avaliaÇÃo do comportamento molecular do …livros01.livrosgratis.com.br/cp021180.pdf · técnicas...
TRANSCRIPT

AVALIAÇÃO DO COMPORTAMENTO MOLECULAR DO POLICARBONATO DE BISFENOL A POR RESSONÂNCIA
MAGNÉTICA NUCLEAR
Pedro Paulo Merat
Dissertação de Mestrado, submetida ao Instituto de Macromoléculas
Professora Eloisa Mano da Universidade Federal do Rio de Janeiro, como parte dos
requisitos necessários para a obtenção do Grau de Mestre em Ciências, em Ciência
e Tecnologia de Polímeros sob orientação da Professora Maria Inês Bruno Tavares
(IMA/UFRJ).
Rio de Janeiro 2005

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

i
Dissertação de Mestrado:
Avaliação do comportamento molecular do policarbonato de bisfenol A por ressonância magnética nuclear
Autor: Pedro Paulo Merat
Orientadora: Maria Inês Bruno Tavares
Data da defesa: 22 de julho de 2005
Aprovada por:
____________________________________ Profa Maria Inês Bruno Tavares - Orientadora UFRJ / IMA ____________________________________ Profa Marcia Christina Amorim Moreira Leite
IQ / UERJ
_____________________________________ Prof. Ricardo Cunha Michel UFRJ / IMA _____________________________________ Profa Rosane Aguiar da Silva San Gil
UFRJ / IQ
Rio de Janeiro
2005

ii
FICHA CATALOGRÁFICA
Merat, Pedro Paulo. Avaliação do comportamento molecular do policarbonato de
bisfenol A por ressonância magnética nuclear. Rio de Janeiro, 2005.
xvii, 83 f.:il.
Dissertação (Mestrado em Ciência e Tecnologia de Polímeros) - Universidade Federal do Rio de Janeiro - UFRJ, Instituto de Macromoléculas Professora Eloisa Mano - IMA, 2005. Orientadora: Maria Inês Bruno Tavares.
1. Policarbonato. 2. Tempo de relaxação. 3. RMN. I. Tavares, Maria Inês Bruno (Orient.) II. Universidade Federal do Rio de Janeiro. Instituto de Macromoléculas Professora Eloisa Mano. III. Título.

iii
Esta Dissertação foi desenvolvida nos
laboratórios do Instituto de Macromoléculas
Professora Eloisa Mano - IMA/UFRJ, e do Núcleo
de Catálise – NUCAT/COPPE/UFRJ.

iv
Dedicatória
Dedico esta Dissertação a
minha família e em especial
à minha esposa Leila, pelo seu
amor e compreensão e às
minhas filhas Laura e Isabella
pelo tempo furtado.

v
Agradecimentos
Agradeço a todos aqueles que participaram direta ou indiretamente desta
Dissertação:
,À minha orientadora Profª Maria Inês Bruno Tavares, pela paciência ٭
compreensão, ensinamentos, dedicação e competência. Amiga, companheira e
incentivadora acima de tudo.
.Aos meus pais pelo apoio e incentivo aos meus estudos ٭
.À Leila, pela ajuda dada nos momentos difíceis e pelo seu companheirismo ٭
,Aos amigos do Nucat: Daniele, Deborah, Sônia, Carlos André, Ricardo, Sidnei ٭
Manoel, Pedro Ivo, Leandro e Bruno pela amizade, ajuda, acolhida e descontração.
Ao lado de vocês a caminhada foi mais fácil. Obrigado por tudo.
Ao Marcos Anacleto, amigo, irmão e companheiro que muito me ajudou, dedicando ٭
o seu pouco tempo no meu estudo. O meu muito obrigado.
A todos do Nucat anexo, pela amizade e proveitosas conversas. Em especial a ٭
Dora pelas fotos e ao amigo Thiago, por ter se empenhado para recuperar os
arquivos do HD do meu computador. Que sufoco!
Aos Amigos Eduardo Miguez e Mônica, pelo tempo dedicado a aquisição dos ٭
espectros de RMN de alto e baixo campo respectivamente.
Às bibliotecárias do IMA Maria das Graças e Solange pelos ensinamentos ٭
passados.
À amiga Eliane, pela amizade e proveitosas conversas nas disciplinas que ٭
cursamos juntos.

vi
À Profª Ângela Coutinho do CEFET Química de Nilópolis pela ajuda na revisão ٭
ortográfica desta dissertação.
.À direção de unidade e geral do CEFET Química pelo apoio e incentivo dado ٭
.A todos os professores e funcionários do CEFET Química ٭
.Aos funcionários do IMA Valdecir, Jorge e Sônia por suas ajudas ٭
.Aos colegas Lúcia, Tavares e Andrea pelo acolhimento ٭
Ao Profº Martin Schmal Coordenador do NUCAT / COPPE pela disponibilização do ٭
Laboratório I - 132 para os meus estudos
.Aos amigos do grupo MIBT pelos bons momentos que passamos juntos ٭
.A Deus ٭

vii
Resumo da Dissertação apresentada no Instituto de Macromoléculas Professora
Eloisa Mano da Universidade Federal do Rio de Janeiro – IMA/UFRJ, como parte
dos requisitos necessários para a obtenção do grau de Mestre em Ciências (MSc)
em Ciência e Tecnologia de Polímeros.
Avaliação do comportamento molecular do policarbonato de bisfenol A por ressonância magnética nuclear
Pedro Paulo Merat
Orientadora: Maria Inês Bruno Tavares
O comportamento molecular do policarbonato de bisfenol A foi avaliado pelas
técnicas de espectrometria de absorção na região do infravermelho, análise térmica
(calorimetria diferencial de varredura e análise termogravimétrica), difração de raios
X, e mais detalhadamente por ressonância magnética nuclear, usando solução e
técnicas de relaxação. Dois tipos de tratamentos foram realizados nas amostras de
policarbonato: indução de cristalinidade por solvente e resfriamento brusco. As
amostras tratadas e não tratadas foram caracterizadas pelas técnicas citadas
anteriormente, usando as mesmas condições de análises. Os resultados mostraram
que o tetracloroetano foi um solvente melhor do que o clorofórmio para a indução de
cristalinidade. A avaliação da indução da cristalinidade por difração de raios X
mostrou que ocorreu formação de diferentes tamanhos de cristais, dependendo do
solvente usado. Embora a espectroscopia de absorção na região do infravermelho
não tenha mostrado mudanças nos espectros das diferentes amostras após os
tratamentos, as outras técnicas mostraram ser adequadas para o entendimento do
comportamento das amostras de policarbonato. Os resultados indicaram uma
reorganização estrutural para os dois processos aplicados. A aplicação da RMN de
baixo campo para estes materiais demonstrou ser eficaz na avaliação das mudanças
observadas com a vantagem de ser mais rápida e precisa.
Rio de Janeiro 2005

viii
Abstract of Dissertation presented to Instituto de Macromoléculas Professora Eloisa
Mano of Universidade Federal do Rio de Janeiro, as partial fulfillment of the
requirement for the degree of Master in Science (MSc), in Science and Technology of
Polymers.
Evaluation of molecular behavior of the bisphenol A polycarbonate by nuclear magnetic resonance
Pedro Paulo Merat
Advisor: Maria Inês Bruno Tavares
We evaluated the dynamical behavior of three bisphenol A polycarbonate
samples by some techniques such as infrared spectrometry, thermal analyses
(differential scanning calorimetry and thermogravimetric analysis), X-ray diffraction
and in more detail by nuclear magnetic resonance (NMR), using solution and
relaxation techniques. Two types of treatment were carried out in the polycarbonate
samples: crystallinity induced by solvent action and quenching. The pellets and the
samples obtained were characterized by all the techniques mentioned before using
the same conditions. The results showed that the tetrachloroethane was a better
solvent than the chloroform for crystallinity induction. The evaluation of crystallinity
induction, carried out by X-ray diffraction showed that, depending on the solvent
used, different crystals size can be obtained. Although infrared spectroscopy did not
show significant change in the samples after the treatments, the other techniques
showed to be useful to the understanding of polycarbonate behavior. The results
indicated a structural reorganization for both processes. Low field NMR demonstrated
to be effective for the evaluation of these polycarbonate samples. This technique was
particularly more accurate and faster to identify the observed changes.
Rio de Janeiro 2005

ix
Parte desta Dissertação de Mestrado foi publicada e apresentada nas seguintes
reuniões científicas:
• VIII Jornada Brasileira de Ressonância Magnética, Rio de Janeiro, AUREMN, julho
de 2004, “Estudo da influência do solvente e da temperatura no assinalamento de
materiais poliméricos à base de policarbonato por RMN em solução policarbonato
por RMN”.
• Colóquio Anual de Engenharia Química 2004, Rio de Janeiro, PEQ/COPPE/UFRJ,
dezembro de 2004, “Estudo da influencia do solvente na dinâmica molécula do
policarbonato”.
• 10th Nuclear Magnetic Resonance Users Meeting
3rd Portuguese – Brazilian NMR Meeting
1st Iberoamerican NMR Meeting, Rio de Janeiro, maio de 2005,”Evaluation of
Proton Relaxation Time for Polycarbonate Films”.

x
Índice Geral 1 - Introdução 1
2 - Revisão Bibliográfica 5
2.1 – Introdução 5
2.2 – Propriedades do policarbonato de bisfenol A 6
2.3 – Estados Físicos dos Polímeros 9
2.3.1 – Estado cristalino 9
2.3.2 – Indução de Cristalinidade 10
2.3.3 – Principais Métodos de Indução de Cristalinidade 11
2.3.3.1 – Indução Térmica 11
2.3.3.2 – Indução Por Meio de Plastificante 11
2.3.3.3 – Indução Por Meio de Solventes 12
2.3.3.4 – Indução em Misturas Poliméricas 12
2.4 – Indução de Cristalinidade do Policarbonato de Bisfenol A 13
2.4.1 – Indução Térmica 13
2.4.2 – Indução Por Meio de Plastificantes 13
2.4.3 – Indução Por Meio de Solvente 13
2.4.4 – Indução do PC em Misturas Poliméricas 14
2.5 – Técnicas de Caracterização Estrutural Aplicada ao Policarbonato 14
de bisfenol A
2.5.1 – Espectroscopia de absorção na Região do Infravermelho 15
com Transformada de Fourier
2.5.2 - Calorimetria Diferencial de Varredura 17
2.5.3 – Análise Termogravimétrica 18
2.5.4 – Difração de Raios X 20
2.5.5 – Ressonância Magnética Nuclear 22
2.5.5.1 - Fatores que influenciam no deslocamento 24
químico de polímeros
2.5.5.2 – Avaliação dos tempos de relaxação em 30
polímeros
2.5.5.2.1 - Relaxação longitudinal 30

xi
2.5.5.2.2 - Relaxação transversal 33
3 - Materiais e Métodos 35
3.1 - Considerações preliminares 35
3.2 - Materiais 35
3.2.1 - Produtos Químicos 35
3.2.2 – Equipamentos 36
3.3 – Métodos 37
3.3.1-Técnicas de Preparação das amostras 37
poliméricas
3.3.2. – Preparação dos filmes de PC em CHCl3 e TCE 38
em solução
3.3.3 - Métodos de Caracterização 38
3.3.4 – Análise Térmica 39
3.3.4.1 – Calorimetria diferencial de varredura (DSC) 39
3.3.4.2 – Termogravimetria (TG) 39
3.3.4.3.-.Espectroscopia de absorção na região 40
do infravermelho com transformada de Fourier-FTIR
3.3.4.4 – Difração de raios X 40
3.3.4.5 – Ressonância Magnética Nuclear 41
4. Resultados e Discussão 45
4.1 – Análises por RMN em solução 45
4.2 – Análise Térmica 49
4.2.1 - Calorimetria Diferencial de Varredura 49
4.2.2 - Análise termogravimétrica 52
4.3 – Análise por Infravermelho 55
4.4 - Difratometria de raios X 56
4.5 – Avaliação por relaxação nuclear 60
4.5.1 - Relaxação spin-rede 60
4.5.2 - Relaxação spin-spin 68
5 - Conclusões 75

xii
6 - Sugestões 76
7 - Bibliografia 77

xiii
Índice de Figuras Figura 1: Formação do policarbonato de Bisfenol-A 2 Figura 2: Estrutura do bisfenol A 5 Figura 3: Curva de tensão versus deformação para o PC de bisfenol A 7 Figura 4: Policarbonato Bisfenol A: a ligação C-O, marcada com um asterisco, 8 indica o ponto de rotação. O anel fenila numerado sofre uma inversão quando a ligação C-O realiza o movimento de rotação Figura 5 – Espectro de FTIR típico de policarbonato de bisfenol A, 16 Duralon A-2600 Figura 6 – Banda de absorção na região o infravermelho das deformações 17 axiais das carbonilas do PC e PCL na mistura 20/80 PC/PCL, a diferentes temperaturas Figura 7 - Cálculo do grau de cristalinidade para diferentes polietilenos: PE- LD –18 polietileno de baixa densidade, PE-LLD – polietileno linear de baixa densidade e PE-HD – polietileno de alta densidade Figura 8: Curvas TG para as membranas de PU com AgSbF6 19 Figura 9:TG e DTG da membrana de PU AgSbF6 20 Figura 10: TG e DTG da membrana 10% AgSbF6 p/p 20 Figura 11:TG e DTG da membrana 20% AgSbF6 p/p 20 Figura 12:TG e DTG da membrana 40% AgSbF6 p/p 20 Figura 13 – Perfil de raios X dos filmes vazados a partir de solução de 22 dioxana do a) PS, b) PS/PC 80/20, c) PS/PC 50/50, d) PS/PC 20/80e) filme de PC, e f) filme de PC obtido a partir do resfriamento brusco a 245ºC Figura 14 – Espectro de RMN de 13C na região do carbono da nitrila da PAN 25 (A) PAN atática; (B) PAN altamente isotática Figura 15 – Espectro de RMN de 13C da polianilina no estado prístina 27 Figura 16 – (A) Parte da cadeia do hidrocarboneto parafínico trans (t) na 28 conformação zigzag planar. (B)Projeção de Newman ilustrando o efeito γ-gauche Figura 17 - Espectro de RMN de 13C do carbono metino do poli(álcool vinílico) 29 obtido em DMSO (A) S-PVA; (B) I-PVA

xiv
Figura 18 - Espectro de RMN de 13C do carbono metino do poli(álcool vinílico) 29 obtido em D2O Figura 19: Perda de coerência dos spins 43 Figura 20: Decaimento exponencial no domínio das freqüências 43 Figura 21 – Espectro de RMN de carbono-13 do PC 4 em CDCl3 46 Figura 22 - Espectro de RMN de carbono-13 do PC 4 em TCEd4 46 Figura 23 - Estrutura da unidade monomérica do policarbonato 47 Figura 24 – Curvas térmicas de DCS das amostras de policarbonato na forma 50 de pelete Figura 25 – Curvas de DSC para as amostras de policarbonato em 51 CHCl3 a 15 % p/v Figura 26 – Curvas de DSC para as amostras de policarbonato em 51 TCE a 30 % pp Figura 27 – Curvas termogravimétricas: a) curva termogravimétrica e a 53 derivada para o pelete de PC 3, b) curva termogravimétrica e a derivada para o pelete de PC 4, c) curva termogravimétrica e a derivada para o pelete de PC 5 e d) curva termogravimétrica para os pelete dos três polímeros Figura 28 - Curvas de DTG dos PCs após indução de cristalinidade em CHCl3: 54 a) PC 3, (b) PC 4, (c) PC 5 e (d) TG dos três PCs Figura 29 - Curvas de DTG dos PCs após indução de cristalinidade em TCE: 55 a) PC 3 (b) PC4 (c) PC 5 e (d) TG dos três PCs Figura 30 – Espectros de Infravermelho para o padrão de policarbonato amorfo 56 e os PC 3, PC 4 e PC 5 após a indução de cristalinidade Figura 31 – Difratograma de raios X do filme de PC 4 obtida por termo 57 Prensagem Figura 32 – Difratograma de raios X do filme de PC 4 obtida por 57 Vazamento em CHCl3 com concentração 15% p/v Figura 33 – Difratograma de raios X do filme de PC 4 obtida por 58 Vazamento em TCE, com concentração de 15% p/v Figura 34 - Difratograma para os filmes de PC 3, PC 4 e PC 5 vazadas 59 por solução a 15% p/v em CHCl3 Figura 35 - Difratograma para os filmes de PC 3, PC 4 e PC 5 vazadas 60 Por solução a 30% p/v em TCE

xv
Figura 36 – Curva de distribuição de domínios de tempo de relaxação 64 spin-rede para o PC 3 após a indução em TCE a 30% p/v, a temperatura de 25°C (a) e 35°C (b) Figura 37 – Curva de distribuição de domínios de tempo de relaxação 66 spin-rede para o PC 4 após termo prensagem, a temperatura de 25°C (a) e 35°C (b) Figura 38 – Curvas de distribuição de domínios de tempo de relaxação 68 spin-rede para o PC5 para o pelete, a temperatura de 25°C (a) e 35°C (b) Figura 39 - Curvas de distribuição de domínios do tempo de relaxação 70 spin-spin para o pelete do PC3 (a) 25°C e (b) 35°C Figura 40 - Curvas de distribuição de domínios do tempo de relaxação 72 spin-spin para o PC4 após a termo prensagem (a) 25°C e (b) 35°C Figura 41 - Curvas de distribuição de domínios do tempo de relaxação 74 spin-spin para o PC5 após a indução de cristalinidade em TCE (a) 25°C e (b) 35°C

xvi
Índice de Tabelas
Tabela 1: Tempo de relaxação T1 e para o hidrogênio da celulose (CEL), 32
PVA e da mistura PVA/CEL
Tabela 2: Tempo de relaxação T1 e para o hidrogênio da celulose 32
Nailon – 6 (Ny6) e da mistura CEL/ Ny6
Tabela 3 – Reagentes empregados nesta Dissertação 36
Tabela 4 – Espectrômetro e condições utilizadas para obtenção dos 42
espectros de RMN de 13C em solução
Tabela 5 - Espectrômetro e condições utilizadas para obtenção do tempo de 44
relaxação longitudinal do núcleo de 1H em baixo campo
Tabela 6 - Espectrômetro e condições utilizadas para obtenção do tempo de 44
relaxação transversal do núcleo de 1H em baixo campo
Tabela 7 - Assinalamentos dos núcleos de carbono-13 policarbonato em 47
CDCl3
Tabela 8 - Assinalamentos dos núcleos de carbono-13 policarbonato em 47
TCEd4
Tabela 9 - Assinalamentos dos núcleos de carbono-13 do PC 4 em CDCl3 48
Tabela 10 - Assinalamentos dos núcleos de carbono-13 do PC 4 em TCEd4 49
Tabela 11 - Resultados de DSC de Tg, Tm, Xc e ∆Hf para as amostras de 50
Policarbonato
Tabela 12 – Principais bandas de absorção na região do infravermelho do 56
policarbonato

xvii
Tabela 13 - Resultados de DRX de 2θ, Xc e tamanho de cristalito 59
Tabela 14 - Tempo de relaxação spin – rede das amostras PC 3, PC 4 e 61
PC 5 na forma de pelete a várias temperaturas
Tabela 15: Tempo de relaxação spin – rede para o PC 3 nas temperaturas 63
de 25 ºC e 35 ºC
Tabela 16: Tempo de relaxação spin – rede para o PC 4 nas temperaturas de 65
25 ºC e 35 ºC
Tabela 17: Tempo de relaxação spin – rede para o PC 5 nas temperaturas 67
de 25 ºC e 35 ºC
Tabela 18: Tempo de relaxação spin – spin para o PC 3 pelete, prensado 69
e após indução de cristalinidade nas temperaturas de 25 ºC e 35 ºC
Tabela 19: Tempo de relaxação spin – spin para o PC 4 pelete, prensado 70
e após indução de cristalinidade nas temperaturas de 25 ºC e 35 ºC
Tabela 20: Tempo de relaxação spin – spin para o PC 5 pelete, prensado 73
e após indução de cristalinidade nas temperaturas de 25 ºC e 35 ºC

1
1 – Introdução
A utilização de produtos poliméricos em substituição aos materiais
convencionais, bem como a busca de novas tecnologias para a redução de custos
e aumento do conforto para a sociedade têm sido fatores determinantes para o
desenvolvimento e introdução de novos materiais poliméricos. A produção de
materiais poliméricos apresentou um crescimento acelerado nos últimos 30 anos
e com a versatilidade, que não é superada por nenhuma outra classe de materiais
garante que os polímeros continuarão a ter uma grande importância no futuro
[1,2]. Com o aumento da demanda e a exigência cada vez maior do mercado, o
desenvolvimento de materiais com propriedades superiores aos já existentes
tornou-se o principal objetivo da indústria polimérica. No entanto, para alcançar
este objetivo, não se faz necessário o desenvolvimento de novos monômeros,
uma vez que o potencial dos monômeros já conhecidos não foi completamente
explorado. Sendo assim, pesquisas vêm sendo realizadas com a finalidade de
modificar as estruturas dos polímeros existentes a partir da síntese de
copolímeros, indução de cristalinidade em materiais amorfos, entre outras
mudanças, de forma a alterar as propriedades dos polímeros, aumentando o
potencial de utilização desses polímeros. Tais fatos explicam o estudo das
modificações estruturais impostas ao policarbonato, neste trabalho [1,3].
A explosão no mercado de discos laser (CD) e a recente introdução no
mercado dos discos de versátil digitais (DVD) estão contribuindo para o aumento
no consumo mundial do policarbonato. Dentre outras aplicações mais comuns do
policarbonato destacam-se as embalagens plásticas, lentes oftálmicas, na
aviação e no setor automotivo, e mais recentemente em vidros a prova de balas,
o que tem incentivado um grande crescimento industrial e de mercado [4].
Policarbonato são polímeros de importante interesse comercial e tecnológico,
devido à combinação única de resistência à distorção pelo calor, ao impacto, ao
escoamento (“creep”), tenacidade, propriedades elétricas, transparência e baixa
absorção de umidade. Eles pertencem a uma classe de poliésteres formada pelas
reações de derivados de ácido carbônico com dióis aromáticos ou alifáticos. Um do

2
policarbonato de grande interesse é o produzido através de reação entre o Bisfenol-
A e o fosgênio (Figura 1)
Figura 1: Formação do policarbonato de Bisfenol-A
O policarbonato oriundo do bisfenol A é normalmente encontrado no estado
amorfo devido à rigidez da cadeia principal, causado pela presença de dois anéis
aromáticos, que diminuem a mobilidade de longas extensões da molécula [5,6].
O estado sólido dos materiais poliméricos pode ser dividido em estado
cristalino e estado amorfo. Os termos cristalino e amorfo são utilizados para indicar
regiões poliméricas ordenadas e desordenadas, respectivamente. Os polímeros
cristalinos são na maioria dos casos semicristalinos, isto é, são sistemas
heterogêneos onde regiões ordenadas estão cercadas por regiões amorfas [7].
O policarbonato pode ter a sua cristalinidade induzida, por exemplo, pelo
aquecimento a 180°C por vários dias, tratamento com solvente, pela ação de
plastificantes ou em misturas poliméricas. A indução térmica somente ocorre no
policarbonato pela exposição excessiva. A exposição realizada ao vapor de solvente
também promove a formação da fase cristalina do policarbonato [8].
A morfologia dos polímeros deve ser estudada para possibilitar o
entendimento de relações entre a estrutura, microestrutura e as propriedades do
material. A estrutura e a morfologia dos polímeros são determinadas por técnicas
diferentes, mas complementares. A estrutura pode ser determinada pela
espectroscopia na região do infravermelho (FTIR) e pela espectroscopia de
ressonância magnética nuclear (RMN) [1]. A RMN está baseada na medida da

3
absorção de radiação eletromagnética na região de radiofreqüência por moléculas
que foram colocadas na presença de um campo magnético forte de 200 até 900
MHz [9,10]. A RMN tem sido bastante difundida na caracterização de polímeros, pois
permite inferir sobre os resultados de reação de polimerização por meio de variações
dos espectros e ainda fornece dados de estrutura de cadeias, mobilidade e
compatibilidade. Dentre os parâmetros da RMN, o deslocamento químico (δ) é a
resposta direta obtida pelo espectro de RMN e sabe-se que fatores podem
influenciar na detecção deste parâmetro, como por exemplo, o solvente, a
temperatura, a interação química, os grupos substituintes e especificamente em
polímeros a massa molar, a ordenação das cadeias e a história térmica [11,12].
A RMN de baixo campo tem sido utilizada na caracterização de polímeros,
empregando-se o estudo da mobilidade molecular, por meio da determinação das
constantes de tempo da relaxação nuclear. Quando os spins são excitados até
um estado de energia mais elevado, pelo uso de radiofreqüência, eles absorvem
energia. Para retornarem ao estado de mais baixa energia, eles emitem esta
energia. O processo pelo qual o núcleo emite essa energia é chamado de
relaxação nuclear, e o tempo em que ocorre este processo é denominado de
tempo de relaxação [13,14]. Dois processos de relaxação ocorrem paralelamente,
um deles é caracterizado como spin-rede ou longitudinal, com constante de tempo
T1, apresentando um caráter mais entálpico e o outro caracterizado por uma troca
de energia entre os spins, sendo chamado de spin-spin ou transversal, mais
entrópico e tem uma constante de tempo T2.
A velocidade com que os núcleos excitados reemitem radiação de
radiofreqüência, em mecanismos de relaxação, está relacionada com a estrutura
molecular e com a dinâmica interna e global da molécula, como por exemplo, com a
sua mobilidade [15]. No tempo de relaxação longitudinal, a energia extra é
transferida para as moléculas das vizinhanças ou rede até que o sistema de spins
restaure o equilíbrio térmico, sendo, portanto um processo de relaxação global da
amostra.

4
O sistema será homogêneo quando possuir apenas um único valor de T1 para
a amostra como um todo, e heterogêneo se possuir mais de um valor. É possível
observar as interações que ocorrem, através do estudo de T1, em função de
modificações impostas ao sistema. O valor de T1 permite avaliar acerca da
ordenação estrutural do sistema polimérico, quanto mais ordenado, menos flexível e
maior será o valor deste parâmetro [15-17].
No tempo de relaxação transversal, a energia excedente é liberada para os
núcleos vizinhos. O T2 corresponde a uma perda de coerência de fase entre os
momentos magnéticos individuais, na sua precessão e, portanto, a um aumento da
entropia.
O T2 é aplicado no estudo de domínios de mobilidades diferentes em sistemas
poliméricos, assim como na heterogeneidade em homopolímeros, fornecendo
informações a respeito da sua compatibilidade, estrutura da molécula e interações.
O T2 é função do decaimento livre de indução (FID), que no estado sólido é
traduzido pela largura do sinal. Para materiais amorfos, o FID cai rapidamente
tornando a linha do espectro mais larga devido a um valor de T2 pequeno. Em
materiais cristalinos o T2 é mediano devido a uma melhor ordenação das cadeias e
em materiais flexíveis o T2 é mais longo, o FID decai mais lentamente gerando sinais
mais finos no espectro [18-21].
O objetivo desta dissertação é estudar a estrutura e dinâmica molecular de policarbonato, na forma recebida e de filmes obtidos por termo prensagem e por vazamento de solução, empregando a RMN em solução, assim como a determinação dos tempos de relaxação, visando o entendimento do comportamento deste polímero quando submetido a tratamentos de indução de cristalinidade.

5
2 – Revisão Bibliográfica
2.1 – Introdução Policarbonatos são uma classe especial de poliésteres resultantes da reação de
derivados do ácido carbônico com compostos dihidroxilados. São essencialmente
polímeros termoplásticos lineares, que podem ser divididos em alifáticos, alifático-
aromáticos e aromáticos.
Os policarbonatos alifáticos têm pouca importância comercial devido a suas
características de baixo ponto de fusão, facilidade de ser hidrolisado e em geral
não formarem fibras. Os primeiros policarbonatos com utilidade tecnológica que
foram preparados foram os aromáticos derivados de bisfenóis. O principal deles é
o policarbonato de bisfenol A (4,4’ - dihidroxi – difenil – 2,2 – propano) (Figura 2),
devido a suas características de alta estabilidade térmica, transparência e
excelentes propriedades mecânicas, entre outras.
Figura 2: Estrutura do bisfenol A
Policarbonatos foram primeiro preparados por Einhorn em 1898 por reação
de dihidroxido-benzeno, com hidroquinona e resorcinol, separadamente, com
fosgênio em solução de piridina.

6
Em 1929, W.H. Carothers e F.J. Natta prepararam vários policarbonatos
alifáticos e alifático-aromáticos usando reações com formação de anel. Esses
materiais não tiveram interesse industrial. Em 1932 Carothers, através de uma
policondensação especial, produziu um grande número de policarbonatos
alifáticos lineares, mas não conseguiu um polímero que formasse fibras. Devido
a isso, Carothers descartou os poliésteres. Contudo, em 1941 Whinfield e
Dickson anunciaram a descoberta de uma fibra de poli(tereftalato de etileno) e,
induzido pelo sucesso desse polímero, Farbenfabriken Bayer iniciou um
programa em busca de outros polímeros úteis contendo anéis aromáticos em
sua cadeia principal. Derivados de ácido carbônico foram reagidos com muitos
compostos dihidroxilados e, um desses, o bisfenol A, produziu um polímero de
muito interesse. Em 1958, os policarbonatos foram introduzidos no mercado
pela Bayer AG com o nome de Makrolon.
Independentemente, na General Electric Co nos Estados Unidos, estava
sendo feita uma pesquisa de resinas térmica e hidroliticamente estáveis, onde
também foram produzidos policarbonatos de bisfenol A, comercializado em
1960 com o nome de Lexan.
Hoje, resinas de policarbonatos comerciais estão sendo vendidas na
Alemanha pela Bayer (Makrolon), nos Estados Unidos pela General Electric
(Lexan), Mobay (Merlon) e pela Dow e no Brasil pela Policarbonato do Brasil S
A. [3, 4, 22,23].
2.2 – Propriedades do policarbonato de bisfenol A
A solubilidade dos policarbonatos aromáticos é dependente do seu estado
cristalino e da natureza dos compostos dihidroxilados aromáticos usados na
sua preparação.

7
As seguintes substâncias são solventes práticos para o policarbonato de
bisfenol A: 1,1,2,2-tetracloroetano, cloreto de metileno, cis-1,2-dicloroetileno,
clorofórmio e 1,1,2-tricloroetano.
As propriedades óticas e mecânicas desses policarbonatos são àquelas
comuns a de polímeros amorfos. O policarbonato de bisfenol A exibe uma
extraordinária estabilidade térmica, que combinada com o seu alto grau de
aromaticidade gera uma característica de ser resistente à chama.
A curva tensão deformação do policarbonato de bisfenol A (Figura 3) é
tipicamente de materiais dúcteis, consistindo de uma extensão linear reversível,
até altos valores de tensão, o que lhe confere uma boa estabilidade
dimensional. Nota-se, por essa região, que o módulo de elasticidade é
relativamente alto. Seguindo essa região observa-se o escoamento, seguido da
deformação plástica até alongamento de 120% antes da ruptura. A área sob
essa curva é uma medida da energia absorvida por unidade de volume, ou seja,
é uma medida da resistência ao impacto.
Figura 3: Curva de tensão versus deformação para o PC de bisfenol A

8
O limitado grau de cristalinidade é um fator que contribui para a alta
resistência ao impacto do policarbonato de bisfenol A. Outro fator importante
para a resistência ao impacto é o volume livre, que são espaços vazios dentro
do material que acomodam movimentos moleculares.
Após várias tentativas para a atribuição desta transição aos movimentos
internos das cadeias e grupos laterais, foi proposto um modelo molecular que
combina movimentos intramoleculares com acoplamento intermolecular [24], como
esquematizado na Figura 4.
Figura 4: Policarbonato Bisfenol A: a ligação C-O, marcada com um asterisco,
indica o ponto de rotação. O anel fenila numerado sofre uma inversão quando a
ligação C-O realiza o movimento de rotação [24]
A cadeia de policarbonato sofre mudanças conformacionais envolvendo
rotações de ligações de dois grupos carbonatos vizinhos, que mudam da
conformação trans-cis para uma trans-trans. Este movimento requer a rotação em
torno da ligação C-O, marcada com asterisco, com uma inversão do grupo fenila

9
em torno do eixo C1C4, resultando em uma flutuação de volume livre como
conseqüência da translação do grupo bisfenol A. Estes processos de relaxação se
difundem ao longo da cadeia com sucessiva mudança conformacional constituindo
um mecanismo de rápida dissipação de tensão e, portanto, rápida absorção de
energia durante o impacto. Este modelo é consistente com os requisitos
geométricos obtidos por ressonância magnética nuclear [24].
2.3 – Estados Físicos dos Polímeros Os polímeros dependendo do empacotamento e mobilidade de suas
moléculas podem existir nos seguintes estados físicos: sólido cristalino,
caracterizado pelo ordenamento tridimensional de longa distância; sólido amorfo,
ou vítreo, com ordenamento de curta distância ambos com baixa mobilidade
molecular; líquido, amorfo com alta mobilidade. Devido ao longo comprimento das
cadeias poliméricas e sua estrutura, somente as que conseguem se posicionar em
um arranjo tridimensional, formam regiões cristalinas. Isto resulta na formação de
polímeros ou totalmente amorfos, semicristalinos ou cristalinos, sendo que nestes
últimos, regiões cristalinas e regiões amorfas coexistem no estado sólido.
Praticamente todas as propriedades físicas e mecânicas são fortemente afetadas
pelo grau de cristalinidade e o estado cristalino é extremamente importante [8, 25].
2.3.1 – Estado Cristalino A cristalização consiste de um processo que envolve a ordenação de
cadeias e conseqüentemente, a passagem para um estado de menor entropia.
Para que esta transformação ocorra, a função termodinâmica de estado, G,
chamada de energia livre de Gibbs, tem que ser negativa. Para uma variação a
temperatura e pressão constantes, a Equação (1) representa a espontaneidade do
processo.
∆G = ∆H - T∆S (Equação 1)

10
onde, ∆H e ∆S são a diferença de entalpia e a diferença de entropia por
unidade repetitiva do polímero, respectivamente [7, 26].
Uma vez que, quando o sistema passa de um estado desordenado, para
um estado mais ordenado, é esperado que cadeias simétricas e cadeias com
fortes interações moleculares cristalizem.
Além dos fatores termodinâmicos, fatores cinéticos relacionados com a
flexibilidade e a mobilidade das cadeias devem ser também considerados. A
criação de uma fase ordenada tridimensional a partir de uma fase desordenada
consiste em um processo de dois estágios. No primeiro estágio ocorre a formação
de núcleos estáveis, pela ordenação paralela das cadeias, estimulada pelas forças
intermoleculares. O segundo estágio, consiste no crescimento de regiões
cristalinas, cujo tamanho é determinado pela adição de outras cadeias aos
núcleos [27]. Os polímeros podem cristalizar basicamente a partir de soluções
diluídas, é possível a obtenção de cristais bem definidos chamados de cristais
únicos, uma vez que a chance de uma cadeia ser incorporada em mais de um
cristal é pequena. Estes cristais apresentam pequenas dimensões e são formados
por lamelas finas, onde as cadeias longas encontram-se dobradas [27]. Na
cristalização a partir de soluções concentradas, a característica básica continua
sendo o cristalito na forma lamelar com superfície ou interfaces amorfas, mas o
modo como são formados pode ser diferente [27].
2.3.2 – Indução de Cristalinidade A indução de cristalinidade de polímeros normalmente amorfos é
importante, pois torna possível a obtenção de materiais poliméricos com elevada
resistência mecânica e química, cuja utilização vai depender da aplicação que se
deseja [28].

11
Os polímeros cristalizáveis podem ser divididos em três classes de acordo
com a facilidade de cristalização em: polímeros que cristalizam espontaneamente;
polímeros que são cristalinos ou amorfos, dependendo do tratamento dado, e
polímeros difíceis de cristalizar e, normalmente, produzidos na forma amorfa,
como o policarbonato do bisfenol A [29].
O policarbonato se encontra em uma classe onde os polímeros são difíceis
de cristalizar. A estrutura do policarbonato apresenta anéis aromáticos na cadeia
principal, o que causa maior rigidez, diminuindo a flexibilidade, desfavorecendo a
cristalização [30].
2.3.3 – Principais Métodos de Indução de Cristalinidade Os principais métodos de indução de cristalinidade são: por meio de
solvente, por meio de plastificante, por meio de mistura ou por meio térmico.
2.3.3.1 – Indução Térmica O tratamento térmico, no qual o polímero é aquecido até uma determinada
temperatura, mantido por algum tempo e então resfriado é chamado de
recozimento. Com o recozimento consegue-se estabilidade dimensional, melhoria
nas propriedades físicas e redução ou remoção de tensões residuais e de
defeitos. Este processo é geralmente empregado em polímeros semicristalinos
gerando uma melhora na perfeição dos cristais. No entanto, alguns polímeros
normalmente amorfos podem desenvolver a cristalinidade após longo período de
recozimento [8, 31].
2.3.3.2 – Indução Por Meio de Plastificante
A cristalização de polímeros normalmente amorfos pode ser facilitada pela
adição de plastificantes, que se dispõem entre as cadeias poliméricas aumentando

12
a sua mobilidade. Com a facilidade de movimentos das cadeias, o impedimento
espacial que restringe o movimento e a reorientação das moléculas do polímero é
menor, permitindo adotar a conformação energeticamente mais favorável,
resultando em estruturas cristalinas [5,32].
2.3.3.3 – Indução Por Meio de Solventes
Um dos métodos mais utilizados na cristalização de polímeros amorfos, que
são capazes de cristalizar, envolve a ação de determinados líquidos orgânicos de
baixa massa molar, como os solventes.
A capacidade de cristalização sob a ação de um solvente, que certos
polímeros apresentam, como o poli(tereftalato de etileno) (PET) e o PC, é
atribuída a habilidade do solvente em aumentar a mobilidade do polímero e com
isso diminuir a temperatura de transição vítrea (Tg). A indução da cristalinidade por
solventes pode ser feita por imersão do polímero no solvente ou por exposição do
polímero ao vapor de solvente [33].
2.3.3.4 – Indução em Misturas Poliméricas
A mistura de dois ou mais componentes poliméricos se tornou uma técnica
bastante importante para a redução de custos e melhoria das propriedades dos
materiais poliméricos. Foi verificado que a indução de cristalinidade pode ocorrer
em um dos componentes da mistura polimérica pela ação do outro polímero. A
adição de um polímero cristalizável pode causar diferentes efeitos na cristalização
do outro polímero presente na mistura, que pode ser amorfo ou semicristalino. A
mistura de maior interesse é a que promove a cristalinidade de um polímero
amorfo como o PC [8].

13
2.4 – Indução de Cristalinidade do Policarbonato de Bisfenol A
A indução da cristalinidade do policarbonato pode ser obtida através de
tratamento térmico, por solventes, por plastificantes e por misturas poliméricas.
2.4.1 – Indução Térmica
A cristalização térmica do policarbonato é muito difícil, sendo necessário um
aquecimento por um longo período. Este método de indução não é econômico
industrialmente e o material polimérico é prejudicado, sofrendo um processo de
degradação devido ao longo tempo de aquecimento necessário para alcançar um
pequeno grau de cristalinidade [5,30].
2.4.2 – Indução Por Meio de Plastificantes
Segundo Gallez [34] e Cheung [35], a ação de plastificantes, como o éster
trideciloctila (TMDO), torna a cinética de cristalização do PC muito mais rápida,
assim como a adição de 15 a 60 % de fosfato de tritolila (TTF), geraram uma
cristalinidade em torno de 20 % no PC.
2.4.3 – Indução Por Meio de Solvente
Ware e colaboradores [33] mostraram que a cristalinidade no PC foi
induzida mais rapidamente quando placas de PC aquecidas a 145ºC foram
imersas em acetona e em tetracloreto de carbono, o que não aconteceu no etanol.
E análises feitas por caloria diferencial de varredura (DCS) mostraram uma
cristalinidade próxima de 17% em clorofórmio e de 20% em acetona.

14
2.4.4 – Indução por meio de misturas poliméricas
Amorim [36] em seu estudo mostrou que a adição de poli(óxido de etileno)
(PEO) ou poli(óxido de propileno) (PPO) ao policarbonato em misturas binárias, no
estado fundido, provoca a indução de cristalinidade do PC. Esta indução pode ser
comprovada através de análises de DSC, quando ao PC foi adicionado o PEO ou
o PPO em teores de 10 a 30%, já que teores menores do que 10% não induziram
a cristalinidade no PC. 2.5 – Técnicas de Caracterização Estrutural Aplicada ao Policarbonato
de bisfenol A
O conhecimento da estrutura química de um material é de grande
importância para o seu estudo no sentido de aumentar sua eficiência e possibilitar
o desenvolvimento de novas tecnologias. No caso da pesquisa de filmes de PC
induzidos a cristalinidade por solvente e termo prensagem é particularmente
importante determinar a mobilidade molecular deste material antes e depois da
indução.
De acordo com o observado na literatura, algumas técnicas são aplicadas
no estudo da caracterização do PC como: a espectroscopia de absorção na região
do infravermelho (FTIR), difração de raios X, análise termogravimétrica (TG),
calorimetria diferencial de varredura (DSC), ressonância magnética nuclear e
outras. A RMN de alto e baixo campos serão utilizadas como ferramentas neste
estudo.

15
2.5.1 – Espectroscopia de absorção na Região do Infravermelho com Transformada de Fourier
A espectroscopia de absorção na região do infravermelho tem sido muito
utilizada na caracterização e identificação de polímeros por ser um método fácil e
rápido de ser realizado. Ela possui a capacidade de determinar: a composição da
estrutura do polímero, os grupos funcionais, as interações com o solvente
podendo também ser usada para a determinação do grau de cristalinidade de
polímeros [37, 38].
Um espectro de emissão ou absorção surge quando moléculas sofrem
transições envolvendo movimentos localizados de grupos de átomos, dando
origem a bandas de absorção em freqüências características, que são tabeladas
de acordo com as características de cada grupo. Embora as faixas sejam bastante
definidas, a freqüência ou comprimento de onda em que um determinado grupo
vai absorver depende de seu ambiente na molécula. A posição exata da banda de
absorção revela detalhes importantes da estrutura, assim como as mudanças nos
contornos das bandas [38].
Delpech e colaboradores [39] analisaram amostras comercias de
policarbonato de bisfenol A e não observaram nenhuma diferença quando
comparados ao Duralon A-2600. Conseqüentemente todos apresentaram o
mesmo espectro de FTIR. A Figura 5 mostra o espectro na região do
infravermelho típico de uma amostra de policarbonato de bisfenol A.

16
4000 3000 2000 1000 0
0
1
2
3
4
Abs
orvâ
ncia
(%)
Número de onda em cm-1
1775 1232
3045
Figura 5 – Espectro de FTIR típico de policarbonato de bisfenol A, Duralon A-2600
[39]
Varnell e colaboradores [40] utilizaram-se da FTIR para acompanhar a
indução da cristalinidade do policarbonato por poli(ε-caprolactona) (PCL) como
mostra a Figura 6 do FTIR na região de absorção da carbonila de filmes de
PC/PCL 20/80 % molar. O PC contido na mistura se encontra no estado amorfo,
evidenciado pela presença de uma banda larga em 1775 cm-1, referente à
deformação axial da carbonila. Utilizando uma célula de aquecimento, as amostras
foram aquecidas da temperatura ambiente até 60ºC. Quando a temperatura é
elevada até 50ºC nenhuma mudança na região da deformação da carbonila é
observada, indicando que o PC continua no estado amorfo. No entanto, entre
50ºC e 55ºC, o PC começa a cristalizar em função da indução causada pelo PCL,
observado pela contribuição a 1768 cm-1, que se torna mais evidente com o
aumento de temperatura até 60ºC. Um comportamento contrário é observado para
o PCL que é cristalino e se torna amorfo com o aumento da temperatura, pelo
deslocamento da banda de 1727 cm-1 para 1724 cm-1 [40].

17
C=O PC
C=O PCL
Figura 6 – Banda de absorção na região do infravermelho das deformações
axiais das carbonilas do PC e PCL na mistura 20/80 PC/PCL, a diferentes
temperaturas [40]
2.5.2 - Calorimetria Diferencial de Varredura
A calorimetria diferencial de varredura é um método dinâmico bastante
utilizado na detecção de transições térmicas que ocorrem durante o aquecimento
e resfriamento de materiais. Na área de polímeros, permite detectar e quantificar a
cristalinidade, a transição vítrea, processo de cristalização entre outros [41, 42].
Como o DSC mede as mudanças de energia calorífica em uma substância,
quando submetida a um programa pré-determinado de temperatura, é possível
detectar e medir transições físicas acompanhadas de modificação na região
calorífica, como a temperatura de transição vítrea (Tg) e a temperatura de fusão
cristalina (Tm) e permite ainda o cálculo do grau de cristalinidade de polímeros por
meio do calor de fusão, ∆Hf [43, 44].

18
A partir dos valores de ∆Hf é possível calcular o grau de cristalinidade (χc)
de vários polímeros, como por exemplo, do polietileno com distintos graus de
cristalinidade (Figura 7). No entanto, algumas dificuldades são encontradas neste
método, como a indefinição da linha base e a possível cristalização do material
durante a corrida de DSC [45].
Figura 7 - Cálculo do grau de cristalinidade para diferentes polietilenos: PE-
LD – polietileno de baixa densidade, PE-LLD – polietileno linear de baixa
densidade e PE-HD – polietileno de alta densidade [45]
2.5.3 – Análise Termogravimétrica (TG)
A técnica da TG consiste na avaliação da perda de massa ocorrida em uma
determinada amostra em função do aumento da temperatura. A TG mede a
quantidade e velocidade de mudanças na massa dos materiais, em função do
tempo e da temperatura, em atmosfera controlada. No caso de polímeros, a TG é
muito utilizada para determinar o grau de umidade, determinação da pureza, teor
de voláteis e resíduos de materiais poliméricos, estabilidade térmica dos polímeros
e no estudo cinético de uma reação.
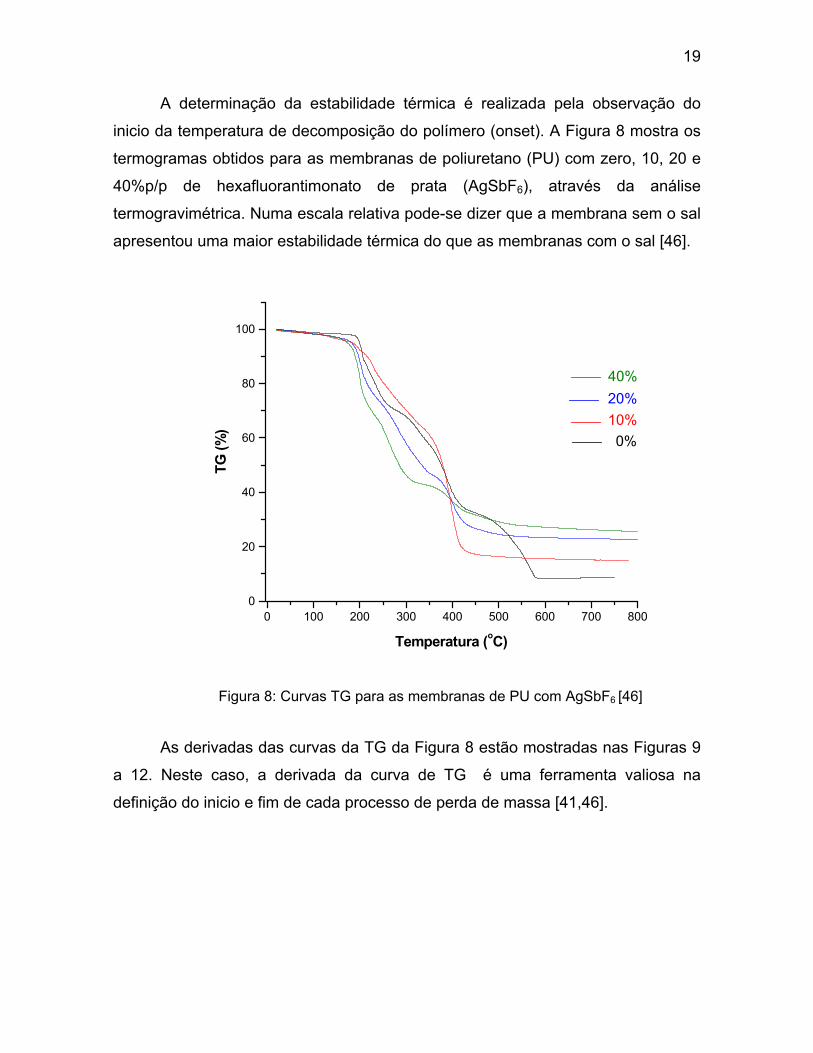
19
A determinação da estabilidade térmica é realizada pela observação do
inicio da temperatura de decomposição do polímero (onset). A Figura 8 mostra os
termogramas obtidos para as membranas de poliuretano (PU) com zero, 10, 20 e
40%p/p de hexafluorantimonato de prata (AgSbF6), através da análise
termogravimétrica. Numa escala relativa pode-se dizer que a membrana sem o sal
apresentou uma maior estabilidade térmica do que as membranas com o sal [46].
0 100 200 300 400 500 600 700 8000
20
40
60
80
100
0%
20%
TG (%
)
Temperatura (oC)
40%
10%
Figura 8: Curvas TG para as membranas de PU com AgSbF6 [46]
As derivadas das curvas da TG da Figura 8 estão mostradas nas Figuras 9
a 12. Neste caso, a derivada da curva de TG é uma ferramenta valiosa na
definição do inicio e fim de cada processo de perda de massa [41,46].

20
0 100 200 300 400 500 600 700 8000
20
40
60
80
100
TG da membrana de PU com 0% p/p de AgSbF6
Temperatura (oC)
TG (%
)
-0,16
-0,14
-0,12
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
0,10
170oC
DTG
0 100 200 300 400 500 600 700 8000
20
40
60
80
100
Temperatura (oC)
TG (%
)
-0,16
-0,14
-0,12
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
0,10
110oC
TG da membrana de PU com 10% p/p de AgSbF6
DTG
Figura 9:TG e DTG do filme PU AgSbF6[46] Figura 10: TG e DTG do filme 10% AgSbF6p/p [46]
0 200 400 600 8000
20
40
60
80
100
TG da membrana de PU com 20% p/p de AgSbF6
Temperatura oC
TG (%
)
-0,16
-0,14
-0,12
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
0,10
150 oC
DTG
0 200 400 600 8000
20
40
60
80
100
TG da membrana de PU com 40% p/p de AgSbF6
Temperatura (oC)
TG (%
)
-0,16
-0,14
-0,12
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
0,10
140oC DTG
Figura 11:TG e DTG do filme 20% AgSbF6 p/p Figura 12:TG e DTG do filme 40% AgSbF6 p/p [46]
2.5.4 – Difração de Raios X A difração de raios X é uma técnica amplamente utilizada para a
caracterização de polímeros, apresentando informações sobre o estado cristalino
e amorfo do material, além de ser o único método de determinação direta da
ordem tridimensional do polímero [47]. Os raios X são fótons de alta energia tendo
comprimento de onda pequeno, que interagem com elétrons. Quando os raios
incidem em uma amostra, alguns elétrons são absorvidos, outros são transmitidos
e outros são espalhados, devido à interação com elétrons do material. Tal
interação é função do ângulo de espalhamento, designado por 2θ [8].

21
A lei de Bragg estabelece a condição essencial para que ocorra a difração e
é dada pela Equação 2 [47]:
nλ = 2d senθ (Equação 2)
onde n é a ordem da reflexão e é igual ao número de comprimentos de
onda contidos na diferença de caminho entre os raios espalhados por planos
adjacentes; λ é o comprimento de onda dos raios X; d é a distância interplanar e θ
é metade do ângulo de espalhamento [47].
O grau de cristalinidade pode ser calculado através da intensidade dos
picos do difratograma (Equação 3) ou pela área sob os picos (Equação 4), como
mostrado abaixo [48]:
Xc = 1 – (Ia / I0a) (Equação 3) e Xc = Ac / (Aa + Ac) (Equação 4)
onde Ia e I0a são, respectivamente, as intensidades dos picos da amostra cristalina
e do polímero totalmente amorfo, Aa e Ac são as áreas correspondentes às frações
cristalina e amorfa do polímero [48].
A análise por raios X da cristalinidade do PC em misturas com PS foi
estudada. A Figura 13 mostra os perfis dos raios X para as misturas de PS (a)
PS/PC (b, c e d) a diferentes proporções, filme de PC (e) e filme de PC obtido por
resfriamento brusco (f). Foi observado que tanto o filme de PC quanto as blendas
de PS/PC nas diferentes proporções apresentaram um pico de cristalinidade em
2θ = 18º. Foi observado, ainda, que o pico de cristalinidade do PC e de suas
misturas com PS tornou-se muito fino com o aumento da fração molar do PC na
mistura, e para a proporção 50/50 o pico apresentou a mesma largura
apresentada para o filme de PC [49].
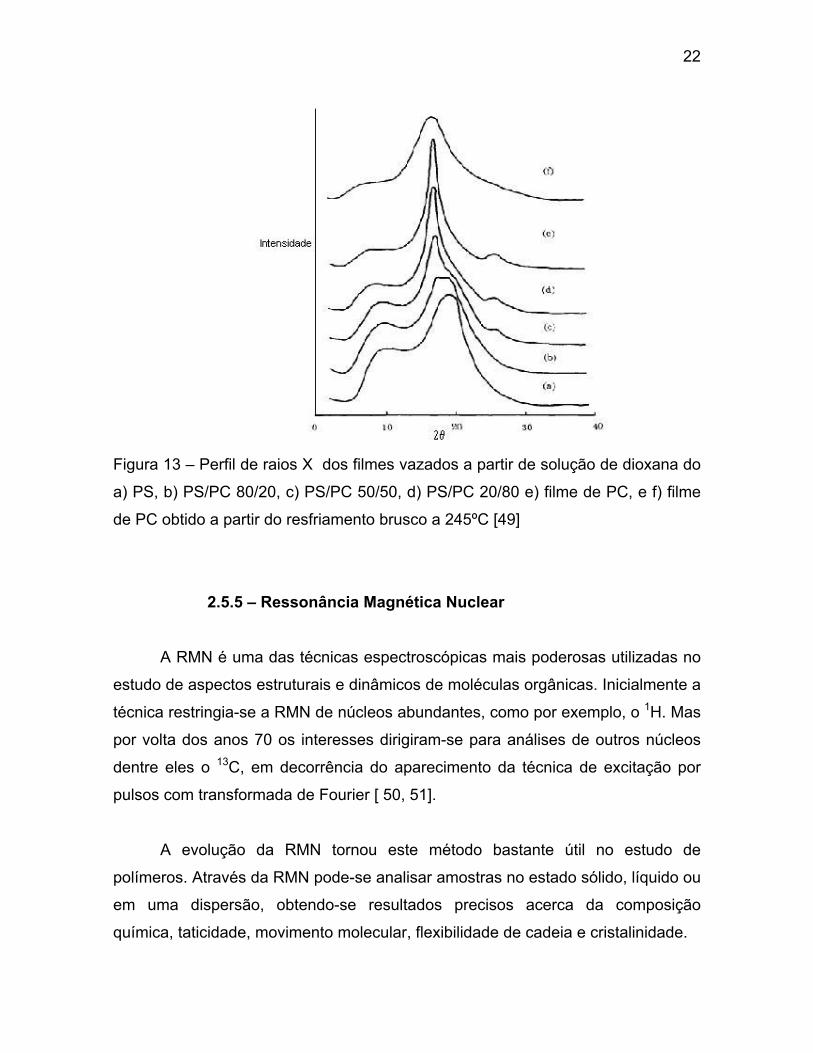
22
Figura 13 – Perfil de raios X dos filmes vazados a partir de solução de dioxana do
a) PS, b) PS/PC 80/20, c) PS/PC 50/50, d) PS/PC 20/80 e) filme de PC, e f) filme
de PC obtido a partir do resfriamento brusco a 245ºC [49]
2.5.5 – Ressonância Magnética Nuclear
A RMN é uma das técnicas espectroscópicas mais poderosas utilizadas no
estudo de aspectos estruturais e dinâmicos de moléculas orgânicas. Inicialmente a
técnica restringia-se a RMN de núcleos abundantes, como por exemplo, o 1H. Mas
por volta dos anos 70 os interesses dirigiram-se para análises de outros núcleos
dentre eles o 13C, em decorrência do aparecimento da técnica de excitação por
pulsos com transformada de Fourier [ 50, 51].
A evolução da RMN tornou este método bastante útil no estudo de
polímeros. Através da RMN pode-se analisar amostras no estado sólido, líquido ou
em uma dispersão, obtendo-se resultados precisos acerca da composição
química, taticidade, movimento molecular, flexibilidade de cadeia e cristalinidade.

23
A RMN é sensível às formas cristalinas e é muito usada nos estudos de
polimorfismos, assim como na transição de fase. A RMN tem um substancial valor
para o estudo de materiais amorfos e heterogêneos, já que esta espectroscopia
compreende diferentes seqüências de pulsos e determinação de relaxação
nuclear que pode discriminar a mobilidade molecular dos componentes em
diferentes domínios. A RMN pode prover, via tempo de relaxação, informações
detalhadas sobre a mobilidade em nível molecular sobre uma ampla faixa de
velocidade de relaxação. O tempo de relaxação longitudinal ou spin- rede (T1) e o
tempo de relaxação transversal ou spin-spin (T2) provém informações sobre a
relaxação em escalas da ordem de milisegundos (ms) a segundos (s) [52, 53].
O fenômeno da RMN fundamenta-se na absorção seletiva de radiação
eletromagnética, na faixa de radiofreqüência, por um núcleo quando ele é
submetido a um campo magnético externo (B0) e nele é aplicada uma freqüência
igual a sua freqüência de precessão, que ao retirá-la, conduz o núcleo ao seu
estado fundamental, emitindo uma energia no domínio da radiofreqüência
(processo de relaxação). O sinal gerado no processo de excitação é armazenado
sob a forma de um decaimento de indução livre (FID).
Na RMN, a condição de ressonância é dada pela equação de Larmor
(Equação 5):
2πν = γB0 (Equação 5)
ν - freqüência de Larmor (faixa de MHz)
γ - constante magnetogírica (rad.T-1.s-1)
B0 - campo magnético externo (Tesla, T)
Todos os núcleos nos experimentos pulsados são excitados ao mesmo
tempo e cada núcleo possui um tempo de relaxação diferente, de modo que o FID

24
é um somatório dos decaimentos de todos os núcleos, ficando praticamente
incompreensível. A compreensão do espectro de RMN é possível quando o
interferograma no domínio do tempo (FID) é transformado para o domínio de
freqüência, por um processo matemático, conhecido como transformada de
Fourier.
Deve ser ressaltado que esse processamento matemático permitiu um
avanço crescente na força do campo magnético externo (B0) e, por conseqüência,
uma melhor resolução espectral, podendo-se assim estudar sólidos, polímeros e
biomoléculas.
Os diferentes núcleos geram uma informação direta no espectro, que está
relacionada com a estrutura química de cada núcleo na molécula, devido à
absorção de freqüência que cada tipo de núcleo apresenta em função do ambiente
químico a que pertence, sendo denominado de deslocamento químico (δ). A
unidade do deslocamento químico é dada em parte por milhão (ppm) e sempre
medida em relação a um padrão [54,55].
2.5.5.1- Fatores que influenciam no deslocamento químico de polímeros
ABRAHAM e colaboradores [56] realizaram estudos sobre o efeito de grupo
substituinte em copolímeros de aril-etér (PEEK) por ressonância do núcleo de 13C. Nestes copolímeros o núcleo de carbono quaternário sofre variação no seu
deslocamento quando grupos substituintes como SO3 e SO3H são introduzidos
nas posições orto ou meta. O grupo SO3 causa variações no deslocamento
químico para freqüências maiores, quando este substituinte está na posição
orto, enquanto que o grupo SO3H causa variação no deslocamento químico
para freqüências menores quando substituído na mesma posição. Entretanto,
quando esses substituintes são introduzidos na posição meta o inverso ocorre.

25
KATSURAYA e colaboradores [57] mostraram que o processo de polimerização
causa efeito na taticidade da poliacrilonitrila (PAN). Quando a polimerização da
PAN é via radicalar, gera apenas polímeros atáticos independentemente da
temperatura e do solvente usado, e na polimerização por radiação γ o material
se torna mais rico na forma isotática. O efeito da polimerização pode gerar
microestruturas diferentes que podem ser detectadas pelas seqüências
adequadas que vão desde tríades até pêntades (Figura 14). A Figura 14 mostra
os espectros da região da nitrila para PAN obtida por tipos de polimerizações
diferentes.
Figura 14 – Espectro de RMN de 13C na região do carbono da nitrila da PAN
(A) PAN atática: onde de 1 a 3 forma isotática, 4 a 6 heterotárica e 7 a 9
sindiotática; (B) PAN altamente isotática [57]

26
HANSEN e colaboradores [58] estudaram o assinalamento do polietileno
(PE), com ênfase na estimativa da distribuição de ramificações. Este foi
polimerizado com um catalisador de Cr(II)/SiO2 modificado com t-butil lítio de
modo a gerar um polímero com ampla faixa de distribuição de massa molar.
Nesta polimerização foram formadas frações de poliolefinas de baixa massa
molar e alto grau de ramificações. O assinalamento dessas ramificações foi
feito de acordo com o descrito por De POOTER e colaboradores [59], o
solvente foi o orto-diclorobenzeno, solvente normalmente empregado, pois
propicia formação de interação dipolar com o polímero e, além disso, seus
sinais estão em região distinta da região do polímero, permitindo assim, um
assinalamento preciso. O sinal em 11,05 ppm é típico de ramificação tipo etil. O
sinal em 40 ppm é devido à ramificação do tipo butil, enquanto que o sinal em
38 ppm é devido à ramificação do tipo hexil. Não foi encontrada ramificação tipo
metil, devido a não detecção de sinal em 19 ppm referente ao grupo metila.
Foram ainda detectados sinais de ramificação de cadeia longa, maiores do que
8 átomos de carbonos não sendo possível distingui-los até o presente
momento.
NI e colaboradores [60] assinalaram o espectro de RMN de 13C da
polianilina em solução de dimetilformamida (DMF) no estado prístina e
detectaram, pelo menos, 25 picos de ressonância que estão resolvidos no
espectro de solução, enquanto que apenas 5 picos aparecem no espectro do
estado sólido. Esta diferença foi atribuída a menor mobilidade molecular da
polianilina no estado sólido, que é resultante da rigidez da cadeia e das fortes
interações intermoleculares. O conjunto de picos de ressonância entre 110 –
123 ppm é muito informativo. O sinal centrado em 124,5 ppm é derivado dos
carbonos hidrogenados no anel benzênico e os sinais 137,7 ppm e 158,6 ppm
são derivados dos carbonos hidrogenados e não hidrogenados do anel quinona,
respectivamente (Figura 15).

27
Figura 15 – Espectro de RMN de 13C da polianilina no estado prístina [60]
Segundo Toneli [61], Hobson e Feast [62], o deslocamento químico em
RMN de 13C é dependente da estereosequência observada em hidrocarbonetos
poliméricos, que pode ser entendido com base no efeito γ. Dentro da
estereoquímica existe um efeito denominado γ-gauche, onde a distância entre o
carbono observado (oC) e o carbono em posição γ (γC) em relação ao carbono
observado, assim como as possíveis orientações do γ-gauche, influirão na
microestrutura e no deslocamento químico do polímero para valores menores,
devido à assimetria da molécula. Portanto, existe uma relação direta entre o
efeito γ substituinte no deslocamento químico da RMN e qualquer variação na
microestrutura do polímero que afete sua conformação local será refletida no
deslocamento químico originado pelo efeito γ-gauche. A Figura 16 mostra a
influência do efeito γ-gauche no estudo da RMN.

28
Sem efeito-γ (φ2= t) Efeito -γ (φ2= g)
CH2CH2
CH2CH2
CH2CH2
CH2γ
β
α
δβ
αφ0 φ1
φ2 φ3φ4
0 1 2 3 4β
A
H CH3
H CH3
H
H
H
H
H
HH
HCH2
CH2γ
φ
H CH3
H CH3
H
H
H
H
H
HH
HCH2
CH2
H
γφ
B
Figura 16 – (A) Parte da cadeia do hidrocarboneto parafínico trans (t) na
conformação zigzag planar e (B)Projeção de Newman ilustrando o efeito γ-
gauche [55,56].
KATSURAYA e colaboradores [63], mostraram no seu trabalho que foi
possível determinar o assinalamento do carbono metino do poli(álcool vinílico)
(PVA) em héxades e héptades respectivamente, sendo os assinalamentos
quantitativos conforme a taticidade do PVA (atático e sindiotático). O PVA
altamente isotático mostrou uma discrepância entre as intensidades observadas
e calculadas, devido à dificuldade de se determinar o mecanismo de
polimerização. A Figura 17 mostra o espectro do carbono metino para os PVAs
medidos em DMSO e a Figura 18 mostra o espectro para o polímero atático em
D2O. Os espectros são divididos em três partes, mm (sinais 1 – 8), mr (sinais 9
– 14) e rr (sinais 15 – 17). Com a melhoria na resolução espectral, os espectros
de RMN de 13C obtidos nesse trabalho incluem as absorções que não tinham
sido detectadas anteriormente.

29
Figura 17 - Espectro de RMN de 13C do carbono metino do poli(álcool vinílico)
obtido em DMSO (A) S-PVA; (B) I-PVA [63]
Figura 18 - Espectro de RMN de 13C do carbono metino do poli(álcool vinílico)
obtido em D2O [63]
NYDEN e colaboradores [64] estudaram as estruturas conformacionais de
cadeias contendo defeito na região cristalina do polipropileno isotático (iPP).
Foram preparados, por recozimento (annealing), polipropilenos com defeitos na
rede cristalina, sendo uns dos defeitos a introdução de um grupo butileno e um
outro foi a geração de um defeito regioespecífico. Foi observado que em todos
os casos a cadeia assumiu a forma da hélice 31, indicando que estes defeitos
não influenciam na formação cristalina da cadeia.

30
2.5.5.2 – Avaliação dos tempos de relaxação em polímeros
2.5.5.2.1 - Relaxação longitudinal
Segundo McCall [65], o tempo de relaxação spin-rede é inversamente
proporcional ao tempo de correlação molecular (τC). O que significa que quando o
τC é curto, devido a uma rápida troca de posição dos núcleos o T1 é longo. O
estudo sobre o T1 de polímeros mostrou que este tempo é dependente da
distribuição do τC. Os valores de T1 variam entre 10-2 a 10 s, e pode ser observado
que movimentos locais e segmentais da cadeia macromolecular freqüentemente
possuem movimentos moleculares significativamente diferentes, propiciando a
detecção de domínios de mobilidade distintas [66].
O estudo sobre a relaxação spin-rede em polímeros vítreos realizados por
Schaefer e colaboradores [67] mostrou que a relaxação spin-rede influencia na
detecção dos núcleos. Foi observado que o carbono não hidrogenado apresenta
T1 longo e, portanto, não são detectados quando o intervalo de tempo usado na
aquisição do espectro é curto, ou seja, apenas uns poucos segundos. Em
materiais amorfos como o poli(metacrilato de metila), poli(cloreto de vinila) e
outros pelo menos dois valores de T1 para os núcleos de 13C e 1H são observados.
Um referente a grupos com maior mobilidade (T1 menor) e o outro a uma fase
mais rígida (T1 maior) devido às ligações físicas entre as moléculas do polímero.
Estudos envolvendo a relaxação polimérica entre a estrutura e a
propriedade feitas por Parker e colaboradores [68] mostra que o tempo de
relaxação T1 informa sobre a morfologia e o movimento molecular dos diferentes
sistemas poliméricos, já que este parâmetro é sensível à variação de mobilidade
em sistemas heterogêneos. Tem sido mostrado que para polímeros amorfos o T1
é usado para medir a heterogeneidade dos movimentos e da morfologia. As
informações obtidas por T1 mostram que domínios de mobilidade diferentes
apresentam tempo de relaxação diferente. Corroborando o trabalho anterior na

31
avaliação de polímeros amorfos e heterogêneos pelo menos dois valores distintos
de T1 são detectados. E qualquer modificação na morfologia do material pode ser
determinada por este parâmetro.
Segundo Vanderhart e colaboradores [69] a heterogeneidade em polímeros
advém das diferenças na organização molecular (cristalinos e não-cristalinos), na
mobilidade (polímeros vítreos e borrachosos) ou até mesmo na composição
(copolímeros e misturas). A determinação do tempo de relaxação T1 para estes
sistemas é usada para monitorar as mudanças de mobilidade em face das
mudanças de reorganização molecular. A formação de estrutura heterogênea
numa faixa de 2 a 50 nm é detectada por T1. A informação morfológica
usualmente deduzida mostra que os valores de T1 curtos apresentam uma alta
eficiência de difusão entre os spins, sendo, portanto atribuído a domínios de maior
mobilidade e o inverso é atribuído a domínios de menor mobilidade.
Estudos da interação de polímeros amorfos e celulósicos realizados por
Masson e colaboradores [70] foram acompanhados pelo tempo de relaxação
spin-rede. É sabido que a relaxação de hidrogênios vizinhos em uma molécula
é praticamente idêntica, devido ao acoplamento dipolar. Em contraste, os
hidrogênios afastados deste ambiente relaxam independentemente um dos
outros. Isto é comum em polímeros heterogêneos. Em uma mistura
homogênea, por exemplo, na escala de caracterização do tempo de relaxação
T1 dos hidrogênios é observada uma média das taxas de relaxação dos
hidrogênios constituintes dos polímeros, assim um único tempo de relaxação é
determinado. Entretanto, para sistemas heterogêneos isto não ocorre. As
Tabelas 1 e 2 mostram os tempos de relaxação para os sistemas PVA /
CELULOSE e NYLON – 6 / CELULOSE, respectivamente. Para o primeiro
sistema observa-se que os tempos de relaxação para ambos os polímeros é o
mesmo, mostrando a homogeneidade do sistema. Ao contrário, o segundo
sistema mostra valores diferentes para os hidrogênios de ambos os polímeros,
evidenciando a heterogeneidade do sistema.

32
Tabela 1: Tempo de relaxação T1 e para o hidrogênio da celulose (CEL), PVA e da
mistura PVA/CEL
T1 (s)
Mistura CEL PVA Teórico
0:100 ----- 1,8 1,8
25:75 1,6 1,7 1,5
50:50 1,3 1,2 1,3
75:25 1,1 1,1 1,1
100:0 0,96 ------ 0,96
Tabela 2: Tempo de relaxação T1 e para o hidrogênio da celulose Nailon – 6 (Ny 6)
e da mistura CEL/ Ny6
T1 (s)
Mistura CEL Ny6 Teórico
0:100 ---------- 0,59 0,59
25:75 0,84 0,60 0,64
50:50 0,94 0,60 0,70
75:25 0,98 0,61 0,80
100:0 0,96 ---------- 0,96
Um outro estudo realizado por Zhao e colaboradores [71], sobre o tempo de
relaxação spin-rede de policarbonatos, mostrou que quando a rotação do grupo
fenileno é lenta, as relaxações advêm do movimento livre deste grupo, que está
sempre presente e é o fator dominante neste processo de relaxação,.o que
corrobora o alto valor da resistência ao impacto deste material. Os valores
determinados para T1 dos hidrogênios do anel aromático foram os mesmos, o que
indica que este grupo controla a relaxação.

33
2.5.5.2.2 - Relaxação transversal
O tempo de relaxação spin-spin revela um movimento de rotação dos spins
no plano XY devido a uma perda de coerência de fase, que fará com que o
decaimento tenha uma constante de tempo caracterizada como T2. De acordo
com a literatura [66], é sabido que o T2 de materiais rígidos ou cristalinos é
muito curto, enquanto que em materiais de maior mobilidade é mais longo. No
caso de sistemas poliméricos este parâmetro pode prover informações sobre a
dinâmica molecular de cadeias poliméricas, na escala de MHz.
No estudo feito por Charleibuy [72] foi predito que o tempo de relaxação T2
é função da massa molar, flexibilidade da cadeia, ligações cruzadas, volume
livre e viscosidade, porque estes fatores afetam profundamente a morfologia e a
mobilidade do polímero. Foi observado que o T2 reflete as características das
ligações cruzadas, devido aos entrelaçamentos físicos que reduzem
drasticamente a mobilidade molecular.
No estudo realizado por Zhang e colaboradores [73], a verificação da
miscibilidade e estrutura de fase de uma mistura do poli(vinil-fenol) (PVPh) e PEO
foi acompanhada pelas avaliações dos tempos de relaxação do hidrogênio. Foi
observado que pelo tempo de relaxação spin-spin o PVPh apresentou um valor de
T2 curto (13 µs), que é condizente com o seu estado vítreo. Já o PEO apresentou
duas componentes para o T2, uma curta (20 µs), referente à região cristalina e a
outra longa (350 µs), referente à região amorfa. As misturas apresentaram dois
valores, um mais curto e que variou de 17 a 22 µs e um mais longo de acordo com
a composição. A interpretação dos resultados de T2 mostrou que nas misturas o
valor mais curto deve-se a interação entre ambos os polímeros e o valor mais
longo devido à parte da mistura mais móvel em função de uma plastificação. A
partir destes dados pode-se concluir que as misturas estudadas apresentaram
miscibilidade na faixa de detecção de T2 entre 200 a 300 Å.

34
Berriot e colaboradores [74] usaram o T2 para a caracterização da
densidade de ligações cruzadas em um sistema formado por elastômeros e carga.
Foi observado que quanto melhor for a dispersão da carga na matriz polimérica,
mais aumenta a interface do polímero com a carga fazendo com que o tempo de
relaxação T2 diminua devido à redução da mobilidade molecular gerada, neste
caso, pelo aumento das densidades de ligações cruzadas. Foi ainda observado
que um aumento da mobilidade molecular da fase amorfa com aumento da
temperatura.
Lequieu e colaboradores [75] utilizaram os tempos de relaxação T1 e T2
para a determinação da morfologia de polímeros segmentados no estado sólido.
Foi observado que ambos os tempos mostraram dois valores de relaxação, sendo
um relativo a região de mobilidade e o outro a domínio rígido. Entretanto, o
comportamento destes materiais pode ser controlado numa ampla faixa por
pequenas mudanças estruturais. Como exemplo, foi verificado que a mobilidade
molecular do poli(tetrahidrofurano)/α,ω - bis – acrilamida foi reduzida quando
comparada a outras redes moleculares, devido a homogeneidade da estrutura da
rede, a relaxação T2 das cadeias é restrita e o tempo de relaxação T1 é longo.
Kolodziejski e colaboradores [76] realizaram análise de polímeros no estado
sólido pelos tempos de relaxação, já que estes são sensíveis às modificações
estruturais como tratamento térmico, resfriamento brusco ou recozimento e por
modificações induzidas ao material. A aplicação do tempo de relaxação spin-spin
mostra o decaimento dos domínios de mobilidades distintas presentes em
homopolímeros ou misturas. Um decaimento rápido, T2 curto, é referente a
domínios de baixa mobilidade, enquanto que um decaimento longo mostra valores
longos, devido à alta mobilidade do domínio ou do grupamento. Um exemplo são
os grupos metil, que devido a sua alta velocidade de rotação apresenta valores de
T2 longo, ao contrário, os carbonos quaternários apresentam constante de tempo
T2 curta (0,1 µs), devido a uma mobilidade restrita. Em particular, o tempo de
relaxação T2 é apropriadamente estabelecido em função do domínio.

35
3 – MATERIAIS E MÉTODOS
3.1 - Considerações preliminares
Neste capítulo serão descritos os materiais utilizados na pesquisa que é
objeto dessa Dissertação; bem como a metodologia empregada para a obtenção
das membranas de policarbonatos (PC) e as técnicas empregadas para
caracterização dos materiais obtidos.
3.2 - Materiais
O polímero usado neste estudo foi o Policarbonato de bisfenol A. Foram
fornecidas 03 amostras de policarbonato, pela Policarbonato S. A. Indústria e
Comércio – Camaçarí/BA e estas foram utilizadas conforme recebidas.
• Policarbonato Durolon A - 2600 massa molecular ponderada média Mw =
44.460 – (amostra PC3)
• Policarbonato Durolon I - 2600 massa molecular ponderada média Mw =
55.600 – (amostra PC4)
• Policarbonato Durolon V - 2700 massa molecular ponderada média Mw =
51.120 – (amostra PC5)
As massas molares não são tão distintas que possam afetar as
propriedades macroscópicas do material, entretanto a escolha se deu no
intuito de avaliar a diferença de mobilidade em nível molecular.
3.2.1 - Produtos Químicos
Os produtos químicos utilizados na parte experimental a ser descrita nesta
Dissertação estão relacionados na Tabela 3, e todos foram usados como
recebidos.

36
Tabela 3 – Reagentes empregados nesta Dissertação
_________________________________________________________________
Reagentes Fórmula Procedência Grau de pureza
__________________________________________________________________
Clorofórmio CDCl3 TediaBrazil P.A.
deuterado
Clorofórmio CHCl3 TediaBrazil P.A.
Tetracloroetano C2D2Cl4 TediaBrazil P.A.
Deuterado (TCE d2)
Tetracloroetano (TCE) C2H2Cl4 TediaBrazil P.A.
__________________________________________________________________
3.2.2 – Equipamentos
Além dos equipamentos normalmente empregados em laboratórios de
pesquisa, também foram utilizados os seguintes aparelhos:
• Difratômetro de raios X Rigaku, Miniflexb;
• Difratômetro de raios X Rigaku, Rix 3100b;
• Calorímetro diferencial de varredura, Rigakub, modelo TAS 100 com acessório
Thermoflex TG 8110 com precisão de + 1°C;
• Espectrômetro de ressonância magnética nuclear Varian, modelo Mercurya;
• Espectrômetro de ressonância magnética nuclear Maran Ultra Resonancea
• Prensa hidráulica, modelo Carvera com aquecimento (temperatura máxima de
260°C),com capacidade de carga de 11 toneladas (24 .000 Libras)
• Analisador Rigaku, modelo Thermoflexb TG 8110 com precisão de + 2°C
Espectrômetro de infravermelho, Perkin-Elmer b, modelo 200 FT-IR
a) IMA/UFRJ
b) PEQ/COPPE/UFRJ

37
3.3 – Métodos
3.3.1-Técnicas de Preparação das amostras poliméricas
Dos polímeros foram preparados filmes vazados, após solubilização
destes nos solventes CHCl3 e TCE e em filmes por termo prensagem.
a) Preparação de filmes vazados
Os filmes dos polímeros foram preparados por meio de moldagem por
prensagem do material no estado fundido. Foi utilizada uma prensa hidráulica
aquecida a 260°C. A prensagem foi realizada por meio da colocação dos peletes
dos polímeros no centro do molde de uma placa de aço inox nas seguintes
dimensões: (180x180x1), sendo os peletes cobertos por uma outra placa da
mesma espécie. Entre essas placas foi colocado um papel de prata boliviana
como agente desmoldante.
Após deixar o policarbonato na estufa por 24h para retirada de umidade, os
peletes foram colocados no molde e este na prensa previamente aquecida por 5
minutos sem carga. Após 5 minutos, foram carregados gradualmente 2 toneladas
por minuto, até alcançar 8 toneladas. No início dos 15 minutos de prensagem, foi
feito o alívio da pressão de 8 para 1 tonelada, repetida por 10 vezes seguidas e,
ao fim dos 15 minutos, os filmes foram imersos em uma cuba com água e gelo.
b) Preparação de filmes via solução vazada
As amostras de policarbonato foram solubilizadas em dois solventes: CHCl3
e TCE, para a formação de filmes para indução de cristalinidade.
b.1) Solução de PC em CHCl3
As soluções de PC em CHCl3 foram preparadas por meio da dissolução da
massa polimérica em erlenmeyer com tampa esmerilhada, pela adição de CHCl3

38
de modo a permitir concentrações de 15% p/v e 30% p/v após a solubilização à
temperatura ambiente, em 5 – 7 dias, com o recipiente fechado, submetido a
agitação. As soluções de PC a 30% p/v apresentaram grumos, evidenciando a não
solubilização completa dessa massa polimérica, sendo descartadas.
b.1.1) Solução de PC em TCE
As soluções de PC em TCE foram preparadas por meio da dissolução da massa
polimérica em erlenmeyer com tampa esmerilhada, pela adição de TCE de modo a
permitir concentrações de 15% p/v e 30% p/v após a solubilização à temperatura
ambiente, em 5 – 7 dias, com o recipiente fechado, submetido a agitação.
3.3.2. – Preparação dos filmes de PC em CHCl3 e TCE em solução
Os filmes poliméricos empregados em todas as análises de caracterização
foram obtidos a partir do vazamento das soluções sob placa de Petri, seguido da
evaporação lenta do solvente, dentro de um dessecador nivelado contendo sílica.
O período para completa evaporação do solvente à temperatura ambiente foi de 1
– 2 semanas. Após este período de evaporação lenta, os filmes foram retirados
das placas de vidro. A retirada do solvente residual das amostras foi realizada por
meio de secagem a vácuo e as mesmas ficaram condicionadas em dessecador
contendo sílica, à temperatura ambiente.
3.3.3 - Métodos de Caracterização
A caracterização de um material polimérico pode ser feita por vários
métodos, dependendo da característica ou propriedade que se quer medir. Dessa
forma, para uma caracterização mais detalhada da fase cristalina ou amorfa do
material foram selecionadas as análises térmicas, por meio da calorimetria
diferencial de varredura (DSC) e a termogravimetria (TG), a espectrometria no
infravermelho com transformada de Fourier (FT-IR), difração de raios X e a
espectroscopia de ressonância magnética nuclear (RMN) em solução (alto campo)
e pela relaxação nuclear (baixo campo).

39
3.3.4 – Análise Térmica
3.3.4.1 – Calorimetria diferencial de varredura (DSC)
A calorimetria diferencial de varredura na área de polímeros permite
detectar e quantificar a cristalinidade, e a temperatura de transição vítrea, dentre
outros estudos. DSC pode ser definida como uma técnica que mede as
temperaturas e o fluxo de calor associados com as transições dos materiais em
função da temperatura e do tempo. Tais medidas fornecem informações sobre a
temperatura de fusão cristalina (Tm), temperatura de transição vítrea (Tg) e ainda a
determinação do grau de cristalinidade do polímero por meio do calor de fusão,
(∆Hf).
As amostras com massa entre 8 e 14 mg foram submetidas a uma
taxa de aquecimento de 10°C/min, na faixa de – 20°C até 190°C, sendo resfriadas
bruscamente até a temperatura inicial. O segundo aquecimento foi conduzido nas
mesmas condições que o primeiro e o resfriamento realizado a uma taxa de
10°C/min até a temperatura de 30°C. As curvas de DSC obtidas registraram a
temperatura de fusão (Tm) e o fluxo de calor para a cristalização da amostra.
A partir dos dados de calor de fusão (∆Hf), calculou-se o grau de
cristalinidade (Xc) da amostra, de acordo com a (Equação 6). O ∆Hf é a entalpia de
fusão da amostra e ∆H100% é a entalpia de fusão do policarbonato hipoteticamente
100% cristalino, cujo valor foi de 147,55 J/g.
Xc = ∆ Hf x 100 (Equação 6) ∆ H100%
3.3.4.2 – Termogravimetria
A termogravimetria é uma técnica muito utilizada na caracterização
do perfil de degradação dos materiais poliméricos. A exposição a temperaturas
elevadas pode, algumas vezes, alterar a estrutura química e, por conseqüência,
as propriedades físicas dos materiais poliméricos. Portanto, a curva de
degradação térmica, em condições não-isotérmicas, mostra o perfil da resistência

40
ou estabilidade térmica que o material apresenta quando submetido a uma
varredura de temperatura. A análise termogravimétrica mede a quantidade e
velocidade de mudanças na perda de massa em função da temperatura e/ou do
tempo em atmosfera controlada de nitrogênio. As análises foram realizadas em
atmosfera inerte de nitrogênio, com uma razão de aquecimento constante de
10ºC/min. A faixa de aquecimento foi de 30 a 300°C.
3.3.4.3.-.Espectroscopia de absorção na região do infravermelho com transformada de Fourier-FTIR
A espectroscopia na região do infravermelho consiste de uma poderosa
ferramenta na caracterização e identificação de polímeros, podendo também ser
utilizada na determinação do grau de cristalinidade de polímeros [48,25].
A análise por FTIR foi realizada nesta dissertação nos filmes poliméricos
obtidos por termo prensagem e por membranas vazadas com TCE e CHCl3, para
investigar a indução da cristalinidade do policarbonato e da completa retirada do
solvente residual.
3.3.4.4 – Difração de Raios X
As análises de raios X foram realizadas com o objetivo de verificar o efeito
do solvente e da temperatura na cristalização do PC. Para tanto, as amostras
foram prensadas na temperatura de 260° C e as soluções vazadas sob placa de
Petri para obtenção dos corpos de prova, que foram colocadas no porta amostra e
analisadas com a emissão de radiação de CuKα (λ = 1,5418 Å), na temperatura
ambiente em 40KV e 30 mA. Os padrões de difração foram coletados numa taxa
de varredura de 2º‹2θ‹50º, por 3 seg, passos de 0,05º.
A difração de raios X é uma técnica muito usada para a caracterização de
polímeros, fornece informações sobre o estado cristalino e amorfo do polímero. Os
raios X são fótons de alta energia com baixos comprimentos de onda (λ ~ 0,5 a
2,5 Å), que interagem com os elétrons. Quando o feixe de raios X atinge uma

41
amostra, alguns fótons são absorvidos, outros são transmitidos e outros são
espalhados, devido à interação com os elétrons da amostra. Essa interação
resulta em um espalhamento que é função de um ângulo, designado ângulo de
espalhamento (2θ), e guarda uma relação com o comprimento de onda dos raios X
(λ) e com a distância entre os planos atômicos, definida pela Equação 2, que
expressa a Lei de Bragg [47].
n λ = 2 d sen θ (Equação 2)
Onde:
n – número inteiro (0, 1, 2 ...);
λ - comprimento de onda dos raios X que é difratado e é tabelado (fonte) (1,5418
Å);
d – distância interplanar;
θ - é a metade do ângulo de espalhamento.
3.3.4.5 – Ressonância Magnética Nuclear
As análises por RMN dos materiais obtidos envolveram estudos em solução
e no estado sólido, empregando diferentes técnicas.
a) RMN em solução
Foram preparadas soluções dos peletes e das membranas em TCE e
CDCl3 na concentração de 15% p/v em tubo de 10 mm. As análises de RMN 13C
em solução foram realizadas à temperatura ambiente da sonda, empregando a
seqüência pulso simples(decaimento de Bloch). Os parâmetros utilizados estão
indicados na Tabela 4.

42
Tabela 4 – Espectrômetro e condições utilizadas para obtenção dos espectros de
RMN de 13C em solução
PARÂMETROS
RMN 13C qualitativo
Espectrômetro Mercury 300
Freqüência de observação (MHz) 75,4
Tempo de aquisição (s) 1,59
Janela espectral (Hz) 18.864
Largura do pulso (π/2, µs) 17,1
Solvente TCE/TCE-d2 ou CDCl3
Intervalo entre os pulsos (s) 1
Modo do desacoplador NNY
Nº de acumulações (transientes) 3000 - 12000
RMN no estado sólido
O estudo por RMN em baixo campo no estado sólido do núcleo 1H foi feito
empregando os tempos de relaxação spin-rede ou longitudinal (T1) e spin-spin ou
transversal (T2) do núcleo de 1 H. Uma das técnicas que podem ser empregadas
para se determinar o valor de T1 é a Inversão-Recuperação: 180° - τ - 90°. Aplica-
se um pulso de 180º para que ocorra a inversão da magnetização e espera-se um
tempo (τ) para que ocorra a recuperação da magnetização. O grau de
recuperação dos spins depende da mobilidade molecular da amostra, após o
intervalo τ um pulso de 90º é aplicado, adquirindo-se em seguida o sinal. Após a
seqüência de pulso é necessário que se espere 5 vezes o maior valor de T1, para
repetir a seqüência de pulsos [18,19].
O T2 corresponde à perda de coerência de fases, ou seja, a magnetização é
espalhada uniformemente no plano XY, ocorrendo uma mudança na entropia do
sistema, como mostra a Figura 19.

43
Figura 19: Perda de coerência dos spins
A constante de tempo T2 é determinada pela seqüência de pulso Car-
Purcell-Meibool-Gill (CPMG), empregando a seguinte seqüência de pulso: 90° - τ -
180°. Como esta pode ser determinada pelo decaimento do FID, tem-se que é
função do comprimento do FID, quanto mais longo melhor, pois se tem uma
melhor resolução espectral. O valor de T2 pode ser obtido pela largura a meia
altura definida pela Equação 7, após a transformada de Fourier de um decaimento
exponencial no domínio das freqüências como mostra a Figura 20.
∆ν1/2 = 1 / T2 (Equação 7)
ω – (1/T2) ω ω + (1/T2)
Figura 20: Decaimento exponencial no domínio das freqüências

44
Para as análises de relaxação, o material prensado e vazado foi cortado em
tiras e colocado em um tubo de 18 mm, sendo este posteriormente colocado na
sonda. As condições de aquisição de T1 e T2 em duas temperaturas (25ºC e 35ºC)
estão descritas nas Tabelas 5 e 6, respectivamente.
Tabela 5 - Espectrômetro e condições utilizadas para obtenção do tempo de
relaxação longitudinal do núcleo de 1H em baixo campo
PARÂMETRO T1 (1H)
Equipamento RESONANCE
Freqüência observação (MHz) 23
Tempo de aquisição (s) 0,024
Janela espectral (Hz) 4999999.9
Largura de pulso (π/2, µs) 4.2
Largura de pulso (π, µs) 8.4
Intervalo entre os pulsos (s) 5
Faixa de τ (µs) 100 – 5000000
Nº de acumulações 4
Números de pontos 20
Tabela 6 - Espectrômetro e condições utilizadas para obtenção do tempo de
relaxação transversal do núcleo de 1H em baixo campo
PARÂMETRO T2 (1H)
Equipamento RESONANCE
Freqüência observação (MHz) 23
Janela espectral (Hz) 4999999.9
Largura de pulso (π/2, µs) 4.2
Largura de pulso (π, µs) 8.4
Intervalo entre os pulsos (s) 5
Valor de τ (µs) 27
Nº de acumulações 9600

45
4. Resultados e Discussão
Aspectos gerais dos policarbonatos
O polímero usado neste estudo foi o policarbonato obtido pela reação do
Bisfenol A com o Fosgênio. Foram recebidas 03 amostras de policarbonato sob a forma
de pelete e estas foram analisadas e caracterizadas empregando as técnicas descritas
no capítulo anterior.
Inicialmente foram realizados testes de solubilidade com bases em dados da
literatura. Acetona, acetato de etila, tetraclorometano, clorofórmio, 1,1,2,2-
tetracloroetano e o acetato de butila podem ser usados como solvente para o
policarbonato [23]. Neste trabalho, após os testes de solubilidade optou-se por trabalhar
com o clorofórmio e o 1,1,2,2-tetracloroetano, que serão utilizados tanto no processo de
indução da cristalinidade do polímero quanto nas análises de caracterização por RMN
de carbono-13 em solução.
4.1 – Análises por RMN em solução
As três amostras de policarbonato foram solubilizados em CDCl3 e TCEd4 e os
espectros de RMN de carbono-13 adquiridos, como podem ser visto nas Figuras 21 e
22, respectivamente. Os sinais detectados por ambos solventes estão listados nas
Tabelas 7 e 8, respectivamente, de acordo com o assinalamento da Figura 23, que
mostra a unidade monomérica do policarbonato.
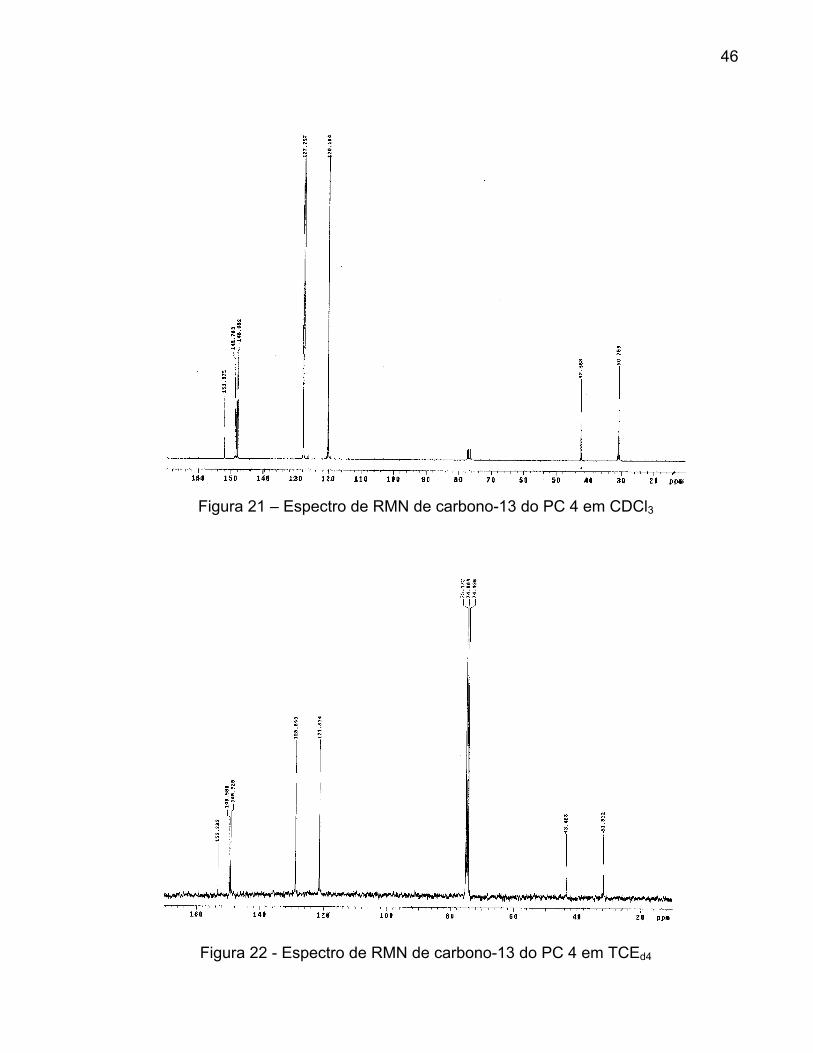
46
Figura 21 – Espectro de RMN de carbono-13 do PC 4 em CDCl3
Figura 22 - Espectro de RMN de carbono-13 do PC 4 em TCEd4

47
OC1
O
. C6
CH37
CH37
23
54
O .
Figura 23 – Estrutura da unidade monomérica do policarbonato
Tabela 7 - Assinalamentos dos núcleos de carbono-13 de policarbonato em CDCl3
Nº Núcleo δ (ppm)
1 C = O 151,9
2 C de aromático 148,0
3 C de aromático 148,7
4 CH de aromático 127,7
5 CH de aromático 120,0
6 C quaternário 42,3
7 CH3 30,7
Tabela 8 - Assinalamentos dos núcleos de carbono-13 de policarbonato em TCEd4
Nº Núcleo δ (ppm)
1 C = O 153,2
2 C de aromático 149,5
3 C de aromático 149,3
4 CH de aromático 128,8
5 CH de aromático 121,3
6 C quaternário 43,4
7 CH3 31,8

48
As três amostras de policarbonato apresentaram os mesmos deslocamentos
químicos nos dois solventes empregados. Entretanto, uma diferença nos valores de
deslocamentos químicos foi detectada quando do uso do TCE. Neste solvente os
deslocamentos químicos foram detectados em valores maiores, o que sugere que a
interação dipolar deste solvente com os policarbonatos é um pouco mais forte do que a
com CDCl3.
Modificações do policarbonato obtido a partir de Bisfenol-A e Fosgênio têm
conduzido a novas aplicações deste polímero nas mais diversas áreas. Sendo assim,
estudos de indução de cristalinidade por ação de solvente e termo prensagem foram
efetuados para verificação de mudanças nos arranjos estruturais deste polímero. Após
as modificações dos materiais obtidos, estes foram solubilizados em CDCl3 e TCEd4, e
os espectros de carbono-13 registrados. As Tabelas 9 e 10 mostram os assinalamentos
para o PC 4 após a indução e termo prensagem em CDCl3 e TCEd4.
Tabela 9 - Assinalamentos dos núcleos de carbono-13 do PC 4 em CDCl3
Nº Núcleo δ (ppm)
Indução
δ (ppm)
Termo prensagem
1 C = O 152,0 152,0
2 C de aromático 148,8 148,7
3 C de aromático 148,1 148,1
4 CH de aromático 127,8 127,8
5 CH de aromático 120,2 120,2
6 C quaternário 42,4 42,4
7 CH3 30,8 30,7
Pelos dados da Tabela 9 pode-se observar que não houve variação no
deslocamento químico em face dos processos de modificação do polímero e os
assinalamentos foram os mesmos para o pelete, o que indica que quando o clorofórmio

49
é usado, o tipo e força de interação do solvente predominam em relação ao tratamento
empregado no polímero.
Tabela 10 - Assinalamentos dos núcleos de carbono-13 do PC 4 em TCEd4
Nº Núcleo δ (ppm)
Indução
δ (ppm)
Termo prensagem
1 C = O 153,2 152,7
2 C de aromático 149,5 150,0
3 C de aromático 149,3 149,1
4 CH de aromático 128,8 128,6
5 CH de aromático 121,3 121,1
6 C quaternário 43,4 43,4
7 CH3 31,8 31,7
De acordo com os dados da Tabela 10 observa-se que houve uma pequena,
porém significativa variação no deslocamento químico em face dos processos de
modificação do polímero. Os assinalamentos dos carbonos da carbonila e do aromático,
que provavelmente participam das interações, tiveram seus valores menores quando a
termo prensagem foi efetuada, comparando-se com os valores do material após
indução de cristalinidade. Em TCE a detecção das mudanças quanto à morfologia do
polímero perante os tratamentos impostos foi evidenciada.
4.2 – Análise Térmica
4.2.1 - Calorimetria Diferencial de Varredura
O estudo através de DSC é importante para mostrar a variação da Tg
relacionada à fase amorfa e a presença ou não de Tm’s relacionadas com as
fases cristalinas do polímero.

50
Na Tabela 11 podem ser observadas as temperaturas de transição vítrea e fusão
cristalina assim como os valores do grau de cristalinidade e o calor latente de fusão
para as amostras de policarbonato estudadas. As amostras de policarbonato amorfos
apresentaram Tg‘s em torno de 150ºC e não foi observada a presença de Tm, o que está
em concordância com os dados da literatura [26,27]. No entanto, pode ser observado o
deslocamento da Tm nas amostras de policarbonato que foram induzidas cristalinidade
por ação de solvente, para valores maiores do que a do PC amorfo. Este deslocamento
pode indicar a formação de um domínio de fase cristalina no PC.
Tabela 11 - Resultados de DSC de Tg, Tm, Xc e ∆Hf para as amostras de
policarbonato
PC em pelete PC em TCE 30% PC em CHCl3 15%
Amostra Tg(°C) Tm(°C) ∆Hf Xc (%) Tg(°C) Tm(°C) ∆Hf Xc (%) Tg(°C) Tm(°C) ∆Hf Xc(%)
PC 3 150 nd nd Nd Nd 244 9,94 28 Nd 238 8,85 25
PC 4 150 nd nd Nd Nd 243 16,86 47,5 Nd 244 7,57 21
PC 5 150 nd nd Nd Nd 246 14,05 39,6 Nd 238 8,40 24
nd – não determinado
Na Figura 24 pode-se observar os valores de Tg’s relacionadas ao PC amorfo,
em torno de 150 ºC e não foi observada a presença de Tm, como relatado na literatura.
100 200 300
-0,1
0,0
0,1
0,2
mca
l/s
PC 3
PC 5
Temperatura / oC
PC 4
Figura 24 – Curvas térmicas de DCS das amostras de policarbonato na forma de
pelete

51
Nas Figuras 25 e 26 a visualização da Tg relacionada aos policarbonatos que
sofreram indução de cristalinidade por TCE e CHCl3 tornou-se difícil, provavelmente
pelo aparecimento de um pico referente à fusão cristalina que variou de 238 ºC a 246
ºC. A temperatura de fusão cristalina dos policarbonatos nos seus respectivos solventes
encontra-se na faixa da temperatura de fusão cristalina do PC, relatada na literatura,
que varia de 230 ºC a 263 ºC. O policarbonato é um polímero normalmente encontrado
no estado amorfo, cristaliza com dificuldade, e neste caso, deve ter cristalizado pela
ação plastificante dos respectivos solventes [5,28,29].
100 150 200 250
PC3
PC5
244 oC
238 oC
mca
l/s
Temperatura / oC
PC4
PC4
Figura 25 – Curvas de DSC para as amostras de policarbonato em CHCl3 a 15 % p/v
150 200 250
243 oC 246 oC
244 oC
m
cal/s
PC3
PC5
Temperatura (oC)
Figura 26 – Curvas de DSC para as amostras de policarbonato em TCE a 30 % p/v

52
4.2.2- Análise termogravimétrica
A eliminação da memória cristalina de um polímero, por tratamento térmico do
material fundido, pode originar variações de massa molecular análogas às
experimentadas em uma análise termogravimétrica. Por isso, essa análise pode
fornecer uma medida quantitativa que permita selecionar o intervalo de temperatura que
assegure a não degradação do material durante o tratamento térmico. A faixa de
temperatura de análise escolhida foi a normalmente utilizada para polímeros, onde foi
possível observar todas as etapas de decomposição [30].
Pela análise das curvas termogravimétricas e derivadas (Figura 27 a, b e c)
pode-se observar que o PC 3 possui o pico máximo de degradação em 513°C,
enquanto que o PC 4 e PC 5 possuem o mesmo valor, 520°C.
O PC amorfo apresenta a temperatura inicial de degradação (onset) em torno de
500 ºC, com um pico na curva de DTG com temperatura máxima de degradação por
volta de 520 ºC e perda de massa próxima a 77%, confirmando a alta estabilidade
térmica que este polímero possui. No entanto, para os PC que sofreram indução de
cristalinidade pelos solventes TCE (Figura 28) e CHCl3 (Figura 29) apresentaram
comportamento diferente do PC amorfo. Este fato pode ser atribuído à existência de
grau de cristalinidade diferente entre as amostras e que a organização estrutural entre
as cadeias poliméricas apresentaram comportamentos diferentes.

53
0 200 400 600 800
20
40
60
80
100
513oC
TG (%
)
Temperatura (oC)
TG da amostra PC 3
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
DTG
0 200 400 600 800
20
40
60
80
100
520oC
TG (%
)
TG da amostra
Temperatura (oC)
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05DTG
PC 4
PC 4
0 200 400 600 800
20
30
40
50
60
70
80
90
100
110 TG da amostra PC 5
Temperatura (oC)
TG (%
)
520oC
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
DTG
0 200 400 600 800
20
40
60
80
100%
TG
Temperatura (oC)
PC 3
PC 5
Figura 27 – Curvas termogravimétricas: a) curva termogravimétrica e a derivada para o
pelete de PC 3, b) curva termogravimétrica e a derivada para o pelete de PC 4, c) curva
termogravimétrica e a derivada para o pelete de PC 5 e d) curva termogravimétrica para
os pelete dos três polímeros

54
0 100 200 300 400 500 600 700-20
0
20
40
60
80
100
TG do PC 3 em CHCl3a 15% p/v
Temperatura (°C)
TG (%
)
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
DTG
0 100 200 300 400 500 600 700
0
20
40
60
80
100
TG DO PC 4 em CHCl3 a 15%
Temperatura (°C)
TG (%
)
-0,5
-0,4
-0,3
-0,2
-0,1
0,0
DTG
0 100 200 300 400 500 600 700
0
20
40
60
80
100
TG do PC 5 em CHCl3 a 15% p/v
Temperatura (°C)
TG (%
)
-0,5
-0,4
-0,3
-0,2
-0,1
0,0D
TG
0 100 200 300 400 500 600 700
0
20
40
60
80
100
TG DE POLICARBONATOS EM CHCl3 A 15% P/V
PC 3
PC 5
Temperatura / oC
TG /
%
PC 4
Figura 28 - Curvas de DTG dos PCs após indução de cristalinidade em CHCl3: a) PC 3,
(b) PC 4, (c) PC 5 e (d) TG dos três PCs

55
0 100 200 300 400 500 600 700-20
0
20
40
60
80
100
TG do PC 3 em TCE a 30%
Temperatra (°C)
TG (%
)
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
DTG
0 100 200 300 400 500 600 700-20
0
20
40
60
80
100
TG do PC 4 em TCE a 30% p/v
Temperatura (°C)
TG (%
)
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
DTG
0 100 200 300 400 500 600 700-20
0
20
40
60
80
100
TG do PC 5 em TCE a 30% p/v
Temperatura (°C)
TG (%
)
-0,4
-0,3
-0,2
-0,1
0,0
DTG
0 100 200 300 400 500 600 700
0
20
40
60
80
100
TG DE POLICARBONATOS EM TCE A 30% P/V
PC 3
PC 5
Temperatura / oC
TG /
%
PC 4
Figura 29 - Curvas de DTG dos PCs após indução de cristalinidade em TCE: a)
PC 3 (b) PC4 (c) PC 5 e (d) TG dos três PCs
4.3 – Análise por Infravermelho
As análises pela espectrometria de infravermelho foram realizadas para
confirmar a não formação de outras substâncias ou espécies químicas geradas quando
dos tratamentos térmicos ou indução realizados nos policarbonatos, assim como a
confirmação da eliminação do solvente residual. Os espectros obtidos na região do
infravermelho para o padrão de policarbonato amorfo, e para os polímeros após a
indução de cristalinidade, não mostraram nenhuma diferença nas bandas de absorção,
confirmando a não formação de novas substâncias e que a eliminação do solvente após
a indução foi efetiva, indicando que os materiais não sofreram nenhuma alteração
quando dos tratamentos a eles impostos. A Figura 30 mostra os espectros de FTIR do
padrão de PC amorfo e dos três tipos de policarbonatos após a indução de

56
cristalinidade em TCE. A Tabela 12 mostra as principais absorções dos grupos
característicos do policarbonato.
4000,0 3000 2000 1500 1000 450,00,00
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
5,5
6,0
6,62
cm-1
A
1775,10
PC3 PC4 PC5 padrão
Figura 30 – Espectros de Infravermelho para o padrão de policarbonato amorfo e os PC
3, PC 4 e PC 5 após a indução de cristalinidade
Tabela 12 – Principais bandas de absorção na região do infravermelho do policarbonato
Número de ondas (cm-1) Banda Modo Vibracional
3045 C-H de anel aromático Deformação axial
2964 C-H do grupo metila Deformação axial simétrica
1773 O-(C=O)-O da carbonila Deformação axial
1479 C=C do anel aromático Deformação axial
1519 C-H do anel aromático Deformação angular no plano
1232 C-O-(C=O)-O-C Deformação axial assimétrica
4.4 - Difratometria de Raios X
A identificação das fases formadas nas membranas vazadas em CHCl3 e TCE, e
em membranas obtidas por termo prensagem foi realizada por difratometria de raios X.

57
Os difratogramas das membranas vazadas em CHCl3 e TCE, com concentração de
15% p/v e por termo prensagem estão mostrados nas Figuras 31, 32 e 33.
10 20 30 40 500
1000
2000
3000
4000
5000
6000
2θ
cps
Ausência de cristalinidade material amorfo
Figura 31 – Difratograma de raios X do filme de PC 4 obtido por termo prensagem.
10 20 30 40 500
1000
2000
3000
4000
5000
6000
cps
2θ
28% de cristalinidade 46Å Tamanho do cristal
Figura 32 – Difratograma de raios X do filme de PC 4 obtido por vazamento em CHCl3
com concentração 15% p/v.

58
10 20 30 40 500
1000
2000
3000
4000
5000
6000
cps
2θ
30% de cristalinidade 76Å Tamanho do cristal
Figura 33 – Difratograma de raios X do filme de PC 4 obtido por vazamento em TCE,
com concentração de 15% p/v
Pelos difratogramas foi confirmado que a termo prensagem gerou, como
esperado, um filme com maior caráter amorfo. Para os filmes obtidos por indução com
os solventes foi observado que, tanto para o clorofórmio quanto para o TCE, o teor de
cristalinidade foi praticamente o mesmo (28 % para CHCl3 e 30% para o TCE).
Entretanto, esses dados mostram que a formação da região cristalina apresentou
tamanho (46 Å para CHCl3 e 76 Å para o TCE), forma e distribuição de cristais
diferentes. O comportamento observado para o TCE indica que a geração de cristais
por esse solvente provavelmente se deu por um menor número de núcleos, esse
solvente permitiu uma melhor solvatação desses núcleos, gerando cristais maiores.
Esses dados confirmam a variação de deslocamentos químicos determinado por RMN
em solução.
O estudo da cristalinidade para os filmes obtidos em concentração máxima nos
solventes empregados (CHCl3 15% p/v e TCE 30% p/v), também foi efetuado. As
membranas obtidas por vazamento de solução a 15% p/v em CHCl3 (Figura 34)
mostrou que os valores de posicionamento do 2θ foram 17,63 para o PC3, 17,65 para o

59
PC 4 e 17,80 para o PC 5. Já para os filmes obtidos por vazamento de solução em TCE
a 30% p/v (Figura 35) os valores de posicionamento do 2θ foram 18,00 para o PC 3,
18,40 para o PC 4 e 18,10 para o PC 5. Essa diferença nos valores de posicionamento
do 2θ indica que os solventes estão atuando de forma diferenciada e o que o TCE
mostrou ser melhor indutor de cristalinidade em relação ao clorofórmio devido ao maior
deslocamento do ângulo 2θ. A Tabela 13 mostra o ângulo 2θ, o grau de cristalinidade e
o tamanho do cristalito para as amostras policarbonato estudadas em concentração
máxima nos solventes empregados.
Tabela 13 - Resultados de DRX de 2θ, Xc e tamanho de cristalito
Amostra PC em CHCL3 15% PC em TCE 30%
2θ Xc Tamanho do cristalito 2θ Xc Tamanho do cristalito
PC3 17,51 35,80 52 18,00 35,60 56
PC4 17,64 34,20 55 18,30 41,20 57
PC5 17,71 36,16 60 18,10 39,95 60
10 20 30 40
0
400
800
1200DRX DE POLICARBONATOS EM CHCl3 A 15% p/v
PC 3
PC 5
cps
2θ
PC 4
Figura 34 - Difratograma para os filmes de PC 3, PC 4 e PC 5 vazados por
solução a 15% p/v em CHCl3

60
10 20 30 40
0
400
800
1200DRX DE POLICARBONATOS EM TCE A 30% p/v
cps
2θ
PC 3
PC 5
PC 4
Figura 35 - Difratograma para os filmes de PC 3, PC 4 e PC 5 vazados por
solução a 30% p/v em TCE
4.5 – Avaliação por relaxação nuclear
Os peletes e os filmes obtidos por termo prensagem e indução de cristalinidade
das três amostras de policarbonato foram analisadas por RMN de baixo campo,
focalizando a determinação dos tempos de relaxação spin-rede (T1) e spin-spin (T2),
para avaliação da dinâmica molecular destes materiais, visando gerarem dados que
possam ser complementares e/ou únicos quando comparados aos métodos
convencionais utilizados.
4.5.1 - Relaxação spin-rede
O primeiro estudo envolveu a análise do tempo de relaxação spin-rede dos
peletes a várias temperaturas, desde a temperatura ambiente (25°C) até 150°C
(temperatura da transição vítrea do policarbonato), com intuito de observar mudanças
neste parâmetro em face das mudanças de mobilidade do material, já que esta
temperatura está na faixa da temperatura de transição vítrea do policarbonato. Os
dados obtidos estão listados na Tabela 14.

61
Tabela 14 - Tempo de relaxação spin – rede das amostras PC 3, PC 4 e PC 5 na forma
de pelete a várias temperaturas
T1 (ms)
T (ºC) PC 3 PC 4 PC 5
25 31,3 32,2 0,6
80,3 115,7 36,5
130,3 116,6
50 40,5 56,8 40,5
155,5 163,1 138,1
70 62,3 57,3 50,3
182,6 167,8 158,9
90 74,7 17,8 53,6
198,7 127,8 173,2
137,5
110 9,2 125,7 49,3
136,4 263,0 189,5
233,5
130 9,4 135,7 139,2
147,7 275,1 258,0
244,9
150 99,4 129,9 101,9
227,4 278,9 250,6

62
Com o aumento da temperatura para o PC 3 , foi observado um aumento do
valor de T1 e nas temperaturas de 110 e 130 ºC três valores foram encontrados para
este parâmetro, indicando que nestas temperaturas, pode estar ocorrendo
reorganização estrutural em função do aumento da mobilidade, já que as temperaturas
estão próximas da Tg. O que nos leva a sugerir que domínios da ordem de 35 nm,
referentes a segmentos menores de cadeia foram detectados, gerando o aparecimento
de um valor de relaxação pequeno em torno de 9 ms. Na temperatura de 150°C, voltou-
se a observar dois valores de T1, sendo o maior valor menor do que os detectados para
as temperaturas de 110 e 130 °C, o que indica que a 150°C esteja ocorrendo mudança
de fase, provavelmente em função desta temperatura ser próxima ou igual a Tg do
polímero.
Para o PC 4 até a temperatura de 90ºC dois valores de T1 foram detectados,
visto que o polímero vem adquirindo um aumento na mobilidade molecular até alcançar
a temperatura onde 3 valores de parâmetro foram detectados, em função da
reorganização estrutural das cadeias poliméricas. Após 90°C dois valores de T1
voltaram a ser detectados. A 150°C foi, então, observada uma mudança de fase, visto
que um dos valores de T1 diminuiu enquanto o outro se manteve praticamente
constante. Como esta temperatura é próxima a Tg, essa mudança de T1 pode ser
associada a esta transição, visto que dois domínios distintos prevalecem.
Já no PC 5 apenas na temperatura de 25°C três valores de T1 foram detectados,
mostrando a heterogeneidade do polímero. A partir desta temperatura até 130°C foi
observado um aumento nos valores de T1, e na temperatura de 150°C houve um
decréscimo de ambos os valores de relaxação, o que sugere a formação de domínios
de mobilidades diferentes.
Para as três amostras de policarbonato foi observado que a mudança de fase
ocorre na faixa de 150°C, polarizando os dois domínios, sendo um referente à região de
maior mobilidade (menor T1) e o outro a região de menor mobilidade (maior T1). Já que
as amostras são de policarbonato e obtidas pelo mesmo tipo de reação, os valores de

63
T1 deveriam ser os mesmos. Entretanto, pode-se observar uma variação deste
parâmetro para as três amostras, sendo esta concernente com a variação da massa
molar desses polímeros. O PC3 possui massa molar menor e os valores de T1 são os
menores, o PC4 possui massa molar maior e os valores de T1 também, enquanto que o
PC5 possui valores de T1 intermediários assim como a sua massa molar.
O estudo seguinte mostra os dados de tempo de relaxação para os polímeros
após a termo prensagem e a indução de cristalinidade por ação de solventes em
comparação com os dos peletes. Nesta etapa as temperaturas escolhidas foram 25°C e
35°C. As Tabelas de 15 a 17 mostram os valores de relaxação para o PC3, PC4 e PC5,
respectivamente.
Tabela 15: Tempo de relaxação spin – rede para o PC 3 nas temperaturas de 25
ºC e 35 ºC
Amostra T1 (25 ºC) T1 (35 ºC)
Pelete 31,3 11,8
80,3 47,2
130,3 137,3
Prensado 38,0 60,9
124,2 155,2
165,9
Induzido –TCE 51,7 33,3
164,0 186,6
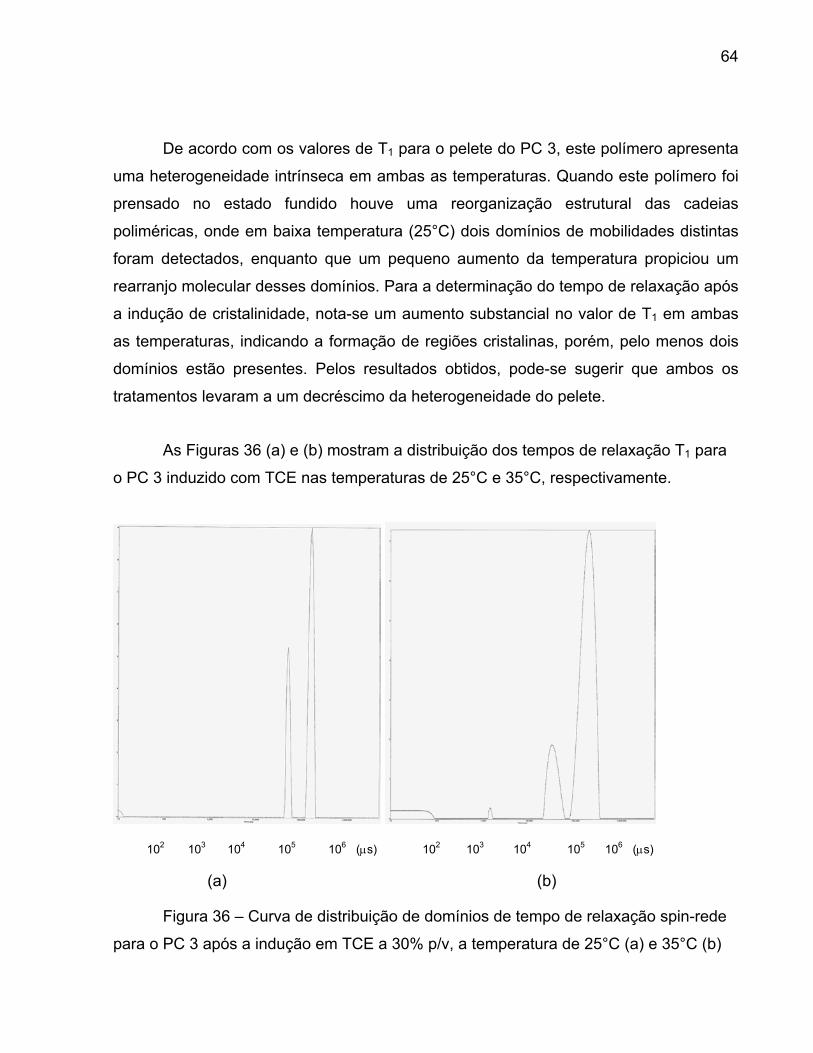
64
De acordo com os valores de T1 para o pelete do PC 3, este polímero apresenta
uma heterogeneidade intrínseca em ambas as temperaturas. Quando este polímero foi
prensado no estado fundido houve uma reorganização estrutural das cadeias
poliméricas, onde em baixa temperatura (25°C) dois domínios de mobilidades distintas
foram detectados, enquanto que um pequeno aumento da temperatura propiciou um
rearranjo molecular desses domínios. Para a determinação do tempo de relaxação após
a indução de cristalinidade, nota-se um aumento substancial no valor de T1 em ambas
as temperaturas, indicando a formação de regiões cristalinas, porém, pelo menos dois
domínios estão presentes. Pelos resultados obtidos, pode-se sugerir que ambos os
tratamentos levaram a um decréscimo da heterogeneidade do pelete.
As Figuras 36 (a) e (b) mostram a distribuição dos tempos de relaxação T1 para
o PC 3 induzido com TCE nas temperaturas de 25°C e 35°C, respectivamente.
102 103 104 105 106 (µs) 102 103 104 105 106 (µs)
(a) (b)
Figura 36 – Curva de distribuição de domínios de tempo de relaxação spin-rede
para o PC 3 após a indução em TCE a 30% p/v, a temperatura de 25°C (a) e 35°C (b)

65
Tabela 16: Tempo de relaxação spin – rede para o PC 4 nas temperaturas de
25ºC e 35ºC
Amostra T1 (25 ºC) T1 (35 ºC)
Pelete 33,2 41,1
115,7 134,3
Prensado 26,3 34,3
92,4 128,8
168,5
Induzido –TCE 41,1 82,5
167,7 175,0
228,6
Para o pelete do PC 4 foram detectados dois domínios de mobilidades diferentes
em função da heterogeneidade deste. Após a termo prensagem no estado fundido,
verificou-se o surgimento de três domínios distintos a baixa temperatura (25°C), devido
a uma maior restrição da mobilidade molecular. Entretanto, a 35°C este domínio não foi
detectado devido ao aumento da mobilidade molecular. Quando a indução de
cristalinidade foi efetuada dois domínios foram detectados a temperatura de 25°C e na
temperatura de 35°C três domínios foram registrados, em face do aumento da
mobilidade, que ocasionou um rearranjo organizacional. De acordo com os dados de
relaxação spin-rede tem-se que para o pelete do PC 4 a temperatura empregada na
termo prensagem não foi suficiente para destruição da história térmica do polímero.
As Figuras 37 (a) e (b) mostram a distribuição dos tempos de relaxação T1 para
o PC 4 após a termo prensagem nas temperaturas de 25°C e 35°C, respectivamente.
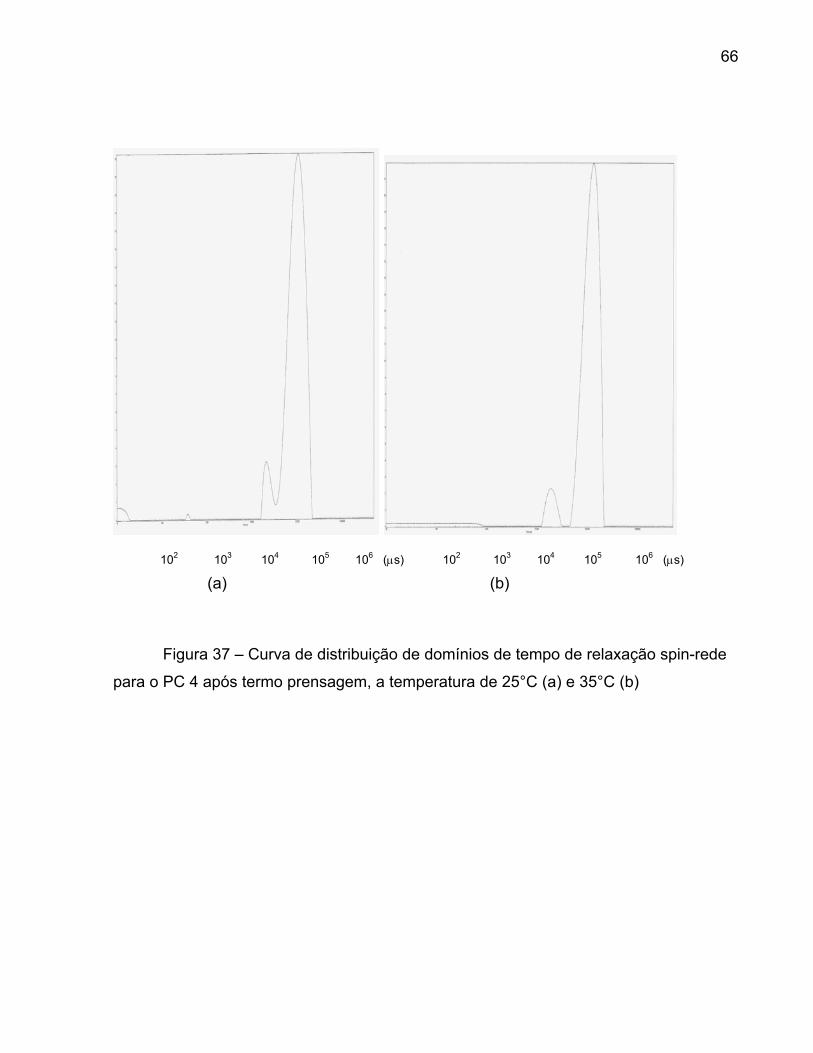
66
102 103 104 105 106 (µs) 102 103 104 105 106 (µs) (a) (b)
Figura 37 – Curva de distribuição de domínios de tempo de relaxação spin-rede
para o PC 4 após termo prensagem, a temperatura de 25°C (a) e 35°C (b)

67
Tabela 17: Tempo de relaxação spin – rede para o PC 5 nas temperaturas de 25
ºC e 35 ºC
Amostra T1 (25 ºC) T1 (35 ºC)
Pelete 0,6 7,6
36,5 56,6
116,6 141,9
Prensado 25,8 31,7
93,5 94,5
141,6 164,1
Induzido –TCE 16,6 25,6
113,3 168,4
263,3
Para o PC 5 na temperatura de 25°C tanto o pelete quanto as membranas três
domínios distintos foram detectados. Na temperatura de 35°C apenas para a membrana
após a indução de cristalinidade dois domínios foram detectados, em face da
reorganização estrutural. Os dados de T1 mostram que tanto a termo prensagem quanto
a indução de cristalinidade não foram efetivas na mudança da heterogeneidade deste
pelete.
As Figuras 38 (a) e (b) mostram a distribuição dos tempos de relaxação T1 para
o PC 5 na forma de pelete nas temperaturas de 25°C e 35°C, respectivamente.

68
102 103 104 105 106 (µs) 102 103 104 105 106 (µs)
(a) (b)
Figura 38 – Curvas de distribuição de domínios de tempo de relaxação spin-rede
para o PC5 para o pelete, a temperatura de 25°C (a) e 35°C (b)
4.5.2 - Relaxação spin-spin
A determinação do tempo de relaxação spin-spin teve por objetivo
confirmar os dados obtidos pelo tempo de relaxação spin-rede, como parte da
metodologia empregada.
Os valores de T2 para as amostras de PC 3, PC 4 e PC 5 estão listados
nas tabelas de 18 a 20, a seguir:

69
Tabela 18: Tempo de relaxação spin – spin para o PC 3 pelete, prensado e após
indução de cristalinidade nas temperaturas de 25 ºC e 35 ºC
Amostra T2 (ms) (25 ºC) T2 (ms) (35 ºC)
Pelete 0,17 0,19
0,25 0,20
0,61
Prensado 0,04 0,19
0,22 0,26
0,23 0,42
Induzido –TCE 0,09 0,19
0,22 0,27
0,28 0,28
Os valores de T2 mostram que houve uma variação significativa na estrutura do
pelete após este ser submetido aos diferentes tratamentos, mostrando que a baixa
temperatura houve a formação de um domínio muito rígido, valores de T2 0,04 a 0,09
ms. Além disso, após a indução de cristalinidade por TCE gerou a formação de três
domínios, em função da reorganização estrutural após o processo. O aumento da
temperatura propiciou uma melhor separação das fases no pelete e no material
prensado, o que não foi observado para o material após a indução de cristalinidade
devido a maior organização estrutural das regiões amorfas e cristalinas.
As curvas de distribuição de domínios do tempo de relaxação spin-spin
para o pelete do PC3 estão mostradas na Figura 39, (a) temperatura de 25°C e (b)
temperatura de 35°C. Pelas curvas pode-se observar que o menor valor de tempo de
relaxação (domínio mais rígido) controla este processo.

70
102 103 104 105 106 (µs) 102 103 104 105 106 (µs) (a) (b)
Figura 39 - Curvas de distribuição de domínios do tempo de relaxação spin-spin
para o pelete do PC3 (a) 25°C e (b) 35°C
Tabela 19: Tempo de relaxação spin – spin para o PC 4 pelete, prensado e após indução de
cristalinidade nas temperaturas de 25 ºC e 35 ºC
Amostra T2 (ms) (25 ºC) T2 (ms) (35 ºC)
Pelete 0,04 1,9
6,9
49,8
Prensado 0,1 0,2
0,4 151,2
196,0
Induzido –TCE 7,4 1,4
73,3 16,4
268,8 79,4
432,0 94,6

71
Os valores de T2 mostram que houve mudanças significativas nas
reorganizações estruturais, quando do tratamento impostos ao pelete, o que advém das
diferentes organizações moleculares geradas nos tratamentos do polímero. Cabe
ressaltar que as intensidades proporcionais dos domínios mostram valores distintos,
como, por exemplo, os valores das intensidades do T2 para a membrana após a termo
prensagem, como pode ser visto na figura da curva de distribuição dos domínios (Figura
40). O comportamento de T2 para as membranas após a indução de cristalinidade
mostrou que um aumento de 10°C na temperatura promoveu uma reorganização
molecular, gerando um material com maior rigidez, visto que os valores de T2 nesta
temperatura são menores que os valores a 25°C.
A curva de distribuição dos valores de T2 mostra apenas um sinal extremamente
largo (Figura 40) para a temperatura de 25°C, enquanto que para 35°C três domínios
de mobilidades distintas foram observados e a intensidade do menor domínio foi
extremamente superior aos outros dois, indicando que este domínio controla o processo
de relaxação do material. O tempo de relaxação de 0,2 ms foi atribuído ao domínio
formado por uma homogeneidade da fase amorfa. Os dois valores de relaxação
maiores foram atribuídos aos pequenos domínios de mobilidade maiores formados por
cadeias de menores comprimentos.

72
102 103 104 105 106 (µs) 102 103 104 105 106 (µs)
(a) (b)
Figura 40 - Curvas de distribuição de domínios do tempo de relaxação spin-spin para o
PC4 após a termo prensagem (a) 25°C e (b) 35°C
Na Figura 40 (a) o perfil da distribuição de domínios do material após a termo
prensagem mostra que o este se encontra completamente amorfo, podendo este dado
ser respaldado pela análise de raio X, como mostrado na figura 31. O que indica que a
técnica de RMN de baixo campo, por meio da determinação de T2, pode gerar o mesmo
tipo de informação que a análise por raio X.

73
Tabela 20: Tempo de relaxação spin – spin para o PC 5 pelete, prensado e após
indução de cristalinidade nas temperaturas de 25 ºC e 35 ºC
Amostra T2 (ms) (25 ºC) T2 (ms) (35 ºC)
Pelete 7,6 6,2
Prensado 0,6 0,2
202,7 196,4
Induzido –TCE 3,0 3,9
55,5 261,0
135,9 302,4
Dos valores de T2 para o PC 5 observa-se que após a prensagem houve a
detecção de dois domínios em ambas as temperatura, em função de uma melhor
separação das diferentes fases geradas pelo tratamento, o valor menor foi atribuído a
região mais rígida, que controla a relaxação e o outro a uma região de maior mobilidade
(menor intensidade). Quanto à indução de cristalinidade por solvente, as mudanças
observadas nos valores de T2 indicam que este sofreu uma reorganização estrutural
gerando tamanhos de cristais grandes com uma fase amorfa intermediária (a 25°C T2 =
55,5 ms e a 35°C T2 = 261,0 ms) e uma região amorfa com maior mobilidade,
apresentando valores de T2 maiores.
As curvas de distribuição dos tempos de relaxação spin-spin para o PC5 após a
indução de cristalinidade em TCE nas temperaturas de 25°C (a) e 35°C (b) estão
mostradas na Figura 41.

74
102 103 104 105 106 (µs) 102 103 104 105 106 (µs)
(a) (b)
Figura 41 - Curvas de distribuição de domínios do tempo de relaxação spin-spin
para o PC5 após a indução de cristalinidade em TCE (a) 25°C e (b) 35°C
As curvas de distribuição de domínios após a indução de cristalinidade e pela
variação de temperatura para o PC5 mostram uma reorganização estrutural, em face
das mudanças sofridas pelo polímero neste tratamento.
As mudanças observadas para as amostras de policarbonato, quando dos
tratamentos devem-se provavelmente as reorganizações estruturais impostas pelo
processo de resfriamento brusco e de indução da cristalinidade pela ação do solvente.
Como esperado, as medidas de T2 para todos os sistemas endossam o comportamento
observado por T1. E ambos complementam os resultados obtidos pela análise térmica
(DSC e TG) e confirmam as modificações estruturais observadas pelas análises de
raios X.

75
5 - CONCLUSÕES
1) Das análises de DSC, por meio da mudança na temperatura de fusão, conclui-se que
tanto a forma quanto o teor de cristalinidade foram modificadas após os tratamentos
aplicados as amostras de policarbonato, sendo o mesmo observado pelas análises de
TG.
2) As análises de raios X evidenciaram as mudanças estruturais e morfológicas geradas
pelos dois tipos de tratamentos aplicados às amostras de policarbonato, que
complementam os dados obtidos pela análise térmica.
3) As determinações dos tempos de relaxação nuclear (T1 e T2) obtidas pela RMN de
baixo campo, foram muito precisas quanto às mudanças causadas pelos dois
tratamentos realizados nas amostras de policarbonato, evidenciando as reorganizações
estruturais e morfológicas em face do aumento da amorficidade e da indução de
cristalinidade nesse polímero. Ampliando as informações obtidas pela análise térmica e
confirmando as do raio X.
4) A RMN de baixo campo mostrou-se sensível na detecção das mudanças de
mobilidade deste polímero mesmo nas amostras com pequenas diferenças na massa
molar, o que não foi possível de ser identificada pelas outras técnicas usadas na
caracterização deste polímero.
5) A RMN de baixo campo mostrou ser uma ferramenta rápida e que pode ser aplicada
em lugar das outras utilizadas, uma vez que as análises são realizadas utilizando o
material na sua forma natural, sem a necessidade de tratamento prévio.

76
6 - SUGESTÕES
1) Realizar análises por RMN no estado sólido, empregando as técnicas de rotação da
amostra segundo o ângulo mágico (MAS) e polarização cruzada e rotação segundo o
ângulo mágico (CPMAS).
2) Investigar o material por infravermelho utilizando os recursos de temperatura.
3) Comparar as mudanças organizacionais causadas pela indução de cristalinidade por
temperatura.
4) Realização dos testes mecânicos tensão versus deformação e impacto.

77
7- BIBLIOGRAFIA 1 – DINIZ, T.M.F.F. Avaliação da miscibilidade e propriedades da mistura ternária de poli(metacrilato de metila) / polivinilpirrolidona e poli(óxido de etileno). 2001. 343 p. Tese de Doutorado – Instituto de Macromoléculas Professora Eloisa Mano, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2001. Orientadora: Maria Inês Bruno Tavares. 2 – WARZELHAN, V. TRENDS in strutural polymer science in industry. Poly. Adv. Technol, V. 8, p. 212-214, 1996. 3 – PASTORINE, M.T, Misturas ABS / policarbonato e mica. Aspecto técnico e de mercado. 1998. 138 p. Dissertação (Mestrado em Ciência e Tecnologia de Polímeros) – Instituto de Macromoléculas Professora Eloísa Mano, Universidade Federal do rio de Janeiro, Rio de Janeiro, 1998. Orientadores: Regina Célia Nunes e Carlos Alberto Hemais Pires Galvão. 4 – O Plástico no Brasil. Plástico em Revista. V. 58, p. 393, 1994. 5 – HORRON, H.R, PRITCHARD,R.G., COPE,B.C., GODDARD, D.T. An atomic force microscope films. J. Polym. Sci. Polym. Phhys, V. B 34, p. 173-180, 1996. 6 - PAKULL, R., GRIGO, U., FREITAS, D. Polycarbonate. Rapra Review Reports – Report, V. 42, 4, 6, p. 3-30, 1991. 7 – ODIAN, G. Principles of Polymerization. New York: Jonh Wiley & Sons. Inc., 1991. 234 p. 8 – BOCAYUVA, L. R. Indução de cristalinidade do policarbonato e avaliação de propriedades de suas misturas físicas. 2000. 248 p. Tese (Doutorado em Ciência e Tecnologia de Polímeros) – Instituto de Macromoléculas Professora Eloísa Mano, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2000. Orientadores: Clara Marize Firemand Oliveira, Márcia Cristina Veiga Amorim e Ailton de Souza Gomes. 9 – BECHER, E. D. High Resolution NMR. 2nd ed. New York: Academic Press, 1992. 409 p. 10 – LANKIN, E. D, FERRARO, R. R, JORNUTOWSKI, R. Spectroscopy, V. 7, p. 18-22, 1992. 11 – MERAT, P.P. Fatores que influenciam o assinalamento estrutural em compostos poliméricos. In: MARQUES, M.F.V., (C00rd.). MMP 751 – Seminário de mestrado. Rio de Janeiro: IMA/UFRJ, 2003. p. 233-258. (Seminários de mestrado IMA/UFRJ, V. 25, p. 2).

78
12 – SOUZA, C.M.G. Emprego de técnicas de RMN na caracterização de polímeros. In: ANDRADE, C.T., (Coord.). MMP 851 – Seminário de doutorado. Rio de Janeiro: IMA/UFRJ, 1997. p. 333-358. (Seminário de doutorado IMA/UFRJ, V. 15, p. 1). 13 – TAVARES, M.I.B. Estudo de sistemas de PVC com plastificante através da medida dos tempos de relaxação dos núcleos de próton e carbono – 13. 1991. 222 p. Tese (Doutorado em Ciência e Tecnologia de Polímeros) – Instituto de Macromoléculas Professora Eloísa Mano, Universidade Federal do Rio de Janeiro, 1991. Orientadora: Elisabeth Ermel da Costa Monteiro 14 – BOVEY, F.A.; MIARU, P.A. NMR of Polymer. New York: Academic Press, 1996. 230 p. 15 – TAVARES, M.I.B., MONTEIRO, C. E. NMR studies of PVC molecular mobility. Polym. Test., V. 14, p. 273-278, 1995. 16 – GUEDES, F. B., AZEVEDO, E.R., BONAGAMBA, T.J. NMR characterization of segmental dynamics in glassy poly(alkyl methacrylate) and poly(methyl α-substituted acrylate)s using cobex and purex. In: VIII Encontro de usuários de ressonância magnética nuclear, resumos, 2001, Rio de Janeiro, AUREMN, 2001. p. 287-289. 17 – BONAGAMBA, T. J., GUEDES, F.B., AZEVEDO, E.R., ROHR, S.K. Slow dynamics of polymers studies by center band – only detection of exchange and pure – exchange solid – state NMR. In: VIII Encontro de usuários de ressonância magnética nuclear / I Encontro luso-brasileiro de ressonância magnética nuclear, resumos, 2001, Rio de Janeiro, AUREMN, 2001. p. 10-11.
18 – MARQUES, R.G.G, TAVARES, M.I.B. Avaliação do sistema PIB/Parafina pela relaxação spin-rede. In: VIII Encontro de usuários de ressonância magnética nuclear / I Encontro luso-brasileiro de ressonância magnética nuclear, resumos, 2001, Rio de Janeiro, AUREMN, 2001. p. 127-128. 19 – FREITAS, J.C.C., BONAGAMBA, T. J. Fundamentos e aplicações da ressonância magnética nuclear. Rio de Janeiro: Ed. Univ., 1999. V. 1, 57 p. 20 – YU,T., GUO, M. Recente developements in 13C solid-state high – resolution NMR of polymer. Prog. Polym. Sci., V. 15, p. 825-908, 1990.

79
21 – LYERLA, J.R., HORIKAWA, T.T. Carbon 13 spin-lattice relation times of isotatic and syndiotatic polymethyl mathacrylate. J. POLYM. Sci. Polym. Lett., V. 14, p. 641-643, 1979. 22 – SCHNELL, H., BAYER, F. Policarbonates in ect. 2 Th Ed. London: Reinhold Publishing Corporation, V. 16. p. 1-6. 1962. 23 – FUCHS, O., SUHR, H.H. Solvents and non-solvents for polymers em polymer handbook. BRANDRUP, J., IMMERGUT, E. H. New York: Eds. J. Wiley & Sons, 1975, Part IV, 241 p. 24 – CASSU, S. N., FELISBERTI, M. I. Comportamento dinâmico – mecânico e relaxações em polímeros e blendas poliméricas. Química Nova., V. 28. Nº 2. 2005.
25 – CALLISTER, W. D. Jr. Material science and engineering – An introduction. New York: John Wiley & Sons, Inc., 1997. 598 p.
26 – FRIED, J. R. Polymer science and technology. New Jersey: Prentice Hall PTR, Englewood Cliffs, 1995. 380 p.
27 – BRANDRUP, J., IMMERGUT, E. H. Polymer Handbook. 3rd Edition. New York: John Wiley & Sons, 1989.
28 – SIMIELLI, E. R. Principais características das blendas poliméricas fabricadas no Brasil. Polímeros: ciência e Tecnologia - ABPOL. 45-49 . jan/mar. 1993.
29 – COWIE, J. M. G., Polymers: Chemistry & Physics of modern materials.New York: Blackie Academic Professional. 1991. 304 p.
30 – TURSKA, E., JANECZEK, H. Liquid – induced crystalization. J. Polym. Sci. Polym. Phys., V. 14. p. 1367-1377. 1979.
31 – NASSAR, T. R., PAUL, D. R., BARLOW, J. W. Polyester-polycarbonate blends. Poly(ethyleneterephthalate). J. Appl. Polim. Sci. V. 23. p. 85-99. 1979.
32 – KALKAR, A. K., ROY, N. R. Thermal and dynamic mechanical properties of polycarbonate / poly(p-t-butylphenol formaldehyde) blends films. Eur. Polym. J., V. 29. p. 1391-1398. 1993.

80
33 – WARE, R. A. , TIRTOWIDJOJO, S., COHEN, C. Diffusion and induced crystalization in polycarbonate. J. Appl. Polym Sci., V. 26. p. 2975-2988. 1981.
34 – CHEUNG, Y. W., STEIN, R. S., Evolution of crystaline structures of poly(ε-caprolactone) / polycarbonate blends. Isothermal crystallization kinetics as probed by synchrotron small-angle X-ray scattering. Macromolecules., V. 27. p. 3589-3595. 1994. 35 – GALLRZ, F., LEGRAS, R., MERCIER, S. P. Some new aspects of the crystallization of bisfenol-A polycarbonate. Polym, Eng. Sci., V. 16. p. 276-283. 1976. 36 – AMORIM, M. C. V., Estudo da influencia de poli(óxido de etileno) e de poli(óxido de propileno) em misturas com poli(metacrilato de metila) e com policarbonato. 1996. 244 p. Tese (Doutorado em Ciência e Tecnologia de polímeros) – Instituto de Macromoléculas Professora Eloísa Mano, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 1996. Orientadora:Clara Marize Firemand Oliveira.
37 – XUE, G., WANG,Y., LU, Y. Rapid crystalization of isotatic polystyrene by shock – cooling and subsequent freeze – dryng of its very dilute solution. Macromolecules.,V. 27. p. 4016 – 4017. 1994.
38 – SILVERSTEIN, R. M., BASSLER, G. C., MORRIL, T. C. Spectrometric identification of organic compounds., 5th Ed. Singapore: John wiley and Sons Inc, 1991. 255 p.
39 – DELPECH, M. C., COUTINHO, F. M. B., HABIBE, M. E. S.Bisphenol A-based polycarbonates: characterization of commercial samples. Polymer Testing., V. 21. p. 155-161. 2002.
40 – VARNELL, D. F., RUNT, J. P., COLEMAN, M. M. Fourier transform infrared studies of polymer blends. 6. Further observations on the poly(bispfenoll-A carconate) – poly(ε-caprolactone) system. Macromolecules., V. 14. p. 1350-1356. 1981.
41 – SEPE, M. P. Thermal analysis of polymers. Rapra Review Reports – Report., V. 95. p. 03-39. 1997. 42 – TURSKA, E., JANECZEK, H. Liquid – induced crystalization. J. Polym. Sci. Polym. Phys., V. 14. p. 1367-1377. 1979.
43 – BLUNDELL, D. J., BECKETT, D. R., WILLCOCKS, P. H. Routine crystallinity measurements of polymers by d.s.c. Polymer, V. 22, p. 704-707. 1981.

81
44 – JI, G., XUE, G., MA, J., DONG, C., GU, X. Concentration dependence of crystallinty of polycarbonate by shock-cooling and subsequent freeze – dryng of its various solutions. Polymer, V. 37, p. 3255-3258. 1996.
45 – BOLETIM DE APLICAÇÃO: análise térmica em polímeros. Disponível em: <http://www.micronal.com.br/artigostecnicos/analise-termica-plimeros> . Acesso em : 14 jun. 2005.
46 - SILVA, M. A., Preparo de membranas com transporte facilitado e caracterização por ressonância magnética nuclear. 2004. 86 p. Dissertação ( Mestrado em Ciência e Tecnologia de Polímeros) – Instituto de Macromoléculas Professora Eloísa Mano, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2004. Orientadoras: Maria Inês Bruno Tavares e Vera Maria Martins Salim.
47 – ALEXANDER, L. E. X- ray diffraction methods in polymer science. New York: Robert E. Krieger Publishing company, 1979. 342 p.
48 – RUNT, J.R. Crystallinity determination. In: Encyclopedia of polymer science and engineering, Editado por MARK,H.M., BIKALES, N.M., OVERBERG, C.G., MENGES, G. New York: Jonh Wiley & Sons. 1985. V. 4, p. 488-509.
49 – BYE, D. J., MILES, I. S. The variation of viscosity with composition for polystyrene/ polycarbonate blends and its dependence on method of sample preparation. European Polymer Journal., V. 22. p. 185-187. 1986. 50 – SOUZA, C. M. G. Emprego de técnicas de ressonância magnética nuclear na caracterização de polímeros. In: MARQUES, M. F. V., (Coord.). MMP 851 – Seminário de doutorado. Rio de Janeiro: IMA/UFRJ, 1997. p. 153-178. (Seminário de doutorado IMA/UFRJ, V. 17, p. 1).
51 – DUARTE, L. T., HABERT, A. C., BORGES, C. P. Preparation and morphological characterization of polyurethane/polyethersulfone composite membranes. Desalination, V. 145, p. 53-59. 2002.
52 – WEBB, G. A., BELTON, P. S., GIL, A. M., DELGADILLO, I., Magnetic resonance in food science a view to the future. Inglaterra: Ed. RS. C. 1º ed. , 2001. 455 p.
53 – GILL, V. M. S., GERALDES, C. F. G. C., Ressonância magnética nuclear-Fundamentos, métodos e aplicações., 2º ed. Lisboa: Fundação Calouste Gulbenkian, 2002. 1012 p.

82
54 –AXELSON, D. E., RUSSEL, K. E.Characterization of polymers by means of 13C NMR spectroscopy. Polym. Sci., V. 11. p.221-282. 1985.
55 – ABRAHAN, J., FISHER, J., LOFTUS, P., Introduction to NMR spectroscopy. New York: John Wiley & Sons, 1991. 322 p.
56 - ABRAHAM, J.R.;HAWORTH,I.S.;BUN,A.;HEARMON,A.R. Substituent effects in the 13C NMR spectra of aryl ether copolymers. 6. Sulphonated aryl ether ketones. Polymer., V. 32. p. 1414-1419. 1991.
57 - KATSURAYA,K.;HATANAKA,K.;MATSUZAKI,K.;MINAGAWA,M. Assignment of finely resolved 13C NMR spectra of polyacrylonitrile. Polymer., V. 42. p. 6323-6326. 2001.
58 - HANSEN, E. W.; BLOM, R.; BADE, O. M. NMR Characterization of polyethylene with emphasis on internal consistency of peak intensities and estimation of uncertainties in derived branch distribution numbers. Polymer., V. 38. p. 4295-4304. 1997.
59 – DEPOOTER, M., SMITH, P. B., DOHRER, K. K., BENNETT, K. F., MEADOWS, M. D., SMITH, C. G., SCHOUWENAARS, H. P., GEERARDS, R. A .J. Appl. Polym Sci., V. 42. p. 399-405. 1991.
60- NI,S.;TANG,J.;WANG,F.; SHEN,L. 13C NMR characterization of soluble polyaniline. Polymer., V. 33. p. 3607-3609. 1992.
61 - TONELLI, A. E. Are the steric effects on the 13C-NMR chemical shifts of hydrocarbon polymers really long range?; Macromolecules., V. 12. p. 255-256. 1979. 62- HOBSON,L.J.;FEAST,W.J. Poly(amindoamine) hyperbranched systems: synthesis, structure and characterization. Polymer., V. 40. p. 1279-1297. 1999.
63 - KATSURAYA, K.; HATANAKA, K.; MATSUZAKI, K. AMIYA, S. Assignment of finely resolved 13C NMR spectra of poly(vinyl alcohol). Polymer., V. 42 . p. 9855-9858. 2001.
64 -NYDEN, M. R.; VANDERHART, D. L. ALAMO, G. The conformational structures of defest-containing chains in the crystalline regions of isotactic polypropylene. Comp. And Theor. Polym.Sci., V. 11. p. 175-189. 2001.
65 - McCALL, D. W. Molecular relaxation in polymers: nuclear magnetic resonance studies of moleculular relaxation mechanisms in polymers. New Jersey: Murray Hill, 1970. 232.

83
66 – BAILEY, R. T., NORTH, A. M., PETHRICK, R. A. Molecular motion in high polymers., Oxford: Clarendon Press. 1981. 285 p.
67 – SCHAEFER, J., STEJSKAL, E. O., BUCHDAHL, R. High-resolution carbon-13 nuclear magnetic resonance study of some solid, glassy polymers. Macromolecules., V. 8. p. 291-296. 1974.
68 – PARKER, A. A., MARCINKO, J. J., SHIEH, Y. T., HEDRICK, D. P., RITCHEY, W. M. A preliminary correlation between macroscopic and microscopic polymer properties: dynamic storge modulos vs. CPMAS NMR cross polarization rates. Journal of applied polymer science., V. 40. p. 1717-1725. 1990.
69 – VANDERHART, D. L., McFADDEN , G. B. Some perspectives on the interpretation of proton NMR spin diffusion data in terms of polymer morphologies. Solid state nuclear magnetic resonance., V. 7. p. 45-66. 1996.
70 – MASSON, J. F., MANLEY, R. S. J. Solid-state NMR of some cellulose / synthetic polymer blends. Macromolecules., V. 25. p. 589-592. 1992.
71 – ZHAO, J., JONES, A. A., INGLEFIELD, P. T., BENDLER, J. T. A spin-lattice relaxation study of dissolved cyclohexyl polycarbonate. Polymer., V. 37. p. 3783-3790. 1996.
72 – ZHANG, X., TAKEGOSHI, K., HIKICHI, K. Composition dependence of the miscibility and phase struture of amorphous /crystalline polymer blends as studied by high-resolution solid-state 13C NMR spectroscopy. Macromolecules., V. 25. p. 2336-2340. 1992.
73 – CHARLESBY, A. Characterization of macromolecular morphology by pulsed NMR spectroscopy. American Chemical Society symposium series., V. 475. p. 193-217. 1991.
74 – BERRIOT, J., MARTIN, F., MONTES, H., MONNERIE, L., SOTTA, P. Reinforcement of model filled elastomers: characterization of the cross-linking density at the filler – elastomer interface by 1H NMR measurements. Polymer., V. 44. 1437-1447. 2003.
75 – LEQUIEU, W., VELDE, P. V., DU PREZ, F. E., ADRIAENSENS, P., STORME, L., GELAN, J. Solid state NMR study of phase morphology via their molecular design. Polymer., V. 45. p. 7943-7951. 2004. 76 – KOLODZIEJSKI, W., KLINOWSKI, J. Kinetics of cross-polarization in solid-state NMR: a guide for chemists. Chem. Rev., V. 102. p. 613-628.

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo