dirac, e as observaveis saorepresentadas por matrizes, p.ex.,
H ≡∑
i,j
|i〉hij〈j| =
:.. hij ..
:
=
:
.. 〈j|H|i〉 ..:
. (1.39)
Os valores possıveis das grandezas fısicas sao as solucoes hn de equacoes de autovalores do tipo,
H|ψn〉 = hn|ψn〉 . (1.40)
Ou seja, a probabilidade de encontrar o valor hn numa medida do operador H e dada porhn = 〈ψn|H|ψn〉.
4Notamos aqui, que saltos quanticos foram observados muito mais tarde!5Vide a apostila do curso Mecanica quantica aplicada do mesmo autor.

14 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
Como o Heisenberg mostrou posteriormente, as duas formulacoes da mecanica quantica saoequivalentes, pois a funcao de onda do Schrodinger e obtida por projecao do estado abstrato doHeisenberg sobre o grau de liberdade sob consideracao, p.ex.,
〈r|ψ(t)〉 = ψ(r, t) . (1.41)
Nao obstante, cada formulacao tem suas vantagens em situacoes diferentes. Aquela do Heisen-berg e bem adaptada para tratar de sistemas discretos e aquela do Schrodinger para tratar desistemas contınuos.
1.3.1 Equacao de Schrodinger
Na maioria dos casos, em fısica atomica e molecular, com a excecao de fenomenos de transicoes,discutiremos situacoes estaveis bem descritas pela equacao de Schrodinger estacionaria,
Hψn(r, t) = Enψn(r, t) , (1.42)
onde H e o hamiltoniano do sistema resumindo a sua energia total. P.ex. para uma partıculaaprisionada num potencial V (r), temos,
H =p2
2m+ V (r) = − ~2
2m∇2 + V (r) . (1.43)
1.3.2 Caracterizacao de sistemas por operadores
Existem grandezas fısicas compatıveis entre si ou nao compatıveis. Lembramos, p.ex., que aposicao e a velocidade de uma partıcula nao podem ser dadas simultaneamente com precisao ar-bitraria, isto e, estas grandezas sao incompatıveis, fato exprimido na famosa relacao de incertezade Heisenberg,
∆px∆x ≥ ~ . (1.44)
Para determinar quais grandezas sao compatıveis, calculamos o comutador que, neste caso devedesaparecer, p.ex.,
[xj , xk] = 0 mas [pj , xk] = −i~δjk . (1.45)
Frequentemente, na fısica atomica, teremos a tarefa de caracterizar um sistemas comple-tamente por um numero mınimo de operadores, o chamado conjunto completo de operadorescomutandos (CCOC). O numero necessario de operadores reflete os graus de liberdade do sis-tema.
1.4 Exercıcios
1.4.1 Modelos do atomo
1.4.1.1 Ex: Analise do espalhamento de Rutherford
a. Quais conclusoes podem ser deduzidas a partir da observacao que a formula de Rutherforddescreve bem o espalhamento de partıcula carregadas na transicao atraves de materia numagrande faixa de parametros?b. Porque observa-se um desvio da formula de Rutherford para grandes energias?c. O espalhamento de protons com a energia E numa fina pelıcula de thorium e bem descritaate energias de E = 4.3 MeV pela formula de Rutherford. Faz para este caso uma estimativa

1.4. EXERCICIOS 15
do alcance das forcas nucleares.d. No espalhamento para pequenos angulos θ observa-se grandes desvios da formula de Ruther-ford. Explique porque? e. Assumindo os atomos de thorium do item (c) distribuıdos numa redeperiodica de espacamento d = 10aB, em qual angulo mınimo θ a formula de Rutherford perde avalidade.
1.4.1.2 Ex: Espalhamento de Rutherford
a. Um feixe de partıculas α de energia Ekin = 3 MeV e fluxo I = 5·103 s−1 impinge numa pelıculade ouro de espessura x = 1 µm. Usando a formula de Rutherford calcule quantas partıculas saoespalhadas em ∆ = 10 minutos dentro do intervalo angular 10 ≤ θ ≤ 30.b. A pelıcula de ouro seja substituıda por uma pelıcula de alumınio com a mesma espessura.Quantas partıculas α sao espalhadas, se as outras condicoes ficam iguais?
1.4.1.3 Ex: Screening dos eletrons
Considere fina camada de carga −Ztge de raio R. Este screening causa um angulo de espalha-mento,
tan θ2 =
D
2b
√1− (b/R)2
1 +D/2R,
com D ≡ 3Ze2
m2v2/2para b < R. Verifique como o screening muda o choque diferencial dσ
dΩ .
1.4.1.4 Ex: Radiacao de um dipolo oscilante
Calcule a distribuicao angular de potencia radiada por um dipolo eletrico ou magnetico osci-lante como extensao da radiacao esferica usando o metodo dos potenciais retardados. Sugestao:Procure as expressoes para os campos eletricos e magneticos emitidos na literatura.
1.4.1.5 Ex: Momentos magneticos
a. Derive a partir da expressao ~µL = 12
∫R3 r × j(r′)d3r′ da eletrodinamica classica e uma para-
metrizacao adequada da densidade de corrente j a relacao entre o momento magnetico dipolar~µ devido a orbita do eletron e o momento angular L.b. O comprimento do vetor do momento angular sendo dado por |L| = ~, calcule o momentomagnetico para um eletron e para um proton.
1.4.1.6 Ex: O atomo de Bohr
Em 1913, Niels Bohr apresentou seu modelo atomico atraves da adaptacao do modelo de Ruther-ford as ideias de quantizacao propostas por Max Planck.a. Imponha a regra de quantizacao para o momento angular (L = n~) para um eletron ao redorde um atomo de numero atomico Z e encontre uma expressao para os raios das orbitas permi-tidas.b. Segundo o modelo de Bohr, a transicao entre diferentes orbitas e acompanhada pela emissao(absorcao) de um foton. Determine a energia do foton emitido devido a transicao entre o pri-meiro estado excitado e o estado fundamental em um atomo de hidrogenio.c. Considere um eletron preso em um poco unidimensional retangular infinito de largura a. De-termine uma expressao para os nıveis de energia eletronicos.d. Qual deveria ser a largura a deste poco, em termos do raio de Bohr, para que um foton

16 CAPITULO 1. REVISAO DA FISICA MODERNA E DA MECANICA QUANTICA
emitido devido a transicao entre o primeiro estado excitado e o estado fundamental se iguale aobtida no item (b)?
1.4.1.7 Ex: O atomo de hidrogenio
O atomo de hidrogenio pode ser visto como um proton puntiforme e um eletron distribuıdocom a densidade de carga ρ = Ae−2r/aB em torno do proton que fica no centro. Aqui A e umaconstante e r a distancia do centro.a. Calcule A considerando o fato que o atomo e eletricamente neutro.b. Calcule a amplitude do campo eletrico no raio r = aB.
1.4.2 Formalismo da mecanica quantica
1.4.2.1 Ex: Atomo excitado
Calcule a evolucao temporal de um atomo com dois nıveis acoplados por um campo de luzusando o hamiltoniano,
H =
(0 1
2~Ω12~Ω ~∆
),
onde ∆ = ω − ω0 e a dessintonizacao entre a frequencia da luz e a frequencia da transicao e Ωa frequencia de Rabi.

Capıtulo 2
Rotacoes / Potenciais centrais
2.1 Partıcula num potencial central
Muitos potenciais nao tem simetria cartesiana. Felizmente, muitos problemas tem algum tipode simetria, cilındrica, esferica ou periodica. Aqueles com simetria cilındrica ou esferica podemser resolvidos por separacao das coordenadas curvilıneas, como mostraremos no seguinte. Par-ticularmente importante sao potenciais esfericos causados por forcas centrais, por exemplo, aforca de Coulomb entre o proton e o eletron no atomo de hidrogenio.
2.1.1 Hamiltoniano em coordenadas esfericas
Podemos reescrever o operador de momento em coordenadas esfericas,
x = r sinϑ cosϕ , y = r sinϑ sinϕ , z = r cosϑ , (2.1)
como
∇2r =
1
r2
∂
∂r
(r2 ∂
∂r
)+
1
r2
L2
~2onde
L2
~2≡ 1
sinϑ
∂
∂ϑ
(sinϑ
∂
∂ϑ
)+
1
sin2 ϑ
∂2
∂ϕ2, (2.2)
e uma abreviacao chamada de legendriano. Para um potencial isotropico, V (r) = V (r), podemostentar o ansatz Ψ(r) = R(r)Y (ϑ, ϕ) para resolver a equacao de Schrodinger (1.42),
r2
R(r)
[− ~2
2m
1
r2
∂
∂r
(r2 ∂
∂r
)+ V (r)− E
]R(r) =
−1
2m
L2Y (ϑ, ϕ)
Y (ϑ, ϕ)= const ≡ − ~2
2m`(`+ 1) , (2.3)
onde escolhemos uma constante de separacao, `(`+ 1), a significacao da qual aprenderemos embreve. Consideramos so a parte angular,
L2Y (ϑ, ϕ) = ~2`(`+ 1)Y (ϑ, ϕ) , (2.4)
e fazemos mais um ansatz de separacao, Y (ϑ, ϕ) = Θ(ϑ)Φ(ϕ),
sin2 ϑ
(1
Θ(ϑ)
1
sinϑ
∂
∂ϑsinϑ
∂
∂ϑΘ(ϑ) + `(`+ 1)
)= − 1
Φ(ϕ)
∂2
∂ϕ2Φ(ϕ) = const ≡ m2 , (2.5)
onde escolhemos uma constante de separacao, m2. Introduzindo outra abreviacao
Lz ≡~i
∂
∂ϕ, (2.6)
a equacao azimutal adota a forma
LzΦ(ϕ) = ~mΦ(ϕ) . (2.7)
17

18 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
Como no caso do potencial cilındrico, a solucao da equacao azimutal e, utilizando a normalizacao
Φ(ϕ) = 1√2πeimϕ , (2.8)
com o numero quantico magnetico m = 0,±1,±2, ...
A equacao polar,
1
Θ(ϑ)
1
sinϑ
∂
∂ϑsinϑ
∂
∂ϑΘ(ϑ) + `(`+ 1) =
m2
sin2 ϑ, (2.9)
se chama equacao diferencial de Legendre e pode ser resolvida por uma serie de potencias emcosk ϑ. Para m = 0, as solucoes sao os polinomios de Legendre, P`(cosϑ) com
P`(z) =1
2``!
d`
dz`[(z2 − 1)`] . (2.10)
Os primeiros polinomios sao,
P0(z) = 1 , P1(z) = z , P2(z) = 12(3z2 − 1) , P3(z) = 1
2(5z3 − 3z) . (2.11)
Para m > 0, as solucoes sao os polinomios associados,
Pm` (z) = (−1)m(1− z2)m/2dm
dzmP`(z) =
(−1)m
2``!(1− z2)m/2
d`+m
dz`+m[(z2 − 1)`] (2.12)
P−m` (z) = (−1)m(`−m)!
(`+m)!Pm` (z) .
A funcao polar ainda deve ser normalizada,
Θm` (ϑ) = Pm` (cosϑ)
√2`+ 1
2
(`−m)!
(`+m)!. (2.13)
As funcoes Y`m(ϑ, ϕ) sao as harmonicos esfericos. Eles formam um sistema ortonormal,
∫ π
0
∫ 2π
0Y ∗`′m′(ϑ, ϕ)Y`m(ϑ, ϕ) sinϑdϑdϕ = δ`′`δm′m . (2.14)
Solucoes finitas so existem, quando o numero quantico do momento angular e ` = 0, 1, .. e para|m| ≤ `.
As solucoes da parte angular da equacao de Schrodinger do atomo de hidrogenio sao final-mente,
Y`m(ϑ, φ) =1√2π
Pm` (cosϑ)
√2`+ 1
2
(`−m)!
(`+m)!eimφ . (2.15)
Os harmonicos esfericos sao simultaneamente autofuncoes dos operadores L2, como pode servisto na Eq. (2.4), e do operador Lz conforme a Eq. (2.7). As quantidades representadas pelosoperadores quanticos H, L2, Lz sao conservadas no sistema do hidrogenio. A conservacao domomento angular deve-se a simetria esferica do potencial de Coulomb.
Verificaremos a paridade dos harmonicos esfericos no Exc. 2.5.1.1.

2.1. PARTICULA NUM POTENCIAL CENTRAL 19
0.5
1
30
210
60
240
90
270
120
300
150
330
180 0
l = 0
0.5
1
30
210
60
240
90
270
120
300
150
330
180 0
l = 1
0.5
1
30
210
60
240
90
270
120
300
150
330
180 0
l = 2
0.5
1
30
210
60
240
90
270
120
300
150
330
180 0
l = 3
Figura 2.1: (Code: AM Hidrogenoido Legendre.m) Funcoes de onda angulares. Mostradas saoos polinomios de Legendre Pml (cosϑ) para ` = 0, 1, 2, 3 e m = 0, .., `. Vermelho: m = 0, verde:|m| = 1, azul: |m| = 2 e magenta: |m| = 3.
2.1.2 Separacao do movimento radial
Na Sec. 2.1.1 derivamos, depois de ter separado o movimento do centro-de-massa (ou seja donucleo pesado) e as coordenadas angulares a equacao radial (2.3) descrevendo a componenteradial do movimento do eletron,
1
R(r)
[− ~2
2m
1
r2
∂
∂r
(r2 ∂
∂r
)+ V (r)− E
]R(r) = − L2
2mr2, (2.16)
Agora fazemos a substituicao R(r) = u(r)/r e a equacao radial fica,
[− ~2
2m
∂2
∂r2+
L2
2mr2+ V (r)
]u(r) = Eu(r) . (2.17)
Essa equacao e bem similar a uma equacao de Schrodinger unidimensional, so que aparece umpotencial adicional que se chama potencial centrifugal,
V`(r) ≡L2
2mr2. (2.18)
Por exemplo, para o potencial de um eletron orbitando um proton temos,
[− ~2
2m
∂2
∂r2− Ze2
4πε0r+
~2`(`+ 1)
2mr2− E
]uE`(r) = 0 . (2.19)
Discutiremos esta equacao intensamente no ambito da discussao do atomo de hidrogenio.

20 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
0 5 10r/a
B
V(r
)
Figura 2.2: (Code: AM Hidrogenoido Centrifugal.m) Soma de um potencial coulombiano e umpotencial centrıfuga para ` = 0 (curva inferior), ` = 1 (curva no meio) e ` = 2 (curva superior).
No Exc. 2.5.1.2 derivamos a equacao de Gross-Pitaevskii radial para um condensado de Bose-Einstein aprisionado num potencial esferico. No Exc. 2.5.1.3 estudaremos partıculas dentro deum potencial central de profundidade nula, nos Excs. 2.5.1.4 e 2.5.1.5 consideramos pocos depotenciais esfericos 3D e no Exc. 2.5.1.6 um potencial esferico harmonico.
Exemplo 2 (Rotor rıgido em coordenadas esfericas): Continuamos a discussao dorotor rıgido, agora em coordenadas esfericas. No caso em que a orbita da partıcula e fixo aum raio R, podemos negligenciar a energia cinetica devido ao movimento radial e o potencial,ambos sendo constantes. Nesse caso a equacao de Schrodinger radial e
[~2`(`+ 1)
2mr2
]uE` = E`uE` .
As energias do rotor rıgido sao
E` =~2`(`+ 1)
2I,
com o momento de inercia I = mR2.
2.2 Tratamento quantico do hidrogenio
Seguinte o modelo planetario do atomo de Rutherford e Bohr podemos imaginar um atomocomo um nucleo muito pesado com carga eletrica positiva cercado por uma nuvem de eletronsmuito leve com carga negativa. Como o nucleo e muito pequeno em comparacao com a nuvemeletronica, tratamo-lhe como uma entidade com a massa M e a carga Ze, onde Z e o numerode protons e corresponde a ordem do elemento no sistema periodico.
O procedimento canonico para calcular todas as propriedades de um atomo e de estabelecero seu hamiltoniano, isto e, determinar as energias cineticas de todos os componentes e todasenergias de interacao entre eles, e de resolver a equacao de Schrodinger. Para cada componenteescrevemos a energia cinetica
Tncl =P 2
2Me Tele =
Z∑
i=1
p2i
2m. (2.20)
Aqui (R,P) sao as coordenadas do nucleo e (ri,pi) aquelas dos eletrons. A energia que corres-ponde as interacoes, isto e, atracao ou repulsao coulombiana, entre as componentes do atomoe
Vncl−ele = −Z∑
i=1
Ze2
4πε0|R− ri|e Vele−ele =
Z∑
i 6=j=1
e2
4πε0|ri − rj |. (2.21)
Tambem existem interacoes devido ao spin das partıculas, que trataremos posteriormente.

2.2. TRATAMENTO QUANTICO DO HIDROGENIO 21
Obviamente, a solucao desse problema de muitos corpos e muito complicado. Por isso, nessecapitulo, baseado na equacao de Schrodinger, calcularemos o espectro completo do atomo maissimples possıvel, o hidrogenio. Esse atomo consiste de um proton e um eletron, so.
Ze-
e-
( -1)Z e-
Figura 2.3: O modelo do hidrogenio se aplica em outros atomos desde que eles tem um eletronde valencia ocupando um espaco tao grande, que ele esta vendo o nucleo e o resto dos eletronsblindando o nucleo como uma unica carga positiva.
2.2.1 O modelo de Bohr
Vamos agora voltar para a parte radial da equacao de Schrodinger descrevendo uma partıculanum potencial radial. Esperamos que as solucoes quanticas para o atomo de hidrogenio saoparecidas as predicoes do modelo de Bohr. Seguinte esse modelo, a orbita e estavel quando aforca de atracao e igual a forca centrıfuga. Mas alem disso, Bohr postulou, que apenas certasenergias sao permitidas. Para o atomo de hidrogenio ele achou
En = −1
2
Ze2
4πε0
1
rn= − Z2~2
2ma2B
1
n2= −Z
2e2
4πε0
1
2aBn2= −Z
2
n213.6 eV , (2.22)
com o raio de Bohr
aB ≡ 4πε0~2
me2. (2.23)
Com essa equacao ele consegui explicar as observacoes espectrais. Os eletrons so podem saltarde um nıvel para um outro, dessa vez emitindo ou absorvendo um foton. As serias observadasno espectro do hidrogenio (En − Em)/~ foram a serie de Lyman (m = 1), de Balmer (m = 2),de Paschen (m = 3) e de Brackett (m = 4).
Ba
llme
r
Pa
sch
en
Lym
an
Bra
cke
tt
Figura 2.4: As transicoes do hidrogenio.
A discussao do atomo de hidrogenio dentro da mecanica quantica pode comecar a partir da

22 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
equacao de Schrodinger radial (2.19) com o potencial de atracao coulombiano,
[− ~2
2m
∂2
∂r2− Ze2
4πε0r+
~2`(`+ 1)
2mr2− E
]uE`(r) = 0 . (2.24)
Para facilitar a comparacao, vamos exprimir a energia em termos da energia de Bohr, E ≡ En =E1/n
2, e escrever o raio em unidade de aB, isto e, r ≡ Zr/aB. Isso da,
u′′n,`(r) +
(−`(`+ 1)
r2+
2
r− 1
n2
)un,`(r) = 0 . (2.25)
Para garantir que para grandes raios, r → ∞, a solucao fica finita, precisamos um comporta-mento assimptotico como un,`(r →∞) = e−r/n. Para garantir que para pequenos raios, r → 0,a solucao fica finita, precisamos un,`(r → 0) = r`+1. Derivamos as solucoes assimptoticas noExc. 2.5.2.1. A equacao diferencial resultante so tem solucoes para um numero quantico principaln inteiro e positivo e quando ` = 0, 1, .., n− 1. Ou seja, na relacao E = E1/n
2 o parametro n einteiro e positivo, tal que os nıveis de energia permanecem degenerados em ` e m. Isso significa,que o postulado de Bohr de nıveis de energias discretos, isto e, quantizados, e valido (uff!)
Figura 2.5: Esquema dos nıveis.
Substituindo o ansatz,
un`(r) = Dn`r`+1e−r/nL(r) , (2.26)
e facil mostrar (vide Exc. 2.5.2.2), que a equacao diferencial (2.25) se reduz para,
rL′′(r) + 2[(`+ 1)− 1
n r]L′(r) + 2
[1− 1
n(`+ 1)]L(r) = 0 . (2.27)
Ainda com a abreviacao ρ ≡ 2r/n = 2Zr/naB o ansatz
un`(ρ) = Dn`ρ`+1e−ρ/2L(ρ) , (2.28)
2.2. TRATAMENTO QUANTICO DO HIDROGENIO 23
leve ate a equacao diferencial 1
ρL′′(ρ) + [2(`+ 1)− ρ]L′(ρ) + [n− `− 1]L(ρ) = 0 . (2.29)
As solucoes desta equacao diferencial, L(2`+1)n−`−1(r), sao os polinomios de Laguerre. Esses po-
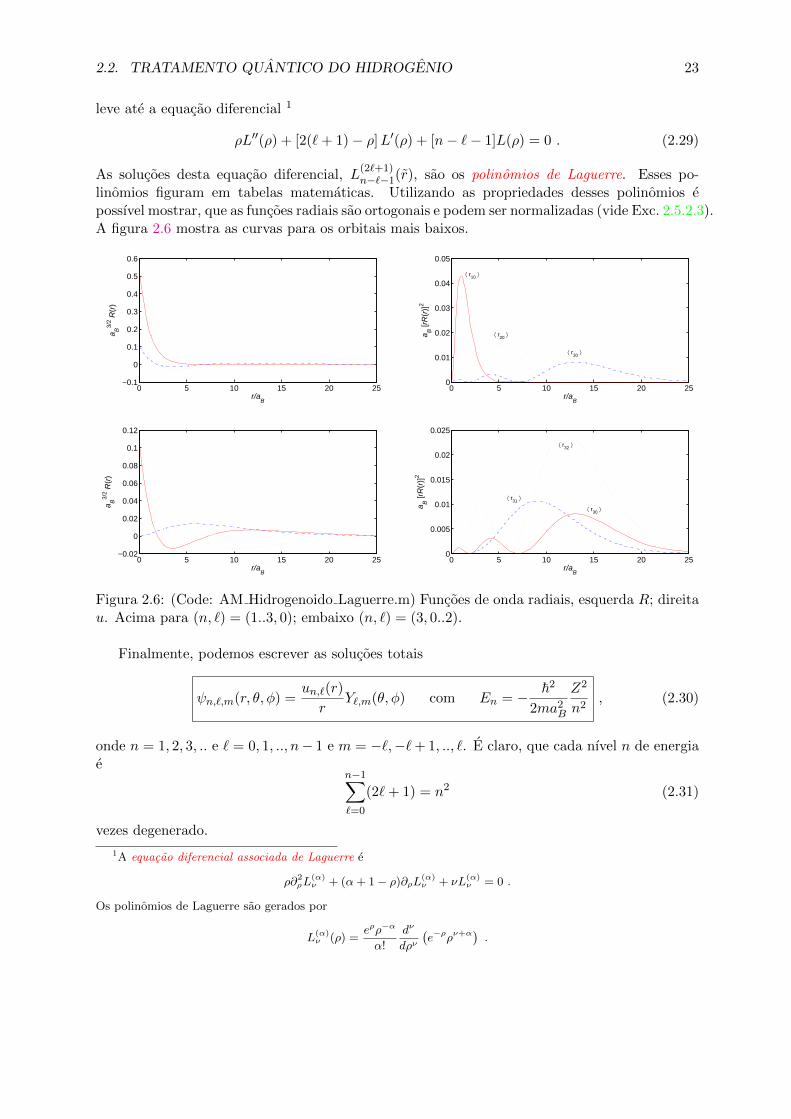
linomios figuram em tabelas matematicas. Utilizando as propriedades desses polinomios epossıvel mostrar, que as funcoes radiais sao ortogonais e podem ser normalizadas (vide Exc. 2.5.2.3).A figura 2.6 mostra as curvas para os orbitais mais baixos.
0 5 10 15 20 25−0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
r/aB
a B3/
2 R(r
)
0 5 10 15 20 250
0.01
0.02
0.03
0.04
0.05
r/aB
a B [r
R(r
)]2
⟨ r10 ⟩
⟨ r20 ⟩
⟨ r30 ⟩
0 5 10 15 20 25−0.02
0
0.02
0.04
0.06
0.08
0.1
0.12
r/aB
a B3/
2 R(r
)
0 5 10 15 20 250
0.005
0.01
0.015
0.02
0.025
r/aB
a B [r
R(r
)]2
⟨ r30 ⟩
⟨ r31 ⟩
⟨ r32 ⟩
Figura 2.6: (Code: AM Hidrogenoido Laguerre.m) Funcoes de onda radiais, esquerda R; direitau. Acima para (n, `) = (1..3, 0); embaixo (n, `) = (3, 0..2).
Finalmente, podemos escrever as solucoes totais
ψn,`,m(r, θ, φ) =un,`(r)
rY`,m(θ, φ) com En = − ~2
2ma2B
Z2
n2, (2.30)
onde n = 1, 2, 3, .. e ` = 0, 1, .., n− 1 e m = −`,−`+ 1, .., `. E claro, que cada nıvel n de energiae
n−1∑
`=0
(2`+ 1) = n2 (2.31)
vezes degenerado.
1A equacao diferencial associada de Laguerre e
ρ∂2ρL
(α)ν + (α+ 1− ρ)∂ρL
(α)ν + νL(α)
ν = 0 .
Os polinomios de Laguerre sao gerados por
L(α)ν (ρ) =
eρρ−α
α!
dν
dρν(e−ρρν+α) .

24 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
Aqui e uma lista das primeiras funcoes do atomo de hidrogenio,
ψ100 = 1√π
(ZaB
)3/2e−r (2.32)
ψ200 = 14√
2π
(ZaB
)3/2(2− r)e−r/2
ψ210 = 14√
2π
(ZaB
)3/2re−r/2 cos θ
ψ21±1 = 18√
2π
(ZaB
)3/2re−r/2 sin θe±iϕ
ψ300 = 181√
3π
(ZaB
)3/2(27− 18r + 2r2)e−r
ψ31±1 =√
281√
3π
(ZaB
)3/2(6− r)re−r/3 sin θe±iϑ
ψ320 = 181√
6π
(ZaB
)3/2r2e−r/3(3 cos2 θ − 1) ,
onde usamos a abreviacao r ≡ Zr/aB. Usando estas funcoes de onda podemos agora calcularvarios valores esperados como, por exemplo,
〈1〉n`m = 1 (2.33)
〈r〉n`m = n2
[1 +
1
2
(1− `(`+ 1)
n2
)]
〈r2〉n`m = n4
[1 +
3
2
(1− `(`+ 1)− 1
3
n2
)]
〈r3〉n`m = n6
[35
8− 35
8n2− 15
4n2(`+ 2)(`− 1) +
3
8n4(`+ 2)(`+ 1)`(`− 1)
]
〈r4〉n`m = n8
[63
8+
35
8n2(2`2 + 2`− 3) +
5
8n45`(`+ 1)(3`2 + 3`− 10) +
12
n8
]
⟨1
r
⟩
n`m
=1
n2
⟨1
r2
⟩
n`m
=1
n3(`+ 12)⟨
1
r3
⟩
n`m
=n
n4`(`+ 12)(`+ 1)
⟨1
r4
⟩
n`m
=32n
2 − 12`(`+ 1)
n5(`+ 32)(`+ 1)(`+ 1
2)`(`− 12)
.
Estes resultados serao importantes posteriormente. No Exc. 2.5.2.4 calcularemos o valor espe-rado 〈r〉 para varios orbitais |Ψn`m〉.
2.2.2 O teorema virial
Originalmente derivado para a mecanica classica, o teorema virial tambem vale para a mecanicaquantica, como mostrado pela primeira vez por Vladimir Aleksandrovich Fock. Avaliamos ocomutador entre o hamiltoniano
H = p2/2m+ V (r) , (2.34)

2.3. MOMENTO ANGULAR 25
e o produto do operador de posicao r com o operador de momento p = −i~∇ da partıcula:
[H, r · p] = [H, r] · p + r · [H, p] = −i~ p2
m+ i~r · ∇V , (2.35)
usando os teoremas de Ehrenfest. Portanto, achamos para o operador Q = r · p o comutador,
i
~[H, Q] = 2Ekin − r · ∇V . (2.36)
O lado esquerda desta equacao e justamente −dQ/dt, seguinte a equacao de movimento de Hei-senberg. O valor esperado 〈dQ/dt〉 da derivada temporal zera no estado estacionario, portantoobtemos o teorema virial,
2〈Ekin〉 = 〈r · ∇V 〉 . (2.37)
Exemplo 3 (Teorema virial aplicado num potencial central): Por exemplo, para umpotencial central V (r) ∝ rs obtemos,
2〈Ekin〉 = 〈r · er∂V
∂r〉 = s〈V 〉 .
No Exc. 2.5.2.5 calcularemos os valores esperados 〈r−1〉 e 〈p2〉 e verificaremos o teoremavirial. Finalmente, no Exc. 2.5.2.6 calculamos elementos da matriz de transicao entre diferentesorbitais.
2.3 Momento angular
2.3.1 Operador do momento angular orbital
A definicao do momento angular orbital e adotada da mecanica classica:
l = r× p = −i~r× ∇ = −i~
∣∣∣∣∣∣
ex ey ezx y z∂x ∂y ∂z
∣∣∣∣∣∣. (2.38)
Para entender melhor as propriedades do operador do momento angular na mecanica quanticaderivaremos nos Excs. 2.5.3.1 e 2.5.3.2 algumas das suas propriedades.
Figura 2.7: Ilustracao do momento angular na mecanica quantica.

26 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
2.3.1.1 Constantes do movimento
O capıtulo anterior dedicou-se a resolucao das equacoes radial e angular no caso de um potencialradial. A equacao radial permitiu calcular as auto-energias do hamiltoniano H,
H|ψ〉 = En`|ψ〉 . (2.39)
Tambem encontramos os autovalores e autofuncoes comuns dos operadores l2 e lz [vide Eqs. (2.4)e (2.7)]. Utilizamos agora a notacao |`,m〉 ≡ Y`m(θ, φ) para as autofuncoes,
l2|`,m〉 = ~2`(`+ 1)|`,m〉 e lz|`,m〉 = ~m|`,m〉 . (2.40)
Com isso temos,
[H, lz]|ψ〉 = H~m|ψ〉 − lzE|ψ〉 = 0 e [H, l2]|ψ〉 = H~2`(`+ 1)|ψ〉 − l2E|ψ〉 = 0 . (2.41)
Portanto, os operadores l2 e lz sao constantes do movimento,
[H, lz] = 0 = [H, l2] . (2.42)
O Exc. 2.5.3.3 pede para mostrar explicitamente no exemplo de um oscilador harmonico tres-dimensional isotropico que l2 e lz sao constantes do movimento.
2.3.2 Algebra SU(2) do momento angular e spin
Ate aqui, resolvemos a equacao angular de autovalores na representacao espacial para um mo-mento angular orbital, l = r × p. Mas nao e claro que cada momento angular tem essa re-presentacao derivada de nocoes classicas. Na verdade, veremos que o eletron tem um spinintrınseco sem cargas orbitandos. O que devemos mostrar agora e, que para um qualquer spin,j, satisfazendo
j× j = i~j , (2.43)
ou [jm, jn] = i~εkmnjk usando o sımbolo de Levi-Civita, obtemos uma algebra consistente.
Como j2 e jz comutam (mostramos isso a partir da Eq. (2.43) no Exc. 2.5.3.4), eles temautofuncoes comuns |j,m〉. Podemos escrever os autovalores assim,
j2|j,m〉 = ~2j(j + 1)|j,m〉 e jz|j,m〉 = ~m|j,m〉 , (2.44)
onde, por enquanto, so sabemos que m e real e j ≥ 0. Mas com 〈j,m|j2|j,m〉 ≥ 〈j,m|j2z |j,m〉 e
claro que j(j + 1) ≥ m2.
2.3.2.1 Operador de subida e descida
Agora introduzimos o operador de subida j+ e o operador de descida j− por
j± ≡ jx ± ijy tais que j− = j†+ .
E facil verificar as seguintes relacoes
[jz, j±] = ±~j± e [j2, j±] = 0 e j∓j± = j2 − j2z ∓ ~jz . (2.45)

2.3. MOMENTO ANGULAR 27
Com isso achamos
jz j±|j,m〉 = ([jz, j±] + j±jz)|j,m〉 = ~(m± 1)j±|j,m〉 (2.46)
j2j±|j,m〉 = j±j2|j,m〉 = ~2j(j + 1)j±|j,m〉 . (2.47)
Isto e, j±|j,m〉 e um autoestado de j2 e jz com os autovalores j e m±1 se j±|j,m〉 6= 0. Portanto,
j+|j,m〉 ∝ |j,m+ 1〉 . (2.48)
Para nao ultrapassar a condicao m2 ≤ j(j + 1) precisamos fixar j±|j,±j〉 = 0. Portanto, paraum j especificado, o m pode ter um dos 2j + 1 valores possıveis m = −j,−j + 1, .., j. Como2j + 1 e um numero inteiro, j so pode ter os valores j = 0, 1
2 , 1,32 , ... Com isso, a equacao de
autovalores das observaveis j2, j e resolvida, pois poderıamos ter escolhido, em vez de jz, cadauma das componentes de j, sabendo que as outras nao comutam com a escolhida.
Todas as componentes do spin jz e do raio j2 so tem autovalores discretos. A menor unidadee ~/2. Com a normalizacao 〈j,m|j′,m′〉 = δj,j′δm,m′ temos
〈j,m|j∓j±|j,m〉 = 〈j,m|(j2 − j2z ∓ ~jz)|j,m〉 = ~2[j(j + 1)−m(m± 1)] , (2.49)
e
j±|j,m〉 = ~√j(j + 1)−m(m± 1)|j,m± 1〉 . (2.50)
No Exc. 2.5.3.5 calculamos a incerteza das componentes do momento angular e no Exc. 2.5.3.6escrevemos o operador jx numa forma matricial.
2.3.3 O spin do eletron
Cada momento angular l gera um momento dipolar ~µ` ∝ l, que interage com campos magneticosexternos, V (B) = ~µ` · B. Campos magneticos inhomogeneos exercem forcas sobre momentosdipolares, F = −∇(~µ` ·B), que sao detectadas pelo experimento de Stern-Gerlach. Esse expe-rimento revela nao so a quantizacao do momento angular, mas tambem a presencia de valoressemi-integrais para o numero quantico magnetico.
Em 1925 Uhlenbeck e Goudsmit propunham que o eletron teria um momento angularintrınseco com o numero quantico s = 1/2. Esse momento angular, chamado spin, nao cor-responde a nenhuma orbita de massas ou de distribuicoes de cargas dentro do raio classicodo eletron do tipo l = r × p. O spin e um fenomeno puramente quantico, pois desaparecequando ~ → 0. Se acredita, hoje em dia, que o eletron e realmente puntiforme sem desviacaodetectavel da lei de Coulomb em todas as distancias. O spin do eletron nao segue da equacao deSchrodinger, mas pode ser incluıdo, ad hoc. E interessante, que e uma consequencia necessariada derivacao estringente relativıstica da mecanica quantica pelo Paul Dirac.
Para caracterizar o spin, podemos usar todo o aparelho formal SU(2) da mecanica quanticado momento angular:
s× s = i~s , (2.51)
e
s2|12 ,±12〉 = ~2 3
4 |12 ,±12〉 , sz|12 ,±1
2〉 = ±~2 |12 ,±1
2〉 , s± = σ± = |12 ,±12〉〈|12 ,∓1
2 | . (2.52)
Os operadores σ± sao as matrizes de Pauli.

28 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
2.4 Acoplamento de momentos angulares
Partıculas orbitandas eletricamente carregadas produzem um campo magnetico. Esse campopode influenciar o movimento de outras partıculas. Do mesmo jeito, o spin de um eletron podeinfluenciar o seu proprio movimento orbital. Isto e, os momentos angulares podem se acoplare interagir de maneira complicada. Mesmo para descrever o comportamento de um atomotao simples como o hidrogenio num campo exterior, precisamos construir os auto-estados domomento angular total resultando de um acoplamento do spin intrınseco do eletron e do seumovimento orbital.
Do outro lado, consideramos ate agora predominantemente hidrogenio e hidrogenoides, istoe, atomos com um nucleo e um unico eletron. Na verdade atomos podem ter ate mais o que 100eletrons, o que complica a descricao exata. Em atomos com muitos eletrons, um dos sistemasde acoplamentos mais comuns e aquele dos momentos angulares de todos os eletrons para ummomento angular total, L =
∑k lk, seguido por um acoplamento de L com o spin total, S =∑
k sk, para formar o momento angular, J = L + S. Designamos momentos totais por letrasmaiusculas.
Adotando uma notacao sem preconceitos estudamos algumas propriedades do momento an-gular total, j ≡ j1 + j2. No Exc. 2.5.4.1 verificamos que a adicao de momentos angulares produzuma grandeza que tambem e um momento angular, mas nao a subtracao.
2.4.1 Sistema de dois eletrons
Nesta secao consideramos primeiramente os estados de spin de dois eletrons, que podem sercombinados em dois grupos com spin bem-definido. Com isso podemos entender o espectro deenergia do helio, que e muito dominado temente pelo principio de Pauli e a estatıstica quantica.Os conceitos introduzidos podem ser estendidos para atomos com muitos eletrons.
O momento angular e um numero quantico importante no tratamento da estrutura internados atomos. Os dois eletrons da camada do helio contribuem cada um spin de 1
2 , que acoplampara um momento angular total 2. Consideramos dois eletrons livres. O estado do sistema deduas partıculas e um elemento do espaco de produto dos dois espacos de Hilbert, nos quais oseletrons individuais sao descritos. Para entender o que e o espaco produto, devemos introduziro produto tensorial de dois vetores (estados),
|α〉 =
(α1
α2
)= (αi)i ∈ A e |β〉 =
(β1
β2
)= (βj)j ∈ B . (2.53)
onde A e B sao os espacos dois-dimensionais dos dois eletrons. O produto tensorial entre vetores(estados) e definido como,
|γ〉 ≡ |α〉 ⊗ |β〉 =
(α1|β〉α2|β〉
)=
α1β1
α1β2
α2β1
α2β2
∈ C , (2.54)
onde k = 1, 2, 3, 4 e identificado com (i, j) = (1, 1), (1, 2), (2, 1), (2, 2). O novo vetor e elementodo espaco vetorial 4-dimensional C. Aqui e importante distinguir de qual espaco o vetor vem.
2As consideracoes se aplicam de maneira analoga para a estrutura hiperfina do hidrogenio, que tambem edevida a interacao de duas partıculas com spin 1
2. Indiretamente ja aprendemos o seguinte na descricao do
positrons e do eletron dentro da teoria de Dirac.

2.4. ACOPLAMENTO DE MOMENTOS ANGULARES 29
Nesta notacao, o vetor antes do simbolo para o produto tensorial (⊗) e o vetor vem do espacoA, e aquele depois do ⊗ vem do espaco B. Para a dimensao do novo espaco C temos,
dim C = dimAdimB , (2.55)
Se |α〉n e |β〉m sao bases nos respetivos espacos A e B, entao |γ〉k e uma base do espacode produto C.
O produto tensorial entre matrizes (operadores) e definido assim:
A⊗B ≡
A11B11 A11B12 A12B11 A11B12
A11B21 A11B22 A12B21 A11B22
A21B11 A11B12 A22B11 A22B12
A21B21 A11B22 A22B21 A22B22
. (2.56)
Com essa definicao podemos verificar que os operadores somente agem sobre os estados respeti-vos:
(A⊗B)(|α〉 ⊗ |β〉) = A|α〉 ⊗B|β〉 . (2.57)
Exemplo 4 (Definicao do produto tensorial): Verificamos,
[(A11 A12
A21 A22
)⊗(B11 B12
B21 B22
)][(α1
α2
)⊗(β1
β2
)]=
A11B11 A11B12 A12B11 A11B12
A11B21 A11B22 A12B21 A11B22
A21B11 A11B12 A22B11 A22B12
A21B21 A11B22 A22B21 A22B22
α11β11
α11β21
α21β11
α21β21
=
A11B11α11β11 +A11B12α11β21 +A12B11α21β11 +A11B12α21β21
A11B21α11β11 +A11B22α11β21 +A12B21α21β11 +A11B22α21β21
A21B11α11β11 +A11B12α11β21 +A22B11α21β11 +A22B12α21β21
A21B21α11β11 +A11B22α11β21 +A22B21α21β11 +A22B22α21β21
=
(A11α1 +A12α2)(B11β1 +B12β2)(A11α1 +A12α2)(B21β1 +B22β2)(A21α1 +A22α2)(B11β1 +B12β2)(A21α1 +A22α2)(B21β1 +B22β2)
=
(A11α1 +A12α2
A21α1 +A22α2
)⊗(B11β1 +B12β2
B21β1 +B22β2
)=
(A11 A12
A21 A22
)(α1
α2
)⊗(B11 B12
B21 B22
)(β1
β2
).
2.4.2 Estados singleto e tripleto
Podemos exprimir o produto tensorial de dois operadores pelas matrizes da identidade I2:
A⊗B = (A⊗ I2)(I2 ⊗A) . (2.58)
Verificaremos isso no 8.4.1.4. O operador A = A ⊗A e a extensao de A agindo no espaco deproduto C.
Aplicaremos agora este formalismo sobre um par de eletrons. Os estados que os dois eletronspodem ocupar sao:
|γ1〉 =
(10
)⊗(
10
)=
1000
≡ | ↑↑〉 , |γ2〉 = | ↑↓〉 , |γ3〉 = | ↓↑〉 , |γ4〉 = | ↓↓〉 . (2.59)

30 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
As matrizes de Pauli agem sobre os spin dos eletrons individuais a e b. Elas podem ser estendidasao espaco C da maneira seguinte,
~2σx ⊗ I2 = ~
2
(0 I2I2 0
), ~
2I2 ⊗ σx = ~2
(σx 00 σx
)(2.60)
~2σy ⊗ I2 = ~
2
(0 iI2−iI2 0
), ~
2I2 ⊗ σy = ~2
(σy 00 σy
)
~2σz ⊗ I2 = ~
2
(I2 00 I2
), ~
2I2 ⊗ σz = ~2
(σz 00 σz
).
Com estes operadores podemos agora construir outros operadores. Consideramos primeiramenteas tres componentes do momento angular total,
Sk = ~2(σk ⊗ I2 + I2 ⊗ σk) tal que (2.61)
Sx = ~2
0 1 1 01 0 0 11 0 0 10 1 1 0
, Sy = i~
2
0 −1 −1 01 0 0 −11 0 0 −10 1 1 0
, Sz = ~
1 0 0 00 0 0 00 0 0 00 0 0 −1
.
O operador para o quadrado do valor absoluto do momento angular total se calcula da seguintemaneira:
S2 = S2x + S2
y + S2z = ~2
2 0 0 00 1 1 00 1 1 00 0 0 2
. (2.62)
Agora procuramos os autovalores do momento angular total. A equacao de autovalores de Sz,
Sz|γk〉 = MS |γk〉 , (2.63)
ja e diagonal na base introduzida γk com os autovalores,
MS = ~, 0, 0,−~ . (2.64)
Para S2 a situacao e mais interessante: Os estados |γ1〉 e |γ4〉 sao autoestados de S2 paraautovalor 2~2, mas os estados |γ2〉 e |γ3〉 nao sao autoestados. Do outro lado, sabemos quea combinacao linear de dois autoestados com o mesmo autovalor tambem e um autoestado.Portanto, os estados,
|γa〉 = 1√2(|γ2〉 − |γ3〉) e |γs〉 = 1√
2(|γ2〉+ |γ3〉) , (2.65)
sao autoestados de Sz, so que eles tambem sao autoestados de S2, pois podemos facilmenteverificar,
S2|γs〉 = 2~2|γs〉 e S2|γa〉 = 0~2|γa〉 , (2.66)
usando as matrizes (2.61). Em resumo, para o autovalor 〈S2〉 = 2~2 existem os seguintes tresestados:
|γ1〉 ms = 1|γ4〉 ms = −1|γs〉 ms = 0
tripleto , s = 1 (2.67)

2.4. ACOPLAMENTO DE MOMENTOS ANGULARES 31
Para 〈S2〉 = 0 somente existe um estado:
|γa〉 ms = 0 singleto , s = 0 . (2.68)
Comutando os dois eletrons, os vetores |γ1〉 e |γ4〉 conservam a sua forma, enquanto os vetoresmistos trocam a sua forma: γ2 ←→ γ3. Sob troca de partıcula |γa〉 inverte seu signo, isto e, eantisimetrico, enquanto |γ1〉, |γ4〉 e |γc〉 conservam seu signa, isto e, sao simetricos.
Em resumo, os estados tripletos tem o numero quantico do momento angular total (como valor esperado para S2 de ~2S(S + 1) = 2~2), e eles sao simetricos a respeito da troca departıculas. O estado singleto tem o numero quantico do momento angular total S = 0, e ele eantisimetrico a respeito da troca de partıculas.
2.4.3 Bases desacopladas e acopladas
Os momentos angulares de dois partıculas ou dois momentos angulares de origens diferentes emuma partıcula representam graus de liberdade independentes, [j1, j2] = 0. Sem interacao entreos momentos angulares os espacos de Hilbert sao ortogonais:
H1 ⊗H2 =
(H1 00 H2
). (2.69)
As autofuncoes agem sobre um espaco da dimensao, dimH1 + dimH2:
|j1,mj1; j2,mj2〉 . (2.70)
Isto e, existe um conjunto completo de operadores comutandos j21, j1z, j
22, j2z. Por isso, po-
demos especificar os numeros quanticos j1, j2, mj1 e mj2 simultaneamente. Do outro lado, oconjunto j2
1, j22, j
2, jz tambem represente um conjunto completo de operadores comutandos,como mostramos no Exc. 2.5.4.2. Ele tem a base
|(j1, j2)j,mj〉 . (2.71)
No Exc. 2.5.4.3 derivamos a representacao matricial de dois spins em bases desacopladas eacopladas.
Para descrever os dois momentos angulares simultaneamente devemos escolher entre a ima-gem desacoplada |j1,mj1; j2,mj1〉 e a imagem acoplada |(j1, j2)j,mj〉. Por enquanto, a escolhada imagem nao faz diferencia, mas veremos mais tarde que pode existir uma energia associadaao acoplamento 3. Neste caso, mostraremos mais tarde, que a escolha da base acoplada e maisnatural, porque a energia comuta como [H, j2] = 0 = [H, jz] mas [H, j2
1] 6= 0 6= [H, j22].
2.4.3.1 Valores permitidos do momento angular total
Como nao especificamos energia de interacao entre os spins ou entre spins e campos externos,todos estados sao energeticamente degenerados. Na imagem desacoplada a degenerescencia efacilmente calculada,
# =
j1∑
mj1=−j1
j2∑
mj2=−j21 = (2j1 + 1)(2j2 + 1) . (2.72)
3Isto e, o hamiltoniano do sistema nao contem termos do tipo j1 · j2, mas sim pode ter termos proporcionaisa j1 + j2.

32 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
Figura 2.8: Ilustracao do acoplamento de dois momentos angulares.
Agora precisamos encontrar os valores possıveis de j e mj na imagem acoplada. Os valoresde mj seguem imediatamente de j1 + j2 = j,
mj = mj1 +mj2 . (2.73)
Com |mj1| ≤ j1 e |mj2| ≤ j2 os valores de mj sao limitados a
|mj | ≤ j1 + j2 . (2.74)
Frequentemente conhecemos os dois momentos angulares j1 e j2 e todas as suas projecoesna base desacoplada,
|mj1| ≤ j1 e |mj2| ≤ j2 . (2.75)
Para achar os numeros quanticos na base acoplada, arranjamos os estados seguinte o numeroquantico magnetico total mj . Podemos, sem restricao da generalidade concentrar na situacaoj1 ≥ j2. A seguinte tabela reproduz as combinacoes possıveis. Os x representam os coeficientesde Clebsch-Gordan.
jmj1 +mj2 = mj
jj
j j − 1j − 1 j − 1
j j − 1 j − 2j − 2 j − 2 j − 2
j j − 1−j + 1 −j + 1
j−j
j1 j2 x
j1 j2 − 1j1 − 1 j2
x xx x
j1 j2 − 2j1 − 1 j2 − 1j1 − 2 j2
x x xx x xx x x
. . .
−j1 + 1 −j2−j1 j2 + 1
x xx x
−j1 −j2 x
Os valores possıveis de j sao todos aqueles permitindo que j ≥ |mj | = |mj1 +mj2|, ou seja,
|j1 − j2| ≤ j ≤ j1 + j2 . (2.76)
Cada valor de j tem a degenerescencia 2j+1. Portanto, como devera ser verificado no Exc. 2.5.4.4,a degenerescencia total e
j1+j2∑
j=|j1−j2|2j + 1 = (2j1 + 1)(2j2 + 1) . (2.77)

2.4. ACOPLAMENTO DE MOMENTOS ANGULARES 33
Exemplo 5 (Acoplamento L · S): Como exemplo consideramos dois eletrons. O primeiro
eletron tem s1 = 12 e `1 = 0, o segundo tem s2 = 1
2 e `2 = 1. Como ilustrado na Fig. 2.9, o
acoplamento da primeiramente S = s1 +s2 = 0, 1 e L = `1 + `2 = 0, 1. Depois determinamos
os valores possıveis do momento angular total J = L + S = 0, 1, 2 dependendo dos valores
de L e S.
Figura 2.9: Acoplamento spin-orbita L · S para dois eletrons.
2.4.4 Coeficientes de Clebsch-Gordan
Por enquanto, so vamos descrever como adicionar dois momentos angulares, j1 e j2. Como elesagem sobre diferentes graus de liberdade,
[~α1 · j1, ~α2 · j2] = 0 , (2.78)
para arbitrarios vetores ~αk. Temos um sistema de autovetores comuns, |η, j1, j2,m1,m2〉, ondeη sao os autovalores de outras observaveis comutando com j1 e j2. Esses autovetores dao osvalores ~2j1(j1 + 1) e ~2j2(j2 + 1) para as observaveis j2
1 e j22 e ~m1 e ~m2 para as observaveis
jz1 e jz2. O numero de estados e (2j1 + 1)(2j2 + 1). Agora queremos construir os autoestadosdo momento angular total j = j1 + j2. Como
[j, j21] = 0 = [j, j2
2] , (2.79)
existem autoestados |j1, j2, j,m〉 comuns para o conjunto de observaveis j21, j2
2, j2 e jz. Essesautoestados sao combinacoes lineares dos estados individuais,
|(j1, j2)j,m〉 =∑
m1,m2
|j1, j2,m1,m2〉〈j1, j2,m1,m2|(j1, j2)j,m〉 (2.80)
=∑
m1,m2
|j1, j2,m1,m2〉(j1 j2m1 m2
∣∣∣∣jm
).
O coeficiente matricial se chama coeficiente de Clebsch-Gordan. Os Clebsch-Gordans desapare-cem se as condicoes 4
|j1 − j2| ≤ j ≤ j1 + j2 e m = −j1 − j2,−j1 − j2 + 1, .., j1 + j2 (2.81)
4Ver [1], p.111 ou [24], p.119. Os Clebsch-Gordans sao relacionados com os sımbolos (3j) de Wigner:(j1 j2m1 m2
∣∣∣∣ jm)
=
(j1 j2 jm1 m2 −m
)= (−1)j1−j2+m
√∆(j1j2j3)×
×√
(j1 +m1)!(j1 −m1)!(j2 +m2)!(j2 −m2)!(j +m)!(j −m)!∑t
(−1)t
t!(m1 − j2 + j + t)!(−j1 −m2 + j + t)!(j1 + j2 − j − t)!(j1 −m1 − t)!(j2 +m2 − t)!
onde ∆(j1j2j) ≡(j1 + j2 − j)!(j1 − j2 + j)!(−j1 + j2 + j)!
(j1 + j2 + j + 1)!.

34 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
nao estao satisfeitas.
As matrizes unitarias de transformacao entre bases desacopladas e acopladas,
|(j1, j2)j,m〉 = UCGC |j1,m1; j2,m2〉 , (2.82)
sao listadas em tabelas dos coeficientes de Clebsch-Gordans.
Exemplo 6 (Clebsch-Gordans para o acoplamento de dois spins 12): Por exemplo,
para o sistema de dois spins 12 temos,
|( 12 ,
12 )1,+1〉
|( 12 ,
12 )1, 0〉
|( 12 ,
12 )0, 0〉
|( 12 ,
12 )1,−1〉
=
1 0 0 0
0√
12
√12 0
0√
12 −
√12 0
0 0 0 1
| 12 ,+ 12 ; 1
2 ,+12 〉
| 12 ,− 12 ; 1
2 ,+12 〉
| 12 ,+ 12 ; 1
2 ,− 12 〉
| 12 ,− 12 ; 1
2 ,− 12 〉
.
Nos Excs. 2.5.4.5 e 2.5.4.6 escrevemos todos estados possıveis de dois momentos angulares embases desacopladas e acopladas. Nos Excs. 2.5.4.7, 2.5.4.8, 2.5.4.9, 2.5.4.10, 2.5.4.11 e 2.5.4.12praticamos a transformacao entre bases desacopladas e acopladas e no Exc. 2.5.4.13 verificamosuma regra garantindo a unitariedade das transformacoes de Clebsch-Gordan.
2.4.4.1 Acoplamento de tres momentos angulares
Tres momentos angulares podem acoplar-se em tres configuracoes diferentes: Primeiro j1 comj2, depois o spin total (j1, j2)j12 com o terceiro j3. Utilizamos a notacao |[(j1, j2)j12, j3]J〉 ou|[(j1, j3)j13, j2]J〉 ou |[(j2, j3)j23, j1]J〉. O reacoplamento de tres spins e descrito por sımbolos6j, por exemplo,
|[(j1, j2)j12, j3]J〉 =∑
j13
6j|[(j1, j3)j13, j2]J〉 . (2.83)
2.4.4.2 Notacao para estados em atomos com acoplamento LS
Num atomo, frequentemente os spins dos eletrons se acoplam para um spin total, S =∑
k sk,e separadamente os momentos angulares orbitais se acoplam para um momento angular orbitaltotal, L =
∑k lk. Esse dois spins totais agora se acoplam para um momento angular total,
J = L + S. Quando esse acoplamento LS acontece, para caracterizar os estados eletronicos ematomos se usa a notacao seguinte:
2S+1LJ . (2.84)
2.4.4.3 Acoplamento jj
Tambem existe o caso que para cada eletron o spin se acopla com o momento orbital, jk = lk+sk,antes de se acoplar os momentos angulares de todos os eletron, J =
∑k jk. Isso se chama
acoplamento jj. O reacoplamento dos quatro spins e descrito por sımbolos 9j =
l1 l2 Ls1 s2 Sj1 j2 J
,
|[(l1, s2)j1, (l2, s2)j2]J〉 =∑
L,S
9j|[(l1, l2)L, (s1, s2)S]J〉 . (2.85)

2.5. EXERCICIOS 35
2.5 Exercıcios
2.5.1 Partıcula num potencial central
2.5.1.1 Ex: Paridade das funcoes harmonicas esfericas
Consideramos a transformacao de paridade P com (x, y, z)P−→ (−x,−y,−z). Use coordenadas
esfericas e mostre, que vale Y`mP−→ (−1)`Y`,m, e portanto, que uma funcao de superfıcie esferica
tem paridade par, quando ` e par, respectivamente impar, quando ` e impar.
2.5.1.2 Ex: Condensado de Bose-Einstein num potencial isotropo
A equacao de Gross-Pitaevskii descrevendo a funcao de onda de um condensado de Bose-Einsteine,
i~∂ψ(r)
∂t=
(− ~2
2m∆ + Vtrp(r) + g|ψ(r)|2
)ψ(r) ,
onde o fator g depende da forca de interacao interatomica e Vtrp e o potencial de aprisionamento
dos atomos. Para V (r) = V (r) a funcao de onda tera simetria radial, ψ(r) = φ(r)r . Reescreve a
equacao de Gross-Pitaevskii para a funcao φ.
2.5.1.3 Ex: Movimento de uma partıcula livre em coordenadas esfericas
Obtenha as autofuncoes da partıcula livre como caso limite do seu movimento num campode forca central com V (r) −→ 0. Compare as autofuncoes assim derivadas - associadas aoconjunto completo de observaveis H, L2 e Lz - aquelas descritas por ondas planas - associadas aomovimento caracterizado pelos observaveis px, py, pz e H = P2/2m -, que igualmente constituemum conjunto completo de observaveis.
2.5.1.4 Ex: Partıcula numa caixa esferica
Encontre os nıveis de energia e as funcoes de onda de uma partıcula confinada em uma caixaesferica descrita pela energia potencial, V (r) = 0 para r < a e V (r) =∞ para r ≥ a considerandoo momento angular ` = 0.
2.5.1.5 Ex: Poco de potencial 3D esferico finito
a. Derive os nıveis de energia possıveis e as funcoes de onda associadas para uma partıculapresa num poco de potencial 3D esferico de profundidade V0 e raio a. Note que este problemae analogico ao espalhamento de Mie com ondas escalares.b. Discute o caso de um poco cercado de paredes infinitas.
2.5.1.6 Ex: Partıcula num potencial harmonico esferico
Uma partıcula quantica de massa m esta sujeita a um potencial
V = 12mω
2(x2 + y2 + z2) .
a. Obtenha os nıveis de energia dessa partıcula. Isto e, determine os autovalores de
− ~2
2m∇2ψ + V ψ = Eψ .

36 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
b. Considere o nıvel fundamental e os dois primeiros nıveis excitados. Monte uma tabela mos-trando para cada um desses tres nıveis, o valor da energia, a degenerescencia e os respectivosestados em termos dos numeros quanticos.c. Utilizando
∇2ψ =
[1
r2
∂
∂r
(r2 ∂
∂r
)− L2
~2r2
]ψ
e lembrando que L2Y`m(θ, φ) = ~2`(`+ 1)Y`m, escreva a equacao diferencial do item (a) para aparte radial da funcao de onda (nao e preciso resolve-la). Identifique nessa equacao o potencialefetivo Vef (r).d. Resolva a equacao diferencial do item anterior para o caso em que ` = 0 e determine oautovalor correspondente. Para isso, admita uma solucao do tipo e−αr
2e determine α.
2.5.2 Tratamento quantico do hidrogenio
2.5.2.1 Ex: Assimptotas dos polinomios de Laguerre
Derive as solucoes assimptoticas das solucoes da equacao (2.25).
2.5.2.2 Ex: Equacao de Laguerre
Mostre que a equacao (2.25) se transforme com o ansatz (2.26) para a equacao (2.27).
2.5.2.3 Ex: Funcoes de Laguerre
Utilizando a relacao de ortogonalidade dos polinomios associados de Laguerre,∫ ∞
0ραe−ρL(α)
n (ρ)L(α)m (ρ)dρ =
Γ(n+ α+ 1)
n!δn,m
∫ ∞
0ρα+1e−ρL(α)
n ρ2dρ =(n+ α)!
n!(2n+ α+ 1) ,
e as formula de recursao,
nL(α+1)n (ρ) = (n− ρ)L
(α+1)n−1 (ρr) + (n+ α)L
(α)n−1(ρ)
ρL(α+1)n (ρ) = (n+ α)L
(α)n−1(ρ)− (n− ρ)L(α)
n (ρ) ,
a. calcule a constante de normalizacao Dn,l para um atomo hidrogenoide com numero atomicoZ;b. calcule o valor medio
〈r〉nlm =n2aBZ
[1 +
1
2
(1− `(`+ 1)
n2
)];
c. calcule o valor medio ⟨1
r
⟩
n`m
=Z
n2aB.
2.5.2.4 Ex: Raios de orbitais no modelo de Bohr
A partir dos resultados acima, obtenha o valor esperado 〈r〉 para os estados Ψ100, Ψ210 e Ψ320
do atomo de hidrogenio. Compare os resultados com aqueles do modelo de Bohr.

2.5. EXERCICIOS 37
2.5.2.5 Ex: Teorema virial e modelo de Bohr
Calcule, para o estado Ψ320 do atomo de hidrogenio, os valores esperados 〈1r 〉 e 〈p2〉.A partir dos resultados, obtenha os valores esperados das energias cinetica e potencial, 〈T 〉 e〈V 〉, e mostre que, de acordo com o teorema virial, 〈T 〉 = −(1/2)〈V 〉. Compare os resultadoscom o modelo de Bohr.
2.5.2.6 Ex: Elementos da matriz de transicao
Usando as seguintes funcoes de onda (nao normalizadas) do hidrogenio, ψ100(r) = e−r, ψ210(r) =re−r/2 cos θ e ψ21±1(r) = re−r/2 sin θe±iφ, calcule os elementos de matriz a. 〈ψ100|z|ψ210〉, b. 〈ψ100|z|ψ211〉,c. 〈ψ100|x− iy|ψ210〉 e d. 〈ψ100|x− iy|ψ211〉. Tente interpretar os resultados. Formulario:
∫ ∞
0x4e−3x/2dx = 256
81 ,
∫ π
0sin3 xdx = 4
3 ,
∫ π
0cosx sin2 xdx = 0 ,
∫ π
0cos2 x sinxdx = 2
3 .
2.5.3 Momento angular
2.5.3.1 Ex: Propriedades do momento angular orbital
Mostre l× l = i~l e [lx, ly] = i~lz.
2.5.3.2 Ex: Tensor de Levi-Civita
Mostre [lk, rm] = i~rnεkmn onde o Levi-Civita tensor e definido por εkmn = 1 quando (kmn) euma permutacao par de (123), εkmn = −1 para uma permutacao impar e εkmn = 0 se dois dosındices sao iguais.
2.5.3.3 Ex: Momento angular orbital de um oscilador harmonico
Mostre para um oscilador harmonico tres-dimensional isotropico [H, l2] = [H, lz] = 0. Fazcalculos explıcitos, isto e, mostre
[p2
2m , lz
]= 0 =
[m2ω2r2, lz
]e
[p2
2m , l2]
= 0 =[m
2ω2r2, l2
].
2.5.3.4 Ex: Comutacao do modulo e das componentes do momento angular orbital
Mostre [j2, j] = 0.
2.5.3.5 Ex: Incerteza das componentes do momento angular
Mostra que se jz e preciso, jx e jy sao imprecisos.
2.5.3.6 Ex: Representacao matricial das componentes do momento angular
Calcule os elementos da matriz de jx e j2x na base, onde jz e observavel.

38 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
2.5.4 Acoplamento de momentos angulares
2.5.4.1 Ex: Adicao/subtracao de momentos angulares
Mostre que j1 + j2 e um momento angular, mas nao j1 − j2.
2.5.4.2 Ex: CCOC de momentos angulares acoplados
Mostre que j21, j
22, j
2, jz e um CCOC; isto e, mostre quea. j2 comuta com j2
1 e j22;
b. j2 nao comute com j1z ou j2z e que nao podemos especificar mj1 ou mj2 junto com j.
2.5.4.3 Ex: Acoplamento spin-orbita
a. Mostre que o operador L · S associado ao acoplamento spin-orbita, satisfaz a relacao L · S =LzSz + (L+S− + L−S+)/2.Obtenha a representacao matricial do operador L · S, considerando as bases:b. |mL〉 ⊗ |mS〉 dos autoestados comuns aos operadores L2, S2, Lz, Sz;c. |J,M〉, associada aos operadores L2, S2, J2, Jz.
2.5.4.4 Ex: Multiplicidade de momentos angular acoplados
Verifique # = (2j1 + 1)(2j2 + 1) dentro da representacao acoplada.
2.5.4.5 Ex: Estados possıveis de dois momentos angulares (des-)acoplados
Ache todos estados possıveis com os momentos angulares j1 = 1 e j2 = 1/2 nas imagens desa-coplados e acoplados.
2.5.4.6 Ex: Estrutura fina e hiperfina do atomo de rubıdio 85Rb
1. O atomo de rubıdio 85Rb tem um eletron de valencia. No primeiro estado excitado esse eletrontem o momento angular orbital, L = 1. Quais sao os estados possıveis?2. No estado fundamental deste atomo o momento angular total dos eletrons, J , acopla com ospin do nucleo, I = 5/2, para formar o momento angular total F = J + I. Determine os valorespossıveis para o momento angular F e o numero quantico magnetico mF .
2.5.4.7 Ex: Expansao da estrutura hiperfina do atomo de rubıdio 87Rb
Determine para os estados S1/2 e P3/2 de um atomo com o spin nuclear I = 3/2 com acoplamento
hiperfino J · I como os autoestados da base acoplada se expandem na base desacoplada. Naoconsideramos campo magnetico externo.
2.5.4.8 Ex: Amplitudes de transicao entre sub-estados Zeeman
a. Consideramos o atomo de 87Rb que tem o momento angular nuclear I = 3/2. Quais saoos estados hiperfinos F possıveis resultando de um acoplamento de I com o momento angulareletronico total do estado fundamental 2S1/2? Quais sao os sub-estados Zeeman possıveis dosF?b. Quais sao os estados hiperfinos F ′ possıveis resultando de um acoplamento de I com omomento angular eletronico total do estado excitado 2P3/2, F ′ = 2? Quais sao os sub-estados

2.5. EXERCICIOS 39
Zeeman possıveis dos F ′?c. Uma transicao entre um estado hiperfino fundamental e um estado hiperfino excitado podeser descrita por um acoplamento do momento angular total F com o momento angular do fotonκ formando o momento angular do estado excitado F ′. Para ver isso, consideramos agora osnıveis F = 1 e F ′ = 2. Expande o momento angular acoplado |(F, κ)F ′,mF ′〉 = |(1, 1)2,mF ′〉numa base desacoplada para cada valor de mF ′ possıvel. Utilize a tabela na Fig. 2.10 paradeterminar os coeficientes de Clebsch-Gordan.Note bem: Os Clebsch-Gordans comparam so as forcas das transicoes entre varios sub-estadosZeeman do mesmo par (F, F ′). Para comparar as forcas entre diferentes pares (F, F ′) e precisocalcular os coeficientes 6j.
2.5.4.9 Ex: Expansao do acoplamento spin-orbit
Considere o problema da adicao do momento angular orbital ` e de um spin 1/2. Obtenha os2l+ 1 estados |`+ 1/2,mj〉, alem dos 2l estados |`− 1/2,mj〉 (que constituem uma base comumaos operadores `21, s2
2, j2, jz), expandidos na base |m1,m2〉 dos autoestados dos operadores l2,s2, `z, sz Voce pode simplificar o procedimento derivando duas relacoes de recorrencia das quaisdecorrem os estados desejados.5
2.5.4.10 Ex: Ginastica de operadores de momento angular
Considere o problema da adicao dos momentos angulares j1 = 1 e j2 = 1/2:a. Quais os possıveis valores de m e j, em que j2|j,m〉 = j(j+ 1)~2|j,m〉 e jz|j,m〉 = m~|j,m〉?b. Quais as degenerescencias gj1,j2(m)?c. Encontre os estados da base |j,m〉, comum aos operadores j2
1, j22, j, jz, expandidos na base
|j1,m1〉 ⊗ |j2,m2〉 dos autoestados de j21, j2
2, j1z, j2z.
2.5.4.11 Ex: Bases (des-)acopladas dos harmonicos esfericos
Expande o estado tripleto 3PJ do estroncio numa base desacoplada e escreve a matriz da trans-formacao entre as bases.
2.5.4.12 Ex: Expansao do acoplamento l · sConsidere o problema da adicao do momento angular orbital l e de um spin 1/2. Obtenha os2l+ 1 estados |l+ 1/2,mj〉, alem dos 2l estados |l− 1/2,mj〉 (que constituem uma base comumaos operadores l21, s2
2, j2, jz), expandidos na base |m, ε〉 dos autoestados dos operadores l2, s2,lz, sz. Voce pode simplificar o procedimento derivando duas relacoes de recorrencia das quaisdecorrem os estados desejados.6
2.5.4.13 Ex: Propriedades dos coeficientes de Clebsch-Gordan
Dado os momentos j1 e j2, e sendo Cm1,m2 os coeficientes de Clebsch-Gordan, prove que∑m1,m2
|Cm1,m2 |2 = 1.
5Veja Cohen-Tannoudji, Vol.2, Complemento A X.6Veja Cohen-Tannoudji, Vol.2, Complemento A X.

40 CAPITULO 2. ROTACOES / POTENCIAIS CENTRAIS
36. Clebsch-Gordan coefficients 1
36. CLEBSCH-GORDANCOEFFICIENTS, SPHERICALHARMONICS,
AND d FUNCTIONS
Note: A square-root sign is to be understood over every coefficient, e.g., for −8/15 read −√8/15.
Y 01 =
√3
4πcos θ
Y 11 = −
√3
8πsin θ eiφ
Y 02 =
√5
4π
(32cos2 θ − 1
2
)
Y 12 = −
√15
8πsin θ cos θ eiφ
Y 22 =
1
4
√15
2πsin2 θ e2iφ
Y −mℓ = (−1)mY m∗
ℓ 〈j1j2m1m2|j1j2JM〉= (−1)J−j1−j2〈j2j1m2m1|j2j1JM〉d ℓ
m,0 =
√4π
2ℓ+ 1Y mℓ e−imφ
djm′,m = (−1)m−m′
djm,m′ = d
j−m,−m′ d 1
0,0 = cos θ d1/21/2,1/2
= cosθ
2
d1/21/2,−1/2
= − sinθ
2
d 11,1 =
1 + cos θ
2
d 11,0 = − sin θ√
2
d 11,−1 =
1− cos θ
2
d3/23/2,3/2
=1 + cos θ
2cos
θ
2
d3/23/2,1/2
= −√31 + cos θ
2sin
θ
2
d3/23/2,−1/2
=√31− cos θ
2cos
θ
2
d3/23/2,−3/2
= −1− cos θ
2sin
θ
2
d3/21/2,1/2
=3 cos θ − 1
2cos
θ
2
d3/21/2,−1/2
= −3 cos θ + 1
2sin
θ
2
d 22,2 =
(1 + cos θ
2
)2
d 22,1 = −1 + cos θ
2sin θ
d 22,0 =
√6
4sin2 θ
d 22,−1 = −1− cos θ
2sin θ
d 22,−2 =
(1− cos θ
2
)2
d 21,1 =
1 + cos θ
2(2 cos θ − 1)
d 21,0 = −
√3
2sin θ cos θ
d 21,−1 =
1− cos θ
2(2 cos θ + 1) d 2
0,0 =(32cos2 θ − 1
2
)
Figure 36.1: The sign convention is that of Wigner (Group Theory, Academic Press, New York, 1959), also used by Condon and Shortley (TheTheory of Atomic Spectra, Cambridge Univ. Press, New York, 1953), Rose (Elementary Theory of Angular Momentum, Wiley, New York, 1957),and Cohen (Tables of the Clebsch-Gordan Coefficients, North American Rockwell Science Center, Thousand Oaks, Calif., 1974).
Figura 2.10: Coeficientes Clebsch-Gordan.

Capıtulo 3
Metodos de aproximacao
Praticamente cada problema indo alem do poco de potencial, do oscilador harmonico ou doatomo de hidrogenio sem spin e campos externos e impossıvel resolver analiticamente. Nessecapitulo vamos falar de tecnicas para resolver aproximadamente problemas em situacoes maisrealısticas. Existem varios metodos dos quais discutiremos os seguintes: 1. O metodo de per-turbacao estacionaria ou de perturbacao dependente do tempo e util para avaliar pequenas per-turbacoes do sistema, por exemplo, por campos eletricos ou magneticos externos; 2. o metodovariacional serve para achar e melhorar chutes para as funcoes de onda motivadas pelas sime-trias do sistema; 3. metodo WKB semiclassico; 4. o metodo de campos auto-consistentes e ummetodo iterativo de resolver a equacao de Schrodinger.
3.1 Perturbacoes estacionarias
3.1.1 Metodo de perturbacao independente do tempo
Introduzimos primeiramente a teoria de perturbacao independente do tempo (TPIT) para siste-mas de muitos nıveis. Separamos o hamiltoniano em uma parte nao perturbada
H(0)|ψ(0)〉 = E(0)|ψ(0)〉 , (3.1)
e perturbacoes proporcionais a pequenos parametros λ,
H = H(0) + λH(1) + λ2H(2) + .. . (3.2)
As funcoes de onda perturbadas sao
|ψ〉 = |ψ(0)〉+ λ|ψ(1)〉+ λ2|ψ(2)〉+ .. , (3.3)
e as energiasE = E(0) + λE(1) + λ2E(2) + .. . (3.4)
As contribuicoes ∝ λn sao as correcoes de ordem n. A equacao que precisamos resolver agora e
H|ψ〉 = E|ψ〉 . (3.5)
Inserindo todas as expansoes acima e segregando todos as ordens de λk, achamos o seguintesistema de equacoes,
H(0)|ψ(0)〉 = E(0)|ψ(0)〉 (3.6)
(H(0) − E(0))|ψ(1)〉 = (E(1) − H(1))|ψ(0)〉(H(0) − E(0))|ψ(2)〉 = (E(2) − H(2))|ψ(0)〉+ (E(1) − H(1))|ψ(1)〉
... .
41

42 CAPITULO 3. METODOS DE APROXIMACAO
3.1.1.1 Correcao de primeira ordem para a energia
Agora consideramos auto-estados |ψ(1)n 〉 do sistema perturbado e expandimos a correcao de
primeira ordem da funcao de onda em uma combinacao linear dos autovetores nao perturbados
|ψ(0)n 〉 ≡ |n〉,
|ψ(1)n 〉 =
∑
m
|m〉〈m|ψ(1)n 〉 . (3.7)
Inserimos esta expansao na segunda equacao (3.6) e multiplicamos com 〈n|
〈n|(H(0) − E(0)n )
∑
m
|m〉〈m|ψ(1)n 〉 = 0 = 〈n|E(1)
n − H(1)|n〉 . (3.8)
Obtemos para a correcao de primeira ordem para a energia de estados nao perturbados
E(1)n = 〈n|H(1)|n〉 . (3.9)
Como primeiro exemplo calcularemos no Exc. 3.3.1.1 a correcao de primeira ordem para aenergia de um poco unidimensional ligeiramente deformado.
3.1.1.2 Correcao de primeira ordem para a funcao de onda
Agora vamos olhar para a correcao de primeira ordem para a funcao de onda considerando asegunda equacao (3.6),
〈m|H(0) − E(0)n |ψ(1)
n 〉 = 〈m|E(1)n − H(1)|n〉 . (3.10)
Quando n = m, o lado esquerdo desta equacao desaparece. Portanto, E(1)n − 〈n|H(1)|n〉 = 0, e
podemos nos restringir aos termos n 6= m descartando os termos em E(1)n ,
〈m|ψ(1)n 〉 =
E(1)n δmn − 〈m|H(1)|n〉
E(0)m − E(0)
n
=〈m|H(1)|n〉E
(0)n − E(0)
m
. (3.11)
Obtemos para a correcao de primeira ordem para a energia dos estados,
|ψ(1)n 〉 =
∑
m
|m〉〈m|ψ(1)n 〉 =
∑
m 6=n|m〉 〈m|H
(1)|n〉E
(0)n − E(0)
m
. (3.12)
Esse procedimento simula a distorcao do estado por misturas com outros estados. A perturbacaoinduz transicoes virtuais para outros estados. A perturbacao e grande quando os nıveis admistossao perto.
Vide Exc. 3.3.1.2. No Exc. 3.3.1.3 calculamos a correcao de primeira ordem devido a extensaofinita do nucleo do hidrogenio. No Exc. 3.3.1.4 tratamos o acoplamento de dos nıveis de energiade um sistema de dois nıveis como perturbacao em primeira ordem, e comparamos o resultadocom a solucao exata. O efeito Stark para um eletron confinado numa caixa pode ser discutido(vide Exc. 3.3.1.5) em primeira ordem TPIT.

3.1. PERTURBACOES ESTACIONARIAS 43
3.1.1.3 Correcao de segunda ordem para a energia
Para calcular a correcao de segunda ordem para a energia expandimos a correcao de segundaordem,
|ψ(2)n 〉 =
∑
m
|m〉〈m|ψ(2)n 〉 , (3.13)
e importamos na terceira equacao (3.6) e multiplicamos com 〈n|,
〈n|(H(0)−E(0)n )
∑
m
|m〉〈m|ψ(2)n 〉 = 〈n|(E(2)
n −H(2))|n〉+〈n|(E(1)n −H(1))
∑
m
|m〉〈m|ψ(1)n 〉 . (3.14)
Agora,
∑
m
〈m|ψ(2)n 〉(E(0)
n − E(0)m )δnm = 0 = E(2)
n − 〈n|H(2)|n〉+∑
m
〈m|ψ(1)n 〉
(E(1)n δnm − 〈n|H(1)|m〉
).
(3.15)O lado esquerdo desta equacao desaparece. Alem disso, no lado direito, para n 6= m, o termo
E(1)n δnm desaparece e para n = m a parentese inteira desaparece. Portanto, podemos descartar
o termo E(1)n inteiramente e nos restringir a soma para termos n 6= m. Inserindo os coeficientes
〈m|ψ(1)n 〉 calculados em (3.11), obtemos finalmente,
E(2)n = 〈n|H(2)|n〉+
∑
m 6=n
〈n|H(1)|m〉〈m|H(1)|n〉E
(0)n − E(0)
m
. (3.16)
O primeiro termo e similar a correcao de primeira ordem; o valor esperado da perturbacao desegunda ordem nos estados nao perturbados. O segundo termo descreve o deslocamento dasenergias atraves de transicoes possıveis temporarias para outros estados.
No Exc. 3.3.1.6 tratamos um sistema de 3 nıveis acoplados ate a segunda ordem perturbativa.O efeito Stark discutido no Exc. 3.3.1.7 precisa do calculo TPIT ate segunda ordem.
3.1.2 TPIT com estados degenerados
Calculos exatos mostram que o efeito de uma perturbacao e maior – porem finito – para estadosdegenerados. Do outro lado, pelas expressoes acima derivadas para as correcoes tanto dasenergias quanto das funcoes de onda, podemos inferir que estas correcoes podem tornar-se muitograndes para pequenas perturbacoes ou mesmo divergir.
Felizmente, o fato que cada combinacao linear de funcoes degeneradas tambem e uma auto-funcao do hamiltoniano nos da a liberdade de escolher a combinacao mais similar a forma finaldas funcoes perturbadas. Por exemplo, considerando uma perturbacao por um campo magneticopode tornar-se vantajoso expandir as funcoes esferica Ylm numa base de coordenadas cilındricas1.Veremos no seguinte que podemos resolver ambos os problemas, a selecao da combinacao iniciale a prevencao de denominadores divergentes em uma vez, sem especificar a expansao explicita-mente.
Consideramos auto-estados |n, ν〉 com a energia E(0)n sendo r vezes degenerada a respeito do
numero quantico ν, onde ν = 1, .., r. Todos estados satisfazem
H(0)|n, ν〉 = E(0)n |n, ν〉 . (3.17)
1Um outro exemplo seria a preferencia para a base acoplada |(l, s)j,mj〉 em comparacao com a base desacoplada|l,ml, s,ms〉 sabendo que a degenerescencia em j e levada quando existe uma energia associada a interacao dosmomentos angulares e a degenerescencia em mj e levada quando aplicamos um campo magnetico.

44 CAPITULO 3. METODOS DE APROXIMACAO
Construımos combinacoes que mais se assemelham aos estados perturbados
|ψ(0)nµ 〉 =
r∑
ν=1
cµν |n, ν〉 . (3.18)
Quando a perturbacao H(1) esta aplicada, supomos que o estado |ψ(0)nµ 〉 e distorcido para o
estado semelhante |ψnµ〉, e a energia muda de E(0)n para Enµ. Precisamos agora do ındice µ para
etiquetar a energia, pois a degenerescencia pode ser removida pela perturbacao. Como antes,escrevemos agora,
H = H(0) + λH(1) + .. (3.19)
|ψnµ〉 = |ψ(0)nµ 〉+ λ|ψ(1)
nµ 〉+ ..
Enµ = E(0)n + λE(1)
nµ + .. .
Substituicao dessas expansoes em H|ψnµ〉 = Enµ|ψnµ〉, e colecao das ordens de λ ate primeiraordem da,
H(0)|ψ(0)nµ 〉 = E(0)
n |ψ(0)nµ 〉 (3.20)
(E(0)n − H(0))|ψ(1)
nµ 〉 = (E(1)nµ − H(1))|ψ(0)
nµ 〉 .
Como antes, tentamos exprimir as correcoes de primeira ordem para as funcoes de onda
atraves das funcoes nao perturbadas degeneradas, |ψ(0)nµ 〉, e nao degeneradas, |ψ(0)
m 〉:2
|ψ(1)nµ 〉 =
∑
ν
bµν |ψ(0)nν 〉+
∑
m
anm|ψ(0)m 〉 . (3.21)
Colocando isso na equacao de primeira ordem (3.20), obtemos,
∑
ν
bµν(E(0)n − E(0)
n )|ψ(0)nν 〉+
∑
m
anm(E(0)m − E(0)
n )|ψ(0)m 〉 = (E(1)
nµ − H(1))|ψ(0)nµ 〉 . (3.22)
O primeiro termo desaparece. Inserindo a expansao (3.18),
∑
m
anm(E(0)m − E(0)
n )|ψ(0)m 〉 = (E(1)
nµ − H(1))∑
ν
cµν |n, ν〉 , (3.23)
e multiplicando os dois lados com 〈n, µ|, obtemos zero na esquerda, pois podemos escolher osestados nao-degenerados ortogonais 〈n, ν|m〉 = δm,n. Portanto,
∑
ν
cµν
[E(1)nµ 〈n, µ|n, ν〉 − 〈n, µ|H(1)|n, ν〉
]= 0 . (3.24)
Essa equacao secular (uma para cada µ), e um conjunto de r equacoes lineares para os coeficientescµν . A condicao para ter solucoes nao-triviais e,
det(〈n, ν|H(1)|n, µ〉 − E(1)
nµ δµ,ν
)µ,ν
= 0 . (3.25)
2Note, que etiquetamos todos os estados que nao sao degenerados com o estado sob investigacao |ψ(1)nµ 〉 com o
ındice m, mesmo se existem degenerescencias entre eles mesmo.

3.1. PERTURBACOES ESTACIONARIAS 45
A solucao dessa determinante secular da as energias E(1)µ procuradas. Depois, a solucao da
equacao secular (3.24) para cada valor de energia da os coeficientes definindo a melhor forma decombinacoes lineares adaptada a perturbacao. Diferentemente aos calculos anteriores com esta-dos degenerados, aqui consideramos combinacoes lineares dos vetores do subespaco degeneradoantes de por a perturbacao.
Na pratica, aplicamos a teoria de perturbacao somente ate a ordem mais baixa relevante.Isto e, somente calculamos a correcao de segunda ordem se a primeira ordem zera. Um exemplofamoso e o efeito Stark quadratico discutido na Sec. 5.3. No caso de autovalores degenerados naausencia de perturbacao a primeira ordem sempre vai produzir uma correcao notavel, como noexemplo do efeito Stark linear, tambem discutido na Sec. 5.3. Por essa razao, nao precisamosdiscutir ordens de perturbacao mais elevados no caso de autovalores degenerados.
Exemplo 7 (Perturbacao num sistema com dois estados degenerados): Como exem-plo consideramos o seguinte hamiltoniano,
H =
(∆ ΩΩ ∆
).
A solucao exata da os autovalores e os autovetores,
E1 = ∆ + Ω , E2 = ∆− Ω , |ψ1〉 = 1√2
(11
), |ψ2〉 = 1√
2
(−11
).
Agora dividimos o hamiltoniano em uma parte nao perturbada e uma perturbacao
H ≡ H(0) + H(1) =
(∆ 00 ∆
)+
(0 ΩΩ 0
).
Obtemos em ordem zero,
E(0)1 = ∆ = E
(0)2 , |1〉 =
(10
), |2〉 =
(01
),
A aplicacao da teoria de perturbacao nao-degenerada em primeira ordem daria,
〈1|H(1)|1〉 = 0 = 〈2|H(1)|2〉 , |ψ(1)1 〉 = |1〉 〈1|H
(1)|2〉E
(0)1 − E(0)
2
→∞← −|ψ(1)2 〉 .
Isto e, a correcao da energia zera em primeira ordem enquanto a correcao da funcao de ondadiverge. Obviamente, a base |ν〉 obtida pela diagonalizacao da matriz H(0) nao e adaptadaao calculo dos elementos da matriz H(1).Agora, aplicando teoria de perturbacao degenerada, obtemos pela determinante secular,
0 = det[〈ν|H(1))|µ〉 − E(1)
µ δµ,ν
]= det
(−E(1)
µ Ω
Ω −E(1)µ
)= (E(1)
µ )2 − Ω2 ,
os autovalores sao E(1)1 = Ω e E
(1)2 = −Ω permitindo estabelecer a equacao secular,
c11
[E
(1)1 − 〈1|H(1)|1〉
]− c12〈1|H(1)|2〉 = c11[Ω− 0]− c12Ω = 0
−c21〈2|H(1)|1〉+ c22
[E
(1)2 − 〈2|H(1)|2〉
]= −c21Ω + c22[−Ω− 0] = 0 .
Obtemos c11 = c12 e c21 = −c22 e com isso,
|ψ(0)1 〉 =
∑
ν
c1ν |ν〉 = c11|1〉+ c12|2〉 = 1√2
(11
), |ψ(0)
2 〉 = c21|1〉+ c22|2〉 = 1√2
(−11
).

46 CAPITULO 3. METODOS DE APROXIMACAO
Assim, podemos verificar que as correcoes para as autoenergias,
E1 = E(0)1 + 〈ψ(0)
1 |H(1)|ψ(0)1 〉 = ∆ + Ω , E2 = E
(0)2 + 〈ψ(0)
2 |H(1)|ψ(0)2 〉 = ∆− Ω ,
coincidem com o calculo exato feito no inıcio. As autofuncoes |ψ(0)1 〉 deveriam ja estar
corrigidas em primeira ordem, por isso,
|ψ(1)1 〉 = |ψ(0)
1 〉〈ψ(0)
1 |H(1)|ψ(0)2 〉
E1 − E2= 0 = |ψ(1)
2 〉 .
No Exc. 3.3.1.8 estudamos um sistema de tres nıveis parcialmente degenerado e a quebra dadegenerescencia por uma perturbacao. No Exc. 3.3.1.9 trataremos uma perturbacao num pococom nıveis de energia degenerados.
3.2 Metodo variacional
3.2.1 A fracao de Rayleigh
Supondo que queremos calcular a energia do estado fundamental Eg de um sistema descritopor um hamiltoniano H, mas nao conhecemos a funcao de onda e nao sabemos como resolver aequacao de Schrodinger. Se pelo menos tivessemos uma boa ideia da forma generica da solucao(gaussiano, sinusoidal,..), poderıamos fazer um chute com um parametro livre e otimizar oparametro minimizando a energia, que deve ser mınima para o estado fundamental. Isso eexatamente a ideia do metodo variacional. Nota bem que o metodo variacional so funciona como estado fundamental.
Para qualquer funcao ψ sabemos que a fracao de Rayleigh E satisfaz,
Eg ≤〈ψ|H|ψ〉〈ψ|ψ〉 ≡ E , (3.26)
nao so quando ψ e a funcao de um estado excitado, mas mesmo quando representa um chute(imperfeito) para o estado fundamental. Assumindo funcoes normalizadas podemos descartaro denominador 〈ψ|ψ〉 = 1. Para verificar o teorema, expandimos a funcao ψ em auto-funcoes(desconhecidas) ortonormais, |ψ〉 =
∑n cn|ψn〉. Como ψ esta normalizada,
1 = 〈ψ|ψ〉 =∑
m,n
〈ψm|c∗mcn|ψn〉 =∑
n
|cn|2 . (3.27)
Do mesmo jeito,
〈ψ|H|ψ〉 =∑
m,n
〈ψm|c∗mHcn|ψn〉 =∑
n
En|cn|2 . (3.28)
Como o estado fundamental e aquele da energia mais baixa, Eg ≤ En, demonstramos a relacao(3.26)
Eg = Eg∑
n
|cn|2 ≤∑
n
En|cn|2 = 〈H〉 . (3.29)
Na pratica, o chute ψα para o estado fundamental permite calcular uma energia que deveser minimaliza por
∂〈ψα|Hα|ψα〉∂α
= 0 . (3.30)
Nos Excs. 3.3.2.1 e 3.3.2.2 nos aproximaremos do estado fundamental, respectivamente, deum potencial quartico e de um oscilador harmonico tentando varios chutes e optimizando osseus parametros livres.

3.3. EXERCICIOS 47
3.2.2 Metodo de Rayleigh-Ritz
Uma modificacao do metodo variacional e o metodo de Rayleigh-Ritz. Aqui, em vez de utili-zar uma funcao ”chute”, utilizamos uma combinacao linear de auto-funcoes com coeficientesvariaveis: |ψ〉 =
∑k ck|k〉. Esses variaveis sao entao otimizadas para minimizar a fracao de
Rayleigh,
Eg ≤∑
k,m ckcm〈k|H|m〉∑k,m ckcm〈k|m〉
= E , (3.31)
onde supomos coeficientes e auto-funcoes reais. Para isso, as derivadas por todos os coeficientesdevem desaparecer:
∂E∂cq
=
∑k ck〈k|H|q〉+
∑m cm〈q|H|m〉∑
k,m ckcm〈k|m〉−∑
k,m ckcm〈k|H|m〉 (∑
k ck〈k|q〉+∑
m cm〈q|m〉)(∑k,m ckcm〈k|m〉
)2
(3.32)
=
∑k ck(〈k|H|q〉 − E〈k|q〉) +
∑m cm(〈q|H|m〉 − E〈q|m〉)∑
k,m ckcm〈k|m〉= 0 ,
utilizando a definicao de E (3.31). A equacao esta satisfeita quando o numerador desaparece:
0 =∑
m
cm(〈q|H|m〉 − E〈q|m〉) . (3.33)
A condicao para a existencia de solucoes e, que a determinante secular desaparece,
0 = det(〈q|H|m〉 − E〈q|m〉) . (3.34)
A solucao dessa equacao leve ate um conjunto de valores E , e o valor mais baixo, Emin, e amelhor aproximacao para a energia do estado fundamental. Os coeficientes da funcao de ondasao obtidos resolvendo a auto-equacao (3.33) com Emin.
No Exc. 3.3.2.3 usaremos o metodo de Rayleigh-Ritz para estimar o efeito da massa nuclearfinita no atomo de hidrogenio sobre as nıveis de energia. No Exc. 3.3.2.4 usaremos o metodode Rayleigh-Ritz para encontrar o numero maximo de atomos permitindo um condensado deBose-Einstein estavel feito de atomos com forca interatomica atraente.
3.3 Exercıcios
3.3.1 Perturbacoes estacionarias
3.3.1.1 Ex: Poco unidimensional com deformacao central
Considere um poco de potencial unidimensional entre −L/2 e L/2 com paredes infinitos. Nocentro do poco seja uma pequena perturbacao
H(1) =
ε para −a2 ≤ x ≤ a
20 fora dessa regiao .
Calcule a correcao para a energia em primeira ordem e discute os limites a L e a→ L.

48 CAPITULO 3. METODOS DE APROXIMACAO
3.3.1.2 Ex: Perturbacao
Demonstre que o produto escalar 〈ψ(0)n |ψ(1)
n 〉 (da correcao de primeira ordem ao estado do sistema”perturbado” com o n-esimo estado do hamiltoniano livre), anula-se quando impomos que o
estado ”perturbado” |ψ(λ)〉 seja normalizado e que o produto 〈ψ(0)n |ψ(λ)〉 seja real.3
3.3.1.3 Ex: Nucleo estendido
A expressao V (r) = −e2/4πε0r para a energia potencial de um eletron no atomo de hidrogenioimplica, que o nucleo (o proton) seja tratamos partıcula puntiforme. Agora supoe que, emcontrario, a carga do proton +e seja distribuıda uniformemente sobre uma esfera de raio R =10−13 cm.a. Da o potencial modificado Vm, que corresponde a esta distribuicao da carga nuclear.b. Supoe que a funcao de onda do atomo de hidrogenio nao muda muito devido ao potencialmodificado. Calcule na ordem mais baixa em R/aB o deslocamento de energia media 〈∆V 〉 paraos estados (n = 1, l = 0,m = 0). Como sera em comparacao o deslocamento de energia para osestados (n = 2, l = 0,m = 0) e (n = 2, l = 1,m = 0)?c. Calcule na mesma maneira 〈∆V 〉 para hidrogenio muonico no estado fundamental.
3.3.1.4 Ex: Perturbacao de sistema de dois nıveis
Consideramos um sistema de dois nıveis. Sem perturbacao o sistema teria o hamiltoniano H(0),
as autoenergias E(0)1,2 e as autofuncoes ψ
(0)1,2. Agora ligamos uma perturbacao estacionaria da
forma H(1) = ε(|1〉〈2|+ |2〉〈1|).a. Calcule as autoenergias resolvendo diretamente a equacao de Schrodinger perturbada.b. Calcule as energias perturbadas usando TPIT e compare com o calculo exato das autoenergias.c. Calcule os autoestados resolvendo diretamente a equacao de Schrodinger perturbada.d. Calcule os estados perturbados usando TPIT e compare com o calculo exato das autofuncoes.
3.3.1.5 Ex: Efeito Stark para um eletron numa caixa
Considere um eletron numa ”caixa unidimensional”, isto e, num poco no intervalo x ∈ [0, a]delimitado por paredes infinitos. Quando um campo eletrico uniforme E e ligado tambem nadirecao x, o eletron experimenta uma forca igual a −eE , sendo −e a carga do eletron, de formaque a energia potencial no interior da caixa torna-se eEx.a. Qual a energia do estado fundamental do eletron (em aproximacao de primeira ordem)?Podemos assumir que eEa seja muito menor que a energia do estado fundamental do eletron nacaixa, na ausencia do campo eletrico.b. Utilize a TPID em primeira ordem para obter uma aproximacao para a funcao de onda doestado fundamental, calculando o primeiro termo da correcao.
3.3.1.6 Ex: Sistema de tres nıveis perturbados ate segunda ordem TPIT
Considere o seguinte hamiltoniano perturbado:
H = H0 +Hλ =
E1 0 00 E2 00 0 E3
+
0 λ 0λ 0 λ0 λ 0
.
3Veja [8], Cap XI, A-2.

3.3. EXERCICIOS 49
a. Determine os autovalores e as autofuncoes perturbados em primeira ordem TPIT.b. Determine os autovalores em segunda ordem TPIT.
3.3.1.7 Ex: Efeito Stark para uma carga num oscilador harmonico
Considere um oscilador harmonico carregado, imerso num campo eletrico uniforme E , descritopelo hamiltoniano H(1) = H + eEx, sendo H = p2/2m+mω2x2/2 o hamiltoniano do osciladorunidimensional livre, e e a carga do oscilador.a. Obtenha, via TPIT, as auto-energias (correcoes de primeira a segunda ordem). Compare osresultados obtidos via TPIT com os analıticos.4
b. Mesma coisa com uma perturbacao da forma ρmω2x2/2.c. Mesma coisa com uma perturbacao σ~ωx3.
3.3.1.8 Ex: Sistema de tres nıveis com degenerescencia
Considere seguinte o hamiltoniano H(0) e sua perturbacao H(1)
H(0) + H(1) =
∆ 0 00 ∆ 00 0 ∆′
+
0 Ω 0Ω 0 Ω0 Ω 0
.
Calcule as correcoes para os autovalores e autofuncoes ate primeira ordem.
3.3.1.9 Ex: Perturbacao num poco 3D com degenerescencia
Seja uma partıcula confinada a um poco cubico tri-dimensional e infinito, descrito pela energiapotencial V (x, y, z) = 0 para 0 < x < a, 0 < y < a e 0 < z < a e V (x, y, z) = ∞ alem da
regiao acima definida. Sabemos que os estados estacionarios da partıcula sao Ψ(0)nx,ny ,nz(x, y, z) =(
2a
)3/2sin(nxπa x)
sin(nyπ
a y)
sin(nzπa z), sendo nx, ny, nz inteiros positivos. As energias associ-
adas sao E(0)nx,ny ,nz = π2~2
2ma2 (n2x + n2
y + n2z). Note que o estado fundamental nao e degenerado
enquanto o primeiro estado excitado e tres vezes degenerado. Considere que a partıcula nestacaixa seja submetida a uma perturbacao da forma H(1) = V0 para 0 < x < a/2 e 0 < y < a/2 eH(1) = 0 alem da regiao acima definida.a. Obtenha a correcao de primeira ordem da energia do estado fundamental.b. Obtenha a correcao de primeira ordem para a energia (degenerada) do primeiro estado exci-tado, alem da base otima (que decorre das combinacoes lineares dos estados degenerados) quemais se aproxima dos estados perturbados.
3.3.2 Metodo variacional
3.3.2.1 Ex: Metodo variacional aplicado um potencial quartico
Determine a energia do estado fundamental do potencial quartico V (x) = bx4 fazendo o ansatzvariacional ψα(x) = (α/π)1/4e−αx
2/2. Formulario:
∫ ∞
−∞e−x
2dx =
√π ,
∫ ∞
−∞x2e−x
2dx = 1
2
√π ,
∫ ∞
−∞x4e−x
2dx = 3
4
√π
4Veja [8], Complemento A XI.

50 CAPITULO 3. METODOS DE APROXIMACAO
3.3.2.2 Ex: Metodo variacional aplicado ao oscilador harmonico
Obtenha, atraves do metodo variacional, a energia do estado fundamental do oscilador harmonicounidimensional, descrito pelo hamiltoniano H = − ~2
2md2
dx2 + 12mω
2x2, e a correspondente funcaode onda, a partir das funcoes tentativasa. ψ(x) = Ae−αx
2sendo α uma constante;
b. ψ(x) = A/(x2 + β2) sendo β uma constante;c. ψ(x) = A cos(πx/a) entre os limites ±a/2 sendo a uma constante.
3.3.2.3 Ex: Efeito da massa nuclear finita no hidrogenio por Rayleigh-Ritz
Use o metodo de Rayleigh-Ritz para estimar o efeito da massa finita do nucleo do atomo dehidrogenio. Para isso, calcule a energia do estado fundamental utilizando o hamiltoniano exato,mas uma base de funcoes de onda assumindo um nucleo infinitivamente pesado. Somente tomaem conta os estados ψ100 e ψ200.
3.3.2.4 Ex: Colapso de um condensado com interacao atrativa
Um condensado de Bose-Einstein de 7Li pode se tornar instavel devido a forca interatomicaatrativa, o comprimento de espalhamento sendo as = −27.3aB. Considere o hamiltoniano deGross-Pitaevskii radial derivado no Exc. 2.5.1.2 com um potencial externo harmonico com afrequencia de oscilacao ωtrp/(2π) = 50 Hz. Usando o metodo variacional determine o numeromaximo de atomos permitindo um condensado estavel. (Note que a condicao de minimizacaoderivada precisa ser avaliada numericamente.)
3.3.2.5 Ex: Oscilacao de Rabi
A populacao seja inicialmente no estado |1〉. Qual deve ser a duracao da perturbacao para deixarum sistema degenerado no estado |2〉?
3.3.2.6 Ex: Oscilador harmonico perturbado
Considere o oscilador harmonico (OH) unidimensional inicialmente preparado (t = −∞) noestado fundamental |0〉 do hamiltoniano nao perturbado H(0) = ~ωa†a, tal que H(0)|n〉 = En|n〉com En = n~ω.a. Atraves da expressao, af (t) ≈ 1
i~∫ tftiWfie
iωfitdt, e do hamiltoniano perturbativo W (t) =
−eExe−t2/τ2(x e o operador posicao do OH), aplicado entre t = −∞ e t = +∞, calcule a
probabilidade de que o sistema esteja no estado excitado |n〉, especificando n, em t = +∞.Analise o resultado.b. Faca o mesmo para uma perturbacao da forma W (t) = Λx2e−t
2/τ2.
3.3.2.7 Ex: Impacto da velocidade de uma perturbacao
Aqui consideramos uma variacao lenta,
W (t) =
0 para t < 0
W0(1− e−γt) para t ≥ 0,
com γ ωfi. Calcule a taxa de transicao para tempos longos, t γ−1.

3.3. EXERCICIOS 51
3.3.2.8 Ex: Efeito fotoeletrico
Um atomo de hidrogenio no estado fundamental 1s e colocado num campo eletrico ~E(t) =~E0 cosωt, tal que W (t) = −er · ~E(t) = W0e
−iωt+W †0 eiωt com W0 = er · ~E0/2. Encontre, via regra
de ouro de Fermi,
R =2π
~|〈f |W (t)|i〉|2ρ(Ef − Ei ∓ ~ω) ,
utilizando a densidade de estados ρ(Ek)dEk = V/(2π)3k2dkdΩ, a probabilidade por unidadede tempo para que o atomo seja ionizado, excitando-se do estado fundamental ψ100(r) =e−r/aB/(πa3
B)1/2 para o estado descrito pela onda plana ψk(r) = e−ik·r/V 1/2. Simplifique o
calculo supondo ~E0 = E0eze k = kez.

52 CAPITULO 3. METODOS DE APROXIMACAO

Capıtulo 4
Subestrutura de atomoshidrogenoides
4.1 Estrutura fina e equacao de Dirac
A estrutura energetica do hidrogenio calculada pelo modelo de Bohr a partir do hamiltonianonao relativıstico,
H =p2
2me− Ze2
4πε0r(4.1)
concorde muito bem com as medidas experimentais. No entanto, em experiencias de alta re-solucao foram observados finos desvios como deslocamentos de energia e desdobramentos daslinhas espectrais chamadas de estrutura fina, que nao eram previstos pela teoria. Isso sugere,que existem efeitos adicionais fracos, que nao afetam fortemente a posicao das linhas espectrais,mas levantam a degenerescencia energetica a respeito do numero quantico orbital `: E = En,`.
Como explicacao possıvel temos o fato que os eletrons apresentam massa e momento re-lativısticos. Para estimar a relevancia das correcoes relativısticas estimamos a velocidade doeletron no estados fundamental do hidrogenio dado por E1 = −~2/2mea
2B. Usando as definicoes
do raio de Bohr, aB = 4πε0~2/(mee2), e da constante da estrutura fina
α ≡ e2
4πε0~c' 1
137, (4.2)
obtemos,
v =
√2E1
me=
~meaB
=e2
4πε0~= αc , (4.3)
o que mostra, que a velocidade do eletron e muito alta e que efeitos relativısticos podem ser naonegligenciaveis.
Outra explicacao pode ser ligada ao spin intrınseco do eletron evidenciado no experimento deStern-Gerlach. Em campos magneticos o spin levou a um desdobramento dos nıveis energeticosdos atomos. Do outro lado sabemos, que a orbita do eletron em torno do nucleo produz umacorrente que pode gerar um campo magnetico com o qual o spin pode interagir 1.
A equacao de onda que satisfaz simultaneamente os requisitos da mecanica quantica e darelatividade se chama equacao de Dirac e foi derivada pelo fısico Paul Dirac em 1928. Noespaco livre incluindo interacoes eletromagneticas ela descreve todas as partıculas massivas despin semi-inteiro com a paridade como simetria, tais como eletrons e quarks. Foi a primeirateoria para explicar inteiramente relatividade especial no contexto da mecanica quantica. A
1O spin do eletron nao gera campo magnetico, em contraste com o seu momento angular. Ele so interage como meio ambiente atraves do requerimento da simetrizacao por ser um fermion.
53

54 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
equacao de Dirac descreve a estrutura fina do espectro do hidrogenio de maneira completamenterigorosa. A equacao tambem implıcita a existencia de uma nova forma de materia, a antimateria,anteriormente insuspeita e nao observada. A equacao tambem justifica a posteriori a introducaode spinores, isto e, de funcoes de onda vetoriais introduzidos por Pauli de maneira heurıstica.
No limite de velocidades altas mas nao-relativısticas, a equacao de Dirac adota a forma deuma equacao de Schrodinger com o seguinte hamiltoniano modificado,
H = H0 + Hrl + H`s + Hdw + Hlamb (4.4)
=
(p2
2me− Ze2
4πε0r
)− p4
8m3ec
2+
1
2m2ec
2
1
r
dV
drl · s +
π~2
2m2ec
2
Ze2
4πε0δ(r) + Hlamb .
Discutiremos os diversos termos nas secoes seguintes.
4.1.1 Correcao para velocidades relativısticas
A primeira correcao na expressao, Hrl na (4.4), vem da expansao da energia relativıstica paravelocidades pequenas ate segunda ordem,
Ekin =√p2c2 +m2
ec4 ' mec
2 +p2
2me− p4
8m3ec
2+ ... . (4.5)
A correcao e da ordem de grandeza,
Hrl
H0=
p4
8m3ec
2
p2
2me
=v2
4c2' α2
4≈ 0.01% . (4.6)
Devido a degenerescencia destes estados, seria apropriado o uso da teoria de perturbacao comestados degenerados. Porem, como Hrl somente depende das coordenadas espaciais comutandocom l e s, a degenerescencia nao e muito importante, pois Hrl ja e diagonal na base |n, `,m〉,isto e, 〈n, `,m|n′, `′,m′〉 = δ``′δmm′ . A partir de,
Hrl = − p4
8m3ec
2= − 1
2mec2
(p2
2me
)2
= − 1
2mec2
(H0 +
Ze2
4πε0r
)2
(4.7)
= − 1
2mec2
(H0 −
2Enn2
r
)2
,
com r ≡ ZraB
e usando como abreviacao as energias do hidrogenio seguinte o modelo de Bohr,
En = 〈n, `|H0|n, `〉 = −Z2e2
4πε0
1
2aBn2= −mec
2
2
Z2α2
n2. (4.8)
Temos
∆Erl = 〈n, `|Hrl|n, `〉 = − 1
2mec2
[〈n, `|H2
0 |n, `〉 − 〈n, `|4Enn
rH0|n, `〉+ 〈n, `|
(2Enn
r
)2
|n, `〉]
=Z2α2
4Enn2
[E2n − 4E2
nn2 1
n2+ 4E2
nn4 1
n3(`+ 12)
], (4.9)
usando os valores esperados calculados em (2.33). Contudo obtemos a seguinte correcao rela-tivıstica,
∆Erl = En(Zα)2
[1
n(`+ 12)− 3
4n2
]. (4.10)
Obviamente, a degenerescencia a respeito do momento angular ` e quebrada por esta correcao.

4.1. ESTRUTURA FINA E EQUACAO DE DIRAC 55
4.1.2 Correcao pelo acoplamento spin-orbita
A segunda correcao, H`s na expressao (4.4), chamada de interacao spin-orbita e uma correcaorelativıstica devido ao fato que o eletron se move rapidamente dentro do campo eletrostatico Edo nucleo. Vamos agora tratar a questao do spin do eletron interagindo com campos magneticos.
4.1.2.1 Momento dipolar do momento angular e do spin
O movimento rotacional de uma carga, −e, cria uma corrente I, correspondendo a uma densidadede corrente,
j(r′) = Ieφδ(r − r′)δ(z′) = −e v
2πrδ(r − r′)δ(z′) . (4.11)
O momento dipolar causado por um movimento circular de um eletron e,
~µl =1
2
∫
Vr× j(r′)d3r′ =
1
4πr×
∫ 2π
0dφ′∫ ∞
−∞dz′∫ ∞
0r′dr′
−evrδ(r − r′)δ(z′) (4.12)
= −12 er× v =
−e2me
l ,
com o momento angular l = r×mev. O quociente γe ≡ −e/2me se chama razao giromagnetica doeletron. Frequentemente usamos o magneton de Bohr, µB ≡ ~e/2me, que representa a unidadeelementaria do spin,
~µl = −µB~gll , (4.13)
onde gl ≡ µl`µB
= 1 e um fator tomando em conta correcoes possıveis entre a derivacao classica ea mecanica quantica.
Do mesmo jeito, poderıamos pensar que o spin do eletron da origem a um momento angular.No entanto, prova-se que a derivacao correta baseada na equacao relativıstica de Dirac da umfator-g, gs ≡ µs
sµB= 2.002319314..,2
~µs =−e2me
gss = −µB~gss . (4.14)
4.1.2.2 Acoplamento spin-orbita
Dentro do sistema de repouso do eletron sendo na posicao x = 0, e o proton que orbita em tornodo eletron. Esta orbita cria uma corrente, −j(r′), que gera um campo magnetico. Seguinte a leide Biot-Savart o potencial vetor e a amplitude do campo sao,
A(x) =µ0
4π
∫
V
−j(r′)d3r′
|x− r′| , (4.15)
B(x) = ∇x ×A(x) =µ0
4π
∫
V
(x− r′)× j(r′)|x− r′|3 d3r′ (4.16)
= −µ0
4π
∫ ∞
−∞dz′∫ ∞
0r′dr′
∫ 2π
0dφ
(x− r′)× v
|x− r′|3e
2πrδ(r − r′)δ(z′) =
eµ0
4π
(x− r)× v
|x− r|3 ,
2O desvio gs − 2 ' απ− 0.164α
2
π2 e devido ao acoplamento do spin as flutuacoes do vacuo eletromagnetico.Precisamos usar metodos da eletrodinamica quantica para calcular as correcoes.

56 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
substituindo a expressao (4.11). Com a expressao para o potencial de Coulomb entre o eletrone o proton e sua derivada radial,
Vcl(r) =−e2
4πε0r,
1
r
dVcl(r)
dr=
e2
4πε0r3, (4.17)
temos na posicao do eletron,
B(0) =eµ0
4π
−r× v
r3= −ε0µ0
e
r× v
r
dVcl(r)
dr= − 1
ec2
r× v
r
dVcl(r)
dr= − 1
emec2r
dVcl(r)
drl . (4.18)
O mesmo resultado pode ser obtido no quadro da eletrodinamica relativıstica, como mostramosno seguinte calculo. Aproximando gs ' 2 obtemos a energia de interacao resultante e
H`s = −~µs ·B(0) ' 1
m2ec
2s · l1
r
dVcl(r)
dr. (4.19)
4.1.2.3 Correcao da energia
Agora so falta transformar de volta para o sistema inercial do nucleo. Esta transformacao,chamada de precessao de Thomas, deve ser feita por uma transformacao de Lorentz, o que nao etrivial com o eletron continuamente acelerado no sua orbita circular. A transformacao traz umfator adicional de 1
2 chamado de fator de Thomas. Assim, temos,
H`s = ξ(r)l · s , (4.20)
com a abreviacao,
ξ(r) ≡ −1
2m2ec
2r
dVcldr
. (4.21)
A vantagem da introducao do potencial Vcl e, que essa expressao tambem vale para atomos maiscomplicados com muitos eletrons, onde o potencial pode desviar consideravelmente do potencialcoulombiano. Note, que o campo magnetico e muito forte, B ' ξ(aB)~/µB ≈ 5 T.
Considerando a orbita fundamental e o fato que os momentos angulares sao da ordem de ~podemos estimar a importancia deste efeito,
H`s
H0=
12m2
ec2
1r
e2
4πε0r2 l · sp2
2me
'1
2m2ec
2e2
4πε01a3B~2
e2
4πε0aB
=1
2m2ec
2
~2
a2B
=α2
2≈ 0.01% . (4.22)
Em princıpio, deverıamos solver de novo a equacao de Schrodinger incluindo a energia V`s.Mas como esse termo e pequeno, e muito mais facil considerar essa energia como perturbacao,e calcular ele utilizando as funcoes de onda nao perturbadas,
∆E`s = 〈n, `, s,m`,ms|V`s|n, `, s,m`,ms〉 = 〈n, `|ξ(r)|n, `〉〈`, s,m`,ms|s · l|`, s,m`,ms〉 . (4.23)
Olhamos primeiro para parte radial. Assumindo um potencial coulombiano,
ξ(r) =−1
2m2ec
2
1
r
dVcldr
= − Ze2
8πε0m2ec
2
1
r3=EnZ
2α2n2
~2
1
r3, (4.24)
com r ≡ Zr/aB e usando as formulas (2.33) a parte radial fica [25],
〈n, `|ξ(r)|n, `〉 =EnZ
2α2n2
~2
1
n3`(`+ 12)(`+ 1)
. (4.25)

4.1. ESTRUTURA FINA E EQUACAO DE DIRAC 57
Usa-se a abreviacao
ζn` ≡ ~2
2 〈ξ(r)〉 . (4.26)
A parte angular e,
s · l = 12(j2 − s2 − l2) . (4.27)
Como os spins precessam um em torno do outro, `z e sz nao sao bons observaveis, a base naoacoplada nao e adaptada. Mas s2, l2 e j2 sao bons observaveis. Na base acoplada n, (`, s)j,mj
〈n, (`, s)j,mj |s · l|n, (`, s)j,mj〉 = ~2
2 [j(j + 1)− `(`+ 1)− s(s+ 1)] . (4.28)
Como j = ` ± 1/2, achamos que cada nıvel se desdobra em dois nıveis com as energiasEn` + `ζn` com a degenerescencia 2`+ 2 e En` − (`+ 1)ζn` com a degenerescencia 2`. Contudotemos uma correcao de energia devido a interacao spin-orbita de,
∆E`s = −En(Zα)2 j(j + 1)− `(`+ 1)− 34
2n`(`+ 1/2)(`+ 1). (4.29)
Note, que o acoplamento l · s leva a degenerescencia de l, mas nao de `z (vide Fig. 4.1). NoExc. 4.4.1.1 verificamos que, no caso de uma energia associada ao acoplamento l·s, so o momentoangular total l + s e uma constante do movimento.
4.1.3 Interacao eletron-nucleo nao-local
Vamos agora discutir a terceira correcao na expressao (4.4). A interacao eletron-nucleo quetemos considerado ate agora e local, isto e, a interacao no ponto r sentido pelo eletron dependeessencialmente do campo naquele ponto do espaco. No entanto, quando a teoria relativıstica ecorretamente aplicada, a interacao eletron-nucleo torna-se nao local, e o eletron e entao afetadopor todos os valores do campo nuclear num domınio ao redor de r. O tamanho deste domınioe da ordem do comprimento de onda Compton do eletron, λC/2π ≡ ~/mec. Esta correcao foiintroduzida por Sir Charles Galton Darwin atraves de uma substituicao na equacao de Diracque resolvia o problema da normalizacao da funcao de onda.
Imaginemos que ao inves do potencial V (r), o potencial do eletron e dado pela integral,
∫f(r′)V (r + r′)d3r′ , (4.30)
onde f(r′) e uma funcao tipo densidade radialmente simetrica e normalizada que assume valoressignificativos somente nas vizinhancas de r dentro de um volume (λC/2π)3 e centrado em r′ = 0.Expandindo o potencial V (r + r′) perto da origem,
V (r + r′) = V (r) + [r′ · ∇r]V (r) + 12! [r′ · ∇r]2V (r) + ... , (4.31)
e inserindo na integral,
∫f(r′)V (r + r′)d3r′ = V (r)
∫f(r′)d3r′ +
∫r′f(r′)d3r′ · ∇rV (r) + 1
2!
∫r′2f(r′)[er′ · ∇r]2d3r′V (r) + ...
= V (r) + 0 + 12!
∫r′2f(r′)d3r′
∂2
∂r2V (r) + ... . (4.32)

58 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
O segundo termo e nulo devido a paridade de f(r′) e o terceiro produz a correcao de Darwinusando V (r) = V (r). Deixando a funcao ser constante dentro do volume, f(r) ' f0, e com anormalizacao,
1 =
∫ ~/2mec
−~/2mec
∫ ~/2mec
−~/2mec
∫ ~/2mec
−~/2mecf(r)dxdydz = 8f0
(~
2mec
)3
, (4.33)
obtemos a integral
∫r2f(r)d3r =
∫ ~/mec
−~/mec
∫ ~/mec
−~/mec
∫ ~/mec
−~/mecf(r)r2dxdydz =
(~
2mec
)2
. (4.34)
Tambem,
∇2V (r) = −e∇2 Ze
4πε0r= −e%(r)
ε0= −Ze
2δ(r)
ε0. (4.35)
Portanto, ∫f(r′)V (r + r′)d3r′ = − Ze2
4πε0r+
π~2
2m2ec
2
Ze2
4πε0δ(r) + ... , (4.36)
o que e justamente a energia eletrostatica com sua correcao de Darwin na expressao (4.4).
Para estimar a importancia deste efeito consideramos o estado fundamental, inserindo a suafuncao de onda (2.32),
〈Hdw〉 =
∫d3rψ∗100(r)
π~2
2m2ec
2
Ze2
4πε0δ(r)ψ100(r) =
π~2
2m2ec
2
Ze2
4πε0|ψ(0)|2 =
π~2
2m2ec
2
Ze2
4πε0
1
πa3B
.
(4.37)Obtemos,
Hdw
H0=
π~2
2m2ec
2Ze2
4πε01
πa3B
e2
4πε0aB
=~2
2m2ec
2
Z
a2B
=α2
2≈ 0.01% . (4.38)
A correcao de Darwin nao depende do momento angular l nem do spin s, tal que fica,
∆Edw = 〈Hdw〉 = −En(Zα)2 . (4.39)
4.1.4 Resumo das correcoes
Combinando as correcoes LS e relativısticas, obtemos,
∆Efs = ∆Erl + ∆E`s + ∆Edw (4.40)
= En(Zα)2
[1
n(`+ 12)− 3
4n2
]− En(Zα)2 j(j + 1)− `(`+ 1)− 3
4
2n`(`+ 12)(`+ 1)
− En(Zα)2
= En(Zα)2
1nj − 3
4n2 − j(j+1)−(j− 12
)(j+ 12
)− 34
2n(j− 12
)j(j+ 12
)− 1 para ` = j − 1
2
1n(j+1) − 3
4n2 − j(j+1)−(j+ 12
)(j+ 32
)− 34
2n(j+ 12
)(j+1)(j+ 32
)− 1 para ` = j + 1
2
= En(Zα)2
[1
n(j + 12)− 3
4n2− 1
].

4.2. ESTRUTURA HIPERFINA 59
Isto e, os nıveis sao agora degenerados em j (vide Fig. 4.1)3. Obviamente os nıveis mais afetadospelas correcoes relativısticas sao aqueles com baixos valores de n e `.
Figura 4.1: Nıveis do hidrogenio.
As energias relativısticas do hidrogenio tambem podem ser obtidas por um calculo exato apartir da equacao de Dirac,
∆Eexatofs = mc2
√√√√√1 +
Zα
n− j − 12 +
√(j + 1
2)2 − Z2α2
2
−mc2 . (4.41)
A expansao deste resultado em potencia de Zα ate segunda ordem reproduz o calculo aproximado(4.40).
4.1.5 Deslocamento de Lamb
So falta discutir a quarta correcao, Hlamb na expressao (4.4). A origem do deslocamento de Lambe na eletrodinamica quantica. Sendo devido a natureza quantica do campo eletromagnetico, estacorrecao nao e prevista dentro da equacao de Dirac.
Podemos imaginar a forca de Coulomb entre partıculas carregadas come sendo mediata porum intercambio continuo de fotons virtuais. Mas cada carga individual tambem emite e reabsorvecontinuamente fotons virtuais, como resultado que a posicao do eletron esta manchada numaregiao de 0.1 fm. Isso reduz a sobreposicao entre as orbitas eletronicas e o nucleo. Por isso, odeslocamento de Lamb causa correcoes que sao mais forte para pequenos n e pequenos `. Porexemplo no hidrogenio, o nıvel 2p1/2 e 4.4 · 10−6 eV = 1 GHz embaixo do 2s1/2 (vide Fig. 4.1).
4.2 Estrutura hiperfina
As medidas do Rutherford sugeriam um nucleo atomico puntiforme e infinitamente pesado. Defato, a massa e finita e a carga nuclear e distribuıda num volume finito e, frequentemente, demaneira nao isotropica, o que leve a interacoes multipolares com os eletrons. Alem disso, muitosnucleos tem um spin que pode interagir com o momento magnetico dos eletrons. As correcoesde energia devido a estes efeitos sao chamadas de estrutura hiperfina 4.
3E interessante, que o tratamento quantico aqui mostrado, incluindo as correcoes relativısticas, por acasocoincide com as correcoes de Arnold Johannes Wilhelm Sommerfeld,
En,j = En
[1 +
α2
n
(1
j + 1/2− 3
4n
)].
4Vide [8] p. 1229 e [27] p. 23 para leitura complementar.

60 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
4.2.1 Acoplamento ao spin do nucleo
4.2.1.1 Momento dipolar do spin nuclear
O nucleo tambem pode ter um momento angular interagindo com o momento angular doseletrons. No entanto, o momento depende inversamente das massas. Isto e, o momento an-gular do nucleo e µN/µB = me/mp ' 10−3 vezes menor, onde µN = ~e/2mp e uma abreviacaochamada de magneton nuclear. Por isso, podemos supor, que a interacao entre o nucleo e osatomos nao vai mexer com o acoplamento L · S entre o momento angular orbital e o spin doseletrons. O spin do nucleo se orientara ao momento total dos eletrons J. No entanto, essainteracao tera a capacidade de quebrar a degenerescencia do hidrogenio, mesmo se o desdobra-mento sera hiperfino. A ordem de grandeza do desdobramento hiperfino e 10−6 eV.
Analogicamente com a equacao (4.13), escrevemos o momento dipolar do nucleo,
~µI =e
2mpgpI =
µN~gpI , (4.42)
com gp ≡ µI/I e, mais uma vez, um fator tomando em conta correcoes possıveis entre a derivacaoclassica e a mecanica quantica 5.
4.2.1.2 Desdobramento hiperfino
O spin nuclear produz na posicao dos eletrons um potencial vetor magnetico,
Adp(r) =µ0
4π
~µI × r
r3, (4.43)
interagindo com o momento angular do eletron L da forma,
HLI =e
meA · p =
e
me
µ0
4πr3(~µI × r) · p (4.44)
=e
me
µ0
4πr3
µN~gp(I× r) · p =
µ0
2πr3
µB~µN~gpL · I ,
usando a definicao de magneton de Bohr.Alem disso, o potencial vetor gerado pelo spin nuclear produz um campo magnetico
B = ∇×A =µ0
4πr3[3(~µI · r)r− ~µI ] , (4.45)
como sera mostrado no Exc. 4.4.2.1 6. Este campo interage com o spin do eletron S da forma,
HSI = −~µS ·B = − µ0
4πr3[3(~µI · r)(~µS · r)− (~µS · ~µI)] (4.46)
=µ0
4πr3
µB~gsµN~gp [3(I · r)(S · r)− (S · I)] ,
usando a equacao (4.14).Combinando os dois termos (4.44) e (4.46), obtemos,
HJI = HLI + HSI =µ0
4πr3
µB~gsµN~gp [3(I · r)(S · r) + L · I− S · I] (4.47)
=µ0
4πr3
µB~gsµN~gpN · I ,
5De fato, o fator g do proton e anormal, gp = 5.58, o que diminuı a razao µl/µI . Para o neutron temos:gp = −3.83
6Apresentamos aqui um calculo simplificado curto-circuitando o termo do contato de Fermi.

4.2. ESTRUTURA HIPERFINA 61
introduzindo
N ≡ 3(S · r)r + L− S . (4.48)
Definir o momento angular total do atomo
F ≡ I + J , (4.49)
e util para poder calcular o acoplamento I · J = 12(F2 − I2 − J2). Agora, como o acoplamento
L · S e forte, projetamos os dois momentos angulares sobre o momento angular total eletronicoJ,
N→ N · J|J|
J
|J| , I→ I · J|J|
J
|J| . (4.50)
Consideramos dois casos:
4.2.1.3 Momentos angulares orbitais L = 0
No caso L = 0 podemos aproximar,
N · S ' S2 , (4.51)
e substituir os J por S nas projecoes (4.50). Com isso o acoplamento entre os spins da camadaeletronica e do nucleo fica,
N · I =(N · S)(I · S)
~2|J|2 =S2(F2 − I2 − S2)
2~2|S|2 . (4.52)
Calculamos
∆E`=0hfs = 〈(S, I)F,mF |HJI |(S, I)F,mF 〉 (4.53)
=µ0
4π
µB~gsµN~gp
⟨N · Ir3
⟩
=µ0
8π
µB~gsµN~gp[F (F + 1)− I(I + 1)− 3
4
] 8π
3
(Z
aB
)3 1
πn3.
Como exemplo consideremos a estrutura hiperfina do estado 1s1/2 do atomo de hidrogenio.
Com J = I = 12 e Z = n = 1 obtemos,
∆EL=0hfs (F = 1)−∆E`=0
hfs (F = 0) =2µ0µBgsµNgp
3πa3B
=2gsgpm
2ec
2
3mpα4 ≈ (2π~) ·1.420 GHz . (4.54)
O valor experimental e 1.4204057518 GHz. Esta frequencia corresponde a linha espectral uti-lizada em radio-astronomia, onde a medida da distribuicao espacial desta radiacao permite omapeamento da distribuicao de hidrogenio interstelar.
4.2.1.4 Momentos angulares orbitais L 6= 0
No caso L 6= 0 obtemos o acoplamento entre os spins da camada eletronica e do nucleo,
N · I =(N · J)(F2 − I2 − J2)
2|J|2 . (4.55)

62 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
Calculamos
∆EL6=0hfs = 〈((L, S)J, I)F,mF |HJI |((L, S)J, I)F,mF 〉 (4.56)
=µ0
4π
µB~gsµN~gp
⟨N · Ir3
⟩
=µ0
4π
µB~gsµN~gp
N · J[F (F + 1)− I(I + 1)− J(J + 1)]
2J(J + 1)
(Z
aB
)3 n
n4L(L+ 12)(L+ 1)
.
Introduzindo o fator de intervalo
AJ ≡µ0
4π
µB~gsµN~gp
(Z
aB
)3 N · J2J(J + 1)
n
n4L(L+ 12)(L+ 1)
, (4.57)
podemos escrever
∆EL6=0hfs = AJ
2 [F (F + 1)− J(J + 1)− I(I + 1)] . (4.58)
Note, que o acoplamento J · I quebra a degenerescencia de J no atomo de hidrogenio, mas naode Jz. Podemos derivar a seguinte regra de intervalo,
∆EF+1 −∆EF = AJ(F + 1) . (4.59)
Alem da interacao magnetica entre os momentos angulares do nucleo e da camada eletronicaexiste uma interacao entre o nucleo, quando nao e esfericamente simetrico e a camada eletronica.Esta interacao causa desvios da regra de intervalo e um desdobramento adicional dos estadoshiperfinos.
4.2.2 Interacao quadrupolar eletrica
O fato do nucleo nao ser perfeitamente esferico, da origem a novas correcoes eletron-nucleo quesao denominadas interacao quadrupolar. A partir de,
Hqud = − 1
4πε0
e2
|re − rN |− 1
4πε0
e2
re, (4.60)
onde re e a coordenada eletronica e rN a coordenada isotomica, ambas tomadas como origemo centro de masso do nucleo. Para re > rN esta interacao pode ser obtida apos varios passosmatematicos como sendo,
Hqud = BJ3(I · J)(2I · J + 1)− 2I2J2
2I(I − 1)2J(J − 1), (4.61)
sendo BJ chamada constante de interacao eletron-nucleo quadrupolar. Com esta expressaopodemos calcular,
∆Equd = 〈IJKmK |Hqud|IJKmK〉 = BJ
32K(K + 1)− 2I(I + 1)J(J + 1)
2I(2I − 1)2J(2J − 1), (4.62)
onde K ≡ 2〈J · I〉 = F (F + 1)− I(I + 1)− J(J + 1). E importante lembrar que um nucleo queapresenta I = 0 ou I = 1
2 nao tem momento quadrupolar, BJ = 0. O mesmo acontece paraJ = 1
2 .

4.3. ATOMOS EXOTICOS 63
Juntando as contribuicoes J ·I da Eq. (4.58) e quadrupolar (4.62), a estrutura hiperfina podeser descrita por,
∆Ehfs = ∆EJI + ∆Equd (4.63)
=AJ2K +
BJ8I(2I − 1)(2J − 1)
[3K(K + 1)− 4I(I + 1)J(J + 1)] ,
onde as constantes AJ e BJ dependem do atomo e do momento angular total eletronico.
Tabela 4.1: Constantes hiperfinas de alguns atomos alcalinos.
atomo n AJ(n2S1/2) AJ(n2P1/2) AJ(n2P3/2) BJ(n2P3/2)
[MHz·h] [MHz·h] [MHz·h] [MHz·h]1H, I = 1
2 1 1420 46.17 −3.07 −0.187Li, I = 3
2 2 401.75 46.17 −3.07 −0.18
3 13.5 −0.9623Na, I = 3
2 3 885.82 94.3 18.65 2.82
4 202 28.85 6.00 0.8685Rb, I = 5
2 5 1011.9 120.7 25.029 26.03
6 239.3 39.11 8.25 8.1687Rb, I = 3
2 5 3417.3 409.1 84.852 12.510
6 809.1 132.5 27.70 3.947
Nos Excs. 4.4.2.2 e 4.4.2.3 determinamos as estruturas hiperfinas de atomo de sodio e rubıdio.
4.3 Atomos exoticos
Atomos ’normais’ consistem de um nucleo de protons e neutrons e de uma casca eletronica. Masoutros sistemas de duas partıculas sao possıveis, p.ex. onde o nucleo ou ou eletron e substituıdopor um outro hadron ou lepton (anti-proton, positron, muon, etc.). Um tal sistema e chamadode atomo exotico. Atomos em estados de Rydberg tambem pertencem nesta categoria.
4.3.1 Positronio e muonio
O positronio (e+e−) e um sistema hidrogenoide constituıdo por leptons, isto e, um eletron e umpositron, que e a antipartıcula do eletron. O muonio (µ+e−) e semelhante ao positronio, so queao inves do positron ha um muon cuja massa e mµ+ = 207me. Leptons sao, de acordo como entendimento atual, partıculas sem estrutura interna. Ambos sistemas sao instaveis: os doispartıculas aniquilam-se uns aos outros produzindo fotons γ. Os nıveis de energia e de orbitadas duas partıculas sao semelhantes a do atomo de hidrogenio. No entanto, devido a massareduzida, as frequencias das linhas espectrais sao menos de metade das linhas de hidrogeniocorrespondentes.
O estado fundamental do positronio, assim como o do hidrogenio, tem duas configuracoespossıveis dependendo das orientacoes relativas dos spins do eletron e do positron. O estadosingleto que com os antiparalelos spins (S = 0, Ms = 0) e conhecido como para-positronio(p-Ps) e denota 1S0. Ele tem uma vida media de
τ =2~
mec2α5= 124.4 ps (4.64)

64 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
e decai preferencialmente em dois raios gamas com energia de 511 keV cada (no centro de massa).O estado tripleto com os spins paralelos (S = 1,Ms = −1, 0, 1) e conhecido como orto-positronio(o-Ps) e denotado como 3S1. Ele tem uma vida media de 138.6 ps, e a forma mais comum dedecaimento produz tres fotons. Outras formas de decaimentos sao negligenciaveis. Por exemplo,o decaimento produzindo cinco fotons e 10−6 vezes menos provavel. Medidas desses tempos devida e dos nıveis de energia do positronio tem sido usados em testes de precisao da eletrodinamicaquantica.
Enquanto que o calculo preciso dos nıveis de energia do positronio e feito baseado na equacaode Bethe-Salpeter, a similaridade entre o positronio e o hidrogenio permite uma estimativaaproximada. Nessa aproximacao, os nıveis de energia sao diferentes daqueles do hidrogeniodevido a diferenca no valor da massa reduzida µ, usada na equacao de energia. Como µ = me/2para o positronio, temos
En = − µq4e
8h2ε20
1
n2= −1
2
meq4e
8h2ε20
1
n2=−6.8 eV
n2. (4.65)
Ja foi observada uma molecula de di-positronio, isto e, de dois atomos de positronio ligados.Positronio em altos estados de energia tem sido previstos para serem a forma dominante demateria atomica no universo em um futuro muito distante, se o decaimento do proton se tornarreal.
4.3.2 Atomos hadronicos
Em contraste com os leptons (tal como o eletron e−, o positron e+ e os muons µ+ e µ−) queparticipam somente em interacoes eletromagneticas e interacoes fracas, os hadrons participamtambem em interacoes fortes (tipo nuclear). Ha dois tipos de hadrons, os barions (como oproton p e antiproton p, o neutron n e antineutron n, hıperons Σ, Ξ,...) que apresentam spinsemi-inteiro comportando-se com fermions e os mesons (como o meson π, o meson K, ...) quetem spin inteiro. Cada hadrons com carga negativa podem formar um atomo hadronico do tipohidrogenoide. Esses sistemas contem um nucleo e um hıperon negativo e sao conhecidos comoatomos hiperonicos. Todos estes sao instaveis e devido ao fato de possuırem um tempo de vidasuficientemente longo algumas de suas linhas espectrais tem sido atualmente observadas.
Uma vez que os hadrons interagem fortemente com o nucleo, a teoria desenvolvida para ossistemas hidrogenoides (em que consiste somente da interacao coulombiana) nao pose ser dire-tamente aplicada. Desta forma os valores mostrados na Tab. 4.2 dao somente uma estimativado ”raio” e do potencial de ionizacao dos atomos hadronicos pπ−, pκ−, pp e pΣ−.
4.3.3 Hidrogenio muonico
A massa do muon e mµ = 207me. Quando um muon e ligado a um proton temos hidrogenio
muonico. O seu tamanho e menor pela razao das massas reduzidas aµ = aB1/me
1/mµ+1/mpe a
energia de ligacao e as energias de excitacao sao maiores pela mesma razao. P.ex. enquantopara H = p+e− a transicao 2S − 2P1/2 fica em 10 eV , 121 nm, para p+µ− fica em 1900 eV.
Atomos muonicos sao interessantes porque eles tem deslocamentos de Lamb, interacoes hiper-finas, e correcoes de eletrodinamica quantica amplificados. Portanto, o deslocamento devido adistribuicao finita da cargas no proton rp = 0.8 fm deve influenciar o espectro. Enquanto emp+e− o 2S e deslocado para cima pelo deslocamento de Lamb shift de um valor 4.4× 10−6 eV,em p+µ− e deslocado para baixo por um valor 0.14 eV. Vide Exc. 4.4.3.1.

4.3. ATOMOS EXOTICOS 65
Tabela 4.2: Principais caracterısticas de alguns atomos exoticos.
sistema massa reduzida raio ”a” Ippe− 1836/1837 ≈ 1 ≈ aB = 1 e2/2aB ≈ 0.5
e+e− 0.5 2 0.25
µ+e− 207/208 ≈ 1 1 0.5
pµ− ≈ 186 5.4 · 10−3 93
pπ− ≈ 238 4.2 · 10−3 119
pκ− ≈ 633 1.6 · 10−3 317
pp ≈ 928 1.1 · 10−3 459
pΣ− ≈ 1029 9.7 · 10−3 515
4.3.4 Atomos de Rydberg
Um atomo excitado para um estado cujo numero quantico principal e muito elevado e chamadode atomo de Rydberg. Tais atomos tem varias propriedades peculiares, incluindo uma grandesensitividade a campos eletricos e magneticos, longos tempos de decaimento e funcoes de ondaque se aproximam a orbitas classicas de eletrons. Os eletron interiores protegem o eletronexterior do campo eletrico do nucleo tal que, visto de longe, o potencial eletrico parece identicodaquele visto pelo eletron de um atomo de hidrogenio.
Apesar de suas falhas, o modelo de Bohr do atomo e util para explicar essas propriedades.No Exc. 1.4.1.6 derivamos a expressao de Bohr para o raio orbital em termos do numero quanticoprincipal n:
r =4πε0n
2~2
e2m. (4.66)
Com isso, fica evidente porque atomos de Rydberg tem propriedades peculiares: o raio vai comon2 (tal que p.ex. o estado com n = 137 do hidrogenio tem um raio de ∼ 1 mm) e a seccaotransversal geometrica vai como n4. Assim, atomos de Rydberg sao extremamente grandes,com eletrons de valencia frouxamente ligados que sao facilmente perturbados ou ionizados porcolisoes ou campos externos.
Como a energia de ligacao de um eletron de Rydberg e proporcional a 1/r e, portanto, caicomo uma 1/n2, o espacamento entre nıveis de energia cai como
∆E =1
(n+ 1)2− 1
n2
n→∞−→ − 2
n3+
3
n4+ ... (4.67)
levando a nıveis cada vez menos espacados. Estes estados de Rydberg formam a serie de Ryd-berg.
4.3.4.1 Producao de atomos de Rydberg
No atomo hidrogenoide so estado fundamental (n = 1) e realmente estavel. Outros estadosdevem ser excitados por varias tecnicas como p.ex. por impacto de eletrons ou por troca decarga. Em contraste com estes metodos, que produzem uma distribuicao de atomos excitadosem varios nıveis, o metodo de excitacao optica permite produzir estados especıficos, mas so emmetais alcalinos cujas transicoes ficam em regimes de frequencia accessıveis por lasers.

66 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
4.3.4.2 Potencial num atomo de Rydberg
O eletron de valencia num atomo de Rydberg com Z protons no nucleo e Z − 1 eletrons emcamadas fechadas ve o potencial Coulomb esfericamente simetrico:
Ucou = − e2
4πε0r. (4.68)
A semelhanca do potencial efetivo ”visto”pelo eletron exterior com o potencial de hidrogeniosugere um tratamento classico dentro do modelo planetario. Existem tres excecoes notaveis:
• Um atomo pode ter dois (ou mais) eletrons em estados altamente excitados com raiosorbitais comparaveis. Neste caso, a interacao eletron-eletron da origem a um desvio signi-ficativo do potencial de hidrogenio. Para um atomo em um estado de Rydberg multiplo otermo adicional, Uee inclui um somatorio de cada par de eletrons altamente excitados:
Uee =e2
4πε0
∑
i<j
1
|ri − rj |. (4.69)
• Se o eletron de valencia tem momento angular muito baixo (interpretado classicamentecomo uma orbita elıptica extremamente excentrica), ele pode passar perto o suficiente donucleo para polarizar-lo, dando origem a um termo adicional
Upol = − e2αd(4πε0)2r4
. (4.70)
• Se o eletron exterior penetra nas camadas eletronicas internas, ele ve mais da carga donucleo e, portanto, sente uma forca maior. Em geral, a modificacao para a energia potencialnao e simples de calcular e deve ser baseado no conhecimento da geometria do nucleo.
No hidrogenio a energia de ligacao e dada por:
EB = −Ryn2
. (4.71)
A energia de ligacao e fraca para altos valores de n, o que explica porque estados de Rydbergsao susceptıveis a ionizacao.
Termos adicionais modificando a energia potencial de um estado de Rydberg requerem aintroducao de um defeito quantico, δl, na expressao para a energia de ligacao:
EB = − Ry(n− δ`)2
. (4.72)
Os longos tempos de vida dos estados de Rydberg com altos momentos angulares orbitaispode ser explicada em termos de uma sobreposicao das funcoes de onda. A funcao de onda deum eletron em um estado com alto ` (grande momento angular, ’orbita circular’) tem poucasobreposicao com as funcoes de onda dos eletrons internos e, portanto, fica relativamente im-perturbavel.

4.4. EXERCICIOS 67
4.3.4.3 Atomos de Rydberg em campos externos
A grande distancia entre o eletron e nucleo ionico em um atomo de Rydberg da jeito a ummomento eletrico dipolar d extremamente grande. Ha uma energia associada com a presenca deum dipolo electrico num campo electrico E , conhecido como uma deslocamento de Stark,
ES = −d · E . (4.73)
Dependendo do sinal da projecao do momento dipolar sobre o vetor do campo electrico local aenergia de um estado aumenta ou diminui com a intensidade do campo. O espacamento estreitoentre nıveis n adjacentes na serie de Rydberg significa que os estados podem se aproximar dadegenerescencia mesmo para campos relativamente fracos. Teoricamente, a forca do campo emque ocorreria uma travessia assumindo que nao ha acoplamento entre os estados e dada pelolimite Inglis-Teller,
FIT =e
12πε0a20n
5. (4.74)
No hidrogenio o potencial Coulombiano puro nao acopla os estados Stark de conjuntos n, o queresulte em cruzamento real. Em outros elementos, os desvios do potencial da forma 1/r idealpermite cruzamento evitado.
4.4 Exercıcios
4.4.1 Estrutura fina e equacao de Dirac
4.4.1.1 Ex: Constantes do movimento no acoplamento l · s
Considere uma partıcula de massa µ descrita pelo hamiltoniano H = − ~2
2µ∇2 +V (r) + ξ(r)L ·S,sendo V (r) um potencial central, L e S os seus momentos angulares orbitais e de spin. Obtenhaas relacoes de comutacao [L, H], [S, H] e [L + S, H] quando consideramos ou nao a interacaospin-orbita ξ(r)L · S introduzida via correcoes relativısticas.
4.4.2 Estrutura hiperfina
4.4.2.1 Ex: Campo de um momento magnetico
a. Calcule o potencial vetor A(r) e o momento dipolar magnetico ~µ produzido por um eletronorbitando numa trajetoria circular pela lei de Biot-Savart usando a expansao de |r − r′|−1 empolinomios de Legendre.b. Calcule o campo magnetico B(r).
4.4.2.2 Ex: Estrutura hiperfina do sodio
Determine a estrutura hiperfina dos estados 2S e 2P do atomo de sodio inclusive os deslocamentosde energia. Consulte a Tab. 6.1 para as constantes hiperfinas AJ e BJ .

68 CAPITULO 4. SUBESTRUTURA DE ATOMOS HIDROGENOIDES
4.4.2.3 Ex: Estrutura hiperfina do Rb
Dados as seguintes distancias energeticas νF,F ′ dos nıveis hiperfinos dos isotopos do rubıdio 87Rbe 85Rb [3],
87Rb, S1/2 se desdobra em ν1,2 = 6834.7 MHz87Rb, P3/2 se desdobra em ν0,1 = 72.3 MHz, ν1,2 = 157.1 MHz, ν2,3 = 267.2 MHz85Rb, S1/2 se desdobra em ν1,2 = 3035.7 MHz85Rb, P3/2 se desdobra em ν1,2 = 29.4 MHz, ν2,3 = 63.4 MHz, ν3,4 = 120.7 MHz ,
calcule as posicoes dos baricentros.
4.4.2.4 Ex: Duas partıculas
Considere um sistema de duas partıculas, de massas µ1 e µ2, submetidas a um potencial centralV (r) e a uma energia potencial de interacao V (|r1−r2|) que depende apenas da distancias entre aspartıculas. O hamiltoniano do sistema na representacao de interacao eH = H1+H2+V (|r1−r2|),com H` = − ~2
2µ`∇2` + V (r`), ` = 1, 2, ... Mostre que os momentos angulares individuais L` nao
sao, em geral, constantes de movimento, diferentemente do momento angular total L = L1 +L2.
4.4.3 Atomos exoticos
4.4.3.1 Ex: Hidrogenio muonico
Hidrogenio muonico consiste em um proton e um muon negativo. Calcula a energia de ligacaodo estado fundamental do hidrogenio muonico em eV e escreve a funcao de onda do estadofundamental.

Capıtulo 5
Atomos com spin em camposexternos
5.1 Partıculas carregada em campo eletromagneticos
5.1.1 Lagrangiano e hamiltoniano de partıculas carregadas
Uma carga sujeita a um campo eletromagnetico sente a forca de Lorentz,
F = qE + qr×B , (5.1)
onde
E = −∇φ− ∂A
∂te B = ∇×A , (5.2)
sendo φ e A respectivamente chamados potenciais escalar e vetorial.Como aprendemos em eletrodinamica e possıvel, derivar esta forca a partir do lagrangiano
do movimento eletronico,
L(ri, ri) =m
2r2 − qφ(r) + qr ·A(r) . (5.3)
Com este objetivo determinamos primeiramente o momento por
pi =∂L∂ri
= mri + qAi , (5.4)
e o hamiltoniano por,
H =∑
ipiri − L(ri, ri) = (mv + qA) · r− m
2r2 + qφ− qr ·A =
m
2v2 + qφ . (5.5)
Ou seja,
H(ri, pi) =1
2m(p− qA)2 + qφ . (5.6)
Valem as equacoes,
ri =∂H∂pi
e pi = −∂H∂ri
. (5.7)
A primeira equacao e facilmente verificada inserindo o hamiltoniano (5.6). A segunda leve aforca de Lorentz,
Fi = mvi = pi − qAi = −∂H∂ri− qAi = qEi + e(v ×B)i , (5.8)
onde o ultimo passo da derivacao sera mostrada no Exc. 5.4.1.1 usando o calibre de Coulomb∇ ·A = 0.
69

70 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS
5.1.2 Acoplamento mınimo
Note, que o mesmo resultado (5.6) pode ser obtido por uma substituicao canonica,
mv −→ p− qA e H −→ H+ qφ . (5.9)
Esta regra de substituicao, chamada de acoplamento mınimo, pode ser aplicada em mecanicaquantica,
mv −→ −i~∇− qA e H −→ H + qφ . (5.10)
No caso do eletron (q = −e) preso no potencial coulombiano central qφ = − Ze2
4πε0re na
presencia de um qualquer potencial magnetico A, obtemos assim,
H =m
2v2 − Ze2
4πε0r=−~2
2m∇2 − i~e
2mA · ∇ − i~e
2m∇ ·A +
e2A2
2m− Ze2
4πε0r. (5.11)
O quarto termo chamado de termo diamagnetico e quadratico em A e geralmente tao pequeno,que pode ser desprezado. O segundo e o terceiro termo descrevem a interacao do eletron atravesdo seu momento p com o potencial vetor A produzido por momentos magneticos no interiordo atomo ou campos magneticos exteriores. Dentro do calibre de Coulomb temos (∇ ·A)ψ =(A · ∇)ψ + ψ(∇ ·A) = (A · ∇)ψ, tal que
Hint = emA · p . (5.12)
5.2 Interacao com campos magneticos
5.2.1 Efeito Zeeman normal da estrutura fina
Os momentos dipolares dos atomos podem interagir com campos magneticos externos. A in-teracao leva a um deslocamento dos nıveis, que depende do numero quantico magnetico. Assim,a ultima degenerescencia na estrutura energetica do atomo esta quebrada. Isso se chama des-dobramento Zeeman. Consideramos um campo magnetico B = Bez uniforme com o potencialvetor,
A = 12B× r = −B
2 (−yex + xey) . (5.13)
Com isso a energia de interacao entre o eletron e o campo e dada pelo hamiltoniano (5.12),
Vzee(B) = − i~em
A · ∇ = − i~e2m
(B× r) · ∇ = − i~e2m
B · (r×∇) (5.14)
= − e2mB · L = −µB
~gLL ·B = −~µL ·B = −µB
~LzB ,
com gL = 1 usando a relacao ~µL = e2mL entre o momento angular do eletron e o momento
magnetico resultante. Essa relacao vale para um atomo sem spin (dois eletrons podem acoplaros seus spins para um estado singleto) e sem estrutura hiperfina (ou uma estrutura hiperfinanao resolvida). As energias sao portanto,
∆Ezee(B) = −µB~B〈n,L,mL|Lz|n,L,mL〉 = −µBmLB . (5.15)
Nos Excs. 5.4.2.1 e 5.4.2.2 representamos a interacao entre um momento angular atomico e umcampo magnetico em diferentes bases caracterizadas por diferentes eixos de quantizacao.

5.2. INTERACAO COM CAMPOS MAGNETICOS 71
5.2.2 Efeito Zeeman anomalo
O efeito Zeeman anomalo ocorre quando o conjunto dos eletrons tem um spin. Utilizando asexpressoes ja conhecidas para os momentos dipolares do momento orbital e do spin do eletron,obtemos para o momento magnetico dipolar,
~µJ = ~µL + ~µS =µB~gLL +
µB~gSS =
µB~
(L + 2S) , (5.16)
com gL = 1 e gS = 2. Podemos ver que o momento dipolar do atomo nao e paralelo ao momentototal, J = L + S.
Quando o campo magnetico e fraco, Vls Vzee(B), o momento total J sera a boa observavel.Portanto, devemos primeiro projetar os momentos L e S sobre J,
L −→(L · J|J|
)J|J| e S −→
(S · J|J|
)J|J| , (5.17)
antes de projetar o resultado sobre o campo B. O potencial fica
Vzee(B) = −~µJ ·B = −µB~
(L + 2S) ·B −→ −µB~
[(L · J|J|
)J|J| ·B + 2
(S · J|J|
)J|J| ·B
]
= − µB~|J|2 [L · J + 2S · J] J ·B = − µB
~|J|21
2
[J2 + L2 − S2 + 2(J2 + S2 − L2)
]J ·B
= −µB~
1
2
3J2 − L2 + S2
|J|2 J ·B . (5.18)
E a energia e,
∆Ezee(B) =
⟨µB~
(1 +
J(J + 1)− L(L+ 1) + S(S + 1)
2J(J + 1)
)J · B
⟩. (5.19)
Introduzindo o fator de Lande,
gJ ≡ 1 +J(J + 1) + S(S + 1)− L(L+ 1)]
2J(J + 1), (5.20)
podemos escrever
∆Ezee(B) = −µB~gJ〈JzB〉 = −µBgJmJB . (5.21)
Esta expressao descreve o efeito Zeeman anomalo, para o qual S 6= 0. Para o efeito Zeemannormal, para o qual o spin e zero, achamos de volta gJ = 1.
Figura 5.1: Acoplamento dos momentos angulares para o efeito (a) Zeeman normal, (b) Zeemananormal, (c) Paschen-Back, (d) Zeeman da estrutura hiperfina e (e) Paschen-Goudsmith.

72 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS
5.2.3 Efeito Paschen-Back e campos magneticos intermediarios
Um campo magnetico externo muito forte (> 1 T), tal que Vls Vzee(B), pode quebrar oacoplamento L · S. Ambos os spins L e S agora se acoplam separadamente ao campo,
L −→(L · B|B|
)B|B| e S −→
(S · B|B|
)B|B| . (5.22)
Por isso,
Vpb(B) = −µB~
(L + 2S) ·B −→ −µB~
[(L · B|B|
)B|B| + 2
(S · B|B|
)B|B|
]·B , (5.23)
tal que,
∆Epb(B) = −µB(mL + 2mS)B . (5.24)
Isso e o efeito Paschen-Back.
As derivacoes que fizemos ate agora se concentraram em situacoes simples bem descritaspor CCOCs em varios esquemas de acoplamento. As projecoes sobre os diferentes eixos dequantizacao [o spin total (5.17) no caso Zeeman ou o campo magnetico aplicado (5.22) nocaso Paschen-Back] garantem que os hamiltonianos Vls e Vzee(B) nestes CCOCs sao descritospor matrizes diagonais. No entanto, em regimes intermediarios entre Zeeman e Paschen-Back,Vls ' Vzee(B), geralmente nao e possıvel achar uma representacao diagonal.
Para calcular o espectro energetico em regimes intermediares devemos, portanto, determinartodas as componentes da matriz
Vls + Vzee(B) = ξ(r)L · S +µB~
(L + 2S) . (5.25)
Utilizando L± ≡ Lx ± iLy e S± ≡ Sx ± iSy, podemos facilmente reescrever a energia na formaseguinte,
Vls + Vzee(B) = ξ(r)(LzSz + 1
2 L+S− + 12 L−S+
)+µB~
(L + 2S) ·B . (5.26)
Esse operador age sobre os estados nao acoplados,
∆Els + ∆Ezee(B) = 〈L′m′L;S′m′S |ξnl(LzSz + 12 L+S− + 1
2 L−S+) + µB(Lz + 2Sz)B|LmL;SmS〉= ~2ξnl
(mLmSδmL,m′LδmS ,m
′S
+ 12L+S−δmL,m′L−1δmS−1,m′S
+ 12L−S+δmL−1,m′L
δmS ,m′S−1
)
+ ~µB(mL + 2mS)BδmL,m′LδmS ,m′S, (5.27)
com as abreviacoes L± ≡√L(L+ 1)−mL(mL ± 1). As energias agora sao os auto-valores
dessa matriz. O fator ξnl e geralmente determinado experimentalmente deixando B = 0. NoExc. 5.4.2.3 calculamos o reacoplamento dos spins de dois eletrons em um campo magneticoexterno.
5.2.4 Efeito Zeeman da estrutura hiperfina
Quando a interacao com o campo magnetico e comparavel com as interacoes hiperfinas, masmuito mais fraco do que as interacoes finas, os campos nao perturbem o acoplamento entre omomento eletronico total J e o spin do nucleo I. Portanto, J, I, F, e mF sao numeros quanticosbons. Portanto, para calcular a energia de interacao,
Vhfs + Vzee(B) = Vhfs − ~µF ·B , (5.28)

5.2. INTERACAO COM CAMPOS MAGNETICOS 73
Figura 5.2: Transicao entre o regime Zeeman e o regime Paschen-Back para o caso de L = 1 eS = 1/2.
projetamos o spin nuclear e o momento eletronico total separadamente na direcao F,
J −→(J · F|F|
)F|F| e I −→
(I · F|F|
)F|F| . (5.29)
O momento magnetico total e,
~µF = ~µJ + ~µI = −µB~gJJ +
µN~gpI . (5.30)
Note o sinal negativo devido a carga negativa do eletron. O fator de Lande gJ e aquele (5.20),causado pelo acoplamento do momento angular orbital L e do spin de eletron S e depende doestado sob consideracao. Com isso,
Vzee(B) =[−µB
~gJ
(J · F|F|
)F|F| +
µN~gp
(I · F|F|
)F|F|
]B (5.31)
=
(− µB~|F|2 gJJ · F +
µN~|F|2 gpI · F
)(B · Fz) .
Usando J · F = 12(F2 + J2 − I2) e I · F = 1
2(F2 − J2 + I2) escrevemos,
Vzee(B) = −µB~gJ
F2 + J2 − I2
2|F|2 BFz + gpµN~
F2 − J2 + I2
2|F|2 BFz , (5.32)
tal que
∆Ehfs + ∆Ezee(B) ' ∆Ehfs + µBgFmFB , (5.33)
usando o fator de Lande gF para o estado F ,
gF ' gJF (F + 1) + J(J + 1)− I(I + 1)
2F (F + 1)− gJ
µNµB
F (F + 1)− J(J + 1) + I(I + 1)
2F (F + 1), (5.34)
onde o segundo termo pode ser desprezado.O desdobramento dos estados eletronicos com o momento F em 2F + 1 subnıveis mF =
−F, .., F e chamado efeito Zeeman da estrutura hiperfina. O resultado (5.31) so vale para camposfracos. Para campos fortes o desdobramento Zeeman muda para o desdobramento Paschen-Backda estrutura hiperfina.

74 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS
5.2.5 Efeito Paschen-Back da estrutura hiperfina
Quando a interacao com o campo magnetico excede a interacao hiperfina, o spin nuclear Ise desacopla do momento total J, e ambos acoplam separadamente com o campo magneticoexterno,
J −→(J · B|B|
)B|B| e I −→
(I · B|B|
)B|B| . (5.35)
O efeito Zeeman da estrutura hiperfina se transforma numa estrutura hiperfina do efeito Zeeman,tambem chamada de efeito Paschen-Back da estrutura hiperfina ou efeito Paschen-Goudsmith.Podemos diagonalizar o potencial numa base, onde I,mI , J, e mJ sao numeros quanticos bons.Usando a expressao (4.63) mas desprezando a contribuicao quadrupolar para a interacao hiper-fina, BJ ' 0, obtemos
Vhfs + Vzee(B) = Vhfs − (~µJ + ~µI) ·B 'AJ~2
J · I + ~µJ ·B (5.36)
−→ AJ~2
(J · B
B
)B
B·(
I · BB
)B
B+ µJzB =
AJ~2JzIz + µJzB ,
onde negligenciamos a interacao do momento dipolar do nucleo com o campo magnetico externo,~µI ' 0. Obtemos,
∆Ehfs + ∆Ezee(B) ' AJmJmI + µBgJmJB . (5.37)
Aqui projetamos o momento angular J e o spin I separadamente sobre a direcao do campomagnetico. O reacoplamento do estado |FmF 〉 para |mImJ〉 em campos magneticos fortes edescrito por coeficientes de Clebsch-Gordan,
|FmF 〉 =∑
mI+mJ=mF|mImJ〉〈mImJ |FmF 〉 . (5.38)
5.2.6 Estrutura hiperfina em regime de campos intermediarios
Sabendo os fatores de intervalo dipolar magnetico AJ e quadrupolar BJ , e possıvel calcularo deslocamento Zeeman da estrutura hiperfina em campos magneticos intermediarios entre osregimes Zeeman e Paschen-Back. Para isso, devemos determinar todas as componentes da matrizVhfs + Vzee(B) e calcular os autovalores. Os termos relevantes das Eqs. (4.63) e (5.35) sao,
Vhfs + Vzee(B) =AJ~2
I · J +BJ~2
6(I · J)2 + 3I · J− 2J2I2
2I(2I − 1)2J(2J − 1)+ gJµBB · J− gIµNB · I . (5.39)
Desenvolvemos a forma matricial completa deste hamiltoniano dentro da base nao acoplada,ondemJ ,mI sao bons numeros quanticos, introduzindo as abreviacoes I± ≡
√I(I + 1)−mI(mI ± 1)
e I±± ≡√I(I + 1)− (mI ± 1)(mI ± 2). A algebra SU(2) fornece expressoes uteis, I · J =
IzJz + 12(I+J− + I−J+). Os elementos da matriz ficam,
〈m′Im′J |Hhfs +HB|mImJ〉 (5.40)
=[AJ + 3BJ
2I(2I−1)2J(2J−1)
]mImJδm′I mI δm
′J mJ + 1
2I+J−δm′I mI+1δm′J mJ−1 + 12I−J+δm′I mI−1δm′J mJ+1
+ 6BJ2I(2I−1)2J(2J−1)〈m′Im′J |
(I·J~)2 |mImJ〉+
[−BJ2I(I+1)J(J+1)
2I(2I−1)2J(2J−1) + (gJmJ − gIµNmI)µBB]δm′I mI δm
′I mI

5.2. INTERACAO COM CAMPOS MAGNETICOS 75
onde
〈m′Im′J |(I·J~)2 |mImJ〉 =
[(mImJ)2 + 1
4I2−J
2+ + 1
4I2+J
2−]δm′I mI δm
′J mJ+ (5.41)
+ 12
(m′Im
′J +mImJ
)I+J−δm′I mI+1δm′J mJ−1+
+ 12
(m′Im
′J +mImJ
)I−J+δm′I mI−1δm′J mJ+1+
+ 14I+J−I++J−−δm′I mI+2δm′J mJ−2+
+ 14I−J+I−−J++δm′I mI−2δm′J mJ+2 .
A matriz 〈m′Im′J |Hhfs + HB|mImJ〉 e dividida em 2F + 1 blocos diagonais, cada um rotuladopor mF . O numero total de nıveis e
∑
F=|I−J |,..,I+J2F + 1 = (2I + 1)(2J + 1) =
∑
mF=|−F,..,F |
∑
mI=|−I,..,I|, mJ=|−J,..,J |, mI+mJ=mF
1
.
Nesta forma a matriz programada, e.g. usando MATLAB, e todos os autovalores do ha-miltoniano para um qualquer estado 2S+1XJ e um spin nuclear de I podem ser calculadosnumericamente. Obviamente, os autovalores seguem da diagonalizacao da matriz e nao depen-dem da base escolhida. A Fig. 5.3 mostra o resultado obtido para o 6Li (I = 3
2) no estado 2P3/2
sabendo, que AJ/h = −1.17 MHz e BJ = 0.
0 2 4-20
-10
0
10
20
B (G)
∆ν
(M
Hz)
Figura 5.3: Estrutura hiperfina e Zeeman do estado 2P3/2 do 6Li.
E interessante analisar os estados chamados de totalmente estirados. Neste caso, o fator deintervalo e A = Vhfs/2. Inserindo F = I + J dentro da primeira formula, obtemos,
Vhfs+Vzee(B) =AJ~2
I·J−BJ3(I + J)
2(2I − 1)(2J − 1)+gJµBB·J−gIµNB·I ≈ AJmJmI+µBgJmJB .
(5.42)O deslocamento de energia dos estados totalmente estirados e linear no campo magnetico. Po-demos tambem olhar para os elementos da matriz I+ = 0 e I− =
√2I e notar que todos os
termos nao diagonais zeram.Quando um dos spins, J ou I, e igual a 1/2 so tem dois estados hiperfinos possıveis: F = I±J ,
que necessariamente sao totalmente estirados. Para este caso existe uma formula analıticaaproximada chamada de formula de Breit-Rabi [2], que sera deduzida no Exc. 5.4.2.4,
∆Ehfs + ∆Ezee(B) = 〈AJ~2 I · J + gJµBB · J− gIµNB · I〉 (5.43)
= −AJ4
+ µNgNmFB ±AJ(I + 1
2)
2
√1 +
4mF
2I + 1x+ x2 ,

76 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS
com a abreviacao x ≡ 2(µBgJ−µNgI)BAJ
.
5.3 Interacao com campos eletricos
5.3.1 Efeito Stark
Campos eletricos interagem com os eletrons do atomo,
Vstark = −ed · E . (5.44)
Isso e o efeito de Stark. Este efeito geralmente e fraco, e sua observacao requer campos fortesou resolucao espectral grande. A teoria de perturbacao estacionaria TPIT da,
E(1)n = 〈ψ(0)
n | − d · E|ψ(0)n 〉 = eEz ·
∫
R3
z|ψ(0)n |2d3r = 0 . (5.45)
Isso so vale, quando os estados tem paridade bem definida e NAO sao degenerados em `. QuandoSAO degenerados a respeito de `, o que e o caso do hidrogenio, os estados nao tem paridadedefinida. Por exemplo, os estados s e p contribuindo no mesmo estado |ψn,j〉 tem paridadesdiferentes. Neste caso, a condicao (5.45) nao precisa ser satisfeita, e a primeira ordem deperturbacao da um valor. E o caso do efeito Stark linear.
Em outros atomos, nao tem esta degenerescencia, e devemos calcular o efeito quadratico deStark em segunda ordem TPIT,
|ψ(1)n 〉 = eEz
∑
n′ 6=n|ψ(0)n′ 〉〈ψ(0)
n′ |z|ψ(0)n 〉
En − En′. (5.46)
e
E(2)n = e2E2
z
∑
n′ 6=n
|〈ψ(0)n′ |z|ψ
(0)n 〉|2
En − En′. (5.47)
Para simplificar os elementos da matriz, separamos a parte radial da parte angular,
〈ψ(0)n′ |z|ψ(0)
n 〉 = 〈n′J ′m′J |z|nJmJ〉 =
∫ ∞
0r3Rn′J ′RnJdr
∫Y ∗J ′m′J
zrYJmJdΩ . (5.48)
A parte radial, escrita como
〈n′JJ ′||z||nJJ〉 ≡∫ ∞
0r3Rn′JJ ′RnJJdr , (5.49)
e chamada de elemento da matriz irreduzıvel, nao depende mais do numero quantico magnetico.Do outro lado, a parte angular pode ser exprimida por coeficientes de Clebsch-Gordan, comosera discutido de maneira mais profunda na Sec. 6.2.2. O resultado e o chamado teorema deWigner-Eckart,
|〈n′JJ ′m′J |z|nJJmJ〉|2∣∣〈n′JJ ′||z||nJJ〉∣∣2 =
1
2J ′ + 1
(J 1 J ′
mJ 0 −m′J
). (5.50)
Com [z, Lz] = 0, o que foi mostrado no Exc. 2.5.3.2 achamos,
0 = 〈J ′m′J |[z, Jz]|JmJ〉 = (mJ −m′J)〈J ′m′J |z|JmJ〉 . (5.51)

5.4. EXERCICIOS 77
Isso significa que para mJ 6= m′J , os elementos da matriz 〈J ′m′J |z|JmJ〉 devem desaparecer.Portanto, a matriz e diagonal em mJ . Consideramos transicoes dipolares com |J − J ′| ≤ 1 1,
(J 1 J + 1mJ 0 −mJ
)=
(J + 1)2 −m2J
(2J + 1)(J + 1), (5.52)
(J 1 JmJ 0 −mJ
)=
m2J
J(J + 1),
(J 1 J − 1mJ 0 −mJ
)=
J2 −m2J
J(2J + 1).
Estados com os mesmos |mJ | levam ao mesmo efeito quadratico de Stark
∆E ∼ A+B|mJ |2 . (5.53)
Os fatores A e B dependem do numero quantico principal n e tambem de L, S, J . Alem disso,dependem da distancia energetica para todos os nıveis contribuintes, por causa do denominadorna equacao de perturbacao (5.46). So os nıveis com paridade diferente (−1)L contribuem. No5.4.3.1 calculamos explicitamente o deslocamento de energia Stark para um atomo de hidrogeniosujeito a um campo eletrico.
5.4 Exercıcios
5.4.1 Partıculas carregada em campo eletromagneticos
5.4.1.1 Ex: Lagrangiano de um eletron em campo eletromagnetico
a. Mostre que o lagrangiano (5.3) reproduz a forca de Lorentz (5.1).b. Mostre que o hamiltoniano (5.5) reproduz a forca de Lorentz (5.1).
5.4.2 Interacao com campos magneticos
5.4.2.1 Ex: Efeito Zeeman com diferentes eixos de quantizacao
O efeito Zeeman pode ser descrito em varias maneiras dependendo da escolha do eixo de quan-tizacao. Considere um campo magnetico B = Bxex e calcule o hamiltoniano de interacaoV (B) = −~µJ ·Ba. escolhendo o eixo de quantizacao ex na direcao do campo magnetico,b. escolhendo o eixo de quantizacao ez perpendicular a direcao do campo magnetico.
5.4.2.2 Ex: Deslocamento Zeeman e eixos de quantizacao
Escolhendo o eixo de quantizacao fixo ez e um campo magnetico B em direcao arbitraria calcule ohamiltoniano de interacao Zeeman com um momento angular J = 1 e mostre, que o deslocamentode energia so depende do valor absoluto |B|.
1Pois e possıvel mostrar que 〈n′JJ ′||z||nJJ〉 = 0 para JJ − J ′| > 1.

78 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS
5.4.2.3 Ex: Acoplamento de dois eletrons
Considere um sistema de dois eletrons.a. Mostre que o operador (~A/~2)s1 · s2 distingue os estados tripletos do singleto.b. Considere agora, que os eletrons sejam expostos a um campo magnetico B aplicado na direcaoez, de forma que adquiram as energias de interacao com o campo (µBB/~)(g1s1z + g2s2z).Obtenha a matriz associada ao hamiltoniano total e demonstre que no regime ~A µBB, arepresentacao que privilegia o momento total e mais adequada.c. Mostre que no regime ~A µBB, e conveniente a utilizacao da representacao que privilegiaos spins individuais do momento total.d. Trata o regime intermediario ~A ' µBB.
5.4.2.4 Ex: Formula de Breit-Rabi
Derive a formula analıtica de Breit-Rabi (5.43) supondo J = 12 .
5.4.2.5 Ex: Poluicao reciproca dos regimes Paschen-Back e Zeeman
a. Determine a matriz de interacao 〈mJmI |Vhfs + Vzee(B)|mJmI〉 de um atomo com spineletronico J e spin nuclear J na base desacoplada sem considerar os termos quadrupolares.b. Determine a matriz de interacao explicitamente para o caso do 6Li (I = 1) no estado funda-mental 2S1/2 (AJ = h · 152.137 MHz) para um campo magnetico de B = 100 G.c. Para o sistema definido em (b) determine os autovalores E(B) da matriz de interacao e osautovetores |α(B)〉 na base desacoplada |mJmI〉.d. Para o sistema definido em (c) determine os autovetores |α(B)〉 na base acoplada |FmF 〉.e. Quao boas sao as regras de selecao para transicoes S1/2 − P3/2 no regime intermediario entreZeeman e Paschen-Back? Comecamos calculando os deslocamentos de Zeeman para ambos osnıveis (s denota a estrutura S1/2, p a estrutura P3/2)
B〈msJm
sI |Hhfs +HB|ms
JmsI〉B = Es(B)
B〈mpJm
pI |Hhfs +HB|mp
JmpI〉B = Ep(B) .
Para o nıvel P3/2 o fator de intervalo e menor. In particular for 6Li e tao pequeno que estamos
imediatamente no regime de Paschen-Back. Isso significa que a matriz ∞〈mpJm
pI |m
pJm
pI〉B =
δmpJ ,mpJδmpI ,m
pI
e diagonal. O elemento da matriz de transicao e entao,
B〈mpJm
pI |T (Eκ)
q |msJm
sI〉B =
∑
msJmsI
∑
mpJmpI
∞〈mpJm
pI |m
pJm
pI〉B ∞〈ms
JmsI |ms
JmsI〉B ∞〈mp
JmpI |T (Eκ)
q |msJm
sI〉∞
=∑
msJmsI
∞〈msJm
sI |ms
JmsI〉B ∞〈mp
JmpI |T (Eκ)
q |msJm
sI〉∞ .
Os elementos da matriz no regime Zeeman puro podem ser expressos por [Deh07, unpublished],
〈F pmpF |T (Eκ)
q |F smsF 〉 =0
⟨mpJm
pI
∣∣T (Eκ)q |ms
JmsI〉0
=
(Js κ Jp
msJ sign(mp −ms) −mp
J
)2Jp Js κF s F p I
2(2F s + 1)(2Jp + 1)(2κ+ 1)
2I + 1.
Que tal o regime puro de Paschen-Back?
∞〈mpJm
pI |T (Eκ)
q |msJm
sI〉∞ =??? .

5.4. EXERCICIOS 79
5.4.3 Interacao com campos eletricos
5.4.3.1 Ex: Efeito Stark no hidrogenio
Considere o atomo de hidrogenio imerso num campo eletrico uniforme E aplicado ao longo dadirecao ez. O termo que corresponde a esta interacao no hamiltoniano total e H(1) = −eEz.Para campos eletricos tıpicos, produzidos em laboratorio, a condicao H(1) H0, que permitea aplicacao da TPIT, e satisfeita. O efeito da perturbacao H(1) denominado efeito Stark, e aremocao da degenerescencia de alguns dos estados do atomo de hidrogenio. Calcule o efeitoStark para o estado n = 2 do atomo de hidrogenio.
5.4.3.2 Ex: Efeito Stark
Derive as Eqs. (5.52) a partir da formula dada na nota de rodape da Sec. 2.4.4.

80 CAPITULO 5. ATOMOS COM SPIN EM CAMPOS EXTERNOS

Capıtulo 6
Interacao de luz com atomosmonoeletronicos
6.1 Transicoes entre estados atomicos
6.1.1 Perturbacao dependente do tempo por uma onda plana
Olhando para o hamiltoniano (5.11) descrevendo a interacao de uma partıcula carregada comum campo eletromagnetico, achamos que o termo A · ∇ ∝ eiωt enquanto o termo A2 ∝ e2iωt.So consideramos o termo de interacao (5.9), que e linear em A, e trataremos este termo comoperturbacao em primeira ordem pela teoria de perturbacao dependente do tempo (TPDT).
Neste ambito separamos o hamiltoniano em uma parte estacionaria e uma parte dependentedo tempo,
H(t) = H(0) + Hint(t) , (6.1)
onde H(0) contem a energia cinetica e o potencial colombiano da Eq. (5.11). Agora inserimos aexpansao,
|ψ〉 =∑
k
ck(t)|k〉e−iEkt/~ , (6.2)
dentro da equacao de Schrodinger e obtemos,
∂
∂t|ψ〉 =
∑
k
ck(t)e−iEkt/~|k〉 − iEk
~∑
k
ck(t)e−iEkt/~|k〉 (6.3)
=1
i~(H(0) + Hint)|ψ〉 =
1
i~∑
k
ck(t)e−iEkt/~(Ek + Hint)|k〉 .
Projetando sobre o estado final 〈f | obtemos,
eiEf t/~〈f | ∂∂t|ψ〉 = cf (t) =
1
i~∑
k
〈f |Hint|k〉ck(t)eiωfkt (6.4)
com ωfk ≡ (Ef − Ek)/~. Ou na versao integral,
cf (t) =1
i~∑
k
∫ t
0ck(t
′)eiωfit′〈f |Hint|k〉dt′ . (6.5)
A aproximacao perturbativa de primeira ordem agora consiste em fixar a condicao inicial ck(t ≤0) = δki e supor, que a probabilidade de encontrar o atomo inicialmente no estado fundamental|i〉 para tempos curtos e 1,
c(1)f (t) ' 1
i~
∫ t
0〈f |Hint|i〉eiωfit
′dt′ . (6.6)
81

82 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
6.1.1.1 Excitacao por ondas planas
Consideramos agora uma perturbacao por uma onda eletromagnetica plana dentro do calibre deCoulomb,
φ = 0 e ∇ ·A = 0 . (6.7)
A solucao da equacao de onda pode ser escrita,
A(r, t) = A∗0(r)eiωt + A0(r)e−iωt . (6.8)
Para onda planas,A0(r) = A0e
ik·r (6.9)
e k = ω/c e k · A0 = 0. Com isso, e possıvel mostrar (vide Exc. 6.4.1.1) que a densidade deenergia e
u(ω) =ε02
E2 +1
2µ0B2 = 2ε0ω
2A20 . (6.10)
Do outro lado, a densidade de energia e proporcional ao numero de fotons N(ω) dentro dovolume V ,
u(ω) =N(ω)~ω
V. (6.11)
A intensidade corresponde a um fluxo de energia,
I(ω) = u(ω)c . (6.12)
Separando a polarizacao ε da amplitude A0,
A = εA0eik·re−iωt + c.c. , (6.13)
e inserindo a perturbacao (5.12) dentro da aproximacao (6.6),
c(1)f (t) = − e
m
∫ t
0dt′〈f |A · ∇|i〉eiωfit′dt′ (6.14)
= − eA0m 〈f |eik·rε · ∇|i〉
∫ t
0dt′ei(ωfi−ω)t′dt′ − eA0
m 〈f |e−ik·rε · ∇|i〉∫ t
0dt′ei(ωfi+ω)t′dt′ .
Qual dos dois processos ocorre depende das energias iniciais e finais. Assim, para Ef = Ei + ~ωo primeiro termo domina descrevendo o processo de absorcao, para Ef = Ei − ~ω o segundotermo descrevendo a emissao prevalece.
6.1.2 Absorcao e emissao estimulada
6.1.2.1 Absorcao
Definimos o elemento de matrix,
Mfi ≡ 〈f |eik·rε · ∇|i〉 , (6.15)
e concentramos no processo de absorcao. Definindo a dessintonizacao por ∆ ≡ ω − ωfi eestimando a integral,
∣∣∣∣∫ t
0e−i∆t
′dt′∣∣∣∣2
=
∣∣∣∣e−i∆t − 1
−i∆
∣∣∣∣2
= 4sin2 ∆t
2
∆2' 2πtδ(∆) , (6.16)

6.1. TRANSICOES ENTRE ESTADOS ATOMICOS 83
para tempos curtos 1, a probabilidade de absorcao fica,
|c(1)f (t)|2 = e2
m2A0(ω)2|Mfi|2∣∣∣∣∫ t
0dt′ei(ωfi−ω)t′dt′
∣∣∣∣2
= e2
m2A0(ω)2|Mfi|22πtδ(∆) . (6.17)
A funcao δ(∆ = 0) simplesmente representa a conservacao de energia. Claro que isso e apenasuma aproximacao nao tomando em conta a largura finita da linha de transicao.
Exprimindo o campo pela intensidade (6.12), obtemos a taxa de transicao para absorcao,
W(ab)fi =
d
dt|c(1)f (t)|2 = 2π
(eA0
m
)2
|Mfi|2δ(ω − ωfi) =πe2
ε0m2c
I(ω)
ω2|Mfi|2δ(ω − ωfi) . (6.18)
Notamos que a taxa de absorcao e proporcional a intensidade da radiacao, o que caracteriza umefeito tipicamente linear.
Se quisermos expressar a taxa de absorcao por atomos em termos de energia, basta multi-plicarmos Wfi por ~ω e, portanto podemos definir a secao de choque para absorcao de energiada radiacao como,
σi→f ≡taxa de absorcao
intensidade incidente=
~ωWfi
I(ω)=
πe2
ε0m2c
~ω|Mfi(ωfi)|2δ(ω − ωfi) . (6.19)
6.1.2.2 Emissao estimulada
Para Ef = Ei − ~ω a equacao descreve o processo de emissao estimulada. Analogicamente aocalculo para absorcao obtemos,
W(st)if =
πe2
ε0m2c
I(ω)
ω2|M∗if |2δ(ω + ωfi) , (6.20)
com M∗fi = 〈f |e−ik·rε · ∇|i〉. E claro, que,
W(st)if = W
(ab)fi . (6.21)
O fato que, num sistema atomo-radiacao em equilıbrio, o campo de radiacao excite o mesmonumero de transicoes na absorcao i→ f como na emissao estimulada f → i se chama princıpiodetalhado de balanco.
Obviamente, a situacao e diferente se ao inves de dois estados temos varios estados quepodem ser excitados pela radiacao ou decair.
6.1.3 Emissao espontanea
Absorcao e a emissao estimulada sao devidos a interacao do atomo com um campo de radiacao.Existe um outro processo de emissao devido a interacao com as flutuacoes do vacuo eletro-magnetico chamado de emissao espontanea. Esta interacao e entendida dentro da eletrodinamicaquantica 2. Aqui vamos adotar um tratamento heurıstico.
Substituindo na Eq. (6.20) a intensidade pelo numero de fotons (6.11) obtemos,
W(st)if =
π~e2N(ω)
ε0m2ωV|Mfi|2δ(ω − ωfi) . (6.22)
1limn→∞nπ
sin2 nx(nx)2
= δ(x).2Vide a apostila do curso Interacao entre luz e materia do mesmo autor.

84 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
O fato de introduzir o conceito de fotons ja implica a quantizacao do campo eletromagnetico.Adicionando ao numero de fotons um foton representando as flutuacoes de vacuo, N(ω) −→N(ω) + 1, conseguimos incluir a emissao espontanea,
W(st)if +W
(sp)if =
π~e2[N(ω) + 1]
ε0m2ωV|Mfi|2δ(ω + ωfi) . (6.23)
Isso significa que, mesmo na ausencia de um campo de radiacao classica, N(ω) = 0, existe uma
probabilidade de emissao. Notamos que W(sp)if depende do volume confinando o atomo, isto e,
a cavidade, pois descreve a transferencia de energia para este volume. Aqui fica claro que aindafalta um argumento, pois a taxa de transferencia deve depender, de alguma forma do numerode estados disponıveis para acomodar o foton emitido, isto e, da densidade dos estados dentroda cavidade. O calculo desta densidade dos estados deveria nos permitir avaliar o volume dequantizacao V .
6.1.3.1 Densidade de estados
Para estimar o numero de modos, isto e, de estados fotonicos dentro de um angulo solido dΩ doespaco livre, consideramos uma caixa de lado L. Os modos dentro deste volume sao impostospelas condicoes de contorno periodicas na parte espacial das ondas planas e−ik·r,
kx,y,z = 2πL nx,y,z , (6.24)
onde (nx, ny, nz) e um conjunto de numeros inteiros que representam os varios estados do foton.No limite de grandes L a variacao entre sucessivos k e muito pequena, tal que podemos trataros numeros como variaveis contınuas. Isto e, o numeros de estados com k entre (kx, ky, kz) e(kx + dkx, ky + dky, kz + dkz) e,
dnxdnydnz = L3
(2π)3dkxdkydkz = V(2π)3k
2dkdΩ = V(2π)3
ω2
c3dωdΩ ≡ ρc(ω)dωdΩ , (6.25)
onde a terceira expressao fica em coordenadas esfericas, a quarta use a relacao ω = ck e a ultimadefine a densidade dos estados,
ρc(ω) = V(2π)3
ω2
c3. (6.26)
Assim, a taxa de emissao espontanea de fotons dentro do angulo solido dΩ e,
W(sp)if dΩ =
(∫
ωW
(sp)if ρc(ω)dω
)dΩ =
∫
ω
π~e2
ε0m2ωV|Mfi|2δ(ω + ωfi)
V
(2π)3
ω2
c3dω dΩ (6.27)
=~e2
8π2ε0m2c3|Mfi|2ωfi dΩ ,
Este tratamento simplificado com somente dois estados atomicos considera a luz como umcampo escalar. De fato, a luz e um campo vetorial e pode ter duas polarizacoes ortogonaisindependentes. A matriz pode depender da polarizacao, tal que,
W(sp)if =
~e2
8π2ε0m2c3
∫ ∑
λ=1,2
|Mλfi|2ωfi dΩ . (6.28)

6.2. TRANSICOES DIPOLARES 85
6.2 Transicoes dipolares
6.2.1 Aproximacao dipolar
Ate agora, utilizamos o elemento de matriz Mλfi(ωfi) sem mencionar como este pode ser calculado
nem quando ele e significativo. Em muitos casos de interesse o calculo deste elemento de matrize consideravelmente simplificado por uma expansao do termo e−ik·r, que faz parte do elementoda matriz (6.15),
e−ik·r = 1− ik · r− 12!(k · r)2 + ... . (6.29)
Esta expansao se justifique no fato que o comprimento de onda e muito maior do que o tamanhodo atomo espalhador, kaB 1. Na aproximacao dipolar supomos
e−ik·r ' 1 , (6.30)
tal que podemos remover a dependencia espacial. Nesta aproximacao so havera interacao docampo eletrico da radiacao com o atomo via um termo de dipolo eletrico d ·E. Desta forma,
Mλfi(ωfi) = 〈f |e−ik·rε · ∇|i〉 ' ε i~〈f |p|i〉 = ε im~ 〈f |r|i〉 . (6.31)
Podemos calcular o valor esperado da velocidade da carga em movimento pela equacao deHeisenberg usando o hamiltoniano nao-perturbado,
Mλfi(ωfi) ' ε im~ 〈f | 1
i~ [r, H0|i〉 = εm~2 〈f |rH0 − H0r|i〉 = εm~2 (Ei − Ef )〈f |r|i〉 . (6.32)
A interpretacao da ultima equacao e, que os estados |i〉 e |f〉 sao conectados atraves de umdeslocamento da nuvem eletronica que, portanto, representa a inducao de um dipolo eletricodurante a transicao eletronica. E conveniente introduzir o momento de dipolo eletrico,
dfi ≡ −e〈f |r|i〉 . (6.33)
Com isso, o elemento de matriz fica,
Mλfi(ωfi) '
mωfie~ ε · dfi (6.34)
e a taxa de absorcao (6.18) fica,
W(dp)fi =
πe2
ε0m2c
I(ωfi)
ω2fi
|Mfi|2δ(ω − ωfi) (6.35)
=π
ε0~2cI(ωfi)|ε · dfi|2δ(ω − ωfi) =
4π2α
~I(ωfi)|ε · rfi|2δ(ω − ωfi) .
usando a definicao da constante da estrutura fina α = e2/4πε0~c.
6.2.1.1 Dependencia da polarizacao
Seguinte a Eq. (6.34) a taxa de absorcao depende da orientacao do momento dipolar a respeitoda polarizacao da luz que, portanto, assume um importante papel nesta transicao. Quando dfientre dois estados e nulo a transicao via dipolo eletrico e proibida. Isso nao quer dizer que naohaja transicao, pois os demais termos da expansao (6.29) nao sao necessariamente nulos e podemexistir transicoes de ordens multipolares superiores. Mesmo o elemento de matriz Mλ
fi(ωfi)

86 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
sendo nulo para transicoes envolvendo um foton ainda existe a possibilidade de transicoes dedois fotons.
Definindo θ como o angulo entre ε e dfi obtemos,
W(dp)fi =
π
ε0~2cI(ωfi)|dfi|2 cos2 θ δ(ω − ωfi) . (6.36)
No caso que a radiacao nao e polarizada (ou aleatoriamente polarizada) podemos substituir adistribuicao angular cos2 θ pelo valor medio,
cos2 θ =1
4π
∫ 2π
0
∫ π
0cos2 θ sin θdθdφ = 1
3 , (6.37)
tal que,
W(dp,no−pol)fi =
π
ε0~2cI(ωfi)|dfi|2δ(ω − ωfi) . (6.38)
Esta expressao tambem representa a taxa de emissao estimulada na aproximacao de dipoloeletrico.
A taxa de emissao espontanea total pode ser obtida a partir da Eq. (6.28) integrando sobretodas as possıveis orientacoes,
W(sp)fi =
~e2
8π2ε0m2c3
∫ ∑
λ=1,2
|Mλfi|2ωfidΩ = 2
~e2
8π2ε0m2c3
∫ 2π
0
∫ π
0
∣∣∣mωfie~
ε · dfi∣∣∣2ωfi sin θdθdφ
=e2
4π2ε0~c3ω3fi|ε · rfi|2
∫ 2π
0
∫ π
0cos2 θ sin θdθdφ =
e2
πε0~c3ω3fi|ε · rfi|2 , (6.39)
tal que,
W(sp)fi =
4α
3c2ω3fi|ε · rfi|2 . (6.40)
6.2.1.2 Probabilidades de transicoes de Einstein
Considerando o problema da transferencia de energia entre o campo eletromagnetico e umaamostra de atomos em equilıbrio termico, Einstein chegou a conclusao que, os processos deabsorcao e de emissao estimulada nao sao suficientes para entender o acoplamento radiativoentre dois nıveis de energia, isto e, o acoplamento nao e corretamente descrito pela regra de ourode Fermi e precisamos introduzir a nocao da emissao espontanea.
Diferentemente da derivacao da secao anterior, Einstein considerou atomos cujas populacoesdos estados de energia ficam em equilıbrio termico com um campo eletromagnetico de um corponegro 3 chegando ao mesmo resultado para a taxa de emissao espontanea (6.38). Os famososcoeficientes de Einstein Afi e Bfi sao dados por,
AfiNf = W(sp)fi e
AfiBfi
=~ω3
fi
π2c3, (6.41)
onde Nf e a populacao do estado excitado. De fato, a emissao espontanea e uma consequencianecessaria da interacao de um atomo com um banho termico (tambem chamado de reservatorio).
3Vide a apostila dos cursos Interacao entre luz e materia e Mecanica quantica do mesmo autor.

6.2. TRANSICOES DIPOLARES 87
6.2.2 Regras de selecao e transicoes eletronicas
As regras de selecao que determinam quais transicoes entre dois conjuntos de numeros quanticosi → f sao permitidas, refletem a propriedades de simetria do sistema, e.g. a conservacao domomento angular (inclusive o spin do foton) ou a mudanca de paridade, que pode ser entendidapelo fato que a emissao de uma foton numa direcao particular deve, de alguma maneira, alterara isotropia espacial do atomo. Note que oscilacoes simetricas da forma da distribuicao da carganao radiam.
Como as transicoes eletronicas via dipolo eletrico sao descritas por |ε · rfi| esperamos umaforte dependencia entre o estado de polarizacao da luz e a existencia de um deslocamento rfina transicao entre estados. Vamos expressar ε e rfi em coordenadas esfericas mais adaptadasao problema 4,
x = r · ex = r sin θ cosφ , y = r · ey = r sin θ sinφ , z = r · ez = r cos θ . (6.42)
Definindoe±1 ≡ − 1√
2(ex ± iey) , e0 ≡ ez . (6.43)
obtemos
r±1 ≡ r · e± = r · 1√2(∓ex − iey) = 1√
2(∓x− iy) = ∓ 1√
2r sin θe±iφ = r
√4π3 Y1,±1(θ, φ) (6.44)
r0 ≡ r · e0 = r · ez = z = r cos θ = r√
4π3 Y1,0(θ, φ) .
Similarmente,
ε±1 ≡ ε · e± (6.45)
ε0 ≡ ε · e0 .
Aplicando a expansao em coordenadas esfericas sobre o elemento da matriz rfi = 〈f |r|i〉 comeq · eq′ = δqq′ e facil verificar,
ε · rfi =∑
q=0,±1
(ε · eq)eq ·∑
q=0,±1
(rfi · eq)eq =∑
q=0,±1
εq〈f |rq|i〉 =√
4π3
∑
q=0,±1
εq〈f |rY1,q|i〉 . (6.46)
Os elementos da matriz sao
〈f |rq|i〉 =√
4π3 〈nf `fmf |rY1,q|ni`imi〉 =
√4π3
∫ ∞
0r3Rnf ,`fRni,`idr
∫Y ∗`f ,mfY1,qY`i,midΩ .
(6.47)
4Geralmente, e util escolher o eixo de quantizacao ao longo do eixo de um campo magnetico,
e0 =B
B.
O segundo eixo pode ser escolhido livremente, por exemplo,
ex =e0 × g
|e0 × g| ,
onde g marca uma direcao arbitraria, por exemplo, da gravidade. O terceiro eixo deve ser perpendicular aos doisprimeiros,
ey =ex × e0
|ex × e0|.

88 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
A integral radial, chamada de elemento de matriz reduzido ou elemento de matriz irreduzıvelcom a notacao,
〈nf `f ||r||ni`i〉 ≡∫ ∞
0r3Rnf ,`fRni,`idr , (6.48)
e sempre nao nula, enquanto a integral angular somente nao e nula se houver um determinadocompromisso entre valores de `i,mi, `f ,mf e q. Sao exatamente isso as regras de selecao. Vale 5,
∫Y ∗`f ,mfYκ,qY`i,midΩ =
√(2`i+1)(2`f+1)
4π(2κ+1)
(`i κ0 0
∣∣∣∣`f0
)(`i κmi q
∣∣∣∣`fmf
), (6.49)
aqui com κ = 1. Definindo o operador tensorial de dipolo eletrico,
Qq1(r) = erq(r) =√
4π3 Y
q1 (θ, φ)er , (6.50)
podemos escrever,
〈nf `fmf |rY1,q|ni`imi〉 = 13
√(2`i + 1)(2`f + 1)〈nf `f ||r||ni`i〉
(`i 10 0
∣∣∣∣`f0
)(`i κmi q
∣∣∣∣`fmf
).
(6.51)Este e o teorema de Wigner-Eckart. O operador do dipolo eletrico e um exemplo mais simples deum operador tensorialQqκ(r) caracterizando a transicao entre estados atomicos. Nos Excs. 6.4.2.1e 6.4.2.2 calculamos explicitamente para um atomo de hidrogenio sujeito a um campo magneticocomponentes do operador do dipolo eletrico.
Regras de selecao podem ser quebradas em ordens superiores, p.ex. por radiacao multipolarcom no caso da transicao dipolar magnetica ou transicao eletrica quadrupolar. Isto e o caso dofenomeno de fosforescencia, que e um tipo de fluorescencia emitida por estados metaestaveis.
6.2.2.1 Paridade
A paridade de um estado foi definida como,
Pψn`m(r) = ψn`m(−r) = (−1)`ψn`m(r) , (6.52)
como mostrado antes. Isto e, estados com ` (im-)par tem paridade (im-)par. Agora a integral(6.51) so nao se anula, quando `i + `f + 1 = par. Portanto, transicoes dipolares devem mudar aparidade do estado. P.ex. transicoes S → P seriam possıveis enquanto S → S seriam proibidas.
6.2.2.2 Momento angular
Na decomposicao (6.51) com κ = 1 o primeiro coeficiente de Clebsch-Gordan so e nao nuloquando |`f − `i| ≤ 1 ≤ `f + `i. Isto e, transicoes dipolares nao podem mudar o momento angularde mais de uma unidade.
5Frequentemente usados sao os sımbolos (3j) conectados aos Clebsch-Gordans por,
〈jimi, jfmf |J,M〉 = (−1)ji−jf+M√
2J + 1
(ji jf Jmi mf −M
),

6.2. TRANSICOES DIPOLARES 89
6.2.2.3 Numero quantico magnetico
Na decomposicao (6.51) com κ = 1 o segundo coeficiente de Clebsch-Gordan so e nao nuloquando |q| ≤ 1. Isto e, transicoes dipolares nao podem mudar o numero quantico magnetico demais de uma unidade. Isso tambem pode ser visto por,
∫Y ∗`f ,mfYκ,qY`i,midΩ ∝
∫ei(mi+q−mf )dΩ ∝ δmi+q,mf . (6.53)
6.2.3 Resumo das regras de selecao inclusive estrutura fina
A estrutura fina e devido a um acoplamento tipo L + S = J. Neste caso,
〈(L, S)JmJ |er|(L′, S′)J ′m′J〉 = (−1)L′+L+2J ′+S+mj−1
√2L+ 1
√2L′ + 1
√2J + 1
√2J ′ + 1
δS′S
L L′ 1J ′ J S
(L′ 1 L0 0 0
)(L′ 1 Lm′ q −m
)〈nL||er||n′L′〉 ,
(6.54)
onde a primeira matriz representa o chamado sımbolo 6j.Transicoes dipolares eletricas sao excitadas por perturbacoes do tipo Stark,
Vstark = −ed · E , (6.55)
onde E = E0 cos(k · r − ωt) e o campo eletrico de uma onda oscilatoria eletromagnetica com apolarizacao E0. Com d = ezez, para determinar quais transicoes dipolares sao possıveis, devemosolhar para a matriz 〈J ′m′J |z|JmJ〉. Aplicando o teorema de Wigner-Eckart (5.50), ja e possıveldeterminar, entre quais numeros quanticos magneticos mJ e m′J transicoes podem ocorrer.
Podemos comparar as amplitudes das varias transicoes entre estados |mJ〉 e |m′J〉 atravesdos coeficientes de Clebsch-Gordan (vide Exc. 5.4.3.2). Transicoes so sao possıveis entre estadospara os quais o coeficiente de Clebsch-Gordan correspondente nao desaparece. Isso se chamaregra de selecao. Olhando nas equacoes (5.52) achamos para transicoes dipolares as seguintesregras de selecao,
∆J = 0,±1 mas (J = 0)→ (J = 0) esta proibido (6.56)
∆mJ = 0,±1 mas (mJ = 0)→ (mJ = 0) esta proibido quando ∆J = 0 .
Alem disso, temos para o acoplamento L · S,
∆S = 0,∆L = ±1 e para o eletron fazendo a transicao ∆l = ±1 . (6.57)
Em presencia de um campo magnetico forte (regime de Paschen-Back) quebrando o acoplamentoL · S as regras de selecao sao,
∆mS = 0,∆mL = 0,±1 . (6.58)
Para acoplamento j · j,
∆j = 0,±1 para um eletron e ∆j = 0 para todos os outros . (6.59)
Para todas transicoes dipolares a paridade deve mudar entre par e impar.
Exemplo 8 (Transicoes permitidas e proibidas na aproximacao dipolar): Exemplos
de transicoes permitidas sao 2S1/2 ↔2 P1/2, 1S0 ↔1 P0. Transicoes proibidas sao 1S0 =3 P1,2S1/2 =2 D3/2, (5s)2 3P0 = (5s6s) 3P0.

90 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
Tabela 6.1: Transicoes permitidas: (1-3) rigorous rules, (4-5) LS coupling, and (6) intermediatecoupling (https://en.wikipedia.org/wiki/Selection rule).
(E1) (M1) (E2) (M2) (E3) (M3)
(1)∆J = 0,±1(∆J = 0 = 0)
∆J = 0,±1,±2
(∆J = 0 = 0, 1; 12
= 12
)
∆J = 0,±1,±2,±3
(0 = 0, 1, 2; 12
= 12, 32
; 1 = 1)
(2) ∆MJ = 0,±1 ∆MJ = 0,±1,±2 ∆MJ = 0,±1,±2,±3(3) πf = −πi πf = πi πf = −πi πf = πi
(4)one e− jump
∆L = ±1no e− jump
∆L = 0; ∆n = 0none or one e− jump
∆L = 0,±2one e− jump
∆L = ±1one e− jump∆L = ±1,±3
one e− jump∆L = 0,±2
(5)if ∆S = 0
∆L = 0,±1(L = 0 = 0)
if ∆S = 0∆L = 0
if ∆S = 0∆L = 0,±1,±2(L = 0 = 0, 1)
if ∆S = 0∆L = 0,±1,±2,±3
(L = 0 = 0, 1, 2; 1 = 1)
(6)if ∆S = ±1
∆L = 0,±1,±2
if ∆S = ±1∆L = 0,±1,±2,±3
(L = 0 = 0)
if ∆S = ±1∆L = 0,±1(L = 0 = 0)
if ∆S = ±1∆L = 0,±1,±2,±3,±4
(L = 0 = 0, 1)
if ∆S = ±1∆L = 0,±1,±2
(L = 0 = 0)
6.2.3.1 Regras de selecao para emissao em certas direcoes
Como mostrado na Eq. (6.46), a taxa de excitacao induzida por um campo de luz depende daorientacao relativa da polarizacao do laser ε e do campo magnetico B. Para tomar em contadessa dependencia, decompomos o vetor de polarizacao (que pode ser linear ou elıptica) numabase de coordenadas como mostrado na Eq. (6.45). Assim, a amplitude relativa das transicoes∆mJ = 0 e proporcional a projecao do vetor de polarizacao no eixo do campo magnetico,ε0 ≡ ε · e0. Para estimar a amplitude das transicoes ∆mJ = ±1, devemos projetar sobre ascoordenadas ε±1 ≡ ε · e±. Note, que a direcao da incidencia do feixe dada pelo vetor de ondak nao influencia a probabilidade de transicao diretamente (ja que a dependencia espacial eik·r
foi removida pela aproximacao dipolar (6.29)), somente atraves do fato, que a polarizacao eperpendicular ao vetor de propagacao, ε ⊥ k.
e-p
-
x
y
z
s
s
+
s+
s-
B
k e
Figura 6.1: Regras de selecao em funcao da polarizacao ε da luz incidente. A projecao destevetor sobre os eixos π = ε · e0 e σ± = ε · e± e proporcional a probabilidade de excitacao (ouigualmente de emissao).
6.3 Linhas espectrais e tempos de vida
6.3.1 Largura natural de uma transicao
Seja Γ ≡ ∑i Sf→i a taxa de decaimento espontaneo do estado f . Isso significa, que a sua
populacao vai diminuindo,
Nf = −ΓNf . (6.60)

6.3. LINHAS ESPECTRAIS E TEMPOS DE VIDA 91
Como Nf = 〈ψf |ψf 〉, temos |ψf (t)〉 = |ψf (0)〉eiωfit−Γt/2. A transformada de Fourier e,
|ξ(ω)〉 =1√2π
∫ ∞
0|ψf (t)〉e−iωtdt =
1√2π
∫ ∞
0eiωfit−iωt−Γt/2dt|ψ0(t)〉 (6.61)
=1√2π
limt→∞
ei(ωfi−ω)t−Γt/2 − 1
i(ωfi − ω)− Γ/2|ψ0(t)〉 =
1√2π
1
i(ω − ωfi) + Γ/2|ψ0(t)〉 .
O espectro,
|ξ(ω)|2 =1
2π
1
(ω − ωfi)2 − Γ2/4, (6.62)
e uma distribuicao de Lorentz. Note, que a largura natural pode ser escondida por efeitos dealargamento de linha, como o efeito Doppler ou colisoes entre atomos.
Estados excitados as vezes podem decair em varios estados de energia inferior. Neste casoa largura de linha e dada pela soma das taxas de decaimento parciais, pois a convolucao dedistribuicoes de Lorentz Lγk com larguras γk tem a largura γ =
∑k γk
6.
6.3.2 Alargamento de linha homogeneo
Distingui-se dois tipos diferentes de alargamento. Os chamados alargamentos homogeneos afetatodos os atomos da mesma maneira independentemente das suas posicoes ou velocidades. Elesgeralmente dao origem a perfis de linha lorentzianos e podem ser incluıdas nas equacoes deBloch. Eles correspondem a uma aceleracao do decaimento. Exemplos sao a largura natural, oalargamento por saturacao e o alargamento por colisoes.
6.3.3 Alargamento de linha inomogeneo
Os chamados alargamentos inomogeneos sao devidos a um deslocamento de nıveis atomicos,que pode ser diferente para cada atomo. Na media sobre uma grande amostra de atomos,os deslocamentos parecem um alargamento geralmente com perfis de linha gaussianos. Elesnao podem ser incluıdas nas equacoes de Bloch, mas so como media sobre todas as trajetoriasdos atomos. Eles nao correspondem a uma aceleracao do decaimento. Eles frequentementesao devidos a perturbacoes exteriores, com p.ex. o alargamento Doppler e o alargamento porflutuacoes temperais ou inomogeneidades espaciais de campos eletricos ou magneticos externos.Resolva os Excs. 6.4.3.1 e 6.4.3.2.
6.3.4 Equacoes de Bloch opticas
Procuramos uma equacao para descrever a evolucao temporal de atomos interagentes com umcampo de radiacao. Entretanto para retratar sistemas que contem processos de excitacao erelaxacao acontecendo simultaneamente, a formulacao usual da mecanica quantica usando aequacao de Schrodinger, ja nao e mais suficiente, pois a mesma somente e capaz de explicarprocessos estimulados como absorcao e uma onda monocromatica. Desta forma, processos maiscomplexos como a emissao espontanea ou qualquer outro processo dissipativo devem ser incluıdosna evolucao temporal do sistema, gerando um carater mais geral no processo de evolucao dosistema.
Assim, ja nao e suficiente representar esta situacao usando somente uma funcao de onda,mas sim por um ensemble delas, onde somos capazes de medir as probabilidades associadasa cada estado do sistema. Para realizar esta tarefa devemos construir um formalismo mais
6Vide a apostila dos cursos Interacao entre luz e materia do mesmo autor.

92 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
abrangente, capaz de englobar casos mais complexos como descritos acima, denominado deformalismo de operador densidade ou simplesmente matriz densidade. Dentro deste contexto, seinseres as equacoes de Bloch opticas [5], capaz de descrever a evolucao temporal dos elementos dematriz do operador densidade, em outras palavras, a evolucao temporal das populacoes, descritopelos termos diagonais da matriz, e das coerencias do sistema, representado pelos termos naodiagonais, conforme veremos a seguir. As equacoes de Bloch foram inicialmente desenvolvidaspara entender fenomenos ressonancia magnetica nuclear (RMN) 7. Resolve o Exc. 6.4.4.1.
6.4 Exercıcios
6.4.1 Transicoes entre estados atomicos
6.4.1.1 Ex: Densidade de energia de ondas planas
Derive o resultado (6.10).
6.4.2 Transicoes dipolares
6.4.2.1 Ex: Estado nao estacionario
Constroi uma funcao de onda de hidrogenio nao-estacionaria com contribuicoes iguais de (n =1, ` = 0,m = 0), (n = 2, ` = 1,m = 1). Calcule os valores esperados 〈|r|〉 e 〈r〉 como funcao dotempo.
6.4.2.2 Ex: Transicoes entre subestados Zeeman
Considere o atomo de hidrogenio imerso num campo magnetico uniforme, descrito pelo hamil-toniano H = H(0) + H(1), sendo H(0) = p2/2m + V (r) e H(1) = −(µB/~)L ·B desprezando ospin.8
a. Dada a funcao inicial, |ψm(0)〉 = cosα|φ000〉+ sinα|φ21m〉, obtenha a sua forma no tempo t.b. Calcule o valor medio 〈D〉m(t) = 〈ψm(t)|D|ψm(t)〉 do operador dipolo eletrico do atomoD = qR.c. Analise as frequencias e polarizacoes da radiacao emitida a partir da transicao dos estadosexcitados |φ21m〉 para o estado fundamental.
6.4.3 Linhas espectrais e tempos de vida
6.4.3.1 Ex: Densidade optica de uma nuvem fria
A secao transversal de um atomo com a frequencia de ressonancia ω0 se movendo com a veloci-dade v e irradiado por um feixe laser de frequencia ω e,
σ(v) =6π
k2
Γ2
4(ω − ω0 − kv)2 + Γ2.
A distribuicao de Maxwell uni-dimensional normalizada e,
ρ(v)dv =
√m
2πkBTe−mv
2/2kBTdv .
7Vide a apostila Interacao luz-materia do mesmo autor.8Veja Cohen-Tannoudji, Complemento D VII

6.4. EXERCICIOS 93
a. Calcule o perfil de absorcao da linha de ressonancia de 461 nm (Γ = (2π) 32 MHz) de um gasde estroncio resfriado ate o limite Doppler (kBTD = ~Γ) desta transicao.b. Calcule o perfil de absorcao da linha de ressonancia de 689 nm (Γ = (2π) 7.6 kHz) de um gasde estroncio resfriado ate o limite Doppler da transicao de 461 nm.c. Compare as densidade opticas em caso de ressonancia.
6.4.3.2 Ex: Espectroscopia de absorcao saturada
A espectroscopia de absorcao saturada e uma tecnica permitindo evitar o alargamento Doppler.O esquema, ilustrado na Fig. ??, consiste em uma celula cheio de um gas de rubıdio (frequencia
de ressonancia ω0 = ck = 2πc/780 nm, taxa de decaimento Γ = (2π) 6 MHz) e dois feixes lasercom a mesma frequencia ω mas contrapropagantes, um chamado de saturacao e outro chamadode prova. A distribuicao das velocidades de Maxwell uni-dimensional e normalizada e,
ρ(v)dv =
√m
2πkBTe−mv
2/2kBTdv .
O gas esta em T = 300 K, onde a pressao parcial do rubıdio fica em torno de P = 10−1 mbar.O comprimento da celula e L = 10 cm. O laser prova tem intensidade abaixo do limite desaturacao, tal que a secao transversal de um atomo se movendo com a velocidade v e,
σ(v) =6π
k2
Γ2
4(ω − ω0 − kv)2 + Γ2.
O laser de saturacao tem alta intensidade. Supomos aqui, Ω ≡ 10Γ, onde Ω e a frequencia deRabi causada pelo feixe de saturacao. Desta maneira ele cria uma populacao Ne de atomos noestado excitado. Como esta populacao falta no estado fundamental, Ng = N −Ne, a absorcaodo feixe prova e diminuıda pelo fator,
Ne
N=
Ω2
4(ω − ω0 + kv)2 + 2Ω2 + Γ2.
Calcule para o laser prova o espectro da densidade optica, OD(ω) = Ln∫∞−∞
Ng−NeN σ(v)ρ(v)dv,
e a intensidade da luz transmitida atraves da celula, II0
= e−OD.
6.4.3.3 Ex: O desacelerador Zeeman
Considere um tubo por onde passa um feixe colimado de atomos, todos inicialmente com velo-cidade v = v0. No sentido contrario ao do movimento dos atomos, incide um feixe luminoso,colimado e monocromatico, com frequencia ω = kc. Conforme foi estudado, a taxa de absorcaode fotons por um atomo e uma lorentziana, podendo ser escrita como:
W (v) =W0
2π
Γ2
(ω − ω0 + kv)2 + (Γ/2)2,
onde Γ e a largura natural da linha espectral em ω0, e W0 e uma constante. Sintoniza-sea frequencia da luz de modo a compensar o efeito Doppler no inıcio do tubo, ou seja, δ =

94 CAPITULO 6. INTERACAO DE LUZ COM ATOMOS MONOELETRONICOS
ω−ω0 = −kv0 (a luz e dessintonizada para o vermelho da ressonancia). Conforme os atomos saodesacelerados, eles deixam de estar em ressonancia com o feixe luminoso, deixando de absorverfotons. Isso poderia ser evitado com a tecnica de resfriamento Zeeman, que compensa o efeitocom campos magneticos. Aqui, vamos ver o que acontece caso essa tecnica nao seja utilizada.a. Para um atomo com velocidade v, escreva uma expressao para a distancia media ∆s(v) que
Paulo Cesar Ventura da Silva
A necessidade do desacelerador Zeeman (2,5 pontos)
Considere um tubo por onde passa um feixe colimado de átomos, todos inicialmente com velocidade
𝑣 = 𝑣0. No sentido contrário ao do movimento dos átomos, incide um feixe luminoso, colimado e
monocromático, com frequência 𝜔 = 𝑘𝑐. Conforme foi estudado, a taxa de absorção de fótons por um
átomo é uma lorentziana, podendo ser escrita como:
𝑊(𝑣) =𝑊0
2𝜋
Γ2
(𝜔 − 𝜔0 + 𝑘𝑣)2 + (Γ 2⁄ )2
Onde Γ é a largura natural da linha espectral em 𝜔0, e 𝑊0é uma constante. Sintoniza-se a frequência
da luz de modo a compensar o efeito Doppler no início do tubo, ou seja, 𝛿 = 𝜔 − 𝜔0 = −𝑘𝑣0 (a luz
é dessintonizada para o vermelho da ressonância). Conforme os átomos são desacelerados, eles
deixam de estar em ressonância com o feixe luminoso, deixando de absorver fótons. Isso poderia ser
evitado com a técnica de resfriamento Zeeman, que compensa o efeito com campos magnéticos. Aqui,
vamos ver o que acontece caso essa técnica não seja utilizada.
a) Para um átomo com velocidade 𝑣, escreva uma expressão para a distância média Δ𝑠(𝑣) que ele
percorre até absorver um fóton, em função dos parâmetros Γ, 𝑣0, 𝑘 e 𝑊0. (O tempo médio que ele leva
para absorver um fóton é 1 𝑊(𝑣)⁄ ). (0,5 pontos)
b) A velocidade do átomo em função do número 𝑛 de fótons absorvidos é 𝑣𝑛 = 𝑣0 − 𝑛Δ𝑣, onde
Δ𝑣 = ℏ𝑘 𝑚⁄ é o recuo devido à absorção de um fóton. A distância total média percorrida por um
átomo após absorver N fótons é estimada por:
𝑆 = ∑ Δ𝑠(𝑣𝑛) ≈ ∫ Δ𝑠(𝑣𝑛)
𝑁
0
d𝑛
𝑁
𝑛=0
Calcule a distância média necessária para que os átomos sejam freados até 𝑣 = 0 (ignore o limite
Doppler). Deixe em função de Γ, 𝑣0, Δ𝑣, 𝑘 e 𝑊0. Dica: faça a mudança de variável 𝑛 → 𝑣 na integral,
isso pode economizar alguns cálculos. (1,5 pontos)
c) Tipicamente, a dessintonia da luz |𝛿| = 𝑘𝑣0 é bem maior do que a largura natural Γ da transição. O
que acontece com 𝑆 no limite em que 𝑘𝑣0 ≫ Γ? Interprete esse resultado, justificando a necessidade
da técnica de resfriamento Zeeman. (0,5 pontos)
ele percorre ate absorver um foton, em funcao dos parametros Γ, v0, k e W0. (O tempo medioque ele leva para absorver um foton e W (v)−1).b. A velocidade do atomo em funcao do numero n de fotons absorvidos e vn = v0 − n~k
m e orecuo devido a absorcao de um foton. A distancia total media percorrida por um atomo aposabsorver N fotons e estimada por:
S =N∑
n=0
∆s(vn) '∫ N
0∆s(vn)dn .
Calcule a distancia media necessaria para que os atomos sejam freados ate v = 0 (ignore o limiteDoppler). Deixe em funcao de Γ, v0, k e W0. Dica: Faca a mudanca de variavel n → v naintegral, isso pode economizar alguns calculos.c. Tipicamente, a dessintonia da luz |δ| = kv0 e bem maior do que a largura natural Γ datransicao. O que acontece com S no limite em que kv0 Γ? Interprete esse resultado, justifi-cando a necessidade da tecnica de resfriamento Zeeman.
6.4.4 Equacoes de Bloch opticas
6.4.4.1 Ex: Esfera de Bloch
Verifique a evolucao temporal da norma do vetor de Bloch definido por ~ρ ≡ (2Reσ+ , 2Imσ− , σz),onde os σk sao as matrizes de Pauli, para um sistema de dois nıveis ressonantemente excitadosem e com emissao espontanea.

Capıtulo 7
Atomos de multiplos eletrons
7.1 Simetrizacao de bosons e fermions
A mecanica quantica deve ser formalizada de tal maneira, que nao preve resultados permi-tindo distinguir partıculas identicas. No entanto, matematicamente e necessario identificar umapartıcula com uma funcao de onda; por exemplo, ψa(x1) seja a funcao de onda a da partıcula1 e ψb(x2) a funcao de onda b da partıcula 2. Na ausencia de interacoes, a funcao de ondatotal, Ψ = ψa(x1)ψb(x2), resolve a equacao de Schrodinger de duas partıculas. Agora, trocandoas coordenadas das partıculas obtemos um estado diferente Ψ′ = ψa(x2)ψb(x1) 1. Isso errada-mente sugere que a funcao de onda de uma partıcula joga o papel de uma etiqueta (ou ”alma”)caracterizando a partıcula alem do conjunto de numeros quanticos. Porque isso representa umproblema, podemos ver no seguinte exemplo.
Exemplo 9 (Indistinguibilidade de partıculas): Consideramos um sistema de duaspartıculas sem spin nao-interagindos num poco de potencial infinito. A funcao de onda totale
Ψ(1,2) ≡ ψa(x1)ψb(x2) = C cosnaπx1
Lcos
nbπx2
L
com a energia
Ea,b =π2n2
a
2mL2+
π2n2b
2mL2.
Para quantidades observaveis, como |Ψ(1,2)|2, precisamos garantir, |Ψ(1,2)|2 = |Ψ(2,1)|2, istoe,
C2 cos2 naπx1
L cos2 nbπx2
L = C2 cos2 naπx2
L cos2 nbπx1
L ,
mas isso nao e valido para na 6= nb. Quando na = nb, temos ψa = ψb. Isto e, as partıculasficam no mesmo estado e nao precisamos nos preocupar com a indistinguibilidade:
Ψ(2,1) = ψa(x2)ψb(x1) = Ψ(1,2) e Ea,b = Eb,a .
No entanto, o fato que este estado nunca e observado com dois eletrons mostra, que a teoria
deve ser corrigida para permitir uma descricao da realidade.
Precisamos construir a funcao de onda total de outra maneira. Utilizamos combinacoeslineares de Ψ(1,2),
ΨS,A ≡ 1√2(Ψ(1,2) ±Ψ(2,1)) = 1√
2[ψa(x1)ψb(x2)± ψa(x2)ψb(x1)] . (7.1)
1Notamos, que os estados sao ortogonais, pois∫Ψ∗(1,2)Ψ(2,1)dx1dx2 =
∫ψ∗a(x1)ψ∗b (x2)ψa(x2)ψb(x1)dx1dx2 =
∫ψ∗a(x1)ψb(x1)dx1
∫ψ∗b (x2)ψa(x2)dx2 = δna,nb .
95

96 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
Essa funcao de onda simetrizada (ou anti-simetrizada) represente um truque para erradicar a eti-queta das partıculas. Pois, sob intercambio de partıculas descrito pelo operador Pxψa(x1)ψb(x2) ≡ψa(x2)ψb(x1), as funcoes (anti-)simetrizadas se comportam como 2,
PxΨS,A = ±ΨS,A enquanto PxΨ(1,2) = Ψ(2,1) 6= ∓Ψ(1,2) . (7.2)
A funcao (anti-)simetrizada resolve a equacao de Schrodinger, tambem. Como [H,Px] = 0,podemos dizer, que o sistema tem a simetria de intercambio ou degenerescencia de intercambiodas partıculas. Observaveis como Ψ∗S,AΨS,A ficam conservadas, por exemplo, a probabilidade
|ΨS,A|2 = 12
[|ψa(x1)ψb(x2)|2 + |ψa(x2)ψb(x1)|2
](7.3)
± 12 [ψ∗a(x1)ψ∗b (x2)ψa(x2)ψb(x1) + ψ∗a(x2)ψ∗b (x1)ψa(x1)ψb(x2)] = Px|ΨS,A|2
nao muda, quando trocamos x1 por x2. Para x1 = x2, observamos,
|ΨS,A|2 = |ψa(x)ψb(x)|2 ± |ψa(x)ψb(x)|2 . (7.4)
Isto e, para um sistema simetrico, a probabilidade de encontrar duas partıculas no mesmo lugare dobrada, enquanto para um sistema anti-simetrico, essa probabilidade e zero.
Wolfgang Pauli mostrou que o carater (anti-)simetrico e relacionado ao spin das partıculas.Partıculas com spin inteiro chamado bosons devem ser simetrizadas. Partıculas com spin semi-inteiro chamado fermions devem ser anti-simetrizadas. Eletrons sao fermions. Por isso, numatomo, eles nao podem ficar no mesmo estado (no mesmo lugar), mas devem se repartir em umacamada complicada de orbitais. Notamos, que isso nao so vale para partıculas elementares, mastambem para partıculas compostas como atomos, por exemplo. Determinaremos no Exc. 8.4.1.4o carater bosonico ou fermionico de varias especies atomicas.
7.1.1 O princıpio de Pauli
Dois eletrons com spins anti-paralelos podem ser separados em campos magneticos inomogeneos,mesmo se eles estao inicialmente no mesmo lugar. Portanto, eles sao distinguıveis e a funcao deonda nao precisa ser anti-simetrica. Mas se trocamos o spin junto com a posicao, as partıculasdevem ser indistinguıveis. Isso deve ser tomado em conta na funcao de onda atribuindo umacoordenada dedicada ao spin, ψa(x1, s1). O operador de intercambio deve, agora, ser generali-zado,
Px,sΨ(1,2) ≡ Px,sψa(x1, s1)ψb(x2, s2) = ψa(x2, s2)ψb(x1, s1) = Ψ(2,1) . (7.5)
Supomos agora, que os eletrons nao so nao interagem entre eles, mas tambem nao existeinteracao entre a posicao e o spin de cada eletron. Isto e, vamos descartar o acoplamentoL · S.3 Podemos entao escrever a funcao total de onda de um eletron como produto de umafuncao espacial, ψ(x), e uma funcao de spin, χ(s) = α ↑ +β ↓, onde α e β sao amplitudes deprobabilidade de encontrar o eletron no estado de spin respetivo, tal que
ψ(x, s) = ψ(x)χ(s) . (7.6)
Para duas partıculas, a funcao de spin total e,
X(1,2) = χa(s1)χb(s2) . (7.7)
2Para garantir |PxΨ(1,2)|2 = |Ψ(1,2)|2 temos que PxΨ(1,2) = eiφΨ(1,2). Daı, PxPxΨ(1,2) = e2iφΨ(1,2) = Ψ(1,2).Portanto, PxΨ(1,2) = ±Ψ(1,2).
3Caso que tem acoplamento L · S, a funcao de onda total nao pode ser escrito como produto das funcoesespaciais e de spin, mas de qualquer jeito deve ser anti-simetrica.

7.1. SIMETRIZACAO DE BOSONS E FERMIONS 97
A versao (anti-)simetrizada e
XS,A = 1√2(X(1,2) ±X(2,1)) = 1√
2[χa(s1)χb(s2)± χa(s2)χb(s1)] . (7.8)
Como so tem duas orientacoes para os spin, existem quatro possibilidades para distribuir osspins ↑ e ↓ para as funcoes χm(sn),
XS =
↑↑ = χ1,11√2
(↑↓ + ↓↑) = χ1,0
↓↓ = χ1,−1
e XA = 1√2
(↑↓ − ↓↑) = χ0,0 (7.9)
Para a funcao de onda total, que deve ser anti-simetrica para eletrons, existem duas possibili-dades,
ΘA =
ΨSXA = 12
(Ψ(1,2) + Ψ(2,1)
) (X(1,2) −X(2,1)
)= 1√
2[ψa(x1)ψb(x2) + ψa(x2)ψb(x1)]χ0,0
ΨAXS = 12
(Ψ(1,2) −Ψ(2,1)
)(X(1,2) +X(2,1)) = 1√
2[ψa(x1)ψb(x2)− ψa(x2)ψb(x1)]
χ1,1
χ1,0
χ1,−1
.
(7.10)
Isto e, os dois eletrons podem estar num estado tripleto com a funcao de onda de spin anti-simetrica, ou num estado singleto com a funcao de onda de spin simetrica 4.
Como generalizar essas consideracoes para N eletrons? As funcoes de onda simetrizadascontem todas as permutacoes da etiquete ak, onde entendemos por ak o conjunto de numerosquanticos especificando sem ambiguidade o estado da partıcula k,
ΘS = N∑
Px,sakψa1(x1)...ψaN (xN ) , (7.11)
com um fator de normalizacao N 5. A funcao de onda (anti-)simetrizada e obtida a partir dadeterminante de Slater,
ΘA = 1N ! detψak(xn) = 1
N !
∣∣∣∣∣∣∣
ψa1(x1) · · · ψa1(xN )...
. . ....
ψaN (x1) · · · ψaN (xN )
∣∣∣∣∣∣∣. (7.12)
Essa funcao satisfazPx,sΘA,(1,..,i,j,..,N) = ΘA,(1,..,j,i,..,N) . (7.13)
A determinante de Slater e zero, quando tem dois conjuntos de numeros quanticos identicos,ai = aj . Por exemplo, para dois eletrons numa camada eletronica, |ni, li,mi, si〉 = |nj , lj ,mj , sj〉.Isso e o princıpio forte de exclusao de Pauli:
A funcao de onda total deve ser antissimetrica com respeito a troca de qualquerpar de fermions identicos e simetrica com respeito a troca de qualquer par de bosonsidenticos.
O princıpio fraco de exclusao de Pauli (em geral suficiente para consideracoes qualitativas) diz,que dois fermions em estados identicos nao podem ocupar a mesma regiao no espaco. Isto e,a onda de Broglie deles interfere destrutivamente, como se o princıpio de Pauli exercesse umainteracao repulsiva sobre as partıculas. Essa ’forca’ tem um grande impacto para a fenomenologiadas ligacoes entre atomos, como discutiremos nas secoes seguintes.
4Na imagem acoplada, o spin total S = s1 + s2 pode ter os valores seguintes S = |s1 − s2|, .., s1 + s2 = 0, 1.No caso S = 0 o numero quantico magnetico so pode ter um valor (singleto), mS = 0. No caso S = 1 ele podeter tres valores mS = −1, 0,+1 (tripleto) (vide Exc. 5.4.2.3).
5E possıvel mostrar N =
√Πm
k=1nk!
N !, onde nk e a populacao do estado ψak , isto e, o numero de partıculas com
o mesmo conjunto de numeros quanticos ak.

98 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
7.1.2 Consequencias para estatıstica quantica
A indistinguibilidade de partıculas quanticas tem consequencias interessantes no comportamentoestatıstico dos bosons e fermions. Isso fica obvio quando consideramos duas partıculas 1 e 2podendo adotar dois estados diferentes a e b. Partıculas distinguıveis podem ficar em um dosquatro seguintes estados,
Ψ = ψa(x1)ψa(x2), ψa(x1)ψb(x2), ψb(x1)ψa(x2), ψb(x1)ψb(x2) (7.14)
com a mesma probabilidade de p = 1/4. Partıculas indistinguıveis bosonicas podem ficar emum dos tres seguintes estados,
Ψ = ψa(x1)ψa(x2), 1√2[ψa(x1)ψb(x2) + ψb(x1)ψa(x2)], ψb(x1)ψb(x2) (7.15)
com a mesma probabilidade de p = 1/3. Finalmente, partıculas indistinguıveis fermionicas sopodem ficar em um estado,
Ψ = 1√2[ψa(x1)ψb(x2)− ψb(x1)ψa(x2)] (7.16)
com a mesma probabilidade de p = 1. Vemos que um simples sistema de duas partıculas jaexibe diferencas qualitativas em seu comportamento estatıstico. Essas diferencas geram fısicasdiferentes quanto tratamos sistemas com numeros grande de partıculas, como podemos ver noscasos de gas de eletrons livres e do condensado de Bose-Einstein 6
Notamos finalmente um resultado do modelo padrao da fısica de partıculas atribuindo umcarater fermionico para todas partıculas constituintes fundamentais da materia enquanto osmediadores de forcas fundamentais sao sempre bosons.
7.2 Helio
O atomo mais simples para discutir o princıpio de Pauli e o helio. O atomo de helio possui umnucleo com carga Ze = +2e e massa mHe ≈ 4mH .
7.2.1 O estado fundamental
O estado fundamental reune os dois eletrons no estado fundamental, isto e, (1s)2. Para trataro atomo de helio podemos, como primeiro chute, descrever o atomo pelo modelo de Bohr,assumindo eletrons independentes. Negligenciando o termo de repulsao eletronica (que dependede r12), podemos separar a funcao de onda total em:
Ψ(r1, r2) = Ψ1(r1)Ψ2(r2) , (7.17)
e ficamos com duas equacoes de Schrodinger, o hamiltoniano sendo igual ao caso de atomoshidrogenoides: [
− ~2
2µ∇2i −
e2
4πε0
Z
ri
]Ψi(ri) = E(i)
n Ψi(ri) , (7.18)
com i = 1, 2. Para atomos hidrogenoides temos,
E = E(1)n + E(2)
n = EBZ2
(1
n21
+1
n22
), (7.19)
6Para uma discussao mais ampla vide a apostila Optica atomica do mesmo autor.

7.2. HELIO 99
com EB = −13.6 eV. Com isso, temos a energia para o estado fundamental:
EHe(1s) = −2Z2EB = −108.8 eV . (7.20)
O valor previsto pelo modelo de Bohr e longe do valor experimental: A energia de ionizacaomedida para o primeiro eletron e 24.6 eV, para o segundo 54.4 eV, dando no total uma energiade ligacao dos dois eletrons de −78.983eV. Isso corresponde a um erro em torno de 38%. Aenergia menor do primeiro eletron e devido a blindagem do nucleo pelo segundo.
7.2.1.1 Perturbacao de primeira ordem na energia
Tratando o termo de repulsao entre os eletrons como uma perturbacao [1] e utilizando as auto-funcoes para atomos hidrogenoides |n, `,m`〉, a funcao de onda total fica |n1, `1,m`1 ;n2, `2,m`2〉,temos como correcao de primeira ordem TPIT para energia:
∆E = 〈n1, `1,m`1 ;n2, `2,m`2 |e2
4πε0r12|n1, `1,m`1 ;n2, `2,m`2〉 . (7.21)
Esta correcao e denominada integral de Coulomb e vale:
∆E =e2
4πε0
∫|Ψn1,`1,m`1
(r1)|2(
1
r12
)|Ψn2,`2,m`2
(r2)|2dV1dV2 . (7.22)
Essa integral e sempre positiva. O termo |Ψn1,`1,m`1(r1)|2dV1 e a probabilidade de encontrar
o eletron no elemento de volume dV1 e, quando multiplicado por −e, da a carga associada a essaregiao. Assim, o integrando representa a energia de interacao coulombiana das cargas confinadasdentro dos dois elementos de volume dV1 e dV2. ∆E e a contribuicao total para energia potencial.Calculando a integral de Coulomb para o estado fundamental, o que sera feito no Exc. 7.5.2.1,obtemos,
∆E =5Z
4
(e2
4πε02a0
)=
5Z
4EB , (7.23)
com a0 o raio de Bohr. ∆E corresponde a 34 eV. Assim, a energia do estado fundamentalfica EHe(1s) = −108.8 eV + 34 eV = −74.8 eV. Comparando com o valor experimental de−78.983 eV temos um erro em torno de 5.3%.
7.2.1.2 Blindagem da carga nuclear
Podemos fazer a aproximacao em que consideramos que cada eletron se move em um potencialcoulombiano, com relacao ao nucleo, blindado pela distribuicao de carga do outro eletron [13].O potencial resultante sera gerado por uma carga efetiva ζe ≡ (Z −B)e. A grandeza B ∈ [0, 1]e chamada constante de blindagem.
O primeiro eletron sente uma carga nuclear total Ze, enquanto o segundo sente uma carganuclear efetiva ζe. Trocamos Z por ζ no termo da energia para atomos hidrogenoides,
En = −ζ EBn2
, (7.24)
e a energia para o estado fundamental fica, supondo uma blindagem total, B = 1,
E = E1 + E2 = −Z2EB − ζ2EB = −4EB − EB = −5EB = −67.5 eV . (7.25)

100 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
Comparando com o valor experimental de −78.983 eV temos um erro em torno de 15%. Parauma constante de blindagem em torno de B = 0.656 o valor experimental e reproduzido. Issosignifica que a carga nuclear efetiva sentida pelo segundo eletron e blindada parcialmente peloprimeiro. No Exc. 7.5.2.2 estudamos a blindagem reciproca dos eletrons no exemplo do ıontipo-helio H−.
Os metodos de TPIT (7.21) e da blindagem (7.24) podem ser combinados num calculovariacional, onde a carga efetiva ζ e o parametro variacional. Isso sera demonstrado no Exc. ??.
7.2.2 Estados excitados
Vamos agora investigar os estados excitados do helio. Os primeiros vao ser aqueles onde somenteum eletron esta excitado, o outro ficando no estado fundamental, (1s)1(2s)1 e (1s)1(2p)1. Aenergia do eletron na camada n = 2 e menor do que previsto pelo modelo de Bohr com Z = 2por causa da interacao com o outro eletron. Tambem, os nıveis (2s) e (2p) nao sao maisdegenerados, porque o potencial eletrostatico nao e mais coulombiano (veja Fig. 7.1).
Figura 7.1: Nıveis do helio e transicoes permitidas de singleto e tripleto. Note que o estado(1s)↑↑ nao existe.
Como ja vimos na discussao da estrutura fina, a energia do acoplamento l · s dada por (4.29)e ∝ En(Zα)2 ∝ Z4. Para helio que ainda tem um Z pequeno, a energia do acoplamento e fraca,e podemos contar com um acoplamento direito dos dois spins. Como as orbitas dos eletrons saoagora diferentes, podemos construir combinacoes de funcoes de onda espaciais ΨS,A simetricasou anti-simetricas, e portanto, combinacoes de spins XA,S anti-paralelos ou paralelos. Quandoos spins sao paralelos (S = 1), a funcao de onda espacial e anti-simetrica, quando sao anti-paralelos (S = 0), e simetrica. Da simetria da funcao de onda depende a energia da interacaocoulombiana intereletronica, pois no estado simetrico a distancia media dos eletrons e muitomenor do que no estado anti-simetrico, onde a funcao espacial total desaparece para distanciazero. Por consequencia, a configuracao (1s)1(2s)1 tem dois estados com S = 0, 1, com energiaES=0 > ES=1. Da mesma forma, todas as configuracoes sao desdobradas, como ilustrado naFig. 7.1. A diferencia de energias (∼ 1 eV) e consideravel e bem superior a energia da interacaofina (∼ 10−4 eV). Isso explica, porque primeiro os dois spins se acoplam para um spin total,s1 + s2 = S, antes ele acoplar-se com o momento angular orbital total, S + L = J. Isso e oacoplamento L · S.

7.2. HELIO 101
7.2.2.1 Energia de troca
A diferencia de energia dos dois estados S = 0, 1 se chama energia de troca. Ela sai de umcalculo de perturbacao de primeira ordem. Por exemplo, para o estado (1s)1(2s)1 escrevemosas funcoes de onda anti-simetrizadas
ΘA± = 1√
2[ψ100(r1)ψ200(r2)± ψ100(r2)ψ200(r1)] · χA,S , (7.26)
onde o signo (+) vale para χA (S = 0) e o signo (−) para χS (S = 1). As energias sao,
∆ES,A = 12
∫dr3
1
∫dr3
2Θ∗A±e2
4πε0|r1 − r2|ΘA,± (7.27)
= 12
∫dr3
1
∫dr3
2
e2
4πε0|r1 − r2|[|ψ100(r1)|2|ψ200(r2)|2 + |ψ100(r2)|2|ψ200(r1)|2]
± 12
∫dr3
1
∫dr3
2
e2
4πε0|r1 − r2|2ψ∗100(r1)ψ∗200(r2)ψ100(r2)ψ200(r1)
≡ ∆Ecoulomb ±∆Eexchange .
A primeira integral,
∆Ecoulomb =
∫dr3
1
∫dr3
2
e2
4πε0|r1 − r2||ψ100(r1)|2|ψ200(r2)|2 , (7.28)
e a energia de Coulomb entre os orbitais eletronicos. Notamos, que essa parte pode ser calculadaa partir do hamiltoniano por orbitais nao simetrizados. A segunda integral,
∆Eexchange =
∫dr3
1
∫dr3
2
e2
4πε0|r1 − r2|ψ∗100(r1)ψ∗200(r2)ψ100(r2)ψ200(r1) , (7.29)
chamada de energia de troca corresponde aos termos de interferencia da simetrizacao e devemser adicionados ou subtraıdos em funcao do caracter de simetria. E interessante notar, que nesteponto, o spin nao entra diretamente no hamiltoniano do helio,
HS,A =p2
1
2m+
p22
2m+ V (r1) + V (r2) + V (|r1 − r2|)±∆Eexchange , (7.30)
mas somente atraves do caracter de simetria da funcao de onda espacial. Do outro lado, numaescala de energia bem menor, o spin entra pela interacao L · S.
O potencial nao e esfericamente simetrico, o termo r12 depende do angulo entre r1 e r2.Assim, a funcao de onda total Ψ(r1, r2) nao e separavel em uma parte radial e outra angular, oque faz com que, diferentemente do hidrogenio, a equacao de Schrodinger com o hamiltoniano(7.20) nao possua solucao analıtica.
Exemplo 10 (TPIT para estados excitados do helio): Consideramos os dois eletrons deum atomo de helio ocupando orbitais diferentes descritos por funcoes de onda denotadas porψa(1) ≡ ψn1,`1,m`1
(r1) e ψb(2) ≡ ψn2,`2,m`2(r2). Se aplicarmos o hamiltoniano sem o termo
de interacao entre eletrons, os estados totais Θ = ψa(1)ψb(2) e ψa(2)ψb(1) possuem mesmaenergia Ea + Eb. Para calcularmos a correcao na energia, utilizamos a TPIT para estados
degenerados. Temos que calcular o determinante secular det(〈n, ν|H(1)|n, µ〉−E(1)n,µδµ,ν). Os

102 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
termos da matriz H(1) ficam:
H(1)11 = 〈ψa(1)ψ(2)| e2
4πε0r12|ψa(1)ψb(2)〉
H(1)22 = 〈ψa(2)ψb(1)| e2
4πε0r12|ψa(2)ψb(1)〉
H(1)12 = 〈ψa(1)ψb(2)| e2
4πε0r12|ψa(2)ψb(1)〉 = H
(1)21 .
Os termos J ≡ H(1)11 = H
(1)22 sao integrais de Coulomb. Ja o termo K ≡ H
(1)12 e denominado
integral de troca:
K =e2
4πε0〈ψa(1)ψb(2)| 1
r12|ψa(2)ψb(1)〉 .
Assim como J e K sao positivos, determinante fica:∣∣∣∣J − E KK J − E
∣∣∣∣ = 0 ,
dando,E(1) = J ±K .
Ou seja, os estados que antes eram degenerados com energia E = Ea +Eb sao quebrados emdois estados com energias E = Ea + Eb + J ±K. E as autofuncoes correspondentes sao:
ΨS,A(1, 2) =1√2
[ψa(1)ψb(2)± ψb(1)ψa(2)] .
Esse resultado mostra que a repulsao entre os dois eletrons quebra a degenerescencia (das
funcoes separaveis em forma de produto) em estados com diferenca de energia 2K. Note que
as autofuncoes estao simetrizadas, o que e abordado na secao seguinte.
7.2.2.2 O espectro do helio
Ate agora vimos que, se os eletrons estao no mesmo orbital, temos um termo para energiaE = 2Ea+J e, quando estao em orbitais diferentes, temos E = Ea+Eb+J±K, com separacaoentre nıveis de 2K.
Na pratica, consideramos apenas a excitacao de um eletron pois a energia para excitar osdois eletrons excede a energia de ionizacao do atomo de helio. Para encontrarmos as regras deselecao para transicao entre estados simetrico e assimetrico, calculamos o momento de dipoloda transicao. Para um sistema de dois eletrons o momento de dipolo vale d = −er1 − er2, quee simetrico pela permutacao de dois eletrons. O momento de dipolo para transicao fica:
d± = −e∫
Ψ∗A(r1, r2)(r1 + r2)ΨS(r1, r2)dV1dV2 . (7.31)
Se permutarmos os eletrons, a integral acima muda de sinal, mas a integral nao pode depender danomenclatura das variaveis de integracao, portanto deve ser nula. Nao pode ocorrer a transicaoentre os estados simetrico e antissimetrico. Considerando a funcao de onda do spin, Θ = ΨSχA
ou ΨAχS , temos que as transicoes so sao permitidas entre estados de singleto ou entre estadosde tripleto. Ou seja, existe uma regra de selecao do spin postulando ∆S = 0 7,8.
Nas primeiras observacoes do espectro do helio, visto as diferencas do espectro de singleto e doespectro de tripleto, acreditava-se que se tratava de diferentes atomos. Porem, analises quımicasmostraram que era o mesmo elemento. Passaram a acreditar que existiam dois tipos de atomosde helio, os quais foram denominados para-helio e orto-helio, como ilustrado na Fig. 7.1.
7Alem disso, transicoes entre os estados 1S0 e 3S1 sao impossıveis, porque violam a regra de selecao do momentoangular, ∆L = ±1.
8Podemos entender as regras de selecao da seguinte maneira: Enquanto a funcao de onda pode ser escrito

7.3. ESTRUTURA DA CASCA ELETRONICA 103
7.3 Estrutura da casca eletronica
A interacao intereletronica e a necessidade de antissimetrizar a funcao de onda dos eletronsambas contribuem para aumentar excessivamente a complexidade de atomos multieletronicos.O Hamiltoniano descrevendo um atomo multieletronico de numero atomico Z,
H = Ekin + Vncl−ele + Vele−ele =
Z∑
i=1
p2i
2m−
Z∑
i=1
Ze2
4πε0|r− ri|+
Z∑
i<j=1
e2
4πε0|ri − rj |, (7.32)
e extremamente complicado para resolvermos, mesmo para o caso mais simples (Z = 2) devemosutilizar metodos de aproximacao.
Note, que caso assumıssemos eletrons independentes (Vele−ele = 0), isto e, cada eletronse move independentemente dos outros dentro do potencial eletrostatico gerado pelo nucleoe os outros Z − 1 eletrons, o problema seria facilmente soluvel: Resolverıamos a equacao deSchrodinger para um estado produto de todas funcoes de onda dos eletrons e conhecerıamos asautofuncoes e autoenergias individuais de cada eletron (tipo atomo de hidrogenio). Em principio,deverıamos usar funcoes de onda anti-simetricas, mas como primeira abordagem poderıamos sorespeitar o princıpio fraco de Pauli, isto e, atribuir um conjunto individual e unico de numerosquanticos para cada eletron. A energia total seria a soma da energia de cada eletron e osautoestados fısicos associados seriam obtidos mediante a antissimetrizacao do produto tensorialdo estado multieletronico.
Exemplo 11 (Gas de Fermi): Consideramos um poco de potencial infinito que nos en-chemos gradualmente com eletrons. O principio de Pauli nos permite colocar no maximodois eletrons em cada orbital,
Ψ = ψ1,↑(x1)ψ1,↓(x2)ψ2,↑(x3)ψ2,↓(x4) · .. .
Essa funcao de onda total satisfaz o princıpio fraco de Pauli, mas obviamente nao e anti-simetrica. A aproximacao e boa, quando a interacao entre os eletrons e desprezıvel. Casocontrario, precisamos considerar os termos de energia de troca.Esse modelo, chamado modelo do gas de Fermi, e frequentemente utilizado para descrever ocomportamento de eletrons que podem mover-se livremente dentro da banda de condutanciade um metal. Seja N o numero de eletrons colocado no potencial sucessivamente enchendotodos os orbitais de numero quantico n baixo para o numero maximo nmax = N/2, se cadaorbital pode aguentar dois eletrons com spins antiparalelos. Para um poco unidimensionalde comprimento L, a energia maxima, chamada de energia de Fermi e,
EF ∝N2
L2.
Para um poco tres-dimensional de volume V ,
EF ∝(N
V
)2/3
.
como um produto, Θ = Ψ(x)χ(s), o caracter de simetria e preservado para as duas funcoes separadamente. Osauto-valores dos operadores Px e Ps sao entao numeros quanticos bons. Mas isso so vale, quando o acoplamentoL · S e fraco. O operador eletrico dipolar de transicao nao age sobre o spin (o que impede o reacoplamentoS = 1 ↔ S = 0 por radiacao E1) e tambem nao age sobre o caracter de simetria dos orbitais (o que impedetransicoes ΨS ↔ ΨA).Em princıpio, isso vale para cada especie de atomos com dois eletrons de valencia. Mas na realidade a influenciado acoplamento L · S vai crescendo com Z, o que renda a interdicao de intercombinacao mais fraca. Nesse caso,so o operador Px,s da bons auto-valores.

104 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
7.3.1 Modelo de Thomas-Fermi
Mesmo se o potencial sentido pelos eletrons e bem diferente do poco tres-dimensional, podemosaproximadamente nos imaginar que o atomo e subdividido em pequenos volumes, todos enchidoscom eletrons seguinte o modelo de gas de Fermi. Disso podemos calcular a distribuicao da cargaeletronica, tal que a energia local media e homogenea e a nuvem eletronica em equilıbrio. Adistribuicao, em torno, serve para determinar a forma do potencial eletrostatico que, quandosubdividido em pequenos volumes cheios de eletrons produz a mesma distribuicao de carga. Esseprincıpio se chama auto-consistencia.
As funcoes de onda (distribuicoes espaciais dos eletrons) determinadas por esse metodofrequentemente servem como ponto inicial para o metodo de Hartree discutido embaixo. Umadas predicoes importantes do modelo de Thomas-Fermi e que o raio medio de um atomo dependeda carga nuclear como R ∝ Z−1/3.
O modelo de Thomas-Fermi permite entender a configuracao eletronica dos estados funda-mentais e fornece a base para o sistema periodico dos elementos. Nesse modelo, os eletronssao tratados como partıculas independentes, de um lado formando um potencial eletrico radialefetivo, do outro lado sendo sujeitos a esse potencial. Em vez de requer anti-simetria da funcaode onda, so e necessario garantir que todos os eletrons se distinguem em pelo menos um numeroquantico. As funcoes de atomos complexos sao parecidos as funcoes do hidrogenio. Podemosutilizar os mesmos numeros quanticos n, `, m` e ms para cada eletron.
No entanto, o potencial radial efetivo depende muito da especies e e bem diferente do po-tencial coulombiano. Portanto, a degenerescencia em ` e quebrada. Em geral, os eletrons compequenos ` sao ligados mais fortemente, porque eles tem uma probabilidade maior de ser pertodo nucleo, onde o potencial e mais profundo (vide Fig. 7.7). O mesmo argumento explique, por-que eletrons com pequenos n sao ligados mais fortemente. Discutiremos estes efeitos com maisprofundidade na Sec. 7.4.2 por uma comparacao dos nıveis de excitacao do eletron de valenciados alcalis.
7.3.1.1 Abordagem TPIT
Assim, como primeira aproximacao utilizamos os autoestados de eletrons individuais (apro-ximacao orbital) e considerar Vele−ele(|ri − rj |) como uma perturbacao fazendo uso da teoriade perturbacao independente do tempo. Entretanto, este termo nao e pequeno o suficiente parajustificar este procedimento, pois aproximando,
Vncl−ele 'Z2e2
aBe Vele−ele '
Z(Z − 1)e2
2aB, (7.33)
percebemos, que Vele−ele/Vncl−ele varia entre 14 para Z = 2 e 1
2 para Z 1/2. Desta forma enecessario o uso de metodos alternativos para descrever atomos multieletronicos.
O modelo de Thomas-Fermi e um modelo semi-classico que visa descrever, de modo aproxi-mado, a energia total dos eletrons como um funcional da densidade de eletrons atomicos/moleculares.Tal modelo e a base para metodos mais sofisticados que visam a obtencao da estrutura eletronica,como a density functional theory (DFT).
7.3.1.2 Gas de eletrons
Dividimos agora o atomo em pequenos volumes de lado L (celulas), contendo N eletrons nao-interagentes uniformemente distribuıdos e cujo numero total e Nt, e analisemos cada celulaindividualmente. O volume pode ser modelado pelo seguinte potencial: V (r) = 0 para 0 ≤

7.3. ESTRUTURA DA CASCA ELETRONICA 105
x, y, x ≤ L e V (r) = ∞ em todos outros lugares. Neste caso reconhecemos os possıveis estados|nx, ny, nz〉 com nx, ny, nz = 1, 2, 3 e as energias de um unico eletron
Enx,ny ,nz =π2~2
2meL2(n2x + n2
y + n2z) . (7.34)
Para N eletrons no estado fundamental cada partıcula ocupa o estados menos energetico dis-ponıvel, obedecendo o princıpio de exclusao de Pauli e considerando-se o spin. A energia total edada pela soma das energias dos N estados menos energeticos, e o estado fısico final e dado pelaantissimetrizacao do estado correspondente. A energia do N -esimo eletron (o mais energetico)e chamada de Energia de Fermi (EF ) 9.
E de interesse calcularmos o numero de estados existentes n(E) cuja energia sao menoresque E. Para isso escrevemos a energia da seguinte forma
Enx,ny ,nz =~2
2mek2nx,ny ,nz , (7.35)
onde knx,ny ,nz = k2x+k2
y +k2z = (nxπL )2 + (
nyπL )2 + (nzπL )2. Cada trinca de valores k = (kx, ky, kz)
corresponde a um possıvel estado acessıvel ao sistema, e cada estado esta associado a um ele-mento de volume (π/L)3 no espaco k. Note que o numero de estados n(E) com energia menorque E e dado pelo numero de estados em que k ≤
√2meE/~2. Isso corresponde a uma esfera
de raio√
2meE/~2 centrada na origem, logo
n(E) = 218
[4π3
(2meE
~2
)3/2]
1
(π/L)3=
L3
3π2
(2meE
~2
)3/2
. (7.36)
A Fig. 7.2 ilustra os possıveis estados no espaco k e a esfera com energia E.
Figura 7.2: Estados disponıveis para uma caixa de potencial.
Com a expressao anterior somos capazes de calcular a energia de Fermi, pois a condicaon(EF ) = N implica,
EF =~2
2me
(3π2 N
L3
)2/3
. (7.37)
E interessante notar que para T = 0 K todos os estados abaixo de EF sao ocupados e todosaqueles acima estao desocupados. Ademais, definimos a densidade de estados ρ(E) onde ρ(E)dEe o numero de estados com energias entre E e E + dE. Assim, a densidade dos estados fica,
ρ(E) =dn(E)
dE=
L3
2π2
(2me
~2
)3/2
E1/2 =3N
2
E1/2
E3/2F
. (7.38)
9Note, entretanto, que para N grande (1 mol de eletrons) esse processo se torna inviavel.

106 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
7.3.1.3 Energia de Thomas-Fermi
A energia cinetica media dos eletrons com o sistema no estado fundamental e,
Ec =
∫ EF
0Eρ(E)dE =
L3
2π2
(2me
~2
)3/2 ∫ EF
0E3/2dE (7.39)
=L3
2π2
(2me
~2
)3/225E
5/2F
=~235/3π4/3
10meL3
(N
L3
)5/3
= CL3ρ5/3 ,
onde ρ ≡ N/L3 e a densidade de eletrons por unidade de volume. Logo,
Nt =
∫ρ(r)d3r . (7.40)
Sendo assim, a densidade de energia cinetica e dada por,
ukin(r) = Cρ5/3 . (7.41)
Portanto, a energia cinetica total dos eletrons na camada eletronica e,
T [ρ] = C
∫ρ5/3d3dr . (7.42)
Os potenciais associados a interacao e−-e− e e−-p+ tambem podem ser escritos como funcionaisda densidade eletronica, tal que,
Vep[ρ] = − Ze2
4πε0
∫ρ(r)
rd3r e Vee[ρ] =
1
2
e2
4πε0
∫ρ(r)ρ(r′)|r− r′| d
3rd3r′ . (7.43)
Logo, a energia total (energia de Thomas-Fermi) pode ser escrita como um funcional da densi-dade eletronica do atomo,
HTH [ρ] = T [ρ] + Vncl−ele[ρ] + Vele−ele[ρ] . (7.44)
7.3.1.4 Densidade eletronica e equacao de Thomas-Fermi
Levando-se em conta o princıpio variacional, estamos interessados na densidade eletronica ρ(r)que minimiza a energia de Thomas-Fermi. Podemos realizar este processo via multiplicadoresde Lagrange, com a restricao de que o numero de eletrons se mantenha constante no atomo.Assim,
δ
HTH [ρ]− µ
(∫ρ(r)d3r −Nt
)= 0 (7.45)
δHTH [ρ]− µδρ(r) = 0 =⇒ δHTH [ρ]
δρ(r)= µ .
Inserindo a energia de Thomas-Fermi (7.44) calculamos,
µ = 53Cρ
2/3 − ξef (r) com ξef (r) =e2
4πε0
Z
r− e2
4πε0
∫ρ(r′)|r− r′|d
3r′ . (7.46)

7.3. ESTRUTURA DA CASCA ELETRONICA 107
Resolvendo pela densidade eletronica,
ρ(r) =
[µ+
3
5Cξef (r)
]3/2
. (7.47)
A expressao anterior descreve a densidade eletronica do atomo no estado fundamental. E inte-ressante notar que, como δHTH [ρ]/δρ(r) = µ, podemos identificar o multiplicador de Lagrange µcomo um potencial quımico. Em particular, para atomos neutros nao-interagentes tem-se µ = 0,assim,
ρ(r) =
(3
5C
)3/2
ξef (r)3/2 . (7.48)
Ademais, como para um atomo tanto o potencial quanto a densidade eletronica deve possuirsimetria esferica, podemos escrever
ξef (r) =Zχ(r)
r. (7.49)
Agora, e comum realizar a transformacao de variavel x = αr (α = 27/3Z1/3/(3π)2/3 = 1.1295Z1/3)e utilizar unidades atomicas, em que ~2/me = 1 tal que C = 35/3π4/3/10 = 2.871 [1]. Assim, a’densidade eletronica’ e,
ρ(x) =Z225
32π3
(χ(x)
x
)3/2
. (7.50)
Alem disso, o potencial deve satisfazer a equacao de Poisson ∇2ξef = 4πρ, resultando na equacaode Thomas-Fermi:
d2χ
dx2=χ3/2
x1/2. (7.51)
E importante notar que a equacao anterior nao depende do parametro Z, sendo assim umresultado geral para qualquer atomo neutro. A funcao χ(x) e determinada numericamente, maspodemos analisar seus valores assintoticos dado o comportamento esperado do potencial efetivoξef (r): para r → 0 espera-se que ξef (r) = Z
r , assim χ(0) = 1; ja para r → ∞ espera-se queξef (r) = 0, assim χ(∞) = 0.
Com χ(x) em maos obtemos a densidade de carga ρ(x) e, portanto, somos capazes de calculara energia total do atomo em questao. Assim, e possıvel mostrar que [1],
HTH [ρ] = −0.7687Z7/3 . (7.52)
E importante ressaltar alguns pontos: (i) este e um resultado para atomos neutros; (ii) nao hacamadas eletronica; (iii) nao e levado em conta o efeito de partıculas identicas. O modelo deThomas-Fermi-Dirac e um modelo mais refinado que lida com o item (iii) e, alem disso, e maisproximo ao DFT.
7.3.2 Metodo de Hartree
Para calcular a maioria das propriedades de um atomo precisamos de potenciais mais realısticos.Os termos mais importantes sao o potencial coulombiano entre o nucleo e os eletrons, Vncl−ele,naturalmente sendo esferico, e os potenciais de interacao entre os eletrons, Vele−ele, que tentamosaproximar por um potencial esferico e tratar os desvios causados pela aproximacao depois.

108 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
Conhecendo o efeito da blindagem do nucleo pelas cargas eletronicas, ja sabemos as assimptotas(vide Fig. 7.7),
V0 = − Ze2
4πε0rpara r → 0 e V0 = − e2
4πε0rpara r →∞. (7.53)
Um potencial efetivo, V0, construıdo para satisfazer esses limites serve como primeiro chute paraestabelecer e resolver numericamente a equacao de Schrodinger para cada eletron independen-temente,
Hi =
(− ~2
2m∇2i + V0
)ψi(ri) = eiψi(ri) . (7.54)
Com isso, calculamos todas as energias e auto-funcoes (so as partes radiais interessam) mini-mizando a energia total e respeitando o princıpio fraco de Pauli, isto e, todos os estados saosucessivamente enchidos com eletrons. Para a funcao de onda total obtemos,
(N∑
i=1
Hi
)ΨN = EnΨN com ΨN = ψ1 · .. · ψN e En =
N∑
i=1
ei . (7.55)
Com as auto-funcoes calculamos as densidades de carga e|ψj(rj)|2. Integramos o campo paraobter um potencial que representa uma estimacao melhorada para o campo eletronico medio,
Vi ←− −Ze2
4πε0ri+∑
j 6=i
∫d3rj
e2
4πε0|ri − rj ||ψj(rj)|2 . (7.56)
Substituımos esse potencial na equacao de Schrodinger, e comecamos todo o processo de novo.Esse metodo auto-consistente se chama metodo de Hartree. Fock melhorou esses calculos usandofuncoes de onda anti-simetricas para os eletrons de valencia. Esse metodo se chama metodo deHartree-Fock. A ideia do metodo de Hartree esta visualizada no seguinte diagrama,
escolhe um potencial comum V (r) paratodos eletrons, p.ex. Thomas-Fermi
↓calcule as auto-funcoes ψ
(0)i com(
−~2
2m ∆i + V0(ri))ψ
(0)i = e
(0)i ψ
(0)i
↓←− ψi
nova entrada←−↓ ↓ ↑↓ classifica seguinte energias crescentes ei ↑↓ satisfazendo o princıpio de Pauli ↑↓ ↓ ↑↓ calcule o potencial medio Vi agindo ↑↓ sobre eletron i para todos i ↑↓ Vi(ri) =
∑j 6=i∫
Ze4πε0rij
|ψi(ri)|2d3rj ↑↓ ↓ ↑↓ calcule novos ψi com esses Vi ↑↓
(−~2
2m ∆i − Ze4πε0ri
+ Vi(ri))ψi = eiψi ↑
↓ ↓ ↑−→ compare com ψi inicial
ruim−→↓ bom
resultado

7.3. ESTRUTURA DA CASCA ELETRONICA 109
7.3.3 Metodo de Hartree Fock
O metodo de Hartree-Fock e um metodo utilizado para tratar sistemas atomicos/moleculares demuitos corpos que visa a obtencao da funcao da onda eletronica do sistema. Ele e um refinamentodo Metodo de Hartree (SCF: Self-Consistent-Field), pois leva em conta a antissimetrizacao dafuncao de onda. De maneira geral o metodo se baseia no princıpio variacional e na suposicaode que podemos escrever a funcao de onda global como um determinante de Slater, sendo quecada eletron ocupa um estado orbital especıfico (spin-orbital) e interage com um potencial efe-tivo oriundo dos eletrons que ocupam outros orbitais. Desta maneira, ao inves de resolvermosa equacao de Schrodinger devemos resolver um sistema de equacoes denominada de equacoesde Hartree-Fock do tipo Fψk(1) = εkψk(1). O metodo e realizado de maneira cıclica ate con-vergencia dos orbitais atomicos e suas respectivas energias, por este motivo e denominado comoauto-consistente: a partir da suposicao inicial da funcao de onda global e calculado o potencialefetivo em cada orbital e e obtido um novo conjunto de funcoes de onda que, por sua vez, ge-ram um novo potencial efetivo; assim, este novo potencial e utilizado em um novo sistema deequacoes de Hartree-Fock. A Fig. 7.3 ilustra a caracterıstica cıclica do metodo.
Aula Estrutura da Camada Eletrônica
2 Método de Hartree-Fock -
2.1 Aspectos Gerais -
O Método de Hartree-Fock é um método utilizado para tratar sistemas atômicos/moleculares de
muitos corpos que visa a obtenção da função da onda eletrônica do sistema. Ele é um refinamento
do Método de Hartree (SCF: Self-Consistent-Field), pois leva em conta a antissimetrização da
função de onda. De maneira geral o método se baseia no princípio variacional e na suposição
de que podemos escrever a função de onda global como um determinante de Slater, sendo que
cada elétron ocupa um estado orbital específico (spin-orbital) e interage com um potencial efetivo
oriundo dos elétrons que ocupam outros orbitais. Desta maneira, ao invés de resolvermos a equação
de Schrödinger devemos resolver um sistema de equações denominada de Equações de Hartree-
Fock do tipo Fψk(1) = εkψk(1). O método é realizado de maneira cíclica até convergência dos
orbitais atômicos e suas respectivas energias, por este motivo é denominado como auto-consistente:
a partir da suposição inicial da função de onda global é calculado o potencial efetivo em cada orbital
e é obtido um novo conjunto de funções de onda que, por sua vez, geram um novo potencial efetivo;
assim, este novo potencial é utilizado em um novo sistema de equações de Hartree-Fock. A Figura
2.1 ilustra a característica cíclica do método.
Definição dosspin-orbitais
Cálculo dos PotenciaisResolução das Equa-ções de Hartree-Fock
Cálculo dos po-tenciais com os
novos spin-orbitaisConvergência?
Fim
Sim
Não
Fig. 2.1: Método de Hartree-Fock.
2.2 Equações de Hartree-Fock -
Vimos que o Hamiltoniano de um átomo multieletrônico é dado por
H =
Z∑
i=1
[p2i
2mi− Ze2
4πε0ri
]+
1
2
∑
i6=j
e2
4πε0|rj − ri|
=
Z∑
i=1
hi +1
2
∑
i6=jVji
8
Figura 7.3: Metodo de Hartree.
7.3.3.1 Equacoes de Hartree-Fock
Vimos que o Hamiltoniano de um atomo multieletronico e dado por,
H =
Z∑
i=1
[p2i
2mi− Ze2
4πε0ri
]+ 1
2
∑
i 6=j
e2
4πε0|ri − rj |=
Z∑
i=1
hi + 12
∑
i 6=jVij , (7.57)
onde hi e o Hamiltoniano de um corpo associado somente ao eletron i e Vij e o termo de interacaoentre os eletrons i e j. Para realizacao do metodo e necessario supor que o estado multieletronicopode ser escrito como o produto dos estados individuais de cada eletron:
Ψ′(1, ..., Z) = ψ1(1)ψ2(2)...ψZ(Z) , (7.58)
onde ψi(1) = φi(r1χ(α) = ψαi (r1) representa o estado spin-orbital do eletron 1, ou seja, a funcaode onda espacial do eletron no estado i e com spin α. Entretanto, devido ao postulado desimetrizacao o estado fısico do sistema deve ser expresso pelo determinante de Slater,
Ψ(1, ..., Z) =1√Z!
det [ψ1(1)ψ2(2)...ψZ(Z)] . (7.59)

110 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
Com isso em maos utilizamos o princıpio variacional para minimizar o valor esperado da energiano estado fundamental mediante alteracao das funcoes ψk(n). Desta forma, os orbitais corretossao aqueles que minimizam a energia. O valor esperado e escrito como,
E = 〈Ψ|H|Ψ〉 = 〈Ψ|Z∑
i=1
hi|Ψ〉+ 〈Ψ|12∑
i 6=jVij |Ψ〉 . (7.60)
E possıvel mostrar que,
〈Ψ|Z∑
i=1
hi|Ψ〉 =Z∑
i=1
〈ψi|hi|ψi〉 e (7.61)
〈Ψ|12∑
i 6=jVij |Ψ〉 = 1
2
Z∑
i,j
[〈ψiψj |Vij |ψiψj〉 − 〈ψjψi|Vij |ψjψi〉] .
Assim,
E =
Z∑
i=1
hi|Ψ〉+ 12
Z∑
i,j
[〈ψiψj |Vij |ψiψj〉 − 〈ψjψi|Vij |ψjψi〉] . (7.62)
A expressao anterior pode ser minimizada via multiplicadores de Lagrange, com o vınculo dadopela ortogonalidade dos estados 〈ψi|ψj〉 = δij ,
δ
〈Ψ|H|Ψ〉 −
∑
i,j
εij [〈ψiψj |Vij |ψiψj〉 − 〈ψjψi|Vij |ψjψi〉]
(7.63)
Assim, obtemos o seguinte conjunto de equacoes de Hartree-Fock:
h1 +
∑
i
(2Ji − Ki)
ψk(1) = εkψk(1) (7.64)
Fψk(1) = εkψk(1) ,
onde, F = h1 +∑
i(2Ji − Ki) e o operador de Fock e εk e a energia associada ao spin-orbital
ψk. O operador Ji, denominado operador de Coulomb, representa o potencial medio sentido pelapartıcula 1 no orbital k devido ao eletron 2 no orbital i:
Jiψk(1) =
∫ψ∗i (2)V12ψi(2)dr2
ψk(1) . (7.65)
Ja o operador Ki, denominado operador de troca, e consequencia do processo de simetrizacao e,portanto, e um efeito puramente quantico, ou seja, sem analogo classico:
Kiψk(1) =
∫ψ∗i (2)V12ψk(2)dr2
ψk(1) . (7.66)
Uma vez com todas as funcoes de onda as energias dos orbitais podem ser obtidas da seguinteforma: ∫
dr1ψ∗k(1)
h1 +
∑
i
(2Ji − Ki)
ψ∗k(1) = εk
∫dr1ψ
∗k(1)ψk(1) = εk , (7.67)

7.3. ESTRUTURA DA CASCA ELETRONICA 111
ou seja,
εk =
∫dr1ψ
∗k(1)h1ψ
∗k(1) +
∑
i
(2Jki − Kki) , (7.68)
onde,
Jki =
∫dr1ψ
∗k(1)Jiψk(1) e a integral de Coulomb (7.69)
Kki =
∫dr1ψ
∗k(1)Kiψk(1) e a integral de troca .
Ja a energia atomica total pode ser calculada por,
E = 2∑
k
εk −∑
k,i
(2Jki − Kki) . (7.70)
Ademais, se assumirmos que se ao tirarmos um eletron do orbital ψk a distribuicao eletronicapermanece inalterada e possıvel associar a energia εk com a energia de ionizacao do eletron nesteorbital. Tal igualdade e conhecida como teorema de Koopman,
Ik ' εk . (7.71)
7.3.3.2 Equacoes de Hartree-Fock-Roothaan
Por fim, vale ressaltar que um refinamento do processo pode ser obtido se expandirmos cadaspin-orbital em uma determinada base de funcoes (nao necessariamente ortogonais), tais que,
ψk(n) =∑
l
Clkφl(n) , (7.72)
onde N e o numero de funcoes na base. Assim, as equacoes de Hartree-Fock podem ser escritascomo,
Fψk(1) = εkψk(1) =⇒ F
N∑
l
Clkφl(1) = εk
N∑
l
Clkφl(1) (7.73)
=⇒N∑
m
Clk
∫dr1φ
∗m(1)F φl(1) = εk
N∑
l
Clk
∫dr1φ
∗m(1)φl(1)
=⇒N∑
m
FmlClk = εk
N∑
l
SmlClk .
Em representacao matricial a expressao anterior e dada pela equacao de Hartree-Fock-Roothaan,
FC = SCε , (7.74)
onde ε e a matriz diagonal contendo as energias orbitais.

112 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
7.4 O sistema periodico dos elementos
Camadas principais cheias n, ` sao isotropicas, ΨN (r) = Ψ(r). Orbitas com pequenos n percebemmenos blindagem, as orbitas deles sao aproximadamente rn ' rH,n/(Z−2). Orbitas com grandesn sao blindadas, as orbitas deles sao aproximadamente rn ' naB. Seguinte o modelo de Bohr, asenergias em potenciais −1/r sao degeneradas a respeito de `. O desvio do potencial coulombianoem comparacao com essa lei, causado por blindagem em sistemas de muitos eletrons, quebraa degenerescencia e diminuı a energia consideravelmente para pequenos `. Para grandes ` otermo centrifuga domina a parte coulombiana e a diminuicao e muito menor. Por exemplo, noatomo de sodio o orbital 3d chega a ser mais baixo do que o 4s. Ao longo do sistema periodicodos elementos, as orbitas sao consecutivamente enchidas com eletrons seguinte essas energiasdeslocadas.
Figura 7.4: Ilustracao da regra de Hund.
E importante distinguir tres sequencias energeticas diferentes: 1. A Tab. 7.6 mostra, paraum dado atomo, as orbitas excitadas do ultimo eletron. 2. A sequencia energetica mostrada naTab. 7.4 fala em qual orbita o proximo eletron sera colocado, quando vamos para o proximoatomo na Tabela periodica 7.10. 3. A sequencia energetica dos eletrons interiores. Enquantopara os eletrons interiores achamos,
En,l < En,l+1 En+1,l , (7.75)
a sequencia esta parcialmente invertida para o ultimo eletron. Mas isso e essencial, porque eo estado dos ultimos eletrons que determinam a reatividade quımica do atomo. A sequencia eilustrada na Fig. 7.8.
Seguinte a regra de Hund o acoplamento L · S e energeticamente favoravel em comparacaocom o acoplamento j · j, o que significa que os spins dos ultimos eletrons, isto e, os eletrons forade sub-camadas (n, `) cheias, preferem orientar os seus spins em paralelo para anti-simetrizar as

7.4. O SISTEMA PERIODICO DOS ELEMENTOS 113
funcoes de onda espaciais e maximizar a distancia entre os eletrons. Cada sub-camada da serieilustrada na Fig. 7.8 deve ser enchida na ordem indicada antes de colocar eletrons na proximacamada.
Gases nobres tem pequenos raios, altas energias de excitacao e energias de ionizacao. Oeletron de valencia deve preencher a lacuna ate numeros quanticos principais mais altos. Ha-logenios tem fortes eletroafinidades, pois a camada eletronica exterior (nmax) e incompleta eportanto maleavel, tal que um eletron se aproximando ao halogenio percebe uma blindagemdo nucleo parcialmente transparente. Alcalis sao semelhantes ao hidrogenio e tem energias deexcitacao opticas. O estado fundamental deles 2S1/2 e determinado por um eletron de valenciaso no orbital ` = 0. Diferentemente do hidrogenio, as energias de excitacoes dependem muitode `, pois orbitas pequenas ` tem mais probabilidade na regiao nao blindadas −Z2e2/r do emorbitas com grandes `, quem demoram mais tempo na regiao blindadas −e2/r. Com a mesmarazao, energias que correspondem a maiores n sao semelhantes do espectro de hidrogenio.
A estrutura de camadas inteiras dos atomos pode ser analisada por espalhamento de raiosX. Eletrons desacelerados por atomos emitem um espectro continuo chamado Bremsstrahlung,mas tambem podem expulsar eletrons das camadas interiores. Quando um buraco e recheadopor cascatas de eletrons vindo de camadas superiores, o atomo emite um espectro de raio Xespecifico (≈ 104 eV). As regras de selecao ∆` = ±1 e ∆j = ±1 desdobram as linhas in doisem dois componentes. Espectros de raios X de elementos vizinhos no sistema periodico doselementos sao muito parecidos, pois as camadas interiores nao sao blindadas com um potencialaproximadamente ∝ Z2/r. Portanto, a dependencia Z das linhas e mais ou menos ω ∝ Z2,como previsto pelo modelo do atomo de Bohr.
7.4.1 Modelo de camadas eletronicas
Para poder dizer algo sobre a estrutura geral de eletrons multiplos, consideramos o hamiltonianode um sistema eletronico multiplo,
Z∑
i
p2i
2m− Ze2
4πε0
1
ri+∑
i>j
e2
4πε0
1
|ri − rj |, (7.76)
consistindo em energia cinetica, energia potencial do nucleo e interacao entre eletrons Hww. Oındice cita os eletrons e corre de 1 ate o numero de carga nuclear Z. Negligenciamos a interacaoeletron-eletron Hww. Entao, cada eletron percebe um atomo de hidrogenio com a solucao αsubstituıda por Zα, porque o potencial de Coulomb do nucleo e agora,
Ze2
4πε0
1
r=Zα~cr
. (7.77)
Para as energias obtemos entao:
E = −Z∑
i
me2 c
2
(Zα
ni
)2
. (7.78)
Cada um dos nıveis de energia nesta soma contem varios estados. Cada um desses estadospode ser ocupado por um unico eletron, de acordo com o princıpio Pauli. Desta forma, obtem-se a configuracao eletronica para os atomos do sistema periodico. Nesta imagem, as energiasdos estados fundamentais dos elementos normalizadas com 1/Z2 pode ser escrita no seguinteesquema:

114 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
N
M
L
K 1s
4 4 4 4
3 3 3
2 2
s p d f
s p d
s p
19 20
11 12 13 14 15 16 17 18
3 4 5 6 7 8 9 10
1 2
K Ca
Na Mg Al Si P S Cl Ar
Li Be B C N O F Ne
H He
Figura 7.5: Ordem periodica.
Isso so funciona para atomos com ate 18 eletrons. Quando o camada 3p e completamentepreenchida, a proxima a ser ocupada nao e 3d mas a 4s. O novo esquema e ilustrado aqui.
As anomalias comecando em Z = 18 surgem devido a interacao eletron-eletron. Para enten-der isso, consideramos um eletron no campo medio de todos os outros eletrons. O eletron estaentao localizado em um potencial efetivo que transita entre dois potenciais de Coulomb paradistancias grandes e pequenas: para pequenas distancias, o eletron ve apenas a carga pontualpositiva do nucleo e o potencial e proporcional a Zα,
V (r) = − Ze2
4πε0
1
r. (7.79)
Para grandes distancias, o nucleo e todos os outros eletrons formam uma pequena fonte de cargaquase esferica chamada ’core’ e podemos aproximar o potencial por
V (r) = − e2
4πε0
1
r. (7.80)
O potencial verdadeiro transita de um para o outro potencial quando aumentamos a distanciado nucleo:
Perto do nucleo, os eletrons blindam o nucleo menos do que para grandes r. Assim, aquelesestados que tem uma baixa probabilidade perto do nucleo sao energeticamente mais altos. Istoe,
E2s < E2p e E6s < E6p < E6d . (7.81)
A degenerescencia do momento angular orbital do modelo de Schrodinger e assim cancelada. Oefeito de blindagem e, como pode ser visto no exemplo dos estados excitados do lıtio, um grandeefeito na gama de alguns eV.
A blindagem conduz de forma semelhante as anomalias de configuracao no sistema periodico,como p.ex. no K ou no Ca. Desde que E4s < E3d, o estado 4s e o primeiro a ser preenchido.Anomalias semelhantes tambem ocorrem em Rb (5s), Cs (6s) e Fr (7s). Nas terras raras o efeitode blindagem e ainda mais pronunciado. Aqui, a energia do estado 6s e abaixo mesmo da energia4f. As camadas 6s, 5s, 5p e 5d protegem assim a camada 4f muito bem 10.
10Um exemplo disto e o Nd:YAG (Neodimio em Yttrium Alumınio Garnet). Neste cristal, as transicoes opticasno Nd dentro da camada 4f podem ser excitadas. No entanto, essas transicoes so sao permitidas por perturbacoesdo campo cristalina. A blindagem muito forte leva a uma longa vida do estado excitado. Como resultado, estecristal e excelente como um material a laser.

7.4. O SISTEMA PERIODICO DOS ELEMENTOS 115
S P D F
4
2
-2
Li Na CsK
S P D F S P D F S P D F S P D F
-1
0
-3
-4
-5
Rb H
55
444
3
3 3
2
4
3
55
45
4
4 3
3
4
4
6
556
4
5
5
3
4
4
6
5
6
8
8
7
7
5
6
4
75
5
6
7
6
5
4
5
2
3
4
5
67
6 7
Figura 7.6: Comparacao das energias de excitacao do eletron de valencia para varios atomosalcalinos.
7.4.2 Alcalinos
Os alcalinos consistem em uma camada de gas nobre completa e um eletron de valencia adicional.O seu espectro e, portanto, fortemente semelhante ao hidrogenio. Uma abordagem empırica eusada para descrever isso,
En,l = −12µEGc
2 c2
n−∆(n, l), (7.82)
onde µEG e a massa reduzida em relacao ao casco de gas nobre e ∆(n, l) e o chamado defeitoquantico. Ele e tabulado para a maioria dos estados alcalinos e e particularmente importantepara estados de baixa energia. Para o sodio, os valores sao, por exemplo:
L n = 3 n = 4 n = 5 n = 6
s 1.37 1.36 1.35 1.34
p 0.88 0.87 0.86 0.86
d 0.10 0.11 0.13 0.11
f - 0.00 -0.01 0.008
Para estados com um grande momento angular, o defeito quantico desaparece. Nesses es-

116 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
0 1 2 3−10
−8
−6
−4
−2
0
r/aB
V(r
) / (
e2 /aB)
Figura 7.7: Potencial exterior (coulombiano blindado) Vcl ∝ e2
r (azul, traco superior), poten-
cial interior (coulombiano nao-blindado) Vbl ∝ Ze2
r (verde, traco inferior) e potencial efetivo(vermelho, traco no meio).
Q 7 7
P
O
N
L
K
s p
6 6 6
5 5 5 5f
4 4 4 4f
M 3 3 3
2 2
1
= 0 1 2 3
s p d
s p d
s p d
s p d
s p
s
l
actinides
lanthanides (terras raras)
elementos de transição
Figura 7.8: Ilustracao da sequencia de preenchimento dos orbitais.
tados, o eletron esta longe do nucleo e o potencial e semelhante daquele do hidrogenio. Osalcalinos sao atualmente muito estudados em laboratorios de otica quantica. Eles sao com-parativamente simples, mas tem estrutura suficiente complexa para serem interessantes. Astransicoes eletronicas situam-se na faixa espectral do infravermelho proximo e visıvel e podemser excitadas com fontes laser comparativamente simples. A vida dos estados excitados estana faixa de 30 ns. Assim, a largura da linha natural e aproximadamente (2π) 25 MHz e, namaioria dos casos, a estrutura de hiperfina dos estados excitados ainda pode ser resolvida. Asintensidades de saturacao sao tipicamente 1− 5 mW/cm−2, o que pode ser bem alcancado comdiodos laser. Os pontos de fusao sao entre 28C para cesio e 180C para lıtio. Em uma celula devapor, uma pressao de vapor de 10−6 mbar e atingida em temperaturas de 307C (Li) e −7C(Cs), respectivamente. Esta pressao corresponde a uma densidade de,
n =p
kT≈ 101atome/cm6 . (7.83)
Os atomos podem ser bem estudados por espectroscopia de saturacao.

7.4. O SISTEMA PERIODICO DOS ELEMENTOS 117
7.4.3 Acoplamento LS e jj
No caso do helio, vimos que o princıpio de Pauli primeiramente determina a orientacao relativados spins dos eletrons. Os spins dos eletrons individuais, portanto, somam para um momentoangular total. Os momentos angulares orbitais tambem adotam uma certa orientacao relativa.Ela e determinada pela interacao de Coulomb residual nao esfericamente simetrica. Uma certacombinacao de momentos angulares orbitais leva a uma certa distribuicao espacial dos eletronse, portanto, a uma certa energia eletrostatica.
O spin total e o momento angular orbital total, em seguida, acoplam para diferentes momen-tos angulares totais muito semelhantes ao acoplamento do spin e do momento angular orbitalpela interacao spin-orbita. Estados com diferentes momentos angulares totais possuem entao asenergias respectivas que o spin total adota no campo do momento angular orbital total.
(3/2,5/2)1,2,3,4
(3/2,3/2)0,1,2,3
(1/2,5/2)2,3
(1/2,3/2)1,2
4321
3210
32
21
(j , j )1 2 J
l =1, l =21 2
S=0
S=0
1F
1P
1D
3F
3P
3D
3P23 1P3 0P
simetria e
energia de troca
interação residual
de Coulomb estrutura fina
energia de
1partícula
Figura 7.9: Hierarquia de energias.
Alem disso, ha as seguintes pequenas contribuicoes:
• ls · lj• ss · sj• correcoes relativısticas
• estrutura hiperfina
Esta descricao e chamada de acoplamento de Russel-Saunders ou acoplamento LS. Funcionaenquanto o acoplamento spin-orbita for pequeno. Neste caso, vale a proibicao de intercom-binacao, isto e, nao pode haver transicoes eletromagneticas entre estados com spin diferente(ver helio metaestavel).
Como ELS ' Zα4 ' Z4, para atomos pesados, o acoplamento do spin de um eletron ao seumomento orbital cresce fortemente com o peso. Para grandes Z, o acoplamento spin-orbita podetornar-se maior, bem como a energia de simetria e de troca orientando mutuamente os spins,bem como a interacao residual de Coulomb, acoplando mutuamente os momentos angularesorbitais. Nesse caso, a orientacao de Li em relacao a Si delivra mais energia do que a energia detroca e a energia residual custam. Portanto, o spin e o momento angular orbital de um eletronse acoplam primeiramente para um momento angular orbital total,
ji = li + si . (7.84)

118 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
Obtemos um novo hamiltoniano de estrutura fina da forma,
HFS = ji · ji . (7.85)
O acoplamento jj puro existe apenas com nucleos muito pesados. Normalmente, temos umchamado acoplamento intermediario, que e uma mistura de acoplamento LS e jj. Isso leva aabolicao da proibicao da intercombinacao. Quando o acoplamento e puro, temos as seguintesregras de selecao dipolar:
acoplamento LS: ∆S = 0, ∆L = ±1, ∆` = ±1
acoplamento jj: ∆j = 0,±1 para um e−, ∆j = 0 para todos os outros
Alem disso temos para os dois acoplamentos: ∆J = 0,±1 mas nao tem J, J ′ = 0, ∆mJ =0,±1 quando ∆J = 0 mas nao tem mJ ,mJ ′ = 0.
7.4.4 Resumo dos graus de liberdade de um atomo
O hamiltoniano total de um unico atomo se compoe da energia cinetica do nucleo e dos eletrons,de varias potenciais de interacao entre o nucleo e os eletrons, de interacoes com varios tipos decampos eletromagneticos externos.
H = − ~2
2m∇2R +
N∑
i=1
(− ~2
2m∇2ri
)+ V (r1, s1, .., rN , sN ) + Vext . (7.86)
E claro que, com a presencia de outros atomos, outras interacoes podem gerar outras contri-buicoes relevantes para o hamiltoniano.
As seguintes interacoes contribuem para o potencial V . As interacoes coulombianas,
Vncl−ele = −Z∑
i=1
Ze2
4πε0|r− ri|e Vele−ele =
Z∑
i<j=1
e2
4πε0|ri − rj |, (7.87)
a antisimetria da funcao de onda, isto e, integrais de intercambio,
Vsym , (7.88)
as energias dos acoplamentos spin-orbita,
Vls = −Z∑
i=1
1
e2m2c2
1
|r− ri|dVcldri
(li · si) , (7.89)
as energias dos acoplamentos spin-spin,
Vss =Z∑
i 6=j=1
e2
m2
[σi · σj|ri − rj |3
− 3[σi · (ri − rj)][σj · (ri − rj)]
(ri − rj)5
], (7.90)
as energias dos acoplamentos orbita-orbita,
Vll =
Z∑
i 6=j=1
cij(li · lj) , (7.91)

7.5. EXERCICIOS 119
interacoes entre o spin dos eletrons e o spin nuclear e entre o momento angular orbital doseletrons e o spin nuclear,
Vhfs =A
~2J · I , (7.92)
correcoes relativısticas,
Vrel , (7.93)
Alem disso, campos externos estaticos deslocam os nıveis de energia e podem influenciar oacoplamento interno dos momentos angulares e dos spins
Vext = −d · ~E , −~µ · ~B . (7.94)
O que sao os numeros quanticos bons depende das amplitudes relativas das interacoes intra-atomicas:Caso 1: Estrutura fina com acoplamento L · S mais desdobramento Zeeman da estrutura hi-perfina: Vncl−ele, V r
ele−ele V aele−ele, Vsym Vls Vhfs VB os numeros quanticos sao
|ni, li,L,S,J,F,mF 〉.Caso 2: Estrutura fina com acoplamento j · j mais desdobramento Zeeman da estrutura hi-perfina: Vncl−ele, V r
ele−ele Vls V aele−ele, Vsym Vhfs VB os numeros quanticos sao
|ni, li, ji,J,F,mF 〉.Caso 3: Estrutura fina com acoplamento L · S mais hiperfina estrutura do desdobramentoZeeman: Vncl−ele, V r
ele−ele V aele−ele, Vsym Vls VB Vhfs os numeros quanticos sao
|ni, li,L,S,J,mJ ,mI〉.Caso 4: Estrutura fina com acoplamento L · S mais desdobramento Paschen-Back da estru-tura fina: Vncl−ele, V r
ele−ele V aele−ele, Vsym VB Vls Vhfs os numeros quanticos sao
|ni, li,L,S,mL,mS ,mI〉.
Vncl−ele desdobramento em nestrutura grossa ↓
Vele−ele desdobramento em l↓
Vsym desdobramento em S ↓ Vl s desdobramento em ji
estrutura fina Vee desdobramento em L ↓↓ Vsym, Vele−ele desdobramento em J
VLS desdobramento em J ↓
estrutura hiperfina Vhfs desdobramento em F↓
efeito Zeeman VLS desdobramento em mF
7.5 Exercıcios
7.5.1 Simetrizacao de bosons e fermions
7.5.1.1 Ex: Atomo de helio
Consultando uma tabela de isotopos determine o carater bosonico ou fermionico das seguintesespecies atomicas: 87Sr, 86Sr, 87Rb, 39K e 40K.

120 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS
7.5.2 Helio
7.5.2.1 Ex: Atomo de helio
Compare a energia de ligacao medida com a predicao do modelo de Bohr considerando a interacaoentre os eletrons ate primeira ordem TPIT.
7.5.2.2 Ex: Blindagem no helio
O atomo de helio (ou atomos tipo-helio como o H−) possuem dois eletrons interagentes em suacomposicao, o que faz com que estes sistemas nao possuam solucao exata. Para contornar esteproblema lancamos mao de uma serie de metodos aproximados com o intuito de calcular os seusauto-estados e suas respectivas auto-energias. Dentre esses metodos, um bastante utilizado,devido principalmente a sua facilidade e praticidade, e o metodo variacional, em que calculamoso estado fundamental de um dado problema atraves de uma funcao teste que nao e solucao doproblema original. Este metodo, quando aplicado em um atomo de helio, utiliza como funcaoteste, a solucao do problema sem interacao coulombiana entre os eletrons, que sentem somentea interacao com a carga original do nucleo. Entretanto, este metodo poderia ser melhoradoainda mais caso considerassemos uma carga efetiva no nucleo, devido a interacao deste com osproprios eletrons, e depois sim obtendo a funcao teste. Obtenha esta correcao no caso do helio.Interprete o resultado. Ajuda:
∫sin θ2√
r21 + r2
2 − 2r21r
22 cos θ2
dθ2 =
√r2
1 + r22 − 2r2
1r22 cos θ2
r1r2e
⟨1
r
⟩=
Z
aB.
7.5.3 Estrutura da casca eletronica
7.5.4 O sistema periodico dos elementos
7.5.4.1 Ex: Nıveis de excitacao eletronica de alcalinos
Explique porquea. o estado [Li] (2s)2SJ tem energia menor do que o [H] (2s)2SJ ;b. o estado [Li] (2s)2SJ tem energia menor do que o [Li] (2p)2PJ ;c. o estado [Na] (3d)2DJ tem energia menor do que o [Na] (4p)2PJ .

7.5. EXERCICIOS 121
3
1s
2s
2Li
Lith
ium
6.9
41
5.3
917
10 N
eN
eon
20.1
797
21.5
646
2
1s2
He
Hel
ium
4.0
0260
24.5
874
9
OO
xyge
n15.9
994
13.6
181
8
FF
luor
ine
18.9
9840
17.4
228
7
NN
itrog
en14.0
0674
14.5
341
6
CC
arbo
n12.0
107
11.2
603
5
1s
2s
2p
22
B Bor
on10.8
11
8.2
980
57 L
aLa
ntha
num
138.9
055
5.5
769
89 A
cA
ctin
ium
(227)
5.1
7
71 L
uLu
tetiu
m174.9
67
5.4
259
103 Lr
Law
renc
ium
(262)
4.9
?
87 [R
n]7
s
Fr
Fra
nciu
m(2
23)
4.0
727
88 R
aR
adiu
m(2
26)
5.2
784
104 R
fR
uthe
rfor
dium
(261)
6.0
?
72 H
fH
afni
um178.4
9
6.8
251
40 Z
rZ
ircon
ium
91.2
24
6.6
339
39
YY
ttriu
m88.9
0585
6.2
171
38 S
rS
tron
tium
87.6
2
5.6
949
56 B
aB
ariu
m137.3
27
5.2
117
73 T
aTa
ntal
um180.9
479
7.5
496
54 X
eX
enon
131.2
9
12.1
298
19 [A
r]4
s
KP
otas
sium
39.0
983
4.3
407
20 C
aC
alci
um40.0
78
6.1
132
21 S
cS
cand
ium
44.9
5591
6.5
615
22 T
iT
itani
um47.8
67
6.8
281
30 Z
nZ
inc
65.3
9
9.3
942
31 G
aG
alliu
m69.7
23
5.9
993
32 G
eG
erm
aniu
m72.6
1
7.8
994
33 A
sA
rsen
ic74.9
2160
9.7
886
34 S
eS
elen
ium
78.9
6
9.7
524
35 B
rB
rom
ine
79.9
04
11.8
138
36 K
rK
rypt
on83.8
0
13.9
996
23
VV
anad
ium
50.9
415
6.7
462
24 C
rC
hrom
ium
51.9
961
6.7
665
25 M
nM
anga
nese
54.9
3805
7.4
340
26 F
eIr
on55.8
45
7.9
024
27 C
oC
obal
t58.9
3320
7.8
810
28 N
iN
icke
l58.6
934
7.6
398
29 C
uC
oppe
r63.5
46
7.7
264
11 [N
e]3
s
Na
Sod
ium
22.9
8977
5.1
391
12 M
gM
agne
sium
24.3
050
7.6
462
13 [N
e]3
s3
p2
Al
Alu
min
um26.9
8154
5.9
858
14 S
iS
ilico
n28.0
855
8.1
517
15
PP
hosp
horu
s30.9
7376
10.4
867
16
S Sul
fur
32.0
66
10.3
600
17 C
lC
hlor
ine
35.4
527
12.9
676
18 A
rA
rgon
39.9
48
15.7
596
1
HH
ydro
gen
1.0
0794
13.5
984
4
1s
2s
22
Be
Ber
ylliu
m9.0
1218
9.3
227
37 [K
r]5
s
Rb
Rub
idiu
m85.4
678
4.1
771
55 [X
e]6
s
Cs
Ces
ium
132.9
0545
3.8
939
42 M
oM
olyb
denu
m95.9
4
7.0
924
41 N
bN
iobi
um92.9
0638
6.7
589
86 R
nR
adon
(222)
10.7
485
74 WT
ungs
ten
183.8
4
7.8
640
43 T
cTe
chne
tium
(98)
7.2
8
75 R
eR
heni
um186.2
07
7.8
335
44 R
uR
uthe
nium
101.0
7
7.3
605
76 O
sO
smiu
m190.2
3
8.4
382
45 R
hR
hodi
um102.9
0550
7.4
589
77
Ir Irid
ium
192.2
17
8.9
670
46 P
dP
alla
dium
106.4
2
8.3
369
78 P
tP
latin
um195.0
78
8.9
587
47 A
gS
ilver
107.8
682
7.5
762
79 A
uG
old
196.9
6655
9.2
255
48 C
dC
adm
ium
112.4
11
8.9
938
80 H
gM
ercu
ry200.5
9
10.4
375
60 N
dN
eody
miu
m144.2
4
5.5
250
62 S
mS
amar
ium
150.3
6
5.6
436
63 E
uE
urop
ium
151.9
64
5.6
704
64 G
dG
adol
iniu
m157.2
5
6.1
501
65 T
bTe
rbiu
m158.9
2534
5.8
638
61 P
mP
rom
ethi
um(1
45)
5.5
82
66 D
yD
yspr
osiu
m162.5
0
5.9
389
67 H
oH
olm
ium
164.9
3032
6.0
215
68 E
rE
rbiu
m167.2
6
6.1
077
69 T
mT
huliu
m168.9
3421
6.1
843
49 In In
dium
114.8
18
5.7
864
50 S
nT
in118.7
10
7.3
439
51 S
bA
ntim
ony
121.7
60
8.6
084
52 T
eTe
lluriu
m127.6
0
9.0
096
53
IIo
dine
126.9
0447
10.4
513
81 T
lT
halli
um204.3
833
6.1
082
82 P
bLe
ad207.2
7.4
167
83 B
iB
ism
uth
208.9
8038
7.2
856
84 P
oP
olon
ium
(209)
8.4
17
?
85 A
tA
stat
ine
(210)
58 C
eC
eriu
m140.1
16
5.5
387
59 P
rP
rase
odym
ium
140.9
0765
5.4
73
70 Y
bY
tterb
ium
173.0
4
6.2
542
90 T
hT
horiu
m232.0
381
6.3
067
92
UU
rani
um238.0
289
6.1
941
93 N
pN
eptu
nium
(237)
6.2
657
94 P
uP
luto
nium
(244)
6.0
262
95 A
mA
mer
iciu
m(2
43)
5.9
738
96 C
mC
uriu
m(2
47)
5.9
915
91 P
aP
rota
ctin
ium
231.0
3588
5.8
9
97 B
kB
erke
lium
(247)
6.1
979
98 C
fC
alifo
rniu
m(2
51)
6.2
817
99 E
sE
inst
eini
um(2
52)
6.4
2
100 Fm
Fer
miu
m(2
57)
6.5
0
101 Md
Men
dele
vium
(258)
6.5
8
102 No
Nob
eliu
m(2
59)
6.6
5
1s
2s
2p
22
21
s2
s2
p2
23
1s
2s
2p
22
41
s2
s2
p2
25
1s
2s
2p
22
6
[Ne
]3s2
[Ne
]3s
3p
22
[Ne
]3s
3p
23
[Ne
]3s
3p
24
[Ne
]3s
3p
25
[Ne
]3s
3p
26
[Ar]
4s2
[Ar]
3d
4s2
[Ar]
3d
4s
22
[Ar]
3d
4s
32
[Ar]
3d
4s
5[A
r]3
d4
s5
2[A
r]3
d4
s6
2[A
r]3
d4
s7
2[A
r]3
d4
s8
2[A
r]3
d4
s1
0[A
r]3
d4
s1
02
[Ar]
3d
4s
4p
10
2[A
r]3
d4
s4
p1
02
2[A
r]3
d4
s4
p1
02
3[A
r]3
d4
s4
p1
02
4[A
r]3
d4
s4
p1
02
5[A
r]3
d4
s4
p1
02
6
[Kr]
5s2
[Kr]
4d
5s2
[Kr]
4d
5s
22
[Kr]
4d
5s
4[K
r]4
d5
s5
[Kr]
4d
5s
52
[Kr]
4d
5s
7[K
r]4
d5
s8
[Kr]
4d
10
[Kr]
4d
5s
10
[Kr]
4d
5s
10
2[K
r]4
d5
s5
p1
02
2[K
r]4
d5
s5
p1
02
3[K
r]4
d5
s5
p1
02
4[K
r]4
d5
s5
p1
02
5[K
r]4
d5
s5
p1
02
6
[Xe
]6s2
[Xe
]5d
6s2
[Xe
]4f5
d6
s2[X
e]4
f6
s3
2[X
e]4
f6
s4
2[X
e]4
f6
s5
2[X
e]4
f6
s7
2[X
e]4
f6
s6
2[X
e]4
f5
d6
s7
2[X
e]4
f6
s9
2[X
e]4
f6
s1
02
[Xe
]4f
6s
11
2[X
e]4
f6
s1
22
[Xe
]4f
6s
13
2[X
e]4
f6
s1
42
[Xe
]4f
5d
6s
14
2
[Xe
]4f
5d
6s
14
22
[Xe
]4f
5d
6s
14
32
[Xe
]4f
5d
6s
14
42
[Xe
]4f
5d
6s
14
52
[Xe
]4f
5d
6s
14
26
[Xe
]4f
5d
6s
14
72
[Xe
]4f
5d
6s
14
9[X
e]4
f5
d6
s1
41
0[X
e]4
f5
d6
s1
41
02
[Hg
]6p
[Hg
]6p
2[H
g]6
p3
[Hg
]6p
4[H
g]6
p5
[Hg
]6p
6
[Rn
]6d
7s2
[Rn
]6d
7s
22
[Rn
]5f
6d
7s
22
[Rn
]5f
6d
7s
32
[Rn
]5f
6d
7s
42
[Rn
]5f
7s
62
[Rn
]5f
7s
72
[Rn
]5f
6d
7s
72
[Rn
]5f
7s
92
[Rn
]5f
7s
10
2[R
n]5
f7
s1
12
[Rn
]5f
7s
12
2[R
n]5
f7
s1
32
[Rn
]5f
7s
14
2[R
n]5
f7
s7
p?
14
2
[Rn
]5f
6d
7s
?1
42
2
[Kr]
4d
5s
5p
10
2
Gro
up
IA
IIA
IIIA
IVA
VA
VIA
VIIA
VIIIA
IBIIB
IIIB
IVB
VB
VIB
VII
B
VII
I
58 C
eC
eriu
m1
40
.11
6
5.5
38
71G
° 4
[Xe
]4f5
d6
s2
Ato
mic
Num
ber
Sym
bol
Nam
e
Gro
und-s
tate
Configura
tion
Gro
und-s
tate
Level
Ioniz
ation
Energ
y(e
V)
105
107
106
108
109
111
110
112
Db
Dub
nium
(262)
Sg
Sea
borg
ium
(263)
Hs
Has
sium
(265)
Bh
Boh
rium
(264)
Mt
Mei
tner
ium
(268)
Uu
nU
nunn
ilium
(269)
Uu
bU
nunb
ium
Uu
uU
nunu
nium
(272)
Period
1 65432 7
PE
RI
OD
IC
TA
BL
E
Ato
mic
Pro
pert
ies
of
the
Ele
men
ts
Fo
ra
de
sc
rip
tio
no
fth
ea
tom
icd
ata
,v
isit
ph
ysic
s.n
ist.
go
v/a
tom
ic
Marc
h1999
Fre
qu
en
tly
us
ed
fun
da
me
nta
lp
hy
sic
al
co
ns
tan
ts
1second
=9
192
631
770
periods
ofra
dia
tion
corr
espondin
gto
the
transitio
n
the
two
hyperf
ine
levels
ofth
egro
und
sta
teof
s
speed
oflig
htin
vacuum
299
792
458
ms
Pla
nck
consta
nt
6.6
261
10
Js
(
ele
menta
rycharg
e1.6
022
10
C
ele
ctr
on
mass
9.1
094
10
kg
pro
ton
mass
1.6
726
10
kg
fine-s
tructu
reconsta
nt
1/1
37.0
36
Rydberg
consta
nt
10
973
732
m
Boltzm
ann
consta
nt
1.3
807
10
JK
c h e m m R k
1
e p
For
the
mos
tacc
urat
eva
lues
ofth
ese
and
othe
rco
nsta
nts,
visi
t
betw
een
C
(exact) /2
)
0.5
110
MeV
3.2
89
84
10
Hz
13.6
057
eV
133
2
27
23
h=
h
mc
Rc
Rhc
e
phys
ics.
nist
.gov
/con
stan
ts
!
! ! ! !
!
!!
H34
19
31
1
15
1
p
H H H
H
H
a
4 4 4
So
lid
s
Art
ific
iall
yP
rep
are
d
Liq
uid
s
Ga
se
s[R
n]7
s2
1s
†B
ased
upon
.(
)in
dic
ate
sth
em
ass
num
ber
ofth
em
oststa
ble
isoto
pe.F
or
adescription
and
the
mostaccura
tevalu
es
and
uncert
ain
ties,see
J.P
hys.C
hem
.R
ef.
Data
,(5
),1239
(1997).
26
12C
Ato
mic
Weig
ht†
U.S
.D
EP
AR
TM
EN
TO
FC
OM
ME
RC
ETechnolo
gy
Adm
inis
tration
NationalIn
stitu
teofS
tandard
sand
Technolo
gy
Ph
ysic
sL
ab
ora
tory
ww
w.n
ist.gov
physic
s.n
ist.gov
ww
w.n
ist.gov/s
rd
Sta
nd
ard
Refe
ren
ce
Data
Pro
gra
m
2S
12/
1S
0
1S
0
3P
2
3P
0
2D
32/
2D
32/
3F
2?
2S
12/
2D
32/
3F
2
1S
0
4F
32/
7S
3
6S
52/
5D
4
4F
92/
3F
4
2S
12/
2S
12/
1S
0
3P
0
1S
0
2S
12/
6D
12/
5D
0
5F
5
5D
4
3D
3
2S
12/
2S
12/
7F
0
3H
6
1S
0
3F
2
2P
° 12/
4S
° 32/
2P
° 32/
2P
° 12/
2P
° 32/
2P
° 12/
2P
° 32/
2P
° 12/
2P
° 12/
1G
° 44- I -° 9
2/
6H
° 52/
8S
° 72/
9D
° 26H
° 152/
2F° 7
2/
2P
° 12?
/
4S
° 32/
3P
2
1S
0
1S
0
3P
0
4S
° 32/
3P
2
1S
0
1S
0
2D
32/
3F
2
7S
3
6S
52/
4F
92/
1S
0
1S
0
3P
0
4S
° 32/
3P
2
2P
° 32/
1S
0
1S
0
3F
2
4F
32/
6S
52/
4F
92/
1S
0
3P
0
4S
° 32/
3P
2
2P
° 32/
1S
0
1S
0
2D
32/
7F
0
8S
° 72/
9D
° 26H
° 152/
3H
6
2F° 7
2/
1S
0
5L° 6
4K
11
2/
6L
11
2/
5- I - 4
4- I -° 1
52/
5- I - 8
5- I - 8
4- I -° 1
52/
2S
12/
2S
12/
2S
12/
Figura 7.10: Tabela periodica.

122 CAPITULO 7. ATOMOS DE MULTIPLOS ELETRONS

Capıtulo 8
Moleculas dimericas
Em sistemas de muitos corpos, as interacoes interatomicas devem ser consideradas. Esses in-teracoes sao geralmente eletrostaticas, mas geralmente nao podem ser dadas em forma de ex-pressoes fechadas. Por exemplo, a colisao de dois atomos pode acontecer em uma multidao decanais, isto e, potenciais de interacao V (ri − rj). As forcas interatomicas nao so governam co-lisoes mas podem sustentar estados ligados moleculares. Isso introduz novos graus de liberdadenos sistemas de muitas partıculas atraves de excitacoes possıveis de movimentos de vibracao oude rotacao.
Figura 8.1: Exemplo de um spaghetti de potenciais interatomico: Os primeiros estados damolecula 85Rb.
Neste curso nao remos alem de dımeres homo- ou heteronucleares, isto e, moleculas queconsistem de dois atomos iguais ou diferentes.
8.1 A ligacao molecular
8.1.1 Ligacao ionica e covalente
Existem duas possibilidades para ligar dois atomos, a ligacao ionica e a ligacao covalente.1 Aligacao ionica e governada pelas grandezas eletroafinidade (EA), eletronegatividade (EN) e aenergia de ionizacao (IE):
1Nao estamos considerando aqui ligacoes metalicas nem de pontos hidrogenicos.
123

124 CAPITULO 8. MOLECULAS DIMERICAS
eq. de Schrödinger
spin
separação do centro das massas
Born-Oppenheimer
coordenada esférica
radial angularMO & VB
mov. translação mov. relativo
espectro rotacional
mov. nuclearmov. eletrônico
espectro vibracionalligação química subestrutura fina & hiperfina
Figura 8.2: Aproximacoes no tratamento de moleculas.
• Energia de ionizacao: Energia necessaria para liberacao de um eletron por um atomoneutro, p.ex. Na + 5.1 eV→ Na+ + e−.
• Eletroafinidade: Energia liberada pela captura de uma eletron por um atomo neutro,p.ex. Cl + e− → Cl− + 3.8 eV.
• Eletronegatividade: Medida para estabilidade de um orbital de valencia, p.ex. aqueledo fluor (3.98) e mais estavel do que aquele do cesio (0.79), tal que o fluor segura melhorseus eletrons do que o cesio.
Em curtas distancias, a troca de um eletron entre os atomos pode diminuir a energia. Aligacao entao se faz pela atracao coulombiana entre dois ioes, e a energia de ligacao pode serestimada atraves da interacao eletrostatica.
Exemplo 12 (Ligacao ionica no NaCl): Por exemplo, um atomo sodio e um de cloroganham energia formando uma molecula,
(8.1)
Na + 5.1 eV→ Na+ + e−
Na+ + Cl− → NaCl + 4.9eV
−−−−−−−−−−−−−−−−−−−Na + Cl→ NaCl + (3.8− 5.1 + 4.9) eV .
As moleculas sao polares e, portanto, tem um momento eletrico dipolar permanente. Aligacao nao tem direcao preferencial, pois cada atomo e perfeitamente isotropico. Portanto, essetipo e bem adaptado para construcao de redes cristalinas.
Para entender a ligacao covalente, consideramos o exemplo H+2 e estimamos a energia para
cada distancia R entre os nucleos. Nesse caso, em contraste aos atomos, a simetria esferica equebrada e, portanto, a degenerescencia energetica a respeito a paridade e abolida. Para asfuncoes de onda ψ(−x) = ±ψ(x) as energias variam diferentemente com R. A funcao de ondapar, que tem uma probabilidade aumentada do eletron de estar entre os nucleos e ligando, afuncao ımpar, que desaparece entre os nucleos, e anti-ligando. De fato, um eletron localizadono centro entre duas cargas positivas pode vencer a repulsao coulombiana entre os nucleos, adistancia recıproca dos quais e o dobre. Obviamente, a energia nao pode cair em baixo daquele

8.1. A LIGACAO MOLECULAR 125
NaCl
Na Cl+ -
1.3 eV
3.6 eV
1 nm
EA = 3.8 eV
IE = 5.1 eV
~1/r
even (binding)
-13.6 eV
-16.3 eV
1 nm
odd (antibinding)
(b)(a)
Figura 8.3: Esquema para (a) ligacao ionica de NaCl e (b) covalente de H2.
do estado fundamental do He+, sendo aproximadamente −4×13.6 eV. Com dois eletrons, comono caso da molecula neutra H2, a orientacao anti-paralela dos spins, ↑↓, permite colocar os doiseletron no mesmo orbital, enquanto para orientacao paralela, ↑↑, leva ate anti-ligacao. Cadaeletron sem parceiro num orbital pode formar uma ligacao covalente, por exemplo, o fosforo[P]=[Ne]3s23p↑↑↑ tem tres orbitais disponıveis que correspondem a diferentes numeros quanticosmagneticos. A ligacao covalente e direcional (hibridizacao sp1, sp2 ou sp3), o que torna-seessencial para estrutura de moleculas como CH4.
8.1.2 Aproximacao de Born-Oppenheimer e a molecula H+2
A aproximacao de Born-Oppenheimer em fısica molecular consiste em considerar, primeiro asposicoes dos nucleos come sendo fixos. Isso nos permite estudar os estados estacionarios doseletrons sujeitos ao potencial criado pelos nucleos. O movimento nao esta tratado ate depois,usando as energias eletronicas. Mudando a distancia internuclear R, as energias eletronicas(computadas para um R fixo) ficam as mesmas por causa da variacao instantanea das funcoesde onda eletronicas. (Essa variacao subita ocorre por causa da massa muito inferior do eletronem comparacao com a massa dos nucleos. As energias eletronicas nao variaveis jogam o papelde energias potenciais das interacoes entre os nucleos [1].
8.1.2.1 Separacao do centro das massas
Consideramos duas massas pesadas, Ma,b = M separadas por uma distancia R e interagindoatraves de um potencial Vnn(R). Tambem tem uma massa leve me interagindo com as outrasmassas atraves do Vne(r). O hamiltoniano e
H =−~2
2M∇2a +−~2
2M∇2b +−~2
2me∇2e + Vnn(|Ra −Rb|) + Vne(|Ra −Re|) + Vne(|Rb −Re|) . (8.2)
Primeiramente, transformamos para o sistema de centro das massas pesadas dado por X ≡MaRa+MbRb
M = Ra + Rb. A distancia das pesadas e R ≡ Ra−Rb, e a coordenada da massa levea contar do centro de massa e r = Ra − 1
2R−Re. Introduzindo a massa reduzida das pesadasMr = M
2 ,
[−~2
2M∇2X +
−~2
2Mr∇2R + Vnn(R) +
−~2
2me∇2r + Vne(|r + 1
2R|) + Vne(|r− 12R|)
]Θ(X)Ψ(R,Re)
= TΘ(X)Ψ(R,Re) . (8.3)

126 CAPITULO 8. MOLECULAS DIMERICAS
Ra
RbX
R
Re
r
Figura 8.4: Sistema de duas massas pesadas e uma leve.
Aqui fizemos o ansatz para a funcao de onda total Ψ = Θ(X)Ψ(R,Re), assumindo que o centrodas massas somente esta influenciado pelas massas pesadas,
−~2
2M∇2XΘ(X) = TΘ(X) (8.4)
[−~2
2Mr∇2R + Vnn(R) +
−~2
2me∇2r + Vne(|r + 1
2R|) + Vne(|r− 12R|)
]Ψ(R,Re) = EΨ(R,Re) .
8.1.2.2 Aproximacao adiabatica
A aproximacao de Born-Oppenheimer agora consiste em assumir, que o movimento de m, des-crito por ψ, nao e afetado pelo potencial de interacao das pesadas. Isso so vale, enquanto aspesadas sao inertes na escala de tempo do movimento dem. Por a mesma razao, o movimento daspesadas e independente da posicao de m, o que nos permite separar, Ψ(R,Re) = ψ(R, r)φ(R)e E = ε(R) + Ec. Portanto, podemos calcular a segunda derivada,
∇2R[ψ(R, r)φ(R)] = φ(R)∇2
Rψ(R, r) + 2[∇Rφ(R)] · [∇Rψ(R, r)] + ψ(R, r)∇2Rφ(R) (8.5)
' ψ(R, r)∇2Rφ(R) ,
e postular, que os dois primeiros termos sejam negligenciaveis em comparacao com o terceiro.Assim, podemos separar a segunda equacao (8.4) em duas partes, a primeira sendo,
[−~2
2me∇2r + Vne(|r + 1
2R|) + Vne(|r− 12R|)
]ψ(r,R) = ε(R)ψ(r,R) . (8.6)
Resolvemos esta equacao para a grau de liberdade eletronico r escolhendo uma distancia inter-nuclear R fixa e substituımos na segunda expressao (8.4), dando,
[−~2
2Mr∇2R + Vnn(R) + ε(R)
]φ(R) = Eφ(R) . (8.7)
Colocando a distancia interatomica R como um parametro fixo, a solucao da Eq. (8.6) forneceos orbitais eletronicos e as suas energias ε(R). O potencial de Born-Oppenheimer se compoedo potencial repulsivo eletrostatico dos nucleos e da energia cinetica do eletron, Vnn + ε(R).Inserindo este potencial interatomico completo na Eq. (8.7), podemos determinar a sua estruturavibracional.
Exemplo 13 (A molecula H2): Para o caso da molecula H2 inserimos o potencial deCoulomb,
Vee(r) = − e2
4πε0re Vne(R) =
e2
4πε0R.
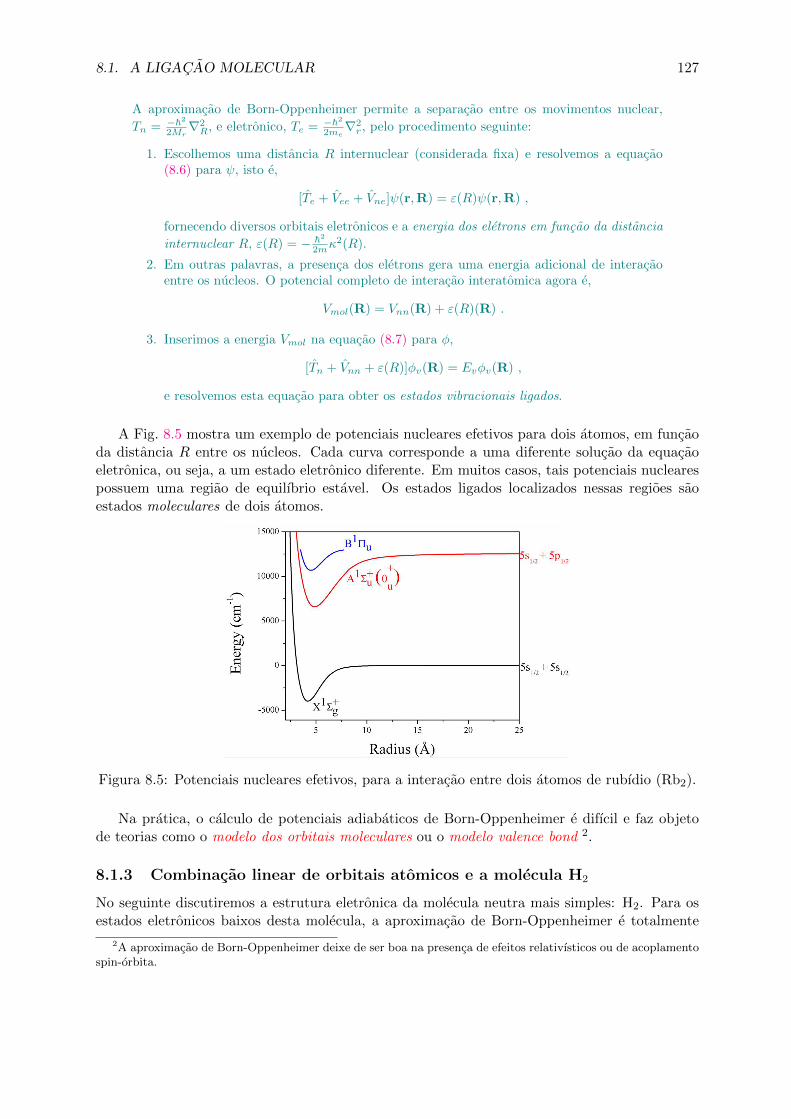
8.1. A LIGACAO MOLECULAR 127
A aproximacao de Born-Oppenheimer permite a separacao entre os movimentos nuclear,
Tn = −~2
2Mr∇2R, e eletronico, Te = −~2
2me∇2r, pelo procedimento seguinte:
1. Escolhemos uma distancia R internuclear (considerada fixa) e resolvemos a equacao(8.6) para ψ, isto e,
[Te + Vee + Vne]ψ(r,R) = ε(R)ψ(r,R) ,
fornecendo diversos orbitais eletronicos e a energia dos eletrons em funcao da distancia
internuclear R, ε(R) = − ~2
2mκ2(R).
2. Em outras palavras, a presenca dos eletrons gera uma energia adicional de interacaoentre os nucleos. O potencial completo de interacao interatomica agora e,
Vmol(R) = Vnn(R) + ε(R)(R) .
3. Inserimos a energia Vmol na equacao (8.7) para φ,
[Tn + Vnn + ε(R)]φv(R) = Evφv(R) ,
e resolvemos esta equacao para obter os estados vibracionais ligados.
A Fig. 8.5 mostra um exemplo de potenciais nucleares efetivos para dois atomos, em funcaoda distancia R entre os nucleos. Cada curva corresponde a uma diferente solucao da equacaoeletronica, ou seja, a um estado eletronico diferente. Em muitos casos, tais potenciais nuclearespossuem uma regiao de equilıbrio estavel. Os estados ligados localizados nessas regioes saoestados moleculares de dois atomos.
Na aproximação de Born-Oppenheimer, resolve-se primeiro a equação eletrônica, que consideraxa a posição dos núcleos (ou seja, o movimento nuclear é desprezado frente ao eletrônico). A soluçãodessa equação fornece diversos estados eletrônicos. Os níveis de energia eletrônicos, que dependemdas posições nucleares R como parâmetro, são então somados à energia de interação entre os núcleos,constituindo um potencial nuclear efetivo:
U(R) = VNN (R) + Eel(R) (5)
Em outras palavras, a presença dos elétrons gera uma energia adicional de interação entre osnúcleos. Na gura 1, mostramos um exemplo de potenciais nucleares efetivos para dois átomos, emfunção da distância R entre os núcleos. Cada curva corresponde a uma diferente solução da equaçãoeletrônica, ou seja, a um estado eletrônico diferente.
Figura 1: Potenciais nucleares efetivos, para a interação entre dois átomos de rubídio (Rb2).
Em muitos casos, tais potenciais nucleares possuem uma região de equilíbrio estável, a exemplodos potenciais do Rb-Rb. Os estados ligados localizados nessas regiões são estados moleculares de doisátomos. Dizemos ainda que os diferentes estados ligados são estados ro-vibracionais de uma molécula.Em geral, pode se mostrar que os níveis de rotação são energeticamente muito mais próximos entre sido que os níveis de vibração; por isso, é comum considerar que a rotação gera um pequeno splitting
dos níveis de vibração. Em geral, podemos tratar as rotações de forma separada das vibrações.Agora que temos uma forma de descrever quanticamente um estado molecular, podemos entender
os mecanismos de transição entre esses estados. As intensidade das possíveis transições molecularessão, qualitativamente, descritas pelo "Princípio de Franck-Condon", cujo enunciado clássico é o que sesegue:
O salto de um elétron durante uma transição molecular ocorre num tempo muito pequeno
comparado com a escala de tempo do movimento nuclear, de modo que, imediatamente
após o salto, os núcleos permanecem praticamente nas mesmas posições e nas mesmas
velocidades relativas de antes do salto. [6]
2
Figura 8.5: Potenciais nucleares efetivos, para a interacao entre dois atomos de rubıdio (Rb2).
Na pratica, o calculo de potenciais adiabaticos de Born-Oppenheimer e difıcil e faz objetode teorias como o modelo dos orbitais moleculares ou o modelo valence bond 2.
8.1.3 Combinacao linear de orbitais atomicos e a molecula H2
No seguinte discutiremos a estrutura eletronica da molecula neutra mais simples: H2. Para osestados eletronicos baixos desta molecula, a aproximacao de Born-Oppenheimer e totalmente
2A aproximacao de Born-Oppenheimer deixe de ser boa na presenca de efeitos relativısticos ou de acoplamentospin-orbita.

128 CAPITULO 8. MOLECULAS DIMERICAS
satisfatoria, isto e, queremos resolver uma equacao de Schrodinger tipo (8.6), mas com doiseletrons. Assim, estamos interessados no Hamiltoniano eletronico,
H = − ~2
2me(∇2
1 +∇22) + e2
4πε0
(1Rab− 1|r1−Ra| −
1|r1−Rb| −
1|r2−Ra| −
1|r2−Rb| + 1
r12
), (8.8)
onde ’1’ e ’2’ denotam os dois eletrons e ’a’ e ’b’ os nucleonsNao e possıvel resolver este problema de forma analıtica. O procedimento padrao comeca com
a escolha de uma para base apropriada, isto e, uma base muito compacta que nao dependesseda configuracao da molecula. Ou seja, queremos a base composta de funcoes que nao dependemda distancia entre os dois nucleos, Rab, para evitar fazer calculos para diferentes comprimentosde ligacao.
As funcoes de base mais naturais sao os orbitais atomicos dos atomos de hidrogenio indi-viduais. Quando o comprimento da ligacao e muito grande, o sistema abordara o limite dedois atomos de hidrogenio nao-interagentes. Neste caso, a funcao de onda eletronica pode seraproximada pelo produto de orbitais atomicos (AO) do atomo ’a’ e do atomo ’b’. Portanto, amenor base que nos da uma imagem realista do estado fundamental da molecula H2 deve conterduas funcoes: |1sa〉 e |1sb〉. Para comprimentos de ligacoes finitas, e aconselhavel permitir queos AOs se polarizem e se deformem em resposta a presenca do outro eletron (e do outro nucleo).No entanto, as funcoes |1sa〉 e |1sb〉 nao precisam ser exatamente as autofuncoes hidrogenicas.E suficiente requerer que elas sejam semelhantes aos orbitais 1s e centradas nos. Uma vez quea forma real dos orbitais ainda nao e fixa, daremos todas as expressoes em forma matricialabstrata, deixando a integracao espacial para quando a forma dos orbitais for especificada. Issoe o metodo de combinacao linear de orbitais atomicos (LCAO).
8.1.4 Teoria dos orbitais moleculares
Estamos agora em condicoes de discutir os princıpios basicos do metodo do orbital molecular(MO), que e o fundamento da teoria da estrutura eletronica das moleculas reais. O primeiropasso em qualquer abordagem do MO consiste na separacao do hamiltoniano em duas partespara descrevendo os eletrons ’1’ e ’2’ separadamente e uma parte contando para a interacaoentre eles:
H = h(1) + h(2) + V12 +e2
4πε0
1
Rabcom (8.9)
h(i) = −~2∇i2me
− e2
4πε0
(1
|ri −Ra|+
1
|ri −Rb|
)e V12 =
e2
4πε0
1
r12,
onde i = 1, 2. Precisamos nos lembrar que, dentro da aproximacao BO, Rab e apenas um numero.Escolhemos os hamiltonianos h(i) como a parte uni-eletronica do hamiltoniano completo emrepresentacao matricial na base mınima:
(〈1sa|h|1sa〉 〈1sa|h|1sb〉〈1sb|h|1sa〉 〈1sb|h|1sb〉
)≡(ε habhab ε
), (8.10)
definindo a energia uni-eletronica media ε ≡ 〈1sa|h|sa〉 e o acoplamento nao-diagonal (frequen-temente chamado de integral de ressonancia) hab ≡ 〈1sa|h|sb〉 = 〈1sa|h|sb〉. Podemos imediata-mente diagonalizar esta matriz, os autovalores o os autoestados sendo:
ε± = ε± hab e |φ±〉 ∝ 12(|sa〉 ± |sb〉) . (8.11)

8.1. A LIGACAO MOLECULAR 129
ha hb
Figura 8.6: Ilustracao dos orbitais atomicos.
Os autoestados do hamiltoniano efetivo uni-eletronico sao chamado de orbitais moleculares(MO). Eles sao funcoes uni-eletronicas delocalizadas nas regioes espaciais da molecula.
Precisamos primeiramente normalizar os MOs, o que e mais complicado do poderia parecer,pois os AOs nao sao ortogonais. Por exemplo, ao se aproximar os AOs dos atomos podem ter aforma ilustrada na Fig. 8.6. No entanto, definindo a integral de sobreposicao por S ≡ 〈1sa|1sb〉,podemos normalizar da maneira seguinte:
|φ±〉 = 1√2(1±S)
(|sa〉 ± |sb〉) (8.12)
com 〈φ±|φ±〉 = 12(〈1sa|sa〉 ± 〈1sa|sb〉 ± 〈1sb|sa〉+ 〈1sb|sb〉) = 1± S .
Estas autofuncoes meramente mostram a simetria da molecula. Os dois atomos de hidrogenio saoequivalentes e, portanto, os auto-orbitais devem dar peso igual a cada orbital 1s. Assim, nossaescolha do hamiltoniano uni-eletronico, na verdade, nao importa muito, pois cada hamiltonianouni-eletronico exibindo a simetria da molecula daria os mesmos orbitais moleculares. Por razoeshistoricas, |φ+〉 e denotado por |σ〉 e |φ−〉 por |σ∗〉.
and this is the MO ground state for H2. How good an approximation is it? Well, we can compute the expectation value of the energy,
σσσσel
H as follows. First, we decompose the wavefunction
into spatial and spin parts and note that the spin part is normalized:
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )21ˆ21
21ˆ21ˆ
σσσσ
σσσσσσσσ
el
spinspinelel
H
HH
=
ΦΦ=
Then, we note that ( ) ( ) ABel RVhhH /1ˆ2ˆ1ˆˆ12 +++= and
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )( ) ( ) ( ) σεσσ
σσσσσσσσ
≡=
=
11ˆ1
2211ˆ1211ˆ21
h
hh
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )( ) ( ) ( ) σεσσ
σσσσσσσσ
≡=
=
22ˆ2
1122ˆ2212ˆ21
h
hh
( ) ( ) ( ) ( ) σσσσσσ JV ≡21ˆ21 12
Taken together, these facts allow us to write:
AB
AB
MOMOMOMOMOMOMOMO
RJ
RVhhh
12
1ˆˆˆˆ12211
++=
+ΨΨ+ΨΨ+ΨΨ=ΨΨ
σσσε
Each of the first two terms is energy of a single electron (either 1 or 2)
in the field produced by the nuclei ( h ) while the third is the average repulsion of the two electrons. Note that the second and third terms are both positive, so binding has to arise from the one-electron piece. This is the MO energy for the ground state of H2. For a reasonable choice of the 1s-like basis functions – it turns out to be more convenient to fit the exponential decay of the hydrogenic orbitals to a sum of Gaussians- we can use a computer to compute the unknowns above ( σε and σσJ )
and plot the total energy as a function of RAB, we get the result pictured at right. The exact adiabatic energy
Figura 8.7: Energia total em funcao da distancia interatomica.
O segunda passo na teoria MO consiste em construir a determinante a partir dos MOscorrespondentes aos estados procurados. Para ilustracao vamos olhar para o estado singletomais baixo construıdo a partir dos orbitais moleculares. Notamos, que hab < 0, tal que |σ〉 temenergia inferior a |σ∗〉. Desprezando a interacao o estados singleto mais baixo,
|ΦMO〉 = |σ〉|σ〉 , (8.13)

130 CAPITULO 8. MOLECULAS DIMERICAS
e o estado molecular fundamental para H2. Para estimar a validade da aproximacao, calculamos
o valor esperado da energia,∣∣∣〈σ|〈σ|H|σ〉|σ〉
∣∣∣, decompondo a funcao de onda em partes espaciais
e de spin notando, que a parte de spin e normalizada:
∣∣∣〈σ|〈σ|H|σ〉|σ〉∣∣∣ = 〈σ(1)|〈σ(2)|H|σ(1)〉|σ(2)〉〈Φspin|Φspin〉 = 〈σ(1)|〈σ(2)|H|σ(1)〉|σ(2)〉 . (8.14)
Assim, com (8.10) e,
〈σ(1)|〈σ(2)|h(2)|σ(1)〉|σ(2)〉 = 〈σ(1)|σ(1)〉〈σ(2)|h(2)|σ(2)〉 = 〈σ(2)|h(2)|σ(2)〉 ≡ εσ〈σ(1)|〈σ(2)|h(1)|σ(1)〉|σ(2)〉 = 〈σ(1)|h(1)|σ(1)〉〈σ(2)|σ(2)〉 = 〈σ(1)|h(1)|σ(1)〉 ≡ εσ〈σ(1)|〈σ(2)|V12|σ(1)〉|σ(2)〉 ≡ Jσσ . (8.15)
Juntando estes fatos, podemos escrever,
〈ΦMO|H|ΦMO〉 = 〈ΦMO|h1|ΦMO〉+ 〈ΦMO|h2|ΦMO〉+ 〈ΦMO|V 12|ΦMO〉+1
4πεRab(8.16)
= 2εσ + Jσσ +1
4πεRab.
Cada um dos dois primeiros termos representa a energia de um unico eletron (ou 1 ou 2) nocampo produzido pelos nucleos (h), enquanto o terceiro e a repulsao media dos dois eletrons.Note, que o primeiro e segundo termo ambos sao positivos, tal que e a ligacao deve vir da parteuni-eletronica. Isto e a energia MO para o estado fundamental do H2. Para um chute maisrazoavel das funcoes de base tipo 1s 3 podemos determinar as grandezas desconhecidas de cima(εσ e Jσσ) numericamente e tracar a energia total como uma funcao de Rab (curva azul pontilhadana Fig. 8.7). A funcao de energia adiabatica exata determinada de dados experimentais (curvapreta solida) concorda bem em baixas energias. Resumindo os resultados com alguns numeros-chave notamos que a teoria MO prediz uma distancia da ligacao de 0.072 nm em concordanciarazoavel com o valor exato de 0.074 nm. Podemos tambem comparar as energias de ligacao,
De = EH2(Re)− eEH . (8.17)
A teoria dos MOs preve uma energia de ligacao de 5.0 eV em comparacao com o valor experi-mental de 4.75 eV. Tendo em visto a simplicidade da funcao de onda e a ausencia de parametrosajustaveis a concordancia nao e tao ruim. Infelizmente, longe da distancia de equilıbrio, temosuma surpresa desagradavel: a molecula nao se dissocia em dois atomos de hidrogenio!
8.1.5 Ligacao de valencia
Para obter uma ideia do que esta acontecendo perto da dissociacao, expandimos o estado MOfundamental em termos de configuracoes AO:
|ΦMO〉 ∝ |σ(1)〉|σ(2)〉|Φspin〉 (8.18)
= 12(1+S)(|1sa(1)〉+ |1sb(1)〉)(|1sa(2)〉+ |1sb(2)〉)|Φspin〉
= 12(1+S)(|1sa(1)〉|1sa(2)〉+ |1sa(1)〉|1sb(2)〉+ |1sb(1)〉|1sa(2)〉+ |1sb(1)〉|1sb(2)〉)|Φspin〉 .
3Acontece que e mais conveniente ajustar a decomposicao exponencial dos orbitais hidrogenicos a uma somade gaussianos.

8.1. A LIGACAO MOLECULAR 131
1s
yb
1s
1s
|y|
1s s1s
s1s*
ya
2
y
ybya y
|y|2
H H H2
E
Figura 8.8: Ilustracao da teoria MO para um dımero de dois atomos iguais com um eletron devalencia no orbital atomico 1s cada um: Ao se aproximar os orbitais atomicos formam novosorbitais moleculares ligantes e anti-ligantes.
Os dois termos no meio da ultima linha, chamados de configuracoes covalentes, sao exatamenteo que esperamos perto da dissociacao: um eletron em cada atomo de hidrogenio. No entanto, oprimeiro e o ultimo termo (que sao chamados de configuracoes ionicas) correspondem a colocardois eletrons em um atomo e nenhum no outro, o que nos da H+ e H− na dissociacao! Umavez que o peso desses termos e fixo, e obvio que obtemos a funcao de onda errada (e, portanto,a energia errada) ao dissociar essa molecula. Perto do ponto de equilıbrio, os termos ionicoscontribuem significativamente para a verdadeira funcao de onda, tal que a teoria MO e boa nesteponto. Mas e sempre terrıvel na dissociacao.
Uma alternativa a teoria MO representa a teoria da ligacao de valencia (VB). Aqui, usa-sesignificativamente mais intuicao fısica e descarta as configuracoes ionicas da funcao de onda MO.Assim, a funcao de onda VB do estado fundamental e:
|Ψ〉 ∝ |1sa(1)〉|1sa(2)〉+ |1sb(1)〉|1sa(2)〉√2
| ↑ (1)〉| ↓ (2)〉+ | ↓ (1)〉| ↑ (2)〉√2
≡ |Ψspace〉|Ψspin〉 .(8.19)
A teoria VB pressupoe, que esta funcao de onda e uma boa aproximacao da funcao de ondaverdadeira para todas as distancias de ligacao e nao somente para grandes distancias Rab. Paraverificar esta aproximacao, podemos calcular a energia media para este estado VB. Primeira-mente, normalizamos a funcao de onda VB,
〈Ψ|Ψ〉 = 〈Ψspace|〈Ψspin|Ψspace〉|Ψspin〉 = 〈Ψspace|Ψspace〉 (8.20)
= 12 (〈1sa(1)|〈1sb(2)|+ 〈1sb(1)|1sa(2)|) (|1sa(1)〉|1sa(2)〉+ |1sb(1)〉|1sa(2)〉)
= 12 (〈1sa(1)|1sb(2)|1sa(1)〉|1sb(2)〉+ 〈1sa(1)|1sb(2)|1sb(1)〉|1sa(2)〉+〈1sb(1)|1sa(2)|1sa(1)〉|1sb(2)〉+ 〈1sb(1)|1sa(2)|1sb(1)〉|1sa(2)〉)
= 12(1 + S2 + S2 + 1) = 1 + S2 .

132 CAPITULO 8. MOLECULAS DIMERICAS
Portanto, a funcao de onda VB corretamente normalizada e:
|ΨV B〉 =1
2√
1 + S2(|1sa(1)〉|1sa(2)〉+ |1sb(1)〉|1sa(2)〉) (| ↑ (1)〉| ↓ (2)〉 − | ↓ (1)〉| ↑ (2)〉) .
(8.21)Agora queremos calcular 〈Hel〉 para este estado. Notamos primeiramente, que a parte do spinnao importa, pois o hamiltoniano e independente do spin:
〈ΨV B|H|ΨV B〉 = 〈Ψspin|〈Ψspace|H|Ψspace〉|Ψspin〉 (8.22)
= 〈Ψspace|H|Ψspace〉〈Ψspin|Ψspin〉 = 〈Ψspace|H|Ψspace〉 .
O unico remanescente do estado de spin e o fato que a funcao de onda espacial e simetrica,o que so e possıvel quando a parte de spin for antissimetrica. Tratando cada termo em 〈H〉separadamente,
〈Ψ|h1|Ψ〉 = 12 (〈1sa(1)|〈1sb(2)|+ 〈1sb(1)|1sa(2)|) h1 (|1sa(1)〉|1sa(2)〉+ |1sb(1)〉|1sa(2)〉) (8.23)
= 12
(〈1sa(1)|1sb(2)|h1|1sa(1)〉|1sb(2)〉+ 〈1sa(1)|1sb(2)|h1|1sb(1)〉|1sa(2)〉
+〈1sb(1)|1sa(2)|h1|1sa(1)〉|1sb(2)〉+ 〈1sb(1)|1sa(2)|h1|1sb(1)〉|1sa(2)〉)
= 12(ε+ SHab + ε+ Shab) =
ε+ Shab1 + S2
.
Como os dois eletrons sao identicos, os elementos da matriz de h2 sao os mesmos como aquelesde h1. O unico termo restante e o valor medio da interacao:
〈Ψ|V12|Ψ〉 = 12 (〈1sa(1)|〈1sb(2)|+ 〈1sb(1)|1sa(2)|) V12 (|1sa(1)〉|1sa(2)〉+ |1sb(1)〉|1sa(2)〉)
(8.24)
= 12
(〈1sa(1)|1sb(2)|V12|1sa(1)〉|1sb(2)〉+ 〈1sa(1)|1sb(2)|V12|1sb(1)〉|1sa(2)〉
+ 〈1sb(1)|1sa(2)|V12|1sa(1)〉|1sb(2)〉+ 〈1sb(1)|1sa(2)|V12|1sb(1)〉|1sa(2)〉).
O segundo e o terceiro termo sao os mesmos. Eles sao chamados de integrais de troca, pois osorbitais ’bra’ tem ordem trocada em comparacao com os ’ket’:
K = 〈1sa(1)|1sb(2)|V12|1sb(1)〉|1sa(2)〉 = 〈1sb(1)|1sa(2)|V12|1sa(1)〉|1sb(2)〉 . (8.25)
O segundo e o terceiro termo tambem sao os mesmos. Eles sao chamados de integrais de Coulomb,pois parecem devido a interacao de Coulomb entre duas densidades de cargas:
J = 〈1sb(1)|1sa(2)|V12|1sa(1)〉|1sb(2)〉 = 〈1sa(1)|1sb(2)|V12|1sb(1)〉|1sa(2)〉 . (8.26)
Portanto, temos o resultado,
〈Ψ|V12|Ψ〉 =J +K
1 + S2. (8.27)
Adicionando todos os termos, obtemos:
〈ΨV B|H|ΨV B〉 = 〈ΨV B|h1|ΨV B〉+ 〈ΨV B|h2|ΨV B〉+ 〈ΨV B|V12|ΨV B〉+1
Rab(8.28)
= 2ε+ Shab1 + S2
+J +K
1 + S2+
1
Rab.

8.1. A LIGACAO MOLECULAR 133
Os termos de Coulomb e de troca sao positivos. A repulsao nuclear e claramente positiva. Assim,os unicos termos que levam a ligacao nesta imagem sao a energia media de um eletron ε e aintegral de ressonancia hab. Se o primeiro termo e dominante, a ligacao se deve a deslocalizacao,uma vez que um eletron localizado em um dos atomo apenas daria o valor atomico para ε,o que nao implica um estado ligado. Se hab for grande, a ligacao envolve algum caracterede ressonancia, que pode ser conectado ao conceito familiar de ressonancia entre diferentesestruturas de pontos de Lewis.
Uma avaliacao numerica de todas as integrais da a curva de potencial apresentada na Fig. 8.7para a teoria do VB. Como esperado, esta funcao de onda VB simples da o limite de dissociacaocorreto, onde a teoria MO falha. Alem disso, a exatidao do resultado VB simples e surpreen-dentemente bom mesmo perto do ponto de equilıbrio: A VB preve uma distancia de ligacaode 0.071 nm (em comparacao com a resposta correta de 0.074 nm) e De = 5.2 eV (em com-paracao com 4.75 eV). Assim, a funcao de onda VB tambem da um bom acordo sem parametrosajustaveis. Mas, o mais importante e, que indica o caminho para melhorar a funcao de ondasempre que constatamos um erro obvio: neste caso, vimos que a descricao da dissociacao erafraca e construımos o ansatz VB para curar o problema. Esta abordagem de VB frequentementee generalizada da seguinte maneira, quando se trata de moleculas poliatomicas. Escrevemos afuncao de onda como um produto de uma parte espacial e uma de spin:
|Ψ〉 = |Ψspace〉|Ψspin〉 . (8.29)
O principal pressuposto na teoria VB e que a parte espacial pode ser bem representada por umproduto de funcoes de tipo atomico. Por exemplo, para a agua, escreverıamos imediatamenteuma parte espacial como:
|Ψspace〉 ' |1sHa〉|1sHb〉|1sO〉|1sO〉|2sO〉|2sO〉|2pxO〉|2pxO〉|2pyO〉|2pyO〉 . (8.30)
No entanto, ha duas coisas erradas com esta funcao de onda. Primeiramente, sabemos que osorbitais atomicos hibridizam em uma molecula. Portanto, precisamos fazer combinacoes linearesapropriadas dos AOs (neste caso hıbridos sp3) para obter os AOs hibridizados. Nesse caso, osquatro hıbridos sp3 podem ser escritos simbolicamente como:
|sp3〉 = cs,i|2s〉+ cx,i|2px〉+ cy,i|2py〉+ cz,i|2pz〉 . (8.31)
e , portanto, uma configuracao espacial mais apropriada e:
|Ψspace〉 ' |1sHa〉|1sHb〉|1sO〉|1sO〉|sp31O〉|sp3
1O〉|sp32O〉|sp3
2O〉|sp33O〉|sp3
4O〉 . (8.32)
O outro problema com este estado e, que falta a simetria adequada para descrever fermions;o estado geral precisa ser antissimetrico. No caso de dois eletrons isso foi facil de aplicar -singletos tem partes espaciais simetricas e tripletos antissimetricas. No entanto, no caso demuitos eletrons, as regras nao sao tao simples; na verdade, o tempo de computacao numericacresce exponencialmente com o numero de eletrons.
Formalmente, deixaremos a derivacao neste ponto, definindo um operador A que ’antisime-triza’ a funcao de onda. Neste caso,
|Ψspace〉 ' A[|1sHa〉|1sHb〉|1sO〉|1sO〉|sp3
1O〉|sp31O〉|sp3
2O〉|sp32O〉|sp3
3O〉|sp34O〉]. (8.33)
Em geral, os resultados da teoria VB sao muito precisos para os pequenos sistemas, onde ela podeser aplicada. Os comprimentos de ligacao sao um pouco curtos, e as energias de ligacao tendem a

134 CAPITULO 8. MOLECULAS DIMERICAS
ser pequenas demais, mas os resultados sao qualitativamente excelentes. Alem disso, os orbitaisatomicos hibridizados corretos caem diretamente fora do calculo, dando uma boa introspeccaoqualitativa. Alem disso, observe que as configuracoes atomicas nao devem mudar (ou muitopouco), quando a geometria da molecula muda (uma vez que os orbitais dependem do atomo enao da estrutura molecular). Portanto, essas funcoes de ondas VB tem uma forte conexao comos estados diabaticos discutidos anteriormente. No entanto, a quantidade exponencial de tempoque se deve investir para fazer esses calculos torna-los impraticaveis para a maioria de moleculasde interesse.
8.2 Estrutura rovibracional dos potenciais moleculares
A separacao do movimento dos nucleos da dinamica eletronica feita na aproximacao de Born-Oppenheimer levou as equacoes (8.6) e (8.7). Na secao precedente analisamos em detalhe aequacao (8.6) no ambito de entender o fenomeno da ligacao molecular.
Na secao seguinte vamos analisar a equacao (8.7), que determina o movimento dos nucleos.Separando as partes radias e angulares do movimento vamos descobrir estados vibracionais erotacionais.
8.2.1 As equacoes radial e angular
A interacao entre dois atomos identicos e descrito pelo seguinte hamiltoniano, onde Mr =(M−1
a +M−1b )−1 = M/2 a massa reduzida dos nucleos,
H =P2
2Mr+ Vmol(R) com Vmol(R) =
e2
4πε0R+ VBO(R) . (8.34)
O potencial de interacao Vmol se compoe de uma forca de Coulomb internuclear repulsiva e deum potencial adiabatico de Born-Oppenheimer devido a interacao dos eletrons entre si e com osdois nucleos 4. A energia cinetica e aquela do movimento relativo (o movimento do centro dasmassas ja foi separado na Sec. 8.1.2, tal que neste sistema inercial, a energia cinetica translacionaldesaparece). Em coordenadas esfericas,
P2
2Mr= − ~2
2Mr
[1
R2
∂
∂R
(R2 ∂
∂R
)+
1
R2
L2
~2
]. (8.35)
A parte angular, que foi discutida na Sec. 2.1.1, descreve uma rotacao rıgida dos atomos homo-nucleares em torno do seu centro de massas com a energia de rotacao,
V`(R) =L2
2MrR2=
~2`(`+ 1)
2MrR2, (8.36)
tambem chamada de barreira centrıfuga. A parte radial,[− ~2
2Mr
∂2
∂R2+ V`(R) + Vcoulomb(R)
]u(R) = Eu(R) , (8.37)
onde u(R) = rR(R) e a funcao de onda radial do movimento nuclear. O potencial interatomicocausa uma movimento de vibracao. Os estados vibracionais do potencial adiabatico sao quan-tizados e caracterizados por uma energia de vibracao bem definida. Discutiremos a estruturaro-vibracional nas secoes seguintes.
4Notamos aqui, que em grandes distancias outras forcas chamadas forcas de van der Waals dominam a interacaointeratomica. Estas serao discutidas na Sec. 8.3.

8.2. ESTRUTURA ROVIBRACIONAL DOS POTENCIAIS MOLECULARES 135
8.2.1.1 Bandas rotacionais e vibracionais
Moleculas tem bem mais graus de liberdade do que atomos. Por exemplo, os atomos de umamolecula dimerica podem vibrar dentro do potencial de interacao mutua. No sistema do centrode massas podemos imaginar estas vibracoes como oscilacoes de um atomo com massa reduzidae com energia quantizada. A molecula pode girar e ter um momento de inercia. Esses grausde liberdade contribuem energias ao hamiltoniano da molecula, ou diretamente ou atraves deinteracoes com outros graus. Por isso, os espectros das moleculas tem uma complexidade bemmaior.
No entanto, os regimes de energias das maiores excitacoes sao bem diferentes. A escalatıpica de energia de ligacao (profundidade do potencial interatomico) e ∆Ep ' 20..200 THz(0.1..1 eV) 5. As excitacoes eletronicas ocorrem no regime de ∆Ee ' 100..1000 THz (1..10 eV).O espacamento entre excitacoes vibracionais tipicamente e de Ev+1 − Ev ' THz (0.01 eV).Finalmente, as excitacoes rotacionais ficam na escala de Ev+1−Ev ' 100 MHz (10−6 eV). Comoem temperatura ambiente (um gas de moleculas em equilıbrio termico T = 300 K) a energia estanuma escala de 2.5×10−2 eV, o grau de liberdade da excitacao eletronica esta gelado, enquantouma distribuicao larga de estados vibracionais e rotacionais pode ser excitada (por exemplo, porcolisoes intermoleculares). A grande diferencia de escalas facilita a separacao delas e, portanto,a identificacao da proveniencia do estados observados em medidas experimentais.
8.2.2 Estados moleculares vibracionais
A energia potencial de uma molecula cresce, quando os nucleos sao deslocados das suas posicoesde equilıbrio. Quando o deslocamento, x ≡ R − Re e pequeno, podemos expandir a energiapotencial,
Vmol(x) = Vmol(0) +dVmol(0)
dxx+
1
2
d2Vmol(0)
dx2x2 + .. . (8.38)
A energia de equilıbrio nao interesse e a primeira derivada desaparece no equilıbrio. Portanto,
Vmol(x) ' 12k
2x2 com k ≡ d2Vmol(0)
dx2. (8.39)
Usando a massa efetiva podemos escrever o hamiltoniano,
Hmol = − ~2
2m1
d2
dx21
− ~2
2m2
d2
dx22
+ 12kx
2 = − ~2
2Mr
d2
dx2+ 1
2kx2 . (8.40)
O espectro de energia desse grau de liberdade, portanto, e
Ev = ~ω(v + 12) . (8.41)
com ω =√k/Mr. Isto e, no fundo de potenciais profundos, os nıveis de energia sao equidistantes.
8.2.2.1 Vibracoes anarmonicas no potencial de Morse
Para maiores deslocamentos nao podemo mais desprezar os termos anarmonicos na expansaode Taylor. Uma melhor aproximacao e o potencial de Morse. Este potencial (azul na Fig. 8.9),
5Estados em potenciais eletronicamente excitados (isto e, um dos eletrons de valencia fica num orbital excitado)sao ligados mais fracamente, porque os eletrons nao sao no orbital mais ligando.
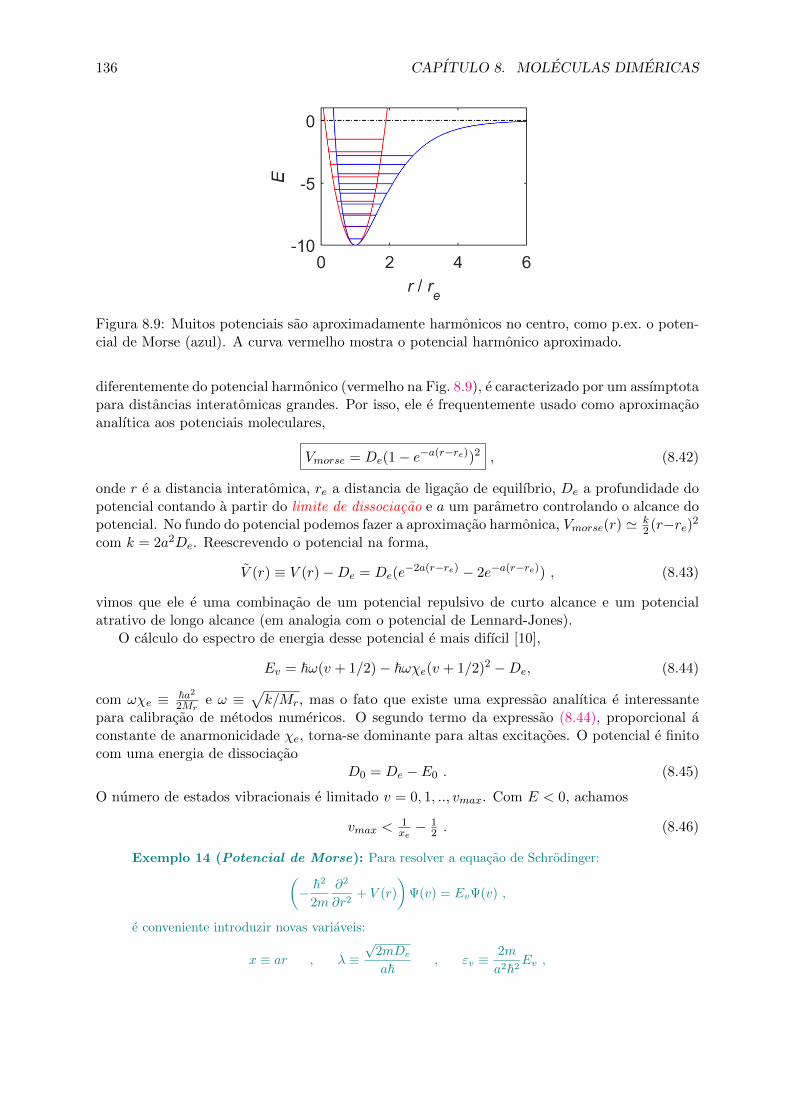
136 CAPITULO 8. MOLECULAS DIMERICAS
Figura 8.9: Muitos potenciais sao aproximadamente harmonicos no centro, como p.ex. o poten-cial de Morse (azul). A curva vermelho mostra o potencial harmonico aproximado.
diferentemente do potencial harmonico (vermelho na Fig. 8.9), e caracterizado por um assımptotapara distancias interatomicas grandes. Por isso, ele e frequentemente usado como aproximacaoanalıtica aos potenciais moleculares,
Vmorse = De(1− e−a(r−re))2 , (8.42)
onde r e a distancia interatomica, re a distancia de ligacao de equilıbrio, De a profundidade dopotencial contando a partir do limite de dissociacao e a um parametro controlando o alcance dopotencial. No fundo do potencial podemos fazer a aproximacao harmonica, Vmorse(r) ' k
2 (r−re)2
com k = 2a2De. Reescrevendo o potencial na forma,
V (r) ≡ V (r)−De = De(e−2a(r−re) − 2e−a(r−re)) , (8.43)
vimos que ele e uma combinacao de um potencial repulsivo de curto alcance e um potencialatrativo de longo alcance (em analogia com o potencial de Lennard-Jones).
O calculo do espectro de energia desse potencial e mais difıcil [10],
Ev = ~ω(v + 1/2)− ~ωχe(v + 1/2)2 −De, (8.44)
com ωχe ≡ ~a2
2Mre ω ≡
√k/Mr, mas o fato que existe uma expressao analıtica e interessante
para calibracao de metodos numericos. O segundo termo da expressao (8.44), proporcional aconstante de anarmonicidade χe, torna-se dominante para altas excitacoes. O potencial e finitocom uma energia de dissociacao
D0 = De − E0 . (8.45)
O numero de estados vibracionais e limitado v = 0, 1, .., vmax. Com E < 0, achamos
vmax <1xe− 1
2 . (8.46)
Exemplo 14 (Potencial de Morse): Para resolver a equacao de Schrodinger:
(− ~2
2m
∂2
∂r2+ V (r)
)Ψ(v) = EvΨ(v) ,
e conveniente introduzir novas variaveis:
x ≡ ar , λ ≡√
2mDe
a~, εv ≡
2m
a2~2Ev ,

8.2. ESTRUTURA ROVIBRACIONAL DOS POTENCIAIS MOLECULARES 137
tal que:
(− ∂2
∂x2+ V (x)
)Ψn(x) = εnΨn(x) com V (x) = λ2
(e−2(x−xe) − 2e−(x−xe)
).
Os autovalores e autofuncoes sao [10]:
εn = 1− 1λ2
(λ− n− 1
2
)2= 2
λ (n+ 12 )2− 1
λ2 (n+ 12 )2 e Ψn(z) = Nnz
λ−n− 12 e−
12 zL(2λ−2n−1)
n (z) ,
onde z = 2λe−(x−xe) e Nn =[n!(2λ−2n−1)
Γ(2λ−n)
] 12
e,
L(α)n (z) =
z−αez
n!
dn
dzn(zn+αe−z
)=
Γ(α+ n+ 1)/Γ(α+ 1)
Γ(n+ 1)1F1(−n, α+ 1, z) ,
e o polinomio generalizado de Laguerre. Os elementos da matriz do operador espacial x e(assumindo m > n e N = λ− 1
2 ),
〈Ψm|x|Ψn〉 =2(−1)m−n+1
(m− n)(2N − n−m)
√(N − n)(N −m)Γ(2N −m+ 1)m!
Γ(2N − n+ 1)n!.
Nas variaveis originais as autoenergias sao:
Ev = ~ω(v + 1/2)− [~ω(v + 1/2)]2
4De,
onde v e o numero quantico vibracional e ω = a√
2De
m . As diferencias de energia de nıveis
adjacentes diminuı com v,
Ev+1 − Ev = ~ω − (~ω)2 v + 1
2De.
Este fato descreve bem a estrutura vibracional em moleculas nao-rotativas. No entanto, aequacao falha em cima de algum valor de vmax, onde Evmax+1−Evmax
fica zero ou negativo,
vmax =2De − ~ω
~ω.
Esta falha e devida ao numero finito vmax de estados ligados no potencial de Morse. Para
energias em cima de vmax todos energias sao possıveis, e a equacao para Ev nao e mais
valida.
8.2.2.2 Regras de selecao vibracionais
Campos eletromagneticos do tipo E1, e.g. a radiacao do corpo negro, podem induzir transicoesentre estados vibracionais e redistribuir as suas populacoes de maneira a estabelecer um equilıbriotermico. No entanto, como as transicoes mais fortes sao induzidas por migracoes dipolares decargas precisamos analisar com mais detalhes as regras de selecao impostas ao momento dipolar〈f |d|i〉.
Os estados relevantes para transicoes vibracionais sao especificados por |ε, v〉, onde ε denotao estado eletronico da molecula, pois o espectro vibracional depende da estrutura eletronica. Aaproximacao de Born-Oppenheimer nos permite considerar as vibracoes lentas separadamente dadinamica dos eletrons. A escala temporal para transicoes eletronicas e 1/∆Ee = 10−16 s−1 e parauma vibracao nuclear 1/∆Ev = 10−13 s−1. Para cada distancia dos nucleos os eletrons formamum estado estacionario adaptado, minimizando a energia para essa distancia. Isso equivale a

138 CAPITULO 8. MOLECULAS DIMERICAS
formacao de um potencial adiabatico de interacao entre os nucleos dentro do qual a distancia dosnucleos pode vibrar. Para achar quais transicoes vibracionais sao possıveis, precisamos calculara matriz,
〈ε′, v′|d|ε, v〉 = 〈v′|dε|v〉 . (8.47)
O momento dipolar, dε = 〈ε|d|ε〉, da molecula depende da distancia dos nucleos, pois asorbitas eletronicas |ε〉 dependem da distancia. Portanto, podemos expandir,
d = d0 +dd0
dxx+
1
2
d2d0
dx2x2 + .. . (8.48)
Portanto, a matriz de transicao e,
〈ε′, v′|d|ε, v〉 = d0δv,v′ +dd0
dx〈v′|x|v〉+
d2d0
dx2〈v′|x2|v〉+ .. . (8.49)
O primeiro termo desaparece, isto e, transicoes so podem acontecer, quando o momento dipolarvaria com a distancia. Por isso, dımeros homonucleares nao fazem transicoes vibracionais.
Para moleculas heteronucleares com cargas eletronicas que nao dependem da distancia dosatomos, o momento dipolar varia linearmente para pequenos deslocamentos. Nesse caso, soprecisamos do segundo termo da expansao. Dentro da aproximacao harmonica, o operador deposicao pode ser exprimido por, x ∝ a+ a†. Portanto, so transicoes ∆v = ±1 sao possıveis. Noentanto, devido a anarmonicidades, termos superiores, xn ∝ (a + a†)n tornam-se influentes, etransicoes com ∆v = ±2,±3, .. ficam possıveis.
Assim, em potenciais anarmonicos, as regras de selecao vibracionais sao substituıdas peloconceito da sobreposicao das funcoes de onda chamada de fator de Franck-Condon.
A espectroscopia Raman e um ferramenta muito util para analisar espectros rovibracionais.Nesse metodo, um espalhamento inelastico da origem a linhas Stokes e anti-Stokes em ∆v =±1,±2. O espectro do estado fundamental e assimetrico por causa da ausencia do estado inferior.Em dımeres homonucleares, os spins nucleares tem um impacto importante sobre os espectrosRaman. Consideracoes de paridade mostram que existem so linhas pares ou impares.
8.2.3 O princıpio de Franck-Condon
As intensidade das possıveis transicoes moleculares sao, qualitativamente, descritas pelo princıpiode Franck-Condon, cujo enunciado classico e o que se segue:
O salto de um eletron durante uma transicao molecular ocorre num tempo muitopequeno comparado com a escala de tempo do movimento nuclear, de modo que, ime-diatamente apos o salto, os nucleos permanecem praticamente nas mesmas posicoese nas mesmas velocidades relativas de antes do salto [15].
Por isso, as transicoes sao desenhadas verticalmente no esquema dos potenciais mostrado naFig. ??. Para dar taxas consideraveis, as transicoes devem ocorrer quando as velocidades nu-cleares nos dois estados acoplados sao identicas, o que e o caso nos pontos de retorno classicos.Nestes pontos, as funcoes de onda sao maximas 6.
Com esse princıpio de Franck-Condon, podemos determinar quais sao as transicoes maisfortes entre nıveis vibracionais de uma molecula, conforme representado na Fig. 8.10(direita).Em particular, devido a compatibilidade com frequencias opticas, nos interessamos por transicoesentre nıveis vibracionais de diferentes estados eletronicos.
6Note, que a presencia de uma estrutura hiperfina pode modificar as regras de selecao.

8.2. ESTRUTURA ROVIBRACIONAL DOS POTENCIAIS MOLECULARES 139
10 15 20 25 30
−12
−10
−8
−6
−4
−2
0
x 10−4
R (aB)
V
(a.u
.)E
R
(b)(a)
(c)
estado fundamental A
estado excitado B
Figura 8.10: (Esquerda, codigo: AM Molecula FourierGrid.m) Funcoes de onda moleculares numpotencial para tres estados vibracionais diferentes. (Direita) Representacao do enunciado classicodo princıpio de Franck-Condon. A transicao (a) possui alta intensidade (ou probabilidade), poisnela tanto a posicao quanto a velocidade relativa dos nucleos nao se altera. Ja as transicoes (b)e (c) sao pouco provaveis, pois exige-se uma mudanca na posicao dos nucleos (caso b) ou navelocidade deles (caso c).
O calculo exato das probabilidades de transicoes se da pelo modulo quadrado do momentode dipolo da transicao (TDM, transition dipole moment). O TDM e um elemento de matriz forada diagonal do operador dipolo eletrico M, dado por:
MAB = 〈Ψ(A)|M|Ψ(B)〉 , (8.50)
sendo |Ψ(A)〉 e |Ψ(B)〉 dois estados moleculares.Ainda dentro da aproximacao de Born-Oppenheimer, podemos quebrar o operador momento
de dipolo em dois termos (nuclear e eletronico), conforme:
M(r,R) = Me(r,R) + Mn(R) . (8.51)
Assim, o TDM fica:
MAB =
∫Ψ(A)∗MΨ(B)dRdr (8.52)
=
∫Meψ
(A)∗e ψ
(B)el ψ(A)∗
n ψ(B)n dRdr +
∫Mnψ
(A)∗n ψ(B)
n
∫ψ
(A)∗el ψ(B)
e drdR .
Como as funcoes eletronicas de estados diferentes sao ortogonais, segue que∫ψ
(A)∗e ψ
(B)e dr = 0,
anulando o segundo termo.Olhando para o primeiro termo, devemos notar que o momento de dipolo eletronico Me(r,R)
depende tambem das coordenadas nucleares como parametro. A formulacao quantica do princıpiode Franck-Condon consiste em afirmar que, num estado molecular, o momento de dipolo eletronicovaria pouco com as coordenadas nucleares. Dessa forma, junto com a condicao da aproximacaode Born-Oppenheimer, podemos quebrar o primeiro termo do TDM em integrais eletronica enuclear:
MAB =
∫Meψ
(A)∗e ψ(B)
e dr
∫ψ(A)∗n ψ(B)
n dR . (8.53)

140 CAPITULO 8. MOLECULAS DIMERICAS
Assim, temos uma expressao comparativa para a probabilidade da transicao, dada por:
PAB ∝ |MAB|2 =
∣∣∣∣∫
Meψ(A)∗e ψ(B)
e dr
∣∣∣∣2 ∣∣∣∣∫ψ
(A)∗N ψ(B)
n dR
∣∣∣∣2
. (8.54)
O segundo fator na equacao (8.54) e o chamado fator de Franck-Condon. Quando estudamosas transicoes entre estados eletronicos distintos, esse fator mede a intensidade da transicaoentre os seus nıveis vibracionais. Ou seja, transicoes entre nıveis vibracionais distintos teraoprobabilidades distintas, e quem mede isso e o fator de Franck-Condon.
Exemplo 15 (Moleculas ultrafrias): Existem diversas propostas para aplicacoes de
moleculas ultrafrias, como espectroscopia de resolucao ultra-alta [20], teste de leis funda-
mentais da fısica [12, 23], computacao quantica [11] e outras [7] 7
Para criar uma amostra de moleculas aprisionadas no estado fundamental de vibracao, um
metodo possıvel consiste em primeiro produzir as moleculas a partir de atomos ultrafrios
utilizando um processo chamado de fotoassociacao, e depois bombear essas moleculas para
o estado fundamental de vibracao.
A fotoassociacao consiste na excitacao de um par de atomos livres para um estado ligado
de um potencial eletronico excitado, por meio da absorcao de um foton. Em seguida, o par
decai por emissao espontanea, podendo retornar novamente a condicao de dois atomos livres
(o que nao e desejavel), ou entao decair para um estado ligado do potencial eletronico funda-
mental. Para moleculas de Rb2, pode-se fazer fotoassociacao com eficiencia em determinadas
frequencias [21], utilizando-se o potencial A1Σ+u como estado excitado (vide Fig. 8.11).
Logo apos serem formadas, as moleculas geralmente estao em nıveis de alta energia vibra-
cional (em torno de ν ≈ 80), pois estes sao os nıveis que melhor se conectam (alto fator de
Franck-Condon) com o estado excitado. E preciso entao transferir uma parcela consideravel
da populacao para o estado fundamental de vibracao, e isso tambem e feito por um metodo
que chamamos de ’bombeamento optico’.
O resfriamento vibracional por bombeamento optico ocorre com a incidencia de uma luz
banda larga na amostra molecular. Essa luz deve conter uma faixa de frequencias que ex-
cite transicoes ate nıveis vibracionais do potencial nuclear excitado. Esses estados serao tais
que, quando as moleculas decaırem novamente por emissao espontanea, os fatores de Franck-
Condon favorecem os estados de menor energia vibracional. Em outras palavras, a molecula
e enviada para um estado excitado e, quando retorna, tem alta probabilidade de ir para um
nıvel de menor energia vibracional. Apos varios desses ciclos de absorcao e emissao, ha um
efeito lıquido de reducao da energia vibracional da amostra (’resfriamento vibracional’), ate
que as moleculas de fato alcancem o estado vibracional fundamental.
8.2.4 Progressao rotacional
Os momentos de inercia nos tres eixos do espaco sao,
Iqq =∑
i
mir2i (q) . (8.55)
A energia cinetica da rotacao e
Erot = 12
∑
q=1,2,3
mqv2q = 1
2
∑
q=1,2,3
Iqqω2q =
J2x
2Ixx+
J2y
2Iyy+
J2z
2Izz, (8.56)
7A maioria dessas aplicacoes exige, no entanto, que a amostra molecular ocupe majoritariamente um unicoestado quantico. Isso constitui um desafio experimental, visto que moleculas possuem mais graus de liberdade doque atomos, como rotacao e vibracao.

8.2. ESTRUTURA ROVIBRACIONAL DOS POTENCIAIS MOLECULARES 141
Figura 3: Esquema da fotoassociação para formar moléculas de Rb2. No processo a), o par de átomoslivres absorve um fóton da radiação incidente, formando um estado ligado no potencial excitado.Em seguida, em b), a molécula recém-formada decai por emissão espontânea para estados ligados dopotencial fundamental, ou ainda pode retornar a um estado de dois átomos livres.
4 Detecção molecular & alguns resultados
O método mais direto que utilizamos para se detectar moléculas se baseia na ionização molecular.Aplica-se um campo elétrico na amostra molecular, e coloca-se um detector de íons na direção docampo. Partículas que se ionizem adquirem uma carga líquida, sendo atraídas pelo campo e caindono detector de íons. Para promover a ionização de moléculas, aplica-se a luz de um laser pulsadona amostra, cuja frequência pode ionizar as moléculas via processo de vários fótons (razão pela quala técnica recebeu o nome de "REMPI- resonantly enhanced multiphoton ionization). As moléculasionizadas pelo laser pulsado caem no detector em um intervalo de tempo bem denido após os pulsosde luz, de forma que conhecendo os instantes em que os pulsos foram emitidos, é possível olhar apenaspara as moléculas e ignorar a maioria dos íons esporádicos que caem no detector.
Outra vantagem desse método de detecção é que a frequência de ionização depende do nível vi-bracional das moléculas que se deseja ionizar. Ou seja, variando-se a frequência do laser ionizante,encontramos diversos picos de intensidade de ionização, referente aos diversos estados vibracionaisem que as moléculas da amostra se encontram. Por essa razão, uma varredura do laser de ionizaçãopode ser chamada de "espectro vibracional", sendo este essencial para o nosso trabalho, pois de fatodesejamos estimar a população de moléculas que está (ou não) no estado fundamental de vibração.
Na gura 5, encontra-se um espectro vibracional numa janela espectral relativamente curta, feito emnosso laboratório. A esta fase, éramos capazes de promover um considerável resfriamento vibracionaldas moléculas, sem contudo alcançar uma população macroscópica no nível fundamental de vibração.Neste espectro, foi capaz de se identicar um pico de ionização partindo do nível vibracional ν = 4 doestado eletrônico fundamental.
5
Figura 8.11: Esquema da fotoassociacao para formar moleculas de Rb2. No processo a), o par deatomos livres absorve um foton da radiacao incidente, formando um estado ligado no potencialexcitado. Em seguida, em b), a molecula recem-formada decai por emissao espontanea paraestados ligados do potencial fundamental, ou ainda pode retornar a um estado de dois atomoslivres.
com o momento angular Jq = Iqqωq.Muitas moleculas tem um eixo de simetria, tal que existem dois momentos de inercia di-
ferentes, I⊥ ≡ Ixx = Iyy e I‖ ≡ Izz. Interpretando os momentos angulares como operadoresquanticos,
H =J2
2I⊥+
(1
2I‖− 1
2I⊥
)J2z . (8.57)
Devemos, primeiramente, considerar a rotacao da molecula em relacao ao eixo de simetriada molecula. Esquecendo-se de campos externos calculamos a energia da molecula associadaas observaveis J2 com o numero quantico J e Jz com o numero quantico K. Achamos osauto-valores,
E(J,K,MJ) =~2J(J + 1)
2I⊥+
(1
2I‖− 1
2I⊥
)~2K2 = BJ(J + 1) + (A−B)K2 , (8.58)
com J = 0, 1, .., K = −J, .., J e MJ = −J, .., J e introduzindo as constantes rotacionais, A ≡ ~2
2I‖
e B ≡ ~2
2I⊥. Em seguida, analisamos esta equacao no contexto da aplicacao de um campo externo
que define tanto a direcao e′z no laboratorio como a projecao do movimento angular J2 nestadirecao, mJ . Isto e, temos dois eixos, o eixo internuclear ez e o eixo de rotacao da molecula e′z.
Cada nıvel J,mJ e 2(2J + 1) vezes degenerado, pois K = −J, .., J e K pode ser positivo ounegativo. Cada nıvel J contem (2J + 1) estados. Note, que para moleculas esfericas, A = B, eo grau de liberdade K desaparece. Vide Exc. 8.4.2.2.
A constante rotacional pode ser aproximada por,
Erot =~2J(J + 1)
2Mr〈R2〉 , (8.59)
onde√〈R2〉 e o valor de esperado para o ’turning point’ exterior do nıvel vibracional. A
constante rotacional para o estado vibracional do 87Rb que e 5.9 cm−1 embaixo do limite de

142 CAPITULO 8. MOLECULAS DIMERICAS
dissociacao e Bv = νJ=1rot − νJ=0
rot = 81 MHz. Para ficar mais preciso precisarıamos calcular〈R2〉v = 〈ψv|R2|ψv〉.
Transicoes vibracionais acompanham-se de transicoes rotacionais simultaneas ∆J = ±1. Porisso, as frequencias de transicoes dependem da constante rotacional Bv, que em torno, dependedo estado vibracional. As energias da molecula sao,
Ev,J = ~ω(v + 1/2)− ~ωxe(v + 1/2)2 + ..+ hcBvJ(J + 1)− hcDvJ2(J + 1)2 + .. . (8.60)
Sob influencia de uma rotacao rapida, os atomos da molecula sao sujeitos a forca centrifuga ese afastam mais 8
Como em temperaturas ambientes muitos nıveis rotacionais sao populados, observamos expe-rimentalmente muitas linhas conhecidas como ramo P quando ∆J = −1, como ramo Q quando∆J = 0 e como ramo R quando ∆J = 1. Vide Exc. 8.4.2.3.
8.2.4.1 Regras de selecao rotacionais
Para transicoes entre estados eletronicos as regras de selecao sao ∆r = 0,±1. Transicoes rotacio-nais podem ocorrer entre nıveis ∆r = ±1. ∆r = 0 nao e permitido, porque violenta a conservacaode paridade. Note tambem, que o isotopo nuclear influencia os nıveis ro-vibracionais via a massareduzida.
Consideramos uma molecula linear no estado |ε, J,MJ〉, onde ε denota o estado eletronico evibracional da molecula. Para achar quais transicoes sao possıveis, precisamos calcular a matriz,
〈ε′, J ′,M ′J |d|ε, J,MJ〉 = 〈J ′,M ′J |dε|J,MJ〉 , (8.61)
com dε = 〈ε|d|ε〉. Aqui, aplicamos a aproximacao de Born-Oppenheimer que permite separara dinamica dos eletrons e tambem as vibracoes da molecula, porque esses movimentos sao taorapidos que sao sempre em estado estacionario, seguindo adiabaticamente o movimento lenta darotacao.
As regras de selecao podem, agora, ser derivadas do teorema de Wigner-Eckart,
〈J ′,M ′J |dε|J,MJ〉|2|〈J ′ ‖ dε ‖ J,MJ〉|2
=1
2J ′ + 1
(J 1 J ′
mJ κ −m′J
). (8.62)
Achamos ∆J = 1 e ∆MJ = 0,±1. Vide Exc. 8.4.2.4.
8.2.5 Computacao dos estados vibracionais
8.2.5.1 Energia de localizacao
Uma consequencia da relacao de incerteza de Heisenberg e, que uma certa energia de localizacaosempre e necessaria para localizar uma partıcula. Como exemplo, consideramos o potencialatrativo,
V = − C
Rn. (8.63)
O espaco disponıvel para a partıcula e limitado pelos pontos de retorno classicos, quem para uma
dada energia e rt =(C|E|
)1/n. O momento correspondente a essa energia e kt =
(2m|E|~2
)1/2. A
8Ver [1], p.326

8.2. ESTRUTURA ROVIBRACIONAL DOS POTENCIAIS MOLECULARES 143
relacao de incerteza de Heisenberg requer ktrt > 2, isto e, pelo menos a metade do comprimentode onda deve caber dentro do potencial (entre 0 e rt) na altura do estado ligado. Portanto,
|E|1−2/n >2~2
mC2/n. (8.64)
Para um potencial coulombiano, com n = 1 e C = e2/4πε0, obtemos a energia do estadofundamental do atomo de hidrogenio,
E > E1 = − e2
4πε02aB, (8.65)
mas nao existe nenhum estado mais alto do que todos os outros. Isto e, todos as energiasEn = E1/n
2 existem.Para n = 2, nao obtemos condicao para a energia. Para o potencial de Casimir-Polder,
n = 3, obtemos
E < − 8~6
m3C2. (8.66)
Isso significa, em contraste com o potencial coulombiano, que a energia de ligacao deve serinferior de um certo limite.
8.2.5.2 Metodo de LeRoy-Bernstein
O metodo de LeRoy-Bernstein permite estimar os nıveis ligados mais altos. So se aplica pertodo limite de dissociacao, onde a formula semiclassica de quantizacao e valida,
v +1
2=
√8Mr
~2
∫ R1
0dR√E(v)− V (R) . (8.67)
Inserindo o potencial
V (R) = De −C
Rn, (8.68)
rende
E(v∗) = De −(
(n− 2)Γ(1 + 1
n
)
2Γ(
12 + 1
n
) (v∗ + vD)
) 2nn−2 ( h2n
(2πMr)nC2
) 1n−2
, (8.69)
onde v∗ e um numero contando os nıveis vibracionais inversamente comecando no limite dedissociacao.
0 2 4-0.5
-0.4
-0.3
-0.2
-0.1
R (µm)
V(R
) (µ
K)
Figura 8.12: Estados vibracionais mais altos obtidos pelo metodo de LeRoy-Bernstein.
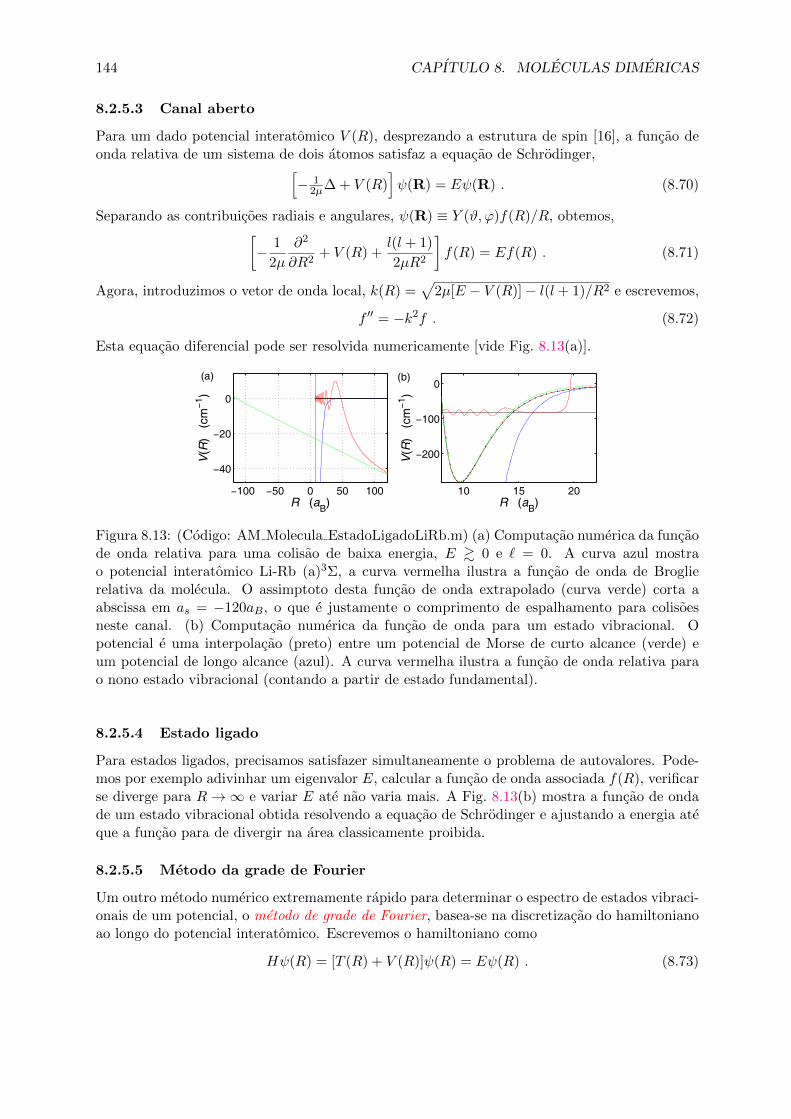
144 CAPITULO 8. MOLECULAS DIMERICAS
8.2.5.3 Canal aberto
Para um dado potencial interatomico V (R), desprezando a estrutura de spin [16], a funcao deonda relativa de um sistema de dois atomos satisfaz a equacao de Schrodinger,
[− 1
2µ∆ + V (R)]ψ(R) = Eψ(R) . (8.70)
Separando as contribuicoes radiais e angulares, ψ(R) ≡ Y (ϑ, ϕ)f(R)/R, obtemos,[− 1
2µ
∂2
∂R2+ V (R) +
l(l + 1)
2µR2
]f(R) = Ef(R) . (8.71)
Agora, introduzimos o vetor de onda local, k(R) =√
2µ[E − V (R)]− l(l + 1)/R2 e escrevemos,
f ′′ = −k2f . (8.72)
Esta equacao diferencial pode ser resolvida numericamente [vide Fig. 8.13(a)].
−100 −50 0 50 100
−40
−20
0
R (aB)
V(R
) (
cm−
1 )
(a)
10 15 20
−200
−100
0
R (aB)
V(R
) (
cm−
1 )
(b)
Figura 8.13: (Codigo: AM Molecula EstadoLigadoLiRb.m) (a) Computacao numerica da funcaode onda relativa para uma colisao de baixa energia, E & 0 e ` = 0. A curva azul mostrao potencial interatomico Li-Rb (a)3Σ, a curva vermelha ilustra a funcao de onda de Broglierelativa da molecula. O assimptoto desta funcao de onda extrapolado (curva verde) corta aabscissa em as = −120aB, o que e justamente o comprimento de espalhamento para colisoesneste canal. (b) Computacao numerica da funcao de onda para um estado vibracional. Opotencial e uma interpolacao (preto) entre um potencial de Morse de curto alcance (verde) eum potencial de longo alcance (azul). A curva vermelha ilustra a funcao de onda relativa parao nono estado vibracional (contando a partir de estado fundamental).
8.2.5.4 Estado ligado
Para estados ligados, precisamos satisfazer simultaneamente o problema de autovalores. Pode-mos por exemplo adivinhar um eigenvalor E, calcular a funcao de onda associada f(R), verificarse diverge para R→∞ e variar E ate nao varia mais. A Fig. 8.13(b) mostra a funcao de ondade um estado vibracional obtida resolvendo a equacao de Schrodinger e ajustando a energia ateque a funcao para de divergir na area classicamente proibida.
8.2.5.5 Metodo da grade de Fourier
Um outro metodo numerico extremamente rapido para determinar o espectro de estados vibraci-onais de um potencial, o metodo de grade de Fourier, basea-se na discretizacao do hamiltonianoao longo do potencial interatomico. Escrevemos o hamiltoniano como
Hψ(R) = [T (R) + V (R)]ψ(R) = Eψ(R) . (8.73)

8.3. FORCAS DE VAN DER WAALS E ACOPLAMENTO AO SPIN 145
Colocamos numa forma matricial usando o conjunto de funcoes da base φi(Rj) = δ(Ri − Rj)com i = 1, .., N , onde Ri = R0 +i(RN−R0)/N . Este problema tem N autovalores Ei. O metodode grade de Fourier agora avalia a energia cinetica em cada ponto da rede. Inserimos os termoslocais Hii = H(Ri) e nao-locais Hij = H(Ri, Rj) dentro do hamiltoniano, bem como as energiaspotenciais Vij = V (Ri)δij . A energia cinetica e a transformada inversa de Fourier do espaco demomento Trs = T (kr)δrs = (k2
r/2µ)δrs e fica [14],
Hij =π2
4µ(RN −R1)2(−1)i−j
(1
sin2 π(i−j)2N
− 1
sin2 π(i+j)2N
)para i 6= j (8.74)
Hij =π2
4µ(RN −R1)2
(2N2 + 1
3− 1
sin2 πi2N
)+ V (Ri) para i = j .
Para melhorar a funcao de onda, podemos interpolar
ψ(q) =∑n
j=1ψ(qj) sinc
π(q − qj)∆q
. (8.75)
O metodo pode ser estendido a canais acoplados σ = A,B via,
Hiσjτ = Tijδστ + Vστ (Ri)δij . (8.76)
O hamiltoniano tem a forma geral
H =
(T 00 T
)+
(VA 00 VB
)+
(WAA WAB
WAB WBB
), (8.77)
onde todas matrizes Vk e Wk sao diagonais 9.
8.3 Forcas de van der Waals e acoplamento ao spin
Os atomos individuais tem uma subestrutura complexa devido aos momentos angulares domovimento eletronico, dos seus spins e do spin nuclear. Todos estes momentos angulares podeminteragir, se acoplar e gerar novos termos de energia, que precisam ser tomadas em conta nahora de calcular os varios potenciais de interacao interatomica,
H =P2
2Mr+ Vcoulomb(R) +
∑
k=1,2
(V
(k)hfs + V (k)
zeeman
)+ Vdipole,spin−spin(R) + Vdipole,spin−orbit(R) .
(8.78)A interacao de Coulomb para gases alcalis interagindos pode ser exprimido como:
Vcoulomb(R) = V S=0coulombPS=0 + V S=1
coulombPS=1 . (8.79)
Os projetores PS=0,1 serao necessarios para expandir o espaco de Hilbert para os graus deliberdade dos spins.
As forcas de van der Waals incluem todas forcas intermoleculares. Sao forcas de longoalcance que ocorrem entre dipolos atomicos permanentes e induzidos ∼ 1/r6 10
9Note, que o metodo da grade de Fourier pode ser melhorado usando uma grade com espacos ajustados aogradiente do potencial [17, 26, 19].
10Elas tambem ocorrem em uma forma pura em ressonadores opticos como efeito de Casimir. Como a frequenciamais baixa numa cavidade e ω =
√2πc/L, as energias do ponto zero dentro e fora da cavidade sao diferentes. Isso
causa uma forca atrativa entre os espelhos da cavidade ∼ 1/r3, 1/r4.
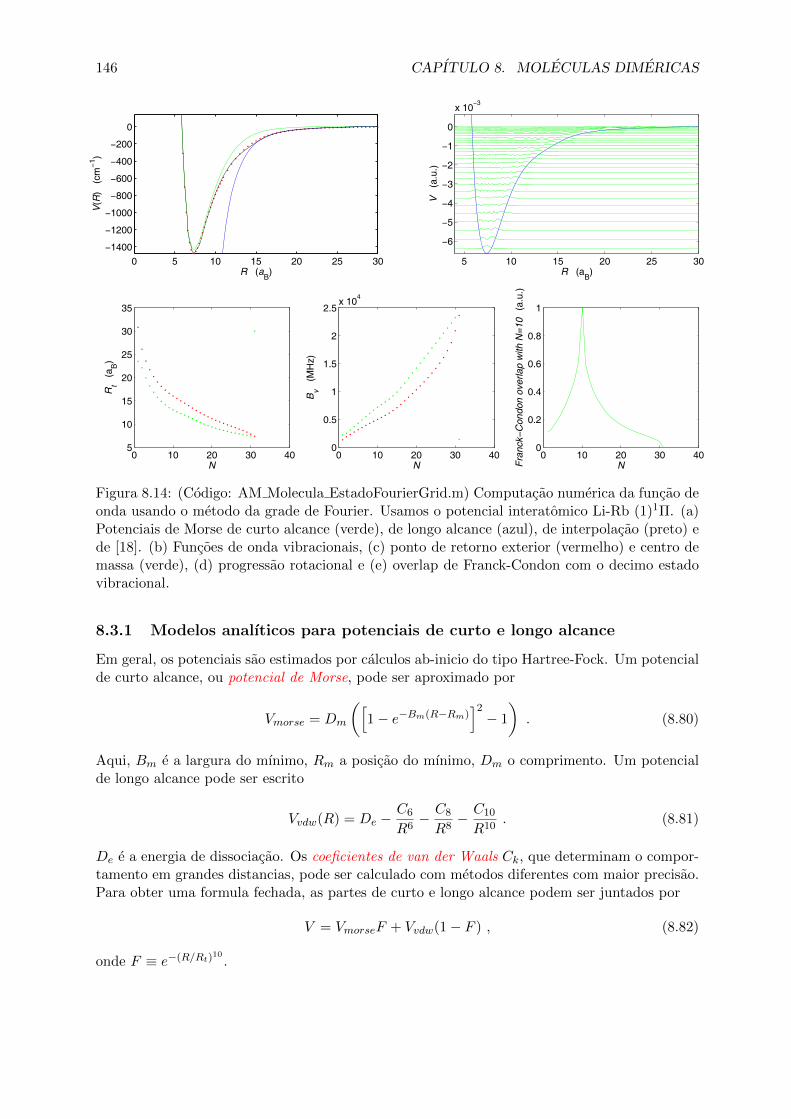
146 CAPITULO 8. MOLECULAS DIMERICAS
0 5 10 15 20 25 30
−1400
−1200
−1000
−800
−600
−400
−200
0
R (aB)
V(R
) (
cm−
1 )
5 10 15 20 25 30
−6
−5
−4
−3
−2
−1
0
x 10−3
R (aB)
V
(a.u
.)
0 10 20 30 405
10
15
20
25
30
35
N
Rt
(aB)
0 10 20 30 400
0.5
1
1.5
2
2.5x 10
4
N
Bv
(MH
z)
0 10 20 30 400
0.2
0.4
0.6
0.8
1
NFra
nck−
Con
don
over
lap
with
N=
10
(a.u
.)
Figura 8.14: (Codigo: AM Molecula EstadoFourierGrid.m) Computacao numerica da funcao deonda usando o metodo da grade de Fourier. Usamos o potencial interatomico Li-Rb (1)1Π. (a)Potenciais de Morse de curto alcance (verde), de longo alcance (azul), de interpolacao (preto) ede [18]. (b) Funcoes de onda vibracionais, (c) ponto de retorno exterior (vermelho) e centro demassa (verde), (d) progressao rotacional e (e) overlap de Franck-Condon com o decimo estadovibracional.
8.3.1 Modelos analıticos para potenciais de curto e longo alcance
Em geral, os potenciais sao estimados por calculos ab-inicio do tipo Hartree-Fock. Um potencialde curto alcance, ou potencial de Morse, pode ser aproximado por
Vmorse = Dm
([1− e−Bm(R−Rm)
]2− 1
). (8.80)
Aqui, Bm e a largura do mınimo, Rm a posicao do mınimo, Dm o comprimento. Um potencialde longo alcance pode ser escrito
Vvdw(R) = De −C6
R6− C8
R8− C10
R10. (8.81)
De e a energia de dissociacao. Os coeficientes de van der Waals Ck, que determinam o compor-tamento em grandes distancias, pode ser calculado com metodos diferentes com maior precisao.Para obter uma formula fechada, as partes de curto e longo alcance podem ser juntados por
V = VmorseF + Vvdw(1− F ) , (8.82)
onde F ≡ e−(R/Rt)10.

8.3. FORCAS DE VAN DER WAALS E ACOPLAMENTO AO SPIN 147
A situacao e diferente para colisoes de atomos identicos em estados excitados, que tem umalcance muito maior por causa da interacao ressonante entre dipolos. Nesse caso, um adicionalpotencial de Movre-Pichler dominado por um coeficiente C3 surge,
V evdw = V e
movre + V edispersion . (8.83)
Em contraste, colisoes em estados excitados de especies diferentes sao puramente de curto al-cance.
8.3.2 Acoplamento de spins em dımeres, numeros quanticos moleculares
Consideramos dois atomos alcalinos interagentes, cada um sendo descrito por um conjunto denumeros quanticos de momentos angulares:
li angular do atomo individual (8.84)
si spin eletronico
ii spin nuclear
li + si = ji momento angular total eletronico
ji + ii = fi momento angular total .
Quando os atomos se aproximam, em distancias intermediarias, eles acoplam seus spins:
` ⊥ ez rotacao molecular (8.85)
Λ ≡ |ML|ez projecao de L sobre o ez interatomico
Σ ≡MS ez projecao de S sobre o ez interatomico
Ω ≡ Λ + Σ projecao de L + S sobre o ez interatomico .
Em curtas distancia, eles formam um dımere molecular descrito pelos numeros quanticos:
L = l1 + l2 momento angular total eletronico (8.86)
S = s1 + s2 spin eletronico
I = i1 + i2 spin nuclear
f = f1 + f2 momento angular total ou (L,S)k + I
J = Ω + `
F = f + ` .
Os numeros quanticos acoplam como,
l1 + l2 = Lez−→ Λ
+ + + +
s1 + s2 = Sez−→ Σ
= = = =
j1 + j2 = jez−→ Ω + ` = J
+ + + +i1 + i2 = I I= = = =f1 + f2 = f + ` = F
(8.87)
As varias opcoes como L, S e j sao projetados sobre o eixo internuclear ou diretamenteacoplam ao momento angular rotacional ` sao tratadas nos casos de Hund (a) ate (e). Oacoplamento de spin e descrito por sımbolos de 9j, como mostrado embaixo.

148 CAPITULO 8. MOLECULAS DIMERICAS
8.3.3 Os casos de acoplamento de Hund
A forca de acoplamento entre spins atomicos depende da distancia entre os atomos. Devido avariedade de spins nos atomos existem muitas possibilidades como eles podem se acoplar. Estasforam classificadas por Hund em cinco casos.
Figura 8.15: Hund’s coupling cases.
8.3.3.1 Caso de Hund (a)
A interacao molecular e tao forte, que L e S acoplam para o eixo ez em vez mutuamente. Estecaso e analogo ao efeito de Paschen-Back,
L→ Λ e S→ Σ (8.88)
((Λ,Σ)Ω, `)J .
Uma notacao comum consiste em rotular os estados Λ = Σ,Π,∆, .... Ou seja, no sımboloX(2S+1ΛΩ)±σ , onde σ = g, u e a simetria de inversao, X,A,B, .. e a, b, .. sao as series singleto etripleto comecando a partir dos nıveis energeticamente mais baixos. Uma notacao alternativaconsiste em assinar rotulos ordenados por energia X = (1), (2), ... Finalmente, ± e a simetriade reflexao. Por exemplo, X1Σ+
g .
8.3.3.2 Caso de Hund (b)
L e projetado sobre eixo ez antes de se acoplar ao `. O momento angular resultante depoisacopla ao S diretamente.
L→ Λ (8.89)
((L, `)k,S)J .
8.3.3.3 Caso de Hund (c)
L e S acoplam juntos em vez de se projetar sobre o eixo ez. Este caso e analogo ao efeitoZeeman,
(L,S)j→ Ω (8.90)
(Ω, `)J .

8.4. EXERCICIOS 149
Uma notacao comum consiste em rotular os estados por Ω = 0, 1, 2, .... Ou seja, no sımboloX(Ω)±s , a letra X = 1, 2, .. e um rotulo ordenado por energia. Por exemplo 2(0−g ).
8.3.3.4 Caso de Hund (d)
L nao e projetado sobre o eixo ez, mas se acopla diretamente ao momento angular rotacional.O momento angular resultante depois somente acopla ao S
((L, `)k, )J . (8.91)
8.3.3.5 Caso de Hund (e)
L e S se acoplam mutuamente, como no caso (c), mas nao sao projetados sobre o eixo ez, masacoplam diretamente com `, o que e quantizado,
((L,S)j, `)J . (8.92)
8.4 Exercıcios
8.4.1 Ligacao molecular
8.4.1.1 Ex: Modelo classico da ligacao covalente
Considere a molecula H+2 com os dois nucleos separados de 1 nm e um eletron localizado no
meio entre os nucleos. Calcule a forca eletrostatica agindo sobre os nucleos.
8.4.1.2 Ex: Modelo classico da ligacao covalente
Calcule as energias do estado fundamental e do estado menos ligado do potencial Vn = − Crn
para qualquer n.
8.4.1.3 Ex: Colisao homonuclear
Consideramos o exemplo de colisoes homonucleares 85Rb. Para colisoes em estados fundamentaisno canal 3Σ+, |f = 2,mf = −2〉, as partes de longo alcance do potencial e fixo por C6 = 4550,C8 = 550600 e C10 = 7.67 × 107 [9, 22], onde Rm = 9.8aB, Dm = 0.13, e Bm = 1/2.5aB. Ospotenciais podem ser mesclados numa dada distancia Rt = 27.6aB. Faz um plot do potencial.
8.4.1.4 Ex: Alem da aproximacao de Born-Oppenheimer para moleculas porCarlos M. de Oliveira Bastos
Para moleculas, a aproximacao de Born-Oppenheimer pode falhar em algumas situacoes. Porisso, e comum a utilizacao de uma outra aproximacao conhecida como Born-Huang. Para ilustraressa aproximacao, consideramos uma molecula diatomica no referencial do laboratorio.a. Escreva o hamiltoniano de muitos corpos, em unidades atomicas, para a molecula.b. Se mudarmos o sistema de coordenadas para a posicao do centro de massa dos nucleos damolecula, eliminamos a dependencia com a translacao global da molecula. O Hamiltoniano passaa ser dado por
− ∇2R
2µAB−∑
i,j
1
2M∇i∇j −
∑
i
∇2i
2+ V

150 CAPITULO 8. MOLECULAS DIMERICAS
sendo que as interacoes coulombianas estao incluıdas no termo V. Escreva a equacao de Schrodin-ger independente do tempo para essa molecula.c. A aproximacao Born-Huang consiste em supor que a funcao de onda total possa ser expandidaem uma base de funcoes de ondas dos nucleos e dos eletrons, ou seja,
Ψ(r,R) =∑
k
|χk(R)〉|φk(r,R)〉 ,
onde χ e φ sao as funcoes de ondas dos nucleos e os eletrons respectivamente. Para a equacao deSchrodinger calculada no item anterior, utilize a aproximacao Born-Huang e obtenha o conjuntode equacoes acopladas
∑
k
[− 1
2µAB
(∇2R + 〈φk|∇2
R|φk〉+ 2〈φk|∇R|φk〉.∇R|)]−
∑
k
1
2M
∑
i,j
〈φi|∇i.∇j |φk〉
+
1
2
∑
i
〈φi|∇2i |φk〉 − 〈φi|V |φk〉|
|χk〉 = E
∑
k
|χk〉
que inclui, ainda que aproximadamente, a energia cinetica dos nucleos e eletrons. Dica: Utilize∇2(αβ) = α∇2β + β∇2α+ 2∇α · ∇β.d. Faca uma breve comparacao entre a aproximacao de Born-Huang (e as equacoes acopladasobtidas na equacao anterior) e a aproximacao de Born-Oppenheimer.
8.4.2 Estrutura rovibracional dos potenciais moleculares
8.4.2.1 Ex: Transicoes entre estados vibracionais
Calcule o momento dipolar entre dois estados vibracionais arbitrarios de (a) um potencialharmonico e (b) um potencial de Morse.
8.4.2.2 Ex: Espectro rotacional de moleculas diatomicas
Calcule o espectro rotacional para uma molecula diatomica.
8.4.2.3 Ex: Espectro ro-vibracional
Determine os espectros de frequencia de transicoes ro-vibracionais para os ramos P , Q e R.
8.4.2.4 Ex: Espectro rotacional
Determine as regras e o espectro de transicoes rotacionais para uma molecula esferica.
8.4.2.5 Ex: Metodo da grade de Fourier
A transformacao de Fourier rapida (FFT) e definida por,
Hn =∑N−1
k=0e−2πink/Nhk =
∑N−1
k=0e−2πink/(N/2)h2k+e
−2πik/N∑N/2−1
k=0e−2πink/(N/2)h2k+1 = even+odd .
a transformada inversa e,
hk =1
N
∑N−1
k=0e2πink/NHn .

8.4. EXERCICIOS 151
A transformada senis de um vetor real sk e,
Sn =2
N
∑N−1
k=1sk sinπnk/N .
Calcule a transformada inversa da matriz Trs = k2rδrs.
8.4.3 Forcas de van der Waals e acoplamento ao spin

152 CAPITULO 8. MOLECULAS DIMERICAS

Capıtulo 9
Colisoes
9.1 Teoria de espalhamento
Neste capıtulo consideramos fenomenos de espalhamento por potenciais independentes do temposatisfazendo rV (r → ∞) → 0, isto e, potenciais de curto alcance. O potencial pode ter regioesatrativas suportando estados ligados com energias E < 0. Aqui, no entanto, so consideramosestados E > 0. Como a situacao e independente do tempo, ∂tH = 0, podemos considerarproblemas independentes do tempo,
Hψk(r) = Ekψk(r) , (9.1)
com H = p2/2m + V (r) e Ek = ~2k2/2m. As condicoes de contorno sao dadas pela geometriado espalhamento, tal que para grandes distancias a funcao de onda se comporta como (videFig. 9.1),
ψk(r) ∼ eik·r + fk(Ω)eiksr
r. (9.2)
Para processos de espalhamento elasticos temos ks = k. A amplitude de espalhamento fk(Ω)depende da energia Ek e do angulo solido de espalhamento. Experimentalmente, espalhamospartıculas individuais descritas por pacotes de ondas. Como a teoria de espalhamento e linear,podemos descrever os pacotes por superposicoes de solucoes estacionarias ψk
1.
Figura 9.1: Espalhamento de luz incidente (vetor de onda k0) por um potencial V .
9.1.1 Equacao de Lippmann-Schwinger
Para considerar p.ex. duas partıculas envolvidas numa colisao podemos ir no sistema do movi-mento relativo (usando as massas reduzidas), fixar a origem do sistema de coordenadas numadas partıculas e analisar a trajetoria da segunda partıcula dentro do potencial.
A teoria de espalhamento sendo baseada no metodo de Green, que nos ja conhecemos daeletrostatica, vamos primeiro lembrar o uso da funcao de Green para resolver problemas ele-trostaticos.
1Note, que k nao e um numero quantico, pois ψk contem componentes de momento 6= k.
153

154 CAPITULO 9. COLISOES
Exemplo 16 (Metodo de Green na eletrostatica): Da terceira equacao de Maxwellobtemos,
∇2φ(r) = −ε−10 ρ(r) .
Sendo definida por,∇2G(r) = δ3(r) ,
a funcao de Green fica,
G(r) =−1
4π
1
|r| .
Com isso, achamos a solucao da equacao de Maxwell,
φ(r) =(−G ? ε−1
0 ρ(r))
(r) = − 1
ε0
∫
V
ρ(r0)G(r− r0)d3r0 =1
4πε0
∫
V
ρ(x)
|r− x|d3x ,
conhecida como lei de Poisson.
9.1.1.1 Metodo de Green na mecanica quantica
O procedimento chamado de metodo de Green pode ser utilizado para resolucao de equacao deSchrodinger com a condicao de contorno (9.2). Comecamos a partir da equacao estacionaria deSchrodinger reduzida (9.1) [24]. A equacao de Schrodinger fica,
(∆ + k2)ψk(r) = 2m~2 V (r)ψk(r) . (9.3)
Esta equacao nao e um problema de autovalores comum, pois cada energia Ek gera uma solucao.A equacao (9.3) e uma equacao diferencial inomogenea parcial com o lado esquerdo descrevendoa propagacao livre e o lado direito descrevendo uma fonte que depende da solucao. Tais equacoesdiferencias geralmente sao resolvidas utilizando funcoes de Green. Escolhemos uma fonte pun-tiforme e resolvemos,
(∆ + k2)G(r, k) = δ3(r) , (9.4)
juntamente com as condicoes de contorno. A solucao adota a forma [4],
G(r, k) = − 1
4π
eik|r|
|r| , (9.5)
tal que,
ψk(r) = eik·r +(G ? 2m
~2 V ψk
)(r) = eik·r + 2m
~2
∫
Vd3r′G(r− r′, k)V (r′)ψk(r′) . (9.6)
A equacao (9.6) se chama equacao de Lippmann-Schwinger. E claro que esta equacao nao resolve,mas so reformula o problema tomando em conta as condicoes de contorno. Ela se presta bempara implementacao de aproximacoes. Vide Exc. 9.3.1.1 e 9.3.1.2.
Agora vamos considerar o campo longe, r →∞, para verificar o comportamento assimptoticoe encontrar uma expressao para fk(Ω) em funcao de V (r). Para r →∞ podemos aproximar
k|r− r′| = kr√
(er − r′/r)2 = kr√
1− 2er · r′/r + (r′/r)2 ' kr − k′ · r′ ' kr , (9.7)
com k′ ≡ ker. Com isso a equacao de Lippmann-Schwinger (9.6) fica,
ψ(r)→ eik·r − 2m
~2
∫
V
1
4π
eik|r−r′|
|r− r′| V (r′)ψ(r′)d3r′ (9.8)
= eik·r − 2m
4π~2
eikr
kr
∫
Ve−ik
′·r′V (r′)ψ(r′)d3r′ ≡ ψin+ fk(Ω)eikr
r,

9.1. TEORIA DE ESPALHAMENTO 155
dando, em comparacao com a expressao (9.2), a amplitude de espalhamento,
fk(Ω) =2m
4π~2
∫
Ve−ik
′·r′V (r′)ψ(r′)d3r′ . (9.9)
A partir das funcoes de onda ψin ≡ eik·r e ψs ≡ fk(Ω)eikr/r podemos calcular as densidadesde corrente,
Jin =~
2mi(ψ†in∇ψin − c.c.) =
~k
m(9.10)
Js =~
2mi(ψ†s∂rψs − c.c.)er =
~k′
m
1
r2|fk(Ω)|2 +O(r−3) .
O numero dI(Ω) de partıculas espalhadas por segundo para o angulo solido dΩ e simplesmentedI(Ω) = |Js|r2dΩ. Com isso podemos calcular a secao eficaz diferencial definida pela razao entredI(Ω) e o numero |Jin| de partıculas incidentes por segundo,
dσ
dΩ≡ dI(Ω)
|Jin|dΩ= |fk(Ω)|2 . (9.11)
Finalmente definimos a secao eficaz total,
σ =
∫dΩ|fk(Ω)|2 . (9.12)
9.1.2 Pacotes de onda
Deixamos incidir o pacote de onda definido num tempo t = t0,
ψ(r, t0) =
∫d3k
(2π)3ake
ik·r , (9.13)
sobre o potencial espalhador. A amplitude ak seja concentrada em torne de k0, tal que o pacotese aproxima do espalhador com a velocidade v0 = ~k0/m. A evolucao temporal da funcao deonda ψ(r, t) determina o sinal medido por um detector num tempo t > t0 posterior. A nossatarefa consiste em determinar ψ(r, t > t0). Os estados espalhados ψk resolvendo a equacao deSchrodinger (9.1) sao completos no espaco das funcoes de onda estendidas, e podemos escrevera evolucao temporal como,
ψ(r, t) =
∫d3k
(2π)3Akψk(r)e−iEk(t−t0)/~ . (9.14)
No tempo t0 os resultados (9.13) e (9.14) devem coincidir. Para verificar isso, escrevemos (9.13)substituindo a onda plana eik·r usando a equacao de Lippmann-Schwinger (9.6) com a funcaode Green (9.5), e depois comparamos os coeficientes,
ψ(r, t0) =
∫d3k
(2π)3ak
[ψk(r) +
m
2π~2
∫d3r′
eik|r−r′|
|r− r′| V (r′)ψk(r′)
]. (9.15)
O processo de espalhamento e ilustrado na Fig.9.2. Para simplificar o calculo do segundo

156 CAPITULO 9. COLISOES
Figura 9.2: Espalhamento de pacotes de onda por um potencial.
termo nesta equacao, supomos que ψk seja suave, isto e, nao haja ressonancias, tal que podemosaproximar, ψk ' ψk0 . Com k ' k · ek0 obtemos,
∫d3k
(2π)3ake
ik|r−r′|ψk(r′) =
∫d3k
(2π)3ake
ik·(ek0|r−r′|)ψk0(r′)
(9.13)= ψ(ek0 |r−r′|, t0)ψk0(r′) . (9.16)
Aqui, ψ(ek0 |r− r′|, t0) e o pacote incidente avaliado para direito, onde, por definicao, e ' 0. Aexpressao (9.15) portanto tem a forma,
ψ(r, t0) =
∫d3k
(2π)3akψk(r) , (9.17)
e a comparacao dos coeficientes com (9.14) da, Ak = ak. Finalmente, avaliamos ψ(r, t) parao tempo de deteccao t > t0 para entender, que a analise estacionaria acima realmente estafisicamente correta. Seguinte (9.14) temos,
ψ(r, t) =
∫d3k
(2π)3Akψk(r)e−iEk(t−t0)/~ (9.8)(9.13)' ψ0(r, t) +
∫d3k
(2π)3akeikr
rfk(Ω)e−iEk(t−t0)/~ .
(9.18)Portanto, ψ0(r, t) descreve a evolucao do pacote de onda sem espalhador,
ψ0(r, t) =
∫d3k
(2π)3ake
ik·r
︸ ︷︷ ︸ψ(r,t0)
e−iEk(t−t0)/~ . (9.19)
Se fk e suave em torno de k = k0, o que nos permite colocar esta amplitude (fk ' fk0) emfrente da integral, e com k ' k · k0 obtemos,
ψ(r, t)t grande
=⇒ ψ0(r, t)︸ ︷︷ ︸pacote nao espalhado
+fk0(Ω)
rψ0(k0r, t)
︸ ︷︷ ︸pacote espalhado
. (9.20)
O processo de espalhamento e mostrado na Fig. 9.2: Seguinte a ultima equacao o processode espalhamento envolve a superposicao do pacote nao espalhado e um pacote espalhado nadirecao Ω. Este ultimo envolve a amplitude Ψ0(k0r, t) de um pacote propagando para direcaopara frente, que so precisa ser avaliado no tempo e na distancia certa. Este pacote sera depoismultiplicado com a amplitude descrevendo a dependencia angula fk0 ; o angulo, portanto, soaparece atraves desta amplitude e nao na funcao de onda ψ0. A analise acima nao pode seraplicada em duas situacoes:
• quando V e de longo alcance, p.ex. , V = 1/r,
• quando a energia incidente Ek e ressonante.

9.1. TEORIA DE ESPALHAMENTO 157
9.1.3 Aproximacao de Born
A equacao de Lippmann-Schwinger sugere a seguinte iteracao perturbativa chamada de serie deBorn [4],
ψ(r) = ψi(r) +(G ? 2m
~2 V ψ)
(r) (9.21)
= ψi(r) + 2m~2 (G ? V ψi)(r) +
(2m~2
)2[G ? V (G ? V ψi)](r)
= ψi(r) + 2m~2
∫
VG(r− r′)V (r′)ψi(r′)d3r′
+(
2m~2
)2∫
VG(r− r′)V (r′)G(r− r′′)V (r′′)ψi(r′′)d3r′d3r′′ .
Na chamada aproximacao de Born consideramos somente a primeira ordem e inserindo umaonda plana, ψi(r) = eikz/(2π)3/2, obtemos,
ψ(r) =eikz
(2π)3/2− m
(2π)3/22π~2
∫
V
eik|r−r′|
|r− r′| V (r′)eikz′d3r′ . (9.22)
O comportamento assimptotico, r r′, segue com (9.7) usando z′ = r′ · ez e definindo ks = kere ki = kez,
ψ(r) ' eikz
(2π)3/2− m
(2π)3/22π~2
∫
V
eik(r−r·r′/r)
rV (r′)eik·r
′d3r′ (9.23)
=eikz
(2π)3/2+
m
(2π)3/22π~2
eikr
r
∫
VV (r′)ei(ki−ks)·r
′d3r′
≡ 1
(2π)3/2
(eikz +
eikr
rf(ki, ks)
),
com
f(ki, ks) ≡m
2π~2
∫
VV (r′)ei(ki−ks)·r
′d3r′ = − m
2π~2〈ks|V |ki〉 .
9.1.4 Potenciais esfericos
Para potenciais de espalhamento esfericamente simetricos, V (r) = V (r), o hamiltoniano H =p2/2m+V (r) comuta com os operadores de rotacao U~ω = e−i~ω·L/~ em torno de qualquer eixo eφ.Portanto, podemos separar o problema angular e decompor o problema de espalhamento seguinteas representacoes irreduzıveis do grupo de rotacao. Esta decomposicao em ondas parciais podeser escrita,
ψk(r) =
∞∑
`=0
(2`+ 1)i`P`(cos θ)Rl(r) , (9.24)
onde o fator (2` + 1)i` e uma convencao facilitando o calculo posteriormente. Inserindo esteansatz de separacao das variaveis radiais e angulares na equacao estacionaria de Schrodinger(9.1), obtemos a equacao de Schrodinger radial,
[∂2
∂r2− `(`+ 1)
r2+ k2
]rR`(r) =
2m
~2V (r)rR` = 0 , (9.25)

158 CAPITULO 9. COLISOES
onde ψk deve satisfazer as condicoes de contorno (9.2). Felizmente, podemos expandir tambema onda incidente por ondas parciais 2,
eikz = eir cos θ =∞∑
`=0
(2`+ 1)i`j`(kr)P`(cos θ) . (9.26)
Usamos agora o resultado (9.26) para encontrar as condicoes de contorno para as ondasradiais R`. No infinito temos rV (r)
r→∞−→ 0. Por isso,
R`(r)r→∞−→ α`[h
(2)` (kr) + s`h
(1)` (kr)] , (9.27)
onde as funcoes de Hankel h(1,2)` (kr) ∼ e±i(ρ−(`+1)π/2) descrevem, respetivamente, ondas esfericas
incidentes (h(2)` ) e saindo (h
(1)` ).
Para determinar os coeficientes α` e s` notamos primeiramente, que sem potencial, V (r) = 0,a solucao da equacao radial (9.25) e conhecida,
R`(r) = j`(kr) = 12 [h
(2)` (kr) + h
(1)` (kr)] , (9.28)
tal que α` = 12 e s` = 1. Para V (r) 6= 0 a onda incidente h
(2)` e a mesma, mais nao a incidente
h(1)` resultando em s` 6= 1. No entanto, a conservacao do numero de partıcula requer, que o
numero de partıculas entrando seja igual ao numero de partıculas saindo. Isto e, o fluxo radialtotal deve estar,
0 = j`r(r) =~
2im[R∗`∂rR` −R`∂rR∗` ] =
~4mkr2
[|s`|2 − 1] , (9.29)
aproximando 2R` ' e−i(kr+w`kr + s`
ei(kr+w`kr . Portanto, |s`| = 1, ou seja,
s` = e2iδ`(k) , (9.30)
onde δ`(k) e a fase do espalhamento. A fase do espalhamento determina a solucao do problemado espalhamento, porque fixa a amplitude de espalhamento: Avaliando a solucao (9.24) noregime assimptotico pela formula (9.26),
ψk(r) ∼ 12
∞∑
`=0
(2`+ 1)i`P`(cos θ)[h(2)` (kr) + e2iδ`h
(1)` (kr)] (9.31)
= eik·r + 12
∞∑
`=0
(2`+ 1)i`P`(cos θ)[e2iδ` − 1]h(1)` (kr) = eik·r + fk(θ)
eikr
r,
obtemos a amplitude de espalhamento na forma 3
fk(θ) = 1k
∑
`
(2`+ 1)P`(cos θ)eiδ` sin δ` . (9.32)
2Para o caso mais geral de vetores arbitrarios k e r, usamos o teorema de adicao para Y`m e exprimimosP`(cos θ) por funcoes esfericas,
eik·r = 4π
∞∑`=0
∑m=−`
i`j`(kr)Y∗`m(Ωk)Y`m(Ωr) .
3Com h(1)` ∼ (−i)`+1 eikr
kr.

9.1. TEORIA DE ESPALHAMENTO 159
Chamamose2iδ` − 1
2ik=eiδ` sin δ`
k≡ f` (9.33)
de amplitude da onda parcial [28, 6].
9.1.5 Fase e comprimento de espalhamento
Resumindo, podemos dentro da aproximacao de Born, exprimir o estado de qualquer tipo departıculas colidindo por potenciais isotropicos como,
ψ(r) ∼ eik·r +eikr
rfk(Ω) . (9.34)
A secao de choque pode ser escrita,
σ =
∫dΩ|fk(Ω)|2 = 1
k2
∫dΩ|
∑
`
(2`+ 1)P`(cos θ) sin δ`|2 (9.35)
=4π
k2
∑
`
(2`+ 1) sin2 δ` = 4π∑
`
(2`+ 1)|f`|2 .
A grandeza
σ` =4π
k2(2`+ 1) sin2 δ` =
4π
k2(2`+ 1)|f`|2 (9.36)
se chama secao eficaz parcial. Obviamente, vale σ` ≤ 4πk2 (2` + 1). O deslocamento de fase e2iδ`
tem uma interpretacao fısica simples: Consideramos a funcao,
eiδ`j`(kr + δ`) = eiδ`2 [h
(2)` (kr + δ`) + h
(1)` (kr + δ`)] (9.37)
∼ eiδ`
2
[(−i)`ei(kr+δ`)
kr + δ`+
(+i)`e−i(kr+δ`)
kr + δ`
]krδ`−→ 1
2 [h(2)` + e2iδ`h
(1)` ] ∼ R` .
Agora comparando o caso V = 0 dando R`(r) = j`(kr) com o caso V 6= 0 dando R`(r) ∼eiδ`j`(kr + δ`), percebemos, que um deslocamento positivo, δ` > 0, puxa a funcao de onda paradentro do potencial, enquanto um deslocamento negativo, δ` < 0, empurra a funcao de ondapara fora, como ilustrado na Fig. 9.3.
V r( )
r
r
| |Y | |Y
0 0
V r( )
para =0V
para =0V
d
para =0V
para =0V
d
Figura 9.3: Deslocamento de fase δ`(k) da funcao de onda espalhada. Um potencial atrativo(esquerda) aumenta a energia cinetica e a funcao de onda oscila mais rapido, o que lida a umdeslocamento de fase positivo. Do outro lado, um potencial repulsivo (direito) desacelera aoscilacao da funcao de onda e produz um δ`(k) negativo.

160 CAPITULO 9. COLISOES
9.1.6 Teorema optico
Considere a amplitude para espalhamento para frente f(0) escrevendo sua parte imaginariacomo,
Im f(0) = 1k
∑
k
(2`+ 1)P`(cos θ) sin2 δ`∣∣θ=0
= 1k
∑
k
(2`+ 1) sin2 δ` ≡k
4πσ . (9.38)
Com isso obtemos o teorema optico,
σ = 4πk Im f(0) . (9.39)
A razao mais profunda do teorema optico e a conservacao do numero de partıculas: O fluxo departıculas espalhadas, (~k/m)σ = Isct, deve ser extraıdo do fluxo incidente I0 por espalhamento,e portanto, falta na direcao para frente. E a interferencia da onda espalhada com a ondaincidente, que diminuı a onda nao espalhada e, portanto, cria uma sombra do espalhador nadirecao a frente. As partıculas faltando na sombra do espalhador sao justamente aqueles queforam espalhadas. E isso a mensagem do teorema optico, que sempre e valido, quando nao temprocessos aprisionando ou transformando partıculas.
9.1.6.1 Aproximacao de Born para a fase de espalhamento
O problema do espalhamento pode ser considerado resolvido, quando conhecemos a amplitude deespalhamento fk(θ), pois essa nos da o fluxo medido pelo detector. E fk(θ) e conhecido, quandosabemos as fases de espalhamento δ`k. Estas geralmente devem ser determinadas por integracaoda equacao radial (9.25). Aqui esperamos, que somente momentos angulares ` < kR0 (R0 eo alcance do potencial) fazem deslocamentos de fase significativos. Partıculas com momentosangulares maiores tem parametros de colisao b ∼ `/k fora do alcance do potencial. Percebemosque ondas parciais s sao sempre espalhadas, enquanto ondas parciais p (ou mais elevadas)sao fracamente espalhadas quando a energia e fraca, E < ~2/2mR2
0. Nestes casos um calculo
aproximativo de δ` e suficiente: Inserimos (9.24) e eikz =∑
`12
√4π(2`+ 1)
∫ 1−1 dzP`(z)e
ikz em(9.9) e a integracao sobre Ω′ da,
fk(θ) =2m
4π~2
∫
Ve−ik
′·r′V (r′)ψ(r′)d3r′ (9.40)
= −2m~2
∞∑
`=0
(2`+ 1)P`(cos θ)
∫ ∞
0dr r2V (r)j`(kr)R`(r) .
Comparando esta formula com (9.32) encontramos,
eiδ` sin δ` = −2mk~2
∫ ∞
0dr r2V (r)j`(kr)R`(r)
R`'j`' −2mk~2
∫ ∞
0dr r2V (r)j2
` (kr) . (9.41)
O resultado (6.58) e a aproximacao de Born para a fase de espalhamento δ`(k). Note que R` ' j`nao e uma boa aproximacao, onde V e grande e R` fortemente suprimido (p.ex., esfera dura).Para ` grande j` ∼ r`, e δ` fica pequeno para um potencial V (r) limitado.
9.1.6.2 Analiticidade de s`(E)
Consideramos um potencial de curto alcance que desaparece para r > R0. A solucao radial forado alcance do potencial entao sera dada por,
R`(r) = 12 [h
(2)` (kr) + s`h
(1)` (kr)] , (9.42)

9.1. TEORIA DE ESPALHAMENTO 161
enquanto para r < R0 a solucao R` deve ser encontrada por integracao da equacao radial (9.25).A fase de espalhamento s` deve ser escolhida de maneira que R` e ∂rR` sejam contınuo em R0.O fator de normalizacao zera na derivada logarıtmica, tal que,
γ` ≡ ∂r lnR`|R−0 =1
R`
∂R`∂r
∣∣∣∣R−0
=∂rh
(2)` + s`∂rh
(1)`
h(2)` + s`h
(1)`
∣∣∣∣∣R+
0
. (9.43)
Agora 4
s` − 1 =2(∂r − γ`)j`(γ` − ∂r)h(1)
`
∣∣∣∣∣R0
(9.44)
ou com s` − 1 = 2icot δ`−i exprimindo δ` por γ`,
cot δ` =(∂r − γ`)n`(∂r − γ`)j`
∣∣∣∣R0
. (9.45)
A secao eficaz parcial e,
σ` =4π
k2(2`+ 1) sin2 δ` =
4π
k2
2`+ 1
1 + cot2 δ`. (9.46)
Analisando as expressoes para s`(cot δ`) e σ`(cot δ`) achamos, que
• para cos δ` = i a fase de espalhamento s` tem um polo e σ` →∞;
• para cos δ` = 0 a fase de espalhamento e s` − 1 e σ` = 4π(2`+ 1)/k2 e maximo.
Os polos de s` sao justamente os estados ligados: Para um estado ligado vale assimptoticamente
R`(r) ∼ h(1)` (iκr) ∝ e−κr com a energia de ligacao EB = −~2κ2/2m. A condicao de continuidade
e dada por,
γ` =∂rh
(1)`
h(1)`
∣∣∣∣∣R0
, (9.47)
e a insercao na condicao de continuidade geral (9.45) da,
cot δ` =h
(1)` ∂rn` − n`∂rh(1)
`
h(1)` ∂rj` − j`∂rh(1)
`
= i . (9.48)
Da mesma maneira os cruzamentos de zero de cot δ` correspondem justamente a ressonanciasde espalhamento. Para ver isso, expandimos em torno de uma ressonancia,
cot δ`(E) ' cot δ`(Er)−1
sin2 δ`
dδ`dE
∣∣∣∣Er
(E −Er) = − dδ`dE
∣∣∣∣Er
(E −Er) ≡ −2
Γr(E −Er) , (9.49)
definindo a largura Γr = 2∂Eδ`
∣∣∣Er
do pico da ressonancia na secao eficaz σ` da forma,
σ` =4π
k2(2`+ 1)
(Γr/2)2
(E − Er)2 + (Γr/2)2. (9.50)
Vide a Fig. 9.4,
s` − 1 =−iΓr
E − (Er − iγr/2). (9.51)
A fase de espalhamento δ` cresce de π. O valor δ`(E = 0) da o numero de estados ligados,δ`(0) = n`ligadoπ.
4Temos para as funcoes de Hankel esfericas: h(1,2)` (x) = j`(x)± iy`(x).

162 CAPITULO 9. COLISOES
6.4. ROTATIONSSYMMETRISCHE POTENTIALE V (R) 175
und einsetzen in die allgemeine Stetigkeitsbedingung (6.62) ergibt
cot !l =h(1)
l &rnl # nl &rh(1)l
h(1)l &rjl # jl &rh
(1)l
h(1)l =jl+inl= i. (6.66)
Ebenso entsprechen die Nullstellen von cot !l gerade den Streuresonanzen:wir entwickeln um die Resonanz herum,
cot !l(E) * cot !l(Er)#1
sin2 !l
d!l
dE
----Er
(E # Er)
= #d!l
dE
-----Er
(E # Er) (6.67)
1 mit: 'r =2
&E!l
---Er
4 # 2'r
(E # Er). (6.68)
Damit erhalten wir einen Resonanzpeak der Breite 'r im partiellen Wir-kungsquerschnitt 0l mit der Form
0l =4$
k2(2l + 1)
('r/2)2
(E # Er)2 + ('r/2)2, (6.69)
vgl. Abb. 6.6. Die Streuamplitude sl(E) hat einen Pol in der 2. Riemann-
! l
( +1)n
EC
E E
"n
"
l#
ErrE
Er
$r$r
$ /2r!i
Abb. 6.6: Resonanzpeak der Breite 'r: Links der Wirkungsquerschnitt 0l(E)und rechts die Streuphase !l(E); in der Mitte ist die Lage des Poles in derkomplexen E-Ebene skizziert.
Ebene der komplexen Energieebene,
sl # 1 =#i'r
E # (Er # i'r/2); (6.70)
Figura 9.4: Pico de ressonancia de largura Γr: (Esquerda) a secao eficaz σ`(E), (direita) a fasede espalhamento δ`(E) e (centro) esquema da posicao do polo no plano complexo de energia E.
9.2 Colisoes de atomos frios
Tecnicas modernas desenvolvidas na area da optica atomica permitem resfriar gases atomicos atetemperaturas bem abaixo de 1 µK. Usamos as expansoes j` ∼ x`/(2`+1)!! e n` ∼ (2`−1)!!/x`+1
na equacao (9.74), e obtemos para kR0 1,
cos δ` '2`+ 1)!!(2`− 1)!!
(kR0)2`+1
`+ 1 +R0α`(E)
`−R0α`(E). (9.52)
Uma aproximacao grossa leva a
cos δ` =cos δ`sin δ`
δ`1' 1
sin δ`' 1
(R0k)2`+1, (9.53)
ou seja,sin δ` ' (R0k)2`+1 , (9.54)
isto e, as fases de espalhamento diminuem, no regime de colisoes frias, rapidamente com `crescentes e colisoes tipo ` = 0 dominam,
k cot δ0α`(E)'α`(0)' −1 +R0α0(0)
R20α`(E)
. (9.55)
O comprimento de espalhamento s definido por,
as ≡R2
0α`(E)
1 +R0α0(0)=
sin δ0
k(9.56)
entao e o unico parametro relevante da colisao. Para R0α0 1 achamos a ' R0. Por exemplo,para uma esfera dura temos R`(R0) = 0, α` = ∞, a = R0 > 0 e cot δ0 = −1/kR0. Parapequenos kR0 obtemos δ0 ' −kR0 < 0 o que corresponde a um deslocamento de fase negativopara o potencial repulsivo, como esperado. A secao eficaz e,
σ0 =4π
k2
1
1 + cot2 δ0' 4π
k2 + 1/a2s
. (9.57)
Em comparacao a secao eficaz para momentos angulares mais elevados, σ` ∝ sin2 δ`k2 se comporta
como,σ` ∝ R2
0(R0k)4` → 0 . (9.58)
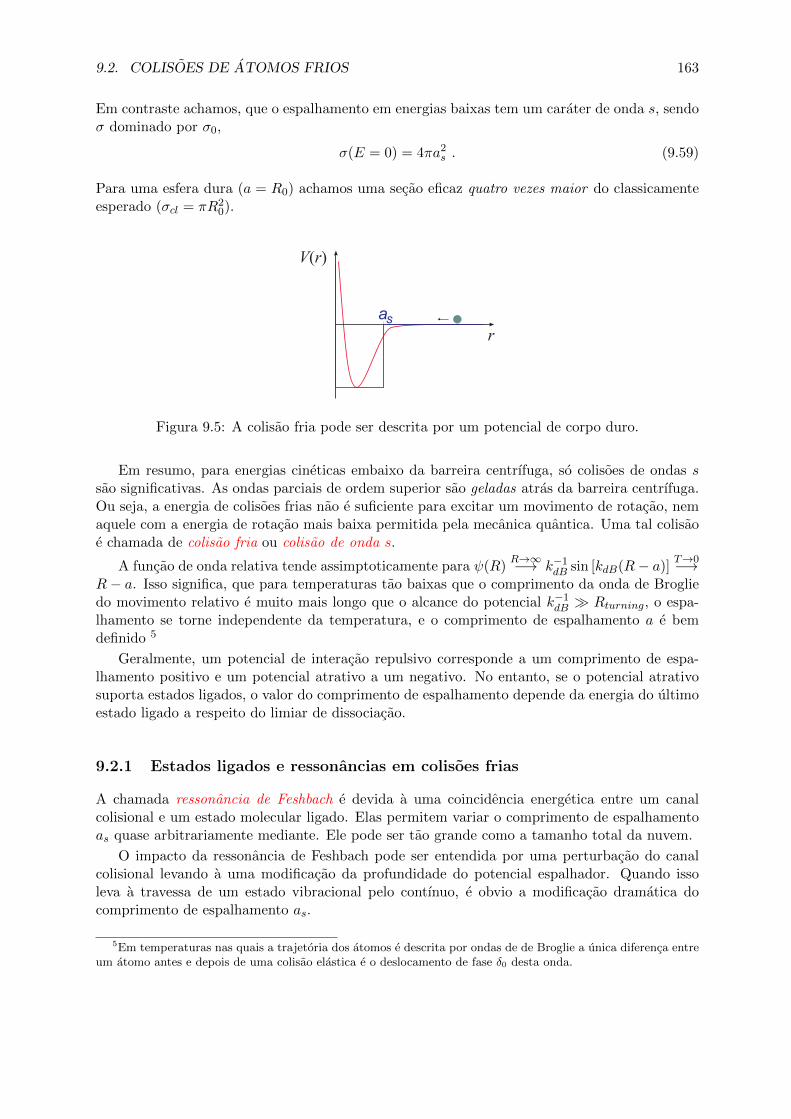
9.2. COLISOES DE ATOMOS FRIOS 163
Em contraste achamos, que o espalhamento em energias baixas tem um carater de onda s, sendoσ dominado por σ0,
σ(E = 0) = 4πa2s . (9.59)
Para uma esfera dura (a = R0) achamos uma secao eficaz quatro vezes maior do classicamenteesperado (σcl = πR2
0).
as
V r( )
r
Figura 9.5: A colisao fria pode ser descrita por um potencial de corpo duro.
Em resumo, para energias cineticas embaixo da barreira centrıfuga, so colisoes de ondas ssao significativas. As ondas parciais de ordem superior sao geladas atras da barreira centrıfuga.Ou seja, a energia de colisoes frias nao e suficiente para excitar um movimento de rotacao, nemaquele com a energia de rotacao mais baixa permitida pela mecanica quantica. Uma tal colisaoe chamada de colisao fria ou colisao de onda s.
A funcao de onda relativa tende assimptoticamente para ψ(R)R→∞−→ k−1
dB sin [kdB(R− a)]T→0−→
R − a. Isso significa, que para temperaturas tao baixas que o comprimento da onda de Brogliedo movimento relativo e muito mais longo que o alcance do potencial k−1
dB Rturning, o espa-lhamento se torne independente da temperatura, e o comprimento de espalhamento a e bemdefinido 5
Geralmente, um potencial de interacao repulsivo corresponde a um comprimento de espa-lhamento positivo e um potencial atrativo a um negativo. No entanto, se o potencial atrativosuporta estados ligados, o valor do comprimento de espalhamento depende da energia do ultimoestado ligado a respeito do limiar de dissociacao.
9.2.1 Estados ligados e ressonancias em colisoes frias
A chamada ressonancia de Feshbach e devida a uma coincidencia energetica entre um canalcolisional e um estado molecular ligado. Elas permitem variar o comprimento de espalhamentoas quase arbitrariamente mediante. Ele pode ser tao grande como a tamanho total da nuvem.
O impacto da ressonancia de Feshbach pode ser entendida por uma perturbacao do canalcolisional levando a uma modificacao da profundidade do potencial espalhador. Quando issoleva a travessa de um estado vibracional pelo contınuo, e obvio a modificacao dramatica docomprimento de espalhamento as.
5Em temperaturas nas quais a trajetoria dos atomos e descrita por ondas de de Broglie a unica diferenca entreum atomo antes e depois de uma colisao elastica e o deslocamento de fase δ0 desta onda.

164 CAPITULO 9. COLISOES
V r( )
r
0 100 200-1000
-500
0
500
1000
B (G)
a s (a
B)
Figura 9.6: Esquerda: A ressonancia de Feshbach e devido a uma coincidencia de canais co-lisionais e ligados. Direita: A ressonancia de Feshbach e sintonizada por campos magneticosaplicados. Variacao do comprimento de espalhamento com a amplitude do campo magnetico.
9.2.2 Colisoes entre partıculas identicas
Consideramos colisoes de duas partıculas identicas. Separamos em coordenadas de centro-de-massa R = r1 + r2 e relativas r = r1− r2. Com isso, R fica simetrico e r antissimetrico em r1 er2. Separamos a funcao de onda em partes orbitais e de spin,
Ψ(x1, x2) = eiP·Rψ(r)χ(s1, s2) . (9.60)
Para partıculas indistinguıveis o resultado do espalhamento tem a forma assimptotica,
ψ(r) ∼ eik·r + f(θ)eikr
r. (9.61)
9.2.2.1 Boson de spin 0
Para boson de spin 0 temos χ = 1 e, por causa da simetria de Ψ, vale ψ(r) = ψ(−r) . Porconsequencia, devemos simetrizar o resultado do espalhamento. Fazemos uso do fato, que atroca das partıculas via r→ −r em coordenadas polares corresponde a transformacao θ → π−θ,r → r,
ψ ∼ (eik·r + e−ik·r) + [f(θ) + f(π − θ)]eikr
r. (9.62)
Para a secao eficaz diferencial obtemos,
dσ
dΩ= |f(θ) + f(π − θ)|2 = |f(θ)|2 + |f(π − θ)|2 + 2<[f∗(θ)f(π − θ)] . (9.63)
Os dois primeiros termos sao classicos. O terceiro termo (de interferencia) ocorre por causa daestatıstica quantica. Os angulos aparecendo em (9.63) sao ilustrados na Fig. 9.7. Para bosons,os termos de interferencia dobram a secao eficaz em comparacao com o caso classico, quandoθ = π/2,
dσ
dΩ= 4|f(π2 )|2 . (9.64)
Para o potencial isotropico V (r) usamos a representacao por ondas parciais,
f(θ) =∑
`
i`f`P`(cos θ) . (9.65)

9.2. COLISOES DE ATOMOS FRIOS 165
314 KAPITEL 12. IDENTISCHE TEILCHEN
Fur den di!erentiellen Wirkungsquerschnitt erhalten wir
d0
d(= |f(() + f($ # ()|2
= |f(()|2 + |f($ # ()|22 34 5klassische Terme
+2Re [f"(()f($ # ()].2 34 5Interferenzterme
(12.80)
Die Interferenzterme erscheinen als Folge der Teilchen-Statistik. Die in(12.80) auftretenden Winkel sind in Abbildung 12.5 dargestellt. Durch dieInterferenzterme verdoppelt sich fur Bosonen im Fall ( = $/2 der Wirkungs-querschnitt gegenuber dem klassischen Resultat,
( =$
2:
d0
d(= 4|f($/2)|2. (12.81)
! " # !
Abb. 12.5: Die Symmetrisie-rung der Streuwellenfunktionerzeugt zwei Trajektorien mitStreuwinkeln ( und $ # ( diekoharent zu addieren sind.
Fur eine zentralsymmetrisches Potential V (r) gehen wir zur Partialwellen-darstellung uber,
f(() =*
l
il(2l + 1)flPl(cos ()
1 Pl(cos () = (#1)lPl(cos($ # ())
f(() + f($ # () = 2*
l gerade
il(2l + 1)flPl(cos (), (12.82)
und finden, dass nur gerade Drehimpulse l vorkommen (fur ungerade l wech-selt das Legendre Polynom das Vorzeichen und die Beitrage vernichten sichgegenseitig).
Spin-1/2 Fermionen: Im Falle der Spin-1/2 Fermionen sind zwei Fallemoglich:
Figura 9.7: A simetrizacao da funcao de onda colisional produz duas trajetorias com os angulosθ e π − θ, que devem ser adicionadas de maneira coerente.
Com P`(cos θ) = (−1)`P`(π − cos θ) obtemos,
f(θ)− f(π − cos θ) = 2∑
`par
i`f`P`(cos θ) , (9.66)
e achamos, que somente momentos angulares pares aparecem 6
9.2.2.2 Fermions de spin 1/2
No caso de fermions de spin-12 duas situacoes sao possıveis 7:
1. O estado de spin singleto χs = 1√2[(| ↑↓〉 − | ↓↑〉] e antissimetrico e consequentemente a
parte orbital,ψ(r) = ψ(−r) (9.67)
deve ser simetrica. A secao eficaz e a mesmo como para bosons de spin 0,
dσ
dΩ
∣∣∣∣s
= |f(θ) + f(π − θ)|2 . (9.68)
2. Os estados de spin tripletos,
χs =
| ↑↑〉1√2(| ↑↓〉+ | ↓↑〉)| ↓↓〉
(9.69)
demandam uma funcao de onda orbital antisimetrica, ψ(r) = −ψ(−r), e obtemos umaamplitude de espalhamento, f(θ) → f(θ) − f(π − θ), que somente contem momentosangulares impares `. Com isso, a secao eficaz fica,
dσ
Ω
∣∣∣∣t
= |f(θ)− f(π − θ)|2 θ=π/2= 0 , (9.70)
Nota, que fermions polarizados somente espalham em canais de momentos angulares im-pares: atomos bosonicos frios mostram um potencial de contato (devido a colisoes de ondas (9.66)), atomos fermionicos frios polarizados somente interagem fracamente no canal p.
6Para ` impar o polinomio de Legendre muda de sinal, e as contribuicoes zeram.7Isso e analogico ao caso do helio, onde a funcao espacial do estado 2s2 ↑↑ e sempre antisimetrizada, mas para
o estado 2s ↑ 2p ↑ existem orbitais espaciais simetricas.

166 CAPITULO 9. COLISOES
No caso de conjunto estatisticamente misturado de fermions nao-polarizados obtemos amedia ponderada,
dσ
dΩ=
3
4
dσ
Ω
∣∣∣∣t
+1
4
dσ
Ω
∣∣∣∣s
= |f(θ)|2 + |f(π − θ)|2 −<[f∗(θ)f(π − θ)] . (9.71)
9.2.2.3 Espectros moleculares
Aqui consideramos espectros rotacionais de baixas energias Erot = ~2`(`+1)/2Θ Eeletronico ∼eV . Em escalas de tempo devagares podemos considerar a casca eletronica como rıgida. Olhamospara dois exemplos de moleculas com nucleos bosonicos e fermionicos:
• Moleculas (C12)2: os nucleos sao bosons de spin 0, por isso somente sao permitidos ` pares.
• Moleculas H2: os nucleos sao fermions de spin 12 , por isso temos para uma funcao de onda
de spin,
χ = χs : ` = par, hidrogenio para, χ = χt : ` = impar, hidrogenio orto. (9.72)
A transformacao de hidrogenio orto em hidrogenio para e difıcil (os nucleos sendo bemblindados), tal que observamos dois tipos de gases com,
Erot,para = 0,3
Θ,10
Θ,21
Θ, ...Erot,orto =
1
Θ,
6
Θ,15
Θ, ... . (9.73)
9.2.3 Colisoes de atomos quentes
Momentos angulares com ` ≤ kR0 deveriam contribuir muito para σ, pois o parametro de colisaofica dentro de R0. Para uma esfera dura temos α` = ∞ e cot δ` = n`(kR0)/j`(kR0). Com asexpressoes assimptoticas de j` e n` obtemos cot δ` ∼ − cot(kR0 − `π/2), isto e, δ∼ − kR0 +`π/2 (+π). Com estas fases de espalhamento podemos calcular a secao eficaz,
σ ' 4πk2
kR0∑
`=0
(2`+ 1) sin2 δ` (9.74)
' 4πk2
kR0∑
`=0
(`+ 1) cos2[kR0 − (`+ 1)π/2] + ` sin2(kR0 − `π/2)
= 4πk2
kR0∑
`=0
`(cos2 + sin2) = 4πk2
kR0(kR0 + 1)
2= 2πR2
0 , (9.75)
o que e o dobre do valor classico.
9.3 Exercıcios
9.3.1 Teoria de espalhamento
9.3.1.1 Ex: Metodo de Green
Mostre que, sabendo a solucao de (9.4), isto e, conhecendo a funcao de Green, podemos escrevera solucao do problema de espalhamento (9.3) como,
ψk(r) = eik·r + 2m~2
∫d3r′G(r− r′, k)V (r′)ψk(r) .

9.3. EXERCICIOS 167
Figura 9.8: (a) Estado ligado para ` = 0. (b) Estado ligado para ` > 0 num potencial incluindoa barreira centrıfuga ~2`(` + 1)/2mr2. (c) Ressonancias para ` = 0 sao largas e eventualmentenao definidas com Γr > Er. Uma ressonancia definida com Γr < Er necessita que |∂Eα0| sejagrande. (d) Para ` > 0 obtemos ressonancias finas chamadas de ressonancia de forma ou shaperesonance, pois o decaimento do estado e suprimida pela barreira centrıfuga.
9.3.1.2 Ex: Funcao de Green
Calcule a funcao de Green da equacao (9.4).
9.3.1.3 Ex: Espalhamento de Rutherford
Considere o espalhamento de uma partıcula da carga Q por uma distribuicao de carga estaticaρ(r) = ρ0e
−αr totalizando a carga totalQ′. Derive a partir de (9.24) a formula (1.13) descrevendoo espalhamento de Rutherford.
9.3.2 Colisoes de atomos frios

168 CAPITULO 9. COLISOES

Referencias Bibliograficas[1] P. W. Atkins and R. S. Friedman, Molecular quantum mechanics, Oxford University Press,
1997.
[2] A. Bambini and S. Geltman, Feshbach resonances in cold collisions of potassium atoms,Phys. Rev. A 65 (2002), 062704, .
[3] G. R. Barwood and et al., Frequency measurements on optically narrowed rb-stabilized laserdiodes at 780 nm and 795 nm, Appl. Phys. B 53 (1991), 142, .
[4] G. Blatter, Quantenmechanik, http://www.itp.phys.ethz.ch/research/condmat/vortex/lectures/qm2/QM.pdf,Zurich, 2005.
[5] Felix Bloch, Nuclear induction, Physical Review 70 (1946), no. 7, 460.
[6] E. Braaten and H.-W. Hammer, Universality in few-body systems with large negative scat-tering length, Phys. Rep. 428 (2006), 259, .
[7] L. D. Carr and et al., Cold and ultracold molecules: science, technology and applications,New J. Phys. 11 (2009), 055009, .
[8] C. Cohen-Tannoudji, B. Diu, and F. Lao, Quantum mechanics, 1, vol. 1 & 2, 1977, Dedalus:530.12 C678q v.1 e.1.
[9] Ph. W. Courteille, R. S. Freeland, D. J. Heinzen, F. A. van Abeelen, and B. J. Verhaar,Observation of a feshbach resonance in cold atom scattering, Phys. Rev. Lett. 81 (1998),69.
[10] J. P. Dahl and M. Springborg, The morse oscillator in position space, momentum space,and phase space, J. Chem. Phys. 88 (1988), 4535.
[11] D. DeMille, Quantum computation with trapped polar molecules, Phys. Rev. Lett. 88 (2002),067901, .
[12] D. Demille, Using molecules to measure nuclear spin-dependent parity violation, Phys. Rev.Lett. 100 (2008), 023003, .
[13] W. Demtroder, Atoms, molecules and photons: An introduction to atomic- molecular- andquantum physics, Springer, 2006.
[14] O. Dulieu and P. S. Julienne, Coupled channel bound states calculations for alkali dimersusing the fourier grid method, J. Chem. Phys. 103 (1995), 60, .
[15] G. Herzberg, Springer, 1950.
[16] P. S. Julienne, K. Burnett, Y. B. Band, and W.C. Stwalley, Stimulated raman moleculeproduction in bose-einstein condensates, Phys. Rev. A 58 (1998), R797, .
[17] V. Kokoouline, O. Dulieu, R. Kosloff, and F. Masnou-Seeuws, Mapped fourier methods forlong-range molecules: Application to perturbations in the Rb2(0+
u ) photoassociation spec-trum, J. Chem. Phys. 110 (1999), 9865, .
169

170 REFERENCIAS BIBLIOGRAFICAS
[18] M. Korek, A. R. Allouche, M. Kobeissi, A. Chaalan, M. Dagher, K. Fakherddin, andM. Aubert-Frecon, Chem. Phys. 256 (2000), 1.
[19] Ch. Lisdat, O. Dulieu, H. Knockel, and E. Tiemann, Inversion analysis of k2 coupled elec-tronic states with the fourier grid method, Eur. Phys. J. D 17 (2001), 319, .
[20] S. Y. T. Meerakker and et. al., Direct measurement of the radiative lifetime of vibrationallyexcited oh radicals, Phys. Rev. Lett. 95 (2005), 013003, .
[21] C. R. Menegatti, Trap loss in a rubidium crossed dipole trap by short-range photoassociation,Phys. Rev. A 87 (2013), 053404, .
[22] J. L. Roberts, N. R. Claussen, J. P. Jr. Burke, C. H. Greene, E. A. Cornell, and C. E.Wieman, Resonant magnetic field control of elastic scattering in cold 85rb, Phys. Rev. Lett.81 (1998), 5109, .
[23] S. Schiller, Hydrogenlike highly charged ions for tests of the time independence of funda-mental constants, Phys. Rev. Lett. 98 (2007), 180801, .
[24] W. R. Theis, Grundzuge der quantentheorie, Teubner Studienbucher, 1985.
[25] L. H. Thomas, Nature 117 (1926), 514, .
[26] E. Tiesinga, C. J. Williams, and P. S. Julienne, Photoassociative spectroscopy of highlyexcited vibrational levels of alkali dimers: Green’s function approach for eigenvalue solvers,Phys. Rev. A 57 (1998), 4257, [Tiesinga98].
[27] J. Vanier and C. Audoin, The quantum physics of atomic frequency standards, Adam Hilger,Bristol and Philadelphia, 1989, Dedalus: 539.7 V258q v.1.
[28] J. Weiner, V. S. Bagnato, S. Zilio, and P. S. Julienne, Experiments and theory in cold andultracold collisions, Rev. Mod. Phys. 71 (1999), 1.

Indice Remissivo6j
sımbolo, 89
absorcao, 82acoplamento jj, 34acoplamento LS, 34acoplamento intermediario, 118acoplamento mınimo, 70alargamento homogeneo, 91alargamento inomogeneo, 91Aristoteles, 4auto-consistencia, 104
barion, 64banho termico, 86Biot-Savart
lei de, 55Bohr
Niels, 12postulados de, 12raio de, 21
Bornaproximacao de, 9, 157serie de, 157
Born-Oppenheimeraproximacao de, 125, 137, 142potencial de, 126
boson, 96Bremsstrahlung, 113
Casimirefeito de, 145
casos de Hund, 148catastrofe de radiacao, 10centrıfuga
barreira, 134Clebsch-Gordan
coeficiente de, 33coeficientes de Einstein, 86colisao de onda s, 163colisao fria, 163combinacao linear de orbitais atomicos, 128Compton
comprimento de onda, 57conjunto completo de operadores comutan-
dos, 14
constante rotacional, 142
constantes rotacionais, 141
contato
potencial de, 165
corpo negro, 86
Coulomb
integral de, 99, 132
operador de, 110
potencial de, 56
covalente
configuracao, 131
cruzamentos evitados, 67
cruzamentos reais, 67
Darwin
Sir Charles Galton, 57
defeito quantico, 66, 115
Democrito, 4
densidade dos estados, 84, 105
density functional theory, 104
descida
operador de, 26
desdobramento hiperfino, 60
determinante de Slater, 97
diamagnetico
termo, 70
dipolar
aproximacao, 85
Dirac
equacao de, 53
Paul, 27, 53
Paul Adrien Maurice, 13
dissociacao
limite de, 136
eletroafinidade, 123
eletrodinamica quantica, 83
eletronegatividade, 123
emissao, 82
emissao espontanea, 83
emissao estimulada, 83
energia de ionizacao, 123
energia de ligacao, 143
energia de localizacao, 142
equacao azimutal, 18
171

172 INDICE REMISSIVO
equacao polar, 18espalhamento
amplitude de, 153, 155comprimento de, 162
estrutura fina, 53constante da, 53
estrutura hiperfinaefeito Paschen-Back da, 74efeito Zeeman da, 73
exoticoatomo, 63
fase do espalhamento, 158fator de intervalo, 62fator de Lande, 73fator-g, 55Fermi
contato de, 60energia de, 103
fermion, 96Feshbach
ressonancia de, 163Fock
Vladimir Aleksandrovich, 24fosforescencia, 88fotoassociacao, 140Fourier
metodo de grade de, 144, 145Franck-Condon
fator de, 138, 140princıpio de, 138
funcao de onda, 13funcional da densidade, 104
gas de Fermimodelo do, 103
GerlachWalter, 12
giromagneticarazao, 55
grau de liberdade, 13Green
funcao de, 153metodo de, 154
hadron, 64helio, 98hadronico
atomo, 64
harmonicos esfericos, 18Hartree
Douglas Rayner, 104metodo de, 108
Hartree-Fockequacoes de, 110metodo de, 108
Hartree-Fock-Roothaanequacao de, 111
HeisenbergWerner, 13
hidrogenio muonico, 64hiperfina
estrutura, 59
integral de ressonancia, 128intercambio
degenerescencia de, 96simetria de, 96
intervaloregra de, 62
irreduzıvelelemento da matriz, 76elemento de matriz, 88
Koopmanteorema de, 111
lepton, 64Laguerre
Edmond, 23equacao diferencial associada de, 23
Lambdeslocamento de, 59Willis Eugene, Jr., 59
Landefator de, 71
Larmorfrequencia de, 11
LegendreAdrien-Marie, 18
LeRoy-Bernsteinmetodo de, 143
ligacao covalente, 124ligacao ionica, 123Lippmann-Schwinger
equacao de, 154Lorentz
distribuicao de, 91

INDICE REMISSIVO 173
LSacoplamento, 117
meson, 64metodo variacional, 46magneton de Bohr, 11, 55magneton nuclear, 60massa reduzida, 134matrix
elemento de, 82mecanica das matrizes, 13mecanica das ondas, 13Mie
espalhamento de, 35moleculares
modelo dos orbitais, 127momento angular orbital, 25momento de dipolo da transicao, 139momento de dipolo eletrico, 85momento de inercia, 20momento dipolar, 27momento magnetico orbital, 10Morse
potencial de, 135, 146Movre-Pichler
potencial de, 147
numero quantico do momento angular, 18numero quantico magnetico, 18numero quantico principal, 22nucleado
modelo, 7
observaveis, 13onda parcial
amplitude da, 159orbital atomico, 128orbital molecular, 129
metodo do, 128orto-helio, 102
pacote de onda, 155para-helio, 102parciais
ondas, 157paridade, 88Paschen-Back
efeito, 72Paschen-Goudsmith
efeito, 74Pauli
matriz de, 27perturbacao dependente do tempo
teoria de, 81perturbacao independente do tempo
teoria de, 41planetario
modelo, 9Poisson
lei de, 154polinomios de Laguerre, 23polinomios de Legendre, 18potencial centrifugal, 19precessao de Thomas, 56princıpio detalhado de balanco, 83princıpio forte de exclusao de Pauli, 97princıpio fraco de exclusao de Pauli, 97
quımicopotencial, 107
quadrupolarconstante de interacao eletron-nucleo, 62interacao, 62
quantizacaoprimeira, 12
Rayleighfracao de, 46John William Strutt, 3. Baron, 46
Rayleigh-Ritzmetodo de, 47
reduzidoelemento de matriz, 88
regra de Hund, 112regra de selecao, 89reservatorio, 86ressonancia de forma, 167rotacao rıgida, 134rotacional
constante, 141rotor rıgido, 20Russel-Saunders
acoplamento de, 117Rutherford
Ernest, 6espalhamento de, 6
Rydbergatomo de, 65

174 INDICE REMISSIVO
serie de, 65
SchrodingerErwin, 13
screening, 9secao eficaz diferencial, 155secao eficaz parcial, 159secao eficaz total, 155secao transversal de espalhamento, 6secular
determinante, 45equacao, 44
selecaoregra de, 102regras de, 88
shape resonance, 167simetrizada
funcao de onda, 96sistema periodico dos elementos, 112Slater
John Clarke, 97Sommerfeld
Arnold, 12Arnold Johannes Wilhelm, 59
spin, 27spin-orbita
interacao, 55Stark
deslocamento de, 67efeito de, 76efeito quadratico de, 76Johannes Nikolaus, 76
Stark linearefeito, 45, 76
Stark quadraticoefeito, 45
SternOtto, 12
Stern-Gerlachexperimento de, 27
subidaoperador de, 26
teorema optico, 160Thomas
fator de, 56Llewellyn, 56
Thomas-Fermienergia de, 106
equacao de, 107modelo de, 104
ThomsonJoseph John, 6
transicao dipolar magnetica, 88transicao eletrica quadrupolar, 88troca
energia de, 101integral de, 102, 132operador de, 110
unidades atomicas, 4
valencialigacao de, 131
valence bondmodelo, 127
van der Waalscoeficientes de, 146forca de, 134, 145
vibracao, 134virial
teorema, 24
WignerEugene Paul, 76
Wigner-Eckartteorema de, 76, 88, 142
Zeemandesdobramento, 70efeito, 11Pieter, 70
Zeeman anomaloefeito, 71
Zeeman normalefeito, 71