faculdade Única de ipatinga quÍmica orgÂnica
TRANSCRIPT

1
FACULDADE ÚNICA
DE IPATINGA

2
Tiago Marcel Oliveira
Técnico em química industrial pelo CEFET/MG (2002), licenciado em Química pelo Centro
Universitário de Caratinga/MG (2005) e Mestre em Engenharia Industrial pelo Centro
Universitário Católica do Leste de Minas Gerais (UnilesteMG) (2010). Trabalha no Governo do
Estado de Minas Gerais como servidor público efetivo lotado na Secretaria de Educação
ministrando a disciplina de química para o ensino médio. Além disso, é Coordenador do
curso de Química Licenciatura Plena da Faculdade Única de Ipatinga na modalidade EaD
e professor adjunto I-A da mesma instituição.
1ª edição
Ipatinga – MG
2021
QUÍMICA ORGÂNICA

3
FACULDADE ÚNICA EDITORIAL
Diretor Geral: Valdir Henrique Valério
Diretor Executivo: William José Ferreira
Ger. do Núcleo de Educação a Distância: Cristiane Lelis dos Santos
Coord. Pedag. da Equipe Multidisciplinar: Gilvânia Barcelos Dias Teixeira
Revisão Gramatical e Ortográfica: Izabel Cristina da Costa
Revisão/Diagramação/Estruturação: Bárbara Carla Amorim O. Silva
Carla Jordânia G. de Souza
Rubens Henrique L. de Oliveira
Design: Brayan Lazarino Santos
Élen Cristina Teixeira Oliveira
Maria Luiza Filgueiras
© 2021, Faculdade Única.
Este livro ou parte dele não podem ser reproduzidos por qualquer meio sem Autorização
escrita do Editor.
T314i
Teodoro, Jorge Benedito de Freitas, 1986 - .
Introdução à filosofia / Jorge Benedito de Freitas Teodoro. – 1. ed. Ipatinga, MG:
Editora Única, 2020.
113 p. il.
Inclui referências.
ISBN: 978-65-990786-0-6
1. Filosofia. 2. Racionalidade. I. Teodoro, Jorge Benedito de Freitas. II. Título.
CDD: 100
CDU: 101 Ficha catalográfica elaborada pela bibliotecária Melina Lacerda Vaz CRB – 6/2920.
NEaD – Núcleo de Educação a Distância FACULDADE ÚNICA
Rua Salermo, 299
Anexo 03 – Bairro Bethânia – CEP: 35164-779 – Ipatinga/MG
Tel (31) 2109 -2300 – 0800 724 2300
www.faculdadeunica.com.br

4
Menu de Ícones
Com o intuito de facilitar o seu estudo e uma melhor compreensão do conteúdo
aplicado ao longo do livro didático, você irá encontrar ícones ao lado dos textos. Eles
são para chamar a sua atenção para determinado trecho do conteúdo, cada um
com uma função específica, mostradas a seguir:
São sugestões de links para vídeos, documentos científico
(artigos, monografias, dissertações e teses), sites ou links das
Bibliotecas Virtuais (Minha Biblioteca e Biblioteca Pearson)
relacionados com o conteúdo abordado.
Trata-se dos conceitos, definições ou afirmações
importantes nas quais você deve ter um maior grau de
atenção!
São exercícios de fixação do conteúdo abordado em cada
unidade do livro.
São para o esclarecimento do significado de determinados
termos/palavras mostradas ao longo do livro.
Este espaço é destinado para a reflexão sobre questões
citadas em cada unidade, associando-o a suas ações, seja
no ambiente profissional ou em seu cotidiano.

5
SUMÁRIO
COMPOSTOS DE CARBONO E LIGAÇÕES QUÍMICAS ............................. 8
1.1 INTRODUÇÃO ......................................................................................................... 8
1.2 BREVE HISTÓRICO DA QUÍMICA ORGÂNICA ...................................................... 9
1.3 REGRA DO OCTETO E LIGAÇÕES QUÍMICAS ..................................................... 10
1.4 REPRESENTAÇÃO DE FÓRMULAS QUÍMICAS E ISOMERIA ................................. 14
1.5 ESTRUTURAS DE LEWIS, CARGA FORMAL E RESSONÂNCIA ............................... 16
1.6 EXCEÇÕES À REGRA DO OCTETO ....................................................................... 20
1.7 ORBITAIS MOLECULARES E HIBRIDIZAÇÃO ......................................................... 21
1.8 GEOMETRIA MOLECULAR E POLARIDADE ........................................................... 24
FIXANDO O CONTEÚDO ...................................................................................... 27
FUNÇÕES ORGÂNICAS E ESPECTROSCOPIA NO INFRAVERMELHO ..... 31
2.1 CLASSIFICAÇÃO DE ÁTOMOS DE CARBONO E CADEIAS CARBÔNICAS ........ 31
2.1.1 Classificação das cadeias abertas ........................................................... 33
2.1.2 Classificação das cadeias fechadas ........................................................ 35
2.1.3 Classificação das cadeias mistas .............................................................. 38
2.2 PRINCIPAIS FUNÇÕES ORGÂNICAS .................................................................... 38
2.3 ESPECTROSCOPIA NO INFRAVERMELHO: FUNDAMENTOS E INSTRUMENTAÇÃO
............................................................................................................................... 45
2.4 APLICAÇÃO DA ESPECTROSCOPIA NO INFRAVERMELHO NA IDENTIFICAÇÃO
DE GRUPOS FUNCIONAIS .................................................................................... 50
2.4.1 Octano ............................................................................................................ 51
2.4.2 Hept-1-Ino ....................................................................................................... 52
2.4.3 Ácido Propanóico ......................................................................................... 53
FIXANDO O CONTEÚDO ...................................................................................... 54
ÁCIDOS E BASES ORGÂNICOS ............................................................... 59
3.1 INTRODUÇÃO ....................................................................................................... 59
3.2 QUEBRA HOMOLÍTICA E HETEROLÍTICA DE LIGAÇÕES ...................................... 59
3.3 ELETRÓFILOS, NUCLEÓFILOS E RADICAIS ............................................................ 59
3.4 CARBOCÁTIONS E CARBÂNIONS ....................................................................... 60
3.5 REPRESENTAÇÃO DE MECANISMOS DE REAÇÃO .............................................. 61
3.6 DEFINIÇÕES DE ÁCIDOS E BASES ........................................................................ 62
3.7 FORÇA RELATIVA DE ÁCIDOS E BASES ............................................................... 64
3.8 EFEITOS QUE AFETAM ACIDEZ E BASICIDADE .................................................... 66
FIXANDO O CONTEÚDO ...................................................................................... 69
ALCANOS E CICLOALCANOS: NOMENCLATURA, CONFORMAÇÕES E
REAÇÕES ................................................................................................. 74
4.1 O PETRÓLEO COMO PRINCIPAL FONTE DE HIDROCARBONETOS ..................... 74
4.2 ALCANOS E CICLOALCANOS: ESTRUTURA E PROPRIEDADES ........................... 75
4.3 NOMENCLATURA DOS ALCANOS E CICLOALCANOS ....................................... 79
4.4 CONFORMAÇÕES E ESTABILIDADE DOS ALCANOS E CICLOALCANOS .......... 82
4.5 CICLOALCANOS DISSUBSTITUÍDOS E ISOMERISMO CIS-TRANS ........................ 85
4.6 SÍNTESE E REAÇÕES DE ALCANOS E CICLOALCANOS ...................................... 86
4.6.1 Reações dos alcanos ................................................................................... 87
4.6.2 Síntese dos alcanos....................................................................................... 88
FIXANDO O CONTEÚDO ...................................................................................... 92
UNIDADE
02
UNIDADE
01
UNIDADE
03
UNIDADE
04

6
ESTEREOQUÍMICA ................................................................................... 96
5.1 SIMETRIA NA NATUREZA ....................................................................................... 96
5.2 ASPECTOS HISTÓRICOS DA ESTEREOQUÍMICA .................................................. 97
5.3 PROPRIEDADES DOS ISÔMEROS ÓTICOS ............................................................ 97
5.4 ENANTIÔMEROS E QUIRALIDADE ........................................................................ 99
5.5 NOMENCLATURA E REPRESENTAÇÃO DE ESTEREOISÔMEROS ........................ 100
5.6 MISTURAS RACÊMICAS E RESOLUÇÃO ............................................................. 103
5.7 MOLÉCULAS QUIRAIS SEM CENTRO QUIRAL .................................................... 104
5.8 FÁRMACOS QUIRAIS .......................................................................................... 105
FIXANDO O CONTEÚDO .................................................................................... 108
HALETOS DE ALQUILA E REAÇÕES DE SUBSTITUIÇÃO NUCLEOFÍLICA E DE
ELIMINAÇÃO ......................................................................................... 112
6.1 ESTRUTURA E NOMENCLATURA DE HALETOS DE ALQUILA ............................... 112
6.2 REAÇÕES DE SUBSTITUIÇÃO NUCLEOFÍLICA .................................................... 113
6.3 MECANISMO E ESTEREOQUÍMICA DAS REAÇÕES SN1 E SN2 ......................... 115
6.4 FATORES QUE INFLUENCIAM AS REAÇÕES SN1 E SN2 ..................................... 118
6.5 REAÇÕES DE ELIMINAÇÃO DE HALETOS DE ALQUILA ..................................... 120
6.6 SUBSTITUIÇÃO VERSUS ELIMINAÇÃO ................................................................ 122
FIXANDO O CONTEÚDO .................................................................................... 125
RESPOSTAS DO FIXANDO O CONTEÚDO ............................................. 129
REFERÊNCIAS ......................................................................................... 130
UNIDADE
05
UNIDADE
06

7
CONFIRA NO LIVRO
Nesta unidade abordaremos a natureza dos compostos orgânicos,
as características do carbono e de suas ligações.
Nesta unidade iremos conhecer as principais categorias de subs-
tâncias orgânicas e seus grupos característicos. Trataremos também
da técnica conhecida como espectroscopia de absorção no
infravermelho e sua utilidade na identificação de grupos funcionais.
A natureza e propriedades de ácidos e bases e alguns aspectos
estruturais que interferem na força ácida ou básica de compostos
orgânicos serão abordados neste capítulo.
Este capítulo trata de alcanos e cicloalcanos, sua estrutura,
principais representantes, suas propriedades e nomenclatura.
Este capítulo trata da estereoquímica dos compostos orgânicos,
com os principais conceitos e nomenclaturas utilizadas neste
campo.
Aqui trataremos dos haletos de alquila e suas reações
características: substituição nucleofílica e eliminação.

8
COMPOSTOS DE CARBONO E
LIGAÇÕES QUÍMICAS
1.1 INTRODUÇÃO
A Química é o estudo da matéria e suas transformações, e tem sido objeto de
investigação desde os primórdios da humanidade, quando os primeiros seres
humanos recorriam a misturas de pigmentos vegetais com carvão e cinzas para a
obtenção de tintas para as pinturas rupestres, obtinham extratos vegetais e, mais
tardiamente, desenvolveram técnicas metalúrgicas. Ao longo dos séculos, numero-
sas substâncias foram isoladas a partir de animais, vegetais e minerais. Os alquimistas
da idade média desenvolveram métodos de purificação e descobriram novos
elementos. Apesar disso, a Química somente obteve o status de ciência ao se
dissociar da Medicina e da Física a partir de meados do século XVII.
A despeito de a Química ser um conjunto único de conhecimentos, modelos
e técnicas, para fins didáticos ela tem sido dividida em campos, ou áreas. A Química
contemporânea é tradicionalmente dividida nas seguintes macroáreas:
Química Inorgânica
Química Orgânica
Físico-Química
Química Analítica
Muitas outras áreas têm sido consideradas devido às suas especifidades,
geralmente associadas a uma das macroáreas citadas. Por exemplo, a Sociedade
Americana de Química (American Chemical Society) adota as seguintes divisões:
• Química inorgânica
• Química Orgânica
• Físico-química
• Química Analítica
• Química de Polímeros
• Química agrícola e de alimentos
• Química computacional
• Nanoquímica
• Química de cosméticos e cuidados
pessoais
• Biotecnologia
• Química de Materiais
• Cristalografia
• Química nuclear
UNIDADE
01

9
• Química forense
• Química de tintas, pigmentos e
recobrimentos
• Astroquímica
• Quimioinformática
• Geoquímica
• Química têxtil
• Engenharia Química ou Química
Industrial
• Química Medicinal
• Química do petróleo
• • Química da água
1.2 BREVE HISTÓRICO DA QUÍMICA ORGÂNICA
Até meados do século XIX, acreditava-se que substâncias consideradas
orgânicas, ou seja, obtidas de organismos (vegetais e animais) eram providas de um
princípio ou “força vital” que as distinguiam das substâncias inorgânicas, que são
obtidas primariamente de minerais. Em 1816, Michel Checreul realizou experimentos
com sabões, e demonstrou que era possível fazer modificações nos óleos e gorduras
utilizados em sua fabricação, para produzir novas substâncias, o que parecia
contradizer a ideia de que não era possível fazer transformações em materiais
orgânicos sem o auxílio da força vital.
Foi Friederich Wöhler que, em 1828, obteve ureia, uma substância obtida da
urina de animais, aquecendo cianato de potássio (NH4(OCN)), que é um sal
inorgânico, demonstrando assim que não há um princípio ou força vital exclusivo de
substâncias provenientes de matéria orgânica.
Os trabalhos de Friedrich August Kekulé e Archibald Scott Couper em 1858
levaram ao conceito moderno de estrutura, com a concepção de que o carbono
era um elemento fundamental na constituição dos compostos orgânicos e de que
estabelecia ligações com os átomos vizinhos seguindo um padrão tetravalente.
Ao longo de todo o século XX até à época atual a síntese de novos compostos
orgânicos tem proporcionado melhorias na qualidade de vida da população
mundial por meio de segmentos como medicamentos, aditivos alimentícios,
defensivos agrícolas e novos materiais.
É curioso o fato de que enquanto são conhecidos atualmente cerca de 500
mil compostos inorgânicos, constituídos de metais (a maior parte dos elementos da

10
Classificação Periódica), ametais e metaloides, já foram identificados ou sintetizados
mais de 20 milhões de compostos orgânicos, constituídos principalmente dos
seguintes elementos: C, H, N, O (e, com menor ocorrência, P, S e halogênios). O que
explica como um número tão pequeno de elementos possa formar um número tão
grande de compostos? A razão está na capacidade que o carbono possui de se
ligar, formando longas cadeias, conforme será abordado mais adiante. De fato, a
Química Orgânica é também chamada de Química dos compostos de carbono.
Embora haja compostos contendo carbono que são considerados inorgânicos
(como o carbonato de cálcio, CaCO3, presente no calcário), este elemento é
tipicamente encontrado nos organismos.
1.3 REGRA DO OCTETO E LIGAÇÕES QUÍMICAS
Os elementos representativos (pertencentes às famílias 1, 2, 13, 14, 15, 16 e 17
da Classificação Periódica dos Elementos) se estabilizam com 8 elétrons na camada
de valência (comportamento que denominamos regra do octeto), embora possa
haver exceções importantes que discutiremos mais adiante. Dependendo do número
de elétrons de valência (Figura 1), cada elemento deverá ganhar ou perder elétrons
de modo a atingir o octeto.
Figura 1: Valência dos elementos representativos.
Disponível em: https://bit.ly/3sJUiE4. Acesso em: 08 ago. 2020.
Metais, sendo pouco eletronegativos, tendem a perder elétrons, e ametais
tendem a ganhar. A ligação entre um metal e um ametal envolverá a formação de
íons (sendo chamada de ligação iônica), como ocorre na reação entre átomos de
potássio e cloro, aqui representados no estado gasoso para efeito de simplicidade:

11
O potássio apresenta configuração eletrônica do tipo [Ar] 4s1, o que significa
que, ao perder 1 elétron, ele adquire a configuração estável do argônio, um gás
nobre. O átomo de cloro tem configuração [Ne] 3s2 3p5, e adquire a configuração
eletrônica de um gás nobre ao ganhar 1 elétron (tornando-se também isoeletrônico
com o argônio).
Quando dois átomos de cloro no estado gasoso se ligam, entretanto, ambos
necessitam ganhar 1 elétron cada um, a fim de se estabilizarem. Para isso, deve
compartilhar seus elétrons de valência, formando uma ligação covalente.
Cada compartilhamento de elétrons pode ser representado por meio de um
traço (cada traço representando dois elétrons que são comuns ao dois átomos da
ligação). Embora seja recomendável representar os elétrons não ligantes em molé-
culas pequenas, estes costumam ser omitidos:
Se a ligação é feita entre dois átomos de oxigênio, que apresentam 6 elétrons
de valência (Figura 1), e portanto precisam de 2 elétrons para atingirem o octeto,
teremos uma ligação dupla.

12
Se dois átomos de nitrogênio (com seus 5 elétrons de valência) se ligam, cada
um precisará de 3 elétrons para atingir o octeto e teremos uma ligação tripla:
É importante frisar que as ligações normalmente não são exclusivamente
iônicas ou covalentes: elas apresentam um caráter iônico maior ou menor,
dependendo da diferença de eletronegatividade entre os átomos que a constituem.
Assim, por exemplo, a ligação entre o átomo de alumínio (um metal) e o cloro (um
ametal) no cloreto de alumínio (AlCl3) guarda muitas semelhanças com as ligações
covalentes, já que o a eletronegatividade do alumínio e do cloro não são muito
diferentes.
Este modelo simplificado das ligações químicas é útil no entendimento das
estruturas em um sentido mais prático, embora haja outras teorias de ligação química
mais adequadas para explicar as propriedades das moléculas. Abordaremos de
maneira breve uma destes teorias (a Teoria do Orbital Molecular) ao final desta
unidade.
Observando a posição do carbono na Classificação Periódica, constatamos
que ele apresenta 4 elétrons de valência, estabilizando-se com mais 4 elétrons, o que
lhe confere a configuração eletrônica do neônio. Assim, ele necessita de quatro
compartilhamentos, o que possibilita quatro formas diferentes para se ligar aos
átomos vizinhos (Figura 2):

13
Figura 2: Padrões de ligação do átomo de carbono
Fonte: Elaborado pelo Autor (2021)
Assim, o carbono apresenta grande versatilidade devido à sua multiplicidade
de formas de se ligar. Uma característica importante das ligações entre carbonos é
o fato de serem muito estáveis, ao contrário das ligações entre o silício, pertencente
à mesma família (Tabela 1).
Tabela 1: Entalpias médias de dissociação a 298 K (DH298) e comprimentos médios (d) de
ligações
Ligação ΔDH298, kJ mol-1 d, pm Ligação ΔDH298, kJ mol-1 d, pm
C–C 347 154 C–F 485 133
C=C 614 134 C–Cl 339 177
C≡C 839 127 C–Br 276 194
C–H 413 109 C–I 240 213
C–O 358 143 Si–Si 340 234
C=O 745 123 Si–O 452 161
C–N 305 147
Fonte: Chemistry LibreTexts (2020). Disponível em: https://bit.ly/3cgZEQX. Acesso em: 13 dez.
2020.
Como se pode observar, as ligações Si – Si são mais fracas (e mais longas) que
C – C, o que inviabiliza a formação de cadeias de silício com a mesma facilidade
com que as cadeias de carbonos se formam; porém, a ligação Si – O é mais forte (e
mais curta) que a ligação Si–Si, existindo cadeias com átomos de Si e O intercalados,
formando o material conhecidos como silicona (ou silicone).
Podemos também observar que quanto maior a multiplicidade da ligação,
mais forte ela se torna. Assim, ligações duplas são mais fortes que as ligações simples

14
entre os mesmos átomos – porém mais fracas que as ligações triplas.
1.4 REPRESENTAÇÃO DE FÓRMULAS QUÍMICAS E ISOMERIA
Os elementos químicos se combinam formando conjuntos ou unidades básicas
que, dependendo da natureza destes mesmos elementos, recebem diferentes
nomes. Conjuntos de átomos de ametais, unidos por ligações covalentes, são
chamados de moléculas. Metais e ametais combinados, contendo pelo menos uma
ligação iônica são pares iônicos. Metais elementares, por outro lado, são constituídos
de átomos individuais como unidades básicas.
Podemos representar estas unidades básicas por meio de diferentes: fórmula
molecular, fórmula mínima (ou empírica), e fórmula centesimal (ou composição).
A fórmula molecular apresenta a quantidade real de elementos de cada tipo
em uma unidade da substância (molécula, par iônico, etc.). A fórmula mínima
apresenta a proporção de cada elemento em uma unidade da substância. A
fórmula centesimal apresenta o percentual em massa de cada elemento em relação
à massa total (soma das massas atômicas na fórmula mínima).
Exemplo:
Fórmula molecular: H2O2
Fórmula mínima: HO
Fórmula centesimal: H5,95%O94,07%
A fórmula centesimal pode ser obtida experimentalmente, por meio de uma
análise elementar. A partir dela, pode-se obter a fórmula mínima dividindo-se o
percentual de cada elemento pela massa atômica e depois dividindo-se o resultado
pelo menor valor, arredondando-se cada índice obtido para valores inteiros.
Exemplo: Glicose – Fórmula centesimal C40,00%H6,71%O53,29%

15
Dividindo-se cada percentual pela massa atômica correspondente, temos:
C(
40,0012,01
)H
(6,71
1,008)O
(53,2916,00
),
resultando em:
C3,331H6,657O3,331
Dividindo-se por 3,331, resulta:
CH2O
A fórmula centesimal pode ser obtida a partir da fórmula mínima multiplican-
do-se cada índice pela massa atômica do elemento, obtendo-se a relação entre as
massas de cada elemento. Dividindo-se cada massa pela soma das massas e
multiplicando-se por 100, chega-se à fórmula centesimal.
Exemplo: Peróxido de hidrogênio – Fórmula centesimal HO
Multiplicando-se cada índice pela massa atômica obtém-se:
H1×1,008O1×16,00
ou
H1,008O16,00
Os valores estão em gramas, e massa total que corresponde a esta fórmula é
1,008 + 16,00 + 16,00 = 17,008. O cálculo dos percentuais será como segue:
H 1,00817,008
×100O 16,00
17,008×100
,
resultando:
H5,95%O94,07%
A análise elementar consiste na combustão completa de uma massa conhecida de uma
substância e conversão dos gases gerados em precipitados que podem ser pesados. O
carbono é convertido em dióxido de carbono, CO2, gerado é absorvido em uma solução
padronizada de hidróxido de bário, formando carbonato de bário, BaCO3. O hidrogênio
é convertido em água, que é absorvida por um agente higroscópico, perclorato de
magnésio, MgClO4. A percentagem de oxigênio é determinada por diferença.

16
1.5 ESTRUTURAS DE LEWIS, CARGA FORMAL E RESSONÂNCIA
Passo 1. Organize os átomos seguindo padrões de conexão comuns. Quando há um
átomo central, geralmente é o elemento menos eletronegativo no composto. O
hidrogênio e os halogênios estão quase sempre conectados a apenas um outro
átomo, então eles são geralmente terminais em vez de centrais. Como o carbono é
um dos elementos com mais possibilidades de ligação (Figura 2), é mais provável
que ele seja o átomo central.
Passo 2. Determine o número total de elétrons de valência na molécula ou íon. Some
os elétrons de valência de cada átomo. Se a espécie for um íon poliatômico,
lembre-se de adicionar ou subtrair o número de elétrons necessários para dar a
carga total no íon. Tomando como exemplo o íon carbonato,𝐶𝑂32−,por exemplo,
adicionamos dois elétrons ao total.
Passo 3. Coloque um par de elétrons compartilhados entre cada par de átomos
adjacentes para formar uma ligação simples. Na molécula de H2O, por exemplo, há
um par de elétrons de ligação entre o oxigênio e cada hidrogênio.
Passo 4. Começando com os átomos terminais, adicione elétrons suficientes a cada
átomo para dar a cada átomo um total de oito elétrons (dois para hidrogênio). Esses
elétrons geralmente serão pares isolados.
Passo 5. Se sobrar algum elétron, coloque-o no átomo central. Um fato que
discutiremos nas próximas seções é o que diversos elementos (sobretudo aqueles
pertencentes ao 3º período da Tabela Periódica em diante) podem comportar mais
de 8 elétrons de valência (chamamos isso de expansão do octeto).
Passo 6. Se o átomo central tiver menos elétrons do que um octeto, use pares
isolados de átomos ligados a ele para formar ligações múltiplas (duplas ou triplas) a
fim se obter um octeto. Isso não mudará o número de elétrons nos átomos terminais.
Vejamos alguns exemplos:
Água (H2O)
Passo 1. Como os átomos de H são quase sempre terminais, o arranjo dentro
da molécula deve ser HOH.
Passo 2. Cada átomo H (grupo 1) possui 1 elétron de valência, e o átomo O
(grupo 16) possui 6 elétrons de valência, resultando em um total de 8 elétrons de
valência.

17
Passo 3. Adicionando um par de elétrons de ligação entre o átomo de O e
cada átomo de H, teremos H:O:H, com 4 elétrons restantes.
Passo 4. Cada átomo de H tem uma camada de valência completa de 2
elétrons.
Passo 5. Adicionando os 4 elétrons restantes ao oxigênio (como dois pares
isolados) obtemos a seguinte estrutura:
Como o octeto do oxigênio e os dois elétrons do hidrogênio estão completos,
não precisamos usar o passo 6.
Íon hipoclorito (OCl-)
Passo 1. Com apenas dois átomos na molécula, não há átomo central.
Passo 2. O oxigênio (grupo 16) tem 6 elétrons de valência e o cloro (grupo 17)
tem 7 elétrons de valência; devemos adicionar mais um para a carga negativa do
íon, resultando em um total de 14 elétrons de valência.
Passo 3. Colocando um par de elétrons entre O e Cl obtemos O:Cl, com 12
elétrons restantes.
Passo 4. Se colocarmos seis elétrons (como três pares solitários) em cada
átomo, obtemos a seguinte estrutura:
Cada átomo agora tem um octeto de elétrons, então as etapas 5 e 6 não são
necessárias. A estrutura do elétron de Lewis é desenhada entre colchetes, como é
usual para um íon, com a carga geral indicada fora dos colchetes, e o par de elétrons
de ligação é indicado por uma linha sólida.
Formaldeído (CH2O)
Passo 1. Como o carbono é menos eletronegativo que o oxigênio e o
hidrogênio normalmente é terminal, C deve ser o átomo central. Um arranjo possível
é o seguinte:

18
Passo 2. Cada átomo de hidrogênio (grupo 1) tem um elétron de valência. O
carbono (grupo 14) tem 4 elétrons de valência e oxigênio (grupo 16) tem 6 elétrons
de valência, resultando em um total de2 × 1 + 4 + 6 = 12elétrons de valência.
Passo 3. Colocando um par de elétrons de ligação entre cada par de átomos
ligados dá o seguinte:
Seis elétrons foram usados e faltam 6.
Passo 4. Adicionando todos os 6 elétrons restantes ao oxigênio (como três pares
isolados) dá o seguinte:
Embora o oxigênio agora tenha um octeto e cada hidrogênio tenha 2 elétrons,
o carbono tem apenas 6 elétrons.
Passo 5. Não há mais elétrons para colocar no átomo central.
Passo 6. Para proporcionar ao carbono o octeto de elétrons, usamos um dos
pares isolados do oxigênio para formar uma ligação dupla carbono-oxigênio:
Assim, todos os átomos têm o octeto completo (e o H tem seus dois elétrons)
Muitas vezes é possível construir mais de uma estrutura de Lewis para o mesmo

19
conjunto de átomos, embora algumas delas sejam menos prováveis que outras. Para
avaliarmos quais estruturas são as mais adequadas, utilizamos as cargas formais dos
átomos. A carga formal de um átomo em uma molécula é a carga hipotética que o
átomo teria se pudéssemos redistribuir os elétrons nas ligações igualmente entre os
átomos. Calculamos a carga formal a partir do número de elétrons de valência de
um átomo neutro, do qual subtraímos os elétrons não ligados e, em seguida,
subtraímos o número de ligações conectadas a esse átomo na estrutura de Lewis.
Carga formal = número de elétrons de valência (átomo livre)
− número de pares de elétrons − 1
2 × elétrons ligantes
A soma das cargas formais dos átomos equivale à carga total da espécie
(sendo zero para moléculas neutras).
Exemplo: Determine as cargas formais de cada átomo da molécula .
Como o átomo de bromo está em menor quantidade, é razoável colocá-lo
como átomo central:
Atribuindo os pares isolados a cada átomo, constatamos que cada átomo de
Cl tem sete elétrons e o átomo de Br tem sete elétrons. Subtraindo este valor dos
elétrons de valência para o átomo neutro resulta:
Br: 7 – 7 = 0
Cl: 7 – 7 = 0
Todos os átomos do BrCl3 têm carga formal zero, e a soma corresponde à
carga zero, tratando-se de uma molécula neutra.
Como as cargas formais podem nos auxiliar a definir qual a melhor estrutura
de Lewis dentre várias alternativas? Alguns princípios podem nos ajudar:
1. Uma estrutura molecular em que todas as cargas formais são iguais a zero é
preferível a outra em que algumas cargas formais não são.
2. Se a estrutura de Lewis deve ter cargas formais diferentes de zero, o arranjo
com o menor número de cargas formais diferentes de zero é preferível.

20
3. As estruturas de Lewis são preferíveis quando as cargas formais adjacentes são
iguais a zero ou com sinais opostos.
4. Quando devemos escolher entre várias estruturas de Lewis com distribuições
semelhantes de cargas formais, a estrutura com as cargas formais negativas
nos átomos mais eletronegativos é a preferível.
Exemplo: Dióxido de carbono (CO2)
Representando as diferentes possibilidades de estruturas do dióxido de car-
bono, com as cargas formais de cada átomo:
Obviamente, a primeira estrutura que tem todas as cargas formais iguais a zero
é a preferível.
1.6 EXCEÇÕES À REGRA DO OCTETO
Dentre as principais exceções à regra do octeto que são importantes para o
estudo da Química Orgânica, mencionamos:
O boro e o alumínio estabilizam-se com 6 elétrons de valência. Compostos
como BCl3 e AlCl3 são chamados ácidos de Lewis (que estudaremos na Unidade 3) e
são utilizados em diversas reações.
Elementos pertencentes ao 3º período da Classificação Periódica em diante,
quando são átomos centrais, podem comportar mais de 8 elétrons de valência (a
isso chamamos expansão do octeto). Por exemplo, a molécula de ácido sulfúrico,
H2SO4 possui um enxofre como átomo central, com 12 elétrons de valência (os pares
isolados dos oxigênios não estão representados):

21
1.7 ORBITAIS MOLECULARES E HIBRIDIZAÇÃO
A Mecânica Quântica lançou uma nova luz sobre a estrutura da matéria e a
interação da matéria com a energia, servindo de base para o entendimento
moderno das moléculas e átomos. O princípio da incerteza de Heisenberg prevê que
é impossível determinar com exatidão e simultaneamente a energia e a posição de
um elétron no átomo. A equação de Schröedinger, que emprega este princípio e
uma equação física denominada equação da onda, permitiu a concepção de
regiões espaciais ao redor do núcleo, com geometrias e orientações específicos,
onde é maior a probabilidade de se encontrar os elétrons (os orbitais atômicos). Esta
equação apresenta valor exato unicamente para o hidrogênio e sistemas
hidrogenóides (isto é, sistemas com apenas um elétron). Sistemas com mais de um
elétron são resolvidos de forma aproximada, por meio de diversos métodos que não
serão discutidos aqui.
A principal característica do elétron no átomo é seu comportamento
ondulatório. Se compararmos com uma onda que se propaga em uma corda,
veremos que ela apresenta regiões de alta e de baixa amplitude (cristas e vales), e
entre cada crista e vale há uma região estacionária (nó). Esta onda é chamada de
estacionária, pois cristas e vales alternam-se ao redor do mesmo ponto (Figura 3).
Figura 3: Onda estacionária em uma corda presa por ambas as extremidades
Fonte: Elaborado pelo Autor (2021)
Orbitais atômicos se apresentam como regiões com formatos bem definidos,
entremeados por nós, nos quais a probabilidade de se encontrar o elétron é nula.
Sem entrarmos em detalhes matemáticos, e considerando-se apenas as situações
mais comuns, podemos prever orbitais do tipo s, p, d e f, que apresentam formatos
variados. A Figura 4 mostra o formato dos orbitais s e p, de maior interesse nos

22
compostos orgânicos.
Figura 4: Representação dos orbitais atômicos s e p
Fonte: CK-12 Chemistry (2010)
O modelo teórico que possibilita o estudo das ligações químicas do ponto de
vista das interações entre orbitais atômicos é chamada Teoria do Orbital Molecular
(TOM), que é estudada comumente na Química Inorgânica. Neste texto nós nos
concentraremos unicamente no conceito de hibridização e geometria molecular,
que são consequências da TOM. Os elementos típicos dos compostos orgânicos
ligam-se por meio de orbitais do tipo s e p da camada de valência. As ligações
químicas covalentes se formam quando orbitais atômicos se sobrepõem, formando
orbitais moleculares. Estes orbitais podem se sobrepor na sua forma pura ou combi-
nados uns com os outros dentro do mesmo átomo, formando orbitais híbridos.
O átomo de carbono isolado, no estado fundamental (isto é, no seu estado de
menor energia), apresenta a seguinte configuração eletrônica:
6C (estado excitado): 1s2 2s1 2p3
Para estabelecer ligações, ocorre a promoção de um dos elétrons que estão
no subnível 2s para o 2p, de modo que teremos 4 orbitais (1 orbital s e 3 orbitais p)
com um elétron cada, capaz de ser compartilhado:
6C (estado excitado): 1s2 2s1 2p3

23
Pode ocorrer a união (“hibridização”) do orbital s com 1 (um) orbital p,
formando dois orbitais iguais chamados sp e deixando os outros dois orbitais p
intactos (“puros”). Os orbitais sp ficam em uma disposição linear, com um ângulo de
180º entre si. Eles se sobrepõem de forma frontal com os orbitais dos átomos ao qual
o carbono está ligado, formando uma ligação chamada (sigma). Os orbitais p se
sobrepõem de forma lateral formam uma ligação chamada (pi).
Esta hibridização pode formar uma ligação simples e uma tripla (no mesmo
carbono) ou duas ligações duplas (Figura 5).
Figura 5: Sobreposição de orbitais híbridos sp.
Fonte: Elaborado pelo Autor (2021)
Se ocorrer a união do orbital s com 2 (dois) orbitais p, formam-se três orbitais
iguais chamados sp2 e um orbital p é deixado intacto.
Os orbitais sp2 formam ligações , enquanto o orbital p forma ligações com
os átomos vizinhos. Estes orbitais ficam em uma disposição trigonal, com um ângulo
de 120º entre si. Esta hibridização forma uma ligação dupla e duas simples no mesmo
carbono (Figura 6).
Figura 6: Sobreposição de orbitais híbridos sp2.
Fonte: Elaborado pelo Autor (2021)

24
Por fim, se ocorrer a união do orbital s com 3 (três) orbitais p, formam-se quatro
orbitais iguais chamados sp3. Estes orbitais ficam em uma disposição tetraédrica, com
um ângulo de 109,5º entre si. Estes orbitais formam quatro ligações . Esta hibridização
forma quatro ligações simples no mesmo carbono (Figura 7).
Figura 7: Sobreposição de orbitais híbridos sp3
Fonte: Elaborado pelo Autor (2021)
A hibridização nos leva à discussão acerca de sua relação com a geometria
das moléculas e como isso nos permite prever sua polaridade e interações.
1.8 GEOMETRIA MOLECULAR E POLARIDADE
A geometria de uma molécula está diretamente relacionada a sua
polaridade. Um dipolo elétrico formado entre duas extremidades com cargas
opostas apresenta um momento de dipolo (𝜇), que é um vetor cujo módulo, expresso
em debye (1 𝐷 = 10−30 𝑐𝑜𝑢𝑙𝑜𝑚𝑏 × 𝑚𝑒𝑡𝑟𝑜)é dado pelo produto da carga (q) pelo
módulo do deslocamento da mesma (𝑑 ) e cujo sentido (segundo as convenções
oficiais) é da extremidade negativa para positiva (embora em muitos livros este
sentido seja o inverso):
μ⃗⃗ = q × d⃗⃗
Moléculas apolares apresentam uma soma vetorial dos dipolos de todas
ligações igual a zero, ao contrário das moléculas polares, em que a soma dos dipolos
é diferente de zero. Este conceito é muito importante na compreensão das

25
propriedades físicas das substâncias, que estão relacionadas às suas interações
intermoleculares.
Exemplo 1: CHCl3 (molécula polar) e CCl4 (molécula apolar)
O carbono central é do tipo sp3 (com quatro ligações simples), e portanto é
tetraédrico. O cloro é mais eletronegativo que o carbono, que por sua vez é mais
eletronegativo que o hidrogênio. Representando-se os momentos de dipolo de cada
ligação e fazendo-se a soma vetorial, conclui-se que os vetores se anulam no caso
do CCl4 e não no caso do CHCl3 (Figura 8). A Figura 9 mostra uma previsão feita por
métodos computacionais dos dipolos.
Figura 8: Momentos de dipolo das ligações e momento de dipolo total do CHCl3
Fonte: Elaborado pelo Autor (2021)
Figura 9: Momentos de dipolo calculados para o CHCl3 e o CCl4. Cálculos gerados pelo
programa Arguslab.
Fonte: Elaborado pelo Autor (2021)

26
É necessário lembrar que o conceito de polaridade é relativo e não absoluto:
não se trata de determinar se determinada molécula é polar ou apolar, mas se uma
molécula é mais polar ou menos polar que a outra (o que dependerá do módulo do
momento de dipolo, 𝜇.
https://bit.ly/3rjYx99. Acesso
em: 13 dez. 2020.

27
FIXANDO O CONTEÚDO
1. O átomo de carbono é capaz de estabelecer _________ compartilhamentos de
elétrons com os vizinhos, e apresenta diversas hibridizações. O carbono que
apresenta uma ligação dupla e duas ligações simples tem geometria __________ e
um ângulo de __________ entre suas ligações.
De acordo com o que foi estudado na Unidade I, marque a opção que melhor
preenche as lacunas acima:
a) três, trigonal, 120º
b) quatro, tetraédrica, 120º
c) quatro, tetraédrica, 109,5º
d) três, tetraédrica, 120º
e) quatro, trigonal, 120º
2. Leia as afirmativas abaixo:
I. A regra do octeto prevê que os átomos se estabilizem com 8 elétrons de
valência e não admite exceções.
II. O caráter iônico de uma ligação depende da diferença de eletronegatividade
entre os átomos que a constituem.
III. Longas cadeias constituídas unicamente de átomos se silício não são estáveis
como as cadeias formadas por átomos de carbono.
De acordo com o que foi estudado na Unidade I, escolha a alternativa que
apresenta as afirmativas corretas:
a) I e III
b) II e III
c) I e II
d) I, II e III
e) Nenhuma das afirmativas está correta.

28
3. De acordo com o que foi estudado na Unidade I, associe a coluna da esquerda
com a da direita:
I. Apresenta a proporção de cada elemento em uma unidade da substância.
II. Apresenta a quantidade real de elementos de cada tipo em uma unidade da
substância (molécula, par iônico, etc.).
III. Apresenta o percentual em massa de cada elemento em relação à massa
total.
( ) Fórmula centesimal
( ) Fórmula molecular
( ) Fórmula mínima
A correta associação entre as duas colunas é dada por:
a) III, I, II
b) I, III, II
c) II, I, III
d) III, II, I
e) I, II, III
4. A carga formal de um átomo é um parâmetro importante para se avaliar se
determinada opção de fórmula estrutural é uma boa representação da mesma.
Dentre os seguintes critérios que envolvem as cargas formais, qual está correto?
a) As estruturas de Lewis são preferíveis quando as cargas formais adjacentes apre-
sentam mesmo sinal.
b) Uma estrutura molecular em que todas as cargas formais apresentam cargas
opostas é preferível a outra em que todas as cargas são iguais a zero.
c) Estruturas contendo cargas negativas em átomos mais eletronegativos são
preferíveis em relação a outras distribuições semelhantes.
d) Se a estrutura de Lewis deve ter cargas formais diferentes de zero, é preferível o
arranjo com o menor número de cargas formais negativas.

29
e) Uma estrutura com cargas formais de maior valor absoluto são preferíveis a
arranjos com cargas formais de valor unitário.
5. Analise as seguintes proposições:
I. A Teoria do Orbital Molecular leva em consideração o comportamento
ondulatório do elétron e prevê a formação de orbitais moleculares a partir da
superposição de orbitais atômicos.
II. A regra do octeto não admite exceções entre os ametais.
III. Ligações com pequena diferença de eletronegatividade entre seus
elementos constituintes apresentam elevado caráter iônico.
Estão corretas as proposições:
a) I apenas.
b) I e II
c) II apenas
d) II e III
e) Todas as alternativas estão corretas.
6. Considere as seguintes estruturas de Lewis
Está(ão) correta(s) a(s) estrutura(s):
a) I e II
b) I e III
c) III apenas
d) I apenas
e) II e III

30
7. A carga formal no átomo de nitrogênio nas espécies NH3, NH4+, NO3
– é,
respectivamente:
a) 0, +1, +1
b) 0, -1, +1
c) +1, 0, -1
d) +1, +1, +1
e) -1, -1, 0
8. Leia as afirmativas abaixo:
I. Os átomos de carbono são capazes de fazer ligações estáveis uns com os ou-
tros, formando cadeias, propriedades esta que não ocorre com outros
elementos da mesma família.
II. Ligações múltiplas são mais fortes que as ligações simples correspondentes.
III. Todas as ligações entre ametais contém pelo menos uma ligação sigma.
Escolha a alterna tiva correta:
a) São verdadeiras apenas as afirmativas I e II.
b) São verdadeiras apenas as afirmativas II e III.
c) É verdadeira apenas a afirmativa II.
d) Nenhuma das afirmativas é verdadeira.
e) Todas as afirmativas são verdadeiras.

31
FUNÇÕES ORGÂNICAS E
ESPECTROSCOPIA NO
INFRAVERMELHO
2.1 CLASSIFICAÇÃO DE ÁTOMOS DE CARBONO E CADEIAS CARBÔNICAS
Um conceito importante no estudo da estrutura de compostos orgânicos é o
de carbonos primários, secundários, terciários e quaternários. Carbonos primários são
aqueles ligados a apenas um carbono; carbonos secundários são ligados a dois
carbonos; carbonos terciários são ligados a três carbonos e carbonos quaternários
são ligados a quatro carbonos (Figura 10).
Figura 10: Exemplo de uma estrutura com carbonos primários, secundários, terciários e
quaternários
Fonte: Elaborado pelo Autor (2021)
Carbonos que não estão ligados a nenhum outro carbono (como no metanol,
CH3OH) não recebem este tipo de classificação, embora alguns autores atribuam a
eles a designação de primários.
Cadeias carbônicas podem ser representadas de diferentes formas. A Figura
11 ilustra a estrutura tridimensional (na forma de bola e bastão), a estrutura de Lewis,
a fórmula completa, a fórmula condensada e a fórmula de traços de algumas
substâncias.
UNIDADE
02

32
Figura 11: Estruturas químicas em suas diversas representações
Fonte: Adaptado de Solomons e Fryhle (2009)
Existem diferentes formas de classificar as cadeias carbônicas, e neste material
adotaremos a seguinte convenção:
Cadeias abertas ou acíclicas
Normais vs ramificadas
Saturadas vs insaturadas
Homogêneas vs heterogêneas
Cadeias fechadas ou cíclicas
Homocíclicas vs heterocíclicas

33
Mononucleares vs polinucleares
Alifáticas (alicíclicas) vs aromáticas
Cadeias mistas
Cadeias abertas apresentam extremidades livres, cadeias fechadas (ou
cíclicas) têm ambas as extremidades ligadas, formando um ciclo ou anel, e cadeias
mistas têm uma parte aberta e uma parte fechada (Figura 12).
Figura 12: Cadeias abertas, fechadas ou mistas
Fonte: Elaborado pelo Autor (2021)
2.1.1 Classificação das cadeias abertas
Cadeias normais apresentam unicamente carbonos primários e secundários.
Cadeias ramificadas, por sua vez, apresentam pelo menos um carbono terciário ou
quaternário (Figura 13).
Figura 13: Cadeias normais ou ramificadas
Fonte: Elaborado pelo Autor (2021)

34
Cadeias saturadas apresentam unicamente ligações simples entre carbonos,
enquanto cadeias insaturadas apresentam pelo menos uma ligação dupla ou tripla
entre carbonos (Figura 14).
Figura 14: Cadeias saturadas ou insaturadas
Fonte: Elaborado pelo Autor (2021)
Cadeias homogêneas não apresentam heteroátomo (átomo diferente de
carbono) separando carbonos. Cadeias heterogêneas apresentam pelo menos um
heteroátomo separando carbonos (Figura 15).
Figura 15: Cadeias homogêneas ou heterogêneas
Fonte: Elaborado pelo Autor (2021)
Finalizada nossa discussão sobre a classificação de cadeias acíclicas, vamos
nos concentrar na classificação de cadeias fechadas (ou cíclicas).

35
2.1.2 Classificação das cadeias fechadas
Cadeias homocíclicas são cadeias fechadas e que não contém heteroátomo
no ciclo. Cadeias heterocíclicas são cadeias fechadas com heteroátomo no ciclo
(Figura 16).
Figura 16: Cadeias homocíclicas ou heterocíclicas
Fonte: Elaborado pelo Autor (2021)
Cadeias mononucleares são cadeias fechadas constituídas de apenas um
ciclo (um núcleo). Cadeias polinucleares são cadeias fechadas constituídas de mais
de um ciclo. Neste caso os anéis podem ser condensados ou isolados (Figura 17).
Figura 17: Cadeias mononucleares ou polinucleares
Fonte: Elaborado pelo Autor (2021)
Cadeias aromáticas são cadeias fechadas que atendem a certas condições
(que examinaremos depois) que permitem que seus elétrons sejam deslocalizados. O
exemplo de referência é o anel do benzeno. A presença deste anel indica que a
cadeia é aromática, embora o conceito seja mais amplo e envolva outros anéis com

36
as mesmas características. Cadeias alicíclicas são cadeias fechadas que não
atende às condições acima.
O benzeno é um anel plano de 6 carbonos com três ligações duplas e três
ligações simples alternadas. Esta configuração permite que seus elétrons fiquem
deslocalizados entre os carbonos, causando maior estabilidade que outros anéis em
um efeito chamado aromaticidade.
Apesar de as ligações duplas serem mais curtas que as ligações simples, sabe-
se que as 6 ligações do anel benzênico têm o mesmo tamanho (cerca de 140 pm).
Podem ser compreendidas como um tipo de ligação intermediário entre uma dupla
e uma simples, na verdade, uma sobreposição lateral entre os orbitais p puros dos
átomos de carbono (Figura 18). A Figura 19 traz alguns exemplos de cadeias
aromáticas (no caso específico que contém o anel benzênico) e cadeias alicíclicas.
Figura 18: Estrutura do benzeno
Fonte: Elaborado pelo Autor (2021)
Figura 19: Algumas cadeias aromáticas (contendo o anel benzênico) e cadeias alicíclicas
Fonte: Elaborado pelo Autor (2021)

37
Cadeias fechadas podem ser saturadas ou insaturadas? Apesar de ser co-
mum, classificar uma cadeia fechada como saturada ou insaturada não é
totalmente correto. Utilizamos aqui um conceito simplificado de
saturação/insaturação, relacionado à presença de ligações duplas ou triplas entre
carbonos.
A ideia de saturação/insaturação é mais ampla e está relacionada à
quantidade de hidrogênios que podem ser ligados à estrutura. Quando uma cadeia
comporta o máximo de átomos de hidrogênio que podem ser ligados, ela é saturada.
Do contrário, é insaturada.
Assim, uma ligação dupla tira a “vaga” de dois hidrogênios. Isso é verdade
mesmo se a ligação dupla for entre átomos diferentes de carbono! (o que contradiz
o conceito mais simples utilizado neste texto). Uma ligação tripla, por sua vez, tira o
lugar de quatro hidrogênios (Figura 20).
Figura 20: Relação entre insaturação e quantidade de hidrogênios em relação à
quantidade de carbonos
Fonte: Elaborado pelo Autor (2021)

38
O mesmo ocorre com um anel (Figura 21). Portanto, se aplicarmos este conceito mais
amplo, todos os anéis são insaturados. Assim, para evitar esta confusão, preferimos
não classificar cadeias fechadas como saturadas ou insaturadas, embora devamos
estar preparados para ver este tipo de classificação em algumas fontes.
Figura 21: Diferentes estruturas insaturadas
Fonte: Elaborado pelo Autor (2021)
Uma vez compreendidos os critérios para classicaçãod e cadeias abertas e
cadeias fechadas, qual seria o procedimento para cassificação de cadeias mistas?
2.1.3 Classificação das cadeias mistas
Cadeias mistas são classificadas separadamente em sua parte aberta e em
sua parte fechada. Considera-se que a cadeia é mista quando a cadeia aberta
ligada ao anel tem no mínimo dois carbonos (Figura 22).
Figura 22: Cadeias mistas
Fonte: Elaborado pelo Autor (2021)
Na próxima seção você será apresentado às principais funções orgânicas,
seus grupos distintivos e a alguns exemplos de sua ocorrência.
2.2 PRINCIPAIS FUNÇÕES ORGÂNICAS
Vimos na Unidade I que as substâncias orgânicas existem em grande número

39
e suas estruturas são muito diversificadas. Devido a esta diversidade, é conveniente
dividi-los em categorias, chamadas funções orgânicas, que podem ser identificadas
de acordo com a presença de certos grupos ou características próprias, que
chamaremos de grupos funcionais.
Hidrocarbonetos. Hidrocarbonetos constituem a função orgânica mais
simples: são os compostos contendo unicamente carbono e hidrogênio. São
divididos em subcategorias, dentre as quais destacamos:
Alcanos: Hidrocarbonetos saturados
Alcenos: Hidrocarbonetos contendo uma ligação C=C
Alcinos: Hidrocarbonetos contendo uma ligação C≡C
Cicloalcanos: Hidrocarbonetos de cadeia fechada sem ligação múltipla
Dienos, trienos e polienos: Hidrocarbonetos contendo duas ou mais ligações
C=C
Hidrocarbonetos aromáticos: Hidrocarbonetos contendo o anel benzênico.
Funções oxigenadas. As principais funções oxigenadas são: álcoois, éteres,
fenóis, enóis e as funções contendo carbonila (cetonas, aldeídos, ácidos carboxílicos
e ésteres).
Álcoois: apresentam um carbono sp³ ligado a OH.
(ou ROH)
(Representa-se uma cadeia carbônica genérica por R; usa-se Ar para especificar cadeias
aromáticas)
Éteres: apresentam oxigênio entre carbonos (cadeia heterogênea). As
cadeias podem ser iguais ou diferentes.
R1 – O – R2, R – O – Ar ou Ar1 – O – Ar2
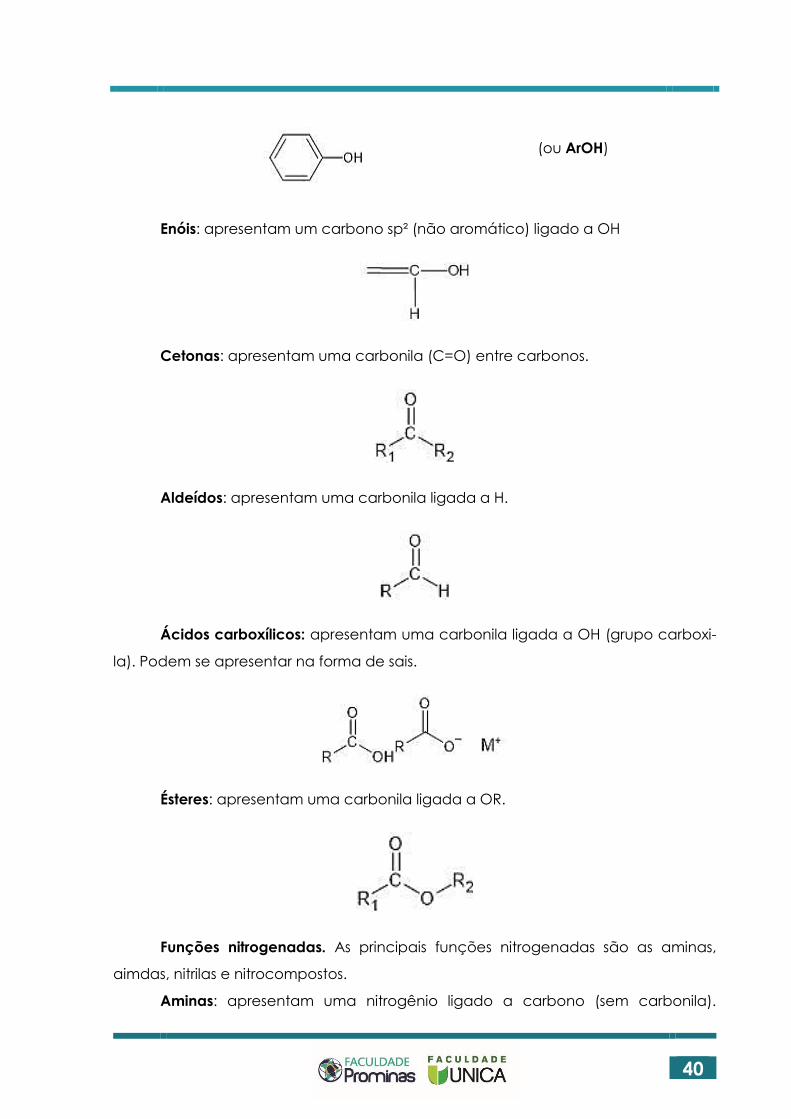
Fenóis: apresentam um anel benzênico ligado a OH
40
(ou ArOH)
Enóis: apresentam um carbono sp² (não aromático) ligado a OH
Cetonas: apresentam uma carbonila (C=O) entre carbonos.
Aldeídos: apresentam uma carbonila ligada a H.
Ácidos carboxílicos: apresentam uma carbonila ligada a OH (grupo carboxi-
la). Podem se apresentar na forma de sais.
Ésteres: apresentam uma carbonila ligada a OR.
Funções nitrogenadas. As principais funções nitrogenadas são as aminas,
aimdas, nitrilas e nitrocompostos.
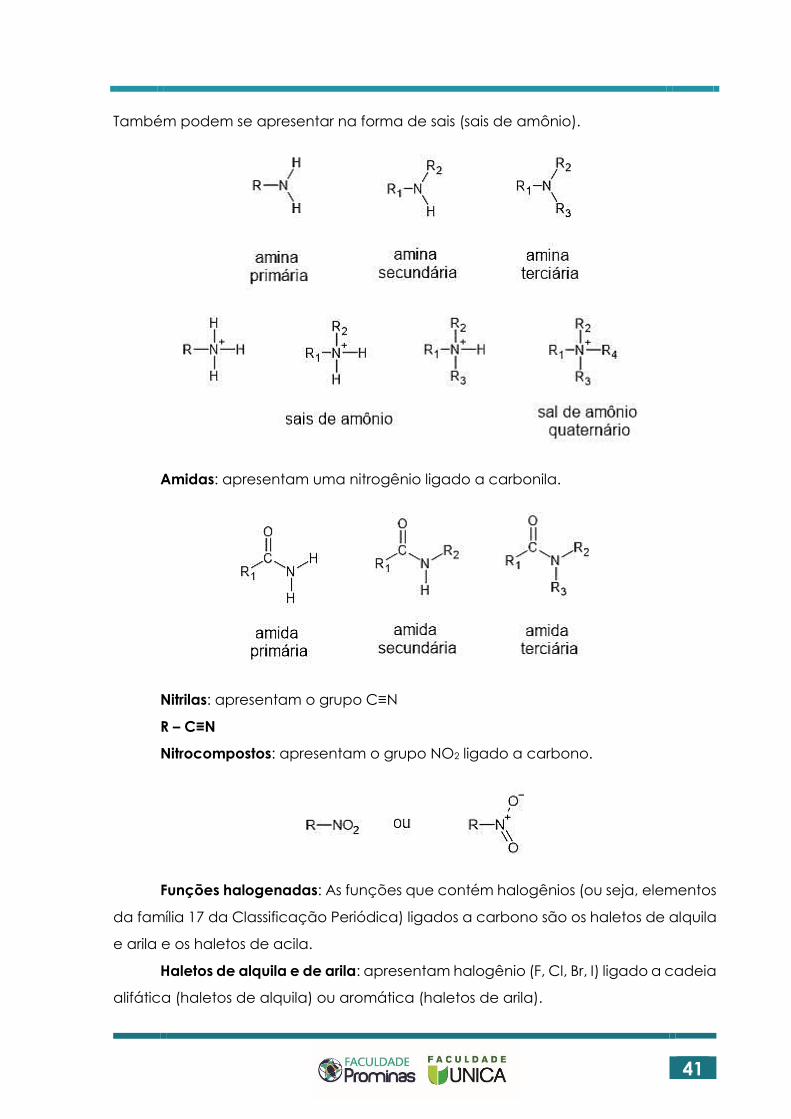
Aminas: apresentam uma nitrogênio ligado a carbono (sem carbonila).
41
Também podem se apresentar na forma de sais (sais de amônio).
Amidas: apresentam uma nitrogênio ligado a carbonila.
Nitrilas: apresentam o grupo C≡N
R – C≡N
Nitrocompostos: apresentam o grupo NO2 ligado a carbono.
Funções halogenadas: As funções que contém halogênios (ou seja, elementos
da família 17 da Classificação Periódica) ligados a carbono são os haletos de alquila
e arila e os haletos de acila.
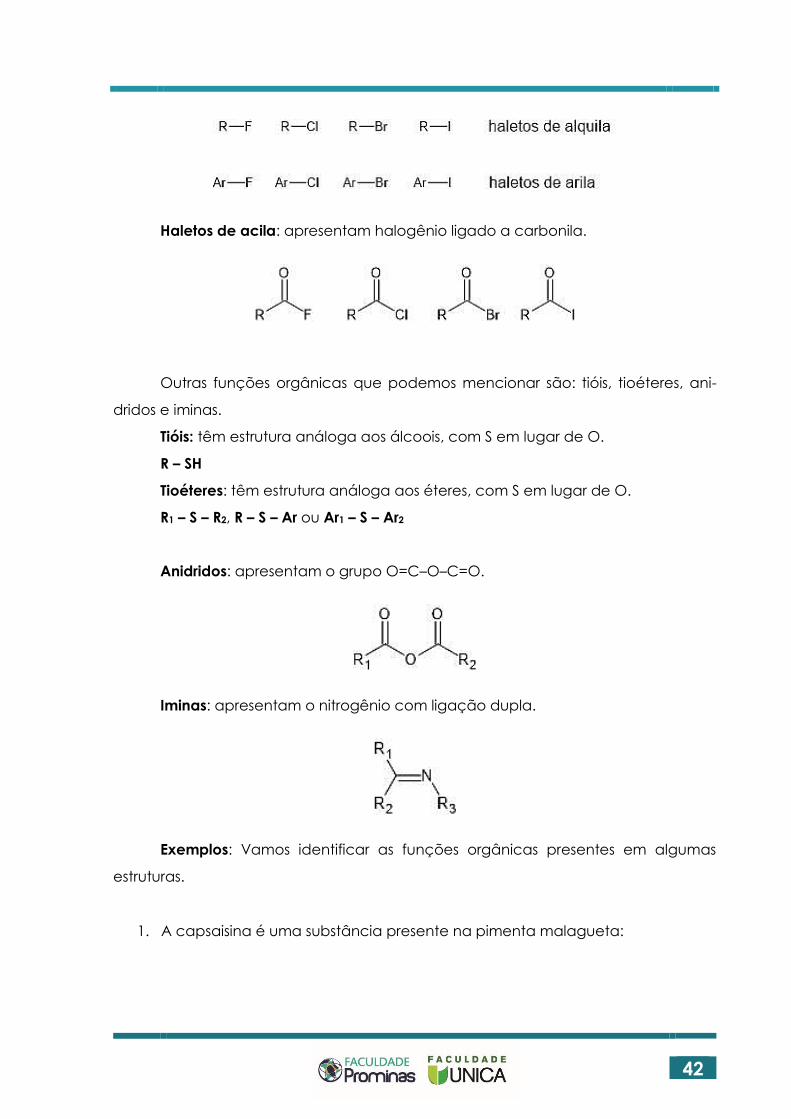
Haletos de alquila e de arila: apresentam halogênio (F, Cl, Br, I) ligado a cadeia
alifática (haletos de alquila) ou aromática (haletos de arila).
42
Haletos de acila: apresentam halogênio ligado a carbonila.
Outras funções orgânicas que podemos mencionar são: tióis, tioéteres, ani-
dridos e iminas.
Tióis: têm estrutura análoga aos álcoois, com S em lugar de O.
R – SH
Tioéteres: têm estrutura análoga aos éteres, com S em lugar de O.
R1 – S – R2, R – S – Ar ou Ar1 – S – Ar2
Anidridos: apresentam o grupo O=C–O–C=O.
Iminas: apresentam o nitrogênio com ligação dupla.
Exemplos: Vamos identificar as funções orgânicas presentes em algumas
estruturas.
1. A capsaisina é uma substância presente na pimenta malagueta:

43
Funções presentes:
2. A epibatidina é isolada do veneno do sapo da espécie Epipedobates antho-
nyi:
Funções presentes:

44
3. O cloreto de tubocurarina, também conhecido como curare, é encontrado
no extrato da planta Chondodendron tomentosum, utilizado pelos índios da
América do Norte para envenenar suas flechas:
Funções presentes:
4. Penicilina V (antibiótico isolado do fungo da espécie Penicilium notatum)
Funções presentes:

45
Do ponto de visa prático, como um profissional da Química pode saber qual
(quais) funções estão presentes em uma determinada substância? Uma técnica
experimental que é rotineira é a espectroscopia de absorção no infravermelho, que
será o tema da próxima seção.
2.3 ESPECTROSCOPIA NO INFRAVERMELHO: FUNDAMENTOS E INSTRUMEN-
TAÇÃO
No dia a dia dos profissionais que atuam na Química, uma atividade
frequente consiste na identificação de substâncias que são sintetizadas ou isoladas
de plantas ou animais. Até meados do século XIX, diversas substâncias químicas
foram identificadas e suas estruturas elucidadas por meio de um processo árduo que
envolvia testes químicos em inúmeras etapas para identificação das funções
presentes. Quantidades muito pequenas de produtos naturais eram obtidas por meio
de métodos cromatográficos, muitas vezes insuficientes para uma identificação
conclusiva.
Foi a partir dos anos 1930 que a interação da luz ultravioleta com a matéria
serviu ao propósito de auxiliar na identificação estrutural, aplicada a compostos
conjugados (ou seja, contendo ligações duplas separadas grupos com pares de
elétrons ou por outras ligações duplas por ligações simples). A espectroscopia no
infravermelho (IV) passou a ser desenvolvida a partir do final dos anos 1940 e
aplicada à identificação de grupos funcionais. Posteriormente, a técnica conhecida
como ressonância magnética nuclear (RMN) trouxe incontáveis contribuições a esta
tarefa, tornando possível o desenvolvimento da indústria farmacêutica, de produtos
46
agrícolas, de novos materiais, dentre outros.
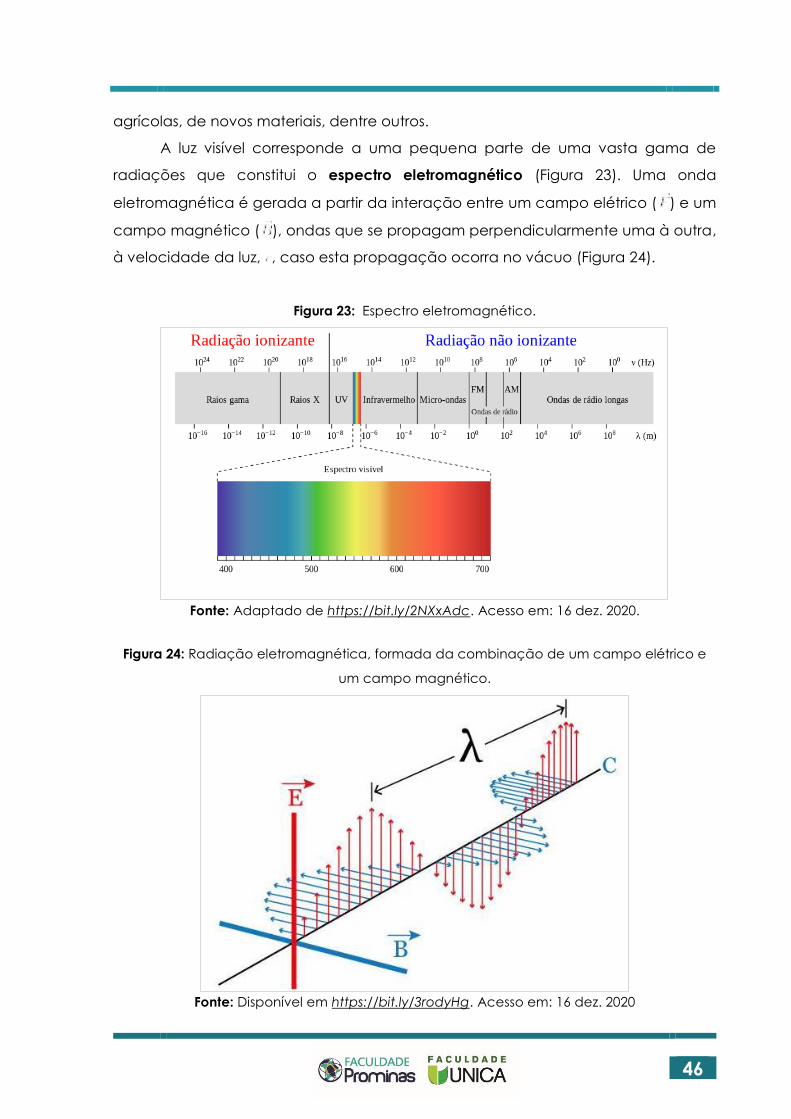
A luz visível corresponde a uma pequena parte de uma vasta gama de
radiações que constitui o espectro eletromagnético (Figura 23). Uma onda
eletromagnética é gerada a partir da interação entre um campo elétrico ( ) e um
campo magnético ( ), ondas que se propagam perpendicularmente uma à outra,
à velocidade da luz, , caso esta propagação ocorra no vácuo (Figura 24).
Figura 23: Espectro eletromagnético.
Fonte: Adaptado de https://bit.ly/2NXxAdc. Acesso em: 16 dez. 2020.
Figura 24: Radiação eletromagnética, formada da combinação de um campo elétrico e
um campo magnético.
Fonte: Disponível em https://bit.ly/3rodyHg. Acesso em: 16 dez. 2020

47
A espectroscopia no IV se baseia no fato de as moléculas estão em um estado
de vibração que é alterado quando interage com a radiação na região do
infravermelho. Uma ligação vibra como uma mola, sofrendo estiramento e
contração (Figura 25).
Figura 25: Vibração de uma ligação sob a ação da radiação no IV.
Fonte: Adaptado de Clayden et al. (2000)
A modelo do sistema massa-mola segue a lei de Hooke, que estabelece que,
quando a mola é deformada de , ela sofre uma força de restauração F, oposta ao
sentido da deformação, que depende da constante de força da mola, k.
F = −k ⋅ x (1)
Se a mola é deformada e liberada pela força que a deformou, ela entra em
vibração, oscilando ao redor de uma posição de equilíbrio com uma frequência
(este sistema em condições em que não há forças dissipativas é chamado de
oscilador harmônico). Suponha que tenhamos o sistema de duas massas m1 e m2 da
Figura 25, que oscila a uma frequência , mais comumente expressa como um
número de onda ν̄ = 1
λ(expresso em cm-1). Pode-se demonstrar que o número de
onda será dado por:
ν̄ = 1
2πc√
k
μ (2)

48
sendo a massa reduzida do sistema, dada por:
μ = m1m2
m1 + m2. (3)
Podemos perceber que, quanto mais forte a ligação (ou seja, quanto maior a
constante de forma k), menor a frequência de vibração. Também pode-se concluir
que, quanto mais pesados os átomos da ligação, maior será o valor de (visto que
o produto de m1 e m2 no numerador da equação anterior aumenta mais
rapidamente que a soma no denominador) e menor a frequência de vibração.
Em uma molécula, além do estiramento e contração das ligações, ocorrem
também outros movimentos de ligações, cujos ângulos aumentam ou diminuem.
Figura 26: Tipos de vibração de ligações no IV
Fonte: Adaptado de Solomons e Fryhle (2009)

49
A Figura 27 mostra o esquema geral de um espectrofotômetro de IV, que
conta com uma fonte (por exemplo, um corpo negro aquecido), um compartimento
para a amostra (geralmente dissolvida em clorofórmio ou compactada em uma
pastilha de brometo de potássio, KBr) e outro para um material de referência, e de
um sistema contendo defletor, detetor, processador do sinal e um display. Um
gráfico de transmitância em função do número de onda é gerado (Figura 27), e é o
ponto de partida para a análise da estrutura da(s) substância(s) presente(s) na
amostra.
Figura 27: Esquema de um espectrofotômetro de IV
Fonte: Elaborado pelo Autor (2021)
Figura 28: Espectro de absorção no IV do formaldeído.
Fonte: Coblentz Society (2018)
Um modelo mais usado de espectrofotômetro, chamado dispersivo, não requer o preparo
da amostra com solvente ou KBr.

50
A absorção de energia é quantizada, ou seja, as substâncias absorvem
energia em determinadas regiões do espectro eletromagnético. Considerando que
o número de onda das ligações depende da massa dos átomos e da natureza das
ligações (força e polaridade), este parâmetro pode ser usado para se estabelecer
uma relação com o tipo de grupos funcionais presentes da estrutura. Como o
movimento das ligações é feito em conjunto, ou seja, quando uma ligação sofre
estiramento ou deformação, todo o esqueleto carbono vibra em outras frequências,
de modo que além do fato de as absorções serem características de cada tipo de
ligação, as frequências (ou números de onda) dependem da vizinhança destes
grupos.
2.4 APLICAÇÃO DA ESPECTROSCOPIA NO INFRAVERMELHO NA
IDENTIFICAÇÃO DE GRUPOS FUNCIONAIS
O Quadro 1 resume os números de onda e as características de diversas
bandas de absorção dos grupos funcionais mais comuns, que utilizamos na análise
estrutural, conforme veremos em alguns exemplos.
Quadro 1: Principais absorções no IR de grupos funcionais
Grupo
funcional Tipo Estrutura
Número de
onda (cm-
1)
Intensidade da
banda
C-H
sp3 R3C-H 2850-3000 média (fina)
sp2 =CR-H 3000-3250 média (fina)
sp C-H 3300 média-forte (fina)
N-H C-H de aldeídos H-(C=O)R 2750, 2850 média (fina)
A espectrofotometria no infravermelho por transformada de Fourier (FTIR) emprega um
método matemático que permite a obtenção dos conhecidos espectros a partir da
incidência de um feixe de luz na região do IV com diversas frequências simultâneas
(simples e combinadas), medindo a absorção ao longo do tempo. Este conjunto de
respostas é processado por meio de um algoritmo matemático, gerando os gráficos de
número de onda por intensidade. Para conhecer mais desta técnica consulte o livro de
“Química Orgânica” (2006) de Paula Yurkanis Bruice a partir da página 479. Disponível em:
https://bit.ly/3bpUfaW. Acesso em: 29 dez. 2020.

51
Grupo
funcional Tipo Estrutura
Número de
onda (cm-
1)
Intensidade da
banda
aminas primárias,
amidas
RN(H)-H,
RCONH2 3300, 3340 forte, forte (larga)
aminas secundárias,
amidas RNR-H, RCONHR 3300-3500 forte (larga)
aminas terciárias,
amidas RN(R3), RCONR2 -
O-H álcoois, fenóis
O-H livre 3620-3580 fraca (fina)
O-H com
ligação de
hidrogênio
3600-3650 forte (larga)
ácidos carboxílicos R(C=O)O-H 3500-2400 forte (larga)
CN nitrilas RCN 2280-2200 forte(fina)
CC alcinos R-CC-R 2260-2180 fraca (fina)
R-CC-H 2160-2100 média (fina)
C=O
aldeídos R(C=O)H 1740-1720 forte (fina)
cetonas R(C=O)R 1730-1710 forte (fina)
ésteres R(CO2)R 1750-1735 forte (fina)
ácidos carboxílicos RCO2H 1720-1680 forte (fina)
amidas
RCONH2,
RCONHR 1670-1640 forte (fina)
RCONH2 1650-1620 forte (fina)
RCONHR 1550 forte (fina)
amidas RCONR2 1650-1620 forte (fina)
anidridos R(CO2CO)R 1820, 1750 forte, forte (fina)
carboxilatos RCO2–, M+ 1600, 1400 forte, forte (fina)
C=C
alcenos (e compostos
contendo ligações
duplas entre
carbonos)
R2C=CR2 1680-1640 fraca (fina)
R2C=CH2 1600-1675 média (fina)
R2C=C(OR)R 1600-1630 forte (fina)
-NO2 grupos nitro RNO2 1550, 1370 forte, forte
(fina)[RHLdO1][CdM2]
Disponível em: https://bit.ly/30oCi6a. Acesso em: 09 mar. 2020.
Vamos aplicar esta abordagem a alguns exemplos.
2.4.1 Octano

52
Figura 29: Espectro de absorção no IV do octano.
Disponível em: https://webbook.nist.gov . Acesso em: 09 mar. 2020.
As bandas A e B indicam o estiramento de ligações C–H (2850-3000 cm-1). A
consulta a tabelas mais completas, permite distinguir a banda A como o estiramento
de ligações C–H de grupos –CH3 e B, este mesmo estiramento em grupos –CH2–. As
bandas C e D indicam, respectivamente, a deformação angular de ligações C–H de
grupos –CH2– e –CH3.
2.4.2 Hept-1-Ino
Figura 30: Espectro de absorção no IV do hept-1-ino
Disponível em: https://webbook.nist.gov. Acesso em: 09 mar. 2020.
A banda A se refere ao estiramento de ligações ≡C–H, B refere-se ao
53
estiramento C–H de grupos –CH2– e –CH3 e C ao estiramento de ligações C≡C. A
banda D é referente à deformação angular de grupos –CH3.
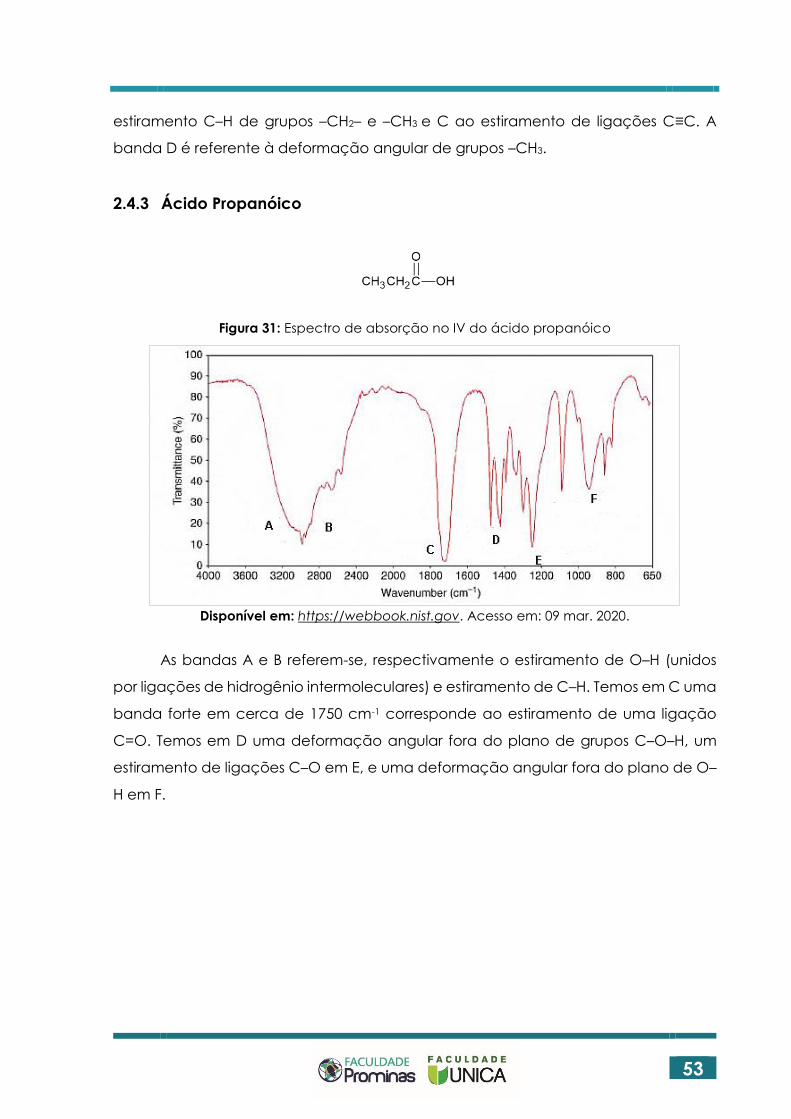
2.4.3 Ácido Propanóico
Figura 31: Espectro de absorção no IV do ácido propanóico
Disponível em: https://webbook.nist.gov. Acesso em: 09 mar. 2020.
As bandas A e B referem-se, respectivamente o estiramento de O–H (unidos
por ligações de hidrogênio intermoleculares) e estiramento de C–H. Temos em C uma
banda forte em cerca de 1750 cm-1 corresponde ao estiramento de uma ligação
C=O. Temos em D uma deformação angular fora do plano de grupos C–O–H, um
estiramento de ligações C–O em E, e uma deformação angular fora do plano de O–
H em F.

54
FIXANDO O CONTEÚDO
1. O taxol é uma substância isolada da casca do teixo do Pacífico (Taxus brevifolia),
com atividade comprovada contra câncer de mama e de ovário.
A alternativa que apresenta apenas funções presentes na estrutura do taxol é:
a) éter, amina, álcool
b) ácido carboxílico, amida, fenol
c) éster, amida, fenol
d) éster, amida, álcool
e) ácido carboxílico, éter, amida
2. Cetonas e aldeídos podem ter aplicações como essências de perfumes e
flavorizantes. Aldeídos de elevado valor agregado podem ser obtidos a parir da
oxidação do limoneno, obtido das cascas de laranjas descartadas em
abundância no Brasil. Alguns produtos da oxidação do limoneno são mostrados
abaixo:

55
Analisando as estruturas e os espectros de absorção no infravermelho do limoneno
e do produto de oxidação mostrado, foram feitas as seguintes afirmativas:
I. Os espectros do carveol e do álcool perílico diferem do 1,2-epoxilimoneno
pela presença de uma banda de intensidade média a alta na região acima
de 3500 cm-1 (ausente no 1,2-epoxilimoneno).
II. A banda de estiramento de ligações C=C em 1680-1640 cm-1 não apresenta
diferença de intensidade quando comparamos o limoneno com o 1,2-
epoxilimoneno.
III. Os produtos de oxidação do limoneno mostrados são éteres e fenóis.
Estão corretas as afirmativas:
a) I apenas.
b) I e II
c) I e III
d) III apenas
e) I, II e III.
3. Vitaminas são compostos multifuncionais que atuam como micronutrientes.
Considere as estruturas das três vitaminas ilustradas:
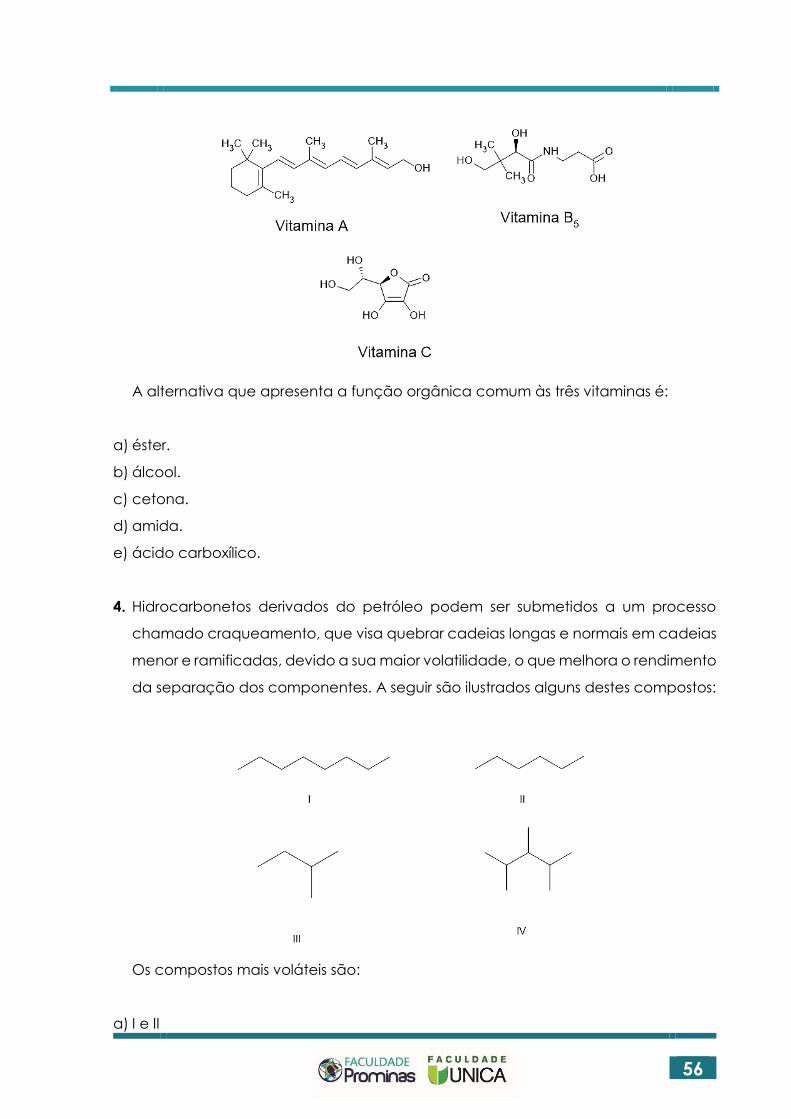
56
A alternativa que apresenta a função orgânica comum às três vitaminas é:
a) éster.
b) álcool.
c) cetona.
d) amida.
e) ácido carboxílico.
4. Hidrocarbonetos derivados do petróleo podem ser submetidos a um processo
chamado craqueamento, que visa quebrar cadeias longas e normais em cadeias
menor e ramificadas, devido a sua maior volatilidade, o que melhora o rendimento
da separação dos componentes. A seguir são ilustrados alguns destes compostos:
Os compostos mais voláteis são:
a) I e II

57
b) I e III
c) II e IV
d) III e IV
e) II e III
5. Compostos poliaromáticos contém diversos anéis benzênicos, e muitos são
conhecidos agentes carcinogênicos, como o fenaleno, o fenantreno e o
antraceno. Muitos destes compostos apresentam a mesma fórmula molecular, ou
seja, são isômeros.
Os compostos citados que são isômeros são:
a) fenaleno e fenantreno
b) fenaleno e antraceno
c) fenantreno e antraceno
d) todos são isômeros um do outro
e) nenhum deles é isômero um do outro
6. O ácido iocetâmico é uma substância utilizada como agente de contraste
A alternativa que apresenta funções a que o ácido iocetâmico pertence é:
a) haleto de arila, amina, amida e ácido carboxílico

58
b) haleto de alquila, amina, cetona e ácido carboxílico
c) haleto de alquila, amida, éster e ácido carboxílico
d) haleto de arila, amida, aldeído e éster
e) haleto de alquila, amina, cetona e aldeído.
7. A preparação em laboratório de um importante analgésico ocorre conforme
ilustrado:
Esta reação envolve as seguintes conversões:
a) um álcool reage com um aldeído formando um éster e um ácido carboxílico
b) um fenol reage com um anidrido formando um éster e um ácido carboxílico
c) um fenol reage com um éster formando um acetona e um ácido carboxílico
d) um álcool reage com um anidrido formando dois ácidos carboxílicos.
e) um fenol reage com um éster formando um aldeído e um ácido carboxílico.
8. O número de onda do estiramento de ligações e da deformação de ângulos com
a absorção no infravermelho depende da natureza dos átomos e ligações
envolvidos. Em relação a esta dependência, pode-se afirmar que:
a) Quanto mais forte a ligação, maior o número de onda.
b) Ligações duplas vibram com maior intensidade que ligações simples.
c) A vibração de uma ligação não afeta a vibração da ligação adjacente.
d) Ligações adjacente sempre vibram de forma simétrica.
e) Quando mais pesados os átomos da ligação, menor o número de onda.

59
ÁCIDOS E BASES ORGÂNICOS
3.1 INTRODUÇÃO
O estudo das substâncias orgânicas, suas propriedades e reações envolve seu
comportamento ácido ou básico, que será objeto de estudo nesta unidade. Estes
conceitos são úteis no entendimento dos mecanismos de reação e na predição dos
produtos.
3.2 QUEBRA HOMOLÍTICA E HETEROLÍTICA DE LIGAÇÕES
Uma ligação química é quebrada mediante ganho de energia. Se uma
ligação covalente apolar é quebrada, o par de elétrons fica distribuído igualmente
entre os dois átomos, e temos uma quebra (ou cisão) homolítica, formando-se
radicais.
Se uma ligação covalente polar é quebrada, o par de elétrons não fica
distribuído igualmente entre os dois átomos, e temos uma quebra heterolítica,
formando-se íons.
3.3 ELETRÓFILOS, NUCLEÓFILOS E RADICAIS
Dependendo de sua função na reação, um reagente pode ser classificado
como eletrofílico, nucleofílico ou radicalar. Um eletrófilo (ou reagente eletrofílico) é
deficiente de elétrons, e se comporta como um ácido de Lewis.
Exemplos: H+, Fe3+, Cl2, Br2, I2
UNIDADE
03

60
Um nucleófilo (ou reagente nucleofílico) é rico em elétrons, e se comporta
como uma base de Lewis.
Exemplos: OH-, CN-, RO-, HSO3-, NH3, H2O, ROH
Um radical (ou reagente radicalar) tem um elétron desemparelhado e pode
se ligar a outros radicais (formando uma ligação) ou a outros átomos (formando ou-
tros radicais).
Exemplos: R•, Cl•, Br•, I•
3.4 CARBOCÁTIONS E CARBÂNIONS
Quando o átomo de carbono sofre uma quebra heterolítica, pode haver a
formação de um átomo de carbono positivo, que é denominado carbocátion, ou
um átomo de carbono negativo, chamado de carbânion.
Por exemplo, a reação entre o cloreto de ter-butila, (CH3)3CCl com NaOH (em
um solvente adequado, que não reaja com nenhum dos dois) ocorrem em duas
etapas, sendo a primeira a saída do cloro com a formação de um carbocátion, e a
segunda a formação de uma ligação do carbono positivo com o íon hidróxido (o
carbono positivo atuando como eletrófilo).
Este carbocátion é térciário, e é mais estável que carbocátions secundários ou
primários:

61
Isso ocorre porque as cadeias carbônicas são doadoras de elétrons e
estabilizam o carbocátion, dispersando sua carga. Esta doação de carga é feita por
meio das ligações e é chamada de efeito indutivo. Quanto mais cadeias
carbônicas ligadas ao carbono positivo, mais estável será o carbocátion.
Um carbânion é uma espécie com um átomo de carbono negativo. Um
exemplo de carbânion que podeoms citar é um reagente Grignard, um composto
contendo uma ligação do carbono com um metal (neste caso, magnésio). Como o
carbono é mais eletronegativo que o metal, a ligação com o magnésio é polar e o
carbono tem uma carga parcial negativa (δ −), e o magnésio tem carga parcial posi-
tiva (δ +)
O carbono negativo atua como nucleófilo, doando sua carga negativa a um
átomo deficiente de elétrons. A estabilidade de carbânions primários, secundários e
terciários é o oposto da dos carbocátions:
3.5 REPRESENTAÇÃO DE MECANISMOS DE REAÇÃO
O conjunto de etapas de uma reação, com seus intermediários e caminhos, é
chamado de mecanismo de reação. É o processo mental pelo qual estudamos um
processo químico, que é essencialmente (na maior parte dos casos) uma reação

62
ácido-base, em que uma espécie doa elétrons para outra.
Como escrevemos um mecanismos de reação? Utilizamos setas curvas para
representar o movimento dos elétrons. As setas indicam o movimentos dos elétrons
(do átomo ou ligação doador para o átomo receptor). As valências dos átomos
devem ser respeitadas, portanto se uma ligação é feita em um átomo com as
valências completas, outra deve ser desfeita (a atribuição de cargas formais
estudada na Unidade I é de grande auxílio aqui). Espécies que funcionam como
catalisadores são introduzidas em certo momento e regeneradas depois.
Exemplo: Equilíbrio entre dois isômeros da propanona, catalisado por ácido
(H+) ou por base (OH-).
3.6 DEFINIÇÕES DE ÁCIDOS E BASES
O conceito de ácido e base, em suas origens, estava relacionado às suas
propriedades organolépticas (ou seja, que afetam os sentidos humanos). Ácidos têm
sabor azedo e muda a cor do papel tornassol azul para vermelho. Bases têm sabor
adstringente e mudam a cor do papel tornassol vermelho para azul.
Conceitos mais precisos forma propostos por Arrhenius (1884), BrØnsted-Lowry
(1923) e de Lewis (1923) proporcionam um entendimento melhor do comportamento
de ácidos e bases e sistemas orgânicos.
Outros modelos de ácido-base, como o de Lux-Flood (1947) e o do sistema-solvente (1925),
também se tornaram populares. No caso de compostos inorgânicos, a teoria de ácidos e
bases duros e macios de Pearson (1963) é muito empregada.

63
De acordo com o conceito de Arrhenius, ácido é uma substância que se
dissocia em água liberando íons hidrônio (H3O+) ou íons hidrogênio (chamados às
vezes simplesmente de prótons, H+). Base é uma substância que se dissocia em água
liberando íon hidróxido (OH-). A reação entre um ácido e uma base é chamada de
neutralização.
𝐇𝐂𝐥(𝒂𝒒) + 𝐍𝐚𝐎𝐇(𝒂𝒒) → 𝐍𝐚𝐂𝐥(𝒂𝒒) + 𝐇𝟐𝐎(𝒍)
Exemplo 2: O conceito de BrØnsted-Lowry se baseia na ideia de protonação-
desprotonação: ácidos são espécies que doam prótons e bases são espécies que
recebem. O ácido que perde um próton se transforma em uma base, e uma base
que ganha um próton se converte em um ácido. Ambos (o ácido e a base resultante,
e a base e o ácido resultante) estabelecem um par ácido-base conjugado.
HCl + H2O → H3O+(𝑎𝑞)
+ Cl−(𝑎𝑞)
O HCl é o ácido que se converte no Cl-, sua base conjugada; a água é a base
que se converte no ácido conjugado H3O+
Exemplo 3: O conceito de Lewis não está relacionado ao movimento de prótons, mas
a pares de elétrons: um ácido recebe pares de elétrons e uma base doa pares de
elétrons. É o conceito mais abrangente dos três, e junto ao conceito de BrØnsted-
Lowry é o mais usado no estudo de mecanismos de reações orgânicas.

64
3.7 FORÇA RELATIVA DE ÁCIDOS E BASES
Ácidos e bases podem ser fortes ou fracos, quando comparados uns aos
outros. Tomando a reação entre HCl e H2O, citada anteriormente, o ácido clorídrico
protona a molécula de água pelo fato de ser o ácido mais forte. Sendo o ácido mais
forte, forma-se a base mais fraca.
O equilíbrio de dissociação de um ácido fraco HA é dado portanto por (4):
HA ↔ H+ + A− (4)
A constante de equilíbrio é chamada constante de acidez, Ka sendo dada
portanto (5):
Ka =
[H+][A−]
[HA] (5)
É usual expressar este valor na forma de logaritmo (6):
pKa = −log(Ka) (6)
Quanto mais forte o ácido, maior a quantidade de ácido dissociado (H+ e A-)
em relação ao ácido associado (HA), maior o Ka e menor o pKa.
Se estamos falando de uma base B: (com um par de elétrons para doar), a
reação com H+ é dada por (7):
B: + H+ → BH+ (7)
A constante deste equilíbrio é a constante de basicidade Kb (8):
Kb =
[BH+]
[B: ][H+] (8)
Para conhecer melhor as diversas teorias ácido-base consulte o livro “Introdução à
Química Orgânica” (2010) de Luiz Cláudio de Almeida Barbosa a partir da página 24
Disponível em: https://bit.ly/3cbd2FV. Acesso em: 17 jan. 2020.

65
E, na forma de logaritmo (9):
pKb = −log(Kb) (9)
A Tabela 2 traz os valores de Ka e pKa de diversos ácidos orgânicos e
inorgânicos. Repare que alguns deles apresentam mais de uma constante por pos-
suírem dois ou mais hidrogênios ionizáveis, como o ácido fosfórico:
H3PO4 → H2PO4− + H+; Ka1
H2PO4− → HPO4
2− + H+; Ka2
HPO42− → PO4
3− + H+; Ka3
Neste caso, note-se que a primeira constante é maior que a segunda, que é
maior que a terceira.
Tabela 2: Valores de Ka e pKa de diversos ácidos.
Ácido há A- Ka pKa
Iodídrico HI I- -10
Bromídrico HBr Br- -9
Perclórico HClO4 ClO4- -7 – -8
Clorídrico HCl Cl- -3 – -7
Clórico HClO3 ClO3- -1 – -1,4
Sulfúrico (1ª ionização) H2SO4 HSO4- -5
Nítrico HNO3 NO3- -1,3
Íon hidrônio H3O+ H2O -1,7
Iódico HIO3 IO3- 1,6 x 10-1 0,80
Oxálico (1ª ionização) H2C2O4 HC2O4- 5,9 x 10-2 1,23
Sulfuroso (1ª ionização) H2SO3 HSO3- 1,54 x 10-2 1,81
Sulfúrico (2ª ionização) HSO4- SO42- 1,2 x 10-2 1,92
Cloroso HClO2 ClO2- 1,1 x 10-2 1,96
Fosfórico (1ª ionização) H3PO4 H2PO4- 7,52 x 10-3 2,12
Arsênico (1ª ionização) H3AsO4 H2AsO4- 5,0 x 10-3 2,30
Cloroacético CH2ClCOOH CH2ClCOO- 1,4 x 10-3 2,85
Cítrico (1ª ionização) H3C6H5O7 H2C6H5O7- 8,4 x 10-4 3,08
Fluorídrico HF F- 7,2 x 10-4 3,14
Nitroso HNO2 NO2- 4,0 x 10-4 3,39
Fórmico HCOOH HCOO- 1,77 x 10-4 3,75
Lático HCH3H5O3 CH3H5O3- 1,38 x 10-4 3,86
Ascórbico (1ª ionização) H2C6H6O6 HC6H6O6- 7,9 x 10-5 4,10
Benzóico C6H5COOH C6H5COO- 6,46 x 10-5 4,19
Por que os valores de Ka1, Ka2 e Ka3 variam desta forma?

66
Ácido há A- Ka pKa
Oxálico (2ª ionização) HC2O4- C2O42- 6,4 x 10-5 4,19
Hidrazóico HN3 N3- 1,9 x 10-5 4,72
Cítrico (2ª ionização) H2C6H5O7- HC6H5O72- 1,8 x 10-5 4,74
Acético CH3COOH CH3COO- 1,76 x 10-5 4,75
Propanóico CH3CH2COOH CH3CH2COO- 1,34 x 10-5 4,87
Íon piridínio C5H4NH+ C5H4N 5,6 x 10-6 5,25
Cítrico (3ª ionização) HC6H5O72- C6H5O73- 4,0 x 10-6 5,40
Carbônico (1ª ionização) H2CO3 HCO3- 4,3 x 10-7 6,37
Sulfuroso (2ª ionização) HSO4- SO42- 1,02 x 10-7 6,91
Arsênico (2ª ionização) H2AsO4- HAsO42- 8 – 9,3 x 10-8 7,10 – 7,03
Sulfídrico H2S HS- 1,0 x 10-7 – 9,1 x 10-8 7 – 7,04
Fosfórico (2ª ionização) H2PO4- HPO42- 6,23 x 10-8 7,21
Hipocloroso HClO ClO- 3,5 – 3,0 x 10-8 7,46 – 7,53
Hipobromoso HBrO BrO- 2 x 10-9 8,70
Cianídrico HCN CN- 6,17 x 10-10 9,21
Bórico (1ª ionização) H3BO3 H2BO3- 5,8 x 10-10 9,23
Íon amônio NH4+ NH3 5,6 x 10-10 9,25
Fenol C6H5OH C6H5O- 1,6 x 10-10 9,80
Carbônico (2ª ionização) HCO3- CO32- 4,8 x 10-11 10,32
Hipoiodoso HIO IO- 2 x 10-11 10,70
Arsênico (3ª ionização) HAsO42- AsO43- 6,0 x 10-10/3,0 x 10-12 9,22 – 11,53
Peróxido de hidrogênio H2O2 HO2- 2,4 x 10-12 11,62
Ascórbico
(2ª ionização) HC6H6O6- C6H6O62- 1,6 x 10-12 11,80
Fosfórico (3ª ionização) HPO42- PO43- 4,8 – 2,2 x 10-13 12,32 – 12,66
Água H2O OH- 1,0 x 10-14 14,0
Fonte: Hall (2002)
3.8 EFEITOS QUE AFETAM ACIDEZ E BASICIDADE
Podemos comparar a força dos ácidos, mesmo sem conhecer seus valores de
pKa, a partir de suas características estruturais. Quanto maior a estabilidade da base
conjugada, mais forte será o ácido. Vamos analisar alguns aspectos que influenciam
na acidez.
Às vezes as tabelas de ácidos contém apenas o Ka ou o pKa. Pode-se, contudo determinar
o Kb e pKb das bases a aprtir da relação:
𝑲𝒂 ⋅ 𝑲𝒃 = 𝑲𝒘 = 𝟏𝟎−𝟏𝟒
ou
𝒑𝑲𝒂 + 𝒑𝑲𝒃 = 𝟏𝟒

67
1. Elementos eletronegativos estabilizam a base conjugada. Comparando o pKa do
CH4, NH3, H2O e HF (48, 33, 16 e 3, respectivamente), observamos que, quanto mais
eletronegativo o átomo ligado ao hidrogênio mais forte o ácido.
2. Deslocalização dos elétrons de uma base por ressonância. Este efeito também
estabiliza a base, tornando o ácido conjugado mais forte.
A base conjugada da primeira estrutura (ácido acético) apresenta estruturas
de ressonância, o que torna a carga deslocalizada (este efeito não ocorre com a
segunda estrutura, o etanol).
3. Efeito indutivo. Este efeito que foi abordado brevemente nesta unidade, e tem
relação com o efeito da atração de elétrons de um átomo mais eletronegativo
por outro menos eletronegativo. Este efeito diminui ao longo das ligações , de
modo que, quanto mais distante o átomo eletronegativo está do centro de carga
negativa da base conjugada, menor a estabilidade desta base e mais fraco será
o ácido.
4. Hibridização do carbono que sustenta a carga negativa na base conjugada.
Carbonos com maior caráter s (ou seja, maior contribuição de orbitais s na
formação dos orbitais híbridos – ver Unidade I) são mais eletronegativos.

68
5. Força da ligação H–A. Quanto mais polar a ligação com o hidrogênio, maior a
tendência de haver quebra heterolítica e mais forte o ácido. A força da ligação
dependerá da eletronegatividade e do tamanho do átomo ligado.
A eletronegatividade diminui do flúor para o iodo, e o tamanho do átomo
ligado ao hidrogênio aumenta.
6. Efeito do solvente. Quanto mais bem solvatada for a base conjugada, maior será
sua estabilidade. Bases conjugadas com mais impedimentos estéricos dificultam a
solvatação.
Ambos os ácidos têm tamanhos semelhantes, mas o segundo apresenta mais
ramificações, o que dificulta a solvatação da base conjugada e torna o ácido mais
fraco.

69
FIXANDO O CONTEÚDO
1. Considere os seguintes derivados halogenados do ácido acético e seus res-
pectivos valores de pKa:
A variação nos valores de pKa estão relacionados ao:
a) efeito estérico causado pelo aumento do volume dos grupos próximos ao
hidrogênio, dificultando sua ionização.
b) efeito indutivo dos átomos de cloro, que estabilizam a base conjugada.
c) efeito eletrônico de conjugação dos pares de elétrons não ligantes do cloro com
o par de elétrons da ligação entre carbono e oxigênio.
d) efeito da mudança de hibridização do carbono ligado aos átomos de cloro.
e) efeito do aumento da eletronegatividade dos carbonos com o aumento do
número de átomos de cloro.
2. Considere os seguintes pares de compostos:

70
A espécie mais ácida em cada par é:
a) B, C, F
b) A, D, E
c) A, C, F
d) B, D, E
e) A, C, E
3. As bases conjugadas das espécies HSO4-, CH3NH2, CH3CH2OH são,
respectivamente:
a) H2SO4, CH3NH3+, CH3CH2O-
b) SO42-, CH3NH3
+, CH3CH2OH2+
c) H2SO4, CH3NH-, CH3CH2O-
d) SO42-, CH3NH-, CH3CH2O-
e) H2SO4, CH3NH3+, CH3CH2OH2
+
4. Considere as seguintes afirmações:
I. Ácidos fracos apresentam concentração maior na forma associada que na
forma dissociada em condições de equilíbrio.
II. Ácidos fortes deslocam íons hidrogênio de bases fracas formando bases
conjugadas fracas e ácidos conjugados fortes, respectivamente.
III. Uma base com pKb maior que a outra retira prótons de uma base com pKb
menor.
Estão corretas as afirmativas:
a) II e III.
b) I e III.

71
c) I e II.
d) I apenas.
e) II apenas.
5. Aminoácidos apresentam um grupo amino básico e um grupo carboxila ácido. O
pH em que as cargas positivas se igualam às cargas negativas é chamado de
ponto isoelétrico (pI). O pI é calculado fazendo-se a média aritmética do pKa dos
grupos ionizáveis.
Considere os três aminoácidos a seguir, com os pKa´s de seus grupos em destaque.
Ao titularmos uma mistura em quantidades iguais (em mol) dos três aminoácidos
com uma solução de NaOH, eles atingirão o pI na ordem:
a) arginina, ácido aspártico, glicina
b) ácido aspártico, glicina, arginina
c) glicina, ácido aspártico, arginina
d) arginina, glicina, ácido aspártico
e) ácido aspártico, arginina, glicina
6. Considere os compostos a seguir:
A ordem crescente de acidez destas substâncias é:
a) I, III, II, IV
b) II, I, IV, III
c) III, I, II, IV

72
d) II, I, III, IV
e) II, I, III, IV
7. Considere as afirmativas:
I. A quebra homolítica de uma ligação leva à formação de radicais.
II. Carbocátions primários são mais estáveis que terciários.
III. Carbânions atuam como nucleófilos.
Estão corretas as afirmativas:
a) I e III
b) I e II
c) II e III
d) I, II e III
e) Nenhuma das alternativas.
8. Compostos conhecidos como clorofluorcarbonos (CFCs) eram utilizados como
propelentes em refrigeradores até 1988, quando foram proibidos devido a seu
efeito deletério na camada de ozônio. Estes compostos sofrem reações sob a luz
ultravioleta, formando radicais que por sua vez reagem com o ozônio (O3), de
acordo com as seguintes etapas:
Considerando todas as quebras homolíticas que ocorrem, pode-se deduzir que a
relação correta de força das ligações mostradas é:
a) C-Cl < C-F

73
b) Cl-O < O-O
c) O=O < O-O
d) O-O < C-Cl
d) O=O < C-Cl

74
ALCANOS E CICLOALCANOS:
NOMENCLATURA,
CONFORMAÇÕES E REAÇÕES
4.1 O PETRÓLEO COMO PRINCIPAL FONTE DE HIDROCARBONETOS
Hidrocarbonetos são compostos contendo carbono e hidrogênio como únicos
elementos. Estão presentes sobretudo nos derivados do petróleo e outros
combustíveis fósseis, embora sejam conhecidos alguns ferormônios que pertencem a
esta categoria (Figura 32).
Figura 32: Alguns feromônios de insetos
Fonte: Blomquist, Tittiger e Jurenka (2020)
UNIDADE
04

75
O petróleo é uma mistura muito complexa de produtos derivados de micro-
organismos primordiais, que após submetidos a condições de elevadas pressões e
temperaturas ao londo das eras geológicas produzem estes compostos. O óleo cru é
drenado no subsolo ou do oceano e submetido a uma destilação fracionada (Figura
33) que separa seus componentes. São recolhidas as frações mais voláteis (hidro-
carbonetos de menor massa molecular) nos compartimentos superiores da coluna de
fracionamento, depois as frações menos voláteis.
Figura 33: Coluna de fracionamento do petróleo
Fonte: Elaborado pelo Autor (2021)
O leque de aplicações dos hidrocarbonetos derivados do petróleo é o mais
diversificado possível. Os compostos mais voláteis (gases e líquidos) são utilizados
como combustíveis (como o butano, presente no gás liquefeito de petróleo, ou o
isooctano, um dos componentes da gasolina). Os componentes menos voláteis têm
aplicações na construção civil, na indústria petroquímica e de química fina, entre
outros.
4.2 ALCANOS E CICLOALCANOS: ESTRUTURA E PROPRIEDADES
Relembrando as funções orgânicas da Unidade II, os hidrocarbonetos podem
ser classificados em alcanos (e cicloalcanos), alcenos (e cicloalcenos), alcinos e
hidrocarbonetos aromáticos. Alcanos são hidrocarbonetos saturados de cadeia
aberta, e cicloalcanos, seus correspondentes de cadeia cíclica. A Tabela 3 traz
exemplos de alcanos e cicloalcanos comuns, com algumas de suas propriedades
físicas.

76
Tabela 3: Alcanos e cicloalcanos comuns.
Obs.: p.f. = ponto de fusão; p.e. = ponto de ebulição. Os valores de densidade referem-se a compostos
que encontram-se na fase líquida a 20 ºC.
Composto
Entalpia de
combustão
(∆𝐂𝐇𝐨, 𝐤𝐉 ∙ 𝐦𝐨𝐥−𝟏)
Fórmula
molecular
p.f.
(ºC)
p.e.
(ºC)
Densidade
(𝐠 ∙ 𝐜𝐦−𝟑)
Alc
an
os
no
rma
is
Metano −75 𝐂𝐇𝟒 −162 −182 -
Etano −1560 𝐂𝟐𝐇𝟔 −89 −183 -
Propano −2220 𝐂𝟑𝐇𝟖 −42 −188 -
Butano −2878 𝐂𝟒𝐇𝟏𝟎 0 −138 -
Pentano −3510 𝐂𝟓𝐇𝟏𝟐 36 −130 0,626
n-Hexano −4180 𝐂𝟔𝐇𝟏𝟒 69 −95 0,659
n-Heptano −4825 𝐂𝟕𝐇𝟏𝟔 98 −91 0,684
n-Octano −5530 𝐂𝟖𝐇𝟏𝟖 126 −57 0,703
n-Nonano −6126 𝐂𝟗𝐇𝟐𝟎 151 −54 0,718
n-Decano −6779 𝐂𝟏𝟎𝐇𝟐𝟐 174 −30 0,730
Alc
an
os
ram
ific
ad
os 2-metilpropano (ou
isobutano)
−2870 𝐂𝟒𝐇𝟏𝟎 –159 –12 -
2,2-dimetilpropano −3515 𝐂𝟓𝐇𝟏𝟐 –17 10 -
2,2,4-trimetilpentano (ou
isooctano)
−5463 𝐂𝟖𝐇𝟏𝟖 –107 99 0,692
Cic
loa
lca
no
s Ciclopropano −2092 𝐂𝟑𝐇𝟔 –128 –33 -
Ciclobutano −2745 𝐂𝟒𝐇𝟖 –91 13 0,720
Ciclopentano −3320 𝐂𝟓𝐇𝟏𝟎 –94 49 0,751
Cicloexano −3954 𝐂𝟔𝐇𝟏𝟐 6-7 81 0,774
Disponível em: https://bit.ly/3kZn0y6. Acesso em: 09 mar. 2020.
Podemos perceber a partir da tabela que há uma relação entre a estrutura e
as propriedades. Hidrocarbonetos são praticamente apolares, tendo em vista que as
eletronegatividades do carbono e do hidrogênio são bastante próximas, o que torna
as ligações apolares.
Por esta razão, as interações intermoleculares que prevalecem nestes
compostos são do tipo dipolo instantâneo-dipolo induzido. Estas interações se tornam
maiores quanto maiores forem as estruturas e quanto maior a área de contato entre
as moléculas. Assim, observa-se um aumento nos pontos de fusão e ebulição dos
alcanos de cadeia normal à medida em que a cadeia se torna mais longa. Esta
tendência fica evidente por meio da Figura 34

77
Figura 34: Variação dos pontos de fusão e de ebulição de alcanos de cadeia aberta
Fonte: Elaborado pelo Autor com dados da Tabela 2 (2021)
Observa-se também a partir da análise da Tabela 3 que alcanos com cadeias
ramificadas apresentam pontos de fusão e ebulição menores que os alcanos de
cadeia normal correspondentes. Este comportamento se explica pela dificuldade de
aproximação entre as moléculas que é imposta pelos grupos laterais (a isso
chamamos de efeito estérico).
Um procedimento rotineiro na indústria do petróleo consiste na conversão de
cadeias normais em ramificadas (ou na conversão de alcanos em alcenos), em um
processo chamado craqueamento. Com isso, obtêm-se compostos com menores
pontos de ebulição, o que melhora o rendimento de sua destilação.
-250
-200
-150
-100
-50
0
50
100
150
200
1 3 5 7 9
Número de carbonos
p.f. (ºC)
p.e. (ºC)

78
Uma das frações do petróleo destilado é a gasolina, que é uma mistura de
alcanos de 5 a 10 átomos de carbono. Motores de centelhamento queimam a gaso-
lina em um processo que envolve o centelhamento por meio de uma descarga
elétrica. A mistura pode sofrer autoignição sob efeito do calor ou da pressão antes
do centelhamento, o que pode causar o efeito de “pancadas no motor”. Este efeito
pode ser reduzido pelo aumento da resistência da gasolina à autoignição, o que se
expressa por sua octanagem, parâmetro que depende das quantidades relativas de
2,2,4-trimetilpentano (ou isooctano) e n-heptano. Quanto maior a quantidade de
isooctano (que é mais ramificada que o n-heptano), maior a octanagem da
gasolina.
Por fim, a Tabela 3 apresenta também as entalpias de combustão, que
dependem do número de átomos de carbono. A combustão completa de um
alcano leva à formação de dióxido de carbono e água, em quantidades que estão
relacionadas à fórmula molecular. Alcanos de cadeia aberta têm fórmula geral
CnH2n+2, enquanto cicloalcanos apresentam fórmula CnH2n (veja o exemplo da Figura
21). Assim, a combustão completa destas duas classes de substâncias pode ser
representada pelas equações (10), para alcanos) e (11), para cicloalcanos:
CnH2n+2 +3n + 1
2O2 → nCO2 + (n + 1)H2O (10)
CnH2n +3n
2O2 → nCO2 + nH2O (11)
O conceito de octanagem não se restringe a hidrocarbonetos, sendo usado como padrão
de comparação entre diferentes combustíveis, como etanol biodiesel.
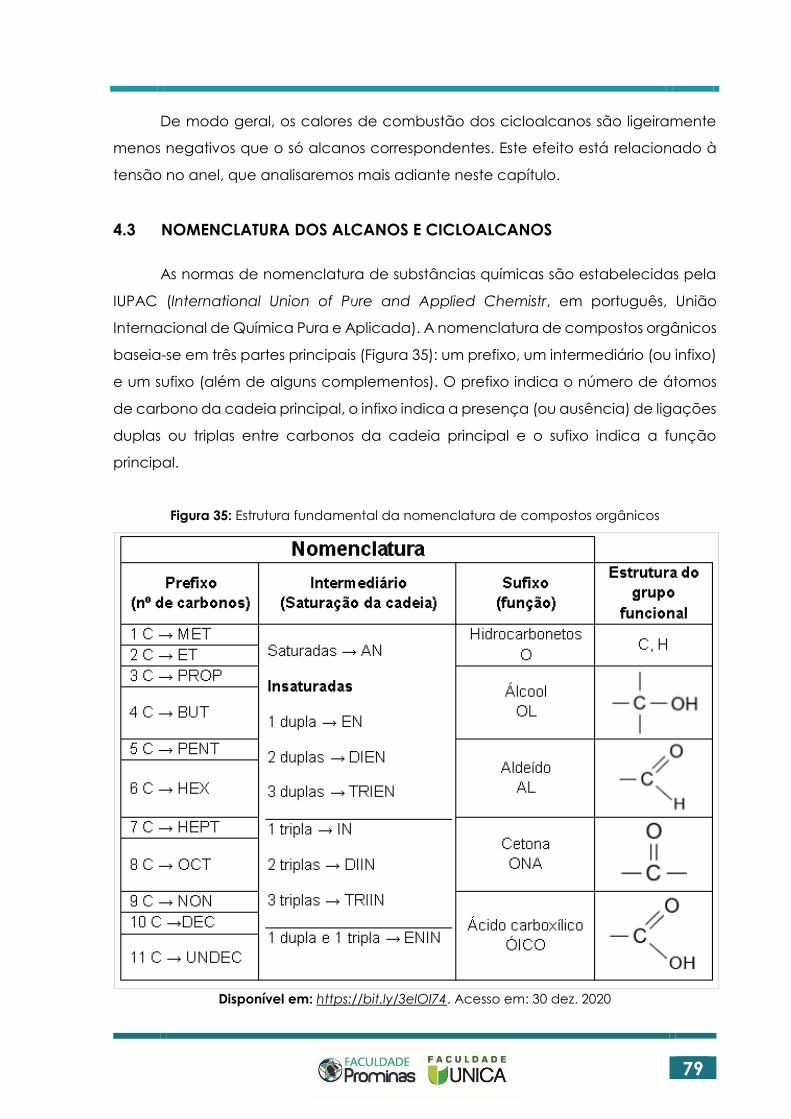
79
De modo geral, os calores de combustão dos cicloalcanos são ligeiramente
menos negativos que o só alcanos correspondentes. Este efeito está relacionado à
tensão no anel, que analisaremos mais adiante neste capítulo.
4.3 NOMENCLATURA DOS ALCANOS E CICLOALCANOS
As normas de nomenclatura de substâncias químicas são estabelecidas pela
IUPAC (International Union of Pure and Applied Chemistr, em português, União
Internacional de Química Pura e Aplicada). A nomenclatura de compostos orgânicos
baseia-se em três partes principais (Figura 35): um prefixo, um intermediário (ou infixo)
e um sufixo (além de alguns complementos). O prefixo indica o número de átomos
de carbono da cadeia principal, o infixo indica a presença (ou ausência) de ligações
duplas ou triplas entre carbonos da cadeia principal e o sufixo indica a função
principal.
Figura 35: Estrutura fundamental da nomenclatura de compostos orgânicos
Disponível em: https://bit.ly/3elOI74. Acesso em: 30 dez. 2020

80
Vamos trabalhar alguns exemplos, começando pelos alcanos de cadeia
normal.
A cadeia apresenta 3 carbonos e o prefixo é PROP-. A ausência de ligações
múltiplas indica que o infixo a ser usado é -AN-. Tratando-se de um hidrocarbonetos,
o sufixo é -O. O nome do composto é então: PROPANO.
Seguindo a mesma lógica, muda-se o prefixo para BUT- (para 4 carbonos) e o
nome será: BUTANO.
Cadeias mais longas podem ser normais ou ramificadas, e para especificar
que se trata de cadeias normais, é usual acrescentar n- no início:
n-PENTANO
n-HEXANO
Quando as cadeias são ramificadas, nomeia-se também os grupos ligados à
cadeia principal, com a terminação -il (Figura 35). A cadeia principal é a mais longa,
e numera-se a partir da extremidade mais próxima da primeira ramificação, como
nos exemplos das Figuras 36 e 3.

81
Figura 36: Grupos orgânicos derivados de hidrocarbonetos
Disponível em: http://bit.ly/3v2AwFY. Acesso em: 30 dez. 2020
Figura 37: Exemplos de alcanos de cadeias ramificadas
Fonte: Elaborado pelo Autor (2021)
Figura 38: Nomenclatura dos alcanos da Figura 34
Fonte: Elaborado pelo Autor (2021)
Observe no último exemplo que quando o prefixo inicia-se com h (ou seja, HEX-
e HEPT-) e é antecedido pelo nome do grupo ligado, pode-se manter o h e adicionar
um hífen ou omitir o h, sem o hífen. Note-se também que, no caso de mais de um

82
grupo ligado à cadeia principal, segue-se a ordem alfabética na nomenclatura (etil
vem antes de metil).
Figura 39: Exemplos de cicloalcanos e suas nomenclaturas
Fonte: Elaborado pelo Autor (2021)
No caso dos cicloalcanos, acrescenta-se o complemento ciclo- ao início do
nome.
4.4 CONFORMAÇÕES E ESTABILIDADE DOS ALCANOS E CICLOALCANOS
Aprendemos na Unidade I que o carbono saturado apresenta geometria te-
traédrica, com ângulos de ligação de cerca de 109,5º. Em estruturas fechadas,
entretanto, há uma tensão que pode ser maior ou menor dependendo do tamanho
do anel. Por exemplo, no ciclopropano o ângulo entre as ligações é de 60º, o que
torna o anel muito tensionado. Uma forma prática de se mensurar esta tensão é
calculando-se a energia de tensão a partir da entalpia padrão de combustão
(∆combHo) dos cicloalcanos (Tabela 4), que pode ser obtida a partir das entalpias de
formação (tabeladas).
Vamos ilustrar este procedimento com o ciclopropano. A combustão de 3 mols
de C com 2 mols de H2 resulta em -487,05 kcal ∙ mol−1, mas a entalpia de combustão
de 1 mol de ciclopropano é de -499,8 kcal ∙ mol−1:
𝐂𝟑𝐇𝟔 + 𝟒𝐎𝟐 → 𝟑𝐂𝐎𝟐 + 𝟑𝐇𝟐𝐎
A diferença, +12,7 kcal ∙ mol−1, corresponde à entalpia de formação do
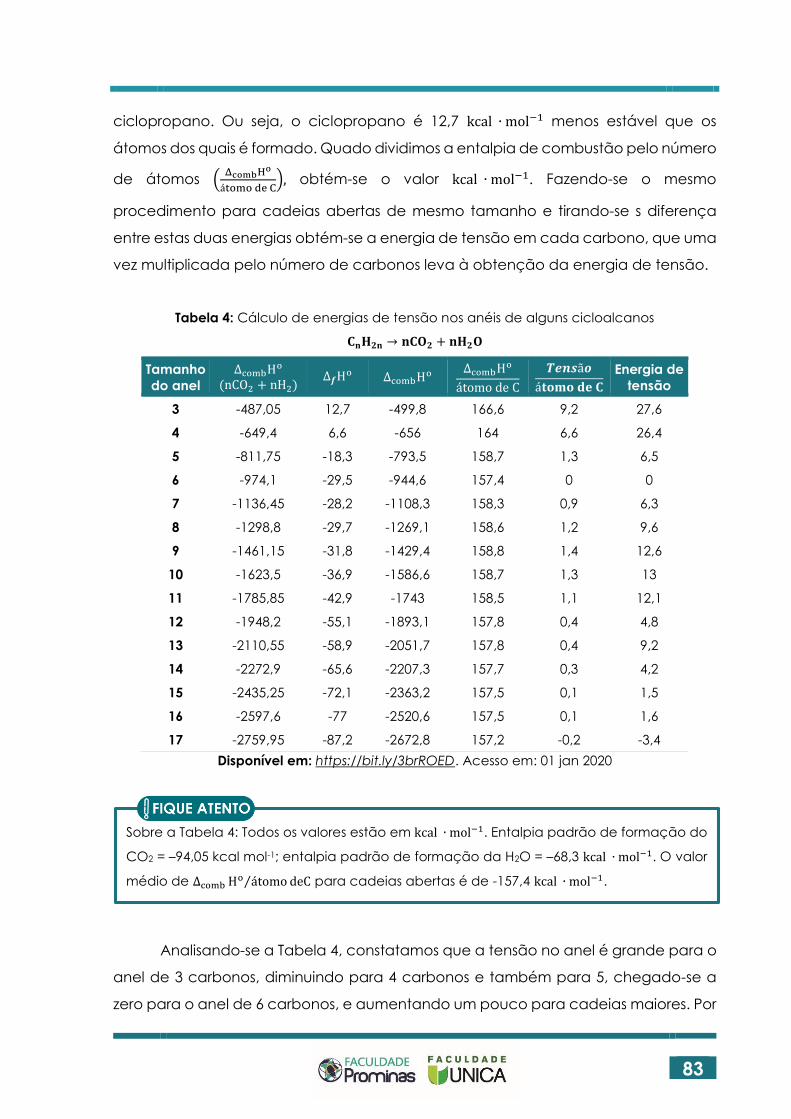
83
ciclopropano. Ou seja, o ciclopropano é 12,7 kcal ∙ mol−1 menos estável que os
átomos dos quais é formado. Quado dividimos a entalpia de combustão pelo número
de átomos (ΔcombHo
átomo de C), obtém-se o valor kcal ∙ mol−1. Fazendo-se o mesmo
procedimento para cadeias abertas de mesmo tamanho e tirando-se s diferença
entre estas duas energias obtém-se a energia de tensão em cada carbono, que uma
vez multiplicada pelo número de carbonos leva à obtenção da energia de tensão.
Tabela 4: Cálculo de energias de tensão nos anéis de alguns cicloalcanos
𝐂𝐧𝐇𝟐𝐧 → 𝐧𝐂𝐎𝟐 + 𝐧𝐇𝟐𝐎
Tamanho
do anel
ΔcombHo (nCO2 + nH2)
Δ𝒇Ho ΔcombHo ΔcombHo
átomo de C
𝑻𝒆𝒏𝒔ã𝒐
á𝐭𝐨𝐦𝐨 𝐝𝐞 𝐂
Energia de
tensão
3 -487,05 12,7 -499,8 166,6 9,2 27,6
4 -649,4 6,6 -656 164 6,6 26,4
5 -811,75 -18,3 -793,5 158,7 1,3 6,5
6 -974,1 -29,5 -944,6 157,4 0 0
7 -1136,45 -28,2 -1108,3 158,3 0,9 6,3
8 -1298,8 -29,7 -1269,1 158,6 1,2 9,6
9 -1461,15 -31,8 -1429,4 158,8 1,4 12,6
10 -1623,5 -36,9 -1586,6 158,7 1,3 13
11 -1785,85 -42,9 -1743 158,5 1,1 12,1
12 -1948,2 -55,1 -1893,1 157,8 0,4 4,8
13 -2110,55 -58,9 -2051,7 157,8 0,4 9,2
14 -2272,9 -65,6 -2207,3 157,7 0,3 4,2
15 -2435,25 -72,1 -2363,2 157,5 0,1 1,5
16 -2597,6 -77 -2520,6 157,5 0,1 1,6
17 -2759,95 -87,2 -2672,8 157,2 -0,2 -3,4
Disponível em: https://bit.ly/3brROED. Acesso em: 01 jan 2020
Analisando-se a Tabela 4, constatamos que a tensão no anel é grande para o
anel de 3 carbonos, diminuindo para 4 carbonos e também para 5, chegado-se a
zero para o anel de 6 carbonos, e aumentando um pouco para cadeias maiores. Por
Sobre a Tabela 4: Todos os valores estão em kcal ∙ mol−1. Entalpia padrão de formação do
CO2 = –94,05 kcal mol-1; entalpia padrão de formação da H2O = –68,3 kcal ∙ mol−1. O valor
médio de Δcomb Ho átomoΤ deC para cadeias abertas é de -157,4 kcal ∙ mol−1.

84
que o anel do cicloexano não apresenta tensões?
O cicloexano não é plano, mas se apresenta no espaço em duas
conformações principais, chamadas cadeira e bote. Conformações são maneiras
como os átomos se distribuem no espaço. As conformações não são fixas, podendo
mudar de uma para a outra. Configurações, por outro lado, são fixas, não se
convertendo uma na outra facilmente.
No cicloexano temos posições aproximadamente perpendiculares ao centro
do anel, chamadas axiais, e posições aproximadamente paralelas ao centro do
anel, chamadas equatoriais (Figura 40).
Figura 40: Conformações do cicloexano
Fonte: Elaborado pelo Autor (2021)
A conformação em cadeira do cicloexano é mais estável que a conformação
em bote, pois há menos repulsões entre os hidrogênios axiais, que estão mais próximos
uns dos outros.
A Figura 41 ilustra também as conformações mais estáveis dos anéis com 7 e 8
carbono. Nela podemos ver que as ligações se ajustam de modo a reduzir os efeitos
de repulsão.
Figura 41: Conformações mais estáveis do cicloeptano e ciclo-octano (os hidrogênios não
estão representados, para fins de simplicidade)
Fonte: Elaborado pelo Autor (2021)

85
Qual o efeito que a presença de substituintes nos anéis dos cicloalcanos
exerce sobre sua estabilidade? Vamos analisar estes efeitos na próxima seção.
4.5 CICLOALCANOS DISSUBSTITUÍDOS E ISOMERISMO CIS-TRANS
Considere o exemplo do cicloexano com suas conformações em cadeira e
bote (Figura 40). Vamos ver o que acontece com a estabilidade de um derivado
substituído em duas posições por outros grupos, como por exemplo o 1,3-
diclorocicloexano (Figura 42).
Neste caso, além das conformações de praxe (bote e cadeira), que se
convertem uma na outra dependendo sobretudo da temperatura do meio, este
composto pode se apresentar na forma de duas configurações (que não são
intercambiáveis). Cada configuração corresponde a um composto diferente de
mesma fórmula molecular (um isômero). Quando os dos grupos iguais estão do
mesmo lado do anel temos o isômero cis, e quando estão em lados opostos temos o
isômero trans.
Figura 42: Conformações do 1,3-diclorocicloexano.
Fonte: Elaborado pelo Autor (2021)
Isômeros cis-trans podem ocorrer também com anéis de outros tamanhos. Por
exemplo, no caso dos derivados do ciclopropano em que há substituição em

86
carbonos vizinhos, haverá isomeria cis-trans se não houver grupos iguais no mesmo
átomo e pelo menos dois grupos dos carbonos vizinhos sejam iguais. Uma extensão
do conceito de isomeria cis-trans é a designação E-Z, que compreende até mesmo
situações em que todos os grupos são diferentes. Esta designação será estudada em
outro contexto (estrutura de alcenos).
Figura 43: Isomeria cis-trans em derivados do ciclobutano
Fonte: Elaborado pelo Autor (2021)
Vamos abordar agora quais são as reações que envolvem alcanos e
cicloalcanos.
4.6 SÍNTESE E REAÇÕES DE ALCANOS E CICLOALCANOS
Reações químicas envolvem a quebra e a formação de ligações. Uma ligação
química é quebrada mediante ganho de energia. Se uma ligação covalente apolar
é quebrada, o par de elétrons fica distribuído igualmente entre os dois átomos, e
temos uma quebra homolítica, formando-se radicais. Se uma ligação covalente
polar é quebrada, o par de elétrons não fica distribuído igualmente entre os dois
átomos, e temos uma quebra heterolítica, formando-se íons (Figura 44).

87
Figura 44: Quebra homolítica e heterolítica de ligações
Fonte: Elaborado pelo Autor (2021)
As principais reações para obtenção de alcanos ou que convertem alcanos
em outras funções são abordadas a seguir.
4.6.1 Reações dos alcanos
Já discutimos anteriormente que hidrocarbonetos são substâncias de baixa
polaridade porque a diferença de eletronegatividade entre carbono e hidrogênio é
pequena. No caso doa alcanos, é mais comum que as ligações sofram quebras
homolíticas formando radicais. Alcenos e alcinos podem também sofrer quebras
heterolíticas.
Reação de alcanos com halogênio: alcanos são pouco reativos, mas podem
sofrer reações de substituição na presença de luz ou aquecimento com halogênios.
Estas reações são radicalares e podem formar múltiplos produtos.
A reação de alcanos com halogênios é radicalar, e envolve uma etapa de
iniciação, uma etapa de propagação e uma de finalização.

88
Reações de combustão: A combustão é a reação com oxigênio (O2). Se um
composto orgânico contendo apenas C, H e O sofre combustão completa, forma-se
dióxido de carbono (CO2) e água. Se a combustão for incompleta, forma-se
monóxido de carbono (CO) ou fuligem (C).
𝐶𝐻4(𝑔)+ 2𝑂2(𝑔)
→ CO2|(𝑔)+ 2𝐻2𝑂(𝑙)
𝐶2𝐻5𝑂𝐻(𝑔) + 3𝑂2(𝑔)→ 2CO2|(𝑔)
+ 3𝐻2𝑂(𝑙)
4.6.2 Síntese dos alcanos
Apesar de os combustíveis fósseis serem a principal fonte industrial de
obtenção de hidrocarbonetos, processos sintéticos de laboratório também podem
ser aplicáveis. Três processos típicos podem ser usados na síntese de alcanos: a
hidrogenação de alcenos e alcinos, a redução de haletos de alquila ou o
acoplamento de compostos organometálicos.
Hidrogenação de alcenos: Alcenos contém ligações duplas entre carbono,
que ao reagirem com hidrogênio na presença de um catalisador metálico finamente
dividido (como Ni, Pd ou Pt) são convertidas em ligações simples. É uma reação de
adição, que se caracteriza pela conversão de uma ligação insaturada em ligação
saturada, com a introdução de dois átomos de hidrogênio do mesmo lado da dupla
(adição sin).
𝐶𝑛𝐻2𝑛(𝑔) 𝐶𝑛𝐻2n+2|(𝑔)
Alceno 𝐻2(1 𝑚𝑜𝑙), 𝑐𝑎𝑡 Alcano

89
Alcinos também podem reagir com hidrogênio mas na proporção de 1 mol do
alcino por 2 mol de H2:
𝐶𝑛𝐻2𝑛−2(𝑔) 𝐶𝑛𝐻2n+2|(𝑔)
Alceno 𝐻2(2 𝑚𝑜𝑙), 𝑐𝑎𝑡 Alcino
O mecanismo de adição sin pode ser exemplificado pela hidrogenação do
1,2-dimetilciclobuteno, que forma o cis-1,2-dimetilciclobutano, com os dois
hidrogênios adicionados do mesmo lado:
Redução de haletos de alquila: A reação entre um haleto de alquila e um
metal (geralmente magnésio, embora outros metais também possam ser usados)
leva à formação de um composto de Grignard, que é um composto organometálico
(substância contendo uma ligação entre carbono e metal). O composto de Grignard
pode ser hidrolizado formando um alcano:
𝑀𝑔 𝐻2𝑂 𝑅𝑋 RMgX RH

90
Outra forma de se reduzir um haleto de alquila a um alcano é pela reação
com um metal (como zinco) em meio ácido:
Acoplamento de reagentes de Grignard: Quando um haleto de alquila reage
com lítio elementar, forma-se um composto alquillítio, que em uma etapa
subsequente reage com um haleto de cobre, formando um dialquilcobre-lítio. Este
composto ao reagir com outro haleto de alquila forma um alcano de cadeia maior
que as cadeias carbônicas originais:
Li CuX RX RLi R2CuLi
R2CuLi R′X R′R′
Reações com compostos organometálicos são de grande interesse sintético
pois possibilitam o aumento da cadeia de carbonos, como neste último exemplo.

91
Para conhecer mais sobre o processo de craqueamento do petróleo e as aplicações dos
produtos da indústria petroquímica, consulte o livro a “Química na Produção do Petróleo”
(2013) de Armando Mateus Pomini. Disponível em: https://bit.ly/38my3wq. Acesso em: 01
jan. 2020

92
FIXANDO O CONTEÚDO
1. Um estudante atribuiu a um composto, erroneamente, a nomenclatura 5-etil-2-
metil-6-butiloctano. A nomenclatura correta desta substância é:
a) 4-etil-7-metil-3-butiloctano
b) 4-etil-3-(3-metilbutil)octano
c) 5,6-dietil-2-metildecano
d) 5,6-dietil-9-metildecano
e) 5-etil-6-(3-metilbutil)octano
2. A maior volatilidade dos alcanos acíclicos ramificados em relação aos alcanos de
cadeia normal se deve a:
a) a diferença no tipo de interação intermolecular.
b) o efeito estérico que dificulta a aproximação das moléculas.
c) ao maior efeito indutivo nas cadeias normais.
d) a maior reatividade das cadeias ramificadas.
e) o número de carbonos terciários e quaternários, que aumentam a tensão
intramolecular.
3. Considere a reação de cloração sob luz do seguinte alcano:
O número de produtos monoclorados distintos que podem ser obtidos é:
a) 1
b) 2
c) 3
d) 4
e) 5

93
4. Um cicloalcano de cadeia mista forma quatro (4) produtos distintos monobro-
mados ao ser submetido à reação com Br2 em presença de luz.
A alternativa que apresenta uma fórmula molecular possível para este cicloalcano
é:
a) C7H14
b) C7H16
c) C8H16
d) C8H18
e) C7H18
5. Hidrocarbonetos derivados do petróleo são usados como combustíveis em
veículos automotores. Uma preocupação constante é em relação à quantidade
de gás carbônico gerada na quaima destes compostos, visto que isto afeta o
efeito estufa. Considere a tabela abaixo, com as entalpias de combustão de
quatro alcanos, em comparação com o etanol:
Substância Fórmula ΔcombH° (kcal mol-1)
Metano CH4(g) -212,8
Etano C2H6(g) -372,8
Propano C3H8(g) -530,6
Butano C4H10(g) -688,0
Etanol C2H5OH(l) -326,5
O composto que produz a maior quantidade de energia por mol de CO2 gerado
é:
a) metano
b) etano
c) propano
d) butano
e) etanol
6. Considere as seguintes afirmativas:

94
I. Alcenos podem ser obtidos a partir da hidrogenação catalítica de alcanos.
II. Dois haletos de alquila podem ser acoplados para obter um alcano de cadeia
maior.
III. Reações de halogenação de alcanos não tem interesse sintético pois geram
múltiplos produtos.
Estão corretas as afirmativas:
a) I apenas
b) II apenas
c) II e III
d) I e II
e) I, II e III
7. Radicais seguem uma ordem de estabilidade semelhante à de carbocátions. Em
uma halogenação, forma-se preferencialmente o produto proveniente do radical
mais estável. Considere os seguintes haletos de alquila, obtidos da hidrogenação:
Considerando estas informações, o produto principal deve ser:
a) I
b) II

95
c) III
d) IV
e) V
8. Relacione a primeira coluna com a segunda:
1. Craqueamento
2. Hidrogenação catalítica
3. Acoplamento
(X) Reação envolvendo compostos organometálicos que resulta na extensão da
cadeia carbônica.
(X) Reação térmica de quebra de cadeias grandes de hidrocarbonetos em
cadeias menores.
(X) Reação de adição que diminui o grau de saturação de um hidrocarboneto.
A relação correta entre as colunas está expressa na alternativa:
a) 1, 2, 3
b) 1, 3, 2
c) 2,1,3
d) 3, 2, 1
e) 3, 1, 2

96
ESTEREOQUÍMICA
5.1 SIMETRIA NA NATUREZA
Simetria é um fenômeno onipresente no mundo natural. Encontramos simetria
na beleza das conchas em espiral, a simetria dos organismos vivos, e até na
repetição de padrões na estrutura quaternária de proteínas (Figura 45), além de
muitos outros sistemas.
Figura 45: Exemplos de sistemas simétricos na natureza
Disponível em: https://bit.ly/3t5RYHX. Acesso em: 03 jan. 2021
Materiais podem apresentar simetria, em geral manifestada por meio de seus
cristais, como nos flocos de neve (Figura 43). A ocorrência destes fenômenos nos
leva a perguntar se a simetria tem origem na estrutura molecular das substâncias e
se há evidências experimentais da mesma. Neste capítulo abordaremos alguns
princípios da assim denominada isomeria ótica em estruturas de moléculas
orgânicas.
Figura 46: Flocos de neve simétricos (Fotografias de Wilson Bentley de 1902)
Fonte: Raviv (2010, p. 19)
A história da estereoquímica pode ser traçada desde o século XIX, tendo sido
importante no desenvolvimento de nossa compreensão acerca da estrutura orânica,
conforme abordaremos agora.
UNIDADE
05

97
5.2 ASPECTOS HISTÓRICOS DA ESTEREOQUÍMICA
A isomeria ótica foi descoberta por Jean-Baptiste Biot em 1815 e estudada
posteriormente por Louis Pasteur (que estudou cristais de ácido tartárico em 1842) e
por Jacobus Henricus van 't Hoff e Joseph Le Bel (que relacionaram este compor-
tamento à presença de carbonos quirais ou assimétricos, em 1874).
Comumente, prefere-se o termo estereoquímica a isomeria ótica.
Estereoquímica provém do grego στερεός (stereos), “sólido”. Quiralidade é outra
palavra de uso corrente neste campo, e também provém do grego χειρ (kheir),
“mão”.
5.3 PROPRIEDADES DOS ISÔMEROS ÓTICOS
Isômeros óticos se diferenciam unicamente na forma como se comportam
diante da luz polarizada (as demais propriedades como PF, PE, etc. permanecem as
mesmas). Para compreendermos esse comportamento, devemos entender como a
luz pode sofrer influência do meio.
A luz natural é uma onda que vibra em todas as direções. Quando uma luz
monocromática passa por um polarizador (por exemplo, um bloco de calcita,
CaCO3, conhecido como prisma de Nicol), apenas uma das direções é mantida e a
luz torna-se polarizada (Figura 47). Certas substâncias (chamada de oticamente
ativas), ao serem colocadas na forma de solução em um tubo pelo qual passa uma
luz polarizada, desviam o plano de vibração desta luz de um determinado ângulo α.
Substâncias que desviam a luz polarizada para direita são dextrógiras e as que
desviam para a esquerda, levógiras.
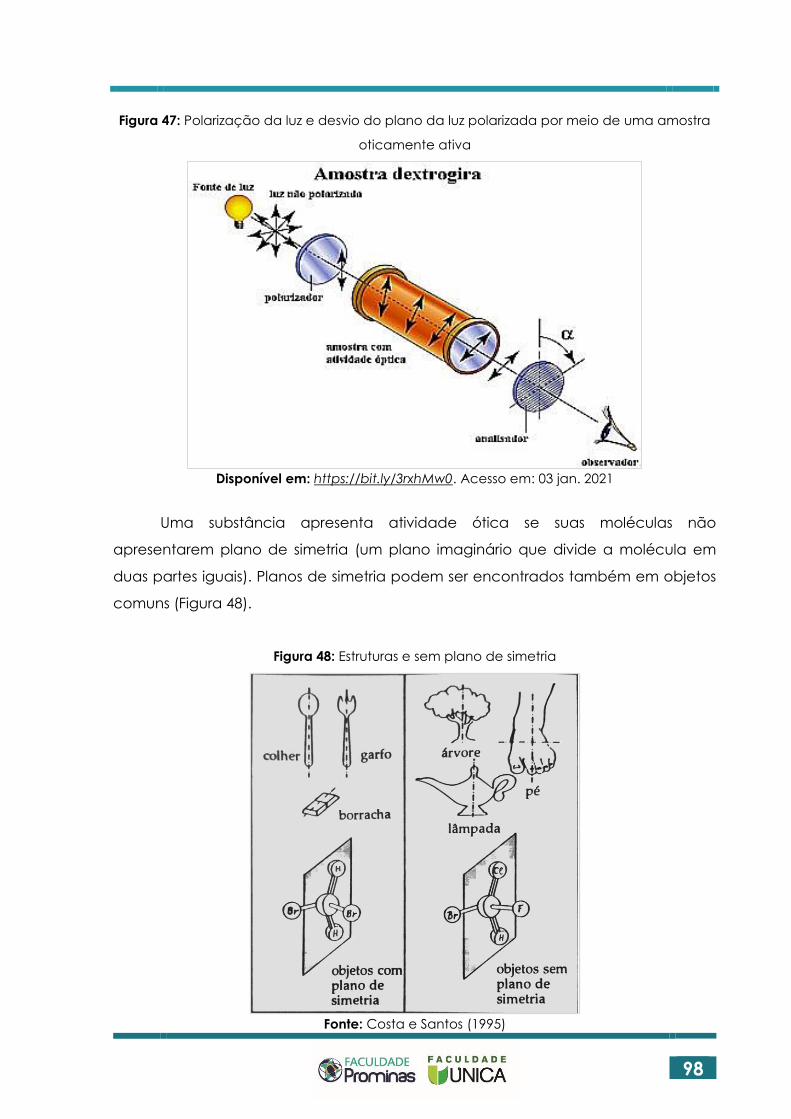
98
Figura 47: Polarização da luz e desvio do plano da luz polarizada por meio de uma amostra
oticamente ativa
Disponível em: https://bit.ly/3rxhMw0. Acesso em: 03 jan. 2021
Uma substância apresenta atividade ótica se suas moléculas não
apresentarem plano de simetria (um plano imaginário que divide a molécula em
duas partes iguais). Planos de simetria podem ser encontrados também em objetos
comuns (Figura 48).
Figura 48: Estruturas e sem plano de simetria
Fonte: Costa e Santos (1995)

99
Vamos definir alguns conceitos fundamentais em estereoquímica a partir da
próxima seção.
5.4 ENANTIÔMEROS E QUIRALIDADE
Um conceito muito importante em estereoquímica é o de centro quiral (ou
centro assimétrico). Embora não seja uma particularidade de compostos de
carbono, aplicaremos a ideia de carbonos quirais, que são carbonos saturados
ligados a quatro grupos (ou vizinhanças) diferentes. Um exemplo que podemos citar
é o 2-cloropropano (que não contém carbono quiral) e o 2-clorobutano (que contém
– Figura 49).
Figura 49: Estruturas com e sem plano de simZetria.
Fonte: Adaptado pelo Autor de Costa e Santos (1995)
Compostos contendo 1 (um) carbono quiral existem como dois isômeros que

100
são como o objeto e a imagem em um espelho, mas diferentes entre si (não se su-
perpõem, assim como a mão direita e a esquerda, como na Figura 50). O par de
estereoisômeros (isômeros óticos) que são como objeto e imagem que não se
superpõem constitui um par de enantiômeros. A Figura 51 ilustra um par de
enantiômeros que foram sobrepostos, demonstrando que não correspondem à
mesma estrutura.
Figura 50: Quiralidade nas mãos direita e esquerda.
Disponível em: https://bit.ly/30lQwVq. Acesso em: 03 jan. 2021
Figura 51: Estereoisômeros que não se superpõem são enantiômeros
Fonte: Elaborado pelo Autor (2021)
Como é feita a nomenclatura de estereoisômeros? A seção a seguir
apresentará o procedimento para isso.
5.5 NOMENCLATURA E REPRESENTAÇÃO DE ESTEREOISÔMEROS
Uma forma útil de representar os estereoisômeros é a projeção de Fischer,
conforme ilustrado na Figura 52.

101
Figura 52: Projeção de Fischer
Fonte: Elaborado pelo Autor (2021)
Logo, se houver um carbono quiral, teremos dois isômeros oticamente ativos. E
se tivermos mais de um carbono quiral? O número de estereoisômeros pode ser
calculado a partir do número de carbonos quirais, n:
número de isômeros oticamente ativos = 2n
Quando há um plano de simetria interno (ou seja, metade na molécula é o
reflexo no espelho da outra), temos um composto meso, que terá um isômero oti-
camente ativo a menos.
número de isômeros oticamente ativos = 2n – 1 (composto meso)

102
Os estereoisômeros que não são enantiômeros (objeto-imagem) são
chamados de diastereoisômeros (Figura 53). Ao contrário dos enantiômeros, estes
têm propriedades físicas diferentes.
Figura 53: Diastereoisômeros
Fonte: Elaborado pelo Autor (2021)
Enantiômeros são designados pela nomenclatura de Cahn-Ingold-Prelog
(sistema R/S). Para se fazer a designação de um centro quiral, atribui-se prioridades
aos grupos ligados a ele com base no número atômico do átomo conectado (grupos
com maior número atômico recebe maior prioridade. Se houver coincidência no
A partir de 2 centros quirais (excetuando-se os compostos meso), temos 2n/2 = 2n-1 pares
de enantiômeros e o restante dos pares de estereoisômeros são diastereoisômeros. Por
exemplo, com 3 centros quirais temos 23 = 8 estereisômeros, sendo 4 pares de enantiômeros
as demais combinações (24 ao todo) de diastereoisômeros.

103
átomo ligado, faz-se a análise com o átomo seguinte. Ligações múltiplas são
contadas como se fossem mais ligações (por exemplo, se um átomo A está ligado a
B por ligação dupla, considera-se como se A estivesse ligado a dois átomos B (Figura
54).
Figura 54: Exemplo da aplicação do sistema de Cahn-Ingold-Prelog
Fonte: Elaborado pelo Autor (2021)
Estabelecidas as prioridades, verifica-se o sentido dos 3 grupos de maior para
menor prioridade; se o sentido for horário, o centro quiral é do tipo R; se for anti-
horário, é do tipo S.
5.6 MISTURAS RACÊMICAS E RESOLUÇÃO
A mistura em partes iguais (equimolares) de dois enantiômeros é chamada de
mistura racêmica ou racemato e é oticamente inativa. Se houver algum excesso de
um dos enantiômeros, teremos atividade ótica, que dependerá da quantidade deste
excesso. O excesso percentual de um enantiômero em relação a outro em uma
mistura de ambos é denominado excesso enantiomérico (e.e.), e pode ser calculado

104
a partir do conhecimento da rotação específica de um enantiômero de referência,
medida a uma temperatura t:
[α]Dt =
α
c ⋅ l (12)
Aqui,[𝛼]𝐷𝑡 corresponde à rotação específica, é rotação observada para a
mistura, l é o comprimento do tubo em dm (Figura 44), c a concentração da solução
ou densidade se for um líquido puro (medidos em g/mL). A letra D indica que se utiliza
a linha D de uma lâmpada de sódio (λ = 589,6 nm). A temperatura geralmente é de
25 ºC. O excesso enantiomérico será dado por:
𝑒. 𝑒.% =𝛼𝑚𝑖𝑠𝑡𝑢𝑟𝑎
[𝛼]𝐷𝑡 × 100 (13)
O processo de purificação de uma mistura de enantiômeros, visando
aumentar o excesso enantiomérico, é chamado de resolução, e envolve uma série
de procedimentos complexos de separação, como o uso de reagentes ou colunas
cromatográficas enantiosseletivos. Obter produtos com e.e. satisfatório é importante
principalmente na indústria farmacêutica, visto que há diferenças às vezes decisivas
na atividade biológica dos enantiômeros, como discutiremos na última sessão deste
capítulo.
5.7 MOLÉCULAS QUIRAIS SEM CENTRO QUIRAL
Apesar de pouco comuns, há exemplos de compostos que apresentam ativi-
dade ótica apesar de não possuírem centros quirais, por não conterem plano de
simetria. Um exemplo clássico é o do propadieno (Figura 55). Considerando a
geometria dos carbonos sp2, nos quais há uma ligação pela sobreposição lateral
de orbitais p puros (conforme estudado na Unidade I), e levando em conta que há
duas ligações duplas no mesmo carbono central (que tem hibridização sp), os pares
de hidrogênios dos carbonos sp2 das extremidades devem estar perpendiculares um
ao outro, tornando a molécula assimétrica.

105
Figura 55: Exemplo uma molécula assimétrica sem centro quiral
Fonte: Elaborado pelo Autor (2021)
5.8 FÁRMACOS QUIRAIS
Foi discutido que isômeros óticos apresentam suas propriedades físicas
idênticas, exceto no seu comportamento diante da luz polarizada. Devemos
acrescentar, entretanto, que sistemas biológicos são capazes de discernir entre os
diferentes enantiômeros, de modo que eles apresentam diferentes comportamentos.
Um exemplo desta disparidade é o limoneno (Figura 56). O enantiômero R é
reconhecido pelos receptores olfativos como odor de laranja, e o S, odor de limão.
Figura 56: Enantiômeros do limoneno
Fonte: Elaborado pelo Autor (2021)

106
Esta diferença de reconhecimento pode ser explicada pelo modelo de in-
teração por 3 pontos (Figura 57). Enantiômeros apresentam atividades biológicas
diferentes devido às diferentes formas como se ligam a receptores biológicos.
Na figura podemos observar que o enantiômero à esquerda interagem bem
com o receptor, ao contrário do enantiômero no centro que, mesmo sendo
rotacionado em uma tentativa de se fazer corresponder os pontos, conforme a
última estrutura, não se liga de forma adequada. Sendo o receptor uma
macromolécula com uma função específica (como uma enzima), sua atividade
muda dependendo da interação. Um dos enantiômeros pode ocasionar uma
determinada resposta biológica, e o outro, uma resposta biológica diferente, como
no exemplo do limoneno.
Figura 57: Modelo da interação por três pontos entre uma molécula quiral e um receptor
Fonte: Elaborado pelo Autor (2021)
Um exemplo trágico envolvendo isômeros óticos é o da talidomida,
medicamento muito utilizado nos anos 1950 durante a gravidez, vendido na forma
de mistura racêmica (Figura 58). O medicamento foi proibido quando foi associado
a problemas de formação em fetos, embora atualmente seja aprovado no
tratamento da hanseníase.

107
Figura 58: Enantiômeros da talidomida
Fonte: Elaborado pelo Autor (2021)
Para conhecer melhor sobre estereoquímica consulte o livro “Química Orgânica” (2020)
de Bianca Sandrino a partir da página 30. Disponível em: https://bit.ly/3qz0TQm. Acesso
em:

108
FIXANDO O CONTEÚDO
1. Relacione a primeira coluna com a segunda:
1. Diastereoisômeros
2. Enantiômeros
3. Compostos meso
(X) Compostos que têm relação objeto-imagem e não são superponíveis
(X) Compostos que não têm relação objeto-imagem.
(X) Compostos contendo um plano de simetria interno.
A ordem correta de correspondência entre as colunas é:
a) 1, 3, 2
b) 2, 1, 3
c) 3, 1, 2
d) 1, 3, 2
e) 2, 3, 1
2. Considere as seguintes afirmativas:
I. Enantiômeros apresentam propriedades físicas diferentes.
II. Compostos com plano de simetria interno podem apresentar atividade ótica.
III. Misturas racêmicas são constituídas de quantidades equimolares de di-
astereoisômeros.
Estão corretas as afirmativas:
a) I e II
b) III apenas
c) II e III
d) Todas as afirmativas
e) Nenhuma das afirmativas

109
3. Aminoácidos (aa´s) são as unidades constituintes das proteínas. É conhecido o fato
de que os aa´s distinguem compostos quirais, e isso se deve em parte à sua própria
quiralidade, devido à presença de um carbono assimétrico. Alguns aa´s
apresentam também outros centros quirais.
Abaixo temos a estrutura de três aa´s essenciais:
O número de estereoisômeros possíveis para os aa´s citados é, respectivamente:
a) 4, 2, 2
b) 2, 2, 4
c) 2, 4, 4
d) 2, 2, 2
e) 4, 4, 2
4. Considere os seguintes compostos: 2-metil-hexano, 1,3-diclorobutano e 2,2-
dimetilpropano.
São oticamente ativos:
a) 2-metil-hexano e 1,3-diclorobutano.
b) 2-metil-hexano e 2,2-dimetilpropano.
c) 1,3-diclorobutano e 2,2-dimetilpropano.
d) os três compostos são oticamente ativos.
e) os três compostos são oticamente inativos.
5. Um composto de fórmula molecular C4H8O2 apresenta no infravermelho uma
banda de absorção em 3600 cm-1 e outra, mais forte, em 1720 cm-1. Este composto
existe em duas formas que desciam o plano da luz polarizada de valores iguais e
sinais opostos, e não reage de forma significativa com NaOH. Foram propostas as
seguintes estruturas para este composto:

110
A estrutura que mais se ajusta às características mencionadas é:
a) I
b) II
c) III
d) IV
e) V
6. A D-tagatose é um carboidrato encontrado em pequenas quantidades em
alimentos, como no leite submetido a fervura, e funciona como adoçante artificial.
Esta substância passa por uma reação de redução não estereosseletiva, ou seja,
forma múltiplos estereoisômeros. As estruturas de Fischer são mostradas a seguir:
O número de estereoisômeros possíveis do reagente e do produto, respecti-
vamente, é:
a) 8 e 16
b) 3 e 4
c) 8 e 64
d) 8 e 15
e) 3 e 6
7. Uma solução aquosa de natureza desconhecida apresenta atividade ótica. Pode-
se afirmar que o conteúdo desta solução:

111
a) contém carbonos quirais.
b) é uma substância pura.
c) apresenta cadeia aberta.
d) contém átomos eletronegativos.
e) tem plano de simetria.
8. Uma substância existe na forma de dois enantiômeros I e II com rotação específica
de +12º e -12º, respectivamente. Se uma solução de um dos componentes (ou de
ambos) apresenta rotação de +3º, pode-se concluir que:
a) a solução contém apenas o enantiômero I, em baixa concentração.
b) a solução contém maior quantidade do componente I que do componente II.
c) a solução é uma mistura racêmica de I e II.
d) a solução contém maior quantidade do componente II que do componente I.
e) a solução contém o componente I ligado ao componente II de forma covalente.

112
HALETOS DE ALQUILA E REAÇÕES
DE SUBSTITUIÇÃO NUCLEOFÍLICA E
DE ELIMINAÇÃO
6.1 ESTRUTURA E NOMENCLATURA DE HALETOS DE ALQUILA
Haletos de alquila e de arila: apresentam halogênio (F, Cl, Br, I) ligado a cadeia
alifática (haletos de alquila) ou aromática (haletos de arila).
Diversos compostos de interesse industrial pertencem a esta classe. O
clorofórmio (CHCl3) é um solvente com ação anestésica, que foi utilizado até meados
do século 20 como anestésico geral em procedimentos cirúrgicos, tendo seu uso
abandonado devido a seu efeito carcinogênico (Figura 59).
Figura 59: (a) Estrutura do clorofórmio. (b) Um inalador de clorofórmio para uso em
cirurgias do início do século 20
Fonte: Elaborado pelo Autor (2021)
Haletos de alquila com mais de um halogênio (sobretudo cloro e flúor, sendo
chamados de CFCs) foram usados no passado como agentes refrigerantes devido
à sua elevada fugacidade, mas devido a seu efeito deletério sobre a camada de
ozônio (que pode ser atribuído à facilidade com que estes compostos formam
radicais sob a ação da luz), forma proibidos para esta finalidade. Atualmente ainda
são empregados em aerossóis (Figura 60).
UNIDADE
06

113
Figura 60: Estrutura de alguns CFCs
Fonte: Elaborado pelo Autor (2021)
As reações de substituição nucleofíica alifática são as principais reações
sofridas pelos haletos de alquila e serão objeto de estudo a partir daqui.
6.2 REAÇÕES DE SUBSTITUIÇÃO NUCLEOFÍLICA
A ligação carbono-halogênio é polar (sendo a polaridade crescente do flúor
para o iodo), e o carbono tem caráter eletrofílico (confira o conceito de eletrófilo e
nucleófilo na seção 3.3). Desta forma, o átomo de carbono pode reagir com um nu-
cleófilo em uma reação de substituição.
Quando um haleto de alquila reage com um nucleófilo, ocorre a substituição
do halogênio, que sai como haleto (sendo chamado de grupo abandonador).
Esta reação é muito útil na obtenção de diversas funções orgânicas, o que a
torna extremamente versátil em sínteses orgânicas (Figura 61).

114
Figura 61: Exemplos de conversões de grupos funcionais por meio de substituição
nucleofílica alifática
Fonte: Elaborado pelo Autor (2021)
Apesar de nossa ênfase ser na substituição de haletos, em que o halogênio é
o grupo abandonador, há outros grupos abandonadores que são úteis, como os íons
sulfonato, ilustrados na Figura 62.
Figura 62: Exemplos de íons sulfonato como bons grupos abandonadores.
Fonte: Elaborado pelo Autor (2021)
Os grupos abandonadores da figura 59 são eficientes por sua estabilidade, conferida pela
deslocalização dos elétrons.

115
6.3 MECANISMO E ESTEREOQUÍMICA DAS REAÇÕES SN1 E SN2
Existem dois mecanismos aceitos para as reações de substituição nucleofílica
alifática, chamados de SN1 e SN2. O primeiro tipo tem mecanismo unimolecular (em
duas etapas, com formação de um intermediário), e o segundo tem mecanismo
bimolecular (em uma etapa, com formação de um estado de transição). A diferença
entre intermediário e estado de transição é que o primeiro é detectável e é possível
isolá-lo, ao contrário do segundo.
Por questões didáticas, abordaremos o mecanismo SN2 primeiramente. A
Figura 63 ilustra como o orbital com um par de elétrons disponível do nucleófilo se
aproxima do carbono tetraédrico no lado oposto ao do halogênio, formando um
estado de transição em que temporariamente o carbono adquire geometria trigonal
plana. Com a saída do grupo abandonador, a configuração se inverte, formando o
carbono tetraédrico invertido.
Figura 63: Mecanismo da SN2
Fonte: Elaborado pelo Autor (2021)
Examinando o gráfico de energia da reação (Figura 64), observamos que o
estado de transição é formado com a aplicação de uma energia de ativação, em
um mecanismo sincronizado (ao mesmo tempo em que o nucleófilo entra o grupo
abandonador sai).

116
Figura 64: Gráfico de energia da reação SN2
Fonte: Elaborado pelo Autor (2021)
A cinética desta reação é bimolecular, visto que a velocidade depende da
concentração do haleto e do nucleófilo:
v = k ⋅ [haleto][Nu] (14)
Quando há um centro quiral, o mecanismo da SN2 leva à inversão da confi-
guração, como no seguinte exemplo:
O 2-bromo-octano (de configuração R, levógiro), ao sofrer substituição
eletrofílica com a formação do octan-2-ol (de configuração S, dextrógiro), ambos

117
com aproximadamente 100% de excesso enantiomérico. Outro exemplo é a reação
do cis-1-cloro-3-metilciclopentano em trans-3-metilciclopentanol pela reação com
base:
O mecanismo de uma reação tipo SN1, por outro lado, ocorre em duas etapas:
na primeira etapa ocorre o rompimento da ligação C–X, formando um carbocátion
intermediário de geometria trigonal plana (Figura 65). Este carbocátion se liga ao
nucleófilo em uma segunda etapa, formando o produto final. Deve-se salientar que,
tendo em vista que o intermediário é plano, o ataque do nucleófilo pode ocorrer a
partir de qualquer um dos lados com igual probabilidade. Forma-se, assim – ao
contrário da SN2 – uma mistura racêmica.
Figura 65: Mecanismo da SN1
Fonte: Elaborado pelo Autor (2021)
Contrariamente às reações SN2, a cinética aqui é unimolecular, a velocidade
dependendo apenas da concentração do haleto:

118
𝑣 = 𝑘 ⋅ [ℎ𝑎𝑙𝑒𝑡𝑜] (15)
Figura 66: Gráfico de energia de uma reação SN1
Fonte: Elaborado pelo Autor (2021)
6.4 FATORES QUE INFLUENCIAM AS REAÇÕES SN1 E SN2
O que determina se uma reação de substituição nucleofílica seguirá o meca-
nismo SN1 ou SN2? Vamos enumerar alguns fatores que podem determinar o curso da
reação.
Efeito da estrutura do haleto. Quando comparamos a probabilidade de
reação nucleofílica SN2 dos haletos dependendo da natureza do carbono central,
percebemos a seguinte ordem:
Velocidade SN2: CH3 > Cprimário > Csecundário ≫ Cterciário
Em contrapartida, a probabilidade de reação SN1 segue a ordem inversa:
Velocidade SN1: CH3 < Cprimário < Csecundário ≪ Cterciário
Por que a probabilidade de ataque do nucleófilo é a mesma em ambos os lados do
carbocátion na SN1? Por que o efeito estérico dos grupos ligados não é determinante
como na SN2?

119
Para explicarmos esta tendência, lembramos que na reação SN2 o estado de
transição deve comportar tanto o nucleófilo quanto o grupo abandonador. Se o
carbono central está muito impedido por grupos volumosos (efeito estérico), a
probabilidade de a reação seguir o curso SN2 é menor, sendo mais provável que o
grupo abandonador saia em uma etapa anterior ao ataque nucleofílico, como é
esperado na reação SN1 (Figura 67).
Figura 67: Efeito estérico nos haletos de alquila em uma substituição nucleofílica
Fonte: Elaborado pelo Autor (2021)
Efeito da concentração e da força nucleofílica. No mecanismo SN1, a
concentração do nucleófilo não afeta a velocidade da reação (o que não ocorre
com a SN2). Em relação à força do nucleófilo, constata-se que nucleófilos com carga
negativa são mais fortes que os nucleófilos neutros. Então OH– é um nucleófilo mais
forte que H2O e RO– mais forte que ROH.
Efeito do solvente. Um solvente contendo um hidrogênio ionizável (como água
e etanol) é chamado solvente prótico e não é adequado a uma reação de
substituição nucleofílica, visto que o solvente obstrui o nucleófilo (e comumente o
protona, doando um íon H+), embora seja também capaz de solvatar o haleto
gerado.
A dependência da natureza do haleto com a influência do solvente prótico é
dada pela relação:
I− > Br− > Cl− > F− (em solventes próticos)
Por outro lado, a força do nucleófilo depende do solvente prótico seguindo a
relação:

120
SH− > CN− > I− > OH− > Br− > CH3CO2− > Cl− > F− > H2O
Solventes apróticos – que não têm hidrogênio ligado a um átomo
eletronegativo – são especialmente úteis nestas reações. Os solventes apróticos mais
utilizados são (Figura 68): N,N-dimetilformamida (DMF), dimetil sulfóxido (DMSO),
dimetilacetamida (DMA) e hexametilfosforamida (HMPA).
Figura 68: Solventes apróticos
Fonte: Elaborado pelo Autor (2021)
Como estes solventes não protonam o nucleófilo, a reatividade dos haletos
neste meio é a inversa daquela observada com solventes próticos:
I− < Br− < Cl− < F− (em solventes apróticos)
6.5 REAÇÕES DE ELIMINAÇÃO DE HALETOS DE ALQUILA
Reações de eliminação são aquelas nas quais uma molécula é eliminada com
a formação de ligações múltiplas. Um exemplo de reação de eliminação é a de-
sidratação de um álcool:

121
Haletos de alquila podem sofrer reações de desidroalogenação, nas quais se
perde um hidrogênio e um átomo de halogênio. Esta reação ocorre na presença de
hidróxido de potássio e álcool, conforme o exemplo abaixo:
Neste exemplo, dois produtos podem ser formados: o but-2-eno e o but-1-eno.
Qual dos dois é formado preferencialmente? Neste caso, aplica-se a regra de
Saytzeff para se prever o produto principal, que pode ser resumida da seguinte forma:
“nas reações de eliminação, retira-se preferencialmente o hidrogênio do carbono
menos hidrogenado.” Entre os carbonos 1 e 3, o menos hidrogenado é o 3. Portanto,
o produto principal é o but-2-eno.
A regra de Saytzeff (ou Zaytsev), do químico russo Aleksander Zaytsev (1841–
1910), pode ser justificada com base na estabilidade do carbocátion que é formado
na primeira etapa da eliminação. O mecanismo da eliminação pode também ser
unimolecular, sendo aqui chamado de E1, ou bimolecular, E2.
No mecanismo E1, a eliminação ocorre em duas etapas: na primeira etapa a
base abstrai um hidrogênio, formando um carbânion, que depois forma uma ligação
dupla com liberação do haleto. No mecanismo E2, a saída do hidrogênio e do
halogênio ocorrem simultaneamente (Figura 69).

122
Figura 69: Mecanismo da desidroalogenação tipo E1 e E2
Fonte: Elaborado pelo Autor (2021)
Reações de substituição nucleofílica competem com reações de eliminação,
dependendo de determinados fatores, conforme será discutido a seguir.
6.6 SUBSTITUIÇÃO VERSUS ELIMINAÇÃO
Visto que o haleto pode sofrer tanto reações de substituição nucleofílica como
eliminação, que fatores são importantes para se determinar o que é mais provável
de acontecer em cada caso?
Um dos fatores é a força da base. Quando o haleto está na presença de uma
base fraca, a tendência maior é que se ocorra substituição do tipo SN1; na presença
de uma base forte, prevalece uma eliminação do tipo E2 (Figura 70). Isso ocorre
porque a base forte tende a remover o hidrogênio vizinho ao carbono contendo o
halogênio, ocorrendo a concomitante formação da ligação dupla, expulsando o
halogênio.
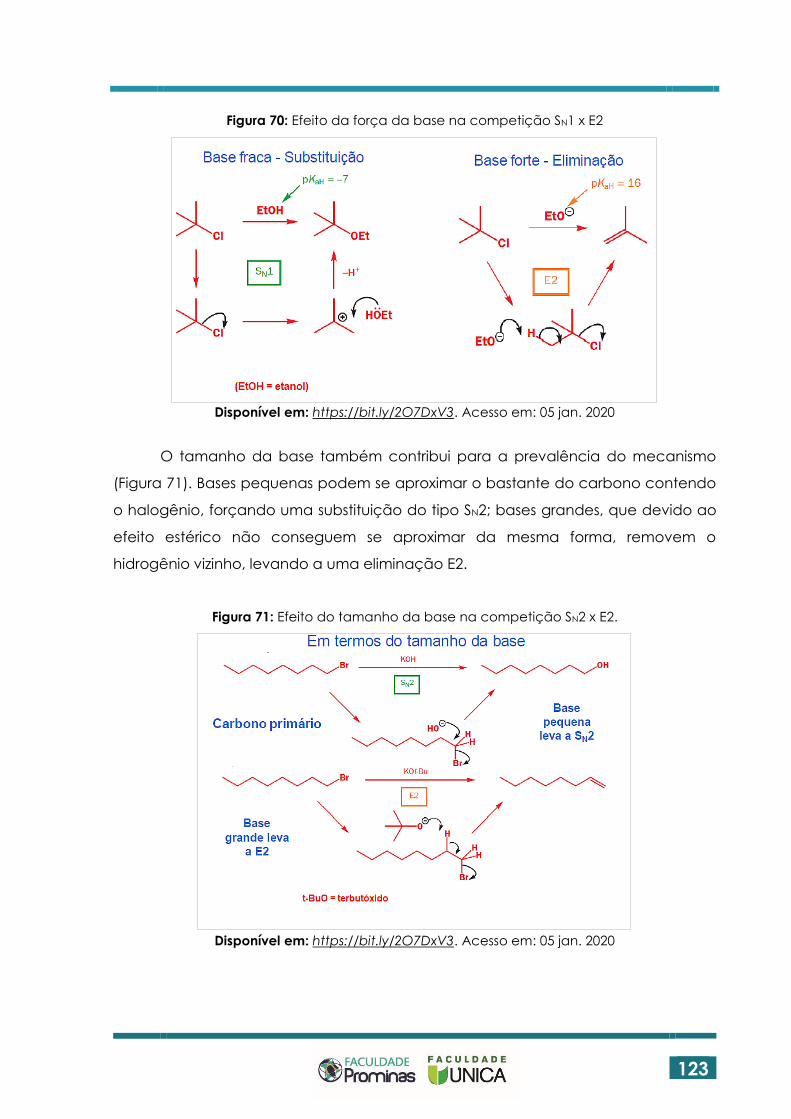
123
Figura 70: Efeito da força da base na competição SN1 x E2
Disponível em: https://bit.ly/2O7DxV3. Acesso em: 05 jan. 2020
O tamanho da base também contribui para a prevalência do mecanismo
(Figura 71). Bases pequenas podem se aproximar o bastante do carbono contendo
o halogênio, forçando uma substituição do tipo SN2; bases grandes, que devido ao
efeito estérico não conseguem se aproximar da mesma forma, removem o
hidrogênio vizinho, levando a uma eliminação E2.
Figura 71: Efeito do tamanho da base na competição SN2 x E2.
Disponível em: https://bit.ly/2O7DxV3. Acesso em: 05 jan. 2020

124
Às vezes uma substituição tipo SN2 ocorre de forma intramolecular, com a formação de
anéis, o que é muito interessante do ponto de vista sintético. Para saber mais, consulte o
livro “Reações de Química Orgânica” (2021) de Bianca Sandrino. Disponível em:
https://bit.ly/2OFeJTS. Acesso em: 05 jan. 2021.

125
FIXANDO O CONTEÚDO
1. Em relação aos mecanismos de substituição nucleofílica, é correto afirmar que:
a) O mecanismo SN2 envolve a formação de um intermediário.
b) A velocidade de reação no mecanismo SN1 depende da concentração do
substrato e do nucleófilo.
c) Solventes apróticos favorecem o mecanismo SN2.
d) Reações do tipo SN1 levam à inversão de configuração.
e) Reações do tipo SN2 levam à formação de produtos sem excesso enantiomérico.
2. A reação de substituição nucleofílica envolvendo um haleto de alquila e um ácido
carboxílico leva à formação de um:
a) álcool
b) éter
c) cetona
d) éster
e) anidrido
3. Considerando os haletos:
CH3CH2Br, (CH3)2CHBr, CH3Br e (CH3)3CBr,
a probabilidade de ocorrência de substituição nucleofílica SN2 segue a seguinte
ordem:
a) (CH3)2CHBr, (CH3)3CBr, CH3CH2Br e CH3Br
b) CH3Br, (CH3)2CHBr, (CH3)3CBr e CH3CH2Br
c) CH3Br, CH3CH2Br, (CH3)2CHBr e (CH3)3CBr
d) (CH3)3CBr, (CH3)2CHBr, CH3CH2Br e CH3Br
e) (CH3)3CBr, (CH3)2CHBr, CH3Br e CH3CH2Br

126
4. A reação mostrada a seguir
leva à formação, como produto principal, de:
5. Reações de eliminação (E1 e E2) competem com reações de substituição. A cada
alternativa, associe E para reações de eliminação e S para substituição.
( ) É favorecida por bases fortes.
( ) É favorecida por bases pequenas.
( ) É desfavorecida por grupos volumosos.
A relação correta é expressa pela alternativa:
a) E, E, S
b) E, S, S
c) S, E, E

127
d) S, S, E
e) E, E, E
6. Em uma reação SN2, ao dobrar-se a concentração do nucleófilo e triplicar-se a
concentração do substrato, a velocidade fica multiplicada por:
a) 5
b) 3
c) 6
d) 9
e) 36
7. Dentre os pares de nucleófilos, escolha qual alternativa representa corretamente
a relação de força:
a) (CH3CH2)2N- é mais forte que (CH3CH2)2NH
b) H2O é mais forte que H2S
c) OH- é mais fraco que H2O
d) PH3 é mais forte que NH3
e) CH3CH2- é mais fraco que CH3O-
8. Considere as seguintes reações:

128
As reações I, II, III e IV podem ser classificadas, respectivamente, como:
a) SN2, E2, SN1, E1
b) SN1, E1, SN2, E2
c) SN2, SN1, E1, E2
d) SN2, E1, SN1, E1
e) SN2, E1, SN1, E2

129
RESPOSTAS DO FIXANDO O CONTEÚDO
UNIDADE 01
UNIDADE 02
QUESTÃO 1 E QUESTÃO 1 D
QUESTÃO 2 B QUESTÃO 2 A
QUESTÃO 3 D QUESTÃO 3 B
QUESTÃO 4 C QUESTÃO 4 D
QUESTÃO 5 A QUESTÃO 5 C
QUESTÃO 6 D QUESTÃO 6 A
QUESTÃO 7 E QUESTÃO 7 B
QUESTÃO 8 E QUESTÃO 8 E
UNIDADE 03
UNIDADE 04
QUESTÃO 1 B QUESTÃO 1 D
QUESTÃO 2 C QUESTÃO 2 B
QUESTÃO 3 D QUESTÃO 3 D
QUESTÃO 4 C QUESTÃO 4 A
QUESTÃO 5 B QUESTÃO 5 A
QUESTÃO 6 E QUESTÃO 6 C
QUESTÃO 7 A QUESTÃO 7 C
QUESTÃO 8 A QUESTÃO 8 E
UNIDADE 05
UNIDADE 06
QUESTÃO 1 B QUESTÃO 1 C
QUESTÃO 2 E QUESTÃO 2 D
QUESTÃO 3 C QUESTÃO 3 D
QUESTÃO 4 A QUESTÃO 4 B
QUESTÃO 5 B QUESTÃO 5 A
QUESTÃO 6 D QUESTÃO 6 C
QUESTÃO 7 D QUESTÃO 7 A
QUESTÃO 8 B QUESTÃO 8 E

130
REFERÊNCIAS
BARBOSA, L. C. D. A. Introdução à Química Orgânica. 2. ed. São Paulo: Pearson, 2010.
362 p.
BLOMQUIST, G. J.; TITTIGER, C.; JURENKA, R. Cuticular Hydrocarbons and Pheromones
of Arthropods. In: WILKES, H. (Ed.). Hydrocarbons, Oils and Lipids: Diversity, Origin,
Chemistry and Fate. New York: Springer, 2020. p. 213-244.
BRUICE, P. Y. Química Orgânica. 4. ed. São Paulo: Pearson, v. I, 2006. 704 p.
CK-12 Chemistry. Palo Alto: CK-12 Foundation, 2010.
CLAYDEN, J. P. et al. Organic Chemistry. 1st. ed. Oxford: Oxford University Press, 2000.
COBLENTZ SOCIETY. Formaldehyde. In: National Institute Of Standards And
Technology. NIST Chemistry WebBook [Online], 2018. Disponível em:
https://bit.ly/3ebl1FH. Acesso em: 16 dez. 2020.
COSTA, M. C.; SANTOS, G. O. Química: a visão do presente. Belo Horizonte: Editora Lê,
1995.
HALL, J. Lab Manual for Zumdahl/Zumdahl’s Chemistry. 6. ed. Totnes: Brooks Cole, 2002.
PEREIRA, C. L. N. A História da Ciência e a Experimentação no Ensino de Química
Orgânica. 2008. 125 f. Dissertação (Mestrado em Ensino de Ciências) - Instituto de
Química, Universidade de Brasília, Brasília, 2008. Disponível em: https://bit.ly/2EHdQFQ.
Acesso em: 16 maio 2020.
PICOLO, K. C. S. D. A. Química Orgânica. São Paulo: Pearson, 2014.
POMINI, A. M. Química na Produção do Petróleo. Rio de Janeiro: InterCiência, 2013.
RAVIV, D. et al. Full and Partial Symmetries of Non-rigid Shapes. Int. J. Comput. Vis.
[online], p. 18–39, 2010. Disponível em: https://bit.ly/2POFPJb. Acesso em: 03 jan. 2021.
SANDRINO, B. Química Orgânica. Curitiba: Intersaberes, 2020.
SANDRINO, B. Reações de Química Orgânica. Curitiba: Intersaberes, 2021.
SOLOMONS, T. W. G.; FRYHLE, C. B. Organic Chemistry. 10th. ed. Hoboken: John Wiley
& Sons, 2009.