anabela cardoso mariano - estudogeral.sib.uc.pt · em 1806 sertϋrner, um farmacêutico alemão,...
TRANSCRIPT

2010
Anabela Cardoso Mariano
DESENVOLVIMENTO DE METODOLOGIAS
ANALÍTICAS PARA A DETERMINAÇÃO DE METADONA,
BUPRENORFINA E SEUS PRINCIPAIS METABOLITOS
EM AMOSTRAS BIOLÓGICAS. APLICAÇÕES EM
CONTEXTO FORENSE.
Dissertação apresentada à Faculdade de Medicina da Universidade de Coimbra
para obtenção do Grau de Mestre em Medicina Legal


Trabalho experimental realizado no
Serviço de Toxicologia Forense da
Delegação do Centro do
Instituto Nacional de Medicina Legal, I.P.
“�Por isso eu tomo ópio. É um remédio.
Sou um convalescente do Momento.
Moro no rés-do-chão do pensamento
E ver passar a Vida faz-me tédio�”
Alvaro de Campos (Fernando Pessoa)


AGRADECIMENTOS
À Dra. Cláudia Margalho, minha orientadora, agradeço pela permanente
disponibilidade, apoio e orientação científica dispensadas durante o desenvolvimento deste
trabalho.
Ao Senhor Professor Doutor Francisco Corte-Real, meu co-orientador, agradeço o
incentivo e facilidades disponibilizadas para a realização deste trabalho de investigação.
Ao Dr. Miguel Franco, Director do Serviço de Toxicologia Forense, que possibilitou a
realização deste trabalho no serviço.
À técnica Alice Castanheira por me ter “aturado” e ajudado sempre que precisei.
Aos meus pais por todo o apoio, força, carinho e por todo o sacrifício que sempre
fizeram por mim, um muito obrigada.
Ao meu irmão e à minha tia Lurdes que também sempre estiveram presentes.
Ao Cláudio que desde o início desta caminhada sempre esteve ao meu lado, apoiando-
me e dando-me toda a força e carinho, sempre acreditando em mim.
Aos meus amigos que continuam a ter paciência para me aturar e que me têm apoiado
incondicionalmente.


SUMÁRIO
Índice de Figuras I
Índice de Tabelas II
Abreviaturas IV
I – Introdução 1
1. Justificação do Tema Escolhido 3
2. Objectivos 5
II – Revisão da Bibliografia 7
1. História do Ópio 9
2. Opiáceos e opióides 13
3. Metadona 17
4. Buprenorfina 21
5. Técnicas para Determinação da Metadona e da Buprenorfina 23
6. Extracção em Fase Sólida 27
7. Derivatização 33
8. Validação de Metodologias Analíticas 37
III – Parte Experimental 45
Materiais e Métodos 47
Resultados e Discussão 59
Conclusões 99
IV – Referências 103


I
ÍNDICE DE FIGURAS
Figura 1. Papaver Somniferum 9
Figura 2. Papaver Bracteatum e Papaver Somniferum 10
Figura 3. Estrutura química da metadona 17
Figura 4. Estrutura química da buprenorfina 21
Figura 5. Coluna SPE 27
Figura 6. Etapas dos procedimentos de SPE 29

II
ÍNDICE DE TABELAS
Tabela 1. Preparação das curvas de calibração para Metadona e EDDP 53
Tabela 2. Preparação das curvas de calibração para Buprenorfina e Norbuprenorfina 53
Tabela 3. Preparação dos CQI para Metadona e EDDP 54
Tabela 4. Preparação dos CQI para Buprenorfina e Norbuprenorfina 54
Tabela 5. Principais parâmetros analíticos da análise instrumental 56
Tabela 6. Sangues usados para construir as misturas ou pools 60
Tabela 7. Tolerância máxima para as áreas relativas dos iões diagnóstico 62
Tabela 8. Estudo da selectividade/especificidade referente à Metadona 63
Tabela 9. Estudo da selectividade/especificidade referente à EDDP 64
Tabela 10. Estudo da selectividade/especificidade referente à Buprenorfina 65
Tabela 11. Estudo da selectividade/especificidade referente à Norbuprenorfina 66
Tabela 12. Resultados obtidos no estudo da especificidade/selectividade 67
Tabela 13. Preparação das curvas de calibração referentes ao estudo dos 69
LD/LQ para a metadona e EDDP
Tabela 14. Preparação das curvas de calibração referentes ao estudo dos 70
LD/LQ para a Buprenorfina e Norbuprenorfina
Tabela 15. Resumo dos valores calculados para os limites de detecção e de 75
quantificação

II
Tabela 16. Resumo dos valores calculados para a eficiência de extracção da 77
Metadona/EDDP
Tabela 17. Resumo dos valores calculados para a eficiência de extracção da 78
Buprenorfina e Norbuprenorfina
Tabela 18. Estudo da linearidade para a metadona e EDDP 80
Tabela 19. Estudo da linearidade para a Buprenorfina e Norbuprenorfina 81
Tabela 20. Resumo dos resultados obtidos para a linearidade e gamas de trabalho 86
Tabela 21. Resumo dos resultados respeitantes à repetibilidade da metadona 87
Tabela 22. Resumo dos resultados respeitantes à repetibilidade da EDDP 88
Tabela 23. Resumo dos resultados respeitantes à repetibilidade da Buprenorfina 88
Tabela 24. Resumo dos resultados respeitantes à repetibilidade da Norbuprenorfina 89
Tabela 25. Curvas de calibração para o estudo da precisão intermédia e exactidão 90
da Metadona e EDDP
Tabela 26. Controlos para o estudo da precisão intermédia e exactidão da 90
Metadona e EDDP
Tabela 27. Curvas de calibração para o estudo da precisão intermédia e 91
exactidão da Buprenorfina e Norbuprenorfina
Tabela 28. Controlos para o estudo da precisão intermédia e exactidão da 91
Buprenorfina e Norbuprenorfina
Tabela 29. Tabela ANOVA (factor único) 92
Tabela 30. Cálculo das estimativas da precisão 93
Tabela 31. Resultados obtidos para a precisão intermédia da Metadona 94

III
Tabela 32 Resultados obtidos para a precisão intermédia do EDDP 94
Tabela 33. Resultados obtidos para a precisão intermédia da Buprenorfina 95
Tabela 34. Resultados obtidos para a precisão intermédia da 95
Norbuprenorfina
Tabela 35. Resultados obtidos para a exactidão da Metadona 97
Tabela 36. Resultados obtidos para a exactidão do EDDP 97
Tabela 37. Resultados obtidos para a exactidão da Buprenorfina 98
Tabela 38. Resultados obtidos para a exactidão da Norbuprenorfina 98

VI
ABREVIATURAS
ANOVA Analysis of Variance (Análise de Variância)
CV Coeficiente de Variação
FDA Food and Drug Administration
GC Gas Chromatography (Cromatografia de gases)
GC/MS Gas Chromatography – Mass Spectrometry (Cromatografia de gases
acoplada a Espectrometria de Massas)
ICH International Conference on Harmonisation
INML Instituto Nacional de Medicina Legal, I. P.
ISO International Standard Organization
IT-STF-C-008 Instrução para a avaliação de ensaios interlaboratoriais em vigor no
Serviço de Toxicologia Forense da Delegação do Centro
LC/MS Liquid Chromatography – Mass Spectrometry (Cromatografia Líquida
acoplada a Espectrometria de Massas)
LLE Liquid-Liquid Extraction (Extracção Líquido-Líquido)
LLOQ Lower Limit of Quantification (Limite Inferior de Quantificação)
LOD Limit of Detection (Limite de Detecção)
LOQ Limit of Quantification (Limite de Quantificação)
MSTFA N-metil-N-(trimetilsilil)trifluoroacetamida
PE-STF-C-205 Procedimento de ensaio de confirmação de metadona e EDDP em vigor
no Serviço de Toxicologia Forense da Delegação do Centro

VI
PE-STF-C-206 Procedimento de ensaio de confirmação de buprenorfina e buprenorfina
em vigor no Serviço de Toxicologia Forense da Delegação do Centro
PE-STF-C-211 Procedimento de ensaio de quantificação de metadona e EDDP em vigor
no Serviço de Toxicologia Forense da Delegação do Centro
PE-STF-C-212 Procedimento de ensaio de quantificação de buprenorfina e
norburenorfina em vigor no Serviço de Toxicologia Forense da Delegação
do Centro
PO-STF-C-006 Procedimento operacional de validação de procedimentos de ensaio
rpm Rotações por minuto
SIM Selected Ion Monitoring (Monitorização Selectiva de Iões)
SNC Sistema Nervoso Central
SPE Solid Phase Extraction (Extracção em fase Sólida)
TMCS Trimetilclorosilano

I - INTRODUÇÃO


Justificação do Tema Escolhido
Anabela Cardoso Mariano Página 3
1. JUSTIFICAÇÃO DO TEMA ESCOLHIDO
A utilização de metadona e buprenorfina como tratamento de substituição na
terapia de reabilitação de toxicodependentes tem vindo a demonstrar-se eficaz e
segura. Existe no entanto a possibilidade da sua utilização ilícita ou interacção com
outras substâncias com possível ocorrência de mortes associadas ao consumo de doses
superiores às terapêuticas.
O interesse pela realização deste trabalho surgiu devido às solicitações para a
realização de análises toxicológicas para as substâncias referidas, e devido ao facto da
metodologia existente no Serviço de Toxicologia Forense da Delegação do Centro do
INML, não se encontrar validada.
Foi escolhida como matriz o sangue total devido a ser a amostra biológica mais
usual e frequentemente a única disponível em toxicologia forense, e por isso aquela
que se afigura como de maior importância. Por outro lado, após pesquisa e estudo
detalhado da bibliografia disponível, são escassos trabalhos em sangue total pos-
mortem.
Considerámos que os resultados obtidos no decurso do trabalho efectuado
justificaram a escolha da temática abordada.


Objectivos
Anabela Cardoso Mariano Página 5
2. OBJECTIVOS
Chega diariamente ao Serviço de Toxicologia Forense um elevado número de
amostras de sangue para pesquisa de substâncias eventualmente presentes,
verificando-se frequentes solicitações para pesquisa de metadona e buprenorfina.
Assim, e no sentido da dar uma resposta cuja interpretação dos resultados
analíticos seja de confiança, é de todo o interesse do serviço desenvolver e validar
metodologias sensíveis para a determinação do maior número de substâncias possível.
A metadona e a buprenorfina usadas cada vez mais para o tratamento de
substituição de drogas opiáceas são muitas vezes usadas de forma ilícita.
Tendo presentes estas situações, o objectivo fundamental do trabalho foi
validar o método em estudo, de modo a obter a máxima confiança na interpretação
dos resultados obtidos. Os parâmetros de validação estudados foram a selectividade,
linearidade, limites de detecção e de quantificação, precisão intermédia, exactidão e
recuperação do processo extractivo.


II – REVISÃO BIBLIOGRÁFICA


História do ópio
Anabela Cardoso Mariano Página 9
1. HISTÓRIA DO ÓPIO
Tão antiga quanto o homem é a busca pela felicidade, mesmo que esta dure
breves instantes. Talvez por isso a primeira droga a ser descoberta tenha sido o ópio. O
ópio é o sumo natural da papoila Papaver Somniferum, do qual 25% do seu peso são
alcalóides (1, 2, 3). É obtido através da realização de uma incisão na cápsula da
papoila, de onde sai um líquido de aspecto leitoso que solidifica com facilidade,
tornando-se acastanhado. Desde o período neolítico que é utilizado para o alívio de
dores e em cerimónias religiosas.
Fig. 1 – Papaver Somniferum
Viajando pelo mundo antigo, encontram-se relatos do uso do ópio em
praticamente todas as civilizações antigas: egípcios, mesopotâmicos, persas, gregos e
romanos. As referências ao seu uso já se encontravam nos escritos de Teofrasto, no
séc. III a.C. (1, 2) e Homero refere o seu uso por Helena de Tróia não só para anular as
dores como também para melhorar o humor. Os físicos arábicos eram especialistas no
uso do ópio, e foram precisamente os comerciantes arábicos que introduziram a droga
no oriente (2, 4). No Papiro de Ebers, o ópio era componente básico em cerca de 700
remédios (2).

História do ópio
Anabela Cardoso Mariano Página 10
Largamente conhecido pelos grandes médicos gregos, como Hipócrates e
Galeno, o ópio era recomendado para a cura de diversas doenças como: epilepsia,
bronquite, asma, pedra dos rins, febre, melancolia, disenteria, diarreia, gota, tétano,
insanidade e até ninfomania e também usado como sedativo e tranquilizante (4).
Para os romanos a papoila era ainda um poderoso símbolo de sono e morte,
utilizada como uma potente arma em suicídios e assassinatos. Em 183 a.C., Aníbal
suicidou-se ao ingerir uma pequena quantidade de ópio que se encontrava no seu anel
e anos mais tarde, Agripina a última mulher do imperador Cláudio, utilizou também o
ópio para envenenar o seu enteado, para que Nero assumisse o império (2, 4).
Na idade média, Avicena considerado o maior médico dessa época, descrevia
no seu livro remédios que misturavam o ópio com nozes, eufórbia e alcaçuz. Acabou
por morrer de overdose de ópio com vinho (4).
Numa família de 28 géneros de papoilas e 250 espécies, apenas 2 delas contêm
uma quantidade razoável de ópio: Papaver Bracteatum e Papaver Somniferum, esta
última cultivada na Ásia Menor.
Fig. 2 – Papaver Bracteatum e Papaver Somniferum
Em 1806 Sertϋrner, um farmacêutico alemão, descreveu o isolamento de uma
substância pura do ópio, a morfina (do grego Morpheus, o Deus dos sonhos) a qual já
tinha sido extraída em 1804 pelo francês Armand Seguin (3, 4).
A morfina é o alcalóide mais activo do ópio, com um teor de 10% do seu peso,
apresenta-se sob a forma de cristais solúveis e serve para a preparação de numerosos
derivados. A descoberta de outros alcalóides do ópio, como a codeína e a papaverina,

História do ópio
Anabela Cardoso Mariano Página 11
rapidamente se seguiu (1).
O ópio foi a única droga a ser motivo de guerra, quando em 1839 o imperador
chinês Ch’ung Ch’en proibiu o consumo desta no seu território, a qual era produzida
em Inglaterra, originando uma guerra de três anos, conhecida como a Guerra do Ópio
(5).
Em 1874 o farmacêutico inglês Alder Wright procurando uma alternativa tão
poderosa quanto a morfina, mas sem o inconveniente da dependência, aqueceu-a com
anidro acético até à ebulição, produzindo a diacetilmorfina, mais conhecida por
heroína (6).
Até 1920 o ópio e seus derivados foram utilizados livremente, no entanto,
devido ao seu uso abusivo e com o aumento de farmacodependentes, estes foram
proibidos em vários países e as convenções internacionais de 1925 e 1931 impuseram
medidas restritas à sua fabricação e exportação (3, 5).
Paralelamente aos efeitos terapêuticos surgiu o conhecimento dos efeitos
tóxicos o que veio estimular a pesquisa de analgésicos opióides sintéticos que tivessem
efeitos tóxicos menos acentuados, surgindo assim a metadona.
A metadona foi desenvolvida no final dos anos 30 na Alemanha Nazi, em
antecipação à possível falta de ópio e seus derivados, uma vez que durante a guerra a
função dos analgésicos é bastante importante para os militares e para a população
civil. A metadona foi testada por médicos profissionais do exército alemão em 1939-
40, mas verificou-se que era demasiado tóxica e com grandes possibilidades de causar
dependência após o seu uso prolongado (7). Em 1950 começou a ser prescrita para o
tratamento dos sintomas associados à dependência de heroína e opióides (8) e em
1964 estudos científicos demonstraram a sua importância no tratamento da
dependência de heroína ao contrariar a síndrome de abstinência e reduzir o consumo
de droga (9). Deste modo a metadona constituiu o primeiro tratamento de
substituição para a dependência crónica de drogas opiáceas.
Mais tarde surge a buprenorfina que foi comercializada pela primeira vez nos
Estados Unidos como analgésico e aprovada pela U.S. Food and Drug Administration
(FDA), em Outubro de 2002, para o tratamento de substituição de drogas opiáceas
(10). O perfil farmacológico da buprenorfina constitui uma alternativa atractiva à
metadona no tratamento da dependência de opióides (11).


Opiáceos e Opióides
Anabela Cardoso Mariano Página 13
2. OPIÁCEOS E OPIÓIDES
Como já referido anteriormente o ópio é o sumo extraído da papoila, do qual
25% do seu peso são alcalóides. Os alcalóides do ópio são de 2 grupos distintos: os
fenantrénicos que têm propriedades analgésicas (morfina, codeína, tebaína) e os
benzilisoquinoleicos que não possuem propriedades analgésicas (papaverina e
noscapina). O ópio e os seus derivados (sintéticos ou não) actuam por estimulação de
receptores opiáceos a nível do sistema nervoso central. As substâncias que possuem
este mecanismo de acção são denominadas por analgésicos opiáceos se forem de
origem natural, ou por analgésicos opióides se forem de origem semi-sintética ou
sintética.
O ópio foi utilizado durante muito tempo devido às suas propriedades
medicinais como analgésico, antitússico e antidiarreico (1).
O comité de taxonomia da Associação Internacional para o Estudo da Dor (IASP)
(126), conceitua a dor como : ‘Uma experiência sensorial e emocional desagradável,
que é associada ou descrita em termos de lesões teciduais.’
A dor pode ser assim definida como uma sensação desagradável, criada por um
estímulo nocivo que atinge o sistema nervoso central por meio de vias específicas.
A origem da dor pode ser central (Sistema nervoso central) ou periférica
(Sistema nervoso periférico). Nesta última são encontradas alterações nos distintos
subtipos de fibras nervosas, enquanto na dor central as alterações são mais complexas
e envolvem vias aferentes, diferentes circuitos cerebrais e a modulação descendente
(12, 13).
A dor é causada pela modificação das condições normais de um organismo
vivo. Esse organismo necessita de apresentar capacidade de resposta com reacções de
adaptação às modificações que ocorrem no meio ambiente.
Os principais processos envolvidos na experiência sensorial da dor são: a
percepção da dor e a reacção à dor.
A percepção da dor envolve mecanismos anátomo-fisiológicos, através dos
quais um estímulo nocivo capaz de gerá-la é criado e transmitido por vias neurológicas
desde os receptores da dor. Os elementos que captam os estímulos a serem
transmitidos ao sistema nervoso central, para uma análise e possível reacção, são

Opiáceos e Opióides
Anabela Cardoso Mariano Página 14
chamados receptores. Os receptores (do latim recipere = receber) são tecidos nervosos
especializados sensíveis a alterações específicas que se produzem no seu meio (1, 2,
12, 13).
As várias modalidades de sensação podem ser percebidas e distinguem-se
umas das outras devido a diferentes tipos de receptores. A pesquisa fisiológica
demonstrou que estímulos específicos são captados por receptores específicos e
assim, por exemplo, os receptores da dor somente respondem com a sensação de dor
a qualquer estímulo que atinja seu limiar de excitação.
Os receptores da dor são, histologicamente, pouco mais que terminações
nervosas livres. Pelo facto de estarem relacionados com estímulos capazes de causar
danos às células, têm importante valor protector, avisando sobre perigos iminentes ou
reais. Como qualquer agente capaz de causar dano é chamado nocivo; os receptores
da dor denominam-se também por nociceptores (1, 2, 12, 13).
A fisiologia da condução de um estímulo nervoso envolve quatro componentes
funcionais que podem transformar os sinais de entrada em libertação de
neurotransmissores (ex. bradicinina, prostaglândinas e substância P) (1, 2).
O primeiro componente funcional é um sinal de entrada (input) que, após fazer
contacto com um receptor dendrítico com suficiente intensidade, induz a geração de
um potencial receptor o que transforma um estímulo sensitivo doloroso em sinal
eléctrico local.
O segundo componente funcional é o sinal de integração. Como o potencial
receptor local não pode por si só gerar um potencial de acção, deve ser modificado por
uma transmissão activa adicional. Caso os potenciais do receptor desenvolvam uma
soma integrada suficientemente excitatória, será iniciado o potencial de acção. Se esse
potencial de acção não se gerar, o sinal de entrada dissipa-se sem que haja resposta
perceptível.
O terceiro componente funcional da condução nervosa é a propagação
continuada do potencial de acção para a medula espinhal. O axónio nervoso é o
componente anatómico do sistema nervoso responsável pela propagação do potencial
de acção.
O quarto componente funcional é a ascenção do estímulo às estruturas do
sistema nervoso central. O corno dorsal é a região da medula espinhal cujo propósito

Opiáceos e Opióides
Anabela Cardoso Mariano Página 15
principal é receber o estímulo aferente (proveniente da região onde ocorreu a lesão),
modificar o sinal de entrada de acordo com as influências descendentes dos centros
cerebrais superiores e transmitir a informação resultante aos centros cerebrais
superiores para continuar o processo de compreensão da dor e do local de lesão (1, 2).
A este sistema de condução ascendente corresponde um outro, descendente, com
origem em vários locais intracranianos e que exerce efeitos de modificação do sinal de
entrada (input) a vários níveis. É o sistema de antinocicepção ou analgesia.
Os opióides endógenos são os mediadores mais importantes da
antinocicepção/analgesia, actuando sobre receptores específicos. Na espécie humana,
são caracterizados 4 tipo de receptores opiáceos endógenos: o receptor µ, ĸ, δ e σ.
Estes receptores encontram-se localizados por todo o sistema nervoso, com diferentes
densidades e zonas de localização, havendo por vezes zonas comuns. Dependendo das
suas localizações assim serão os seus efeitos fisiológicos; por essa razão é que alguns
apresentam maior analgesia e dependência que outros (1, 2, 3).
Os mediadores endógenos dos receptores opiáceos são polipeptídeos de 3
famílias diferentes: encefalinas, endorfinas e dinorfinas, que apresentam afinidades
distintas para com os diferentes receptores. As encefalinas apresentam maior
afinidade para o receptor δ, enquanto as endorfinas β e as dinorfinas para o receptor µ
e receptor ĸ, respectivamente (1, 3). O receptor σ não é estimulado por nenhuma
família de ligandos endógenos conhecida, no entanto, ocorre estimulação por
substâncias exógenas (1). Como tal, são nestes receptores que os analgésicos
opiáceos/opióides actuam. Os efeitos farmacológicos dos opióides podem ser úteis ou
adversos, conforme a dose e a situação. Cada fármaco pode produzir efeitos de
intensidade diferente conforme a sua especificidade para uns ou outros receptores.
Deste modo, da estimulação dos receptores opiáceos pode resultar não só o efeito de
analgesia mas um conjunto de outros efeitos (por exemplo, euforia e estados
hipnóticos) aliados a uma forte componente de dependência física e psíquica. Os
opióides apresentam duas características que os tornam drogas de abuso
particularmente perigosas, produzem euforia e bem-estar, e a sua acção necessita de
doses cada vez maiores para manter o mesmo efeito -fenómeno de tolerância (1,3).
Actualmente o opióide de abuso mais usado é a heroína, um derivado da
morfina com praticamente os mesmos efeitos mas com maior solubilidade aquosa, o

Opiáceos e Opióides
Anabela Cardoso Mariano Página 16
que facilita o seu consumo abusivo.
O tratamento de substituição é, por definição, uma forma de tratamento
médico para os dependentes de opiáceos (em especial os da heroína) baseado na
utilização de uma substância com acção semelhante à droga normalmente consumida
(17).
A metadona e a buprenorfina são duas das substâncias que podem ser
utilizadas no tratamento de substituição, em pessoas dependentes de opiáceos (17).
As substâncias de substituição podem ser agonistas — substâncias que activam os
receptores de opiáceos, no cérebro, desencadeando o efeito do consumo de droga —
ou agonistas-antagonistas — substâncias que simultaneamente activam os receptores
de opiáceos, no cérebro, e limitam ou eliminam os efeitos de outros opiáceos ou
opióides.

Metadona
Anabela Cardoso Mariano Página 17
3. METADONA
a) Propriedades físico-quimicas
A metadona (6-dimetilamino-4,4-difenil-3-heptanona - C21H27NO) é o primeiro
opióide de síntese a perder o anel piperidínico; é usada sob a forma de cloridrato
racêmico porque possui dois isômeros: a L-metadona, responsável pela acção
analgésica e sedativa e a D-metadona que tem actividade antitússica. O isómero L-
metadona é o mais activo e é composto por 2 enantiómeros: R-metadona e S-
metadona (14, 15). Apresenta um peso molecular de 309.44 g/mol, um ponto de fusão
de 241ºC, é solúvel em água, álcool e clorofórmio, e é praticamente insolúvel em éter
e glicerol (16).
Fig. 3 – Estrutura química da metadona
b) Grupo Terapêutico e mecanismo de acção
A metadona é uma difenilpropilamina sintética usada em tratamentos de
desintoxicação e manutenção temporária da dependência de narcóticos e também no
tratamento da dor aguda e crónica. É um agonista dos receptores opióides µ, embora
também tenha efeito no receptor NMDA agindo aí como um antagonista do
glutamato, e parece bloquear a recaptação de serotonina e noradrenalina (18, 19, 20,
21). A metadona tem muitas das propriedades farmacológicas da morfina, sendo a sua
potência analgésica semelhante. Ao contrário da morfina, a administração repetida de
metadona provoca efeitos sedativos marcados devido à sua acumulação no organismo.

Metadona
Anabela Cardoso Mariano Página 18
c) Toxicocinética
A metadona é metabolizada pelas enzimas CYP3A4, CYP2B6 e CYP2D6 (14, 22,
25), as quais têm uma grande variabilidade entre indivíduos. A síndroma do desmame
da metadona é qualitativamente semelhante ao da morfina, diferindo no facto de ser
menos intensa e de se desenvolver de uma forma mais lenta, gradual e prolongada.
Por estas razões, a metadona é usada na gestão da dependência de narcóticos,
porventura eliminando a necessidade de drogas opiáceas ilícitas. É administrada
intramuscularmente para fins analgésicos e oralmente para terapia de manutenção. A
metadona é rapidamente absorvida ao nível do trato gastro-intestinal, aparecendo os
seus primeiros efeitos após 30 a 60 minutos. O tempo médio de pico plasmático é 2,5
horas para a metadona em solução e 3 horas para a metadona em comprimidos. Após
a sua administração repetida, a duração e a semi-vida aumentam em proporção (23).
Após ingestão este fármaco é bem absorvido pelo aparelho gastrointestinal, como já
referido, e largamente distribuído pelo fígado, pulmões, rins, baço, sangue e urina (18).
O facto da metadona se ligar em grande extensão às proteínas dos tecidos (86%) pode
explicar os seus efeitos cumulativos. A metadona tem um metabolismo lento e uma
elevada lipossolubilidade, fazendo com que o seu tempo de semi-vida seja longo.
Apesar desta longa semi-vida, o efeito analgésico geralmente dura de 6 a 8 horas. A
sua metabolização ocorre principalmente no fígado através de um processo de mono e
di-N-desmetilação e os metabolitos são inactivos (26, 27). Após ingestão, a metadona é
rapidamente metabolizada no seu metabolito primário, a normetadona. No entanto, a
normetadona raramente se detecta, por se desidratar rapidamente dando origem a
outro composto o 2-etilideno-1,5-dimetil-3,3-difenilpirrolidina (EDDP). O EDDP é
posteriormente desmetilado para formar a 2-etil-5-metil-3,3-difenil-1-pirrolina (EMDP)
(26).
d) Toxicidade
Devido à sua acumulação no organismo, a metadona tem efeitos mais
prolongados neste que a morfina, podendo originar uma maior depressão respiratória.
A sua toma contínua pode originar uma acentuada sedação.

Metadona
Anabela Cardoso Mariano Página 19
e) Doses terapêuticas e tóxicas
A concentração plasmática em doses terapêuticas é de 0,05 a 0,5 mg/L,
podendo chegar até 1 mg/L. A concentração plasmática analgésica é de 0,1 a 0,3 mg/L
e em terapêutica de substituição a concentração varia entre 0,2 a 0,75 mg/L. A
concentração tóxica para não consumidores será de 0,2 mg/L, para consumidores é
superior a 0,75 mg/L e a dose letal situa-se entre 0,2 e 1 mg/L (46).


Buprenorfina
Anabela Cardoso Mariano Página 21
4. BUPRENORFINA
a) Propriedades físico-quimicas
A buprenorfina (C29H41NO4) é um derivado semi-sintético da tebaína com
elevada lipofilia. Apresenta um peso molecular de 467.64 g/mol, um ponto de fusão de
218 ºC, é pouco solúvel em água, muito solúvel em acetona e solúvel em álcool (16).
Fig. 4 – Estrutura química da buprenorfina
b) Grupo terapêutico e mecanismo de acção
A buprenorfina actua como agonista parcial dos receptores opióides µ e como
antagonista dos receptores k.
A sua actividade no tratamento de manutenção da dependência dos opiáceos é
atribuída à sua ligação lenta e reversível aos receptores µ induzindo uma redução da
necessidade de consumo de drogas por parte do doente toxicodependente, durante
um período de tempo prolongado (10).
c) Toxicocinética
Esta substância é metabolizada no fígado em N-dialquilbuprenorfina
(norbuprenorfina) e metabolitos conjugados glucurónicos. O pico das concentrações
plasmáticas é atingido 90 minutos após a administração sublingual. A absorção da

Buprenorfina
Anabela Cardoso Mariano Página 22
buprenorfina é seguida de uma fase de distribuição rápida (semi-vida de distribuição
de 2 a 5 horas). Cerca de 96% da buprenorfina circulante é ligada às proteínas (2).
A buprenorfina é metabolizada por 14-N-desalquilação e glucuroconjugação da
molécula original e do metabolito desalquilado. Os dados clínicos confirmam que a
CYP3A4 é responsável pela N-desalquilação da buprenorfina (24).
A eliminação da buprenorfina é bi- ou tri-exponencial e a semi-vida plasmática
média é de 32 horas. A buprenorfina é eliminada nas fezes por excreção biliar dos
metabolitos glucuroconjugados (70 %), sendo a restante fracção eliminada na urina
(24).
d) Toxicidade
Tal como sucede com outros opiáceos potentes, pode ocorrer depressão
respiratória, mesmo com doses pertencentes aos limites terapêuticos recomendados.
A buprenorfina é metabolizada pelo fígado e a sua eliminação está relacionada com a
circulação sanguínea hepática. A redução do metabolismo do fármaco em indivíduos
com doença hepática grave pode potenciar os efeitos do fármaco, na dose terapêutica
recomendada (24).
e) Doses terapêuticas e tóxicas
A concentração plasmática em doses terapêuticas é de 0,001 a 0,01 mg/L. A
concentração plasmática tóxica é de 0,2 mg/L e a dose letal 1,1 mg/L (46).

Técnicas para determinação da metadona e buprenorfina
Anabela Cardoso Mariano Página 23
5. TÉCNICAS PARA DETERMINAÇÃO DA METADONA E BUPRENORFINA
Ao longo dos anos têm sido publicados diversos trabalhos que descrevem
diferentes metodologias para a determinação de metadona, EDDP, buprenorfina e
norbuprenorfina, em diversas matrizes biológicas. Na pesquisa bibliográfica realizada
não foi encontrado nenhum estudo que combine a análise em sangue total pos-
mortem, utilizando a técnica de extracção em fase sólida descrita neste trabalho e a
cromatografia de gases com detector de massas. Em Toxicologia Forense, sendo o
sangue total o tipo de matriz mais frequente e muitas vezes o único disponível, e a
extracção em fase sólida combinada com cromatografia de gases serem técnicas cuja
utilização é vantajosa devido à sua rapidez e eficácia, será de todo o interesse a
realização de um trabalho que combine estes três factores.
Os procedimentos sistemáticos para o estudo das substâncias em causa,
nomeadamente na monitorização e controlo de doses terapêuticas, no controlo de
adesão à terapêutica e na determinação das concentrações sanguíneas pos-mortem
são: a cromatografia de gases acoplada a espectrometria de massas (GC-MS) (73, 74,
78, 80-86, 91, 93, 96) por impacto electrónico (78, 89) ou por ionização química ião-
positivo (85), a cromatografia líquida acoplada a espectrometria de massas (LC-MS)
(77, 82, 88, 103-105), e técnicas de imunoensaios (75, 77, 86, 88).
Em relação às matrizes biológicas utilizadas elas são diversas, havendo
referências que incluem urina (45, 75-77, 80-83, 86-89, 93, 97, 99-101, 116, 118),
plasma (45, 47, 74, 79, 88, 91, 92, 110-114), cabelo (45, 88, 97, 102-104, 106-109),
sangue total (45, 73, 85, 87, 99, 102-105), suor (88, 95, 96), tendo sido também
encontrados alguns trabalhos em leite materno (13, 78, 88), placenta (84), fluido oral
(119) e cordão umbilical (74, 88). A amostra mais estudada é a urina sobre a qual
existem muitos trabalhos de screening (77, 84, 88, 89, 93, 102, 104) e alguns de
quantificação (79, 83, 85). No entanto existem também trabalhos de screening em
plasma (94) e quantificação em plasma (47,74,91,110,112) e cordão umbilical (88).
Foram também encontrados estudos farmacocinéticos em plasma de rato (91) e de
plasma humano (110-114).
Relativamente aos métodos extractivos usados para o isolamento das
substâncias da matriz biológica o resultado da pesquisa bibliográfica foi: a extracção

Técnicas para determinação da metadona e buprenorfina
Anabela Cardoso Mariano Página 24
em fase sólida (SPE) e a extracção líquido-líquido (LLE) precedidas ou não por hidrólise
enzimática, no caso da buprenorfina e norbuprenorfina (75, 77, 86, 88). A hidrólise
enzimática tem sido aplicada a amostras de plasma e urina (47). A técnica extractiva
mais encontrada é a extracção em fase sólida (SPE) (74-76, 78, 80, 83, 84, 86-88) no
entanto a extracção líquido-líquido (LLE) (81, 95, 99, 101) geralmente também
consegue uma extracção bastante eficiente (48, 97). Em relação às técnicas extractivas
encontradas existem algumas que extraem a metadona, buprenorfina e respectivos
metabolitos em simultâneo, e existem trabalhos onde a técnica extractiva descrita é
utilizada apenas para uma das substâncias e respectivo metabolito. De acordo com as
técnicas extractivas descritas os solventes usados para a extracção dessas substâncias
em fluidos biológicos incluem: tampão carbonato e acetato de etilo (44),
clorofórmio/2-propanol/n-heptano (45), hidrogenofosfato de sódio e acetato n-butilo
(73), diclometano/metanol/hidróxido de amónia (78), metanol/hidróxido de amónia
(76), diclorometano/2-propanol/amónia (80), acetato de butilo (82),
clorofórmio/isopropanol (81, 95), diclorometano/isopropanol/hidróxido de amónia
(97); cloridrato de metileno/isopropanol/hidróxido amónia (83, 88, 92), cloridrato
metileno/2-propanol/hidróxido amónia (96), metanol/hidróxido amónia/isopropanol
(84), dietileter (85), acetato de etilo/hidróxido de amónia (86), metanol (87),
metanol/ácido acético (90), cloridrato de butilo (100), triclorometano (101).
Os adsorventes mais usados em SPE foram: troca catiónica com colunas OASIS®
MCX (76, 97); colunas de fase reversa Microsorb-MV C8 (89); octilo - colunas Bond Elut
C8 (87, 90); colunas C18 (Cat.No.1211-3024) (47); colunas mistas de troca catiónica-
resina hidrofóbica OASIS® HCX (74, 78, 83, 94); colunas Clean Screen® ZSDAU020 (96);
colunas poliméricas STRATATM-X-C (84, 88); ciclohexilo (89); colunas
hidrofílicas/lipofílicas OASIS® HLB (87); colunas troca de catiónica/resina lipofílica (87).
Segundo alguns estudos as colunas octilo C8 foram as que obtiveram melhores
resultados quando comparadas com as colunas OASIS® HLB que obtiveram
concentrações muito baixas dos analitos, com as colunas mistas de troca
catiónica/resina lipofílica nas quais foram encontradas interferentes severos, e por fim
com ciclohexilo com as quais surgiram interferentes e baixa concentração extraída de
padrão interno (87).

Técnicas para determinação da metadona e buprenorfina
Anabela Cardoso Mariano Página 25
Após tratamento da amostra pelos diversos métodos usados, a técnica
cromatográfica mais utilizada para analisar a metadona, buprenorfina e respectivos
metabolitos é a análise por cromatografia de gases acoplada a detector de massas
(GC/MS), embora a cromatografia líquida acoplada a detector de massas (LC-MS)
também seja utilizada. A metadona e o EDDP quando analisados por qualquer um dos
métodos atrás referidos não necessitam de derivatização para serem
cromatografados. Quanto à buprenorfina e à norbuprenorfina, quando analisada por
cromatografia de gases acoplada a detector de massas (GC/MS) a derivatização torna-
se indispensável. Vários têm sido os procedimentos de derivatização estudados para
tornar as moléculas mais aptas para análise por cromatografia de gases, como seja o
caso da derivatização por alquilação, acilação e sililação (115-117, 119, 128).


Derivatização
Anabela Cardoso Mariano Página 27
6. EXTRACÇÃO EM FASE SÓLIDA
O processo extractivo dos analitos que estão presentes numa amostra é um dos
passos mais importantes no procedimento sistemático de análises toxicológicas. A
presença de interferentes na matriz pode dificultar, ou mesmo impedir a análise dos
componentes em estudo. Assim sempre que possível é necessário minimizar, ou
mesmo eliminar essas interferências. Os dois objectivos fundamentais na preparação
de amostras são: a purificação e a concentração dos extractos obtidos.
A purificação é necessária, tal como já foi referido, para que o analito em
estudo consiga ser analisado, e por outro lado, para que não prejudiquem o tempo de
vida das colunas cromatográficas.
Em relação à concentração da amostra, é frequente que os analitos em estudo
se encontrem nas amostras em baixos níveis de concentração, necessitando de ser
concentrados para que possam ser detectados.
Entre as várias técnicas usadas neste procedimento destacam-se como sendo
as mais comuns a extracção líquido-líquido (LLE), a centrifugação, a precipitação e a
extracção em fase sólida (SPE).
A extracção SPE surge nos anos 70 como uma técnica rápida e eficaz na
preparação de amostras para determinações analíticas. O conceito de SPE é similar à
cromatografia líquida de baixa pressão. Consiste em pequenas colunas de extracção
descartáveis com uma grande variedade de enchimentos (adsorventes) disponíveis.
Actualmente as colunas de SPE são de polipropileno ou de polietileno com material de
enchimento constituído por diferentes grupos funcionais, com partículas de cerca de
40 µm e poros de aproximadamente 60 Ǻ de diâmetro (50).
Fig. 5 – Coluna de SPE
Reservatório para amostra
Disco poroso
Camada adsorvente
Disco poroso

Derivatização
Anabela Cardoso Mariano Página 28
A SPE é cada vez mais uma técnica útil na preparação das amostras, onde
muitos dos problemas associados à extracção líquido/líquido tais como: separações de
fase incompleta, recuperações quantitativas menores, uso de material quebrável
(tubos de vidro) e utilização de grandes quantidades de solventes orgânicos poderão
ser evitados. A SPE é uma técnica mais eficiente, que tem associado um maior
rendimento quantitativo de extracção, mais fácil e rápida de executar, elimina a
ocorrência de emulsão dos extractos e tem possibilidade de automatização (51, 52).
Por outro lado esta técnica tem disponíveis inúmeros adsorventes que permitem
interacções intermoleculares do tipo: interacções polares, hidrofóbicas e iónicas;
enquanto a extracção líquido/líquido se limita a equilíbrios de partição na fase líquida
(50, 53).
A SPE é um processo de separação pela qual os compostos que estão
dissolvidos ou suspensos numa mistura líquida são separados nos restantes compostos
da mistura de acordo com suas propriedades físicas e químicas. A extracção em fase
sólida pode ser usada para isolar os analitos de interesse de uma grande variedade de
matrizes tais como: urina, sangue, tecidos biológicos, água, bebidas, solo e tecido
animal. É usada na maioria das vezes para preparar amostras líquidas e extrair analitos
voláteis ou semi-voláteis, podendo também ter como objectivo a concentração do
analito na amostra, como já foi referido anteriormente.
A extracção SPE utiliza a afinidade do soluto dissolvido ou suspenso num
líquido (fase móvel) para um sólido através do qual a amostra passa (fase estacionária)
para separar os diferentes componentes da mistura. A fracção que passa através da
fase estacionária é recolhida ou descartada, dependendo se contém os analitos
desejados ou indesejados. Se a fracção retida na fase estacionária incluir os analitos
desejados, estes podem ser removidos da fase estacionária através de uma etapa
adicional – a lavagem da fase estacionária com um eluente adequado.

Derivatização
Anabela Cardoso Mariano Página 29
Recolha do analito
Remoção de interferentes
Fig. 6 Etapas do procedimento de SPE
O processo de extracção em fase sólida envolve quatro etapas básicas: -
acondicionamento da coluna com um solvente não-polar ou ligeiramente polar
seguido de água ou de uma solução tampão que vai embeber a fase sólida e solvatar
os grupos funcionais das suas partículas constituintes. Assim o ar que se encontrava
entre as partículas da coluna é eliminado e substituído pelo solvente ocorrendo a
activação da coluna, permitindo os mecanismos de adsorção entre o adsorvente da
fase estacionária e a amostra aquosa (fase móvel). É fundamental não deixar secar o
adsorvente para que a recuperação não fique comprometida (51); - seguidamente a
amostra é adicionada à coluna e ao passar através da fase estacionária, os analitos da
amostra vão interagir com a coluna e ficar retidos (concentrados). Por vezes, é
necessário ajustar as características da matriz tratando previamente a amostra (ajuste
de pH, polaridade ou viscosidade); - a coluna é lavada com tampão ou solvente para
remover as impurezas retidas no adsorvente, sem no entanto remover o analito; por
fim, o analito é eluído com um solvente não-polar ou um tampão de pH adequado
capaz de romper as ligações entre o analito e o adsorvente resultando na sua eluição,
sem no entanto remover as outras substâncias que possam também estar retidas no
Metanol
H2O LAVAGEM AMOSTRA ELUIÇÃO

Derivatização
Anabela Cardoso Mariano Página 30
adsorvente (51).
A fase estacionária utilizada nestas colunas consiste normalmente em sílicas
com características de retenção modeladas por incorporação dum revestimento
superficial constituído por grupos de estrutura seleccionada que se unem por ligações
covalentes ao gel de sílica. Em função do mecanismo de retenção que proporcionam,
as fases quimicamente ligadas a este suporte podem agrupar-se em três grupos
principais: fase normal, fase reversa e troca iónica.
A fase normal é a fase padrão utilizada pela cromatografia líquida clássica. É
classicamente utilizada para separar os compostos orgânicos neutros, cuja natureza
varia de hidrofóbico a moderadamente polar. O mecanismo de retenção primário é
devido a interacções entre os grupos funcionais polares do analito e os grupos polares
na superfície do adsorvente. Estas, incluem ligações de hidrogénio, interacções dipolo-
dipolo e dipolo-dipolo induzido. Compostos adsorvidos por estes mecanismos são
eluídos por um solvente que perturba o mecanismo de ligação, geralmente um
solvente que é mais polar que o original da matriz da amostra. Os tipos de fases não
ligadas usadas neste modo de adsorção são a sílica (a mais usada), a albumina e o
silicato de magnésio. No entanto a utilização de sílicas de fases ligadas tem vindo a
aumentar uma vez que estas não formam locais de adsorção fortes e irreversíveis
induzidos pelos grupos silanol livres. A SPE em fase normal é tipicamente usada para
limpeza de extractos orgânicos, sendo também utilizada para isolamento de analitos
provenientes de líquidos orgânicos (51, 52).
A fase reversa em SPE separa analitos com base na sua polaridade. É
denominada por fase reversa por ser o oposto da fase normal. As separações em fase
reversa envolvem uma amostra polar (geralmente aquosa) ou moderadamente polar
(fase móvel) e uma fase estacionária apolar (52).
O processo de isolamento consiste em interacções não polares por forças de
Van der Waals, ou forças de dispersão ou de partição. O mecanismo de partição é um
processo de baixa energia sendo análogo à remoção de uma molécula a partir de uma
amostra de água por LLE. Esta partição consiste na interacção do analito com as
cadeias da fase estacionária ligada. Este processo depende da diferença de
solubilidade do analito nas duas fases, pelo que a sua polaridade assume um papel de
grande importância na estimativa da eficiência da fase sólida no isolamento do analito.

Derivatização
Anabela Cardoso Mariano Página 31
A eluição dos analitos é um processo simples, que consiste na escolha de um solvente
não polar capaz de romper as forças de Van der Waals que retêm o analito. Assim a
amostra pode ser eluída por lavagem da coluna com um solvente não-polar, que
interrompe a interacção do analito e a fase estacionária. Os solventes compatíveis com
os adsorventes usados na fase reversa são o metanol, o acetonitrilo e o acetato de
etilo, por serem capazes de estabelecer ligações de hidrogénio com os grupos silanol
livres da superfície da sílica. Estes solventes têm capacidade para dissolver a água
residual que se possa encontrar retida na fase ligada, rompendo assim facilmente as
interacções de Van der Waals. No entanto existem analitos hidrofóbicos que não são
eficientemente eluídos com estes solventes, nestes casos, utiliza-se uma mistura de
solventes, por exemplo, diclorometano e acetato de etilo (1:1).
A troca iónica consiste na separação de um analito com base em interacções
electrostáticas entre o analito de interesse e os grupos carregados positivamente na
fase estacionária. Para que ocorra a troca iónica, tanto a fase estacionária como a
amostra devem estar a um pH a que ambos se encontrem carregados. Uma solução
em que o pH neutralize os grupos funcionais do analito ou os grupos funcionais do
adsorvente é usada para eluir o analito de interesse. Quando um destes grupos
funcionais é neutralizado, a força electrostática que liga o analito e o adsorvente é
quebrada e o analito é eluído. Por outro lado uma solução que tenha elevada força
iónica ou que contenha espécies iónicas que desliguem o analito do adsorvente pode
ser usada para a sua eluição (52). Os adsorventes de troca iónica tanto podem ter
grupos funcionais aniónicos como catiónicos, fortes e fracos. Os adsorventes de troca
aniónica forte contêm grupos amina quaternários, que possuem uma carga positiva
permanente em soluções aquosas, e os adsorventes de troca aniónica fraca contêm
grupos amina primários, secundários e terciários nos quais o sítio trocador pode ou
não encontrar-se carregado dependendo do pH que se pode encontrar acima ou
abaixo do pKa. Os adsorventes de troca aniónica forte são úteis porque as impurezas
fortemente ácidas na amostra ligam-se ao adsorvente e, geralmente, não serão eluídos
com o analito de interesse.
Os adsorventes de troca catiónica forte contêm grupos de ácido sulfónico que
estão sempre carregados negativamente em solução aquosa, e os adsorventes de
troca catiónica fraca contêm ácidos carboxílicos cujos sítios de troca, tal como nos

Derivatização
Anabela Cardoso Mariano Página 32
adsorventes de troca aniónica fraca, estão dependentes do pH.
Verifica-se assim que a SPE tem um âmbito de aplicação muito alargado pelo
facto de dispormos de diversos adsorventes cujas características divergem bastante. A
selectividade dos procedimentos a usar para determinada extracção sólida é assim
determinada pela natureza do adsorvente e pelos solventes usados nos procedimentos
de adsorção, lavagem e eluição. Assim na escolha do procedimento é necessário ter
em consideração todas as características do analito em questão, para que o
procedimento escolhido seja o mais adequado e eficaz.
A técnica extractiva SPE tem um vasto campo de aplicação, nomeadamente, na
área das análises forenses, podendo ser utilizada na determinação de várias
substâncias em fluidos biológicos (54-77).

Derivatização
Anabela Cardoso Mariano Página 33
7. DERIVATIZAÇÃO
A derivatização é uma técnica usada em química que transforma um composto
químico num produto de estrutura química similar, derivado.
Existem compostos que devido ao facto de possuírem uma composição química
desfavorável à sua detecção por esta técnica analítica têm de ser modificados (por
exemplo, compostos pouco voláteis). Muitos materiais que sejam termicamente
instáveis nas condições da técnica utilizada também se enquadram nesta categoria. O
processo de derivatização química é assim um processo que modifica a estrutura dos
compostos em estudo de forma a que estes possam ser analisados através de GC-MS.
Geralmente, um determinado grupo funcional do composto participa na
reacção de derivatização e transforma o composto num produto derivado de
solubilidade, ponto de ebulição, ponto de fusão, estado de agregação ou composição
química diferente da que inicialmente tinha, o que permite que este seja devidamente
cromatografado por GC/MS. A análise directa de muitos compostos e misturas de
compostos é difícil devido às interacções entre os compostos e a coluna ou entre os
diferentes compostos da mistura. Estas interacções podem levar a uma resolução de
picos pobre e/ou a picos assimétricos, o que torna o pico desadequado para a
identificação do composto. Por outro lado a derivatização também é utilizada para
aumentar a detecção de compostos. Esta detecção pode ser reforçada através do
aumento do volume da substância ou pela introdução de átomos ou grupos funcionais
que interagem fortemente com o detector.
É pré-requisito para uma derivatização bem sucedida a formação rápida,
reprodutível e quantitativa de um único derivado.
No sangue temos os compostos inerentes à própria matriz que possuem grupos
polares e que, por essa razão, não permitem um comportamento cromatográfico
razoável sem derivatização. Existem muitos reagentes adequados para derivatizar
grupos funcionais polares tornando desta maneira a molécula adequada para
determinações por cromatografia de gases. Os grupos funcionais típicos existentes nos
compostos desta natureza, e que podem sofrer derivatização são os grupos hidroxilo,
as acetonas, os ácidos carboxílicos, as aminas e as amidas.
De um modo geral a derivatização analítica é usada por duas razões: permitir a

Derivatização
Anabela Cardoso Mariano Página 34
análise de compostos que são inadequados para a análise directa e para melhorar o
processo de análise (72)
Por outro lado a necessidade de cada vez mais se determinar pequenas
quantidades de composto nas amostras biológicas, torna importante o alargamento do
intervalo de detectibilidade dos compostos. Este aumento consegue-se através do
aumento do tamanho do composto e pela introdução de átomos ou de grupos
funcionais que interajam fortemente com o detector (121, 122).
Os derivados também são usados para estabilizar os iões formados no
espectrómetro de massas favorecendo a informação estrutural dos modos de
fragmentação molecular.
A maior parte das reacções de derivatização de análise química utilizados em
cromatografia gasosa (GC) são de cinco tipos: sililação, alquilação, acilação, formação
de derivados cíclicos e derivatização quiral (72, 90).
Os derivados silil (sililação) são provavelmente os mais amplamente utilizados
para aplicações de GC. São geralmente formados pela substituição dos protões activos
nos grupos –OH, -SH e –NH, por um grupo alquilsilil, sendo estes derivados mais
voláteis, menos polares e termicamente mais estáveis do que os compostos que lhes
deram origem. A trimetilsililação é a reacção mais usada no procedimento de
derivatização da sililação.
A reacção de sililação é a seguinte:
Amostra-OH + R3Si-X Amostra-O-SiR3 + HX
Este mecanismo caracteriza-se por um ataque nucleofílico sobre o átomo de
silício. No entanto a preparação destes derivados obedece a condições especiais como
seja a ausência de humidade.
A alquilação é usada em derivatização para GC, e consiste na substituição de
um hidrogénio activo por um grupo R-COOH, R-OH, R-SH, R-NH e R-NH2, com um
grupo alquilo ou por vezes arilo. Esta técnica é usada para modificar os compostos
contendo hidrogénios acídicos, tais como ácidos carboxílicos e fenóis. A principal
finalidade cromatográfica desta reacção é a conversão de ácidos orgânicos em ésteres
que produzem cromatogramas de melhor qualidade do que os ácidos livres. As

Derivatização
Anabela Cardoso Mariano Página 35
reacções de alquilação também podem ser usadas para preparar éteres, tioéteres e
tioésteres, N-alquilaminas, amidas e sulfonamidas. Como a acidez do hidrogénio
activo diminui, a força do reagente de alquilação deve aumentar. Como os reagentes e
as condições se tornam mais severas, a selectividade e aplicabilidade do método
torna-se mais limitada. Em geral, os produtos da alquilação são menos polares que as
matérias-primas, uma vez que o hidrogénio activo foi substituído por um grupo alquilo.
Provavelmente, a maior aplicação da alquilação na derivatização analítica é a
conversão de ácidos orgânicos em ésteres, em especial ésteres metílicos. Embora os
derivados de ácidos carboxílicos-TMS sejam facilmente formados, esses compostos
sofrem de estabilidade limitada. Por outro lado, os ésteres alquilo, oferecem
excelente estabilidade e podem ser isolados e armazenados por longos períodos, se
necessário.
A principal reacção na preparação deste tipo de derivados é a substituição
nucleofílica do tipo:
R’-X
Amostra-OH Amostra-O-R’ + HX
Base
Onde X representa um halogénio ou outro grupo de substituição. A alquilação
de grupos acídicos fracos necessita de catalisadores básicos fortes enquanto que
grupos mais acídicos necessitam de catalisadores básicos mais fracos.
A acilação é outra forma de derivatização utilizada em GC-MS e consiste
na conversão de compostos com hidrogénio activo em ésteres, tioésteres e amidas,
respectivamente e na adição de funcionalidades halogenadas. Estes derivados acil
formam-se com reagentes acil anidrido, acil halido e acil amidas activadas. Estes
reagentes são sensíveis à presença de humidade formando produtos ácidos os quais
necessitam de ser removidos imediatamente antes da análise por GC.
Na formação de derivados cíclicos os reagentes mais usados são os que
derivatizam um amplo espectro de grupos funcionais e os quais sejam muito selectivos
para grupos funcionais ou compostos específicos. A grande estabilidade destes
compostos em relação à fragmentação molecular origina espectros de massas com

Derivatização
Anabela Cardoso Mariano Página 36
massas mais elevadas e iões de maior abundância. Possuem como desvantagem a
possibilidade de formação de outros derivados indesejados devido a serem sensíveis à
humidade.
Em relação à derivatização quiral esta pode ocorrer por conversão em
diastereomeros por reacção com reagentes opticamente puros e separação em fases
cromatográficas aquirais; e por separação directa em fases estacionárias quirais sem
necessidade da derivatização quiral (72).
As reacções de derivatização analítica constituem uma forma de síntese em
química orgânica a qual tem como principal objectivo obter um rendimento próximo
dos 100% originando um único derivado e não permitindo reacções secundárias.

Validação
Anabela Cardoso Mariano Página 37
8. VALIDAÇÃO
A confiança na interpretação dos resultados analíticos obtidos é um aspecto
extremamente relevante na área da toxicologia forense. Assim torna-se imperativo
que os métodos utilizados sejam rigorosos e fiáveis, e para tal requerem a sua
validação. A validação de métodos é um aspecto vital da garantia da qualidade
analítica. Os métodos de ensaio usados para avaliar a conformidade dos resultados
com especificações estabelecidas devem atingir padrões adequados de exactidão,
precisão e confiabilidade. Assim a validação do método analítico é a confirmação por
análise e fornecimento de evidência objectiva de que os requisitos específicos para um
determinado uso pretendido são atendidos. A validação do método analítico permite
demonstrar que o método é adequado ao uso pretendido. Os procedimentos devem
ser claros, objectivos e completos. A escolha de uma metodologia analítica adequada é
de fundamental importância para o procedimento do controle de qualidade.
Num laboratório de toxicologia forense é de extrema importância que todos os
métodos sejam validados, ou pelos menos que estejam em fase de validação, uma vez
que se torna mais seguro apresentar resultados finais, visto que estão de acordo com
um conjunto de normas e critérios estabelecidos. Por outro lado, caso seja necessário
justificá-los em tribunal, existe um suporte de elevada confiança que os corrobora.
A validação de métodos analíticos desenvolvidos no laboratório é efectuada
após selecção, desenvolvimento e optimização dos métodos (45).
Baseando-nos em recomendações e critérios de organizações internacionais de
grande relevância científica (28, 99), os parâmetros considerados mais significativos e
que devem ser estudados num processo de validação de procedimentos analíticos para
determinações quantitativas de um ou mais analitos numa determinada matriz
biológica são: selectividade/especificidade, modelo de calibração, precisão, exactidão,
LOD e LOQ, estabilidade, recuperação e robustez (29-47).
Especificidade do método analítico refere-se à capacidade de determinar com
exactidão o analito de interesse em presença de outros componentes ou interferentes
que possam estar presentes na matriz da amostra. Diz-se que o método é específico
para determinado analito se for selectivo para esse analito na presença de outros. Para
estudar estes parâmetros utilizam-se amostras sem analito (amostras brancas) e

Validação
Anabela Cardoso Mariano Página 38
amostras às quais se adiciona analito em quantidades conhecidas dentro do intervalo
da gama de trabalho usada. Pelo menos, devem ser estudadas 6 amostras de origens
diferentes da mesma matriz (30, 31).
Escolher um modelo de calibração adequado é determinante para obter
quantificações fiáveis. Como tal, deve ser estudada a relação que existe entre a
concentração do analito da amostra e a resposta correspondente do detector. Este
estudo é feito a partir de amostras brancas fortificadas com calibradores em
concentrações que abranjam toda a gama de trabalho. As curvas obtidas são
posteriormente avaliadas através de métodos gráficos ou matemáticos.
Linearidade é a habilidade do método analítico de produzir resultados que são
directamente, ou por uma transformação matemática bem definida, proporcionais à
concentração do analito dentro de uma dada gama de trabalho.
Como gama de trabalho considera-se o intervalo de concentração do analito na
matriz, entre o valor mais baixo e o mais alto, para o qual se demonstrou que o
procedimento era preciso, exacto e linear. Este intervalo deve conter, no mínimo, seis
níveis de concentração equidistantes, no qual cada padrão de calibração deve ser
estudado em duplicado.
Precisão do método analítico é o grau de concordância entre resultados de
medidas independentes em torno de um valor central, efectuadas várias vezes numa
amostra homogénea, sob condições pré-estabelecidas. Precisão é expressa em termos
de desvio padrão e desvio padrão relativo. Este parâmetro pode ser avaliado através
da repetibilidade, precisão intermédia e reprodutibilidade. A repetibilidade ou variação
intra-dia, é efectuada no mesmo laboratório, mesmo analista, mesma instrumentação
em curtos intervalos de tempo. A precisão intermédia ou variação inter-dia, é feita em
dias diferentes, diferentes analistas e equipamentos, em sucessivas determinações. Já
a reprodutibilidade expressa a precisão entre diferentes laboratórios, sendo necessário
a sua avaliação se o método for para aplicar em diferentes laboratórios. A precisão de
um procedimento analítico é habitualmente expressa em termos de variabilidade,
desvio padrão ou coeficiente de variação de uma série de medidas:
CV = s/x
Sendo CV, o coeficiente de variação, obtido pela relação entre o desvio padrão
relativo (s) e a média (x).

Validação
Anabela Cardoso Mariano Página 39
Os valores de precisão calculados em cada nível de concentração não devem
exceder 15% do coeficiente de variação (CV), excepto para o LLOQ, onde não deve
exceder 20%. Os mesmos critérios devem ser aplicados a cada tipo de medição de
precisão (intra-ensaio, inter-ensaio) (28).
Exactidão do método analítico é o grau de concordância entre o valor médio
obtido de uma série de resultados e o valor de referência aceite, sendo afectada pelos
componentes do erro sistemático (bias) e do erro aleatório (28). Os valores médios do
bias devem situar-se dentro de ±15% relativamente ao valor real (valor teórico),
excepto para o valor mais baixo de quantificação (LLOQ) em que um valor de ±20% é
aceitável (28). Exactidão pode ser demonstrada pela comparação dos resultados
obtidos com material de referência certificado ou com outro método validado cujo
erro sistemático é sabidamente não significativo. Outra forma de investigação é
comparar a média dos resultados obtidos com a média obtida do programa inter-
laboratorial, ou ainda por meio de estudos de recuperação de quantidades conhecidas
do analito adicionado na matriz isenta de analito ou ainda na matriz da amostra. As
fortificações devem ser efectuadas, no mínimo em triplicado.
Limite de detecção (LD) é a menor concentração do analito numa amostra, que
pode ser detectada, mas não necessariamente quantificada, sob determinadas
condições experimentais. Existem muitos critérios para definição do limite de detecção
(26-29, 33-36, 39-41):
LOD = t x s
Onde t é um parâmetro estatístico que dá indicação da variabilidade de s para
determinados níveis de confiança e n-1 graus de liberdade e s é o desvio padrão
associado às varias leituras do padrão;
LOD = 3,3 x Sy/x/b
Onde Sy/x e b são respectivamente o desvio padrão residual e o declive da recta
de calibração; ou pode ser estimado a partir da concentração que dá origem a uma
resposta superior ao ruído cromatográfico,
LOD = 3 x Sinal/Ruído
Podendo também ser calculado pela média da abundância do ruído x e o
respectivo desvio padrão SD, pela seguinte fórmula
LOD = x + 3 SD

Validação
Anabela Cardoso Mariano Página 40
Limite de quantificação (LQ) do método analítico é a menor concentração do
analito que pode ser determinada e quantificada com precisão e exactidão, aceitáveis,
sob determinadas condições experimentais, podendo ser calculado de várias formas:
LOQ = padrão mais baixo da curva de calibração ou valor bastante aproximado,
que possa ser quantificado com uma percentagem de erro e coeficiente de variação
inferiores a 20%; ou através da fórmula,
LOQ = 10 x Sy/x/b
Onde Sy/x e b são respectivamente o desvio padrão residual e o declive,
da recta de calibração; ou pela razão entre o sinal e o ruído através da expressão,
LOD = 10 x Sinal/Ruído
Podendo ainda ser usada a média da abundância do sinal do ruído x e o
respectivo desvio padrão SD, pela seguinte fórmula,
LOQ = x + 6 SD
O analito na amostra biológica deve apresentar estabilidade em função das
condições e sistemas de armazenamento utilizadas, das propriedades químicas da
substância e da própria matriz. No estudo da estabilidade devem ser reproduzidas ao
máximo as condições de uma amostra real e analisadas em várias vertentes, desde a
colheita e manipulação, períodos de armazenamento longos e curtos períodos à
temperatura ambiente, até os ciclos de congelação e descongelação (31).
O rendimento da extracção é avaliado pelo parâmetro da recuperação, que
pode ser calculado através da relação percentual entre a resposta do detector obtida a
partir de uma quantidade de analito adicionada e posteriormente extraída da matriz
biológica, com a resposta do detector para uma concentração real da substância de
referência. No caso de haver derivatizações na amostra, o cálculo da recuperação
absoluta pode ser dispensado, uma vez que os derivados dificilmente se encontram
disponíveis como substâncias de referência.
A robustez do método é a medida de capacidade que o método apresenta em
se manter inalterável através de pequenas, mas deliberadas modificações nos seus
parâmetros e fornecer indicações de segurança durante o uso normal (49).
A validação é assim, um processo dinâmico e constante que começa nas fases
de selecção, desenvolvimento e optimização do método e na qualificação dos
instrumentos, materiais e pessoas e continua na fase experimental e de transferência

Validação
Anabela Cardoso Mariano Página 41
do método. Um processo de validação bem definido e documentado fornece evidência
objectiva de que o sistema e o método são adequados ao uso pretendido.


III – PARTE EXPERIMENTAL


MATERIAS E MÉTODOS


Material e Métodos
Anabela Cardoso Mariano Página 47
1. AMOSTRA BIOLÓGICA
Para a validação dos métodos analíticos foram usadas amostras biológicas
obtidas por vias institucionais junto do Instituto Português do Sangue, I.P. As referidas
amostras consistiam de sangue humano provenientes de bolsas de dadores cujo prazo
de validade já havia expirado.
Foram ainda usadas 75 amostras de sangue (relativas ao ano de 2004),
negativas para opiáceos após análise por ensaio imuno-enzimático. Estas amostras
resultaram de colheitas efectuadas em indivíduos vivos (obtidas no âmbito do Código
da Estrada e do Serviço da Clínica Forense) e no decorrer de autópsias (realizadas no
Serviço de Patologia Forense da Delegação de Centro e nos Gabinetes Médico-Legais
sob a sua dependência).
Todas estas amostras foram mantidas à temperatura de congelação (-20ºC) até
ao momento da sua utilização. Antes do procedimento extractivo foram retiradas do
congelador e deixadas atingir a temperatura ambiente.
2. REAGENTES E MATERIAL
2.1 Reagentes
2.1.1 2-Propanol (isopropanol) p.a.;
2.1.2 Água desionizada;
2.1.3 Amónia 25% p.a.;
2.1.4 Diclorometano p.a.;
2.1.5 Dihidrogenofosfato de potássio p.a.;
2.1.6 Metanol p.a.;
2.1.7 MSTFA (Sigma-Aldrich);
2.1.8 n-Hexano p.a.;

Material e Métodos
Anabela Cardoso Mariano Página 48
2.1.9 TMCS (Macherey-Nagel).
2.1.10 Ácido cloridríco p.a;
2.1.11 Acetato de etilo p.a;
2.1.12 2-propanol p.a;
2.2 Material
2.2.1 Balões volumétricos de 2, 5, 10, 250, 500 e 1000 mL;
2.2.2 Copos de vidro de 250 e 500 mL;
2.2.3 Colunas para extracção em fase sólida (Waters Oasis MCX®, 3 mL);
2.2.4 Frascos de vidro âmbar de 4 e 12 mL;
2.2.5 Frascos de vidro de 250 e 1000 mL;
2.2.6 Suporte para tubos;
2.2.7 Suporte para vials;
2.2.8 Tubos de plástico de 10 mL com tampa;
2.2.9 Tubos de vidro de 10 mL com tampa de rosca;
2.2.10 Vials de 300 µL.
2.3 Preparação de soluções
2.3.1 Diclorometano / 2-Propanol / Amónia (78:20:2, v/v/v)
Num frasco de volume apropriado juntar 78 mL de diclorometano, 20 mL de 2-
Propanol e 2 mL de amónia (25%). Agitar até homogeneizar.
Manter esta solução refrigerada (2-8ºC).
2.3.2 MSTFA/TMCS (95:5, v/v)
Num frasco de vidro âmbar juntar 9,5 mL de MSTFA e 0,5 mL de TMCS. Inverter
o frasco sucessivas vezes para homogeneizar. Manter esta solução refrigerada (2-8ºC).

Material e Métodos
Anabela Cardoso Mariano Página 49
2.3.3 Tampão fosfato 0,1M
Pesar 13,61 g de dihidrogenofosfato de potássio e dissolver em água desionizada
até perfazer um volume de 1000 mL.
2.3.4 Solução de ácido cloridríco 0,1M
Dissolver-se 2,47 mL de ácido cloridríco concentrado em água desionizada até
perfazer um volume de 250 mL.
2.3.5 Soluções de padrões primários a 1 mg/mL
Na forma líquida, transferir o conteúdo para frascos de vidro âmbar de 4 mL.
Na forma sólida, pesar 10 mg do padrão de referência e dissolver em metanol até
perfazer um volume de 10 mL.
2.3.6 Mistura de armazenamento de padrões primários a 100 µg/mL
Diluir em metanol 500 µL das soluções descritas em 2.3.5 até perfazer um
volume de 5 mL.
2.3.7 Mistura de armazenamento de padrões primários a 50 µg/mL
Diluir em metanol 250 µL das soluções descritas em 2.3.5 até perfazer um
volume de 5 mL.
2.3.8 Mistura de trabalho de padrões primários a 5 µg/mL
Diluir em metanol 500 µL das soluções descritas em 2.3.7 até perfazer um
volume de 5 mL.

Material e Métodos
Anabela Cardoso Mariano Página 50
2.3.9 Mistura de trabalho de padrões primários a 0,5 µg/mL
Diluir em metanol 500 µL das soluções descritas em 2.3.8 até perfazer um
volume de 5 mL.
2.3.9 Solução de trabalho do padrão primário deuterado a 2 µg/mL
Diluir em metanol 200 µL das soluções descritas em 2.3.6 até perfazer um
volume de 10 mL.
2.3.10 Mistura de trabalho de padrões primários a 5 µg/mL
Diluir em metanol 25 µL das soluções descritas em 2.3.5 e 250 µL das soluções
descritas em 2.3.6 até perfazer um volume de 5 mL.
2.3.11 Mistura de trabalho de padrões primários a 0,1 µg/mL
Diluir em metanol 100 µL das soluções descritas em 2.3.10 até perfazer um
volume de 5 mL.
2.3.12 Solução de trabalho do padrão primário deuterado a 0,5 µg/mL
Diluir em metanol 50 µL das soluções descritas em 2.3.6 até perfazer um volume
de 10 mL.

Material e Métodos
Anabela Cardoso Mariano Página 51
3. EQUIPAMENTOS
3.1 Aparelhos de uso comum
3.1.1 Balança de precisão (resolução de 0,0001 g);
3.1.2 Banho seco;
3.1.3 Banho ultra sons;
3.1.4 Centrífuga;
3.1.5 Equipamento para capsular/descapsular vials;
3.1.6 Equipamento de extracção em fase sólida;
3.1.7 Evaporador de solventes (com azoto);
3.1.8 Homogeneizador de amostras;
3.1.9 Hotte (com filtros próprios para solventes);
3.1.10 Vortex.
3.2 Pipetas e doseadores
3.2.1 Pipetas automáticas de 20-200 µL;
3.2.2 Pipeta de volume fixo de 25 µL;
3.2.3 Pipeta automática variável de 1000 µL;
3.2.4 Doseador (e.g. Eppendorf Multipette 4780 com combitips de 50 mL)
3.3 Instrumentos analíticos
3.3.1 GC-MS
Componente Principais características
Injector automático Injecção em modo split/splitless
GC Coluna analítica: HP-5MS 30 m x 0,32 mm x 0,25 µm (Agilent
19091S-413, ou equivalente)
MS Quadrupolo

Material e Métodos
Anabela Cardoso Mariano Página 52
4. PROCEDIMENTO
4.1 Homogeneização das amostras primárias
As amostras primárias devem ser homogeneizadas através de agitação suave, até
atingirem uma temperatura próxima da ambiente.
4.2 Identificação das amostras de ensaio
Durante a realização do ensaio todas as amostras de ensaio devem estar
claramente identificadas (e.g. código interno da amostra).
4.3 Diluição das amostras de ensaio e adição de padrão interno
Para tubos de 10 mL adicionar:
� 500 µL de amostra;
� 8 mL de tampão fosfato (ver 2.3.3);
� 25 µL de solução de trabalho de padrão primário deuterado PI (2.3.9 ou 2.3.12).
Homogeneizar através de movimentos de rotação/inversão durante cerca de 10
minutos e centrifugar a 3000 rpm durante 10 minutos.
4.4 Preparação da curva de calibração
A curva de calibração permite calcular a concentração das substâncias cuja
presença nas amostras foi confirmada através da aplicação de um método de ensaio
de confirmação qualitativo.
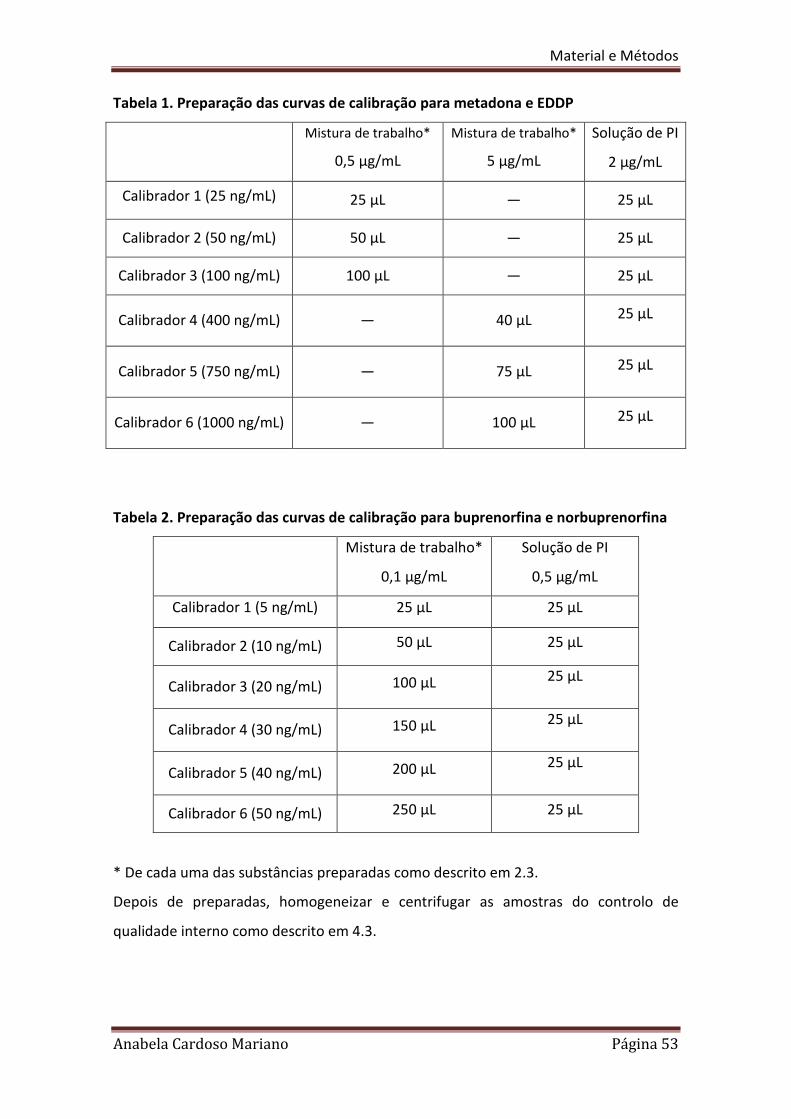
Os calibradores são preparados tal como foi descrito no ponto 2.3 (utilizando
amostras brancas) e fortificados de acordo com a tabela 1 e 2.
Material e Métodos
Anabela Cardoso Mariano Página 53
Tabela 1. Preparação das curvas de calibração para metadona e EDDP
Mistura de trabalho*
0,5 µg/mL
Mistura de trabalho*
5 µg/mL
Solução de PI
2 µg/mL
Calibrador 1 (25 ng/mL) 25 µL ― 25 µL
Calibrador 2 (50 ng/mL) 50 µL ― 25 µL
Calibrador 3 (100 ng/mL) 100 µL ― 25 µL
Calibrador 4 (400 ng/mL) ― 40 µL 25 µL
Calibrador 5 (750 ng/mL) ― 75 µL 25 µL
Calibrador 6 (1000 ng/mL) ― 100 µL 25 µL
Tabela 2. Preparação das curvas de calibração para buprenorfina e norbuprenorfina
Mistura de trabalho*
0,1 µg/mL
Solução de PI
0,5 µg/mL
Calibrador 1 (5 ng/mL) 25 µL 25 µL
Calibrador 2 (10 ng/mL) 50 µL 25 µL
Calibrador 3 (20 ng/mL) 100 µL 25 µL
Calibrador 4 (30 ng/mL) 150 µL 25 µL
Calibrador 5 (40 ng/mL) 200 µL 25 µL
Calibrador 6 (50 ng/mL) 250 µL 25 µL
* De cada uma das substâncias preparadas como descrito em 2.3.
Depois de preparadas, homogeneizar e centrifugar as amostras do controlo de
qualidade interno como descrito em 4.3.
Material e Métodos
Anabela Cardoso Mariano Página 54
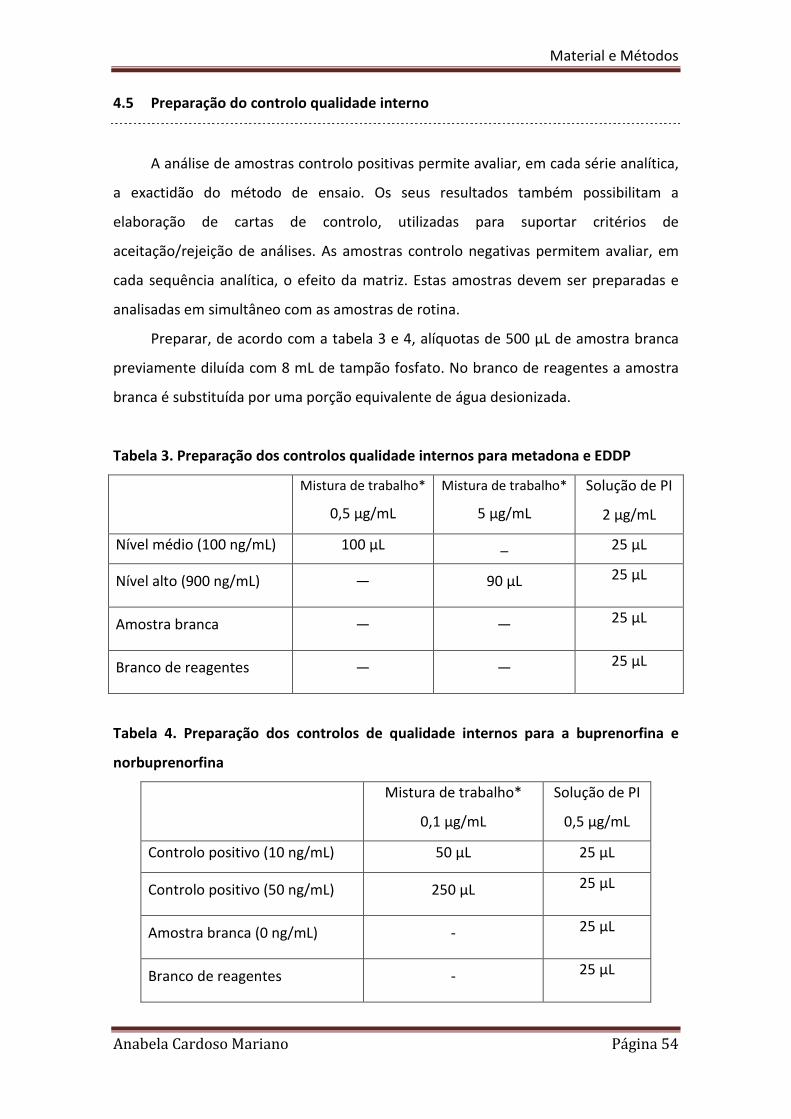
4.5 Preparação do controlo qualidade interno
A análise de amostras controlo positivas permite avaliar, em cada série analítica,
a exactidão do método de ensaio. Os seus resultados também possibilitam a
elaboração de cartas de controlo, utilizadas para suportar critérios de
aceitação/rejeição de análises. As amostras controlo negativas permitem avaliar, em
cada sequência analítica, o efeito da matriz. Estas amostras devem ser preparadas e
analisadas em simultâneo com as amostras de rotina.
Preparar, de acordo com a tabela 3 e 4, alíquotas de 500 µL de amostra branca
previamente diluída com 8 mL de tampão fosfato. No branco de reagentes a amostra
branca é substituída por uma porção equivalente de água desionizada.
Tabela 3. Preparação dos controlos qualidade internos para metadona e EDDP
Mistura de trabalho*
0,5 µg/mL
Mistura de trabalho*
5 µg/mL
Solução de PI
2 µg/mL
Nível médio (100 ng/mL) 100 µL _ 25 µL
Nível alto (900 ng/mL) ― 90 µL 25 µL
Amostra branca ― ― 25 µL
Branco de reagentes ― ― 25 µL
Tabela 4. Preparação dos controlos de qualidade internos para a buprenorfina e
norbuprenorfina
Mistura de trabalho*
0,1 µg/mL
Solução de PI
0,5 µg/mL
Controlo positivo (10 ng/mL) 50 µL 25 µL
Controlo positivo (50 ng/mL) 250 µL 25 µL
Amostra branca (0 ng/mL) - 25 µL
Branco de reagentes - 25 µL

Material e Métodos
Anabela Cardoso Mariano Página 55
* De cada uma das substâncias preparadas como descrito em 2.3.
Depois de preparadas, homogeneizar e centrifugar as amostras do controlo de
qualidade interno como descrito em 4.3.
4.6 Extracção em fase sólida
Através das colunas de extracção Waters Oasis MCX®, passar as seguintes soluções:
� 2 mL de metanol Acondicionamento
� 2 mL de água desionizada
� O sobrenadante obtido em 4.3 (cerca de 1 mL/min) Aplicação da
amostra
� 2 mL de água desionizada
Lavagem � 2 mL de HCl 0,1N
� 2 mL de metanol
� 2 mL de n-Hexano
� Secar as colunas sob vácuo (cerca de 15 min) 1 Secagem das
colunas
� 2 mL de diclorometano/2-Propanol/Amónia (78:20:2,
v/v/v) (7.3.1) Eluição
1Se necessário, este tempo deverá ser aumentado até à secagem total das colunas.
4.7 Derivatização química
� Recolher o eluído obtido em 4.6 para um tubo de vidro e secar sob corrente de
azoto a cerca de 40ºC;
� Adicionar ao extracto seco 60 µL de MSTFA/TMCS (2.3.2) e aquecer durante 30
min a cerca de 80ºC;
� Transferir o extracto derivatizado para um vial de 300 µL.
Material e Métodos
Anabela Cardoso Mariano Página 56
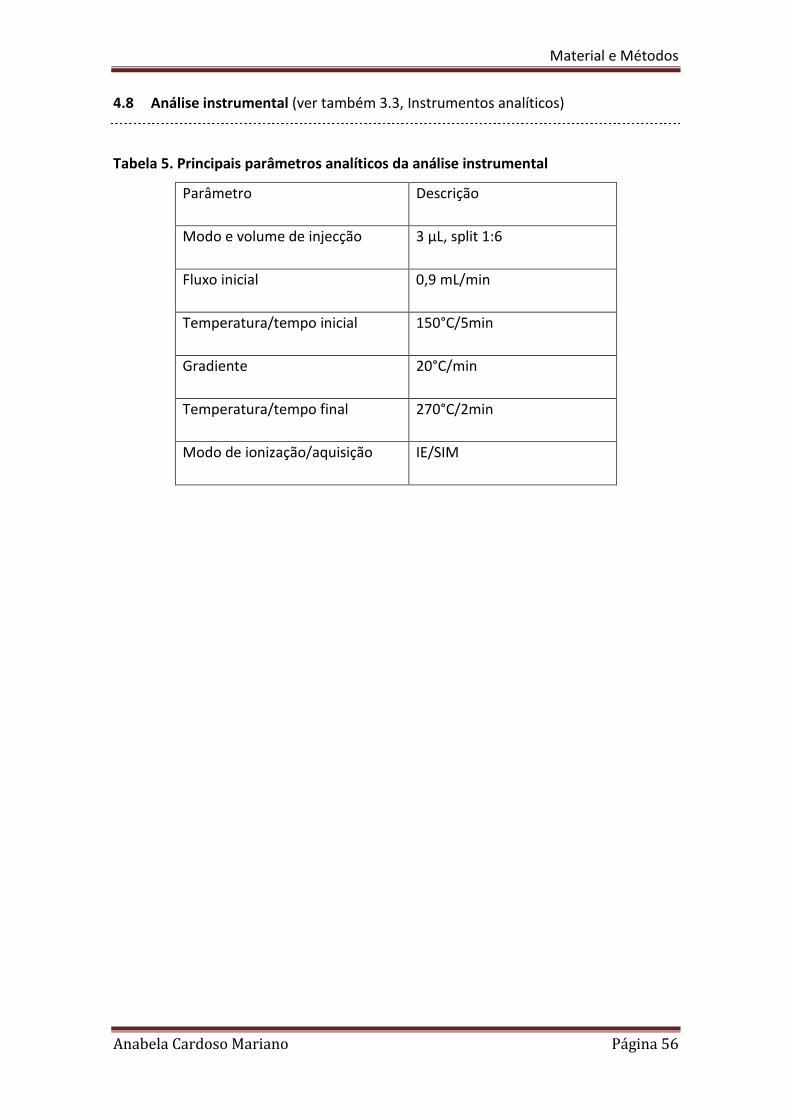
4.8 Análise instrumental (ver também 3.3, Instrumentos analíticos)
Tabela 5. Principais parâmetros analíticos da análise instrumental
Parâmetro Descrição
Modo e volume de injecção 3 µL, split 1:6
Fluxo inicial 0,9 mL/min
Temperatura/tempo inicial 150°C/5min
Gradiente 20°C/min
Temperatura/tempo final 270°C/2min
Modo de ionização/aquisição IE/SIM

RESULTADOS E DISCUSSÃO


Resultados e Discussão
Anabela Cardoso Mariano Página 59
RESULTADOS E DISCUSSÃO
No presente estudo de validação foi aplicada a metodologia genérica
documentada no procedimento PO-STF-006 Rev00 (validação de procedimentos de
ensaio), tendo sido avaliados os parâmetros seguintes:
� Especificidade/Selectividade;
� Capacidade de Identificação;
� Linearidade/Gama de trabalho;
� Arrastamento (carryover);
� Limite de Detecção (LD) e Limite de Quantificação (LQ);
� Eficiência da Extracção;
� Repetibilidade;
� Precisão Intermédia;
� Exactidão;
� Robustez.
Especificidade/Selectividade
Utilizaram-se 40 amostras, escolhidas aleatoriamente do universo das 75 já
referidas. Das 40 amostras escolhidas misturaram-se, também de forma aleatória,
quatro a quatro amostras (2 mL de cada uma delas) constituindo-se assim 10 misturas
ou pools. Foram medidas 2 alíquotas de 500 µL de cada uma das misturas constituídas
conforme indicado na tabela 6. A um dos dois grupos de 10 alíquotas foram
adicionados, os padrões internos (metadona-d3, EDDP-d3; e buprenorfina-d4) e os
padrões das substâncias em estudo (metadona, EDDP, buprenorfina e
norbuprenorfina), a uma concentração de 100 ng/mL para a metadona e EDDP, e
concentração de 10 ng/mL para a buprenorfina e norbuprenorfina, ao outro grupo de
10 alíquotas nada foi adicionado. Foi aplicado o procedimento de ensaio a todas as
amostras.
Resultados e Discussão
Anabela Cardoso Mariano Página 60
Tabela 6. Sangues usados para constituir as misturas ou pools
Processo Proveniência Sexo Idade Origem
(causa de morte ou pessoa viva) Amostra Opiáceos
Misturas
ou
Pools
37 Tanatologia M 40 Desconhecida Sangue Neg 48 Leiria M 73 Desconhecida Sangue Neg 1 53 Leiria M 53 Desconhecida Sangue Neg 115 Leiria M 71 Enforcamento Sg. Periférico Neg 127 Tanatologia M 55 Desconhecida Sg. Cardíaco Neg 183 Viseu M 44 Acidente de viação Sangue Neg 2 340 Viseu M 14 Morte Súbita Sangue Neg 354 Leiria M 54 Desconhecida Sg. Periférico Neg 355 Leiria M 53 Desconhecida Sangue Neg 357 Leiria M 43 Desconhecida Sangue Neg 419 Leiria M 52 Desconhecida Sangue Neg 3 538 Tanatologia F 60 Desconhecida Sg. Cardíaco Neg 602 Leiria M 40 Acidente de trabalho Sangue Neg 650 Leiria F Desc. Vivo Sangue Neg 651 Leiria M 34 Acidente de viação Sangue Neg 4 652 Leiria M 31 Morte Súbita Sangue Neg 655 Leiria M 20 Acidente de viação Sangue Neg 657 Leiria M 34 Acidente de viação Sangue Neg 670 Fig. da Foz F 30 Suspeita de intoxicação Sangue Neg 5 676 Ponta Delgada M 41 Acidente de viação Sg. Femoral Neg 694 Viseu M 26 Desconhecida Sangue Neg 698 Viseu M 52 Desconhecida Sangue Neg 6 699 Viseu M 69 Enforcamento Sangue Neg 700 Viseu M 48 Morte Súbita Sangue Neg 910 Tanatologia M 22 Afogamento Sg. Torácico Neg 1004 Viseu M 52 Desconhecida Sangue Neg 1005 Viseu M 28 Morte Súbita Sangue Neg 7 1013 Guarda M 40 Acidente de trabalho Sg. Periférico Neg 1155 A. do Heroísmo M 37 Acidente de trabalho Sg. Periférico Neg 1202 Tanatologia M 33 Acidente de viação Sg. Torácico Neg 8 1283 Leiria M 55 Acidente de viação Sangue Neg 1307 Fig. da Foz M 61 Morte Súbita Sg. Femoral Neg 1625 GNR Fundão Vivo Sg. Periférico Neg 1783 GNR Viseu Vivo Sg. Periférico Neg 9 2409 GNR Mealhada Vivo Sg. Periférico Neg 2652 PSP Covilhã Vivo Sg. Periférico Neg 2663 PSP T. Novas Vivo Sg. Periférico Neg 2941 PSP Fig.Foz Vivo Sg. Periférico Neg 80 81
PSP P. Delgada Vivo Sg. Periférico Neg 10 GNR Pataias Vivo Sg. Periférico Neg
Foram usadas soluções de trabalho nas concentrações de 0,05, 0,1, 0,5, 1,0 e 5,0
µg/mL. Para a preparação dessas soluções utilizaram-se soluções de armazenamento a 1
mg/mL.
A positividade/negatividade das amostras foi avaliada com base nos critérios de
identificação de substâncias, documentados no procedimento operacional PO-STF-006 Rev00
(procedimento interno do Serviço de Toxicologia Forense da Delegação do Centro): tempo de

Resultados e Discussão
Anabela Cardoso Mariano Página 61
retenção relativo (TRR), relações iónicas dos iões estudados e razão sinal/ruído. Para o
controlo positivo fortificámos uma alíquota de 500 µL de sangue branco pertencente à pool
10, com 100 µL da solução de trabalho da mistura de padrões de metadona e EDDP 0,5 µg/mL,
no caso da metadona e com 10 µL da solução de trabalho do padrão de buprenorfina e
norbuprenorfina 0,1 µg/mL no caso da buprenorfina, e com 25 µL de metadona-d3 e EDDP-d3
a 2,0 µg/mL, no primeiro caso, e 25 µL de buprenorfina-d4 a 0,5 µg/mL no segundo caso.
Critério de Tempo de Retenção Relativo (TRR)
O TRR da substância pesquisada na amostra não deve diferir mais do que 1% ou ±0,2
minutos (o que for menor) do TRR da mesma substância na amostra controlo analisada
contemporaneamente e nas mesmas condições. Seguidamente apresenta-se a forma de
calcular a diferença relativa de TRR:
∆TRR=
TRRSubst.Pesquisada - Tempo de retenção relativo da substância pesquisada na amostra
TRRSubst.Controlo - Tempo de retenção relativo da substância na amostra controlo
∆TRR - Diferença relativa de TRR
Critério das relações iónicas dos iões
As áreas relativas dos iões diagnóstico da substância pesquisada na amostra devem
cumprir os critérios definidos na tabela 7, relativamente às áreas relativas dos mesmos iões
diagnóstico na amostra controlo.
TRRSubst.Pesquisada __TRRSubst.Controlo
TRRSubst.Controlo

Resultados e Discussão
Anabela Cardoso Mariano Página 62
Tabela 7. Tolerância máxima para as áreas relativas dos iões diagnóstico
Abundância relativa (% do pico base) Tolerância máxima
>50% ± 10% (absoluta)
> 25 % e <50% ± 20% (relativa)
<25% ± 5% (absoluta)
Critério do sinal/ ruído (S/N)
O valor da razão sinal/ruído para os três iões diagnóstico da substância pesquisada na
amostra deve ser superior a 3.
Seguidamente, apresentam-se os resultados obtidos na avaliação de cada um dos critérios de
positividade anteriormente descritos.
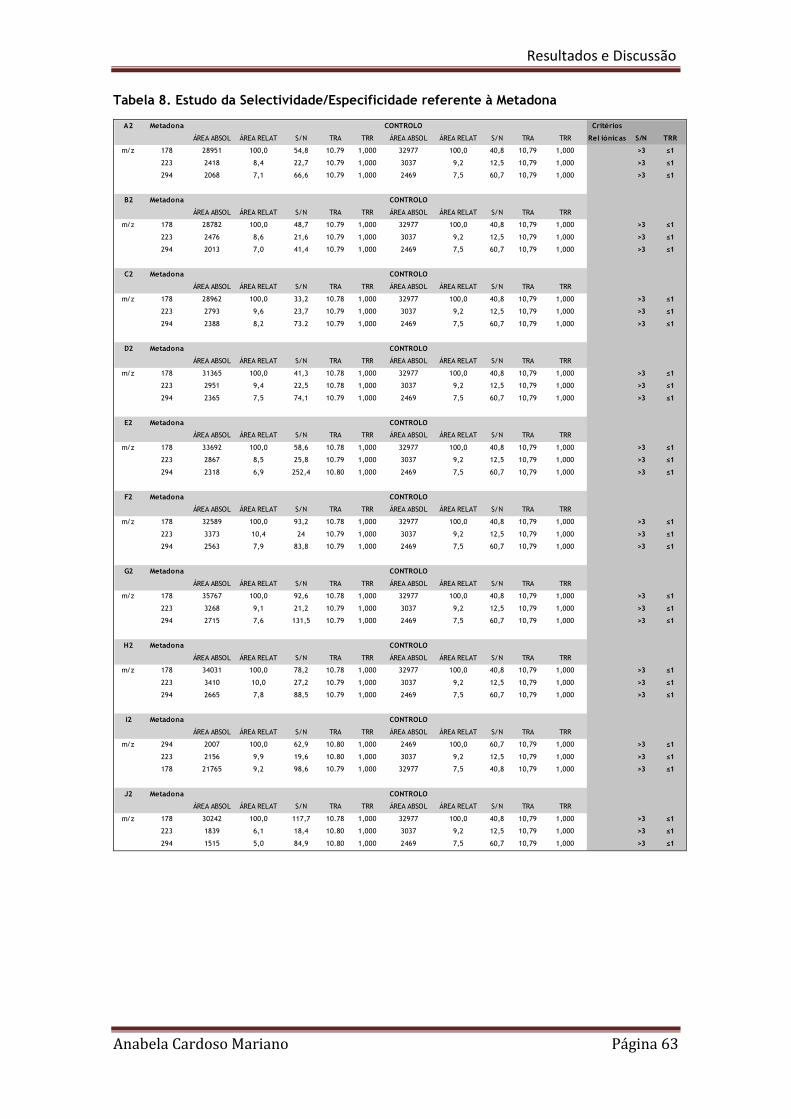
Resultados e Discussão
Anabela Cardoso Mariano Página 63
Tabela 8. Estudo da Selectividade/Especificidade referente à Metadona
A2 Metadona CONTROLO Critérios
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR Rel iónicas S/N TRR
m/z 178 28951 100,0 54,8 10.79 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 2418 8,4 22,7 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2068 7,1 66,6 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
B2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 28782 100,0 48,7 10.79 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 2476 8,6 21,6 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2013 7,0 41,4 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
C2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 28962 100,0 33,2 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 2793 9,6 23,7 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2388 8,2 73.2 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
D2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 31365 100,0 41,3 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 2951 9,4 22,5 10.78 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2365 7,5 74,1 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
E2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 33692 100,0 58,6 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 2867 8,5 25,8 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2318 6,9 252,4 10.80 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
F2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 32589 100,0 93,2 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 3373 10,4 24 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2563 7,9 83,8 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
G2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 35767 100,0 92,6 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 3268 9,1 21,2 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2715 7,6 131,5 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
H2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 34031 100,0 78,2 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 3410 10,0 27,2 10.79 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 2665 7,8 88,5 10.79 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
I2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 294 2007 100,0 62,9 10.80 1,000 2469 100,0 60,7 10,79 1,000 >3 ≤1
223 2156 9,9 19,6 10.80 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
178 21765 9,2 98,6 10.79 1,000 32977 7,5 40,8 10,79 1,000 >3 ≤1
J2 Metadona CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 178 30242 100,0 117,7 10.78 1,000 32977 100,0 40,8 10,79 1,000 >3 ≤1
223 1839 6,1 18,4 10.80 1,000 3037 9,2 12,5 10,79 1,000 >3 ≤1
294 1515 5,0 84,9 10.80 1,000 2469 7,5 60,7 10,79 1,000 >3 ≤1
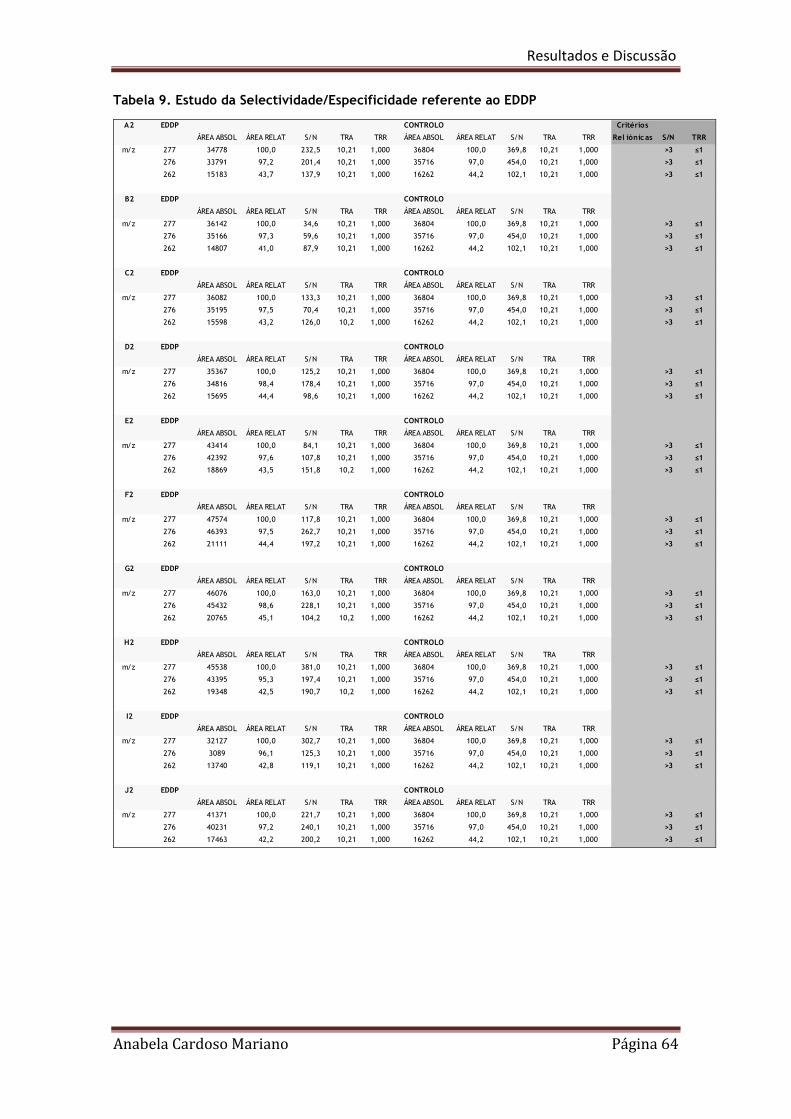
Resultados e Discussão
Anabela Cardoso Mariano Página 64
Tabela 9. Estudo da Selectividade/Especificidade referente ao EDDP
A2 EDDP CONTROLO Critérios
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR Rel iónicas S/N TRR
m/z 277 34778 100,0 232,5 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 33791 97,2 201,4 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 15183 43,7 137,9 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
B2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 36142 100,0 34,6 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 35166 97,3 59,6 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 14807 41,0 87,9 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
C2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 36082 100,0 133,3 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 35195 97,5 70,4 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 15598 43,2 126,0 10,2 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
D2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 35367 100,0 125,2 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 34816 98,4 178,4 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 15695 44,4 98,6 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
E2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 43414 100,0 84,1 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 42392 97,6 107,8 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 18869 43,5 151,8 10,2 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
F2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 47574 100,0 117,8 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 46393 97,5 262,7 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 21111 44,4 197,2 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
G2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 46076 100,0 163,0 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 45432 98,6 228,1 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 20765 45,1 104,2 10,2 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
H2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 45538 100,0 381,0 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 43395 95,3 197,4 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 19348 42,5 190,7 10,2 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
I2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 32127 100,0 302,7 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 3089 96,1 125,3 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 13740 42,8 119,1 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1
J2 EDDP CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 277 41371 100,0 221,7 10,21 1,000 36804 100,0 369,8 10,21 1,000 >3 ≤1
276 40231 97,2 240,1 10,21 1,000 35716 97,0 454,0 10,21 1,000 >3 ≤1
262 17463 42,2 200,2 10,21 1,000 16262 44,2 102,1 10,21 1,000 >3 ≤1

Resultados e Discussão
Anabela Cardoso Mariano Página 65
Tabela 10. Estudo da Selectividade/Especificidade referente à buprenorfina
A2 Buprenorfina CONTROLO Critérios
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR Rel iónicas S/N TRR
m/z 450 4365 100,0 27,8 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1307 29,9 10,5 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 609 14,0 6,4 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
B2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 7359 100,0 55,5 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1973 26,8 16,3 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1418 19,3 9,4 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
C2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 4251 100,0 25,9 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1486 35,0 9,9 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 851 20,0 4,9 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
D2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 6085 100,0 46,3 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 2010 33,0 19,6 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1031 16,9 8,6 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
E2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 6660 100,0 58,3 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1821 27,3 17,2 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1348 20,2 8,3 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
F2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 6326 100,0 62,2 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1900 30,0 14,3 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1123 17,8 8,5 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
G2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 7162 100,0 54,7 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 2033 28,4 22,1 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1210 16,9 10,4 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
H2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 7206 100,0 44,6 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 2325 32,3 21,9 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1191 16,5 13,5 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
I2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 6644 100,0 63,9 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1937 29,2 19,8 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1351 20,3 9,1 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1
J2 Buprenorfina CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 450 6414 1000,0 48,3 10,45 1,000 5808 100,0 36,5 10,45 1,000 >3 ≤1
482 1710 26,7 14,7 10,45 1,000 1807 31,1 18 10,45 1,000 >3 ≤1
506 1245 19,4 11,6 10,45 1,000 915 15,8 16,6 10,45 1,000 >3 ≤1

Resultados e Discussão
Anabela Cardoso Mariano Página 66
Tabela11. Estudo da Selectividade/Especificidade referente à norbuprenorfina
A2 Norbuprenor. CONTROLO Critérios
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR Rel iónicas S/N TRR
m/z 468 5577 100,0 53,8 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 506 9,1 8,6 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 432 7,7 6,2 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
B2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 10445 100,0 81,3 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 981 9,4 17,6 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 886 8,5 13,6 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
C2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 4889 100,0 36,4 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 566 11,6 8,1 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 450 9,2 6,2 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
D2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 8771 100,0 104 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 955 10,9 14,2 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 871 9,9 10,5 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
E2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 11832 100,0 92,3 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 1155 9,8 10,7 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 1135 9,6 21,4 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
F2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 9346 100,0 87,9 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 792 8,5 28,7 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 757 8,1 10,2 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
G2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 9443 100,0 73,4 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 999 10,6 10,8 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 925 9,8 17,9 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
H2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 9682 100,0 60,6 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 948 9,8 16 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 921 9,5 17,4 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
I2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 10395 100,0 95,5 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 931 9,0 16,3 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 898 8,6 19,9 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
J2 Norbuprenor. CONTROLO
ÁREA ABSOL ÁREA RELAT S/N TRA TRR ÁREA ABSOL ÁREA RELAT S/N TRA TRR
m/z 468 8559 100,0 61,9 8,64 1,000 6970 100,0 67,4 8,64 1,000 >3 ≤1
524 838 9,8 13,9 8,64 1,000 686 9,8 7,7 8,64 1,000 >3 ≤1
500 725 8,5 14,9 8,64 1,000 536 7,7 11,5 8,64 1,000 >3 ≤1
Foram cumpridos os três critérios de positividade (relações iónicas, sinal/ruído e
tempo de retenção relativo) para a metadona, EDDP, buprenorfina e norbuprenorfina nas
diferentes amostras de sangue analisadas.
A análise dos resultados confirmou que todas as amostras de sangue às quais não
foram adicionadas drogas evidenciaram resultados negativos. Nas amostras às quais foram
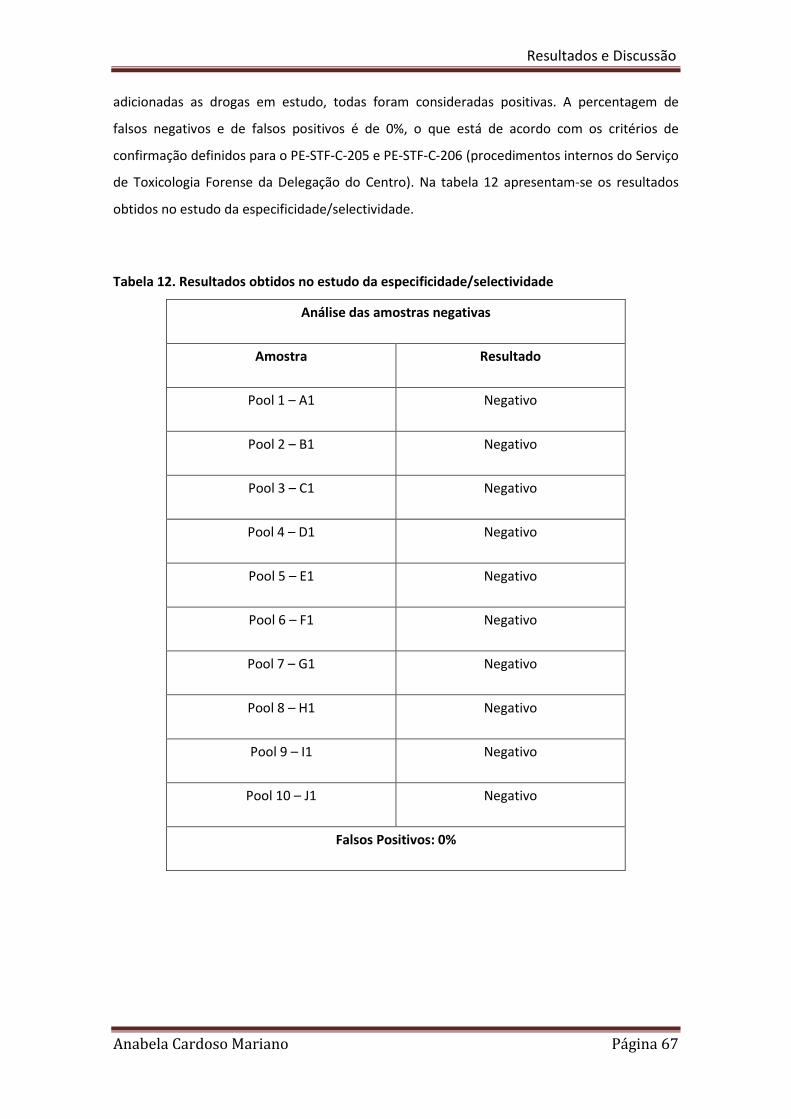
Resultados e Discussão
Anabela Cardoso Mariano Página 67
adicionadas as drogas em estudo, todas foram consideradas positivas. A percentagem de
falsos negativos e de falsos positivos é de 0%, o que está de acordo com os critérios de
confirmação definidos para o PE-STF-C-205 e PE-STF-C-206 (procedimentos internos do Serviço
de Toxicologia Forense da Delegação do Centro). Na tabela 12 apresentam-se os resultados
obtidos no estudo da especificidade/selectividade.
Tabela 12. Resultados obtidos no estudo da especificidade/selectividade
Análise das amostras negativas
Amostra Resultado
Pool 1 – A1 Negativo
Pool 2 – B1 Negativo
Pool 3 – C1 Negativo
Pool 4 – D1 Negativo
Pool 5 – E1 Negativo
Pool 6 – F1 Negativo
Pool 7 – G1 Negativo
Pool 8 – H1 Negativo
Pool 9 – I1 Negativo
Pool 10 – J1 Negativo
Falsos Positivos: 0%
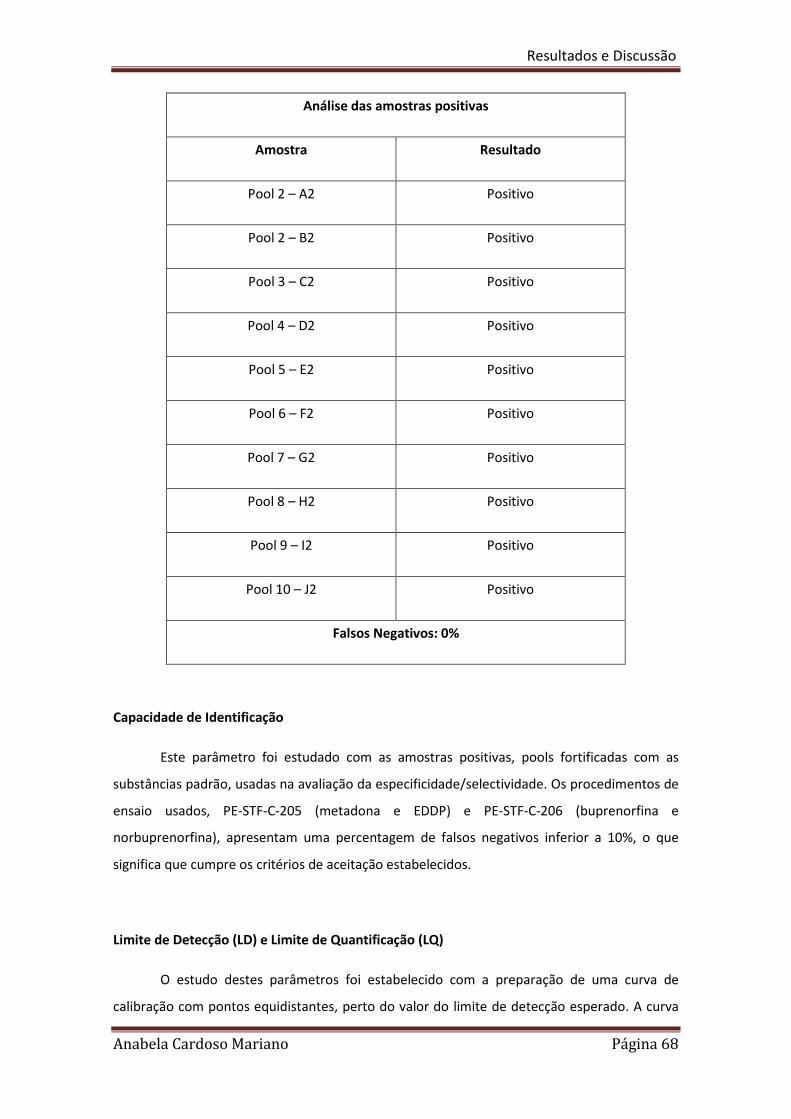
Resultados e Discussão
Anabela Cardoso Mariano Página 68
Análise das amostras positivas
Amostra Resultado
Pool 2 – A2 Positivo
Pool 2 – B2 Positivo
Pool 3 – C2 Positivo
Pool 4 – D2 Positivo
Pool 5 – E2 Positivo
Pool 6 – F2 Positivo
Pool 7 – G2 Positivo
Pool 8 – H2 Positivo
Pool 9 – I2 Positivo
Pool 10 – J2 Positivo
Falsos Negativos: 0%
Capacidade de Identificação
Este parâmetro foi estudado com as amostras positivas, pools fortificadas com as
substâncias padrão, usadas na avaliação da especificidade/selectividade. Os procedimentos de
ensaio usados, PE-STF-C-205 (metadona e EDDP) e PE-STF-C-206 (buprenorfina e
norbuprenorfina), apresentam uma percentagem de falsos negativos inferior a 10%, o que
significa que cumpre os critérios de aceitação estabelecidos.
Limite de Detecção (LD) e Limite de Quantificação (LQ)
O estudo destes parâmetros foi estabelecido com a preparação de uma curva de
calibração com pontos equidistantes, perto do valor do limite de detecção esperado. A curva
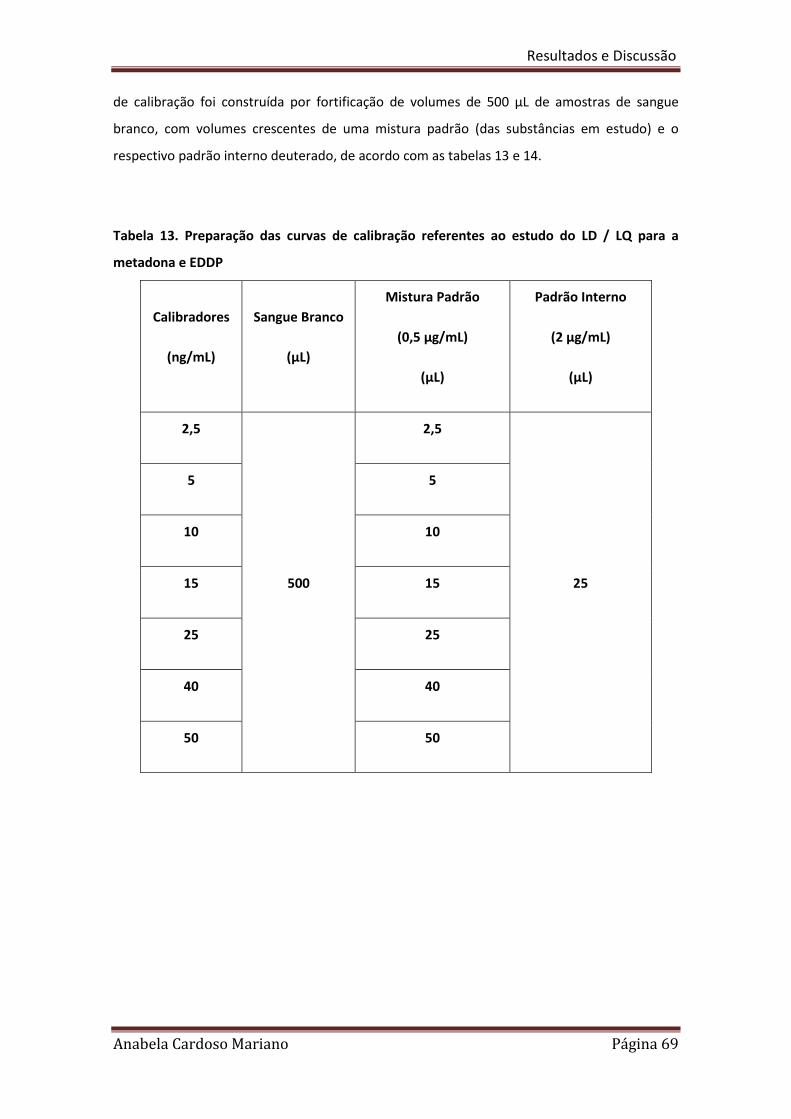
Resultados e Discussão
Anabela Cardoso Mariano Página 69
de calibração foi construída por fortificação de volumes de 500 µL de amostras de sangue
branco, com volumes crescentes de uma mistura padrão (das substâncias em estudo) e o
respectivo padrão interno deuterado, de acordo com as tabelas 13 e 14.
Tabela 13. Preparação das curvas de calibração referentes ao estudo do LD / LQ para a
metadona e EDDP
Calibradores
(ng/mL)
Sangue Branco
(µL)
Mistura Padrão
(0,5 µg/mL)
(µL)
Padrão Interno
(2 µg/mL)
(µL)
2,5
500
2,5
25
5 5
10 10
15 15
25 25
40 40
50 50
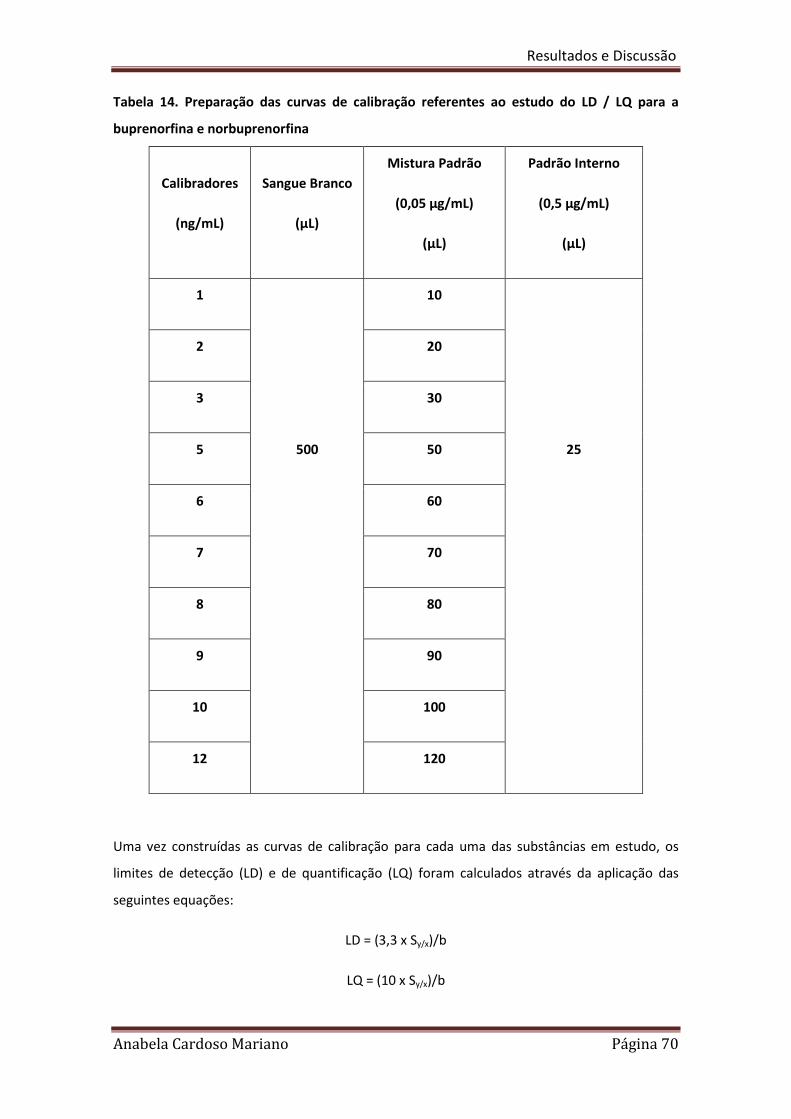
Resultados e Discussão
Anabela Cardoso Mariano Página 70
Tabela 14. Preparação das curvas de calibração referentes ao estudo do LD / LQ para a
buprenorfina e norbuprenorfina
Calibradores
(ng/mL)
Sangue Branco
(µL)
Mistura Padrão
(0,05 µg/mL)
(µL)
Padrão Interno
(0,5 µg/mL)
(µL)
1
500
10
25
2 20
3 30
5 50
6 60
7 70
8 80
9 90
10 100
12 120
Uma vez construídas as curvas de calibração para cada uma das substâncias em estudo, os
limites de detecção (LD) e de quantificação (LQ) foram calculados através da aplicação das
seguintes equações:
LD = (3,3 x Sy/x)/b
LQ = (10 x Sy/x)/b
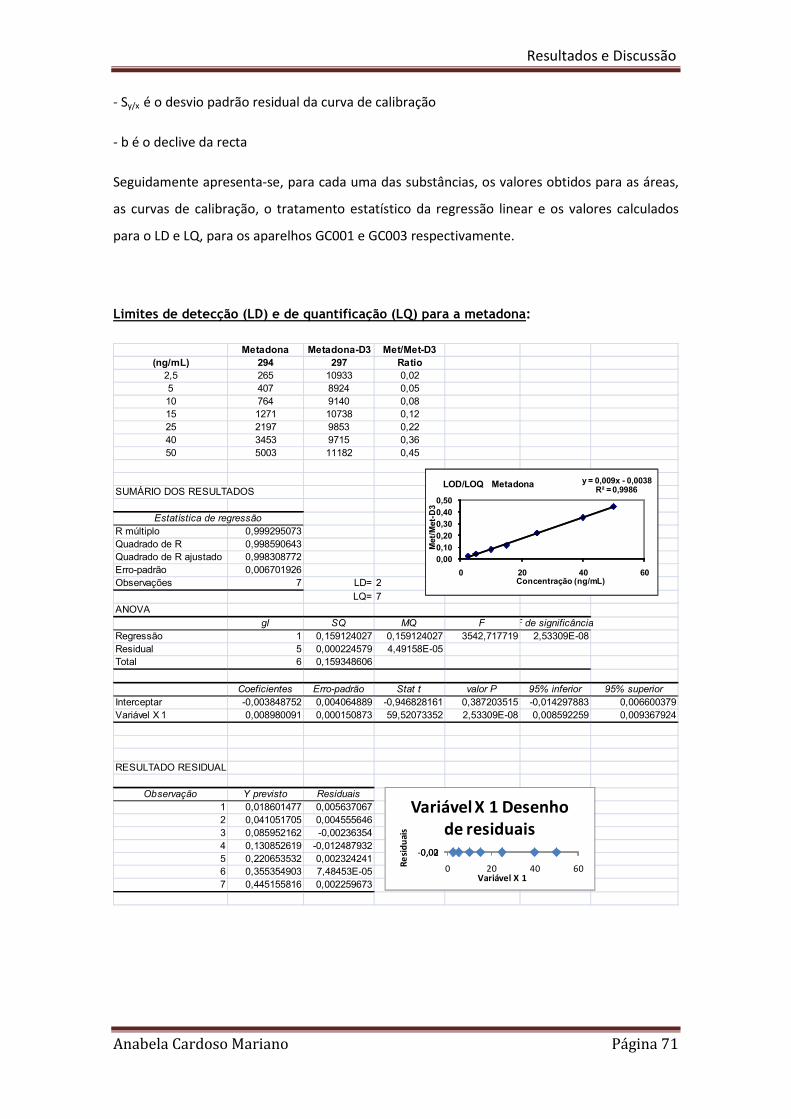
Resultados e Discussão
Anabela Cardoso Mariano Página 71
- Sy/x é o desvio padrão residual da curva de calibração
- b é o declive da recta
Seguidamente apresenta-se, para cada uma das substâncias, os valores obtidos para as áreas,
as curvas de calibração, o tratamento estatístico da regressão linear e os valores calculados
para o LD e LQ, para os aparelhos GC001 e GC003 respectivamente.
Limites de detecção (LD) e de quantificação (LQ) para a metadona:
Metadona Metadona-D3 Met/Met-D3
(ng/mL) 294 297 Ratio
2,5 265 10933 0,025 407 8924 0,0510 764 9140 0,0815 1271 10738 0,1225 2197 9853 0,2240 3453 9715 0,3650 5003 11182 0,45
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,999295073Quadrado de R 0,998590643Quadrado de R ajustado 0,998308772Erro-padrão 0,006701926Observações 7 LD= 2
LQ= 7ANOVA
gl SQ MQ F F de significância
Regressão 1 0,159124027 0,159124027 3542,717719 2,53309E-08Residual 5 0,000224579 4,49158E-05Total 6 0,159348606
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar -0,003848752 0,004064889 -0,946828161 0,387203515 -0,014297883 0,006600379Variável X 1 0,008980091 0,000150873 59,52073352 2,53309E-08 0,008592259 0,009367924
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,018601477 0,0056370672 0,041051705 0,0045556463 0,085952162 -0,002363544 0,130852619 -0,0124879325 0,220653532 0,0023242416 0,355354903 7,48453E-057 0,445155816 0,002259673
y = 0,009x - 0,0038R² = 0,9986
0,00
0,10
0,20
0,30
0,40
0,50
0 20 40 60
Met/Met-D3
Concentração (ng/mL)
LOD/LOQ Metadona
-0,0200,02
0 20 40 60Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
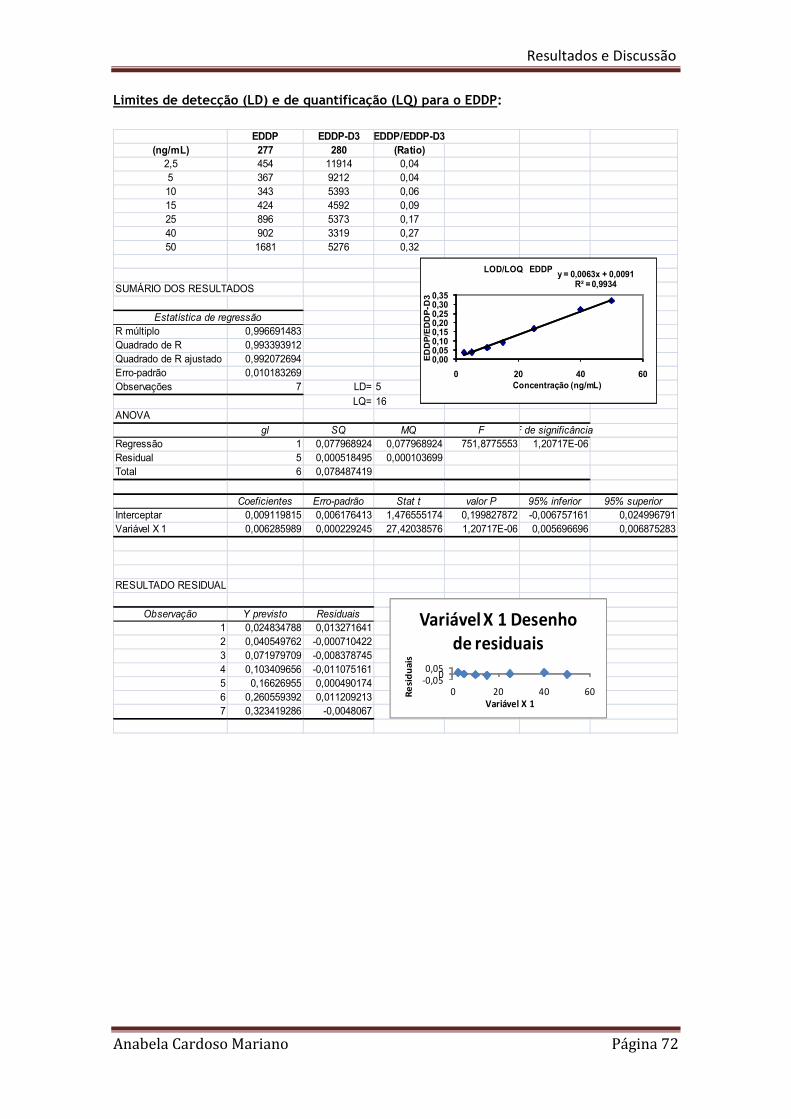
Resultados e Discussão
Anabela Cardoso Mariano Página 72
Limites de detecção (LD) e de quantificação (LQ) para o EDDP:
EDDP EDDP-D3 EDDP/EDDP-D3
(ng/mL) 277 280 (Ratio)
2,5 454 11914 0,045 367 9212 0,0410 343 5393 0,0615 424 4592 0,0925 896 5373 0,1740 902 3319 0,2750 1681 5276 0,32
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,996691483Quadrado de R 0,993393912Quadrado de R ajustado 0,992072694Erro-padrão 0,010183269Observações 7 LD= 5
LQ= 16ANOVA
gl SQ MQ F F de significância
Regressão 1 0,077968924 0,077968924 751,8775553 1,20717E-06Residual 5 0,000518495 0,000103699Total 6 0,078487419
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar 0,009119815 0,006176413 1,476555174 0,199827872 -0,006757161 0,024996791Variável X 1 0,006285989 0,000229245 27,42038576 1,20717E-06 0,005696696 0,006875283
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,024834788 0,0132716412 0,040549762 -0,0007104223 0,071979709 -0,0083787454 0,103409656 -0,0110751615 0,16626955 0,0004901746 0,260559392 0,0112092137 0,323419286 -0,0048067
y = 0,0063x + 0,0091R² = 0,9934
0,000,050,100,150,200,250,300,35
0 20 40 60
EDDP/EDDP-D3
Concentração (ng/mL)
LOD/LOQ EDDP
-0,0500,05
0 20 40 60Re
sid
ua
is
Variável X 1
Variável X 1 Desenho de residuais
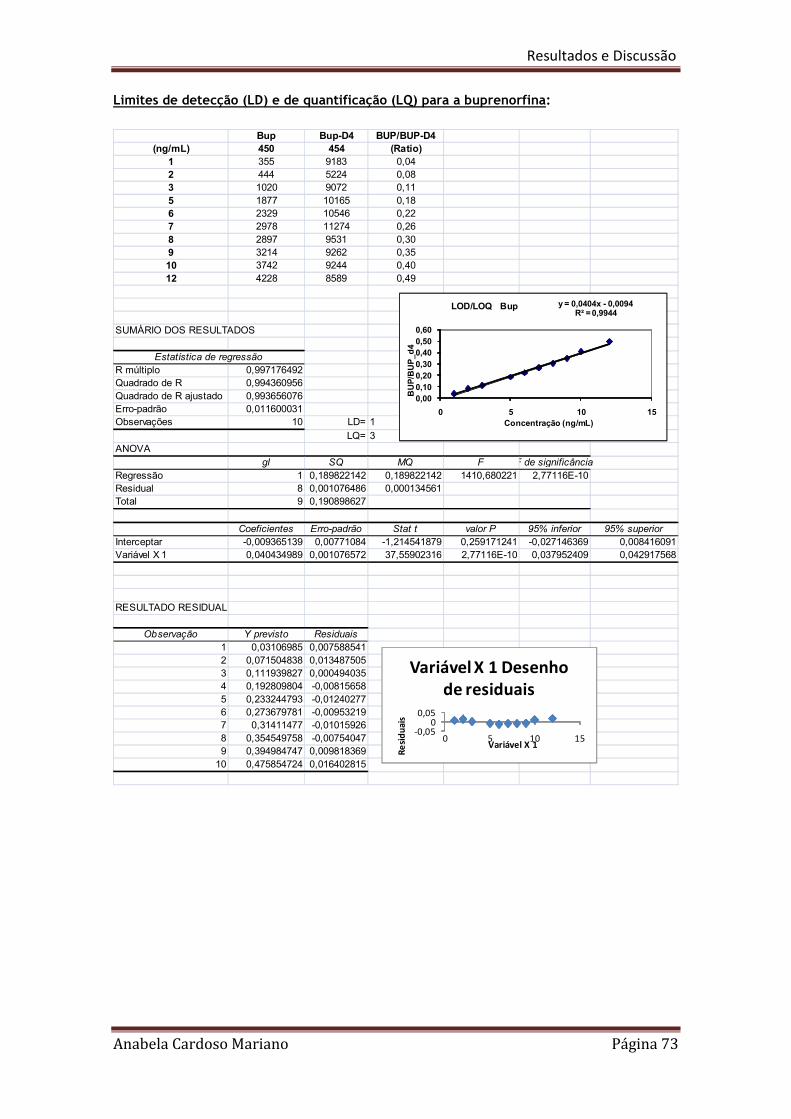
Resultados e Discussão
Anabela Cardoso Mariano Página 73
Limites de detecção (LD) e de quantificação (LQ) para a buprenorfina:
Bup Bup-D4 BUP/BUP-D4
(ng/mL) 450 454 (Ratio)
1 355 9183 0,042 444 5224 0,083 1020 9072 0,115 1877 10165 0,186 2329 10546 0,227 2978 11274 0,268 2897 9531 0,309 3214 9262 0,3510 3742 9244 0,4012 4228 8589 0,49
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,997176492Quadrado de R 0,994360956Quadrado de R ajustado 0,993656076Erro-padrão 0,011600031Observações 10 LD= 1
LQ= 3ANOVA
gl SQ MQ F F de significância
Regressão 1 0,189822142 0,189822142 1410,680221 2,77116E-10Residual 8 0,001076486 0,000134561Total 9 0,190898627
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar -0,009365139 0,00771084 -1,214541879 0,259171241 -0,027146369 0,008416091Variável X 1 0,040434989 0,001076572 37,55902316 2,77116E-10 0,037952409 0,042917568
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,03106985 0,0075885412 0,071504838 0,0134875053 0,111939827 0,0004940354 0,192809804 -0,008156585 0,233244793 -0,012402776 0,273679781 -0,009532197 0,31411477 -0,010159268 0,354549758 -0,007540479 0,394984747 0,009818369
10 0,475854724 0,016402815
y = 0,0404x - 0,0094R² = 0,9944
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0 5 10 15BUP/BUP_d4
Concentração (ng/mL)
LOD/LOQ Bup
-0,050
0,05
0 5 10 15
Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
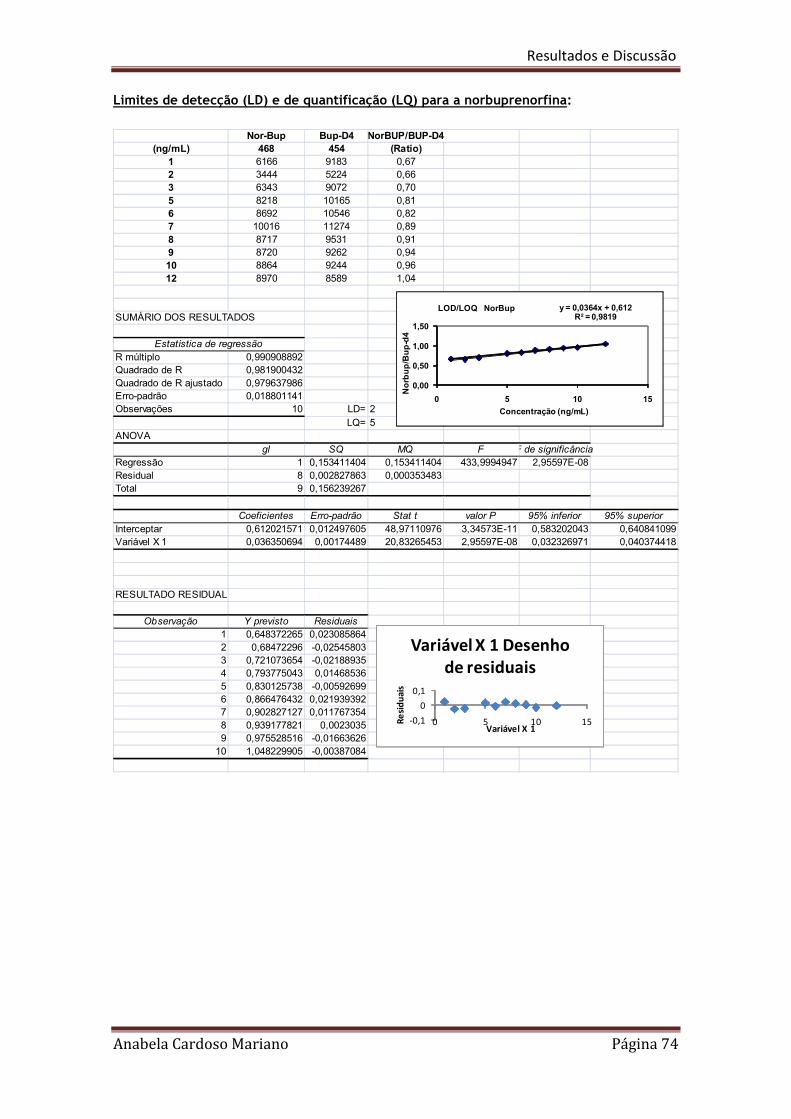
Resultados e Discussão
Anabela Cardoso Mariano Página 74
Limites de detecção (LD) e de quantificação (LQ) para a norbuprenorfina:
Nor-Bup Bup-D4 NorBUP/BUP-D4
(ng/mL) 468 454 (Ratio)
1 6166 9183 0,672 3444 5224 0,663 6343 9072 0,705 8218 10165 0,816 8692 10546 0,827 10016 11274 0,898 8717 9531 0,919 8720 9262 0,9410 8864 9244 0,9612 8970 8589 1,04
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,990908892Quadrado de R 0,981900432Quadrado de R ajustado 0,979637986Erro-padrão 0,018801141Observações 10 LD= 2
LQ= 5ANOVA
gl SQ MQ F F de significância
Regressão 1 0,153411404 0,153411404 433,9994947 2,95597E-08Residual 8 0,002827863 0,000353483Total 9 0,156239267
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar 0,612021571 0,012497605 48,97110976 3,34573E-11 0,583202043 0,640841099Variável X 1 0,036350694 0,00174489 20,83265453 2,95597E-08 0,032326971 0,040374418
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,648372265 0,0230858642 0,68472296 -0,025458033 0,721073654 -0,021889354 0,793775043 0,014685365 0,830125738 -0,005926996 0,866476432 0,0219393927 0,902827127 0,0117673548 0,939177821 0,00230359 0,975528516 -0,01663626
10 1,048229905 -0,00387084
y = 0,0364x + 0,612R² = 0,9819
0,00
0,50
1,00
1,50
0 5 10 15Norbup/Bup-d4
Concentração (ng/mL)
LOD/LOQ NorBup
-0,1
0
0,1
0 5 10 15Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
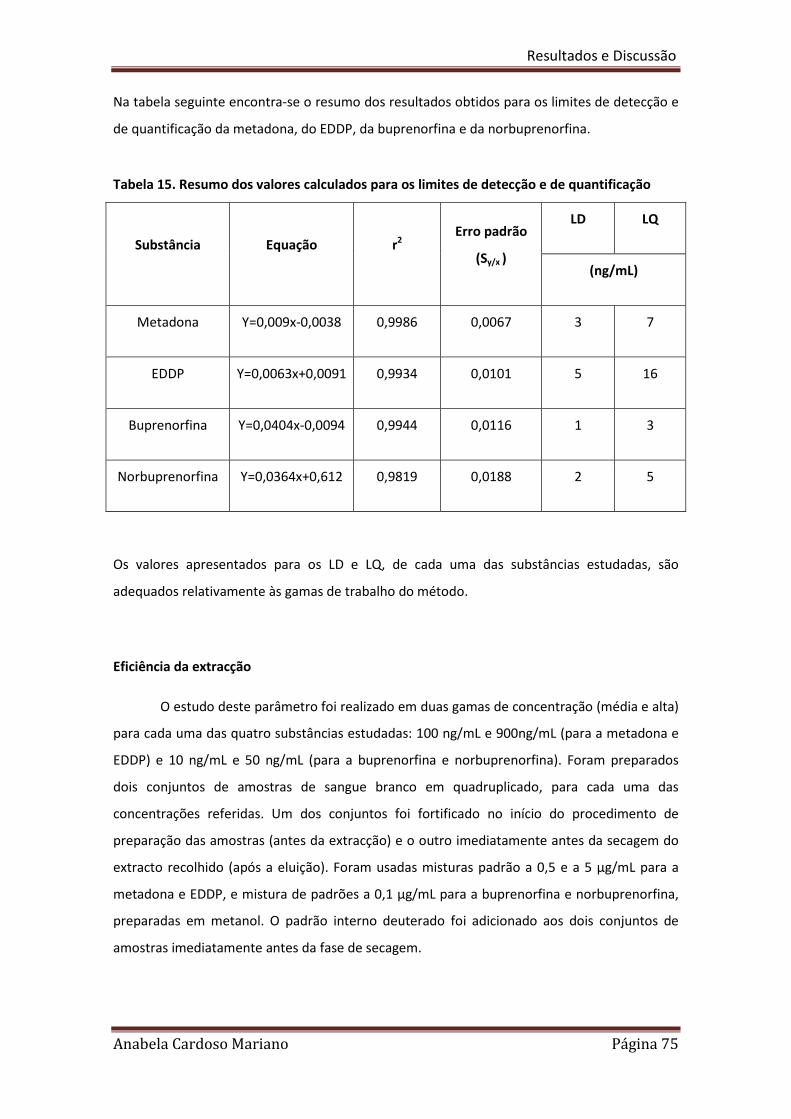
Resultados e Discussão
Anabela Cardoso Mariano Página 75
Na tabela seguinte encontra-se o resumo dos resultados obtidos para os limites de detecção e
de quantificação da metadona, do EDDP, da buprenorfina e da norbuprenorfina.
Tabela 15. Resumo dos valores calculados para os limites de detecção e de quantificação
Substância Equação r2 Erro padrão
(Sy/x )
LD LQ
(ng/mL)
Metadona Y=0,009x-0,0038 0,9986 0,0067 3 7
EDDP Y=0,0063x+0,0091 0,9934 0,0101 5 16
Buprenorfina Y=0,0404x-0,0094 0,9944 0,0116 1 3
Norbuprenorfina Y=0,0364x+0,612 0,9819 0,0188 2 5
Os valores apresentados para os LD e LQ, de cada uma das substâncias estudadas, são
adequados relativamente às gamas de trabalho do método.
Eficiência da extracção
O estudo deste parâmetro foi realizado em duas gamas de concentração (média e alta)
para cada uma das quatro substâncias estudadas: 100 ng/mL e 900ng/mL (para a metadona e
EDDP) e 10 ng/mL e 50 ng/mL (para a buprenorfina e norbuprenorfina). Foram preparados
dois conjuntos de amostras de sangue branco em quadruplicado, para cada uma das
concentrações referidas. Um dos conjuntos foi fortificado no início do procedimento de
preparação das amostras (antes da extracção) e o outro imediatamente antes da secagem do
extracto recolhido (após a eluição). Foram usadas misturas padrão a 0,5 e a 5 µg/mL para a
metadona e EDDP, e mistura de padrões a 0,1 µg/mL para a buprenorfina e norbuprenorfina,
preparadas em metanol. O padrão interno deuterado foi adicionado aos dois conjuntos de
amostras imediatamente antes da fase de secagem.

Resultados e Discussão
Anabela Cardoso Mariano Página 76
A eficiência da extracção calcula-se através da seguinte expressão:
% R = A rel método extractivo x 100
A rel sem extracção
onde:
A rel método extractivo – razão das áreas (substância/padrão interno) após aplicação do
método extractivo.
A rel sem extracção – razão das áreas (substância/padrão interno) sem aplicação do método
extractivo.
Os resultados obtidos referentes a este parâmetro analítico encontram-se apresentados nas
tabelas 16 a 17.
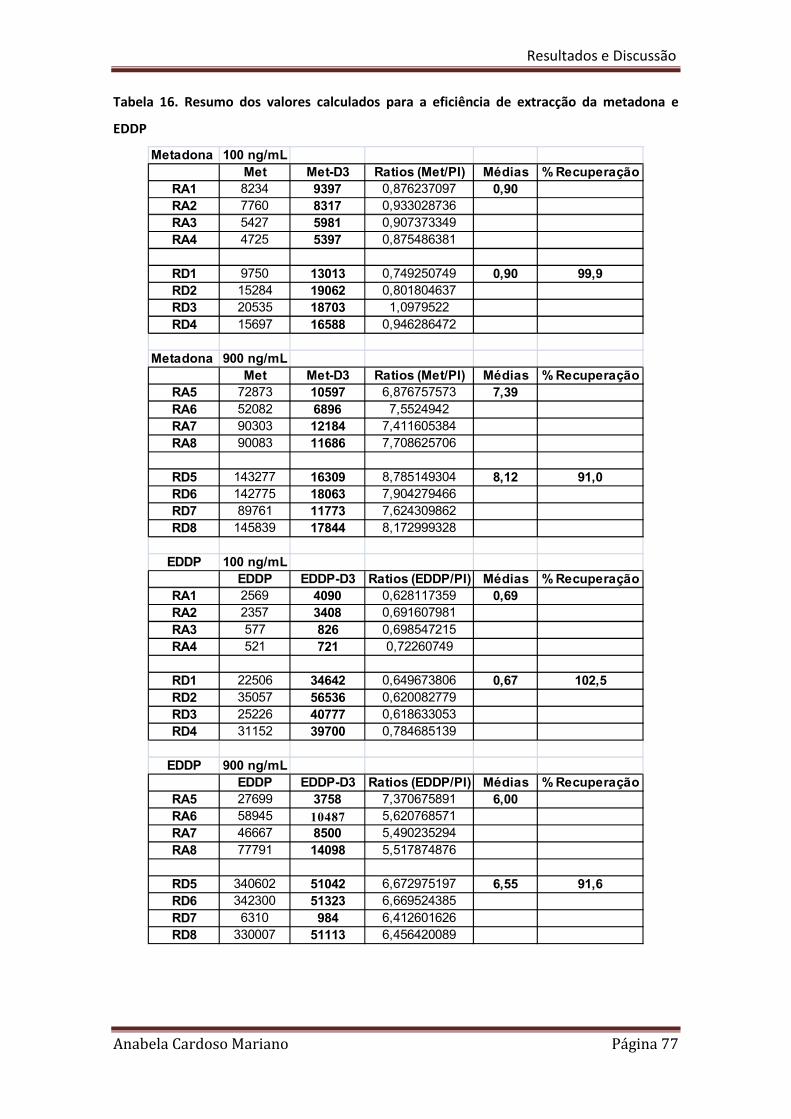
Resultados e Discussão
Anabela Cardoso Mariano Página 77
Tabela 16. Resumo dos valores calculados para a eficiência de extracção da metadona e
EDDP
Metadona 100 ng/mL
Met Met-D3 Ratios (Met/PI) Médias % Recuperação
RA1 8234 9397 0,876237097 0,90
RA2 7760 8317 0,933028736RA3 5427 5981 0,907373349RA4 4725 5397 0,875486381
RD1 9750 13013 0,749250749 0,90 99,9
RD2 15284 19062 0,801804637RD3 20535 18703 1,0979522RD4 15697 16588 0,946286472
Metadona 900 ng/mL
Met Met-D3 Ratios (Met/PI) Médias % Recuperação
RA5 72873 10597 6,876757573 7,39
RA6 52082 6896 7,5524942RA7 90303 12184 7,411605384RA8 90083 11686 7,708625706
RD5 143277 16309 8,785149304 8,12 91,0
RD6 142775 18063 7,904279466RD7 89761 11773 7,624309862RD8 145839 17844 8,172999328
EDDP 100 ng/mL
EDDP EDDP-D3 Ratios (EDDP/PI) Médias % Recuperação
RA1 2569 4090 0,628117359 0,69
RA2 2357 3408 0,691607981RA3 577 826 0,698547215RA4 521 721 0,72260749
RD1 22506 34642 0,649673806 0,67 102,5
RD2 35057 56536 0,620082779RD3 25226 40777 0,618633053RD4 31152 39700 0,784685139
EDDP 900 ng/mL
EDDP EDDP-D3 Ratios (EDDP/PI) Médias % Recuperação
RA5 27699 3758 7,370675891 6,00
RA6 58945 10487 5,620768571RA7 46667 8500 5,490235294RA8 77791 14098 5,517874876
RD5 340602 51042 6,672975197 6,55 91,6
RD6 342300 51323 6,669524385RD7 6310 984 6,412601626RD8 330007 51113 6,456420089
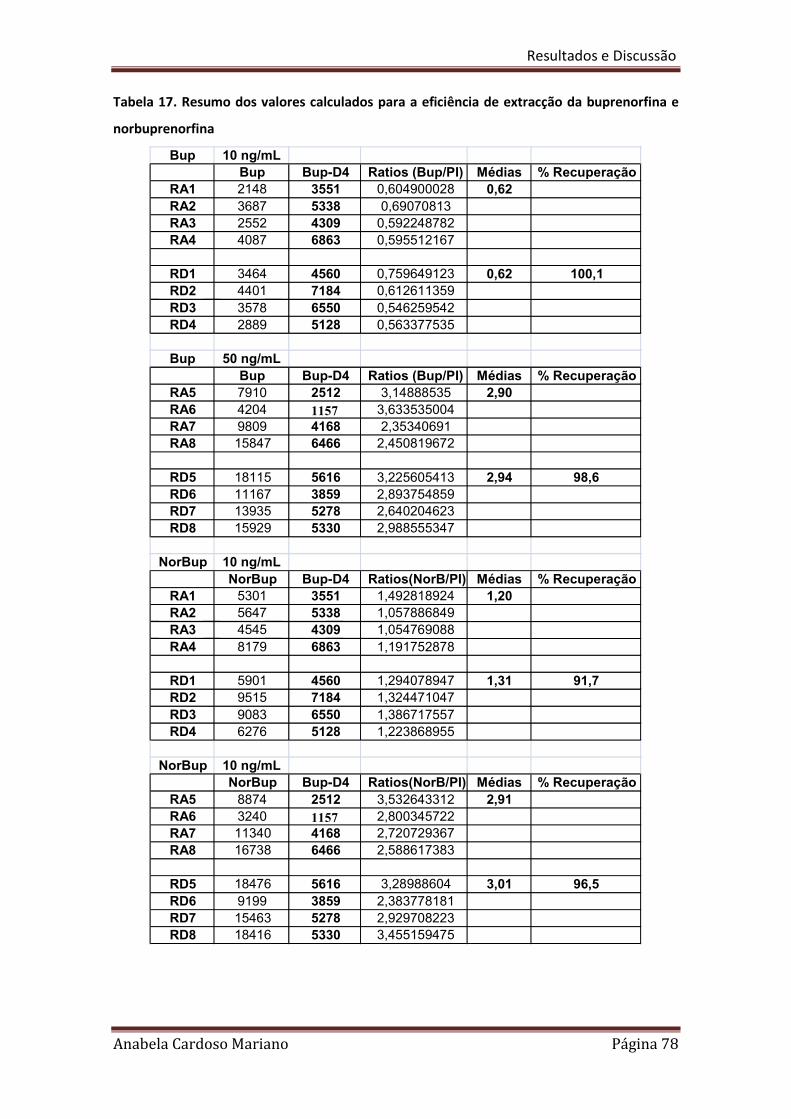
Resultados e Discussão
Anabela Cardoso Mariano Página 78
Tabela 17. Resumo dos valores calculados para a eficiência de extracção da buprenorfina e
norbuprenorfina
Bup 10 ng/mL
Bup Bup-D4 Ratios (Bup/PI) Médias % Recuperação
RA1 2148 3551 0,604900028 0,62 RA2 3687 5338 0,69070813 RA3 2552 4309 0,592248782RA4 4087 6863 0,595512167
RD1 3464 4560 0,759649123 0,62 100,1
RD2 4401 7184 0,612611359RD3 3578 6550 0,546259542RD4 2889 5128 0,563377535
Bup 50 ng/mL
Bup Bup-D4 Ratios (Bup/PI) Médias % Recuperação
RA5 7910 2512 3,14888535 2,90 RA6 4204 1157 3,633535004RA7 9809 4168 2,35340691 RA8 15847 6466 2,450819672
RD5 18115 5616 3,225605413 2,94 98,6
RD6 11167 3859 2,893754859RD7 13935 5278 2,640204623RD8 15929 5330 2,988555347
NorBup 10 ng/mL
NorBup Bup-D4 Ratios(NorB/PI) Médias % Recuperação
RA1 5301 3551 1,492818924 1,20 RA2 5647 5338 1,057886849RA3 4545 4309 1,054769088RA4 8179 6863 1,191752878
RD1 5901 4560 1,294078947 1,31 91,7
RD2 9515 7184 1,324471047RD3 9083 6550 1,386717557RD4 6276 5128 1,223868955
NorBup 10 ng/mL
NorBup Bup-D4 Ratios(NorB/PI) Médias % Recuperação
RA5 8874 2512 3,532643312 2,91 RA6 3240 1157 2,800345722RA7 11340 4168 2,720729367RA8 16738 6466 2,588617383
RD5 18476 5616 3,28988604 3,01 96,5
RD6 9199 3859 2,383778181RD7 15463 5278 2,929708223RD8 18416 5330 3,455159475

Resultados e Discussão
Anabela Cardoso Mariano Página 79
As percentagens de recuperação das substâncias estudadas situam-se entre 40 e 120%,
cumprindo os critérios em vigor, descritos no PO-STF-006 Rev00.
Linearidade/Gama de trabalho
Com o estudo da linearidade/gama de trabalho procura-se avaliar se, para um
determinado intervalo de concentrações, os sinais iónicos são directamente proporcionais às
concentrações usadas.
Foi preparada uma curva de calibração, de acordo com o PT-STF-C-211 para a
metadona e EDDP e de acordo com o PT-STF-C-212 para a buprenorfina e norbuprenorfina
(procedimentos internos do Serviço de Toxicologia Forense da Delegação do Centro), por
fortificação de alíquotas de 500 µL de sangue branco com soluções de trabalho a 0,5 e 5
µg/mL, entre 25 e 1000 ng/mL para a metadona e EDDP e com soluções de trabalho a 0,1
µg/mL, entre 5 e 50 ng/mL para a buprenorfina e norbuprenorfina. A todas as amostras foi
adicionado 25 µL de padrão interno a 2 µg/mL no caso da metadona e EEDP e a 0,5 µg/mL para
a buprenorfina e norbuprenorfina. Para os calibradores da recta de calibração foram
escolhidas concentrações equidistantes e distribuídas uniformemente entre o valor mais baixo
e o mais elevado do intervalo de concentrações de cada substância estudada (Tabela 18 e 19).
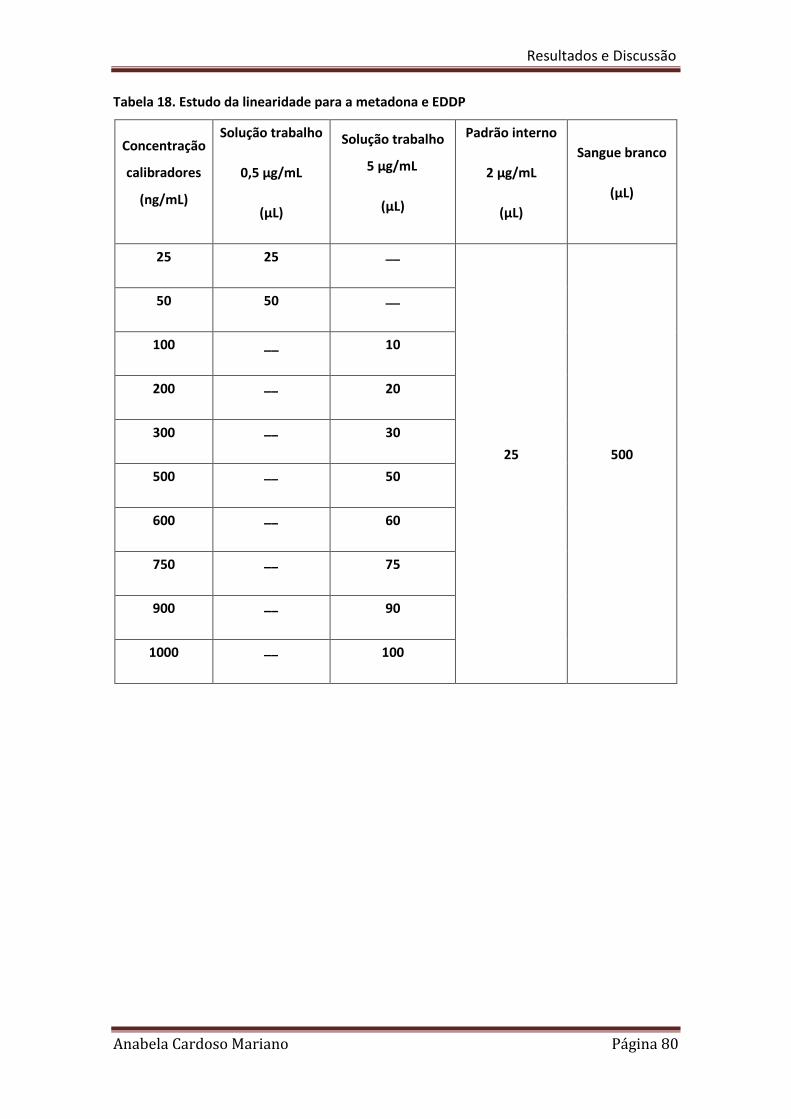
Resultados e Discussão
Anabela Cardoso Mariano Página 80
Tabela 18. Estudo da linearidade para a metadona e EDDP
Concentração
calibradores
(ng/mL)
Solução trabalho
0,5 µg/mL
(µL)
Solução trabalho
5 µg/mL
(µL)
Padrão interno
2 µg/mL
(µL)
Sangue branco
(µL)
25 25 __
25 500
50 50 __
100 __ 10
200 __ 20
300 __ 30
500 __ 50
600 __ 60
750 __ 75
900 __ 90
1000 __ 100
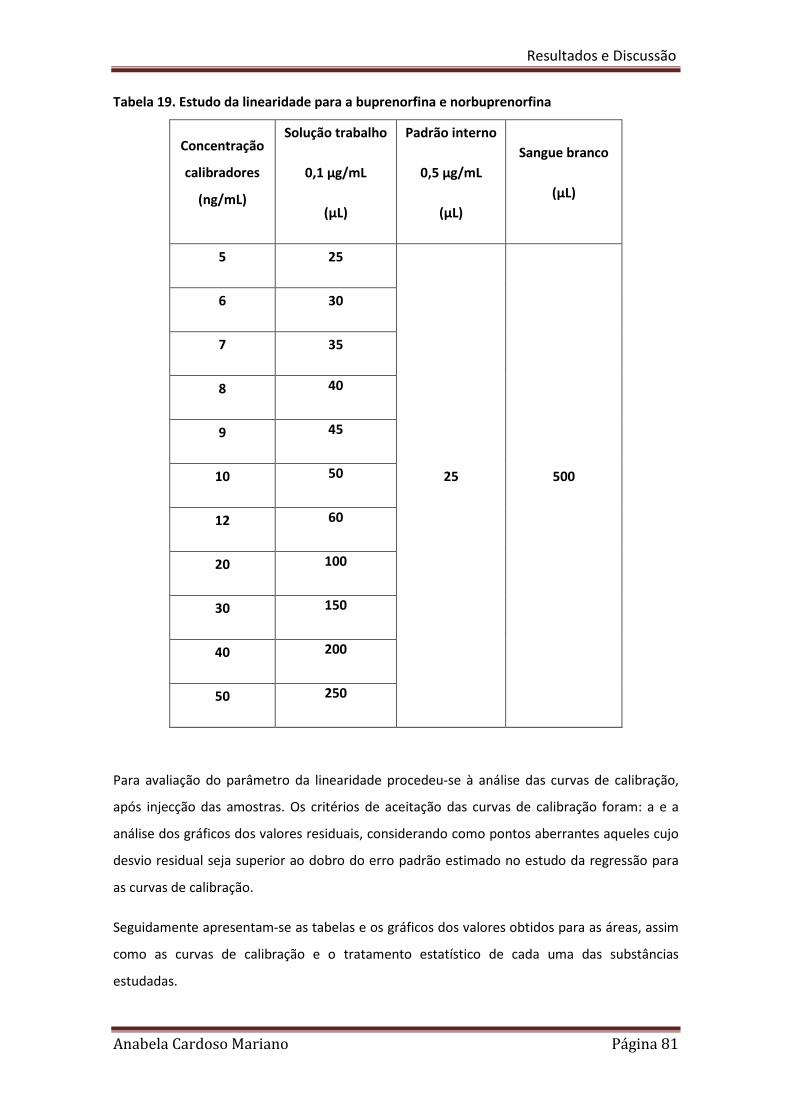
Resultados e Discussão
Anabela Cardoso Mariano Página 81
Tabela 19. Estudo da linearidade para a buprenorfina e norbuprenorfina
Concentração
calibradores
(ng/mL)
Solução trabalho
0,1 µg/mL
(µL)
Padrão interno
0,5 µg/mL
(µL)
Sangue branco
(µL)
5 25
25 500
6 30
7 35
8 40
9 45
10 50
12 60
20 100
30 150
40 200
50 250
Para avaliação do parâmetro da linearidade procedeu-se à análise das curvas de calibração,
após injecção das amostras. Os critérios de aceitação das curvas de calibração foram: a e a
análise dos gráficos dos valores residuais, considerando como pontos aberrantes aqueles cujo
desvio residual seja superior ao dobro do erro padrão estimado no estudo da regressão para
as curvas de calibração.
Seguidamente apresentam-se as tabelas e os gráficos dos valores obtidos para as áreas, assim
como as curvas de calibração e o tratamento estatístico de cada uma das substâncias
estudadas.
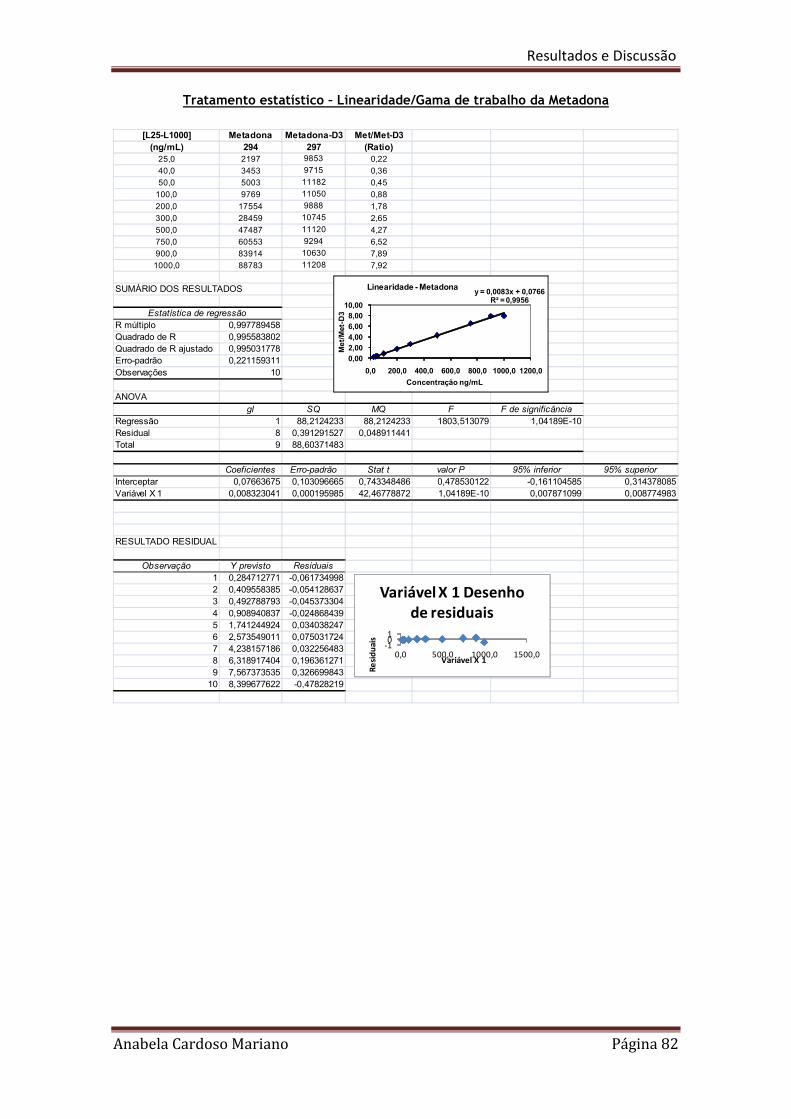
Resultados e Discussão
Anabela Cardoso Mariano Página 82
Tratamento estatístico – Linearidade/Gama de trabalho da Metadona
[L25-L1000] Metadona Metadona-D3 Met/Met-D3
(ng/mL) 294 297 (Ratio)
25,0 2197 9853 0,22
40,0 3453 9715 0,36
50,0 5003 11182 0,45
100,0 9769 11050 0,88
200,0 17554 9888 1,78
300,0 28459 10745 2,65
500,0 47487 11120 4,27
750,0 60553 9294 6,52
900,0 83914 10630 7,89
1000,0 88783 11208 7,92
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,997789458Quadrado de R 0,995583802Quadrado de R ajustado 0,995031778Erro-padrão 0,221159311Observações 10
ANOVAgl SQ MQ F F de significância
Regressão 1 88,2124233 88,2124233 1803,513079 1,04189E-10Residual 8 0,391291527 0,048911441Total 9 88,60371483
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar 0,07663675 0,103096665 0,743348486 0,478530122 -0,161104585 0,314378085Variável X 1 0,008323041 0,000195985 42,46778872 1,04189E-10 0,007871099 0,008774983
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,284712771 -0,0617349982 0,409558385 -0,0541286373 0,492788793 -0,0453733044 0,908940837 -0,0248684395 1,741244924 0,0340382476 2,573549011 0,0750317247 4,238157186 0,0322564838 6,318917404 0,1963612719 7,567373535 0,326699843
10 8,399677622 -0,47828219
y = 0,0083x + 0,0766R² = 0,9956
0,00
2,00
4,00
6,00
8,00
10,00
0,0 200,0 400,0 600,0 800,0 1000,0 1200,0
Met/Met-D3
Concentração ng/mL
Linearidade -Metadona
-101
0,0 500,0 1000,0 1500,0
Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
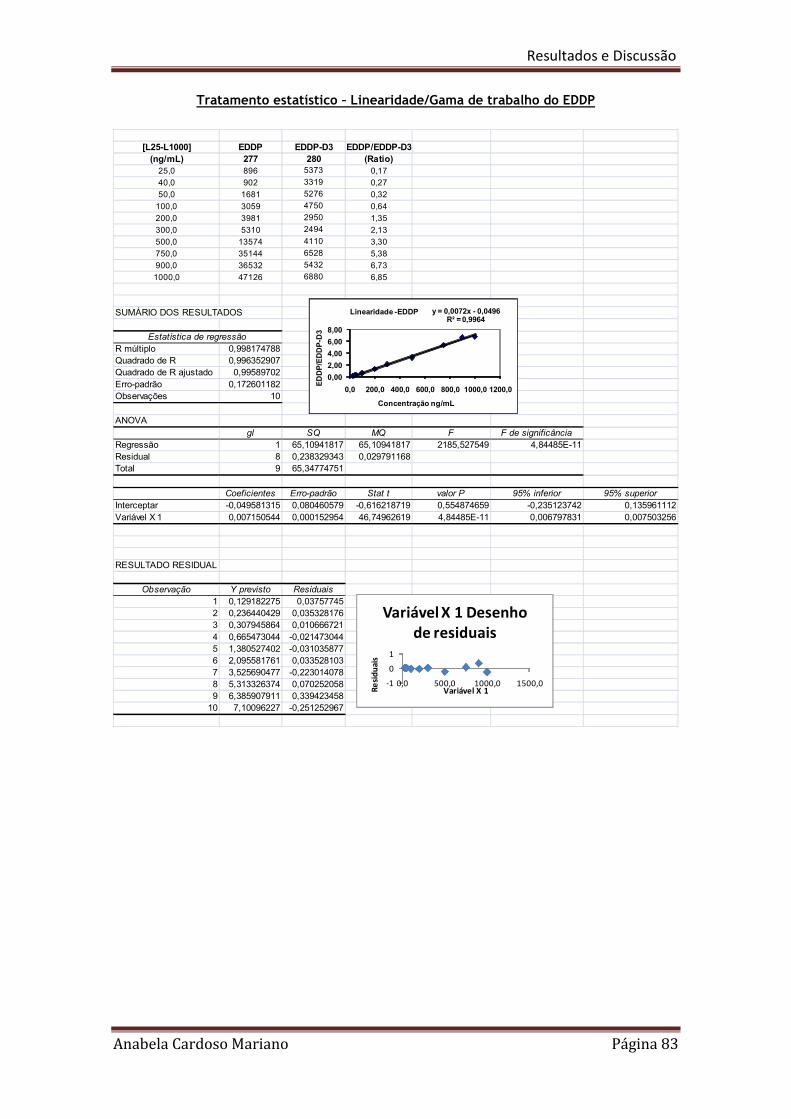
Resultados e Discussão
Anabela Cardoso Mariano Página 83
Tratamento estatístico – Linearidade/Gama de trabalho do EDDP
[L25-L1000] EDDP EDDP-D3 EDDP/EDDP-D3
(ng/mL) 277 280 (Ratio)
25,0 896 5373 0,17
40,0 902 3319 0,27
50,0 1681 5276 0,32
100,0 3059 4750 0,64
200,0 3981 2950 1,35
300,0 5310 2494 2,13
500,0 13574 4110 3,30
750,0 35144 6528 5,38
900,0 36532 5432 6,73
1000,0 47126 6880 6,85
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,998174788Quadrado de R 0,996352907Quadrado de R ajustado 0,99589702Erro-padrão 0,172601182Observações 10
ANOVAgl SQ MQ F F de significância
Regressão 1 65,10941817 65,10941817 2185,527549 4,84485E-11Residual 8 0,238329343 0,029791168Total 9 65,34774751
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar -0,049581315 0,080460579 -0,616218719 0,554874659 -0,235123742 0,135961112Variável X 1 0,007150544 0,000152954 46,74962619 4,84485E-11 0,006797831 0,007503256
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,129182275 0,037577452 0,236440429 0,0353281763 0,307945864 0,0106667214 0,665473044 -0,0214730445 1,380527402 -0,0310358776 2,095581761 0,0335281037 3,525690477 -0,2230140788 5,313326374 0,0702520589 6,385907911 0,339423458
10 7,10096227 -0,251252967
y = 0,0072x - 0,0496R² = 0,9964
0,00
2,00
4,00
6,00
8,00
0,0 200,0 400,0 600,0 800,0 1000,0 1200,0
EDDP/EDDP-D3
Concentração ng/mL
Linearidade -EDDP
-1
0
1
0,0 500,0 1000,0 1500,0
Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
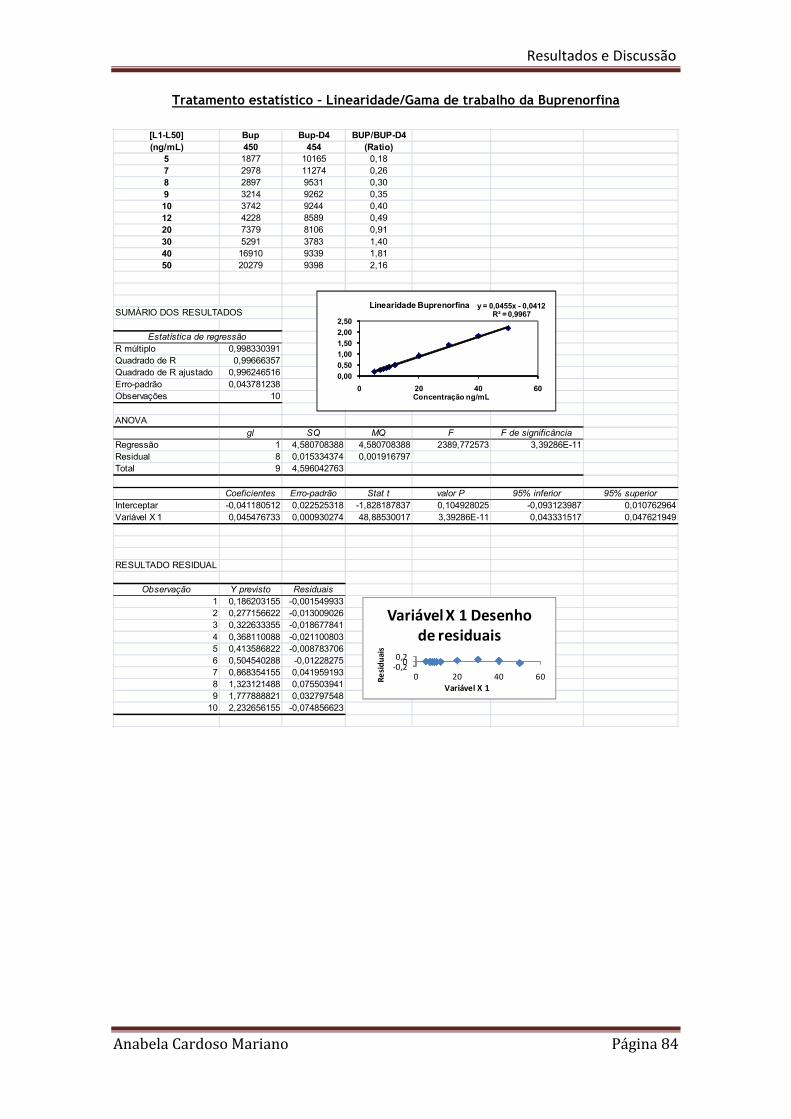
Resultados e Discussão
Anabela Cardoso Mariano Página 84
Tratamento estatístico – Linearidade/Gama de trabalho da Buprenorfina
[L1-L50] Bup Bup-D4 BUP/BUP-D4
(ng/mL) 450 454 (Ratio)
5 1877 10165 0,187 2978 11274 0,268 2897 9531 0,309 3214 9262 0,3510 3742 9244 0,4012 4228 8589 0,4920 7379 8106 0,9130 5291 3783 1,4040 16910 9339 1,8150 20279 9398 2,16
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,998330391Quadrado de R 0,99666357Quadrado de R ajustado 0,996246516Erro-padrão 0,043781238Observações 10
ANOVAgl SQ MQ F F de significância
Regressão 1 4,580708388 4,580708388 2389,772573 3,39286E-11Residual 8 0,015334374 0,001916797Total 9 4,596042763
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar -0,041180512 0,022525318 -1,828187837 0,104928025 -0,093123987 0,010762964Variável X 1 0,045476733 0,000930274 48,88530017 3,39286E-11 0,043331517 0,047621949
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,186203155 -0,0015499332 0,277156622 -0,0130090263 0,322633355 -0,0186778414 0,368110088 -0,0211008035 0,413586822 -0,0087837066 0,504540288 -0,012282757 0,868354155 0,0419591938 1,323121488 0,0755039419 1,777888821 0,032797548
10 2,232656155 -0,074856623
y = 0,0455x - 0,0412R² = 0,9967
0,00
0,50
1,00
1,50
2,00
2,50
0 20 40 60Concentração ng/mL
Linearidade Buprenorfina
-0,200,2
0 20 40 60Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
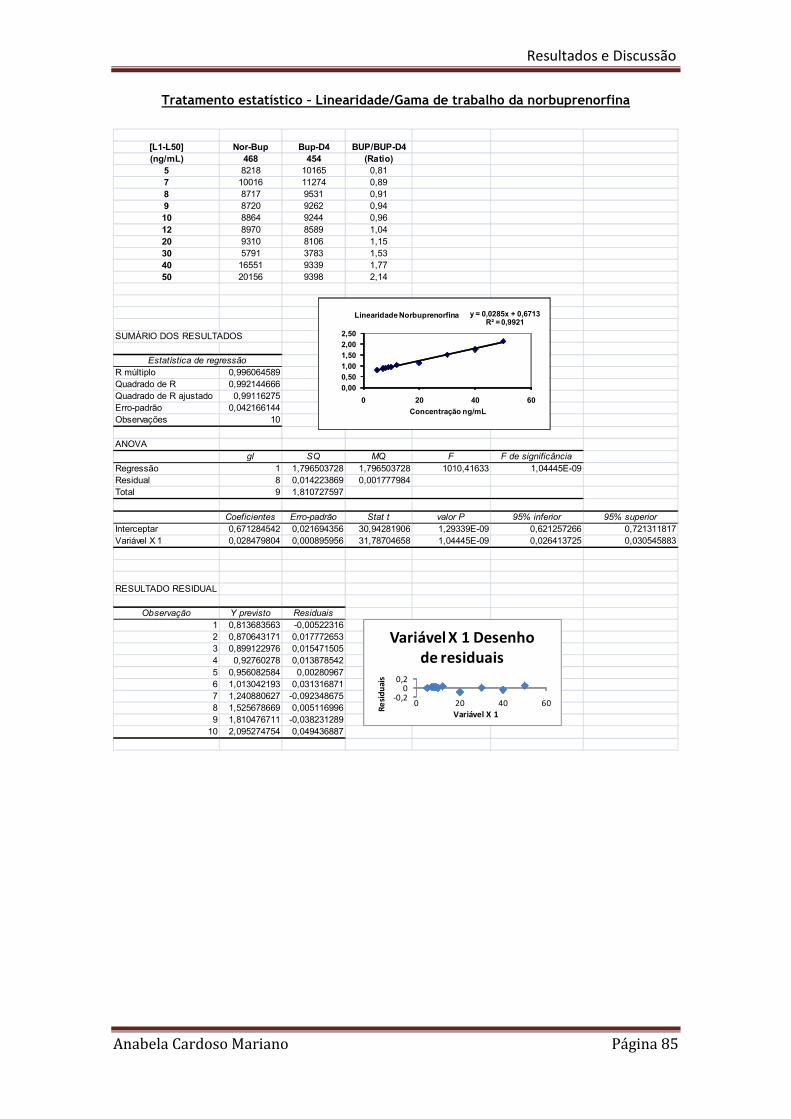
Resultados e Discussão
Anabela Cardoso Mariano Página 85
Tratamento estatístico – Linearidade/Gama de trabalho da norbuprenorfina
[L1-L50] Nor-Bup Bup-D4 BUP/BUP-D4
(ng/mL) 468 454 (Ratio)
5 8218 10165 0,817 10016 11274 0,898 8717 9531 0,919 8720 9262 0,9410 8864 9244 0,9612 8970 8589 1,0420 9310 8106 1,1530 5791 3783 1,5340 16551 9339 1,7750 20156 9398 2,14
SUMÁRIO DOS RESULTADOS
Estatística de regressão
R múltiplo 0,996064589Quadrado de R 0,992144666Quadrado de R ajustado 0,99116275Erro-padrão 0,042166144Observações 10
ANOVAgl SQ MQ F F de significância
Regressão 1 1,796503728 1,796503728 1010,41633 1,04445E-09Residual 8 0,014223869 0,001777984Total 9 1,810727597
Coeficientes Erro-padrão Stat t valor P 95% inferior 95% superior
Interceptar 0,671284542 0,021694356 30,94281906 1,29339E-09 0,621257266 0,721311817Variável X 1 0,028479804 0,000895956 31,78704658 1,04445E-09 0,026413725 0,030545883
RESULTADO RESIDUAL
Observação Y previsto Residuais
1 0,813683563 -0,005223162 0,870643171 0,0177726533 0,899122976 0,0154715054 0,92760278 0,0138785425 0,956082584 0,002809676 1,013042193 0,0313168717 1,240880627 -0,0923486758 1,525678669 0,0051169969 1,810476711 -0,038231289
10 2,095274754 0,049436887
y = 0,0285x + 0,6713R² = 0,9921
0,00
0,50
1,00
1,50
2,00
2,50
0 20 40 60
Concentração ng/mL
Linearidade Norbuprenorfina
-0,20
0,2
0 20 40 60
Re
sid
uai
s
Variável X 1
Variável X 1 Desenho de residuais
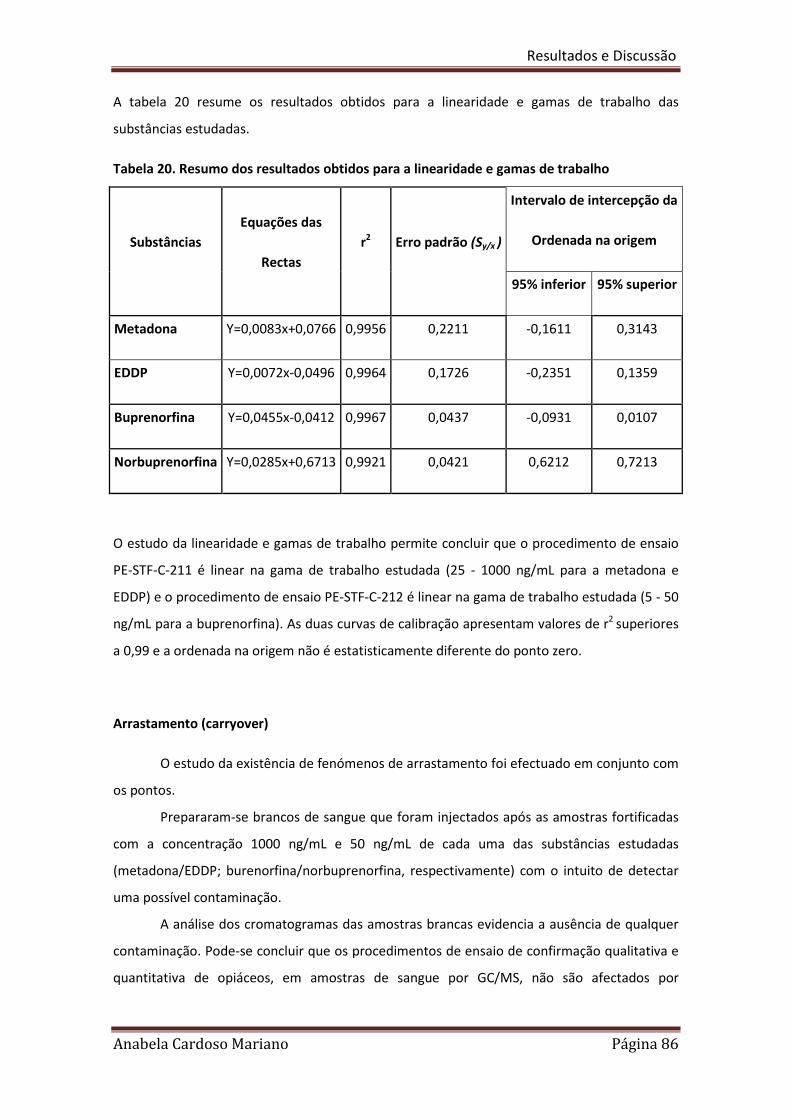
Resultados e Discussão
Anabela Cardoso Mariano Página 86
A tabela 20 resume os resultados obtidos para a linearidade e gamas de trabalho das
substâncias estudadas.
Tabela 20. Resumo dos resultados obtidos para a linearidade e gamas de trabalho
Substâncias
Equações das
Rectas
r2 Erro padrão (Sy/x )
Intervalo de intercepção da
Ordenada na origem
95% inferior 95% superior
Metadona Y=0,0083x+0,0766 0,9956 0,2211 -0,1611 0,3143
EDDP Y=0,0072x-0,0496 0,9964 0,1726 -0,2351 0,1359
Buprenorfina Y=0,0455x-0,0412 0,9967 0,0437 -0,0931 0,0107
Norbuprenorfina Y=0,0285x+0,6713 0,9921 0,0421 0,6212 0,7213
O estudo da linearidade e gamas de trabalho permite concluir que o procedimento de ensaio
PE-STF-C-211 é linear na gama de trabalho estudada (25 - 1000 ng/mL para a metadona e
EDDP) e o procedimento de ensaio PE-STF-C-212 é linear na gama de trabalho estudada (5 - 50
ng/mL para a buprenorfina). As duas curvas de calibração apresentam valores de r2 superiores
a 0,99 e a ordenada na origem não é estatisticamente diferente do ponto zero.
Arrastamento (carryover)
O estudo da existência de fenómenos de arrastamento foi efectuado em conjunto com
os pontos.
Prepararam-se brancos de sangue que foram injectados após as amostras fortificadas
com a concentração 1000 ng/mL e 50 ng/mL de cada uma das substâncias estudadas
(metadona/EDDP; burenorfina/norbuprenorfina, respectivamente) com o intuito de detectar
uma possível contaminação.
A análise dos cromatogramas das amostras brancas evidencia a ausência de qualquer
contaminação. Pode-se concluir que os procedimentos de ensaio de confirmação qualitativa e
quantitativa de opiáceos, em amostras de sangue por GC/MS, não são afectados por
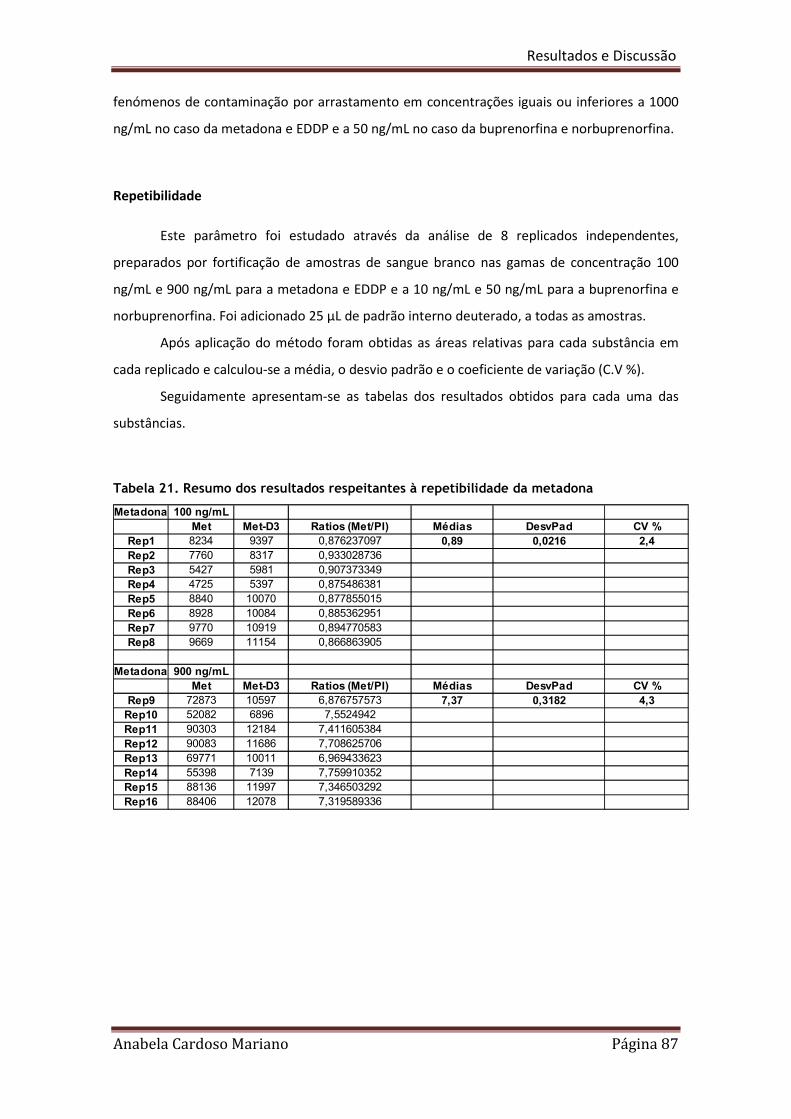
Resultados e Discussão
Anabela Cardoso Mariano Página 87
fenómenos de contaminação por arrastamento em concentrações iguais ou inferiores a 1000
ng/mL no caso da metadona e EDDP e a 50 ng/mL no caso da buprenorfina e norbuprenorfina.
Repetibilidade
Este parâmetro foi estudado através da análise de 8 replicados independentes,
preparados por fortificação de amostras de sangue branco nas gamas de concentração 100
ng/mL e 900 ng/mL para a metadona e EDDP e a 10 ng/mL e 50 ng/mL para a buprenorfina e
norbuprenorfina. Foi adicionado 25 µL de padrão interno deuterado, a todas as amostras.
Após aplicação do método foram obtidas as áreas relativas para cada substância em
cada replicado e calculou-se a média, o desvio padrão e o coeficiente de variação (C.V %).
Seguidamente apresentam-se as tabelas dos resultados obtidos para cada uma das
substâncias.
Tabela 21. Resumo dos resultados respeitantes à repetibilidade da metadona
Metadona 100 ng/mL
Met Met-D3 Ratios (Met/PI) Médias DesvPad CV %
Rep1 8234 9397 0,876237097 0,89 0,0216 2,4
Rep2 7760 8317 0,933028736Rep3 5427 5981 0,907373349Rep4 4725 5397 0,875486381Rep5 8840 10070 0,877855015Rep6 8928 10084 0,885362951Rep7 9770 10919 0,894770583Rep8 9669 11154 0,866863905
Metadona 900 ng/mL
Met Met-D3 Ratios (Met/PI) Médias DesvPad CV %
Rep9 72873 10597 6,876757573 7,37 0,3182 4,3
Rep10 52082 6896 7,5524942Rep11 90303 12184 7,411605384Rep12 90083 11686 7,708625706Rep13 69771 10011 6,969433623Rep14 55398 7139 7,759910352Rep15 88136 11997 7,346503292Rep16 88406 12078 7,319589336
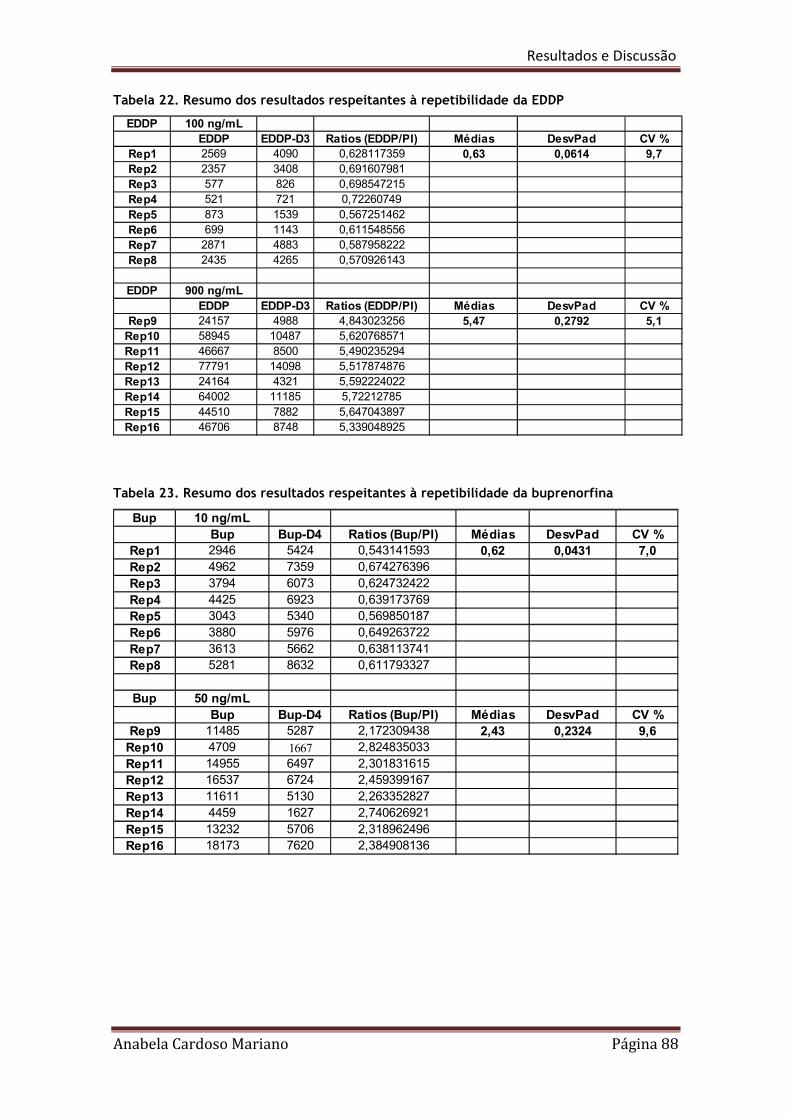
Resultados e Discussão
Anabela Cardoso Mariano Página 88
Tabela 22. Resumo dos resultados respeitantes à repetibilidade da EDDP
EDDP 100 ng/mL
EDDP EDDP-D3 Ratios (EDDP/PI) Médias DesvPad CV %
Rep1 2569 4090 0,628117359 0,63 0,0614 9,7
Rep2 2357 3408 0,691607981Rep3 577 826 0,698547215Rep4 521 721 0,72260749Rep5 873 1539 0,567251462Rep6 699 1143 0,611548556Rep7 2871 4883 0,587958222Rep8 2435 4265 0,570926143
EDDP 900 ng/mL
EDDP EDDP-D3 Ratios (EDDP/PI) Médias DesvPad CV %
Rep9 24157 4988 4,843023256 5,47 0,2792 5,1
Rep10 58945 10487 5,620768571Rep11 46667 8500 5,490235294Rep12 77791 14098 5,517874876Rep13 24164 4321 5,592224022Rep14 64002 11185 5,72212785Rep15 44510 7882 5,647043897Rep16 46706 8748 5,339048925
Tabela 23. Resumo dos resultados respeitantes à repetibilidade da buprenorfina
Bup 10 ng/mL
Bup Bup-D4 Ratios (Bup/PI) Médias DesvPad CV %
Rep1 2946 5424 0,543141593 0,62 0,0431 7,0
Rep2 4962 7359 0,674276396Rep3 3794 6073 0,624732422Rep4 4425 6923 0,639173769Rep5 3043 5340 0,569850187Rep6 3880 5976 0,649263722Rep7 3613 5662 0,638113741Rep8 5281 8632 0,611793327
Bup 50 ng/mL
Bup Bup-D4 Ratios (Bup/PI) Médias DesvPad CV %
Rep9 11485 5287 2,172309438 2,43 0,2324 9,6
Rep10 4709 1667 2,824835033Rep11 14955 6497 2,301831615Rep12 16537 6724 2,459399167Rep13 11611 5130 2,263352827Rep14 4459 1627 2,740626921Rep15 13232 5706 2,318962496Rep16 18173 7620 2,384908136
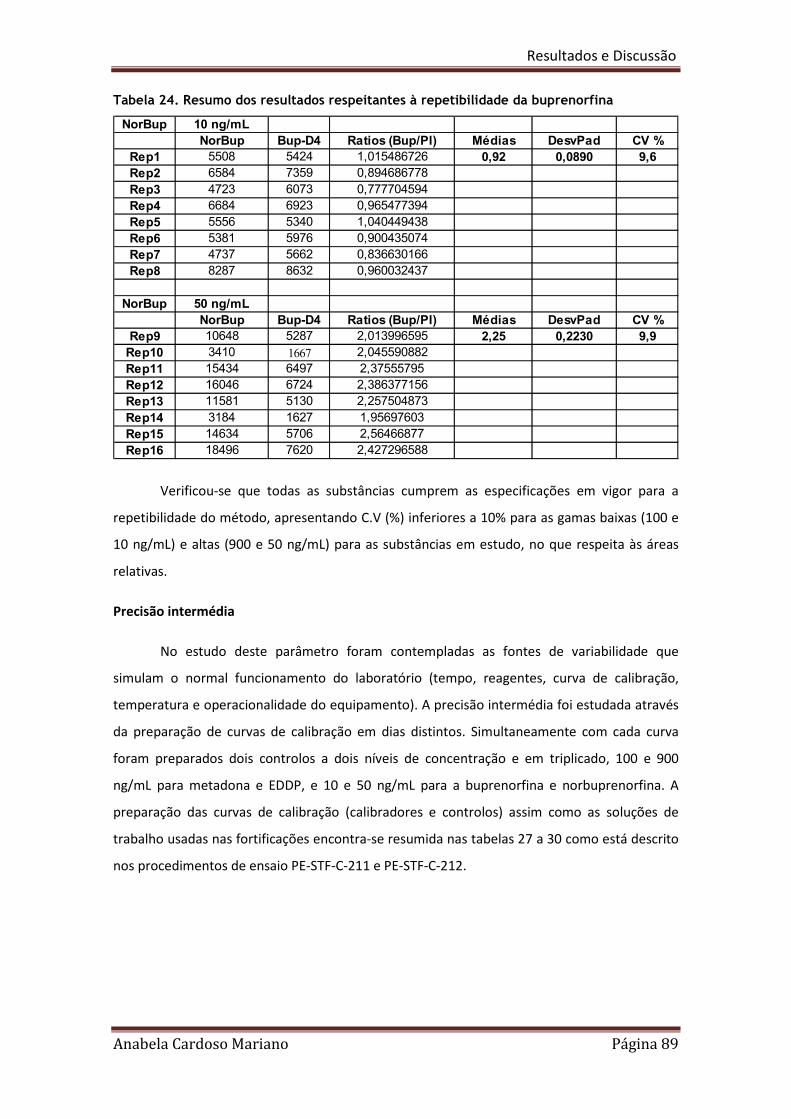
Resultados e Discussão
Anabela Cardoso Mariano Página 89
Tabela 24. Resumo dos resultados respeitantes à repetibilidade da buprenorfina
NorBup 10 ng/mL
NorBup Bup-D4 Ratios (Bup/PI) Médias DesvPad CV %
Rep1 5508 5424 1,015486726 0,92 0,0890 9,6
Rep2 6584 7359 0,894686778Rep3 4723 6073 0,777704594Rep4 6684 6923 0,965477394Rep5 5556 5340 1,040449438Rep6 5381 5976 0,900435074Rep7 4737 5662 0,836630166Rep8 8287 8632 0,960032437
NorBup 50 ng/mL
NorBup Bup-D4 Ratios (Bup/PI) Médias DesvPad CV %
Rep9 10648 5287 2,013996595 2,25 0,2230 9,9
Rep10 3410 1667 2,045590882Rep11 15434 6497 2,37555795Rep12 16046 6724 2,386377156Rep13 11581 5130 2,257504873Rep14 3184 1627 1,95697603Rep15 14634 5706 2,56466877Rep16 18496 7620 2,427296588
Verificou-se que todas as substâncias cumprem as especificações em vigor para a
repetibilidade do método, apresentando C.V (%) inferiores a 10% para as gamas baixas (100 e
10 ng/mL) e altas (900 e 50 ng/mL) para as substâncias em estudo, no que respeita às áreas
relativas.
Precisão intermédia
No estudo deste parâmetro foram contempladas as fontes de variabilidade que
simulam o normal funcionamento do laboratório (tempo, reagentes, curva de calibração,
temperatura e operacionalidade do equipamento). A precisão intermédia foi estudada através
da preparação de curvas de calibração em dias distintos. Simultaneamente com cada curva
foram preparados dois controlos a dois níveis de concentração e em triplicado, 100 e 900
ng/mL para metadona e EDDP, e 10 e 50 ng/mL para a buprenorfina e norbuprenorfina. A
preparação das curvas de calibração (calibradores e controlos) assim como as soluções de
trabalho usadas nas fortificações encontra-se resumida nas tabelas 27 a 30 como está descrito
nos procedimentos de ensaio PE-STF-C-211 e PE-STF-C-212.
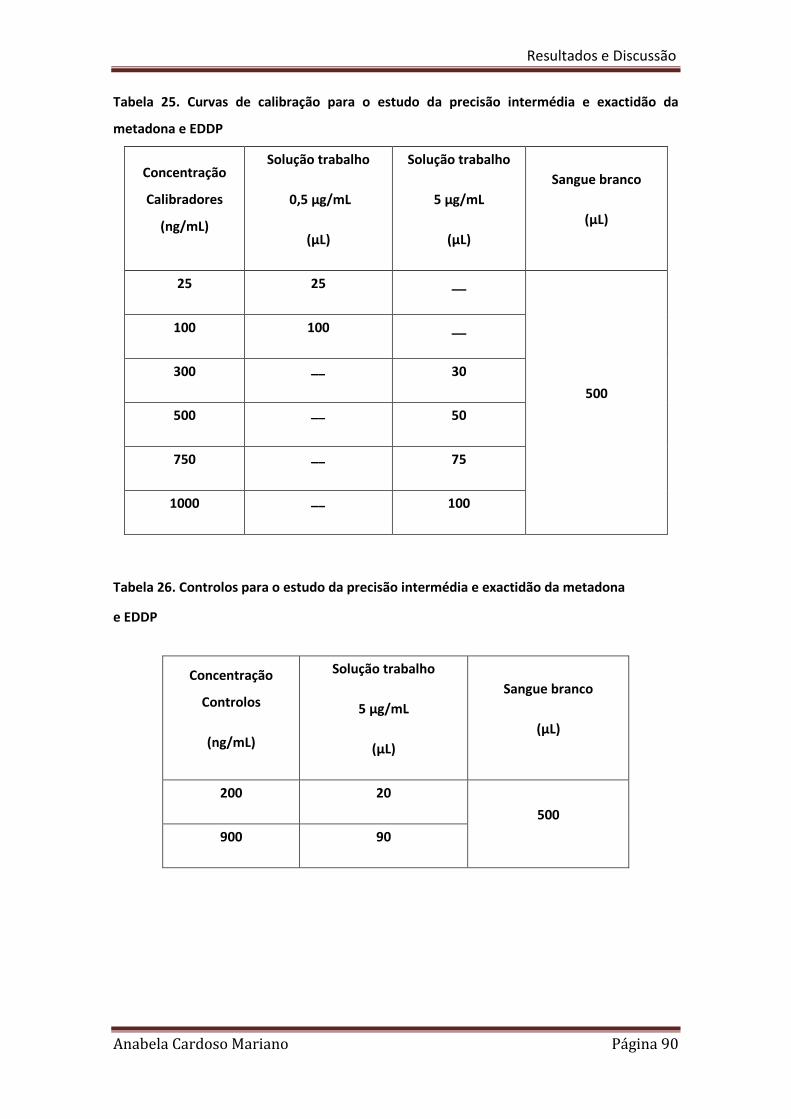
Resultados e Discussão
Anabela Cardoso Mariano Página 90
Tabela 25. Curvas de calibração para o estudo da precisão intermédia e exactidão da
metadona e EDDP
Concentração
Calibradores
(ng/mL)
Solução trabalho
0,5 µg/mL
(µL)
Solução trabalho
5 µg/mL
(µL)
Sangue branco
(µL)
25 25 __
500
100 100 __
300 __ 30
500 __ 50
750 __ 75
1000 __ 100
Tabela 26. Controlos para o estudo da precisão intermédia e exactidão da metadona
e EDDP
Concentração
Controlos
(ng/mL)
Solução trabalho
5 µg/mL
(µL)
Sangue branco
(µL)
200 20
500
900 90
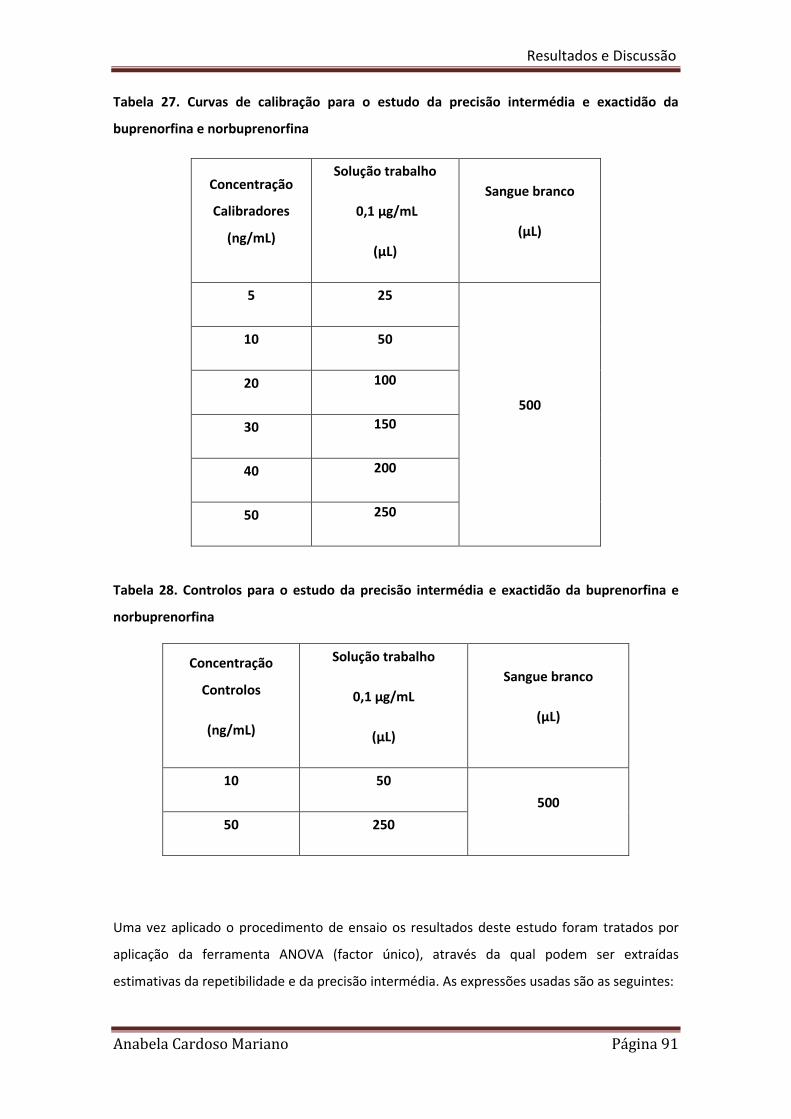
Resultados e Discussão
Anabela Cardoso Mariano Página 91
Tabela 27. Curvas de calibração para o estudo da precisão intermédia e exactidão da
buprenorfina e norbuprenorfina
Tabela 28. Controlos para o estudo da precisão intermédia e exactidão da buprenorfina e
norbuprenorfina
Uma vez aplicado o procedimento de ensaio os resultados deste estudo foram tratados por
aplicação da ferramenta ANOVA (factor único), através da qual podem ser extraídas
estimativas da repetibilidade e da precisão intermédia. As expressões usadas são as seguintes:
Concentração
Calibradores
(ng/mL)
Solução trabalho
0,1 µg/mL
(µL)
Sangue branco
(µL)
5 25
500
10 50
20 100
30 150
40 200
50 250
Concentração
Controlos
(ng/mL)
Solução trabalho
0,1 µg/mL
(µL)
Sangue branco
(µL)
10 50
500
50 250

Resultados e Discussão
Anabela Cardoso Mariano Página 92
Tabela 29. Tabela ANOVA (factor único)
Fonte Média Quadrática (MQ) Graus de liberdade
Entre grupos MSrun =
( )1
1
2
−
−∑=
p
xxnp
i
i
p-1
Dentro de grupo MSr =
)1(
)(1
2
1
−
−∑∑= =
np
xxp
i
iij
n
j
p(n-1)
Total
pn-1
Onde:
p é o número de sequências de análises para cada nível de concentração (uma sequência por
cada dia);
n é o número de replicados em cada sequência;
xij representa um replicado individual (replicado j) obtido na sequência i;
xi representa a média de n replicados obtidos na sequencia i ;
x é a média das médias de p sequências.

Resultados e Discussão
Anabela Cardoso Mariano Página 93
Tabela 30. Cálculo das estimativas da precisão
Precisão Expressão
Repetibilidade (Sr) Sr = SrM
Between-run precision
(Srun)
Srun = n
MSrMSrun −
Precisão intermédia (S1) S1 = ( ) ( )22
runrSS +
Foi estudado o comportamento do desvio padrão e do coeficiente de variação ao
longo das gamas de trabalho.
Se p desvio padrão for aproximadamente constante ao longo da gama de
concentração, pode ser estimado o desvio padrão como valor calculado por análise de todos
os resultados obtidos ao longo da gama de trabalho. É aplicado o teste F para avaliar a
homogeneidade de variâncias.
Se o coeficiente de variação for aproximadamente constante ao longo da gama de
trabalho, é aplicada a equação seguinte para calcular um coeficiente de variação ponderado:
CVpool =( ) ( )
( ) ( ) ...11
...11
21
2
22
2
1
+−+−
+−+−
nn
CVnCVn xx
Se, simultaneamente, o desvio padrão e o coeficiente de variação não forem constantes ao
longo da gama de trabalho, deve ser reportado, para cada gama de trabalho o respectivo
desvio padrão.
Seguidamente apresentam-se as tabelas dos resultados obtidos relativamente ao cálculo da
precisão intermédia para cada uma das substâncias individualmente.
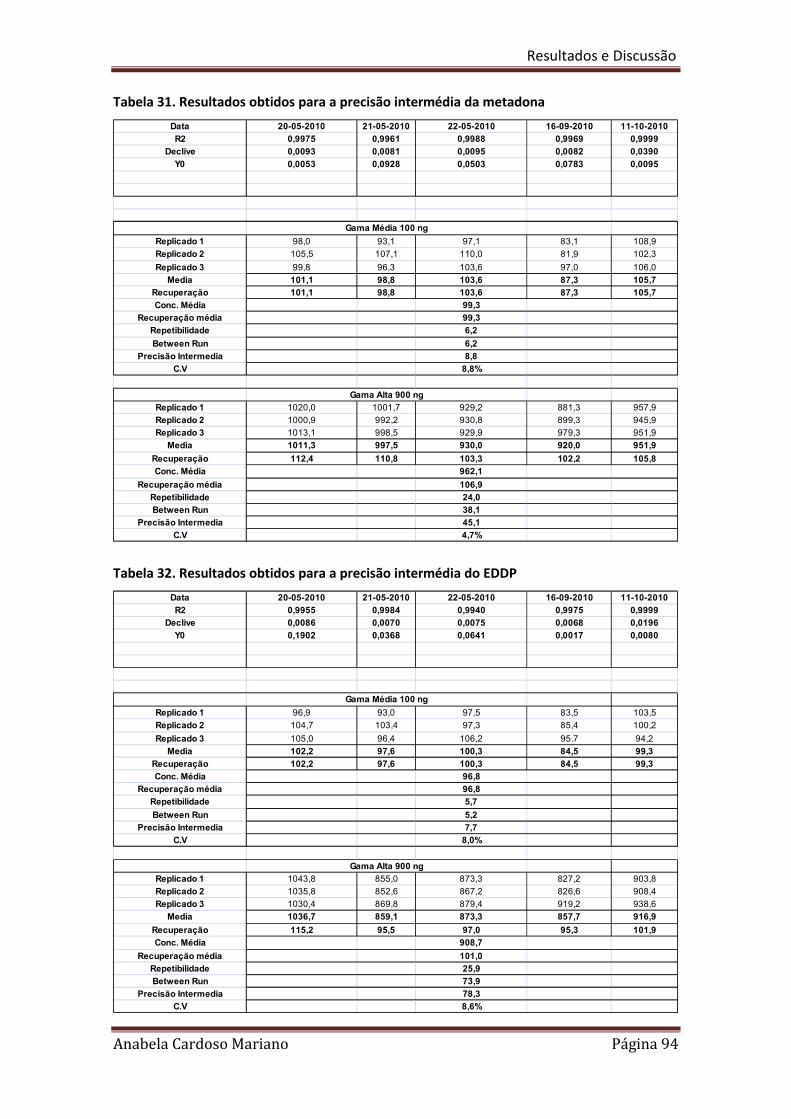
Resultados e Discussão
Anabela Cardoso Mariano Página 94
Tabela 31. Resultados obtidos para a precisão intermédia da metadona
Data 20-05-2010 21-05-2010 22-05-2010 16-09-2010 11-10-2010
R2 0,9975 0,9961 0,9988 0,9969 0,9999
Declive 0,0093 0,0081 0,0095 0,0082 0,0390
Y0 0,0053 0,0928 0,0503 0,0783 0,0095
Gama Média 100 ng
Replicado 1 98,0 93,1 97,1 83,1 108,9
Replicado 2 105,5 107,1 110,0 81,9 102,3
Replicado 3 99,8 96,3 103,6 97,0 106,0
Media 101,1 98,8 103,6 87,3 105,7
Recuperação 101,1 98,8 103,6 87,3 105,7
Conc. Média 99,3
Recuperação média 99,3
Repetibilidade 6,2
Between Run 6,2
Precisão Intermedia 8,8
C.V 8,8%
Gama Alta 900 ng
Replicado 1 1020,0 1001,7 929,2 881,3 957,9
Replicado 2 1000,9 992,2 930,8 899,3 945,9
Replicado 3 1013,1 998,5 929,9 979,3 951,9
Media 1011,3 997,5 930,0 920,0 951,9
Recuperação 112,4 110,8 103,3 102,2 105,8
Conc. Média 962,1
Recuperação média 106,9
Repetibilidade 24,0
Between Run 38,1
Precisão Intermedia 45,1
C.V 4,7%
Tabela 32. Resultados obtidos para a precisão intermédia do EDDP
Data 20-05-2010 21-05-2010 22-05-2010 16-09-2010 11-10-2010
R2 0,9955 0,9984 0,9940 0,9975 0,9999
Declive 0,0086 0,0070 0,0075 0,0068 0,0196
Y0 0,1902 0,0368 0,0641 0,0017 0,0080
Gama Média 100 ng
Replicado 1 96,9 93,0 97,5 83,5 103,5
Replicado 2 104,7 103,4 97,3 85,4 100,2
Replicado 3 105,0 96,4 106,2 95.7 94,2
Media 102,2 97,6 100,3 84,5 99,3
Recuperação 102,2 97,6 100,3 84,5 99,3
Conc. Média 96,8
Recuperação média 96,8
Repetibilidade 5,7
Between Run 5,2
Precisão Intermedia 7,7
C.V 8,0%
Gama Alta 900 ng
Replicado 1 1043,8 855,0 873,3 827,2 903,8
Replicado 2 1035,8 852,6 867,2 826,6 908,4
Replicado 3 1030,4 869,8 879,4 919,2 938,6
Media 1036,7 859,1 873,3 857,7 916,9
Recuperação 115,2 95,5 97,0 95,3 101,9
Conc. Média 908,7
Recuperação média 101,0
Repetibilidade 25,9
Between Run 73,9
Precisão Intermedia 78,3
C.V 8,6%
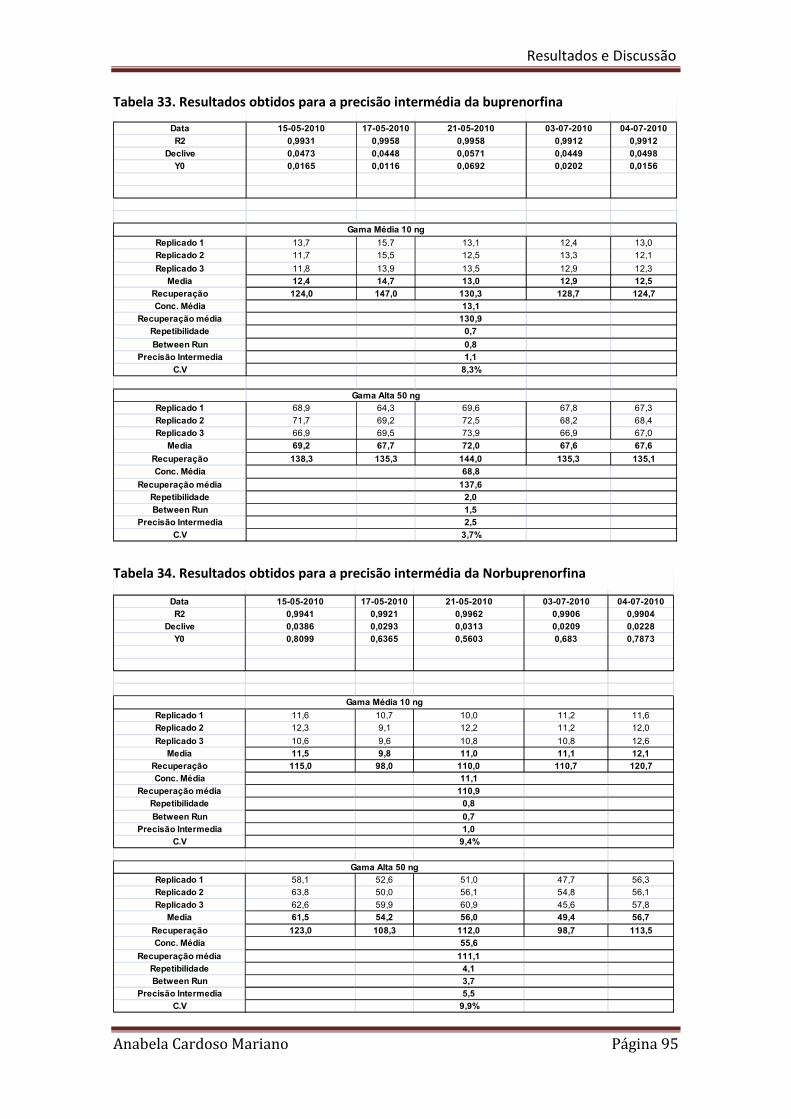
Resultados e Discussão
Anabela Cardoso Mariano Página 95
Tabela 33. Resultados obtidos para a precisão intermédia da buprenorfina
Data 15-05-2010 17-05-2010 21-05-2010 03-07-2010 04-07-2010
R2 0,9931 0,9958 0,9958 0,9912 0,9912
Declive 0,0473 0,0448 0,0571 0,0449 0,0498
Y0 0,0165 0,0116 0,0692 0,0202 0,0156
Gama Média 10 ng
Replicado 1 13,7 15.7 13,1 12,4 13,0
Replicado 2 11,7 15,5 12,5 13,3 12,1
Replicado 3 11,8 13,9 13,5 12,9 12,3
Media 12,4 14,7 13,0 12,9 12,5
Recuperação 124,0 147,0 130,3 128,7 124,7
Conc. Média 13,1
Recuperação média 130,9
Repetibilidade 0,7
Between Run 0,8
Precisão Intermedia 1,1
C.V 8,3%
Gama Alta 50 ng
Replicado 1 68,9 64,3 69,6 67,8 67,3
Replicado 2 71,7 69,2 72,5 68,2 68,4
Replicado 3 66,9 69,5 73,9 66,9 67,0
Media 69,2 67,7 72,0 67,6 67,6
Recuperação 138,3 135,3 144,0 135,3 135,1
Conc. Média 68,8
Recuperação média 137,6
Repetibilidade 2,0
Between Run 1,5
Precisão Intermedia 2,5
C.V 3,7%
Tabela 34. Resultados obtidos para a precisão intermédia da Norbuprenorfina
Data 15-05-2010 17-05-2010 21-05-2010 03-07-2010 04-07-2010
R2 0,9941 0,9921 0,9962 0,9906 0,9904
Declive 0,0386 0,0293 0,0313 0,0209 0,0228
Y0 0,8099 0,6365 0,5603 0,683 0,7873
Gama Média 10 ng
Replicado 1 11,6 10,7 10,0 11,2 11,6
Replicado 2 12,3 9,1 12,2 11,2 12,0
Replicado 3 10,6 9,6 10,8 10,8 12,6
Media 11,5 9,8 11,0 11,1 12,1
Recuperação 115,0 98,0 110,0 110,7 120,7
Conc. Média 11,1
Recuperação média 110,9
Repetibilidade 0,8
Between Run 0,7
Precisão Intermedia 1,0
C.V 9,4%
Gama Alta 50 ng
Replicado 1 58,1 52,6 51,0 47,7 56,3
Replicado 2 63,8 50,0 56,1 54,8 56,1
Replicado 3 62,6 59,9 60,9 45,6 57,8
Media 61,5 54,2 56,0 49,4 56,7
Recuperação 123,0 108,3 112,0 98,7 113,5
Conc. Média 55,6
Recuperação média 111,1
Repetibilidade 4,1
Between Run 3,7
Precisão Intermedia 5,5
C.V 9,9%

Resultados e Discussão
Anabela Cardoso Mariano Página 96
Exactidão
Neste parâmetro, a grandeza principal a determinar é o desvio entre o valor obtido e o
valor teórico aceite como exacto, de modo a investigar a existência de erros sistemáticos,
definir factores de correcção e se relevante estimar a incerteza associada à recuperação do
método U R como dado de entrada do procedimento de cálculo de incertezas. Através dos
resultados obtidos a partir da experiência de avaliação da precisão intermédia, pode-se
proceder ao cálculo de U R utilizando as seguintes equações:
Ri = Fortf
Obsi
C
C
R = p
Rp
i
i∑=1
tExp = RU
R
%
1−
U%R = P
SObs U R =
R
U R%
U R = R
)R -(1+)(U 22 R%
Onde,
Ri é a recuperação média de n replicados obtidos em condições de repetibilidade;
ObsS é o desvio padrão de p valores de Ri obtidos em condições de precisão intermédia;
tcrit é o valor valor crítico bi-caudal para p-1 graus de liberdade num intervalo de confiança de
95%.
Para cada nível foi aplicado um teste estatístico de forma a averiguar se a recuperação
média é significativamente diferente de 100%:
Se tExp < tcrit, então R não é significativamente diferente de 100% e U R é calculado de acordo
com a segunda equação. Neste caso, a incerteza é aumentada de forma a contemplar este
factor adicional de incerteza.
Nas tabelas seguintes apresenta-se os resultados obtidos relativos ao estudo da
exactidão.
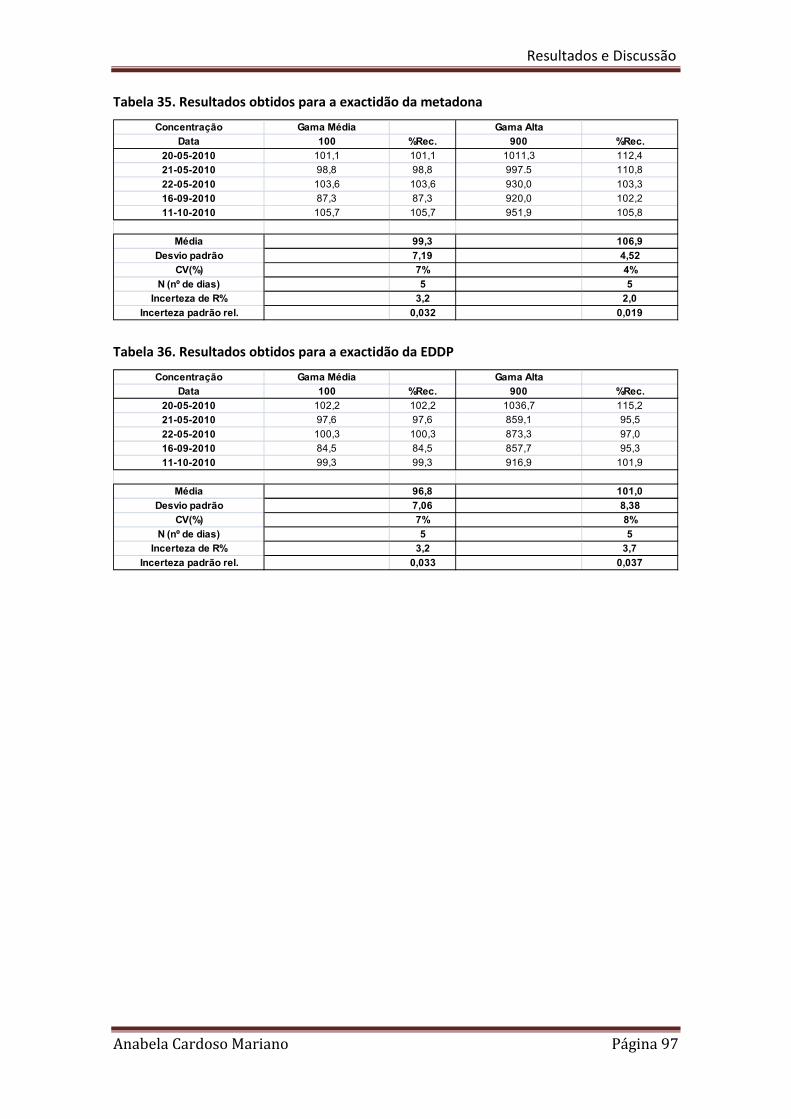
Resultados e Discussão
Anabela Cardoso Mariano Página 97
Tabela 35. Resultados obtidos para a exactidão da metadona
Concentração Gama Média Gama Alta
Data 100 %Rec. 900 %Rec.
20-05-2010 101,1 101,1 1011,3 112,4
21-05-2010 98,8 98,8 997.5 110,8
22-05-2010 103,6 103,6 930,0 103,3
16-09-2010 87,3 87,3 920,0 102,2
11-10-2010 105,7 105,7 951,9 105,8
Média 99,3 106,9
Desvio padrão 7,19 4,52
CV(%) 7% 4%
N (nº de dias) 5 5
Incerteza de R% 3,2 2,0
Incerteza padrão rel. 0,032 0,019
Tabela 36. Resultados obtidos para a exactidão da EDDP
Concentração Gama Média Gama Alta
Data 100 %Rec. 900 %Rec.
20-05-2010 102,2 102,2 1036,7 115,2
21-05-2010 97,6 97,6 859,1 95,5
22-05-2010 100,3 100,3 873,3 97,0
16-09-2010 84,5 84,5 857,7 95,3
11-10-2010 99,3 99,3 916,9 101,9
Média 96,8 101,0
Desvio padrão 7,06 8,38
CV(%) 7% 8%
N (nº de dias) 5 5
Incerteza de R% 3,2 3,7
Incerteza padrão rel. 0,033 0,037
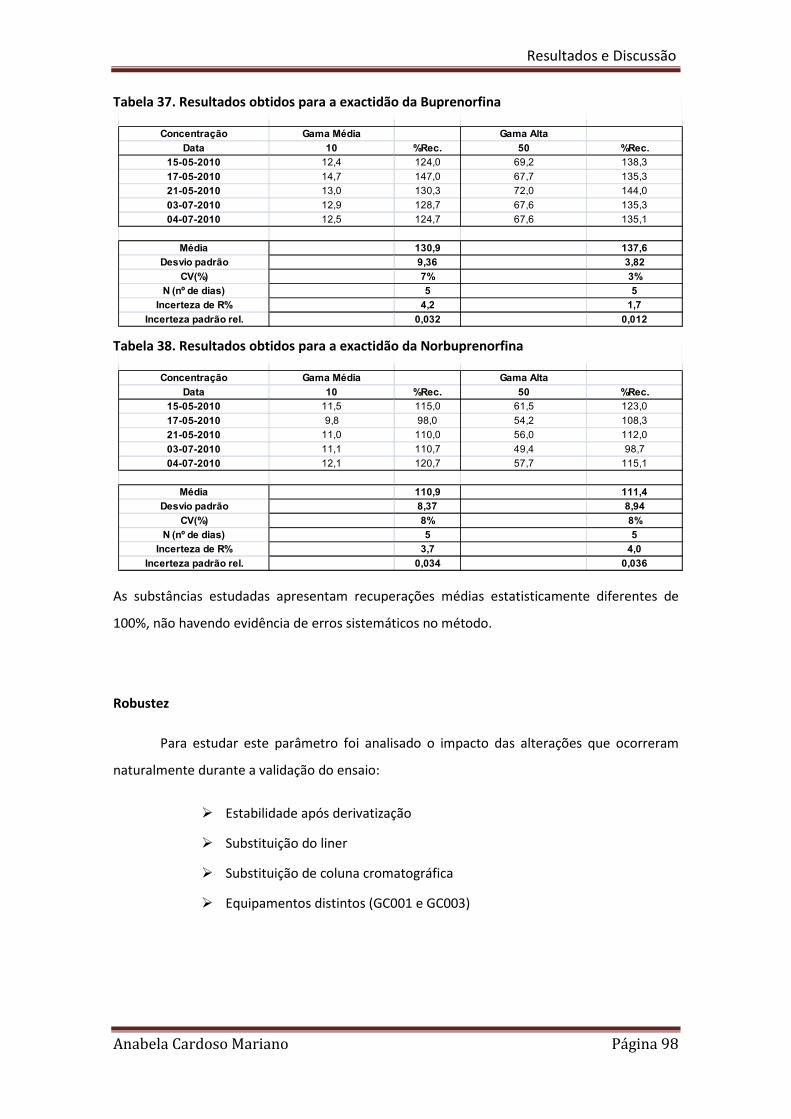
Resultados e Discussão
Anabela Cardoso Mariano Página 98
Tabela 37. Resultados obtidos para a exactidão da Buprenorfina
Concentração Gama Média Gama Alta
Data 10 %Rec. 50 %Rec.
15-05-2010 12,4 124,0 69,2 138,3
17-05-2010 14,7 147,0 67,7 135,3
21-05-2010 13,0 130,3 72,0 144,0
03-07-2010 12,9 128,7 67,6 135,3
04-07-2010 12,5 124,7 67,6 135,1
Média 130,9 137,6
Desvio padrão 9,36 3,82
CV(%) 7% 3%
N (nº de dias) 5 5
Incerteza de R% 4,2 1,7
Incerteza padrão rel. 0,032 0,012
Tabela 38. Resultados obtidos para a exactidão da Norbuprenorfina
Concentração Gama Média Gama Alta
Data 10 %Rec. 50 %Rec.
15-05-2010 11,5 115,0 61,5 123,0
17-05-2010 9,8 98,0 54,2 108,3
21-05-2010 11,0 110,0 56,0 112,0
03-07-2010 11,1 110,7 49,4 98,7
04-07-2010 12,1 120,7 57,7 115,1
Média 110,9 111,4
Desvio padrão 8,37 8,94
CV(%) 8% 8%
N (nº de dias) 5 5
Incerteza de R% 3,7 4,0
Incerteza padrão rel. 0,034 0,036
As substâncias estudadas apresentam recuperações médias estatisticamente diferentes de
100%, não havendo evidência de erros sistemáticos no método.
Robustez
Para estudar este parâmetro foi analisado o impacto das alterações que ocorreram
naturalmente durante a validação do ensaio:
� Estabilidade após derivatização
� Substituição do liner
� Substituição de coluna cromatográfica
� Equipamentos distintos (GC001 e GC003)

CONCLUSÕES


Conclusões
Anabela Cardoso Mariano Página 101
CONCLUSÕES
1. O procedimento para a obtenção de produtos derivados revelou ser bastante
apropriado, uma vez que obtivemos um derivado único e de formação rápida
não se tendo verificado reacções secundárias.
2. A metodologia analítica usada apresentou-se robusta.
3. O procedimento de validação adoptado na determinação das drogas de
substituição estudadas em sangue total, demonstrou ser eficaz uma vez que
obtivemos valores considerados aceitáveis em todos os parâmetros estudados.
4. O método desenvolvido mostrou-se adequado à determinação das substâncias
em estudo, tendo sido obtidos resultados aceitáveis (Z-Score <3) de acordo
com IT-STF-C-008, nas participações no programa de controlo de qualidade
externo: STM – Repalcement Drugs in serum and urine 2010.


REFERÊNCIAS


Referências
Anabela Cardoso Mariano Página 105
(1) Osswald W, Guimarães S. Bases farmacológicas, 4ª Edição. Porto Editora, 2001
(2) Gilman AG, Goodman LS, Gilman A: The Pharmacological Basis of Therapeutics: Opioid
Analgesics and Antagonists, 6th Edition. New York, Macmillan, 1980
(3) Brownstein, M J. A brief history of opiates, opioid peptides, and opiod receptors. Proc.
Natl. Acad. Sci. USA. Jun 1993; Vol 90, pp 5391-5393
(4) Booth, M. Opium – A History. St Martin’s Press. New York, 1998
(5) Golding, A M B. Two hundred years of drug abuse. Journal of royal society of medicine.
May 1993; vol 86 pp 282-286
(6) Gordon, N. O físico. Tradução: Aulyde Soares Rodrigues. Ed. Rocco. Rio de Janeiro 2000
(7) Methadone treatment of opiate addiction - Medical Staff Conference,University of
California, San Francisco. Calif Med 116:44-48, Feb 1972
(8) Shieds L B E, Hunsaker C, Corey T S, Ward M K, Stewart D. Methadone toxicity fatalities:
a review of medical examiner cases in a large metropolitan area. J Forensic Sci, 2007; 52:
1389–1395.
(9) Mercolini L, Mandrioli R, Conti M, Leonardi C, Gerra G, Raggi M A. Simultaneous
determination of methadone, buprenorphine and norbuprenorphine in biological fluids
for therapeutic drug monitoring purposes. Journal of Chromatography B, 2007; 847: 95–
102.
(10) Hendrée E. Jones, Practical Considerations for the Clinical Use of Buprenorphine. Science
& practice perspectives—August 2004.
(11) Polettini A, Huestis M A. Simultaneous determination of buprenorphine,
norbuprenorphine, and buprenorphine-glucuronide in plasma by liquid
chromatography-tandem mass spectrometry. Journal of Chromatography B, 2001;
754:447-459.
(12) Benjamin, L. Neurophysiological aspects. California medicine. San Francisco, 1957; vol
86
(13) Scott, J W. The physiological basis of pain. Canad. M A J. Jan 1959; vol 80
(14) Buchard A, Linnet K, Johansen S S, Munkholm J, Fregerslev M, Morling N. Postmortem
Blood Concentrations of R- and S-Enantiomers of Methadone and EDDP in Drug Users:
Influence of Co-Medication and P-glycoprotein Genotype J Forensic Sci, 2010 1-7.
(15) Armstrong S C, Wynn G H, Sandson N B. Pharmacokinetic Drug Interactionsof Synthetic
Opiate Analgesics. Psychosomatics 2009; 50:169–176.

Referências
Anabela Cardoso Mariano Página 106
(16) Sweetman S, editor. Martindale: The complete drug reference. 34th ed. London:
Pharmaceutical Press; 2004
(17) Substitution treatment; EMCDDA 2000 selected issue In EMCDDA 2000 Annual report on
the state of the drugs problem in the European Union [document online consultado em
2010 Jun 25] Disponível em: www.emcdda.europa.eu
(18) Garrido MJ, Troconiz IF - Methadone: a review of its pharmacokinetic/pharmacodynamic
properties. J Pahrmacol Toxicol, 1999; 42:61-66.
(19) Codd E, Shank R - Serotonin and norepinephrine uptake inhibiting activity of centrally
acting analgesics : structural determinants and role in antinociception. J Pharmacol Exp
Ther, 1995; 274:1263-1270.
(20) Pereira J, Lawlor P, Vigano A et al - Equianalgesic dose ratios for opioids. A critical review
and proposals for long-term dosing. J Pain Symptom Manage, 2001; 22:672-687.
(21) Kristensen K, Blemmer T, Angelo HR et al – Stereoselective pharmacokinetics of
methadone in chronic pain patients. Ther Drug Monit, 1996; 18:221-227.
(22) Crettol S, D_glon JJ, Besson J, Croquette-Krokkar M, Gothuey I, H_mmig R, et al.
Methadone enantiomer plasma levels, CYP2B6, CYP2C19, and CYP2C9 genotypes, and
response to treatment. Clin Pharmacol Ther 2005;78:593–604.
(23) Inturrisi, C E. Pharmacology of methadone and its isomers. Minerva anestesiol. 2005; 71;
453-7.
(24) Resumo das Características do Medicmento - Buprenorfina. [document online
consultado em 2010 Abr 13] Disponível em: www.emea.europa.eu
(25) Kharasch E D, Hoffer C, Whittington D, Walker A, Bedynek P S. Methadone
Pharmacokinetics Are Independent of Cytochrome P4503A (CYP3A) Activity and
Gastrointestinal Drug Transport. Anesthesiology 2009; 110:660–72.
(26) Choo R E, Jansson L M, Scheidweiler K, Huestis M A. Validated Liquid Chromatography
Atmospheric Pressure Chemical Ionization-Tandem Mass Spectrometric Method for the
Quantification of Methadone, 2-Ethylidene-1,5-dimethyl-3,3- diphenylpyrrolidine
(EDDP), and 2-Ethyl-5-methyl-3,3- diphenylpyroline (EMDP) in Human Breast Milk. J Anal
Toxicol. 2007 June ; 31(5): 265–269.
(27) Inturrisi CE, ColburnWA, Kaiko RF et al - Pharmacokinetics and pharmacodynamics of
methadone in patients with chronic pain. Clin Pharmacol Ther, 1987; 41:392-401.

Referências
Anabela Cardoso Mariano Página 107
(28) International Organization for Standardization. ISO 5725: 1994. Accuracy (trueness and
precison) of measurement method and results:
ISO 5725-1 Part 1: General principles and definitions.
ISO 5725-2 Part 2: Basic method for determination of repeatability and reproducibility
of a standard measurement method.
ISO 5725-3 Part 3: Intermediate measures of the precision of a standard measurement
method.
(29) Hartmann C, Smeyers-Verbeke J, Massart D L,McDowall R D. Validation of bioanalytical
chromatographic methods. J Pharm Biomed Anal 1998; 17: 193-218.
(30) Peters F T, Maurer H H. Bioanalytical method validation and its implications for forensic
and clinical toxicology – A review. Accredit Qual Assur 2002; 7: 441-449.
(31) Peters F T, Maurer H H. Bioanalytical method validation – How, how much and why? Bull
Int Assoc Forensic Toxicologists 2002; 32 (1): 16-23.
(32) Miller J C, Miller J N. Basic Statistical Methods for Analytical Chemistry. Part I. Statistics
of Repeated Measurements: a review. Analyst 1988; 113: 1351-1356.
(33) Rudy J L. Differentiating between sensitivity and limit of detection. Clin Chem 1989; 35:
509.
(34) Anderson D J, Rudy J L. Determination of the lower limit of detection. Clin Chem 1989;
35(10): 2152-2153.
(35) Nix A B J, Wilson D W. Assay detection limits: concept, definition, and estimation. Eur J
Pharmacol 1990; 39: 203-206.
(36) Needleman S B, Romberg R W. Limits of linearity and detection for some drugs of abuse.
J Anal Toxicol 1990; 14: 34-38
(37) Miller J N. Basic Statistical Methods for Analytical Chemistry. Part 2. Calibration and
Regression Methods. A Review. Analyst 1991;116: 3-59
(38) Karnes H T, Shiu G, Shah P V.Validation of Bioanalytical Methods. Pharm Research 1991;
8(4): 421-426.
(39) Lawson G M. Defining limit of detection and limit of quantification as applied to drug of
abuse testing: Striving for a consensus. Clin Chem 1994; 40(7): 1233-1238.
(40) Armbruster A, Tillman M D, Hubbs L M. Limit of detection (LOD)/Limit of quantification
(LOQ): Comparison of the empirical and the statistical methods exemplified with GC-MS
assays of abuse drugs. Clin Chem 1994; 40(7): 1233-1238

Referências
Anabela Cardoso Mariano Página 108
(41) Underwood P J, Kananen G E, Armitage E K. A pratical approach to determination of
laboratory GC-MS limits of detection. J Anal Toxicol 1997; 21: 12-16.
(42) Miller J N, Miller J C. Statistics and Chemometrics for Analytical Chemistry. 4th ed.
London: Prentice Hall; 2000 (ISBN: 0-130-22888-5)
(43) Jiménez C, Ventura R, Segura J. Validation of qualitative chromatograpic methods:
strategy in antidoping control laboratories. J Chromatogr B Analyt Technol Biomed Life
Sci 2002; 767: 341-351.
(44) Taracha E, Mierzejewski P, Lehner M, Chrapusta S J, Kata M, Lechowicz W, Hamed A,
Skórzewska A, Kostowski W, PlaŸnik A. Stress-opioid interactions: a comparison of
morphine and methadone. Pharmacological reports, 2009; 61: 424-435.
(45) Swartz, M. & Krull, I. (Ed.) Analytical Method Development. Marcel Dekker, NY. 1997
(46) The International Association of Forensic Toxicologists. TIAFT net – Therapeutic and
Toxic Drug Concentration List [Documento online] (consultado em 2010 Set 7)
www.tiaft.org
(47) Stout PR, Farrel LJ. Opioids – Effects on human performance and behaviour. Forensic Sci
Ver. 2002; 15:29
(48) Pirnay S, Borron SW, Giudicelli CP, Tourneau J, Baud FJ, Ricordel I. A critical review of the
causes of death among post-mortem toxicological investigations: analysis of 34
buprenorphine-associated and 35 methadone-associated deaths. Addiction. 2004;
99(8):978-88.
(49) United States Pharmacopeia XXIV, 200
(50) Morris Z, Ruthann K. Solid Phase Extraction for Sample Preparation. J.T. Baker. P. 3-42.
(51) Thurman E M, Mills M S. Solid-Phase Extraction: Principles and Practice. New York: John
Wiley & Sons, Inc., 1998.
(52) Sigma-Aldrich Co. Supleco. Guide to Solid Phase extraction. Bulletin 910; 1998.
(53) McDowall R D. Sample preparation of biomedical analysis. J Chromatograph 1989; 49: 3-
58.
(54) Russel G M F, Tan Y, Meijel J J M V, Gribnau F W J, Ginneken C A M V. Solid-Phase
Extraction of furosemide from plasma and urine and subsequent analysis by high-
performance liquid chromatography. J Chromatogr 1989; 496: 234-241.

Referências
Anabela Cardoso Mariano Página 109
(55) Franke J P, Zeeuw R A. Solid-phase extraction procedures in systemic toxicological
analysis. J Chromatogr B Biomed Sci Appl 1998; 713: 51-59
(56) Chen X-H, Franke J-P, Ensing K, Wijsbeek J, Zeeuw R A. Pitfalls and solutions in th
development of a fully automated solid-phase extraction method for drug screening
urposes in plasma and whole blood. J Anal Toxicol 1993; 17: 421-426.
(57) Zweipfening P G M, Wilderink A H C M, Horstihuis P, Franke J-P, Zeeuw R A. Toxicological
analysis of whole blood samples by means of Bond-Elut Certify columns and gas
chromatography with nitrogen-phosphorus detection J Chromatogr A 1994; 674: 87-95.
(58) Gerostamoulos J, Drummer O H. Solid phase extraction of morphine and its metabolites
from postmortem blood. Forensic Sci Int 1995; 77: 53-63.
(59) Herráez-Hernández R, Campíns-Falcó P, Sevillano-Cabeza A. Liquid Chromatographic
analysis of amphetamine and related compounds in urine using solid phase extraction
and 3,5-dinitrobenzoyl chloride for derivatzation. J Forensic Sci 1997; 35: 169-175.
(60) Jones C W, Chaney G, Mastorides S. Simultaneous analysis of opiates in urine by SPE and
GC-MS with stabilization of keto-opiates via conversion to oxime derivative. Abstract J
Anal Toxicol 1997; 21: 86.
(61) Allen D L, Scott K S, Oliver J S. Comparison of solid-phase extraction and supercritical
fluid extraction for the analysis of morphine in whole blood. J Anal. Toxicol 1999; 23:
216-218.
(62) Rashed A M, Anderson R A, King L A. Solid-phase Extraction for profiling of ecstasy
tablets. J Forensic Sci 2000; 45: 413-417.
(63) Weinmann W, Renz M, Vogt S, Pollak S. Automated solid-phase extraction and two-step
derivatization for simultaneous analysis of basic illicit drugs in serum by GC/MS. Int J
Legal Med 2000; 113: 229-235.
(64) Soriano T, Jurado C, Menéndez M, Reppeto M. Improved solid-phase extraction method
for systematic toxicological analysis in biological fluids. J Anal Toxicol 2001; 25: 137-143.
(65) Boland D M, Burke M F, Mitchell T, Madley P. Development of a generic method to the
solid-phase extraction of acidic compounds from complex matrices. J Anal Toxicol 2001;
25: 602-606.

Referências
Anabela Cardoso Mariano Página 110
(66) Martinez M A, Sánchez C T, Almarza E. Simultaneous determination of vilo9xazine,
imipramine, desipramine, sertraline and amoxapine in whole blood: Comparison of two
extraction/cleanup procedures for capillary gás chromatography with nitrogen-
phosphoru detection. J Anal Toxicol 2002; 276: 296-302.
(67) Stout P R, Gehlhausen J M, Horn C K, Klette K L. Evaluation of a solid-phase extraction
method for benzoylecgonine urine analysis in a high-thoughput forensic urine drug-
testing laboratory. J Anal Toxicol 2002; 26: 401-405.
(68) Stout P R, Horn C K, Klette K L. Rapid simultaneous determination of amphetamine,
methamphetamine, 3,4-methylenedioxyamphetamine, 3,4-
methylenedioxyethymethamphetamine and 3,4- ethylenedioxyethyamphetamine in
urine by solid phase extraction and GC-MS: A method optimized for high-volume
laboratories. J Anal Toxicol 2002; 26: 253-261.
(69) Martinez M A, Sánchez C T, Almarza E. A comparative solid-phase extraction study for
the simultaneous determination of fluoxetine, amitriptyline, nortriptyline, trimipramin,
maprotiline, clomipramine and trazodone in whole blood by capillary gás-liquid
chromatography with Nitrogen-Phosphorus detection. J Anal Toxicol 2003; 27: 353-358.
(70) Schütz H, Erdmann F, Verhoff M A, Wiler G. Pitfalls of toxiclogical analysis. Leg Med.
2003; 5: S6-S19.
(71) Cock K J S, Delbeke F T, De Boer D, Eenoo P V, Roels K. Quantification of 11-nor-Δ9-
tetrahydrocannabinol-9-carboxilic acid with GC-MS in urine collected for doping
analysis. J Anal Toxicol 2003; 27: 10-109.
(72) Knapp P K. Handbook of Analytical Derivatization reactions. New York: John Wiley &
Sons, In; 1979.
(73) Gunnar T, Eskole T, Lillsunde P. Fast gas chromatography/mass spectrometric assay for
the validated quantitative determination of methadone and the primary metabolite
EDDP in whole blood. Rapid Commun. Mass Spectrom. 2006; 20: 673–679.
(74) Nikolaou P D, Papoutsis I I, Atta-Politou J, Athanaselis S A, Spiliopoulou C A, Calokerinos
A C, Maravelias C P. Validated method for the simultaneous determination of
methadone and its main metabolites (EDDP and EMDP) in plasma of umbilical cord
blood by gas chromatography–mass spectrometry. Journal of Chromatography B, 2008;
867: 219–225.

Referências
Anabela Cardoso Mariano Página 111
(75) Kacinko S L, Jones H, Johnson R E, Choo R E, Concheiro-Guisan M, Huestis M A. Urinary
Excretion of Buprenorphine, Norbuprenorphine, Buprenorphine-Glucuronide, and
Norbuprenorphine- Glucuronide in Pregnant Women Receiving Buprenorphine
Maintenance Treatment. Clinical Chemistry, 2009; 55:61177–1187.
(76) Picard N, Cresteil T, Djebli N, Marquet P. In vitro metabolism study of buprenorphine:
evidence for new metabolic pathways. The American Society for Pharmacology and
Experimental Therapeutics, 2005; 33: 689–695.
(77) Fox E J, Tetlow V A, Allen K R. Quantitative Analysis of Buprenorphine and
Norbuprenorphine in Urine using Liquid Chromatography Tandem Mass Spectrometry.
Journal of Analytical Toxicology, 2006; 30: 238-244.
(78) Nikolaou P D, Papoutsis I I, Maravelias C P, Spiliopoulou C A, Pistos C M, Calokerinos A C,
Atta-Politou J. Development and Validation of an EI-GC–MS Method for the
Determination of Methadone and its Major Metabolites (EDDP and EMDP) in Human
Breast Milk. Journal of Analytical Toxicology, 2008; 32: 478-484.
(79) Virk M S, Arttamangkul S, Birdsong W T, Williams J T. Buprenorphine Is a Weak Partial
Agonist That Inhibits Opioid Receptor Desensitization. The Journal of Neuroscience, June
3, 2009; 29 (22):7341–7348.
(80) Galloway J H, Ashford M, Marsh I D, Holden M, Forrest A RW. A method for the
confirmation and identification of drugs of misuse in urine using solid phase extraction
and gas-liquid chromatography with mass spectrometry. Clin Pathol 1998; 51: 326-329.
(81) Larson M E M, Richards T M. Quantification of a Methadone Metabolite (EDDP) in Urine:
Assessment of Compliance. Clinical Medicine & Research, 2009; 7 - 4: 134-141.
(82) Gergov M, Nokua P, Vuori E, Ojanpera I. Simultaneous screening and quantification of
25 opioid drugs in post-mortem blood and urine by liquid chromatography–tandem
mass spectrometry. Forensic Science International, 2009; 186: 36–43.
(83) Moore C, Guzaldo F, Hussain M J, Lewis D. Determination of methadone in urine using
ion trap GC/MS in positive ion chemical ionization mode. Forensic Science International,
2001; 119: 155–160.
(84) Joya X, Pujadas M, Falcon M, Civit E, Garcia-Algar O, Vall O, Pichini S, Luna A, Torre R.
Gas chromatography–mass spectrometry assay for the simultaneous quantification of
drugs of abuse in human placenta at 12th week of gestation. Forensic Science
International 2010; 196: 38–42.

Referências
Anabela Cardoso Mariano Página 112
(85) Paterson S, Cordero R, Burlinson S. Screening and semi-quantitative analysis of post
mortem blood for basic drugs using gas chromatography/ion trap mass spectrometry.
Journal of Chromatography B, 2004; 813: 323–330.
(86) Wang G, Vincent M, Rodrigues W, Agrawal A, Moore C, Barhate R, Abolencia E, Coulter
C, Soares J, Zheng Y-F, Taylor C, Morjana N. Development and GC–MS Validation of a
Highly Sensitive Recombinant G6PDH-Based Homogeneous Immunoassay for the
Detection of Buprenorphine and Norbuprenorphine in Urine. Journal of Analytical
Toxicology, 2007; 31: 377-382.
(87) Yawney J, Treacy S, Hindmarsh KW, Burczynski FJ. A general screening method for acidic,
neutral and basic drugs in whole blood using the Oasis MCX column. J. Anal. Toxicol.
2002; 325-332.
(88) Concheiro M, Shakleya D M, Huestis M A. Simultaneous Quantification of
Buprenorphine, Norbuprenorphine, Buprenorphine Glucuronide and
Norbuprenorphine-Glucuronide in Human Umbilical Cord by Liquid Chromatography
Tandem Mass Spectrometry. Forensic Sci Int. 2009; 188(1-3): 144–151.
(89) Lisi AM, Kazlauskas R, Trout GJ. Gas chromatographic-mass spectrometric quantitation
of urinary buprenorphine and norbuprenorphine after derivatization by direct extractive
alkylation. J Chromatogr B Biomed Sci Appl. 1997 Apr 25; 692(1):67-77. (90) Regis Tecnologies, Inc. Chromatography GC Derivatization Agents. 2000 [document
online] (consultado em 2010 Mai 18) http://www.registech.com/Library/gcderrev.pdf
(91) Pirnay S, Bouchonnet S, Hervé F, Libong D, Milan N, Athis P, Baud F, Ricordel I.
Development and validation of a gas chromatography-mass spectrometry method for
the simultaneous determination of buprenorphine, flunitrazepam and their metabolites
in rat plasma: application to the pharmacokinetic study. Journal of Chromatography B,
2004; 807: 335–342.
(92) Juhascik M, Habbel S, Barron W, Behonick G. Validation of an ELISA method for screenin
methadone in postmortem blood. Journal of Analyticl Toxicology, 2006; 30: 617–620.
(93) Cheng P-S, Lee C-H, Liu C, Chien C-S. Simultaneous determination of ketamine, tramadol,
methadone, and their metabolites in urine by gas chromatography-mass spectrometry.
Journal of Analytical Toxicology, 2008; 32: 253–259.

Referências
Anabela Cardoso Mariano Página 113
(94) Oechsler S, Skopp G. Buprenorpine and major metabolites in blood specimens collected
for drug analysis in law enforcement purposes. Forensic Science International, 2010;
195: 73–77.
(95) Fucci N, Giovanni N, Scarlata S. Sweat testing in addicts under methadone treatment: an
Italian experience. Forensic Science International, 2008; 174: 107–110.
(96) Brunet B R, Barnes A J, Scheidweiler K B, Mura P, Huestis M A. Development and
validation of a solid-phase extraction gas chromatography-mass spectrometry method
for the simultaneous quantification of methadone, heroin, cocaine and metabolites in
sweat. Anal Bioanal Chem, 2008; 392: 115-127.
(97) Barroso M, Dias M, Vieira D N, López-Rivadulla M, Queiroz J A. Simultaneous
quantitation of morphine, 6-acetylmorphine, codeine, 6-acetylcodeine and tramadol in
hair using mixed-mode solid-phase extraction and gas chromatography–mass
spectrometry. Anal Bioanal Chem 2010; 396: 3059–3069
(98) Drummer O H. Postmortem toxicology of drugs of abuse. Forensic Science International,
2004; 142: 101–113.
(99) International Conference on Harmonization (ICH). Validation of analytical procedures:
Text and methodology ICH Q2 (R1). 1995. [document online consultado em 2010 Jun 30]
Disponível em: http://www.eudra.org/emea.html.
(100) Drummer O H, Opeskin K, Syrjanen M, Cordner S M. Methadone toxicity causing death
in ten subjects starting on a methadone maintenance program. The American Journl of
Forensic Medicine and Pathology, 1992; 13(4): 346-350.
(101) Musshoff F, Lachenmeieir D W, Madea B. Methadone substitution: medicolegal
problems in Germany. Forensic Science International, 2003; 133: 118–124.
(102) Cirimele V, Etienne S, Villain M, Ludes B, Kintz P. Evaluation of the One-Step ELISA kit for
the detection of buprenorphine in urine, blood, and hair specimens. Forensic Sci Int
2004; 143: 153–156.
(103) Tracqui A, Kintz P, Mangin P. HPLC-MS determination of buprenorphine and
norbuprenorphine in biological fluids and hair samples. J Forensic Sci 1997; 42(1): 111–
114.

Referências
Anabela Cardoso Mariano Página 114
(104) Favretto D, Frison G, Vogliardi S, Ferrara S D. Potentials of ion trap collisional
spectrometry for liquid chromatography/electrospray ionization tandem mass
spectrometry determination of buprenorphine and nor-buprenorphine in urine, blood
and hair samples. Rapid Commun Mass Spectrom 2006; 20: 1257–1265.
(105) Hoja H, Marquet P, Verneuil B, Lotfi H, Dupuy J L, Lachatre G. Determination of
buprenorphine and norbuprenorphine in whole blood by liquid chromatography-mass
spectrometry. J Anal Toxicol 1997; 21: 160–165.
(106) Goodwin R S, Wilkins D G, Averin O, Choo R E, Schroeder J R, Jasinski D R, Johnson R E,
Jones H E, Huestis M A. Buprenorphine and Norbuprenorphine in Hair of Pregnant
Women and Their Infants after Controlled Buprenorphine Administration. Clin Chem
2007; 53: 2136–2143.
(107) Wilkins D G, Rollins D E, Valdez A S, Mizuno A, Krueger G C, Cone E J. A retrospective
study of buprenorphine and norbuprenorphine in human hair after multiple doses. J
Anal Toxicol 1999; 23: 409–415.
(108) Kintz P. Determination of buprenorphine and its dealkylated metabolite in human hair. J
Anal Toxicol 1993; 17: 443–444.
(109) Thieme D, Sachs H, Thevis M. Formation of the N-methylpyridinium derivative to
improve the detection of buprenorphine by liquid chromatography-mass spectrometry.
J Mass Spectrom 2008; 43: 974–979.
(110) Crettol S, Déglon J J, Besson J, Croquette-Krokkar M, Gothuey I, Hämmig R, Monnat M,
Hüttemann H, Baumann P, Eap C B. Methadone enantiomer plasma levels, CYP2B6,
CYP2C19, and CYP2C9 genotypes, and response to treatment. Clin Pharmacol Ther. 2005
Dec; 78(6): 593-604.
(111) Eap C B, Broly F, Mino A, Hämmig R, Déglon J J, Uehlinger C, Meili D, Chevalley A F,
Bertschy G, Zullino D, Kosel M, Preisig M, Baumann P. Cytochrome P450 2D6 genotype
and methadone steady-state concentrations. J Clin Psychopharmacol. 2001 Apr; 21(2):
229-34.
(112) Eap C B, Bourquin M, Martin J, Spagnoli J, Livoti S, Powell K, Baumann P, Déglon J.
Plasma concentrations of the enantiomers of methadone and therapeutic response in
methadone maintenance treatment. Drug Alcohol Depend. 2000 Dec 22; 61(1): 47-54.

Referências
Anabela Cardoso Mariano Página 115
(113) Ansermot N, Albayrak O, Schläpfer J, Crettol S, Croquette-Krokar M, Bourquin M, Déglon
J J, Faouzi M, Scherbaum N, Eap C B. Substitution of (R,S)-methadone by (R)-methadone:
Impact on QTc interval. Arch Intern Med. 2010 Mar 22; 170(6): 529-36.
(114) Elkader A, Sproule B. Buprenorphine: clinical pharmacokinetics in the treatment of
opioid dependence. Clin Pharmacokinet. 2005; 44(7): 661-80.
(115) Wang Y S, Lin D L, Yang S C, Wu M Y, Liu R H, Su L W, Cheng P S, Liu C, Fuh M R. Issues
pertaining to the analysis of buprenorphine and its metabolites by gas chromatography-
mass spectrometry. J Chromatogr A. 2010; 1217(10): 1688-94.
(116) Fuller D C. A simple gas chromatography-mass spectrometry procedure for the
simultaneous determination of buprenorphine and norbuprenorphine in human urine. J
Anal Toxicol. 2008; 32(8): 626-30.
(117) Wu C H, Yang S C, Wang Y S, Chen B G, Lin C C, Liu R H. Evaluation of various
derivatization approaches for gas chromatography-mass spectrometry analysis of
buprenorphine and norbuprenorphine. J Chromatogr A. 2008; 1182(1): 93-112.
(118) Liu D X, He J N. A study in extraction method of buprenorphine in urine. Fa Yi Xue Za Zhi.
2007 Aug; 23(4): 292-4.
(119) Gunnar T, Ariniemi K, Lillsunde P. Validated toxicological determination of 30 drugs of
abuse as optimized derivatives in oral fluid by long column fast gas
chromatography/electron impact mass spectrometry. J Mass Spectrom. 2005; 40(6):
739-53.
(120) Jones G. Postmortem Toxicology. In: Moffat A C, Osselton M D, Widdop B, editors.
Clarke’s Analysis of Drug and Poison. 3 rd ed. London: Pharmaceutical Press, 2004. Vol. 1.
P. 94-108.
(121) Ehrsson H. Quantitative gas chromatography determination of carboxylic acids and
phenols after derivatization with pentafluorobenzyl bromide. Act Pharm Suec 1971; 8:
113-118.
(122) Koshy K T, Kaiser D G, Silk Van Der AL. O-(2,3,4,5,6-Pentafluorobenzyl) hydroxylamine
Hydrochloride as a sensitive derivatizing agent for the electron capture gas liquid
chromatographic analysis of keto steroids. J Chromatogr Sci 1975; 13: 97-103.
(123) Norn S, Kruse P R, Kruse E. History of opium poppy and morphine. Dan Medicinhist
Arbog. 2005; 33:171-84.
(124) Duarte D F. Opium and opioids: a brief history. Rev Bras Anestesiol. 2005; 55(1):135-46.

Referências
Anabela Cardoso Mariano Página 116
(125) Klockgether-Radke A P. F. W. Sertürner and the discovery of morphine. 200 years of pain
therapy with opioids. Anasthesiol Intensivmed Notfallmed Schmerzther. 2002;
37(5):244-9.
(126) Merskey H, Bogduk. Classification of chronic Pain. 2º Edition. IASP, Task Force on
Taxonomy. 1994.
(127) FDA’s Draft Guidance for Industry. Analytical Procedures and Methods Validation. 2000.
(Center for Drug Evaluation and Research. CDER).
(128) Wu C-H, Yang S-C, Wang Y-S, Chen B-G, Lin C-C, Liu R H. Evaluation of various
derivatization approaches for gas chromatography-mass spectrometry analysis of
buprenorphine and norbuprenorphine. Journal of Chromatography A. 2008; 1182: 93-
112.
(129) PG-STF-C-014 (Recomendações de segurança) (Procedimento interno do Serviço de
Toxicologia da Delegação do Centro)
(130) Baselt RC. Disposition of toxic drugs and chemicals in man. 7th ed. Foster City:
Biomedical Publications; 2004
(131) PE-STF-C-205 (Procedimento de ensaio de confirmação de metadona e EDDP)
(Procedimento interno do Serviço de Toxicologia da Delegação do Centro)
(132) PE-STF-C-206 (Procedimento de ensaio de confirmação de buprenorfina e buprenorfina)
(Procedimento interno do Serviço de Toxicologia da Delegação do Centro)
(133) PE-STF-C-211 (Procedimento de ensaio de quantificação de metadona e EDDP)
(Procedimento interno do Serviço de Toxicologia da Delegação do Centro)
(134) PE-STF-C-212 (Procedimento de ensaio de quantificação de buprenorfina e
norburenorfina) (Procedimento interno do Serviço de Toxicologia da Delegação do
Centro)
(135) PO-STF-C-006 (Procedimento operacional de validação de procedimentos de ensaio)
(Procedimento interno do Serviço de Toxicologia da Delegação do Centro)
(136) IT-STF-C-008 (Instrução para a avaliação de ensaios interlaboratoriais) (Procedimento
interno do Serviço de Toxicologia Forense da Delegação do Centro)
Referências
Anabela Cardoso Mariano Página 117
Referências elaboradas de acordo com:
United States National Library of Medicine: Bibliographic Services Division.
International Committee of Medical Journal Editors (Vancouver Group). Uniform
requirements for manuscripts submitted to biomedical journals: samples references
[documento online] [consultado em 2010 Set 7] Disponível em:
http://www.nlm.nih.gov/bsd/uniform_requirements.html