dissertação de mestrado uso de métodos semiempíricos para
TRANSCRIPT

UFS
UNIVERSIDADE FEDERAL DE SERGIPE Centro de Ciências Exatas e Tecnologia Departamento de Química Programa de Pós-Graduação em Química
Dissertação de Mestrado
Uso de Métodos Semiempíricos para o Estudo
da Aplicação de Redes Híbridas de
Coordenação como Carreadores de Fármacos
e Dispositivos Luminescentes.
Nailton Martins Rodrigues
Aracaju-SEBrasil
Março/ 2013

UFS
UNIVERSIDADE FEDERAL DE SERGIPE CENTRO DE CIÊNCIAS EXATAS E TECNOLOGIA DEPARTAMENTO DE QUÍMICA PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
Uso de Métodos Semiempíricos para o Estudo
da Aplicação de Redes Híbridas de
Coordenação como Carreadores de Fármacos
e Dispositivos Luminescentes.
Nailton Marins Rodrigues*
Dissertação apresentada ao Programa de
Pós-Graduação em Química da UFS
como parte dos requisitos para a
obtenção do título de Mestre em
Química.
Orientador: Prof. Dr. Ricardo Oliveira Freire Co-orientador: Prof.Dr. Nivan Bezerra da Costa Júnior
*Bolsista CAPES
Aracaju-SEBrasil
Março / 2013

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
ii
FICHA CATALOGRÁFICA ELABORADA PELA BIBLIOTECA CENTRAL UNIVERSIDADE FEDERAL DE SERGIPE
R696u
Rodrigues, Nailton Martins Uso de métodos semiempíricos para o estudo da aplicação de redes híbridas de coordenação como carreadores de fármacos e dispositivos luminescentes / Nailton Martins Rodrigues ; orientador Ricardo Oliveira Freire. – São Cristóvão, 2013.
128 f. : il.
Dissertação (mestrado em Química) –Universidade Federal de Sergipe, 2013.
1. Química quântica. 2. Metal Organic Frameworks – MOFs. 3. Simulação (Computadores). 4. Carreadores de fármacos. 5. Luminescência. I. Freire, Ricardo Oliveira, orient.. II. Título.
CDU 544.183.26

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
iii

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
iv
AGRADECIMENTOS
Primeiramente agradeço a Deus por ter me dado o dom da vida.
Aos meus pais, que sempre estiveram ao meu lado, me ensinaram todos os valores da vida e deram todo o suporte para que eu conseguisse chegar até aqui.
A minha noiva Edna da Silva Machado, por todo carinho e pelo incentivo incondicional aos meus estudos.
Ao professor Dr. Ricardo Oliveira Freire, pela amizade, pela orientação e por ter me dado essa oportunidade.
Ao professor Dr. Nivan Bezerra da Costa Jr. pela amizade, coorientação e pelos ensinamentos.
A professora Dra. Viviane Costa Felicíssimo, pela amizade, conselhos e sugestões no exame de qualificação.
A professora Dra. Iara de Fátima Gimenez, pela amizade, ensinamentos e sugestões no exame de qualificação.
Aos amigos de laboratório Edna Machado, Danylo Alves, Silvando, Alexandre Santos, Danilo Rodrigues, Manoel,Luciano, Cristiane, Ricardo Andrade, Aloísio Santana, Júlio Gomes e José Diogo Dutra (desenvolvedor do LUMPAC junto com Prof. Ricardo Freire). Estes que de uma forma ou de outra contribuíram para este trabalho.
Aos meus amigos que sempre torceram por mim, e a todos que de forma direta ou indireta, contribuíram para a conclusão deste trabalho.
A CAPES pelo suporte financeiro.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
v
ÍNDICE Lista de Figuras....................................................................................................... viii
Lista de Tabelas....................................................................................................... xi
Lista de Siglas.......................................................................................................... xiii
Resumo..................................................................................................................... xiv
Abstract................................................................................................................... xvi
Capítulo 1 – Introdução e Objetivos..................................................................... 18
1.1 - Introdução........................................................................................................ 18
1.2 - Objetivos.......................................................................................................... 19
Referências............................................................................................................... 21
Capítulo 2 – Redes Metal-Orgânica..................................................................... 22
2.1 - Características das MOFs................................................................................ 22
2.2 - Síntese.............................................................................................................. 25
2.3 - Aplicações....................................................................................................... 27
2.3.1 - Catálise................................................................................................. 27
2.3.2 - Molde para síntese de nanotubos de carbono....................................... 27
2.3.3 - Nanorrastreadores fotônicos................................................................. 28
2.3.4 - Armazenamento e transporte de gás..................................................... 29
2.3.5 - Carreador de fármaco............................................................................ 30
2.4 - Simulações computacionais envolvendo MOFs.............................................. 32
Referências............................................................................................................... 34
Capítulo 3 – Métodos de Química Teórica e Aplicações..................................... 36
3.1 - A química computacional................................................................................. 36
3.2 - Métodos de química teórica............................................................................. 37
3.2.1 - Método de Hartree-Fock........................................................................ 38
3.2.2 - Teoria do Funcional da Densidade........................................................ 41
3.2.2.1 - Funções de Bases........................................................................ 43
3.2.3 - Métodos Semiempíricos........................................................................ 45
3.2.3.1 - Método AM1............................................................................... 47
3.2.3.2 - Método PM3............................................................................... 47

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
vi
3.2.3.3 - Método PM6................................................................................ 48
3.2.3.4 - Método RM1................................................................................ 49
3.3 - Descrevendo cálculos em fase sólida para uma MOF...................................... 50
3.3.1–Critérios básicos para a escolha de um cluster............................................. 51
Referências............................................................................................................... 52
Capítulo 4 – Cálculos em Fase Sólida.................................................................... 53
4.1 - Métodos e Objetivos: Cálculos semiempíricos em fase sólida......................... 53
4.1.1 - Metodologia dos cálculossemiempíricosdas MOFs em fase sólida 54
4.2 - Resultados e Discussões................................................................................... 55
4.2.1 - Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
para o cálculo de redes de coordenação contendo os metais Al, Zn, Pb, Hg e Ba.... 55
4.2.2 - Avaliação da capacidade de predição dos métodos Sparkle/AM1,
Sparkle/PM3 e Sparkle/PM6 para redes de coordenação contendo íons lantanídeos
como centros metálicos............................................................................................ 59
4.2.3 - Avaliação do método PM6 na predição de redes de coordenação
contendo centros metálicos do tipo: Mn, Fe, Co, Ni, Cu, Y, Ag, Cd....................... 61
4.2.4 - Casos Especiais.................................................................................... 63
4.2.4.1 - Caso 1: ZnBDC........................................................................... 63
4.2.4.2 - Caso 2: [Ba(C2O4)0.5(HC2O4)(H2O)]........................................... 67
4.2.4.3 - Caso 3: Elemento Sr (Estrôncio)................................................. 69
4.2.4.4 – Caso 4: Ga(OH)(BPDA)............................................................. 72
4.2.4.5 – Caso 5: Mg(C10H16O4)2(H2O).................................................... 73
Referências............................................................................................................... 76
Capítulo 5 – Aplicação de MOFs como Carreadores de Fármacos................... 78
5.1 – Introdução e Objetivos..................................................................................... 78
5.1.1 – Metodologia dos cálculos para os fármacos avaliados.......................... 80
5.1.2 – Metodologia dos cálculos de interação MOF-Fármaco........................ 81
5.2 – Resultados e Discussões................................................................................. 81
5.2.1 – Avaliação do poder de predição dos métodos semiempíricosAM1, PM3,
PM6 e RM1 para os fármacos selecionados.................................................... 81

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
vii
5.2.1.1 –Otimização dos fármacos utilizando a teoria do funcional da
densidade (DFT).................................................................................... 82
5.2.1.2 – Otimização dos fármacos utilizando métodos
semiempíricos........................................................................................................... 82
5.2.3 – Estudo da interação entre doxorrubicina e a ZIF-8........................... 88
Referências............................................................................................................... 91
Capítulo 6 – Estudo das propriedades Luminescentes da MOF EuMell 1, com
Simulação da Variação de temperatura............................................................... 93
6.1 – Introdução e Objetivos..................................................................................... 93
6.2 – Metodologia..................................................................................................... 95
6.3 – Resultados e discussão..................................................................................... 96
6.3.1 – Resultados da descrição estrutural para a EuMell 1..................................... 96
6.3.2 – Estudo luminescente com a EuMell 1, comparativo entre cálculos
utilizando condições de periodicidade e no cálculo no vácuo................................. 98
Referências............................................................................................................... 106
Capítulo 7 – Conclusões e Perspectivas................................................................ 107
7.1 – Conclusões...................................................................................................... 107
7.2 – Perspectivas..................................................................................................... 109
Apêndice 1................................................................................................................ 110
Apêndice 2................................................................................................................ 126

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
viii
LISTA DE FIGURAS
Figura 2.1. MOF-5 e suas unidades de construção..................................................... 23
Figura 2.2. Alguns exemplos da série IRMOFs......................................................... 24
Figura 2.3. Formação de uma MOF em diferentes dimensões.................................. 25
Figura 2.4. Possíveis coordenações geométricas dos íons de metais transição.......... 26
Figura 2.5. Tipos de emissão e aplicações relatadas para lantanídeos....................... 28
Figura 2.6. Representação das MOFs MIL-100 e MIL-101, esferas em amarelo
representam o volume do poro, triângulos representam ligações entre as unidades de
construção ................................................................................................................... 31
Figura 3.4. Elementos parametrizados para o AM1................................................... 47
Figura 3.5. Elementos parametrizados para o PM3.................................................... 48
Figura 3.6. Elementos parametrizados para o PM6................................................... 48
Figura 3.7. Elementos parametrizados para o RM1................................................... 49
Figura 3.8. Descrição espacial da MOF-5.................................................................. 50
Figura 4.1. Comparação do RMS médio (RMSM) obtidos a partir das redes de
coordenação contendo Al, Zn, Hg, Pb e Ba, otimizadas com os métodos AM1, PM3
e PM6........................................................................................................................... 56
Figura 4.2. Comparação do RMS médio (RMSM) obtidos a partir das redes
de coordenação contendo Al, Zn, Hg e Pb, otimizadas com os métodos AM1, PM3
e PM6.......................................................................................................................... 57
Figura 4.3. Comparação do RMS médio (RMSM) obtidos a partir das redes de
coordenação contendo íons lantanídeos trivalentes, otimizadas com os métodos
AM1, PM3 e PM6...................................................................................................... 59
Figura 4.4. RMS médio (RMSM) obtidos a partir das redes de coordenação
contendo Mn, Fe, Co, Ni, Cu, Y, Ag, Cd, otimizadas com o método PM6................ 62
Figura 4.5. Sobreposição da estrutura cristalográfica (em cinza) da MOF ZnBDC
com a estrutura otimizada (em azul) com o método AM1.( RMS=0,1732)............... 65
Figura 4.6. Sobreposição da estrutura cristalográfica (em cinza) da MOF ZnBDC
com a estrutura otimizada (em azul) com o método PM3. (RMS=0,1190)................ 65

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
ix
Figura 4.7. Sobreposição da estrutura cristalográfica (em cinza) da MOF ZnBDC
com a estrutura otimizada (em azul) com o método PM6. (RMS=0,0132)................ 66
Figura 4.8. Sobreposição da estrutura cristalográfica (em cinza) da MOF
[Ba(C2O4)0.5(HC2O4)(H2O)] com a estrutura otimizada (em azul) com o método
PM6. (RMS=0,0883)..................................................................................................
68
Figura 4.9. Sobreposição da estrutura cristalográfica (em cinza) da MOF
Sr[C2H4(SO3)2] com a estrutura otimizada (em azul) com o método PM6 (Sr)......... 70
Figura 4.10. Sobreposição da estrutura cristalográfica (em cinza) da MOF
Sr[C2H4(SO3)2] com a estrutura otimizada (em azul) com o método PM6 –
fragmento (1,2,1)...................................................................................................... 71
Figura 4.11. Sobreposição da estrutura cristalográfica (em cinza) da MOF
Ga(OH)(BPDA) com a estrutura otimizada (em azul) com o método AM1............ 72
Figura 4.12. Sobreposição da estrutura cristalográfica (em cinza) da MOF
Ga(OH)(BPDA) com a estrutura otimizada (em azul) com o método PM6.............. 73
Figura 4.13. Sobreposição da estrutura cristalográfica (em cinza) da
MOFMg(C10H16O4)2(H2O)2 com a estrutura otimizada (em azul) com o método
PM6...... 74
Figura 5.1 – Estrutura tridimensional cristalográfica da MOF ZIF-8...................... 79
Figura 5.2 - Determinação teórica das dimensões da molécula do fármaco
ibuprofeno.................................................................................................................... 83
Figura 5.3.Estrutura da molécula de Ibuprofeno....................................................... 85
Figura 5.4.Estrutura da molécula de diclofenaco...................................................... 85
Figura 5.5.Estrutura da molécula de Fluoracil.......................................................... 85
Figura 5.6.Estrutura da molécula de Doxorubicina................................................... 85
Figura 5.7.Estrutura da molécula de ciclofosfamida................................................. 85
Figura 5.8.Estrutura da molécula de mercaptopurina................................................ 85
Figura 5.9.Estrutura da molécula de raloxifeno......................................................... 86
Figura 5.10.Estrutura da molécula de fulvestrante.................................................... 86
Figura 5.11.Estrutura da molécula de tamoxifeno..................................................... 86
Figura 5.12.Estrutura da molécula de toremifeno...................................................... 86

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
x
Figura 5.13.Estrutura da molécula de cisplatina........................................................ 86
Figura 5.14.Estrutura da molécula de carboplatina................................................... 86
Figura 5.15.Estrutura da molécula de betalapachona................................................ 86
Figura 5.16.Estrutura da molécula de oxaliplatina.................................................... 86
Figura 5.17.Possibilidade1 para adsorção da doxorrubicina com a superfície da
ZIF8............................................................................................................................. 89
Figura 5.18.Possibilidade2 para adsorção da doxorrubicina com a superfície da
ZIF8............................................................................................................................. 89
Figura 5.19.Possibilidade3 para adsorção da doxorrubicina com a superfície da
ZIF8............................................................................................................................. 89
Figura 5.20.Possibilidade4 para adsorção da doxorrubicina com a superfície da
ZIF8............................................................................................................................. 89
Figura 5.21.Possibilidade5 para adsorção da doxorrubicina com a superfície da
ZIF8............................................................................................................................. 90
Figura 6.1. Comparação entre a estrutura cristalográfica e a estrutura calculada com
o modelo Sparkle/AM1............................................................................................... 97
Figura 6.2. Comparação entre a estrutura cristalográfica e a estrutura calculada com
o modelo Sparkle/PM3................................................................................................ 97
Figura 6.3. Comparação entre a estrutura cristalográfica e a estrutura calculada com
o modelo Sparkle/PM6............................................................................................... 97
Figura 6.4. Fragmento contendo o poliedro de coordenação da EuMell1................ 99
Figura 6.5. Análise do aquecimento sobre os parâmetros de intensidade Ω2 e Ω4 103
Figura 6.6. Análise do aquecimento sobre os parâmetros de intensidade Arad e Anrad 104

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xi
LISTA DE TABELAS
Tabela 3.1.Tipos e quantidades de funções utilizadas na descrição de orbitais s, p, d
e f............................................................................................................................. 43
Tabela 3.2. Aplicação de diferentes conjuntos de bases ao sistema H2O utilizando o
Gaussian-03W.............................................................................................................. 44
Tabela 4.1.RMS médio (RMSM) obtidos a partir das redes de coordenação
contendo Al, Zn, Hg e Pb, otimizadas com os métodos AM1, PM3 e PM6............... 58
Tabela 4.2.RMS médio (RMSM) obtidos a partir das redes de coordenação
contendo íons lantanídeos trivalentes, otimizadas com os métodos AM1, PM3 e
PM6............................................................................................................................. 60
Tabela 4.3.RMS médio (RMSM) obtidos a partir das redes de coordenação
contendo Mn, Fe, Co, Ni, Cu, Y, Ag, Cd, otimizadas com o método PM6................ 63
Tabela 4.4.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
para um clusterde ZnBDC......................................................................................... 63
Tabela 4.5.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
para um cluster (1,2,1) de ZnBDC............................................................................. 64
Tabela 4.6.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
docluster (1,1,1) da MOF [Ba(C2O4)0.5(HC2O4)(H2O)]............................................. 69
Tabela 4.7.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 do
cluster (2,1,2) da MOF [Ba(C2O4)0.5(HC2O4)(H2O)]............................................. 68
Tabela 4.8.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 do
cluster (1,1,1) da MOF Sr[C2H4(SO3)2]................................................................. 69
Tabela 4.9.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 dada em valores da célula unitária, para (1,2,1) da MOF Sr[C2H4(SO3)2] ................
70
Tabela 4.10.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
do cluster (1,1,1) da MOF [Sr2(1,3-pdta)(H2O)6].H2O........ ...................................... 71
Tabela 4.11.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
do cluster (1,1,1) da MOF Ga(OH)(BPDA)............................................................... 72
Tabela 4.12.Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6
do cluster (1,1,1) da MOF Mg(C10H16O4)2(H2O)2...................................................... 74
Tabela 5.1. Fármacos e suas respectivas aplicações clinicas..................................... 83

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xii
Tabela 5.2. Erro médio absoluto (UME) para os fármacos otimizados com os
métodos semiempíricos AM1, PM3, PM6 e RM1...................................................... 87
Tabela 6.1. Resultados para [Eu2(Mell)(H2O)6], utilizando os modelos
Sparkle/AM1, Sparkle/PM3 e Sparkle/PM6.............................................................. 96
Tabela 6.2. Dados experimentais e calculados no vácuo e em condições de
periodicidades para EuMell 1..................................................................................... 99
Tabela 6.3. Resultados para [Eu2(MELL)(H2O)6], utilizando os Sparkles AM1
(vácuo) e AM1 (periódico)........................................................................................ 100
Tabela 6.4. Dados experimentais e calculados no vácuo e em condições de
periodicidades para EuMell 1.................................................................................... 101
Tabela 6.5. Estudo do sistema [Eu2(MELL)(H2O)6] simulando aquecimento de
25ºC a 250 °C. Os dados teóricos foram calculados a partir da estrutura calculada
com o modelo Sparkle/PM3 em fase sólida............................................................... 102
Tabela 6.6. Taxas de transferências e retro transferências para 25, 150 e 200 °C..... 102

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xiii
LISTA DE SIGLAS
AM1 – Austin Model1
B3LYP – Funcional usado em cálculos DFT
CCDC - Cambridge Crystalographic Data Centre
DFT – Density Functional Theory
DMCLs – Dispositivos Moleculares Conversores de Luz
GTO – Orbitais do tipo Gaussianas
HF – Hartree-Fock
IRMOF – Isoreticular MOF
LED – Diodo Emissor de Luz
MIL – Materiais do Instituto Lavoisier
MNDO – Modified Negect of Diatômic Overlap
MOF – Metal-Orgânic Framework
MP2 – Moller-Plesset perturbation theory
NDDO – Neglect of Diatomic Differential Overlap
PM3 – Parametric Model 3
PM4 – Parametric Model 4
PM5 – Parametric Model 5
PM6 – Parametric Model 6
RHF – Hartree-FockRestrito
RM1 – Recife Model 1
RMS - valor quadráticomédio, do inglêsRoot Mean Square
ROHF - Hartree-Fock restrito de Camada Aberta
SBU – Secondary Building Units
STO – Orbitais do tipo Slater
UHF - Hartree-Fock não Restrito
1D, 2D, 3D – Uma, Duas e Três Dimensões

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xiv
RESUMO
Relatos na literatura envolvendo asMOFs (Metal Orgânic Framework),
têmaumentado consideravelmente nos últimos anos, no entanto, trabalhos envolvendo
métodos teóricos computacionais com estas estruturas ainda são escassos.
O presente trabalho inicialmente avalia a capacidade de predição das estruturas em
fase sólida de MOFs através dos métodos semiempíricos AM1, PM3 e PM6. Foram
consideradas 82 diferentes redes de coordenação compostas por 26 diferentes centros
metálicos, retiradas de artigos publicados nos últimos cinco anos. Com relação às MOFs
formadas pelos metais (Al, Zn, Hg, Pb, Ba, Mn, Fe, Co, Ni, Cu, Y, Ag, Cd, Sr, Ga, Mg) os
resultados obtidos sugerem que o método PM6 mostrou-se o mais exato apresentando
resultados em grande concordância com as respectivas estruturas experimentais. Com
relação às MOFs compostas por íons lantanídeos trivalentes os resultados obtidos sugerem
que a utilização do modelo Sparkle/PM3 permite o cálculo em fase sólida de MOFs dos
mais variados tipos com elevada exatidão.
O uso do método semiempírico se justifica pelo fato das MOFs serem estruturas
cuja célula unitária geralmente possui mais que 100 átomos, o que torna o método o mais
ideal para sua aplicação neste estudo, pois o uso dos métodos semiempíricos apresentam
bons resultados e requerem menores custo computacional quando comparado com as
metodologias DFT (Teoria do Funcional da Densidade) e HF( Hartree-Fock)).
Com base nos resultados obtidos com as otimizações, constatou-se que o método
semiempírico Sparkle/PM6 é omais indicado para estudar as interações entre o fármaco
Doxorrubicina e a MOF ZIF-8, quando esta é utilizada como veículo carreador da
Doxorrubicina. Os resultados teóricos sugerem que ocorre a adsorção do fármaco com a
superfície da ZIF-8 e o estudo de docking forneceu cinco possibilidades de interação via
adsorção.
Os estudos de comparação entre os diferentes modelos Sparkle considerando
cálculos em fase sólida e no vácuo indicaram o cálculo em fase sólida como o mais

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xv
indicado paraotimização estrutural desse tipo de sistema, e o método Sparkle/PM3 se
sobressaiu como o mais eficiente para este fim. O cálculo da estrutura com simulação da
variação de temperatura, resultou em dados luminescentes em grande acordo com os
valores obtidas experimentalmente. A MOF denominada deEuMell 1, foi a utilizada nestes
estudos.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xvi
ABSTRACT
Releases on literature involving MOFs (Metal Organic Framework), has undergone
significant growth on last years, Although applications using theoretical methods with these
structures are still scant.
The present work initially evaluated the predictive ability of the MOFs structures in
the solid phase through the semiempirical AM1, PM3 and PM6 methods. We considered 82
different coordination networks composed of 26 different metal centers, drawn from
articles published in the last five years. With relation to MOFs formed by metals (Al, Zn,
Hg, Pb, Ba, Mn, Fe, Co, Ni, Cu, Y, Ag, Cd, Sr, Ga, Mg) the results suggest that the PM6
method is the most accurate results in close agreement with the respective experimental
structures. Regarding MOFs that consist of trivalent lanthanide ions, our results suggest
that the use of Sparkle/PM3 model premises the solid phase calculation of MOFs of various
types with high accuracy.
The use of semiempirical method is justified by the fact that MOFs are structures
whose unit cell usually has more than 100 atoms, which makes the method the most ideal
for their application in this study. As is known the use of semiempirical methods shows
good results and requires a littler computational cost when compared with DFT (Density
Functional Theory) and HF (Hartree-Fock) methodologies.
Based on these results, it was found that the Sparkle/PM6
semiempirical method is the most suitable for studying the chemical interactions between
the drug Doxorubicin and ZIF-8 MOF, when this one is used as Doxorubicin carrier. The
teoric results suggest that occurs adsorption of the drug with the surface of ZIF-8 and
docking study provided five different interactions by adsorption.
The studies comparing different Sparkle models considering
calculations at solid phase and vacuum indicated the solid phase
calculation as the most appropriate for this type of study, and the
Sparkle/PM3 method stood out as the most efficient for this purpose. The simulation of the

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
xvii
structure with variation in temperature resulted in convincing luminescence data with those
obtained experimentally. The MOF named EuMell1, was the used in these studies.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
18
11 INTRODUÇÃO & OBJETIVOS
1.1 – Introdução
As MOFs, vem sendo objeto de pesquisa há algunsanos, e ao longo desses anos vários
grupos foram designando diferentes nomes para esses materiais, mas que possuem o
mesmo significado. Na literatura podemos encontrar diversos trabalhos que apresentam
nomes como: polímero de coordenação, hibrido orgânico-inorgânico, zeólita orgânica e o
termo que será adotado nesse trabalho para nomeá-las, redes metal-orgânica, que pode ser
abreviada como MOF que é derivada do termo em inglês metal-organic frameworks. [1]
De forma resumida, MOFs são estruturas porosas que têm o seu esqueleto formado
por um íon metálico coordenado a partes orgânicas e que se estendem em uma, duas ou três
dimensões. Essas duas partes, o íon metálico e o ligante orgânico, são conhecidas como
unidades de construção secundária ou SBUs (SecondaryBuildingUnits) [2].
O grande atrativo para a pesquisa e desenvolvimento de novas MOFs está na
possibilidade de se obter várias estruturas com diferentes propriedades como, por exemplo:

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
19
(i) diversidade em tamanhos de poro o que possibilita controlar seu volume e área
superficial, (ii) estrutura estável, flexível e funcional. [1]
Existe uma ampla gama de aplicações já bem estudadas e outras que surgem como
boa alternativa e que requerem mais atenção. O estudo de MOFs como carreador de
fármacos e como dispositivos eletroluminescentes são exemplos e serão estudados
teoricamente nesse trabalho.
Convencionalmente estudos envolvendo materiais desta natureza são realizados
experimentalmente. Poucos são os relatos na literatura envolvendo o uso de ferramentas
teóricas, principalmente as baseadas em métodos de mecânica-quântica. Provavelmente,
devido ao grande número de átomos presentes nessas redes de coordenação, o uso de
métodos quânticos tradicionais seja considerado inadequado. Métodos híbridos são uma
alternativa, mas a dificuldade de tratar a região de fronteira dificulta a sua aplicação. Os
métodos semiempíricos surgem como candidatos naturais para serem aplicados a este tipo
de estudo, principalmente com a recente metodologia proposta pelo Prof. James Stewart
para calcular estruturas em fase sólida [3]. Contudo seria inevitável nos questionar se os
métodos semiempíricos atuais aliados a essa nova metodologia proposta pelo Prof. James
Stewart apresentam exatidão satisfatória para tratar tais sistemas. Deste modo, além de
estudar teoricamente a aplicação de algumas MOFs como sistemas carreadores de fármacos
e no desenvolvimento de dispositivos luminescentes, este trabalho buscará responder a esta
questão.
1.2 – Objetivos
Esta dissertação possui três objetivos distintos:
(i) A avaliação da descrição de estrutura de MOFs com os diferentes métodos
semiempíricos AM1 (Austin Model1) [4], PM3 (ParametricModel 3) [5] e PM6
(ParametricModel 6) [6]. Para tanto, analisaremos MOFs que contenham diferentes íons
metálicos em sua estrutura. Também buscaremos avaliar a exatidão do método PM6
quando apenas este método apresentar parâmetros para o metal em questão.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
20
(ii) Estudo da interação hóspede-hospedeiro envolvendo a rede de coordenação ZIF-8,
e o fármaco doxorrubicina objetivando o design de novos carreadores de fármacos.
(iii) Estudo das propriedades luminescentes da MOFEuMell 1, contendo íon
lantanídeo Eu3+, para o design de eficientes Dispositivos Nanométricos Conversores de Luz
(DNCLs).
A divisão dessa dissertação se encontra em capítulos, que abordam temas diferentes,
temas que estão dispostos como uma forma crescente e evolutiva de conceitos, para o bom
entendimento do estudo realizado.
No capítulo 2 abordaremos toda a base necessária para entender como são
constituídas as MOFs, o que delineia suas propriedades, suas aplicações e um pouco dos
estudos teóricos que vêm sendo relatados com MOFs na literatura.
Em seguida, no capítulo 3, discutiremos sobre os métodos quânticos. Apresentaremos
um pouco da base dos métodos quânticos ab initio e discorreremos sobre os métodos
semiempíricos empregados neste trabalho.
No capítulo 4, apresentaremos a metodologia e discutiremos os resultados obtidos do
estudo de avaliação da capacidade de predição estrutural dos métodos semiempíricos
disponíveis considerando uma grande variedade de MOFs. Os resultados obtidos nesse
capítulo proporciona suporte e perspectivas aos estudos de aplicação que serão
apresentados nos capítulos a seguir.
O estudo teórico da aplicação de MOFs como carreadores de fármacos será
apresentado no capítulo 5.
No capitulo 6, é apresentado a parte referente ao estudo das propriedades
luminescente de uma MOF contendo o íon lantanídeo Eu3+ como centro metálico, bem
como a analise das mudanças das propriedades luminescentes frente a variação da
temperatura. E por fim, o capítulo 7 apresentará as conclusões e perspectivas deste trabalho.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
21
Referências
[1] Rowsell, J. L. C.; Yaghi, O. M., Microporous and Mesoporous Materials.73(2004) 3.
[2] Yaghi, O. M.; O Keeffe, M.; Ockwing, N. W.; Chae, H. K.; Eddaoudi, M. Kim,J.,
Nature.423 (2003) 705.
[3] James J. P, Stewart.,J. Mol. Model. 14 (2008) 499.
[4]Dewar, M.J.S.; Zoebish, E.G.; Healy, E.F.; e Stewart, J.J.P., J. Am. Chem. Soc.107
(1985) 3902.
[5]Stewart, J.J.P., J. Comp. Chem.10 (1989) 209.
[6] Stewart, J.J.P., J. Mol. Model.13(2007) 1173.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
22
22 REDES METAL-ORGÂNICAS
2.1 – Características das MOFs
Redes metal-orgânicas ou MOFs (Metal Organic Frameworks) são estruturas
porosas que tem o seu esqueleto formado por um íon metálico coordenado a partes
orgânicas. Como foi relatado no capítulo 1, essas duas partes, o íon metálico e o ligante
orgânico, são denominadas de unidades de construção secundária ou SBUs
(SecondaryBuildingUnits) [1].
As propriedades das MOFs serão determinadas pelo material usado para a síntese
bem como, o tamanho e espaçamento dos poros são determinados pelas unidades de
construção orgânicas (espaçadores). Os ligantes orgânicos irão se conectar com as
chamadas unidades inorgânicas (centros metálicos) formando a MOF [2].
Na figura 2.1 podemos observar um encaixotamento contendo parte da MOF-5
destacando as suas SBUsconstituintes, do qual a esfera em amarelo representa o volume do
poro para este fragmento.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
23
Figura 2.1. MOF-5 e suas unidades de construção. Figura adaptada da referência [3].
Da figura 2.1, podemos observar a subunidade orgânica constituída por carbono em
cinza escuro e seus oxigênios (em vermelho) que formam as bases do prisma. Os triângulos
representam ligações dos oxigênios da unidade orgânica com o íon zinco (unidade
inorgânica), os triângulos que constituem o poliedro de coordenação tem objetivo visual de
ilustrar a repetição geométrica a que constitui a formação estrutural de uma MOF como um
todo.
Quando tratamos de MOFs, um exemplo que não pode deixar de ser citado é a
MOF-5 (Figura 2.1), pois esta é a mais famosa e mais investigada. Além disso, é um
excelente exemplo de uma MOF com poros em 3D [3].
A MOF-5 foi a primeira a manter a sua estrutura porosa denominada de rede aberta,
por permitir a passagem de espécies químicas em suas cavidades, sendo bastante estável
após a remoção ou troca de moléculas hóspedes. Estudo computacional com a MOF-5 pode
ser encontrado na literatura usando cálculos DFT [4].
A partir da topologia reticular cristalina da MOF-5, Yaghi e colaboradores
projetaram uma nova série chamada de IRMOFs (IsoreticularMOF). Nessa nova série
existe somente a modificação das unidades de construção orgânicas, e em consequência
disso, a mudança das propriedades específicas. Pelo fato desta nova série ser derivada da

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
24
MOF-5, esta ficou conhecida como IRMOF-1 ou MOF-5, dois termos para a mesma
estrutura [3], [5]– [6]. Na figura 2.2 temos alguns exemplos de MOFs dessa série, na qual
podemos perceber que a diferença característica entre elas esta na unidade orgânica.
Figura 2.2. Alguns exemplos da série IRMOFs. Figura retirada da referência [7].
As MOFs possuem uma grande variedade de tamanho de poros que mudam de
acordo com tamanho de seus espaçadores, sendo uma das principais causas na mudança de
suas propriedades. Temos também que ressaltar outra característica dessas estruturas, que é
a possibilidade de obtenção de estruturas com uma dimensão (1D), duas dimensões (2D) e
em três dimensões (3D), que são derivados de diferentes arranjos espaciais das unidades de
construção, que podem possuir interações do tipo: ligações coordenadas, ligações de
hidrogênio, interações de empilhamento π- π em aromáticos como forças de van der Waals
[8].

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
25
2.2 – Síntese
A síntese das MOFs pode ser discutida como sendo uma auto-organização dos
materiais de partida. Sais metálicos e moléculas orgânicas (moléculas com carboxilas são
bastante utilizadas por facilmente se coordenar ao metal) são adicionados em um recipiente
formando o meio reacional, no qual ocorrerá um rearranjo entres as unidades primárias de
construção inorgânica e orgânica [9] que podem resultar em estruturas nas dimensões já
citadas anteriormente. As MOFs são conhecidas por apresentarem simetria ao longo de sua
estrutura, onde parcelas mínimas (células unitárias) se repetem, sendo consideradas como
um exemplo de material cristalino.
Não iremos adentrar em detalhes referentes a síntese dessas estruturas, pois não é
parte de nossos objetivos realizá-las. No entanto, apresentamos na figura 2.3,o esquema
representando o processo de reorganização já citado para melhor esclarecimento. Na figura
2.3vemos as unidades primárias de partida utilizadas na síntese de algumas MOFs, que irão
se reorganizar e gerar as estruturas com diferentes arranjos espaciais (1D, 2D ou 3D).
Figura 2.3. Formação de uma MOF em diferentes dimensões. Adaptada das referências [9] e [10].

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
26
Além da grande variedade de tamanhos de poros que podem ser obtidos, também
podemos citar a grande variedade de formas fazem das MOFs um atrativo campo de
pesquisa repleto de possibilidades. O fato das possíveis geometrias que os íons de metais de
transição são capazes de aderir em suas interações com outros átomos, possibilita essa
variedade de formas. Na figura 2.4 temos as possíveis coordenações geométricas que os
metais (constituinte da unidade inorgânica) de transição apresentam, as quais dependem da
valência do elemento metal.
Outro aspecto que pode ser explorado nas MOFs são as suas propriedades
espectroscópicas. Sobre isso podemos destacar MOFs contendo íons lantanídeos as quais
podem apresentar emissão em diferentes regiões do espectro visível. Podemos citar como
exemplo MOFscontendo o íon Eu(III) que emitem na região do vermelho e MOFs
compostas por Tb(III) que emitem na região do verde.
MOFs contendo íons lantanídeos, além de possibilitar novas aplicações a essas
estruturas, também podem apresentar uma grande variedade de formas, como acontece com
as MOFs de metais de transição [11].
Figura 2.4. Possíveis coordenações geométricas dos íons de metais transição, adaptada da referência [11].

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
27
2.3 – Aplicações
O grande interesse demonstrado por vários grupos de pesquisa na obtenção de
sistemas dessa natureza pode ser explicado pela vasta gama de aplicações envolvendo
MOFs.
Dentre as aplicações dessas estruturas podemos citar: o uso em catálise [12]-[14],
como molde para síntese de nanotubos de carbono[15], nanorastreadoresfotônicos
[16],[17], no armazenamento e transporte de gás [18] e como carreadores de fármacos [19]-
[20].
2.3.1 – Catálise
A possibilidade de organização e funcionalização de sítios ativos em nano escala
promove a base do desenvolvimento de novas MOFs com propriedades funcionalizadas que
podem ser aplicadas em catálise, tornando processos específicos de interesse, muito mais
rápidos e eficientes. Devido à possibilidade de obter MOFs com grande tamanho de poro e
variados sítios ativos, colaboram para o seu uso como, por exemplo, catalisadores em
reações de alquilação por combinações de poliaromáticos extensos [12].
Reações como isomerização de terpenos e hidrogenação de alquenos por meio do
uso de MOFs como catalisadores têm sido relatadas nos últimos anos [13], bem como
reações debenzilação de Friedel-Crafits foi reportada para a MIL-101 com Fe e Cr (MIL=
Materiais do Instituto Lavoisier) [14].
2.3.2 – Molde para síntese de nanotubos de carbono
As MOFs também podem ser utilizadas como uma interessante forma de realizar
síntese de nanotubos de carbono de tamanhos específicos como foi relatado o uso da MOF-
5 para esse fim[15]. Este trabalho expõe uma nova técnica na qual os poros da MOF-5 são
usados como template para a formação dos nanotubos de carbono.
A técnica utilizada no referido trabalho consistiu em realizar uma
polimerizaçãodentro do poro da MOF-5 e, na sequência, realizar uma carbonização sob
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
28
temperatura de 530 a 1000 °C durante 8 horas, sendo obtidos nanotubos de carbono na
dimensão do poro, ao qual seriam empregados na pesquisa de supercapacitores [15].
2.3.3 – Nanorastreadoresfotônicos
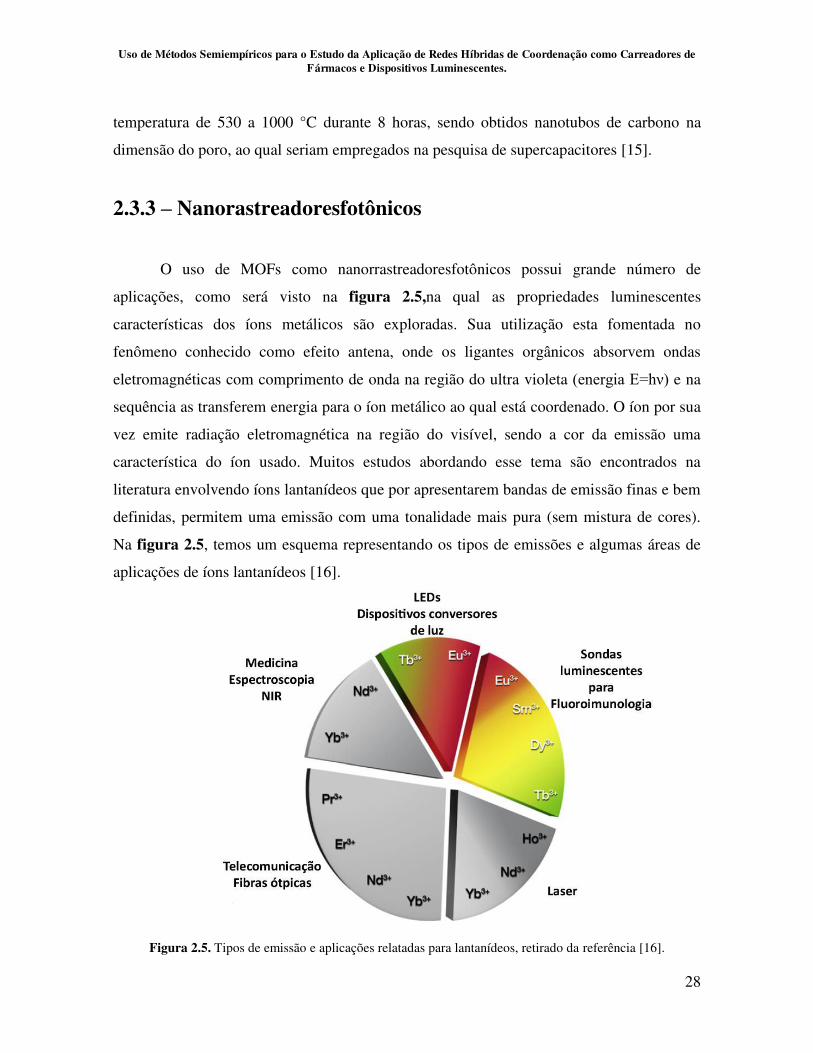
O uso de MOFs como nanorrastreadoresfotônicos possui grande número de
aplicações, como será visto na figura 2.5,na qual as propriedades luminescentes
características dos íons metálicos são exploradas. Sua utilização esta fomentada no
fenômeno conhecido como efeito antena, onde os ligantes orgânicos absorvem ondas
eletromagnéticas com comprimento de onda na região do ultra violeta (energia E=hν) e na
sequência as transferem energia para o íon metálico ao qual está coordenado. O íon por sua
vez emite radiação eletromagnética na região do visível, sendo a cor da emissão uma
característica do íon usado. Muitos estudos abordando esse tema são encontrados na
literatura envolvendo íons lantanídeos que por apresentarem bandas de emissão finas e bem
definidas, permitem uma emissão com uma tonalidade mais pura (sem mistura de cores).
Na figura 2.5, temos um esquema representando os tipos de emissões e algumas áreas de
aplicações de íons lantanídeos [16].
Figura 2.5. Tipos de emissão e aplicações relatadas para lantanídeos, retirado da referência [16].

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
29
Uma recente aplicação que se utiliza de propriedades características de MOFs como
estabilidade térmica, estabilidade química e propriedades luminescentes, foi relatada como
uma proposta do uso dessas estruturas como um rastreador luminescente para armas de
fogo. No trabalho a MOF usada foi aplicada em cápsulas de armamento, que ao ser
disparado impregna o armamento, a mão e roupas de quem realizou o disparo e o seu alvo,
no alvo usado após 30 meses ainda foram encontrados vestígios [17].
O resíduo de pólvora com MOF pode ser facilmente identificado com o uso de
radiação ultravioleta, podendo assim identificar a fonte do disparo, ou diferenciar munições
de militares de civis, as possibilidades são muitas. Nesse trabalho foram usadas 2MOFs
com os íons lantanídeos Eu(III) e Tb(III) [17].
2.3.4 – Armazenamento e transporte de gás
Podemos citar como um dos motivos para o grande interesse nessa área de pesquisa,
que é o perigo em armazenar e, principalmente, transportar gases tão reativos quanto o gás
hidrogênio, sendo eminente o risco de explosão, que é um fator bastante preocupante.
Essa área que recentemente entrou no campo de aplicação das MOFs, é sustentada
pelo aumento crescente do desenvolvimento e fabricação de carros que são movidos por
eletricidade. A eletricidade que impulsiona o carro é provinda das chamadas células
combustíveis, que necessitam de hidrogênio armazenado (em recipientes sob alta pressão)
para o seu funcionamento.
Os carros movidos à célula combustível possuem como justificativa de uso, a
geração de energia elétrica sem a poluição do meio ambiente, uma vez que os reagentes da
utilizados para a reação química são: hidrogênio armazenado em cilindros a alta pressão
com o oxigênio do ar; e como produto obtém-se: corrente elétrica e água.
A partir da justificativa acima, podemos encontrar pesquisas científicas que usam
MOFs funcionalizadas como uma possibilidade de armazenamento de gases como o

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
30
hidrogênio por meio de adsorção [18], que tem como objetivo tornar mais seguro o
armazenamento não só de hidrogênio como também de outros gases.
2.3.5 – Carreador de fármaco
Este é um ponto importante para o entendimento de uma das propostas de nosso
trabalho, que é referente à aplicação de MOFs como carreador de fármacos. O uso da MOF
para este fim pode assegurar uma evolução no tratamento de doenças, onde um dos
principais fatores que desfavorecem ou até interrompem o seu diagnóstico ou tratamento,
são efeitos colaterais indesejados.
Técnicas terapêuticas tradicionais estão limitadas pela distribuição não específica
de fármaco no corpo, o que conduz a um alto nível de concentração do mesmo no
plasma[19]. Esse nível que geralmente ultrapassa a faixa terapêutica e entra na faixa tóxica
diminui a eficácia do tratamento e produz efeitos colaterais que podem provocar o fim do
tratamento antes mesmo que a droga tenha a chance de erradicar a doença.
A utilização de MOFs como sistemas carreadores de fármacos pode possibilitar uma
liberação contínua da droga no meio plasmático dentro da faixa terapêutica aumentando a
atividade das moléculas do fármaco, amenizando possíveis efeitos colaterais e melhorando
o bem estar do paciente durante o tratamento. Geralmente as MOFs não são metabolizadas
pelo organismo, sendo totalmente excretadas pelo paciente.
Entre os grupos de MOFs existentes, o primeiro a ser investigado como um
potencial carreador de fármaco foi a família MIL que possuem como característica grandes
poros (25-34Å) e grande área superficial (3100-5900m²/g). Estas características
possibilitam tanto uma inserção de fármacos em seus poros como também adsorção do
mesmo em sua superfície.
O trabalho pioneiro que abriu esta nova área de aplicações foi realizado por Gérard
Férey e colaboradores, que estudaram o armazenamento e a liberação de ibuprofeno nas
MOFs MIL-100 e MIL-101. Nesse estudo os autores concluíram que ambos os materiais
possuíam um alto poder de armazenamento da droga, com um armazenamento de 0,347g de
ibuprofeno/g de MOF para a MIL-100 e 1,367g de ibuprofeno/g de MOF para a MIL-

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
31
101[19],[20].Embora as MOFs MIL-101 e MIL-100 (figura 2.6) tenham sido capazes de
armazenar uma boa quantidade de droga por grama de MOF, os autores observaram uma
diferença de capacidade de armazenamento das mesmas, sendo a MIL-101 a mais eficiente
nesse aspecto. No trabalho este fato foi explicado pela diferença nos tamanhos dos poros
das MOFs, sendo que a MIL-101 com poros de dimensões maiores (12700 e 20600 Å) e a
MIL-100, na qual foi observada menor rendimento, com dimensões de poro de 8200 e
12700 Å.
Figura 2.6. Representação das MOFs MIL-100 e MIL-101, esferas em amarelo representam o volume do
poro, triângulos representam ligações entre as unidades de construção [21].
O estudo da cinética de liberação do fármaco foi realizado suspendendo o conjunto
ibuprofeno-carreador em fluido do corpo simulado (SBF-SimulatedBodyFluid) a uma
temperatura de 37°C. Foi observada uma liberação total do fármaco em um intervalo de três
dias para a MIL-100 e seis dias para a MIL-101. Notou-se um crescimento na taxa de
liberação durante as oito primeiras horas, com uma completa liberação após os dias já
citados. Apesar da grande eficiência observada para essas estruturas, existe um agravante
que impossibilita o uso das mesmas como possível carreador, que é o fato da toxidade do
cromo contido em ambas estrutura.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
32
2.4 – Simulações computacionais envolvendo MOFs
Com uma ampla gama de aplicações, as MOFs se tornaram um atrativo para a
descoberta de novos materiais que possuam propriedades para aplicações específicas. Um
dos meios para conseguir um aprimoramento nas propriedades destas estruturas, seria o uso
do processo de química combinatória, que é um caminho dereagentes com elevado custo
para tantas sínteses e de grande dificuldade de caracterização de todos os complexos
sintetizados simultaneamente. Também deve-se destacar o elevado tempo necessário na
realização da síntese de tantas estruturas diferentes bem como o tempo necessário nos testes
e análises das propriedades intrínsecas de cada estrutura. Todos esses fatores tornam esse
processo inviável.
Nesse aspecto, o uso de simulação computacional para o estudo das estruturas
existentes mas pouco exploradas e de novas MOFs com diversos tamanhos de poro bem
como o estudo das possíveis e desejadas interações, tem como vantagem, o baixo custo
financeiro e de tempo, que são fatores determinantes no andamento de pesquisas.
A aplicação da química computacional no estudo dessas estruturas (MOFs), está
presente na literatura como uma forma de entender melhor e explicar os fenômenos já
citados acima e que a cada dia ganha novas extensões com atrativas aplicações [22].
Como exemplo de tentativas de se caracterizar detalhadamente a forma estrutural,
fazendo uma junção teoria e experimento, podemos citar o trabalho de Valenzano e
colaboradores [23]. Nesse trabalho encontra-se uso do método de Teoria Funcional da
Densidade (DFT- do inglês, DensityFunctionalTheory) como um suporte para explicar ou
justificar algumas propriedades eletrônicas, vibracionais, magnéticas ou paramagnéticas.
Visando explicar o comportamento da MOF CuBTC diante de moléculas que
possam interagir com a sua estrutura, através do uso de ferramentas computacionais,
podemos citar o trabalho de Grajciar et. al [24]. Nesse trabalho foi realizado um estudo
computacional, utilizando o método DFT, objetivando entender a adsorção de água em
sítios insaturados (metal com carga positiva) dessa MOF. Vale salientar que apenas uma
pequena parcela contendo dois átomos de cobre foi otimizada computacionalmente. Este
procedimento justifica-se uma vez que métodos quânticos ab initio ou DFT possuem um

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
33
elevado custo computacional, não sendo, portanto indicados para serem utilizados em
estudos envolvendo grandes fragmentos de estrutura, como é o caso da CuBTC cuja célula
unitária é constituída por 768 átomos.
Ainda relativo à MOF CuBTC temos um estudo realizado como forma de entender a
adsorção de CO2 e identificar sítios insaturados com uso do método DFT. Neste trabalho,
isotermas de adsorção obtidas experimentalmente foram utilizadas como suporte no estudo
[25].
Abordando ainda o estudo das interações entre MOFs e moléculas hóspedes temos o
trabalho publicado por Watanabe e colaboradores no qual foram analisadas as possíveis
interações entre H2O, CO, NO, piridina, C2H2, H2S e NH3, com os sítios de insaturação da
MOF CuBTC [26]. Neste trabalho também foi empregado o método DFT.
Como podemos observar a maioria dos trabalhos que se utilizam de métodos
computacionais para estudar sistemas envolvendo redes de coordenação fazem uso do
método DFT. Mesmo sendo um método de reconhecida eficiência do ponto de vista de
previsão de estruturas, o fato das redes de coordenação apresentarem-se geralmente em três
dimensões e a sua células unitárias podem ser constituídas por centenas de átomos, limita a
aplicação do método DFT a pequenos fragmentos da estrutura. O uso desse pequeno
fragmento pode omitir detalhes importantes do sistema estudado levando a conclusões
incompletas ou não precisas. Também vale ressaltar que não há na literatura estudos
validando a qualidade das geometrias dos fragmentos otimizados quando comparadas com
os respectivos dados cristalográficos.Vale salientar que a gama de estudos hoje é imensa,
expomos as que achamos mais interessantes e que vem despertando o maior interesse da
comunidade científica.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
34
Referências
[1] Yaghi, O. M.; O Keeffe, M.; Ockwing, N. W.; Chae, H. K.; Eddaoudi, M. Kim,J.,
Nature, 423 (2003) 705.
[2] Natarajan, S.; Mahata, P., Chemical Society Reviews, 38(2009) 2304.
[3] figuracontida pertencente a referência [1] deste capitulo que foi obtida no endereço
eletrônico:http://www.nature.com/nature/journal/v423/n6941/fig_tab/nature01650_
F1.html.
[4] Kleist, W.; Maciejewski, M.; Baiker, A., ThermochimicaActa , 499(2010) 71.
[5] Tafipolsky, M.; Amirjalayer, S.; Schmid, R., Journal of Computational Chemistry,
28 (2007) 1167.
[6] Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.
M., Science, 295 (2002) 469.
[7] Pawsey, S.; Moudrakovski, I.; Ripmeester, J.; Wang, L. Q.; Exarhos, G. J.; Rowsell,
J. L. C, Yaghi, O. M., Journal Physical Chemical C, 111 (2007) 6060.
[8] Ye, B. H.; Tong, M. L.; Chen, X. M., Coordination Chemistry Reviews,249(2005)
545.
[9] Vilela, S. M. F., Síntese e Caracterização de Polímeros de Coordenação com
Lantanídeos. UNIVERSIDADE DE TRÁS-OS-MONTES E ALTO DOURO,
(2009).
[10] Zhu, L. C.; Zhao, Y.; Yu, S. J.; Zhao, M. M., Inorganic Chemistry
Communications, 13 (2010) 1299.
[11] Noro, S. I.; Kitagawa, S.; Akutagawa, T.; Nakamura, T., Progress in Polymer
Science, 34 (2009) 240.
[12] Ravon, U.; Savonnet, M.; Aguado, S.; Domine, M. E.; Janneau, E.; David, F.,
Microporous and Mesoporous Materials,129 (2010) 319.
[13] Alaerts, L.; Seguin, E.;Poelman, H.;Thibault-Starzyk, F.; Jacobs, P. A.; De Vos, D.
E.; Chemistry – A European Journal, 12 (2006) 7353.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
35
[14] Horcajada, P.; Surble, S.; Serre, C.; Hong, D. Y.; Seo, Y. K.; Chang, J. S..;
Greneche, J. M.; I, Margiolaki.; G, Ferey.; Chemical Communications, (2007)
2820.
[15] Liu, B.; Shioyama, H.; Jiang, H.; Zhang, X.; Xu, Q.; Carbon, 48 (2010) 456.
[16] Armelao, L.; Quici, S.; Barigelletti, F.; Accorsi, G.; Bottaro, G.; Cavazzini, M.;
Tondello, E., Coordination Chemistry Reviews, 254 (2010) 487.
[17] Weber, I. T.; de Melo, A, J. G.; de Melo Lucena, M. A.; Rodrigues, M. O.; Junior,
S. A., AnalyticalChemistry, 83 (2011) 4720.
[18] Xiao, B.; Wheatley, P. S.; Zhao, X.; Fletcher, A. J.; Fox, S.; Rossi, A. G .;
Megson, I. L.; Bordiga, S.; Regli, L.; Thomas, K. Mark.; Morris, R. E., Journal
American Chemistry Society,129 (2007) 1203.
[19] Huxford, R. C.; Rocca, J. D.; Lin, W., Current Opinion in Chemical Biology
(2010) 14262.
[20] Ma, Z.; Moulton, B., Coordination Chemistry Reviews, 255 (2011) 1623.
[21] O’ Keeffe, M., Materials Research Bulletin, 41 (2006) 911.
[22] Rodrigues, M. O.; Almeida Paz, F. A.; Freire, R. O.; de Sá, G. F.; Galembeck, A.;
Montenegro, M.C. B. S. M.; Araujo, A. N.; Alves-Jr, S., J. Phys. Chem. B, 113
(2009) 12181.
[23] Valenzano, L.; Civalleri, B.; Chavan, S.; Bordiga, S.; Nilsen, M. H.; Jakobsen, S.;
Lillerud, K. P.; Lamberti, C., Chemistry of Materials,23 (2011) 1700
[24] Grajciar, L.; Bludsky, O.; Nachtigall, P., Journal of Physical Chemistry Letters, 1
(2010) 3354.
[25] Grajciar, L.; Wiersum, A. D.; Llewellyn, P. L.; Chang, J. S.; Nachtigall, P., The
Journal of Physical Chemistry C, 115 (2011) 17925.
[26] Watanabe, T.; Sholl, D. S., The Journal of Chemical Physics, 133 (2010) 094509.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
36
33 MÉTODOS DE QUÍMICA
TEÓRICA E APLICAÇÕES
3.1- A Química computacional
O advento dos computadores possibilitou o crescimento de uma área que surgiu
timidamente e que tratava apenas pequenos sistemas, pois, nos primórdios da utilização de
métodos teóricos em computadores, estes possuíam baixo poder de processamento que
refletia em um grande tempo computacional. O fato da entrada de dados no sistema ser
feito através de cartões perfurados também era um fator limitante e que tornava o
procedimento muito complicado.
O desenvolvimento de computadores cada vez mais robustos, e com preços mais
acessíveis, bem como o desenvolvimento de métodos que podem ser aplicados a sistemas
cada vez maiores, tem contribuído para tornar a área da química teórica computacional cada

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
37
vez mais importante e de utilidade para solução e análise de fenômenos, com um baixo
custo financeiro e de tempo.
Em vista aos avanços no ramo da química teórica computacional nos deparamos,
hoje, com uma vasta gama de trabalhos utilizando os métodos de química teórica, de uma
forma que fica visível a importânciado uso dessas ferramentas para facilitar e fortalecer
nossas análises, obtenção de dados, e como forma de explanar resultados que, com o
desenvolvimento já citado, se tornam cada vez mais confiáveis.
3.2- Métodos de química teórica
No âmbito das simulações computacionais nos deparamos com vários métodos de
cálculo de estrutura eletrônica. A questão de qual método utilizar pode ser respondida com
outra questão: Que sistema e quais propriedades eu preciso necessários?
Métodos pós Hartree-Fock(HF) como MP2 (Teoria da Perturbação de Segunda
ordem, do inglês, SecondOrderMollerPlessetPertubationTheory) são extremamente
indicados para o estudo de pequenas moléculas cujas propriedades precisam ser preditas
com elevada exatidão. À medida que o sistema a ser estudado cresce precisamos fazer uso
de métodos menos exatos, mas com eficiência computacional maior.Sendo assim, sistemas
com dezenas ao até pouco mais de uma centena de átomos podem ser perfeitamente
estudados utilizando a teoria do funcional da densidade (DFT), apresentando ainda uma
elevada exatidão. Quando os sistemas de interesse possuem centenas ou até milhares de
átomos, os métodos semiempíricos, que são métodos químico-quânticos aproximados, são
uma excelente opção. A depender do método pode-se obter resultados com exatidão
bastante satisfatória. Por fim, quando é preciso tratar sistema contendo centenas de milhares
de átomos, os métodos quânticos não são mais factíveis, mesmo com o elevado poder de
processamento que grandes clusters de computadores apresentam hoje. Nestes casos é
indicado o uso de métodos clássicos que podem apresentar elevada exatidão quando a
propriedade de interesse é de natureza física. Contudo, tais métodos são incapazes de
estudar propriedades de natureza química.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
38
Dos métodos quânticos computacionais existentes, podemos citar os mais importantes
com relação a sua grande aplicação em trabalhos científicos, que são:
Hartree-Fock: O método Hartree-Fock possui grande exatidão sendo utilizado
puramente os postulados da mecânica quântica em sua abordagem matemática salvo
algumas aproximações para tornar o método aplicável [1]-[3].
DFT: A metodologia DFT usa a densidade eletrônica como a variável fundamental na
descrição de um sistema eletrônico [1]-[3].
Semiempírico: Os métodos semiempíricos são derivados do método Hartree-Fock-
Roothaan e possuem parâmetros obtidos por ajuste numérico ou derivados de
resultados experimentais para tornar mais rápidaa resolução de integrais que possuem
grande custo computacional [1]-[3].
3.2.1 - Método de Hartree-Fock
O método Hartree-Fock nos fornece boa solução para cálculos de estrutura
eletrônica em sistemas envolvendo um grande número de elétrons. Em sua abordagem são
feitas algumas restrições e aproximações matemáticas para a obtenção da solução da
equação de Schrödinger [1],[2]. Uma aproximação utilizada nestes métodos é inicialmente
desconsiderar os efeitos relativísticos que ocorrem com os elétrons e aplicar aproximação
de Born-Oppenheimer, que consiste em separar os movimentos dos núcleos dos
movimentos dos elétrons [1]-[5].
Sabendo que a massa dos elétrons é cerca de 1800 vezes menor que a dos prótons e
nêutrons, podemos esperar que a velocidade (movimento) dos elétrons seja muito maior
que o movimento do núcleo sendo assim podemos supor que os elétrons se movem num
campo de núcleos fixos [1],[2].
Da equação de Schrödinger temos: (3.1)

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
39
sendor as coordenadas dos elétrons e Ra coordenada dos núcleos. Da equação 3.1
temos o operador Hamiltoniano H em unidades atômicas, ou seja, a distância é dada em
raios Bohr e a energia em Hartree, que é descrito como:
(3.2)
Onde:
N é o número de elétrons
M é o número de núcleos
MA é a massa do núcleo
ZA é a carga do núcleo
Podemos escrever abreviadamente o Hamiltoniano:
H= TN + Te + VNe + Ve + VN (3.3)
A equação 3.2 pode ser dividida em cinco partes diferentes que são:
: Energia cinética nuclear (TN);
: Energia cinética dos elétrons (Te);
: Atração elétron-núcleo (VNe);
:Energia potencial de repulsão elétron-elétron (Ve);
: Energia potencial repulsiva núcleo-núcleo (VN).

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
40
Aplicando a aproximação de Born-Oppenheimer, ou seja, considerando os núcleos
fixos obtemos o Hamiltoniano total:
HT= Te + VNe + Ve + VN (3.4)
Que pode ser escrito como a soma do hamiltoniano eletrônico mais o operador de
energia potencial repulsiva núcleo-núcleo:
HT= Hele + VN (3.5)
Onde:
Hele=Te + VNe + Ve (3.6)
Da equação acima podemos dividi-la em 2 partes:
(3.7)
ondeO1 só contem operadores de 1 elétron:
(3.8)
sendo:
(3.9)
eO2o operador de 2 elétrons.
(3.10)
Não iremos abordar aqui todo manejo matemático do método Hartree-Fock, sendo
que o enfoque matemático anteriormente exposto é de fundamental importância no
tratamento e abordagem dos métodos utilizados neste trabalho, e também, se trata de uma

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
41
das aproximações fundamentais para o tratamento de sistemas por meio dos métodos
quânticos. A ideia central do método Hartree-Fock, que não podemos deixar de citar é a
combinação do princípio variacional com a descrição de um único determinante, conhecido
como determinante de Slater para as funções spins orbitais [1]-[5].
O método Hartree-Fock está dividido em:
Hartree-Fock Restrito (RHF do inglês, Restricted Hartree-Fock): utilizado em sistema
com número par de elétrons que possuem camada fechada, sendo feita restrição de que
em cada orbital existem dois elétrons com spins opostos [1].
Hartree-Fock não Restrito (UHF do inglês, Unrestricted Hartree-Fock): caracterizado
por não fazer restrição aos orbitais. É indicado para tratar sistemas de camadas abertas
[1].
Hartree-Fockrestrito de Camada Aberta (ROHF): nele, o tratamento da camada aberta é
feita considerando duas partes: uma relativa aos orbitais de camada fechada e a outra
relativa aos orbitais de camadas abertas. Também é indicado para o tratamento de
sistemas de camada fechada.
Os métodos de Hartree-Fock são indicados para o uso em sistemas pequenos, pois nos
deparamos com a relação custo computacional (tempo), sendo o método mais rigoroso na
descrição por meio de teoria quântica. A resolução de um grande número de integrais
requer um maior tempo computacional que é relativamente semelhante a descrição por
meio de Funcional da Densidade, mas que é muito maior que o tempo computacional
necessário para a descrição por meio do uso de métodos semiempíricos.
3.2.2 – Teoria do Funcional da Densidade
Um funcionalexiste quando uma função depende de outra função, neste caso, temos
a energia do sistema como sendo uma função que depende da densidade eletrônica. Este

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
42
método descreve as energias de um sistema no estado fundamental como um funcional da
densidade eletrônica.
A densidade eletrônica descreve o número de elétrons bem como o potencial
externo em consequência, obtemos o Hamiltoniano do sistema, que pode ser utilizado para
resolver a equação de Schrödinger. A solução da equação resultará em energia igual ou
superior a energia total do sistema.Esses teoremas foram propostos por Hohemberg-Kohn e
podem ser observados mais detalhadamente nas referências [1],[2],[6].
Uma diferença fundamental do método DFT para o Hartree-Fock esta na descrição
do Hamiltoniano eletrônico. Como visto anteriormente,na equação (3.3),o Hamiltoniano
eletrônico é constituído por 3 operadores.
Na descrição do DFT,o termo atração elétron-núcleo ganha uma nova roupagem
com a evolução do tratamento matemático, ganhando a forma de um funcional da
densidade eletrônica. No DFT não existe mais a função de spin, ou seja, agora o problema
só consiste nas 3 coordenadas espaciais.
Para o cálculo de estruturas precisamos fornecer no arquivo de entrada (a depender
do software)um funcional para o tratamento do sistema, existe uma grande gama de
funcionais que são utilizados em DFT, mas a construção dos funcionais não é de grande
importância em nosso trabalho e, portanto não será discutido aqui. O funcional que será
utilizado nesse trabalho será B3LYP que é o funcional mais comumente utilizado.
Cálculos DFT ganharam grande prestígio no meio científico por sua exatidão
quando se compara as propriedades preditas com os respectivos valores obtidos
experimentalmente. Tanto para o DFT como para o método Hartree-Fock, a boa descrição
do sistema depende do conjunto de bases escolhidas para o tratamento do sistema,
remontandonuma tarefa difícil para o iniciante na área de química computacional a escolha
desse conjunto.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
43
3.2.2.1 – Funções de Bases
As funções de bases estão juntas na ideia de que os orbitais moleculares são
formados pela combinação linear de orbitais atômicos que serão descritos e tratados por
funções de base.
Funções de bases são funções matemáticas que são usadas na descrição da função
de onda e as principais são do tipo Gaussianas (GTOs) e de Slater (STO).
Por exemplo, se quisermos representar o átomo de Si usando bases mínimas teremos
funções do tipo 1s, 2s, 2px, 2py, 2pz, 3s, 3px, 3py, 3pz para o de Li teremos 1s, 2s agora se
quisermos descrever um sistema constituído por uma molécula de O2, então teremos
funções do tipo 1s, 2s e 2px, 2py, 2pz, ou seja, a formação do orbital molecular será feito
pela combinação linear de 10 orbitais atômicos (10 funções de base) 10 de um oxigênio e
mais 10 do outro oxigênio [3],[6].
Para cada tipo de função temos um múltiplo para a sua descrição que é dada na
tabela 3.1:
Tabela 3.1. Tipos e quantidades de funções utilizadas na descrição de orbitais s, p, d e f.
Tipo de Função Numero de funções Esféricas Numero de funções Gaussianas
s 1 1
p 3 3
d 5 6
f 7 10
Conjunto de base é o termo que engloba as funções de base que serão usadas no
tratamento do sistema, que pode ser separada em caroço e camada de valência ou não, a
depender da nossa necessidade.
Os principais conjuntos de base são:
Base mínima: STO-nG, onde n=2, 3, 4, 5 ou 6;
Base Split Valence: n-21G, n=3,6 e n-31G com n=4, 5 e 6, no qual a camada de
valência é separada em2 partes a externa e a interna;

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
44
Base Triple Valence: n-311G, n=6,da qual a camada de valência é separada em 3
partes;
Base Double Zetae Triple Zeta: na qual todos os orbitais são separados.
Agora como exemplo digamos que se queira tratar a molécula de H2O usando o
conjunto de base 3-21G onde o numero 3 significa que o caroço será tratado com 3 funções
gaussianas (GTOs primitivas), o 2 representa o tratamento da camada interna de valência
que utilizará 2 funções gaussianas e 1 representa que iremos tratar a camada de valência
externa com 1 função gaussiana. Colocando em números de funções usadas temos para o
“O” funções 1s, βs, βpx, 2py, 2pz, para “H1” 1s e para “Hβ” 1s. [γ],[6].
Podemos estender as bases com o acréscimo do sinal “*” depois do “G” para a
inclusão de funções de polarização (d) se incluirmos um segundo “*” significa a inclusão
também de funções do tipo (p), podemos após o “G” escrever “**” ou (d, p) e tais funções
de polarização descrevem melhor as ligações, como em 6-31G** ou 6-31G (d, p). Também
podemos adicionar “+” antes do “G” para a inclusão de funções difusas tipo s e p para
átomos pesados, a inclusão de funções difusas é importante para a descrição de ligações
fracas e uma melhor descrição da camada de valência. O uso de “++” representa que são
incluídas gaussianas para todos os átomos pesados e também para os hidrogênio, como em
6-21++G. [3]
Sendo assim, podemos construir uma tabela com referência ao uso de diferentes
funções de base para comparar em que irá refletir a escolha das bases e suas extensões, com
referência à energia RHF para a nossa molécula de H2O.
Tabela 3.2. Aplicação de diferentes conjuntos de bases ao sistema H2O utilizando o Gaussian-03W.
Base usada Nº de Funções esféricas Nº Funções de Gaussianas Energia RHF
3-21G 13 21 -75.5859596632
6-21G 13 24 -75.8884340586
6-31G 13 30 -75.9853589489
6-31G* 19 36 -76.0107465000
6-31G** 25 42 -76.0236150192
6-31+G** 22 46 -76.0312305235
6-31++G** 31 48 -76.0313090366

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
45
Da tabela 3.2, temos que quanto maior o conjunto de bases utilizadas, menor a
energia encontrada para o sistema H2O no estado fundamental. Para o sistema estudado por
ser constituído por apenas 3 átomos com 10 elétrons no total, não encontramos diferenças
significativas referente ao tempo computacional, por este motivo os mesmos não expostos
na tabela, mas para sistemas maiores nos deparamos com um problema, quanto maior a
base maior o tempo computacional para a realização dos cálculos, visto que serão usados
um número bem maior de funções de base e de gaussianas.
Então, para a realização de cálculos do tipo HF e DFT, deve-se ficar atento para a
escolha de bases que sejam apropriadas para o tratamento do sistema de estudo, e também
ter o conhecimento do que elas irão diferenciar uma da outra nos cálculos.
3.2.3 – Métodos Semiempíricos
Há 60 anos atrás, os computadores estavam surgindo e era muito complicada a
execução de cálculos químico-quânticos. Na tentativa de poder estudar sistemas de
relevância, cientistas buscaram aproximar as metodologias quânticas existentes através do
ajuste de resultados teóricos a dados experimentais. Este procedimento foi utilizado com
objetivo de gerar parâmetros que pudessem substituir o cálculo de integrais complexas para
reduzir o tempo necessário para executar tais cálculos. Os métodos gerados a partir deste
procedimento ficaram conhecidos como métodos semiempíricos [1].
Já é de nosso entendimento que métodos como HF e DFT, apesar de descreverem
com grande precisão propriedades eletrônicas experimentais, possuem uma limitação com
respeito ao tamanho dos sistemas a serem calculados. À medida que as estruturas de análise
vão ficando maiores, o poder de processamento necessário para executar tais cálculos
cresce consideravelmente e mesmo com computadores de alto desempenho disponíveis
atualmente, sistemas com centenas de átomos podem consumir meses de processamento.
Os métodos semiempíricos surgiram numa época onde as tecnologias de hardwares
bem como seus algoritmos estavam “engatinhando”. Assim, os métodos semiempíricos

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
46
foram fruto do desejo de se obter um método que retratasse dados experimentais com pouca
margem de erro e, que necessitassem de um menor poder de processamento para serem
realizados, quando sistemas de interesse real fossem estudados.
Os primeiros métodos semiempíricos surgiram no início da década de 50. Tratava-
se de métodos qualitativos e semiquantitativos para orbitais moleculares π. A teoria PMO,
desenvolvida em 1952 por Dewarpode ser considerado o método mais importante deles[8].
Apesar da limitação dos computadores da época, a teoria PMO foi um sucesso e
pôde ser aplicada para um amplo grupo de sistemas. A sua precisão era limitada,contudo,
apresentava melhores resultados que outras metodologias da época.
Em seu processo evolutivo, os métodos semiempíricos foram cada vez mais se
sofisticando em relação às correções, aproximações e melhoras no processo de
parametrização, fato que os tornaram uma poderosa ferramenta na descrição por meio de
química quântica, de sistemas e propriedades com precisão, permitindo tratar
quanticamente sistemas com milhares de átomos [9], que é uma tarefa impossível de ser
realizada utilizando outras metodologias quânticas.
Neste contexto, além de poder ser aplicado para macromoléculas,formados por
milhares de átomos, os métodos semiempíricos também são candidatos naturais quando é
preciso calcular inúmeras estruturas ao mesmo tempo, ou ainda, no cálculo de várias
estruturas sequenciais como acontece no processo de design de novas moléculas, utilizando
achamada Química Teórica Combinatória. [10]
Os métodos semiempíricos tentam resolver de forma autoconsistente a equação de
Schöndinger contendo mais restrições matemáticas que em métodos ab initio, mas que são
corrigidas por meio do uso de parâmetros ajustáveis, que proporcionam a redução do custo
computacional [3].
A seguir, descreveremos características importantes dos métodos semiempíricos
usados no nosso trabalho, que são: AM1, PM3, PM6 e RM1.
3.2.3.1 – Método AM1

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
47
O método AM1 (do inglês, Austin Model1) foi desenvolvido de modo semelhante à
descrição matemática do seu antecessor MNDO (do inglês,
ModifiedNegectofDiatômicOverlap), que por sua vez, possuía uma tendência errônea em
descrever interações repulsivas núcleo-núcleo. Como exemplo, átomos distantes cerca 2 a
3Å, possuíam alta repulsão. Sendo assim, o método AM1 trouxe em sua formulação até 4
funções gaussianas esféricas por átomo, como meio de corrigir erros nas integrais de
repulsão caroço-caroço [1],[6],[7].
Uma importante evolução do método AM1 quando comparado com seus
antecessores foi a melhora na descrição da força das ligações de hidrogênio [7]. Os átomos
parametrizados para este método são apresentados na figura 3.1.
Figura 3.1. Elementos Parametrizados para o AM1. [11]
3.2.3.2 – Método PM3
A formulação matemática do método PM3 (do inglês, ParametricModel3) é
semelhante à usada na construção do método AM1.Uma das poucas diferenças está no
tratamento das integrais de repulsão caroço-caroço a qual foram acrescentadas até 2
funções gaussianas esféricas por átomo. Além disso, o processo de parametrização foi
muito mais aprimorado e contou com a inclusão de orbitais dna sua formulação [1],[3],[6].
Os átomos parametrizados para este método são:
Metais de Transição

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
48
Figura 3.2. Elementos parametrizados para o PM3. [11]
3.2.3.3 – Método PM6
O método semiempírico PM6 (do inglês, ParametricModel6) é um resultado da
modificação da parcela de interações caroço-caroço feita no método NDDO (do inglês,
NeglectofDiatomicDifferentialOverlap) que é antecessor do MNDO. O nome PM6 surgiu
de seus antecessores não publicados o PM4 e PM5 [12]. Ele possui uma grande gama de
átomos parametrizados (Figura 3.3).
Figura 3.3. Elementos parametrizados para o PM6. [11].
Como podemos observar comparando os elementos parametrizados para os métodos
AM1, PM3 e PM6, vemos a possibilidade de realizar estudos com metais de transição
utilizando o método PM6. Este fato é muito importante do ponto de vista do trabalho
apresentado nesta dissertação, pois uma boa parte das MOFs de interesse para o nosso
grupo contém metais de transição o que muitas vezes inviabiliza a escolha de um método
diferente do PM6 para tratar o sistema.
Metais de Transição
Metais de Transição

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
49
Uma importante novidade no procedimento de parametrização do método PM6 foi a
inclusão estrutura de cristais (inclusão de sistemas periódicos) no conjunto de
parametrização. Entretanto, algumas estruturas utilizadas no banco de dados foram obtidas
através de cálculos teóricos de DFT com o uso do funcional B3LYP [12].
3.2.3.4 – Método RM1
O método RM1 (do inglês, Recife Model1) foi desenvolvido a partir de uma parceria
do grupo de Química Teórica e Computacional da UFPE (Universidade Federal de
Pernambuco) com o desenvolvedor dos métodos AM1, PM3 e PM6 (métodos já
abordados), Prof. James J. P. Stewart. O RM1 possui a mesma formulação do método
AM1, contudo, foi gerado a partir de um procedimento de parametrização muito mais
elaborado [1],[3].
Embora esteja parametrizado apenas para 10 átomos, do ponto de vista biológico, os
elementos parametrizados no RM1 constituem a maioria das moléculas que são
responsáveis pela vida em nosso planeta. Em seu processo de parametrização, os
parâmetros atômicos foram ajustados considerando dados experimentais de calor de
formação, momento de dipolo, potencial de ionização e parâmetros geométricos como
distâncias e ângulos de ligação. O conjunto de parametrização contou com 1736 moléculas
representativas da bioquímica e química orgânica. [1].Os átomos parametrizados para este
método são apresentados na figura 3.4.
Figura 3.4. Elementos parametrizados para o RM1. [11]
Metais de Transição

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
50
3.3 – Descrevendo Cálculos em Fase Sólida para uma MOF
Uma das principais características das MOFs é possuir uma estrutura com uma
simetria regular repetitiva em todas as direções (sólido cristalino), diferente de um sólido
amorfo em que o arranjo se encontra desordenado. Por meio do uso da mecânica quântica
podemos descrever esse sistema e obter propriedades de interesse, mas para isso é preciso
fazer alguns tratamentos que tornem possível simplificar ao máximo esse sistema tão
complexo e de caráter estrutural infinito.
Os sólidos cristalinos podem ser classificados em, moleculares, reticulares,
metálicos e iônicos. As MOFs geralmente são sólidos moleculares (estrutura mantida por
forças interatômicas), sólidos reticulares (formado por ligações covalentes) ou ainda um
misto das duas.
Observando uma MOF a exemplo da MOF-5 (Figura 5), é possível observar que a
estrutura se repete regularmente, mas se separarmos o fragmento mínimo que se repete ao
longo da estrutura chegaremos à chamada célula unitária, tal que possui um empacotamento
cúbico no chamado retículo de Bravais.
Figura 3.5. Descrição espacial da MOF-5.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
51
Da figura acima temos os termos a, b e c que representam as dimensões e α, e
que representam os ângulos da célula unitária. No caso da MOF-5, os valores de a, b e c
são iguais e tem valor de 25.89 Å, α, e também possuem mesmo valor que é de 90°. O
softwareMOPAC não utiliza somente a célula unitária da estrutura a depender do caso
podemos utilizar um fragmento que pode ser a célula unitária duplicada, triplicada ou
maior, a depender da necessidade, e este fragmento é chamado de cluster.
3.3.1 – Critérios básicos para a escolha de um cluster.
Para a realização de cálculos semiempíricos em fase sólida, devemos escolher um
cluster grande o suficiente para que um orbital de um átomo que esteja no centro não
apresente sobreposição com átomos da extremidades do cluster ,em outras palavras,
teremos que ter um cluster em que os valores de a, b e c estejam entre 7 a 8 Å para
estruturas em que a união dos seus átomos sejam por ligações covalentes. Para estruturas
unidas por empilhamento π ou por outra interação fraca, recomenda-se a utilização de um
cluster com os valores de a, b e c da ordem de 15 a 20 Å.
Estes dados são introduzidos no MOPAC2009 por meio de vetores de translação
onde os valores de a, b, c, α, e estão incluídos nele. No processo de otimização da
estrutura os vetores de translação podem relaxar e geralmente seus valores se tornam
diferentes dos iniciais, a grandeza da mudança está associada ao método utilizado e ao
gradiente de normalização exigido no cálculo [11].
No capítulo a seguir, iremos abordar estruturas que chamaremos de “casos
especiais”, as quais, exemplificam o critério de escolha do cluster, por meio de resultados
com um cluster igual a célula unitária, e na sequência um cluster seguindo o ajustes dos
valores de a, b e c, conforme a interação inter-atômica presente na estrutura.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
52
REFERÊNCIAS
[ 1 ] Morgon,N. H..; Coutinho,K., Métodos de Química Teórica e Modelagem Molecular .
Editora Livraria de Física, São Paulo – 2007.
[ 2 ] Viana,J. D. M..;Fazzio,A.; Canuto,S., Teoria Quântica de Moléculas e Sólidos:
Simulação Computacional. Editora Livraria da Física, São Paulo – 2004.
[ 3 ] Trsic,M.; Pinto,M. F. S., Química Quântica: Fundamentos e Aplicações. Manole,
Barueri – 2009.
[ 4 ] Laschuk,E. F., Novo Formalismo Semi-Empírico para Cálculos Químico-
Quântico.Tese de Doutorado submetida na Universidade Federal do Rio Grande do Sul,
Porto Alegre – 2005
[ 5 ] Rocha,G. B.;Simas, A. M., Metodossemi-empíricos em química quântica:
formalismos, novos desenvolvimentos, aplicações recentes e perspectivas.
[ 6 ] Alcácer,L., Introdução à química quântica Computacional. IST Press, Lisboa – 2007
[ 7 ] Jensen F., Introduction to Computional Chemistry. John Wiley & Sons, London – 2001
[ 8] Dewar, M. J. S., J. Am. Chem. Soc., 74, (1952) 3341
[ 9 ] Anikin, N. A.; Anisimov, V. M.; Bugaenko, V. L.; Bobrikov, V. V.; Adreyev, A. M. J.
Chem. Phys. 121 (2004) 1266
[ 10 ] Freire, R.O.; Albuquerque, R.Q.; Alves Jr, S.; Rocha, G.B.; de Mesquita, M.E. Chem.
Phys. Lett.405 (2005) 123
[ 11 ] http://openmopac.net
[ 12 ] Stewart,J. J. P.,J Mol Model, 13 (2007) 1173.
[13] Yaghi, O. M.; O’Keeffe, M.; Ockwig, N. W.; Chae, H. K.; Eddaoudi, M.; Kim, J.,
Nature 423 (2003) 705 figura obtida no endereço eletrônico:
http://www.nature.com/nature/journal/v423/n6941/fig_tab/nature01650_F1.html.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
53
4 CÁLCULOS EM FASE SÓLIDA
4.1 – Métodos e Objetivos: Cálculos semiempíricos em fase
sólida
Neste capítulo apresentaremos um estudo sistemático com o objetivo avaliar a
capacidade de predição de estruturas de redes de coordenação através dos métodos
semiempíricos AM1[1], PM3 [2] e PM6[3] que são as metodologias mais utilizadas na
atualidade. Para tanto, utilizaremos a metodologia proposta pelo Prof. Stewart [4] para
cálculo de sólidos. Vimos no capítulo 3, que o método PM6, diferente dos métodos AM1 e
PM3,contou com sólidos no seu conjunto de parametrização. Além disso, para algumas
estruturas, apenas o PM6 poderá ser avaliado, uma vez que os métodos AM1 e PM3 não
foram parametrizados para todos os metais de transição.
Nesta etapa iremos usar como estrutura de entrada a célula unitária contida e
descrita pelo arquivo com extensão .cif, pois ao final da otimização, a estrutura obtida será
comparada com a cristalográfica. Todos os resultados de parâmetros de rede obtidos nas

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
54
otimizações estruturais para os métodos AM1, PM3 e PM6 estão apresentados no apêndice
1.
4.1.1 – Metodologia utilizada nos cálculos semiempíricos das
MOFs em fase sólida
Inicialmente foi realizada uma pesquisa na literatura buscando artigos publicados
nos últimos cinco anos que apresentam dados cristalográficos de redes de coordenação
contendo como centro metálico íons lantanídicos ou metais de transição. Geralmente
artigos com dados desta natureza depositam as estruturas cristalográficas no Cambridge
Crystalographic Data Centre (CCDC), e relatam na respectiva publicação o número de
deposição. De posse destes números solicitamos cada uma das estruturas no site
www.ccdc.cam.ac.uk/data_request/cif. Alguns trabalhos, apesar de relatar dados
cristalográficos não depositaram as respectivas estruturas no CCDC, o que inviabilizou a
obtenção das estruturas em questão. Contudo, através deste procedimento foi possível
coletar os dados cristalográficos de 45 redes de coordenação contendo metais de transição e
37 redes de coordenação contendo íons lantanídeos.
No arquivo .cif enviados pelo CCDC podemos encontrar a geometria da célula
unitária das redes de coordenação. O software Mercury [5]foi utilizado para visualizar cada
uma das estruturas. Em seguida, o programa GaussView versão 4.1 [6] foi utilizado para a
construção do arquivo de entrada (construção do cluster) para o MOPAC2009 [7]. Ainda
utilizando o GaussView editamos algumas poucas estruturas adicionando os hidrogênios de
moléculas de água coordenadas aos centros metálicos bem como apagado falhas em
estruturas que apareciam duplicadas no arquivo .cif.
Após preparar o arquivo de entrada para a realização do cálculo considerando
condições periódicas, editamos os inputs adicionando as seguintes palavras chave:
- AM1, PM3 ou PM6: método escolhido;
- MERS=(a,b,c): tamanho do fragmento, para uma célula unitária usa-se (1,1,1);
- GNORM=1: quando a gradiente for menor que 1 o cálculo termina;

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
55
- AUX= gera o arquivo contendo a geometria inicial e a final para visualização no
MERCURY.
Após o cálculo em fase sólida ter sido concluído para cada uma das redes de
coordenação utilizando os métodos AM1, PM3 e PM6 realizamos a comparação da
estrutura otimizada com a respectiva estrutura cristalográfica. Para tanto, utilizamos o
programa Cache versão 6.01 [8]que realiza a sobreposição das estruturas e calcula o RMS
(valor quadrático médio, do inglês root meansquare) para cada uma das estruturas
otimizadas (equação (4.1)). Para esse cálculo é necessário a escolha de três átomos da
estrutura para que o programa realize a sobreposição. Buscamos escolher átomos
localizados nos extremos das estruturas ou centros metálicos, nunca átomos de hidrogênio.
Os átomos escolhidos para o dado cristalográfico foram os mesmos escolhidos para as
estruturas otimizadas com os métodos AM1, PM3 e PM6.
4.2 – RESULTADOS E DISCUSSÕES.
4.2.1 – Avaliação da capacidade de predição dos métodos AM1,
PM3 e PM6 para o cálculo de redes de coordenação contendo os
metais Al, Zn, Pb, Hg e Ba.
Inicialmente vamos avaliar a capacidade de predição dos métodos AM1, PM3 e
PM6 para redes de coordenação que possuem centros metálicos de Al, Zn, Pb, Hg e Ba.
Usaremos como medida de comparação o RMS que é obtido por meio da sobreposição da
estrutura cristalográfica frente à estrutura otimizada. Após a obtenção do RMS para cada
estrutura, utilizamos a equação (4.1) para o cálculo do erro RMS médio (RMSM) para
redes de coordenação que possuem o mesmo centro metálico. O RMS médio nada mais é
que a média dos RMSs para MOFs com centros metálicos semelhantes.
( 4.1 )
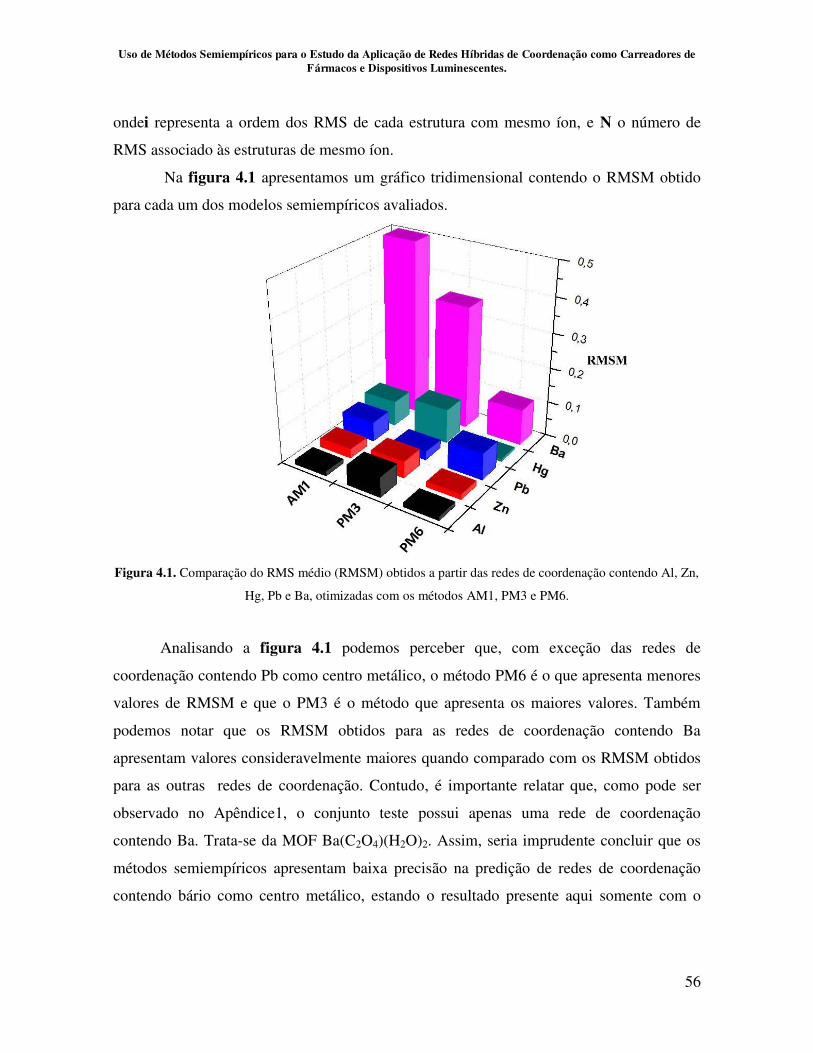
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
56
ondei representa a ordem dos RMS de cada estrutura com mesmo íon, e N o número de
RMS associado às estruturas de mesmo íon.
Na figura 4.1 apresentamos um gráfico tridimensional contendo o RMSM obtido
para cada um dos modelos semiempíricos avaliados.
Figura 4.1. Comparação do RMS médio (RMSM) obtidos a partir das redes de coordenação contendo Al, Zn,
Hg, Pb e Ba, otimizadas com os métodos AM1, PM3 e PM6.
Analisando a figura 4.1 podemos perceber que, com exceção das redes de
coordenação contendo Pb como centro metálico, o método PM6 é o que apresenta menores
valores de RMSM e que o PM3 é o método que apresenta os maiores valores. Também
podemos notar que os RMSM obtidos para as redes de coordenação contendo Ba
apresentam valores consideravelmente maiores quando comparado com os RMSM obtidos
para as outras redes de coordenação. Contudo, é importante relatar que, como pode ser
observado no Apêndice1, o conjunto teste possui apenas uma rede de coordenação
contendo Ba. Trata-se da MOF Ba(C2O4)(H2O)2. Assim, seria imprudente concluir que os
métodos semiempíricos apresentam baixa precisão na predição de redes de coordenação
contendo bário como centro metálico, estando o resultado presente aqui somente com o

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
57
objetivo de comparar os erros maiores e menores. Uma análise mais detalhada do RMSM
pode ser feita desconsiderando a rede de coordenação contendo bário (ver figura 4.2).
Figura 4.2. Comparação do RMS médio (RMSM) obtidos a partir das redes de coordenação contendo Al, Zn,
Hg e Pb, otimizadas com os métodos AM1, PM3 e PM6.
Analisando a Figura 4.2, é possível notar que o método PM6 apresentou um baixo
valor de RMSM para a rede de coordenação contendo Hg como centro metálico. Contudo,
assim como no caso do bário, o conjunto possui apenas uma rede de coordenação contendo
mercúrio como centro metálico, o que nos impede de concluir que o PM6 apresenta elevada
precisão na predição destaMOFs, lembrando que o número de estruturas foi fomentado
segundo a disposição das estruturas na literatura e que estavam disponíveis para solicitação.
Analisando a figura 4.2 também é possível confirmar que o método PM3 apresentou os
resultados não tão exatos, salvo para Pb,do qualeste método foi o melhor. O AM1
apresentou resultados inferiores ao PM6 para Hg e Zn mas para Al os dois métodos
apresentam poder de predição semelhante. Uma análise da tabela 4.1 deixa claro que o
PM6 apresenta o menor RMSM para as MOFs contendo Al como centro metálico.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
58
Contudo, mais uma vez, apenas uma MOF contendo Al como centro metálico foi
considerada.
Tabela 4.1. RMS médio (RMSM) obtidos a partir das redes de coordenação contendo Al, Zn, Hg e Pb,
otimizadas com os métodos AM1, PM3 e PM6.
Metal
Número de
MOFs
utilizadas
Erro RMS médio
AM1
PM3
PM6
Al 1 0,0136 0,0583 0,0125
Zn 9 0,0256 0,0471 0,0149
Pb 3 0,0562 0,0246 0,0762
Hg 1 0,0710 0,0983 0,0066
Ba 1 0,4994 0,3528 0,1017
Na tabela 4.1, observamos os resultados obtidos para RMSM e a quantidade de
MOFs utilizadas no conjunto teste considerado nesse estudo. Amostragem como a do Zn
(Zinco) com um total de 9 estruturas reforçam a eficácia do método PM6 e possibilita a
recomendação do método para o tratamento de MOFs contendo zinco, já para o Pb
(chumbo) com 3 estruturas, o PM6, foi o método que apresentou maiores valores de
RMSM, assim, para estudar redes de coordenação contendo Pb como centro metálico, é
mais recomendado utilizar o método PM3. Como podemos observar na tabela A1, contida
no Apêndice1, este método é o que apresenta menor RMS para duas das três redes de
coordenação contendo Pb avaliadas.
Devemos ressaltar o resultado obtido com o PM6 para a rede de coordenação
contendo Hg com erro de 0,0066, veremos à frente que esse valor é excelente e remonta
mínima diferença entre o cristalográfico e o teórico. Uma análise dos parâmetros de rede
apresentados na tabela A1 (Apêndice1) confirma este excelente resultado. Para a estrutura
cristalográfica os valores dos parâmetros a, b, c, α, e são, respectivamente, 14,47; 20,05;
8,74; 90,00; 95,31 e 90,00. Os valores obtidos com o PM6 foram, 14,49; 20,05; 8,78;
89,99; 95,14 e 89,96, respectivamente.
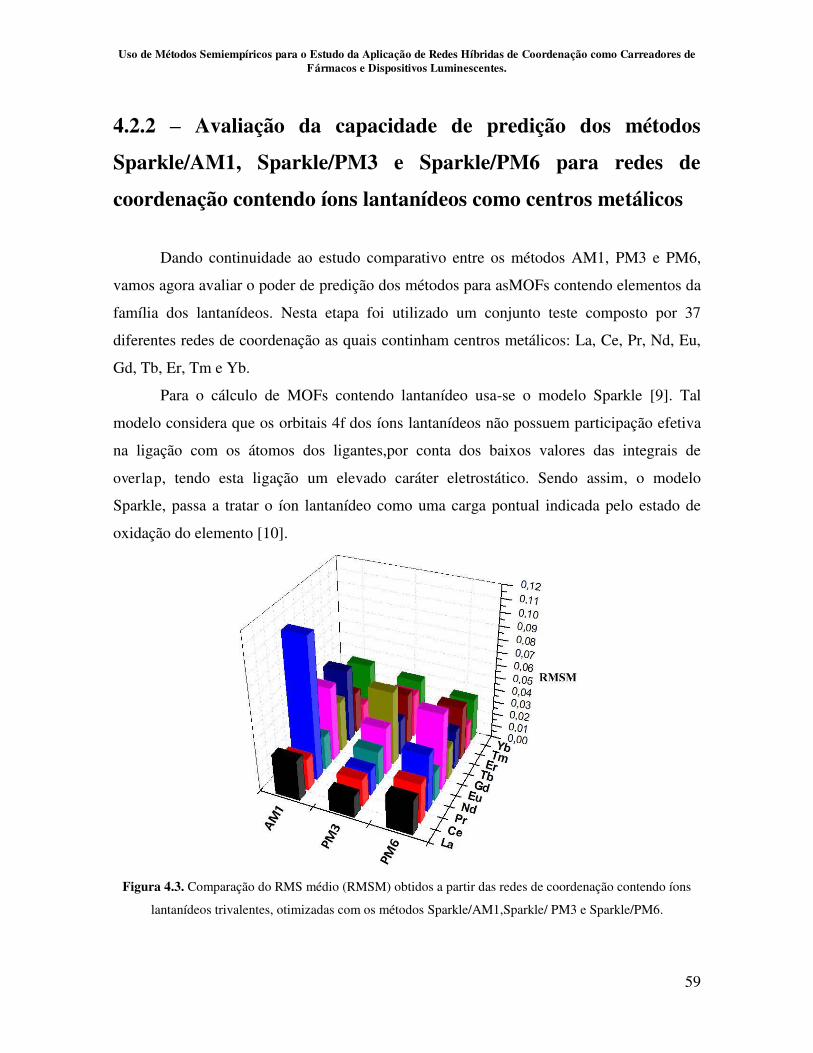
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
59
4.2.2 – Avaliação da capacidade de predição dos métodos
Sparkle/AM1, Sparkle/PM3 e Sparkle/PM6 para redes de
coordenação contendo íons lantanídeos como centros metálicos
Dando continuidade ao estudo comparativo entre os métodos AM1, PM3 e PM6,
vamos agora avaliar o poder de predição dos métodos para asMOFs contendo elementos da
família dos lantanídeos. Nesta etapa foi utilizado um conjunto teste composto por 37
diferentes redes de coordenação as quais continham centros metálicos: La, Ce, Pr, Nd, Eu,
Gd, Tb, Er, Tm e Yb.
Para o cálculo de MOFs contendo lantanídeo usa-se o modelo Sparkle [9]. Tal
modelo considera que os orbitais 4f dos íons lantanídeos não possuem participação efetiva
na ligação com os átomos dos ligantes,por conta dos baixos valores das integrais de
overlap, tendo esta ligação um elevado caráter eletrostático. Sendo assim, o modelo
Sparkle, passa a tratar o íon lantanídeo como uma carga pontual indicada pelo estado de
oxidação do elemento [10].
Figura 4.3. Comparação do RMS médio (RMSM) obtidos a partir das redes de coordenação contendo íons
lantanídeos trivalentes, otimizadas com os métodos Sparkle/AM1,Sparkle/ PM3 e Sparkle/PM6.
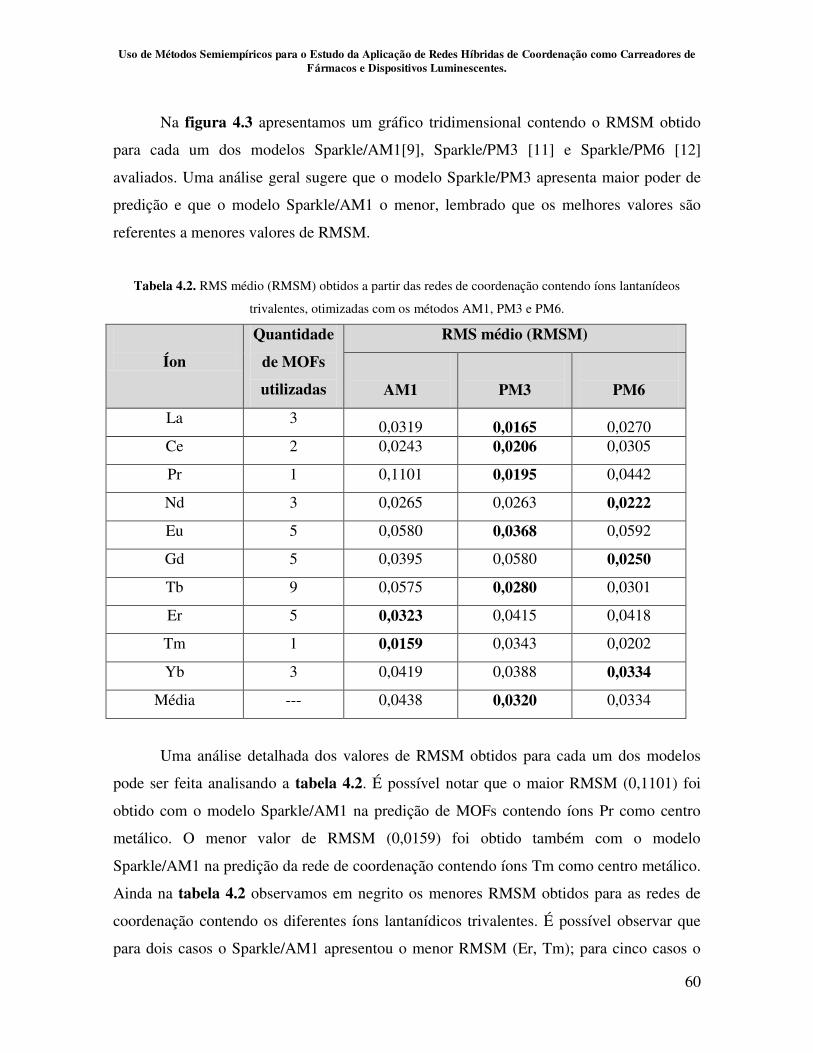
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
60
Na figura 4.3 apresentamos um gráfico tridimensional contendo o RMSM obtido
para cada um dos modelos Sparkle/AM1[9], Sparkle/PM3 [11] e Sparkle/PM6 [12]
avaliados. Uma análise geral sugere que o modelo Sparkle/PM3 apresenta maior poder de
predição e que o modelo Sparkle/AM1 o menor, lembrado que os melhores valores são
referentes a menores valores de RMSM.
Tabela 4.2. RMS médio (RMSM) obtidos a partir das redes de coordenação contendo íons lantanídeos
trivalentes, otimizadas com os métodos AM1, PM3 e PM6.
Íon
Quantidade
de MOFs
utilizadas
RMS médio (RMSM)
AM1
PM3
PM6
La 3 0,0319 0,0165 0,0270
Ce 2 0,0243 0,0206 0,0305
Pr 1 0,1101 0,0195 0,0442
Nd 3 0,0265 0,0263 0,0222
Eu 5 0,0580 0,0368 0,0592
Gd 5 0,0395 0,0580 0,0250
Tb 9 0,0575 0,0280 0,0301
Er 5 0,0323 0,0415 0,0418
Tm 1 0,0159 0,0343 0,0202
Yb 3 0,0419 0,0388 0,0334
Média --- 0,0438 0,0320 0,0334
Uma análise detalhada dos valores de RMSM obtidos para cada um dos modelos
pode ser feita analisando a tabela 4.2. É possível notar que o maior RMSM (0,1101) foi
obtido com o modelo Sparkle/AM1 na predição de MOFs contendo íons Pr como centro
metálico. O menor valor de RMSM (0,0159) foi obtido também com o modelo
Sparkle/AM1 na predição da rede de coordenação contendo íons Tm como centro metálico.
Ainda na tabela 4.2 observamos em negrito os menores RMSM obtidos para as redes de
coordenação contendo os diferentes íons lantanídicos trivalentes. É possível observar que
para dois casos o Sparkle/AM1 apresentou o menor RMSM (Er, Tm); para cinco casos o

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
61
Sparkle/PM3apresentou o menor RMSM (La, Ce, Pr, Eu, Tb) e para três casos o
Sparkle/PM6 apresentou o menor RMSM (Nd, Gd, Yb). Contudo, apesar do Sparkle/PM3
mostrar-se melhor para a maioria dos casos, analisando a média geral na última linha da
tabela 4.2podemos verificar que os modelos Sparkle/PM3 e Sparkle/PM6 apresentam
valores praticamente iguais (0,032 e 0,033 respectivamente). Assim podemos afirmar que
os dois modelos apresentam poder de predição semelhante.
Na tabela A2, presente no Apêndice1, podemos encontrar os valores de RMS
obtidos com os três modelos Sparkle para cada uma das 37 redes de coordenação
consideradas. Nesta tabela também está apresentado os valores cristalográficos e teóricos
dos seis parâmetros de rede de cada uma das MOFs bem como os valores cristalográficos e
teóricos das densidades.
Uma análise detalhada dos parâmetros de rede a, b, c, α, e contidos nesta tabela
confirma que os modelos Sparkle/PM3 e Sparkle/PM6 apresentam elevada precisão nas
predições das estruturas de redes de coordenação.
4.2.3 – Avaliação do método PM6 na predição de redes de
coordenação contendo centros metálicos do tipo: Mn, Fe, Co, Ni,
Cu, Y, Ag, Cd.
Nas secções anteriores analisamos a capacidade de predição dos métodos AM1,
PM3 e PM6 no cálculo de redes de coordenação contendo metais do tipo Al, Zn, Pb, Hg e
Ba ou contendo íons lantanídeos (modelo Sparkle). Contudo, os métodos AM1 e PM3 não
possuem parâmetros para a maioria dos metais de transição, assim nesta seção iremos
avaliar apenas o método PM6, por ser o único que contem parâmetros para os metais Mn,
Fe, Co, Ni, Cu, Y, Ag e Cd.
Embora a homepage do MOPAC [13] informe que tanto o método AM1 quanto o
PM3 encontra-se parametrizado para o cádmio (Cd), por algum motivo, os parâmetros não
estão implementados no software MOPAC versões 2007 e 2009. Por esta razão, apenas o
método PM6 foi considerado.
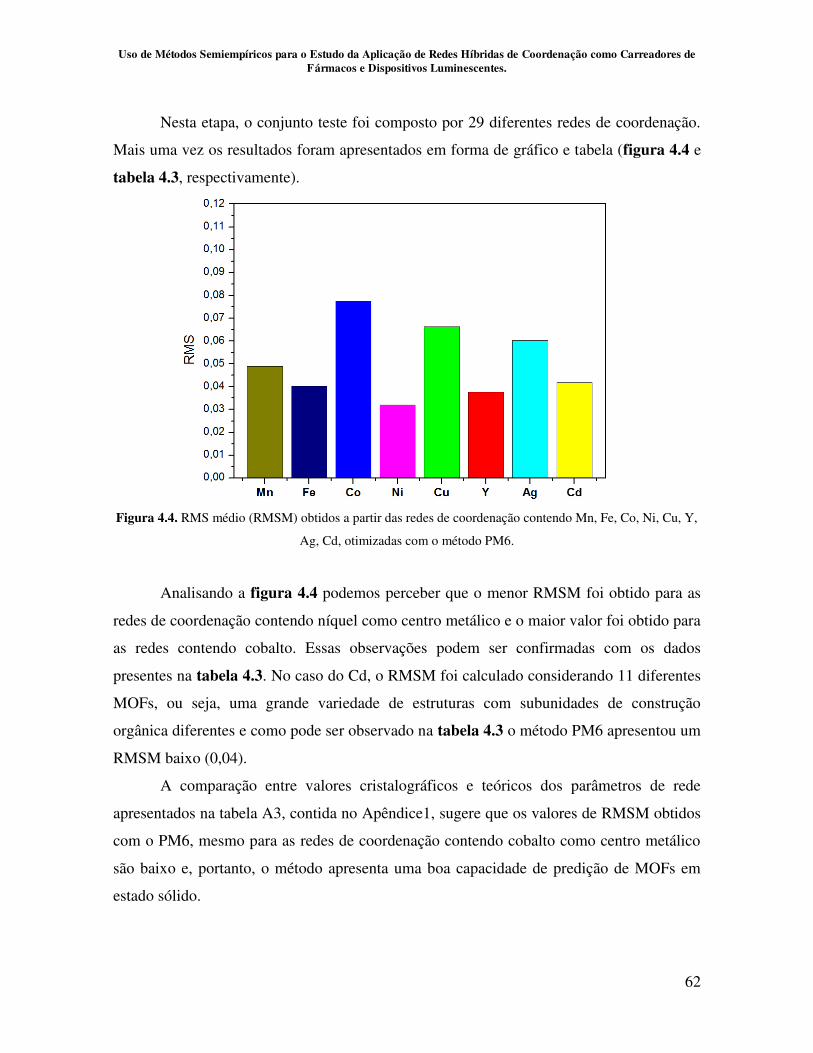
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
62
Nesta etapa, o conjunto teste foi composto por 29 diferentes redes de coordenação.
Mais uma vez os resultados foram apresentados em forma de gráfico e tabela (figura 4.4 e
tabela 4.3, respectivamente).
Figura 4.4. RMS médio (RMSM) obtidos a partir das redes de coordenação contendo Mn, Fe, Co, Ni, Cu, Y,
Ag, Cd, otimizadas com o método PM6.
Analisando a figura 4.4 podemos perceber que o menor RMSM foi obtido para as
redes de coordenação contendo níquel como centro metálico e o maior valor foi obtido para
as redes contendo cobalto. Essas observações podem ser confirmadas com os dados
presentes na tabela 4.3. No caso do Cd, o RMSM foi calculado considerando 11 diferentes
MOFs, ou seja, uma grande variedade de estruturas com subunidades de construção
orgânica diferentes e como pode ser observado na tabela 4.3 o método PM6 apresentou um
RMSM baixo (0,04).
A comparação entre valores cristalográficos e teóricos dos parâmetros de rede
apresentados na tabela A3, contida no Apêndice1, sugere que os valores de RMSM obtidos
com o PM6, mesmo para as redes de coordenação contendo cobalto como centro metálico
são baixo e, portanto, o método apresenta uma boa capacidade de predição de MOFs em
estado sólido.
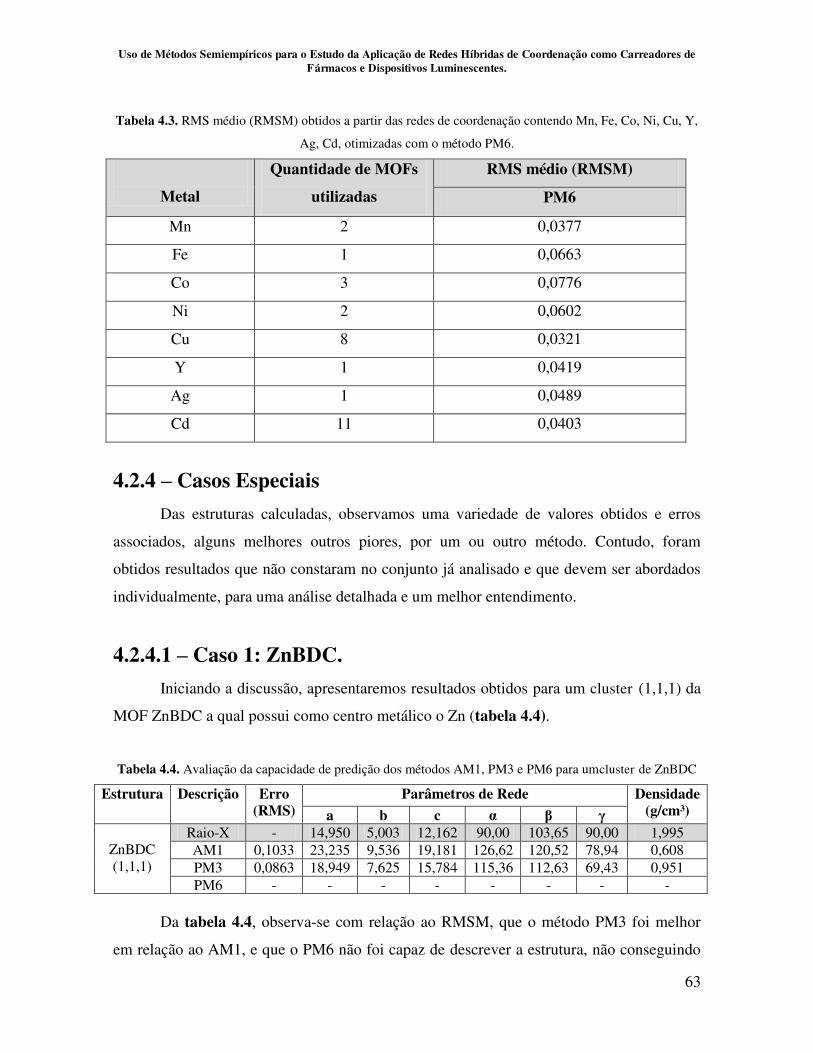
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
63
Tabela 4.3. RMS médio (RMSM) obtidos a partir das redes de coordenação contendo Mn, Fe, Co, Ni, Cu, Y,
Ag, Cd, otimizadas com o método PM6.
Metal
Quantidade de MOFs
utilizadas
RMS médio (RMSM)
PM6
Mn 2 0,0377
Fe 1 0,0663
Co 3 0,0776
Ni 2 0,0602
Cu 8 0,0321
Y 1 0,0419
Ag 1 0,0489
Cd 11 0,0403
4.2.4 – Casos Especiais
Das estruturas calculadas, observamos uma variedade de valores obtidos e erros
associados, alguns melhores outros piores, por um ou outro método. Contudo, foram
obtidos resultados que não constaram no conjunto já analisado e que devem ser abordados
individualmente, para uma análise detalhada e um melhor entendimento.
4.2.4.1 – Caso 1: ZnBDC.
Iniciando a discussão, apresentaremos resultados obtidos para um cluster (1,1,1) da
MOF ZnBDC a qual possui como centro metálico o Zn (tabela 4.4).
Tabela 4.4. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 para umcluster de ZnBDC
Estrutura
Descrição Erro (RMS)
Parâmetros de Rede Densidade (g/cm³) a b c α
ZnBDC (1,1,1)
Raio-X - 14,950 5,003 12,162 90,00 103,65 90,00 1,995 AM1 0,1033 23,235 9,536 19,181 126,62 120,52 78,94 0,608 PM3 0,0863 18,949 7,625 15,784 115,36 112,63 69,43 0,951 PM6 - - - - - - - -
Da tabela 4.4, observa-se com relação ao RMSM, que o método PM3 foi melhor
em relação ao AM1, e que o PM6 não foi capaz de descrever a estrutura, não conseguindo
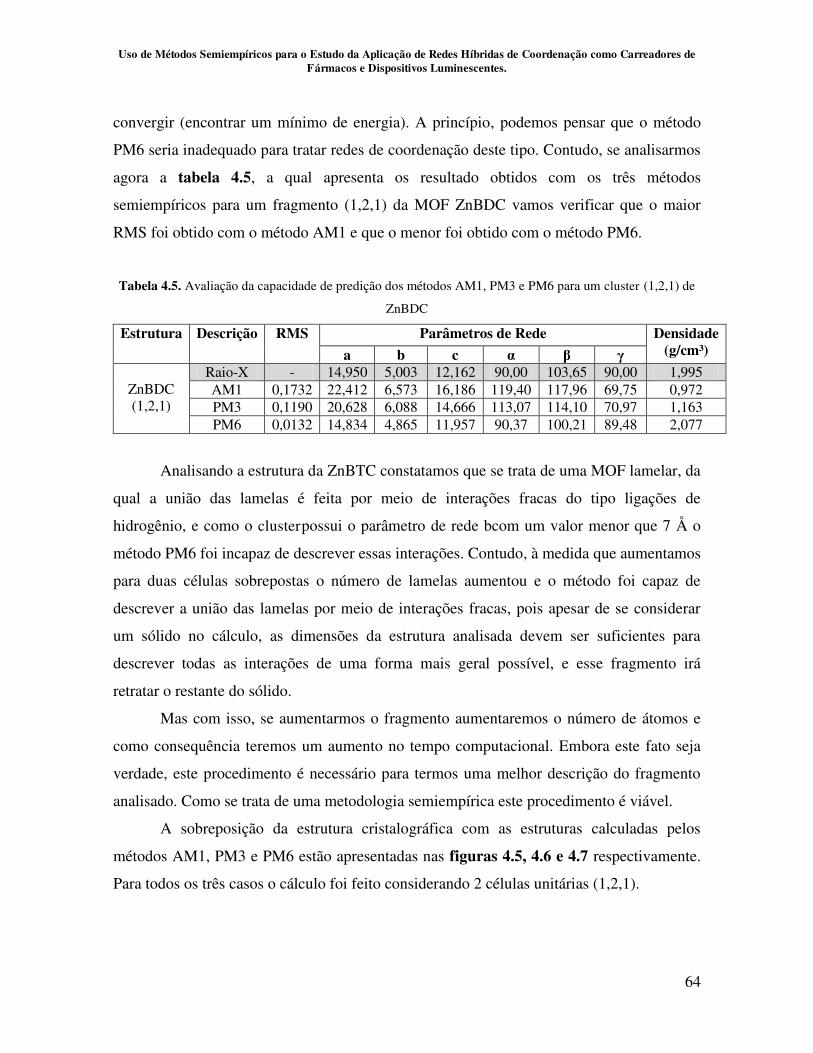
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
64
convergir (encontrar um mínimo de energia). A princípio, podemos pensar que o método
PM6 seria inadequado para tratar redes de coordenação deste tipo. Contudo, se analisarmos
agora a tabela 4.5, a qual apresenta os resultado obtidos com os três métodos
semiempíricos para um fragmento (1,2,1) da MOF ZnBDC vamos verificar que o maior
RMS foi obtido com o método AM1 e que o menor foi obtido com o método PM6.
Tabela 4.5. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 para um cluster (1,2,1) de
ZnBDC
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
ZnBDC (1,2,1)
Raio-X - 14,950 5,003 12,162 90,00 103,65 90,00 1,995 AM1 0,1732 22,412 6,573 16,186 119,40 117,96 69,75 0,972 PM3 0,1190 20,628 6,088 14,666 113,07 114,10 70,97 1,163 PM6 0,0132 14,834 4,865 11,957 90,37 100,21 89,48 2,077
Analisando a estrutura da ZnBTC constatamos que se trata de uma MOF lamelar, da
qual a união das lamelas é feita por meio de interações fracas do tipo ligações de
hidrogênio, e como o clusterpossui o parâmetro de rede bcom um valor menor que 7 Å o
método PM6 foi incapaz de descrever essas interações. Contudo, à medida que aumentamos
para duas células sobrepostas o número de lamelas aumentou e o método foi capaz de
descrever a união das lamelas por meio de interações fracas, pois apesar de se considerar
um sólido no cálculo, as dimensões da estrutura analisada devem ser suficientes para
descrever todas as interações de uma forma mais geral possível, e esse fragmento irá
retratar o restante do sólido.
Mas com isso, se aumentarmos o fragmento aumentaremos o número de átomos e
como consequência teremos um aumento no tempo computacional. Embora este fato seja
verdade, este procedimento é necessário para termos uma melhor descrição do fragmento
analisado. Como se trata de uma metodologia semiempírica este procedimento é viável.
A sobreposição da estrutura cristalográfica com as estruturas calculadas pelos
métodos AM1, PM3 e PM6 estão apresentadas nas figuras 4.5, 4.6 e 4.7 respectivamente.
Para todos os três casos o cálculo foi feito considerando 2 células unitárias (1,2,1).
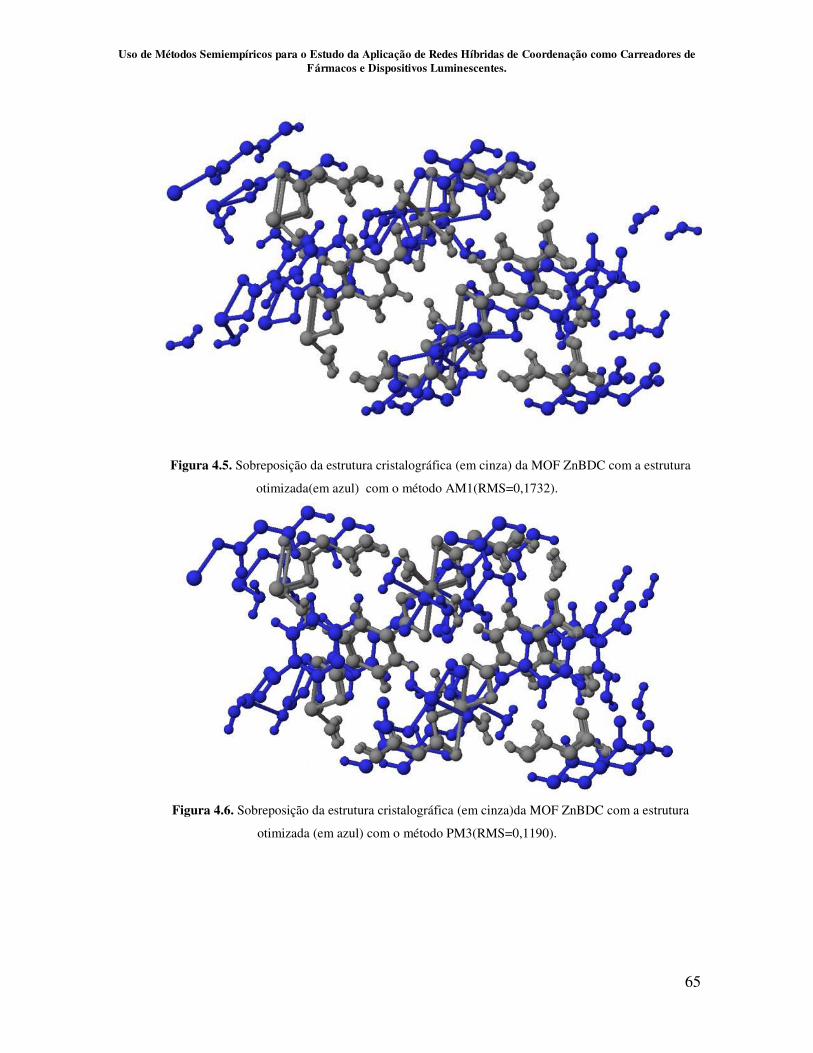
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
65
Figura 4.5. Sobreposição da estrutura cristalográfica (em cinza) da MOF ZnBDC com a estrutura
otimizada(em azul) com o método AM1(RMS=0,1732).
Figura 4.6. Sobreposição da estrutura cristalográfica (em cinza)da MOF ZnBDC com a estrutura
otimizada (em azul) com o método PM3(RMS=0,1190).

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
66
Figura 4.7. Sobreposição da estrutura cristalográfica (em cinza) da MOF ZnBDC com a estrutura
otimizada (em azul) com o método PM6(RMS=0,0132).
Com o auxílio das figuras 4.5, 4.6 e 4.7, podemos observar as distorções e ter uma
noção de quanto que os valores RMS retratam o comparativo entre a estrutura
cristalográfica e a estrutura calculada. Para esta MOF que possui ligações fracas unindo
seus fragmentos, temos que valores de RMS acima de 0,15 sugerem grandes distorções na
estrutura calculada e que as ligações de hidrogênio que unem os fragmentos não são
retratadas pelo cálculo. Para valores próximos de 0,10 (caso do PM3) observou-se que ele
descreveu bem ligações de hidrogênios para as águas que estavam coordenadas ao zinco
contido na célula unitária. Contudo, as moléculas de água que se coordenam ao zinco do
fragmento vizinho (água da interface) não tiveram suas ligações de hidrogênio bem
descritas o que resultou em uma grande distorção do fragmento.
O PM6 foi capaz de descrever fielmente as ligações hidrogênios e não foram
observadasgrandes distorções em sua estrutura. Assim, podemos afirmar que o PM6
mostrou-se o melhor método para a descrição e realização de estudos com a MOF ZnBDC.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
67
4.2.4.2 – Caso 2:[Ba(C2O4)0.5(HC2O4)(H2O)]
Novamente iniciaremos expondo os resultados obtidos com um cluster (1,1,1) da
MOF [Ba(C2O4)0.5(HC2O4)(H2O)] (tabela 4.6). Neste caso temos o bário como centro
metálico[14].
Tabela 4.6. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 docluster(1,1,1) da MOF
[Ba(C2O4)0.5(HC2O4)(H2O)].
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
(1,1,1)
Raio-X - 5,731 18,033 6,577 90,00 98,04 90,00 2,846 AM1 - - - - - - - - PM3 0,2866 15,475 21,898 13,591 85,35 82,23 86,17 0,422 PM6 0,2810 7,217 9,356 7,949 104,77 64,98 93,84 4,077
Analisando os dados contidos na tabela 4.6, vemos que o método AM1 foi incapaz
de descrever a estrutura para o cluster(uma célula unitária) e que os métodos PM3 e PM6
chegaram a resultados com RMS praticamente iguais. Basicamente a diferença entre as
estruturas obtidas com os métodos PM3 e PM6 foi que o PM3 expandiu a estrutura (como
pode ser visto pelo valor de sua densidade) gerando átomos de bário isolados e várias
moléculas de CO2 provenientes de carbonos e oxigênios que antes faziam parte dos
ligantes, e o PM6 fez o contrário, pois comprimiu a estrutura da MOF (densidade 4,077
g/cm³).
Na tabela 4.7 apresentamos os resultados obtidos para o cálculo de um fragmento
com quatro células unitárias (2,1,2). Analisando os resultados é possível notar que o AM1
chegou a um resultado melhor que o PM3. Mais uma vez o PM3 expandiu o fragmento da
MOF e o RMS obtido foi semelhante ao obtido para o fragmento anterior (ver tabela 4.6).
Analisando o resultado obtido com o método PM6 observa-se que novamente a estrutura da
MOF foi comprimida entretanto o RMS obtido para a otimização deste fragmento foi de
0,0883, ou seja, consideravelmente menor que o obtido na otimização do fragmento
composto por apenas uma célula unitária.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
68
Tabela 4.7. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 docluster(2,1,2) da MOF
[Ba(C2O4)0.5(HC2O4)(H2O)].
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
(2,1,2)
Raio-X - 11,463 18,033 13,154 90,00 98,05 90,00 2,846 AM1 0,1381 13,244 18,427 13,924 75,08 93,91 77,70 2,409 PM3 0,2813 15,772 38,515 36,664 78,98 94,98 83,69 0,355 PM6 0,0883 10,069 18,125 10,220 91,01 107,59 85,65 4,322
Figura 4.8. Sobreposição da estrutura cristalográfica (em cinza) da MOF
[Ba(C2O4)0.5(HC2O4)(H2O)] com a estrutura otimizada (em azul) com o método PM6(RMS=0,0883)
Observando a figura 4.8, podemos ter a noção de quanto uma estrutura que
apresenta RMS próximo a 0,09 se distorce em relação à cristalográfica. Observa-se que a
mesma contem ligações de hidrogênio com comprimento de 1,903 Å, as moléculas de água

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
69
desta MOF fazem duas interações (ligações) distintas, sendo elas uma ligação de
hidrogênio com um oxigênio do ligante orgânico e a outra uma ligação coordenada com íon
metálico. Podemos notar que o método PM6, quando empregado para calcular o fragmento
(2,1,2), conseguiu descrever as ligações entre as moléculas de água coordenadas com o
centro metálico.Contudo, as moléculas de água passaram a coordenar com dois centros
metálicos diferentes resultando em uma compressão do fragmento da MOF. As ligações de
hidrogênio não foram descritas pelo método PM6 que apresentou hidrogênios soltos, tais
que antes estavam ligados a átomos de oxigênio
4.2.4.3 – Caso 3: Elemento Sr (Estrôncio)
Para o terceiro caso, analisaremos duas MOFs contendo Sr (estrôncio - elemento
que ainda não foi avaliado). A primeira possui fórmula estrutural Sr[C2H4(SO3)2] [15].
Nesta MOF átomos de enxofre encontram-se ligados a três diferentes átomos de oxigênio e
formando um conjunto em ressonância com três metais.
Tabela 4.8. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 do cluster(1,1,1) da MOF
Sr[C2H4(SO3)2].
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
(1,1,1)
Raio-X - 8,318 5,442 14,978 90,00 93,63 90,00 2,707 AM1 - - - - - - - - PM3 - - - - - - - - PM6 0,0703 7,476 5,335 16,341 90,03 89,28 90,07 2,811
Observando a tabela 4.8 é possível notar que as estruturas calculadas com os
métodos AM1 e PM3 não convergiram. O PM6 descreveu o sistema com um RMS de
0,0703, e para termos noção de quanto um valor de RMS deste reflete na estrutura podemos
observar a sobreposição apresentada na figura 4.9.
Analisando a figura 4.9 podemos notar que o método PM6 conseguiu descrever
muito bem as ligações, mais houve algumas distorções nos oxigênios e hidrogênios que
fazem ligações com as células unitárias vizinhas (não explícitas no cálculo). De forma
geral, o método conseguiu predizer muito bem a estrutura.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
70
Figura 4.9. Sobreposição da estrutura cristalográfica (em cinza) da MOF Sr[C2H4(SO3)2] com a
estrutura otimizada (em azul) com o método PM6 (Sr).
Agora vamos apresentar os resultados obtidos para um fragmento maior (1,2,1)
(b=10,884). Os valores da tabela 4.9 são correspondentes a esse fragmento.
Tabela 4.9. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 dada em valores da célula
unitária,para (1,2,1) da MOF Sr[C2H4(SO3)2].
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
(1,2,1)
Raio-X - 8,318 5,442 14,978 90,00 90,00 90,00 2,707 AM1 - - - - - - - - PM3 0,1045 7,831 5,149 18,331 89,38 94,78 90,55 2,487 PM6 0,0216 7,571 5,298 15,561 89,79 93,88 90,00 2,942
Analisando os dados presentes na tabela 4.9, observamos que novamente o AM1 foi
incapaz de descrever a estrutura da MOF contendo estrôncio. Embora a estrutura calculada
com o PM3 tenha convergido o RMS obtido foi maior que 0,1 sugerindo grandes distorções
quando comparamos a estrutura otimizada com a estrutura cristalográfica.
Mais uma vez o método PM6 mostrou-se o mais exato. Comparando os resultados
obtidos para o fragmento (1,1,1) (tabela 4.8) com os resultados obtidos para o fragmento
(1,2,1) (tabela 4.9) observamos que o RMS foi reduzido de 0,0703 para 0,0216. Na figura

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
71
4.10 podemos observar a sobreposição da estrutura cristalográfica com a estrutura calculada
com o método PM6. Fica evidente o excelente resultado obtido pelo método PM6.
Figura 4.10. Sobreposição da estrutura cristalográfica (em cinza) da MOF Sr[C2H4(SO3)2] com a estrutura
otimizada (em azul) com o método PM6 – fragmento (1,2,1),(RMS=0,0216)
Agora vamos analisar a segunda MOF contendo Sr que é a [Sr2(1,3-
pdta)(H2O)6].H2O, (pdta=propanediaminetetraacetate) [16]. Os resultados obtidos estão
apresentados na tabela 4.10. É possível notar que a estrutura calculada com o método AM1
não convergiu. Mais uma vez o PM6 foi o método que apresentou o menor valor de RMS.
Tabela 4.10. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 da célula unitária (1,1,1)
da MOF [Sr2(1,3-pdta)(H2O)6].H2O
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
Sr2 (1,1,1)
Raio-X - 11,983 13,635 13,767 90,00 90,00 90,00 1,782 AM1 - - - - - - - - PM3 0,0776 18,354 19,736 13,998 86,67 93,54 94,93 0,796 PM6 0,0402 11,023 13,277 14,117 90,86 95,17 94,40 1,955

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
72
4.2.4.4 – Caso 4:Ga(OH)(BPDA).
Agora iremos avaliar a descrição dos métodos para redes de coordenação contendo
para outro elemento ainda não avaliado, o Ga (Gálio). Na tabela 4.11 apresentamos os
resultados obtidos com a célula unitária. A MOF em questão é a Ga(OH)(BPDA) (BPDA=
C14O4H8)[17].
Tabela 4.11. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 da célula unitária (1,1,1)
da MOF Ga(OH)(BPDA).
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
(1,1,1)
Raio-X - 18,006 6,769 10,957 90,00 92,32 90,00 1,628 AM1 0,0604 19,201 7,015 11,145 91,75 98,61 88,07 1,458 PM3 - - - - - - - - PM6 0,0027 17,954 6,940 10,958 90,03 91,85 90,02 1,591
Na tabela 4.11 observa-se que a estrutura calculada com o método PM3 não
convergiu. A estrutura obtida com o método AM1 apresentou RMS de 0,0604 e a
sobreposição da estrutura AM1 com a estrutura cristalográfica pode ser observada na
figura 4.11.
Figura 4.11. Sobreposição da estrutura cristalográfica (em cinza) da MOF Ga(OH)(BPDA) com a
estrutura otimizada(em azul) com o método AM1.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
73
A figura 4.11 mostra que valores de RMS próximos a 0,0604 remonta pequenas
distorções nas subunidades de construção orgânica, pequenas distorções que ao longo da
estrutura causa desacordos visuais na sobreposição, mas que ainda torna um resultado
muito bom, pois como trata-se de um resultado obtido com uma metodologia semiempírica,
requer um tempo computacional muito curto comparado com um cálculo DFT, por
exemplo.
Figura 4.12. Sobreposição da estrutura cristalográfica (em cinza) da MOF Ga(OH)(BPDA) com a
estrutura otimizada (em azul) com o método PM6.
A figura 4.12 mostra a sobreposição da estrutura cristalográfica com a estrutura
otimizada com o método PM6. É possível notar a elevada capacidade de predição do
método PM6 para esta estrutura, sendo o melhor resultado obtido entre todas as MOFs
analisadas neste estudo (ver tabelas apresentadas no apêndice 1).
4.2.4.5 – Caso 5: Mg.
Rede de coordenação contendo Mg como centro metálico, ainda não avaliada até
este momento, será avaliada agora. A MOF Mg(C10H16O4)2(H2O)2 (Bis-aqua-
sebacatomagnesium) [18] será utilizada para isso.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
74
Os resultados obtidos estão apresentados na tabela 4.12. Observa-se que também,
neste caso, a estrutura otimizada com o método PM3 não convergiu. O PM6 apresentou o
menor valor de RMS, contudo o valor encontrado para a estrutura AM1 foi muito próximo.
Outro ponto importante a ser destacado é que a estrutura obtida com o método PM6
apresenta uma densidade maior que o valor da estrutura cristalográfica.
Tabela 4.12. Avaliação da capacidade de predição dos métodos AM1, PM3 e PM6 do cluster (1,1,1)
da MOF Mg(C10H16O4)2(H2O)2.
Estrutura
Descrição RMS Parâmetros de Rede Densidade (g/cm³) a b c α
ZnBDC (1,1,1)
Raio-X - 17,336 7,456 9,573 90,00 96,43 90,00 1,386 AM1 0,0293 21,260 7,570 9,740 90,39 102,90 89,56 1,115 PM3 - - - - - - - - PM6 0,0172 15,656 7,674 10,049 91,56 96,42 102,35 1,456
Na figura 4.13, podemos observar a sobreposição da estrutura cristalográfica com a
estrutura otimizada com o método PM6. Mais uma vez fica evidente a elevada exatidão do
método em predizer estruturas cristalográficas de redes de coordenação.
Figura 4.13. Sobreposição da estrutura cristalográfica (em cinza) da MOF Mg(C10H16O4)2(H2O)2 com a
estrutura otimizada (em azul) com o método PM6.
Os resultados analisados, demonstram o quanto as estruturas se destorcem em
comparativo com o método utilizado, e também, a importância da escolha do cluster. Para

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
75
ligações covalente um cluster pequeno proporciona resultados satisfatórios, já para uma
estrutura que contenham interações mais fracas, a escolha de um cluster maior vê-se
necessária.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
76
REFERÊNCIAS
[1] Dewar, M.J.S.; Zoebish, E.G.; Healy, E.F.; e Stewart, J.J.P., J. Am. Chem. Soc,107
(1985) 3902.
[2] Stewart, J.J.P., J. Comp. Chem,10(1989)209-220.
[3] Stewart, J.J.P., J. Mol. Model,13(2007)1173.
[4] Stewart J. J. P.., J. Mol. Model,14 (2008) 499
[5] Mercury CSD 2.0 - New Features for the Visualization and Investigation of Crystal
Structures, C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E.
Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl.
Cryst., 41 (2008) 466.
[6] GaussView 4.1; Gaussian, Inc., 340 Quinnipiac St Bldg 40 Wallingford, CT 06492 USA, 2006 [7] Stewart; J.J.P. MOPAC2009, Stewart Computational Chemistry, Colorado
Springs, USA, 2009
[8] CAChe 6.0—Fujitsu, Ltd, Chiba, Japan.
[9] Freire,R. O..;Rocha,G. B.; Simas,A. M., InorganicChemistry, 44 (2005) 3299
[10] de Andrade,A. V. M.; da Costa Jr,N. B.; Simas,A. M..;de Sá,G, F., Chem. Phys. Lett
227 (1994) 349.
[11]Freire,R. O.; Rocha,G. B.; Simas,A. M., journal of the Brazilian Chemical Society 20
(2009) 1638
[12] Freire,R.O.; Simas,A. M., Journal of Chemical Theory and Computational 13 (2010)
2019
[13] www.openmopac.netacessado no dia 29 de maio de 2011.
[14] Borel,C.; Grazzali, M.; Langer,V.; Öhrström,L,.Inorganic Chemistry Communications,
12 (2009) 105
[15] Salami, T. O.; Patterson,S.N,.;Jones, V, D.; Masello, A.; Abboud,K, A,. Inorganic
Chemistry Communications, 12 (2009) 1150.
[16] Rychlewska,U.; Warzajtis,B.; Radanovic,D. D.; Draskovic,N. S.; Stanojevic,I. M.;
Djuran,M. I, ,. Polyhedron 30 (2011) 983.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
77
[17] Zhou,G.; Yang,Y.; Fan,R,.Inorganic Chemistry Communications 16 (2012) 17.
[18] Mesbah, A.; Aranda, L.; Steinmetz, J.;Rocca,E.; Françõis,M,. SolidStateSciences, 13
(2011) 1438.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
78
55 APLICAÇÃO DE MOFs COMO
CARREADORES DE FÁRMACOS
5.1 – Introdução e Objetivos
Neste capítulo utilizaremos ferramentas teóricas para estudar a interação hóspede
(fármaco) – hospedeiro (MOF) visando entender melhor o processo de encapsulamento
para o desenvolvimento de carreadores de fármacos mais eficientes. O objetivo final é
entender como o fármaco escolhido interage com a MOF ZIF-8. Buscaremos responder
questões do tipo: O fármaco fica alojado dentro dos poros da MOF ou adsorvido na
superfície? Que tipo de interação química ocorre entre o fármaco e a MOF esteja ele dentro
do poro ou adsorvido na superfície? A MOF escolhida realmente é a melhor para servir
como hospedeiro para o fármaco de interesse?
É importante destacar que o estudo teórico que será apresentado é parte de um projeto
maior que conta com toda a parte de síntese e estudo de liberação que está sendo
desenvolvido pela aluna de doutorado Iane Bezerra Vasconcelos, orientanda do professor
Severino Alves Jr., no programa de Pós-Graduação em Ciências de Materiais da UFPE.
Desta forma, como as duas partes (teórica e experimental) iniciaram juntas algumas

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
79
propostas deste estudo não podem ser incorporada no projeto geral. Contudo,
apresentaremos todas as conclusões de nosso, estudo independente se foram aplicadas ou
não do ponto de vista experimental.
Assim, iniciamos o estudo avaliando a capacidade de predição estrutural de diversos
fármacos utilizando os métodos semiempíricos AM1, PM3, PM6 e RM1. Como não
dispomos das estruturas cristalográficas dos fármacos estudados, as estruturas
semiempíricas foram comparadas a estruturas otimizadas com a metodologia DFT/6-31G*.
A razão pela qual buscamos elucidar qual dos métodos semiempíricos é o mais apropriado
para prever a estrutura dos fármacos pode ser explicada pela necessidade de otimização do
conjunto MOF-fármaco, uma vez que a MOF será calculada usando o mesmo procedimento
discutido no capítulo 4 desta dissertação.
Por razões experimentais, afinal de contas este estudo é parte de um projeto maior, a
MOF escolhida foi a ZIF 8, cuja estrutura tridimensional pode ser observada na figura 5.1.
Figura 5.1 – Estrutura tridimensional cristalográfica da MOF ZIF-8.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
80
Como é possível observar na figura 5.1, a ZIF8 possui como centros metálicos
átomos de zinco (azul escuro),e apresenta dois poros distintos, um maior com 5.6 Å de
diâmetro e outro menor com 2.8 Å de diâmetro.
Esta MOF foi objeto do estudo apresentado no capítulo anterior e como pode ser
observado na tabela A1, presente no Apêndice1, apesar do PM6 ter apresentado o menor
RMS, podemos notar pelos parâmetros de rede a, b, c, α, e que os métodos AM1 e PM3
também fornecem excelentes resultados. Assim, o modelo semiempírico que será escolhido
para estudar o sistema MOF-fármaco dependerá dos resultados apresentados por cada
modelo na predição estrutural dos fármacos.
5.1.1 – Metodologia dos cálculos para os fármacos avaliados
Inicialmente foi feita uma pesquisa na literatura os fármacos mais vendidos ou os
mais utilizados no tratamento de câncer, uma vez que o objetivo do projeto geral é o
desenvolvimento de carreadores para o tratamento de câncer. Tendo estes dados em mãos,
as estruturas tridimensionais de cada um dos fármacos foram geradas utilizando o programa
Hyperchem versão 8.0 [1]. Em seguida o softwareGaussView versão 4.1[2].foi utilizado
para gerar a partir do arquivo de saída do Hyperchem os arquivos de entrada (input) para o
cálculo das estruturas no programa Gaussian versão 2003 [3].
Os arquivos de saída do Hyperchem também foram utilizados para gerar os arquivos
de entrada do MOPAC [4],em que foram realizados os cálculos de otimização de geometria
dos fármacos utilizando os métodos semiempíricos AM1, PM3, PM6 e também o RM1. O
RM1 foi utilizado nesse comparativo, contudo, ele não será utilizado no estudo da interação
MOF-fármaco, uma vez que o RM1 ainda não possui parâmetros para metais.
Depois de finalizados todos os cálculos de otimização de geometria, calculamos as
dimensões de cada fármaco otimizado por cada método e comparamos os resultados com as
dimensões obtidas a partir da otimização com a metodologia B3LYP/6-31G*. A
comparação foi feita com base nas medidas tridimensionais uma vez que um dos objetivos
desta etapa é avaliar quais fármacos podem ser inseridos dentro dos poros da MOF ZIF8.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
81
5.1.2 – Metodologia dos cálculos de interação MOF-fármaco
Inicialmente é preciso esclarecer que o grupo experimental do professor Severino A.
Jr. optou em trabalhar com o fármaco doxorrubicina, assim como veremos ainda neste
capítulo, os resultados teóricos indicam que devido ao tamanho da doxorrubicina a mesma
não pode ser incorporada dentro dos poros da MOF ZIF8. Desta forma, se há algum tipo de
interação entre o fármaco e a MOF esta interação ocorre na superfície. Dados experimentais
também corroboram com esta observação [6].
Visando entender como ocorre a adsorção da doxorrubicina com a superfície da ZIF8
foi realizado um estudo de docking buscando possíveis pontos de adsorção. Estes estudos
foram realizados com o programa Autodock versão 4.2.3 [5], pela Profa. Thereza Soares do
Departamento de Química Fundamental/UFPE. Vale salientar que a profa. Thereza
participa conosco do projeto em questão.
As estruturas MOF-fármaco propostas pelo estudo de docking foram utilizadas para
construir os arquivos de entrada para a otimização utilizando o método semiempírico
escolhido na etapa anterior.
5.2 – RESULTADOS E DISCUSSÕES.
5.2.1 – Avaliação do poder de predição dos métodos semiempíricos AM1,
PM3, PM6 e RM1 para os fármacos selecionados
Nesta fase, o principal objetivo foi a avaliação da capacidade de predição dos
principais métodos semiempíricos frente aos resultados obtidos com a teoria do funcional
da densidade.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
82
5.2.1.1 – Otimização dos fármacos utilizando a teoria do funcional da
densidade (DFT)
A grande vantagem do uso dos métodos semiempíricos sobre as metodologias ab
initio é o seu tempo computacional reduzido. Contudo, geralmente, as metodologias ab
initio ou DFT possibilitam resultados mais exatos que os obtidos a partir de métodos
semiempíricos. Após revisão da literatura escolhemos a teoria do funcional da densidade
[7] que, de forma comparativa, relata melhores resultados que a metodologia Hartree-
Fock[8]. O funcional escolhido foi o B3LYP e a base escolhida foi a 6-31G*[9].
5.2.1.2 – Otimização dos fármacos utilizando métodos semiempíricos
Todos os fármacos tiveram suas geometrias do estado fundamental calculadas
utilizando os métodos AM1 [10]; PM3 [11],[12]; RM1[13] e PM6 [14]. A otimização foi
realizada utilizando o programa MOPAC versão 2009 [4] e as palavras chave utilizadas
foram:
PRECISE – concede um aumento de 100 vezes no critério de convergência;
XYZ – realiza a otimização da geometria em coordenadas cartesianas;
GRAPHF – gera o arquivo de saída com extensão .mgf, que é arquivo de entrada para
o software Jmol;
GNORM=0.01 – finaliza o processo de otimização quando a norma do gradiente
estiver abaixo de 0,01;
AUX – gera o arquivo de saída com extensão .aux, que fornece as geometrias de pré e
pós otimização, tais que são visualizadas facilmente no software Mercury.
De posse das geometrias otimizadas com todos os quatro métodos semiempíricos e
com o método DFT, foram realizadas as medidas das dimensões nos eixos X, Y e Z. As
dimensões de todos os fármacos com o método DFT e também com os métodos
semiempíricos AM1, PM3, PM6 e RM1 podem ser encontradas no Apêndice 2, juntamente
com o erro associado as medidas do semiempírico quando comparado com o DFT. Na

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
83
figura 5.2 podemos ver as dimensões do fármaco ibuprofeno que é um fármaco utilizado
no tratamento de inflamações e contra a dor causada por esse processo inflamatório.
Figura 5.2 - Determinação teórica das dimensões da molécula do fármaco ibuprofeno.
Conhecendo as dimensões da cada um dos fármacos e conhecendo os diâmetros de
poro da(s) MOF(s) de interesse podem-se ter uma ideia de quais fármacos podem ser
realmente incorporados. Entretanto, algumas MOFs, durante o processo de incorporação
podem se expandir permitindo a entrada de “hóspedes” ligeiramente maiores que seus
diâmetros de poro.
Na tabela 5.1 encontramos a lista com os fármacos selecionados para este estudo
pelo critério de serem os mais vendidos ou serem os mais aplicados no tratamento de
câncer.
Tabela 5.1. Fármaco e sua respectiva aplicação clínica.
FÁRMACO APLICAÇÃO CLÍNICA
IBUPROFENO Utilizado no tratamento de inflamação e contra a dor causada por esse
processo.
DICLOFENACO Analgésico, antinflamatório e antireumático.
5-FLUORACIL Tratamento de câncer de pele.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
84
DOXORRUBICINA Carcinomas de mama, de ovário, endometrial, de próstata, de testículo,
de bexiga, de esôfago, de cabeça e pescoço, de pulmão, de garganta e
boca, sarcomas, linfomas, neuroblastoma, linfocítica.
CICLOFOSFAMIDA Carcinomas de mama, endometrial, colon de próstata, de testículos, de
garganta e boca, sarcoma, linfomas, neuroblastoma.
MERCAPTOPURINA Mielocítica, mielomonocítica.
RALOXIFENO Antiosteoporótico.
FULVESTRANTE Antiestrógeno.
TAMOXIFENO Tratamento de câncer de mama com linfonodo axilar negativo em
mulheres após mastectomia total ou mastectomia segmentar,
linfadenectomia axilar e radioterapia.
TOREMIFENO Prevenção do câncer próstata.
CISPLATINA Carcinoma da cabeça e do pescoço, da próstata e da bexiga, é ativa
também nos sarcomas, linfomas, câncer pulmonar, câncer esofagiano,
câncer da tireoide, neuroblastoma e melanoma maligno.
CARBOPLATINA Usada principalmente em carcinoma do ovário, pulmão, cabeça e
pescoço, câncer.
BETALAPACHONA Tratamento de células cancerígenas.
OXALIPLATINA Para o tratamento adjuvante de câncer colorretal em pacientes que
sofreram ressecção completa do tumor primário, reduzindo o risco de
recidiva tumoral.
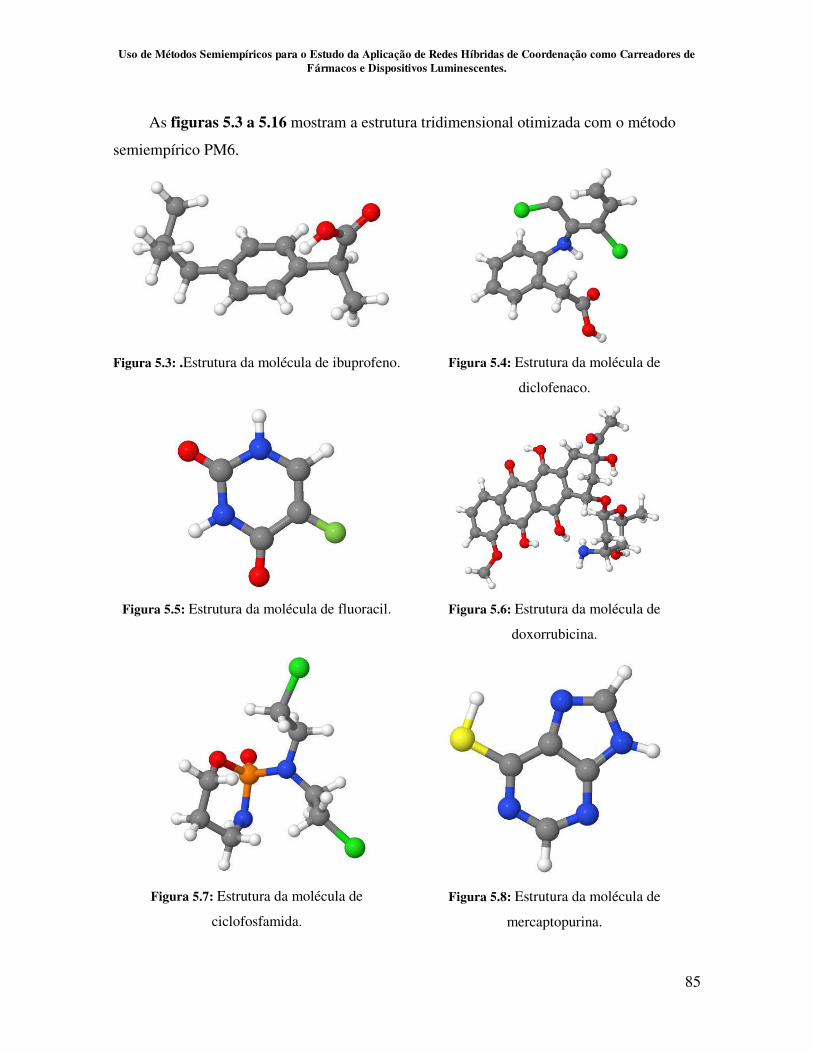
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
85
As figuras 5.3 a 5.16 mostram a estrutura tridimensional otimizada com o método
semiempírico PM6.
Figura 5.3: .Estrutura da molécula de ibuprofeno.
Figura 5.4: Estrutura da molécula de
diclofenaco.
Figura 5.5: Estrutura da molécula de fluoracil.
Figura 5.7: Estrutura da molécula de
ciclofosfamida.
Figura 5.6: Estrutura da molécula de
doxorrubicina.
Figura 5.8: Estrutura da molécula de
mercaptopurina.
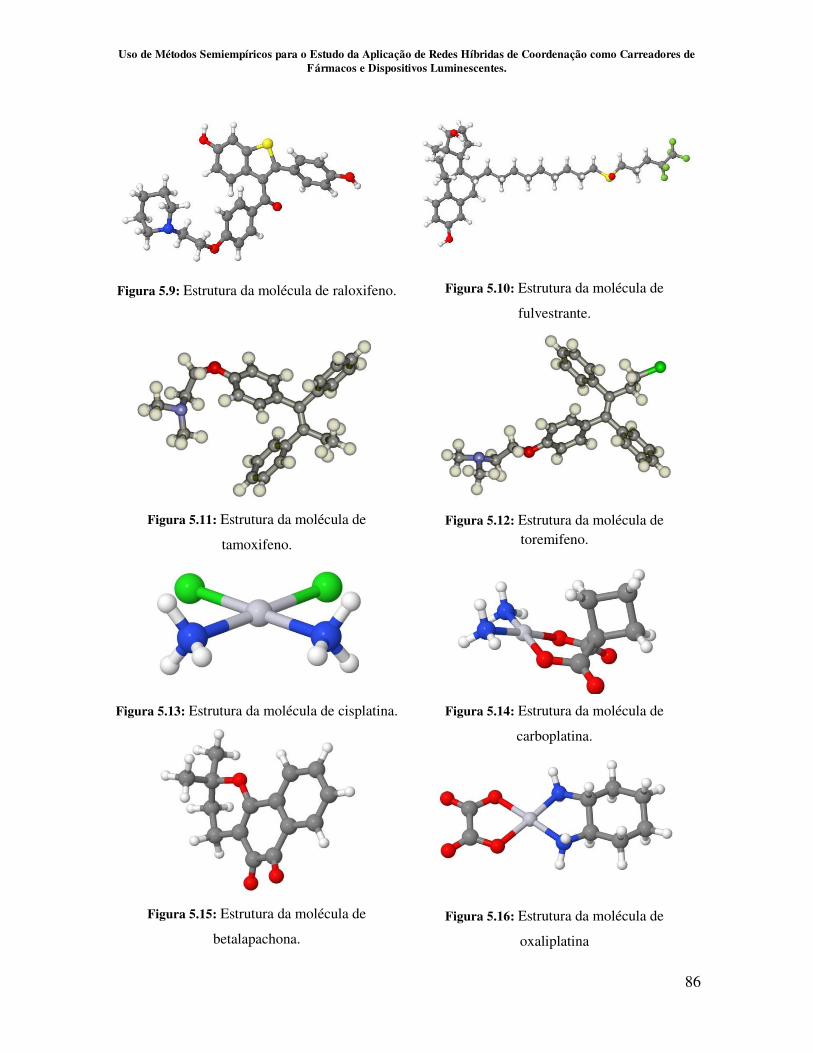
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
86
Figura 5.9: Estrutura da molécula de raloxifeno.
Figura 5.10: Estrutura da molécula de
fulvestrante.
Figura 5.11: Estrutura da molécula de
tamoxifeno.
Figura 5.12: Estrutura da molécula de toremifeno.
Figura 5.13: Estrutura da molécula de cisplatina.
Figura 5.15: Estrutura da molécula de
betalapachona.
Figura 5.14: Estrutura da molécula de
carboplatina.
Figura 5.16: Estrutura da molécula de
oxaliplatina
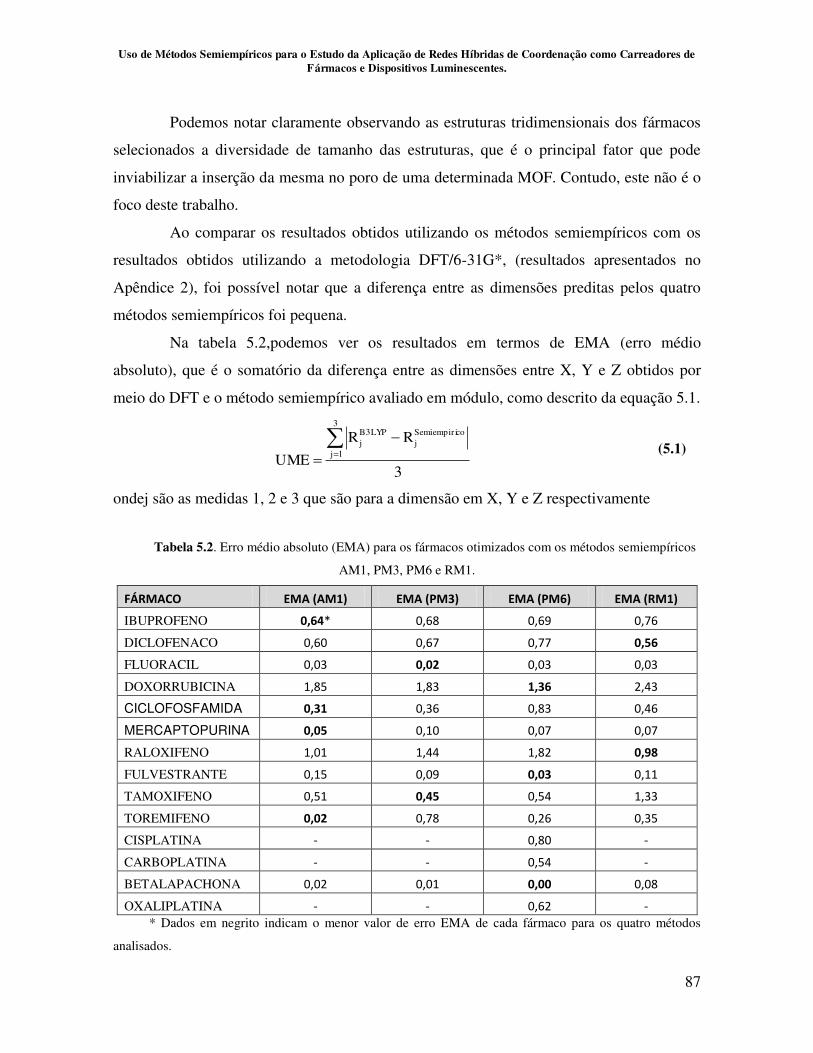
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
87
Podemos notar claramente observando as estruturas tridimensionais dos fármacos
selecionados a diversidade de tamanho das estruturas, que é o principal fator que pode
inviabilizar a inserção da mesma no poro de uma determinada MOF. Contudo, este não é o
foco deste trabalho.
Ao comparar os resultados obtidos utilizando os métodos semiempíricos com os
resultados obtidos utilizando a metodologia DFT/6-31G*, (resultados apresentados no
Apêndice 2), foi possível notar que a diferença entre as dimensões preditas pelos quatro
métodos semiempíricos foi pequena.
Na tabela 5.2,podemos ver os resultados em termos de EMA (erro médio
absoluto), que é o somatório da diferença entre as dimensões entre X, Y e Z obtidos por
meio do DFT e o método semiempírico avaliado em módulo, como descrito da equação 5.1.
3
3
1
3 j
coSemiempir ij
LYPBj RR
UME (5.1)
ondej são as medidas 1, 2 e 3 que são para a dimensão em X, Y e Z respectivamente
Tabela 5.2. Erro médio absoluto (EMA) para os fármacos otimizados com os métodos semiempíricos
AM1, PM3, PM6 e RM1.
FÁRMACO EMA (AM1) EMA (PM3) EMA (PM6) EMA (RM1)
IBUPROFENO 0,64* 0,68 0,69 0,76
DICLOFENACO 0,60 0,67 0,77 0,56
FLUORACIL 0,03 0,02 0,03 0,03
DOXORRUBICINA 1,85 1,83 1,36 2,43
CICLOFOSFAMIDA 0,31 0,36 0,83 0,46
MERCAPTOPURINA 0,05 0,10 0,07 0,07
RALOXIFENO 1,01 1,44 1,82 0,98
FULVESTRANTE 0,15 0,09 0,03 0,11
TAMOXIFENO 0,51 0,45 0,54 1,33
TOREMIFENO 0,02 0,78 0,26 0,35
CISPLATINA - - 0,80 -
CARBOPLATINA - - 0,54 -
BETALAPACHONA 0,02 0,01 0,00 0,08
OXALIPLATINA - - 0,62 -
* Dados em negrito indicam o menor valor de erro EMA de cada fármaco para os quatro métodos
analisados.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
88
Observando a tabela 5.2 notamos que não constam valores para a cisplatina,
carboplatina e oxaliplatina, exceto para o PM6, isso se explica pelo fato de os métodos
AM1, PM3 e RM1 não possuírem parâmetros para o elemento platina que está contido nos
três fármacos.
Uma análise dos resultados contidos na tabela 5.2 mostra uma boa concordância entre
os resultados obtidos pelos métodos semiempíricos e DFT. É possível perceber também que
para a doxorrubicina, fármaco escolhido para a realização dos estudos de interação, o
método semiempíricoPM6foi o que apresentou menor erro quando comparado com os
resultados obtidos com DFT.
Outro detalhe que chama atenção são os resultados obtidos para o betalapachona
(Figura 5.15) do qual os valores de erros foram baixíssimos, mas o melhor resultado foi
obtido com o PM6, quepara duas casas decimais não há diferença entre o resultado PM6 e o
B3LYP/6-31G*.
5.2.3 – Estudo da interação entre a Doxorrubicina e a ZIF8.
Neste ponto já identificamos o método semiempírico PM6 como sendo o mais
indicado tanto para o cálculo da geometria da doxorrubicina como para o cálculo em fase
sólida da MOF ZIF8. Os estudos de docking sugerem cinco possibilidades de interação
entre a superfície da ZIF8 com a doxorrubicina, como podemos observar nas figuras5.17-
5.21.
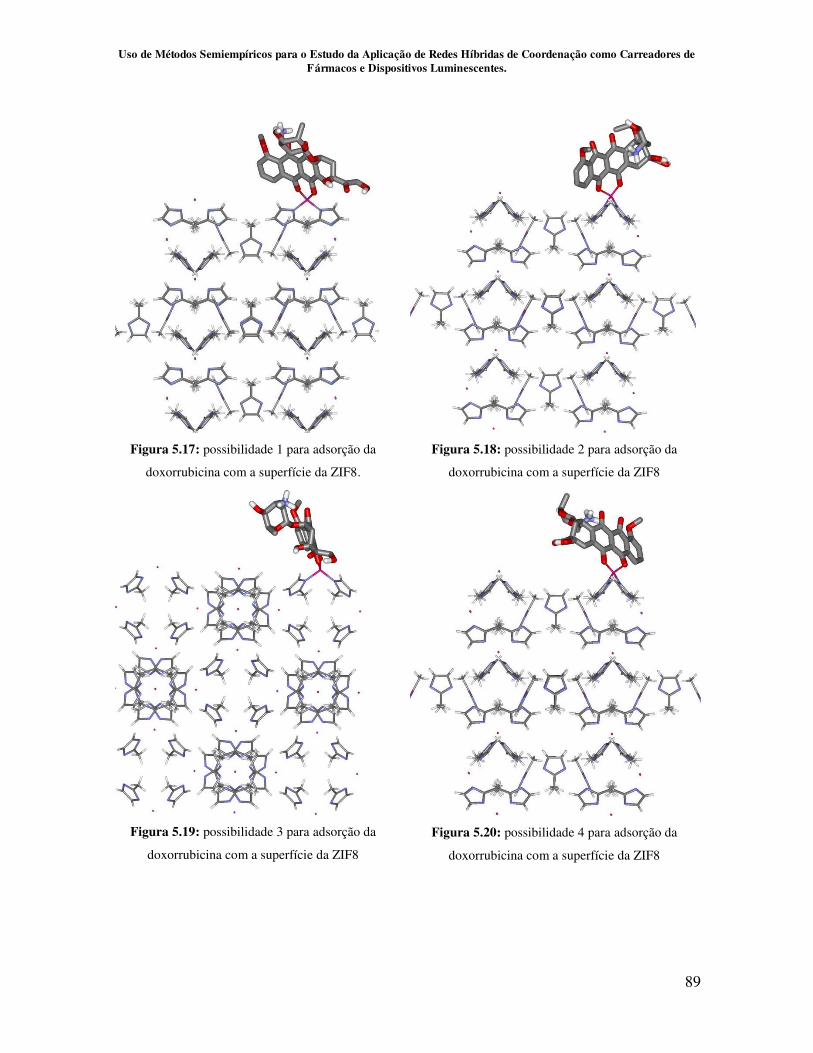
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
89
Figura 5.17: possibilidade 1 para adsorção da
doxorrubicina com a superfície da ZIF8.
Figura 5.18: possibilidade 2 para adsorção da
doxorrubicina com a superfície da ZIF8
Figura 5.19: possibilidade 3 para adsorção da
doxorrubicina com a superfície da ZIF8
Figura 5.20: possibilidade 4 para adsorção da
doxorrubicina com a superfície da ZIF8

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
90
Figura 5.21: possibilidade 5 para adsorção da doxorrubicina com a superfície da ZIF8sugerida através
do estudo de docking.
Como podemos observar na figuras 5.17a 5.21, as possibilidades de interação entre a
doxorrubicina e a superfície da ZIF8 acontece com a coordenação do fármaco ao centro
metálico MOF que é oátomo de zinco através de dois oxigênios do fármaco. Estas
possibilidades sugeridas a partir do docking, submetidas à otimização utilizando o método
PM6, podem nos proporcionar um melhor entendimento de como ocorre a interação visto
que o tratamento viria a ser por método quântico.
Através das energias obtidas por cálculos PM6 espera-sereforçar como ocorre a
adsorção da doxorrubicina na superfície da ZIF8 e realizar a análise das interações que
ocorrem entre as duas espécies. Atualmente os cálculos estão em processamento, e esta
disposta nas perspectivas de trabalhos futuros, da qual a gama de fármacos e MOFs
utilizadas pode ser maior.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
91
Referências
[1] HyperChem(TM) Professional, version 8; Hypercube Inc., Gainesville, Florida,
USA, 2008
[2] GaussView 4.1; Gaussian, Inc., 340 Quinnipiac St Bldg 40 Wallingford, CT 06492
USA, 2006
[3] Gaussian 03, Revision A.1.; M. J. Frisch.; G. W. Trucks.; H. B. Schlegel.; G. E.
Scuseria.; M. A. Robb.; J. R. Cheeseman.; J. A. Montgomery.; Jr. T. Vreven.; K. N.
Kudin.; J. C. Burant.; J. M. Millam.; S. S. Iyengar.; J. Tomasi.; V. Barone.; B.
Mennucci.; M. Cossi.; G. Scalmani.; N.Rega.; G. A. Petersson.; H. Nakatsuji.; M.
Hada.; M. Ehara.; K. Toyota.; R. Fukuda.; J. Hasegawa.; M. Ishida.; T. Nakajima.;
Y. Honda.; O. Kitao.; H. Nakai.; M. Klene.; X. Li.; J. E. Knox.; H. P. Hratchian.; J.
B. Cross.; C. Adamo.; J. Jaramillo.; R. Gomperts.; R. E. Stratmann.; O. Yazyev.; A.
J. Austin.; R. Cammi.; C. Pomelli.; J. W. Ochterski.; P. Y. Ayala.; K. Morokuma.;
G. A. Voth.; P. Salvador.; J. J. Dannenberg.; V. G. Zakrzewski.; S. Dapprich.; A. D.
Daniels.; M. C. Strain.; O. Farkas.; D. K. Malick.; A. D. Rabuck.; K. Raghavachari.;
J. B. Foresman.; J. V. Ortiz.; Q. Cui.; A. G. Baboul.; S. Clifford.; J. Cioslowski.; B.
B. Stefanov.; G. Liu.; A. Liashenko.; P. Piskorz.; I. Komaromi.; R. L. Martin.; D. J.
Fox.; T. Keith.; M. A. Al-Laham.; C. Y. Peng.; A. Nanayakkara.; M. Challacombe.;
P. M. W. Gill.; B. Johnson.; W. Chen.; M. W. Wong.; C. Gonzalez.; and J. A.
Pople.; Gaussian, Inc., Pittsburgh PA, 2003.Gaussian
[4] Stewart; J.J.P. MOPAC2009, Stewart Computational Chemistry, Colorado
Springs, USA, (2009)
[5] Trott,O.;Olson,A. J.;AutoDockVina: improving the speed and accuracy of docking
with a new scoring function, efficient optimization and multithreading, Journal of
Computational Chemistry 31 (2010) 455.
[6] Vasconcelos,I. B.; da Silva,T. G.; MilitãoG. C. G.; Soares, T. A.; Rodrigues,N. M.;
Rodrigues,M. O.;da Costa Jr,N. B..; Freire, R. O.; Junior,S. A.,RSC Advances, 2
(2012) 9437.
[7] Hohenberg, P.; Kohn,W,. "Inhomogeneous electron gas". Physical
Review,136(1964)(3B): B864–B871

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
92
[8] Hartree,D.R., Proc. Camb. Phil. Soc., 24 (1928)89; V. Fock In: Foundations of
Quantum Mechanics (1931)
[9] Davidson,E.;Feller,D., "Basis set selection for molecular calculations". Chemical
Reviews 86(1986) (4): 681–696
[10] Dewar, M.J.S.; Zoebish, E.G.; Healy, E.F.; e Stewart, J.J.P., J. Am. Chem. Soc,
107 (1985) 3902.
[11] Stewart, J.J.P., J. Comp. Chem, 10 (1989) 209.
[12] Stewart, J.J.P., J. Comp. Chem, 10 (1989) 221.
[13] Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P.,J Comp.
Chem,27(2006)1101.
[14] Stewart, J.J.P., J. Mol. Model,13 (2007) 1173.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
93
6 ESTUDO DAS PROPRIEDADES LUMINESCENTES DA
MOF EuMell 1, COM SIMULAÇÃO DA VARIAÇÃO DE
TEMPERATURA
6.1 – Introdução e Objetivos
Neste capítulo, será apresentadoo estudode propriedades de suma importância na
aplicação de MOFs como rastreadores fotônicos, que são as propriedades luminescentes.
Tais sistemas apresentam maior eficiência de emissão devido ao fenômeno conhecido como
efeito antena, no qual os ligantes orgânicos absorvem energia com comprimento de onda na
região do ultravioleta e, na sequência, transfere para o íon metálico ao qual esta
coordenado, no caso dos lantanídeos a transferência, é feita para os níveis 4f [1]. O íon, por
sua vez, emite radiação eletromagnética na região do visível, sendo a cor da emissão uma
característica do íon lantanídeo usado. Estruturas contendo íons lantanídeos são muito
utilizadas para tais dispositivos, por razão destes íons apresentarem bandas de emissão finas
e bem definidas, o que permite uma emissão com maior pureza (sem mistura de cores).

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
94
Neste capítulo será estudada uma MOF contendo como centro metálico o íon európio,
com isso é necessário (como visto no capitulo 4) o uso dos modelos Sparkle/AM1,
Sparkle/PM3 ou Sparkle/PM6 para a otimização da geometria do estado fundamental.
Omodelo Sparkle trata o íon lantanídeo como uma carga pontual +3e, pois geralmente é o
estado de oxidação apresentado por estes íons [2].
A partir da estrutura otimizada é possível extrair características intrínsecas, como por
exemplo, as propriedades luminescentes de uma estrutura contendo como centro metálico o
íon Eu3+.
Para o cálculo das energias do estado excitado singleto e tripleto, que são dados
necessários para o cálculo das propriedades luminescentes, o método semiempírico
INDO/S-CIS (IntermediateNeglectofDifferentialOverlap/SpectroscopicSingle
ConfigurationInteration) [3] implementado no programa ZINDO [4] pode ser utilizado. O
método INDO/S-CIS foi parametrizado para o cálculo de estados excitados. No presente
estudo foram consideradas excitações simples para uma janela de 60 orbitais ocupados e 60
orbitais não ocupados.
A teoria de Judd–Ofelt [5],[6] e a teoria das transições f-f [7] são o suporte necessário
para o cálculo das propriedades luminescentes. Dos dados obtidos com a aplicação destas
teorias temos como foco:
Parâmetros de intensidade (Ω2, Ω4 e Ω6): Ω2 reflete o grau de ligação covalente
existente, a polarizabilidade e o comportamento hipersensível da transição 5D0→7F2, Ω4 esta relacionado com a rigidez do meio.
Decaimento radiativo (Arad): decaimento que geralmente emite radiação eletromagnética no visível, que a exemplo do íon Eu3+ emite radiação na região do vermelho;
Decaimento não radiativo (Anrad): desfavorece o rendimento de luminescência, da qual a energia é dissipada por outro meio, por exemplo, por vibração na molécula;
Eficiência quântica (η): faz uma referência ao percentual de decaimento radiativo frente ao valor da quantidade de energia que decai radiativamente e não radiativamente, é expressa pela equação 6.1.
(6.1)

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
95
- Rendimento quântico (q): é expresso em porcentagem e é a razão entre o número de
fótons emitidos e o número defótons absorvidos pelo ligante [1].
Não iremos abordar a descrição matemática contida na teoria de Judd–Ofelt bem
como a proposta por Malta e colaboradores, tais questões estão contidas no software
LUMPAC, com isso, é indicado ao leitor as referências [5], [6] que contem a descrição da
teoria de Judd–Ofelt, como também a referência [7]que contem o método desenvolvido por
Malta e colaboradores para o cálculo das transferências de energia.
Partindo para os objetivos deste capítulo, de forma clara, temos:
Avaliar o método mais eficiente para a descrição estrutural em fase sólida para uma
MOF contendo íon európio como centro metálico;
Obter as propriedades espectroscópicas de nosso interesse entre elas os parâmetros
de intensidade (Ω2, Ω4 e Ω6), eficiência quântica (η) e o rendimento quântico (q);
Comparar as propriedades espectroscópicas obtidas com as estruturas calculadas em
fase sólida e no vácuo;
Com o melhor método realizar um estudo simulando o aquecimento da estrutura
(perda de água de coordenação) e obter suas propriedades luminescentes.
6.2 – Metodologia
Para este estudo, optou-se por trabalhar exclusivamente com a MOF
[Eu2(Mell)(H2O)6] [8] a qual chamaremos aqui deEuMell 1. Esta MOF contém como centro
metálico o íon Eu3+. Os métodos químicos quânticos utilizados na otimização foram os
semiempíricos Sparkle/AM1 [9], Sparkle//PM3 [10] e Sparkle//PM6 [11], todos contidos
no pacote computacional MOPAC2009 [12].
O cálculo da MOF EuMell 1 foi realizado com um fragmento (1.2.1), o qual possui
176 átomos e que foi obtido pela simples duplicação da célula unitária cristalográfica com
88 átomos. Este fragmento é consideravelmente pequeno comparado as grandes estruturas
utilizadas até o momento, mais pelo fato de ter sido observado um bom resultado, e por

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
96
questões de tempo decidiu-se trabalhar com um fragmento menor. Contudo, como vimos no
capítulo 4, que um maior fragmento proporciona uma descrição mais exata da estrutura.
Do fragmento otimizado foi realizado o estudo espectroscópico e para isso foram
utilizados o programa ZINDO para o cálculo dos estados excitados, e o pacote
computacional LUMPAC para todos os cálculos referentes à luminescência. O LUMPAC
(LuminescencePackage) foi desenvolvido no laboratório Pople (Pople
ComputationalChemistryLaboratory) da Universidade Federal de Sergipe.
6.3 – RESULTADOS E DISCUSSÃO.
6.3.1 – Resultados da descrição estrutural para a EuMell 1.
Na tabela 6.1 apresentamos o comparativo para a descrição das estruturas feita para
um clustercom dimensões (1,1,1) (uma célula unitária) utilizando os métodos avaliados.
Tabela 6.1. Resultados para [Eu2(Mell)(H2O)6], utilizando os modelos Sparkle/AM1, Sparkle/PM3 e
Sparkle/PM6.
Estrutura Erro RMS
a b C α Volume
Cristalográfica - 13,436 6,625 10,142 90,00 90,00 90,00 1805,49 Sparkle/AM1 0,0355 14,438 6,392 9,659 90,00 90,00 89,92 1782,89 Sparkle/PM3 0,0331 14,719 6,529 9,780 90,08 89,98 89,98 1879,75 Sparkle/PM6 0,0630 13,520 4,759 10,317 96,75 89,49 91,83 1317,72
Conforme dados contidos na tabela 6.1, podemos observar que o método
semiempírico PM3 possui o menor erro RMS entre os 3 métodos analisados e método
Sparkle/AM1 proporcionou resultado em patamar semelhante mais com erro um pouco
maior. Para fazer uma avaliação visual podemos observar as figuras 6.1, 6.2 e 6.3, o qual
em cinza temos a estrutura cristalográfica e em azul temos a calculada para o fragmento
utilizado.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
97
Figura 6.1.Comparação entre a estrutura cristalográfica e a estrutura calculada com o modelo Sparkle/AM1.
Figura 6.2. Comparação entre a estrutura cristalográfica e a estrutura calculada com o modelo Sparkle/PM3.
Figura 6.3.Comparação entre a estrutura cristalográfica e a estrutura calculada com o modelo Sparkle/PM6.
Das figuras acima, observa-se visualmente que as figura 6.1 e 6.2 confirmam as
semelhanças nos resultados expostos na tabela 6.1 em que o método Sparkle/PM3 foi o que
melhor reproduziu a estrutura cristalográfica. Vale também ressaltar que na análise do
fragmento, notou-se para o Sparkle/PM6 uma diferente conformação frente ao dado
cristalográfico, do qual um dos oxigênios do ligante passou a interagir com dois íons
lantanídeos distintos, e a esfera de coordenação que para o dado cristalográfico é composta
por nove átomos ligantes passou a ter onze átomos, fato que explica a redução do volume
da estrutura devido ao encurtamento da distância entre íons európio.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
98
6.3.2 – Estudo luminescente com a EuMell 1, comparativo entre
cálculos utilizando condições de periodicidade e no cálculo no
vácuo.
Em um artigo publicado em 2009 Rodrigues e colaboradores [8] realizaram além do
estudo sobre síntese e caracterização do material, um estudo luminescente da
[Eu2(Mell)(H2O)6] designada aqui como EuMell 1.
Uma parte do estudo foi realizada por meio de cálculos quânticos computacionais,
no qual o modelo Sparkle/AM1 foi o escolhido para a otimização no vácuo de uma porção
da estrutura. A partir da estrutura calculada no vácuo foi realizado o cálculo das
propriedades luminescentes que são:
Os parâmetros de intensidade (Ω2, Ω4 e Ω6);
Decaimento radiativo (Arad);
Decaimento não radiativo (Anrad);
Eficiência quântica (η);
Rendimento quântico (q);
Taxas de transferência e retrotransferência de energia (τ);
Com o desenvolvimento em 2008 da metodologia para cálculos das estruturas em
fase sólida proposta por Stewart [12] que foi implementada no MOPAC2009 [13], surgiu
uma nova janela para a obtenção de propriedades de estruturas periódicas que antes eram
otimizadas no vácuo, o qual é negligenciada a periodicidade do sistema.
Neste contexto serão expostos os resultados para a EuMell 1 calculada no vácuo e
emcondições de periodicidadeutilizando o Sparkle/AM1 que foi o método utilizando na
publicados em 2009 e os resultados obtidos com vácuo e emcondições de periodicidade
para o Sparkle/PM3. Considerando cálculos com condições de periodicidade, sabemos que
o Sparkle/PM3 é o mais exato entre os três modelosSparkle. Mesmo Assim, reportaremos
aqui o comparativo entre os resultados dos cálculosno vácuo e em condições de
periodicidade para os modelosSparkle/AM1 e Sparkle/PM3. O estudo será realizado
utilizando os novos dados experimentais obtidos em 2013 pelo grupo do Prof. Severino

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
99
Alves Júnior, utilizando de equipamentos mais sofisticados que os utilizados na obtenção
dos dados publicados em 2009 [8].
A EuMell 1 possui íon európio (Eu3+) com número de coordenação igual a nove.Das
nove ligações, três são realizadas por moléculas de água, e as outras seis restantes são
realizadas pelos quatro ligantes orgânicos que o circundam da seguinte maneira: dois
ligantes fazem duas ligações com o íon e os outros dois somente uma, conforme a figura
6.4.
Figura 6.4. Fragmento contendo o poliedro de coordenação da EuMell 1.
Na tabela 6.2, podemos observar o resultado deste estudo comparativo entre dados
obtidos a partir da estrutura calculada no vácuo e com condições de periodicidade. Pode-se
observar que os ajustes dos Ω2 e Ω4, obtido da estrutura calculada com Sparkle/AM1 com
condições de periodicidade,possui os valores mais próximos aos obtidos experimentalmente
(13,67 e 3,35 respectivamente).
Tabela 6.2. Dados experimentais e calculados no vácuo e em condições de periodicidades paraEuMell 1.
Ω2 Ω4 Ω6 Arad Anrad τ η q
Experimental 13.69 3,47 - 554,0 3701,3 0,23 13,0 ---
Sparkle/AM1 (Vácuo) 12,91 2,24 0,12 473,27 3874,56 - 10,89 10,78
Sparkle/AM1 (Periódico) 13,67 3,35 0,70 513,62 3834,21 - 11,81 11,70

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
100
Para Arad, Anrad, η e q, os dados obtidos com o Sparkle/AM1 considerando condições
periódicas, foram melhores que os com Sparkle/AM1 no vácuo, quando comparados com
os respectivos valores experimentais.
Com relação as taxas de transferência (TE) e retrotransferência (RTE) de energia
que são quantidades difíceis de serem medidas experimentalmente, pois suas medidas
necessitariam de um aparato experimental muito sofisticado, a comparação foi realizada
tomando como base os valores calculados a partir da estrutura cristalográfica. (ver tabela
6.3).
Tabela 6.3. Resultados para [Eu2(MELL)(H2O)6], utilizando os Sparkle/AM1 (vácuo) e Sparkle/AM1
(periódico).
Geometria Singleto→5D4 Tripleto→5D1 Tripleto→5D0
TE (s-1)a RTE (s-1)b TE (s-1)a RTE (s-1)b TE (s-1)a RTE (s-1)b
Raio-X 4,48x100 2,31x10-22 6,83x109 1,15x106 7,01x109 2,38x102
Sparkle/AM1 (Vácuo) 2,11x104 2,54x10-5 7,84x109 1,31x106 8,05x109 2,72x102
Sparkle/AM1 (Periódico) 6,57x100 1,44x10-24 6,92x109 9,95x105 7,06x109 2,05x102
Analisando os resultados obtidos para transferência Singleto→5D4 pode-se notar
que o valor da taxa de TE calculada a partir da estrutura otimizada no vácuo é quatro
ordens de grandeza maior que o calculado a partir da estrutura cristalográfica, contudo o
valor da TE calculado a partir da estrutura calculada considerando condições de
periodicidade é da mesma ordem de grandeza. O mesmo pode ser observado com relação as
taxas de retrotransferência de energia. Para as transferências e retrotransferências
Tripleto→5D1os resultados obtidos com ambas as estruturas (vácuo e considerando
condições de periodicidades) reproduzem bem os valores obtidos a partir da estrutura
cristalográfica.
Vimos nos resultados obtidos na otimização das estruturas, que o modelo
Sparkle/PM3 foi o que melhor retratou a estrutura da EuMell 1 comparado ao dado
experimental. Desta forma iremos expor na tabela 6.4 resultados referentes ao comparativo
entre cálculos no vácuo e em condições de periodicidade para o Sparkle/PM3.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
101
Tabela 6.4. Dados experimentais e calculados no vácuo e em condições de periodicidades paraEuMell 1.
Ω2 Ω4 Ω6 Arad Anrad τ η q
Experimental 13.69 3,47 - 554,0 3701,3 0,23 13,0 ---
Sparkle/PM3 (Vácuo) 13,58 2,59 0,36 499,0 3848,8 - 11,48 11,36
Sparkle/PM3 (Periódico) 13,69 3,47 0,12 515,6 3832,3 - 11,86 11,74
Analisando os resultados da tabela 6.2 e 6.4, podemos observar que os resultados
com Sparkle/PM3 para vácuo e para condições de periodicidade são ligeiramente melhores
que os resultados com Sparkle/AM1. Também pode-se notar na tabela 6.4, que os
resultados para a estrutura otimizada utilizando condições de periodicidade são novamente
melhores que a os resultados obtidos para uma estrutura otimizada no vácuo.
De forma geral, os dados presentes nastabelas6.2, 6.3 e 6.4 sugerem que os
resultados obtidos a partir da estrutura calculada, considerando condições de periodicidade,
são um pouco mais exatos que os obtidos no vácuo.
Uma vez que os cálculos em condições de periodicidade apresentam resultados
melhores que os obtidos a partir de estruturas otimizadas no vácuo e, baseado nos
resultados apresentados no tópico 6.3.1 e na tabela 6.4, que mostrou o Sparkle/PM3 como
sendo o melhor para a descrição estrutural da MOF EuMell 1,optamos em realizar cálculos
em fase sólida utilizando o modelo Sparkle/PM3 para o estudo das propriedades
luminescentes a partir da simulação do aquecimento da referida MOF. Os dados
experimentais utilizados para comparação foram obtidos em 2012 e 2013pelo grupo do
Prof. Severino Alves Júnior (UFPE).
A simulação da variação de temperatura considerou a partir de dados experimentais
(por análise de curva termogravimétrica) que, na temperatura de 25 oC o centro metálico da
MOF EuMell 1 contem três moléculas de água coordenadas. Na temperatura de 150 °C o
poliedroapresenta 2 águas coordenadas e a200 oCpossivelmente não há mais água de
coordenação.
Na tabela 6.5, pode-se observar os resultados obtidos a partir das estruturas calculas
seguindo a descrição feita anteriormente, para o critério da simulação da perda de água no
poliedro a partir do aumento de temperatura.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
102
Tabela 6.5.Estudo do sistema [Eu2(MELL)(H2O)6] simulando aquecimento de 25ºC a 200 oC. Os dados
teóricos foram calculados a partir da estrutura calculada em fase sólida com o modelo Sparkle/PM3.
25 oC Ω2 Ω4 Ω6 Arad Anrad τ η q
Experimental 13,69 3,47 - 554,00 3701,30 0,23 13,00 ---
Sparkle/PM3 13,69 3,47 0,12 515,57 3832,26 - 11,9 11,6
150 oC
Experimental 16,43 3,34 - 645,90 2802,40 0,29 18,7 ---
Sparkle/PM3 16,44 3,33 0,34 596,46 2851,70 - 17,3 17,1
200oC
Experimental 12,95 3,89 - 549,2 4450,80 0,20 11,0 ---
Sparkle/PM3 18,52 1,05 0,17 624,4 4375,60 - 12,5 11,4
Pelos resultados contidos na tabela 6.5, podemos observar que a medida que a
temperatura aumenta até 150 ºC, o valor do rendimento quântico q aumenta
gradativamente. Esse fato é explicado pela perda de molécula de água, tal que resulta na
redução da taxa não radiativa,essa redução é favorecida pelas perdas de energia por
vibração na molécula de água. Contudo, quando a temperatura aumenta de 150 para 200
°C, observa-se uma redução no valor do rendimento quântico, que é proveniente do grande
aumento dos valores da taxa não radiativa que pode ser explicado a partir dos dados
presentes na tabela 6.6. a qual contém os valores de transferência e de retro transferênciade
energia entre o metal e a parte orgânica da MOF.
Tabela 6.6. Taxas de transferências e retro transferências para 25, 150 e 200 °C.
Temperatura Singleto→5D4 Tripleto→5D1 Tripleto→5D0
TE (s-1)a RTE (s-1)b TE (s-1)a RTE (s-1)b TE (s-1)a RTE (s-1)b
25 1,7x101 0,0 1,0x1010 2,0x109 1,5x1010 5,8x105
150 2,1x101 0,0 1,1x1010 2,6x108 1,4x1010 2,72x104
200 6,2x104 0,0 7,5x109 1,5x1010 1,2x1010 5,0x106
Observando a tabela 6.6, nota-se que para as taxas de transferência e retro
transferências para as transições Tripleto→5D1 eTripleto→5D0, até 150 °C estão em um
patamar semelhante. Entretanto os valores das taxas mudam consideravelmente quando
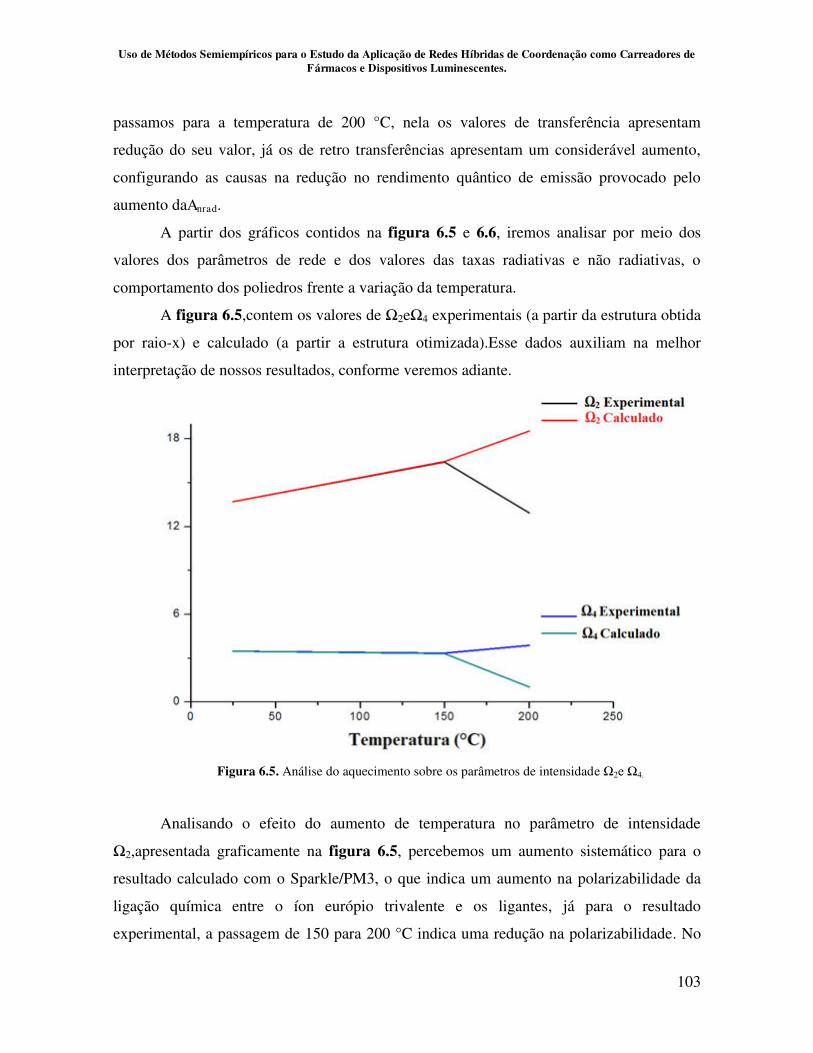
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
103
passamos para a temperatura de 200 °C, nela os valores de transferência apresentam
redução do seu valor, já os de retro transferências apresentam um considerável aumento,
configurando as causas na redução no rendimento quântico de emissão provocado pelo
aumento daAnrad.
A partir dos gráficos contidos na figura 6.5 e 6.6, iremos analisar por meio dos
valores dos parâmetros de rede e dos valores das taxas radiativas e não radiativas, o
comportamento dos poliedros frente a variação da temperatura.
A figura 6.5,contem os valores de Ω2eΩ4 experimentais (a partir da estrutura obtida
por raio-x) e calculado (a partir a estrutura otimizada).Esse dados auxiliam na melhor
interpretação de nossos resultados, conforme veremos adiante.
Figura 6.5. Análise do aquecimento sobre os parâmetros de intensidade Ω2e Ω4.
Analisando o efeito do aumento de temperatura no parâmetro de intensidade
Ω2,apresentada graficamente na figura 6.5, percebemos um aumento sistemático para o
resultado calculado com o Sparkle/PM3, o que indica um aumento na polarizabilidade da
ligação química entre o íon európio trivalente e os ligantes, já para o resultado
experimental, a passagem de 150 para 200 °C indica uma redução na polarizabilidade. No
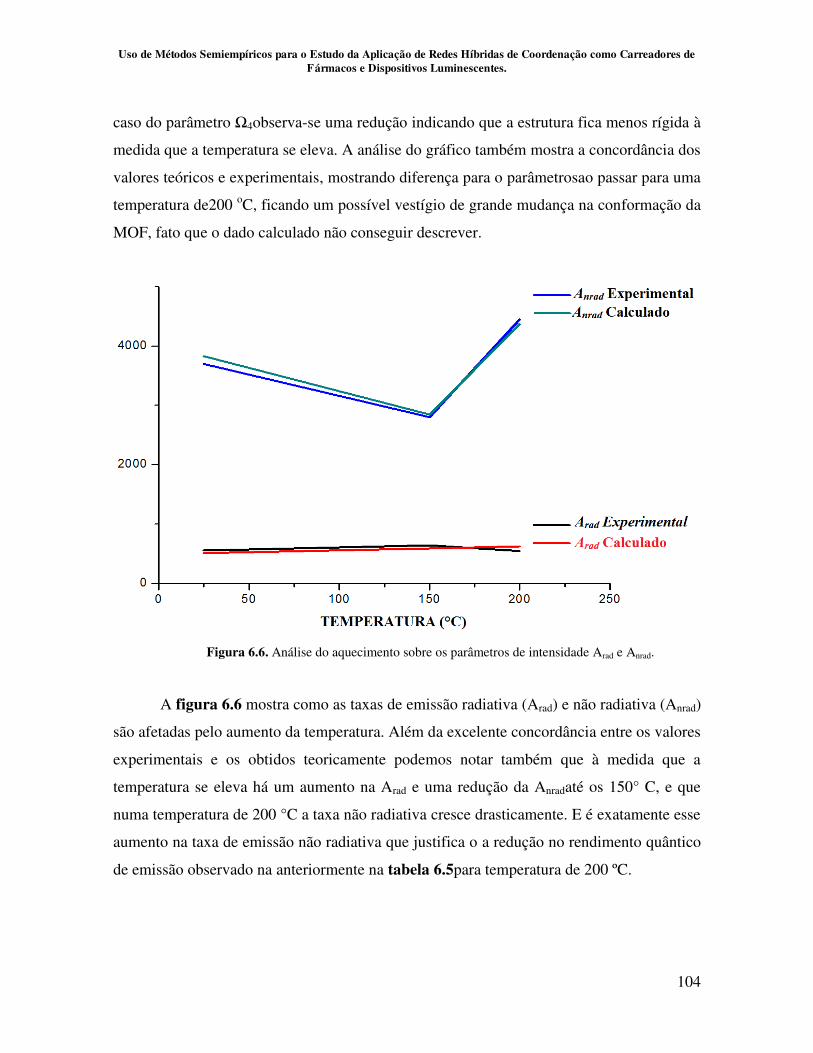
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
104
caso do parâmetro Ω4observa-se uma redução indicando que a estrutura fica menos rígida à
medida que a temperatura se eleva. A análise do gráfico também mostra a concordância dos
valores teóricos e experimentais, mostrando diferença para o parâmetrosao passar para uma
temperatura de200 oC, ficando um possível vestígio de grande mudança na conformação da
MOF, fato que o dado calculado não conseguir descrever.
Figura 6.6. Análise do aquecimento sobre os parâmetros de intensidade Arad e Anrad.
A figura 6.6 mostra como as taxas de emissão radiativa (Arad) e não radiativa (Anrad)
são afetadas pelo aumento da temperatura. Além da excelente concordância entre os valores
experimentais e os obtidos teoricamente podemos notar também que à medida que a
temperatura se eleva há um aumento na Arad e uma redução da Anradaté os 150° C, e que
numa temperatura de 200 °C a taxa não radiativa cresce drasticamente. E é exatamente esse
aumento na taxa de emissão não radiativa que justifica o a redução no rendimento quântico
de emissão observado na anteriormente na tabela 6.5para temperatura de 200 ºC.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
105
Por fim podemos concluir que os resultados dos estudos espectroscópicos foram
muito satisfatórios e que o protocolo semiempírico aplicado é bastante indicado para o
estudo de sistemas luminescentes envolvendo MOFs contendo íons lantanídeos.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
106
Referências
[1] Lima, P. P.; Malta, O. L.; Júnior, S. A., Química Nova. 28 (2005) 805.
[2] de Andrade,A. V. M.; da Costa Jr,N. B.; Simas, A. M.; G, F. de Sá.,Chem. Phys. Lett,
227 (1994) 349.
[3] Ridley, J.E.; Zerner, M.C.,Theor. Chim.Acta, 42, (1976) 223-236.
[4] Zerner, M. C. ZINDO manual, QTP; University of Florida:Gainesville, FL, 1990.
[5] Judd, B. R., Phys. Rev, 127 (1962) 750.
[6] Ofelt, G.S., J. Chem. Phys, 37 (1962) 551.
[7] Malta,O.L; Silva, F.R.G. Spectrochim. Acta Part A, 54 (1998)1593
[8] Rodrigues, M. O.; Paz, F. A. A.; Freire, R. O.; de Sá, G. F.; Galembeck, A.;
Montenegro, M. C. B. S. M.; Araújo, A. N.; Júnior, S . A., J. Phys. Chem. B, 113 (2009)
12181
[9] Freire, R. O.;Rocha, G. B.; Simas, A. M., InorganicChemistry, 44 (2005) 3299
[10] Freire,R. O.; Rocha,G. B.; A. M. Simas., journal of the Brazilian Chemical Society, 20
(2009) 1638
[11] R.O. Freire.; A. M. Simas., Journal of Chemical Theory and
Computational,13(2010)2019
[12] Stewart, J. J. P., J Mol Model. 14 (2008) 499
[13] Stewart, J.J.P.,MOPAC2009, Stewart Computational Chemistry, Colorado
Springs, USA, 2009

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
107
77 CONCLUSÕES E PERSPECTIVAS
7.1 – CONCLUSÕES
Nesta dissertação apresentamos inicialmente um estudo sobre a capacidade de
predição dos principais métodos semiempíricos no cálculo em fase sólida de redes de
coordenação (MOFs). O estudo contou comMOFs formadas pelos mais variados tipos de
ligantes orgânicos e de centros metálicos.
Com relação às MOFs, formadas pelos metais (Al, Zn, Hg, Pb, Ba, Mn, Fe, Co, Ni,
Cu, Y, Ag, Cd, Sr, Ga, Mg), o método PM6 mostrou-se o mais exato, apresentando
resultados em concordância com as respectivas estruturas experimentais. Foi possível notar
também que, algumas vezes se faz necessário aumentar o tamanho do fragmento a ser
otimizado visando alcançar resultados mais exatos. Contudo, o PM6 é um método
semiempírico e, portanto, apresenta tempos de otimização consideravelmente reduzidos
quando comparamos com otimizações realizadas utilizando métodos ab initio ou DFT e,
por este motivo, o aumento do fragmento a ser otimizado não inviabiliza o cálculo do
sistema com o método PM6.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
108
A análise realizada nas MOFs que contém íons lantanídeos trivalentes como centros
metálicos sugere que a utilização do modelo Sparkle/PM3 permite o cálculo em fase sólida
de MOFs dos mais variados tipos com elevada exatidão. Os resultados apresentados pelos
modelos Sparkle/AM1 e Sparkle/PM6 também apresentam boa concordância com as
respectivas estruturas cristalográficas.
O estudo realizado no capítulo 5 mostrou a importância de ferramentas teóricas no
projeto de carreadores de fármacos mais eficientes. A simples medida teórica das
dimensões dos fármacos de interesse e dos diâmetros de poro das MOFssugere se haverá ou
não incorporação do fármaco na MOF em questão. Vale salientar que o grupo do Prof.
Severino Alves Jr., que realizou a parte experimental deste estudo, escolheu estudar a
incorporação do fármaco doxorrubicina na MOF ZIF-8. Assim, constatou-se que o fármaco
não caberia no poro da MOF, desta formadirecionamos nossos esforços para tentar entender
como ocorrea adsorção da doxorrubicina na superfície da ZIF-8, uma vez que pelas
dimensões da doxorrubicina (15,16 Å, 10,00 Å e 5,86 Å) já sabíamos que a mesma não
poderia ser incorporada dentro dos poros da ZIF-8 com diâmetros de 5,6 Å e 2,8 Å.
Através de cálculos de docking, cinco possíveis tipos de interação (adsorção) entre a
doxorrubicina e a superfície da ZIF-8 foram identificados e que podem ser fortalecidas pelo
uso do método quântico computacional PM6 já avaliado. Através das energias que podem
ser obtidas pelo uso do PM6 pode-se sugerir como ocorre a adsorção da doxorrubicina na
superfície da ZIF-8 e realizar a análise das interações que ocorrem entre as duas espécies.
As simulações com a MOF EuMell 1 mostraram os cálculos em fase sólida com o
método Sparkle/PM3 como o mais exato. Na comparação entre as metodologias vácuoe
levando em conta a periodicidade para a obtenção das propriedades luminescentes, o
cálculo em condições de periodicidade mostrou resultados mais próximos aos
experimentais. Resultados referentes ao estudo de variação da temperatura se mostraram de
acordo com os dados obtidos experimentalmente.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
109
7.2 – PERSPECTIVAS
Cálculos dos cinco diferentes sistemas de interação DOXO – ZIF8, serão realizados
como forma de sugerir por meio de cálculo químico quântico como ocorre a adsorção, e
analisar as interações entre as duas espécies.
Também procederemos com o estudo de MOFs compostas por íons lantanídios, mais
especificamente o íon Eu3+ na busca de eficientes dispositivos moleculares conversores de
luz que possam ser aplicados como marcadores luminescentes.
Por fim,iremos escrever e submeter os artigos referentes aos estudos apresentados no
capítulo 4 e 6 uma vez que um dos trabalhos relativos ao estudo apresentado no capítulo 5
foi publicado na revista RSC Advances.
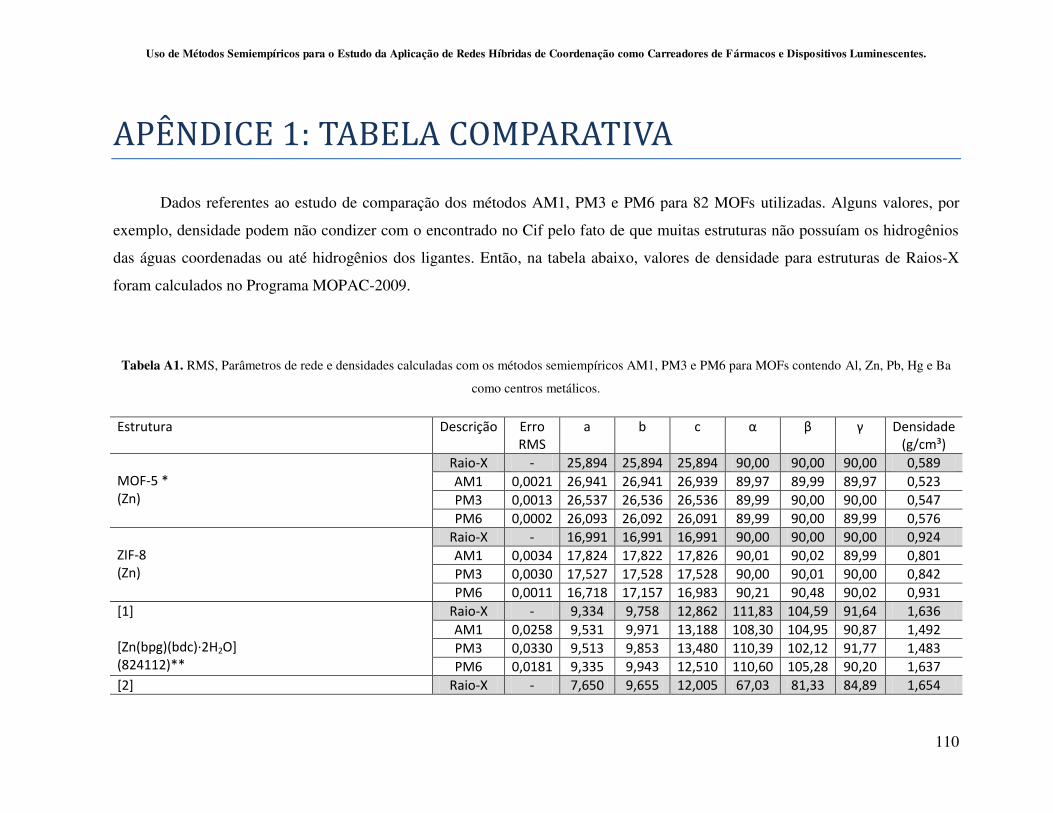
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
110
APÊNDICE 1: TABELA COMPARATIVA
Dados referentes ao estudo de comparação dos métodos AM1, PM3 e PM6 para 82 MOFs utilizadas. Alguns valores, por
exemplo, densidade podem não condizer com o encontrado no Cif pelo fato de que muitas estruturas não possuíam os hidrogênios
das águas coordenadas ou até hidrogênios dos ligantes. Então, na tabela abaixo, valores de densidade para estruturas de Raios-X
foram calculados no Programa MOPAC-2009.
Tabela A1. RMS, Parâmetros de rede e densidades calculadas com os métodos semiempíricos AM1, PM3 e PM6 para MOFs contendo Al, Zn, Pb, Hg e Ba
como centros metálicos.
Estrutura
Descrição Erro
RMS
a b c α β γ Densidade
(g/cm³)
MOF-5 *
(Zn)
Raio-X - 25,894 25,894 25,894 90,00 90,00 90,00 0,589
AM1 0,0021 26,941 26,941 26,939 89,97 89,99 89,97 0,523
PM3 0,0013 26,537 26,536 26,536 89,99 90,00 90,00 0,547
PM6 0,0002 26,093 26,092 26,091 89,99 90,00 89,99 0,576
ZIF-8
(Zn)
Raio-X - 16,991 16,991 16,991 90,00 90,00 90,00 0,924
AM1 0,0034 17,824 17,822 17,826 90,01 90,02 89,99 0,801
PM3 0,0030 17,527 17,528 17,528 90,00 90,01 90,00 0,842
PM6 0,0011 16,718 17,157 16,983 90,21 90,48 90,02 0,931
[1]
[Zn(bpg)(bdc)·2H2O]
(824112)**
Raio-X - 9,334 9,758 12,862 111,83 104,59 91,64 1,636
AM1 0,0258 9,531 9,971 13,188 108,30 104,95 90,87 1,492
PM3 0,0330 9,513 9,853 13,480 110,39 102,12 91,77 1,483
PM6 0,0181 9,335 9,943 12,510 110,60 105,28 90,20 1,637
[2] Raio-X - 7,650 9,655 12,005 67,03 81,33 84,89 1,654
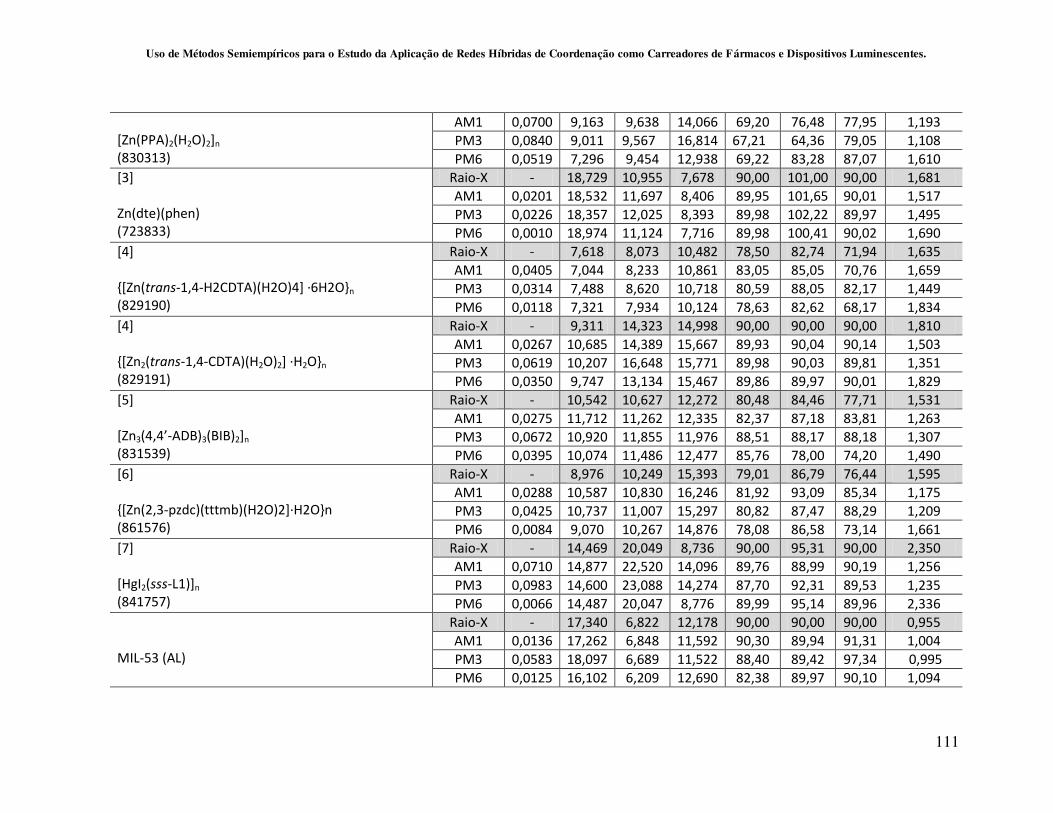
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
111
[Zn(PPA)2(H2O)2]n
(830313)
AM1 0,0700 9,163 9,638 14,066 69,20 76,48 77,95 1,193
PM3 0,0840 9,011 9,567 16,814 67,21 64,36 79,05 1,108
PM6 0,0519 7,296 9,454 12,938 69,22 83,28 87,07 1,610
[3]
Zn(dte)(phen)
(723833)
Raio-X - 18,729 10,955 7,678 90,00 101,00 90,00 1,681
AM1 0,0201 18,532 11,697 8,406 89,95 101,65 90,01 1,517
PM3 0,0226 18,357 12,025 8,393 89,98 102,22 89,97 1,495
PM6 0,0010 18,974 11,124 7,716 89,98 100,41 90,02 1,690
[4]
[Zn(trans-1,4-H2CDTA)(H2O)4] ·6H2On
(829190)
Raio-X - 7,618 8,073 10,482 78,50 82,74 71,94 1,635
AM1 0,0405 7,044 8,233 10,861 83,05 85,05 70,76 1,659
PM3 0,0314 7,488 8,620 10,718 80,59 88,05 82,17 1,449
PM6 0,0118 7,321 7,934 10,124 78,63 82,62 68,17 1,834
[4]
[Zn2(trans-1,4-CDTA)(H2O)2] ·H2On
(829191)
Raio-X - 9,311 14,323 14,998 90,00 90,00 90,00 1,810
AM1 0,0267 10,685 14,389 15,667 89,93 90,04 90,14 1,503
PM3 0,0619 10,207 16,648 15,771 89,98 90,03 89,81 1,351
PM6 0,0350 9,747 13,134 15,467 89,86 89,97 90,01 1,829
[5]
[Zn3 4,4’-ADB)3(BIB)2]n
(831539)
Raio-X - 10,542 10,627 12,272 80,48 84,46 77,71 1,531
AM1 0,0275 11,712 11,262 12,335 82,37 87,18 83,81 1,263
PM3 0,0672 10,920 11,855 11,976 88,51 88,17 88,18 1,307
PM6 0,0395 10,074 11,486 12,477 85,76 78,00 74,20 1,490
[6]
[Zn(2,3-pzdc)(tttmb)(H2O)2]·H2On
(861576)
Raio-X - 8,976 10,249 15,393 79,01 86,79 76,44 1,595
AM1 0,0288 10,587 10,830 16,246 81,92 93,09 85,34 1,175
PM3 0,0425 10,737 11,007 15,297 80,82 87,47 88,29 1,209
PM6 0,0084 9,070 10,267 14,876 78,08 86,58 73,14 1,661
[7]
[HgI2(sss-L1)]n
(841757)
Raio-X - 14,469 20,049 8,736 90,00 95,31 90,00 2,350
AM1 0,0710 14,877 22,520 14,096 89,76 88,99 90,19 1,256
PM3 0,0983 14,600 23,088 14,274 87,70 92,31 89,53 1,235
PM6 0,0066 14,487 20,047 8,776 89,99 95,14 89,96 2,336
MIL-53 (AL)
Raio-X - 17,340 6,822 12,178 90,00 90,00 90,00 0,955
AM1 0,0136 17,262 6,848 11,592 90,30 89,94 91,31 1,004
PM3 0,0583 18,097 6,689 11,522 88,40 89,42 97,34 0,995
PM6 0,0125 16,102 6,209 12,690 82,38 89,97 90,10 1,094
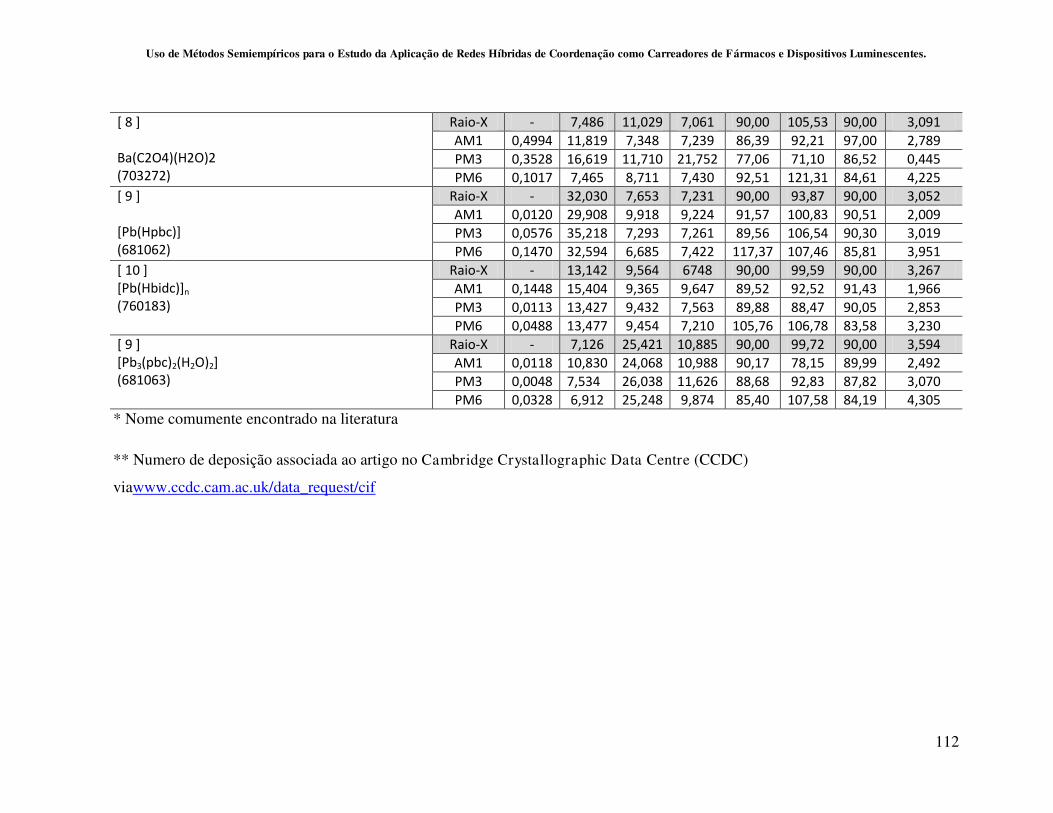
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
112
[ 8 ]
Ba(C2O4)(H2O)2
(703272)
Raio-X - 7,486 11,029 7,061 90,00 105,53 90,00 3,091
AM1 0,4994 11,819 7,348 7,239 86,39 92,21 97,00 2,789
PM3 0,3528 16,619 11,710 21,752 77,06 71,10 86,52 0,445
PM6 0,1017 7,465 8,711 7,430 92,51 121,31 84,61 4,225
[ 9 ]
[Pb(Hpbc)]
(681062)
Raio-X - 32,030 7,653 7,231 90,00 93,87 90,00 3,052
AM1 0,0120 29,908 9,918 9,224 91,57 100,83 90,51 2,009
PM3 0,0576 35,218 7,293 7,261 89,56 106,54 90,30 3,019
PM6 0,1470 32,594 6,685 7,422 117,37 107,46 85,81 3,951
[ 10 ]
[Pb(Hbidc)]n
(760183)
Raio-X - 13,142 9,564 6748 90,00 99,59 90,00 3,267
AM1 0,1448 15,404 9,365 9,647 89,52 92,52 91,43 1,966
PM3 0,0113 13,427 9,432 7,563 89,88 88,47 90,05 2,853
PM6 0,0488 13,477 9,454 7,210 105,76 106,78 83,58 3,230
[ 9 ]
[Pb3(pbc)2(H2O)2]
(681063)
Raio-X - 7,126 25,421 10,885 90,00 99,72 90,00 3,594
AM1 0,0118 10,830 24,068 10,988 90,17 78,15 89,99 2,492
PM3 0,0048 7,534 26,038 11,626 88,68 92,83 87,82 3,070
PM6 0,0328 6,912 25,248 9,874 85,40 107,58 84,19 4,305
* Nome comumente encontrado na literatura
** Numero de deposição associada ao artigo no Cambridge Crystallographic Data Centre (CCDC)
viawww.ccdc.cam.ac.uk/data_request/cif
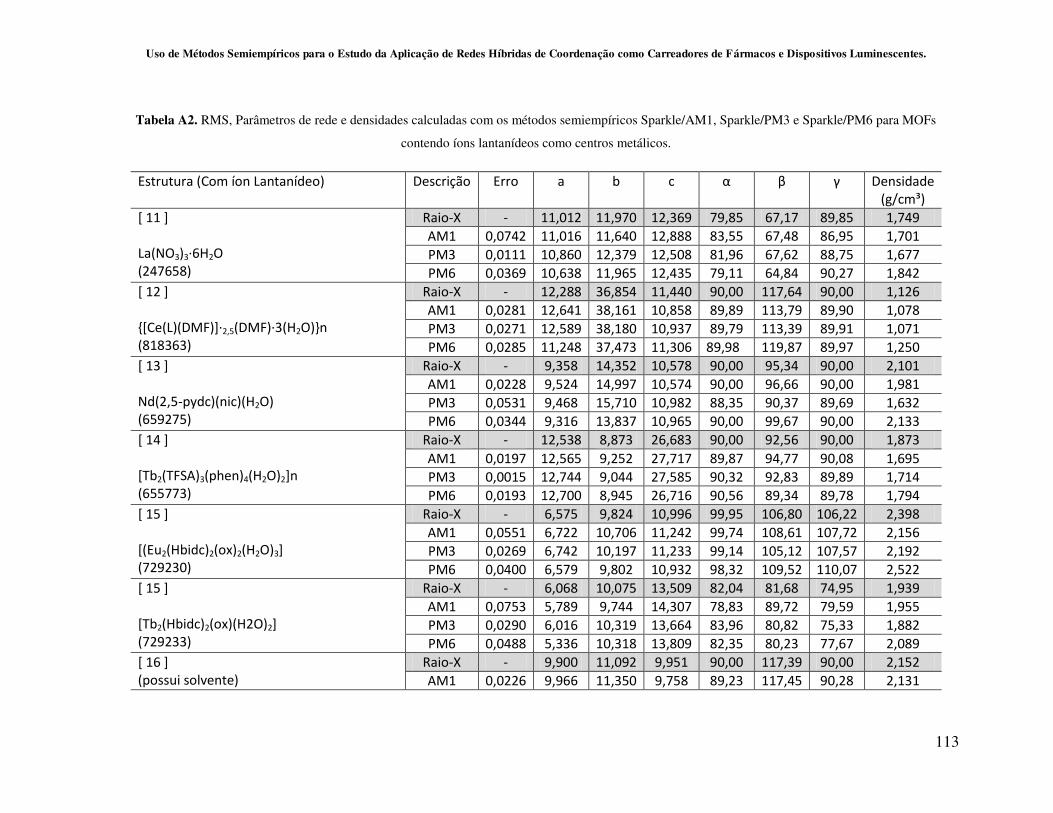
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
113
Tabela A2. RMS, Parâmetros de rede e densidades calculadas com os métodos semiempíricos Sparkle/AM1, Sparkle/PM3 e Sparkle/PM6 para MOFs
contendo íons lantanídeos como centros metálicos.
Estrutura (Com íon Lantanídeo)
Descrição Erro a b c α β γ Densidade
(g/cm³)
[ 11 ]
La(NO3)3·6H2O
(247658)
Raio-X - 11,012 11,970 12,369 79,85 67,17 89,85 1,749
AM1 0,0742 11,016 11,640 12,888 83,55 67,48 86,95 1,701
PM3 0,0111 10,860 12,379 12,508 81,96 67,62 88,75 1,677
PM6 0,0369 10,638 11,965 12,435 79,11 64,84 90,27 1,842
[ 12 ]
[Ce(L)(DMF)]·2,5(DMF)·3(H2O)n
(818363)
Raio-X - 12,288 36,854 11,440 90,00 117,64 90,00 1,126
AM1 0,0281 12,641 38,161 10,858 89,89 113,79 89,90 1,078
PM3 0,0271 12,589 38,180 10,937 89,79 113,39 89,91 1,071
PM6 0,0285 11,248 37,473 11,306 89,98 119,87 89,97 1,250
[ 13 ]
Nd(2,5-pydc)(nic)(H2O)
(659275)
Raio-X - 9,358 14,352 10,578 90,00 95,34 90,00 2,101
AM1 0,0228 9,524 14,997 10,574 90,00 96,66 90,00 1,981
PM3 0,0531 9,468 15,710 10,982 88,35 90,37 89,69 1,632
PM6 0,0344 9,316 13,837 10,965 90,00 99,67 90,00 2,133
[ 14 ]
[Tb2(TFSA)3(phen)4(H2O)2]n
(655773)
Raio-X - 12,538 8,873 26,683 90,00 92,56 90,00 1,873
AM1 0,0197 12,565 9,252 27,717 89,87 94,77 90,08 1,695
PM3 0,0015 12,744 9,044 27,585 90,32 92,83 89,89 1,714
PM6 0,0193 12,700 8,945 26,716 90,56 89,34 89,78 1,794
[ 15 ]
[(Eu2(Hbidc)2(ox)2(H2O)3]
(729230)
Raio-X - 6,575 9,824 10,996 99,95 106,80 106,22 2,398
AM1 0,0551 6,722 10,706 11,242 99,74 108,61 107,72 2,156
PM3 0,0269 6,742 10,197 11,233 99,14 105,12 107,57 2,192
PM6 0,0400 6,579 9,802 10,932 98,32 109,52 110,07 2,522
[ 15 ]
[Tb2(Hbidc)2(ox)(H2O)2]
(729233)
Raio-X - 6,068 10,075 13,509 82,04 81,68 74,95 1,939
AM1 0,0753 5,789 9,744 14,307 78,83 89,72 79,59 1,955
PM3 0,0290 6,016 10,319 13,664 83,96 80,82 75,33 1,882
PM6 0,0488 5,336 10,318 13,809 82,35 80,23 77,67 2,089
[ 16 ]
(possui solvente)
Raio-X - 9,900 11,092 9,951 90,00 117,39 90,00 2,152
AM1 0,0226 9,966 11,350 9,758 89,23 117,45 90,28 2,131
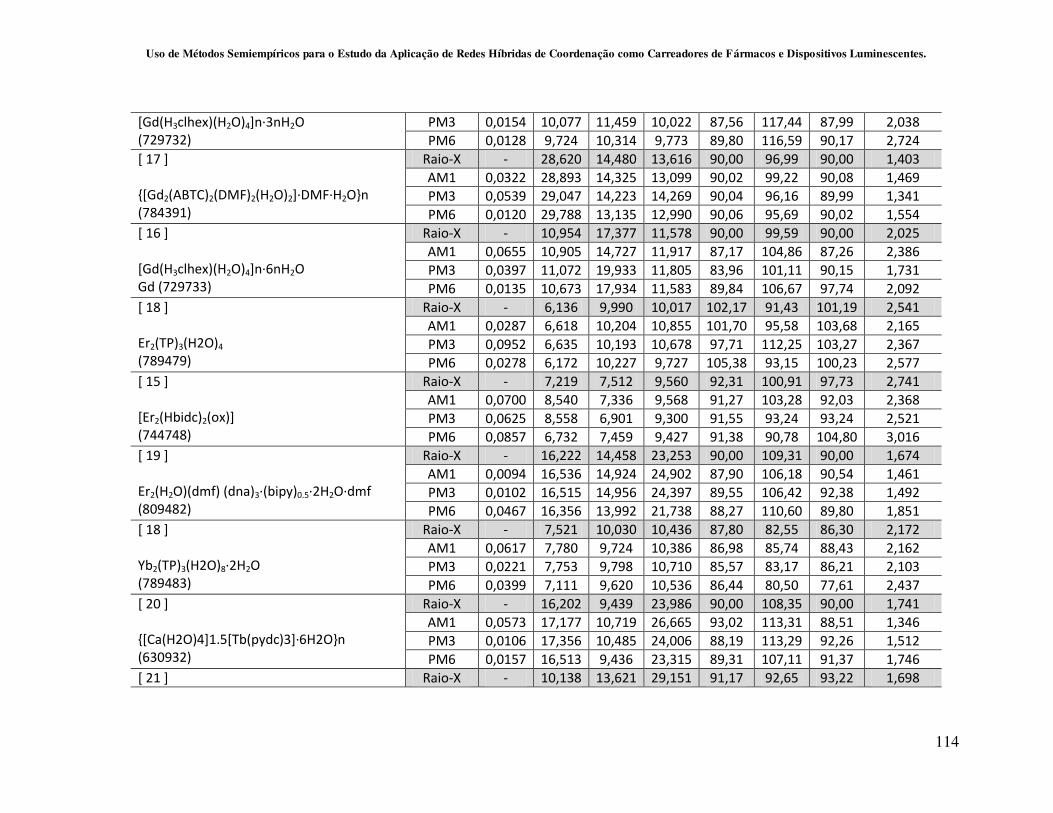
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
114
[Gd(H3clhex)(H2O)4]n·3nH2O
(729732)
PM3 0,0154 10,077 11,459 10,022 87,56 117,44 87,99 2,038
PM6 0,0128 9,724 10,314 9,773 89,80 116,59 90,17 2,724
[ 17 ]
[Gd2(ABTC)2(DMF)2(H2O)2]·DMF·H2On
(784391)
Raio-X - 28,620 14,480 13,616 90,00 96,99 90,00 1,403
AM1 0,0322 28,893 14,325 13,099 90,02 99,22 90,08 1,469
PM3 0,0539 29,047 14,223 14,269 90,04 96,16 89,99 1,341
PM6 0,0120 29,788 13,135 12,990 90,06 95,69 90,02 1,554
[ 16 ]
[Gd(H3clhex)(H2O)4]n·6nH2O
Gd (729733)
Raio-X - 10,954 17,377 11,578 90,00 99,59 90,00 2,025
AM1 0,0655 10,905 14,727 11,917 87,17 104,86 87,26 2,386
PM3 0,0397 11,072 19,933 11,805 83,96 101,11 90,15 1,731
PM6 0,0135 10,673 17,934 11,583 89,84 106,67 97,74 2,092
[ 18 ]
Er2(TP)3(H2O)4
(789479)
Raio-X - 6,136 9,990 10,017 102,17 91,43 101,19 2,541
AM1 0,0287 6,618 10,204 10,855 101,70 95,58 103,68 2,165
PM3 0,0952 6,635 10,193 10,678 97,71 112,25 103,27 2,367
PM6 0,0278 6,172 10,227 9,727 105,38 93,15 100,23 2,577
[ 15 ]
[Er2(Hbidc)2(ox)]
(744748)
Raio-X - 7,219 7,512 9,560 92,31 100,91 97,73 2,741
AM1 0,0700 8,540 7,336 9,568 91,27 103,28 92,03 2,368
PM3 0,0625 8,558 6,901 9,300 91,55 93,24 93,24 2,521
PM6 0,0857 6,732 7,459 9,427 91,38 90,78 104,80 3,016
[ 19 ]
Er2(H2O)(dmf) (dna)3·(bipy)0.5·2H2O·dmf
(809482)
Raio-X - 16,222 14,458 23,253 90,00 109,31 90,00 1,674
AM1 0,0094 16,536 14,924 24,902 87,90 106,18 90,54 1,461
PM3 0,0102 16,515 14,956 24,397 89,55 106,42 92,38 1,492
PM6 0,0467 16,356 13,992 21,738 88,27 110,60 89,80 1,851
[ 18 ]
Yb2(TP)3(H2O)8·2H2O
(789483)
Raio-X - 7,521 10,030 10,436 87,80 82,55 86,30 2,172
AM1 0,0617 7,780 9,724 10,386 86,98 85,74 88,43 2,162
PM3 0,0221 7,753 9,798 10,710 85,57 83,17 86,21 2,103
PM6 0,0399 7,111 9,620 10,536 86,44 80,50 77,61 2,437
[ 20 ]
[Ca(H2O)4]1.5[Tb(pydc)3]·6H2On
(630932)
Raio-X - 16,202 9,439 23,986 90,00 108,35 90,00 1,741
AM1 0,0573 17,177 10,719 26,665 93,02 113,31 88,51 1,346
PM3 0,0106 17,356 10,485 24,006 88,19 113,29 92,26 1,512
PM6 0,0157 16,513 9,436 23,315 89,31 107,11 91,37 1,746
[ 21 ] Raio-X - 10,138 13,621 29,151 91,17 92,65 93,22 1,698

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
115
[Zn2Er(L)2(Py)2(NO3)2]·[ZnEr(L)(Py)
(NO3)3(H2O)]·NO3·mMeOH·nS
(733875)
AM1 0,0426 11,156 13,980 33,781 86,42 85,70 91,25 1,300
PM3 0,0318 11,629 14,768 32,889 98,71 83,55 100,97 1,248
PM6 0,0045 10,185 13,278 29,876 89,33 95,47 95,35 1,702
[ 21 ]
[Zn2Gd(L)2(Py)2(NO3)2]·[ZnGd(L)
(Py)(NO3)3(H2O)]·NO3·mMeOH·nS
(734050)
Raio-X - 10,146 13,592 29,126 91,16 92,76 93,24 1,669
AM1 0,0103 11,206 14,988 31,614 94,77 92,12 83,20 1,272
PM3 0,0868 12,297 14,709 31,464 94,77 93,41 89,00 1,180
PM6 0,0271 9,948 13,548 28,728 91,77 92,35 88,61 1,728
[ 22 ]
[La(BDOA)1,5(H2O)]·H2On
(614802)
Raio-X - 12,158 16,915 8,932 90,00 108,88 90,00 1,954
AM1 0,0270 13,281 17,832 9,804 89,83 116,78 89,78 1,639
PM3 0,0288 13,549 16,862 9,658 90,03 116,16 90,03 1,714
PM6 0,0236 12,606 16,883 9,213 90,08 111,18 90,15 1,857
[ 22 ]
[Nd(BDOA)1,5(H2O)]·H2On
(639026)
Raio-X - 12,220 16,734 8,953 90,00 110,01 90,00 1,994
AM1 0,0350 12,236 17,272 9,156 90,00 111,38 89,91 1,904
PM3 0,0192 13,284 16,650 9,580 90,03 116,73 90,04 1,813
PM6 0,0264 12,203 16,760 8,870 90,04 110,38 89,91 2,017
[ 22 ]
[Tm(BDOA)1,5(H2O)]n
(630419)
Raio-X - 6,047 10,651 13,492 111,95 96,82 97,78 2,214
AM1 0,0159 6,569 10,957 13,660 111,60 93,37 98,20 1,935
PM3 0,0343 6,311 11,526 13,881 115,08 97,38 97,22 1,956
PM6 0,0202 6,364 10,527 13,363 112,78 96,69 98,98 2,173
[ 22 ]
[Yb(BDOA)1,5(H2O)]n
(641231)
Raio-X - 6,049 10,626 13,484 111,90 96,75 97,71 2,235
AM1 0,0356 6,424 10,826 13,657 112,17 94,81 100,12 2,051
PM3 0,0643 6,235 11,582 13,246 112,55 96,12 97,16 2,027
PM6 0,0469 6,219 10,455 13,118 112,85 97,80 95,88 2,282
[ 23 ]
[Eu(PYDC)(C2O4)0.5(H2O)2]·H2O
(668575)
Raio-X - 6,070 10,163 11,329 105,68 100,94 105,13 2,211
AM1 0,0936 5,984 9,749 12,550 109,04 106,78 101,24 2,196
PM3 0,1152 6,049 10,269 12,458 109,71 102,57 103,16 2,052
PM6 0,1400 5,293 10,002 12,795 104,64 118,99 99,30 2,553
[ 23 ]
[Gd(PYDC)(C2O4)0.5(H2O)2]·H2O
(668576)
Raio-X - 6,059 10,135 11,304 105,61 100,85 105,17 2,253
AM1 0,0668 6,380 9,929 11,900 110,53 106,81 105,53 2,267
PM3 0,0943 6,206 10,300 12,210 102,34 98,04 108,56 1,982
PM6 0,0596 5,524 10,097 12,070 106,68 106,29 103,94 2,408
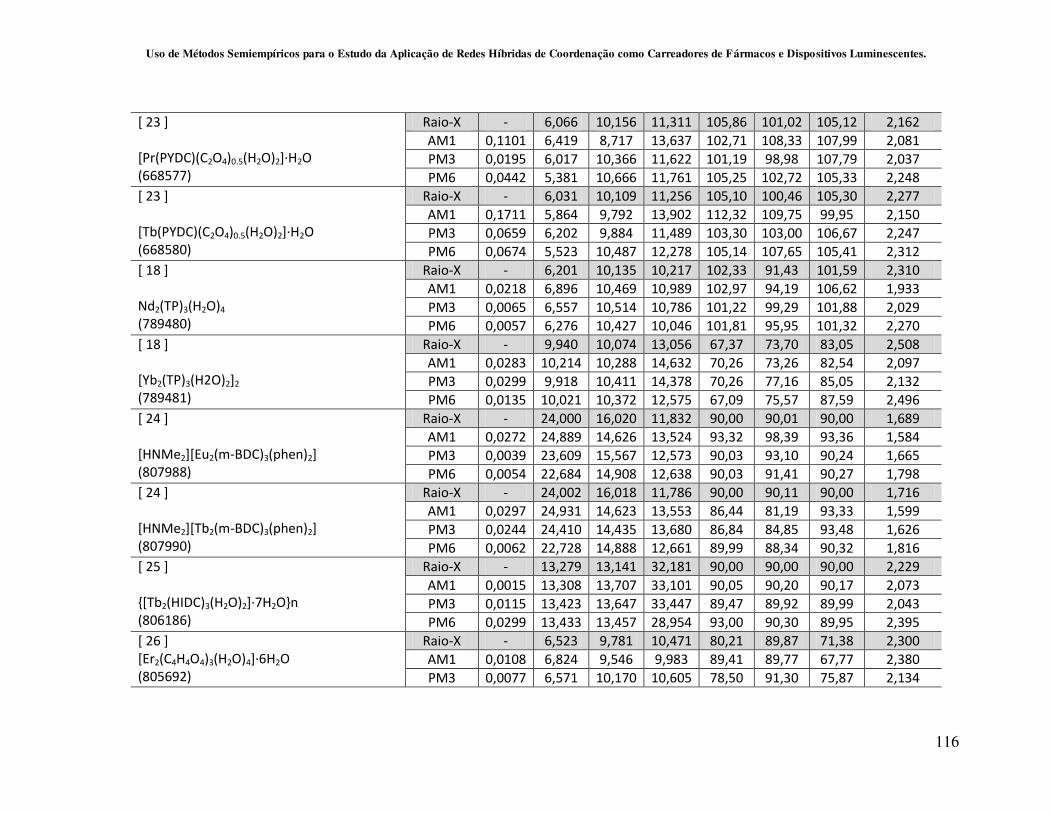
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
116
[ 23 ]
[Pr(PYDC)(C2O4)0.5(H2O)2]·H2O
(668577)
Raio-X - 6,066 10,156 11,311 105,86 101,02 105,12 2,162
AM1 0,1101 6,419 8,717 13,637 102,71 108,33 107,99 2,081
PM3 0,0195 6,017 10,366 11,622 101,19 98,98 107,79 2,037
PM6 0,0442 5,381 10,666 11,761 105,25 102,72 105,33 2,248
[ 23 ]
[Tb(PYDC)(C2O4)0.5(H2O)2]·H2O
(668580)
Raio-X - 6,031 10,109 11,256 105,10 100,46 105,30 2,277
AM1 0,1711 5,864 9,792 13,902 112,32 109,75 99,95 2,150
PM3 0,0659 6,202 9,884 11,489 103,30 103,00 106,67 2,247
PM6 0,0674 5,523 10,487 12,278 105,14 107,65 105,41 2,312
[ 18 ]
Nd2(TP)3(H2O)4
(789480)
Raio-X - 6,201 10,135 10,217 102,33 91,43 101,59 2,310
AM1 0,0218 6,896 10,469 10,989 102,97 94,19 106,62 1,933
PM3 0,0065 6,557 10,514 10,786 101,22 99,29 101,88 2,029
PM6 0,0057 6,276 10,427 10,046 101,81 95,95 101,32 2,270
[ 18 ]
[Yb2(TP)3(H2O)2]2
(789481)
Raio-X - 9,940 10,074 13,056 67,37 73,70 83,05 2,508
AM1 0,0283 10,214 10,288 14,632 70,26 73,26 82,54 2,097
PM3 0,0299 9,918 10,411 14,378 70,26 77,16 85,05 2,132
PM6 0,0135 10,021 10,372 12,575 67,09 75,57 87,59 2,496
[ 24 ]
[HNMe2][Eu2(m-BDC)3(phen)2]
(807988)
Raio-X - 24,000 16,020 11,832 90,00 90,01 90,00 1,689
AM1 0,0272 24,889 14,626 13,524 93,32 98,39 93,36 1,584
PM3 0,0039 23,609 15,567 12,573 90,03 93,10 90,24 1,665
PM6 0,0054 22,684 14,908 12,638 90,03 91,41 90,27 1,798
[ 24 ]
[HNMe2][Tb2(m-BDC)3(phen)2]
(807990)
Raio-X - 24,002 16,018 11,786 90,00 90,11 90,00 1,716
AM1 0,0297 24,931 14,623 13,553 86,44 81,19 93,33 1,599
PM3 0,0244 24,410 14,435 13,680 86,84 84,85 93,48 1,626
PM6 0,0062 22,728 14,888 12,661 89,99 88,34 90,32 1,816
[ 25 ]
[Tb2(HIDC)3(H2O)2]·7H2On
(806186)
Raio-X - 13,279 13,141 32,181 90,00 90,00 90,00 2,229
AM1 0,0015 13,308 13,707 33,101 90,05 90,20 90,17 2,073
PM3 0,0115 13,423 13,647 33,447 89,47 89,92 89,99 2,043
PM6 0,0299 13,433 13,457 28,954 93,00 90,30 89,95 2,395
[ 26 ]
[Er2(C4H4O4)3(H2O)4]·6H2O
(805692)
Raio-X - 6,523 9,781 10,471 80,21 89,87 71,38 2,300
AM1 0,0108 6,824 9,546 9,983 89,41 89,77 67,77 2,380
PM3 0,0077 6,571 10,170 10,605 78,50 91,30 75,87 2,134
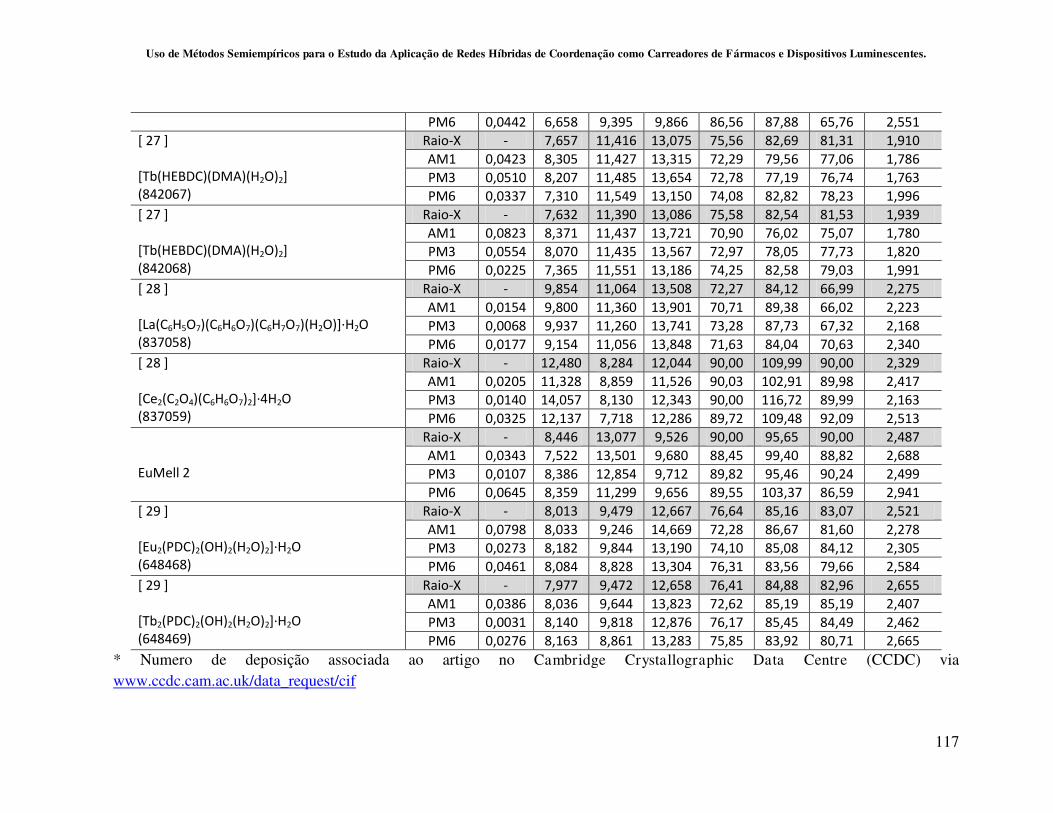
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
117
PM6 0,0442 6,658 9,395 9,866 86,56 87,88 65,76 2,551
[ 27 ]
[Tb(HEBDC)(DMA)(H2O)2]
(842067)
Raio-X - 7,657 11,416 13,075 75,56 82,69 81,31 1,910
AM1 0,0423 8,305 11,427 13,315 72,29 79,56 77,06 1,786
PM3 0,0510 8,207 11,485 13,654 72,78 77,19 76,74 1,763
PM6 0,0337 7,310 11,549 13,150 74,08 82,82 78,23 1,996
[ 27 ]
[Tb(HEBDC)(DMA)(H2O)2]
(842068)
Raio-X - 7,632 11,390 13,086 75,58 82,54 81,53 1,939
AM1 0,0823 8,371 11,437 13,721 70,90 76,02 75,07 1,780
PM3 0,0554 8,070 11,435 13,567 72,97 78,05 77,73 1,820
PM6 0,0225 7,365 11,551 13,186 74,25 82,58 79,03 1,991
[ 28 ]
[La(C6H5O7)(C6H6O7)(C6H7O7)(H2O)]·H2O
(837058)
Raio-X - 9,854 11,064 13,508 72,27 84,12 66,99 2,275
AM1 0,0154 9,800 11,360 13,901 70,71 89,38 66,02 2,223
PM3 0,0068 9,937 11,260 13,741 73,28 87,73 67,32 2,168
PM6 0,0177 9,154 11,056 13,848 71,63 84,04 70,63 2,340
[ 28 ]
[Ce2(C2O4)(C6H6O7)2]·4H2O
(837059)
Raio-X - 12,480 8,284 12,044 90,00 109,99 90,00 2,329
AM1 0,0205 11,328 8,859 11,526 90,03 102,91 89,98 2,417
PM3 0,0140 14,057 8,130 12,343 90,00 116,72 89,99 2,163
PM6 0,0325 12,137 7,718 12,286 89,72 109,48 92,09 2,513
EuMell 2
Raio-X - 8,446 13,077 9,526 90,00 95,65 90,00 2,487
AM1 0,0343 7,522 13,501 9,680 88,45 99,40 88,82 2,688
PM3 0,0107 8,386 12,854 9,712 89,82 95,46 90,24 2,499
PM6 0,0645 8,359 11,299 9,656 89,55 103,37 86,59 2,941
[ 29 ]
[Eu2(PDC)2(OH)2(H2O)2]·H2O
(648468)
Raio-X - 8,013 9,479 12,667 76,64 85,16 83,07 2,521
AM1 0,0798 8,033 9,246 14,669 72,28 86,67 81,60 2,278
PM3 0,0273 8,182 9,844 13,190 74,10 85,08 84,12 2,305
PM6 0,0461 8,084 8,828 13,304 76,31 83,56 79,66 2,584
[ 29 ]
[Tb2(PDC)2(OH)2(H2O)2]·H2O
(648469)
Raio-X - 7,977 9,472 12,658 76,41 84,88 82,96 2,655
AM1 0,0386 8,036 9,644 13,823 72,62 85,19 85,19 2,407
PM3 0,0031 8,140 9,818 12,876 76,17 85,45 84,49 2,462
PM6 0,0276 8,163 8,861 13,283 75,85 83,92 80,71 2,665
* Numero de deposição associada ao artigo no Cambridge Crystallographic Data Centre (CCDC) via www.ccdc.cam.ac.uk/data_request/cif

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
118
Tabela A3. RMS, Parâmetros de rede e densidades calculadas com o método semiempírico PM6 para MOFs contendo Mn, Fe, Co, Ni, Cu, Y, Ag e Cd como
centros metálicos.
Estrutura
Numero de
deposição*
Descrição Erro a b c α β γ Densidade
(g/cm³)
[30]
[Ni(PDA)(BPE)]
(786118) Raio-X - 10,130 14,417 13,377 90,00 102,79 90,00 1,517
PM6 0,0457 9,593 17,156 12,989 90,41 94,93 89,95 1,357
[ 31 ]
[Co(L1)2(H2O)4]
(616704) Raio-X - 13,113 9,826 8,053 90,00 106,25 90,00 1,631
PM6 0,1196 16,609 13,597 14,355 65,57 64,70 79,19 0,609
[ 31 ]
[Cu(L1)2(H2O)(Py)2]
(616706) Raio-X - 15,075 5,967 30,824 90,00 90,00 90,00 1,450
PM6 0,0068 14,877 5,365 31,694 90,09 98,68 89,94 1,589
[ 31 ]
[Cu(L3)(H2O)Cl]·H2O
(616708) Raio-X - 7,413 18,289 21,406 90,00 90,00 90,00 1,631
PM6 0,0096 7,423 17,619 21,476 89,84 89,86 89,86 1,685
[ 31 ]
[Mn(L1)2(H2O)4]
(616709) Raio-X - 13,256 9,869 8,100 90,00 106,48 90,00 1,586
PM6 0,0124 13,088 9,967 7,849 90,40 108,70 89,52 1,662
[ 32 ]
[Cu2(bpp)(μ-CH3COO)4]·2H2O
(680505) Raio-X - 13,023 28,433 8,819 90,00 90,00 90,00 1,215
PM6 0,0190 13,251 28,107 8,579 89,90 89,73 90,45 1,242
[ 33 ]
[Cu(PDCO)·(H2O)]n
(679651) Raio-X - 11,429 6,871 19,916 90,00 90,00 90,00 2,240
PM6 0,0212 11,966 7,643 19,283 89,94 89,93 90,06 1,986
[ 34 ]
Y2(C4H4O4)3(H2O)4·6H2O
(735614) Raio-X - 6,533 9,783 10,489 80,21 89,83 71,38 1,876
PM6 0,0419 6,694 8,204 8,490 71,81 85,82 66,14 2,900
[ 35 ]
[Ni(deen)(μ1,5-dca)2]n
(748445) Raio-X - 15,590 8,162 10,789 90,00 90,00 90,00 1,485
PM6 0,0747 15,685 8,896 10,877 89,84 89,70 90,06 1,344
[4]
[CoII(trans-1,4-
H2CDTA)(H2O)4]·6H2On
(756613) Raio-X - 7,603 8,192 10,557 77,89 83,10 72,13 1,586
PM6 0,0187 7,430 7,747 10,454 81,05 86,59 75,90 1,681
[7]
[CoCl2(ass-L1)(EtOH)]2
(841758) Raio-X - 8,332 13,665 13,752 63,44 79,63 81,47 1,485
PM6 0,0944 11,693 16,962 16,652 60,63 93,46 83,24 0,721
[ 36 ] (792204) Raio-X - 18,120 18,120 7,518 90,00 90,00 120,00 2,223

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
119
[Cu4I4(dach)2] PM6 0,0932 14,371 15,826 9,723 89,55 90,80 125,05 2,626
[ 37 ]
[MnL2(CH3OH)]n
(835967) Raio-X - 9,219 12,257 13,587 69,01 72,41 82,80 1,375
PM6 0,0630 9,229 12,717 13,685 70,01 72,15 83,00 1,307
[ 37 ]
[AgL]∞
(835965) Raio-X - 11,075 11,191 19,833 90,00 96,13 90,00 1,887
PM6 0,0489 22,258 11,822 18,654 91,05 92,48 90,89 0,941
[ 38 ]
[Fe 4,4’-ipy)(H2O)4]·(C5O5)·(H2O)2n
(617098) Raio-X - 6,770 11,437 12,383 90,00 103,73 90,00 1,541
PM6 0,0663 7,175 10,133 13,133 88,86 109,52 92,60 1,700
CuBTC ------------ Raio-X - 26,343 26,343 26,343 90,00 90,00 90,00 0,958
PM6 0,0044 26,021 26,017 25,895 90,24 89,65 89,73 0,999
[ 39 ]
Cu6 μ3-O μ3-OH)(pz)6(btc)
(847915) Raio-X -
PM6 0,0171 20,126 20,683 19,757 88,94 88,57 90,03 1,653
[ 36 ]
[Cu4l4(bpp)2]
(792205) Raio-X - 19,559 36,445 10,438 90,00 90,00 90,00 2,097
PM6 0,0718 23,188 34,879 10,091 90,28 90,00 90,22 1,912
[ 40 ]
[Cu0.5(btrb)0.5Cl]n
(800362) Raio-X - 7,517 9,397 8,942 90,00 96,97 90,00 1,730
PM6 0,0309 7,292 9,193 8,728 89,62 94,91 90,15 1,861
Estrutura
Numero de
deposição*
Descrição Erro a b c α β γ Densidade
(g/cm³)
[41]
[Cd(μ3-HIDC)(bbi)0,5]n
(608891) Raio-X - 11,913 10,147 11,913 90,00 115,70 90,00 1,851
PM6 0,0480 11,932 9,511 13,136 78,87 121,06 109,11 1,991
[33]
[Cd(PDCO)(bipy)· (H2O)·5H2O]n
(679652) Raio-X - 7,704 8,821 15,558 83,06 77,10 77,23 1,848
PM6 0,0210 8,438 8,416 15,138 76,49 72,07 75,87 1,896
[1]
[Cd(bpg)(bdc)2H2O]
(824111) Raio-X - 9664 9,922 12,934 111,11 105,50 93,35 1,599
PM6 0,1461 10,813 7,954 14,674 101,34 120,60 96,40 1,703
[37]
[CdL2](CH3OH)(H2O)n
(835966) Raio-X - 9,690 13,157 13,669 117,76 94,75 102,80 1,443
PM6 0,0516 8,401 12,132 14,952 117,82 100,94 96,04 1,643
[7]
[CdCl2(aas-L1)]n
(841756) Raio-X - 8,741 10,086 13,318 82,14 87,82 89,04 1,777
PM6 0,0175 8,088 9,499 13,995 82,25 87,22 88,84 1,941
[42]
[Cd(pmida)H2O]·1,8 H2O]n
(846089) Raio-X - 8,395 9,562 16,475 90,00 90,00 90,00 2,012
PM6 0,0403 8,029 9,634 15,762 90,45 94,04 89,01 2,188
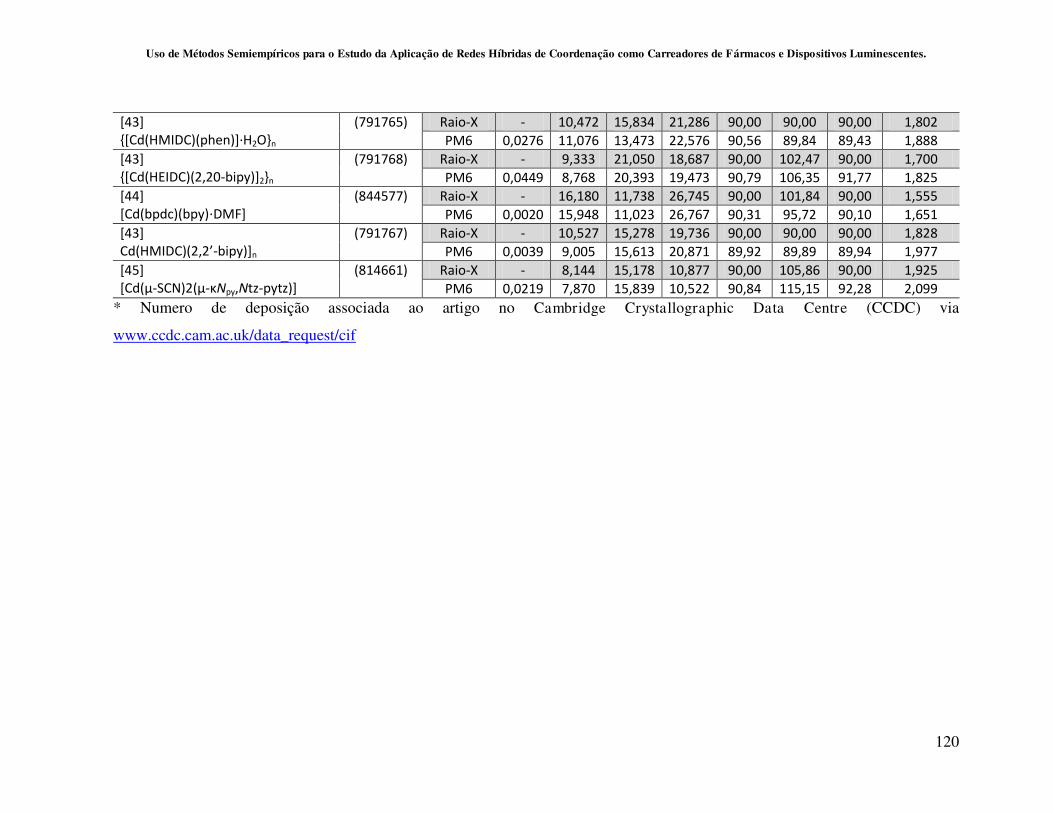
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
120
[43]
[Cd(HMIDC)(phen)]·H2On
(791765) Raio-X - 10,472 15,834 21,286 90,00 90,00 90,00 1,802
PM6 0,0276 11,076 13,473 22,576 90,56 89,84 89,43 1,888
[43]
[Cd(HEIDC)(2,20-bipy)]2n
(791768) Raio-X - 9,333 21,050 18,687 90,00 102,47 90,00 1,700
PM6 0,0449 8,768 20,393 19,473 90,79 106,35 91,77 1,825
[44]
[Cd(bpdc)(bpy)⋅DMF]
(844577) Raio-X - 16,180 11,738 26,745 90,00 101,84 90,00 1,555
PM6 0,0020 15,948 11,023 26,767 90,31 95,72 90,10 1,651
[43]
Cd HMIDC 2,2’-bipy)]n
(791767) Raio-X - 10,527 15,278 19,736 90,00 90,00 90,00 1,828
PM6 0,0039 9,005 15,613 20,871 89,92 89,89 89,94 1,977
[45]
[Cd(μ-SCN)2(μ-κNpy,Ntz-pytz)]
(814661) Raio-X - 8,144 15,178 10,877 90,00 105,86 90,00 1,925
PM6 0,0219 7,870 15,839 10,522 90,84 115,15 92,28 2,099
* Numero de deposição associada ao artigo no Cambridge Crystallographic Data Centre (CCDC) via
www.ccdc.cam.ac.uk/data_request/cif

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
121
Na tabela A4 temos dados para as estruturas que contêm íon metálico e íon lantanídeo com o centro metálico, não
discutimos no capítulo de resultado os dados obtidos e os mesmos estão aqui somente para análise do leitor.
Tabela A4. RMS, Parâmetros de rede e densidades calculadas com os métodos semiempíricos PM6 e Sparkle/PM6 para MOFs contendo Sm, Cu, Co, Yb e Eu
como centros metálicos.
Estrutura
Descrição Erro a b c α β γ Densidade
(g/cm³)
1 [SmCu2K(BTC)2(H2O)4]·2H2O
(798913)
Raio-X - 14,268 12,075 8,623 90,00 121,64 90,00 2,313
PM6 14,137 13,052 8,538 90,19 122,65 90,68 2,206
2 [CoHo2(Himdc)2(SO4)2(H2O)4]·H2On
(776857)
Raio-X - 9,055 10,697 12,913 93,02 96,86 108,65 2,776
PM6 9,519 11,249 13,019 90,29 98,94 110,92 2,533
3 [CoYb2(Himdc)2(SO4)2(H2O)4]·H2On
(776858)
Raio-X - 9,030 10,649 12,850 92,91 97,28 108.54 2,858
PM6 9,632 11,191 12,976 90,43 99,01 112,22 2,592
4 (NH4)4[Eu2(L)2(H2O)9(H2W12O40)]·nH2O
(800868)
Raio-X - 12,940 25,082 21,341 90,00 97,093 90,00 3,742
PM6 13,879 28,830 21,836 87,70 93,13 90,08 2,951

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
122
REFERÊNCIAS
[ 1 ] Wei, Y.; LI, J.; Song, W.; Zang, S,. Inorganic Chemistry Communications, 15 (2012) 16.
[ 2 ] Zhou, Y.; Xu, W.; Wu, M.; Lan, A.; Feng, L. R.; Jiang, F.; Hong, M,. Inorganic Chemistry Communications, 15 (2012) 140.
[ 3 ] Hu, T.; Liu, L.; Lv, X.; Chen, X.; He, H.; Daí, F.; Zhang, G.; Sun, D,. Polyhedron, 13 (2010) 296.
[ 4 ] Bakkali, H. E.; Choquesillo-Lazart, D.; Domínguez-Martín, A.; Brandi-Blanco, M. del P.; Castiñeiras, A.; Niclós-Gutiérrez, J,. Polyedron, 31 (2012) 463.
[ 5 ] Liu,Y.; Yue, K.; Shan, B.; Xu, L.; Wang, C.; Wang Y., inorganic Chemistry Communications, 17 (2012) 30.
[ 6 ] Wang, X.; Bai, Y.; Xing, F.; Shao, M.; Zhu, S,. Inorganic Chemistry Communications, Accepted Manuscript,. DOI: 10.1016/j.inoche.2012.02.031
[ 7 ] Wu, J.; Wang, P.; Huang, C.; Fu, L.; Song, F.; Hou, H.; Chang, J,. Inorganic Chemistry Communications, 15 (2012) 301.
[ 8 ] Borel, C.; Ghazzali, M. M.; Langer, V.; Öhrstöm, L ,. Inorganic Chemistry Communications, 12 (2009) 106.
[ 9 ] Chen, C. F. Z.; Liu, X.; Yang, Y.; Deng, M.; Weng, L.; Jia, Y.; Zhou, Y,. Inorganic ChimicaActica,362 (2009) 2101.
[ 10 ] Wang, W.; Sun, J.; Zhang, D.; Song, T.; Song, W.; Zhang, L.; Chen, Y.; Fan, F.; Zhang, P,. Inorganic ChimicaActicaI,384 (2012) 105.
[ 11 ] Neogi, S.; Bharadwaj, P. K,. Polyhedron 25 (2006) 1491.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
123
[ 12 ] Ma, R.; Chen, C.; Sun, B.; Zhao, X.; Zhang, N,. Inorganic Chemistry Communications,14 (2011) 1532.
[ 13 ] Zhang, D.; Song, T.; Wang, L.; Shi, L.; Xu, J.; Wang, Y.; Ma, K.; Yin, W.; Zhang, L.; Fan, Y,. InorganicChimicaActica,362 (2009) 299.
[ 14 ] Li, X.; Zhang, Y.; Shi, M.; Li, P,. Inorganic Chemistry Communications,11 (2008) 869.
[ 15 ] Sun, Y.; Yu, W.; Wang, L.; Rong, S.; Wu, Y.; Ding, F.; Zhang, W.; Gao, E,. Inorganic Chemistry Communications,13 (2010) 479.
[ 16 ] Delgado, L.; Fabelo, O.; Pasán, J.; Julve, M.; Lloret, F.; Ruiz-Pérez, C,. Polyhedron 29 (2010) 188.
[ 17 ] Zhang, L.; Lu, C.; Chen, S.; Yu, F.; Li, X.; Tan, J.; Yang, X,. Inorganic Chemistry Communications, 14 (2011) 143.
[ 18 ]Zehnder, R. A.; Renn, R. A.; Pippin, E.; Zeller, M.; Wheeler, K. A.; Carr, J. A.; Fontaine, N.; McMullen, N. C.; Journal of Molecular Structure, 985 (2011) 109.
[ 19 ] Zhang, G.; Zhao, X.; Daí, F.; Sun, D,. Inorganic Chemistry Communications, 14 (2011) 948.
[ 20 ] Li, M.; Yuan, L.; Li, H.; Sun, J,. Inorganic Chemistry Communications, 10 (2007) 1281.
[ 21 ] Wei, T.; Zhao, S.; Bi, W.; Lü, X.; Hui, Y.; Song, J.; Wong, W.; Jones, R. A,.Inorganic Chemistry Communications,12 (2009) 1216.
[ 22 ] Qiu, Y.; Daiguebonne, C.; Liu, J.; Zeng, R.; Kerbellec, N.; Deng, H.; Guillou, O,. InorganicChimica Acta,360 (2007) 3265.
[ 23 ] Zhu, X.; Gao, S.; Yang, S.; Li, G.; Xu, B.; Cao, R,. Journal of Solid State Chemistry, 182 (2009) 421.
[ 24 ] Zhang, H.; Peng, Y.; Shan, X.; Tian, C.; Ping-Lin.; Du, S,. Inorganic Chemistry Communications, 14 (2011) 1165.
[ 25 ] Zheng, S.; Cai, S.; Yang, Q.; Xiao, T.; Fan, J.; Zhang, W,. Inorganic Chemistry Communications, 14 (2011) 826.
[ 26 ] Bernini, M, C.;Brusau, E, V.; Narda, G, E.; Echeverria, G.; Fantoni, A.; Punte, G.; Ayala, A. P,. Polyedron 31 (2012) 729.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
124
[ 27 ] Zheng, B.; Zhang, D.; Peng, Y.; Huo, Q.; Liu, Y,. Inorganic Chemistry Communications,16 (2012) 70.
[ 28 ] Weng, S.; Wang, Y.; Lee, C,. Journal of Solid State Chemistry, 188 (2012) 77.
[ 29 ] Song, H.; Li, Y,. InorganicaChimica Acta, 361 (2008) 1421.
[ 30 ] Lan, A.; Chen, L.; Yuan, D.; Huang, Y.; Hong, M.; Wang, X,. Polyhedron 30 (2011) 47.
[ 31 ] Pramanik, A.; Abbina, S.; Das, G,. Polyhedron, 26 (2007) 5225.
[ 32 ] Wang, X.; Chen, B.; Lin, H,. Transition Met Chem,34 (2009) 307.
[ 33 ] Xu, Y.; Lin, J.; Yao, J.; Gao, S.; Zhu, H.; Meng, Q,. Inorganic Chemistry Communications, 11 (2008) 422.
[ 34 ] Amghouz, Z.; Roces, L.; García-Granda, S.; García, J. R.;Souhail, B.; Mafra, L.; Shi, F.; Rocha, J,. Journal of Solid State Chemistry, 182 (2009) 3365.
[ 35 ] Mal, D.; Sem, R.; Rentschler, E.; Okamoto, K.; Miyashita, Y.; Koner, S,. InorganicaChimica Acta, 385 (2012) 27.
[ 36 ] Hou, Q.; Jia, M.; Zhao, J.; jin, J.; Yu, J.; Xu, J,. InnorganicaChimica Acta,384 (2012) 287.
[ 37 ] Han, S. H.; Lee, S. W,. Polyhedron, 31 (2012) 255.
[ 38 ] Manna, S. C.; Ghosh, A. K.; Zangrando, E.; Chaudhuri, N. R,. Polyhedron, 26 (2007) 1105.
[ 39 ] Lu, H. Z. Y.; Zhang, Z.; Wang, E,.Inorganic Chemistry Communications,17 (2012) 9.
[ 40 ] Tian, L.; Chen, Z,. Inorganic Chemistry Communications, 14 (2011) 1302.
[ 41 ] Liu, W.; Ye, L.;Liu, X.; Yuan, L.; Lu, X.; Jiang, J,. Inorganic Chemistry Communications, 11 (2008) 1250.
[ 42 ] Hou, C.; Liu, Q.; Lu, Y.; Okamura, T.; Wang, P.; Chen, M.; Sun, W,. MicroporousandMesoporousMaterials, 152 (2012) 96.

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
125
[ 43 ] Li, Z.; Luo, X.; Gao, Y.; Lu, H.; Li, G,. InorganicaChimica Acta, 384 (2012) 352.
[ 44 ] Wang, S.; Ma, J.; Li, L.; Chen, T.; Sun, Z.; Luo, J,. Inorganic Chemistry Communications, 16 (2012) 65
[ 45 ] Tahli, A.; Maclaren, J.K.; Boldog, I.; Janiak, C,. InorganicaChimica Acta, 374 (2011) 506
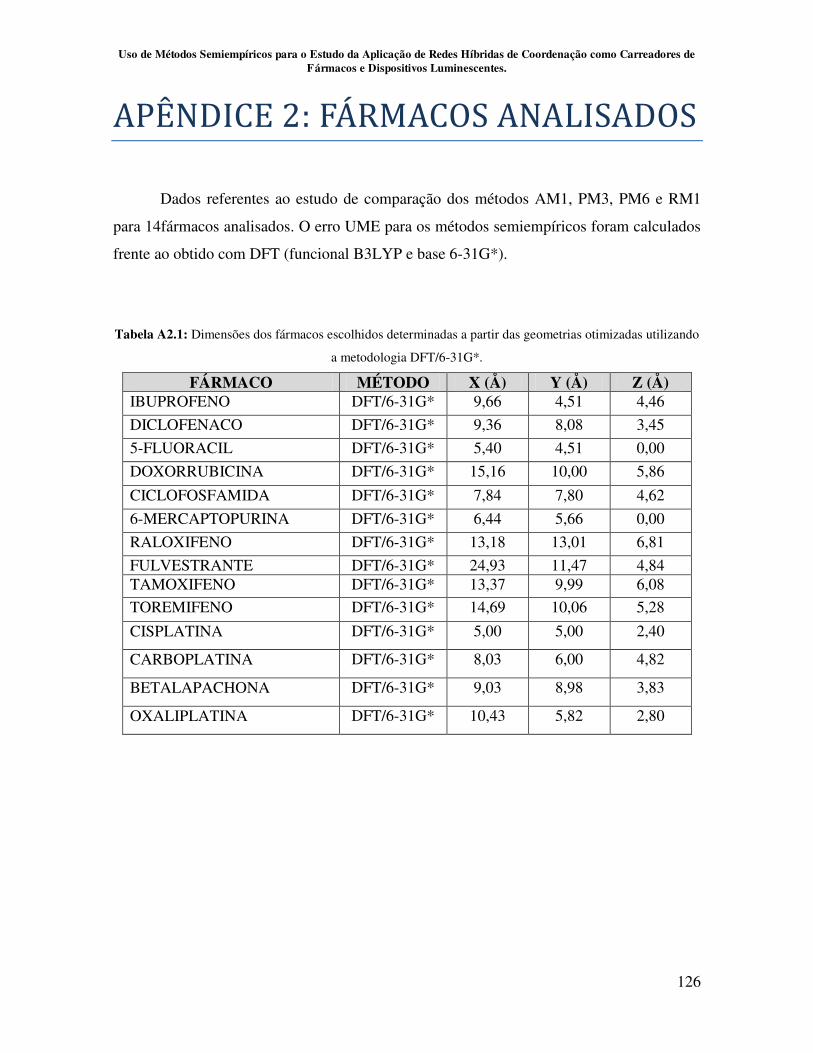
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
126
APÊNDICE 2: FÁRMACOS ANALISADOS
Dados referentes ao estudo de comparação dos métodos AM1, PM3, PM6 e RM1
para 14fármacos analisados. O erro UME para os métodos semiempíricos foram calculados
frente ao obtido com DFT (funcional B3LYP e base 6-31G*).
Tabela A2.1: Dimensões dos fármacos escolhidos determinadas a partir das geometrias otimizadas utilizando
a metodologia DFT/6-31G*.
FÁRMACO MÉTODO X (Å) Y (Å) Z (Å) IBUPROFENO DFT/6-31G* 9,66 4,51 4,46
DICLOFENACO DFT/6-31G* 9,36 8,08 3,45
5-FLUORACIL DFT/6-31G* 5,40 4,51 0,00
DOXORRUBICINA DFT/6-31G* 15,16 10,00 5,86
CICLOFOSFAMIDA DFT/6-31G* 7,84 7,80 4,62
6-MERCAPTOPURINA DFT/6-31G* 6,44 5,66 0,00
RALOXIFENO DFT/6-31G* 13,18 13,01 6,81
FULVESTRANTE DFT/6-31G* 24,93 11,47 4,84 TAMOXIFENO DFT/6-31G* 13,37 9,99 6,08
TOREMIFENO DFT/6-31G* 14,69 10,06 5,28
CISPLATINA DFT/6-31G* 5,00 5,00 2,40
CARBOPLATINA DFT/6-31G* 8,03 6,00 4,82
BETALAPACHONA DFT/6-31G* 9,03 8,98 3,83
OXALIPLATINA DFT/6-31G* 10,43 5,82 2,80

Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
127
Tabela A2.2: Dimensões dos fármacos escolhidos determinadas a partir das geometrias otimizadas utilizando o método semiempírico AM1.
FÁRMACO MÉTODO X (Å) Y (Å) Z (Å) UME IBUPROFENO AM1 9,50 6,26 4,45 0,64
DICLOFENACO AM1 8,90 7,74 4,45 0,60
FLUORACIL AM1 5,42 4,58 0,00 0,03
DOXORRUBICINA AM1 14,64 10,02 6,90 1,85
CICLOFOSFAMIDA AM1 7,93 7,58 4,00 0,31
MERCAPTOPURINA AM1 6,55 5,71 0,00 0,05
RALOXIFENO AM1 14,37 11,31 6,96 1,01
FULVESTRANTE AM1 24,73 11,49 4,61 0,15 TAMOXIFENO AM1 13,02 9,62 5,28 0,51
TOREMIFENO AM1 14,66 10,06 5,30 0,02
CISPLATINA AM1 - - - -
CARBOPLATINA AM1 - - - -
BETALAPACHONA AM1 9,02 8,97 3,79 0,02
OXALIPLATINA AM1 - - - -
- Método sem parâmetros para platina.
Tabela A2.3: Dimensões dos fármacos escolhidos determinadas a partir das geometrias otimizadas utilizando o método semiempírico PM3.
FÁRMACO MÉTODO X (Å) Y (Å) Z (Å) UME IBUPROFENO PM3 9,42 6,19 4,58 4,58
DICLOFENACO PM3 8,54 7,87 4,42 4,42
FLUORACIL PM3 5,39 4,56 0,00 0,00
DOXORRUBICINA PM3 14,89 10,12 7,22 7,22
CICLOFOSFAMIDA PM3 7,84 7,28 4,05 4,05
MERCAPTOPURINA PM3 6.61 5,80 0,00 0,00
RALOXIFENO PM3 14,52 11,44 5,42 5,42
FULVESTRANTE PM3 24,96 11,52 5,04 5,04 TAMOXIFENO PM3 13,45 10,98 6,34 6,34
TOREMIFENO PM3 15,70 11,21 5,46 5,46
CISPLATINA PM3 - - - -
CARBOPLATINA PM3 - - - -
BETALAPACHONA PM3 9,02 8,98 3,83 3,83
OXALIPLATINA PM3 - - - -
- Método sem parâmetros para platina.
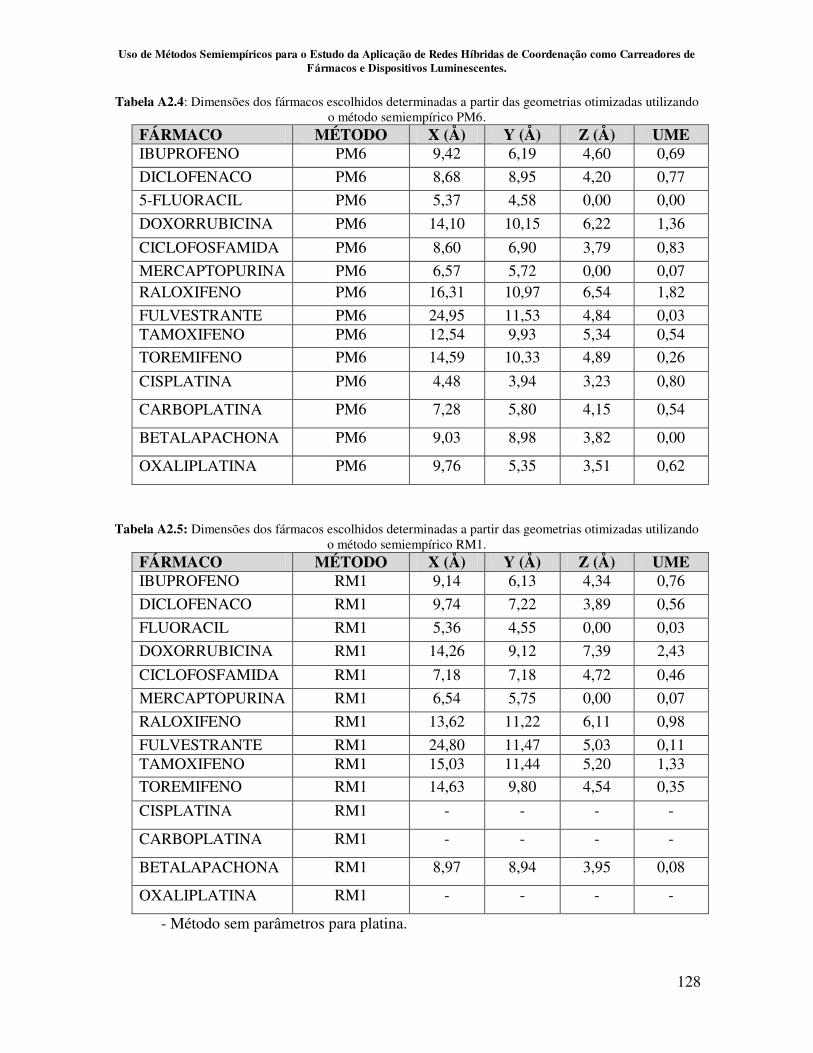
Uso de Métodos Semiempíricos para o Estudo da Aplicação de Redes Híbridas de Coordenação como Carreadores de Fármacos e Dispositivos Luminescentes.
128
Tabela A2.4: Dimensões dos fármacos escolhidos determinadas a partir das geometrias otimizadas utilizando o método semiempírico PM6.
FÁRMACO MÉTODO X (Å) Y (Å) Z (Å) UME IBUPROFENO PM6 9,42 6,19 4,60 0,69
DICLOFENACO PM6 8,68 8,95 4,20 0,77
5-FLUORACIL PM6 5,37 4,58 0,00 0,00
DOXORRUBICINA PM6 14,10 10,15 6,22 1,36
CICLOFOSFAMIDA PM6 8,60 6,90 3,79 0,83
MERCAPTOPURINA PM6 6,57 5,72 0,00 0,07 RALOXIFENO PM6 16,31 10,97 6,54 1,82
FULVESTRANTE PM6 24,95 11,53 4,84 0,03 TAMOXIFENO PM6 12,54 9,93 5,34 0,54
TOREMIFENO PM6 14,59 10,33 4,89 0,26
CISPLATINA PM6 4,48 3,94 3,23 0,80
CARBOPLATINA PM6 7,28 5,80 4,15 0,54
BETALAPACHONA PM6 9,03 8,98 3,82 0,00
OXALIPLATINA PM6 9,76 5,35 3,51 0,62
Tabela A2.5: Dimensões dos fármacos escolhidos determinadas a partir das geometrias otimizadas utilizando
o método semiempírico RM1. FÁRMACO MÉTODO X (Å) Y (Å) Z (Å) UME IBUPROFENO RM1 9,14 6,13 4,34 0,76
DICLOFENACO RM1 9,74 7,22 3,89 0,56
FLUORACIL RM1 5,36 4,55 0,00 0,03
DOXORRUBICINA RM1 14,26 9,12 7,39 2,43
CICLOFOSFAMIDA RM1 7,18 7,18 4,72 0,46
MERCAPTOPURINA RM1 6,54 5,75 0,00 0,07
RALOXIFENO RM1 13,62 11,22 6,11 0,98
FULVESTRANTE RM1 24,80 11,47 5,03 0,11 TAMOXIFENO RM1 15,03 11,44 5,20 1,33
TOREMIFENO RM1 14,63 9,80 4,54 0,35
CISPLATINA RM1 - - - -
CARBOPLATINA RM1 - - - -
BETALAPACHONA RM1 8,97 8,94 3,95 0,08
OXALIPLATINA RM1 - - - -
- Método sem parâmetros para platina.