daniele gomes müller ppgqta rio grande, rs - brasil · iii universidade federal do rio grande...
TRANSCRIPT

i
FURG
Dissertação de Mestrado
Obtenção de copolímero EPS-Lignina por via
fotoquímica
___________________________________
Daniele Gomes Müller
PPGQTA
Rio Grande, RS - Brasil
2017

ii
Obtenção de copolímero de EPS-Lignina por via fotoquímica
por
DANIELE GOMES MÜLLER
Dissertação apresentada ao Programa de Pós-Graduação
em Química Tecnológica e Ambiental da Universidade
Federal do Rio Grande (RS), como requisito parcial para
obtenção do título de MESTRE EM QUÍMICA.
PPGQTA
Rio Grande, RS - Brasil
Março de 2017

iii
Universidade Federal do Rio Grande Escola de Química e Alimentos
Programa de Pós-Graduação em Química Tecnológica e Ambiental
A Comissão Examinadora abaixo assinada aprova a Dissertação de Mestrado
Obtenção de copolímero de EPS-lignina por via fotoquímica
elaborada por
DANIELE GOMES MÜLLER
Como requisito parcial para a obtenção do título de
Mestre em Química
COMISSÃO EXAMINADORA
Prof. Dr. Alex Fabiani Claro Flores (FURG- RS)
Prof. Dr. Tito Roberto Sant’Anna Cadaval Junior (FURG – RS)
Prof. Dr. Daniel Eduardo Weibel (UFRGS - RS)
Rio Grande, 03 de março de 2017.

iv
AGRADECIMENTOS
Primeiramente, à Deus pela vida, por ouvir todas as minhas preces e atendê-las
na medida do possível, me dando força e clareza.
A minha família, em especial minha mãe, meu pai (que mesmo em outro plano
está sempre olhando por mim), meu irmão e meu “namorido” Samuel. Agradeço por
toda a paciência, pelas palavras de motivação por sempre estarem comigo, me
apoiando incondicionalmente, e entendendo minhas ausências.
Ao meu orientador e ao meu co-orientador, que me deram liberdade e ao mesmo
tempo direção para finalizar esse trabalho.
Aos colegas do grupo de pesquisa em Fotoquímica e Química de Materiais
(FQMAT) por todo auxílio desde o começo dessa jornada, principalmente ao Marcelo e
a Jessica.
Aos colegas do grupo NEESH.
Aos colegas do Laboratório de Físico-Química Aplicada e Tecnológica (LAFQAT)
pela parceria.
Aos IC’s que passaram esse período comigo, em especial à Evelyn. Muito
obrigada pela parceria desde o início até o final dessa jornada.
Aos laboratórios de Operações Unitárias (LOU) do prof. Pinto e Tecnologia de
Alimentos (LTA) do Prof. Carlos pelas análises de DSC e Ensaios Mecânicos.
Ao Centro de Microscopia Eletrônica da instituição (CEME-SUL) pelas análises
de MEV e EDS, em especial ao Alisson pela possibilidade de troca de informações e
conhecimento.
A Central Integrada de Análise (CIA-FURG) pelas análises de RMN.
A UFRGS pelas análises de GPC.
Aos amigos como um todo que compreenderam minhas ausências em alguns
momentos e me mandaram boas vibrações, mesmo de longe.

v
“Para realizar grandes conquistas, não devemos apenas agir, mas sonhar;
não apenas planejar, mas também acreditar.” (Anatole France)

vi
SUMÁRIO
LISTA DE FIGURAS ..................................................................................................... viii
LISTA DE TABELAS ...................................................................................................... xi
LISTA DE ABREVIATURAS E SÍMBOLOS ....................................................................xii
RESUMO....................................................................................................................... xiii
ABSTRACT ...................................................................................................................xiv
1. INTRODUÇÃO.............................................................................................................1
2. OBJETIVOS.................................................................................................................4
2.1Objetivo geral..............................................................................................................4
2.2 Objetivos específicos.................................................................................................4
3. REVISÃO DA LITERATURA........................................................................................5
3.1 Polímeros - aspectos gerais.......................................................................................5
3.2 Síntese de polímeros.................................................................................................8
3.3 Copolímeros por enxertia.........................................................................................13
3.4 Poliestireno expandido.............................................................................................15
3.5 Polímeros lignocelulósicos - a lignina.......................................................................17
3.6 Fotoquímica como ferramenta de modificação da matéria.......................................21
3.6.1 Interações entre a luz e a matéria e transições eletrônicas........................21
3.6.2 Reações fotoquímicas.................................................................................24
3.7 Caracterização molecular / estrutural de materiais poliméricos...............................28
3.7.1 Análises térmicas........................................................................................28
3.7.2 Análises morfológicas.................................................................................29
3.7.3 Análises espectroscópicas..........................................................................30
3.7.4 Análise cromatográfica................................................................................32
3.7.5 Ensaio mecânico de tração.........................................................................33
3.8 Modificação em poliestireno expandido....................................................................34
3.9 Modificação em lignina.............................................................................................35
4. MATERIAIS E MÉTODOS.. ....................................................................................... 38
4.1 Extração da lignina pelo método Organossolve.......................................................38
4.2 Matriz polimérica, carga e promotores da reação....................................................38

vii
4.3 Caracterização fotoquímica dos precursores...........................................................39
4.4 Preparação dos copolímeros EPS-Lignina..............................................................40
4.5 Purificação dos copolímeros EPS-Lignina...............................................................42
4.6 Caracterizações.......................................................................................................43
4.6.1 Análise térmica...........................................................................................43
4.6.2 Análises morfológicas.................................................................................44
4.6.3 Análises espectrocópicas...........................................................................44
4.6.4 Análise de cromatografia de permeação em gel........................................45
4.6.5 Ensaio mecânico de tração........................................................................46
5. APRESENTAÇÃO E DISCUSSÃO DOS RESULTADOS..........................................48
5.1 Preparação e purificação dos copolímeros..............................................................48
5.2 Análises espectrocópicas.........................................................................................51
5.2.1 Espectroscopia UV-Vis...............................................................................51
5.2.2 Espectroscopia de infravermelho com transformada de Fourrier...............52
5.2.3 Espectroscopia de ressonância magnética................................................58
5.2.4 Espectroscopia de energia dispersiva........................................................63
5.3 Análises morfológicas...............................................................................................64
5.3.1 Microscopia eletrônica de varredura...........................................................64
5.3.2 Ângulo de contato.......................................................................................68
5.4 Análise térmica.........................................................................................................70
5.5.Cromatografia de permeação em gel.......................................................................72
5.6 Ensaio mecânico de tração......................................................................................76
5.7 Discussão geral dos resultados................................................................................81
6. CONCLUSÕES ..................................................................................................... ....84
7. REFERÊNCIAS BIBLIOGRÁFICAS... ....................................................................... 85
Anexos............................................................................................................................89

viii
LISTA DE FIGURAS
Figura 1. Classificação polimérica em função da organização molecular dos
monômeros.....................................................................................................................5
Figura 2. Etapas de polimerização por adição...............................................................9
Figura 3. Esquema de ramificação de cadeia na polimerização................................. .10
Figura 4. Reação de policondensação do Náilon 6,6....................................................11
Figura 5. Reação de policondensação em aminoácidos (A); possível ciclização
intermolecular (B)...........................................................................................................11
Figura 6. Tipos de copolimerização em bloco...............................................................12
Figura 7. Diferentes estratégias de copolimerização por enxertia................................13
Figura 8. Representação simplificada da estrutura vegetal..........................................17
Figura 9. Álcoois Precursores das unidades Fenil-propanóides Guaiacila (G), Siringila
(S) e p-hidroxifenila (H)..................................................................................................18
Figura 10. Estrutura da lignina......................................................................................19
Figura 11. Tipos de ligações entre as unidades fenilpropanóides................................20
Figura 12. Diagrama de Jablonski................................................................................22
Figura 13. Espectro eletromagnético............................................................................22
Figura 14. Representação da formação de orbitais moleculares (ligantes e antiligantes)
a partir dos orbitais atômicos.........................................................................................23
Figura 15. Esquema representativo das reações fotoquímicas....................................24
Figura 16. Exemplo de fotocicloadição (2+2)...............................................................25
Figura 17. Exemplo de formação de anéis com heteroátomos via fotoquímica...........25

ix
Figura 18. Fotoenxertia de MMA em PS (a) e modificação de PS (b) via reação
fotoquímica....................................................................................................................26
Figura 19. Copolimerização de poli(arilsulfonas) com monômeros vinílicos ...............26
Figura 20. Reações fotoquímicas usuais envolvendo compostos carbonílicos...........27
Figura 21. Ilustração explicativa da técnica de DSC...................................................29
Figura 22. Esquema representativo do funcionamento do MEV..................................29
Figura 23. Princípio da análise de ângulo de contato..................................................30
Figura 24. Fundamentos da técnica de RMN...............................................................31
Figura 25. Esquema do espectrômetro de dispersão de energia.................................32
Figura 26. Comportamento mecânico para materiais poliméricos................................34
Figura 27. Possíveis reações fotoquímicas para o EPS...............................................35
Figura 28. Possíveis reações fotoquímicas da LO........................................................36
Figura 29. Reações de LO com monômero bifuncional................................................37
Figura 30. Monômero encontrado no poliestireno........................................................39
Figura 31. Diácidos carboxílicos como moléculas de sacrifício....................................39
Figura 32. Esquema de obtenção dos copolímeros EPS-Lignina.................................42
Figura 33. Purificação dos polímeros com ácido adípico..............................................48
Figura 34. Copolímeros com ácido adípico pós processo de purificação.....................49
Figura 35. Purificação dos polímeros com ácido itacônico...........................................50
Figura 36. Copolímeros com ácido itacônico pós processo de purificação..................50
Figura 37. Espectro de UV-Vis dos copolímeros com ácido adípico............................51
Figura 38. Espectro de UV-Vis dos copolímeros com ácido itacônico.........................52

x
Figura 39. Espectro de IV dos materiais de partida. Em (A) EPS; (B) LO; (C) Ácido
itacônico; (D) Ácido adípico............................................................................................53
Figura 40. Espectro de IV dos copolímeros com ácido itacônico. (A) EPS; (B) AI2; (C)
AI5; (D) AI10; (E) AI20; (F) AI50.....................................................................................55
Figura 41. Espectro de IV dos copolímeros com ácido adípico. (A) EPS; (B) AA2; (C)
AA5; (D) AA10; (E) AA20; (F) AA50...............................................................................56
Figura 42. Espectros de RMN ¹H para LO (a) e EPS (b)...............................................58
Figura 43. Espectro de RMN ¹H do copolímero AA10 (a) e RMN ¹³C do AA20 (b).......61
Figura 44. Espectros HSQC para os copolímeros com ácido itacônico em (a) AA2, (b)
AA5, (c) AA10, (d) AA20, (e) AA50.................................................................................62
Figura 45. Imagens de microscopia eletrônica dos materiais de partida.......................64
Figura 46. Imagens de MEV para copolímeros com ácido itacônico. Em (A) AI2; (B)
AI5; (C) AI10; (D) AI20; (E) AI50 nas ampliações 10.000 e 40.000,
respectivamente.............................................................................................................65
Figura 47. Imagens de MEV para copolímeros com ácido adípico. Em (A) AA2; (B)
AA5; (C) AA10; (D) AA20; (E) AA50 nas ampliações 10.000 e 40.000,
respectivamente.............................................................................................................66
Figura 48. Imagens de gotas na superfície. Em A representa imagem do copolímero
AI50 e em B do EPS......................................................................................................70
Figura 49. Análise de DSC dos copolímeros com ácido adípico..................................70
Figura 50. Análise de DSC dos copolímeros com ácido itacônico...............................71
Figura 51. Cromatogramas para os copolímeros com ácido adípico...........................73
Figura 52. Cromatogramas para os copolímeros com ácido itacônico........................73
Figura 53. Cromatograma para LO..............................................................................74
Figura 54. Gráfico Tensão X Elongação para os copolímeros com ácido adípico.......77
Figura 55. Gráfico Tensão X Elongação para os copolímeros com ácido itacônico....79
Figura 56. Mecanismo sugerido para a obtenção dos copolímeros.............................82
C

xi
LISTA DE TABELAS
Tabela 1 – Relação de massa de lignina e volume de solvente....................................41
Tabela 2 – Massas de amostras para análise por DSC.................................................43
Tabela 3 – Fator de diluição para cada copolímero.......................................................45
Tabela 4 – Massas de amostra para GPC.....................................................................46
Tabela 5 – Massas dos copolímeros c4m ácido adípico pré e pós-purificação.............49
Tabela 6 – Massa dos copolímeros com ácido itacônico pré e pós-purificação............50
Tabela 7– Análise de EDS para os copolímeros obtidos...............................................63
Tabela 8 – Ângulos de contato médios para os copolímeros com ácido itacônico........68
Tabela 9 – Ângulos de contato médios para os copolímeros com ácido adípico..........69
Tabela 10 – Copolímeros obtidos e respectivas temperaturas de transição vítrea.......71
Tabela 11 – Massas molares médias para os copolímeros com ácido adípico.............74
Tabela 12 – Massas molares médias para os copolímeros com ácido itacônico..........75
Tabela 13 – Ensaios mecânicos para os copolímeros com ácido adípico....................76
Tabela 14 – Ensaios mecânicos para os copolímeros com ácido itacônico..................79

xii
LISTA DE ABREVIATURAS E SÍMBOLOS
Significado [do inglês] Sigla Significado [do inglês]
Sigla
ARGET Transferência de elétrons por ativador regenerado [activators regenerated by electron transfer]
O Oxigênio
ASTM Sociedade Americana de testes e materiais [American Society for Testing and Materials]
PDI Polidispersividade
ATR Refletância total atenuada, [attenued total refletance] PE Polietileno
ATRP polimerização radicalar com transferência de átomos, [atom transfer radical polymerization]
PEG Poli(etilenoglicol)
CEME-SUL Centro de Microscopia Eletrônica Sul PEO Poli(oxietileno)
CIA-FURG Centro Integrado de Análises da FURG PET Poli(tereftalato de etileno)
CONAB Companhia Nacional de Abastecimento PHBHV Poli(3-hidroxibutirato-co-3-hidroxivalerato)
DEPT 135
PLA Poli(ácido lático)
DMAc Dimetilacetamida PMDI Poli(metileno difenil diisocianato)
DMF Dimetilformamida PP Polipropileno
DSC Calorimetria diferencial de varredura [differential scanning calorimetry]
PPO Poli(oxipropileno)
DVB Divinilbenzeno PS Poliestireno
EDS Espectroscopia de energia dispersiva PSBA Poli(estireno-co-butilacrilato)
EPS Poliestireno expandido [expanded polystyrene] PVC Cloreto de polivinila
FAO Organização das Nações Unidas para a Alimentação e a Agricultura, [Food and Agriculture Organization]
PVP Poli(vinilpirrolidinona)
G Unidades guaiacila da lignina RMN Espectroscopia de ressonância magnética
GPC Cromatografia de permeação em gel S Unidade siringila da lignina
H Unidades p-hidroxifenila da lignina SEBS Borracha etireno-etileno-butileno-estireno [styrene-ethylene-butylene-styrene]
HSQC Espectrocopia de coerência quântica heteronuclear única [heteronuclear single quantum coherence spectrocopy]
Tc Temperatura de cristalização
IV Espectroscopia de infravermelho Tf Temperatura de fusão
LAFQAT Laboratório de Físico-Química Tecnológica e Aplicada Tg Temperatura de transição vítrea
LDPE Polietileno de baixa densidade [low-density polyethylene] THF Tetrahidrofurano
LED Diodo emissor de luz [light emittior diode] UFRGS Universidade Federal do Rio Grande do Sul
LO Lignina organossolve UV Ultravioleta
MEV Microscopia eletrônica de varredura UV-Vis Espectroscopia de ultravioleta-visível
MMA Metilmetacrilato WCA Ângulo de contato em água, [water angle contact]
MME Ministério de Minas e Energia λ Comprimento de onda
Mn Peso molecular numérico médio n→π* Transição eletrônica entre os elétrons livres e o orbital pi- anti-ligante
Mw Peso molecular ponderal médio π→π* Transição eletrônica entre o orbital pi-ligante e o orbital pi- anti-ligante
Mz Peso molecular médio das cadeias maioes σ→σ* Transição eletrônica entre o orbital sigma ligante e o orbital sigma anti-ligante
N Nitrogênio δ Deslocamento químico
NAFTA Tratado de Livre Comércio da América do Norte [North AmericanFree Trade Agreement]
ν Estiramentos no espectro de infravermelho

xiii
RESUMO
Título: Obtenção de copolímero EPS-lignina por via fotoquímica
Autor: Daniele Gomes Müller
Orientador: Prof. Dr. Paulo Henrique Beck
Co-orientador: Prof. Dr. Felipe Kessler
A busca por matérias-primas a partir de fontes renováveis aliadas com a
biomassa e a reutilização de materiais residuais são os principais desafios na obtenção
de processos sustentáveis. O presente trabalho traz uma aplicação para a lignina
extraída da casca de arroz pelo método Organossolve. Esta foi empregada como carga
enxertada na matriz de poliestireno expandido pós-consumo para a obtenção de
copolímero EPS-Lignina. Dois diferentes diácidos carboxílicos (ácido adípico e ácido
itacônico) foram usados como promotores da reação. O sistema pós solubilização foi
exposto à radiação UV de média pressão como fonte de energia promotora da reação.
Variou-se a concentração da carga para avaliar seu efeito no material final. Este foi
caracterizado a partir de análises espectroscópicas, morfológicas, térmica,
cromatográfica e ensaios mecânicos. Foi possível observar diferença nas transições
eletrônicas entre materiais de partida e copolímeros e o desaparecimento das bandas
de OH e aparecimento das bandas C-O de éter sugerindo a presença de LO enxertada.
Além disso, foi possível visualizar ligeira redução do %C e a aumento do %O com o
acréscimo de carga. As análises morfológicas mostraram que mesmo com grandes
ampliações, os filmes apresentaram homogeneidade e a superfície possui caráter mais
hidrofílico. A análise térmica mostrou que não houveram mudanças relevantes
indicando que o copolímero manteve comportamento térmico semelhante à matriz. Já a
cromatográfica aponta a fotoenxertia pelo aumento no número de macromoléculas
mediante ligeira quebra das cadeias. Os ensaios mecânicos mostraram redução da
tensão de ruptura com aumento da carga e aumento do módulo de elasticidade. A
partir de todas as análises realizadas, o mecanismo sugerido parte da abstração de H
da cadeia alifática do EPS com enxertia da lignina. O uso de biomassa e polímero pós-
consumo em conjunto com a radiação UV vem como proposta de reaproveitamento de
resíduos de baixo custo que vai ao encontro dos princípios de Química Verde.
Palavras-chave: lignina, poliestireno expandido, radiação UV, copolimerização

xiv
ABSTRACT
Title: Photochemical EPS-lignin copolymer
Author: Daniele Gomes Müller
Advisor: Prof. Dr. Paulo Henrique Beck
Co-advisor: Prof. Dr. Felipe Kessler
The search for raw materials from renewable sources combined with biomass
and a reuse of waste materials are the main challenges in achieving sustainable
processes. The present work presents an application for a lignin extracted from the rice
husk by the method Organossolve. This was used as a load on a post-consumer
expanded polystyrene matrix for obtaining EPS-Lignin copolymer. Two different
carboxylic diacids (adipic acid and itaconic acid) were used as promoters of the
reaction. The solubilization system was exposed to medium pressure UV radiation as a
source of energy that promotes the reaction. A concentration of the load was varied to
evaluate its effect on no final material. This was characterized from spectroscopic
analyzes, morphological, thermal, chromatographic and mechanical tests. It was
possible to observe the difference between the electronic transitions between the
starting materials and the copolymers and the disappearance of the OH bands and the
appearance of the bands. In addition, it was possible to visualize slight reduction of %C
and increase of %O with increase of load. As morphological analyzes showed that even
with large extensions, the films presented homogeneity and a more hydrophilic
character surface. Thermal analysis showed no significant changes indicating that the
copolymer maintained the thermal behavior of the matrix. Already a chromatography
points a photoenxertia by increasing non-number of macromolecules through the light
breaking of the chains. The mechanical tests showed a reduction in tensile strength with
increasing load and increasing modulus of elasticity. From all the analyzes performed,
the suggested mechanism starts from the EPS H abstraction with lignin grafting. The
use of biomass and post-consumption polymer in conjunction with UV radiation comes
as a proposal for the reuse of low cost waste that meets the principles of Green
Chemistry.
Key words: lignin, expanded polystyrene, UV radiation, copolymerization

1
1. INTRODUÇÃO
O caminho para o desenvolvimento sustentável de forma integrada nas esferas
econômica, social e ambiental ainda contém grandes desafios. Segundo a AGENDA
2030 da Organização das Nações Unidas, para se ter um mercado sustentável é
preciso processos que aumentem a competitividade utilizando de forma eficiente os
recursos e a energia, reduzindo o impacto ao meio ambiente. Nesse contexto, os
princípios referentes à sustentabilidade e Química Verde emergem como potenciais
alternativas. Utilizar fontes renováveis, reciclagem, aumento da eficiência energética ,
evitar o uso de substâncias persistentes, bioacumulativas e tóxicas são algumas ideias-
chaves dos doze princípios da Química Verde1.
Popularmente conhecido como “plástico”, materiais poliméricos são
indispensáveis na vida moderna. Tamanha é a versatilidade dos polímeros que
resultam em infinitas aplicações. A produção mundial de plásticos vem crescendo de
forma contínua. No ano de 2014, foram produzidos 260 milhões de toneladas a nível
mundial. O maior produtor foi a China responsável por 26% desse total, seguido da
Europa (20%) e o Bloco Econômico NAFTA (19%)2. Na Europa, o plástico mais
utilizado foi o polipropileno correspondendo a 19,2%. No Brasil, esse mesmo material
correspondeu a 19,4% da demanda total. Outro polímero que merece destaque é o
EPS (poliestireno expandido, do inglês, expanded polystyrene) já que é apontado pelo
Plano Nacional de Resíduos Sólidos como material reciclável. Na Europa seu
reaproveitamento correspondeu a 7% do total de plásticos enquanto que no Brasil
correspondeu a 2,3%, em 2014. Esses números demostram que há grande produção
de plásticos, mas seu reaproveitamento pós-consumo ainda é pequeno. Por essa
razão, o principal destino final desses materiais, aqui no Brasil, são lixões e aterros
sanitários3.
A maior parte dos materiais poliméricos de uso comercial tem como origem os
derivados de petróleo. As reservas mundiais comprovadas de petróleo do mundo, em
2015, estavam em 1,492.677 milhões de barris enquanto que a produção mundial foi
de 75.079,8 mil barris por dia. No Brasil, as reservas são de aproximadamente 16,194
bilhões de barris com produção de 2.433,7 mil barris por dia4. Apesar dessa produção,
boa parte do petróleo é destinada para a geração de energia e cerca de 36,20% foi
destinada à Brasken S.A., empresa do segmento petroquímico, que produz eteno,
polietileno, polipropileno e PVC5.

2
Outra fonte já consolidada para geração de energia é a biomassa. Essas
matérias orgânicas renováveis que podem ser transformadas em energia mecânica,
térmica ou elétrica podem ser de origem florestal, agrícola (principalmente nos cultivos
de soja, milho e arroz) ou até mesmo rejeitos urbanos e industriais. Estima-se que a
quantidade de biomassa na Terra seja de 1,8 trilhões de toneladas6. Isso vai ao
encontro dos dados do Balanço Energético Nacional do Ministério de Minas e Energia
(MME)7, de 2014, que aponta a biomassa, em geral, como a fonte renovável de maior
participação no mundo. Projeções do MME para 2020 indicam que a participação da
biomassa na matriz energética do país será maior que 30%.
Outras aplicações para a biomassa de origem vegetal podem ser dadas dentro
de biorrefinarias. Essa conversão da biomassa em biocombustíveis, insumos químicos,
materiais, alimentos e energia tem como principal finalidade a maximização do uso dos
recursos naturais disponíveis aliado a minimização de efluentes agregando valor ao
produto final. Já que esta é constituída basicamente de polissacarídeos (celulose e
hemicelulose), constituintes inorgânicos e ligninas, estima-se que 50 milhões de
toneladas de lignina são geradas somente da indústria de papel8, onde é empregada,
basicamente, para a geração de energia. As ligninas são macromoléculas aromáticas
organizadas de forma randômica constituídas de unidades fenil-propanóides
apresentando inúmeros anéis aromáticos além de grupos hidroxilas e metoxilas. Tendo
em vista que tamanha diversidade e complexidade química deve ser aproveitada,
especialmente em processos que envolvam reações químicas, o presente trabalho
procurou estruturar-se de forma a agregar valor utilizando essa matéria-prima
renovável em grande disponibilidade respeitando as premissas de sustentabilidade e
Química Verde.
Este crescente interesse pela Química Verde, principalmente nas últimas duas
décadas, tem levado ao desenvolvimento de novas abordagens que atendam essa
finalidade. O uso de reações sem solventes9, 10, sínteses mediadas por micro-ondas11,
12 e o emprego de biocatalisadores13, 14 são exemplos. Nesse contexto, destaca-se o
emprego de radiação ultravioleta como alternativa para reações químicas15. As reações
fotoquímicas desempenham um papel importante no planeta Terra sendo responsáveis
por exemplo pela fotossíntese. Assim, usar energia solar de forma eficiente é
considerada uma questão-chave para o desenvolvimento sustentável.
Há inúmeras vantagens em empregar a fotoquímica conforme princípios da
Química Verde. Uma delas está associada ao uso de condições mais brandas já que

3
as reações iniciadas por radiação UV são caracterizadas pela sua seletividade. Pode
haver até mesmo a economia de átomos, ou seja, maximizar a incorporação de todos
os compostos de partida no produto final reduzindo a formação de subprodutos. Isso é
possível já que as reações fotoquímicas podem ocorrer pela ativação de um grupo
funcional específico sem a necessidade de outros reagentes promotores da reação16.
Geralmente, são reações que não precisam da adição de catalisadores, são mais
rápidas, de baixo custo e de menor complexidade experimental.
Esta dissertação traz os resultados obtidos para produção de copolímeros de
EPS reciclado e lignina extraída da casca de arroz pelo método Organossolve por
fotoenxertia utilizando radiação ultravioleta (UV) como fonte de energia. Foram usados
como promotores da reação o ácido itacônico e o ácido adípico. O tema busca a
produção de novos materiais a partir de resíduos, casca de arroz e EPS, agregando
valor aos mesmos. As análises espectroscópicas, morfológicas, térmica,
cromatográficas e mecânicas indicam a produção dos copolímeros. Discussão
referente aos possíveis caminhos reacionais responsáveis pelas características
observadas dos novos materiais foram realizadas.

4
2. OBJETIVOS
2.1 Objetivo Geral
Estudar o processo de obtenção de novos materiais poliméricos a partir
de EPS-lignina através de reações fotoquímicas.
2.2 Objetivos Específicos
Avaliar o efeito da solubilidade da lignina para diferentes solventes;
Avaliar as variações ocorridas no sistema (lignina, solvente e diácido
carboxílico fotoiniciador da reação) quando submetidos à radiação ultravioleta;
Estudar a influência da concentração de lignina nas propriedades dos
filmes obtidos;
Produzir filmes de poliestireno expandido pós-consumo, com lignina por
via fotoquímica, e somente de EPS, para comparação;
Caracterizar o copolímero obtido utilizando as técnicas espectroscópicas
de infravermelho com transformada de Fourier (IV), Ultravioleta-visível (UV-
VIS), ressonância magnética nuclear de hidrogênio e carbono (RMN de 1H e
RMN de ¹³C) e de energia dispersiva (EDS); cromatográficas através da
cromatografia de permeação em gel (GPC); morfológicas através da
microscopia eletrônica de varredura (MEV) e ângulo de contato em água
(WCA); térmica através da calorimetria diferencial de varredura (DSC) e
ensaios mecânicos.

5
3. REVISÃO DA LITERATURA
3.1 Polímeros – aspectos gerais
A síntese do acetato de celulose, na segunda metade do século XIX, assim
como da borracha vulcanizada e a fabricação do papel foi um marco na química de
polímeros. Embora sejam de fontes renováveis, décadas se passaram para que seus
detalhes fossem completamente aceitas e compreendidas pela comunidade científica17.
A revolução na síntese de polímeros ocorreu, primeiramente a partir das
transformações do carvão, e, posteriormente, com o uso de fontes fósseis. Embora
haja inúmeras questões ambientais envolvidas desde a perfuração do poço de petróleo
até sua extração e refino, , ainda há grande dependência desses materiais poliméricos
oriundos de fontes fósseis tornando-os muito importantes na vida das pessoas18.
Estas macromoléculas de elevados pesos moleculares (de 20.000 a 2000000
Da) são constituídas por unidades que se repetem ao longo da cadeia (monômeros)
interligados por ligações covalentes através de reações de polimerização19. Usa-se a
expressão homopolímero quando há apenas um único tipo de monômero presente. Já
copolímeros são designados pela presença de mais de um tipo de monômero que
podem estar organizados de forma randômica, de forma alternada, em blocos ou até
mesmo graftizados ou enxertados. Nesse caso particular, ramificações poliméricas
partem da cadeia principal da macromolécula (Figura 1).
Fonte: https://materiaisplasticos.wordpress.com/
Figura 1. Classificação polimérica em função da organização molecular dos
monômeros

6
Os polímeros também podem ser classificados quanto à sua origem em
sintéticos e naturais. Poliestireno (PS), Polietileno (PE), Poli (1,1,2,2-Tetrafluor-etileno)
comercialmente conhecido como Teflon, Poli(hexametilenoadipamida) comercialmente
denominado como Náilon dentre outros são sintéticos provenientes de fontes fósseis17,
enquanto que a lignina, a celulose, o amido e a seda são naturais. Outra classificação é
em função do comportamento desses materiais frente à elevação na temperatura20.
Aqueles que apresentam fusão quando aquecidos e podem retornar à forma original
quando resfriados são considerados termoplásticos. Cabe ressaltar que esse
comportamento está diretamente relacionado à estrutura molecular com cadeias
totalmente lineares ou com ramificações flexíveis. Já os termofixos, são polímeros que
permanecem rígidos durante a sua formação e não sofrem fusão com o aquecimento.
Isso se dá devido a presença de ligações cruzadas e em rede entre as cadeias que
impedem os movimentos de vibração e rotação ocasionado pelo aumento da
temperatura.
A estrutura química dos polímeros é um dos fatores mais importantes no que diz
respeito a cristalinidade. Em função do tamanho da cadeia e suas complexidades, os
polímeros normalmente são semicristalinos, ou seja, regiões cristalinas dispersas no
material amorfo. Dessa forma, cadeias lineares favorecem a ordenação enquanto que
cadeias mais ramificadas ou organizadas em rede previnem a cristalização tornando-o
um polímero amorfo. No caso de copolímeros, quanto mais irregulares e aleatoriedades
existentes, maior a tendência de desenvolver um material amorfo20.
Outra forma de classificar os polímeros é de acordo com suas propriedades
mecânicas. Assim, há os materiais poliméricos plásticos que apresentam alguma
rigidez estrutural quando submetidos a uma carga; os elastômeros que por
apresentarem ligações cruzadas são mais flexíveis e suportam temperatura mais
elevada e as fibras que são capazes de ser estirados em longos filamentos com razão
entre comprimento e diâmetro de 100:1 cm20.
Os polímeros podem ser classificados considerando o seu método de
preparação. Há uma série de características, tais como: tipo de reação, mecanismo e
velocidade de crescimento da cadeia que são importantes para a constituição desses
materiais. É tamanha a dependência por materiais poliméricos que em 2014, foram
produzidos 260 milhões de toneladas de resinas termoplásticas. O Brasil contribuiu
com cerca de 2,5% esse montante. Com relação ao consumo, os países desenvolvidos
consomem cerca de 100 kg/hab enquanto que no Brasil o consumo per capita gira em

7
torno de 35 kg/hab. No que tange ao destino final desses materiais plásticos pós-
consumo, 80,3% ainda são dispostos em aterros sanitários e lixões e desse total 13,5%
são plásticos3. Ou seja, uma fonte química preciosa é descartada.
Uma alternativa viável é a reciclagem desses materiais. A reciclagem como
conhece-se usualmente envolve o reprocessamento de materiais poliméricos
termoplásticos em produtos similares ao de origem do resíduo ou em aplicações menos
nobres, como a produção de embalagens plásticas para acondicionamento de resíduos
domésticos, lixeiras, baldes etc. Porém neste tipo de reciclagem, a estrutura química do
polímero não é alterada. Neste tipo de reprocessamento uma desvantagem é a
contaminação do resíduo polimérico o que pode inviabilizar a reciclagem, sendo
necessário utilizar grandes volumes de água para a lavagem deste material ou mesmo
rejeição da matéria-prima devido inadequação, contribuindo para o baixo percentual de
reciclagem.
Há inúmeros trabalhos na literatura que buscam o reaproveitamento desses
materiais. Um deles trata do reaproveitamento de poliestireno expandido reciclado na
produção de compósitos com lignina obtida pelo método Kraft esterificada com anidrido
malêico. Através de análises reométricas avaliou-se a variação da concentração de
lignina em 2, 5 e 10% (em massa). Os autores observaram um decréscimo da
estabilidade térmica com 10% de carga e quanto maior a quantidade de lignina, mais
homogêneos foram os filmes21. Svinterikos et al. relatam a produção de nanofibras de
carbono a partir de garrafas PET e lignina Kraft. Através do emprego do ácido
trifluoracético usando o método eletrospinning o material obtido foi avaliado térmico,
estrutural e morfologicamente. As análises estruturais revelaram mudanças na
estrutura da lignina devido a forças intermoleculares com a cadeia polimérica e as
análises térmicas mostram boa miscibilidade entre o PET e a lignina22. Já Bonelli et al.
avaliaram o comportamento térmico, mecânico e morfológico do compósito de
polietileno de alta densidade proveniente de embalagens de óleo lubrificante pós-
consumo e fibras de “piaçava” naturais. As fibras foram tratadas de duas formas
diferentes: somente com 3-metacriloxipropil-trimetoxi-silano e também este composto
em metanol. Foi observado melhor comportamento mecânico no compósito com fibra
tratada somente com silano devido melhor adesão carga-matriz. Com relação a
estabilidade térmica dos compósitos, não houve grandes alterações23. O efeito de
lignina extraída de eucalipto pelo método Kraft nas propriedades termo-mecânicas e
morfológicas de poliestireno reciclado foram relatas por Pérez-Guerrero et al. A lignina

8
foi esterificada com anidrido acético e anidrido malêico e cargas de 5 e 15% foram
misturadas ao polímero empregando reômetro. Foi feito filme de poliestireno reciclado
e comercial para comparação. Embora os autores tenham observado o aumento da
estabilidade térmica das amostras quando a carga de lignina esterificada foi de 5%,
houve diminuição os valores de torque24.
Independente da metodologia empregada, os artigos mencionados acima tratam
da transformação e do reaproveitamento de polímeros reciclados e biomassa a base de
lignina. O emprego desses materiais de partida vem como alternativa a síntese de
novos materiais. As análises mostram que a inserção de carga em uma matriz
polimérica é viável e alterações nas propriedades termo-mecânicas e morfológicas
podem ser controladas em função de um objetivo final.
3.2 Síntese de polímeros
Polimerização é o nome dado a reação química que, partindo de unidades
menores (denominadas monômeros) forma cadeias longas e até mesmo redes
poliméricas. Essa ligação pode se dar de forma direta no qual os monômeros vão
sendo consumidos do meio para a formação imediata da cadeia. Também pode haver
a formação de dímeros, trímeros, oligômeros em geral para posterior constituição da
cadeia polimérica. Um tipo de reação de polimerização são as reações de adição (ou
reação em cadeia) no qual cada monômero liga-se, um de cada vez, à cadeia principal,
ou seja, o polímero formado tem a mesma composição dos monômeros. O consumo
inicial destes é lento, entretanto, esse tipo de reação apesenta alto grau de
polimerização já no começo da reação. O mecanismo das reações de poliadição
envolvem três etapas: iniciação, propagação e terminação via cisão homolítica ou
heterolítica obtendo-se no final um único produto de reação20. Um exemplo de polímero
que possui esse mecanismo de reação é o poliestireno, mas o polietileno,
poliisobutileno, polivinilacetato e polimetilmetacrilato também são polímeros obtidos por
adição.
A etapa de Iniciação ocorre a partir da formação de uma espécie química
(radical livre, íon ou complexo de coordenação) dependendo do monômero de partida.
Tanto agentes físicos (calor, radiações eletromagnéticas de baixa e alta energia)
quanto químicos (peróxidos, azoderivados, ácidos e bases de Lewis e o emprego de
catalisadores) podem provocar o início da polimerização19. A fase de propagação
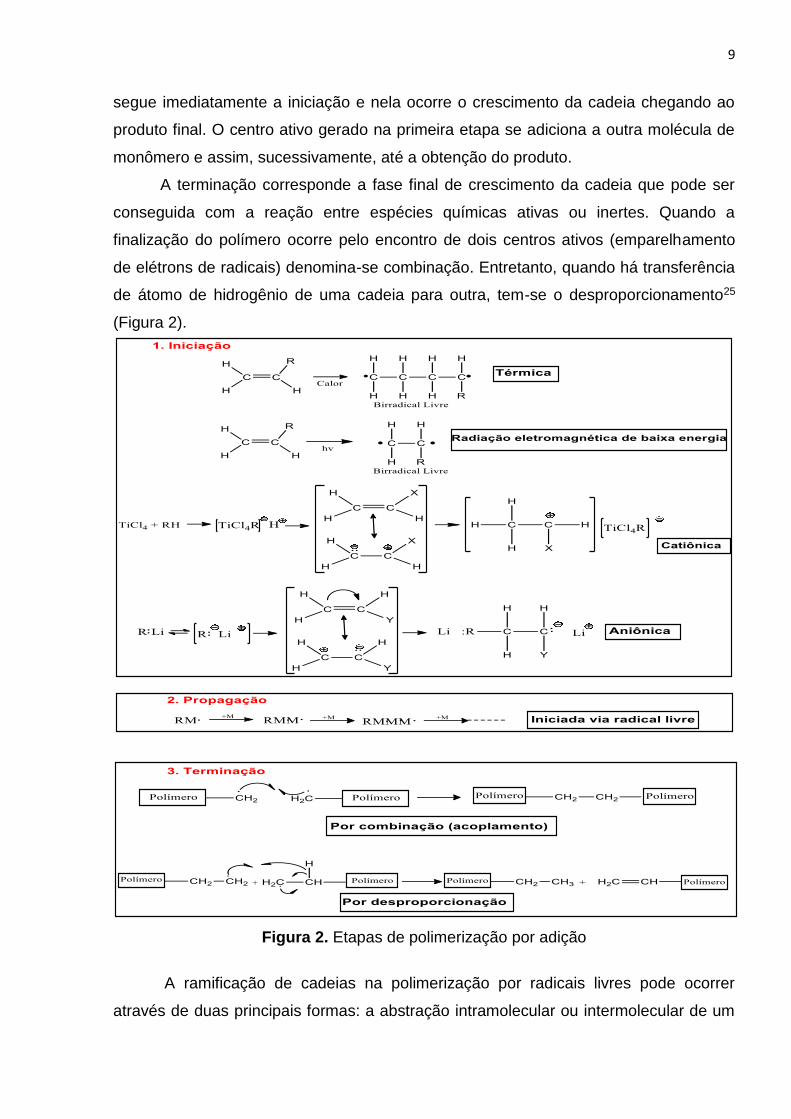
9
segue imediatamente a iniciação e nela ocorre o crescimento da cadeia chegando ao
produto final. O centro ativo gerado na primeira etapa se adiciona a outra molécula de
monômero e assim, sucessivamente, até a obtenção do produto.
A terminação corresponde a fase final de crescimento da cadeia que pode ser
conseguida com a reação entre espécies químicas ativas ou inertes. Quando a
finalização do polímero ocorre pelo encontro de dois centros ativos (emparelhamento
de elétrons de radicais) denomina-se combinação. Entretanto, quando há transferência
de átomo de hidrogênio de uma cadeia para outra, tem-se o desproporcionamento25
(Figura 2).
Figura 2. Etapas de polimerização por adição
A ramificação de cadeias na polimerização por radicais livres pode ocorrer
através de duas principais formas: a abstração intramolecular ou intermolecular de um

10
átomo de hidrogênio. Na primeira forma, a abstração de um átomo de hidrogênio de
dentro da molécula leva a formação de um radical secundário mais estável que o
primário. Dessa forma, o crescimento da cadeia se dará a partir desse novo radical
formado26. Já a subtração intermolecular ocorre entre uma cadeia em crescimento e
uma terminada. Assim, a cadeia antes terminada passa ser ativa e vice-versa, Esse
mecanismo é chamado de transferência de cadeia levando não somente à ramificação,
mas também favorecendo a disparidade no comprimento de cadeias27 (Figura 3).
Figura 3. Esquema de ramificação da cadeia na polimerização
Outro tipo de reação para a obtenção de polímeros é a policondensação. Este
tipo de polimerização apresenta velocidade de reação lenta para a constituição da
cadeia principal, mas com rápido consumo de monômeros. Neste caso, os monômeros
reagem entre si formando dímeros, trímeros, oligômeros em geral que se
intercondensam para a formação do polímero final. Usualmente, as policondensações
empregam dois tipos de monômeros bifuncionais que permitem o crescimento da
cadeia por meio dos grupamentos terminais na molécula (Figura 4). Com o
esgotamento de um desses monômeros, cessa o crescimento da macromolécula19.

11
Figura 4. Reação de policondensação do Náilon 6,6.
Outro tipo de polimerização por condensação emprega um único monômero com
dois grupos funcionais (Figura 5-A). Neste caso poderá haver a competição entre a
polimerização linear e as reações de ciclização intramolecular (figura 5-B). Para que
isso ocorra é necessário controle do equilíbrio químico e cinético, do tamanho do anel
do possível produto cíclico e das condições reacionais26.
Figura 5. Reação de policondensação em aminoácidos (A); possível ciclização
intermolecular (B)
A modificação de polímeros também é possível a fim de introduzir
funcionalidades desejadas na cadeia. Esta técnica é conhecida como copolimerização
e pode se dar de várias formas. Tem-se copolímeros em blocos no qual os monômeros
podem formar diblocos, triblocos, multiblocos e formato estrela (Figura 6). Esse tipo de
copolimerização permite ação compatibilizante podendo ser usado na interface entre
polímeros imiscíveis28. A possibilidade do formato estrela permite que esse tipo de

12
arranjo polimérico possa ser empregado como surfactante. Nesse caso, um bloco
possui caráter hidrofílico e outro hidrofóbico29.
Adaptado de Oliveira, M.A.M. et al.
Figura 6. Tipos de copolimerização em bloco
Já na copolimerização alternada, os monômeros estão organizados de forma
ordenada e sequencial. Um exemplo comum é o poli(tereftalato de etileno) – PET – que
possui característica parcialmente cristalina com inúmeras aplicações. A
copolimerização de forma aleatória é caracterizada pela organização randômica dos
monômeros, independente de sua distribuição estatística. Dessa forma, utilizando uma
mistura adequada de monômeros com diferentes grupos funcionais, é possível obter
esse tipo de material26. Nesse caso as resinas fenólicas merecem destaque. A
copolimerização multi-cadeia é possível quando um ou mais monômeros com mais de
dois grupos funcionais por moléculas estão presentes. A presença dessas ramificações
na cadeia principal, por sua vez, pode levar a formação de uma estrutura em rede.
Nesse caso, até mesmos as cadeias inseridas podem ser atacadas26.
Todas essas técnicas de modificação adicionam propriedades desejáveis de
mais de um homopolímero em um único material. Os copolímeros randômicos tem
menor cristalinidade em função de sua desorganização estrutural. Já os alternados
mudam seu caráter cristalino, quando comparado ao homopolímero, se um dos
monômeros for muito flexível ou rígido. Já na copolimerização em bloco as mudanças
são mais perceptíveis já que cada bloco monomérico apresenta comportamentos
distintos. Pode-se afirmar que todas as formas de modificação polimérica visam
agregar propriedades que podem aumenta o valor comercial ou industrial do polímero
de origem. Entretanto, a copolimerização por enxertia é um vasto campo a ser
explorado já que altera não somente propriedades térmicas, mas também mecânicas.
Destacam-se os polímeros randômicos altamente ramificados, que em larga escala são

13
mais viáveis economicamente e os dendrímeros (com ramificações mais controladas e
de síntese mais complexa).
3.3 Copolimerização por enxertia
A copolimerização por enxertia consiste na adição de um ou mais monômeros
diferentes ao longo da cadeia principal, organizados de forma regular ou não30. Neste
método de modificação de polímeros é possível obter a propriedade que se deseja
variando o grupo funcional enxertado.
Basicamente, há três estratégias de síntese de polímeros via enxertia: “Grafting
to”, “Grafting from” e “Grafting through” (Figura 7).
Fonte: Banerjee, S. et a.l, Polymer Chemistry (2014)
Figura 7. Diferentes estratégias de copolimerização por enxertia
A primeira estratégia consiste na formação de uma ligação covalente na cadeia
principal através da interação entre os grupos funcionais da matriz e da carga a ser
enxertada26. É mais empregada quando se tem matriz de cadeia plana e partículas
maiores para serem anexadas e ciclos de lavagens sucessivas, como pós-tratamento,
são capazes de remover polímeros não enxertados31. A produção de novos
precursores a partir de estireno enxertado em matriz de lignina Kraft foi possível
empregando radiação gama (entre 5 – 50 kGy)32. Foi observado redução no peso
molecular em 10kGy seguido de aumento do peso molecular por ligações cruzadas e
diminuição da quantidade de grupos hidroxilas presentes na lignina. O emprego de

14
nanocristais de celulose modificados com PEO e PEG como reforço em
nanocompósitos de poliestireno através de reações de carboxilação e amidação pode
ser encontrado na literatura33. Os resultados mostram dispersão homogênea e boa
compatibilidade entre os nanocristais modificados e a matriz de PS. A modificação de
superfícies de membranas empregadas para microfiltração de proteínas usando
tribloco PEO-PPO-PEO e celulose esterificada pode ocorrer através da abordagem
“grafting to” via fotoquímica34. Chen et al.35, empregando abordagem semelhante
modificou a hidrofobicidade superficial de polietileno de baixa densidade (LDPE) com
2,2,2-trifluoretil-metacrilato por radiação UV. As diferentes morfologias superficiais
foram destacadas pelo autor. Em todos os casos o ângulo de contato da superfície
modificada com a água foi maior que o filme de LDPE confirmando a fotoenxertia.
Grafting from envolve dois passos principais: o primeiro consiste na inserção de
um iniciador na cadeia polimérica para que em uma segunda etapa ocorra o
crescimento da ramificação a partir do iniciador enxertado. Este tipo de enxertia é
comumente utilizado para inserção de proteínas, peptídeos e enzimas36.
Esta técnica de enxertia é bem diversa e levas a várias rotas de
copolimerização, como por exemplo, nanopartículas de ouro foram inseridas em matriz
de poli(etilenoglicol) com a enzima glucose oxidase por via fotoquímica utilizando a
antraquinona como iniciador para constituição de sistema modelo de biosensor37. Outro
estudo promoveu a foto-enxertia de ácido polimetacrílico na superfície da matriz de
PHBHV para posterior complexação de nanopartículas de prata formando material com
boa atividade anti-bacteriana38. Empregando baixas concentrações de catalisador
fotorredox e diodo emissor de luz (LED) foi possível obter polímeros-proteínas
bioconjugados tanto em solventes orgânicos, água quanto em meio biológico mantendo
a atividade enzimática da proteína através da abordagem “grafting from”39. A obtenção
de nanocompósito com matéria-prima de fonte renovável foi possível pela inserção de
nanopartículas de celulose em matriz de PLA pela polimerização com abertura de anel
iniciada pelos grupos hidroxílicos empregando tolueno e catalisador a 80ºC40. Pela
combinação de polimerização aniônica e enxertia catiônica fotoinduzida, Kahveci et al.
com estruturas poliéteres complexas. A síntese do poli (óxido de etileno)-graft-poli (éter
vini-lbutílico) ocorreu via abordagem “grafting-from”. As análises de RMN de H¹ e GPC
confirmaram a estrutura do copolímero41.
Grafting through envolve a polimerização de um macromonômero, normalmente
de origem vinílica, mas depende de sua síntese26. Alguns exemplos na literatura

15
podem ser citados: Benzofenona e éter etil-benzóico foram usados como
fotossensibilizadores no “grafting through” de superfícies de PP e PS via radiação
ultravioleta agregando características de ambas as macromoléculas em um novo
material42; Em outro trabalho a inserção de estireno em moléculas de celulose de
algodão utilizando éter etil-benzóico e nitrato de urânio como fotossensibilizadores se
deu via fotoquímica obtendo um material com maior estabilidade térmica43. Durmaz et
al., empregando metilmetacrilato como monômero vinílico, sintetizaram copolímeros
com poliestireno-co-clorometilestireno via radiação UV. As análises de GPC e DSC
confirmaram a enxertia com aumento de Mn e Tg, respectivamente44.
Copolímeros graftizados podem ser sintetizados com diferentes densidades de
ramificações anexados na cadeia polimérica principal, não necessariamente durante a
polimerização, mas sim como uma forma de modificação. Altas densidades são obtidas
através de “Grafting to” e “Grafting from”. Destaca-se a segunda abordagem já que a
ramificação vai ao longo da reação se constituindo enquanto que “Grafting to” pode ter
problemas de impedimentos estéricos em função da ramificação enxertada45. Já o
método “Grafting through” possui densidade de ramificações controlada pela taxa de
transformação do macromonômero em co-monômero.
3.4 Poliestireno expandido
Uma das comodites poliméricas mais produzidas no mundo e muito empregada
atualmente é o poliestireno expandido. Ele é obtido a partir do poliestireno com
emprego de agente expansivo (como o pentano e butano) e é composto de 98% de ar
e 2% de poliestireno. Devido sua composição química, é um material versátil que pode
ser moldado de várias formas e espessuras com inúmeras aplicações. Dentre elas
destaca-se o uso em embalagens de produtos para transporte, alimentos, caixas
térmicas, na construção civil (estradas, pontes, ferrovias, prédios) dentre outras. Esta
vasta aplicação só é possível pelas características presentes no EPS tais como:
resistência mecânica, isolante térmico, leveza, resistência química e à humidade.
Avaliando o ciclo de vida dos materiais poliméricos, sabe-se que os plásticos,
em geral, levam cerca de 450 anos para se decompor. Dentre esses materiais, o
poliestireno expandido pós consumo tem como principal destino final aterros e lixões.
Estas montanhas de lixo quando descartadas a céu aberto podem causar inúmeros
danos tanto ao meio ambiente quanto à saúde pública. Além da poluição do solo, em

16
alguns casos esses materiais pós-consumo são descartados em rios e oceanos,
poluindo não somente os ambientes marinhos, mas também afetando a fauna local
através da persistência e bioacumulação desses materiais. Do Sul et al. relacionam a
presença de microplásticos encontrados no plâncton, em sedimentos, em animais
vertebrados e invertebrados e até mesmo interagindo com outros poluentes químicos
com o risco eminente aos ecossistemas marinhos46. As autoras destacam que as
principais questões que ainda prevalecem dizem respeito ao controle da fonte desses
descartes e como esse passivo ambiental gerado nos últimos 60 anos vai ser
endereçado. Já Gewert et al. destacam que 60% do dendritos flutuantes encontrados
nos oceanos são plásticos. Estes, quando expostos às condições climáticas
principalmente radiação UV proveniente do sol, podem sofrer processos oxidativos
foto-iniciados conduzindo a cisões das cadeias ligações cruzadas e rearranjos. Dentre
as olefinas, o PS se destaca pelo seu comportamento fotorreativo à radiação UV.
Entretanto a adição de anti-oxidantes e estabilizadores UV reduzem a degradabilidade
desse material47.
Tanto a incineração para geração de energia quanto a reciclagem mecânica de
poliestireno pós-consumo são abordagens de gerenciamento desse resíduo. A queima
pode gerar produtos perigosos tais como cianetos, ftalatos, compostos organo-
estanhos, bisfenol A e metais pesados (como chumbo e cádmio) além de dioximas e
furanos. Já a reciclagem, considerando que as políticas públicas de coleta seletiva não
são efetivas e que o custo de transporte desse material é alto (devido ao grande
volume ocupado), além dos altos volumes de água de lavagem gastos, ainda não é
economicamente viável48.
Na busca por reuso alternativo, Shin et al. utilizando o solvente natural d-
limoneno sugerem uma nova técnica de reciclagem através do processo eletrospinning.
Variando a proporção de EPS em 8 a 35% (em massa), os autores produziram fibras
poliméricas de EPS/THF, EPS/DMF e EPS/DMAc com diâmetro entre 200-500nm e
EPS/d-limoneno com diâmetro variando entre 300-900nm49. Produção de nano e
micropartículas de poliestireno pós-consumo a partir de uma reciclagem verde foi
realizada por Mangalara et al.48. Foi empregado d-limoneno como solvente verde, água
como não-solvente e PVA como estabilizante e dispersante de partículas. O tamanho
da partícula foi dependente da concentração de PVA e o d-limoneno foi recuperado ao
final do processo por destilação. Um potencial material de construção obtido a partir de
EPS reciclado e resíduo de óleo de cozinha é relatado por Sarmiento et al. A mistura

17
dos dois componentes foi aquecida até 165ºC e ainda quente foram produzidas fibras,
folhas e blocos desse material para posterior resfriamento a temperatura ambiente. As
análises estruturais mostraram a formação de um novo material pós processamento
com características mecânicas de compressão semelhantes ao concreto e azulejos de
cerêmica50. Estes trabalhos mostram a versatilidade do EPS pós-consumo para
produção de novos materiais com aplicações em diversas áreas.
3.5 Polímeros lignocelulósicos - a lignina
Dentre os matérias poliméricos que se encontram na natureza, os
lignocelulósicos se destacam principalmente pela sua abundância correspondendo a
aproximadamente 50% da biomassa do mundo51. Basicamente a estrutura dos
vegetais é constituída de polissacarídeos (como a celulose e a hemicelulose), as
ligninas e constituintes inorgânicos, como a sílica. A proporção desses constituintes se
modifica em função da espécie do vegetal, sendo que a quantidade de lignina varia
entre 15-30%52 e a celulose pode corresponder a 50%53. A casca de arroz, resíduo
gerado a partir da agroindústria, é um exemplo de biomassa vegetal que pode ser
reaproveitada. No mundo, cerca de 740 milhões de toneladas de arroz foram
produzidos em 2014 (FAO). O Brasil contribuiu com cerca de 12 milhões de toneladas
e o RS é o maior produtor no país com 8,2 milhões de toneladas (CONAB).
As ligninas, segundo polímero mais abundante na natureza, forma junto com a
hemicelulose uma matriz amorfa no qual as fibras de celulose são incorporadas e
protegidas contra microorganismos53 (Figura 8). Além disso, tem como outras funções
dar resistência mecânica à parede celular do vegetal e ser responsável pelo transporte
de nutrientes e metabólitos54.
Fonte: http://pt.slideshare.net/JuNNioRe/madeira-propriedades-processos-de-fabricao-e-aplicaes
Figura 8. Representação simplificada da estrutura vegetal

18
Quanto a sua característica estrutural, a lignina é uma macromolécula aromática
que possui unidades fenilpropanóides arranjadas de forma randômica e tridimensional.
Sua estrutura depende do tipo de vegetal (podendo variar até mesmo dentro do mesmo
vegetal analisando partes diferentes do mesmo), estação do ano, diferentes condições
de solo, de clima, processo de extração52. É formada pela polimerização
desidrogenativa de três álcoois precursores principais: álcool coniferílico, sinpílico e p-
cumarílico55 (Figura 9) pela ação enzimática da peroxidade e lacase.
Figura 9. Álcoois Precursores das unidades Fenil-propanóides Guaiacila (G),
Siringila (S) e p-hidroxifenila (H).
Dependendo da biomassa, a proporção desses precursores é diferente. Em
gramíneas, predominam as unidades guaiacila (G) e siringila (S) e em menor
quantidade as unidades fenilpropanóides (H). Já em coníferas, há mistura de G e S
enquanto que em plantas folhosas há o predomínio da unidade G56. Na casca de arroz
há o predomínio da unidade guaiacila57. A união desses precursores tanto por ligações
a partir do anel quanto pela cadeia alifática dão origem à estrutura complexa da lignina
(Figura 10). Os tipos de ligações entre esses precursores dão à lignina diferentes
características e propriedades (Figura 11) assim como a presença de grupos
metoxílicos e carbonílicos.

19
Figura 10. Estrutura da lignina

20
Figura 11. Tipos de ligações entre as unidades fenilpropanóides.
Em consonância com a tendência mundial de aproveitamento máximo de
recursos, diversos trabalhos buscam novas aplicações além da queima para os
resíduos da biomassa. Pode-se citar a produção de resinas fenol-formaldeído58,
adesivos59, tintas e vernizes60, fabricação de cimento61, como precursor em fibras de
carbono62, dentre outras. Nos últimos anos, há uma tendência de utilizar a lignina como
carga em matriz polimérica21, 63, 64. Dessa forma, aproveitando-se das características
naturais da lignina que oferece resistência e estruturação na planta.
Há também diversas formas de modificação da lignina. Uma delas é por reações
químicas para obtenção de inúmeros produtos da Química Fina como a vanilina,
dimetilsulfóxido, fenóis, cresóis dentre outros65. É possível encontrar na literatura
trabalhos que modificam a estrutura da lignina, por exemplo através de esterificação,
para posterior utilização21. As modificações podem ser tanto no sentido de criar novos
centros ativos na molécula (como a aminação, nitração) quanto na funcionalização dos
grupos hidroxilas (através da esterificação, fenolação, dentre outros)66. A
biotransformação de ácido ferúlico em vanilina a partir da lignina pode se dar através

21
da descarboxilação, redução e por reações bioquímicas empregando enzimas67. As
reações fotoquímicas também são empregadas para modificar a estrutura da lignina.
Um exemplo é a produção de azo-polímeros a base de lignina. A partir da lignina álcali
e de sais de diazônio sintetizados, foi possível obter compostos com diferentes grupos
azo-cromóforos sendo um deles com significativo efeito fotocrômico. Esse exemplo de
azo-polímero a base de lignina oferece um método econômico de produção de azo
compostos com potencial aplicação como material fotossensível68. Outro exemplo é a
produção de copolímeros termoplásticos a base de lignina, poliestireno e acriloil-
benzofenona como fotossensibilizador por via fotoquímica. Esse copolímero foi
sintetizado via transferência de elétron por ativador regenerado (ARGET) e também via
polimerização radicalar com transferência de átomos (ATRP)69.
3.6 Fotoquímica como ferramenta de modificação da matéria
3.6.1 Interações entre a luz e a matéria e transições eletrônicas
A luz corresponde a uma radiação eletromagnética que se propaga no espaço e
possui comportamento de partícula e de onda. Dessa forma, ela pode ser entendida em
função do seu comprimento de onda e frequência quando se comporta como onda e na
forma de “pacotes de energia” quando partícula. No momento em que a radiação
interage com a matéria, vários processos podem ocorrer, tais como: reflexão,
espalhamento, fluorescência, fosforescência, absorção, reações fotoquímicas, etc
(Figura 12). Dentre esses fenômenos, cabe destacar a fluorescência e a
fosforescência. Em ambos os processos ocorre, primeiramente, a absorção de luz
levando a molécula a um estado excitado (de maior energia). Na fluorescência, de
acordo com a Regra de Kasha, a molécula passa a emitir fótons, ou seja, fluorescer, a
partir do menor nível de energia do estado excitado, S1 (singleto) ou T1 (tripleto),
dependendo da multiplicidade. Essa emissão ocorre em comprimentos de ondas
maiores daquele absorvido. Esse fenômeno ocorre em curto espaço de tempo, na
ordem de 10-9s e enquanto há absorção de energia. Já na fosforescência, a molécula
no estado excitado sofre inversão do spin do elétron ao efetuar o cruzamento
intersistemas singleto-tripleto. Dessa forma, a molécula no estado excitado leva muito
mais tempo para retornar ao estado fundamental devido a inversão no spin do
elétron70. Dependendo do comprimento de onda empregado, pode-se ter radiações de
alta energia, como os raios-γ, passando pela radiação ultravioleta (entre 400 – 200nm),

22
até ondas de rádio, de acordo com o espectro eletromagnético (Figura 13). Apenas
uma pequena parcela é visível ao olho humano (entre 400 – 800nm).
Fonte: Sotomayor, M. D. P. T. et a.l, Química Nova (2008)
Figura 12. Diagrama de Jablonski.
Figura 13. Espectro eletromagnético
O emprego de radiação ultravioleta baseia-se na excitação dos elétrons do
estado fundamental para um estado excitado, ou seja, essas transições, que ocorrem
em níveis eletrônicos, acontecem quando um elétron é promovido de um orbital
ocupado para um desocupado de maior energia potencial71. Em termos de orbitais,
sabe-se são os espaços mais prováveis de encontrar o elétron. Estão relacionados a
uma função de onda, com energia definida. A união de dois orbitais atômicos da

23
camada de valência dos dois átomos ligantes origina dois orbitais moleculares (ligante
e anti-ligante) que redistribuem a nuvem eletrônica. Assim como os orbitais, as funções
de onda são combinadas para dar origem às funções de onda dos orbitais moleculares
(Figura 14). Essa combinação pode gerar interferência construtiva na qual a
sobreposição de orbitais em fase leva a formação do orbital molecular ligante (σs, σx e
πyz) ou interferência destrutiva originando o orbital molecular anti-ligante (σs*, σx* e πyz*).
Dependendo do valor da energia fornecida ao sistema, os elétrons excitados podem
ser provenientes de ligações simples (transições σ→σ*), de ligações duplas (transições
π→π*) além de pares de elétrons não ligantes de átomos como oxigênio, nitrogênio e
enxofre (transições n→σ* ou n→π*). Este último tipo de transição mencionada, são
conhecidas como “proibidas” ou “não-permitidas” por simetria. Entretanto, observa-se
sua ocorrência devido a questões espaciais já que os elétrons não ligantes dos átomos
de O e N estão espacialmente muito próximos dos orbitais moleculares antiligantes,
diminuindo a barreira energética e efetuando a ligação química.
Figura 14. Representação da formação de orbitais moleculares (ligantes e antiligantes)
a partir dos orbitais atômicos (1s, 2px e 2py)

24
3.6.2 Reações fotoquímicas
O esquema representativo abaixo mostra como as reações fotoquímicas
ocorrem (Figura 15). Ao incidir radiação eletromagnética, os elétrons que estão no
estado fundamental (área inferior azul) passam para o estado excitado (área
intermediária). Nesta etapa, os éxcitons formados (centros energeticamente ativados)
possuem propriedades físicas e químicas diferentes das encontradas no estado
fundamental. Dependendo das condições de reação fornecidas ao sistema,
principalmente no que diz respeito a solvente, pH e concentração dos materiais de
partida, pode haver rearranjos, quebras de ligações e até formação de novas
moléculas. Se maior energia é fornecida ao sistema, pode haver a formação de radicais
(área superior em vermelho)72, 73.
Figura 15. Esquema representativo das reações fotoquímicas
Os compostos orgânicos e inorgânicos, são capazes de absorver radiação
eletromagnética desde que possuam elétrons de valência que possam ser excitados
para níveis de maior energia. Os compostos orgânicos com ligações saturadas
precisam de uma energia relativamente alta para se excitarem o que resulta em
absorções na chamada região do ultravioleta no vácuo (λ < 185nm). Dificuldades
experimentais, principalmente com o vácuo, tornam inviáveis as aplicações nessa
região do espectro eletromagnético. A maioria das aplicações está em torno de
compostos orgânicos que apresentem transições do tipo π→π* e n→π* já que as

25
energias necessárias para essas transições levam a bandas de absorção para a região
do ultravioleta-visível (200 a 800nm)74.
Ao traçar um paralelo entre as reações fotoquímicas e as reações químicas
clássicas, é possível perceber que o uso da radiação eletromagnética é mais seletivo,
pois determinado comprimento de onda excita grupos cromóforos específicos nas
moléculas75, 76. Já nas reações orgânicas convencionais, a energia térmica excita todos
os grupos existentes na molécula. Sendo assim, as reações orgânicas originam os
produtos termodinamicamente estáveis, restringindo diferentes conformações que
podem ser obtidas fotoquimicamente. Além disso, as reações fotoquímicas estão
aliadas aos princípios da Química Verde e sustentabilidade77. Essa afirmativa é
coerente já que é uma via reacional rápida, de baixo custo e complexidade
experimental16, 72, 78. Além disso, o processo pode ocorrer à temperatura ambiente
dispensando etapas sucessivas de síntese e purificação com solventes com alta
toxicidade.
Devido essa vantagem frente às reações orgânicas clássicas, a fotoquímica
pode ser empregada para inúmeras reações. Dentro das adições empregando radiação
UV, cabe destacar as fotocicloadições (2+2) de cetonas e ésteres α,β-insaturados. É
possível obter um único composto cíclico de quatro carbonos a partir de reação de
apenas um passo (figura 16). Reações fotoquímicas também pode ocorrer formando
anéis com heteroátomos79 (Figura 17).
Figura 16. Exemplo de fotocicloadição (2+2)
Figura 17. Exemplo de formação de anéis com heteroátomos via fotoquímica

26
Durmaz et al., relatam a copolimerização em bloco fotoinduzida de
metilmetacrilato em PS terminados com íon N-alcóxi piridinio por via fotoquímica, pós
modificação da cadeia Figura 18a)80. A modificação de polímeros empregando a
radiação UV também é encontrada na literatura44 (Figura 18b). Kilduff et al. propõem
mecanismo para copolimerização por enxertia via radiação UV de membranas de
poli(arilsulfonas) com monômero vinílico81 (Figura 19).
Figura 18. Fotoenxertia de MMA em PS (a) e modificação de PS (b) via reação
fotoquímica
Figura 19. Copolimerização de poli(arilsulfonas) com monômeros vinílicos

27
Quando se deseja atribuir novas características a um determinado material de
forma específica, a inserção de uma cadeia em outra por via fotoquímica surge como
potencial alternativa. Para que a fotofuncionalização de materiais ocorra é necessário
que o polímero ou a cadeia que se quer enxertar possua sítios ativos excitáveis pela
radiação eletromagnética selecionada. Ajustando parâmetros reacionais tais como
tempo de reação, concentração, solvente e conhecendo a estrutura dos polímeros é
possível controlar tanto a formação das espécies ativas quanto o produto formado.
Dependendo desse sistema, possíveis reações fotoquímicas podem ocorrer.
Sabe-se que a lignina além de centros carbonílicos, possui ligações C-O em sua
estrutura. Com o intuito de compreender o mecanismo de reação que possa estar
ocorrendo e estimar os possíveis sítios de reação, alguns tipos de reações serão
mencionados a seguir.
A reação fotoquímica pode começar a partir do centro carbonílico da lignina.
Dessa forma, destacam-se as reações Norrish Tipo I e II. A reação de Norrish Tipo I
consiste na clivagem da ligação α-carbonila podendo formar dois radicais. Já a reação
Norrish Tipo II se dá pela quebra da ligação β-carbonila levando a formação de ligação
C=C. Ela pode ocorrer tanto no estado singlete, levando à fragmentação quanto no
estado triplete levando à cicloadição82. Pode também ocorrer a abstração de hidrogênio
da posição ϒ-carbonila (Figura 20). Em seguida ocorre a propagação da reação
fotoquímica.
F
Figura 20. Reações fotoquímicas usuais envolvendo compostos carbonílicos.

28
Além disso, reações de abstração de hidrogênio intra e intermolecular tanto da
cadeia principal do poliestireno, da parte alifática da estrutura da lignina quanto dos
anéis aromáticos são passíveis de ocorrer. Cabe salientar que além da formação de
éxcitons (estados excitados sem formação de radical), pode estar havendo a formação
de radicais quando os níveis energéticos são maiores. A fim de compreender as
mudanças ocorridas após a fotoreação, é necessário o emprego de técnicas de
caracterização. Cabe ressaltar que as modificações podem ser estruturais,
morfológicas, térmicas e/ou mecânicas.
3.7 Caracterizações molecular / estrutural de materiais poliméricos
3.7.1 Análises Térmicas
Um fator muito importante para determinar as propriedades de um polímero é a
estrutura macromolecular. Essa propriedade é função da disposição ordenada dos
monômeros nas macromoléculas. Dessa forma, podem existir polímeros no estado
amorfo (disposição desordenada de moléculas) ou no estado cristalino (com
ordenamento molecular), sendo que, na maioria dos casos, os polímeros apresentam
uma estrutura parcialmente amorfa, semi-cristalina ou cristalina. O grau de
cristalinidade de um polímero é dependente da estrutura química, peso molecular e
tratamento físico (temperatura e forças que o material é submetido)8.
Há vários métodos analíticos para determinação de cristalinidade de polímeros
tais como: difração de raio X, espectroscopia de ressonância magnética nuclear no
estado sólido, calorimetria diferencial de varredura (DSC) dentre outros. Um dos mais
amplamente usados, o DSC é uma técnica versátil já que além do grau de
cristalinidade, fornece informações sobre a ordem de reação, energia de ativação e
energia de polimerização. O grau de cristalização em função do tempo relaciona o calor
gerado no tempo t (numerador) e o calor gerado durante todo processo de cristalização
(denominador) (Equação 1). Dentre as informações mais relevantes fornecidas pelo
DSC, cabe destacar a temperatura de cristalização (Tc) temperatura de fusão (Tf)
(temperatura pela qual o material passa do estado cristalino ou semi-cristalino para o
estado líquido), as capacidades caloríficas e temperatura de transição vítrea (Tg) que
corresponde a temperatura que o material passa do estado mais rígido para o mais
flexível83 (Figura 21).

29
(1)
Fonte: http://images.treccani.it/enc/media/share/images/orig/system/galleries/NPT/VOL_X/IMMAGINI/termico_01.jpg
Figura 21 - Ilustração explicativa da técnica de DSC
3.7.2 Análises Morfológicas
O microscópio eletrônico de varredura (MEV) que utiliza feixes de elétrons para
explorar a superfície da amostra é um dos mais versáteis instrumentos disponíveis para
observação e análise das características microestruturais de matérias sólidos. A
maioria dos instrumentos usa como fonte de elétrons uma lâmpada de tungstênio
aquecido que opera numa faixa de tensão de aceleração de 1 a 50 kV. O sinal de
imagem que é gerado pelo equipamento resulta da interação do feixe de elétrons
incidido com a superfície da amostra (Figura 22).
Fonte: http://www.degeo.ufop.br/laboratorios/microlab/figuras/esquema_mev.gif
Figura 22. Esquema representativo do funcionamento do MEV
Essa técnica permite visualizar a interação entre a carga de minerais
incorporados em uma matriz polimérica. O desempenho mecânico de muitos polímeros

30
está associado à dispersão de forma homogênea dessa carga na matriz ou a
homogeneidade de fases na matriz. Cabe ressaltar que o preparo da amostra através
do recobrimento com ouro é relevante já que aumenta a interação da amostra com o
feixe de elétrons84.
Outra análise superficial de materiais poliméricos é o ângulo de contato. Esta
técnica determina o grau de molhabilidade de uma superfície através da interação de
uma gota de água quando depositada na superfície de um material. Dessa forma,
mede-se o ângulo formado entre um plano tangente a uma gota do líquido e um plano
contendo a superfície do material85 (Figura 23). Essa técnica caracteriza o material
quanto a hidrofilicidade (ângulo menor que 90º) ou hidrofobicidade (ângulo maior que
90º). Embora simples, toda mudança química ou mofológica na superfície do polímero
reflete no seu valor de WCA.
Fonte: http://www.alfaconnection.pro.br/images/FQM010111a.gif
Figura 23. Princípio da análise de ângulo de contato
3.7.3. Análises Espectroscópicas
Dentre as análises espectroscópicas, destaca-se a espectroscopia de UV-VIS.
Nesta técnica, moléculas com diferentes grupos cromóforos se comportam de maneira
diferenciada em função da radiação UV incidida. Assim, é possível acompanhar a
absorção dessa energia eletromagnética com relação ao comprimento de onda através
de um espectro gerado71.
Outra técnica muito empregada é a FTIR. Ela possibilita a identificação de um
composto orgânico considerando duas etapas. A primeira delas consiste na
determinação dos constituintes da cadeia polimérica no que diz respeito ao percentual
de grupos metoxílicos e demais grupos funcionais além os tipos de ligações
predominantes. Já a segunda etapa corresponde à comparação detalhada do espectro
do composto desconhecido com o espectro do composto puro que contém todos os
grupos funcionais encontrados na primeira etapa.

31
Para compreender o processo de enxertia via radiação UV e a estrutura química
molecular do copolímero, emprega-se a espectroscopia de ressonância magnética
nuclear. Esta técnica consiste na absorção de energia eletromagnética na região de
radiofrequência regida pelas características da amostra, sob condições apropriadas em
um campo magnético. Sabe-se que os elétrons são partículas carregadas capazes de
girar em dois estados de spin: +1/2 e -1/2. Alguns núcleos também possuem essa
propriedade, como ¹H e ¹³C o que permite o estudo por RMN. Quando uma amostra é
colocada em um determinado campo magnético, o núcleo gira e tende a se alinhar a
favor (estado de spin α) ou contra (estado de spin β) o campo de maior magnetismo
sendo o alinhamento a favor menos energético (Figura 24). A diferença de energia
entre os estados de spin α e β depende da força do campo elétrico aplicado. Quando
essa variação de energia é alcançada, o núcleo no estado de spin α é excitado para o
estado de spin β. Quando os núcleos sofrem relaxamento, são emitidos sinais
eletromagnéticos cuja frequência corresponde à variação de energia absorvida. Busca-
se com essa técnica a detecção desses sinais e o registro gráfico da frequência do
sinal versus suas intensidades, principalmente no que diz respeito à posição dos
hidrogênios (RMN ¹H) e à posição dos carbonos (RMN ¹³C).
Fonte: http://docs.fct.unesp.br/docentes/dfqb/eduardoperez/Instrumental2011/Espectroscopia%20RMN.pdf
Figura 24. Fundamentos da técnica de RMN
Por fim, a espectroscopia de energia dispersiva (EDS) corresponde a um
sistema acoplado à microscopia eletrônica de varredura. É uma ferramenta capaz de
determinar de forma qualitativa e semi-quantitativa a composição química a partir da
emissão de raio X característico de cada elemento químico da amostra. Com esta
emissão, os raios X são detectados, amplificados (Figura 25) para gerar ao final da
análise um espectro com os elementos presentes além de uma tabela com as
proporções de cada componente.

32
Fonte: https://repositorio.ufsc.br/bitstream/handle/123456789/105176/Priscila_Klauss.pdf?sequence=1&isAllowed=y
Figura 25. Esquema do espectrômetro de dispersão de energia.
3.7.4 Análise Cromatográfica
Verifica-se que polímeros de origem natural têm pesos moleculares muito mais
elevados que os obtidos de forma sintética. Cabe ressaltar que os pesos moleculares
dos polímeros podem diminuir ou aumentar por meio de tratamentos físicos e químicos,
quando necessários à processabilidade ou a sua utilização. Na literatura, há menção
de alguns tipos de pesos moleculares: peso molecular numérico médio, peso molecular
ponderal médio, peso molecular médio mais elevado e peso do pico molecular.
O peso molecular numérico médio (Mn) depende do número de moléculas de
polímeros presentes na solução, qualquer que seja sua estrutura ou tamanho (Equação
2). Já o peso molecular ponderal médio (Mw) depende do número e do peso médio das
moléculas presentes na solução, qualquer que seja sua estrutura e tamanho (Equação
3). O peso molecular médio das cadeias maiores (Mz) (Equação 4) são mais sensíveis
para polímeros com alto peso molecular. Através dos valores de Mw e Mn é possível
determinar a polidispersão, que será tão maior quanto mais heterogêneos forem os
pesos moleculares. Em polímeros industriais, a polidispersão é próxima de 2, mas pode
atingir valores maiores19.

33
Eq. (2) Onde: Ni = número de moléculas de classe i
Mi = peso molecular de moléculas da classe i
Mn = peso molecular numérico médio
Eq. (3) Mw = peso molecular ponderal médio
Eq (4) Na eq. (4) n = 1 então M= Mw
n = 2 então M = Mz
n = 3 então M = Mz+1
A Cromatografia de Permeação em Gel (GPC) ou também conhecida como
cromatografia de exclusão de tamanho que consiste na separação de moléculas
dissolvidas com base no seu peso molecular, conforme o tamanho de cada molécula.
Introduz-se a solução de amostra em uma coluna contendo como recheio partículas
porosas de estrutura rígida e semi-rígida, de tamanho de poros bem controlado, com
base em sílica ou poliméricas. Se um dado soluto (polímero) é maior que o tamanho
dos poros maiores, ele será excluído e deverá passar todo seu tempo na fase móvel,
ou seja, será eluido primeiro86. Caso contrário, terá um tempo de retenção maior.
3.7.5 Ensaio Mecânico de Tração
As propriedades mecânicas de polímeros pelos seguintes parâmetros: módulo
de elasticidade, limite de escoamento e limite de resistência à tração. Um ensaio de
tensão-deformação nos permite obter essas informações que são influenciadas pela
taxa de deformação, pela temperatura e pela estrutura química do polímero20.
Dentre os parâmetros obtidos, o limite de escoamento corresponde ao máximo
escoamento ainda na deformação elástica. O limite de resistência à tração corresponde
à máxima tensão que o material suporta e o módulo de elasticidade é a razão da
tensão e a deformação elástica longitudinal do material.
Basicamente são encontrados três comportamentos para materiais poliméricos
(Figura 26). Em A podemos visualizar o comportamento tensão-deformação de um
polímero frágil que rompe enquanto se deforma. Em B tem-se o comportamento de um
material plástico: apresenta deformação inicial elástica seguida de escoamento e
deformação plástica. Já em C, é possível observar o comportamento de um material
completamente elástico (grande deformação ao empregar baixa tensão).
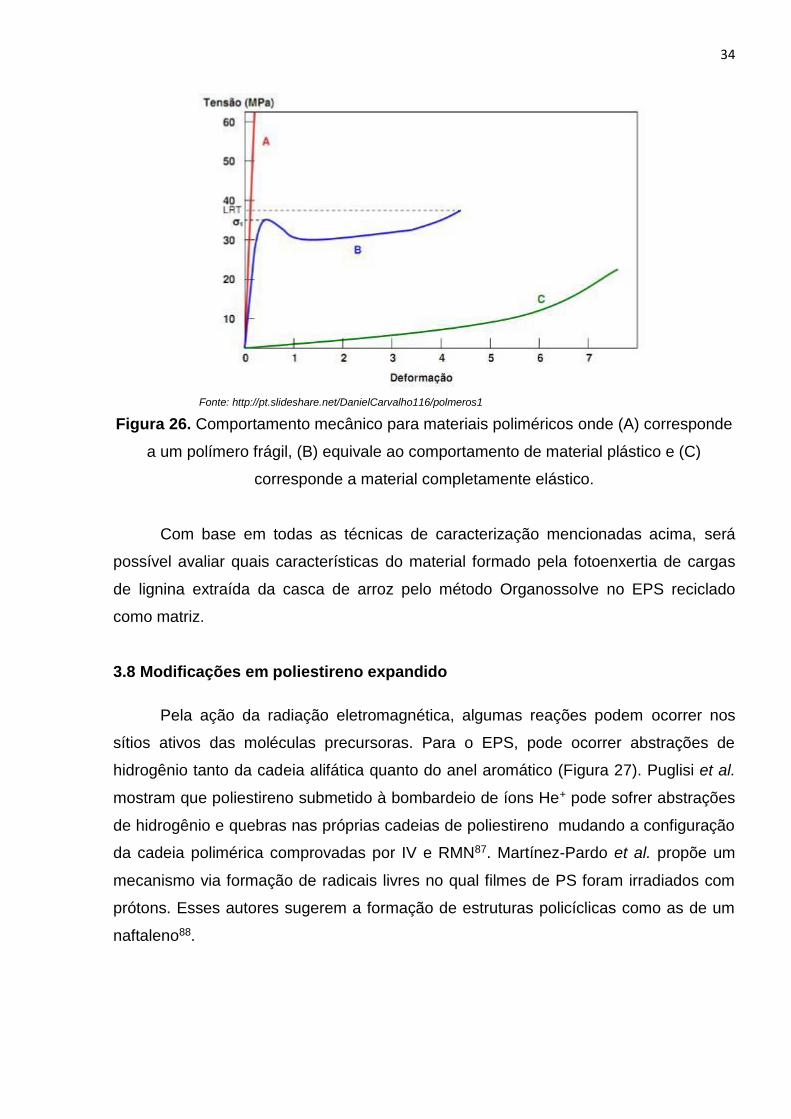
34
Fonte: http://pt.slideshare.net/DanielCarvalho116/polmeros1
Figura 26. Comportamento mecânico para materiais poliméricos onde (A) corresponde
a um polímero frágil, (B) equivale ao comportamento de material plástico e (C)
corresponde a material completamente elástico.
Com base em todas as técnicas de caracterização mencionadas acima, será
possível avaliar quais características do material formado pela fotoenxertia de cargas
de lignina extraída da casca de arroz pelo método Organossolve no EPS reciclado
como matriz.
3.8 Modificações em poliestireno expandido
Pela ação da radiação eletromagnética, algumas reações podem ocorrer nos
sítios ativos das moléculas precursoras. Para o EPS, pode ocorrer abstrações de
hidrogênio tanto da cadeia alifática quanto do anel aromático (Figura 27). Puglisi et al.
mostram que poliestireno submetido à bombardeio de íons He+ pode sofrer abstrações
de hidrogênio e quebras nas próprias cadeias de poliestireno mudando a configuração
da cadeia polimérica comprovadas por IV e RMN87. Martínez-Pardo et al. propõe um
mecanismo via formação de radicais livres no qual filmes de PS foram irradiados com
prótons. Esses autores sugerem a formação de estruturas policíclicas como as de um
naftaleno88.

35
Figura 27. Possíveis reações fotoquímicas para o EPS
3.9 Modificações em lignina
Já para LO, no que diz respeito a possíveis reações fotoquímicas que podem
ocorrer, Haifeng et al. sugerem que o emprego de H2O2 e FeCl2 permitem a formação
de radicais hidroxilas que podem estar envolvidos na formação de radicais na lignina
promovendo a reação de enxertia. A terminação da copolimerização pode se dar pelo
encontro de dois radicais ou pela transferência de cadeia63. Meister et al. empregando
H2O2 e CaCl2 promoveram a copolimerização de 1-feniletileno e lignina. Os autores
trazem como sugestão de mecanismo, primeiramente a formação do radical hidroxila
seguida de abstração de hidrogênio da lignina para posterior copolimerização com o
outro polímero89. Como a LO é rica em grupos carbonílicos, este sítio também pode
estar sendo afetado pela radiação UV (Figura 28).

36
Figura 28. Possíveis reações fotoquímicas da LO.
O emprego de outros promotores da reação, como moléculas orgânicas
bifuncionalizadas, como diácidos carboxílicos, permitem que o copolímero tenha mais
de um sítio ativo para crescimento. Essas moléculas podem ser empregadas devido a
presença de grupos cromóforos sensíveis à radiação UV que agem como moléculas de
sacrifício promotoras das reações fotoquímicas. Duval et al. afirmam que dependendo
dos sítios reativos desses precursores pode haver a formação de cadeias lineares ou
ramificadas e até mesmo a formação de redes poliméricas (Figura 29).

37
Fonte: Antoine Duval et al., Reactive and Funcional Polymer (2014)
Figura 29. Reação da lignina com monômero bifuncional. Em (a) tem-se a
formação de cadeias lineares normais e ramificadas e em (b) a formação de rede
polimérica
A matriz polimérica é constituída de uma parte alifática tendo como ramificação
um anel aromático. Já a carga corresponde a uma macromolécula complexa que
apresenta grupos cromóforos sensíveis à radiação UV. Em alguns casos, o emprego
de moléculas de sacrifício vem como alternativa na promoção da reação via radiação
UV. Da união desses precursores pode surgir um copolímero com características
diferenciadas. As reações fotoquímicas vêm como potencial alternativa não somente
pela seletividade e baixa complexidade experimental, mas também por ir ao encontro
dos princípios de sustentabilidade e Química Verde

38
4. MATERIAIS E MÉTODOS
4.1 Extração da Lignina pelo método Organossolve
A lignina utilizada neste trabalho foi extraída pelo método Organossolve
utilizando a casca de arroz como biomassa, já consolidada no nosso grupo de
trabalho90. Este consiste de duas etapas de preparação da casca: lavagem com água
corrente seguida de secagem por 48h à 60ºC e a extração de extrativos utilizando
etanol e hexano por 48h sob refluxo seguida de filtração e secagem. Neste momento a
casca está adequada para a etapa de extração da lignina.
Utilizando 5g de casca pré-tratada, 100 mL de etanol e 1 mL de H2SO4, a lignina
é separada dos demais polissacarídeos empregando refluxo por 24h à 100ºC e
pressão atmosférica. Ao final da extração, é feita a primeira filtração para separar a
lignina (em solução) da casca de arroz. Em seguida, a solução é concentrada por meio
da evaporação do solvente e resfriada. Adiciona-se água à solução em uma proporção
de 3x o volume inicial para precipitar a lignina. Deixa-se a solução em repouso.
Realiza-se a segunda filtração seguida de sucessivas lavagens com água destilada até
pH neutro. Por fim, para garantir a total separação da lignina dos demais constituintes
vegetais, o filtrado é lavado com água, sob agitação, por 2h a 70ºC. Faz-se a última
filtração e obtém-se um sólido marrom que é seco até massa constante.
4.2 Matriz polimérica, carga e promotores da reação
Foi empregado como matriz polimérica o poliestireno expandido (EPS) reciclado,
vulgarmente conhecido por ISOPOR, marca registrada da empresa KNAUF.
Quimicamente, o EPS pode ser obtido a partir da polimerização do estireno em água,
seguido de expansão com vapor, podendo chegar a 50X o seu tamanho original. Este
polímero é caracterizado pela presença de inúmeros anéis aromáticos que conferem
ação protetiva frente processos fotoquímicos88. Dessa forma, o poliestireno é um
polímero que absorve radiação eletromagnética por apresentar um grupo cromóforo,
mas consegue estabilizar bem essa espécie ativa gerada pelo efeito de deslocalização
de elétrons (Figura 30).

39
Figura 30. Monômero encontrado no poliestireno
O polímero empregado como carga foi a lignina, de origem natural. De acordo
com a sua estimativa de estrutura (Figura 10), é possível perceber a presença de
inúmeras ligações metoxilícas e hidroxílicas além da presença de anéis aromáticos.
Esses grupos cromóforos também são sensíveis à radiação eletromagnética.
Como moléculas de sacrifício foram utilizados dois diácidos carboxílicos: ácido
adípico e ácido itacônico (Figura 31). Estas moléculas são conhecidos como
antioxidantes, de forma a prevenir os processos de degradação91. Porém espera-se
que a presença de grupos oxigenados na duas extremidades sejam úteis na formação
de espécies reativas para a construção do copolímero, atuando como molécula de
sacrifício, ou seja atuando como interface entre o polímero e a lignina.
Figura 31. Diácidos carboxílicos como moléculas de sacrifício
4.3 Caracterização Fotoquímica dos Precursores
A primeira etapa do trabalho correspondeu a escolha do solvente já que o
sistema, em reações fotoquímicas, pode influenciar na reação. Realizou-se teste de
solubilidade da lignina sulfonato comercial para alguns solventes tais como álcool
isopropropílico, acetona, DMF, THF e dioxano 50% (em proporção 1:1 de água).
Variou-se a agitação (através de ultrassom ou magnética) por tempo de 15min sempre
a temperatura ambiente. Observou-se solubilidade parcial de lignina sulfonato
comercial em THF, DMF e dioxano 50% com agitação magnética. O uso do ultrassom

40
foi descartado já que esse tipo de energia estava elevando a temperatura do sistema
podendo levar a quebras de cadeias da lignina. Em seguida, testou-se moléculas de
sacrifício bifuncionalizadas, como o ácido oxálico, um diácido carboxílico a fim de
compreender seu comportamento gente à radiação UV. O tempo de reação variou,
inicialmente, de 10 min até 60 min. Estudando o comportamento dos sistemas através
de espectros de UV-Vis, comparando antes e após a exposição, otimizou-se o tempo
em 10 min.
A fonte de radiação UV é composta de um refletor parabólico e uma lâmpada de
vapor de mercúrio de média pressão (400W). Testes na fonte foram feitos tais como:
ajuste do foco, altura para exposição do reator e testes de temperatura em distâncias
variáveis da fonte. O reator do sistema sempre foi um béquer mantido a distâncias
variáveis em função da temperatura de ebulição do solvente. A faixa de comprimentos
de onda da lâmpada empregada na região do UV-Vis variou de 270 – 436nm.
Reais dificuldades de solubilidade com a lignina sulfonato comercial levaram ao
emprego da lignina Organossolve. Esta se mostrou mais solúvel principalmente em
dioxano 50%, etanol e acetato de etila. A fim de promover a formação dos copolímeros
de LO em matriz de EPS, optou-se pelo emprego de diácidos carboxílicos de cadeias
maiores como o ácido itacônico e o ácido adípico. Primeiramente foi testado seu
comportamento fotoreativo para a seguir testar o sistema como um todo.
4.4 Preparação dos Copolímeros EPS-Lignina
Os copolímeros obtidos empregaram dois diferentes materiais poliméricos
oriundos de resíduos: poliestireno expandido reciclado (EPS) e lignina extraída da
casca de arroz pelo método Organossolve. Dessa forma, a matriz polimérica é
constituída de EPS reciclado, um polímero proveniente de fonte fóssil que possui
caráter cristalino com unidade monomérica que se repete (Figura 30). A lignina foi
utilizada como carga enxertada na cadeia principal de forma a mudar as características
do material.
Em função da solubilidade dos dois polímeros, foram testados solventes com
diferentes polaridades. Somente para a lignina, foi testado álcool isopropílico, álcool
etílico, acetona, dimetilformamida (DMF), tetrahidrofurano (THF), acetato de etila e
dioxano (em solução 50% v /v). O melhor solvente nesse caso foi a solução de dioxano

41
e água, mas o álcool etílico e o acetato de etila também se mostraram bons solventes.
Somente para o EPS foram testados os seguintes solventes: acetato de etila, dioxano,
tolueno e acetona. Para o sistema lignina-EPS foi usado o acetato de etila P.A. da
marca Vetec que foi o melhor solvente para o sistema. Dentre os solventes que
poderiam ser usados, ele é um dos menos tóxicos, de menor custo e pode ser
considerado um solvente verde já que é obtido a partir da esterificação do ácido acético
com etanol.
Para a produção do copolímero foi utilizado 1g de EPS reciclado. Variou-se a
concentração de lignina e volume do solvente segundo Tabela 1. Foram utilizados dois
diácidos carboxílicos como promotores da reação: ácido itacônico (0,02 g) e ácido
adípico (0,005g). Para ambas as moléculas de sacrifício foram utilizadas as mesmas
condições de concentração de lignina e volume de solvente.
Tabela 1 – Relação de massa de lignina e volume de solvente
Entrada Massa de Lignina (g) Volume do Solvente (mL)
1 0,020 50
2 0,050 75
3 0,10 100
4 0,20 100
5 0,50 150
O sistema lignina + EPS + molécula de sacrifício + solvente foi deixado sob
agitação magnética por 30 minutos a temperatura ambiente. Em seguida, expôs-se o
sistema à radiação UV por 10 minutos também sob agitação. Após o tempo de reação,
deixou-se o sistema agitando por mais 10 minutos e fez-se o filme em placa de petri
deixando a temperatura ambiente para evaporação. Todas as amostras foram
evaporadas controladamente, reduzindo a área de escape de vapores da placa de
petri. Além disso, filme de 1g de EPS em 50 mL de acetato de etila foi produzido para
comparação. Deixou-se sob agitação de 30 minutos e verteu-se a solução em em placa
de petri. Cabe ressaltar que a identificação dada aos copolímeros está relacionada à
molécula de sacrifício e a percentagem de carga de LO usada. Dessa forma, o

42
copolímero denominado AI2, por exemplo, foi produzido usando ácido itacônico como
molécula de sacrifício e 2% de carga de LO (em massa). Já, AA5, por exemplo, é
constituído de ácido adípico como promotor da reação e 5% de LO. Cabe ressaltar que
esta metodologia de preparo dos copolímeros foi realizada inúmeras vezes por mais de
um operador. Dessa forma, os produtos obtidos possuem as características de
repetitividade e reprodutibilidade.
4.5 Purificação dos copolímeros de EPS-Lignina
Como é de grande interesse para o trabalho procurar compreender o
mecanismo de enxertia observado, os polímeros foram purificados para eliminar
resíduos de lignina que possam não ter reagido.
Todos os dez copolímeros produzidos, sendo cinco utilizando o ácido adípico
como molécula de sacrifício e cinco utilizando o ácido itacônico, foram pesados e
ressolubilizados em 50mL de clorofórmio, pois a LO não é solúvel em clorofórmio. Esse
novo sistema foi mantido sob agitação magnética a temperatura ambiente por 1h.
Deixou-se o sistema em repouso para decantar durante 1 dia. Após, fez-se novamente
os filmes em placa de petri sem misturar o sobrenadante (excesso de LO) e deixou-se
secar a temperatura ambiente. Pesou-se os filmes pós-purificação. De maneira geral, a
produção dos copolímeros EPS-Lignina pode ser sintetizada no esquema abaixo
(Figura 32).
Figura 32. Esquema de obtenção dos copolímeros EPS-Lignina

43
4.6 Caracterização
Os copolímeros formados e o filme-controle de EPS foram caracterizados
através de técnicas espectroscópicas de UV-Vis, IV, EDS, RMN ¹H e ¹³C e análises
morfológicas empregando MEV e WCA. Além disso, os materiais foram caracterizados
termicamente através da técnica de DSC, GPC e ensaios mecânicos de tração. Essas
técnicas irão indicar a existência do copolímero além de orientar para possível
aplicação do novo material obtido.
4.6.1 Análise térmica
Para a análise por DSC a massa de amostra utilizada variou entre 4 a 6 mg
(Tabela 2) e foi feita uma amostra de cada copolímero. Estas foram colocadas em
cadinho metálico contra outro de referência e levadas a aquecimento em sistema
fechado com fluxo de nitrogênio de 50mL.min-1. O comportamento da amostra em
função das taxas de aquecimento e do tempo é fornecido pelo computador através de
um gráfico. A faixa de aquecimento variou de 30 a 200ºC e a taxa de aquecimento foi
de 5ºC.min-1.
Tabela 2 – Massas de amostras para análise por DSC
Promotor da reação Amostras Massa (g)
ÁCIDO ITACÔNICO
AI2 0,0044
AI5 0,0054
AI10 0,0044
AI20 0,0047
AI50 0,0060
ÁCIDO ADÍPICO
AA2 0,0053
AA5 0,0060
AA10 0,0045
AA20 0,0043
AA50 0,0043
Poliestireno Expandido 0,0058

44
4.6.2 Análises morfológicas
As análises de microscopia eletrônica de varredura foram realizadas em
microscópio JEOL JSM-6610LV (Japão) e foram feitas imagens com ampliação de
1.000, 10.000 e 40.000 com tensão de aceleração de 10 kV. As amostras sofreram o
processo de metalização com ouro utilizando o equipamento Sputtering (Denton
Vaccum – USA) do Centro de Microscopia Eletrônica – CEME-SUL.
No que diz respeito às análises de ângulo de contato, os copolímeros
fotofuncionalizados foram analisados a temperatura ambiente. Uma gota de água
destilada foi depositada na superfície por meio de uma seringa. Um microscópio foi
empregado para observação e ajuste de foco e a aquisição da imagem da gota foi
obtida por câmera com controle computacional. Foram tiradas cinco fotos de gotas
diferentes para cada copolímero. Os ângulos de contato correspondem a média de
cinco medidas de cada gota. Esses valores foram obtidos empregando o software
Surftens v. 3.0.
4.6.3 Análises espectroscópicas
As análises de espetroscopia de UV-Vis foram realizadas em um equipamento
Shimadzu UV-2500PC com velocidade de leitura média, abertura de fenda 1,0 nm e
faixa de comprimento de onda de 200 – 800 nm. Para todas as amostras foram
pesados 2mg e diluídos em clorofórmio de acordo com a Tabela 3. Foi utilizado
clorofórmio como branco.

45
Tabela 3 – Fator de diluição para cada copolímero
Promotor da reação Amostras Volume de
solvente (mL)
ÁCIDO ITACÔNICO
AI2 10
AI5 10
AI10 10
AI20 10
AI50 30
ÁCIDO ADÍPICO
AA2 15
AA5 15
AA10 15
AA20 15
AA50 20
Para a análise de infravermelho com transformada de Fourrier, foi utilizado um
equipamento Shimadzu – IR PRESTIGE 21 com velocidade de análise de 24 scans,
Foi usado o modo percentagem de transmitância (%T) e em uma faixa de número de
onda de 4000-400 cm-1. As análises foram feitas no módulo de ATR.
Já para análise da estrutura do copolímero formado foi empregado equipamento
de RMN Bruker modelo Ascend 400 MHz da Central Integrada de Análises (CIA-
FURG). O solvente empregado foi clorofórmio deuterado. Para a amostra de LO usou-
se acetona deuterada. A massa das amostras variou entre 25 – 40 mg. Foi empregado
para tratamento dos dados o software TOP SPIN 3.5pl5.
Para a análise de EDS, uma pequena amostra de cada copolímero e do filme
somente de EPS foi recoberta com ouro. A partir do microscópio eletrônico da marca
JEOL JSM-6610LV (Japão), do CEME-SUL, obteve-se a imagem analisada (com
magnificação de 1000 e aceleração de 15 keV), a composição elementar e a
quantidade de cada elemento.
4.6.4 Análise de cromatografia de permeação em gel
As análises de cromatografia de permeação em gel foram realizadas na Central
Analítica da UFRGS em cromatógrafo modelo Viscotek VE 2001 com detector triplo

46
(índice de refração, viscosimétrico e por espalhamento de luz) Viscotek TDA 302.
Usou-se como eluente solvente orgânico CHCl3 a um fluxo de 1mL.min-1 e colunas de
PS-DVB a 45ºC. Usaram-se as massas de amostras vide Tabela 4.
Tabela 4 – Massas de amostra para GPC
Promotores da reação Amostras Massa (g)
ÁCIDO ITACÔNICO
AI2 0,052
AI5 0,051
AI10 0,050
AI20 0,054
AI50 0,054
ÁCIDO ADÍPICO
AA2 0,054
AA5 0,054
AA10 0,053
AA20 0,058
AA50 0,051
Poliestireno Expandido 0,053
4.6.5 Ensaio mecânico de tração
Os copolímeros foram testados segundo a norma ASTM D638. A fim de
investigar seu comportamento mecânico com relação à tensão-deformação, usou-se
cinco amostras de cada copolímero. Para cada tira da amostra (100mm x 25mm),
foram feitas 15 medidas aleatórias de espessura utilizando o micrômetro da marca
Mitutoyo (0,001mm 0-25mm) para cálculo da área da secção transversal. Para
obtenção dos parâmetros, utilizou-se um texturômetro modelo TA.XT plus produzido
pela Stable Micro Systems (Reino Unido). As amostras foram fixadas em duas garras
distantes 50mm entre si. A velocidade empregada para ruptura do material foi de
2mm.min-1 e foi informado, ao final, a força máxima no momento da ruptura assim
como a distância de separação.
De posse dessas informações e após o cálculo da área transversal, foi possível
calcular a tensão de ruptura (Eq.5) e a elongação na ruptura (Eq.6).
A
FT m
R eq.(5) 100xd
dE
i
r eq.(6)
Onde: TR = Tensão de ruptura ou resistência máxima à tração (Mpa)

47
Fm = Força máxima no momento da ruptura do filme (N)
A = área da seção transversal (m²)
E = elongação (%)
di = distância inicial de separação (cm)
dr = distância no momento da ruptura (cm) (diferença entre a distância de
separação no momento da ruptura e a distância inicial

48
5. APRESENTAÇÃO E DISCUSSÃO DOS RESULTADOS
5.1 Preparação e purificação dos copolímeros
Durante o preparo, para o copolímero com ácido adípico, inicialmente tentou-se
empregar a proporção 1:1 (em massa) de ácido e EPS. Entretanto, ficou evidente o
excesso de ácido no momento da secagem do filme já que o mesmo apresentou
cristais do ácido na superfície. O mesmo ocorreu quando usado na proporção de 1:1
(em massa) de ácido e LO. Dessa forma, otimizou-se 1g de EPS para 0,005g de ácido
adípico variando a concentração de carga de LO. Nessa proporção os filmes se
formaram em aproximadamente 24h de secagem a temperatura ambiente. Dos cinco
copolímeros, dois apresentaram muitos orifícios superficiais devido a evaporação do
solvente: AA20 e AA50. Dessa forma, eles foram pesados e ressolubilizados em
150mL de acetato de etila, adquirindo, pós processo de secagem, aspecto uniforme,
mas com excesso de LO.
Em todos os copolímeros foi necessária a purificação para retirada do excesso
de LO (Figura 33). A fase líquida é solúvel em clorofórmio, entretanto, a LO não é. A
partir dessa diferença de solubilidade, optou-se por esse solvente para purificação.
Nesse processo, os filmes foram pesados antes e depois, segundo Tabela 5.
Figura 33. Purificação dos polímeros com ácido adípico. Em (a) o excesso de lignina
no sobrenadante e em (b) o polímero funcionalizado na fase líquida
a
b
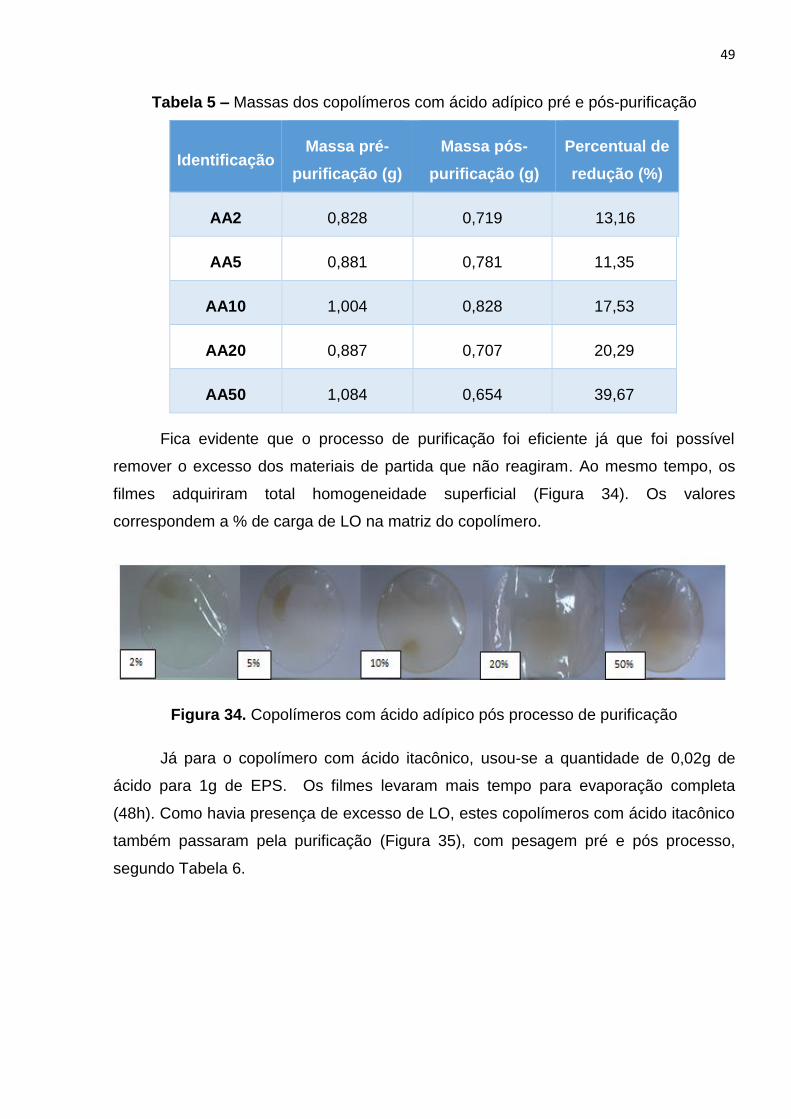
49
Tabela 5 – Massas dos copolímeros com ácido adípico pré e pós-purificação
Identificação Massa pré-
purificação (g)
Massa pós-
purificação (g)
Percentual de
redução (%)
AA2 0,828 0,719 13,16
AA5 0,881 0,781 11,35
AA10 1,004 0,828 17,53
AA20 0,887 0,707 20,29
AA50 1,084 0,654 39,67
Fica evidente que o processo de purificação foi eficiente já que foi possível
remover o excesso dos materiais de partida que não reagiram. Ao mesmo tempo, os
filmes adquiriram total homogeneidade superficial (Figura 34). Os valores
correspondem a % de carga de LO na matriz do copolímero.
Figura 34. Copolímeros com ácido adípico pós processo de purificação
Já para o copolímero com ácido itacônico, usou-se a quantidade de 0,02g de
ácido para 1g de EPS. Os filmes levaram mais tempo para evaporação completa
(48h). Como havia presença de excesso de LO, estes copolímeros com ácido itacônico
também passaram pela purificação (Figura 35), com pesagem pré e pós processo,
segundo Tabela 6.

50
Figura 35. Purificação dos polímeros com ácido itacônico. Em (a) o excesso de lignina
no sobrenadante e em (b) o polímero funcionalizado na fase líquida
Tabela 6 – Massa dos copolímeros com ácido itacônico pré e pós-purificação
Identificação Massa pré-
purificação (g)
Massa pós-
purificação (g)
Percentual de
redução (%)
AI2 1,131 0,673 40,50
AI5 0,972 0,643 33,85
AI10 0,804 0,694 13,68
AI20 0,925 0,547 40,86
AI50 0,728 0,730 0,27
Novamente o processo de purificação foi satisfatório já que foi possível remover
o excesso dos materiais de partida não reagidos presente nos copolímeros. Estes,
também apresentaram ótima homogeneidade superficial (Figura 36).
Figura 36. Copolímeros com ácido itacônico pós processo de purificação
a
b
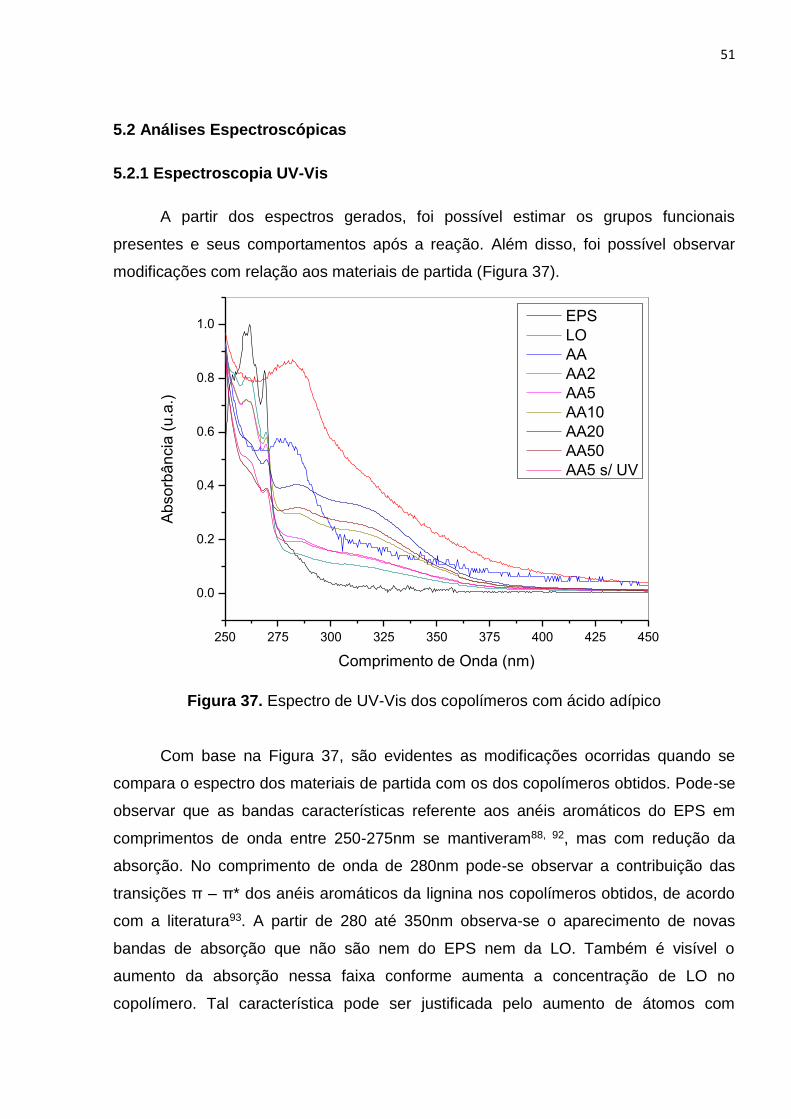
51
250 275 300 325 350 375 400 425 450
0.0
0.2
0.4
0.6
0.8
1.0
Ab
so
rbâ
ncia
(u
.a.)
Comprimento de Onda (nm)
EPS
LO
AA
AA2
AA5
AA10
AA20
AA50
AA5 s/ UV
5.2 Análises Espectroscópicas
5.2.1 Espectroscopia UV-Vis
A partir dos espectros gerados, foi possível estimar os grupos funcionais
presentes e seus comportamentos após a reação. Além disso, foi possível observar
modificações com relação aos materiais de partida (Figura 37).
Figura 37. Espectro de UV-Vis dos copolímeros com ácido adípico
Com base na Figura 37, são evidentes as modificações ocorridas quando se
compara o espectro dos materiais de partida com os dos copolímeros obtidos. Pode-se
observar que as bandas características referente aos anéis aromáticos do EPS em
comprimentos de onda entre 250-275nm se mantiveram88, 92, mas com redução da
absorção. No comprimento de onda de 280nm pode-se observar a contribuição das
transições π – π* dos anéis aromáticos da lignina nos copolímeros obtidos, de acordo
com a literatura93. A partir de 280 até 350nm observa-se o aparecimento de novas
bandas de absorção que não são nem do EPS nem da LO. Também é visível o
aumento da absorção nessa faixa conforme aumenta a concentração de LO no
copolímero. Tal característica pode ser justificada pelo aumento de átomos com

52
250 275 300 325 350 375 400 425 450
0.0
0.2
0.4
0.6
0.8
1.0
Ab
so
bâ
ncia
(u
.a.)
Comprimento de Onda(nm)
EPS)
LO
AI
AI2
AI5
AI10
AI20
AI50
AI5 s/ UV
elétrons não ligantes junto a anéis aromáticos. Assim, as interações entre os elétrons n
e π causam deslocamentos batocrômicos nas bandas de absorção em direção a
comprimentos de onda maiores71. Ao comparar o copolímero AA5, com radiação UV e
sem (teste em branco), é possível observar modificações. Sugere-se que a radiação
UV seja a responsável por tais mudanças. Também foi realizada análise
espectroscópica UV-Vis para os copolímeros com ácido itacônico (Figura 38).
Figura 38. Espectro de UV-Vis dos copolímeros com ácido itacônico
O comportamentamento dos copolímeros com ácido itacônico é similar ao com
ácido adípico. Neste caso, as absorções dessas novas bandas a partir de 280 até
350nm são menos intensas quando comparada com a anterior, o que também pode ser
visto na literatura94. Com excessão da amostra AI50 onde as bandas de absorção
demonstram um comportamente diferente dos demais. Novamente é possível visualizar
que há grandes modificações quando compara-se o copolímero AI5 com e sem
radiação UV.
5.2.2 Espectroscopia de infravermelho com transformada de Fourier
A fim de caracterizar os materiais de partida, foi necessária uma análise prévia
dos espectros dos mesmos por espectroscopia de infravermelho com transformada de
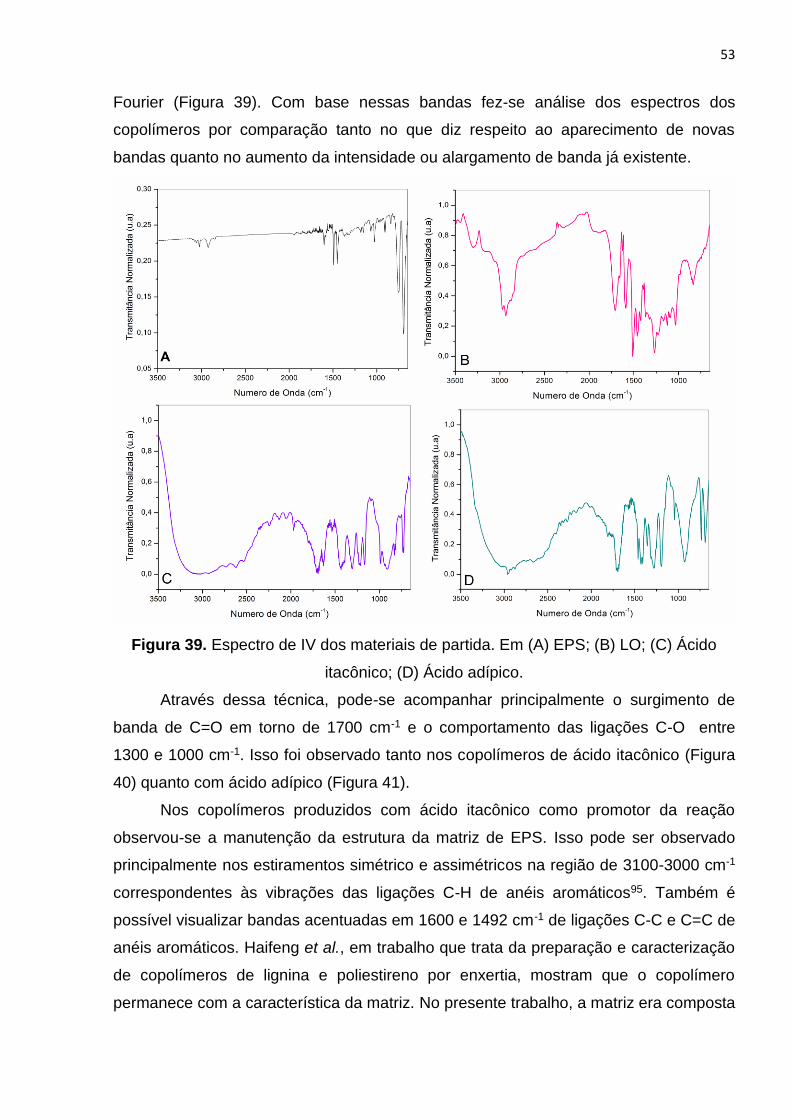
53
Fourier (Figura 39). Com base nessas bandas fez-se análise dos espectros dos
copolímeros por comparação tanto no que diz respeito ao aparecimento de novas
bandas quanto no aumento da intensidade ou alargamento de banda já existente.
Figura 39. Espectro de IV dos materiais de partida. Em (A) EPS; (B) LO; (C) Ácido
itacônico; (D) Ácido adípico.
Através dessa técnica, pode-se acompanhar principalmente o surgimento de
banda de C=O em torno de 1700 cm-1 e o comportamento das ligações C-O entre
1300 e 1000 cm-1. Isso foi observado tanto nos copolímeros de ácido itacônico (Figura
40) quanto com ácido adípico (Figura 41).
Nos copolímeros produzidos com ácido itacônico como promotor da reação
observou-se a manutenção da estrutura da matriz de EPS. Isso pode ser observado
principalmente nos estiramentos simétrico e assimétricos na região de 3100-3000 cm-1
correspondentes às vibrações das ligações C-H de anéis aromáticos95. Também é
possível visualizar bandas acentuadas em 1600 e 1492 cm-1 de ligações C-C e C=C de
anéis aromáticos. Haifeng et al., em trabalho que trata da preparação e caracterização
de copolímeros de lignina e poliestireno por enxertia, mostram que o copolímero
permanece com a característica da matriz. No presente trabalho, a matriz era composta

54
por lignina e a carga enxerta, por polimerização radicalar empregando FeCl2 e H2O2
como co-iniciadores, foi o poliestireno. A enxertia foi confirmada, dentre outras
análises, pela presença de duas bandas de anel aromático monossubstituído
característico da carga63.
Diferente da matriz de EPS, surgiram bandas entre 1328, 1266 e 1216 cm-1 que
correspondem aos estiramentos de C-O da unidade siringila e guaiacila e C-C e C=C
em unidades G96. A partir do copolímero AI5 começou a surgir uma banda em 1508
cm-1 que refere-se ao estiramento C=C de unidade de lignina (guaiacila e siringila).
Essas bandas sugerem que a reação fotoquímica de enxertia foi alcançada. Outra
forma de evidenciar a fotoenxertia é a presença de carbonila na região de 1719 cm-1
que pode ser tanto uma contribuição da LO quanto do ácido promotor. Rossberg et al.
mostram, em trabalho sobre separação e caracterização de ligninas de diferentes
resíduos agrícolas, que a lignina obtida pelo método organossolve possui banda de
carbonila mais acentuada em 1710 cm-1.97 Cabe destacar que essa banda começou a
aparecer no copolímero AI5 e o AI50 foi o que apresentou o pico mais pronunciado
devido excesso de LO. Foi realizado um teste em branco para o copolímero AI10, ou
seja, sem a exposição dos constituintes do sistema à radiação UV. Ao comparar os
espectros desses copolímeros com e sem exposição à radiação UV (espectro em
anexo) é possível visualizar grande redução na intensidade do pico em torno de 1710
cm-1. Isso sugere que a reação fotoquímica possa ter ocorrido nos centros carbonílicos.
Nas bandas referentes C-H alifático, assimétrico e simétrico, em torno de 2920 e
2854 cm-1, respectivamente, observou-se deslocamentos nos estiramentos em todos
os copolímeros com ácido itacônico quando comparado ao EPS. Esse comportamento
pode ser explicado pela presença de grupo retirador de elétron como o oxigênio,
presente em metoxilas da lignina, indicando a reação98. Também sugere que o
mecanismo de fotoenxertia pode ter acontecido a partir da abstração de hidrogênio da
cadeia alifática da matriz. As alterações nessa região, acompanhadas de redução na
intensidade de transmitância, de acordo com Jairam et al., podem ser atribuidas a
quebras da cadeia polimérica após a exposição à radiação UV99. Outros resultados tem
sido reportados para sistemas com acrilatos que sofrem degradação pela
fotoirradiação100, 101. Além disso, observa-se nos copolímeros AI2 e AI50 o
aparecimento de uma banda em torno de 2968 cm-1 que corresponde ao estiramento
C-H assimétrico de metila presente na lignina.
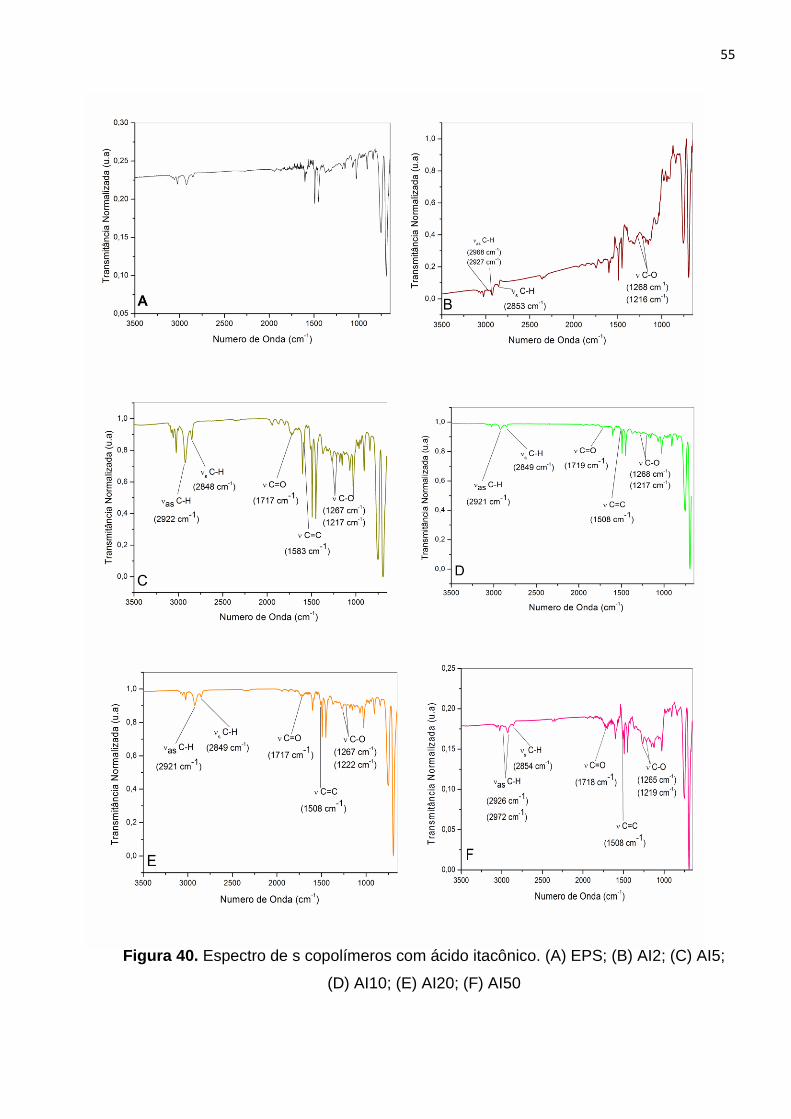
55
Figura 40. Espectro de s copolímeros com ácido itacônico. (A) EPS; (B) AI2; (C) AI5;
(D) AI10; (E) AI20; (F) AI50
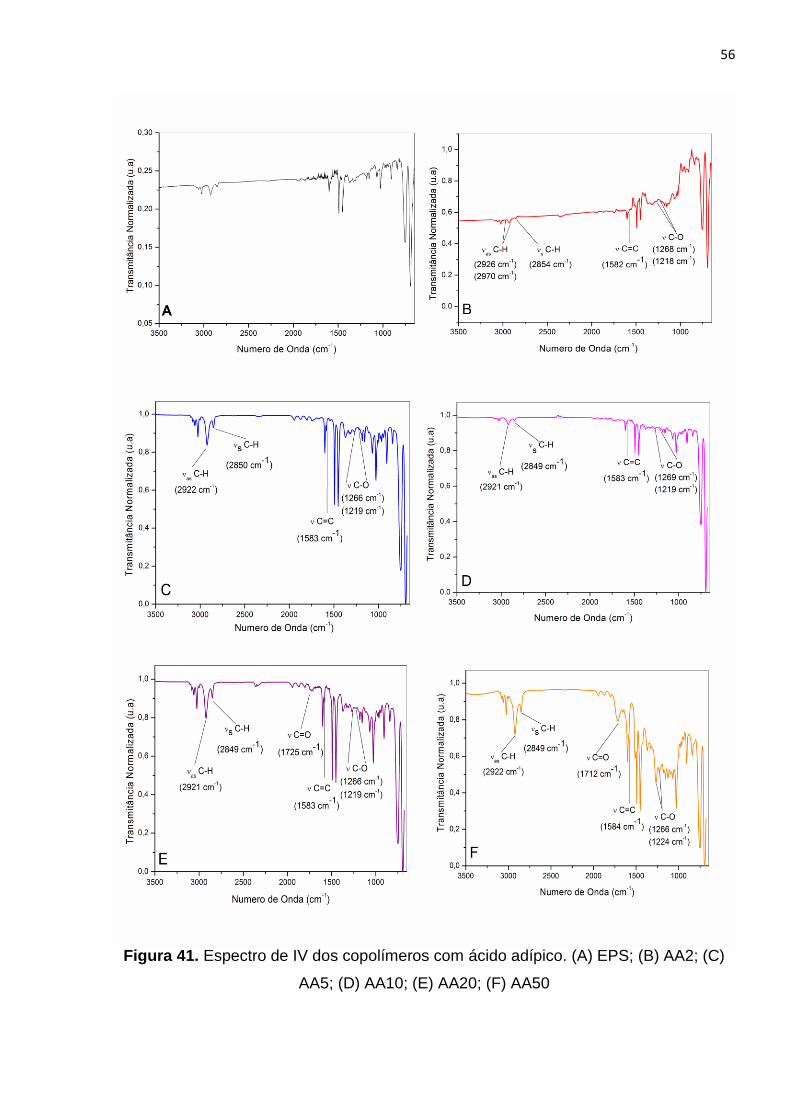
56
Figura 41. Espectro de IV dos copolímeros com ácido adípico. (A) EPS; (B) AA2; (C)
AA5; (D) AA10; (E) AA20; (F) AA50

57
A partir das análises dos espectros dos copolímeros feitos com ácido adípico,
foram feitas comparações com os materiais de partida. É possível perceber que os
copolímeros mantiveram a estrutura da matriz de EPS. Dessa forma, observou-se o
desaparecimento das bandas entre 3441-3298 cm-1 referentes à deformação axial da
ligação O-H da lignina. Na literatura é encontrado um trabalho sendo a lignina Kraft a
matriz e poliestireno a carga. Neste, os autores encontraram estiramentos entre 3400 –
3200 cm-1 correspondentes a vibração da ligação C-OH alifático e aromático63. Isso
ressalta que nesse tipo de enxertia os copolímeros obtidos tendem a manter a estrutura
da carga.
Para os estiramentos correspondentes a ligação C-H de carbono sp² em torno
de 3082-3000 cm-1 houve poucas modificações o que está condizente com a
literatura87. Para a deformação axial de C-H de carbono alifático assimétrico e
simétrico, em torno de 2921 e 2854 cm-1, houve deslocamentos de bandas, quando
comparado ao EPS, assim como para os copolímeros com ácido itacônico. Novamente,
isso pode sugerir quebras na cadeia devido à radiação UV e também a presença de
grupos oxigenados. Para o copolímero denominado AA2 houve o aparecimento de uma
banda em 2970 cm-1 que corresponde a cadeia alifática da lignina.
Foram encontrados bandas em torno de 1582 cm-1, em todos os copolímeros,
correspondentes a anéis aromáticos em unidades guaiacilas e siringilas sendo
contribuição da LO102. Além disso, foram encontradas bandas entre 1328, 1266 e 1218
cm-1 correspondentes aos estiramentos C-O de unidade siringila e guaiacila e
estiramentos C-C e C-O de unidades guaicila. Também em todos os copolímeros, em
torno de 1127 cm-1 foram encontradas bandas correspondentes a anéis aromáticos de
unidades guaiacilas e em torno de 880-800 cm-1 estiramentos equivalentes a ligações
C-H fora do plano de unidades G e S103. Além disso, para copolímeros com ácido
adípico encontrou-se banda de carbonila, em 1712 cm-1, que pode ser tanto
contribuição do ácido adípico quanto da LO. Isso se deve ao excesso de carga que
provalvelmente não reagiu com a matriz. Dessa forma, o que sugere a formação de um
copolímero é a presença de grupos oxigenados enxertados na matriz polimérica aliada
aos deslocamentos presentes na cadeia alifática do EPS. Ao comparar o teste em
branco com o copolímero AA5, ficou evidente a redução de intensidade em banda em
torno de 1712 cm-1. Sugere-se, dessa forma, reação fotoquímica em centro carbonílico
(espectro em anexo).

58
5.2.3 Espectroscopia de ressonância magnética nuclear
Afim de compreender como a reação ocorre, fez-se necessário o emprego da
espectroscopia de ressonância magnética nuclear. Considerando que a estrutura da
lignina é constituída por unidades formadoras (guaiacila, siringila e p-cumarílica),
organizadas de forma randômica, a interpretação dos espectros de RMN para os
copolímeros se deu de forma comparativa. Dessa forma, os espectros obtidos para os
copolímeros foram comparados com os materiais de partida. A Figura 42 traz os
espectros de RMN ¹H para a LO (a) e para o EPS (b). Em (a) a região que compreende
δ 0,8 – 1,3 ppm correspondem a prótons em grupos alifáticos; em δ 2,10 – 2,28 ppm e
em torno de δ 3,83 ppm correspondem a prótons próximos a grupos metoxílicos; entre
δ 6 – 8 ppm são hidrogênios de anéis aromáticos. Já em (b) é possível identifcar a
região dos hidrogênios da cadeia alifática (δ 1,29 – 2,08 ppm) e dos hidrogênios
aromáticos (δ 6,41 – 7,12 ppm).

59
Figura 42. Espectros de RMN ¹H para LO (a) e EPS (b), em CDCl3
Os copolímeros com ácido adípico, de maneira geral, se comportaram de forma
semelhante. Houve o aparecimento de novas bandas nos espectros de RMN ¹H tanto
nos hidrogênios alifáticos quanto nos aromáticos (Figura 43a). Como ambos os
materiais de partida possuem anéis aromáticos, não há como especificar qual
deslocamento químico refere-se à LO e ao EPS, entretanto, é perceptível o acréscimo
de sinais com o aumento de carga de LO. Já para a região dos deslocamentos
químicos de hidrogênios alifáticos é possível perceber variação nos valores. Há o
aparecimento de bandas em δ 2,10 ppm e δ 3,88 ppm que são referentes à H
próximos a metoxilas. Essa informação vai ao encontro dos espectros de IV que
mostraram a presença de ligações C-O de éteres, ou seja, uma clara contribuição da
lignina. Sivasankarapillai et al. encontraram comportamento semelhante para
copolímeros de lignina e poli(amina-amida). Além disso, os autores relatam que não
foram encontradas muitas diferenças nos espectros pós copolimerização o que indica a
formação de redes complexas resultando em várias organizações estrutrais104. Outro
dado relevante é a ausência de picos de hidrogênios fenólicos entre δ 7,5 – 4,5 ppm105.
Esta constatação também pode ser verificada nos espectros de IV. Ayoub et al. relata
que em condições de irradiação, radicais fenóxidos podem ser gerados a partir da

60
abstração de hidrogênio. Esses radicais, por sua vez, interagem com outras
ramificações e iniciam a modificação química da molécula32. Ren et al., em trabalho
que trata da síntese de copolímeros de lignina e PLA, relatam que após a
copolimerização, houve o desaparecimento dos grupos hidroxílicos da lignina e o
aparecimento de grupos metoxilicos de baixa intensidade106.
Os espectros de RMN ¹³C mostraram a presença de carbono alifático de metila
(em δ 40,40 ppm) confirmado pelo DEPT 135. Além disso, a partir do copolímero AA10
até AA50 houve o aparecimento de um pico, de baixa intensidade, de carbono
metilênico em δ 29,71 ppm. Para os copolímeros com maior carga (AA20 e AA50)
houve o aparecimento de um pico em δ 145,5 ppm (figura 43b). Esse valor pode
corresponder ao carbono benzílico do EPS (δ 148 ppm) segundo a literatura105. Li et al.
relatam a existência de sinal em δ 144,5 ppm referente ao carbono benzílico para
copolímero poliestireno isotático e polietilenoglicol107. Garcia et al., relatam que
carbonos aromáticos das unidades guaiacila e siringila localizam-se de δ 153 – 134ppm
e δ 147 – 120 ppm respectivamente108. Novamente não há como distinguir se o
carbono aromático é contribuição da lignina ou do EPS. Além disso, é possível
visualizar a presença de carbonos de anéis aromáticos entre δ 125 – 127 ppm. De
maneira geral, os espectros de RMN ¹³C vão ao encontro dos espectros de IV que
também mostram a presença de ligações C=C de aromático tanto da lignina quando do
EPS. Entretanto, foi observado a ausência de carbonos das ligações C-O alifáticos em
δ 89 – 58ppm e de C=O em δ 185 – 160 ppm. Lin et al. sugerem que as ligações
denominadas β-O-4 podem ser degradadas quando expostas a radiação UV109.
Lanzalunga et al. sugerem a abstração de hidrogênio das ligações β-O-4 após a
radiação UV que podem levar a quebras e rearranjos na estrutura da lignina110.

61
Figura 43. Espectro de RMN ¹H do copolímero AA10 (a) e RMN ¹³C do AA20 (b), em
CDCl3
Ao observar os espectros de RMN ¹H, os copolímeros com ácido itacônico
também se comportaram de forma semelhante, ou seja, houve o aparecimento de
novas bandas tanto na região correspondente aos H alifáticos quanto nos H
aromáticos. Isso também fica evidente ao analisar os espectros de HSQC (Figura 44).

62
Nessa figura, é possível observar o aparecimento de sinal em δ 29,71 ppm a partir do
copolímero AI5 ficando mais intenso para AI50 que corresponde a carbono metilênico
(segundo DEPT135). Além disso, há sinais entre δ 40-43 ppm referentes a carbonos na
região alifática e na região dos aromáticos entre δ 125-145ppm. Pode-se verificar que
tanto os copolímeros com ácido adípico quanto os com ácido itacônico mostraram
comportamento semelhante. Devido ao aumento de sinais da região alifática, acredita-
se que a enxertia tenha se dado por abstração de H da parte linear da cadeia do EPS.
Para a lignina, Lanzalunga sugere a ocorrência de abstração de H do carbono β da
ligação β-O-4 levando não somente a quebras da cadeia, mas também possíveis
rearranjos110. Doherty et al. também destacam esse aspecto111. Duval et al. ressaltam
que não fica claro onde pode ocorrer enxertia porque os radicais formados são
estabilizados por ressonância em diferentes posições dos anéis aromáticos112.
Figura 44. Espectros HSQC para os copolímeros com ácido itacônico em (a) AA2, (b)
AA5, (c) AA10, (d) AA20, (e) AA50, em CDCl3
(a) (b)
(e)
(d) (c)

63
5.2.4 Espectroscopia de energia dispersiva
A partir de pequena quantidade de amostra dos copolímero, foi possível
determinar quais elementos estão presentes e principalmente estipular, mesmo que de
forma semi-quantitativa, a percentagem dos mesmos. A técnica mostrou a presença de
carbono e oxigênio presentes nas amostras, como já era esperado. Alguns picos do
elemento ouro foram quantificados. Isso se deve ao recobrimento feito previamente às
amostras para realização da análise. Cabe destacar que essa comparação é possível
devido LO e EPS possuirem diferentes razões entre %C e %O. A Tabela 7 sintetiza a
%C e %O tanto para os copolímeros com ácido adípico, ácido itacônico quanto para a
matriz de EPS.
Tabela 7- Análise de EDS para os copolímeros obtidos
Identificação %C Erro
Relativo %O
Erro Relativo
AA2 98,91 ±1,0 1,09 ± 2,0
AA5 88,65 ±1,0 11,35 ± 2,0
AA10 97,76 ±1,0 2,24 ± 2,0
AA20 96,67 ±1,0 3,33 ± 2,0
AA50 85,33 ±1,0 14,67 ± 2,0
AA50' 50,34 ±1,0 49,66 ± 2,0
AI2 90,25 ±1,0 9,75 ± 3,0
AI5 95,29 ±1,0 4,71 ± 2,0
AI10 71,56 ±1,0 28,44 ± 2,0
AI20 78,32 ±1,0 21,68 ± 2,0
AI50 96,49 ±1,0 3,51 ± 2,0
EPS 97,75 ±1,0 2,25 ± 2,0
LO 50,65 ±1,0 49,35 ± 1,0
Analisando a tabela acima, é possível perceber acréscimo da percentagem de
oxigênio para o copolímero AA5. Isso indica que nessa proporção de carga a
fotoenxertia foi mais efetiva. Esse resultado vai ao encontro da análise de GPC já que
esse copolímero foi o que aumentou o número de macromoléculas na cadeia
polimérica. Além disso, o AA5 foi o que apresentou melhores propriedades mecânicas
quando comparado aos demais e em comparação à matriz de EPS. Cabe destacar que
durante o processamento do EPS há a adição de CO2 o que justifica o baixo %O.
Outra informação importante retirada da tabela é a %C e %O do copolímero
AA50. Ao observar as imagens de microscopia eletrônica, é possível observar a

64
presença de domínios circulares tanto na ampliação de 10.000 quanto de 40.000. A fim
de compreender esse fenômeno, realizou-se duas análises: uma no exterior do domínio
circular (AA50) e outra no se interior (AA50’). Dessa forma, é visível que a %C, no
interior do círculo, é menor enquanto que a %O sobe drasticamente. Ao comparar essa
amostra com a de LO, é possível sugerir que há domínios de lignina na matriz do EPS
funcionalizada já que no exterior dos domínios circulares as %O retornam para valor
esperado. Com relação aos copolímeros com ácido itacônico, observou-se que todos
os copolímeros apresentaram %O maior que a matriz. percebeu-se que a maior %O é
no copolímero AI10. Novamente isso vai ao encontro de outras análises, tais como
GPC e ensaios mecânicos.
5.3 Análises Morfológicas
5.3.1 Microscopia eletrônica de varredura
É comum encontrar trabalhos que trazem o comportamento morfológico via
imagens de MEV quando se obtém copolímeros enxertados. Esta técnica mostra a
característica superficial indicando homogeneidade. Primeiramente foram obtidas as
imagens do EPS e da LO, nas ampliações 20.000 e 2.500 respectivamente (Figura 45).
Em seguida, foram obtidas imagens da superfície com ampliação de 10.000 e 40.000
tanto para o copolímero com ácido itacônico (Figura 46) quanto para o com ácido
adípico (Figura 47).
Figura 45. Imagens de MEV dos materiais de partida. Em (A) EPS em (B) LO
C
A
B

65
Figura 46. Imagens de MEV para copolímeros com ácido itacônico. Em (A) AI2; (B)
AI5; (C) AI10; (D) AI20; (E) AI50 nas ampliações 10.000 e 40.000, respectivamente.
E
A

66
Figura 47. Imagens de MEV para copolímeros com ácido adípico. Em (A) AA2; (B)
AA5; (C) AA10; (D) AA20; (E) AA50 nas ampliações 10.000 e 40.000, respectivamente.
Visualizando as imagens é possível concluir que todos os copolímeros obtidos
apresentam característica superficial homogênea. Algumas partículas começam a
aparecer em ampliações maiores mostrando que ainda pode haver excesso de lignina

67
no produto final como, por exemplo, no AI10 (ampliação 40.000). E essas mesmas
partículas aparecem conforme aumenta-se a carga de lignina como em AI20 e no
copolímero denominado AI50 ambos na ampliação 10.000. Além disso, orifícios,
devido marcas de evaporação, podem ser observados nos copolímeros AI50. Essa
homogeneidade na produção de novos materiais é encontrada na literatura. Liu et al.,
em estudo sobre as propriedades e estrutura de blendas de PVP-lignina mostram que a
mistura lignina proveniente do bambu e PVP em DMF resulta em filmes transparentes e
homogêneos. Variando-se a proporção de lignina de 5 a 20% foi possível perceber a
ausência de separação de fases sugerindo que a blenda PVP-Lignina é miscível98.
Wang et al., na produção de copolímeros enxertados a partir de resíduo lignocelulósico
de butanol também obteve filme de característica homogênea e transparente. Com a
síntese do macromonômero a partir da lignina proveniente de biobutanol e cloreto de
acriloílo, foi possível sintetizar a blenda usando o macromonômero, metil-metacrilato e
butil-metacrilato. Os autores destacam que não há a separação de fase porque o
macromonômero foi completamente envolto pelas cadeias enxertadas94. Pouteau et al.
ressalta a compatibilidade de blenda entre lignina e poliestireno quando comparado a
outros polímeros sintéticos113.
Cabe também destacar, a partir da análise das imagens, a existência de partes
escuras presentes, principalmente, em imagens na ampliação 40.000. Para os
copolímeros com ácido itacônico esse fato aparece no AI2, AI20 e AI50. Já para os
copolímeros com ácido adípico aparece em AA2, AA10, AA20 E AA50. Essas partes
escuras precisam de maior investigação podendo ser usada a microscopia confocal ou
até mesmo pela microscopia de energia dispersiva. Acredita-se que sejam domínios de
copolímero enxertado e o restante seja a matriz de poliestireno expandido. Lisperguer
et al. menciona a existência de domínios de lignina esterificada com anidrido malêico
em matriz de poliestireno expandido. A diferença entre fases na matriz pode ser
encontrada em diversos trabalhos na literatura23, 63, 114-116.
É importante salientar também o papel de moléculas como o ácido adípico e
itacônico que atuam como facilitadoras da reação e seus efeitos na morfologia do
copolímero. Barzegari et al. mostram a importância do uso de moléculas capazes de
melhorar a adesão interfacial entre a carga e a matriz. Essa melhora na dispersão da
carga se dá pela interação entre os grupos funcionais da carga, matriz e
compatibilizador117. Sahoo et al. também ressaltam isso em estudo que trata da

68
melhora das propriedades de copolímeros biodegradáveis a base de lignina. Os
autores destacam que quando foi empregado 1% de PMDI como compatibilizador
aumentou a aderência entre a carga e a matriz o que também refletiu nas propriedades
mecânicas118. Sendo assim uma possibilidade de melhora de características mecânicas
destes materiais, será no futuro adicionar um compatibilizador ou também plastificante
à formulação.
5.3.2 Ângulo de contato
As análises de ângulo de contato indicam se a superfície do copolímero presenta
um caráter hidrofílico. Sabendo que o ângulo de contato para o EPS foi em torno de
72º (Tabela 8), ângulos de contato com valor maior correspondem a uma superfície
mais hidrofóbica. Já a redução do ângulo de contato indica caráter hidrofílico. Esse
ângulo para os copolímeros com ácido itacônico são encontradas na Tabela 8, que
relaciona a média dos WCA para cinco gotas de cada copolímero com ácido itacônico.
Tabela 8– Ângulos de contato médios para os copolímeros com ácido itacônico
Ângulo de Contato Médio (em graus)
Copolímero AI2 AI5 AI10 AI20 AI50 EPS
Nº
da
Go
ta 1 70,73 77,37 75,24 74,57 63,26 75,39
2 73,95 74,60 69,42 71,53 71,13 73,74
3 72,46 69,74 76,12 70,26 60,80 67,65
4 68,64 74,57 71,62 68,52 66,26 70,69
5 66,43 71,44 72,16 66,68 67,26 71,76
Média 70,44 73,54 72,91 70,31 65,74 71,85
Desvio Padrão
± 3,0 ± 3,0 ± 3,0 ± 3,0 ± 4,0 ± 3,0
Como pode ser observado, o ângulo de contato do filme de poliestireno ficou em
torno de 72º o que difere da literatura que menciona em torno de 83º32. Essa diferença
ocorre por se tratar de filme de poliestireno expandido proveniente de material reciclado
que passou por processamentos e tratamentos químicos com aditivos. Esse tipo de
tratamento tende a reduzir o ângulo de contato. De acordo com a Tabela 8, fica
evidente que quando a carga de lignina é máxima (AI50), o ângulo de contato diminui.
Isso se deve a presença dos grupos hidroxilas que dão caráter mais hidrofílico para
esse copolímero, mesmo assim o carácter hidrofílico é uma surpresa, pois a lignina
demonstra ser hidrofóbica.

69
Os copolímeros com ácido itacônico apresentaram valores de ângulos de
contato ligeiramente inferiores ao filme padrão de EPS. Isso se deve não só a presença
da lignina, mas também pela estrutura do ácido itacônico que além de ser um diácido
carboxílico com quatro carbonos possui uma instauração. Esse diácido por apresentar
três sítios ativos possibilitou maior interação entre a carga e a matriz tornando o
copolímero mais hidrofílico.
Ao comparar a média dos ângulos de contato dos copolímeros de ácido adípico
com o ângulo de contato do filme somente de poliestireno expandido é possível
observar que não houve grandes alterações. As análises de ângulo de contato para os
copolímeros com ácido adípico são mostradas na Tabela 9. É possível perceber que
houve redução em todos os ângulos de contato quando comparado ao EPS. Ayoub et
al. estudaram o efeito da radiação gama nas propriedades de lignina enxertada em
poliestireno. Os autores também observaram a redução no ângulo de contato após a
enxertia e alegam que isso ocorreu devido a reação dos grupos polares resultando em
material mais hidrofóbico32. Xiaoning et al. observaram maior redução no ângulo de
contato em estudo sobre melhoria do desempenho de material formado por espuma de
poliestireno com solução de tanino e lignina119. Imagens de gotas na superfície dos
copolímeros também podem ser visualizadas (Figura 48).
Tabela 9 - Ângulos de contato médios para os copolímeros com ácido adípico
Ângulo de Contato Médio
Copolímero AA2 AA5 AA10 AA20 AA50 EPS
Nº
da
Go
ta
1 61,83 67,72 63,06 63,76 61,67 75,39
2 70,85 65,31 63,33 65,79 63,20 73,74
3 67,13 67,64 71,71 68,67 64,84 67,65
4 65,09 63,29 65,25 65,71 64,97 70,69
5 68,59 65,56 72,18 63,33 67,31 71,76
Média 66,69 65,90 67,10 65,45 64,39 71,85
Desvio Padrão
± 3,0 ± 2,0 ± 4,0 ± 2,0 ± 2,0 ± 3,0
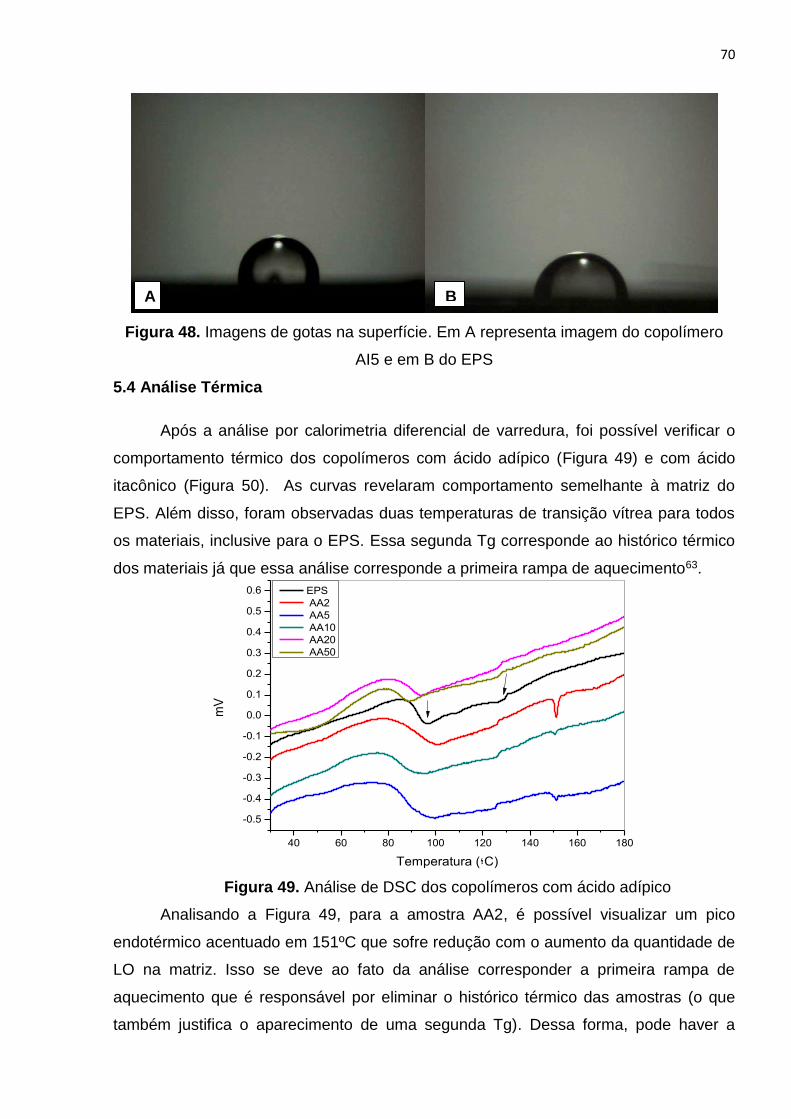
70
40 60 80 100 120 140 160 180
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
mV
Temperatura (؛C)
EPS
AA2
AA5
AA10
AA20
AA50
40 60 80 100 120 140 160 180
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
mV
Temperatura (؛C)
EPS
AI2
AI5
AI10
AI20
AI50
Figura 48. Imagens de gotas na superfície. Em A representa imagem do copolímero
AI5 e em B do EPS
5.4 Análise Térmica
Após a análise por calorimetria diferencial de varredura, foi possível verificar o
comportamento térmico dos copolímeros com ácido adípico (Figura 49) e com ácido
itacônico (Figura 50). As curvas revelaram comportamento semelhante à matriz do
EPS. Além disso, foram observadas duas temperaturas de transição vítrea para todos
os materiais, inclusive para o EPS. Essa segunda Tg corresponde ao histórico térmico
dos materiais já que essa análise corresponde a primeira rampa de aquecimento63.
Figura 49. Análise de DSC dos copolímeros com ácido adípico
Analisando a Figura 49, para a amostra AA2, é possível visualizar um pico
endotérmico acentuado em 151ºC que sofre redução com o aumento da quantidade de
LO na matriz. Isso se deve ao fato da análise corresponder a primeira rampa de
aquecimento que é responsável por eliminar o histórico térmico das amostras (o que
também justifica o aparecimento de uma segunda Tg). Dessa forma, pode haver a
A B

71
40 60 80 100 120 140 160 180
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
mV
Temperatura (؛C)
EPS
AI2
AI5
AI10
AI20
AI50
presença de gases adsorvidos na superfície do copolímero ou até mesmo lignina em
excesso justificando esse pico. O mesmo é observado na Figura 44 para a amostra AI2
e AI10 com picos em 165ºC e 164ºC, respectivamente.
Figura 50. Análise de DSC dos copolímeros com ácido itacônico
A Tabela 10 traz a análise das mudanças ocorridas nas temperaturas de
transição dos copolímeros obtidos. A primeira Tg corresponde a primeira inflexão
indicada nas Figuras 49 e 50 e a segunda Tg equivale a segunda inflexão nas curvas.
Tabela 10 – Copolímeros obtidos e respectivas temperaturas de transição vítrea
Molécula de Sacrifício Identificação 1ª Tg (ºC) 2ª Tg (ºC)
Ácido Adípico
AA2 99 126
AA5 92 125
AA10 93 126
AA20 93 127
AA50 87 127
Ácido Itacônico
AI2 100 122
AI5 95 123
AI10 93 124
AI20 92 126
AI50 84 127
Poliestireno Expandido (EPS) 95 128

72
Com base na tabela anterior, é possível observar que as temperaturas de
transição vítrea não tiveram mudanças relevantes com a inserção da LO na matriz
polimérica de EPS. A não alteração da Tg de um polímero indica que a matriz
polimérica está mantendo as características iniciais. Na literatura há trabalhos que
também relacionam a enxertia de carga de lignina (modificada ou não) em matriz
polimérica e que também apresentam comportamento semelhante69, 120. Ayoub et al.
mostra decréscimo da Tg quando é empregado poliestireno como carga em matriz de
lignina sob irradiação32. Ainda afirma que a diminuição da Tg com o aumento da
radiação são consistentes com a substituição de grupos OH da lignina no monômero
do estireno diminuindo as forças intermoleculares e permitindo maior mobilidade. Há
também relato de ligeiro aumento da Tg pós enxertia, mas mantendo a estabilidade
térmica33.
Ainda analisando a tabela 10, é possível observar que quando o acréscimo de
LO na matriz é de 50% (AA50 e AI50) obtém-se o menor valor de Tg. Isso indica que a
presença de LO não reagida no produto final tende a reduzir o valor da temperatura de
transição vítrea devido seu caráter amorfo.
5.5 Cromatografia de permeação em gel
Os cromatogramas para os copolímeros obtidos apesentaram comportamento
similar para os volumes de retenção de cada amostra com pouca variação entre si e
também quando comparado ao do EPS. Isso pode ser visualizado tanto para os
copolímeros com ácido adípico (Figura 51) e com ácido itacônico (Figura 52). Além
disso, é possível observar que de maneira geral tanto a Figura 51 quanto a 52 são bem
uniformes, apresentando um único pico Gaussiano, sem ombros ou novos picos,
evidenciando que não há produtos de menor peso molecular, ou seja as cadeias
mantém sua integridade e homogeneidade. Pode-se notar que à medida que aumenta-
se a quantidade de LO na matriz de EPS há um deslocamento do máximo para
menores volumes de retenção significando um acréscimo de massa molecular.
Argyropoulos et al., mostraram comportamento semelhante ao copolimerizar lignina
Kraft solúvel em acetona com difuorodifenil sulfona. Neste trabalho ficou evidente que
conforme aumenta-se o grau de metilação a distribuição de pesos moleculares é mais
homogênea e estreita121. Comportamento semelhante foi relatado por Wang et al.
Neste trabalho, os autores observaram que o aumento na proporção de estireno e
acriloilbenzofenona (como compatibilizador), reduziu a faixa de distribuição de peso

73
20 25 30 35 40
0.0
0.2
0.4
0.6
0.8
1.0
Re
sp
osta
do
de
tecto
r (m
V)
Volume (mL)
EPS
AA2
AA5
AA10
AA20
AA50
20 25 30 35 40
0.0
0.2
0.4
0.6
0.8
1.0
Re
spo
sta
do
De
tect
or
(m
V)
Volume (mL)
EPS
AI2
AI5
AI10
AI20
AI50
molecular indicando que o copolímero formado entre lignina-Br e estireno foi mais
homogênea28, 69.
Figura 51. Cromatogramas para os copolímeros com ácido adípico
Figura 52. Cromatogramas para os copolímeros com ácido itacônico
A partir do volume de retenção 37 mL há a presença de picos largos referentes à
LO que não reagiu. Este pico se confirma e aumenta a partir dos copolímeros AA10 e
AI10 sendo mais pronunciado nos copolímeros AA50 e AI50. Isso reforça o
comportamento observado na etapa de pré-purificação onde resíduo de LO era visível
nos filmes. Afirma-se que o excesso corresponde há LO já que esta apresenta pico
máximo em 37,5 mL (Figura 53)122.

74
30 33 36 39 42
0.0
0.2
0.4
0.6
0.8
1.0
Re
sp
osta
do
De
tecto
r (m
V)
Volume (mL)
Figura 53. Cromatograma para LO
As massas molares média (Mn) e ponderal (Mw), assim como as massas
molares das cadeias maiores (Mz) e a polidispersividade (Mn / Mw) para os
copolímeros com ácido adípico foram calculados a partir dos cromatogramas conforme
Tabela 11 e para o ácido itacônico na Tabela 12.
Tabela 11 - Massas molares médias para os copolímeros com ácido adípico
Copolímeros com ácido adípico
AA2 AA5 AA10 AA20 AA50 EPS LO
Mn (Da) 89.06 97.87 79.82 82.30 92.51 93.13 1.22
Mw (Da) 216.42 208.66 186.09 199.86 219.12 221.06 2.07
Mz (Da) 447.73 421.17 404.31 449.40 427.08 453.58 3.31
PDI 2,43 2,13 2,33 2,43 2,37 2,37 1,60
% redução
(Mw) 2% 5,6% 15,8% 10% 0,87% - -
É possível visualizar a partir da Tabela 11 os pesos moleculares do EPS pós-
consumo e da LO. Pode-se perceber que os valores encontrados para a LO são cerca
de 100 vezes menores quando comparados ao EPS. Acredita-se que esse tamanho da
cadeia da LO seja responsável por promover melhor interação com as cadeias
poliméricas. De acordo com a Tabela 11, o valor de Mn aumenta para o copolímero
denominado AA5 quando comparado à matriz do EPS. Nesse caso a reação de
fotoenxertia promoveu aumento no número de macromoléculas na cadeia sem ocorrer
total quebra da mesma, pois, para o restantes das amostras há uma diminuição do
valor de Mn em comparação com o valor original de EPS. O aumento do número de
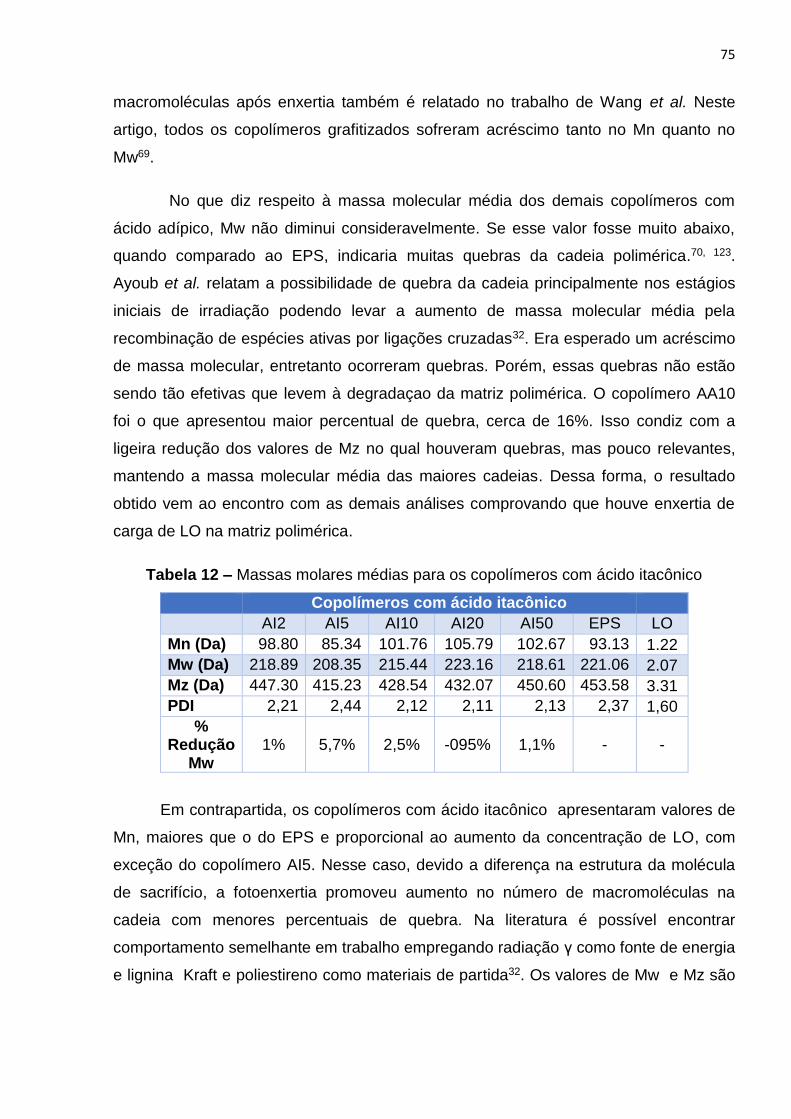
75
macromoléculas após enxertia também é relatado no trabalho de Wang et al. Neste
artigo, todos os copolímeros grafitizados sofreram acréscimo tanto no Mn quanto no
Mw69.
No que diz respeito à massa molecular média dos demais copolímeros com
ácido adípico, Mw não diminui consideravelmente. Se esse valor fosse muito abaixo,
quando comparado ao EPS, indicaria muitas quebras da cadeia polimérica.70, 123.
Ayoub et al. relatam a possibilidade de quebra da cadeia principalmente nos estágios
iniciais de irradiação podendo levar a aumento de massa molecular média pela
recombinação de espécies ativas por ligações cruzadas32. Era esperado um acréscimo
de massa molecular, entretanto ocorreram quebras. Porém, essas quebras não estão
sendo tão efetivas que levem à degradaçao da matriz polimérica. O copolímero AA10
foi o que apresentou maior percentual de quebra, cerca de 16%. Isso condiz com a
ligeira redução dos valores de Mz no qual houveram quebras, mas pouco relevantes,
mantendo a massa molecular média das maiores cadeias. Dessa forma, o resultado
obtido vem ao encontro com as demais análises comprovando que houve enxertia de
carga de LO na matriz polimérica.
Tabela 12 – Massas molares médias para os copolímeros com ácido itacônico
Copolímeros com ácido itacônico
AI2 AI5 AI10 AI20 AI50 EPS LO
Mn (Da) 98.80 85.34 101.76 105.79 102.67 93.13 1.22
Mw (Da) 218.89 208.35 215.44 223.16 218.61 221.06 2.07
Mz (Da) 447.30 415.23 428.54 432.07 450.60 453.58 3.31
PDI 2,21 2,44 2,12 2,11 2,13 2,37 1,60
% Redução
Mw 1% 5,7% 2,5% -095% 1,1% - -
Em contrapartida, os copolímeros com ácido itacônico apresentaram valores de
Mn, maiores que o do EPS e proporcional ao aumento da concentração de LO, com
exceção do copolímero AI5. Nesse caso, devido a diferença na estrutura da molécula
de sacrifício, a fotoenxertia promoveu aumento no número de macromoléculas na
cadeia com menores percentuais de quebra. Na literatura é possível encontrar
comportamento semelhante em trabalho empregando radiação γ como fonte de energia
e lignina Kraft e poliestireno como materiais de partida32. Os valores de Mw e Mz são

76
ainda mais próximos do EPS indicando que houve quebras, entretanto manteve-se, em
média, a massa molecular das maiores cadeias
Para os dois copolímeros os valores de PDI fica em torno de 2,0 o que coincide
com os valores encontrados na literatura. Botaro et al. encontraram comportamento
semelhante em trabalho que trata da modificação química de materiais
lignocelulósicos. Os valores de PDI e Mw sofreram ligeira diminuição após a irradiação
com laser pulsado. Os autores explicam tal comportamento devido a presença de
fragmentos de baixo peso molecular e a degradação parcial de fragmentos de alto peso
molecular124. O trabalho de Wang et al., em uma amostra de poliestireno grafitado com
lignina e em outra com o uso de menor quantidade de compatibilizador encontraram
PDI em torno de 2,069.
5.6 Ensaio mecânico de tração
Os ensaios mecânicos de tração trazem informações referentes à tensão de
ruptura, ou seja, a máxima força que o material suporta antes de romper e a sua
capacidadade de deformação representada pelo Módulo de Young. Neste trabalho,
foram produzidos cinco amostras de cada copolímero, tanto para o ácido adípico
quanto para o ácido itacônico além das cinco amostras do filme de EPS. O resultado
pode ser sintetizado na Tabela 13 e na Figura 54, para o ácido adípico.
Tabela 13 – Ensaios mecânicos para os copolímeros com ácido adípico
Copolímero com ácido adípico
AA2 AA5 AA10 AA20 AA50 EPS
Tensão de Ruptura (MPa)
18,65 21,00 19,65 21,63 6,18 21,78
Módulo de Elasticidade
(GPa)
1,83 2,57 2,46 1,96 2,45 2,33

77
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
Te
nsa
o (
MP
a)
Elongaçao (%)
EPS
AA2
AA5
AA10
AA20
AA50
Figura 54. Gráfico Tensão X Elongação para os copolímeros com ácido adípico
Como pode ser observado na Tabela 13, o aumento da carga de lignina nos
copolímeros reduziu a tensão de ruptura. Isso já era esperado visto que a estrutura da
lignina possui rigidez característica. Os copolímeros AA5 e AA20 possuem os valores
de tensão de ruptura mais próximos da matriz. Essas foram as amostras que quando
submetidas a uma força mostraram maior resistência. Com base no gráfico tensão X
elongação, a amostra que mais se deformou elasticamente até a ruptura foi o AA2
apresentando comportamento superior à amostra. Com relação ao módulo de Young, a
amostra AA5 apresentou o maior valor. Isso traz evidências que a fotoenxertia
conseguiu, a partir da formação de redes, melhorar a capacidade de ditribuição da
energia ao longo do material quando sujeito a um esforço. AA50 foi o copolímero mais
frágil com menor tensão de ruptura. A presença do excesso de lignina que não reagiu
pode ser a responsável por tal comportamento. Em contrapartida,esse excesso foi
capaz de melhorar a distribuição de força no material melhorando o módulo de Young
quando comparado à matriz. Essa obsevação não vai ao encontro do estudo de Jairam
et al. Neste trabalho, os autores variaram a carga de lignina-saponita nanohíbrida em 1,
2 e 5% em matriz de PSBA via polimeriação por miniemulsão. Após a produção da
blenda, foi estudado o efeito da exposição do sistema à radiação UV (em 0,6,12,18 e
24h) nas propriedades mecânicas. Observou-se aumento da tensão de ruptura com o
aumento da carga de lignina e maior tempo de exposição à radiação UV. Já no módulo

78
de elasticidade não houve grandes alterações. O autor sugere que ocorreram inúmeras
clivagens C-H ao longo da cadeia polimérica que causa uma redução local no seu
comprimento. Essa redução no comprimento da cadeia pode melhor redistribuir a carga
aplicada sobre os emaranhados de lignina permitindo que o compósito resista a uma
deformação maior e conduza, consequentemente, a um módulo e resistência maior à
tração. O melhor resultado foi com 5% de carga99.
Entretanto, é possível encontrar na literatura comportamento parecido com o
que foi encontrado para os copolímeros com ácido adípico. O trabalho de Toriz et al.,
na produção de compósitos de lignina kraft e PP, sendo duas amostras modificadas
com anidrido malêico, os autores relatam que a tensão de ruptura diminuiu conforme
aumentou a carga de lignina. Isso é justificado pela fraca adesão entre carga e matriz
aumentando a chance de falhas. Os autores ressaltam ainda que a força é transferida
para a lignina de forma pobre e as partículas não são muito fortes sendo suscetíveis a
falha a menor força125. Por outro lado, os autores reforçam a ideia que as partículas da
lignina são mais rígidas o que justifica o aumento no módulo de elasticidade
encontrado nesse trabalho. Os valores referentes ao módulo de elasticidade estão de
acordo com Sahoo et al. Neste trabalho, para blendas de Lignina-PBS os valores
variaram de 1,1 – 3,3 GPa118. Comportamento semelhante é encontrado para blendas
de lignina-PE variando o tratamento dado à lignina e o pH do processo. Aleckina et al.
mostraram que as blendas produzidas com lignina tratadas em meio ácido (pH = 5,0)
possuem valores de módulo de elasticidade entre 1,5 – 2,5 GPa126.
O estudo do efeito da lignina de eucalipto modificada nas propriedades
morfológicas e termo-mecânicas, de Pérez-Guerrero et al., mostra comportamento
semelhante24. Foi utilizado lignina modificada com anidrido acético e malêico como
carga, variando a proporção de 5 a 15% em massa, em matriz de EPS reciclado
fazendo comparação com filme somente de EPS reciclado e PS puro. Os resultados
mostraram um efeito plastificante da lignina com a redução no valor de torque sendo a
proporção de 5% de carga de lignina o melhor resultado. Cabe ressaltar que esse
trabalho mostra que filmes de EPS reciclado perdem cerca de 34% da resposta ao
torque. Isso justifica o baixo valor para a tensão de ruptura encontrado para a matriz.
Aradoaei et al. apresentam resultado semelhante127. O copolímero AA5 foi um dos que
apresentou a tensão de ruptura mais próxima ao EPS e módulo de elasticidade maior

79
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
Te
nsa
o (
MP
a)
Elongaçao (%)
EPS
AI2
AI5
AI10
AI20
AI50
que a matriz. Isso vai ao encontro da análise de massa molecular no qual este
copolímero foi o o que apresentou maior Mn e Mw próxima ao valor do EPS.
O resultado dos ensaios mecânicos para os copolímeros com ácido itacônico
são sintetizados na Tabela 14 e Figura 55.
Tabela 14 - Ensaios mecânicos para os copolímeros com ácido itacônico
Copolímero com ácido itacônico
AI2 AI5 AI10 AI20 AI50 EPS
Tensão de Ruptura (MPa)
17,76 16,93 16,32 17,06 6,56 21,78
Módulo de Elasticidade
(GPa)
2,41 1,82 2,49 2,30 1,55 2,34
Figura 55. Gráfico Tensão X Elongação para os copolímeros com ácido itacônico
Novamente pode ser observado que o aumento da carga de lignina reduziu a
tensão de ruptura dos filmes quando comparado à matriz. Entretanto, os valores de
tensão para os copolímeros com carga que varia entre 2 – 20% foram semelhantes
entre si. Além disso, a figura 54 mostra que AI50 é o copolímero mais frágil, com menor
tensão de ruptura e Módulo de Young. Já AI5 foi o copolímero que mais se deformou
elasticamente quando submetido a uma tensão. Com relação ao módulo de
elasticidade, AI10 apresentou os maiores valores. O estudo de propriedades e
estrutura de blendas na forma de filmes de PVP-lignina proveniente de bamboo traz
resultado próximo ao que foi encontrado para os copolímeros com ácido itacônico98. Os
autores assinalam que com 10% de carga houve o maior módulo de elasticidade. A

80
partir dessa informação, concluem que em cargas maiores que 15% há a existência de
micro-domínios de lignina que fazem reduzir a tensão de ruptura, como se esse
excesso de partícula não conseguisse transferir a força submetida ao longo do
material. Alexy et al. em trabalho sobre a preparação de blendas de lignina-PP e
lignina-PE observou redução na tensão de ruptura com aumento da carga de lignina.
Além disso, com carga de 10% de lignina obteve os melhores resultados128. Para
blendas de PS-lignina e PS-vanilina, El-Zawawy et al. observaram tanto menor tensão
de ruptura quanto menor módulo de elasticidade quando comparado ao PS puro. Já o
complexo lignina-Co(II) e vanilna-Co(II) aumentaram as propriedades de tensão
mostrando ser mais compatível com a matriz de PS120.
Barzegari et al., na produção de blenda de lignina kraft como carga, PS como
matriz, e o copolímero SEBS (estireno-etileno-butileno) modificado com anidrido
malêico também observaram pequena perda das propriedades de tensão com o
aumento da quantidade de lignina. É destaca que todas as amostras apresentaram um
comportamento frágil o que é típico de polímeros vítreos sob tensão. Uma razão
mencionada para essa redução nas propriedades de tensão pode ser o grosseiro e não
uniforme molde das partículas de lignina que podem produzir concentração de força na
matriz vítrea de PS. Além disso, uma pequena força mecânica pode iniciar a quebra da
matriz vítrea com propagação ao longo de interfaces. Consequentemente um menor
valor de força é suficiente para causar uma falha mecânica permitindo uma menor
tensão de ruptura117. Isso foi observado tanto para os copolímeros com ácido adípico
quanto para o ácido itacônico.
Ácido adípico e itacônico foram usados como promotores da reação fotoquímica
entre a lignina extraída da casca de arroz pelo método Organossolve e o poliestireno
expandido reciclado. O uso de compatibilizadores para a produção de blendas e
copolímeros é relatado na literatura. Barzegari et al. utilizaram SEBS (estireno-etileno-
butileno) modificado com anidrido malêico em três diferentes concenrações: 1, 2 e 5%
em massa. As propriedades mecânicas e morfológicas melhoraram com o aumento da
concentração de compatibilizador. Isso pode ser interpretado como melhor adesão das
partículas da lignina com a matriz de PS, permitindo uma melhor transferência de força
na interface, principalmente em matrizes hidrofóbicas117. Sahoo et al. incorporaram
PMDI como compatibilizador aos filmes de lignina e PE. Também foi relatado melhora
nas propriedades mecânicas de tensão aprimorando a adesão interfacial no

81
compósito118. Toriz et al. relatam que a lignina não parecem ter muitas interações por
isso o uso de compatibilizadores que melhoram as propriedades mecânicas do produto
final125.
5.7 Discussão geral dos resultados
A partir de todas as análises realizadas, pode-se sugerir a obtenção de
copolímero EPS-Lignina através de reações fotoquímicas em 10min. O emprego de
dois diácidos carboxílicos permitiu a produção de novos materiais com características
distintas. As reações em branco, ou seja, sem a exposição do sistema à radiação UV,
evidenciaram as modificações ocasionadas pela presença da luz. Independente do
diácido usado, foi possível observar pelos espectros de UV-Vis mudanças nas
transições eletrônicas do novo material quando comparado aos materiais de partida e
ao teste em branco. As principais mudanças foram nas transições π->π* de ligações
C=C e C=O, em 1508 cm-1 e 1712 cm-1, respectivamente, indicando o sítio reacional.
Alterações nas bandas referentes a esses sítios reacionais também são mostradas
pelos espectros de IV. A análise destes indica bandas de ligação C-O características
da LO na região de 1328, 1266 e 1218 cm-1. Também em todos os copolímeros, em
torno de 1127 cm-1 foram encontradas bandas correspondentes a anéis aromáticos. Os
copolímeros com ácido adípico, de maneira geral, possuem banda de C=O, já os
formados com ácido itacônico apresentaram essas bandas, embora discreta mas com
acréscimo de intensidade proporcional ao acréscimo de massa de LO na reação. Os
sítios carbonílicos são sensíveis à radiação UV podendo levar à reações do tipo Norish
Tipo I e Tipo II82. Há evidência de que o mecanismo de enxertia difere de uma série de
amostras para outra, ou seja, variando-se a estrutura do promotor da reação, tem-se
um mecanismo diferenciado de reação, resultando em modificação diferenciada na
cadeia polimérica. Outras alterações nas bandas de C-H tanto de anel aromático
quanto de cadeia alifática indicam possível abstração de hidrogênio também já
estudada anteriormente87. Essas alterações, com acréscimo de sinais tanto na parte
alifática quanto aromática pode ser observado nos espectros de RMN ¹H e ¹³C. A
ausência de sinais de carbonos de carbonila indica possível descarboxilização dos
diácidos empregados podendo ser estes responsáveis pela abstração de hidrogênio do
sistema assim como a radiação UV. Dessa forma, sugere-se que o mecanismo da
reação inicie da abstração de H da cadeia principal do EPS ou pela nas ligações β-O-4
da lignina (Figura 56).

82
Figura 56. Mecanismo sugerido para a obtenção dos copolímeros
A partir das análises de EDS, em combinação com as demais, é possível
observar algumas relações. Os copolímeros que apresentaram menor %O (AA2, AA10
AI5 e AI50), quando comparados ao EPS, apresentaram, de maneira geral, redução de
Mn, Mw, tensão de ruptura e módulo de elasticidade e aumento do valor de PDI. O
menor %O pode estar relacionado com menor quantidade de cadeias de lignina
enxertadas. Provavelmente, o número de domínios de lignina enxertadas não foi
suficiente para estabilizar a lignina não-enxertada presente nos copolímeros. Para
essas amostras pode haver a formação de microdomínios de lignina pura, conduzindo
a separação de fases, como observado nas análises de MEV
Apesar da presença de domínios de lignina, as análises morfológicas
mostraram, de maneira geral, filmes homogêneos em ampliações de até 40.000. A

83
análise de ângulo de contato, mostra um decréscimo do caráter hidrofóbico do material
refletindo uma modificação do EPS e da lignina. Quanto às análises térmicas, é
possível verificar que, independente do diácido empregado, as alterações na
temperatura de transição foram pequenas. Isso indica que os copolímeros mantiveram
a característica térmica da matriz polimérica.
A cromatografia de permeação em gel reforça a possibilidade de fotoenxertia já
que traz a informação de aumento do número de macromoléculas na cadeia polimérica
(para AA5 e copolímeros com ácido itacônico) mediante ligeira quebra da cadeia. Esse
percentual de quebra da cadeia foi maior para os copolímeros com ácido adípico que
para o ácido itacônico. Os ensaios mecânicos estão de acordo com as demais
análises. Os copolímeros com maior carga de lignina mostraram-se mais frágeis, com
menor tensão de ruptura. Em alguns casos o módulo de elasticidade foi maior que o da
matriz mostrando que as cadeias enxertadas de lignina possuem capacidade de
distribuir melhor a energia ao longo do material.
Além disso, pode-se afirmar que o método empregando radiação UV possui
baixa complexidade experimental, baixo custo, utiliza condições mais brandas de
reação e é mais rápido, quando comparado com outras reações de copolimerização.
Essa potencial alternativa aos métodos convencionais vai ao encontro das premissas
de Química Verde e sustentabilidade.

84
6 CONCLUSÕES
A copolimerização de lignina extraída da casca de arroz pelo método
Organossolve e EPS reciclado via radiação UV foi bem sucedida. Os copolímeros
foram solubilizados e expostos à fonte de radiação UV por 10 min para posterior
confecção dos filmes a partir da evaporação do solvente. Estes filmes, por sua vez,
passaram por processo de purificação. As análises espectroscópicas de UV-Vis
mostraram diferenças nas transições eletrônicas dos copolímeros quando comparados
aos materiais de partida e ao teste em branco. Isso vai ao encontro dos espectros de
infravermelho que mostraram mudanças nos perfis espectroscópicos configurando
novo arranjo polimérico entre matriz e carga sugerindo a reação fotoquímica. As
análises de RMN ¹³C e ¹H indicam possível abstração de H da cadeia alifática do EPS
com posterior inserção da carga de lignina. Também concordando com as demais
análises, o EDS mostrou aumento do %O após a reação fotoquímica. As análises
morfológicas mostraram filmes bem homogêneos quando comparados ao de EPS.
Algumas diferenças em altas cargas de LO para alguns copolímeros foram encontradas
em ampliação de 40.000, o que condiz com a literatura. Quanto às análises de ângulo
de contato, independente do ácido promotor da reação, os ângulos de contato foram
inferiores ao do EPS (72º). Isso se deve à enxertia de LO à matriz dando um caráter
mais hidrofílico aos copolímeros. As análises térmicas revelaram duas Tg tanto para os
copolímeros quanto para o EPS. Esta 2º Tg corresponde ao histórico térmico dos
materiais. Além disso, que os copolímeros apresentam Tg próxima ao do EPS (em
torno de 99ºC) e em altas cargas de LO (AA50 e AI50) a Tg diminui consideravelmente
indicando excesso de LO não reagida. O GPC mostrou aumento do número de
macromoléculas para o copolímero denominado AA5 e ligeira redução de Mw.
Enquanto que para os copolímeros produzidos com ácido itacônico, devido a estrutura
do ácido promotor, a formação de ligações cruzadas permitiu valores mais próximos do
EPS para Mn e Mw. A quebras das cadeias poliméricas seguida de rearranjos é
evidente. Os ensaios de tração mostraram que o material obtido apresenta tensão de
ruptura inferior ao EPS, mas, em alguns casos, capacidade de distribuição de carga
melhor que a matriz. Este trabalho mostrou uma alternativa viável para
reaproveitamento de resíduos aliada à fonte de radiação UV como promissora
ferramenta ambientalmente adequada e de baixo custo.

85
7 REFERÊNCIAS BIBLIOGRÁFICAS
1. E. J. Lenardao, R. A. Freitag, M. J. Dabdoub, A. C. F. Batista and C. D. Silveira, Quimica Nova, 2003, 26, 123-129.
2. A. o. P. Manufaturers and E. A. o. P. R. a. R. Organisations, Journal, 2015, 1-30. 3. A. B. d. Plásticos, Perfil 2015, 2014. 4. O. o. t. P. E. Countries, Journal, 2016, 22-28. 5. P. BR, Journal, 2015. 6. A. N. d. E. Elétrica, Journal, 2008, 1-12. 7. E. d. P. Energética, Journal, 2015, 1-291. 8. Y. Wu, S. Z. Zhang, X. Y. Guo and H. L. Huang, Bioresource Technology, 2008, 99, 7709-7715. 9. G. Imanzadeh, A. Banaei, T. Fathi and Z. Soltanzadeh, Green Chemistry Letters and Reviews,
2017, 10, 1-9. 10. Y. Y. Li, J. L. Huang, X. J. Hu, F. L. Y. Lam, W. J. Wang and R. Luque, Journal of Molecular Catalysis
a-Chemical, 2016, 425, 61-67. 11. M. A. P. Martins, P. Muraro, P. Beck, P. Machado, C. P. Frizzo, N. Zanatta and H. G. Bonacorso,
Synthetic Communications, 2008, 38, 3465-3476. 12. Y. M. Li, B. Z. Li, F. L. Du, Y. Wang, L. X. Pan and D. Chen, Journal of Applied Polymer Science,
2017, 134. 13. G. O. Ferrero, H. J. Rojas, C. E. Argarana and G. A. Eimer, Journal of Cleaner Production, 2016,
139, 495-503. 14. T. J. Lawton and A. C. Rosenzweig, Current Opinion in Chemical Biology, 2016, 35, 142-149. 15. E. Coyle, Phd, Dublin City University, 2010. 16. D. M. He, H. Susanto and M. Ulbricht, Progress in Polymer Science, 2009, 34, 62-98. 17. A. Gandini, Macromolecules, 2008, 41, 9491-9504. 18. J. V. Kurian, Journal of Polymers and the Environment, 2005, 13, 159-167. 19. E. B. Mano and L. C. Mendes, Introdução a Polímeros, Blucher, São Paulo, 2ª edn., 1999. 20. W. D. Callister and D. G. Rethwisch, Ciência e Engenharia de Materiais - Uma Introdução, LTC,
Rio de Janeiro, 8ª edn., 2013. 21. J. Lisperguer, C. Nunez and P. Perez-Guerrero, Journal of the Chilean Chemical Society, 2013, 58,
1937-1940. 22. E. Svinterikos and I. Zuburtikudis, Journal of Applied Polymer Science, 2016, 133, 12. 23. C. M. C. Bonelli, A. Elzubair and J. C. M. Suarez, Journal, 2005, 15, 256-260. 24. P. Perez-Guerrero, J. Lisperguer, J. Navarrete and D. Rodrigue, Bioresources, 2014, 9, 6514-6526. 25. F. A. Carey, Química Orgânica, McGraw-Hill, 7ª edn., 2011. 26. G. Odian, Principles of Polimerization, 4ª edn., 2004. 27. R. L. Adelman and R. C. Ferguson, Journal of Polymer Science Part a-Polymer Chemistry, 1975,
13, 891-911. 28. S.-H. Wang, F. M. B. Coutinho, G. Galli and E. Chiellini, Journal, 1996, 38-44. 29. D. Myers, Surfactant Science and Technology, New Jersey, 3ª edn., 2006. 30. D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chemical Society Reviews, 2009, 38, 2046-
2064. 31. S. Banerjee, T. K. Paira and T. K. Mandal, Polymer Chemistry, 2014, 5, 4153-4167. 32. A. Ayoub, R. A. Venditti, H. Jameel and H. M. Chang, Journal of Applied Polymer Science, 2014,
131. 33. N. Lin and A. Dufresne, Macromolecules, 2013, 46, 5570-5583. 34. S. Rajam and C. C. Ho, Journal of Membrane Science, 2006, 281, 211-218.

86
35. Y. N. Chen, D. Chen, Y. H. Ma and W. T. Yang, Journal of Polymer Science Part a-Polymer Chemistry, 2014, 52, 1059-1067.
36. H. F. D. Oliveira, A. A. Weiner, A. Majumder and V. P. Shastri, Journal of Controlled Release, 2008, 126, 237-245.
37. D. Odaci, M. U. Kahveci, E. L. Sahkulubey, C. Ozdemir, T. Uyar, S. Timur and Y. Yagci, Bioelectrochemistry, 2010, 79, 211-217.
38. D. L. Versace, J. Ramier, D. Grande, S. A. Andaloussi, P. Dubot, N. Hobeika, J. P. Malval, J. Lalevee, E. Renard and V. Langlois, Advanced Healthcare Materials, 2013, 2, 1008-1018.
39. J. T. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chemical Science, 2014, 5, 3568-3575. 40. A. L. Goffin, J. M. Raquez, E. Duquesne, G. Siqueira, Y. Habibi, A. Dufresne and P. Dubois,
Biomacromolecules, 2011, 12, 2456-2465. 41. M. U. Kahveci, C. Mangold, H. Frey and Y. Yagci, Macromolecular Chemistry and Physics, 2014,
215, 566-571. 42. Y. H. Li, J. M. Desimone, C. D. Poon and E. T. Samulski, Journal of Applied Polymer Science, 1997,
64, 883-889. 43. S. R. Shukla and A. R. Athalye, Polymer, 1992, 33, 3729-3733. 44. Y. Y. Durmaz, G. Yilmaz and Y. Yagci, Macromolecular Chemistry and Physics, 2007, 208, 1737-
1743. 45. D. Bontempo, N. Tirelli, K. Feldman, G. Masci, V. Crescenzi and J. A. Hubbell, Advanced
Materials, 2002, 14, 1239-+. 46. J. A. I. do Sul and M. F. Costa, Environmental Pollution, 2014, 185, 352-364. 47. B. Gewert, M. M. Plassmann and M. MacLeod, Environmental Science-Processes & Impacts,
2015, 17, 1513-1521. 48. S. C. H. Mangalara and S. Varughese, Acs Sustainable Chemistry & Engineering, 2016, 4, 6095-
6100. 49. C. Y. Shin and G. G. Chase, Polymer Bulletin, 2005, 55, 209-215. 50. A. M. Sarmiento, H. L. Guzman, G. Morales, D. E. Romero and A. Y. Pataquiva-Mateus, Waste
and Biomass Valorization, 2016, 7, 1245-1254. 51. F. V. Silva, Journal, 2012, 13. 52. E. d. O. S. Saliba, N. M. Rodriguez, S. A. L. d. Morais and D. Piló-Veloso, Journal, 2001, 31, 917-
928. 53. A. T. Martinez, M. Speranza, F. J. Ruiz-Duenas, P. Ferreira, S. Camarero, F. Guillen, M. J.
Martinez, A. Gutierrez and J. C. del Rio, International Microbiology, 2005, 8, 195-204. 54. G. S. Hall, Nature, 1984, 310, 521-521. 55. K. Freudenberg, Science, 1965, 148, 595-+. 56. E. Ten and W. Vermerris, Polymers, 2013, 5, 600-642. 57. A. Salanti, L. Zoia, M. Orlandi, F. Zanini and G. Elegir, Journal of Agricultural and Food Chemistry,
2010, 58, 10049-10055. 58. N. S. Cetin and N. Ozmen, International Journal of Adhesion and Adhesives, 2002, 22, 477-480. 59. D. Stewart, Industrial Crops and Products, 2008, 27, 202-207. 60. M. N. Belgacem, A. Blayo and A. Gandini, Industrial Crops and Products, 2003, 18, 145-153. 61. X. P. Ouyang, L. X. Ke, X. Q. Qiu, Y. X. Guo and Y. X. Pang, Journal of Dispersion Science and
Technology, 2009, 30, 1-6. 62. F. Souto, V. Calado and N. J. Pereira, Journal, 2015, 20, 100-114. 63. H. Li, Q. Zhang, P. Gao and L. Wang, Journal of Applied Polymer Science, 2015, 132. 64. R. A. Perez-Camargo, G. Saenz, S. Laurichesse, M. T. Casas, J. Puiggali, L. Averous and A. J.
Muller, Journal of Polymer Science Part B-Polymer Physics, 2015, 53, 1736-1750. 65. J. E. Holladay, J. F. White, J. J. Bozell and J. D., Top Value-Added Chemicals from Biomass -
Results of Screening for Potencial Candidates from Biorefinarey Lignin, Pacific Northwest National Laboratory, United States of American, 2007.
66. S. Laurichesse and L. Averous, Progress in Polymer Science, 2014, 39, 1266-1290.

87
67. H. Priefert, J. Rabenhorst and A. Steinbuchel, Applied Microbiology and Biotechnology, 2001, 56, 296-314.
68. Y. Deng, Y. Liu, Y. Qian, W. Zhang and X. Qiu, Acs Sustainable Chemistry & Engineering, 2015, 3, 1111-1116.
69. C. Wang and R. A. Venditti, Acs Sustainable Chemistry & Enguneering, 2015, 1-26. 70. P. Atkins and J. d. Paula, Physycal-Chemistry, Oxford University Press, Great Britain, 8ª edn.,
2006. 71. D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introdução à Espectroscopia,
Washington, USA, 4ª edn., 2010. 72. F. Kessler, S. Kuhn, C. Radtke and D. E. Weibel, Polymer International, 2013, 62, 310-318. 73. F. Kessler and D. E. Weibel, Doutor em Química, Universidade Federal do Rio Grande do Sul,
2014. 74. J. F. Holler, D. A. Skoog and S. R. Crouch, Princípios de Análise Insrumental, 2009. 75. S. Dadashi-Silab, S. Doran and Y. Yagci, Chemical Reviews, 2016, 116, 10212-10275. 76. M. D. Karkas, J. A. Porco and C. R. J. Stephenson, Chemical Reviews, 2016, 116, 9683-9747. 77. M. Oelgemoller, Chemical Reviews, 2016, 116, 9664-9682. 78. M. Chen, M. J. Zhong and J. A. Johnson, Chemical Reviews, 2016, 116, 10167-10211. 79. N. Hoffmann, Journal, 2008, 108, 1052-1103. 80. Y. Y. Durmaz, G. Yilmaz and Y. Yagci, Journal of Polymer Science Part a-Polymer Chemistry, 2007,
45, 423-428. 81. J. E. Kilduff, S. Mattaraj, M. Y. Zhou and G. Belfort, Journal of Nanoparticle Research, 2005, 7,
525-544. 82. N. Hoffmann, Chemical Reviews, 2008, 108, 1052-1103. 83. Y. Kong and J. N. Hay, Polymer, 2002, 43, 3873-3878. 84. C. R. Chinaglia and C. A. Correa, Journal, 1997, 19-23. 85. D. J. Shaw, Introdução à química dos colóides e de superfícies, 1975. 86. F. M. Lanças, Cromatografia Líquida Moderna - HPLC/CLAE, 1ª edn., 2009. 87. O. Puglisi, A. Licciardello, S. Pignataro, L. Calcagno and G. Foti, Radiation Effects and Defects in
Solids, 1986, 98, 161-170. 88. M. E. Martinez-Pardo, J. Cardoso, H. Vazquez and M. Aguilar, Nuclear Instruments & Methods in
Physics Research Section B-Beam Interactions with Materials and Atoms, 1998, 140, 325-340. 89. J. J. Meister and M. J. Chen, Macromolecules, 1991, 24, 6843-6848. 90. M. P. Rosa, P.H. Beck, D. G. Müller, J. B. Moreira, J. S. da Silva, A. M. M. Durigon, Biological and
Chemical Research, 2017, 4, 87-98. 91. A. Chapiro, Nuclear Instruments & Methods in Physics Research Section B-Beam Interactions
with Materials and Atoms, 1988, 32, 111-114. 92. A. Licciardello, O. Puglisi, L. Calcagno and G. Foti, Nuclear Instruments & Methods in Physics
Research Section B-Beam Interactions with Materials and Atoms, 1989, 39, 769-772. 93. H. X. Zhang, X. F. Ding, X. Chen, Y. J. Ma, Z. C. Wang and X. Zhao, Journal of Hazardous Materials,
2015, 291, 65-73. 94. X. H. Liu, J. F. Wang, S. H. Li, X. W. Zhuang, Y. Z. Xu, C. P. Wang and F. X. Chu, Industrial Crops and
Products, 2014, 52, 633-641. 95. L. C. d. A. Barbosa, Espectroscopia no Infravermelho na Caracterização de Compostos Orgânicos,
Editora UFV, Viçosa, MG, Brasil, 1ª edn., 2007. 96. A. Tejado, C. Pena, J. Labidi, J. M. Echeverria and I. Mondragon, Bioresource Technology, 2007,
98, 1655-1663. 97. C. Rossberg, M. Bremer, S. Machill, S. Koenig, G. Kerns, C. Boeriu, E. Windeisen and S. Fischer,
Industrial Crops and Products, 2015, 73, 81-89. 98. C. H. Liu, C. B. Xiao and H. Liang, Journal of Applied Polymer Science, 2005, 95, 1405-1411. 99. S. Jairam, R. Bucklin, M. Correll, T. S. Sakthivel, S. Seal, J. Truett and Z. H. Tong, Materials &
Design, 2016, 90, 151-156. 100. D. K. Chattopadhyay, S. S. Panda and K. Raju, Progress in Organic Coatings, 2005, 54, 10-19.

88
101. R. Yang, P. A. Christensen, T. A. Egerton and J. R. White, Polymer Degradation and Stability, 2010, 95, 1533-1541.
102. J. Lisperguer, P. Perez and S. Urizar, Journal of the Chilean Chemical Society, 2009, 54, 460-463. 103. R. J. Sammons, D. P. Harper, N. Labbe, J. J. Bozell, T. Elder and T. G. Rials, Bioresources, 2013, 8,
2752-2767. 104. G. Sivasankarapillai, A. G. McDonald and H. Li, Biomass & Bioenergy, 2012, 47, 99-108. 105. R. M. Silverstein, F. X. Webster and D. J. Kiemle, Identificação Espectroscópica de Compostos
Orgânicos, LTC - Livros Técnicos e Científicos LTDA, Rio de Janeiro, 7ª edn., 2012. 106. W. Ren, X. Y. Pan, G. Wang, W. R. Cheng and Y. Liu, Green Chemistry, 2016, 18, 5008-5014. 107. Z. Y. Li, R. Liu, B. Y. Mai, S. Feng, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, Polymer Chemistry,
2013, 4, 954-960. 108. A. Garcia, M. G. Alriols, G. Spigno and J. Labidi, Biochemical Engineering Journal, 2012, 67, 173-
185. 109. Y. Lin, J. Y. King, S. D. Karlen and J. Ralph, Biogeochemistry, 2015, 125, 427-436. 110. O. Lanzalunga and M. Bietti, Journal of Photochemistry and Photobiology B-Biology, 2000, 56,
85-108. 111. W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Industrial Crops and Products, 2011, 33,
259-276. 112. A. Duval and M. Lawoko, Reactive & Functional Polymers, 2014, 85, 78-96. 113. C. Pouteau, S. Baumberger, B. Cathala and P. Dole, Comptes Rendus Biologies, 2004, 327, 935-
943. 114. R. Pucciariello, V. Villani, C. Bonini, M. D'Auria and T. Vetere, Polymer, 2004, 45, 4159-4169. 115. A. Hambardzumyan, L. Foulon, N. B. Bercu, M. Pernes, J. E. Maigret, M. Molinari, B. Chabbert
and V. Aguie-Beghin, Chemical Engineering Journal, 2015, 264, 780-788. 116. W. Deoliveira and W. G. Glasser, Journal of Wood Chemistry and Technology, 1994, 14, 119-126. 117. M. R. Barzegari, A. Alemdar, Y. L. Zhang and D. Rodrigue, Polymer Composites, 2012, 33, 353-
361. 118. S. Sahoo, M. Misra and A. K. Mohanty, Composites Part a-Applied Science and Manufacturing,
2011, 42, 1710-1718. 119. X. N. Lu, K. Yue, J. Q. Cui, S. G. Han and X. L. Ding, Cellulose Chemistry and Technology, 2012, 46,
467-470. 120. W. K. El-Zawawy, M. M. Ibrahim, M. N. Belgacem and A. Dufresne, Materials Chemistry and
Physics, 2011, 131, 348-357. 121. D. S. Argyropoulos, H. Sadeghifar, C. Z. Cui and S. Sen, Acs Sustainable Chemistry & Engineering,
2014, 2, 264-271. 122. M. P. d. Rosa, 2015. 123. J. F. Rabek, Polymer Photodegradation: mechanims and experimental methods, Chapman & Hall,
1º edn., 1995. 124. V. R. Botaro, C. G. dos Santos, G. Arantes and A. R. da Costa, Applied Surface Science, 2001, 183,
120-125. 125. G. Toriz, F. Denes and R. A. Young, Polymer Composites, 2002, 23, 806-813. 126. M. Alekhina, J. Erdmann, A. Ebert, A. M. Stepan and H. Sixta, Journal of Materials Science, 2015,
50, 6395-6406. 127. S. Aradoaei, R. Darie, G. Constantinescu, M. Olariu and R. Ciobanu, Journal of Non-Crystalline
Solids, 2010, 356, 768-771. 128. P. Alexy, B. Kosikova and G. Podstranska, Polymer, 2000, 41, 4901-4908.

89
Anexos
Tabela 1. Análise IV para EPS.......................................................................................91
Tabela 2. Análise IV para LO.........................................................................................91
Tabela 3. Análise IV para ácido adípico.........................................................................92
Tabela 4. Análise IV para ácido itacônico......................................................................92
Tabela 5. Análise IV para o copolímero AA2.................................................................92
Tabela 6. Análise IV para o copolímero AA5.................................................................93
Tabela 7. Análise IV para o copolímero AA10...............................................................93
Tabela 8. Análise IV para o copolímero AA20...............................................................94
Tabela 9. Análise IV para o copolímero AA50...............................................................94
Tabela 10. Análise IV para o copolímero AI2.................................................................95
Tabela 11. Análise IV para o copolímero AI5.................................................................95
Tabela 12. Análise IV para o copolímero AI10...............................................................96
Tabela 13. Análise IV para o copolímero AI20...............................................................97
Tabela 14. Análise IV para o copolímero AI50...............................................................98
Figura 1. Espectro RMN ¹H do copolímero AA2, em CDCl3..........................................99
Figura 2. Espectro de RMN ¹³C e DEPT 135 do copolímero AA2, em CDCl3.............100
Figura 3. Espectro HSQC do copolímero AA2, em CDCl3...........................................101
Figura 4. Espectro RMN ¹H do copolímero AA5, em CDCl3........................................102
Figura 5. Espectro RMN ¹³C e DEPT 135 do copolímero AA5, em CDCl3..................103
Figura 6. Espectro RMN HSQC do copolímero AA5, em CDCl3.................................104
Figura 7. Espectro RMN ¹H do copolímero AA10, em CDCl3......................................105
Figura 8. Espectro RMN ¹³C e DEPT 135 do copolímero AA10, em CDCl3................106
Figura 9. Espectro RMN HSQC do copolímero AA10, em CDCl3...............................107
Figura 10. Espectro RMN ¹H do copolímero AA20, em CDCl3....................................108
Figura 11. Espectro RMN ¹³C e DEPT 135 do copolímero AA20, em CDCl3..............109
Figura 12. Espectro RMN HSQC do copolímero AA20, em CDCl3.............................110
Figura 13. Espectro RMN ¹H do copolímero AA50, em CDCl3....................................111
Figura 14. Espectro RMN ¹³C e DEPT 135 do copolímero AA50, em CDCl3..............112
Figura 15. Espectro RMN HSQC do copolímero AA50, em CDCl3.............................113
Figura 16. Espectro RMN ¹H do copolímero AI2, em CDCl3.......................................114
Figura 17. Espectro RMN ¹³C e DEPT 135 do copolímero AI2, em CDCl3..................115
Figura 18. Espectro RMN HSQC do copolímero AI2, em CDCl3.................................116

90
Figura 19. Espectro RMN ¹H do copolímero AI5, em CDCl3.......................................117
Figura 20. Espectro RMN ¹³C e DEPT 135 do copolímero AI5, em CDCl3..................118
Figura 21. Espectro RMN HSQC do copolímero AI5, em CDCl3.................................119
Figura 22. Espectro RMN ¹H do copolímero AI10, em CDCl3.....................................120
Figura 23. Espectro RMN ¹³C e DEPT 135 do copolímero AI10, em CDCl3................121
Figura 24. Espectro RMN HSQC do copolímero AI10, em CDCl3...............................122
Figura 25. Espectro RMN ¹³C e DEPT 135 do copolímero AI20, em CDCl3................123
Figura 26. Espectro RMN HSQC do copolímero AI20, em CDCl3...............................124
Figura 27. Espectro RMN ¹³C e DEPT 135 do copolímero AI50, em CDCl3................125
Figura 28. Espectro RMN HSQC do copolímero AI50, em CDCl3...............................126
Figura 29. Espectro IV em (a) copolímero AA5 com e sem exposição à radiação e em
(b) copolímero AI10 com e sem exposição..................................................................127
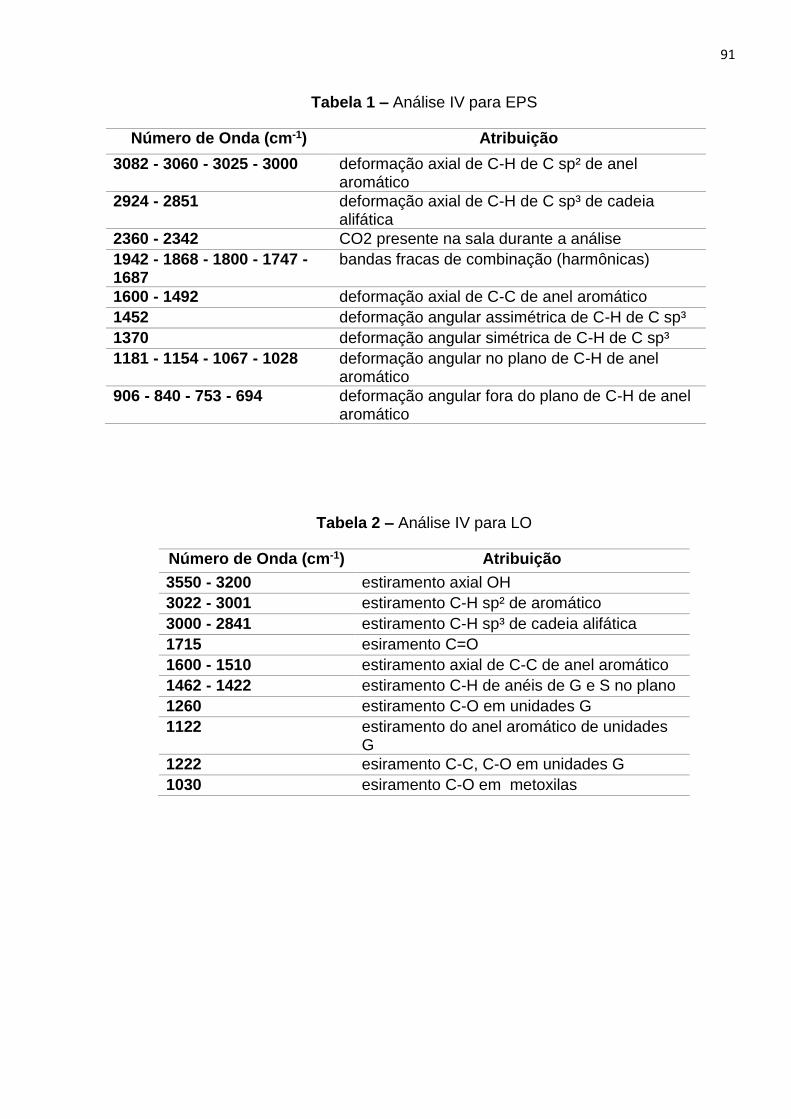
91
Tabela 1 – Análise IV para EPS
Número de Onda (cm-1) Atribuição
3082 - 3060 - 3025 - 3000 deformação axial de C-H de C sp² de anel aromático
2924 - 2851 deformação axial de C-H de C sp³ de cadeia alifática
2360 - 2342 CO2 presente na sala durante a análise
1942 - 1868 - 1800 - 1747 - 1687
bandas fracas de combinação (harmônicas)
1600 - 1492 deformação axial de C-C de anel aromático
1452 deformação angular assimétrica de C-H de C sp³
1370 deformação angular simétrica de C-H de C sp³
1181 - 1154 - 1067 - 1028 deformação angular no plano de C-H de anel aromático
906 - 840 - 753 - 694 deformação angular fora do plano de C-H de anel aromático
Tabela 2 – Análise IV para LO
Número de Onda (cm-1) Atribuição
3550 - 3200 estiramento axial OH
3022 - 3001 estiramento C-H sp² de aromático
3000 - 2841 estiramento C-H sp³ de cadeia alifática
1715 esiramento C=O
1600 - 1510 estiramento axial de C-C de anel aromático
1462 - 1422 estiramento C-H de anéis de G e S no plano
1260 estiramento C-O em unidades G
1122 estiramento do anel aromático de unidades G
1222 esiramento C-C, C-O em unidades G
1030 esiramento C-O em metoxilas

92
Tabela 3 – Análise IV para ácido adípico
Número de Onda (cm-1) Atribuição
3300 - 2500 estiramento da ligação O-H
2961 - 2947 - 2926 - 2918
estiramento da ligação C-H (sp³) simétrico e assimétrico
1714 estiramento C=O
1460 - 1429 - 1406 banda de combinação do estiramento C-O e a deformação angular OH para o dímero
1311 - 1279 estiramento C-O como dupleto para o dímero
925 deformação angular for do plano para OH
Tabela 4 – Análise IV para ácido itacônico
Número de Onda (cm-1) Atribuição
3300 - 2500 estiramento da ligação O-H
2965 - 2945 - 2934 - 2925
estiramento da ligação C-H (sp³) simétrico e assimétrico
1713 estiramento C=O
1451 - 1440 - 1433 banda de combinação do estiramento C-O e a deformação angular OH para o dímero
1335 - 1306 estiramento C-O como dupleto para o dímero
909 deformação angular for do plano para OH
Tabela 5 – Análise IV para copolímero AA2
AA2
Número de Onda (cm-1) Atribuição
3101 - 3081 - 3059 - 3024 - 2998
deformação axial de C-H de C sp² de anel aromático
2970 - 2926 - 2854 deformação axial de C-H de C sp³ de cadeia alifática
2359 CO2 presente na sala durante a análise
1942 - 1867 - 1801 - 1748 - 1681
bandas fracas de combinação (harmônicas) as duas últimas deslocadas
1600 - 1582 - 1556 - 1490 - 1474
deformação axial de C-C de anel aromático
1451 - 1403 deformação angular assimétrica de C-H de C sp³
1367 - deformação angular simétrica de C-H de C sp³
1328 - 1311 - 1268 - 1218 1328 estiramento C-O de S, 1268 cm-1 estiramento C-O em G e 1218cm-1 estiramento C-C, C-O e C=O em G
1180 - 1154 - 1127 - 1066 - 1050
deformação angular no plano de C-H de anel aromático
907 - 883 - 841 - 824 - 801 - 752 - 694
deformação angular fora do plano de C-H de anel aromático

93
Tabela 6 – Análise IV para copolímero AA5
AA5
Número de Onda (cm-1) Atribuição
3104 - 3082 - 3061 - 3026 - 3001 deformação axial de C-H de C sp² de anel aromático
2922 - 2850 deformação axial de C-H de C sp³ de cadeia alifática
2357 - 2340 CO2 presente na sala durante a análise
1942 - 1869 - 1803 - 1744 - 1667 bandas fracas de combinação (harmônicas)
1602 - 1583 - 1542 - 1493 deformação axial de C-C de anel aromático
1452 deformação angular assimétrica de C-H de C sp³
1372 deformação angular simétrica de C-H de C sp³
1348 - 1328 - 1313 - 1267 - 1219 1328 estiramento C-O de S, 1267 cm-1 estiramento C-O em G e 1219cm-1 estiramento C-C, C-O e C=O em G
1182 - 1154 - 1113 - 1069 - 1028 - 1003
deformação angular no plano de C-H de anel aromático
906 - 842 - 750 - 696 deformação angular fora do plano de C-H de anel aromático
Tabela 7 – Análise IV para copolímero AA10
AA10
Número de Onda (cm-1) Atribuição
3082 - 3060 - 3025 - 3002 deformação axial de C-H de C sp² de anel aromático
2921 - 2849 deformação axial de C-H de C sp³ de cadeia alifática
2347 - 2334 CO2 presente na sala durante a análise
1946 - 1872 - 1801 - 1739 - bandas fracas de combinação (harmônicas)
1600 - 1582 - 1542 - 1493 deformação axial de C-C de anel aromático
1451 deformação angular assimétrica de C-H de C sp³
1372 deformação angular simétrica de C-H de C sp³
1329 - 1313 - 1269 - 1219 1329 estiramento C-O de S, 1269 cm-1 estiramento C-O em G e 1219 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1155 - 1113 - 1068 - 1028 - 1001
deformação angular no plano de C-H de anel aromático
906 - 872 - 840 - 820 - 813 deformação angular fora do plano de C-H de anel aromático

94
Tabela 8 – Análise IV para copolímero AA20
AA20
Número de Onda (cm-1) Atribuição
3103 - 3082 - 3060 - 3025 - 3001
deformação axial de C-H de C sp² de anel aromático
2921 - 2849 deformação axial de C-H de C sp³ de cadeia alifática
2360 - 2338 CO2 presente na sala durante a análise
1943 - 1869 - 1802 - 1730 - bandas fracas de combinação (harmônicas)
1601 - 1583 - 1540 - 1514 - 1492
deformação axial de C-C de anel aromático
1452 deformação angular assimétrica de C-H de C sp³
1372 deformação angular simétrica de C-H de C sp³
1350 - 1328 - 1312 - 1267 - 1236 - 1219
1328 estiramento C-O de S, 1267 cm-1 estiramento C-O em G e 1219 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1154 - 1113 - 1068 - 1028 - 1003
deformação angular no plano de C-H de anel aromático
907 - 841 - 751 - 696 deformação angular fora do plano de C-H de anel aromático
Tabela 9 – Análise IV para copolímero AA50
AA50
Número de Onda (cm-1) Atribuição
3104 - 3082 - 3060 - 3025 - 3001
deformação axial de C-H de C sp² de anel aromático
2922 - 2849 deformação axial de C-H de C sp³ de cadeia alifática
2365 - 2338 CO2 presente na sala durante a análise
1943 - 1871 - 1804 - 1655 - bandas fracas de combinação (harmônicas)
1712 estiramento C=O que pode ser tanto do ácido promotor da reação quanto da LO
1602 - 1582 - 1511 - 1492 deformação axial de C-C de anel aromático
1462 - 1451 - 1422 deformação angular assimétrica de C-H de C sp³
1369 deformação angular simétrica de C-H de C sp³
1328 - 1309 - 1266 - 1224 - 1328 estiramento C-O de S, 1266 cm-1 estiramento C-O em G e 1224 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1154 - 1124 - 1094 - 1068 - 1027
deformação angular no plano de C-H de anel aromático
907 - 842 - 752 - 696 deformação angular fora do plano de C-H de anel aromático

95
Tabela 10 – Análise IV para copolímero AI2
AI2
Número de Onda (cm-1) Atribuição
3103 - 3082 - 3059 - 3025 – 3001
C-H de C sp² de anel aromático
2968 - 2927 - 2853 C-H de C sp³ de cadeia alifática
2360 - 2342 CO2 presente na sala durante a análise
1942 - 1867 - 1802 - 1747 – 1697
bandas fracas de combinação (harmônicas)
1601 - 1583 - 1558 - 1492 deformação axial de C-C de anel aromático, em 1583 e 1558 corresponde a anel aromático de G e S
1452 deformação angular assimétrica de C-H de C sp³
1365 deformação angular simétrica de C-H de C sp³
1328 - 1312 - 1216 1328 estiramento C-O de S, 1268 cm-1 estiramento C-O em G e 1216 cm-1 estiramento C-C, C-O e C=O em G
1181 - 1154 - 1126 - 1067 – 1030
deformação angular no plano de C-H de anel aromático e em 1126 deformação de anel aromático de G
909 - 841 - 754 - 694 deformação angular fora do plano de C-H de anel aromático
Tabela 11 – Análise IV para copolímero AI5
AI5
Número de Onda (cm-1)
Atribuição
3103 - 3081 - 3058 - 3025 - 3000
deformação axial de C-H de C sp² de anel aromático
2922 - 2848 deformação axial de C-H de C sp³ de cadeia alifática
2362 - 2337 CO2 presente na sala durante a análise
1943 - 1869 - 1802 bandas fracas de combinação (harmônicas)
1717 Estiramento C=O que pode ser da LO ou do ácido itacônico (1712 cm-1)
1600 - 1583 - 1508 - 1492
deformação axial de C-C de anel aromático, em 1583 e 1508 corresponde a anel aromático de G e S
1451 deformação angular assimétrica de C-H de C sp³
1369 deformação angular simétrica de C-H de C sp³

96
1328 - 1310 - 1267 - 1217
1328 estiramento C-O de S, 1267 cm-1 estiramento C-O em G e 1217 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1153 - 1126 - 1068 - 1028
deformação angular no plano de C-H de anel aromático e em 1126 deformação de anel aromático de G
906 - 841 - 750 - 694 deformação angular fora do plano de C-H de anel aromático
Tabela 12 – Análise IV para copolímero AI10
AI10
Número de Onda (cm-1) Atribuição
3104 - 3083 - 3060 - 3025 - 3001
deformação axial de C-H de C sp² de anel aromático
2920 - 2850 deformação axial de C-H de C sp³ de cadeia alifática
2368 - 2346 CO2 presente na sala durante a análise
1945 - 1871 - 1802 bandas fracas de combinação (harmônicas)
1719 Estiramento C=O que pode ser da LO ou do ácido itacônico (1712 cm-1)
1602 - 1582 - 1560 - 1542 – 1535 - 1508 - 1492
deformação axial de C-C de anel aromático e na faixa de 1500 corresponde a anel aromático de G e S
1452 deformação angular assimétrica de C-H de C sp³
1371 deformação angular simétrica de C-H de C sp³
1326 - 1310 - 1268 - 1217 1326 estiramento C-O de S, 1268 cm-1 estiramento C-O em G e 1217 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1153 - 1117 - 1069 - 1027
deformação angular no plano de C-H de anel aromático e em 1117 deformação de anel aromático de G
906 - 841 - 751 - 695 deformação angular fora do plano de C-H de anel aromático

97
Tabela 13 – Análise IV para copolímero AI20
AI20
Número de Onda (cm-1) Atribuição
3104 - 3080 - 3058 - 3023 - 3000
deformação axial de C-H de C sp² de anel aromático
2921 - 2849 deformação axial de C-H de C sp³ de cadeia alifática
2366 - 2343 CO2 presente na sala durante a análise
1942 - 1868 - 1800 - 1733 bandas fracas de combinação (harmônicas)
1717 Estiramento C=O que pode ser da LO ou do ácido itacônico (1712 cm-1)
1600 - 1583 - 1559 - 1541 - 1508 - 1491
deformação axial de C-C de anel aromático e na faixa de 1500 corresponde a anel aromático de G e S
1465 - 1451 deformação angular assimétrica de C-H de C sp³
1371 deformação angular simétrica de C-H de C sp³
1328 - 1311 - 1267 - 1222 1328 estiramento C-O de S, 1267 cm-1 estiramento C-O em G e 1222 cm-1 estiramento C-C, C-O e C=O em G
1181 - 1153 - 1126 - 1093 - 1068 - 1026
deformação angular no plano de C-H de anel aromático e em 1126 deformação de anel aromático de G
906 - 841 - 751 - 694 deformação angular fora do plano de C-H de anel aromático

98
Tabela 14 – Análise IV para copolímero AI50
AI50
Número de Onda (cm-1) Atribuição
3101 - 3081 - 3059 - 3024 - 3000
deformação axial de C-H de C sp² de anel aromático
2972- 2926 - 2854 deformação axial de C-H de C sp³ de cadeia alifática
2365 - 2344 CO2 presente na sala durante a análise
1944 - 1869 - 1800 - 1735
bandas fracas de combinação (harmônicas)
1718 Estiramento C=O que pode ser da LO ou do ácido itacônico (1712 cm-1)
1599 - 1570 - 1560 - 1544 - 1533 - 1508 - 1492
deformação axial de C-C de anel aromático e na faixa de 1500 corresponde a anel aromático de G e S
1473 - 1466 - 1451 - 1420
Deformação de anéis de G e S no plano
1367 deformação angular simétrica de C-H de C sp³
1328 - 1308 - 1266 - 1219 -
1328 estiramento C-O de S, 1266 cm-1 estiramento C-O em G e 1219 cm-1 estiramento C-C, C-O e C=O em G
1180 - 1154 - 1124 - 1094 - 1067 - 1029
deformação angular no plano de C-H de anel aromático e em 1124 deformação de anel aromático de G
906 - 883 - 840 - 823 - 811 - 750 - 694
deformação angular fora do plano de C-H de anel aromático