Área de ciÊncias tecnolÓgicas curso de mestrado em...
TRANSCRIPT

CENTRO UNIVERSITÁRIO FRANCISCANO
PRÓ-REITORIA DE PÓS-GRADUAÇÃO, PESQUISA E EXTENSÃO
ÁREA DE CIÊNCIAS TECNOLÓGICAS
Curso de Mestrado em Nanociências
FABRICIO ANDRE DUTRA
MODELAGEM AB INITIO DA CISTEÍNA ADSORVIDA EM GRAFENO
Santa Maria, RS
2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

FABRICIO ANDRE DUTRA
MODELAGEM AB INITIO DA CISTEÍNA ADSORVIDA EM GRAFENO
Dissertação apresentada ao Curso de
Mestrado em Nanociências do Centro
Universitário Franciscano de Santa Maria-
RS como requisito parcial para obtenção
do título de Mestre em Nanociências.
Orientador(a): ProfaDra SOLANGE BINOTTO FAGAN
Santa Maria, RS
2010


Ficha Catalográfica
D978m
Dutra, Fabricio Andre Modelagem AB Initio da cisteína adsorvida em grafeno / Fabricio Andre Dutra ; orientação Solange Binotto Fagan. – Santa Maria, 2010.
66f. : il. Dissertação (Mestrado em Nanociências) – Centro Universitário Franciscano.
1. Grafeno 2. Aminoácidos 3. Adsorção I. Fagan, Solange Binotto. II. Título.
CDU 546.26:62-181.4
Elaborada pela Bibliotecária Zeneida Mello Britto CRB10/1374

DEDICATÓRIA
Para a Aline pelo imenso carinho e apoio e para meus pais,
Pedro (in memorian) e Cerlei.

AGRADECIMENTOS
Dedico meus agradecimentos às todos que de alguma forma colaboraram com a
realização deste trabalho:
– a Deus, sou grato por todas as oportunidades que me foram dadas;
– à Profa. Dra. Solange B. Fagan, pela orientação, tolerância, paciência, e pelos
ensinamentos.
– à Profa. Dra. Ivana Zanella, pela co-orientação neste trabalho;
– ao Prof. Dr. Leandro Barros da Silva, pela grande ajuda e colaboração neste
trabalho;
– aos colegas de pós-graduação da UNIFRA, pelo incentivo, troca de saberes e
amizade; em especial à Elenice (Nena), ao André, ao Heleno (Neno) e ao Paulo pelo
companheirismo principalmente em meus primeiros passos neste curso;
- à Vivian, colega da UFSM, pela ajuda na fase final;
– aos demais professores do Mestrado em Nanociências, que, de alguma forma,
colaboraram na minha formação acadêmica;
– ao CENAPAD – SP, pelo espaço disponível para a realização dos cálculos e
suporte técnico;
- à todas as pessoas que compreenderam o quão difícil foi conciliar o trabalho e o
curso;
– aos meus familiares, pelo apoio incondicional nas minhas decisões, em especial à
Aline pelo companheirismo, amor e incentivo constantes.

RESUMO
O grafeno consiste numa rede bidimensional, composta unicamente de
átomos de carbono dispostos em anéis hexagonais; sendo recentemente
caracterizado como “material mais fino do universo”, o qual exibe alta qualidade
cristalina e transporte elétrico balístico em baixas temperaturas. Neste material as
propriedades eletrônicas e a grande área superficial são algumas das propriedades
mais interessantes de modo que podem ser adsorvidos ou substituídos aminoácidos,
visando aumentar a hidrofilicidade da estrutura originalmente constituída apenas de
átomos de carbono. Neste trabalho visamos avaliar as propriedades estruturais e
eletrônicas resultantes da adsorção ou substituição de átomos de carbono pelo
aminoácido cisteína, bem como investigar o tipo de interação estabelecida entre este
e a monocamada de grafeno perfeita ou com defeito estrutural. Avaliamos estas
propriedades por meio de simulação computacional de primeiros princípios utilizando
a Teoria do Funcional da Densidade. Indicamos as propriedades eletrônicas e
estruturais de aminoácidos substituindo átomos de carbono na estrutura do grafeno
ou adsorvendo na superfície. Constatamos que a energia de ligação entre o
aminoácido e a monocamada de grafeno varia, sendo totalmente dependente do
sítio reacional que aproximamos, sendo que dentre os três sítios considerados
(carboxila, amino e tiol), tanto na aproximação da camada de grafeno perfeita quanto
da camada com defeito estrutural, o grupo tiol foi o que demonstrou uma interação
mais forte com o grafeno, além de apresentar também uma menor distância de
ligação entre a monocamada e a molécula de aminoácido. Nas estruturas de
bandas, demonstramos a pouca interação entre os sistemas, pelo fato de os níveis
dos sistemas isolados aparecerem claramente nas estruturas dos sistemas
interagentes, o que evidencia a baixa energia de ligação encontrada. Desta forma,
este trabalho demonstrou, de forma inédita, as propriedades associadas com a
interação da cisteína adsorvida no grafeno fomentando o potencial de aplicação
tecnológica deste material inovador como sensor de moléculas mais complexas.
Palavras-chave: grafeno, aminoácidos, adsorção.

ABSTRACT
The graphene is a two-dimensional network composed of carbon atoms
arranged in hexagonal rings; characterized as "thinner material of the universe",
which exhibits high crystal quality and ballistic electrical transport at low
temperatures. This material presents electronic properties and large surface
area that are interesting to adsorb or substitut aminoacids, to increase the
hydrophilicity of the structure originally constituted only of carbon atoms and
then enable the development of new sensors with high molecular sensitivity.
This work aim to evaluate the structural and electronic properties of the
grapheme interacting with the aminoacid cysteine, and the type of interaction
established between this and monolayer graphene perfect or structural defect.
These properties are investigated through computer simulation from first
principles methods using the density functional theory that indicate the
electronic and structural properties of the resulting systems. We found that the
binding energy between the amino and graphene monolayer varies, being
totally dependent on the reaction site and among the three sites considered
(carboxyl, amine and thiol), both, in considering the perfect layer of graphene or
with as structural defect, present the thiol group with a stronger interaction. In
the electronic properties, we demonstrate little interaction between the systems,
because the levels of isolated systems appear clearly in the structures of
interacting systems, which demonstrates the low binding energy found. Thus,
this study demonstrated, the properties associated with the interaction of amino
acids adsorbed on graphene encouraging potential technological application of
this innovative material as a sensor of more complex molecules.
Keywords: graphene, aminoacids, adsorption.

SUMÁRIO
1. INTRODUÇÃO .......................................................................................... 8
2. REVISÃO DE LITERATURA .................................................................... 10
2.1. CARBONO ............................................................................................... 10
2.2. HIBRIDIZAÇÃO DO CARBONO ............................................................... 10
2.3. ALÓTROPOS DO CARBONO ................................................................. 15
2.3.1. Grafite ....................................................................................................... 16
2.3.2. Diamante .................................................................................................. 17
2.3.3. Fulerenos .................................................................................................. 18
2.3.4. Nanotubos de Carbono ............................................................................. 19
2.3.5. Grafeno ..................................................................................................... 20
2.4. CISTEÍNA ................................................................................................ 27
2.5. GRAFENO COMO SENSOR DE MOLÉCULAS ...................................... 30
3. METODOLOGIA ....................................................................................... 32
3.1. APROXIMAÇÃO DE BORN-OPPENHEIMER .......................................... 33
3.2. TEORIA DO FUNCIONAL DA DENSIDADE ............................................ 34
3.2.1. Teoremas de Hohenberg e Kohn ............................................................. 36
3.2.2. Equações de Kohn-Sham ......................................................................... 37
3.2.3. Termo de Troca e Correlação ................................................................... 38
3.3. PSEUDOPOTENCIAL .............................................................................. 40
3.4. ORBITAIS ATÔMICOS ............................................................................ 42
4. RESULTADOS ......................................................................................... 45
4.1. PROCEDIMENTOS DE CÁLCULO .......................................................... 45
4.2. GRAFENO PERFEITO, GRAFENO COM DEFEITO E CISTEÍNA........... 46
4.3 ADSORÇÃO DA CISTEÍNA EM GRAFENO PERFEITO .......................... 49
4.4 ADSORÇÃO DA CISTEÍNA EM GRAFENO COM DEFEITO ................... 55
5. CONCLUSÃO ........................................................................................... 60
REFERÊNCIAS BIBLIOGRÁFICAS ........................................................ 62

8
1. INTRODUÇÃO
Atualmente, o interesse em materiais nanoestruturados vem crescendo de
forma extraordinária devido a sua potencial aplicação em diversas áreas científicas e
tecnológicas, tais como catálise, sensores químicos e biológicos, materiais
optoeletrônicos e materiais magnéticos. Dentre estes, o grafeno é um dos materiais
bi-dimensionais que promete grandes revoluções na área de nanodispositivos e
nanomateriais.
O grafeno é formado por uma única camada de grafite e foi recentemente
obtido experimentalmente como uma forma bi-dimensional de carbono, o qual exibe
alta qualidade cristalina e transporte elétrico balístico em baixas temperaturas
(NOVOSELOV et al , 2005; ZHANG et al, 2005), e recentemente foi caracterizado
como o “material mais fino do universo” (GEIM e NOVOSELOV, 2007). Neste
material as propriedades eletrônicas e a grande área superficial são umas das
propriedades mais interessantes de modo que podem ser adsorvidos ou substituídos
grupamentos funcionais orgânicos, visando aumentar a hidrofilicidade da estrutura
do grafeno (MEYER et al, 2007). Desta forma, pode-se acarretar uma maior
compatibilidade e seletividade com determinados sistemas biológicos, bem como
utilizar o grafeno para ancorar outros átomos ou moléculas (NOVOSELOV et al,
2008; ZANELLA et al, 2008).
Estudos recentes sobre a monossubstituição de grupos arila na camada do
grafeno constataram que depois de feita uma segunda substituição por outro
carbono do grafeno, do mesmo hexágono em posição “para-dissubstituído (1,4)”, ou
seja, dois substituintes em posição 1 e 4 no anel aromático, aumentaram a
intensidade desta ligação (JIANG, SUMPTER E DAI, 2006), bem como Pei e
colaboradores (2010) relatam que dependendo da posição e da quantidade de
grupos (grupamentos metila) adsorvidos no grafeno, as propriedades mecânicas são
radicalmente alteradas.
O estudo das propriedades dos nanotubos com grupamentos orgânicos
substitucionais e adsorvidos, demonstrou relevantes alterações eletrônicas em
relação à estrutura primária (VELOSO et al, 2006), o que motivou esta análise
análoga aplicada ao grafeno, uma vez que estudos nessa linha foram recentemente
desenvolvidos utilizando carbono amorfo (CHUTIA et al, 2008), átomos alcalinos e

9
halogênios (SILVA, 2008) e vários grupamentos orgânicos como dopantes isolados
(BOUKHVALOV E KATSNELSON, 2008).
Portanto, neste trabalho, realizamos simulações de primeiros princípios com o
aminoácido cisteína a fim de avaliar seu potencial como possível adsorvente numa
monocamada de grafeno, sem e com defeitos estruturais, para sua posterior
utilização como sensor de moléculas orgânicas e/ou inorgânicas.
Ressalta-se que não seria possível pensar neste trabalho de forma disciplinar,
por que houve necessidade da colaboração de várias áreas do conhecimento para
sua construção e concretização. Destaca-se a área da química que contribuiu com a
avaliação das possíveis estruturas a serem escolhidas para interagirem, a física que
fundamentou a metodologia do trabalho, a informática com o suporte computacional,
bem como todas as outras áreas que ajudaram a definir os objetivos deste trabalho,
que se fosse visto de forma disciplinar, não alcançaria os mesmos resultados.
Da mesma forma, a área da nanotecnologia, tem se desenvolvido mais
rapidamente nos últimos anos devido, em grande parte, ao avanço das técnicas de
análise estrutural e microscópica da matéria. Já há bastante tempo, estudos teóricos
na área de materiais esbarravam na impossibilidade de reproduzir, confirmar ou
refutar resultados teóricos na prática, hoje, em grande parte das situações, já
podemos testar em laboratório, in vitro ou in vivo, os resultados teóricos ou
simulados. Com isto, já existe uma gama de produtos nanoestruturados no mercado,
tanto no mercado restrito aos centros acadêmicos, quanto ao mercado varejista
acessível a qualquer pessoa. Nesse ínterim este trabalho vem a contribuir no sentido
em que dá suporte para a compreensão de como um aminoácido interage com esta
nova estrutura carbonácea que é o grafeno.
Tendo sido definida a motivação e os objetivos deste trabalho, no Capítulo 2
são apresentados os referenciais teóricos sobre o carbono e seus alótropos,
destacando-se o grafeno e suas propriedades estruturais, e também as principais
características da cisteína. No Capítulo 3 são descritos os fundamentos dos cálculos
de primeiros princípios, com ênfase na Teoria do Funcional da Densidade. No
Capítulo 4 são apresentados os procedimentos de cálculo e são apresentados e
discutidos os resultados obtidos. Finalmente, no Capítulo 5, são analisados os
resultados e apresentadas as conclusões finais.

10
2. REVISÃO DE LITERATURA
2.1. CARBONO
O carbono é um elemento químico de símbolo “C”, número atômico 6 (6
prótons e 6 elétrons), massa atômica 12u e sólido à temperatura ambiente.
Dependendo das condições de formação, pode ser encontrado na natureza em
diversas formas alotrópicas: carbono amorfo e cristalino, em forma de grafite,
diamante ou ainda nanotubos (IIJIMA, 1991) e grafeno (NOVOSELOV et al, 2004).
Pertence ao grupo (ou família) 14.
O carbono se destaca dentre os elementos químicos uma vez que forma um
grande número de compostos, mais do que todos os outros elementos combinados.
Este elemento, é o pilar básico da química orgânica, é o quarto elemento mais
abundante em massa no universo e se conhecem cerca de 10 milhões de
compostos de carbono e forma parte de todos os seres vivos. Mesmo o carbono
sendo capaz de formar esta grande variedade de compostos, a maioria desses são
pouco reativos sob condições normais de temperatura e pressão.
A singularidade do carbono se justifica por meio da análise dos diferentes
tipos de hibridização que seus orbitais da camada de valência compõem. A seguir
apresentamos as principais características das diferentes hibridizações do carbono
(SOLOMONS e FRYHLE, 2001).
2.2. HIBRIDIZAÇÃO NO CARBONO
O átomo de carbono possui número atômico 6 (Z=6) e portanto 6 elétrons,
distribuídos nos orbitais atômicos 1s2,2s2 e 2p2. Os elétrons do subnível 1s estão
fortemente ligados ao núcleo e não participam das ligações químicas. Os elétrons
responsáveis pelas ligações químicas são os elétrons da última camada, ou camada
de valência, no caso do carbono os elétrons dos subníveis 2s e 2p.
No estado fundamental, o carbono possui elétrons de valência distribuídos
nos orbitais 2s, 2px e 2py, e nesta situação o átomo de carbono não poderia atingir a
estabilidade eletrônica, pois desta forma só poderia haver a formação de duas
ligações química covalentes, uma vez que estas se formam através de

11
emparelhamento de elétrons com spins contrários. A seguir apresentamos como o
carbono consegue formar as quatro ligações químicas necessárias para atingir a
estabilidade eletrônica clássica (8 elétrons na camada de valência) (SOLOMONS e
FRYHLE, 2001).
O carbono ao formar uma ligação química passa por três etapas
energeticamente distintas:
Estado Fundamental 1s2 2s2 2p2
Estado Excitado 1s2 2s1 2p3
Estado Hibridizado sp3 – 1s2 2sp3 2sp3 2sp3 2sp3
sp2 – 1s2 2sp2 2sp2 2sp2 2p (puro)
sp – 1s2 2sp 2sp 2p 2p
Na Figura 2.1 estão representados os diferentes estados do carbono,
fundamental e excitado.
Figura 2.1: Diagrama energético das diferentes fases do carbono durante o
processo de ativação ou excitação (SOLOMONS e FRYHLE, 2001).
A formação dos orbitais híbridos sp, sp2
e sp3
no carbono é devido a
sobreposição dos orbitais s e p da camada de valência (por estarem em níveis
energéticos próximos) para formar novos orbitais.
No carbono sp3, todos os quatro elétrons de valência encontram-se
igualmente distribuídos em quatro orbitais híbridos degenerados do tipo sp3. Nesta
configuração, o átomo de carbono central possui seus 4 elétrons localizados nos
vértices de um tetraedro regular. As ligações formadas são do tipo σ, e o ângulo
formado entre estas ligações é de 109°28’.

12
A Figura 2.2 apresenta um esquema energético do carbono normal ou
fundamental ao carbono sp3.
Figura 2.2: Diagrama energético das diferentes fases do carbono durante o
processo de hibridização sp3 (SOLOMONS e FRYHLE, 2001).
Como principal exemplo de estrutura que apresenta o carbono sp3, temos a
estrutura do gás metano (CH4), Figura 2.3.
Figura 2.3: Gás metano (SOLOMONS e FRYHLE, 2001).
Para o carbono sp2, 3 elétrons encontram-se igualmente distribuídos em três
orbitais híbridos degenerados e simétricos (ligações σ) e o quarto elétron restante no
orbital p (puro) é capaz de formar uma ligação π (p-p) com outro orbital p. A
configuração estrutural (geometria molecular) apresentada neste caso é do tipo
trigonal plana (120°).
A Figura 2.4 apresenta um esquema energético do carbono normal ou
fundamental ao carbono sp2.

13
Figura 2.4: Diagrama energético das diferentes fases do carbono durante o
processo de hibridização sp2 (SOLOMONS e FRYHLE, 2001).
Como principal exemplo de estrutura que apresenta o carbono sp2, temos a
estrutura do gás eteno (C2H4), Figura 2.5.
Figura 2.5: Gás eteno (SOLOMONS e FRYHLE, 2001).
Já no carbono sp, dois elétrons formam dois orbitais híbridos simétricos
(ligações σ) e os dois elétrons restantes nos orbitais p (puros), formam duas ligações
do tipo π (p-p). A configuração estrutural para a hibridização sp é linear, com um
ângulo de 180°
entre as ligações.
A Figura 2.6 apresenta um esquema energético do carbono normal ou
fundamental ao carbono sp.

14
Figura 2.6: Diagrama energético das diferentes fases do carbono durante o
processo de hibridização sp (SOLOMONS e FRYHLE, 2001).
Como principal exemplo de estrutura que apresenta o carbono sp, temos a
estrutura do gás etino (C2H2), Figura 2.7.
Figura 2.7: Gás etino ou acetileno (SOLOMONS e FRYHLE, 2001).
A Tabela 2.1 resume as principais características estruturais de cada estado
híbrido do carbono.
Na próxima seção, discutiremos as diferentes substâncias simples (alótropos)
que podem ser formadas pela união dos átomos de carbono a partir de suas
diferentes hibridizações.

15
Tabela 2.1: Síntese das propriedades acerca das diferentes hibridizações
(SOLOMONS e FRYHLE, 2001).
Hibridização N° de
orbitais híbridos
Tipo de Interação entre os orbitais
Tipo de ligação
Forma espacial dos
orbitais
Ângulo entre as ligações dos orbitais
híbridos
sp3 4 4σ 4 ligações
simples tetragonal 109° 28’
sp2 3 3σ + 1π 2 ligações
simples + 1 dupla
trigonal planar
120°
sp 2 2σ + 2π
1 ligação simples + 1 tripla ou 2
duplas
linear 180°
2.3. ALÓTROPOS DO CARBONO
Alotropia é a propriedade que alguns elementos químicos apresentam e
consiste na possibilidade destes elementos formarem diferentes substâncias
simples, no entanto, o carbono é o único elemento capaz de formar alótropos com
dimensionalidades diferentes, tais como 0D (fulerenos), 1D (nanotubos), 2D
(grafenos) e 3D (grafite) e ainda formar estruturas amorfas (SAITO et al., 1998). O
que justifica esta versatilidade é o fato de o carbono como já visto anteriormente,
interagir com outros átomos através dos mais diferentes orbitais híbridos (sp, sp2 e
sp3, nas ligações sigma (σ)), bem como realizar ligações múltiplas (uma ou duas
ligações pi (π)), nos orbitais “p” puros restantes (não híbridos).
De forma resumida, a Tabela 2.2 apresenta as principais propriedades dos
alótropos e das nanoestruturas do carbono, distribuídos a partir de sua
dimensionalidade; dimensão zero (0D), unidimensional (1D), bidimensional (2D) e
tridimensional (3D).

16
Tabela 2.2: Exemplos de estruturas alotrópicas do carbono com diferentes
dimensionalidades (SAITO et al, 1998).
Dimensão 0D 1D 2D 3D
Alótropo C60
Fulerenos Nanotubo Grafeno
Diamante, Grafite,
C-amorfo
Hibridização sp2 sp2(sp) sp2 sp3
Densidade (g/cm3)
1,72 1,20 - 2,00 2,68 - 3,13
2,26 ~2,00
3,52 2-3
Comprimento da ligação
(Å)
1,40 (C=C) 1,46 (C-C)
1,44 (C=C) 1,42 (C=C) 1,44 (C-C)
1,52 (C-C)
Propriedades eletrônicas
Semicondutor Egap=1,90 eV
Metal ou semicondutor Egap~variável
Semicondutor ou condutor
com Efeito Hall Quântico
Isolante, não-metal Egap=5,47
eV
A partir de agora, estudaremos as diferentes propriedades de cada um dos alótropos do carbono. 2.3.1. Grafite
Também chamada chumbo negro ou plumbagina, o grafite tem múltiplas e
importantes aplicações industriais, embora seja mais conhecida popularmente por
sua utilização como mina do lápis. (SOLOMONS e FRYHLE, 2001).
No grafite cada átomo de carbono está unido a outros três em um plano
composto de células hexagonais. Neste estado, 3 elétrons se encontram em orbitais
híbridos planos sp2 e o quarto em um orbital p (Figura2.8). São conhecidas duas
formas de grafite, uma é denominada α (hexagonal), tendo uma estrutura do tipo
ABAB... e a outra é conhecida como β (romboédrica), com estrutura do tipo
ABCABC...
Mineral de variadas propriedades físicas, o grafite tem numerosas aplicações
industriais. É mole, facilmente desgastável, untuosa e de boa condutibilidade
elétrica. Naturalmente encontra-se em três formas, que determinam o emprego
industrial: amorfa, cristalina e em lâminas. A forma amorfa formou-se por intrusões
ígneas em leitos de carvão, que se calcinou, convertendo-se em grafita, cuja pureza
raramente é superior a 85%. A forma cristalina ocorre em grupos maciços de cristais

17
de brilho argênteo e sua pureza supera 99%. O grafite em escamas, a mais rara e
em alguns casos a mais valiosa, encontra-se disseminada em rochas que
experimentaram alto grau de metamorfose local.( (SOLOMONS e FRYHLE, 2001).
É utilizada na fabricação de cadinhos refratários para as indústrias do aço, do
latão e do bronze sendo usado, também, como lubrificante. Misturado com argila
muito fina forma a mina do lápis; empregando-se ainda largamente na fabricação de
tinta para proteção de estruturas de ferro e de aço.
Figura 2.8: Grafite (SOLOMONS e FRYHLE, 2001).
2.3.2. Diamante
No diamante, cada átomo de carbono encontra-se covalentemente ligado a
quatro outros átomos de carbono localizados nos vértices de um tetraedro regular,
com uma distância interplanar de 1,54 Å, apresentando uma hibridização do tipo sp3
(Figura 2.9).
O diamante é um material isolante com band gap igual a 5,5 eV. O diamante
natural é um material bastante raro. As impurezas presentes na estrutura do
diamante são muito importantes, uma vez que tais impurezas provocam mudanças
na condutividade térmica e elétrica deste material. Estas modificações nas
propriedades encontram aplicações principalmente em processos industriais.
Somente algumas espécies químicas são capazes de entrarem como substituintes
no retículo do diamante, devido a isto, os melhores diamantes naturais contém
impurezas com concentrações de aproximadamente 0,1 ppm (SOLOMONS e
FRYHLE, 2001).

18
Figura 2.9: Diamante. (SOLOMONS e FRYHLE, 2001).
2.3.3. Fulerenos
Os fulerenos são moléculas ocas de carbono, formadas por uma superfície
curva com ligações do tipo sp2. Podem possuir diferentes quantidades de átomos de
carbono, sendo formados por 20, 60, 70, 100, 180, 240 e até 540 átomos de carbono
(sempre números pares de átomos de carbono). O C60, o mais conhecido e
abundante dos fulerenos, possui simetria icosaédrica, consistindo de 20 anéis
hexagonais e 12 pentagonais, parecendo uma de bola de futebol com 7Å de
diâmetro. A molécula C60 é conhecida como buckyball e sua descoberta rendeu a
Kroto, Smalley e Curl (KROTO et al., 1985), o prêmio Nobel de Química, em 1996.
Mas para chegar até aí, não foi fácil, afinal eles precisaram imaginar como os 60
átomos de carbono poderiam se arranjar para formar um composto estruturalmente
estável, um cluster contendo apenas átomos de carbono, baseado no que se
conhecia até então a respeito de carbono não seria possível formar uma esfera oca
constituída apenas por átomos de carbono. Imaginaram então construir uma
estrutura com ligações entre orbitais híbridos sp2 e desta forma uma estrutura
esferoidal poderia satisfazê-lo, desde que não fosse constituída apenas de
hexágonos, assim sendo, basearam-se nos estudos do arquiteto americano R.
Buckminster Fuller e propuseram um icosaedro truncado, constituído de 20
hexágonos e 12 pentágonos onde cada vértice representaria um átomo de carbono
com o aspecto típico de uma bola de futebol (Figura 2.10).

19
Figura 2.10: Fulereno. (SOLOMONS e FRYHLE, 2001).
2.3.4. Nanotubos de Carbono
Nanotubos de carbono (NTC) são materiais cuja estrutura química básica é
formada por uma folha de grafeno enrolada, em dimensões nanométricas, podendo
suas extremidades ser fechadas, por uma espécie de “abóboda” de grafite,
formando uma cavidade interna oca. Em 1991, Sumio Iijima (IIJIMA, 1991) observou
a existência de compostos formados de múltiplas camadas de folhas de grafeno
enrolados de forma cilíndrica (Figura 2.11), através da microscopia eletrônica. Era a
descoberta de uma nova família de alótropos do carbono, os chamados nanotubos,
devido à sua morfologia tubular com dimensões nanométricas. Esses nanotubos de
múltiplas camadas, MWCNs (multiwall carbon nanotubes), foram sintetizados
utilizando técnicas similares às utilizadas para a produção de fulerenos. Dois anos
depois, foram sintetizados os nanotubos de uma única camada, os SWCNs (single-
wall carbon nanotubes), gerados de uma única folha de grafeno enrolada de forma
cilíndrica (IIJIMA e ICHIHASHI, 1993).
Figura 2.11: Imagens obtidas por Iijima em um microscópio eletrônico de
transmissão de nanotubos de carbono de múltiplas camadas (a-d) e de uma camada
(e) (adaptada de IIJIMA, 1991).

20
Os nanotubos de carbono possuem propriedades mecânicas e eletrônicas
notáveis, devido à combinação de sua dimensionalidade (grande razão
comprimento/diâmetro), estrutura e topologia, e são os compostos mais rígidos (na
direção perpendicular ao seu eixo), flexíveis (na direção normal ao eixo) e
resistentes às tensões que já foram produzidas (DRESSELHAUS et al., 1996).
2.3.5. Grafeno
O grafeno, o qual é a mais recente de todas as formas alotrópicas do
carbono, foi sintetizado apenas em 2004, sendo constituído por uma das folhas
isoladas do grafite, apresentando, assim, hibridização sp2. Por outro lado, físicos
teóricos presumiam que um filme de carbono com um átomo de espessura (grafeno)
jamais poderia ser estável, não podendo existir no estado livre (ASCROFT e
MERMIN, 1976).
Porém, em 2004, o grupo de André Geim e Kostya Novoselov (NOVOSELOV
e GEIM, 2004) conseguiu isolar um único plano do grafite por esfoliação mecânica
do grafite de um lápis através de uma fita adesiva. Assim, foi obtido pela primeira
vez o grafeno, material estável e flexível que promete uma revolução em aplicações
na indústria de semicondutores. Na próxima seção discutiremos de forma mais
aprofundada as propriedades desta nova forma alotrópica de carbono
(NOVOSELOV et al, 2005).
Obtenção do Grafeno
Em 2004 a equipe de Geim e Novoselov preparou filmes de grafeno por meio
de esfoliação mecânica, obtendo de forma termodinamicamente estável, as
primeiras estruturas isoladas bidimensionais de carbono (NOVOSELOV et al, 2005).
Inicialmente eles utilizaram uma plaqueta de 1mm de espessura de HOPG (Highly
Ordered Pyrolytic Graphite). Usando a técnica de gravura seca com plasma de
oxigênio produziram blocos de grafite com 5μm de profundidade no topo da plaqueta
(mesas quadradas de comprimentos variando de 20μm a 2mm). A superfície
estruturada foi então prensada contra um fotoresistor disposto sobre um substrato
de vidro. Após a secagem, as bases ficam presas a camada de fotoresistor,
possibilitando a sua clivagem do resto da amostra de HOPG. Com uma fita adesiva

21
os pesquisadores iniciaram uma esfoliação repetida de fragmentos de grafite destas
amostras. Os finos fragmentos que restaram no foto resistor foram colocados em
acetona. Quando a amostra de Si foi mergulhada na solução e então lavada com
água e propanol, alguns fragmentos foram capturados na superfície do substrato.
Após isso, foi feito uma limpeza, eliminando a maioria dos fragmentos mais
espessos. Os fragmentos mais delgados (d < 10nm) foram encontrados adsorvidos
no SiO2, devido a forças de van der Waals ou de capilaridade.
Figura 2.12: Filmes de grafeno. (A) Espectroscopia óptica de uma lâmina de
múltiplas camadas de grafeno com espessura 3 nm no topo do óxido de Si. (B) Imagem de um microscópio de força atômica [AFM] da amostra próxima à borda. A
cor marrom escura [laranja] é a superfície do SiO2 [3 nm acima do óxido]. (C) imagem de AFM de uma única camada de grafeno. A cor marrom escura é a superfície de SiO2.(D) Imagem de microscópio eletrônico de varredura de um dispositivo experimental com filme de grafeno de poucas camadas (E) Figura
esquemática do dispositivo (D) (NOVOSELOV et al, 2004).
Avaliando a espessuras do grafeno, os pesquisadores usaram uma
combinação de microscópios óticos, de feixe eletrônico e de força atômica (Figura
2.12). Filmes grafíticos com espessura menor que 50 nm, que são transparentes à
luz visível, entretanto sobre a superfície de SiO2 eles são facilmente visíveis devido à
adição de caminho ótico que altera a cor da interface. O SiO2, com uma espessura
de 300 nm tem cor violeta, a espessura extra, proveniente do filme delgado de
grafite provoca sua mudança para o azul. À medida que a espessura da folha de
grafeno tende para uma única camada, ou seja, quando se tem filmes de grafeno

22
com poucas camadas ela vai ficando invisível para o microscópio óptico.
Embora folhas de grafeno com espessuras menores que nmd 5,1 não sejam
visíveis através de microscópio ótico podem ser vistas claramente por microscópios
de alta resolução de varredura MEV (Microscópio eletrônico de varredura). A forma
mais fácil de isolar o grafeno é por meio de esfoliação química. Para isso a amostra
de grafite é inicialmente intercalada, de forma que as folhas de grafeno ficam
separadas por uma camada de átomos ou moléculas. Esse procedimento em geral
cria um novo composto tridimensional (3D). Mas em alguns casos é possível
introduzir moléculas grandes de forma a forçar espaçamentos entre os planos de
grafeno, resultando assim em grafeno isolado envolvido numa matriz em três
dimensões. O inconveniente desta técnica é que é comum por meio de uma reação
química a intercalação ser desfeita formando folhas de grafeno enroladas e re-
empilhadas (DRESSELHAUS et al, 2002).
Para obtenção de melhor qualidade na produção de grafeno, os
pesquisadores têm usado amostras obtidas pela técnica de clivagem
micromecânica, a mesma usada no primeiro isolamento do grafeno (NOVOSELOV
et al, 2004). Após um refinamento da técnica, consegue-se produzir cristais de
grafeno de alta qualidade com mais de 100 μm, sendo suficiente para a maioria das
pesquisas propostas.
Atualmente, diversas técnicas têm sido desenvolvidas e aprimoradas para
obtenção de grafeno de camadas isoladas buscando uma qualidade cada vez
melhor para aplicação no desenvolvimento de novas tecnologias (GEIM e
NOVOSELOV, 2007).
Propriedades do Grafeno
Grafeno é o nome dado a uma monocamada planar de átomos de carbono
dispostos em uma rede bidimensional (2D) hexagonal, e é base para estruturas
grafíticas de outras dimensões, como mostra a Figura 2.13.

23
Figura 2.13: Plano de grafeno originando diferentes estruturas alotrópicas do carbono, em (a) fulereno em (b) nanotubo e em (c) grafite (GEIM et al, 2007).
O grafeno é formado por átomos de carbono organizados em uma estrutura
hexagonal. A estrutura atômica do grafeno não é uma rede de bravais, mas pode ser
visto como uma rede triangular com uma base de dois átomos por célula unitária. Os
vetores da rede direta do grafeno podem ser escritos como:
, (2.1)
onde a = 2,40 Å é o parâmetro de rede da estrutura hexagonal. A rede recíproca é
dada pelos vetores:
, (2.2)
A Figura 2.14 mostra a estrutura hexagonal na rede direta e recíproca com os
vetores correspondentes.

24
Figura 2.14: À esquerda, estrutura de grafeno feito de duas redes
triangulares (1
a
e 2
a
são os vetores unitários da rede). À direita, corresponde a zona
de Brillouin (CASTRO et al, 2007).
Devido a simetria da estrutura hexagonal do grafeno alguns pontos de
simetria da zona de Brillouin são necessários para descrever as propriedades
eletrônicas deste material, como os pontos , K, K´ e M. Os pontos K e K´ também
são chamados de pontos de Dirac e suas posições são dadas por:
aaK
33
2,
3
2,
aaK
33
2,
3
2' . (2.3)
Fazendo uso de um modelo de tight-binding para o grafeno e considerando no
Hamiltoniano que esses elétrons possuem interação somente com seus primeiros e
segundos vizinhos, temos:
(2.4)
onde destrói (cria) elétrons com spin no sítio i
R da sub-
rede A. Uma equivalente definição é usada na sub-rede B, t = 2,8 eV e t=0,1 eV são
as energias para os elétrons saltarem para os primeiros e segundos vizinhos,
respectivamente (REICH et al, 2002). As bandas de energia deste Hamiltoniano
devem ter a forma (WALLACE, 1947):

25
, (2.5)
, (2.6)
onde o sinal positivo representa a banda de energia superior (π) e o sinal negativo a
banda inferior (π*).
A Figura 2.15 apresenta o espectro de energia para toda a zona de Brillouin
calculada usando a equação (2.5). A estrutura de bandas próxima aos pontos de
Dirac tem uma dispersão cônica:
, (2.7)
onde q
é o momento medido a partir dos pontos de Dirac e F é a velocidade de
Fermi dada 23 /taF
, com um valor por s/mxF
6101 (WALLACE, 1947).
A diferença entre esse resultado e o do caso usual é que ,
onde m é a massa do elétron e q a velocidade de Fermi na equação (2.7) a qual não
depende da energia ou momento.
Figura 2.15: À esquerda, bandas de energia de valores finitos de t e t’, com
eVt 7,2 e tt 2,0' . À direita, ampliação em um dos pontos de Dirac (CASTRO et al,
2007).
A dispersão de energia a partir da equação (2.7) tem semelhança com a
energia de partículas sem massa chamadas de ultra-relativísticas; a energia destas
partículas é descrita quanticamente pela equação de Dirac.

26
A densidade de estados eletrônica por célula unitária do grafeno, derivada
pela equação (2.5), é mostrada na Figura 2.16, 0't e 0't , mostrando nos casos
um comportamento semi-metálico (BENA et al, 2005). Perto do ponto de Dirac a
dispersão é dada, aproximadamente, pela equação (2.7) e a expressão da
densidade de estados por célula unitária é dada por:
onde c
A é a área da célula unitária dada por 233 2 /aAc
. Note que a densidade
de estados do grafeno é muito diferente da densidade de estados dos nanotubos de
carbono (SAITO et al, 1992a).
Figura 2.16: Densidade de energia dos estados por célula unitária computado
para a dispersão de energia usando a equação (2.5), 0't em (a) e (b) e 0't em (c)
e (d) (adaptado de CASTRO et al, 2007).
Aplicações do Grafeno
A síntese de grafite de monocamada (grafeno) e a observação experimental
dos portadores de carga de Dirac neste sistema (NOVOSELOV et al, 2005; ZHANG

27
et al, 2005) despertaram um enorme interesse do ponto de vista de aplicações deste
material bidimensional. As propriedades incomuns de portadores de carga no
grafeno são consequências do pequeno gap e da dispersão aproximadamente linear
de elétrons nas imediações do nível de Fermi a dois pontos não equivalentes da
zona de Brillouin. No limite de baixas energias, as quasi-partículas neste sistema
são descritas em termos de férmions relativísticos governados pela equação de
Dirac.
As propriedades interessantes de sensores de nanotubos de carbono já são
conhecidas há algum tempo (KONG et al, 2000) mas, recentemente, a possibilidade
para uso de grafeno como um sensor de gás altamente sensível também foi relatada
(SHEDIN et al, 2007). Foi mostrado que no grafeno o aumento da concentração de
portadores de carga induzida por adsorção de moléculas de gás pode ser usado
para fazer sensores altamente sensíveis, até mesmo com a possibilidade de
detectar moléculas individuais. A sensibilidade está baseada em mudanças na
resistividade devido às moléculas absorvidas na folha de grafeno que atuam como
doadores ou aceitadores de carga.
Outra possibilidade seria o uso do pó do grafeno em baterias elétricas que já
é um dos principais mercados para o grafite. A razão de superfície por volume e a
alta condutividade do grafeno podem conduzir para significantes melhorias na
eficiência de baterias. Os nanotubos de carbono foram cogitados para aplicação em
baterias, mas a produção de pó de grafeno é muito mais barata (STANKOVICH et al,
2006).
Desta forma, torna-se essencial o conhecimento das propriedades resultantes
do grafeno com moléculas adsorvidas que podem ser doadoras ou aceitadoras de
carga, tornando o grafeno com propriedades ainda mais interessantes do ponto de
vista de aplicações. Neste estudo, especificamente, avaliaremos as propriedades do
grafeno quando interagindo com a molécula do aminoácido cisteína, a qual
descreveremos suas propriedades quando isolada na seção a seguir. Esta molécula
tem um interesse especial devido a seu comportamento anfótero, uma vez que todos
aminoácidos possuem grupos ácidos (COOH) e básicos (NH2).

28
2.4. CISTEÍNA
A cisteína é um dos aminoácidos codificados pelo código genético, sendo
portanto um dos componentes das proteínas dos seres vivos. Sua fórmula molecular
é C3H7NO2S sendo um aminoácido relativamente simples, possui em sua estrutura
os grupos amino (NH2), carboxila (COOH) e tiol (SH). A cisteína apresenta-se na
natureza sob a forma de dois isômeros espaciais, cisteína-D e cisteína-L, sendo que
para este trabalho foi utilizada a cisteína-L, o isômero mais abundante na natureza.
O seu nome tem origem na palavra grega kustis, significando "bexiga", pois foi
isolada inicialmente a partir de cálculos renais (sob a forma de cistina). A cisteína
(Figura 2.17) possui um grupo tiol na sua cadeia lateral e é, principalment,e
encontrada em proteínas e no tripeptídeo glutationa. Quando exposto ao ar, e sob
determinadas condições fisiológicas (incluindo no interior de proteínas), a cisteína
oxida-se formando cistina, composta por duas cisteínas unidas por uma ligação
dissulfeto (LEHNINGER, 2002).
Figura 2.17: Molécula do aminoácido cisteína.
O grupo tiol possui caráter nucleofílico (doador de elétrons). Como o pKa
deste grupo é de 8,3, a sua atividade química pode ser regulada pelo ambiente em
que se enquadra.
As moléculas de cisteína têm um papel fundamental na manutenção da

29
estrutura terciária de proteínas. Ao formarem ligações dissulfeto entre os seus
grupos tiol, aumentam a estabilidade molecular e a resistência à proteólise. A
insulina é um exemplo deste tipo de ligações, pois é formada por dois peptídeos
ligados por duas destas ligações dissulfeto.
Alimentos ricos em cisteína incluem pigmentos vermelhos, alho, cebola,
bróculis, couve-de-bruxelas, aveia e gérmen de trigo. Não é, contudo, um
aminoácido essencial: é sintetizado no organismo humano se existir uma quantidade
disponível suficiente de metionina.
Embora muitos aminoácidos estejam disponíveis já há bastante tempo
através da fermentação, só em 2001 uma companhia alemã apresentou uma via
fermentativa para a produção de L-cisteína, sem o recurso a produtos animais ou
humanos (LEHNINGER, 2002).
A cisteína (particularmente a L-cisteína) é usada não só em investigação
laboratorial mas também como suplemento alimentar, em produtos farmacêuticos e
de cuidado pessoal. Também é usada industrialmente em pastelaria e padaria, em
doses que não excedem as dezenas de ppm, para amaciar a massa, reduzindo o
seu tempo de processamento.
O derivado N-acetilcisteína é usado como medicamento contra a tosse, pois
interfere com a formação de ligações dissulfeto em proteínas do muco, liquefazendo-
o e tornando mais simples a sua expulsão. A N-acetilcisteína é usada como
suplemento alimentar tal como a cisteína (LEHNINGER, 2002).
O gado ovino necessita de cisteína para produzir lã, sendo nestes organismos
um aminoácido essencial. As ovelhas adquirem este aminoácido ao ingerir erva;
como tal, a produção de lã cai ou para totalmente em períodos de baixa
disponibilidade de alimentos (como secas prolongadas). Foram desenvolvidas
ovelhas transgênicas que conseguem produzir cisteína, obviando a necessidade
permanente da sua ingestão.
Neste trabalho, utilizamos a cisteína devido á presença de grupos doadores e
receptores de carga em uma molécula de peso molecular relativamente baixo.
2.5. GRAFENO COMO SENSOR DE MOLÉCULAS
O estudo das propriedades dos nanotubos com grupamentos orgânicos

30
substitucionais e adsorvidos, demonstrou relevantes alterações eletrônicas em
relação à estrutura primária (VELOSO et al, 2006), o que motivou esta análise
análoga aplicada ao grafeno, uma vez estudos nessa linha foram recentemente
desenvolvidos utilizando carbono amorfo (CHUTIA et al, 2008) e átomos alcalinos e
halogênios (SILVA, 2008).
A funcionalização de estruturas carbonáceas como fulerenos e nanotubos
pelos mais diferentes substituintes e utilizando as mais variadas rotas reacionais, é
uma linha de pesquisa que tem despertado interesse de pesquisadores de diversos
grupos dos mais diversos institutos de pesquisa do mundo; a dificuldade em
sintetizar, caracterizar e controlar a produção destas estruturas tem sido um
empecilho bastante sério para a pesquisa nesta área. Nesse ínterim, o grafeno
chega como uma alternativa rápida e fácil para possíveis estudos de
funcionalização, uma vez que sua síntese já demonstrou ser bem mais simples que
a das outras nanoestruturas de carbono.
A simulação da possível funcionalização de nanofitas de grafeno com
carboxilas substituídas em anéis contendo defeitos do tipo “stone-wales” (anéis com
cinco ou sete membros na rede); foi recentemente realizada, e a posterior análise
dos resultados demonstrou que a mono e dupla adsorção de carboxilas, modifica
consideravelmente as propriedades eletrônicas do grafeno, aumentando sua
condutibilidade eletrônica, fazendo com que um sistema que inicialmente é
caracterizado como semicondutor, passe a ter comportamento metálico podendo
assim ser utilizado como sensor químico ou ser componente de dispositivos
eletrônicos baseados em nanofitas de grafeno.
Neste material as propriedades eletrônicas e a grande área superficial são
algumas das propriedades mais interessantes de modo que como poderemos ver a
seguir, são adsorvidos ou substituídos grupamentos funcionais orgânicos e diversos
outros átomos e moléculas, visando controlar ou modificar as propriedades
eletrônicas e estruturais do grafeno.
Adsorvendo óxido de cromo (III), (ZANELLA et al, 2008), mudaram a
configuração eletrônica da folha de grafeno de semicondutor para semi-metal e
finalmente para metal.
Utilizando átomos de hidrogênio e flúor (ZHOU et al, 2009), foram feitas
modificações na superfície do grafeno, adsorvendo os átomos de forma diferente,

31
“afinaram” as propriedades eletrônicas e magnéticas, de grafeno puro passaram
para grafano, o qual consistia em uma superfície de grafeno com seus dois lados
totalmente hidrogenados. Após foram retirados os hidrogênios de um dos lados, o
que chamaram de grafono, e posteriormente recobriram os dois lados novamente
com átomos de flúor, alterando significativamente as propriedades eletrônicas do
sistema, uma vez que o sistema passou de metálico para semicondutor e de não
magnético para magnético, modificações estas que dependeram somente das
espécies atômicas e do tipo de cobertura que foi realizada na superfície do grafeno.
Levando estes trabalhos em consideração, abrem-se novos ramos para
funcionalização da monocamada de grafeno, em que poderão ser utilizados grupos
orgânicos presentes nos aminoácidos.
Os aminoácidos são monômeros naturais, os quais originam as proteínas
através de uma reação de polimerização por condensação em que se unem “n”
moléculas de aminoácidos para originar as proteínas, que são polímeros naturais
diversas funções biológicas, tais como catalisadores e tecidos de construção
biológicos.
Desta forma, podemos perceber que tanto o grafeno puro tanto o grafeno
funcionalizado possuem a capacidade de reter moléculas em sua superfície, no caso
do grafeno puro, ou em seus diferentes sítios de adsorção, no caso das moléculas
ou átomos que estarão ligados ao grafeno, por esta razão, escolhemos o
aminoácido cisteína para realizar uma análise da possibilidade da utilização do
grafeno como sensor de proteínas e aminoácidos, uma vez que a principal
característica de um sensor deve ser de indicar a presença de determinada
substância em um determinado meio analisado.
No próximo capítulo descreveremos a metodologia utilizada neste trabalho
para a modelagem da molécula da cisteína interagindo com o grafeno.

32
3. METODOLOGIA
Uma descrição microscópica dos materiais requer o estudo da estrutura e
dinâmica de sistemas de muitos elétrons e núcleos. Em se tratando de sistemas
interagentes que possuam moléculas orgânicas, aminoácidos no caso, os desafios
são ainda maiores, afinal em uma pequena molécula estão presentes alguns grupos
concentradores de carga, como grupo amino, grupo carboxila, além de outros
possíveis grupos como oxidrila e sulfidrila.
Nestes tipos de sistema interagente o conhecimento de sua estrutura
termodinamicamente mais estável é fundamental. A estabilidade de um sistema em
que estão interagindo grafeno e aminoácido, depende basicamente da troca, ou não,
de cargas entre as duas partes. O balanço energético decorrente da soma do
potencial atrativo dos núcleos sobre os elétrons, dos potenciais repulsivos elétron-
elétron e núcleo-núcleo e das energias cinéticas dos elétrons e dos núcleos
determina a estrutura de mínima energia, ou estado fundamental do sistema.
Uma vez conhecida a estrutura do estado fundamental, pode-se determinar
sua estrutura de bandas e energia de ligação, o que irá nos informar se o sistema
está interagindo forte ou fracamente, ou seja, se a ligação formada entre o grafeno e
o aminoácido é de caráter físico ou químico.
Nos casos em que o potencial é independente do tempo, a parte espacial,
chamada de equação de Schrödinger independente do tempo, e dada pela Equação
3.1:
(3.1)
onde, H é o operador hamiltoniano, ),( Rr
é a função de onda e E é a energia total
do sistema.
O grafeno, assim como sólidos e as moléculas, é formado por um conjunto de
N elétrons e M núcleos que interagem coulombianamente entre si. O hamiltoniano
para estes sistemas pode ser escrito como (FERNANDES, 2006):
ˆ ˆ ˆ ˆ ˆ ˆ
n e nn ee enH T T V V V (3.2)
),(),(ˆ RrERrH

33
onde, o primeiro termo é o operador energia cinética dos núcleos, o segundo é o
operador energia cinética dos elétrons, o terceiro operador é o potencial de interação
coulombiana núcleo-núcleo, o quarto operador corresponde a interação coulombiana
elétron-elétron, e o último corresponde a interação coulombiana elétron-núcleo.
Neste trabalho utilizamos o Programa SIESTA (Spanish Initiative for
Electronic Simulations with Thousand of Atoms), o qual utiliza outras aproximações
(pseudopotencial, supercélula e funções de base) para resolver as equações de
Kohn-Sham de forma autoconsistente (SOLER et al., 2002).
A resolução da equação (3.2) depende diretamente do número de partículas
envolvidas, pois matematicamente sistemas a partir do átomo de hidrogênio não
possuem solução analítica exata. Desta forma, as aproximações tornam-se
essenciais para a descrição das propriedades físicas e químicas dos sistemas. A
seguir apresentamos a aproximação de Born-Oppenheimer que reduz
consideravelmente o número de partículas envolvidas no cálculo das propriedades
do sistema, uma vez que desacopla (individualiza) o movimento dos elétrons do
movimento dos núcleos.
3.1. APROXIMAÇÃO DE BORN-OPPENHEIMER
Uma vez que a massa nuclear é muitas vezes maior que a massa dos
elétrons, a velocidade dos elétrons é muito maior que a dos núcleos. Então, para
qualquer movimento dos núcleos há uma reação quase que instantânea de rearranjo
por parte dos elétrons. Assim, com núcleos em posições fixas, os elétrons movem-
se em um campo de núcleos estáticos. O termo do hamiltoniano que corresponde à
energia cinética nuclear (Tn) pode ser desprezado, e o termo devido à repulsão
núcleo-núcleo (Vnn) é constante (FAGAN, 2003).
Desta forma, o sistema de interesse pode ser pensado como um conjunto de
N elétrons, em movimento, interagindo uns com os outros, com energia cinética (T) e
interação coulombiana (U), numa região onde existe um campo externo (V) gerado
pelos núcleos fixos. Nesta aproximação, o hamiltoniano eletrônico para o sistema
particular poderá ser escrito como:
(3.3) VUTH ˆˆˆˆ

34
Os termos T e U são universais, isto é, têm a mesma forma qualquer que
seja o sistema de muitos elétrons em questão:
N
i
r
eim
T
1
22
2ˆ
, é o operador energia cinética de interação dos elétrons;
ji
N
ij
N
ijo rr
eU
1
4ˆ
)(11
2
, é o operador energia de interação elétron-elétron;
iA
AN
i
M
Ao rR
ZeV
11
2
4ˆ
, é o operador potencial de interação íon-
elétron.
A informação que especifica as características particulares de um sistema
está contida inteiramente em V , que representa a energia de interação entre os
elétrons e os núcleos.
Para calcularmos esta energia, fazemos uso de um método que reduz
bastante o custo de cálculo devido ao fato de que ele trata da interação de cada
átomo do sistema com uma densidade de carga composta das energias de todos os
átomos restantes no sistema, este é o princípio da teoria do funcional da densidade.
3.2. TEORIA DO FUNCIONAL DA DENSIDADE
O método da teoria do funcional da densidade (DFT) tem sido considerado,
na física do estado sólido, como uma ferramenta bem sucedida entre os métodos ab
initio (primeiros princípios) devido à alta eficiência computacional e aos bons
resultados fornecidos (KOHN, 1999).
A partir da mecânica quântica sabemos que a função de onda total, que
depende das coordenadas dos N elétrons e dos M núcleos, é o objeto fundamental
de estudo e pode gerar todas as propriedades do sistema. Existe outra maneira de
resolver o problema, em que o objeto fundamental é a densidade eletrônica total n(r)

35
(THOMAS, 1927; FERMI, 1928), ou seja, a grandeza fundamental não é a função de
onda e sim a densidade. A equação de N elétrons com a função de onda de 3N
variáveis (se não considerarmos o spin) pode ser escrita como uma equação da
densidade eletrônica com somente três variáveis (x, y, z).
Thomas (1927) e Fermi (1928) (TF), independentemente, desenvolveram um
procedimento simplificado para determinar densidade eletrônica do sistema quando
o potencial varia suavemente com r
. Baseando-se nas seguintes suposições:
a) As correções relativísticas são desprezíveis;
b) No átomo há um campo efetivo dado por um potencial v, que dependente
somente da distância r ao núcleo de carga nuclear Z, tal que v → 0 quando r
→ ∞ e v → Ze quando r → 0;
c) Os elétrons estão distribuídos uniformemente num espaço de fase de
dimensão 6, onde cada par de elétrons ocupa um volume de h3, sendo h a
constante de Planck;
d) O potencial v é determinado por si mesmo, pela carga nuclear e por sua
distribuição eletrônica.
O modelo de Thomas-Fermi é uma aproximação que considera somente a
energia cinética de forma quântica, enquanto as contribuições das interações
núcleo-elétron e elétron-elétron recebem um tratamento clássico, esta formulação
não leva em conta cálculos variacionais, bem como não trata o termo de correlação
eletrônica, sendo que a parte de troca e correlação foi adicionada à teoria de
Thomas-Fermi por Dirac em 1931. Mesmo com o tratamento de Exchange essa
formulação não é adequada para cálculos de energia total, pois não leva em conta o
uso da autoconsistência no cálculo da energia total.
3.2.1. Teoremas de Hohenberg-Kohn
A DFT esta baseada em dois teoremas propostos em 1964 por Hohenberg e
Kohn (HK) (HOHENBERG e KOHN, 1964). Neste caso, podemos considerar um
sistema com N partículas e escrevemos o hamiltoniano na forma geral abaixo
(equação 3.4), bem como na equação. 3.2:

36
. (3.4)
O primeiro teorema diz que a densidade de carga do estado fundamental
n(r
) é determinada de modo unívoco, a menos de uma constante aditiva, a partir do
potencial externo )(rVext
.
Para um sistema de N elétrons, o potencial externo )(rVext
define a estrutura
nuclear do sistema e fixa completamente o hamiltoniano eletrônico. Se conhecermos
N e )(rVext
podemos, portanto, determinar as propriedades do estado fundamental
(não degenerado), tais como, energia, força, autovalores, etc. O número de elétrons
é definido como um funcional da densidade eletrônica:
(3.5)
Ao invés de usar N (elétrons) e )r(Vext
, o primeiro teorema de HK impõe o uso
da densidade n(r
) como uma variável básica. Temos então o corolário 1.
O segundo teorema de HK baseia-se no princípio variacional da energia,
sendo escrito como o valor mínimo do funcional da energia ]n[E é a energia do
estado fundamental e a densidade com a qual se obtém esse mínimo é a densidade
exata de uma partícula no estado fundamental.
Assim podemos ressaltar que estes dois teoremas nos garantem que
podemos encontrar a densidade eletrônica do sistema no estado fundamental
resolvendo-se as equações de Kohn-Sham, as quais serão descritas a seguir.
3.2.2. Equações de Kohn-Sham
Foram Kohn e Sham (1965) quem introduziram a idéia fundamental de utilizar
um conjunto de orbitais para representar a densidade eletrônica e tornar o cálculo
autoconsistente.
De acordo com Kohn e Sham (KS) a energia cinética ][nT pode ser
rdrnnN
)(][

37
decomposta em uma parte que representa a energia cinética de um sistema de
partículas não-interagentes ][nTS , e outra que contêm o resto da energia cinética
][nTC , devida a interação elétron-elétron:
(3.6)
E o potencial de interação elétron-elétron, ][nVee , pode ser representado por uma
parte clássica conhecida como potencial de Hartree, ][nVH , e uma contribuição não-
clássica, ][nVxc , que contêm os efeitos da consideração da auto-interação e de
correlações eletrônicas (coulombiana) e de troca (exchange):
(3.7)
A partir destas considerações, o funcional de HK pode ser descrito por:
(3.8)
onde ][nTS representa a energia cinética de um sistema de partículas não
interagente, ][nVH é o potencial elétron-elétron (potencial de Hartree), e
][][][ nVnTnE XCCXC inclui todas as correções.
A função de onda exata para o sistema é obtida pelo produto antissimetrizado
das N funções de onda de um elétron, representado pelo determinante de Slater,
onde as funções )(ri
são os N orbitais de Kohn-Sham.
Os orbitais são, portanto, obtidos por meio de uma equação de autovalores
para um elétron.
3.2.3. Termo de Troca-Correlação
Uma vez obtidas às equações de Kohn-Sham, a aplicabilidade da DFT
depende diretamente da escolha de uma boa aproximação (que implica em
][][][][ nEnVnTnF XCHS
][][][ nTnTnT cs
][][][ nVnVnV xcHee

38
simplicidade e precisão tão grande quanto possíveis) para o termo de troca-
correlação. Não se conhece a expressão exata do funcional da energia de troca e
correlação. Dentre as aproximações utilizadas para se calcular o termo de troca e
correlação estão a LDA do inglês: Local Density Aproximation e os vários tipos de
GGA do inglês: Generalized Gradient Aproximation.
A aproximação mais simples para o termo de troca-correlação é a LDA. Nesta
aproximação o sistema não homogêneo é dividido em pequenos volumes, que
chamamos de células e dentro destas células a energia é calculada considerando a
densidade como sendo aquela de um gás homogêneo, ou seja, a densidade é
uniforme na região considerada.
Assim, ao somarmos sobre todas as células teremos uma aproximação para o
termo de troca-correlação do sistema como um todo. Matematicamente tem-se:
, (3.9)
onde (3.10)
representando a energia de troca e correlação por partícula de um sistema
homogêneo e,
(3.11)
é a densidade eletrônica de cada célula.
Desta forma, para Ni → 0, Vi → 0 e, conseqüentemente, ni → n. Pode-se
também escrever que:
(3.12)
Na expressão 3.10, a função Vxc[n(r)] é tratada separadamente, ou seja,
i
i
i
xcLDA
xcV
NnvrnV ][)]([
N
nVnv xc
xc
][][
hom
i
ii
V
Nn

39
Vxc[n(r)] = Vx[n(r)] + Vc[n(r)].
A contribuição do termo de troca (exchange) é bem conhecida e dada por,
, (3.13)
que é a energia de exchange para um gás de elétrons homogêneo. O termo de
correlação é mais complexo e não pode ser determinado exatamente nem mesmo
para o caso particular de um gás de elétrons homogêneo.
Com isso, aproximações para este termo devem ser efetuadas, e uma das
mais utilizadas é a parametrização de Perdew e Zunger (1981) e Perdew e Wang
(1991) para um gás de elétrons homogêneo. Nos nossos cálculos, esta será a
aproximação utilizada.
A princípio poderia se pensar que esta aproximação deveria ser válida
somente para sistemas onde a densidade eletrônica não varia muito, porém de
maneira surpreendente ela descreve satisfatoriamente sistemas atômicos,
moleculares e cristalinos onde a densidade de partículas varia rapidamente com a
posição. Empiricamente, são conhecidas determinadas tendências para LDA:
subestima o valor da constante de rede entre 1-3%, os bulk modulus são
superestimados entre 8 e 18% e não é capaz de reproduzir com precisão as
energias de coesão devido ao erro cometido ao calcular a energia por átomo como
um sistema homogêneo. Tanto LDA e GGA subestimam o tamanho do gap nos
semicondutores e isolantes na ordem de 50%. Como citado anteriormente, o GGA
corrige alguns problemas em relação à LDA, mas isso não é válido necessariamente
para todos os sistemas (FUCHS et al, 1998).
A aproximação LDA é anterior a DFT, pois nos cálculos de Thomas-Fermi
esta aproximação já foi utilizada para descrever a energia cinética.
A aproximação da densidade local pode ser generalizada de forma a incluir o
spin (LSDA - Local Spin Density Approximation),
, (3.14) rdrnrnvrnrnnnV xcxc
)](),([)()(],[

40
onde, como é convencional, n ↑ é a densidade eletrônica para spin up e n ↓ a
densidade eletrônica para spin down.
3.3. PSEUDOPOTENCIAL
O pseudopotencial foi introduzido originalmente para simplificar o cálculo da
estrutura eletrônica pela eliminação dos estados eletrônicos de caroço.
Um átomo é constituído pelo núcleo positivo (prótons e nêutrons) e pela
eletrosfera negativa (elétrons). Grande parte das propriedades dos sistemas físicos
e químicos depende quase que unicamente dos elétrons de valência. Então, além de
reduzir os cálculos computacionais, costuma-se tratar o átomo como sendo
constituído por um caroço interno acrescido dos elétrons de valência. Este caroço é
formado pelo núcleo atômico e pelos elétrons internos.
Este tratamento reduz em muito o custo computacional dos cálculos, já que
somente os elétrons de valência são tratados explicitamente; ao conjunto elétrons
internos + núcleo - o caroço - atribui-se um pseudopotencial. Este pseudopotencial
deve reproduzir os estados de valência do átomo real.
Portanto os elétrons em um sólido dividem-se basicamente em dois tipos:
i) Os elétrons próximos aos núcleos atômicos que sentem um forte potencial
atrativo, tendo pouca participação nas ligações químicas. A região que é formada
pelo núcleo atômico e os elétrons mais internos denominados de caroço. A principal
contribuição das funções de onda dos elétrons do caroço para a ligação química é
forçar as funções de onda dos elétrons de valência a serem ortogonais aos auto-
estados de caroço;
ii) Os elétrons de valência têm uma grande participação nas ligações
químicas, determinando, portanto a maior parte das propriedades físicas de um
sólido ou de uma molécula. Esses elétrons de valência sentem um potencial bem
menos atrativo e seus orbitais apresentam formas mais suaves.
O método do pseudopotencial, em sua proposta inicial, consiste em simplificar
cálculos muito trabalhosos no procedimento de ortogonalização de ondas planas
com as funções de estado de caroço. Os pseudopotenciais ab initio de norma
conservada são os mais utilizados nos cálculos com a DFT. Os pseudopotenciais de

41
norma conservada possuem as seguintes propriedades (TROULLIER e MARTINS,
1991):
a) os autovalores de energia obtidos para os estados de valência atômicos
devem, ser por construção, iguais aos autovalores obtidos com o
pseudopotencial;
b) as autofunções relativas à “solução exata” e à solução obtida com o
pseudopotencial devem ser iguais para distancias r acima de um raio de
corte rc devidamente escolhido;
c) as integrais de 0 a r, r < rc das densidades de carga da “solução exata”
devem ser iguais às das soluções com pseudopotenciais;
d) A pseudofunção de onda obtida através do pseudopotencial não pode
conter nodos, garantindo assim uma configuração suave para o
pseudopotencial.
A Figura 3.1 a seguir traz um exemplo de pseudopotencial:
Figura 3.1: Função de onda real e pseudofunção para o átomo de carbono (2s e 2p,
linha preta e vermelha, respectivamente). As funções AE são representadas pelas
linhas cheias e PS pelas linhas tracejadas (FAGAN, 2003).

42
3.4. ORBITAIS ATÔMICOS
Para resolvermos a equação de Kohn-Sham (KOHN e SHAM, 1965) é
necessário utilizar uma base para descrever os orbitais i
r
. O programa SIESTA
(Spanish Initiative for Electronic Simulations with Thousand of Atoms) faz uso, como
base, funções de onda atômicas localizadas, as quais possuem dois parâmetros
importantes, número de orbitais por átomo e alcance desses orbitais (ARTACHO et
al, 1999). As bases são uma boa escolha para um reduzido custo computacional e
cálculos precisos. O ajuste das bases para cada átomo deve-se ter um cuidado,
pois há neste método a desvantagem da convergência.
Em nosso trabalho utilizamos apenas bases numéricas estritamente
localizadas. Os orbitais de Kohn-Sham, podem ser expandidos em funções de base
, que são os pseudoorbitais atômicos. Por sua vez, estes orbitais são expandidos
em outras funções, as funções gaussianas que são representadas pelos seus
expoentes zeta (ζ). Cada orbital é expandido em uma combinação linear de ζ, o que
nos fornece uma maior liberdade variacional para o problema.
Assim, o número de ζ dá nome a base atômica, no SIESTA. Uma única
função ζ constitui uma SZ (single zeta), duas funções ζ constituem uma DZ (dupla
zeta), e assim sucessivamente. As bases menos rigorosas (por exemplo, SZ) tornam
os cálculos mais rápidos. Bases mais rigorosas demandarão um esforço
computacional maior, porém a precisão é melhorada.
Funções que correspondem a um momento angular superior podem ser
adicionadas às funções de base, para garantir flexibilidade aos orbitais dos elétrons
de valência quando estes formam ligações químicas. Estas funções são chamadas
de funções de polarização. As bases utilizadas nesta dissertação são do tipo DZ
(dupla zeta) e também uma dupla zeta com uma função de polarização (DZP).
O problema para as bases estritamente localizadas é encontrar uma maneira
sistemática de definir todos os raios das funções de base. O esquema proposto, no
qual todos os raios são definidos em função de um só parâmetro, é a correção na
energia (energy shift), isto é um incremento na energia que sofre o orbital quando
está confinado (SOLER et al, 2002). Este processo cresce a curvatura do orbital e,
portanto sua energia cinética.

43
Uma das grandes vantagens de escolhermos orbitais atômicos localizados
(orbitais que se anulam acima de um determinado raio de corte) é que as interações
estendem-se a um alcance finito de camadas de vizinhos reduzindo
consideravelmente o custo computacional das simulações.
Figura 3.2: Em (a) observamos a pseudofunção de onda, e a primeira e segunda ζ
para o orbital 2s do C e em (b) a PS e as funções base para o orbital 2p do C
(FAGAN, 2003).
A Figura 3.2 mostra os pseudo-orbitais atômicos para os dois orbitais de
valência do carbono. Observe que a segunda ζ tem um raio RDZ menor que o raio de
corte rc da primeira ζ (FAGAN, 2003).
No próximo Capítulo apresentaremos os resultados obtidos para a molécula
de cisteína interagindo com a monocamada de grafeno, fazendo uso da metodologia
explicitada neste Capítulo.

44
4. RESULTADOS
Neste Capítulo será descrito o estudo da interação da molécula de cisteína
em uma monocamada de grafeno perfeita e com defeito estrutural via cálculos de
primeiros princípios. Inicialmente apresentaremos os procedimentos de cálculo, a
seguir as propriedades do grafeno isolado e finalmente os resultados com a
adsorção do aminoácido no grafeno perfeito e no grafeno com defeito, avaliando as
propriedades estruturais e eletrônicas.
4.1. PROCEDIMENTOS DE CÁLCULO
O estudo das propriedades estruturais e eletrônicas foi realizado por cálculos
de primeiros princípios que foram executados, fazendo uso da teoria do funcional da
densidade (HOHENBERG e KOHN, 1964; KOHN e SHAM, 1965), utilizando o
código computacional SIESTA (SOLER et al, 2002). As equações de Kohn-Sham
(KS) foram resolvidas de forma autoconsistente usando pseudo-orbitais atômicos
numéricos como base. Em todas as simulações foram utilizadas bases numéricas do
tipo double-ζ (DZ) sendo que o alcance da base, definido pelo energy shift, foi de
0,02 eV para os orbitais atômicos de C, H, N, O, e S.
Os termos de troca e correlação foram descritos pela aproximação LDA,
(modelo proposto por Perdew e Zunger (PERDEW e ZUNGER, 1981) para a
molécula de aminoácido adsorvida em uma monocamada de grafeno. Para
descrever o grafeno utilizamos uma supercélula quadrada contendo átomos de
carbono distribuídos hexagonalmente contendo 48 átomos (Figura 4.1).
A interação entre o grafeno e os aminoácidos pode ser analisada em duas
linhas principais de interesse avaliadas a partir de resultados experimentais
preliminares: via adsorção física, com fraca interação entre a folha de grafeno e o
agente adsorvedor, ou via adsorção química, neste caso há formação de ligações
covalentes usualmente a partir da funcionalização de uma das estruturas (grafeno
ou aminoácido).
O pseudopotencial suave, não-local, de norma conservada utilizado para
descrever a interação entre elétron-íon foi o de Troullier e Martins (TROULLIER E
MARTINS, 1991).
Os cálculos foram realizados com um parâmetro de convergência, de que as

45
forças sobre cada coordenada de todos os átomos fossem menores que 0,05 eV/Å.
Para os cálculos da energia de ligação utilizamos a equação:
EB = - { E[A+B] – E[A] – E[B] } (4.1)
onde Eb a energia de ligação, E[A] a energia total do grafeno puro, E[B] a energia total
da molécula e E[A+B] a energia total do grafeno com a molécula interagindo.
Foram analisadas três configurações para a molécula de cisteína,
aproximando a partir do grupo carboxila (COOH), do grupo amino (NH2) e do grupo
tiol (SH).
A seguir apresentaremos os principais resultados da simulação ab initio para
o grafeno perfeito e com o defeito estrutural (falta de um carbono na rede), para
efeitos de comparação com a cisteína adsorvida no grafeno.
4.2. GRAFENO PERFEITO, GRAFENO COM DEFEITO E CISTEÍNA
Apresentaremos as principais características do grafeno puro e com defeito,
bem como a cisteína isolada. A supercélula usada para a simulação de uma folha de
grafeno infinita contém 48 átomos de carbono, constituída de uma rede quadrada
bidimensional (Figura 4.1 (a)). O plano descrito encontra-se a uma distância de,
aproximadamente, 15 Å em relação aos planos de grafeno vizinhos. A distância
média de ligação entre átomos de carbono após a otimização da geometria é de
1,44 Å, valor similar ao encontrado na literatura (CASTRO et al, 2007).
A estrutura eletrônica de bandas do grafeno puro, após a otimização (Figura
4.1 (b)) mostra uma região de energia proibida (gap) próximo a energia de
Fermitendendo a zero no ponto k, o que dá ao grafeno o caráter semi-metálico
conforme descrito também na literatura (CASTRO et al, 2007).

46
Figura 4.1: (a) Supercélula de grafeno com 48 átomos de carbono. (b) Estrutura de
bandas do grafeno puro. A linha horizontal tracejada corresponde a energia de
Fermi.
A supercélula usada para a simulação de uma folha de grafeno infinita
contendo defeitos estruturais contém 47 átomos de carbono, constituída de uma
rede quadrada bidimensional (Figura 4.2 (a)).
A estrutura eletrônica de bandas do grafeno com defeito, após a otimização
(Figura 4.2 (b)) mostra os níveis eletrônicos cortando o nível de Fermi (-4,7 eV)
evidenciando o caráter metálico devido à presença de defeitos.
(a)
(b)
Figura 4.2: (a) Supercélula de grafeno defeituoso com 47 átomos de carbono. (b)
Estrutura de bandas do grafeno defeituoso. A linha horizontal tracejada (~-4,7 eV)
corresponde a energia de Fermi.

47
A estrutura da cisteína com seus átomos indicados e de seus níveis
eletrônicos ocupados e desocupados está demonstrada na Figura 4.3.
a)
b)
Figura 4.3: (a) Estrutura da molécula da cisteína, (b) níveis eletrônicos, níveis
ocupados localizados abaixo, (cheios) e os níveis desocupados localizados acima,
(vazios).
Podemos verificar na Figura 4.3 (b), a diferença energética entre o HOMO
(Highest Occupied Molecular Orbital) o qual corresponde ao orbital molecular
ocupado de mais alta energia que tem valor aproximado de -5,2 eV, e o LUMO
(Lowest Unoccupied Molecular Orbital) que é o orbital molecular desocupado de
menor energia com valor aproximado de -1,7 eV.
As distâncias de ligação entre os átomos da molécula de cisteína otimizada
estão descritos na Tabela 4.1.
Quando a cisteína for colocada para interagir com o grafeno, a alteração nos
valores dos níveis energéticos e nos valores das distâncias entre os átomos indicará
a intensidade da interação ocorrida, demontrando seu caráter físico ou químico.

48
Tabela 4.1: Distâncias de ligação entre os átomos na molécula de cisteína.
Átomos da ligação Distância entre os átomos (Å)
S – H5 1,40
C3 – S 1,88
C3 – C2 1,57
C3 – H6 1,11
C3 – H7 1,11
C2 – H1 1,11
C2 – N 1,44
N – H2 1,03
N – H3 1,03
C2 – C1 1,52
C1 = O1 1,23
C1 – O2 1,34
O2 – H4 1,01
4.3. ADSORÇÃO DA CISTEÍNA EM GRAFENO PERFEITO
Nesta seção descreveremos os resultados do potencial de adsorção entre o
aminoácido estudado (cisteína) e a monocamada de grafeno perfeito.
Propriedades Estruturais
Analisamos três possíveis posições de aproximação para a cisteína:
aproximação pelo grupo carboxila (COOH), pelo grupo amino (NH2) e pelo grupo tiol
(SH), a Figura 4.4 (a), (c) e (e) apresenta as configurações iniciais para os sistemas
descritos, respectivamente.

49
Cisteína adsorvida pelo grupo COOH (carboxila)
a)
b)
Cisteína adsorvida pelo grupo NH2 (amino)
c)
d)
Cisteína adsorvida pelo grupo SH (tiol)
e)
f)
Figura 4.4: Cisteína interagindo com o grafeno pelo grupo carboxila: a)vista superior
b) vista lateral; cisteína interagindo com o grafeno pelo grupo amino: c)vista superior
d) vista lateral; cisteína interagindo com o grafeno pelo grupo tiol: e)vista superior f)
vista lateral;

50
Realizamos cálculos preliminares estáticos (sem otimização de geometria),
visando definirmos a melhor distância inicial entre a superfície de grafeno e a
molécula do aminoácido para que o sistema fosse posteriormente completamente
otimizado.
A molécula de cisteína foi aproximada pelo grupo COOH. Analisamos a
distância entre a molécula do aminoácido a partir de 1,35 até 4,40 Å (Figura 4.5 (a)),
de forma a avaliar a energia mínima do sistema.
A partir dos valores calculados para a energia mínima do sistema, definimos a
distância ideal de aproximadamente 3,0 Å para então liberar o aminoácido
inicialmente estático e avaliar a configuração mais estável e assim calcular a energia
de ligação e definir a estrutura de bandas do sistema.
A Figura 4.5 (b) demonstra a estrutura final da molécula de cisteína adsorvida
no grafeno pelo grupo carboxila com a geometria completamente relaxada. Observa-
se que a menor distância entre o plano de grafeno e o átomo mais próximo é de 2,9
Å e o valor da energia de ligação obtida através da equação (4.1) foi de 0,07 eV,
bem como não houve mudanças nos comprimentos das ligações 2 e 4, indicadas na
figura (4.5 (b)). Uma pequena alteração de 0,01 Å no comprimento da ligação 3, foi
observada indicando uma fraca interação entre a molécula e o plano de grafeno.
a)
b)
Figura 4.5: a) Gráfico preliminar da energia do sistema em função da distância entre
a molécula de cisteína e o plano de grafeno. b) estrutura final da cisteína adsorvida
no grafeno pelo grupo COOH.
A molécula de cisteína também foi avaliada interagindo com o grafeno por
meio do grupo NH2. A distância entre a molécula do aminoácido foi avaliada a partir

51
de 1,5 até 4,40 Å (Figura 4.6 (a)), de forma a avaliar a energia mínima do sistema
quando este está estático.
A partir dos valores calculados para a energia mínima do sistema definimos a
distância ideal de aproximadamente 3,2 Å para avaliar a configuração mais estável e
assim obter as energias de ligação e a estrutura de bandas do sistema.
A Figura 4.6 (b), demonstra a estrutura final da molécula de cisteína adsorvida
no grafeno pelo grupo NH2 com a geometria completamente relaxada, observa-se
que a menor distância entre o plano de grafeno e o átomo mais próximo é de 2,8 Å e
o valor da energia de ligação obtida é de 0,09 eV, um valor bastante reduzido,
indicando uma interação fraca, a qual pode ser evidenciada também pela
manutenção dos comprimentos das ligações indicadas, as quais permaneceram com
os mesmos valores encontrados nos sistemas isolados.
a)
b)
Figura 4.6: a) Gráfico preliminar da energia do sistema em função da distância entre
a molécula de cisteína e o plano de grafeno. b) estrutura final da cisteína adsorvida
no grafeno pelo grupo NH2.
De modo a obter a configuração de mínima energia, consideramos a cisteína
aproximando-se do grafeno pelo grupo tiol. A distância entre a molécula do
aminoácido e o grafeno foi considerada partir de 1,45 até 4,00 Å (Figura 4.7 (a)), e
chegamos ao valor da distância em que a energia final do sistema era mínima.
A partir dos valores calculados para a energia mínima do sistema, definimos a
distância ideal de aproximadamente 2,6 Å para que o programa encontrasse a
configuração mais estável deste e assim nos fornecesse as energias de ligação e a
estrutura de bandas do sistema.

52
a)
b)
Figura 4.7: a) Gráfico preliminar da energia do sistema em função da distância entre
a molécula de cisteína e o plano de grafeno. b) estrutura final da cisteína adsorvida
no grafeno pelo grupo SH.
A Figura 4.7 (b), demonstra a estrutura final da molécula de cisteína
adsorvendo no grafeno pelo grupo tiol com a geometria completamente relaxada,
observa-se que a menor distância entre o plano de grafeno e o átomo mais próximo
é de 1,93 Å, assim o valor da energia de ligação 0,3 eV é maior que para as outras
configurações, indicando ser esta a forma preferencial de aproximação do
aminoácido frente ao plano de grafeno perfeito; os comprimentos de ligação não
sofreram alteração significativa.
Tabela 4.2: Molécula de cisteína adsorvida em uma monocamada de grafeno
através da COOH, NH2 e SH. Onde Eb(eV) é a energia de ligação do sistema e DG-at
é a distância entre o grafeno e o átomo da molécula mais próximo à superfície (em
ângstrons).
Cisteína + grafeno Eb (eV) DG-at (Å)
COOH 0,07 2,9
NH2 0,09 2,8
SH 0,30 1,93
Resumidamente a Tabela 4.2 apresenta os resultados calculados a partir da
equação 4.1, para as energias de ligação e distâncias de ligação para a cisteína

53
adsorvida em uma monocamada de grafeno.
Propriedades Eletrônicas
Avaliamos as propriedades eletrônicas como estrutura de bandas para os
sistemas cisteína + grafeno perfeito, os quais possuem energias mais estáveis,
conforme as Figuras 4.5, 4.6 e 4.7, respectivamente.
Na Figura 4.8 estão apresentadas as estruturas de banda do grafeno puro (a)
interagindo com a cisteína pelos grupos b) COOH, c) NH2 e d)SH.
Figura 4.8: Estruturas de banda dos sistemas interagentes grafeno@cisteína,
representando as aproximações pelos grupos b) COOH, c)NH2 e d)SH.
Através da Figura 4.8, percebe-se claramente uma maior mistura nos níveis
da cisteína e do grafeno quando se aproxima pelo grupo NH2 Figura 4.8 (c), e
também o caráter semimetálico do grafeno, pois há um gap energético tendendo a
zero no ponto K.

54
Na Figura 4.8 (b), (c) e (d), identificamos os níveis de energia próximos de -
5,5 eV praticamente flats, sem muita modificação em relação ao seu estado inicial, o
que evidencia a fraca interação entre os grupos estudados e a monocamada de
grafeno.
4.4. ADSORÇÃO DA CISTEÍNA EM GRAFENO COM DEFEITO
Nesta seção descreveremos os resultados do potencial de adsorção entre o
aminoácido estudado (cisteína) e a monocamada de grafeno com defeito.
Analisamos as três possíveis posições de aproximação para a cisteína, de
forma análoga à feita anteriormente para o grafeno perfeito.
A cisteína foi aproximada pelo grupo carboxila (COOH), pelo grupo amino
(NH2) e pelo grupo sulfidrila (SH), a Figura 4.9 (a), (c) e (e) apresenta as
configurações iniciais para os sistemas descritos, respectivamente.
Utilizamos os mesmos valores para a distância inicial entre a camada de
grafeno e o aminoácido para as três configurações analisadas, para aproximação
pelo grupo COOH 3,0 Å, na aproximação pelo grupo NH2 3,2 Å e para aproximação
pelo grupo SH é 2,60 Å.
A Figura 4.10, demonstra a estrutura final da molécula de cisteína adsorvida
no grafeno pelo grupo carboxila com a geometria completamente relaxada, observa-
se que a menor distância entre o plano de grafeno e o átomo mais próximo é de 2,7
Å e o valor da energia de ligação obtida através da equação (4.1) foi de 0,13 eV
(Tabela 4.3), indicando uma interação fraca entre a molécula e o plano de grafeno,
sendo que este valor de energia de ligação é quase o dobro do que o valor
encontrado para a aproximação da molécula no plano de grafeno perfeito.

55
Cisteína adsorvida pelo grupo COOH (carboxila)
a)
b)
Cisteína adsorvida pelo grupo NH2 (amino)
c)
d)
Cisteína adsorvida pelo grupo SH (tiol)
e)
f)
Figura 4.9: Cisteína interagindo com o grafeno com defeito pelo grupo COOH.
a)vista superior b) vista lateral; com o grupo NH2: c)vista superior d) vista lateral; e
interagindo pelo grupo SH: e)vista superior f) vista lateral;

56
Figura 4.10: Estrutura final da cisteína adsorvida no grafeno com defeito pelo grupo
COOH.
A Figura 4.11, demonstra a estrutura final da molécula de cisteína adsorvida
no grafeno pelo grupo NH2 com a geometria completamente relaxada, observa-se
que a menor distância entre o átomo mais próximo da molécula e o plano de grafeno
é de 2,3 Å e o valor obtido para a energia de ligação é de 0,27 eV, com energia três
vezes maior em relação ao sistema puro.
Figura 4.11: Estrutura final da cisteína adsorvida no grafeno com defeito pelo grupo
NH2.
A Figura 4.12, demonstra a estrutura final da molécula de cisteína adsorvida
no grafeno pelo grupo tiol com a geometria completamente relaxada, onde se

57
observa que a distância entre o átomo mais próximo da molécula e o plano de
grafeno é de 2,1 Å e o valor da energia de ligação obtida através da fórmula (4.1) foi
de 0,38 eV (Tabela 4.3) indicando a interação mais forte encontrada neste estudo
entre a molécula de cisteína e o plano de grafeno.
Figura 4.12: Estrutura final da cisteína adsorvida no grafeno com defeito pelo SH.
Tabela 4.3: Molécula de cisteína adsorvida em uma monocamada de grafeno
perfeito e de grafeno com defeito, através da COOH, NH2 e SH. Onde Eb(eV) é a
energia de ligação do sistema e DG-at é a distância entre o grafeno e o átomo da
molécula mais próximo à superfície (em ângstrons).
Cisteína +
grafeno
perfeito
Eb (eV) DG-at (Å)
Cisteína +
grafeno
com
defeito
Eb (eV) DG-at (Å)
COOH 0,07 2,9 COOH 0,13 2,7
NH2 0,09 2,8 NH2 0,27 2,3
SH 0,30 1,93 SH 0,38 2,1
A Tabela 4.3 apresenta um estudo comparativo entre as energias de ligação e
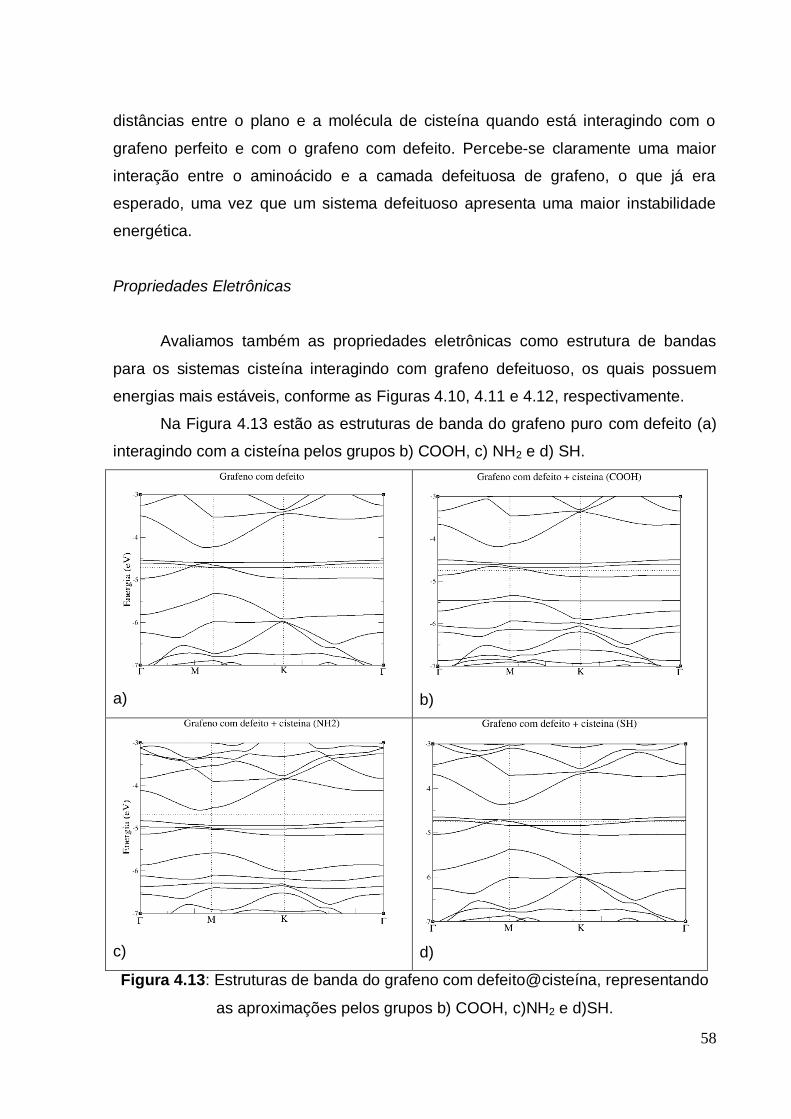
58
distâncias entre o plano e a molécula de cisteína quando está interagindo com o
grafeno perfeito e com o grafeno com defeito. Percebe-se claramente uma maior
interação entre o aminoácido e a camada defeituosa de grafeno, o que já era
esperado, uma vez que um sistema defeituoso apresenta uma maior instabilidade
energética.
Propriedades Eletrônicas
Avaliamos também as propriedades eletrônicas como estrutura de bandas
para os sistemas cisteína interagindo com grafeno defeituoso, os quais possuem
energias mais estáveis, conforme as Figuras 4.10, 4.11 e 4.12, respectivamente.
Na Figura 4.13 estão as estruturas de banda do grafeno puro com defeito (a)
interagindo com a cisteína pelos grupos b) COOH, c) NH2 e d) SH.
a)
b)
c)
d)
Figura 4.13: Estruturas de banda do grafeno com defeito@cisteína, representando
as aproximações pelos grupos b) COOH, c)NH2 e d)SH.

59
Através da Figura 4.13 (a), percebemos o caráter metálico do grafeno com
defeito, pois há cruzamento destes com a energia de Fermi localizada pela linha
tracejada em aproximadamente -4,75 eV. Na Figura 4.13 (b), identificamos os níveis
de energia abaixo de -5,5 eV praticamente flats, sem muita modificação em relação
ao seu estado inicial, o que evidencia a fraca interação entre o grupo COOH e a
monocamada de grafeno. Na Figura 4.13 (c) percebemos uma maior alteração na
característica dos níveis do sistema sendo que as propriedades resultam em um gap
de energia caracterizando um material metálico. Na Figura 4.13 (d) percebe-se
claramente uma intensa mistura nos níveis da cisteína (abaixo do nível de Fermi) e
do grafeno quando se aproxima pelo grupo SH, o que corrobora o maior valor
encontrado para a energia de ligação do sistema.

60
5. CONCLUSÃO
Neste trabalho, foram avaliadas as interações do aminoácido cisteína com a
monocamada de grafeno perfeito e com a monocamada de grafeno com um defeito
estrutural, em que estava faltando um átomo de carbono na rede hexagonal. As
aproximações do aminoácido foram realizadas, através de três possíveis sítios de
adsorção diferentes, grupo carboxila, grupo amino e grupo tiol, usando simulações
de primeiros-princípios baseada na teoria funcional da densidade. Cálculos de
energia de ligação, distância ótima de ligação e estrutura eletrônica foram também
realizados. Também foram realizados cálculos para determinar a estrutura eletrônica
final dos sistemas.
Observou-se que há uma adsorção fraca quando o aminoácido interage pelos
grupos carboxila e amino com energias de ligação entre 0,07 e 0,09 eV, enquanto
que a energia de ligação entre o aminoácido e o grafeno perfeito quando este é
aproximado pelo grupo tiol é mais alta (0,30 eV).
Já no caso da interação com o grafeno defeituoso, as energias encontradas
demonstraram uma maior afinidade da cisteína por este sistema, uma vez que os
valores de distância entre o aminoácido e o grafeno são menores, e os valores das
energias de ligação para os três sistemas avaliados são maiores, 0,13 e 0,27 eV
para os grupos carboxila e amino respectivamente e 0,38 eV para o grupo tiol, o que
evidencia a maior intensidade da interação destes sistemas com a monocamada do
grafeno com defeito estrutural, e dentre os sítios avaliados, nos dois casos a
interação do grupo tiol com o grafeno foi maior.
A partir das estruturas de banda dos sistemas pudemos perceber claramente
a maior interação entre o aminoácido e a camada de grafeno defeituoso, o qual já
apresentava características metálicas, a maior intensidade da interação ficou
evidenciada pela alta mistura entre as bandas do grafeno puro defeituoso e os níveis
eletrônicos da molécula da cisteína.
Através deste estudo ficou demonstrado a alta seletividade e variabilidade dos
sítios de adsorção molecular e também a possibilidade da utilização deste como
sensor de moléculas, uma vez que moléculas com diferentes sítios ativos poderão
ser adsorvidas e retiradas do sistema com relativa facilidade bem como ser
desenvolvido uma nova classe de dispositivo, fazendo uso das possíveis moléculas

61
que estarão adsorvidas no sistema.
Para trabalhos futuros, poderão ser avaliadas as propriedades estruturais e
eletrônicas do grafeno interagindo com um maior número de moléculas de um
mesmo aminoácido ou moléculas de diferentes aminoácidos, ou ainda a interação
de mais de uma camada de grafeno, visando compreender o comportamento destes
diferentes sistemas para que futuramente possamos escolher dentre eles qual será
usado para cada finalidade.

62
REFERÊNCIAS BIBLIOGRÁFICAS
ARTACHO, E.; SÁNCHEZ-PORTAL, D.; ORDEJÓN, P.; GARCIA A.; SOLER, J. M.
Linear-scaling ab initio calculations for large and complex systems Phys. Stat.
Sol. (b), 215, 809 (1999).
ASCROFT, N. W.; MERMIN, N. D. Solid State Physics, Saunders College,
Philadelphia, PA, (1976).
BENA, C.; KIVELSON, S. A. Quasiparticle scattering and local density of states
in graphite, Phys. Rev, B 72, 125432, (2005).
BOUKHVALOV, D. W.; KATSNELSON, M. I. Chemical functionalization of
graphene with defects. Nano Letters, 8, 12, (2008).
BOUKHVALOV, D. W.; KATSNELSON, M. I. Chemical functionalization of
Graphene. Jour. Phys.-Cond. Matt., 21, 34, (2009).
BOUKHVALOV, D. W.; KATSNELSON, M. I. Tuning the gap in bilayer graphene
using chemical functionalization: Density functional calculations. Phys. Rev. B,
78, 8, (2008).
CASTRO, H. N.; GUINEA, F.; PERES, N. M. R.; NOVOSELOV, K. S.; GEIM, A. K.
Electronic properties of bilayer and multilayer graphene. Phys. Rev. B, 78, 4
(2007).
CHUTIA, A.; CIMPOESU, F.; TSUBOI, H.; MIYAMOTO, A. Influence of Organic
Functional Groups on the Electrical Properties of Carbon Black: A Theoretical
Study. Jap. Jou. Of Ap.Phys., 47, 4, 3147 (2008).
DRESSELHAUS, M. S.; DRESSELHAUS, G.; EKLUND, P. C. Science of
Fullerenes and Carbon Nanotubes. New York, NY, San Diego, CA: Academic
Press (1996).
DRESSELHAUS, M. S.; DRESSELHAUS, G. Intercalation compounds of
graphite. Adv. Phys., 51, 1 (2002).

63
FAGAN, S. B. Funcionalização de nanotubos de carbono: uma abordagem de
primeiros princípios. Tese de Doutorado em Física, Universidade Federal de Santa
Maria, UFSM (2003).
E. Fermi, Z. Physik, 48, 73 (1928).
FERNANDES, M. Estudo de vacâncias e falhas de empilhamento em ZnO
wurtzita. Dissertação de Mestrado em Física, Universidade Federal de Uberlândia,
UFU (2006).
FUCHS, M.; BOCKSTEDTE, M.; PEHLKE, E.; SCHEFFLER, M. Pseudopotential
study of binding properties of solids within generalized gradient
approximations: The role of core-valence exchange correlation. Phys. Rev. B,
57, 2134 (1998).
GEIM, A. K.; NOVOSELOV, K. S. The rise of graphene. Nature. Mater., 6, 183
(2007).
GREENWOOD, N. N.; EARNSHAW, A. Chemistry of the Elements, 2nd Edition,
Oxford:Butterworth-Heinemann (1997).
HOHENBERG, P.; KOHN, W. Inhomogeneous Electron Gas. Phys. Rev. 136B,
864 (1964).
IIJIMA, S. Helical microtubules of graphitic carbon. Nature, 354, 56 (1991).
IIJIMA, S.; ICHIHASHI, T. Single-shell carbon nanotubes of 1-nm diameter.
Nature 363, 603 (1993).
JIANG, D.E.; SUMPTER, B.G.; DAI, S. How do aryl groups attach to a graphene
sheet? J. of Phys. Chem. B, 110, 47, 23628 (2006).
KOHN, W.; SHAM, L. J. Self-consistent equations including exchange and
correlation effects. Phys. Rev.140A,1133 (1965).
KOHN, W. Nobel lecture: Electronic structure of matter - Wave functions and
density functional; Rev. Mod. Physics, 71, 1253 (1999).

64
KONG, J.; FRANKLIN, N. R.; ZHOU, C.; CHAPLINE, M. G.; PENG, S.; CHO, K.;
DAI, H. Nanotube molecular wires as chemical sensors; Science, 287, 622
(2000).
KROTO, H. W. ; HEATH, J. R. ; O’BRIEN, S. C. ; CURL, R. F. ; SMALLEY, R. E. C60
- buckminsterfullerene. Nature, 318, 162 (1985).
LEHNINGER; N., D. L.; COX, M. Princípios de Bioquímica. 3ed. São Paulo:
Sarvier, (2002).
MARTIN, R. M. Electronic Structure: Basic Theory and Practical Methods,
Cambridge University Press, Urbana-Champaign (2004).
MEYER, J. C.; GEIM, A. K.; KATSNELSON, M. I.; NOVOSELOV, K.S.; BOOTH, T.
J.; ROTH, S. The structure of suspended graphene sheets. Nature, 446, 7131,
60, (2007).
NOVOSELOV, K. S. ; GEIM, A. K. ; MOROZOV, S. V. ; JIANG, D. ; ZHANG, Y. ;
DUBONOS, S. V. ; GRIGORIEVA, I. V. ; FIRSOV, A. A. Electric Field Effect in
Atomically Thin Carbon Films. Science, 306, 666 (2004).
NOVOSELOV, K. S. ; JIANG, D. ; SCHEDIN, F. ; BOOTH, T. J. ; KHOTKEVICH, V.
V. ; MOROZOV, S. V. ; GEIM, A. K. Two-dimensional atomic crystals. American
Proc. Natl. Acad. Sci. U. S. A., 102, 10451 (2005).
NOVOSELOV, K. S. ; WEHLING, T. O. ; MOROZOV, S. V. ; VDOVIN, E. E. ;
KATSNELSON, M. I. ; GEIM, A. K. ; LICHTENSTEIN, A. I. Molecular Doping of
Graphene. Nano Letters, 8, 1, 173, (2008).
PEI, QX.; ZHANG, Y. W.; SHENOY, V. B. Mechanical properties of methyl
functionalized graphene: a molecular dynamics study. Nanotechnology. 21,
11,115709 (2010).
PERDEW, J. P.; WANG, Y. Accurate and simple analytic representation of the
electron-gas correlation energy. Phys. Rev. B, 45,13244 (1991).
PERDEW, J. P.; ZUNGER, A. Self-interaction correction to density-functional

65
approximations for many electrons systems. Phys. Rev. B, 23,10 (1981).
REICH, S.; MAULTZSCH, J.; THOMSEN, C.; ORDEJÓN, P. Tight-binding
description of grapheme. Phys. Rev. B 66, 035412 (2002).
SAITO, R.; FUJITA, M.; DRESSELHAUS, G.; DRESSELHAUS, M. S. Electronic-
structure of chiral graphene tubules. Appl. Phys. Lett. 60, 2204 (1992a).
SAITO, R.; FUJITA, M.; DRESSELHAUS, G.; DRESSELHAUS, M. S. Electronic
structure of graphene tubules based on C60. Appl. Phys. Rev. B 46, 1804 (1992b).
SCHEDIN, F.; GEIM, A. K.; MOROZOV, S. V.; HILL, E. W.; BLAKE, P.;
KATSNELSON, M. I. NOVOSELOV, K. S. Detection of individual gas molecules
adsorbed on grapheme. Nat. Mater. 6, 652 (2007).
SILVA, J. J. A. Adsorção de átomos alcalinos e halogênios em uma superfície
de grafeno: um estudo de primeiros princípios. Dissertação de Mestrado
apresentada ao programa de pós-graduação do Departamento de Física da
Universidade Federal do Ceará, UFC, (2008).
SOLER, J. M. ; ARTACHO, E. ; GALE, J. D. ; GARCIA, A. ; JUNQUERA, J.
ORDEJON, P. ; SANCHEZ-PORTAL, D. The siesta method for ab initio order N
materials simulation. J. Phys: Condens. Matter. 14, 2745 (2002).
SOLOMONS, G.; FRYHLE, C. Organic Chemistry, 7th edition, vol. 1, LTD, (2001).
STANKOVICH, S.; DIKIN, D. A.; DOMMETT, G. H. B.; KOHLHAAS, K. M.; ZIMNEY,
E. A.; STACH, E. A.; PINER, R. D.; NGUYEN, S. T.; RUOFF, R. S. Graphene-based
composite materials, Nature 442, 282 (2006).
THOMAS L.H., Proc. Camb. Phil. Soc. 23, 542 (1927);
TROULLIER, N.; MARTINS, J. L. Efficient pseudopotentials for plane-wave
calculations. Phys. Rev. B, 43,1993 (1991).
VELOSO, M. V. ; FILHO, A. G. S. ; FILHO, J. M. ; FAGAN, S. B. ; MOTA, R. Ab
initio study of covalently functionalized carbon nanotubes. Chem. Phys. Lett.,
430, 71, (2006).

66
WALLACE, P. R. The band theory of graphite, Phys. Rev. 71, 622 (1947).
ZANELLA, I.; GUERINI, S.; FAGAN, S. B.; FILHO, J. M.; FILHO, A. G. S. Chemical
doping-induced gap opening and spin polarization in graphene. Phys. Rev. B,
77, 7,073404, (2008).
ZHANG, Y., TAN, Y.-W., STORMER, H. L. and KIM, P. Experimental observation
of the quantum Hall effect and Berry's phase in graphene. Nature 438, 201
(2005).
ZHOU, J., WU, M. M., ZHOU, X., SUN, Q. Tuning electronic and magnetic
properties of graphene by surface modification. Appl. Phys. Lett. 95, 103108
(2009).

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo