tallita cruz lopes tavares - repositorio.ufc.br · preocupa em perguntar como é que vão viver os...
TRANSCRIPT

TALLITA CRUZ LOPES TAVARES
ESTRUTURA E DIVERSIDADE TAXONÔMICA E FUNCIONAL DAS
ASSEMBLEIAS DE ARCHAEA E BACTERIA ASSOCIADAS A SEDIMENTOS
PORTUÁRIOS DA PLATAFORMA CONTINENTAL DO CEARÁ (NE - BRASIL)
FORTALEZA
2014
Tese de Doutorado apresentada ao Curso de Pós-
graduação em Ciências Marinhas Tropicais do Instituto de
Ciências do Mar da Universidade Federal do Ceará como
requisito parcial para obtenção do título de Doutor em
Ciências Marinhas Tropicais.
Orientadora: Profª Drª Vânia Maria Maciel Melo

Dados Internacionais de Catalogação na Publicação
Universidade Federal do Ceará
Biblioteca Rui Simões de Menezes
T233e Tavares, Tallita Cruz Lopes.
Estrutura e diversidade taxonômica e funcional das assembléias de archaea e bactéria associadas
a sedimentos portuários da plataforma continental do Ceará (NE – Brasil) / Tallita Cruz Lopes
Tavares. – 2014.
152f.: il. color., enc. ; 30 cm.
Tese (doutorado) – Universidade Federal do Ceará, Instituto de Ciências do Mar, Programa de
Pós-Graduação em Ciências Marinhas Tropicais, Fortaleza, 2014.
Área de Concentração: Utilização e Manejo de Ecossistemas Marinhos e Estuarinos.
Orientação: Profª. Drª. Vânia Maria Maciel Melo.
1. Portos marítimos. 2. Sedimentos. 3. Archaea. I. Título.
CDD 387.1


A minha família, dedico com amor.

AGRADECIMENTOS
Gostaria de agradecer a todos que de alguma forma contribuíram com este trabalho, não apenas
nesses quatro anos de aprendizado, sacrifício e desafios, mas também os que me levaram a
querer sempre mais da vida e a desejar me tornar uma Doutora.
Primeiramente, agradeço a meus pais. Um deles me olha com olhos que não posso mais ver,
apenas sentir, meu pai, João Eudes Vieira Tavares. Herdei seu espírito sonhador e vontade de
querer sempre mais da vida, a que lhe serei para sempre grata. Meu amor e saudade são
imensuráveis. Minha mãe me olha de perto com os olhos de uma guerreira capaz dos maiores
sacrifícios pela felicidade de sua família. Espero ter herdado uma parte, por menor que seja, de
sua bondade e vontade de ver um mundo onde reine a paz.
Agradeço também às minhas queridas irmãs, Thaís e Tainan Tavares, por quem tenho tanto
amor e zelo. Saibam que desejo mais bonanças para vocês do que sempre desejei para mim e
que essa minha conquista é nossa. São as brigas com a irmã estressada, o monopólio do
computador, as ausências em momentos de lazer... Espero que nossa união se preserve ao longo
dos anos.
Sou grata também a meu amigo e esposo, Leonardo Normando, a quem amo tanto. Pelos anos
de amadurecimento compartilhados e pela família que iniciamos. Você me faz querer ser
sempre melhor. Agradeço também pela assessoria 24 h desse “Mago do Excel e do Illustrator”
em assuntos ligados à edição de imagens, planilhas do Excel, Ecologia e correlatos.
Agradeço a minha orientadora, Dra. Vânia Melo, pela orientação desde os tempos de IC. Todas
as lições aprendidas me foram e sempre serão muito úteis. Por sempre me inspirar com todo
seu conhecimento e olhar sensível pela ciência.
Agradeço à Dra. Vanessa Nogueira, minha co-orientadora também dos tempos de IC, pelos
tempos de pesquisa com a dactylomelina-P, pela amizade e pela ajuda prestada sempre que
possível e necessária. Agradeço também por ter aceitado participar da defesa de minha tese.
Agradeço aos demais membros da banca por aceitarem participar dessa fase de minha jornada
rumo ao título de Doutora em Ciências Marinhas Tropicais.
Aos professores que me inspiraram desde os tempos de colégio, durante a graduação e pós-
graduação, meu muito obrigada. Seus modelos foram essenciais para o meu crescimento.
Alguns deles cito aqui: Prof. Gerardo Furtado, Prof. Cleudo, Prof. Nilo Aragão, Profa. Helena
Mathews-Cascon, Prof. Paulo Cascon, Profa. Ana de Fátima Urano, Prof. Rodrigo Maggione,
Profa. Cristina Rocha-Barreira, Prof. Tito Lotufo e Profa. Letícia Costa-Lotufo.
Aos amigos e companheiros que fiz no Instituto Federal de Educação, Ciência e Tecnologia do
Ceará, pela jornada compartilhada. Em especial aos Professores do Curso de Ciências
Biológicas do IFCE-Acarau e aos amigos do Lopes Residence Club, Amaurícia Lopes,
Amauricio Lopes e Ermelinda Lopes.

Aos meus alunos do Curso de Ciências Biológicas do IFCE-Acarau, pelo muito que me
ensinaram durante nossas aulas. Boa sorte nas suas jornadas.
Ao meu amigo do doutorado, Elthon Ferreira, o Engenheiro de Pesca mais biólogo que conheci.
Obrigada pelo carinho e por partilhar essa luta que é ser um estudante de pós-graduação nesse
país.
Aos amigos do Lembiotech, de hoje e do passado, e da Biotrends (Alysson Lira, Bárbara
Cibelle, Camila Porfírio, Denise Hissa, Geórgia Barguil, Gustavo Amaral, Hortência Barroso,
Júlio Ximenes, Kizeane Fajardo, Laís Feitosa, Lara Azevedo, Leonardo Normando, Lidianne
Leal, Luína Benevides, Luís Ribeiro, Lyanderson Aquino, Natália Falcão, Rafaela Barreto,
Samantha Pinheiro, Samuel Araujo, Sasha Gabriele, Tatiana Bomfim, Vanessa Câmara,
Vanessa Nogueira, Walderly Melgaço), agradeço pela ajuda, pela companhia, pela
compreensão, pela amizade, pelas discussões filosóficas e científicas e pelo apoio. Desejo a
vocês uma vida plena e muita força para seguir no caminho que escolherem.
Aos amigos das Ciências Biológicas e as minhas borboletas (Juliana Lucena, Tatyane Bandeira,
Mariana de Lima, Lívia Mendes e Kézia Lacerda), meu grande agradecimento pelos momentos
compartilhados de alegria (e também pelos de desespero diante de algumas provas!) Hoje está
todo mundo meio espalhado pelo mundo afora, em diferentes áreas da Biologia, da Economia,
do Direito ou da vida, mas para mim vocês sempre serão aquele povo da Biologiaufc2003.1 por
quem tenho tanto carinho.
Aos meus amigos de longa data e agregados, da Escola de Ballet Madiana Romcy para a vida
– Maria Lina Carvalho e Pedro Thomé; do Colégio GEO – Bruna Sales, Romário Fernandes,
Carolina Parente, Diego e Kassiane Maia, Igor Vieira, Mirella Parente, Paulo Eliézer e Rodrigo
Faria; e do Floresta - Juliana Brilhante, Luisa de Matos e Samara Lima, meus sinceros
agradecimentos por fazerem parte da minha vida há tanto tempo. Conhecer pessoas por longos
períodos tem suas desvantagens (MAIA, 2002), mas as vantagens são tantas que me faz querê-
los sempre por perto.
Aos órgãos financiadores sem os quais essa pesquisa não teria se concretizado e que
financiaram meus passos como pesquisadora em formação desde 2004 – FUNCAP, CNPq e
CAPES.
Ao financiamento proporcionado pelo Projeto CAPES-Ciências do Mar (Processo 0532/09).

"Estamos a destruir o planeta, e o egoísmo de cada geração não se
preocupa em perguntar como é que vão viver os que virão depois. A
única coisa que importa é o triunfo do agora. É a isto que eu chamo
a «cegueira da razão»."
José Saramago (El Cronista, Buenos Aires, 11 de Setembro de 1998)
Que meu triunfo presente não cegue meu futuro vindouro.

RESUMO
Este trabalho descreve a estrutura, diversidade e função das assembleias de Bacteria e Archaea
associadas aos sedimentos de dois portos na Plataforma Continental do Estado do Ceará e a
avaliação dos efeitos das diferenças arquitetônicas e operacionais portuárias sobre essas
comunidades. A estrutura das assembleias de Archaea e de Bacteria foram estudadas por
Eletroforese em Gel de Gradiente Desnaturante (DGGE), utilizando amostras obtidas em 10
estações de coleta localizadas no Porto do Pecém (São Gonçalo do Amarante) e 15 no Porto do
Mucuripe (Fortaleza). Após essa avaliação preliminar, as amostras foram reunidas em dois
pools e comparados com um pool de amostras de uma região sem atividade portuária (Icapui).
Foram analisados também a granulometria e teores de carbonato, matéria orgânica, nitrito,
sulfato, bifenilas, metais e hidrocarbonetos policíclicos aromáticos (HPAs) dos sedimentos. Os
perfis de DGGE foram comparados com base no índice de similaridade de Jaccard, estimadores
de riqueza e medidas de beta diversidade. As diversidades taxonômica e funcional associadas
aos sedimentos foram estudadas por pirossequencimento dos metagenomas e análises de
bioinformática. Os dados mostraram que os sedimentos diferiram quanto aos teores de
carbonato, matéria orgânica, metais (Cu, Cr, Ni, Zn e Fe), nitrito, nitrato e sulfato, que foram
mais altos no Porto do Pecém, e de carbono orgânico total, mais alto no Porto do Mucuripe. As
estruturas das assembleias de Archaea e de Bacteria foram diferentes nos dois portos, assim
como nas estações de coleta em cada porto, sendo a maior riqueza encontrada no Porto do
Mucuripe. As estimativas de beta diversidade mostraram que o Porto do Mucuripe é
espacialmente mais heterogêneo do que o Porto do Pecém. A comparação dos pools de amostras
portuárias com uma região não portuária sugere a seleção de unidades taxonômicas
operacionais peculiares em cada porto. A análise metagenômica mostrou a predominância de
Deltaproteobacteria e Actinobacteria no Porto do Pecém e Cyanobacteria, Bacteroidetes e
Chloroflexi no Porto do Mucuripe, e que ambos os portos não diferem funcionalmente. Para o
Domínio Archaea, destacou-se a dominância de Thaumarchaeota no Porto do Pecém. A peculiar
composição taxonômica do Porto do Mucuripe sugere que alterações decorrentes de impactos
acumulados por mais de 50 anos de funcionamento levaram à seleção dos táxons dominantes,
Synechococcus, Rhodothermus e Sphaerobacter, os quais podem vir a ser avaliados como
potenciais bioindicadores de sedimentos portuários cronicamente contaminados.
Palavras-chave: Portos marítimos. Sedimentos. DGGE. Metagenoma. Bacteria. Archaea.
Bioindicadores.

ABSTRACT
This research describes the structure, diversity and function of Bacteria and Archaea
assemblages associated to sediments from two seaports in the Continental shelf of Ceará state
and the evaluation of the effects of architectural and operational differences on those
communities. Structure and richness of Archaea and Bacteria assemblages were studied by
Denaturing Gradient Gel Electrophoresis (DDGE), using 10 samples obtained at Port of Pecém
and 15 at Port of Mucuripe. After this preliminary evaluation, the samples were pooled and
compared against an external pool from a region without seaport activities. The sediments also
had its granulometry, carbonate, organic matter, nitrite, nitrate, sulfate, metals, biphenyls and
polycyclic aromatic hydrocarbons (PAHs) contents analyzed. The DGGE profiles were
compared based on Jaccard similarity index, richness estimators and measures of beta diversity.
Finally, the taxonomic and functional diversities associated with the sediments were accessed
by pyrosequencing of the metagenomes. The sediments differed in relation to its amounts of
carbonate, organic matter, metals (Cu, Cr, Ni, Zn and Fe), nitrate, nitrite and sulfate, more
concentrated in Port of Pecém, as well as total organic carbon, more concentrated in Port of
Mucuripe. The structure of Archaea and Bacteria assemblages were different in both ports as
well as the sampling stations in each port, with a more elevate richness found on Port of
Mucuripe. Besides, beta diversity measures pointed Port of Mucuripe as a more heterogenic
environment than Port of Pecém. The comparison of the pooled samples suggested the selection
of peculiar operational taxonomic units in each port. The metagenomic analysis showed the
prevalence of Deltaproteobacteria and Actinobacteria in Port of Pecém, and Cyanobacteria,
Bacteroidetes and Chloroflexi in Port of Mucuripe, and that both ports did not differed
functionally. In Port of Pecém, the dominance of Thaumarchaeota stand out. The peculiar
taxonomic composition of Port of Mucuripe suggested that changes caused by the impacts
accumulated in more than 50 years of operation lead to the selection of dominant taxa,
Synechococcus, Rhodothermus and Sphaerobacter, which could be evaluated as potential
bioindicators of chronically contaminated sediments from ports.
Keywords: Seaports. Sediments. DGGE. Metagenome. Bacteria. Archaea. Bioindicators.

LISTA DE FIGURAS
FIGURA 1 - SUBDIVISÕES DO AMBIENTE MARINHO EM FUNÇÃO DA TOPOGRAFIA DOS FUNDOS, DA PROFUNDIDADE E
DA LUMINOSIDADE. ADAPTADO DE GARRISON (2011). ................................................................................. 28
FIGURA 2 - MAPA DA AMÉRICA DO SUL DEFININDO OS CINCO GRANDES ECOSSISTEMAS MARINHOS (LME):
PACÍFICO TROPICAL LESTE, EM AZUL; SISTEMA DA CORRENTE DE HUMBOLDT, EM LILÁS; ATLÂNTICO OESTE
TROPICAL, EM LARANJA; PLATAFORMA BRASILEIRA, EM VERDE; E PLATAFORMA DA PATAGÔNIA, EM ROSA.
(FONTE: MILOSLAVICH ET AL., 2011). .......................................................................................................... 32
FIGURA 3 – COMPOSIÇÃO DA COMUNIDADE MICROBIANA DE ACORDO COM OS TIPOS DE AMBIENTES. EM AZUL,
OBSERVA-SE O AMBIENTE PELÁGICO, E EM AMARELO O AMBIENTE BENTÔNICO. (A) FREQUÊNCIAS MÉDIA DAS
SEQUÊNCIAS PARA AS DEZ CLASSES MAIS ABUNDANTES. (B) PROPORÇÕES MÉDIAS DOS PRINCIPAIS GRUPOS
MICROBIANOS. O PREFIXO B REPRESENTA OS AMBIENTES BENTÔNICOS; E P, AMBIENTES PELÁGICOS. FONTE:
ZINGER ET AL., 2011. .................................................................................................................................. 35
FIGURA 4 - PROCESSOS GEOQUÍMICOS EM SEDIMENTOS COSTEIROS. O PERFIL VERTICAL MOSTRA UM GRADIENTE
ENTRE UMA ZONA OXIDADA E UMA ZONA REDUZIDA DETERMINADOS PELA DISPONIBILIDADE DE ACEPTORES
DE ELÉTRONS. AS ZONAS GEOQUÍMICAS NÃO SÃO MOSTRADAS COM ESCALA PARA MELHOR VISUALIZAÇÃO,
MAS REPRESENTAM O MÁXIMO DE 1 METRO DE PERFIL DE SEDIMENTO. FONTE: KIRCHMAN, 2012; ACOSTA-
GONZÁLEZ, 2013. ......................................................................................................................................... 39
FIGURA 5 - ESQUEMA CONCEITUAL DOS EFEITOS DE DISTÚRBIOS AMBIENTAIS SOBRE AS COMUNIDADES
MICROBIANAS MARINHAS. ADAPTADO DE NOGALES ET AL. (2011). .............................................................. 50
FIGURA 6 - REGIÕES CONSERVADAS E HIPERVARIADAS DO GENE DO RNA RIBOSSOMAL 16S. AS REGIÕES
CONSERVADAS (C1-C9) SÃO MOSTRADAS EM ROSA; E AS HIPERVARIADAS (V1-V9), EM DIFERENTES CORES.
EM DETALHE, PODE-SE OBSERVAR O TAMANHO DE UMA DAS REGIÕES HIPERVARIADAS (V4) COM SEUS SÍTIOS
DE LIGAÇÃO DE INICIADORES (FONTE: PETROSINO ET AL., 2009). ................................................................. 53
FIGURA 7 - DESENHO ESQUEMÁTICO ILUSTRANDO COMO O PROCEDIMENTO DE ELETROFORESE EM GEL DE
GRADIENTE DESNATURANTE (DGGE) DESDE A OBTENÇÃO DO DNA TOTAL DA COMUNIDADE ATÉ A
EXCISÃO DE BANDAS PARA SEQUENCIAMENTO APÓS A CORRIDA DO GEL DE DESNATURAÇÃO (FONTE:
ANGELIM, 2012). .......................................................................................................................................... 57
FIGURA 8 - ESQUEMA ILUSTRANDO COMO OCORRE A IMOBILIZAÇÃO DOS DNA MOLDE E SUA AMPLIFICAÇÃO NA
TECNOLOGIA DE SEQUENCIAMENTO DE NOVA GERAÇÃO ROCHE/454. (FONTE: METZKER, 2010). ................ 64
FIGURA 9 - LOCALIZAÇÃO DAS ESTAÇÕES DE COLETA NO PORTO DO PECÉM (MUNICÍPIO DO SÃO GONÇALO DO
AMARANTE, CE) E NO PORTO DO MUCURIPE (MUNICÍPIO DE FORTALEZA, CE), LOCALIZADOS NO ESTADO
DO CEARÁ, BRASIL. AS ESTAÇÕES DE COLETA SÃO INDICADAS NA FIGURA COMO P1 A P10 (PORTO DO PECÉM)
E M1 A M15 (PORTO DO MUCURIPE). ............................................................................................................. 76
FIGURA 10 - ASPECTO DOS SEDIMENTOS DO PORTO DO PECÉM (A; TRÊS PLACAS SUPERIORES) E DO PORTO DO
MUCURIPE (B; TRÊS PLACAS INFERIORES) APÓS LAVAGEM PARA RETIRADA DA FRAÇÃO SILTE-ARGILA,
EVIDENCIANDO A MAIOR QUANTIDADE DE PARTÍCULAS MAIORES NO PORTO DO PECÉM. ............................. 87

FIGURA 11 - CONCENTRAÇÃO DOS METAIS PESADOS MENSURADOS NO ESTUDO EM COMPARAÇÃO COM OS
CRITÉRIOS DE QUALIDADE (CQ) TRATADOS NA RESOLUÇÃO CONAMA Nº 344, DE 2004, E COM A
PUBLICAÇÃO DE BURUAEM ET AL. (2012; PEC* E MUC*), A QUAL TAMBÉM ANALISOU OS SEDIMENTOS
DOS PORTOS ESTUDADOS. CQ1 E CQ2 REPRESENTAM, RESPECTIVAMENTE, UM LIMIAR ABAIXO DO QUAL SE
PREVÊ POUCA PROBABILIDADE DE EFEITOS ADVERSOS À BIOTA (CQ1) E UM LIMIAR ACIMA DO QUAL SE
PREVÊ UM PROVÁVEL EFEITO ADVERSO SOBRE A BIOTA (CQ2). ................................................................... 87
FIGURA 12 - CONCENTRAÇÃO DOS HIDROCARBONETOS POLICÍCLICOS AROMÁTICOS (HPAS) MENSURADOS NOS
SEDIMENTOS DO PORTO DO PECÉM (PEC) E DO PORTO DO MUCURIPE (MUC) EM COMPARAÇÃO COM OS
CRITÉRIOS DE QUALIDADE TRATADOS NA RESOLUÇÃO CONAMA 344, DE 2004. A RESOLUÇÃO TRAZ DOIS
NÍVEIS DE QUALIDADE, CQ1 E CQ2, OS QUAIS REPRESENTAM, RESPECTIVAMENTE, UM LIMIAR ABAIXO DO
QUAL SE PREVÊ POUCA PROBABILIDADE DE EFEITOS ADVERSOS À BIOTA (CQ1 – NÍVEL 1) E UM LIMIAR
ACIMA DO QUAL SE PREVÊ UM PROVÁVEL EFEITO ADVERSO SOBRE A BIOTA (CQ2 – NÍVEL 2). ∑HPAS
REPRESENTA O SOMATÓRIO DAS CONCENTRAÇÕES INDIVIDUAIS DE HPAS PREVISTA NA RESOLUÇÃO
CONAMA 344, DE 2004, ENQUANTO ∑HPAS* REFERE-SE AO SOMATÓRIO TOTAL DE HPAS OBSERVADA
NOS PORTOS ESTUDADOS CONSIDERANDO TRÊS HPAS NÃO CONSTANTES NA REFERIDA RESOLUÇÃO,
BENZO(B)FLUORANTENO, BENZO(G,H,I)PERILENO E BENZO(K)FLUORANTENO. ............................................. 88
FIGURA 13 – ANÁLISE DE AGRUPAMENTO DOS PERFIS DE DGGE PARA O DOMÍNIO BACTERIA (A) E DOMÍNIO
ARCHAEA (B) ASSOCIADOS AOS SEDIMENTOS DO PORTO DO PECÉM. PARA COMPARAÇÃO DOS GRUPOS, FOI
UTILIZADO O ÍNDICE DE JACCARD NO PROGRAMA PAST V2.17C. ................................................................. 90
FIGURA 14 - ANÁLISE DE AGRUPAMENTO DOS PERFIS DE DGGE PARA O DOMÍNIO BACTERIA (A) E DOMÍNIO
ARCHAEA (B) ASSOCIADOS AOS SEDIMENTOS DO PORTO DO MUCURIPE. PARA COMPARAÇÃO DOS GRUPOS,
FOI UTILIZADO O ÍNDICE DE JACCARD NO PROGRAMA PAST V2.17C. ........................................................... 91
FIGURA 15 - ANÁLISE DE AGRUPAMENTO EM CONJUNTO DAS AMOSTRAS OBTIDAS NO PORTO DO PECÉM (PEC 1 A
PEC 10) E NO PORTO DO MUCURIPE (MUC 1 A MUC 15) PARA O DOMÍNIO BACTERIA. PARA COMPARAÇÃO
DOS GRUPOS, FOI UTILIZADO O ÍNDICE DE JACCARD NO PROGRAMA PAST V2.17C. ...................................... 94
FIGURA 16 - ANÁLISE DE AGRUPAMENTO EM CONJUNTO DAS AMOSTRAS OBTIDAS NO PORTO DO PECÉM (PEC 1 A
PEC 10) E NO PORTO DO MUCURIPE (MUC 1 A MUC 15) PARA O DOMÍNIO ARCHAEA. PARA COMPARAÇÃO
DOS GRUPOS, FOI UTILIZADO O ÍNDICE DE JACCARD NO PROGRAMA PAST V2.17C. ...................................... 95
FIGURA 17 – DIAGRAMA DE VENN EVIDENCIANDO O NÚMERO DE UTOS DO DOMÍNIO BACTERIA (A) E DO
DOMÍNIO ARCHAEA (B) PRESENTES EM CADA UM DOS AMBIENTES ESTUDADOS, PORTO DO PECÉM (PEC),
PORTO DO MUCURIPE (MUC) E PRAIA DE REDONDA (ICA). ........................................................................ 97
FIGURA 18 - ANÁLISE DE AGRUPAMENTO DOS PERFIS DE DGGE PARA O DOMÍNIO BACTERIA (A) E DOMÍNIO
ARCHAEA (B) ASSOCIADOS AOS POOLS DE SEDIMENTOS DO PORTO DO PECÉM (PECA, PECB E PECC),
PORTO DO MUCURIPE (MUCA, MUCB E MUCC) E ICAPUI (ICAA, ICAB E ICAC). PARA COMPARAÇÃO DOS
GRUPOS, FOI UTILIZADO O ÍNDICE DE JACCARD NO PROGRAMA PAST V2.17C. ............................................. 98
FIGURA 19 - CURVAS DE RAREFAÇÃO DOS METAGENOMAS SEQUENCIADOS DESCREVENDO O CRESCIMENTO DO
NÚMERO DE ESPÉCIES ENCONTRADAS COMO FUNÇÃO DO NÚMERO DE SEQUÊNCIAS SEQUENCIADAS. PEC
REPRESENTA A CURVA DE RAREFAÇÃO PARA O METAGENOMA DO PORTO DO PECÉM (TOTAL DE SEQUÊNCIAS

= 330.470) E MUC REPRESENTA A CURVA DE RAREFAÇÃO PARA O METAGENOMA DO PORTO DO MUCURIPE
(TOTAL DE SEQUÊNCIAS = 146.724). ........................................................................................................... 113
FIGURA 20 - CLASSIFICAÇÃO DAS SEQUÊNCIAS METAGENÔMICAS OBTIDAS A PARTIR DO PORTO DO PECÉM (PEC) E
DO PORTO DO MUCURIPE (MUC) PARA O DOMÍNIO ARCHAEA. OS VALORES APRESENTADOS NO GRÁFICO
REPRESENTAM O NÚMERO DE SEQUÊNCIAS CLASSIFICADAS NO BANCO DE DADOS M5NR PARA O NÍVEL DE
FILO. ........................................................................................................................................................... 116
FIGURA 21 - CLASSIFICAÇÃO DAS SEQUÊNCIAS METAGENÔMICAS OBTIDAS A PARTIR DO PORTO DO PECÉM (PEC) E
DO PORTO DO MUCURIPE (MUC). À ESQUERDA É MOSTRADA A AFILIAÇÃO TAXONÔMICA BASEADA NOS
BANCOS DE DADOS REFSEQ, GENBANK E M5NR PARA O NÍVEL DE FILO. OS VALORES APRESENTADOS
ACIMA DO GRÁFICO REPRESENTAM O NÚMERO DE SEQUÊNCIAS CLASSIFICADAS EM CADA UM DOS BANCOS DE
DADOS. POSICIONADA À DIREITA, É MOSTRADA A CLASSIFICAÇÃO TAXONÔMICA BASEADA NO BANCO DE
DADOS M5NR PARA O NÍVEL DE CLASSE. ................................................................................................... 117
FIGURA 22 - ESQUEMA COMPARATIVO DOS PERFIS TAXONÔMICOS EM NÍVEL DE ORDEM PARA O PORTO DO PECÉM
(PEC) E PORTO DO MUCURIPE (MUC). OS PERFIS TAXONÔMICOS PARA PEC (COLORIDO EM VERDE) E MUC
(COLORIDO EM LARANJA) FORAM COMPUTADOS USANDO A PLATAFORMA MG-RAST V3.0 E O PROGRAMA
STAMP V2.0.0. O DIÂMETRO DAS ESFERAS MOSTRADAS NA FIGURA FORAM CALCULADOS COM BASE NAS
ABUNDÂNCIAS RELATIVAS DE CADA TÁXON. O SÍMBOLO * DESIGNA ORDENS QUE APRESENTARAM BAIXAS
ABUNDÂNCIAS RELATIVAS, PORÉM OCORRERAM EM APENAS UM DOS PORTOS. A DISTRIBUIÇÃO DAS ORDENS
APRESENTADA NÃO ESTÁ CORRELACIOADA A SUA POSIÇÃO NOS PORTOS, MAS REPRESENTA APENAS UMA
ORGANIZAÇÃO DAS MESMAS EM ORDEM DE MAIOR PARA MENOR ABUNDÂNCIA RELATIVA. ....................... 118
FIGURA 23 - COMPARAÇÃO DOS PERFIS TAXONÔMICOS EM NÍVEL DE ORDEM (A) E EM NÍVEL DE ESPÉCIE (B) PARA
O PORTO DO PECÉM (PEC) E PORTO DO MUCURIPE (MUC). OS PERFIS TAXONÔMICOS PARA PEC (COLORIDO
EM VERDE) E PARA MUC (COLORIDO EM LARANJA) FORAM COMPUTADOS USANDO A PLATAFORMA MG-
RAST V3.0 E O PROGRAMA STAMP V2.0.0. OS PONTOS MOSTRADOS EM CADA LADO A LINHA DE
TENDÊNCIA TRACEJADA ENCONTRAM-SE ENRIQUECIDOS EM UMA DAS AMOSTRAS ANALISADAS. OS PONTOS
MAIS DISTANTES DESSA LINHA INDICAM QUE TAIS TÁXONS POSSUEM MAIOR DIFERENÇA PROPORCIONAL
ENTRE OS DOIS METAGENOMAS E FORAM IDENTIFICADOS NA FIGURA. UM FILTRO FOI APLICADO PARA
REMOVER PONTOS COM P > 0,05. ................................................................................................................ 119
FIGURA 24 - CURVA DE DISTRIBUIÇÃO DAS ABUNDÂNCIAS DOS GÊNEROS ASSOCIADOS AOS SEDIMENTOS DO PORTO
DO PECÉM. .................................................................................................................................................. 120
FIGURA 25 - CURVA DE DISTRIBUIÇÃO DAS ABUNDÂNCIAS DOS GÊNEROS ASSOCIADOS AOS SEDIMENTOS DO PORTO
DO MUCURIPE. ............................................................................................................................................ 121
FIGURA 26 - COMPARAÇÃO DOS PERFIS METAGENÔMICOS FUNCIONAIS OBTIDOS PARA O PORTO DO PECÉM (PEC) E
PARA O PORTO DO MUCURIPE (MUC) PARA AS CATEGORIAS DO KEGG DE METABOLISMO ENERGÉTICO (A),
METABOLISMO DO METANO (B) E METABOLISMO E DEGRADAÇÃO DE XENOBIÓTICOS (C), E PARA A
CATEGORIA DO SEED DE VIRULÊNCIA (D). OS PERFIS FUNCIONAIS DO PEC (COLORIDO EM VERDE) E DO
MUC (COLORIDO EM LARANJA) FORAM COMPUTADOS USANDO A PLATAFORMA MG-RAST V3.0 E O
PROGRAMA STAMP V2.0.0. OS PONTOS MOSTRADOS EM CADA LADO DA LINHA DE TENDÊNCIA TRACEJADA

ENCONTRAM-SE ENRIQUECIDOS EM UMA DAS AMOSTRAS ANALISADAS. OS PONTOS MAIS DISTANTES DESSA
LINHA INDICAM QUE TAIS TÁXONS POSSUEM MAIOR DIFERENÇA PROPORCIONAL ENTRE OS DOIS
METAGENOMAS E FORAM IDENTIFICADOS NA FIGURA. UM FILTRO FOI APLICADO PARA REMOVER PONTOS
COM P > 0.05. ............................................................................................................................................. 123
FIGURA 27 - ANÁLISE DE AGRUPAMENTO DOS PERFIS TAXONÔMICOS EM NÍVEL DE CLASSE PARA OS 13
METAGENOMAS DE ECOSSISTEMAS MARINHOS ANALISADOS, INCLUINDO O PORTO DO PECÉM (PEC) E PORTO
DO MUCURIPE (MUC). A FREQUÊNCIA DOS GRUPOS TAXONÔMICOS EM CADA METAGENOMA FOI EXTRAÍDA
DA PLATAFORMA MG-RAST V3.0 E COMPARADA NO PROGRAMA PAST V2.17C USANDO O ÍNDICE DE
SIMILARIDADE DE BRAY-CURTIS................................................................................................................ 127

LISTA DE TABELAS
TABELA 1 - TAMANHO DAS PARTÍCULAS FORMADORAS DOS SEDIMENTOS. ADAPTADO DE GARRISON (2011)....... 29
TABELA 2 - CLASSIFICAÇÃO DOS ORGANISMOS QUANTO A SUA FONTE DE ENERGIA, ELÉTRONS E CARBONO.
ADAPTADO DE KARL (2007). ...................................................................................................................... 37
TABELA 3 - SUMÁRIO DOS RISCOS IMPOSTOS POR ATIVIDADES ANTROPOGÊNICAS AOS ECOSSISTEMAS MARINHOS.
..................................................................................................................................................................... 41
TABELA 4 - COMPARAÇÃO DAS TÉCNICAS DE SEQUENCIAMENTO DE NOVA GERAÇÃO E SEQUENCIAMENTO DE
SANGER UTILIZADAS EM PROJETOS DE METAGENÔMICA. .............................................................................. 63
TABELA 5 - LISTAGEM DE BANCOS DE DADOS DE REFERÊNCIA PARA PROTEÍNAS OU ÁCIDOS NUCLÉICOS
UTILIZADOS PARA CLASSIFICAÇÃO DE SEQUÊNCIAS METAGENÔMICAS. ........................................................ 65
TABELA 6 – LOCALIZAÇÃO DAS ESTAÇÕES DE COLETA E CARACTERÍSTICAS GERAIS DAS AMOSTRAS DE SEDIMENTO
OBTIDAS. ...................................................................................................................................................... 78
TABELA 7 - CARACTERIZAÇÃO DAS AMOSTRAS DE DNA EXTRAÍDAS DOS SEDIMENTOS ORIUNDOS DO PORTO DO
PECÉM (P1-P10) E DO PORTO DO MUCURIPE (M1-M15). PMÉDIA E MMÉDIA REPRESENTAM AS MÉDIAS DAS
CONCENTRAÇÕES DE DNA, RELAÇÕES 260/280 E RELAÇÕES 260/230 DAS AMOSTRAS OBTIDAS NO PORTO DO
PECÉM E NO PORTO DO MUCURIPE, RESPECTIVAMENTE. .............................................................................. 84
TABELA 8 - ATIVIDADE PORTUÁRIA EM TONELADAS DE CARGA TRANSPORTADA E PARÂMETROS FÍSICOS E
QUÍMICOS DO PORTOS DO PECÉM (PEC) E PORTO DO MUCURIPE (MUC) ..................................................... 86
TABELA 9 – RIQUEZA DE UTOS ENCONTRADAS NOS PERFIS DE DGGE PARA OS DOMÍNIOS BACTERIA E ARCHAEA
DO PORTO DO PECÉM (P1 A P10) E DO PORTO DO MUCURIPE (M1 A M15). ...................................................... 92
TABELA 10 – ESTIMADORES DE RIQUEZA CALCULADOS A PARTIR DAS MATRIZES BINÁRIAS REPRESENTATIVAS DOS
PERFIS DE DGGE OBTIDOS PARA O DOMÍNIO BACTERIA E ARCHAEA DE AMBOS OS PORTOS EM ESTUDO,
PORTO DO PECÉM E PORTO DO MUCURIPE, NO PROGRAMA PAST V2.17C. ................................................... 93
TABELA 11 - MEDIDAS DE BETA DIVERSIDADE CALCULADAS A PARTIR DAS MATRIZES BINÁRIAS REPRESENTATIVAS
DOS PERFIS DE DGGE OBTIDOS PARA O DOMÍNIO BACTERIA E ARCHAEA DE AMBOS OS PORTOS EM ESTUDO,
PORTO DO PECÉM E PORTO DO MUCURIPE, NO PROGRAMA PAST V2.17C. ................................................... 93
TABELA 12 – RIQUEZAS DE UTOS ENCONTRADAS NOS PERFIS DE DGGE DOS POOLS DE SEDIMENTOS PARA O
DOMÍNIO BACTERIA E ARCHAEA DOS PORTOS EM ESTUDO, PORTO DO PECÉM (PEC), PORTO DO MUCURIPE
(MUC) E PRAIA DE REDONDA (ICA). ........................................................................................................... 96
TABELA 13 - ESTIMADORES DE RIQUEZA CALCULADOS A PARTIR DOS PERFIS DE DGGE DOS POOLS DE SEDIMENTOS
OBTIDOS PARA O DOMÍNIO BACTERIA E ARCHAEA DOS PORTOS EM ESTUDO, PORTO DO PECÉM (PEC), PORTO
DO MUCURIPE (MUC) E PRAIA DE REDONDA (ICA). .................................................................................... 97
TABELA 14 - CLASSIFICAÇÃO TAXONÔMICA DAS SEQUÊNCIAS METAGENÔMICAS OBTIDAS OBTIDAS NO PORTO DO
PECÉM (PEC) E NO PORTO DO MUCURIPE (MUC) RELACIONADAS A ALGUMAS CATEGORIAS DA
CLASSIFICAÇÃO FUNCIONAL PELO BANCO DE DADOS KEGG. ..................................................................... 125

SUMÁRIO
1. INTRODUÇÃO .................................................................................................................. 24
2. REVISÃO DA LITERATURA ......................................................................................... 26
2.1. O AMBIENTE MARINHO ..................................................................................................... 26
2.2. OS SEDIMENTOS MARINHOS ............................................................................................. 28
2.3. OS SEDIMENTOS NERÍTICOS .............................................................................................. 29
2.3.1. Ambiente marinho sul-americano ............................................................................ 30
2.4. DIVERSIDADE PROCARIÓTICA EM SEDIMENTOS MARINHOS .............................................. 32
2.5. METABOLISMO MICROBIANO EM SEDIMENTOS MARINHOS ............................................... 36
2.6. INFLUÊNCIAS ANTRÓPICAS SOBRE O AMBIENTE MARINHO ................................................ 40
2.6.1. Os impactos decorrentes do transporte marítimo e operação de portos ................. 42
2.7. EFEITOS DOS CONTAMINANTES SOBRE A BIOTA MARINHA ................................................ 47
2.7.1. Efeitos sobre a microbiota ........................................................................................ 49
2.8. FERRAMENTAS DE ESTUDO DA DIVERSIDADE MICROBIANA .............................................. 51
2.9. COMPARAÇÃO DE SEQUÊNCIAS METAGENÔMICAS ........................................................... 61
2.9.1. Tecnologias de sequenciamento .............................................................................. 61
2.9.2. Classificação ............................................................................................................. 64
2.9.3. Análises estatísticas .................................................................................................. 68
2.10. ARMAZENAMENTO E COMPARTILHAMENTO DOS DADOS .................................................. 69
3. OBJETIVO GERAL .......................................................................................................... 71
3.1. OBJETIVOS ESPECÍFICOS .................................................................................................. 71
4. CAPÍTULO 1 - ESTRUTURA E RIQUEZA LOCAL DAS ASSEMBLEIAS DE
BACTERIA E ARCHAEA ASSOCIADAS A SEDIMENTOS MARINHOS SOB
INFLUÊNCIA DE REGIÕES PORTUÁRIAS ............................................................... 72
4.1. INTRODUÇÃO ................................................................................................................... 72
4.2. HIPÓTESE ......................................................................................................................... 74
4.3. MATERIAIS E MÉTODOS ................................................................................................... 74
4.3.1. Área de estudo .......................................................................................................... 74
4.3.2. Coleta das amostras ................................................................................................. 76
4.3.3. Caracterização física e química dos sedimentos estudados ..................................... 77
4.3.4. Extrações de DNA ..................................................................................................... 79
4.3.5. Eletroforese em Gel de Gradiente Desnaturante por meio de Reação em Cadeia de Polimerase com iniciadores para o gene rRNA 16S .................................................. 80
4.3.6. Análise dos géis de DGGE ......................................................................................... 82
4.4. RESULTADOS ................................................................................................................... 83
4.4.1. Caracterização das amostras de DNA ...................................................................... 83

4.4.2. Caracterização física e química dos sedimentos ...................................................... 84
4.4.3. Perfis de DGGE das assembleias de Bacteria e Archaea associadas aos sedimentos oriundos de diferentes estações de coleta no Porto do Pecém e no Porto do Mucuripe .................................................................................................................. 88
4.4.4. Comparação das estruturas das assembleias de Bacteria e Archaea do Porto do Pecém e do Porto do Mucuripe com a de uma região sem atividade portuária ...... 96
4.5. DISCUSSÃO ...................................................................................................................... 99
4.6. CONCLUSÕES ................................................................................................................. 106
5. CAPÍTULO 2 - COMPOSIÇÃO TAXONÔMICA E FUNCIONAL DO
PATRIMÔNIO MICROBIANO REGIONAL DE SEDIMENTOS MARINHOS SOB
INFLUÊNCIA DE REGIÕES PORTUÁRIAS ............................................................. 107
5.1. HIPÓTESE ....................................................................................................................... 108
5.2. MATERIAIS E MÉTODOS ................................................................................................. 108
5.2.1. Locais de Estudo ..................................................................................................... 108
5.2.2. Coleta das amostras e processamento ................................................................... 109
5.2.3. Extração do DNA ambiental ................................................................................... 109
5.2.4. Pirosequenciamento para análise metagenômica ................................................. 110
5.2.5. Classificação taxonômica das sequências ambientais ............................................ 110
5.2.6. Análise funcional utilizando os bancos de dados KEGG e SEED .............................. 111
5.2.7. Comparação estatística dos perfis metagenômicos ............................................... 111
5.2.8. Classificação taxonômica dos principais grupos procarióticos envolvidos em rotas metabólicas do KEGG ............................................................................................. 111
5.2.9. Comparação dos metagenomas de sedimentos dos portos com outros metagenomas de ecossistemas marinhos .............................................................. 112
5.3. RESULTADOS ................................................................................................................. 112
5.3.1. Pirosequenciamento dos metagenomas dos portos ............................................... 112
5.3.2. Perfil taxonômico dos metagenomas dos portos ................................................... 114
5.3.3. Perfil funcional dos metagenomas dos portos ....................................................... 122
5.3.4. Classificação taxonômica dos principais grupos procarióticos envolvidos em rotas metabólicas do KEGG ............................................................................................. 125
5.3.5. Comparação com perfis metagenômicos de outros hábitats marinhos ................. 126
5.4. DISCUSSÃO .................................................................................................................... 128
5.5. CONCLUSÕES ................................................................................................................. 136
6. CONSIDERAÇÕES FINAIS E PERSPECTIVAS FUTURAS .................................... 137
7. REFERÊNCIAS ............................................................................................................... 138

24
1. INTRODUÇÃO
Palco de grandes inovações adaptativas ao longo da evolução da vida na Terra, o
ambiente costeiro é hoje cenário da luta pela sobrevivência de milhares de espécies ameaçadas
pelo contato frequente com o estilo de vida da espécie humana. Dentre os variados tipos de
intervenções antrópicas nos ambientes costeiros, os portos marítimos e atividades afins
destacam-se por constituírem ambientes com elevado potencial poluidor, capazes de liberar
grandes quantidades de xenobióticos no ambiente marinho, em decorrência de acidentes ou
atividades rotineiras. Esses poluentes podem induzir alterações nas comunidades presentes na
coluna d’água e nos sedimentos, tanto nas zonas portuárias como nas regiões costeiras
adjacentes. Entretanto, pouco se conhece a respeito de seus efeitos, particularmente sobre os
micro-organismos.
Apesar de parecer tarefa fácil apontar as fontes de impactos antrópicos, tal
empreitada é na verdade bem complexa, pois qualquer ação humana tem o potencial de causar
alterações ambientais, com maior ou menor gravidade. Os portos e transportes marítimos,
particularmente, são capazes de causar desde alterações na transparência da coluna d´água até
a introdução de poluentes prioritários, como é o caso dos hidrocarbonetos policíclicos
aromáticos (HPAs). Contudo, as práticas atuais de monitoramento ambiental estão focadas nos
poluentes químicos e em nutrientes capazes de causar enriquecimento orgânico, conforme
tratado na Resolução nº 344 do Conselho Nacional do Meio Ambiente (CONAMA), de 25 de
março de 2004. A análise apenas desses parâmetros fornece uma imagem inacabada da real
situação local, pois, a depender do composto químico e das características locais, diferentes
efeitos, agudos ou crônicos, podem ser observados. Tais análises deixam de lado, ainda, os
efeitos sobre a biota, os quais podem ser bastante variados. Além disso, os portos, como locais
que entram em contato com embarcações e resíduos originados em outras localidades do
planeta, são sítios de entrada de espécies exóticas, para as quais falta um monitoramento
adequado. Portanto, como a legislação ambiental é omissa em vários aspectos, depende-se hoje
de um comprometimento particular de cada autoridade portuária em melhorar sua qualidade
ambiental, em apontar riscos e vulnerabilidades em diferentes áreas e em dar tratamento aos
seus resíduos, resultando em uma redução do passivo ambiental.
Cada vez mais espécies são descritas como passíveis de serem utilizadas no
monitoramento ambiental de regiões portuárias, como é o caso de várias espécies de moluscos

25
e poliquetas estudados como bioinvasores, de crustáceos utilizados em testes de ecotoxicidade
e de micro-organismos capazes de prosperar em ambientes contaminados. Nesse contexto, a
microbiota, essencial para todos os ecossistemas, destaca-se, pois alterações na mesma podem
ter sérias consequências sobre os serviços prestados por ecossistemas costeiros, além de seus
membros constituírem excelentes ferramentas para o biomonitoramento e/ou biorremediação
ambiental.
Felizmente, com o conhecimento que se tem atualmente sobre as mudanças
sucessionais que ocorrem em comunidades microbianas expostas a outras intervenções
antrópicas, como liberação de esgotos ou derramamento de petróleo, pode-se hipotetizar que os
membros das comunidades associadas aos sedimentos sob influência de estruturas portuárias
podem ser afetados, resultando em modificações em sua estrutura de comunidade e composição.
Ademais, especula-se se estruturas portuárias diferenciadas resultam em respostas diversas por
parte da comunidade, resultando em estruturas e riquezas próprias, as quais representam a
adaptação de seus membros às condições vigentes em cada tipo de estrutura portuária.
Dessa forma, com o objetivo de avaliar tais hipóteses, foram comparados dois
portos marítimos do Estado do Ceará, o Porto do Mucuripe e o Porto do Pecém. Estes portos
possuem características estruturais e operacionais distintas, destacando-se a arquitetura e o
tempo de operação como principais divergências. O Porto do Mucuripe, em operação desde
1953, é um terminal portuário convencional, construído com um canal principal e berços de
atracação protegidos por moles para redução da hidrodinâmica local. Já o Porto do Pecém, cujas
operações foram iniciadas em 2001, possui uma arquitetura mais moderna do tipo offshore, ou
seja, fora da costa, sendo protegido por um quebra-mar em ‘L’, localizado mais distante da
linha de costa, e construído sobre pilastras para não impedir o transporte de sedimentos pelas
correntes oceânicas. Além disso, o Porto do Pecém faz parte de um complexo industrial e
portuário em instalação. Quando estiver em plena operação, possivelmente resultará em
significante aumento no fluxo de embarcações e resíduos associados à operação das indústrias
e atividades correlatas, o que poderá levar a grandes mudanças no ecossistema costeiro no local,
inserindo um caráter histórico nesse estudo. Dessa forma, hipotetizou-se que a estrutura e a
diversidade das assembleias de Bacteria e Archaea seriam diferenciadas de acordo com as
características ambientais em cada porto, o que foi investigado por meio de duas técnicas
independentes de cultivo, a Eletroforese em Gel de Gradiente Desnaturante (DGGE) e a
metagenômica.

26
2. Revisão da literatura
2.1. O ambiente marinho
O oceano pode ser definido como uma grande massa de água salina que ocupa as
depressões da superfície terrestre. Este contém mais de 97% da água disponível no planeta e
cobre cerca de 70% da superfície da Terra. Numa escala humana, o oceano é considerado
imenso, cobrindo 361 milhões de Km2, com profundidade média de 3682 m, volume de 1,33
bilhões de Km3 e temperatura média de 3,9 °C (GARRISON, 2011). Apesar de possuir
salinidade relativamente constante em toda a sua extensão, a temperatura pode variar
enormemente, atingindo extremos nas regiões polares e nas chaminés submarinas. O ambiente
marinho constitui o maior hábitat da Terra, os quais variam de poças tropicais às misteriosas
fossas oceânicas há mais de 11.000 m de profundidade e com pressões de mais de 100 Mpa
(KENNEDY et al., 2010).
Embora não possua barreiras rígidas, o oceano constitui um mosaico de hábitats
semi-isolados estabelecidos e mantidos por processos de escala global, como as correntes e as
interações entre atmosfera e oceano, e de escala regional/local, como os aportes continentais
(KARL, 2007). Esse ambiente de alta heterogeneidade abriga uma rica diversidade de espécies,
além de ser essencial para a vida dos organismos terrestres (ORCUTT et al., 2011). Sua
importância reside, primeiramente, no fato de ter sido o berço da vida e cenário de grande parte
de sua evolução e irradiação, além de ser o maior hábitat da Terra. Em segundo lugar, por
estabilizar condições climáticas que possibilitam a manutenção da maioria das formas de vida.
Com a intensificação dos efeitos do aquecimento global, os oceanos passaram a ser vistos com
grande interesse por ser um importante local de transformação e sequestro de carbono orgânico
e inorgânico, com enorme capacidade de absorção do dióxido de carbono atmosférico
principalmente devido a fotossíntese desempenhada pelos organismos fotoautotróficos que
habitam os ecossistemas costeiros, como manguezais e marismas, depósitos de carbono
conhecidos como carbono azul (MCLEOD et al., 2011).
Devido ao seu tamanho, o oceano pode ser dividido verticalmente e
horizontalmente em zonas, conforme pode-se observar na Figura 1. Além disso, pode ser
dividido geograficamente em cinco oceanos –Atlântico, Pacífico, Índico, Ártico e Antártico.
Verticalmente, o oceano é dividido em domínio pelágico, correspondente à coluna
d’água, e domínio bentônico, correspondente ao assoalho e sedimentos sobrejacentes. Por sua

27
vez, o domínio pelágico pode ser dividido horizontalmente em duas províncias: a província
nerítica, referente à coluna d’água sobre a plataforma continental; e a província oceânica,
correspondente às águas de oceano aberto. O domínio pelágico pode ser dividido ainda em
subpartes de acordo com a penetração de radiação solar em zona eufótica ou epipelágica, onde
há chegada de radiação solar para sustentar a produtividade primária em excesso; zona disfótica
ou mesopelágica, de transição com entrada de luz suficiente para a visão, mas não para saldo
de produtividade primária; e zona afótica, onde não há entrada de luz. A última zona, de acordo
com a profundidade, pode ser subdividida em batipelágica, abissopelágica e hadopelágica
(GARRISON, 2011).
Por sua vez, fazem parte do domínio bentônico a plataforma continental,
correspondente ao fundo marinho localizado sob a zona nerítica na região costeira até 200 m
de profundidade; o talude continental ou zona batial, com profundidade de até 4000 m; a
planície abissal, com profundidades entre 4000 e 6000 m; e a zona hadal, correspondente à
região das fossas submarinas, com profundidades entre 6000 e 11000 m (GARRISON, 2011).
Em regiões próximas ao continente, correspondentes à plataforma continental (de
10 a 200 Km de distância), os hábitats marinhos são, em geral, mais rasos, sendo influenciados
principalmente por processos continentais. A afluência de materiais terrígenos, como
sedimentos, água doce, carbono orgânico e nutrientes, determina características influenciadas
fortemente por fatores locais, aumentando sua variabilidade e produtividade e as diferenciando
de regiões marinhas de maior profundidade, caracterizadas por uma maior estabilidade (KARL,
2007). Além disso, há uma influência antrópica aumentada devido à maioria das grandes
metrópoles mundiais estar localizada em áreas fronteiriças com regiões oceânicas, de forma
que aproximadamente metade da população mundial vive a pelo menos 100 Km da linha de
costa (GARRISON, 2011). Portanto, a região costeira refere-se ao local onde oceano e
continente se encontram, envolvendo toda a zona afetada por este encontro, como praias
arenosas, manguezais, dunas, bancos de areia, dentre outros, sofrendo influências de eventos
naturais e processos comuns ao continente e ao oceano, abrangendo um total de 440 mil Km de
costa no mundo todo (GARRISON, 2011).

28
Figura 1 - Subdivisões do ambiente marinho em função da topografia dos fundos, da profundidade e da
luminosidade. Adaptado de Garrison (2011).
2.2. Os sedimentos marinhos
Após a coluna d’água oceânica, os sedimentos marinhos constituem o segundo
maior hábitat do mundo, correspondendo a toda a área que se encontra sob a mesma, como
sedimentos costeiros, leito oceânico e fontes hidrotermais (ORCUTT et al., 2011). Sedimentos
são partículas de matéria orgânica ou inorgânica que se acumulam de forma inconsolidada. Tais
partículas originam-se a partir do intemperismo e erosão das rochas, da atividade dos
organismos vivos, de erupções vulcânicas, de processos químicos que ocorrem na própria
coluna d’água ou mesmo vindos do espaço. Na verdade, a maior parte do assoalho oceânico
está sendo lentamente coberta por um grande volume de sedimentos cuja taxa de acumulação
varia a depender das características locais. Como exemplos de sedimentos marinhos pode-se
citar a areia das praias, lamas de baías e manguezais e mesmo acúmulos de conchas e silte na
plataforma continental (GARRISON, 2011).

29
Os sedimentos podem ser classificados de acordo com o tamanho das partículas que
o formam, conforme pode-se observar na Tabela 1. Apesar de pedregulhos e seixos poderem
ocorrer nos oceanos, a maior parte dos sedimentos marinhos é composta de partículas mais
finas, como areia, silte e argila. Em geral, quanto menor o tamanho da partícula constituinte,
mais facilmente esta pode ser transportada por ondas e correntes (GARRISON, 2011).
Outra maneira de classificar os sedimentos é de acordo com a origem das fontes de
deposição. Esse esquema de classificação os separa em quatro categorias: biogênicos,
hidrogênicos ou autigênicos, cosmogênicos, e litogênicos ou terrígenos. Os sedimentos
terrígenos possuem origem no continente, em ilhas, em erupções vulcânicas ou poeira e são os
mais abundantes em volume, cobrindo 45% do assoalho marinho. Os sedimentos biogênicos
originam-se a partir da incorporação de material silicoso e calcário em estruturas de organismos
vivos, como esqueletos calcários e conchas, sendo o segundo tipo mais abundante,
principalmente em áreas de alta produtividade próximas aos continentes ou em áreas de
ressurgência. Já os sedimentos hidrogênicos são oriundos da própria água do mar, acumulando-
se como resultado da precipitação, e os cosmogênicos têm origem extraterrestre, constituindo
o tipo de sedimento menos abundante (GARRISON, 2011).
Tabela 1 - Tamanho das partículas formadoras dos sedimentos. Adaptado de Garrison (2011).
Tipo de partícula Diâmetro (mm)
Bloco > 256
Seixo 64 - 256
Cascalho 2 - 64
Areia 0,062 - 2
Silte 0,004 - 0,062
Argila < 0,004
2.3. Os sedimentos neríticos
Os sedimentos que se acumulam na plataforma continental são chamados de
sedimentos neríticos ou costeiros e consistem, primariamente, de depósitos litogênicos
(ACOSTA-GONZÁLEZ, 2013). Em geral, as correntes distribuem a areia e partículas maiores
ao longo da costa, e as ondas carreiam siltes e argilas para profundidades maiores. Já em locais

30
protegidos ou profundos, livres da ação das ondas, os sedimentos mais finos conseguem
assentar-se. Os sedimentos neríticos quase sempre contêm material biológico em adição ao
terrígeno como resultado da alta produtividade das águas costeiras, que permitem o acúmulo de
restos de organismos em meio aos depósitos litogênicos (GARRISON, 2011).
As plataformas continentais constituem 9% da área do oceano e contêm 15% do
volume total de sedimentos marinhos (GARRISON, 2011). Atualmente, 40% da população
mundial habita áreas continentais distantes até 100 Km da linha de costa, com importantes
consequências sobre os ecossistemas que se estabelecem. Em geral, o depósito de matéria
orgânica e inorgânica da coluna d’água em direção ao sedimento ocorre por sedimentação
gravitacional, atividades de filtração bentônica e agregação na interface água-sedimento. Na
região oceânica, a maior parte do material que afunda da coluna d’água é decomposto antes de
atingir os sedimentos, sendo de apenas 1% a proporção de matéria orgânica da coluna d’água
que atinge a superfície dos sedimentos. Contudo, as regiões costeiras constituem uma exceção
devido à menor profundidade, ocasionando padrões de chegada, reação e deposição do carbono
orgânico de origem continental não uniformes que podem diferir cerca de 30 vezes dos padrões
de mar aberto. Considerando que os sedimentos costeiros possuem, em sua maior parte, origem
litogênica, as atividades antropogênicas têm o potencial de aumentar este influxo. Tal tendência
é mais acentuada em regiões cujos rios possuem pequena quantidade de barramentos físicos e
que, portanto, transportam grande quantidade de material erodido com potencial de contribuir
com o aumento da quantidade de sedimentos finos na região costeira. Em ambos os casos, o
impacto sobre as comunidades bentônicas é significante, sendo exacerbado pela possibilidade
de transporte de contaminantes de origem continental para o ambiente marinho (ACOSTA-
GONZÁLEZ, 2013).
2.3.1. Ambiente marinho sul-americano
As áreas marinhas da América do Sul incluem mais de 30 mil quilômetros de costa,
reunindo três domínios oceânicos diferentes – o do Caribe, do Pacífico e do Atlântico, desde
12°N a 55°S de latitude. Essa vasta extensão está distribuída em cinco Grandes Ecossistemas
Marinhos (LME; do inglês, Large Marine Ecosystems): Pacífico Tropical Leste; Sistema da
Corrente de Humboldt; Atlântico Tropical Oeste; Plataforma Brasileira; e Plataforma da
Patagônia, conforme pode-se observar na Figura 2. Na região caribenha destacam-se as costas
continentais da região sul da América Central, norte da América do Sul e ilhas oceânicas. No

31
Pacífico, tem-se a costa peruana, limite da distribuição de manguezais do Pacífico, e a Costa
Chilena, uma das maiores (4500 Km de extensão) e mais ricas em biodiversidade devido às
condições oceanográficas únicas que levam à ocorrência de ressurgência e heterogeneidade
costeira. Por sua vez, a costa Atlântica é bem diferente da Pacífica. Ela é profundamente
influenciada por três grandes rios, Orinoco, Amazonas e La Plata, que descarregam enormes
quantidades de água doce e sedimentos para o oceano, resultando em uma extensa plataforma
continental. As regiões costeiras da Argentina e do Uruguai constituem a Região Patagônica,
sob forte influência da descarga do Rio La Plata e de correntes frias austrais. Ao norte, sob
influência dos rios Amazonas e Orinoco, tem-se as costas de Suriname, Guiana Francesa,
Guiana e Venezuela, conhecida como Atlântico Tropical Oeste (MILOSLAVICH et al., 2011).
A Plataforma Brasileira corresponde à maior linha de costa da América do Sul,
estendendo-se por 7491 Km. Além do Mar territorial e da Zona Econômica Exclusiva, que se
estendem até 12 e 200 milhas náuticas, respectivamente, o Brasil obteve uma extensão 900
Km2, de forma que sua zona costeira conta com 4,5 milhões de Km2, tendo sido designada como
“Amazônia Azul”. A Plataforma Continental Brasileira é bastante heterogênea, sendo mais
estreita na região Nordeste (~ 8Km) e mais ampla na região onde deságua o Rio Amazonas
(~300 Km) e na costa do Rio Grande do Sul (246 Km). Além disso, também varia em
profundidade. Na região Norte, atinge 80 - 100 m; na região Nordeste e ao Norte da Cadeia
Vitória-Trindade, atinge 60 – 70 m; e na região Sul e ao sul da Cadeia Vitória-Trindade, atinge
de 160 – 200 m. Infelizmente, devido a sua vastidão, grandes áreas da costa brasileira
permanecem inexploradas (MILOSLAVICH et al., 2011).

32
Figura 2 - Mapa da América do Sul definindo os cinco Grandes Ecossistemas Marinhos (LME): Pacífico Tropical
Leste, em azul; Sistema da Corrente de Humboldt, em lilás; Atlântico Oeste Tropical, em laranja; Plataforma
Brasileira, em verde; e Plataforma da Patagônia, em rosa. (Fonte: Miloslavich et al., 2011).
2.4. Diversidade Procariótica em sedimentos marinhos
O estudo da diversidade da vida é um desafio persistente em Biologia.
Particularmente na Microbiologia, essa tarefa é mais complicada já que os objetos de estudo
não são visíveis a olho nu e são mensurados em números superiores a 1030 indivíduos. Isso
exige que os micro-organismos sejam estudados por meio de análises indiretas que permitam
enxergar além das limitações impostas pelas técnicas de cultivo.
O ecologista Robert May afirmou, em 1992, que ‘catalogamos todos os corpos
celestes que nossos instrumentos podem detectar no Universo, mas ignoramos quantos
organismos vivos compartilham a Terra conosco’. Segundo Pedrós-Alió (2006), essa
ignorância é particularmente maior nos oceanos. Nesses ambientes, o número total de células

33
procarióticas foi estimado em 1030. Entretanto, não se sabe exatamente quantos táxons tal
abundância representa. A resposta para tal pergunta pode contribuir para um melhor
entendimento de questões essenciais atualmente. Primeiramente, tal diversidade esconde
metabolismos novos que forçam uma reavaliação dos fluxos de carbono e energia nos oceanos,
como é o caso do foto-heterotrofismo, recentemente descoberto. Em segundo lugar, os micro-
organismos constituem o maior reservatório potencial de genes para a medicina e a
biotecnologia. Por fim, o conhecimento da diversidade microbiana é essencial para se entender
a evolução das formas de vida (PEDRÓS-ALIÓ, 2006).
Os sedimentos contêm aproximadamente 85% do total estimado de células
procarióticas presentes no ambiente marinho, sendo considerados os ambientes mais povoados
da Terra (ACOSTA-GONZÁLEZ, 2013). Eles podem abrigar diferentes tipos de micro-
organismos (bactérias, arqueias, protozoários e vírus), porém os procariotos constituem a maior
parte da biomassa e atividade química, constituindo o segundo grupo biológico mais abundante
após os vírus (BREITBART, 2012). Atualmente, há mais de 10 mil espécies de procariotos
nomeadas (http://www.bacterio.net/-number.html#rhodococcus; acessado em 07/07/2014).
Isso representa apenas a pequena porção dos micro-organismos que conseguiram ser cultivados
em culturas puras (PEDRÓS-ALIÓ, 2006). Além disso, os procariotos são ubíquos, fato devido
a sua elevada capacidade de dispersão assim como a sua versatilidade metabólica e capacidade
de resistir a condições consideradas hostis para a maioria dos organismos (ACOSTA-
GONZÁLEZ, 2013).
Essa grande parcela de diversidade desconhecida vem sendo estudada por meio de
técnicas moleculares. Estas vêm revelando que as comunidades microbianas são formadas por
poucos táxons muito abundantes, os quais constituem o “microbioma fundamental”, ou seja,
bem adaptado a um determinado ecossistema, e por um grande número de táxons pouco
abundantes (raros). Aqueles que constituem o microbioma fundamental tomam parte
ativamente nos processos de predação e lise viral, alimentando o fluxo de carbono e energia.
Por outro lado, os táxons de baixa abundância fazem parte do chamado “banco de sementes”,
cujas espécies proliferam de acordo com as condições locais de forma ocasional. É importante
destacar que, a depender das condições físico-químicas vigentes, um micro-organismo pode
habitar a porção fundamental ou rara da comunidade (PEDRÓS-ALIÓ, 2006). Na verdade, a
chamada “biosfera rara” constitui uma rica fonte de diversidade genética da qual dependem a
resistência e a resiliência das comunidades (KARL, 2007). A célebre hipótese de Bas Becking
poderia, nesse caso, ser complementada da seguinte forma: “Tudo está em todo lugar, mas o
ambiente seleciona” quem serão os grupos abundantes.

34
É importante ressaltar que um problema associado aos táxons raros é que muitas
vezes são ignorados nos levantamentos utilizando técnicas dependentes de PCR. Alguns
estudos mostraram que apenas táxons com mais de 1% de abundância podem ser detectados
por tais técnicas, com os táxons com menos de 0,1% sendo de difícil detecção (CASAMAYOR
et al., 2000).
Alguns estudos vêm investigando a diversidade microbiana e a estrutura de
comunidades associadas a sedimentos marinhos. Eles revelaram que tanto os ambientes de
sedimento marinho como de solo possuem uma diversidade procariótica mais elevada que a
encontrada em ambientes aquáticos (TORSVIK; ØVREÅS, 2002; ZINGER et al., 2011). Um
estudo do Censo Internacional de Micro-organismos Marinhos (ICoMM) revelou a estrutura de
diversas comunidades, bentônicas ou pelágicas, oriundas de diversos ambientes marinhos,
evidenciando que a composição das comunidades microbianas varia vertical e horizontalmente
de acordo com variáveis, como produtividade e distribuição espacial (ZINGER et al., 2011).
Os autores também mostraram que as comunidades bentônicas exibiram maior dissimilaridade
com o aumento da distância, se comparadas às comunidades pelágicas, sugerindo que as forças
locais de mistura física dos sedimentos podem contribuir para mudanças nos padrões de
distribuição dos procariotos marinhos.
Alguns estudos com assembleias procarióticas associadas a sedimentos marinhos
vêm evidenciando um padrão geral de filos e principais classes presentes nos mesmos. Um
esboço geral desses achados é apresentado na Figura 3. Os autores vêm observando elevadas
frequências de sequências pertencentes ao filo Proteobacteria, o qual constitui o táxon
dominante, sendo composto principalmente de Gammaproteobacteria e Deltaproteobacteria
(ZINGER et al., 2011), incluindo espécies com grandes diferenças morfológicas, fenotípicas e
metabólicas.
Os dados relativos a outros filos abundantes são mais variáveis. Em sedimentos do
Oceano Ártico, foram encontrados elevados teores de Gammaproteobacteria e
Deltaproteobacteria (47%), Bacteroidetes (9%), Actinobacteria, Acidobacteria e
Verrucomicrobia, cada qual com cerca de 5% de abundância relativas (JACOB et al., 2013). Já
sedimentos marinhos da costa africana do Oceano Atlântico Tropical foram dominados por
Bacteria da classe Gammaproteobacteria, que perfizeram 40% das sequências. Além disso,
apresentaram quantidades significativas de sequências pertencentes às famílias
Enterobacteriaceae, Alteromonadaceae, Oceanospirillaceae e Legionellaceae. A classe
Deltaproteobacteria aparece secundariamente com 14% das sequências, sendo seguida de
Bacteroidetes, Planctomycetes e Acidobacteria (SCHAUER et al., 2010). Também Xiong et al.

35
(2014) confirmaram o padrão geral observado, com a presença dos táxons
Gammaproteobacteria, Deltaproteobacteria, Planctomycetes, Bacteroidetes e Chloroflexi como
dominantes em sedimentos poluídos do Mar da China. Particularmente as
Gammaproteobacteria da família Xanthomonadaceae pareceram diminuir ao longo de um
gradiente de concentração decrescente, mostrando que Gammaproteobacteria heterotróficas
proliferam rapidamente em condições de altos teores de nutrientes. Tais táxons podem ser
utilizados para indicar os níveis de contaminantes costeiros (XIONG et al., 2014).
Figura 3 – Composição da comunidade microbiana de acordo com os tipos de ambientes. Em azul, observa-se o
ambiente pelágico, e em amarelo o ambiente bentônico. (a) Frequências média das sequências para as dez classes
mais abundantes. (b) Proporções médias dos principais grupos microbianos. O prefixo B representa os ambientes
bentônicos; e P, ambientes pelágicos. Fonte: ZINGER et al., 2011.

36
Os procariotos que habitam sedimentos marinhos participam de uma variedade de
rotas bioquímicas envolvendo compostos orgânicos e inorgânicos, além de participarem dos
principais ciclos biogeoquímicos, como o do enxofre, do nitrogênio e do ferro. Em geral, as
condições geoquímicas prevalentes nos sedimentos, como disponibilidade de aceptores de
elétrons e a quantidade e qualidade de matéria orgânica, determinam a estrutura e dinâmica das
assembleias procarióticas. É interessante que a análise das comunidades microbianas de vários
sedimentos em diferentes partes do mundo, como Mar do Norte, Oceano Ártico e Antártico,
Baía de Tóquio e Puget Sound (Oceano Pacífico, costa dos EUA) revelaram a predominância
de muitos grupos filogenéticos similares, como Gammaproteobacteria e Deltaproteobacteria,
além de Flavobacteria e Plantomycetes. No Mar Mediterrâneo, outros grupos foram observados
além de Deltaproteobacteria, Gammaproteobacteria e Planctomycetes; estes foram:
Betaproteobacteria, Acidobacteria, Bacteroidetes e Firmicutes (KÖCHLING et al., 2011;
ZINGER et al., 2011; GOBET, BÖER, HUSE, 2012; ACOSTA-GONZÁLEZ, ROSSELÓ-
MÓRA, MARQUÉZ, 2013). Já o Domínio Archaea tem suas assembleias dominadas pelos filos
Euryarchaeota e Crenarchaeota, associados a processos de metanogênese e oxidação anaeróbica
do metano (ACOSTA-GONZÁLEZ, 2013).
A maioria dos estudos vem mostrando que tais padrões de diversidade podem ser
explicados por uma combinação de efeitos da distância espacial e outras variáveis físicas,
químicas ou biológicas. No caso da estrutura de comunidades bentônicas de bactérias, variações
de profundidade da água e na produtividade na superfície da água se mostraram grandes
direcionadores de diversidade (ZINGER et al., 2011). Particularmente os padrões dependentes
de distância geográfica já foram observados em ambientes marinhos do Atlântico Sul, até
18.000 Km de distância, e do Ártico, em amostras de sedimento superficiais (de 40 a 447 m) e
profundas (até 3.850 m) (JACOB et al., 2013).
2.5. Metabolismo microbiano em sedimentos marinhos
Durante metade dos 4,5 bilhões de anos que a Terra possui, um conjunto de
processos metabólicos mudou profundamente muitas características físicas e químicas do
planeta. Tais processos ocorrem apenas em micro-organismos e são responsáveis por manter o
fluxo de nutrientes e energia, sustentando várias outras formas de vida que evoluíram
subsequentemente. Recentemente, dados oriundos do sequenciamento de vários genomas
microbianos tornaram claro que os micro-organismos se tornaram os engenheiros

37
biogeoquímicos da vida na Terra. Tal biodiversidade é baseada em reações de oxidação e
redução, ou seja, sucessivas transferências de elétrons, entre um número limitado de elementos
químicos - Hidrogênio, Carbono, Nitrogênio, Oxigênio, Enxofre e Fósforo (FALKOWSKI et
al., 2008).
Os micro-organismos são bastante diversos metabolicamente, variando suas fontes
de energia, além de fontes de elétrons e carbono, conforme pode-se observar na Tabela 2
(KARL, 2007). Contudo, todos requerem acesso a fontes de energia e carbono, as quais variam
de acordo com o ambiente em que habitam (ORCUTT et al., 2011).
Tabela 2 - Classificação dos organismos quanto a sua fonte de energia, elétrons e carbono. Adaptado de KARL
(2007).
Fonte de
Energia
Fonte de
elétrons Fonte de carbono
Classificação do
organismo Exemplos
Radiação
solar (foto-)
Composto
inorgânico
(-lito)
CO2 (-autotrófico) Fotolitoautotrófico
Bactéria verde
sulfurosa
Chlorobium
phaeovibrioides
Composto
orgânico
(-organo)
Composto Orgânico (-heterotrófico) Foto-
organoheterotrófico
Cenarchaeum
symbiosum
Energia
química
(quimio-)
Composto
inorgânico
(-lito)
CO2 (-autotrófico) Quimiolitoautotrófico Nitrosomonas
crytolerans
Composto
orgânico
(-organo)
Composto Orgânico (-heterotrófico) Quimio-
organoheterotrófico Syntrophobacter
Decaimento
radioativo
(-radio)
Composto
inorgânico
(-lito)
CO2 (-autotrófico) Radiolitoautotrófico
Cladosporium
sphaerospermum Composto
orgânico
(-organo)
Composto Orgânico (-heterotrófico) Radio-
organoheterotrófico

38
Muito antes do aparecimento dos organismos aeróbicos, o metabolismo anaeróbico
dominava a biosfera. Atualmente, ainda existem muitos hábitats expostos a condições de
anoxia, como é o caso dos sedimentos marinhos. A anoxia pode ter várias causas, dentre elas a
elevada demanda por oxigênio para realização de processos de degradação aeróbica. Como
resultado desse processo, o processo de hipóxia estende-se sobre os ecossistemas marinhos
como uma consequência da eutrofização costeira e do aquecimento global (ACOSTA-
GONZÁLEZ, 2013).
Os sedimentos costeiros são caracterizados por importantes processos de entrada de
matéria orgânica de origem continental. Em oceano aberto, apenas uma pequena parte dessa
matéria orgânica acumula-se no leito submarino devido aos processos de remineralização que
ocorrem enquanto decantam na coluna d’água. Já nos rasos ambientes costeiros, o
processamento da matéria orgânica na coluna d’água é limitado, o que resulta em sua
acumulação no sedimento de forma não degradada. Consequentemente, nas regiões costeiras
os processos de remineralização ocorrem nos sedimentos. São os micro-organismos que
dominam tais transformações, balanceando e mantendo os ciclos biogeoquímicos globais
(ORCUTT et al., 2011).
Uma vez nos sedimentos, a matéria orgânica é oxidada principalmente por bactérias
quimio-organoheterotróficas, as quais causam, em consequência, a redução de diferentes
aceptores de elétrons disponíveis (LLOBET-BROSSA et al., 2002). Em sedimentos marinhos,
o aceptor final de elétrons mais abundante é o sulfato, presente na água do mar. Outro aceptor
comum é o oxigênio, assim como também os óxidos de ferro e manganês, nitrato e dióxido de
carbono. Em um perfil de sedimento típico, uma sucessão entre os aceptores de elétrons é
observada. Primeiramente, os micro-organismos consomem o oxigênio que penetra nos
primeiros milímetros ou centímetros de sedimento, e, uma vez consumido o oxigênio,
condições anóxidas são estabelecidas. Abaixo a zona de influência do oxigênio, os processos
anóxidos são estimulados se houver matéria orgânica biodegradável presente. Nesse caso,
nitrato, manganês, ferro e sulfato, se presentes, podem ser utilizados no processo de respiração
anaeróbica. São formadas, portanto, diferentes zonas redox que podem ser diferenciadas por
uma simples inspeção a olho nu: (1) uma camada superior oxidada de coloração marrom; (2)
uma zona de transição anóxida onde nitrato e óxidos de ferro e/ou manganês são reduzidos; e
(3) uma camada sulfídica reduzida de coloração verde escura, onde a redução de sulfato
predomina (MOXLEY, SCHMIDT, 2012). A ordem de utilização dos diferentes aceptores de
elétrons pode ser explicada pela tendência desses compostos em aceitar elétrons, ou seja, pela
eletronegatividade dos elementos que os formam. Dessa forma, o oxigênio, mais eletronegativo,

39
é o primeiro a ser deplecionado enquanto o dióxido de carbono é o último (KIRCHMAN, 2012;
ACOSTA-GONZÁLEZ, 2013), conforme pode-se observar na Figura 4. Em sedimentos
marinhos, o sulfato, aceptor presente em maior concentração na água do mar, é o aceptor de
elétrons dominante no metabolismo anaeróbico. Particularmente em sedimentos costeiros ricos
em matéria orgânica, populações de bactérias heterotróficas deplecionam o sulfato e outros
aceptores rapidamente, permitindo a oxidação dessa matéria orgânica e sustentando populações
microbianas abundantes (TESKE; SØRENSEN, 2008).
A metanogênese também pode ocorrer nos sedimentos dependendo da interação dos
metanogênicos com os redutores de sulfato, pois são mutuamente excludentes na maior parte
dos ambientes ao competirem por hidrogênio e acetato. É importante destacar que tanto
organismos quimiolitoautotróficos como processos abióticos podem oxidar os compostos
reduzidos pelos organismos quimio-organoheterotróficos, contribuindo para a reciclagem dos
elementos nos sedimentos (ACOSTA-GONZÁLEZ, 2013).
Figura 4 - Processos geoquímicos em sedimentos costeiros. O perfil vertical mostra um gradiente entre uma zona
oxidada e uma zona reduzida determinados pela disponibilidade de aceptores de elétrons. As zonas geoquímicas
não são mostradas com escala para melhor visualização, mas representam o máximo de 1 metro de perfil de
sedimento. FONTE: Kirchman, 2012; Acosta-González, 2013.

40
2.6. Influências antrópicas sobre o ambiente marinho
Os ecossistemas marinhos proporcionam uma variedade de serviços, incluindo
fornecimento de alimentos, regulação climática, produtividade primária e reciclagem de
nutrientes. Entretanto, muitos deles se encontram em um estado de degradação tal que
compromete os serviços que podem desempenhar. Portanto, atualmente, um dos desafios da
ecologia marinha é relacionar a natureza e magnitude dos serviços ecossistêmicos prestados
pelas regiões costeiras aos hábitats e comunidades biológicas que as habitam, à diversidade que
contêm e aos tipos e níveis de distúrbios que suportam. A principal causa desse fenômeno, o
aumento da população humana, foi acompanhada de um crescimento na diversidade e na
intensidade dos impactos ambientais de origem antrópica, afetando todos os tipos de
ecossistemas em tamanha intensidade que podem tornar os esforços de manejo e conservação
tardios (ELLIS et al., 2011).
As zonas costeiras são extremamente diversas e muito importantes, cobrindo cerca
de 20% da superfície terrestre, constituindo rotas de transporte e parques industriais, áreas de
habitação, recreação, turismo e fonte de minerais, mas também áreas com ecossistemas ricos
em biodiversidade, os quais funcionam como importantes consolidadores de sedimentos e
tampões para as mudanças que ocorrem no mar ou no continente. Os ecossistemas costeiros
constituem os sistemas mais diversos e produtivos do planeta. Tão expressiva biodiversidade é
essencial para seu funcionamento adequado e para importantes propriedades, como alta
estabilidade (resistência e resiliência) a perturbações naturais e antropogênicas (JOHNSTON;
ROBERTS, 2009). Entretanto, considerando que a humanidade vem utilizando as regiões
costeiras de todo o mundo por séculos, as influências de origem humana certamente vêm
ocorrendo. Isso significa que as zonas costeiras entraram em contato com grandes quantidades
de substâncias naturais e xenobióticas oriundas, principalmente, nas diversas atividades
antrópicas. Algumas dessas ações incluem a pesca e a aquicultura, o escoamento superficial, as
atividades de extração, transporte e refino de petróleo, engenharia costeira, transporte marítimo,
turismo, mineração e lançamento de efluentes industriais (NOGALES et al., 2011), cujas
distribuição e magnitude são alvo de considerável interesse investigativo (PORT et al., 2012).
Grande parte de tais ações ocorreu devido a busca por recursos originados nos oceanos, os quais
podem ser categorizados em recursos físicos, energéticos, biológicos e não extrativos; este
último tipo referindo-se à utilização do oceano para transporte, recreação e despejo de resíduos
(GARRISON, 2011).

41
Um estudo visando mapear os impactos humanos sobre os ecossistemas marinhos
analisou uma variedade de ecossistemas (recifes de coral, áreas rasas, áreas de plataforma
continental e talude, águas pelágicas e de elevada profundidade) e encontrou que todos os
ecossistemas oceânicos analisados poderiam ser considerados antropogenicamente impactados,
mesmo que em diferentes graus. Os ambientes costeiros e áreas da plataforma continental, com
profundidades de até 200 metros, mostraram ser os mais impactados. Estas constituem as áreas
mais intensamente exploradas e que recebem o maior impacto das atividades continentais
(SPALDING et al., 2007; HALPERN et al., 2007). Os autores revelaram também que, em
escala global, a navegação comercial e as atividades derivadas da pesca possuem o maior grau
de ameaça. A Tabela 3 sumariza os riscos impostos por algumas atividades antropogênicas aos
ecossistemas marinhos. Em geral, os contaminantes advindos de tais ações afetam a coluna
d’água e os sedimentos, onde podem se acumular. Além disso, há a troca bidirecional entre
atmosfera e superfície da água por meio de deposição de compostos do ar ou sua liberação a
partir da água (NOGALES et al., 2011).
Tabela 3 - Sumário dos riscos impostos por atividades antropogênicas aos ecossistemas
marinhos.
Atividade humana Poluentes Impactos e riscos associados
Agricultura e
pecuária
Fertilizantes, pesticidas,
antibióticos e estrume
Enriquecimento com nutrientes, hipóxia/anoxia,
desenvolvimento de blooms algais tóxicos,
bioacumulação, patógenos e difusão de resistência a
antibióticos
Desenvolvimento
urbano
Resíduos domésticos, esgotos e
resíduos de queima de
combustíveis fósseis
Enriquecimento orgânico, eutrofização, patógenos,
hipóxia/anoxia, desenvolvimento de blooms algais
tóxicos, toxicidade por compostos químicos e metais
pesados, perda de valor recreacional, acúmulo de
resíduos sólidos
Indústrias
Resíduos industriais,
xenobióticos, metais pesados,
radionuclídeos e resíduos de
queima de combustíveis fósseis
Toxicidade, bioacumulação, aumento da deposição
atmosférica
Transporte marítimo
Xenobióticos, agentes anti-
incrustação, metais pesados,
água de lastro e resíduos
sólidos
Toxicidade, bioacumulação, introdução de espécies
exóticas e patógenos

42
Extração e refino de
óleo
Combustão de
combustíveis fósseis
Hidrocarbonetos, metais
pesados, calor e dióxido de
carbono
Toxicidade, bioacumulação, aumento da deposição
atmosférica, aquecimento
Turismo (incluindo
atividades náuticas
recreativas)
Resíduos sólidos, resíduos
fecais, nutrientes e xenobióticos
Morte de membros da fauna, acumulação de
sedimentos, hipóxia/anoxia, perda de valor recreacional,
enriquecimento com nutrientes, desenvolvimento de
blooms algais tóxicos e patógenos
Dragagem Partículas, nutrientes, poluentes
orgânicos, metais pesados
Turbidez, diminuição da penetração de luz,
enriquecimento com nutrientes, toxicidade e
bioacumulação
Pesca Peixes descartados, patógenos,
xenobióticos
Destruição de hábitats, decréscimos dos estoques,
patógenos, toxicidade e bioacumulação
Aquicultura Input orgânico, resíduos fecais,
antibióticos
Enriquecimento orgânico, hipóxia/anoxia nos
sedimentos, patógenos, introdução de espécies exóticas,
disseminação de resistência a antibióticos
FONTE: NOGALES et al., 2011.
2.6.1. Os impactos decorrentes do transporte marítimo e operação de portos
As pessoas vêm utilizando os oceanos para o transporte há milhares de anos.
Entretanto, diferente do que ocorria há algumas décadas, os navios agora transportam
principalmente bens em vez de pessoas em decorrência do crescimento do comércio global.
Cerca de 8,4 bilhões de toneladas são transportadas por mar todo ano, correspondendo a cerca
de 90% do comércio mundial (GRECH et al., 2013). Atualmente, particularmente o transporte
de petróleo por meio de petroleiros representa a maior porção de carga transportada, um total
de 341 milhões de toneladas (65% do total). Em segundo lugar aparece o transporte de minérios
de ferro, carvão e grãos (24%) (GARRISON, 2011).
As inovações foram essenciais para aumentar a importância desse tipo de
transporte. O tamanho, design, especialização e velocidade dos navios foram campos de
grandes novidades nos anos recentes. Entretanto, apesar de reduzirem os custos e tempo de
transporte, melhorias nas embarcações necessitam de desenvolvimentos compatíveis das
estruturas portuárias. As autoridades portuárias devem responder através da expansão da
infraestrutura e da melhoria do acesso ao porto. Soma-se a isso o fato de a carga não ser mais
embarcada e desembarcada uma a uma, mas sim de forma automatizada em contêineres,
permitindo rapidez e eficiência na circulação de mercadorias. O maior porto do mundo, o Porto

43
de Singapura, processou em 2009 cerca de 29 milhões de TEUs (unidades equivalentes a 20
pés, medida padrão para calcular o volume de um contêiner), correspondente a mais de 500
milhões de toneladas de cargas transportadas (DIAS, 2013).
Apesar de representarem um bom indicador da economia de um local devido ao
grande fluxo de transações comerciais ocorrentes, os portos constituem o tipo de ambiente
costeiro mais alterado e estressado, caracterizando-se por considerável poluição da água, ar e
terra, pela redução nos níveis de oxigênio dissolvido, pelas altas concentrações de
contaminantes e pela baixa diversidade da comunidade bentônica em geral (INGOLE et al.,
2009). Alguns poluentes comumente encontrados em portos são: hidrocarbonetos, surfactantes
e compostos anti-incrustantes (principalmente metais pesados) (NOGALES et al., 2011).
Portanto, juntamente com o crescimento das estruturas portuárias cresceram também os
impactos negativos ou externalidades sobre o meio ambiente. Os efeitos ambientais dos portos
podem ser diretos, i.e., ocorrem na área do porto, ou indiretos, como resultado dos movimentos
das embarcações ou pelo uso de outros tipos de veículos para escoar os produtos (OECD, 2011).
Alguns impactos diretos específicos incluem (TROZZI; VACCARO, 2000):
Efeitos sobre ecossistemas costeiros sensíveis;
Efeitos sobre a circulação oceânica;
Contaminação de sedimentos;
Poluição do ar e da água;
Derramamentos de óleo;
Descarga de água de lastro e introdução de espécies exóticas;
Poluição por resíduos e compostos originados de produtos anti-incrustação;
Contaminação com resíduos de dragagem;
Ruídos, odores e poluição visual;
Perigos de contaminação e acidentes durante a recepção de cargas
perigosas.
As embarcações e portos, nas extremidades das rotas de navegação marítima,
representam papéis significativos e crescentes para a conservação da biodiversidade marinha e
costeira. Entretanto, dentre as fontes de poluição do ar e da água, a navegação marítima é uma
das mais difíceis de regular. As fontes de contaminação podem ser várias, tanto resultantes de
acidentes como de atividades rotineiras, possuindo graves consequências para a biodiversidade.

44
Houve algum progresso para reduzir os impactos dos portos e da navegação com a implantação
de normas regionais e internacionais, como é o caso da Convenção Internacional para
Prevenção da Poluição por Navios – MARPOL, de 1972, que visa atingir a completa eliminação
da poluição global do ambiente marinho por petróleo e outras substâncias perigosas assim como
dos riscos de derrames acidentais. Contudo, estes não foram suficientes para balancear o grande
aumento no tráfego de navios e no desenvolvimento de portos desde os anos 70 (GRECH et al.,
2013).
Commendatore e Esteves (2007) relataram que, na Patagônia argentina, o aumento
do tráfego marítimo devido ao aumento da importância turística local, da pesca e das atividades
mercantes marítimas, em uma infraestrutura portuária ineficiente, contribuiu bastante para a
disposição e manejo inadequado dos resíduos e combustíveis. As violações à Convenção
MARPOL durante atividades rotineiras, como carga, descarga e lavagem de navios, impedem
que a atividade de transporte marítimo seja realizada de forma sustentável, constituindo um
sério problema mundial. Segundo Ball, (1999), 80% dos derramamentos de petróleo e derivados
ocorrem em águas portuárias. Trata-se de um dado preocupante, pois a entrada de
contaminantes, principalmente derivados do petróleo, em áreas portuárias de baixa energia pode
resultar em contaminação crônica, afetando as áreas em si e regiões adjacentes. A extensão de
tal dano dependerá de fatores importantes que governam a distribuição de contaminantes no
ambiente, como as correntes marítimas, ventos predominantes, características sedimentares,
morfologia da costa e comportamento dos organismos marinhos presentes no local
(COMMENDATORE; ESTEVES, 2007).
A contaminação por hidrocarbonetos é um problema ambiental altamente
difundido, especialmente em áreas costeiras expostas aos efeitos das atividades antrópicas.
Dentre esses, os portos se destacam por serem sistemas, em geral, cronicamente contaminados
com hidrocarbonetos oriundos de derrames de óleo, escoamento superficial urbano, navegação
marítima e atividades industriais associadas. As elevadas concentrações de hidrocarbonetos se
acumulam nos sedimentos superficiais e sub-superficiais, impondo riscos ambientais e de saúde
pública. Os problemas são intensificados quando os sedimentos sofrem ressuspensão natural ou
por atividades de dragagem, pois há a formação de plumas de contaminação que se espalham
para as áreas adjacentes; nesse caso, os sedimentos funcionam como fontes secundárias de
contaminação. Além disso, devido aos altos níveis de contaminação, muitas vezes os
sedimentos dragados não podem ser reutilizados. Dessa forma, há uma necessidade crescente
de se reduzirem as concentrações de contaminantes por meio de redução de sua liberação e
biorremediação de sedimentos contaminados (DELL’ANNO et al., 2012).

45
No Ceará, o último derramamento de óleo de grandes proporções foi registrado em
março de 2008 devido a uma colisão entre um rebocador e um navio cargueiro de 140 m de
extensão, o Chembulk Shangai, de Cingapura. Ambos navegavam na área do Porto do
Mucuripe, em Fortaleza (Ceará, Brasil), tendo o vazamento liberado cerca de três toneladas de
óleo combustível. Os efeitos foram sentidos no Litoral Oeste do estado, principalmente na Praia
de Icaraí, no município de Caucaia, distante 20 Km do local do acidente (DIÁRIO DO
NORDESTE, 2008). Neste mesmo ano, mais três acidentes envolvendo vazamento de
derivados de petróleo ocorreram no mesmo porto (IBAMA, 2009).
É importante ressaltar que a contribuição dos grandes acidentes de derramamento
de petróleo para o total de contaminação por hidrocarbonetos no mar é muito baixa. No Mar
Mediterrâneo, por exemplo, 80 mil toneladas de óleo foram despejadas em acidentes entre 1999
e 2005. Entretanto, 250 mil toneladas são despejadas, por ano, como resultado das operações
rotineiras de navios (i.e. lançamento de água de lastro, lavagem de tanques e resíduos de salas
de máquinas) e 120 mil toneladas, também por ano, de oleodutos e operações no continente
(EEA, 2006).
Uma vez na coluna d’água, os hidrocarbonetos, particularmente os HPAs,
particionam nas partículas suspensas ou nos sedimentos de fundo, resultando em uma
concentração de HPAs nos sedimentos cerca de 1000 vezes maior que na coluna d’água
(HAYES; LOVLEY, 2002), o que previne sua utilização pelo bacterioplâncton e promove sua
acumulação em fase sólida (JOHNSEN, WICK, HARMS, 2005; PAISSÉ et al., 2008). Além
disso, os sedimentos contaminados podem agir como reservatórios e fontes secundárias de
contaminação através da liberação dos contaminantes a depender das características e condições
ambientais locais. Portanto, há uma necessidade urgente de estudos com foco detalhado em
sedimentos de áreas portuárias e no planejamento de estratégias adequadas para a proteção das
zonas costeiras para que não sejam afetadas as funções do próprio ecossistema, além das
atividades relacionadas ao turismo e à pesca (COMMENDATORE; ESTEVES, 2007).
Os HPAs, poluentes ubíquos e de difícil degradação, constituem um sério problema,
especialmente nos ecossistemas marinhos devido a contaminação resultante de derramamentos
de óleo e problemas de vazamento de tubulações. Ademais, muitos deles, principalmente os de
maior peso molecular, são identificados como carcinogênicos e mutagênicos. As concentrações
de HPAs em sedimentos marinhos podem variar de 0,32 a 170.000 ng g-1 ao redor do mundo
(LATIMER, ZHENG, 2003). As maiores concentrações são encontradas geralmente em portos
e estaleiros próximo a centros urbanos (GRECH et al., 2013).

46
No ambiente marinho, em geral, esses compostos estarão disponíveis para
degradação sob condições aeróbicas, o que tem sido extensivamente documentado (ATLAS et
al., 1991). Entretanto, grande parte do metabolismo da matéria orgânica em sedimentos
marinhos é, na verdade, anaeróbica. De fato, nas últimas décadas, alguns estudos mostraram o
potencial de degradação anaeróbica de hidrocarbonetos em sedimentos marinhos costeiros,
tendo os processos ocorridos sob condições de redução de sulfato (WIDDEL; RABUS, 2001),
e indicado o metabolismo dos HPAs acoplado à redução do sulfato como um dos mais
relevantes dentre os processos anaeróbicos. Esses dados são interessantes porque até
recentemente se pensava que os HPAs não poderiam ser degradados sob condições anaeróbicas.
Coates et al. (1997) relataram a degradação de naftaleno, fenantreno, fluoreno, fluoranteno e
metilnaftaleno em sedimentos marinhos com o sulfato servindo como aceptor final de elétrons.
Esses dados são significantes, pois sugerem que, diferentemente do que se pensava antes, os
HPAs podem não persistir sob condições anaeróbicas de redução de sulfato, tornando muito
relevante o potencial biorremediação dos sedimentos de portos (HAYES, NEVIN, LOVLEY,
1999).
A redução dissimilatória de sulfato (SRD) é a principal via para mineralização de
carbono orgânico em sedimentos costeiros marinhos, sendo responsável por cerca de 50% da
mineralização de carbono. Os procariotos redutores de sulfato são componentes diversos e
ubíquos das comunidades bacterianas (MIYATAKE, MACGREGOR, BOSCHKER, 2009), já
tendo sido investigados através do uso de bibliotecas de rRNA 16S, de genes para redução
dissimilatória do sulfato ou por técnicas de Hibridização de Fluorescência In vitro (FISH –
Fluorescence In Vitro Hybridization) (HAYES; LOVLEY, 2002). Outro estudo recente sobre
a composição de assembleias redutoras de sulfato em sedimentos portuários encontrou
sequências relacionadas a redutores de sulfato cultiváveis capazes de metabolizar compostos
aromáticos, como benzoato e naftaleno (CHIN et al., 2008).
A descoberta de tais micro-organismos capazes de metabolizar xenobióticos em
condições consideradas hostis impulsionou a pesquisa por processos de biorremediação
baseados no estímulo da microbiota autóctone. A biorremediação é uma estratégia
ambientalmente limpa que ganha popularidade atualmente devido a seu potencial de limpar
ambientes contaminados. Dessa forma, tem se procurado estratégias e tecnologias
ambientalmente seguras para serem aplicadas em processos de remediação de sedimentos
contaminados por hidrocarbonetos. Dentre essas, biotecnologias baseadas no bio-estímulo da
comunidade autóctone para acelerar os processos de biodegradação de poluentes orgânicos são
de particular relevância (BEOLCHINI et al., 2010).

47
Contudo, a estratégia mais efetiva para contribuir com uma melhoria na qualidade
ambiental em qualquer ambiente é a redução. Dessa forma, numa tentativa de combater a
liberação dos contaminantes, a Organização Europeia de Portos Marítimos (ESPO) criou em
1994 uma rede de portos credenciados, chamada ECOPORTS, a qual visa a identificação de
riscos ambientais e o estabelecimento de ações para combatê-los ou controlá-los, integrando o
gerenciamento ambiental ao gerenciamento portuário, tendo como resultado final a proteção
ambiental. Trata-se de uma iniciativa única, contando atualmente com 69 portos europeus e 1
porto no Oriente Médio (ESPO, 2013). Espera-se que adquira bastante signatários, de modo a
permitir uma operação mais ambientalmente segura das estruturas portuárias, tão importantes
mundialmente.
2.7. Efeitos dos contaminantes sobre a biota marinha
As preocupações com os ecossistemas costeiros de todo o mundo têm estimulado
muitas pesquisas cujo foco é o efeito de poluentes sobre a biota marinha (NIPPER, 2000). Nesse
contexto, o Brasil, como um país de dimensões continentais com uma das maiores costas do
mundo (~7500 km), se destaca por ter grande parte de sua costa desconhecida e por abrigar
algumas indústrias e portos marítimos de grandes proporções. Os portos, em particular, são
intervenções antrópicas capazes de causar sérias alterações nos ecossistemas costeiros via
dragagem, operações de manutenção de navios e derrames acidentais (NIPPER, 2000).
Portanto, constituem ambientes muito alterados e estressados com níveis significantes de
poluentes na água, no ar e nos sedimentos (INGOLE et al., 2009).
Segundo Ager et al. (2010), atualmente, um dos maiores desafios científicos é
mitigar as ameaças antropogênicas contra os ambientes naturais e a diversidade. Para superar-
se tal desafio, a detecção e a quantificação da biodiversidade é fundamental, levando a um
melhor entendimento dos ecossistemas e dos serviços que são capazes de realizar, bem como
dos efeitos das atividades antrópicas e de seu potencial ecossistêmico de recuperação. Deste
modo, o estudo da biodiversidade apresenta-se como chave para entender verdadeiramente os
complexos processos que ocorrem nos ecossistemas e que necessitam ser considerados quando
da tomada de decisões relacionadas ao manejo ambiental e à conservação de recursos naturais
Ademais, considerando que muitos organismos podem ser afetados por variadas mudanças
ambientais e que as modificações induzidas podem ser bastante sutis, uma avaliação mais global

48
e detalhada das comunidades naturais pode permitir o acesso a informações muito importantes
a seu respeito (KISAND et al., 2012).
A poluição marinha refere-se à introdução no oceano pelo homem de substâncias e
energias capazes de modificar a qualidade da água ou afetar as características físicas, químicas
ou biológicas do ambiente. A identificação de um poluente não é tarefa fácil, pois este pode ser
originado em grandes quantidades em processos naturais, como, por exemplo, uma erupção
vulcânica, capaz de liberar imensas quantidades de dióxido de carbono, metano, compostos
sulfurosos e óxidos de nitrogênio. Contudo, as atividades antrópicas produzem tais substâncias
em excesso, intensificando seus efeitos sobre os ecossistemas. Infelizmente, não se sabe em que
extensão o homem contaminou os oceanos, pois quando as mensurações de poluentes
começaram a ser feitas os efeitos de vários deles já haviam ocorrido, de forma que é possível
encontrar-se traços de compostos sintéticos em praticamente qualquer ponto do ambiente
marinho. O próprio conhecimento acerca de condições pristinas dos oceanos é muito limitado,
sendo restrito a amostras de água de blocos de gelo polares ou de bolhas aprisionadas em
glaciares. Não é possível afirmar que ainda existam locais não perturbados ou biota não afetada
a serem estudados. Dentre os principais poluentes podemos citar compostos orgânicos, metais
pesados, plásticos e outras formas de resíduos sólidos (GARRISON, 2011).
No caso dos resíduos sólidos, somente de plásticos, das 120 milhões de toneladas
métricas que são produzidas anualmente, 10% acaba no oceano, sendo que apenas 15% deste
percentual tem origem em navios e plataformas petrolíferas; grande parte provém do continente.
Os mesmos atributos que os fazem úteis, sua durabilidade e estabilidade, são as mesmas que os
tornam problemáticos no ambiente marinho. Os danos que podem causar se dão principalmente
devido à ingestão de resíduos ou aprisionamento nos mesmos. Recentemente, foi descoberta
uma mancha de resíduos sólidos do tamanho do Estado americano do Texas no Giro subtropical
do Pacífico Norte, a qual ficou conhecida, dentre outras denominações, como “Vórtex de lixo”
(HOWELL et al., 2012).
As pressões sobre a biota impostas por atividades portuárias e afins são múltiplas.
A seguir são expostas algumas pressões sobre a biodiversidade resultantes de construção de
portos e da atividade de navegação marítima (GRECH et al., 2013):
Remoção de organismos associados aos sedimentos durante as operações de
dragagem;
Sufocamento causado pelo despejo de material dragado;

49
Erosão costeira devido às mudanças no hidrodinamismo com consequentes
alterações nos hábitats;
Perda direta de hábitat causada pela construção e desenvolvimento da
estrutura portuária;
Danos aos hábitats e à biota causados pelo despejo de lixo;
Danos físicos causados pelo impacto das embarcações e âncoras nos
hábitats bentônicos;
Turbulência causada pelo movimento das embarcações;
Contaminação causada pela liberação de contaminantes;
Distúrbio no fundo causando remobilização de contaminantes sintéticos,
como hidrocarbonetos, resíduos de carvão e metais pesados, com efeitos
sobre a biota;
Descarga de óleo das embarcações e equipamentos durante operações
rotineiras e acidentes;
Introdução de espécies exóticas por meio de dragagem, descarga de água de
lastro e bioincrustação;
Efeitos da poluição sonora e visual sobre e sob as águas.
2.7.1. Efeitos sobre a microbiota
Quando se trata particularmente de micro-organismos, observam-se efeitos bastante
complexos das perturbações antrópicas sobre as comunidades microbianas marinhas, conforme
mostrado no esquema apresentado no Figura 5, adaptado de Nogales et al. (2011). Em geral,
estresses múltiplos, de origem antropogênica e naturais, coexistem em uma área, onde há uma
combinação de riscos de poluição. Esses estresses podem interagir, podendo ter efeitos
sinergéticos ou antagonistas, o que proporciona uma ideia da complexidade das possíveis
respostas ecossistêmicas a perturbações. Por outro lado, é importante considerar a magnitude e
a frequência dos impactos e o fato de que em ecossistemas costeiros a magnitude do impacto
nem sempre é proporcional à quantidade de poluente. Na verdade, o impacto em ecossistemas
marinhos também é dependente de atributos locais, como energia das marés, ventos, correntes,
batimetria, tempo de residência da água, condições meteorológicas e climáticas (NOGALES et
al., 2011).

50
Quando uma comunidade microbiana não muda após um distúrbio, pode-se dizer
que ela é resistente. Já se a comunidade mudar sua composição e recuperar-se, retornando à
composição original, é considerada resiliente. Por outro lado, mesmo que sejam verificadas
mudanças na composição da comunidade, pode-se não observar alterações funcionais, o que é
explicado pelo conceito de redundância funcional. Segundo este conceito, táxons diferentes
podem desempenhar as mesmas funções nos ecossistemas. Contudo, se a alteração for capaz de
modificar a funcionalidade da comunidade, mudanças nos serviços ecossistêmicos também
ocorrerão, implicando em efeitos negativos na saúde do ecossistema e em seu valor econômico
e social (MARTINY et al., 2011). Há, portanto, importantes atributos a serem considerados
para estabelecer a ligação entre distúrbios ambientais e padrões na diversidade e funcionamento
de comunidades microbianas, assim como com seus efeitos sobre o funcionamento dos
ecossistemas (BÖER et al., 2009).
Figura 5 - Esquema conceitual dos efeitos de distúrbios ambientais sobre as comunidades microbianas marinhas.
Adaptado de Nogales et al. (2011).
A presença de contaminantes especificamente em sedimentos marinhos pode ter um
grande impacto sobre as comunidades microbianas associadas através de sua toxicidade e
efeitos sobre a estrutura e funcionamento das mesmas. Por sua vez, os micro-organismos podem
desempenhar papéis importantes sobre o comportamento dos contaminantes através de efeitos

51
diretos, via metabolização de contaminantes orgânicos e remoção destes compostos do sistema,
e indiretos, por meio do aumento da estabilidade do meio e consequente redução na
ressuspensão e redistribuição dos contaminantes (SLATER et al., 2008).
Portanto, os micro-organismos podem ser muito úteis no controle de alterações
ambientais. Além de serem capazes de viver em variados tipos de hábitats, influenciam muitos
processos globais, como os ciclos biogeoquímicos do carbono, do nitrogênio e do enxofre
(KISAND et al., 2012), exercendo importantes papéis como recicladores primários de matéria
orgânica (ZINGER et al., 2011) e como responsáveis por 98% da produtividade marinha
(KENNEDY et al., 2010).
Apesar de até recentemente pouca atenção ter sido dispensada à avaliação
sistemática da diversidade microbiana devido ao vasto número de micro-organismos não
cultiváveis existentes, esse cenário vem mudando. O advento das recentes técnicas de
sequenciamento de nova geração, em paralelo com a metagenômica, vêm sendo de fundamental
importância para permitir o acesso à grande parcela de micro-organismos não cultiváveis. Esses
novos métodos expandiram e continuam a fazê-lo rapidamente, se tornando mais baratos e
permitindo uma transformação na nossa visão acerca do mundo microbiano (WOOLEY et al.,
2010).
Apesar do advento de tais técnicas ser recente, seu impacto pode ser sentido em
muitas áreas das ciências da saúde e ambientais (ARNDT et al., 2012). Atualmente, é possível
identificar o microbioma fundamental de cada ambiente, ou seja, os micro-organismos que são
críticos para o seu funcionamento apropriado, o que é essencial para desvendar a ecologia de
comunidades microbianas afetadas por atividade antropogênicas. A identificação desse
microbioma fundamental é o primeiro passo para que se possa, futuramente, definir o status de
saúde de um ambiente ou para prever as respostas das comunidades frente às mudanças
ambientais (SHADE; HANDELSMAN, 2012).
2.8. Ferramentas de estudo da diversidade microbiana
A microbiologia passou por muitas mudanças nos últimos 30 anos que
transformaram a forma como os microbiologistas viam os micro-organismos. Dentre tais
alterações, a constatação de que a maioria não pode ser cultivada em culturas puras foi a maior
e fez com que se questionasse o que se sabia até então sobre o mundo microbiano e sobre a
melhor forma de estudá-lo (HANDELSMAN, 2004).

52
Nos anos seguintes, os microbiologistas dedicaram intensos esforços para descrever
a diversidade filogenética dos mais variados ambientes, desde superfícies oceânicas às
profundezas dos mares, conseguindo descrever muitas linhagens novas não cultiváveis. O
desafio seguinte foi elucidar as funções desses filotipos e determinar se representavam novas
espécies, gêneros ou filos procarióticos. Em questão de poucos anos, o estudo de micro-
organismos não cultiváveis evoluiu tanto que se expandiu além da pergunta ‘Quem está aqui?’
para tentar responder também à ‘O que estão fazendo?’ (HANDELSMAN, 2004).
Apesar dos avanços proporcionados pelos estudos baseados na caracterização de
culturas puras, a descoberta de que o cultivo não era capaz de capturar o espectro total da
diversidade microbiana trouxe um novo olhar para a microbiologia. A ‘Grande Anomalia da
Contagem em Placas’ (STALEY, KONOPKA, 1985), capaz de demonstrar a discrepância entre
os tamanhos de população obtidos via contagem em placas e por microscopia, representou um
dos indícios mais importantes de que muita coisa não estava sendo considerada. Os autores
mostraram que, em ambientes oligotróficos (águas oceânicas e sedimentos profundos) e
mesotróficos (sedimentos costeiros e lagos), menos que 1% das bactérias viáveis totais podem
ser enumeradas ou isoladas por diferentes métodos de cultivo. Em seguida, observou-se em
solos que apenas 0.1 a 1% das bactérias são prontamente cultiváveis em meios convencionais
e condições padrão (TORSVIK; ØVREÅS, 2002).
Nesse contexto histórico, surgiram um conjunto de técnicas que conseguiam ir além
das limitações do cultivo, as técnicas independentes de cultivo. Essas metodologias
proporcionaram grandes oportunidades para superar a necessidade de cultura de micro-
organismos e aumentar nosso entendimento a respeito da diversidade microbiana e de sua
funcionalidade no ambiente. Tais técnicas baseiam-se na caracterização de constituintes
celulares extraídos diretamente de amostras ambientais sem a necessidade de cultivá-las
(MALIK et al., 2008).
Existe uma grande variedade de técnicas independentes de cultivo. Cada qual tem
características próprias que as tornam adequadas para diferentes estudos, com possibilidade de
responder a variadas perguntas (CARDENAS; TIEDJE, 2008). Podemos dividi-las em métodos
moleculares ou bioquímicos com base na utilização ou não de ácidos nucleicos,
respectivamente. Particularmente as técnicas moleculares vêm permitindo a realização de
investigações independentes de cultivo nos mais diversos ambientes.
Grande parte dos estudos moleculares foi realizada com sequências do gene da
subunidade menor do RNA ribossomal (SSU RNA ou 16S rRNA), tendo se popularizado desde
que Pace e colaboradores (1985) propuseram a clonagem direta de DNA ambiental para tais

53
estudos. Recentemente, alguns genes codificadores de proteínas passaram a ser utilizados, a
exemplo do gene da ribulose-1,5-bifosfato carboxilase/oxigenase (RuBisCO), da amônia
monoxigenase (amoA), tioesterase/tiohidrolase do sulfato (soxB) e da redutase da metil-
coenzima M (mcrA) (SU et al., 2012). Contudo, a comparação de rRNAs constitui a primeira
descrição taxonômica consistente para micro-organismos. Consequentemente o rRNA se
tornou a medida de diversidade microbiana prevalente (SPIEGELMAN, WHISSEL, GREER,
2005). Entretanto, evidências recentes sugerem que grupos com microdiversidade ribotípica,
como os víbrios, podem ser subestimados, sugerindo-se a utilização de genes constitutivos,
como o gene da subunidade beta da RNA polimerase, como alternativas (CASE et al., 2007).
Há três tipos de rRNA nos ribossomos de procariotos e eucariotos: 5S, 16S e 23S
em procariotos e 5.8S, 18S e 28S em eucariotos, sendo 16S e 18S chamados de subunidades
menores (SSU; do inglês, small subunit) e 23S e 28S de subunidades maiores (LSU; do inglês,
large subunit). O que torna tais sequências tão importantes é que seus genes codificadores são
altamente conservados, presentes em altas quantidades de forma ubíqua, além de serem
essenciais para todas as funções celulares (WOESE, 1987).
Inicialmente, as análises de diversidade microbiana consistiam da extração direta,
purificação e sequenciamento do rRNA 5S. Entretanto, apesar dos avanços proporcionados por
sua utilização, possuem apenas 120 nucleotídeos e pequeno número de posições nucleotídicas
variáveis, o que limitou seu uso para estudar relações filogenéticas mais distantes. Por sua vez,
o rRNA 16S possui aproximadamente 1500 nucleotídeos, tamanho grande o suficiente para ser
usado para inferências filogenéticas. Curiosamente, o rRNA 23S contém o dobro do tamanho
do 16S e poderia proporcionar muitas oportunidades para o estudo de filogenias. Contudo, o
16S tornou-se o gene de referência devido à facilidade de sequenciá-lo (SPIEGELMAN,
WHISSEL, GREER, 2005).
Este gene possui regiões conservadas e hipervariadas, das quais as hipervariadas
constituem os marcadores genéticos mais utilizados em estudos de ecologia microbiana,
variando de 50 a 100 bases em tamanho. Na Figura 6, pode-se observar a quantidade de regiões
hipervariadas presentes no rRNA 16S e como estão localizadas junto às regiões conservadas,
Figura 6 - Regiões conservadas e hipervariadas do gene do RNA ribossomal 16S. As regiões conservadas (C1-
C9) são mostradas em rosa; e as hipervariadas (V1-V9), em diferentes cores. Em detalhe, pode-se observar o
tamanho de uma das regiões hipervariadas (V4) com seus sítios de ligação de iniciadores (Fonte: Petrosino et
al., 2009).

54
assim como detalhes do tamanho e de locais de ligação de iniciadores (PETROSINO et al.,
2009).
2.8.1.1. Métodos de ribotipagem
Dentre as técnicas independentes de cultivo destacam-se as dependentes de PCR,
as quais buscam separar o material genético amplificado originário de diferentes genes
presentes em uma amostra comunitária. Por meio delas, são produzidos perfis das comunidades
analisadas baseados em padrões de separação dos fragmentos de DNA amplificados, os
chamados fingerprintings ou ribotipagens. A partir da seleção de iniciadores adequados
desenhados para serem complementares a genes ou regiões gênicas específicas, é possível
amplificar exponencialmente o gene ou região gênica visada, o que torna a PCR uma técnica
simples, rápida, confiável e barata para obter-se quantidades suficientes de genes alvo
(SPIEGELMAN, WHISSEL, GREER, 2005).
Entretanto, a técnica de PCR possui limitações que podem resultar em dados
tendenciosos ou de baixa qualidade. Tais restrições iniciam-se na manipulação das amostras e
em seu armazenamento, passando por cada etapa de extração do DNA e pela ação de compostos
inibidores da DNA polimerase possivelmente co-extraídos. Outra fonte de entraves é a escolha
dos iniciadores, os quais devem possibilitar a amplificação de toda a região de interesse,
evitando o anelamento preferencial a sequências particulares em detrimento das sequências alvo
e a formação de estruturas secundárias, como grampos (SPIEGELMAN, WHISSEL, GREER,
2005). A presença de múltiplas cópias de algumas sequências e a possibilidade de formação de
quimeras, fitas de DNA derivadas de diferentes organismos, constituem ainda dificuldades
adicionais (THIES, 2008).
As técnicas de ribotipagem são utilizadas para diferenciar a estrutura de
comunidades microbianas. A vantagem dessas técnicas é que o balanço entre custo e benefício
oferecido, pois são mais rápidas e baratas. Além disso, possibilitam a comparação de um grande
número de amostras. As técnicas de ribotipagem mais utilizadas atualmente para caracterizar a
estrutura de comunidades microbianas são a Eletroforese em Gel de Gradiente Desnaturante
(DGGE; do inglês, Denaturing Gradient Gel Electrophoresis) ou a Eletroforese em Gel de
Gradiente de Temperatura (TGGE; do inglês, Temperature Gradient Gel Electrophoresis)
(MUYZER; SMALLA, 1998), a Análise de Polimorfismo do Tamanho do Fragmento de

55
Restrição ou Análise de Polimorfismo do Tamanho do Fragmento de Restrição Terminal (RFLP
ou T-RFLP; do inglês, Terminal-Restriction Fragment Length Polymorphism) (LAGUERRE
et al., 1994; DUNBAR, TICKNOR, KUSKE, 2000) e a Análise do Polimorfismo
Conformacional de Fita Simples (SSCP; do inglês, Single Strand Conformational
Polymorphism) (SCHWIEGER; TEBBE, 1998). Todas essas técnicas se baseiam na separação
de moléculas de DNA amplificadas, os amplicons, de tamanhos semelhantes em diferentes
bandas ou Unidades Taxonômicas Operacionais (UTO ou OTU, do inglês, Operational
Taxonomic Units), que serão utilizadas para a comparação das comunidades estudadas (THIES,
2008).
A DGGE, uma das técnicas utilizadas nesse trabalho, separa os amplicons com base
em um gradiente de condições de desnaturação ao longo de um gel de poliacrilamida. Os
amplicons de fita dupla são adicionados a poços presentes no topo do gel e, conforme o DNA
penetra no gel e começa a migrar, vão atingindo gradualmente porções do gel com maiores
condições de desnaturação conferidas por desnaturantes químicos, tipicamente uréia e
formamida. Como as moléculas de DNA amplificadas têm fita dupla, que possui uma estrutura
bastante compacta, tendem a migrar mais rápido do que fitas de DNA que sofrem desnaturação
devido ao gradiente de desnaturação presente no gel. Os amplicons que sofrem desnaturação
têm a estrutura de dupla hélice desfeita, e as fitas só não se separam por completo devido à
adição de um iniciador contendo um grampo com guanina e citosina (grampo GC) que mantém
as fitas unidas. As fitas que sofrem desnaturação mais rápido abrem-se e ficam restritas às
porções superiores do gel, enquanto as fitas mais resistentes à desnaturação atingem porções do
gel mais inferiores. Percebe-se, portanto, que o que determina o quanto um amplicon irá
desnaturar é sua sequência nucleotídica; quanto maior o conteúdo de bases guanina e citosina,
que formam três pontes de hidrogênio entre si, mais os amplicons suportam desnaturação e
conseguem migrar no gel. O resultado é um gel com várias bandas em cada raia de corrida
características de cada amostra de DNA. Entretanto, não há uma correspondência direta entre
cada banda no DGGE e a riqueza exata de organismos da amostra, pois sequências amplificadas
do DNA de diferentes organismos podem ter propriedades de desnaturação semelhantes,
ocupando a mesma altura no gel. Dessa forma, as bandas são referidas como UTOs e serão
essas a serem analisadas na comparação de diferentes comunidades utilizando matrizes de
similaridade e estatísticas multivariadas (NAKATSU, 2007). Uma grande vantagem da corrida
em gel é a possibilidade de excisar bandas de interesse após a corrida para reamplificação,
clonagem e sequenciamento (Figura 7). As mesmas, contudo, têm o tamanho restrito até 600
pb devido a limitações de separação no gel (THIES, 2008).

56
É importante ressaltar que, como não há banco de dados para dados de DGGE, a
única maneira de obter informações filogenéticas dessas abordagens é utilizar métodos
adicionais ou a excisão de bandas para posterior clonagem e sequenciamento. Contudo, pode-
se comparar vários perfis de comunidades obtidos entre si ou mesmo com os perfis obtidos para
culturas de isolados de referência. Uma grande desvantagem de tais técnicas é que
proporcionam a comparação apenas dos membros mais abundantes das comunidades, com os
resultados sendo limitados a apenas os 50 organismos de maior frequência presentes, o que
representa uma séria limitação para o estudo de comunidades mais complexas (SPIEGELMAN,
WHISSEL, GREER, 2005).

57
Figura 7 - Desenho esquemático ilustrando como o procedimento de Eletroforese em Gel de Gradiente
Desnaturante (DGGE) desde a obtenção do DNA total da comunidade até a excisão de bandas para sequenciamento
após a corrida do gel de desnaturação (Fonte: Angelim, 2012).

58
2.8.1.2. Metagenômica
Dentre as técnicas idealizadas para se ter acesso à fisiologia e à genética de micro-
organismos não cultiváveis, a metagenômica desempenha papel central. O desenvolvimento da
metagenômica, na verdade, é considerado o evento mais marcante no campo da ecologia
microbiana nos últimos dez anos (THOMAS, GILBERT, MEYER, 2012). Trata-se da análise
genômica de uma comunidade de organismos baseada no isolamento direto de fragmentos de
DNA oriundos de amostras ambientais, sobrepondo a necessidade de isolamento e cultivo de
espécies individuais. Este termo for proposto por Handelsman e colaboradores (1998) e trouxe
novas perspectivas ao estudo da Ecologia Microbiana ao tornar possível análises de amostras
ambientais de solo, de água ou mesmo associadas a hospedeiros eucariontes (HANDELSMAN,
2004), colocando sob foco de estudo os aproximadamente 99% das espécies microbianas que
não podem ser cultivadas. Outros termos também são utilizados para descrever essa
metodologia, como: genômica ambiental, ecogenômica, genômica de comunidades e
megagenômica, todos intercambiáveis (CHISTOSERDOVA, 2010).
A metagenômica confere acesso à composição taxonômica e funcional de genes de
comunidades microbianas inteiras, revelando informações genéticas de enzimas em potencial,
ligações existentes entre função e filogenia de exemplares não cultiváveis e perfis de estrutura
e função de diversas comunidades microbianas. Atualmente, com a redução dos custos do
sequenciamento de DNA, houve um grande desenvolvimento de estudos metagenômicos
baseados em análises de sequências. De fato, o número de sequências desse tipo geradas nos
últimos anos aumentou bastante. Dessa forma, especula-se que no futuro a metagenômica será
utilizada para descrever perfis de comunidades microbianas de modo similar aos métodos de
ribotipagem baseados no rRNA 16S, tornando-se, possivelmente, uma ferramenta padrão para
muitos pesquisadores da área de Ecologia Microbiana (THOMAS, GILBERT, MEYER, 2012).
É importante destacar que as técnicas que utilizam ácidos nucléicos tratadas
previamente não são consideradas metagenômica, mas métodos que utilizam o DNA ou RNA
metagenômico, ou seja, conjunto de material genético total da comunidade. A metagenômica
diferencia-se por proporcionar acesso à grande diversidade de genes presentes em amostras
ambientais sem selecionar alvos específicos para avaliação, garantindo que se vislumbre a
proporção real em que tais componentes se fazem presentes no ambiente.
Inicialmente, a metagenômica baseava-se na clonagem de DNA ambiental, tendo sido
seguida de análise funcional através de avaliação de expressão de clones e complementada por
sequenciamento direto aleatório de fragmentos de DNA ambiental (THOMAS, GILBERT,

59
MEYER, 2012). Os clones derivados de amostras ambientais podem ser processados utilizando
tanto abordagens baseadas em sequências como funcionais. A análise baseada em sequências é
fundamentada na procura por informações genéticas ou genes de interesse em fragmentos de
DNA sequenciados, enquanto a análise funcional baseia-se na expressão heteróloga e
identificação de clones capazes de expressar funções de interesse (CHEN; PACHTER, 2005).
O sucesso requer a transcrição bem sucedida, assim como a tradução do gene de interesse,
enovelamento proteico e a secreção do produto gênico quando se tratar de um produto
extracelular. Como tal abordagem não depende do reconhecimento de marcadores filogenéticos
previamente conhecidos, tem o potencial de identificar novas classes de genes para funções
novas ou já conhecidas, como antibióticos, genes de resistência ou novas enzimas catalíticas
(HANDELSMAN, 2004; KAKIRDE, PARSLEY, LILES, 2010).
O sequenciamento aleatório de clones é uma abordagem alternativa à utilização de
marcadores filogenéticos e proporciona importantes observações, especialmente quando
conduzido em larga escala. Alguns exemplos recentes incluem os enormes esforços de
sequenciamento para reconstrução de genomas de organismos não cultiváveis presentes em
efluentes oriundos de mineração (TYSON et al., 2004) e do Mar do Sargasso (VENTER et al.,
2004), ilustrando o poder dos esforços de sequenciamento em larga escala para ampliar nosso
entendimento das comunidades de organismos não cultiváveis. Esses estudos permitiram que
se vislumbrassem novas ligações entre filogenia e função, além de terem indicado a
surpreendente abundância de alguns tipos de genes e contribuído para a reconstrução de
genomas de organismos não cultiváveis (HANDELSMAN, 2004).
2.8.1.2.1. Sequenciamento do Genoma Completo
Recentemente, o sequenciamento aleatório do genoma completo (WGS, do inglês
Whole Genome Sequencing) emergiu como uma terceira forma de abordagem para a
metagenômica. Diferentemente das abordagens baseadas em sequência e em função, que
geralmente focam em um único gene ou em genomas individuais, oferece uma vista mais
global, permitindo melhor acesso a diferentes níveis de diversidade filogenética, o estudo de
genes e rotas metabólicas da comunidade e, em alguns casos, a reconstrução de genomas quase
completamente (CHEN; PACHTER, 2005). Ademais, a abordagem de WGS permite identificar
e anotar diversos genes microbianos codificando para várias funções bioquímicas ou
metabólicas (PETROSINO et al., 2009).

60
O sequenciamento do gene do RNA ribossomal 16S diretamente de amostras
ambientais contribuiu enormemente para revelar nas últimas duas décadas uma surpreendente
diversidade microbiana. Contudo, os iniciadores universais utilizados nas amplificações foram
desenhados com base em grupos de organismos já conhecidos que compõem as sequências de
referência presentes na maioria dos bancos de dados, o que proporcionou uma vista
relativamente tendenciosa do mundo microbiano. Além disso, o sequenciamento baseado no
gene do rRNA é capaz de detectar os membros predominantes das comunidades, ignorando os
membros raros (PETROSINO et al., 2009). Portanto, o WGS tem o potencial de revelar uma
diversidade mais próxima do real sem depender de iniciadores pré-determinados. Tal
abordagem se mostrou também essencial para avançar no estudo dos componentes virais, tão
abundantes nas comunidades microbianas. Anteriormente ao WGS, era complicado estudar os
vírus, pois estes não possuem um gene universal análogo ao 16S para ser investigado (CHEN;
PACHTER, 2005).
As sequências metagenômicas podem ser classificadas usando uma abordagem
baseada em similaridade ou em composição. A primeira abordagem envolve classificar
fragmentos de DNA com base em homologias de sequências, determinadas através da busca
em bancos de dados de referência utilizando ferramentas de alinhamento, como o BLAST (do
inglês, Basic Local Alignment Search Tool), uma ferramenta de busca por alinhamento básico
local entre sequências. Em contraste, a abordagem baseada em composição analisa
características intrínsecas das sequências, como conteúdo CG e frequências oligonucleotídicas,
comparando tais caracteres com características de sequências genômicas de referência com
origem taxonômica conhecida (SIMON e DANIEL, 2011).
Considerando os problemas relacionados a abordagens dependentes de PCR, como
iniciadores tendenciosos e baixa cobertura, o sequenciamento completo se destaca como uma
ferramenta livre de tais deficiências capaz de revelar a complexa diversidade das comunidades
microbianas. É possível através de tal abordagem identificar e anotar diversos genes que
codificam para variadas funções bioquímicas e metabólicas, além de novos genes e funções
descobertos em um grande volume de dados. Contudo, há alguns desafios e limitações a serem
superados pela abordagem de sequenciamento total, como as quantidades relativamente grandes
de material inicial necessárias, o potencial de contaminação de amostras com material genético
de hospedeiros e o grande número de genes com função desconhecida ou sem anotação de
qualidade encontrados (PETROSINO et al., 2009).

61
2.9. Comparação de sequências metagenômicas
O desenvolvimento de técnicas independentes de cultivo mais modernas permitiu
análises em larga escala de comunidades microbianas, resultando em novas aplicações, como a
metagenômica de comunidades, além da metatranscriptômica e da metaproteômica. A
combinação de conjuntos de dados de qualidade derivados dessas várias abordagens em
combinação com dados de parâmetros ambientais poderão nos permitir desvendar complexas
funções ecossistêmicas desempenhadas por comunidades microbianas (SIMON; DANIEL,
2011). Para tanto, foi essencial a modernização das tecnologias disponíveis de sequenciamento,
seguida de sua popularização, o que permitiu o acesso a uma grande quantidade de informações
genéticas de variadas amostras, assim como o desenvolvimento de métodos de análise
computacionais, sem os quais seria impossível retirar conhecimentos de grandes volumes de
informações no formato de enormes sequências nucleotídicas, na maioria das vezes.
2.9.1. Tecnologias de sequenciamento
Desde 1977, quando foram introduzidas as técnicas de sequenciamento do dideoxi
de Sanger (SANGER, NICKLEN E COULSON, 1977) e de degradação química de Maxam e
Gilbert (MAXAM, GILBERT, 1977), as técnicas de sequenciamento de DNA vem evoluindo
para cobrir uma variedade cada vez maior de tipos de sequências assim como para tornarem-se
mais acessíveis. Após a automação do sequenciamento de DNA por meio da detecção em
fluorescência em 1986, o processo evoluiu bastante, tendo passado pelo desenvolvimento da
eletroforese capilar e de enzimas mais efetivas até chegar às técnicas de sequenciamento de alto
desempenho, também conhecidas como técnicas de sequenciamento de nova geração (NGS; do
inglês, next generation sequencing). Ocorreu praticamente uma troca da aplicação do
sequenciamento automatizado de Sanger, considerado como uma tecnologia de ‘primeira
geração’, para a utilização das novas tecnologias, as NGS (METZKER, 2010). Tais técnicas,
além de serem rápidas e eficientes, permitem o sequenciamento sem a necessidade de clonagem
e a produção de um enorme volume de dados de forma mais econômica (BAZINET;
CUMMINGS, 2012).
Essa mudança fez com que, do trato gastrointestinal humano às profundezas
oceânicas, houvesse uma revolução na nossa maneira de entender a estrutura, a diversidade e a
função das comunidades microbianas. Apesar de o sequenciamento de Sanger ainda ser

62
considerado o ‘padrão ouro’ para sequenciamentos por sua baixa taxa de erro e maiores
tamanhos de sequências geradas (em média, maiores que 700 pb), as tecnologias NGS, como
454, Illumina e SOLiD, foram peças chave nessa revolução. Elas proporcionam uma alta taxa
de cobertura de comunidades microbianas, tornando possível avaliar quantitativamente o
impacto de várias atividades antrópicas no ambiente (LUO et al., 2012). Dentre as metodologias
de NGS, tanto o pirosequenciamento pela plataforma 454 quanto o Illumina têm sido aplicados
extensivamente (THOMAS, GILBERT, MEYER, 2012). Eles são capazes de gerar milhões de
sequências de pequeno tamanho, variando de dezenas de pares de bases a cerca de 700 pb, cada
vez se aproximando mais do ideal representado pelo tamanho médio de um gene bacteriano
(~950 pb) ou pelo tamanho das sequências obtidas pelo método de Sanger (até 1000 pb) (LUO
et al., 2012).
2.9.1.1. Pirosequenciamento 454 (Roche)
Este foi o primeiro sequenciador de nova geração disponibilizado no mercado
(ANSORGE, 2009). O sistema 454, da empresa Roche Applied Sciences (Basel, Suíça),
emprega a reação em cadeia de polimerase de emulsão (ePCR) para amplificar clonalmente
fragmentos aleatórios de DNA. Estes são ligados a esferas microscópicas através de
adaptadores que causam a ligação de cada tipo de fragmento de DNA a uma única esfera (Figura
8a). Cada microesfera é, então, depositada em poços presentes em placas de picotitulação
contendo os reagentes necessários para a amplificação imersos em óleo, sendo, em seguida,
pirosequenciados individualmente e em paralelo. O processo de pirosequenciamento em si
envolve a adição sequencial de desoxinucleosídeos trifosfatados, os quais, se complementares
à fita molde de DNA, serão incorporados pela DNA polimerase. A cada ciclo de adição de
nucleotídeos, é adicionada a enzima apirase para degradar nucleotídeos não incorporados,
preparando o poço para um novo ciclo de adição sequencial de desoxinucleosídeos. A
incorporação de nucleotídeos provoca a liberação de pirofosfato, que, por sua vez, leva à
produção de ATP pela enzima ATP sulfurilase. A energia do ATP será então utilizada para
dirigir a conversão enzimática, mediada pela luciferase, de luciferina em oxiluciferina, levando
à emissão de fótons. Durante o sequenciamento, a luz produzida por 1,2 milhões de reações é
detectada em paralelo por um sensor de captação de imagens (CCD, um dispositivo de carga
acoplada; – do inglês, charge-coupled device) e convertida na informação da real sequência do
DNA molde (THIES, 2008).

63
Faz-se necessário ressaltar dois aspectos importantes desse sistema quando
aplicados à metagenômica. Primeiramente, mostrou-se que a ePCR produz sequências
replicadas artificialmente, o que pode influenciar nas estimativas de abundância gênica
(GOMEZ-ALVAREZ, TEAL, SCHMIDT, 2009). Portanto, entender a quantidade de
sequências replicadas artificialmente é essencial para assegurar a qualidade dos dados gerados
neste tipo de sequenciamento de modo que se possa identificar tais sequências e filtrá-las
através de ferramentas de bioinformática. O segundo ponto diz respeito à intensidade de luz
produzida quando a DNA polimerase lê um homopolímero (i.e. três ou mais bases idênticas em
sequência no DNA). Nessa situação, é complicado correlacionar o real número de posições
nucleotídicas com a produção de luz detectada, resultando em erros de inserção ou deleção e
em consequentes trocas de janelas de leitura (QUINCE et al., 2009). Entretanto, esse tipo de
erro pode ser incorporado em modelos de predição e resultar em alta, porém não perfeita,
acurácia (THOMAS, GILBERT, MEYER, 2012).
Apesar dessas desvantagens, o sistema 454 tem um custo reduzido de cerca de 10
dólares por megabase (Mpb), o que contribuiu para torná-lo um método popularmente escolhido
para se trabalhar com sequenciamento de metagenomas. Além disso, o sistema 454 produz
sequências de tamanho médio de 600-800 pb, o que é suficiente para causar somente perdas
pequenas no número de sequências que podem ser classificadas. A própria preparação das
amostras foi melhorada, permitindo que nanogramas de DNA sejam suficientes para o
sequenciamento. Outra vantagem do método diz respeito a sua capacidade de realizar
‘multiplexing’, permitindo que até 12 amostras sejam analisadas em uma única corrida de 500
Mpb (THOMAS, GILBERT, MEYER, 2012). Quando comparado a outras plataformas de nova
geração e ao sequenciamento de Sanger, o 454 destaca-se devido aos grandes tamanhos de
fragmentos gerados e ao tempo e custo de corrida reduzidos, conforme pode-se observar na
Tabela 4.
Tabela 4 - Comparação das técnicas de sequenciamento de nova geração e sequenciamento de
Sanger utilizadas em projetos de metagenômica.
Características 454 Illumina/Solexa Sanger
Química de sequenciamento Pirosequenciamento Sequenciamento
por síntese
Sequenciamento baseado
em terminação da cadeia
Tamanho do fragmento ~700 pb 50-300 pb 400-900 pb
Acurácia 99% 98% 99.9%
Sequências/corrida 1 milhão 3 bilhões N/A
Tempo/corrida 24 h 1-10 diasa 20 min a 3 h
Custo/1 Mb (US$) $ 10 $ 0.05 a $ 0.15 $ 2400
FONTE: Quail et al., 2012 e Liu, Li e Li, 2012.

64
Figura 8 - Esquema ilustrando como ocorre a imobilização dos DNA molde e sua amplificação na tecnologia de
sequenciamento de nova geração Roche/454. (Fonte: Metzker, 2010).
2.9.2. Classificação
Após o sequenciamento, as sequências obtidas poderão passar pelos passos de
montagem e predição de genes, caso o objetivo do estudo seja reconstrução de genomas.
Contudo, em caso de ter-se como objetivo estudos de comunidades, passa-se para o passo de
classificação.
A anotação ou classificação de dados de sequências metagenômicas é feita em dois
passos. Primeiramente, caracteres de interesse (genes) são identificados, processo que se
conhece como predição de caracteres. Em seguida, prováveis funções gênicas e parentes
taxonômicos são identificados, passo chamado de anotação funcional (THOMAS, GILBERT,
MEYER, 2012).
A predição de caracteres é o processo inicial de marcar sequências como genes ou
elementos genômicos, sendo baseado na identificação de sequências codificadoras de proteínas
(CDS; do inglês, coding sequences). Já a anotação funcional representa um grande desafio
computacional para a maioria dos projetos metagenômicos. Atualmente, estima-se que apenas
20 a 50% das sequências possam ser anotadas, havendo uma grande proporção de sequências
cuja importância e função ainda são desconhecidas. Esse passo não é feito de novo, mas sim
com base em genes e proteínas previamente descritos depositados em bancos de dados de forma
manual, quando o conjunto de dados tem menos de 10.000 sequências, ou automaticamente
para conjuntos de dados maiores (THOMAS, GILBERT, MEYER, 2012).
2.9.2.1. Bancos de dados de referência
Com o desenvolvimento e redução dos custos das novas tecnologias de
sequenciamento, o sequenciamento de DNA deixou de ser uma atividade desempenhada apenas
por grandes centros para se tornar mais distribuída, democrática e descentralizada.

65
Concomitantemente, um volume de dados gigantesco vem sendo gerado, o que requer o
desenvolvimento de ferramentas de análise mais eficientes e capazes de incluir a grande parcela
da comunidade científica com recursos computacionais limitados (WILKE et al., 2012).
Para estudar tais dados, um passo crucial é a classificação das sequências, feitas em
geral através de buscas por similaridade (WILKE et al., 2012). Para tanto, as sequências obtidas
são comparadas com sequências pertencentes a uma variedade de bancos de dados. Estes são
repositórios biológicos digitais compostos de sequências de ácidos nucléicos, proteínas ou outro
polímero, armazenadas em um computador. A Tabela 5 traz a listagem de alguns bancos de
dados bem estabelecidos utilizados para classificação de sequências metagenômicas.
Tabela 5 - Listagem de bancos de dados de referência para proteínas ou ácidos nucléicos
utilizados para classificação de sequências metagenômicas.
BANCO DE DADOS TIPO DE SEQUÊNCIA GESTOR
GenBank DNA International Nucleotide Sequence Database
Collaboration - INSDC
SILVA RNA ribossomal Grupo de pesquisa sobre Genômica Microbiana
e Bioinformática do Instituto Max Planck para
Microbiologia Marinha
RDP – Ribosomal Database
Project RNA ribossomal
Centro de Ecologia Microbiana da Michigan
State University
Greengenes RNA ribossomal Centro de Biotecnologia Ambiental do
Lawrence Berkeley National Laboratory
RefSeq DNA, RNA e proteínas International Nucleotide Sequence Database
Collaboration - INSDC
KEGG (Kyoto Encyclopedia of
Genes and Genomes)
Sequências gênicas
integradas a vias
metabólicas
Kanehisa Laboratories em colaboração com o
Centro de Bioinformática do Instituto de
Pesquisa Química da Kyoto University e com o
Centro Genoma Humano do Instituto de
Ciências Médicas da University of Tokyo
SEED
Sequências gênicas
integradas a subsistemas ou
a papéis funcionais
Sociedade para Interpretação de Genomas –
FIG (do inglês, Fellowship for Interpretation of
Genomes)
COG (Clusters of Orthologous
Groups of proteins)
Classificação filogenética
das proteínas codificadas
em genomas completos
Centro Nacional de Informações
Biotecnológicas (NCBI; do inglês, National
Center for Biotechnological Information)
M5NR Proteínas Divisão de Matemática e Ciências da
Computação, Argonne National Laboratory

66
2.9.2.2. Plataformas de análise de metagenomas
Classificar taxonomicamente ou funcionalmente sequências derivadas de amostras
ambientais constitui um grande problema. A maior parte dos estudos que utiliza sequências
metagenômicas é de cunho ecológico; portanto, busca caracterizar estatisticamente e
dinamicamente as comunidades em termos de composição, estrutura, abundância, demografia
ou sucessão, muitas vezes considerando vários fatores abióticos e bióticos. Portanto, trata-se de
dados de alta complexidade que necessitam de acurácia e precisão na classificação das
sequências metagenômicas envolvidas. A estratégia básica de classificação de tais sequências
de origem desconhecida e de tamanhos variáveis envolve sua comparação com sequências
anotadas em bancos de dados públicos, como o M5NR (WILKE et al., 2012). Com base em
tais comparações, pode-se dizer com certa confiabilidade que determinada sequência pertence
a um táxon definido em uma hierarquia taxonômica que vai de Domínio à Espécie. Entretanto,
algumas vezes a sequência alvo de classificação não encontra semelhante nos bancos de dados,
o que pode ser problemático em qualquer um dos métodos utilizados (BAZINET;
CUMMINGS, 2012).
Considerando que nenhum banco de dados é capaz de cobrir todas as funções
biológicas, a habilidade de visualizar e copilar informações obtidas em diferentes bancos de
dados em um único local é essencial e foi implementada nas versões mais recentes de algumas
plataformas de comparação de metagenomas (THOMAS, GILBERT, MEYER, 2012). Tais
plataformas tornam possível depositar, processar e analisar conjuntos de dados de
metagenomas, sendo o MG-RAST (GLASS et al., 2010), o IMG/M (MARKOWITZ et al.,
2008) e o CAMERA (SUN et al., 2011) os três sistemas proeminentes.
O MG-RAST (Metagenomics Rast - Rapid Analysis using Subsystems Technology;
em português: Análise Rápida usando Tecnologia de Subsistemas) é um repositório de dados
gratuito que também funciona como plataforma de análise de dados por meio da comparação
com bancos de dados de proteínas e de nucleotídeos (MEYER et al., 2008). É totalmente
automatizado, permitindo fazer controle de qualidade de sequências, predição de caracteres e
anotação funcional. Ademais, foi otimizado para garantir um balanço benéfico entre acurácia e
alta eficiência computacional para anotação e análise de sequências de metagenomas através
do uso do BLAT, do inglês Basic Local Alignment Tool, ferramenta básica de alinhamento local
(KENT, 2002). Os resultados gerados são expressos na forma de perfis de abundância de táxons
ou de grupos funcionais específicos. O MG-RAST suporta comparações utilizando perfis
taxonômicos do NCBI baseado em rRNA 16S ou dados de sequenciamento aleatório do genoma

67
completo (WGS) e comparações de abundância relativa utilizando o KEGG, COG e SEED,
dentre outros bancos de dados, em vários níveis de resolução. Podem-se baixar todos os
produtos gerados nas análises realizadas nessa plataforma, além de poder-se compartilhar tais
dados com outros usuários ou mesmo torná-los públicos. A plataforma possui uma interface na
internet que permite comparações utilizando uma variedade de técnicas estatísticas e a
incorporação de metadados, como características físico-químicas, nas estatísticas. Em julho de
2014, o MG-RAST contava com mais de 7.000 usuários e mais de 125.000 metagenomas
analisados, desses sendo mais de 18.000 públicos. Tais números podem representar uma
tendência na comunidade científica de centralização e padronização de recursos e sistemas de
análise de dados (THOMAS, GILBERT, MEYER, 2012).
Similarmente ao MG-RAST, o IMG/M também proporciona uma plataforma
padronizada. Contudo esta possui maior sensibilidade, pois utiliza o modelo oculto de Markov
(do inglês, hidden Markov model – HMM) e buscas através de BLASTX, ou seja, buscas por
proteínas a partir de sequências nucleotídicas traduzidas, consequentemente, com alto custo
computacional. Diferentemente do MG-RAST, as comparações no IMG/M não são baseadas
em tabelas de abundância, mas sim em comparações de todos versus todos os genes, integrando
todos os bancos de dados em uma única abstração no nível de proteínas. Da mesma forma que
o MG-RAST, IMG/M pode armazenar os resultados para comparações, permitindo que novos
metagenomas sejam comparados com um rico conjunto de outros metagenomas sem que seja
necessário ao usuário prover os meios computacionais para reanalisar todos os metagenomas
envolvidos no estudo (THOMAS, GILBERT, MEYER, 2012).
Outro sistema que estava disponível até recentemente é o CAMERA (do inglês,
Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis) –
Infraestrutura Cibernética Comunitária para Pesquisa e Análise de Ecologia Microbiana
Avançadas), que oferecia um esquema de anotação bastante flexível. Essa plataforma requeria,
para fins de comparação de vários metagenomas, que todos fossem analisados em conjunto, o
que aumentava bastante os custos computacionais (THOMAS, GILBERT, MEYER, 2012). Em
maio de 2014, a iniciativa anunciou o encerramento do portal CAMERA em razão do término
do apoio da Fundação Gordon e Betty Moore.
O MEGAN (do inglês, MEtaGenome ANalyzer – Analisador de metagenomas) é
outra ferramenta utilizada para comparar todas as sequências obtidas no sequenciamento com
um ou mais bancos de dados de referência utilizando BLAST. O programa utiliza o resultado
da comparação como a entrada e gera dendogramas taxonômicos ou funcionais (Huson et al.,
2007), o que torna a comparação de diferentes grupos taxonômicos e funcionais visualmente

68
mais fácil, contribuindo para a interpretação dos dados (THOMAS, GILBERT, MEYER,
2012). Além disso, o programa permite uma exploração interativa dos metagenomas, tanto em
níveis altos como em níveis mais detalhados. A plataforma utiliza o algoritmo LCA (do inglês,
Lowest Common Ancestor, o Último Ancestral Comum), o qual considera o último ancestral
entre duas sequências comparadas para escolher a classificação mais adequada (MITRA,
KLAR, HUSON, 2009). Contudo, este tipo de análise requer maior poder de processamento
computacional para processar novamente a comparação por BLAST sempre que for necessário
mudar os parâmetros de comparação escolhidos.
2.9.3. Análises estatísticas
Recentemente, os estudos de metagenômica deixaram de ter um caráter meramente
descritivo e passaram a tentar resolver questões fundamentais de Ecologia Microbiana. Tal
conquista foi possível devido à redução dos custos de sequenciamento e da proposição de
desenhos experimentais cada vez mais robustos baseados na riqueza de informações que a
metagenômica pode oferecer. Para que tal objetivo fosse atendido, foi necessária uma mudança
na forma com que as amostras eram obtidas, de forma a assegurar repetição adequada, e na
análise estatística bem fundamentada dos dados (THOMAS, GILBERT, MEYER, 2012).
Por muito tempo, o aspecto estatístico foi uma questão que preocupou muitos
ecólogos microbianos por não se saber se os métodos disponíveis eram adequados para analisar
comunidades microbianas. A razão para tal incerteza é que tais comunidades constituíram por
muito tempo uma grande caixa preta para a ecologia, de tal forma que grande parte dos modelos
estatísticos foi desenhada originalmente para macro-organismos. Contudo, atualmente, sabe-se
que muitas abordagens e estratégias derivados de décadas de pesquisas de ecologia quantitativa
com macro-organismos podem ser utilizadas com sucesso para a análise de micro-organismos
(THOMAS, GILBERT, MEYER, 2012).
Primeiramente, como os demais dados de Ecologia quantitativa, os dados de
sequenciamento de metagenomas podem ser reduzidos a tabelas, onde as colunas representam
amostras; as linhas, grupos taxonômicos ou funcionais; e os campos contêm dados de
abundância ou presença/ausência. Portanto, muitas das ferramentas estatísticas disponíveis para
identificar correlações e padrões estatisticamente significantes se mostraram transferíveis para
a análise de micro-organismos. Entretanto, como as matrizes de espécie-amostra de

69
metagenomas normalmente contêm mais grupos taxonômicos ou funcionais que amostras
obtidas, é preciso que correções apropriadas para testes de hipóteses múltiplas sejam
implementados, como, por exemplo, correção de Bonferroni para análises baseadas no teste-t
(THOMAS, GILBERT, MEYER, 2012). Outra consequência do volume de dados é a
necessidade de avaliá-los através de estatísticas multivariadas.
Alguns pacotes estatísticos desenvolvidos para macro-organismos vêm sendo
utilizados com sucesso, como é o caso do Primer-E, que permite análises estatísticas
multivariadas, incluindo escalonamento multidimensional (MDS), análise de similaridade
(ANOSIM) e identificação de grupos taxonômicos ou funcionais que contribuem para a
diferença entre duas amostras (SIMPER) (PROSSER, 2010). Recentemente, passou-se a
utilizar preferencialmente outros pacotes baseados em dados taxonômicos desenvolvidos
especialmente para metagenomas, como é o caso do MetaStat (WHITE, NAGARAJAN, POP,
2009), uma ferramenta de internet capaz de revelar com alta confiabilidade funções
discriminatórias entre metagenomas. Além desse, pode-se utilizar um pacote do ambiente
estatístico R, o ShutgunFunctionalizeR (KRISTIANSSON, HUGENHOLTZ E DALEVI,
2009), que provê vários procedimentos estatísticos para analisar diferenças entre amostras
metagenômicas (THOMAS, GILBERT, MEYER, 2012).
Outra ferramenta estatística bastante útil, o STAMP (do inglês Statistical Analysis
of Metagenomic Profiles; Parks, Beiko, 2010), foi desenvolvido para comparar estatisticamente
perfis metagenômicos de forma a identificar diferenças biologicamente significativas entre os
metagenomas e a diferenciar influências ecológicas de artefatos de amostragem ou das técnicas
utilizadas, tarefa complicada de ser feita manualmente devido ao volume de dados a ser
analisado (PARKS; BEIKO, 2010).
2.10. Armazenamento e compartilhamento dos dados
A capacidade de processamento das amostras e sequências obtidas via
sequenciamento de larga escala é outra questão a ser considerada quando se trabalha com uma
quantidade de dados muito superior ao pequeno número de sequências de clones com as quais
se trabalhava anteriormente. Os metagenomas apresentam tamanho médio que varia de 100-
500 mil pares de bases por amostra, podendo atingir mais de 1 bilhão de bases em projetos com
maior número de amostras. Esse enorme volume de dados gerados requer que programas e

70
profissionais de bioinformática sejam capazes de processá-los com acurácia a um custo de
processamento reduzido (KAKIRDE, PARSLEY, LILES, 2010).
O compartilhamento de dados metagenômicos vem sendo defendido atualmente em
prol da formação de um corpo amplo de conhecimentos em ecologia microbiana e de formar
um conjunto de dados passível de ser utilizado por qualquer estudioso em qualquer lugar do
mundo. Para tanto, faz-se necessário um novo nível de organização e colaboração para prover,
além de dados de sequências e resultados analisados, também os metadados. Para poder-se
disponibilizar os resultados analisados, alguns aspectos das plataformas de análise precisam ser
coordenados. Para tanto, o Consórcio dos Padrões Genômicos (do inglês, Genome Standards
Consortium - GSC) propôs uma série de formatos padrão para gravar dados ambientais e
experimentais (metadados), as Informações Mínimas sobre qualquer (‘x’) Sequência (do inglês,
Minimum Information about any (x) Sequence – MixS). MIxS é um termo geral para descrever:
Informações Mínimas sobre uma Sequência Genômica (do inglês, Minimum Information about
a Genome Sequence – MIGS), Informações Mínimas sobre uma Sequência Metagenômica (do
inglês, Minimum Information about a Metagenome Sequence – MIMS) e Informações Mínimas
sobre uma Sequência Marcadora (do inglês, Minimum Information about a MARKer Sequence
– MIMARKS) (YILMAZ et al., 2011).
Contudo, uma questão complicada que emerge dessa proposição de centralização é
“quem paga pelo armazenamento?” O Centro Nacional de Informações Biotecnológicas dos
EUA (NCBI, do inglês The US National Center for Biotechnology Information) tem a obrigação
de armazenar todos os dados metagenômicos, entretanto, o volume bruto de dados sendo
gerados significa que há uma necessidade urgente de novas maneiras de armazenar
apropriadamente vastas quantidades de sequências. Na verdade, enquanto os custos de
sequenciamento continuam a cair, os custos de análise e armazenamento se mantêm constantes,
o que é bastante preocupante. A questão de onde irá ser armazenado tal volume crescente de
dados, se na forma biológica (amostra que foi sequenciada) ou digital em arquivos centralizados
ou descentralizados emerge com força nesse contexto (THOMAS, GILBERT, MEYER, 2012).
Acima de tudo, tal paradoxo demonstra a necessidade urgente da formação de mais recursos
humanos dedicados a se estudar tal volume crescente de informações.

71
3. OBJETIVO GERAL
O objetivo deste trabalho foi investigar se a arquitetura e o tempo de operação de
dois portos marítimos do Estado do Ceará, o Porto do Mucuripe (município de Fortaleza) e o
Porto do Pecém (município de São Gonçalo do Amarante), podem influenciar os atributos de
estrutura, riqueza e diversidade taxonômica e funcional das assembleias de Archaea e Bacteria
associadas aos sedimentos dessas áreas, resultando em padrões de diversidade próprios, os quais
representam a adaptação de seus membros às condições vigentes em cada tipo de estrutura
portuária.
3.1. Objetivos Específicos
Descrever as características físicas e químicas dos portos estudados e dos sedimentos
constituintes;
Caracterizar as estruturas e riquezas das assembleias de Archaea e de Bacteria
associadas aos sedimentos de cada estação de coleta em ambos os portos por meio de
Eletroforese em Gel de Gradiente Desnaturante (DGGE);
Caracterizar as estruturas e riquezas das assembleias de Archaea e de Bacteria
associadas aos sedimentos na forma de pools das estações de coleta de cada porto por
meio de DGGE;
Avaliar se as DGGE cobriram a diversidade de Archaea e de Bacteria associadas aos
sedimentos portuários;
Relacionar as variações de estrutura e riqueza observadas com as características físicas
e químicas dos portos;
Sequenciar os metagenomas originados dos sedimentos portuários por
pirosequenciamento 454 (Roche);
Classificar taxonomicamente e funcionalmente as sequências obtidas por meio de
pirosequenciamento;
Descrever a composição taxonômica das assembleias de Archaea e de Bacteria
associadas aos sedimentos portuários;
Descrever a composição funcional dos sedimentos portuários;
Relacionar a composição taxonômica e funcional com as características físicas e
químicas de cada porto.

72
4. CAPÍTULO 1 - ESTRUTURA E RIQUEZA LOCAL DAS ASSEMBLEIAS DE
BACTERIA E ARCHAEA ASSOCIADAS A SEDIMENTOS MARINHOS SOB
INFLUÊNCIA DE REGIÕES PORTUÁRIAS
4.1. Introdução
A ecologia microbiana tem três objetivos principais: (1) definir as dinâmicas de
populações microbianas em comunidades; (2) definir as características físico-químicas dos
ambientes onde vivem os micro-organismos; e (3) entender os processos metabólicos
desempenhados por estes em hábitats específicos. Ela procura, portanto, responder a três
perguntas principais: que organismos ocorrem em cada tipo de hábitat (‘quem está ali?’), que
processos realizam nestes hábitats (‘o que estão fazendo?’) e como esses convivem entre si e
com o ambiente (‘como o fazem?’) (ATLAS, BARTHA, 1998).
Buscando atingir o primeiro objetivo, os principais atributos das comunidades
biológicas devem ser investigados: estrutura, diversidade, dominância, estabilidade, estrutura
trófica e estrutura física. A diversidade é composta por três elementos principais, a composição,
a riqueza e a equabilidade. A composição diz respeito à identidade dos táxons observados
enquanto a riqueza diz respeito à quantidade desses táxons. Por sua vez, a equabilidade refere-
se à distribuição dos indivíduos dentre os táxons, sendo calculada com base na abundância de
cada táxon. A partir da abundância, pode-se estudar a dominância dos grupos encontrados, ou
seja, que grupos possuem maior abundância em relação aos demais (abundância relativa).
Compilando-se informações desses atributos, pode-se inferir a respeito da estabilidade da
comunidade estudada, ou seja, de sua resistência às mudanças e de sua capacidade de retornar
ao um estado anterior não perturbado (resiliência). Por fim, estudos de estrutura trófica
requerem um conhecimento mais profundo dos membros da comunidade e de como interagem
entre si em teias tróficas assim como da quantidade de energia que flui através do ecossistema
do qual faz parte a comunidade. Já a estrutura física refere-se à distribuição no espaço dos
membros da comunidade (BEGON, THOUSEND, HARPER, 2007). É importante ressaltar que
o parâmetro de diversidade de uma comunidade influencia em sua estabilidade de forma que,
em geral, comunidades muito diversas não têm sua estrutura afetada pela saída de uma
população. Entretanto, não é claro que nível de diversidade é necessário para manter a
estabilidade de uma comunidade (ATLAS, BARTHA, 1998). Essa relação é muito importante,
pois indica que a partir de estudos de diversidade pode-se inferir a respeito da estabilidade de

73
uma comunidade. Considerando o elevado grau de exposição dos diferentes ecossistemas
marinhos a perturbações de origem antrópica, estudos que indicam a estabilidade de uma
comunidade e sua consequente capacidade de adaptar-se por meio de resistência ou resiliência
se revelam essenciais.
A comparação de comunidades microbianas por meio da técnica de DGGE é
considerada uma abordagem bem reconhecida, capaz de processar um grande número de
amostras em um curto período de tempo. Dessa forma, desde que foi trazida para a área da
microbiologia (MUYZER, DE WAAL, UITTERLINDEN, 1993) já foi utilizada em estudos
com objetivos variados e em vários hábitats, permitindo obter-se informações realistas sobre os
micro-organismos presentes nos ambientes naturais e antropizados (MUYZER, SMALLA,
1998). Antes do advento das tecnologias de sequenciamento de nova geração, a DGGE e outras
técnicas de ribotipagem, como o T-RFLP, constituíam as metodologias capazes de fornecer a
melhor visão possível da diversidade, estrutura e dinâmica dos micro-organismos disponíveis
(CLEARY et al., 2012).
Atualmente, mesmo com a riqueza de informações trazidas pelas novas tecnologias
de sequenciamento, alguns estudos que compararam as duas técnicas mostraram que resultados
semelhantes podem ser obtidos por DGGE e pirosequenciamento. Os autores conseguiram
demonstrar que ambas as técnicas são compatíveis, com o DGGE sendo sugerido com uma
excelente metodologia para explorar a diversidade microbiana de diferentes amostras com a
vantagem de ter um custo mais barato e menor tempo de processamento (POMMIER et al.,
2010; CLEARY et al., 2012). Entretanto, um problema das abordagens de ribotipagem baseadas
em iniciadores universais é que revelam apenas os táxons mais abundantes presentes em um
conjunto de amostras. Tal problema pode ser minimizado com a utilização de iniciadores mais
específicos para cada táxon em conjunto com nested PCR. Quando comparados a dados obtidos
por pirosequenciamento, o DGGE parece, em geral, capaz de revelar os mesmos grupos
principais. Contudo, as tecnologias de avaliação de diversidade mais abrangentes, como é o
caso do pirosequenciamento, são capazes de mostrar uma comunidade mais diversa, com uma
maior representatividade de táxons de menor frequência (ROH et al., 2010; CLEARY et al.,
2012).
Apesar das desvantagens apresentadas, trata-se de uma técnica bastante utilizada
nos mais diversos ambientes. Particularmente no ambiente marinho, o DGGE já foi utilizado,
por exemplo, para detectar mudanças na comunidade microbiana presente em amostras de água
de lastro (TOMARU et al., 2010) ou ainda para investigar a capacidade de micro-organismos
associados a sedimentos marinhos de degradar poluentes e sobre os efeitos dos contaminantes

74
sobre as estruturas das comunidades microbianas (WANG et al., 2010; KÖCHLING et al.,
2011; WANG; TAM, 2011).
Neste trabalho a técnica de DGGE foi utilizada com o objetivo de investigar a
influência da arquitetura portuária e do tempo de operação sobre a estrutura espacial e a riqueza
das assembleias dos Domínios Bacteria e Archaea associadas aos sedimentos de dois sistemas
portuários localizados no Estado do Ceará; o Porto do Mucuripe e o Porto do Pecém,
representando o primeiro estudo de comunidades microbianas associadas aos sedimentos de
regiões portuárias realizado para a Plataforma Continental Brasileira.
Os resultados dessa investigação poderão vir a auxiliar na tomada de decisões a
respeito de ações de dragagens e expansão dos píers, além de constituir um registro histórico
dos padrões dessas comunidades. Para o Porto do Pecém, localizado na região onde está sendo
instalado o Complexo Industrial e Portuário do Pecém, o registro histórico é sobretudo
importante para acompanhar futuramente os efeitos das mudanças que possivelmente ocorrerão
em decorrência da instalação de indústrias e de seus respectivos sistemas de escoamento de
produção e de efluentes.
4.2. Hipótese
Regiões portuárias são tipicamente impactadas por poluentes característicos de suas
atividades. Entretanto, possuem particularidades relacionadas ao tempo de operação,
hidrodinamismo e geomorfologia local, localização, arquitetura e volume de carga transportada.
Tais características podem influenciar na qualidade do sedimento subjacente e,
consequentemente, na microbiota associada. Hipotetiza-se, portanto, que os perfis de estrutura
espacial e riqueza das assembleias de Bacteria e Archaea associadas aos sedimentos superficiais
de regiões portuárias podem variar em função da arquitetura e do tempo de operação do porto.
4.3. Materiais e Métodos
4.3.1. Área de estudo
O estudo foi realizado em duas áreas portuárias, o Porto do Pecém (PEC)
(03°32’52.03’’S; 38°48’46.14’’O) e o Porto do Mucuripe (MUC) (03°42’45.55’’S;
38°28’26.80’’O), ambos localizados no Estado do Ceará, Brasil (Figura 9). O Porto do

75
Mucuripe está localizado na Enseada do Mucuripe, em Fortaleza, capital do Estado. O Porto
está em operação desde 1953, contando atualmente com grande movimentação de granéis
líquidos de derivados de petróleo, gasolina e Gás Liquefeito de Petróleo (GLP). O Porto do
Pecém está posicionado na Ponta do Pecém, na região destinada à instalação do Complexo
Industrial e Portuário do Pecém, distante 60 Km da região metropolitana. O Porto do Pecém
constituirá um concorrente ao Porto do Mucuripe no setor de transporte de combustíveis, pois
o armazenamento desses produtos passará a ser feita no Pecém, e não mais na cidade de
Fortaleza. Além disso, o terminal do Pecém possui perspectivas de ampliação da movimentação
de combustíveis e derivados do petróleo devido a um projeto da Petrobrás de instalação de uma
nova refinaria no Complexo, obra esta prevista para 2017, com capacidade de 300 mil barris de
petróleo por dia (GIACOBO et al., 2012). Além de diferenças de funcionamento e cargas
transportadas, tais portos diferem em relação ao seu tempo de operação, com PEC estando ativo
há 13 anos e MUC há mais de 50 anos. Outra diferença refere-se a suas infraestruturas, pois
PEC é um porto moderno do tipo offshore, construído a 2000 m da linha de costa e conectado
à terra através de uma ponte sobre pilastras. O objetivo de tal ponte foi preservar a linha de
costa e minimizar o impacto negativo associado com as arquiteturas portuárias tradicionais, que
afetam as correntes costeiras e o transporte de sedimentos (BURUAEM et al., 2012). Por sua
vez, MUC é um porto marítimo artificial construído entre dois espigões tradicionais. Nesse
porto, ao longo dos anos de funcionamento, a estrutura portuária gerou uma forte alteração no
balanço sedimentar regional com interrupção do transporte de sedimentos e consequente
deposição de sedimentos finos na área portuária. Por essa razão, atividades de dragagem são
efetuadas periodicamente no porto, objetivando a manutenção de seu calado e do canal de
navegação (Maia et al., 1998).
As áreas portuárias foram comparadas com uma região considerada livre de
impactos relacionados à atividade portuária. Esta foi a Praia de Redonda, localizada no
município de Icapuí (Ceará, Brasil) (4°37'41.5"S; 37°27'23.8"O), caracterizada pela
conservação de seus atributos naturais e pela atividade de pesca da lagosta.

76
Figura 9 - Localização das estações de coleta no Porto do Pecém (Município do São Gonçalo do Amarante, CE)
e no Porto do Mucuripe (Município de Fortaleza, CE), localizados no Estado do Ceará, Brasil. As estações de
coleta são indicadas na figura como P1 a P10 (Porto do Pecém) e M1 a M15 (Porto do Mucuripe).
4.3.2. Coleta das amostras
No Porto do Pecém, a amostragem foi realizada em Novembro do ano de 2011 em dez
diferentes estações (P1-P10) enquanto no Porto do Mucuripe as amostras foram coletadas em
Dezembro do mesmo ano em 15 diferentes estações (M1-M15) (Figura 9). Ambas as coletas
foram realizadas em embarcações pesqueiras com auxílio de equipamento de GPS para
distribuição das estações de coleta dentro de cada área portuária. As distâncias entre as estações
de coleta em ambos os portos foram de cerca de 500 m, de modo a cobrir toda a área de operação
dos portos estudados. Na Praia de Redonda (área sem atividade portuária), a amostragem foi
realizada em julho de 2011 em um barco lagosteiro, cobrindo pontos distantes cerca de 500 m
entre si e distantes dois quilômetros da linha de costa. Foram obtidas cinco subamostras, as
quais foram reunidas para formar uma amostra composta.
A amostragem foi conduzida com o pegador de fundo de aço inoxidável do tipo Van
Veen em profundidades medindo entre 10 e 30 m. Após a coleta, as amostras obtidas em cada
estação foram transferidas para recipientes plásticos esterilizados e transportados sob
refrigeração para o Laboratório de Ecologia Microbiana e Biotecnologia (Lembiotech),

77
localizado no Campus do Pici da Universidade Federal do Ceará. A profundidade e a
transparência da água, medida com disco de Sechii, foram mensuradas in situ, enquanto a
salinidade e o pH foram mensurados após a chegada ao laboratório em refratômetro e
potenciômetro, respectivamente. A profundidade das coletas variou de 15 a 20 m, com pH de
7,5-7,6, salinidade de 40 e transparência da água de 1,9 a 2,9 m, conforme pode-se observar na
Tabela 6.
4.3.3. Caracterização física e química dos sedimentos estudados
Os sedimentos em estudo foram analisados para determinação de sua
granulometria, teor de matéria orgânica e carbonato e sua composição química. Para a
caracterização dos sedimentos, as amostras obtidas nas estações de coleta do Porto do Mucuripe
e do Porto do Pecém foram reunidas para formar amostras compostas dos sedimentos de cada
porto, as quais foram analisadas em triplicata.
A análise da distribuição do tamanho das partículas que compunham os sedimentos
foi realizada por peneiramento a seco em uma série de peneiras de diferentes aberturas de malha
(SUGUIO, 1973) após separação prévia das frações de silte e argila por peneiramento úmido.
O teor de matéria orgânica foi medido de acordo com o protocolo de Schulte e Hopkins (1996)
através da medida da perda de peso após combustão em mufla. A quantidade de matéria
orgânica foi calculada pela equação: MO (g/Kg) = [(Mi-Mf)/Mi]* 1000, na qual MO representa
a matéria orgânica; Mi, o peso inicial; e Mf, o peso final após a perda de matéria orgânica por
combustão. O teor de carbonato foi estimado por digestão dos sedimentos com HCl 4N, seguida
de várias lavagens para eliminar o HCl e gravimetria (GROSS, 1971). Estas análises foram
realizadas no Laboratório de Ecologia Microbiana e Biotecnologia, na UFC.
Também foram analisadas as quantidades de metais tóxicos, Hidrocarbonetos
Policíclicos Aromáticos (HPAs), hidrocarbonetos BTEX (Benzeno, Tolueno, Etil-benzeno e
Xilenos), Bifenilas, Carbono Orgânico Total (COT), nitrato, nitrito e sulfato, de acordo com os
procedimentos propostos pela Agência de Proteção Ambiental Americana (EPA, EUA). Tais
análises foram realizadas na Empresa EP - Engenharia do Processo, localizada em Guarulhos,
São Paulo), para onde as amostras conservadas foram enviadas.

78
Tabela 6 – Localização das estações de coleta e características gerais das amostras de sedimento obtidas.
Estação de coleta Coordenadas de GPS Profundidade
(m) Sechii (m)
Aspecto visual
do sedimento
Porto do Pecém
P1 S3 31.874 O38 47.649 17 2.4 lamoso
P2 S3 31.963 O38 47.734 16 2.7 lamoso
P3 S3 31.867 O38 47.785 17 2.3 lamoso
P4 S3 31.710 O38 47.788 19 2.5 lamoso
P5 S3 31.835 O38 47.858 22 2.2 lamoso
P6 S3 31.886 O38 47.908 21 2.5 lamoso
P7 S3 31.973 O38 47.978 25 2.5 cascalho
P8 S3 32.085 O38 47.887 27 2.5 lamoso
P9 S3 32.023 O38 47.794 24 2.9 lamoso
P10 S3 31.993 O38 47.878 23 2.9 lamoso
Porto do Mucuripe
M1 S3 42.827 O38 28.678 10 2.0 lamoso
M2 S3 42.792 O38 28.643 12 1.9 lamoso
M3 S3 42.724 O38 28.629 12 2.2 lamoso
compactado
M4 S3 42.648 O38 28.612 10 2.0 lamoso
M5 S3 42.568 O38 28.595 9 2.4 lamoso
M6 S3 42.520 O38 28.696 12 2.1 lamoso
M7 S3 42.604 O38 28.730 13 2.2 lamoso
M8 S3 42.706 O38 28.732 16 2.1 lamoso
M9 S3 42.770 O38 28.764 16 2.1 lamoso
M10 S3 42.805 O38 28.785 18 1.9 lamoso
M11 S3 42.756 O38 28.897 17 2.4 lamoso
M12 S3 42.653 O38 28.924 15 2.6 lamoso
M13 S3 42.553 O38 28.915 15 3.0 lamoso
M14 S3 42.416 O38 28.938 15 3.0 lamoso
M15 S3 42.245 O38 28.993 14 2.8 lamoso

79
4.3.4. Extrações de DNA
A extração de DNA total de cada uma das amostras de sedimento obtidas nas estações
de coleta do Porto do Pecém e do Porto do Mucuripe foi realizada após descongelamento
seguido de homogeneização das amostras utilizando o kit de extração comercial Power Soil
DNA Isolation (MoBio™, EUA), seguindo o protocolo do fabricante.
Em seguida, as amostras de DNA total extraído foram quantificadas e avaliadas quanto
a sua pureza por espectrofotometria (NanoDrop Technologies, EUA). Para tanto, obteve-se a
quantidade de nanogramas de DNA por L de amostra e mediu-se a proporção entre as
absorbâncias obtidas a 260 nm e a 280 nm (relação 260/280) para avaliar a pureza das amostras
de DNA quanto a presença de proteínas, fenóis e outros contaminantes que absorvem
fortemente a 280 nm. Secundariamente, também como forma de avaliar a pureza das amostras
de DNA extraído, avaliou-se a proporção entre as absorbâncias obtidas a 260 nm e a 230 nm
(relação 260/230). A 230 nm absorvem contaminantes como o fenol, compostos aromáticos,
trizol e alguns outros reagentes utilizados na extração.
Após a extração, as amostras foram concentradas por meio da adição de uma solução de
NaCl 5M e etanol P.A. subsequentemente, seguida da centrifugação das amostras, despejo do
sobrenadante e evaporação do precipitado. Ao final, o pellet contendo o DNA purificado e
concentrado foi ressuspenso em água MilliQ estéril. Em seguida, os parâmetros de concentração
e qualidade das amostras de DNA foram novamente aferidos.
As cinco subamostras obtidas na Praia de Redonda, no município de Icapuí, foram
homogeneizadas e combinadas para formar uma amostra composta, representativa da região e
destinada a funcionar como uma área não impactada por atividades portuárias. Esta amostra foi
extraída da mesma forma que a amostras originadas nas estações de coleta do Porto do Pecém
e do Porto do Mucuripe.
Para comparar as assembleias de Bacteria e Archaea presentes nos portos em estudo
com as da área sem atividade de portos, após a extração de DNA, as soluções de DNA oriundas
das dez subamostras de sedimento do Porto do Pecém foram combinadas para formar uma
amostra composta representativa do Porto do Pecém. O mesmo procedimento foi feito com as
extrações de DNA obtidas das 15 subamostras do Porto do Mucuripe.

80
4.3.5. Eletroforese em Gel de Gradiente Desnaturante por meio de Reação em Cadeia de
Polimerase com iniciadores para o gene rRNA 16S
Para proceder a avaliação dos perfis estruturais das assembleias de Bacteria e
Archaea presentes nos sedimentos dos portos estudados por DGGE, as amostras de DNA
obtidas tiveram sua região V3 do gene codificador do rRNA 16S amplificada por meio de PCR
(termociclador de modelo Mastercycler EP Gradient S, Eppendorf, EUA). Foi utilizado o
método de duas reações de amplificação sequenciais (nested PCR) que consistiu de uma reação
inicial com iniciadores gerais para o rRNA 16S, seguida de reação para a região V3.
Na primeira reação do Domínio Bacteria foi utilizado um volume final para uma
reação de 25 L com 1 L da amostra de DNA (10 ng/L) servindo de molde, 2,5 L de cada
iniciador (5 M), 0,5L da solução de desoxirribonucleotídeos trifosfatados (dNTPs; 10 mM),
1,0 L de MgCl2 (50 mM), 5,0 L de tampão de reação 5X, 0,2L de Gotaq DNA polimerase
(5U/mL) e água MilliQ ultrapura para completar o volume da reação. Os iniciadores utilizados
foram o 63F (5' -CAG GCC TAA CAC ATG CAA GTC -3') e 1389R (5'- ACG GGC GGT
GTG TAC AAG -3') (MARCHESI et al., 1998). O programa de corrida consistiu de
desnaturação inicial a 95°C por 2 min, seguida de 30 ciclos consistindo de desnaturação a 95°C
por 1 min, anelamento a 55°C por 0,5 min e 72° C de extensão por 1 min, seguidos de um passo
de extensão final a 72°C por 10 min.
Na segunda reação do Domínio Bacteria, 1 L do produto de PCR obtido na
primeira reação foi utilizado como molde. Foi utilizado o par de iniciadores 338F GC (5'-CGC
CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG
CAG CAG -3') e 518R (5’ - ATT ACC GCG GCT GCT GG -3’) (MUYZER, DE WAAL,
UITTERLINDEN, 1993). As condições da segunda reação, de volume final de 30 L,
consistiram de 3,0 L de cada iniciador (5 M), 0,6 L da solução de desoxirribonucleotídeos
trifosfatados (dNTPs; 10mM), 1,8 L de MgCl2 (50 mM), 6,0L de tampão de reação 5X, 0,2
L de Gotaq DNA polimerase (5U/mL) e água MilliQ ultrapura para completar o volume da
reação. O programa de corrida consistiu de desnaturação inicial a 95°C por 5 min, seguida de
30 ciclos consistindo de desnaturação a 94°C por 1 min, anelamento a 55°C por 1 min e 72° C
de extensão por 2 min, seguidos de um passo de extensão final a 72°C por 5 min.
Na primeira reação do Domínio Archaea foi utilizado um volume final para uma
reação de 30 L com 1 L da amostra de DNA (10 ng/L) servindo de molde, 1,8 L de cada
iniciador (5 M), 0,6L da solução de desoxirribonucleotídeos trifosfatados (dNTPs; 10 mM),

81
1,2 L de MgCl2 (50 mM), 0,24 L de gelatina 1%, 6,0 L de tampão de reação 5X, 0,2L de
Gotaq DNA polimerase (5U/mL) e água MilliQ ultrapura para completar o volume da reação.
Os iniciadores utilizados foram o 20F (5’ – TTC CGG TTG ATC CYG CCRG – 3’) e 958R (5’
– YCC GGC GTT GAM TCC AATT – 3’) (DELONG, 1992). O programa de corrida consistiu
de desnaturação inicial a 94°C por 5 min, seguida de 30 ciclos consistindo de desnaturação a
94°C por 0,5 min, anelamento a 55°C por 1 min e 72° C de extensão por 1 min, seguidos de um
passo de extensão final a 72°C por 10 min.
Na segunda reação do Domínio Archaea, 1 L do produto de PCR obtido na
primeira reação foi utilizado como molde. Foi utilizado o par de iniciadores 340F GC (5’ - CGC
CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC CTA CGG GGY
GCA SCA - 3’) e 519R (5’ - TTA CCG CGG CKG CTG – 3’) (OVREÅS et al., 1997). As
condições da segunda reação, de volume final de 30 L, consistiram de 3,0 L de cada iniciador
(5 M), 0,6 L da solução de desoxirribonucleotídeos trifosfatados (dNTPs; 10 mM), 1,8 L
de MgCl2 (50 mM), 6,0L de tampão de reação 5X, 0,2 L de Gotaq DNA polimerase (5U/mL)
e água MilliQ ultrapura para completar o volume da reação. O programa de corrida consistiu
de desnaturação inicial a 92°C por 2 min, seguida de 35 ciclos consistindo de desnaturação a
92°C por 1 min, anelamento a 53.5°C por 0,5 min e 72° C de extensão por 1 min, seguidos de
um passo de extensão final a 72°C por 6 min.
Os produtos de PCR amplificados foram visualizados em um fotodocumentador
com iluminação ultravioleta após eletroforese em gel de agarose a 2% (m/v) adicionado do
corante SyBr-Safe DNA em tampão Tris-Borato-EDTA 0,5X (45 mM Tris Base, 45 mM de
ácido bórico e 1 mM de EDTA pH 8,0) (Life Technologies, EUA). Um marcador molecular de
1 Kb foi utilizado como padrão de peso molecular. Quando todas as amostras de DNA de cada
estação de coleta eram amplificadas na ausência de amplificação do controle negativo,
procedia-se o procedimento de DGGE.
Após a amplificação das amostras de DNA obtidas das dez estações de coleta do
Porto do Pecém e das 15 estações de coleta do Porto do Mucuripe, assim como de seus
respectivos pools de DNA e da amostra da Praia de Redonda, foram realizadas corridas de
DGGE conforme descrito por Heuer et al. (1997) com o equipamento BioRad Dcode-System
(BioRad, EUA). Em cada corrida, foram utilizadas as dez amostras de DNA originadas dos
sedimentos do Porto do Pecém e as 15 amostras de DNA originadas dos sedimentos do Porto
do Mucuripe em conjunto com um marcador de 50 pb.

82
Foram realizadas também corridas contendo os pools de DNA do Porto do Pecém
e do Porto do Mucuripe em comparação com a região da Praia de Redonda. As amostras foram
analisadas em triplicatas originadas em reações de polimerização independentes.
Os géis de poliacrilamida a 8% (m/v) foram preparados a partir de soluções de
concentração 0% e 100% de desnaturação (7 M de uréia e 40% de formamida), as quais foram
combinadas para formar um gradiente de 35% a 65% de desnaturação. A eletroforese foi
realizada a uma temperatura constante de 60°C e a uma voltagem de 200V por quatro horas em
tampão Tris-Acetato-EDTA 0.5X (20 mM de Tris, 20 mM de acetato e 1 mM de EDTA pH
8.0). Após a eletroforese, o gel foi retirado e corado com SyBr-Green I (Life Technologies,
EUA) por uma hora. Em seguida, foi visualizado e fotografado em um fotodocumentador com
iluminação ultravioleta.
4.3.6. Análise dos géis de DGGE
Os perfis de DGGE obtidos foram analisados e comparados no programa
Bionumerics versão 7.0 (Applied Maths, Bélgica). No programa, a imagem de cada gel foi
normalizada e avaliada de forma padronizada para identificar as bandas presentes em cada
amostra, sendo em seguida convertida em matrizes de presença e ausência de bandas (matrizes
binárias). Tais matrizes foram utilizadas em análises qualitativas e quantitativas da estrutura
das assembleias de Bacteria e Archaea presentes nos portos.
As análises de agrupamento por similaridade das diferentes amostras foram
realizadas no próprio programa Bionumerics, permitindo distinguir as estações de coleta quanto
a sua composição de UTOs. A partir das matrizes binárias obtidas, foram calculados os
estimadores de riqueza e beta diversidade que utilizam apenas dados de presença e ausência.
Os estimadores de riqueza utilizados foram: Chao-2, Jackknife de primeira ordem (Jackknife
1), e bootstrap, utilizados para a estimativa da riqueza de espécies para várias parcelas
amostradas de igual tamanho, tendo sido calculados a partir de 1000 reamostragens aleatórias.
As medidas de beta diversidade, capazes de evidenciar a taxa de mudança das espécies entre
diferentes unidades amostrais, utilizadas foram: βw (WHITTAKER, 1960) e βt (WILSON,
SHMIDA, 1984). Os índices e estimadores de diversidade que necessitam de dados de
abundância relativa para o seu cálculo não foram utilizados.
Em seguida, as matrizes binárias de cada porto para os Domínios Bacteria e
Archaea, respectivamente, foram reunidas e utilizadas para gerar agrupamentos hierárquicos a

83
partir da utilização do coeficiente de similaridade ou índice qualitativo de Jaccard, o qual, com
base na matriz de presença e a ausência de bandas, avalia o número de espécies encontradas em
duas amostras diferentes, sendo calculado sempre para um par de amostras. A partir da mesma
matriz também foram avaliadas as riquezas das assembleias de Bacteria e Archaea associadas
aos sedimentos de cada porto, construídos Diagramas de Venn para detalhar o
compartilhamento e exclusividade de UTOs. As análises estatísticas descritas foram realizadas
no programa PAST versão 2.17c (HAMMER, HARPER, RYAN, 2001).
4.4. Resultados
4.4.1. Caracterização das amostras de DNA
As amostras de sedimentos de ambos os portos não apresentaram diferenças na
concentração média de DNA extraído (14,5 ng/µL), nem nas médias das razões das
absorbâncias em 260/280 (1,7) e 260/230 de (1,3), que expressam a qualidade do DNA extraído.
A amostra de DNA da Praia de Redonda, referência não portuária, apresentou concentração de
4,27 ng/µL, relação 260/280 de 2,32 e relação 260/230 de 0,27.
Apesar das concentrações médias de DNA terem sido semelhantes para ambos os
portos, as estações de coleta dentro de cada porto variaram bastante, destacando-se as estações
P7, M2, M3 e M12 , que apresentaram concentrações muito baixas de DNA, enquanto P5, P10,
M1, M13 e M14, renderam concentrações mais altas (Tabela 7). Esse primeiro resultado aponta
para a heterogeneidade espacial dessas regiões portuárias e sugere a existência de comunidades
procarióticas variadas, possivelmente modeladas pelas características peculiares de cada
estação.

84
Tabela 7 - Caracterização das amostras de DNA extraídas dos sedimentos oriundos do Porto
do Pecém (P1-P10) e do Porto do Mucuripe (M1-M15). Pmédia e Mmédia representam as médias das
concentrações de DNA, relações 260/280 e relações 260/230 das amostras obtidas no Porto do
Pecém e no Porto do Mucuripe, respectivamente.
Amostras Concentração de DNA
(g/L)
Relação
260/280
Relação
260/230
P1 10,2 ± 1,06 1,60 ± 0,11 1,40 ± 0,11
P2 10,50 ± 3,39 1,80 ± 0,06 2,00 ± 0,59
P3 17,30 ± 0,64 1,70 ± 0,08 1,10 ± 0,05
P4 15,90 ± 1,84 1,80 ± 0,02 1,50 ± 0,09
P5 23,20 ± 3,11 1,80 ± 0,13 1,60 ± 0,01
P6 19,70 ± 1,77 1,80 ± 0,14 1,70 ± 0,04
P7 3,00 ± 0,42 1,50 ± 0,11 1,20 ± 0,03
P8 9,70 ± 0,0 1,60 ± 0,14 1,00 ± 0,01
P9 14,4 ± 1,20 1,70 ± 0,01 1,10 ± 0,02
P10 20,7 ± 0,21 1,80 ± 0,04 1,50 ± 0,01
Pmédia 14,40 ± 0,13 1,70 ± 0,04 1,40 ± 0,07
M1 19,50 ± 0,71 1,70 ± 0,06 1,30 ± 0,08
M2 6,60 ± 1,34 1,70 ± 0,11 1,30 ± 0,04
M3 2,60 ± 0,57 1,60 ± 0,24 0,50 ± 0,06
M4 15,80 ± 1,70 1,80 ± 0,03 1,40 ± 0,04
M5 13,30 ± 0,14 1,60 ± 0,00 1,00 ± 0,01
M6 13,90 ± 0,42 1,50 ± 0,07 1,00 ± 0,01
M7 15,40 ± 0,28 1,70 ± 0,06 1,40 ± 0,01
M8 12,80 ± 0,35 1,70 ± 0,05 1,30 ± 0,02
M9 7,50 ± 0,07 1,60 ± 0,06 1,10 ± 0,01
M10 8,20 ± 0,71 1,60 ± 0,24 1,00 ± 0,07
M11 12,60 ± 0,35 1,60 ± 0,12 1,00 ± 0,05
M12 6,40 ± 0,57 1,30 ± 0,03 0,80 ± 0,01
M13 41,40 ± 1,20 1,80 ± 0,06 1,70 ± 0,01
M14 38,80 ± 0,28 1,80 ± 0,01 1,70 ± 0,01
M15 8,50 ± 0,64 1,60 ± 0,02 1,00 ± 0,01
Mmédia 14,90 ± 0,01 1,60 ± 0,04 1,20 ± 0,00
4.4.2. Caracterização física e química dos sedimentos
As variáveis ambientais analisadas juntamente com algumas informações
relacionadas à operação dos portos estão apresentadas na Tabela 8. Exceto pela profundidade,
salinidade, transparência da água, Hidrocarbonetos Policíclicos Aromáticos (HPAs) totais,
compostos BTEX (Benzeno, Tolueno, Etilbenzeno e Xileno) e teores de chumbo, arsênio,
cádmio e mercúrio, todos os demais parâmetros físico-químicos diferiram estatisticamente entre
os portos (p < 0.05).

85
Com relação ao total de carga transportada em cada porto, observa-se uma diferença
de mais de 780 mil toneladas de carga, evidenciando a maior atividade do Porto do Mucuripe
no ano de 2010.
Os sedimentos de ambos os portos foram dominados por silte, argila e areia fina,
porém o Porto do Pecém apresentou um teor mais elevado de cascalho e areia de muito grossa
a média do que o Porto do Mucuripe (Figura 10). Outras diferenças encontradas foram o teor
de carbonato, mais elevado no Porto do Pecém, e matéria orgânica, igualmente superior no
Pecém. Já o Carbono Orgânico Total (COT) foi mais elevado no Porto do Mucuripe.
As concentrações de metais pesados, arsênio, PCBs e HPAs encontradas em ambos
os portos encontram-se de acordo com valores de referência constantes na Resolução do
Conselho Nacional do Meio Ambiente (CONAMA) nº 344, de 25 de março de 2004, conforme
pode-se conferir nas Figuras 11 para os metais e Figura 12 para HPAs. Contudo, é importante
destacar que, dentre os HPAs quantificados, os compostos benzo(b)fluoranteno e
benzo(k)fluoranteno representaram mais de 50% dos teores observados em ambos os portos.
Esses compostos apresentaram concentrações bem acima do somatório total de HPAs apontado
na legislação, conforme pode-se observar na Figura 12. Entretanto, as quantidades críticas para
ambos não são tratadas na referida Resolução.

86
Tabela 8 - Atividade portuária em toneladas de carga transportada e parâmetros físicos e
químicos do Portos do Pecém (PEC) e Porto do Mucuripe (MUC)
PEC MUC
Profundidade (m) b * 21,33 13,67
Salinidade b * 41 40
pH b * 7,51 7,60
Transparência da água b * 2,53 2,40
Atividade dos portos (ton) (carga
transportada em 2010) a 3.565.476 4.349.022
Granulometria b
Cascalho 2,40%
Areia muito grossa 3,90%
Areia grossa 8,10%
Areia média 9,99%
Areia fina 5,25%
Areia muito fina 8,10%
Silte-argila 62,28%
Cascalho 0,10%
Areia muito grossa 0,10%
Areia grossa 0,63%
Areia média 1,61%
Areia fina 7,62%
Areia muito fina 23,96%
Silte-argila 66,01%
Carbonato (g Kg-1) b 4036,73 ± 197,76 1741,40 ± 225,66
Matéria Orgânica (g Kg-1) b 150,46 ± 5,27 94,29 ± 30,45
COT (mg Kg-1) b ¥ 75,77 ± 0,06 130,73 ± 0,80
Nitrato (mg Kg-1) b 10,06 ± 0,21 9,17 ± 0,11
Nitrito (mg Kg-1) b c * < 1 < 1
Sulfato (mg Kg-1) b 443,96 ± 2,77 375,37 ± 4,98
Bifenilas totais (mg Kg-1) b c * < 9,50 < 9,50
HPAs totais (mg Kg-1) b * 40,82 ± 0,21 39,15 ± 1,16
BTEX total (mg Kg-1) b c * < 0,07 < 0,07
As (mg Kg-1) b c * < 0,50 < 0,50
Cd (mg Kg-1) b c * < 0,50 < 0,50
Pb (mg Kg-1) b * 3,70 ± 0,10 3,47 ± 0,35
Cu (mg Kg-1) b 4,77 ± 0,15 3,00 ± 0,10
Cr (mg Kg-1) b 15,60 ± 0,20 10,80 ± 0,22
Hg (mg Kg-1) b c * < 0,10 < 0,10
Ni (mg Kg-1) b 4,47 ± 0,06 3,20 ± 0,20
Zn (mg Kg-1) b 17,70 ± 0,04 11,90 ± 0,10
Fe (mg Kg-1) b 9445,77 ± 80,78 7600,63 ± 65,91
a ANTAQ – Análise da quantidade de carga transportada em portos organizados e terminais privativos (2010). b Dados obtidos no presente estudo. c Medidas abaixo do mínimo detectável pelo método de mensuração. * Parâmetros que não apresentam significativas entre os portos estudados. ¥ Carbono Orgânico Total.
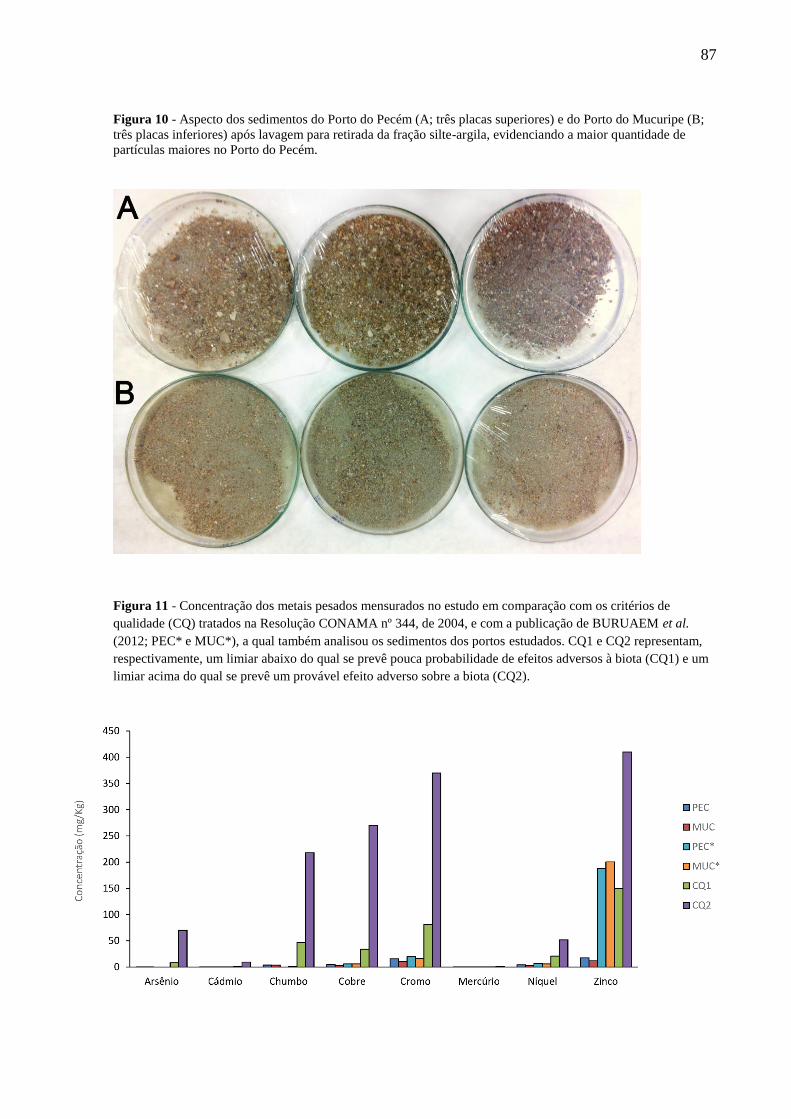
87
Figura 10 - Aspecto dos sedimentos do Porto do Pecém (A; três placas superiores) e do Porto do Mucuripe (B;
três placas inferiores) após lavagem para retirada da fração silte-argila, evidenciando a maior quantidade de
partículas maiores no Porto do Pecém.
Figura 11 - Concentração dos metais pesados mensurados no estudo em comparação com os critérios de
qualidade (CQ) tratados na Resolução CONAMA nº 344, de 2004, e com a publicação de BURUAEM et al.
(2012; PEC* e MUC*), a qual também analisou os sedimentos dos portos estudados. CQ1 e CQ2 representam,
respectivamente, um limiar abaixo do qual se prevê pouca probabilidade de efeitos adversos à biota (CQ1) e um
limiar acima do qual se prevê um provável efeito adverso sobre a biota (CQ2).
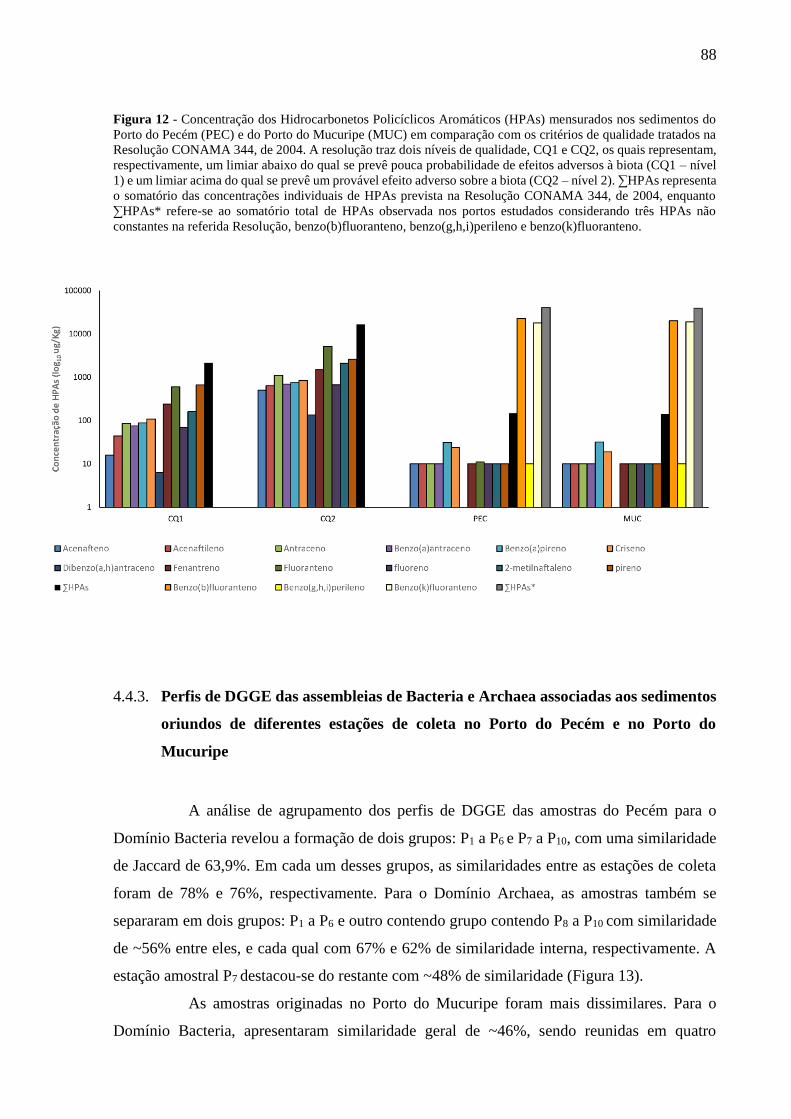
88
Figura 12 - Concentração dos Hidrocarbonetos Policíclicos Aromáticos (HPAs) mensurados nos sedimentos do
Porto do Pecém (PEC) e do Porto do Mucuripe (MUC) em comparação com os critérios de qualidade tratados na
Resolução CONAMA 344, de 2004. A resolução traz dois níveis de qualidade, CQ1 e CQ2, os quais representam,
respectivamente, um limiar abaixo do qual se prevê pouca probabilidade de efeitos adversos à biota (CQ1 – nível
1) e um limiar acima do qual se prevê um provável efeito adverso sobre a biota (CQ2 – nível 2). ∑HPAs representa
o somatório das concentrações individuais de HPAs prevista na Resolução CONAMA 344, de 2004, enquanto
∑HPAs* refere-se ao somatório total de HPAs observada nos portos estudados considerando três HPAs não
constantes na referida Resolução, benzo(b)fluoranteno, benzo(g,h,i)perileno e benzo(k)fluoranteno.
4.4.3. Perfis de DGGE das assembleias de Bacteria e Archaea associadas aos sedimentos
oriundos de diferentes estações de coleta no Porto do Pecém e no Porto do
Mucuripe
A análise de agrupamento dos perfis de DGGE das amostras do Pecém para o
Domínio Bacteria revelou a formação de dois grupos: P1 a P6 e P7 a P10, com uma similaridade
de Jaccard de 63,9%. Em cada um desses grupos, as similaridades entre as estações de coleta
foram de 78% e 76%, respectivamente. Para o Domínio Archaea, as amostras também se
separaram em dois grupos: P1 a P6 e outro contendo grupo contendo P8 a P10 com similaridade
de ~56% entre eles, e cada qual com 67% e 62% de similaridade interna, respectivamente. A
estação amostral P7 destacou-se do restante com ~48% de similaridade (Figura 13).
As amostras originadas no Porto do Mucuripe foram mais dissimilares. Para o
Domínio Bacteria, apresentaram similaridade geral de ~46%, sendo reunidas em quatro

89
subgrupos distintos; um contendo apenas as estações de coleta M1 e M2 com 60% de
similaridade, outro contendo M12 a M15 com aproximadamente 63% de similaridade e um
terceiro com as amostras dos pontos M4 a M11 com ~60% de similaridade. A estação de coleta
M3 destacou-se das demais, apresentando 47% de similaridade. Para o Domínio Archaea,
observou-se uma similaridade entre todas as estações de coleta de 47%, com exceção de M3,
que divergiu das demais. As estações restantes foram reunidas em cinco grupos; um contendo
apenas M15, outro contendo M4 e M5, um com M6 a M11 compartilhando 84% de similaridade,
um com M13 e M14 com 81% de semelhança e outro contendo M1, M2 e M12 com
aproximadamente 53% de similaridade (Figura 14).
Portanto, nota-se que os menores índices de similaridade foram encontrados entre
as estações de coleta do Porto do Mucuripe, com o Domínio Archaea exibindo também menores
similaridades de Jaccard em ambos os portos, sugerindo uma maior variabilidade, conforme
pode-se observar nas Figuras 13 e 14.

90
Figura 13 – Análise de agrupamento dos perfis de DGGE para o Domínio Bacteria (A) e Domínio Archaea (B)
associados aos sedimentos do Porto do Pecém. Para comparação dos grupos, foi utilizado o índice de Jaccard no
programa PAST v2.17c.
A
B
65% 35%
65% 35%

91
Figura 14 - Análise de agrupamento dos perfis de DGGE para o Domínio Bacteria (A) e Domínio Archaea (B)
associados aos sedimentos do Porto do Mucuripe. Para comparação dos grupos, foi utilizado o índice de Jaccard
no programa PAST v2.17c.
A
B
65% 35%
65% 35%

92
No total foram encontradas para o Domínio Bacteria, 42 UTOs no Porto do Pecém
e 53 no Porto do Mucuripe, as quais foram distribuídas nas estações de coleta em uma média
de 28,20 ± 1,03 e 27,40 ± 2,97 UTOs, respectivamente. Para o Domínio Archaea, encontrou-se
um total de 47 UTOs no Porto do Pecém e 56 no Porto do Mucuripe, tendo sido estas
distribuídas nas estações de coleta, em média de 26,20 ± 2,82 e 28,00 ± 3,14 UTOs,
respectivamente, conforme pode-se observar na Tabela 9.
As estimativas do número total de espécies presentes em cada assembleia,
calculadas pelos estimadores Chao 2, Jackknife 1 e bootstrap, baseados na presença de grupos
raros, resultaram em valores próximos ao total de UTOs encontradas por DGGE (Tabela 10),
evidenciando que o método cobriu bem a diversidade de cada um dos portos e que o Porto do
Mucuripe possui maior riqueza para ambos os domínios estudados.
Tabela 9 – Riqueza de UTOs encontradas nos perfis de DGGE para os Domínios Bacteria e
Archaea do Porto do Pecém (P1 a P10) e do Porto do Mucuripe (M1 a M15).
Riqueza (S) Riqueza (S)
Amostras Bacteria Archaea Amostras Bacteria Archaea
P1 28 29 M1 27 30
P2 30 28 M2 21 27
P3 28 25 M3 29 18
P4 29 28 M4 22 29
P5 29 28 M5 28 29
P6 27 25 M6 31 28
P7 28 28 M7 29 29
P8 27 21 M8 30 28
P9 29 22 M9 31 28
P10 27 28 M10 29 31
M11 28 31
M12 29 29
M13 26 29
M14 25 29
M15 26 25
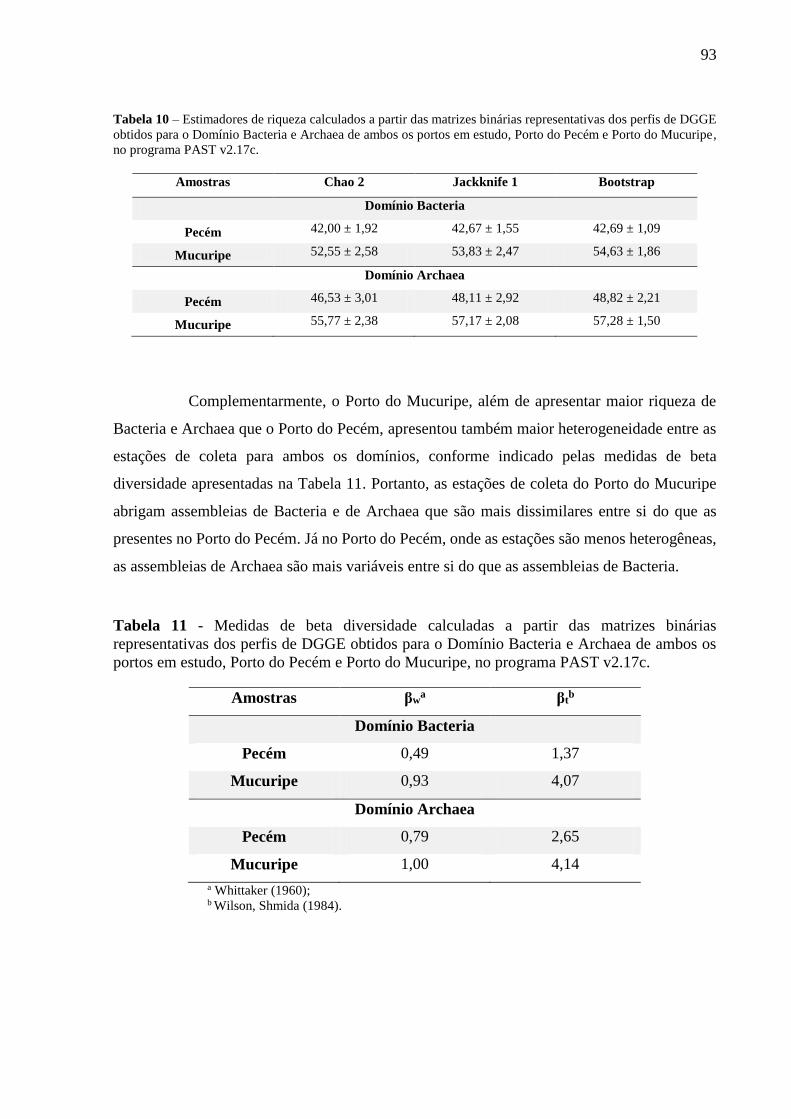
93
Tabela 10 – Estimadores de riqueza calculados a partir das matrizes binárias representativas dos perfis de DGGE
obtidos para o Domínio Bacteria e Archaea de ambos os portos em estudo, Porto do Pecém e Porto do Mucuripe,
no programa PAST v2.17c.
Amostras Chao 2 Jackknife 1 Bootstrap
Domínio Bacteria
Pecém 42,00 ± 1,92 42,67 ± 1,55 42,69 ± 1,09
Mucuripe 52,55 ± 2,58 53,83 ± 2,47 54,63 ± 1,86
Domínio Archaea
Pecém 46,53 ± 3,01 48,11 ± 2,92 48,82 ± 2,21
Mucuripe 55,77 ± 2,38 57,17 ± 2,08 57,28 ± 1,50
Complementarmente, o Porto do Mucuripe, além de apresentar maior riqueza de
Bacteria e Archaea que o Porto do Pecém, apresentou também maior heterogeneidade entre as
estações de coleta para ambos os domínios, conforme indicado pelas medidas de beta
diversidade apresentadas na Tabela 11. Portanto, as estações de coleta do Porto do Mucuripe
abrigam assembleias de Bacteria e de Archaea que são mais dissimilares entre si do que as
presentes no Porto do Pecém. Já no Porto do Pecém, onde as estações são menos heterogêneas,
as assembleias de Archaea são mais variáveis entre si do que as assembleias de Bacteria.
Tabela 11 - Medidas de beta diversidade calculadas a partir das matrizes binárias
representativas dos perfis de DGGE obtidos para o Domínio Bacteria e Archaea de ambos os
portos em estudo, Porto do Pecém e Porto do Mucuripe, no programa PAST v2.17c.
Amostras βwa βt
b
Domínio Bacteria
Pecém 0,49 1,37
Mucuripe 0,93 4,07
Domínio Archaea
Pecém 0,79 2,65
Mucuripe 1,00 4,14
a Whittaker (1960); b Wilson, Shmida (1984).

94
Quando as estações de coleta de ambos os portos foram analisadas em conjunto,
foi possível separar perfeitamente as estações de coleta do Porto do Pecém das do Porto do
Mucuripe para ambos os domínios (Figuras 15 e 16). Além disso, as similaridades encontradas
entre as amostras foram próximas às observadas quando as mesmas foram analisadas de forma
separada, tendo sido de cerca de 50% entre as amostras do Pecém e aproximadamente 30% para
as estações do Mucuripe, para o Domínio Bacteria. Para Archaea, encontrou-se 40% de
similaridade entre as amostras do Pecém e 30% entre as do Mucuripe. Os padrões de
agrupamento das amostras de cada um dos portos foi mantido em sua maioria, preservando a
divisão do Porto do Pecém entre P1-P6 e P7-P10 para Bacteria e a estação P7 separada desses
grupos para Archaea. Apenas no Porto do Mucuripe foram observadas algumas disparidades
dentro dos agrupamentos contendo M4-M11 e M12-M15, onde alguns subgrupos formados foram
diferentes dos obtidos previamente.
Figura 15 - Análise de agrupamento em conjunto das amostras obtidas no Porto do Pecém (PEC 1 a PEC 10) e
no Porto do Mucuripe (MUC 1 a MUC 15) para o Domínio Bacteria. Para comparação dos grupos, foi utilizado
o índice de Jaccard no programa PAST v2.17c.
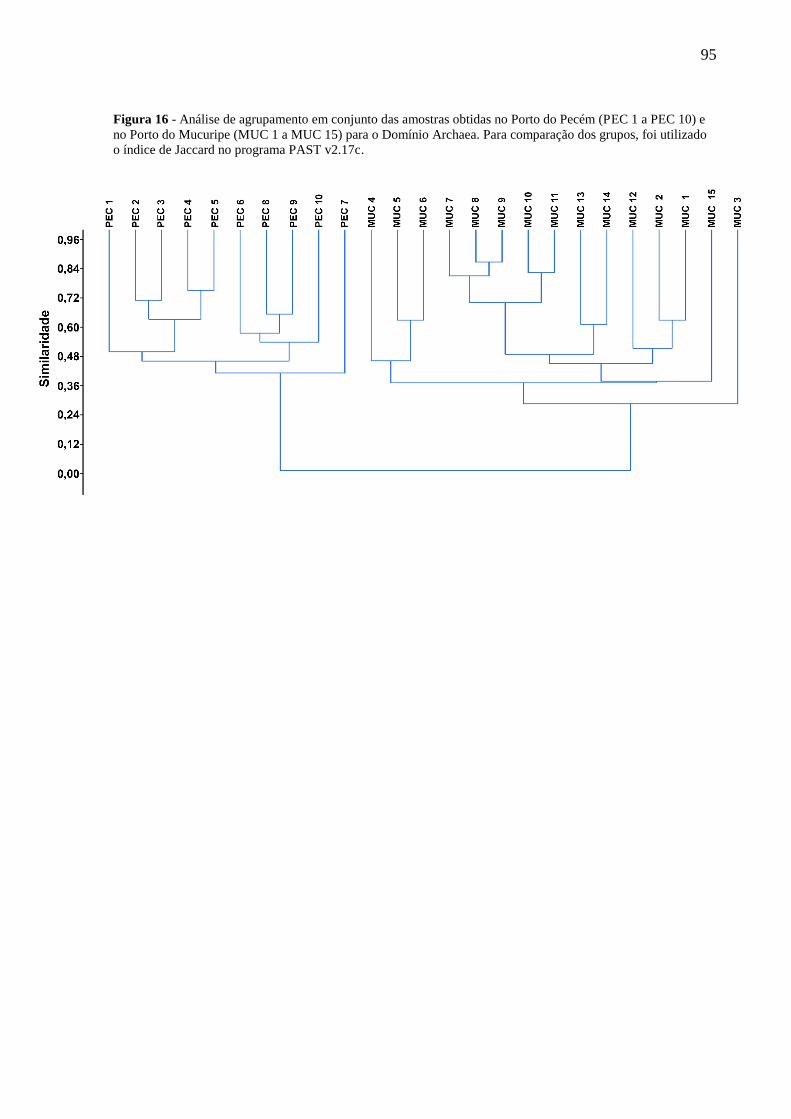
95
Figura 16 - Análise de agrupamento em conjunto das amostras obtidas no Porto do Pecém (PEC 1 a PEC 10) e
no Porto do Mucuripe (MUC 1 a MUC 15) para o Domínio Archaea. Para comparação dos grupos, foi utilizado
o índice de Jaccard no programa PAST v2.17c.

96
4.4.4. Comparação das estruturas das assembleias de Bacteria e Archaea do Porto do
Pecém e do Porto do Mucuripe com a de uma região sem atividade portuária
Com o objetivo de obter um perfil das amostras obtidas em cada região de coleta para
podermos compará-las quanto a sua estrutura e riqueza dos Domínios Bacteria e Archaea com
uma região onde a atividade portuária é ausente, a Praia de Redonda (município de Icapuí,
Ceará), foram realizadas corridas de DGGE contendo estas três amostras.
As análises dos perfis de DGGE para o Domínio Bacteria revelaram 32 UTOs no Porto
do Mucuripe (MUC), 29 no Porto do Pecém (PEC) e 31 na região da Praia de Redonda, em
Icapui (ICA) (Tabela 12). Destas UTOs, 11 foram compartilhadas pelos três locais e quatro
delas foram exclusivas do Mucuripe; outras quatro, exclusivas do Pecém; e ainda sete,
peculiares a ICA, conforme pode ser visto no Diagrama de Venn apresentado na Figura 17a.
Ademais, pode-se observar que foram encontradas 9 UTOs compartilhadas apenas entre os
portos. Para o Domínio Archaea, foram encontradas 32 UTOs no Porto do Mucuripe, 31 no
Porto do Pecém e apenas 10 em ICA (Tabela 12). Deste montante, foram compartilhadas cinco
UTOs entre as três áreas, e a quantidade de táxons exclusivos foi de 12 para o Mucuripe, 11
para o Pecém e três para ICA. Um total de 14 UTOs foi compartilhado apenas entre os portos,
conforme pode ser observado na Figura 17b.
O cálculo dos estimadores de riqueza resultou em valores mais elevados do que o
número de UTOs revelado por DGGE para os Domínios Bacteria e Archaea do Porto do
Mucuripe e para o Domínio Bacteria do Porto do Pecém em menor proporção. Isso significa
que existem no ambiente números maiores de UTOs do que os reduzidos valores encontrados
através da análise dos pools, os quais foram incapazes de cobrir bem a diversidade dos portos
estudados, com exceção do Domínio Archaea para o Porto do Pecém.
Tabela 12 – Riquezas de UTOs encontradas nos perfis de DGGE dos pools de sedimentos para o Domínio Bacteria
e Archaea dos portos em estudo, Porto do Pecém (PEC), Porto do Mucuripe (MUC) e Praia de Redonda (ICA).
Riqueza (S)
Amostras Domínio
Bacteria Archaea
MUC 32 32
PEC 29 31
ICA 31 10

97
Tabela 13 - Estimadores de riqueza calculados a partir dos perfis de DGGE dos pools de sedimentos obtidos para
o Domínio Bacteria e Archaea dos portos em estudo, Porto do Pecém (PEC), Porto do Mucuripe (MUC) e Praia
de Redonda (ICA).
Amostras Chao 2 Jackknife 1 Bootstrap
Domínio Bacteria
PEC 33,45 ± 1,06 34,18 ± 1,59 34,67 ± 1,27
MUC 40,08 ± 5,76 43,52 ± 7,38 46,33 ± 5,91
ICA 32,51 ± 1,21 33,31 ± 1,78 32,80 ± 1,40
Domínio Archaea
PEC 31,70 ± 0,46 32,16 ± 0,77 32,33 ± 0,61
MUC 36,66 ± 3,45 39,31 ± 4,57 40,56 ± 3,76
ICA 11,51 ± 1,04 12,26 ± 1,56 12,67 ± 1,25
Figura 17 – Diagrama de Venn evidenciando o número de UTOs do Domínio Bacteria (A) e do Domínio Archaea
(B) presentes em cada um dos ambientes estudados, Porto do Pecém (PEC), Porto do Mucuripe (MUC) e Praia de
Redonda (ICA).
A B

98
As análises de beta diversidade com o coeficiente de similaridade de Jaccard
mostraram que, para o Domínio Bacteria, as amostras dos portos e da Praia de Redonda (ICA)
compartilham menos de 60% de similaridade. Internamente, cada porto reuniu as suas
respectivas triplicadas, que foram quase 100% idênticas; apenas uma das replicatas das
amostras do Porto do Pecém apresentou similaridade em torno de 70%. Já para o Domínio
Archaea, a área da Praia de Redonda (ICA) apresentou menos de 40% de similaridade com os
portos. Estes formaram dois grupos, cada qual composto por suas respectivas triplicatas, as
quais foram quase 100% idênticas no Mucuripe. Dentre as triplicatas do Porto do Pecém, apenas
uma divergiu das demais, com cerca de 70% de similaridade, conforme pode-se observar na
Figura 18.
Figura 18 - Análise de agrupamento dos perfis de DGGE para o Domínio Bacteria (A) e Domínio Archaea (B)
associados aos pools de sedimentos do Porto do Pecém (PECa, PECb e PECc), Porto do Mucuripe (MUCa, MUCb
e MUCc) e Icapui (ICAa, ICAb e ICAc). Para comparação dos grupos, foi utilizado o índice de Jaccard no
programa PAST v2.17c.
65% 35%
65% 35%
A
B

99
4.5. Discussão
O Estado do Ceará conta com dois grandes portos marítimos, o Porto do Pecém e o
Porto do Mucuripe. Estes, entretanto, são bastante distintos. Diferem primeiramente em relação
a sua localização, já que o Mucuripe localiza-se em uma área industrial e dentro da capital do
Estado, enquanto o Pecém localiza-se em uma área em processo de industrialização, distante
60 Km da capital. Também são diferentes as arquiteturas de cada porto, pois o Porto do
Mucuripe possui um desenho tradicional, com os berços de atracação protegidos por moles para
redução da hidrodinâmica local, e o Porto do Pecém segue uma concepção mais moderna, do
tipo offshore, ou seja, fora da linha de costa (há dois quilômetros desta), e sobre pilastras para
não impedir o transporte de sedimentos pelas correntes oceânicas. Completando a lista de
diferenças, aparecem ainda o tempo de funcionamento, já que o Mucuripe conta com mais de
50 anos de operação (desde 1953) enquanto o Pecém possui apenas 13 anos de funcionamento
(desde 2001), e o volume de carga transportado, maior no Porto do Mucuripe. O Mucuripe
possui ainda uma área retroportuária bem desenvolvida onde estão instalados três moinhos de
trigo, uma fábrica de margarina, nove distribuidoras de combustíveis e a refinaria Lubnor da
Petrobrás, em contraposição à área do Complexo Industrial e Portuário do Pecém ainda em fase
de instalação.
Com base nessas características gerais dos dois portos e nas potenciais fontes
poluidoras, alguns parâmetros físico-químicos foram escolhidos para complementar essa
caracterização. Foram analisados: granulometria, profundidade, transparência da água, pH,
matéria orgânica, carbonato, COT, HPAs totais, BTEX e os metais As, Cr, Cd, Cu, Fe, Hg, Ni,
Pb e Zn. Essas análises ajudaram a entender melhor o biótopo dos portos, tendo destacado-se
como principais divergências as concentrações de carbonato e matéria orgânica, que foram mais
de duas vezes maiores no Porto do Pecém, e o teor de COT, que foi cerca de duas vezes maior
no Porto do Mucuripe. Também diferiram estatisticamente as concentrações de Cu, Cr, Ni, Zn
e Fe, conforme pode-se conferir na Tabela 8 (seção 4.4.2). Os portos foram similares apenas
em relação à profundidade (~ 20 m), salinidade, transparência da água, teor de HPAs totais,
BTEX, Pb, As, Cd e Hg.
A caracterização físico-química per se foi capaz de mostrar como os dois ambientes
são diferentes, porém as perturbações ambientais induzidas pelas atividades antrópicas são
capazes de ir além, alterando profundamente a biota de um ecossistema; com destaque para a
microbiota, hábil em responder rapidamente a modificações ambientais. Há vários trabalhos
mostrando que as comunidades microbianas podem ser afetadas por atividades antropogênicas,

100
resultando em alterações em sua estrutura detectáveis e indicativas das condições prevalentes
em um ecossistema. Com base nessa premissa, investigamos a estrutura espacial e a riqueza das
assembleias de Archaea e de Bacteria associadas aos sedimentos desses dois portos por meio
de DGGE visando averiguar se a arquitetura e o tempo de funcionamento diferenciados, assim
como as demais diferenças reveladas nesse trabalho, poderiam influenciar em tais atributos de
comunidade.
A respeito das concentrações de carbonato e matéria orgânica de sedimentos da costa
nordeste do Brasil, Freire et al. (2004) discutem que os sedimentos da região Nordeste possuem
uma composição principalmente biogênica devido a uma combinação de vários fatores, como
clima semiárido, pequena drenagem fluvial e baixo índice pluviométrico, contribuindo para
uma baixa entrada de sedimentos terrestres e uma plataforma rica em carbonato de cálcio
(NASCIMENTO, FREIRE, MIOLA, 2010). Os substratos da costa do Ceará são derivados de
algas calcárias e contribuem com 75-95% da deposição de carbonato de cálcio, com as
concentrações de matéria orgânica variando de 23.6% a 46%. Já os substratos terrígenos,
possuem teor de carbonato variando de 0.2% a 95% e de matéria orgânica entre 0.76% e 38.9%.
Em geral, o tipo de sedimentação ocorrente em determinada área, terrígena ou carbonácea,
controla a concentração de carbonatos e matéria orgânica, os quais são negativamente
correlacionados; quanto maior a influência continental, menor o teor de carbonatos e matéria
orgânica nos sedimentos (KNOPPERS et al., 1999).
No Porto do Pecém e no Porto do Mucuripe, foram encontradas baixas quantidades de
carbonato e matéria orgânica, o que também foi observado por Buruaem et al. (2012).
Conforme discutido por Knoppers et al. (1999), isso pode indicar uma maior influência
continental, evidenciando uma modificação do padrão regional local, ou seja, altas
concentrações de carbonato e matéria orgânica. Quando os dois portos são comparados entre
si, observa-se quantidades menores de carbonato e matéria orgânica no Porto do Mucuripe,
indicando uma maior influência continental neste porto devido à diminuição da hidrodinâmica
local e consequente acúmulo de sedimentos finos (GARRISON, 2011).
Com relação às concentrações de HPAs, é importante ressaltar que todas as moléculas
analisadas constantes na Resolução nº 344 do CONAMA se encontram em quantidades abaixo
dos níveis de referência da legislação, assim como seu somatório. Porém, há dois dos HPAs
estudados que não constam na referida Resolução, benzo(b)fluoranteno e benzo(k)fluranteno,
os quais estão presentes em quantidades muito elevadas e representam mais de 50% do total de
HPAs investigados. Tratam-se de HPAs de elevado peso molecular contendo cinco anéis
aromáticos, o que dificulta sua solubilidade e degradabilidade. São perigosos poluentes

101
carcinogênicos cuja concentração nos ambientes aquáticos vem aumentando recentemente
(PAN et al., 2005), sendo considerados poluentes prioritários pela Agência de Proteção
Ambiental dos EUA (EPA). Além do despejo de efluentes, esses compostos oriundos de
processos de combustão incompleta atingem a água na forma de poeira e por precipitação a
partir da atmosfera. Quando na água, adsorvem em matéria particulada na coluna d’água ou no
sedimento (IRWIN et al., 1997). Hidrocarbonetos policíclicos aromáticos, como os
benzofluorantenos, já foram previamente relacionados a alterações em comunidades
microbianas. Edlund e Jansson (2006), estudando sedimentos altamente poluídos na costa da
Suécia, observaram alterações na comunidade microbiana reversíveis após a dragagem das
camadas superficiais dos sedimentos e redução dos níveis de HPAs, PCBs e mercúrio.
As amostras obtidas nos portos resultaram em concentrações médias de DNA maiores
que as amostras obtidas na região sem atividade portuária. Isso ocorreu, possivelmente, devido
à granulometria dos sedimentos de Icapui, que apresenta maior teor de areia e cascalho
(OLIVEIRA, 2012). Alguns estudos mostraram que quanto menor os tamanhos das partículas
de sedimento, maior a diversidade bacteriana da comunidade associada (JACKSON; WEEKS,
2008). A razão para o aumento nas quantidades de micro-organismos é a maior disponibilidade
de área superficial para colonização nas partículas menores (GUILBEAU, 2003). Analisando
também o rendimento das extrações de DNA de cada um dos pontos de coleta, percebe-se que
há alguns pontos com concentrações bem elevadas no Porto do Pecém e no Porto do Mucuripe,
e outros com concentrações bem abaixo da média, como é o caso de P7 no Porto do Pecém e
M3 no Porto do Mucuripe. Quando as características particulares de cada uma dessas estações
de coleta são consideradas, observa-se que são justamente as que se diferenciaram das demais
estações em relação às características dos sedimentos – no caso, P7, com grande quantidade de
cascalho, e M3, com sedimento mais compactado. Reforça-se, portanto, como o tipo de
sedimento pode influenciar a biota associada. Na verdade, as concentrações de DNA obtidas
nessas estações de coleta foram comparáveis à concentração observada na Praia de Redonda,
em Icapui, região sem influência da instalação e operação de portos.
A análise dos perfis de DGGE revelou riquezas mais elevadas no Porto do
Mucuripe, tanto para Bacteria como para Archaea. Considerando que esse porto é ativo há mais
de 50 anos, a maior riqueza no Mucuripe pode ser relacionada às assembleias terem entrado em
contato com um maior grau de perturbação ao longo dos anos, tendo resultado em um maior
número de nichos ecológicos, conforme proposto pela Hipótese do Distúrbio Intermediário e o
Mecanismo de Particionamento de Nichos (MA et al., 2012). De fato, os sedimentos constituem

102
ambientes que, diferentemente da água do mar, propiciam a formação de vários nichos
ecológicos (BOLHUIS et al., 2013).
Uma revisão da resposta de comunidades microbianas de distintos ambientes a
quatro diferentes perturbações em mais de 100 trabalhos publicados mostrou que, na maioria
dos casos, a perturbação causou mudanças na estrutura das comunidades (ALONSO-
GUTIÉRREZ et al., 2009). Os autores relataram que eventos de contaminação aguda causam
redução na diversidade microbiana rapidamente devido ao desaparecimento de certos grupos e
seleção de resistentes. Em contraste, a diversidade microbiana é usualmente alta em ambientes
cronicamente poluídos, como o Porto do Mucuripe, como resultado da adaptação dos
organismos às novas características do ecossistema.
Analisando-se o número de UTOs encontradas em cada estação de coleta, nota-se que
possuem número total similar, em torno de 27 UTOs, tanto para o Domínio Bacteria como
Archaea, em ambos os portos. A estação que apresentou menor quantidade de UTOs foi a M3,
amostrada no Porto do Mucuripe, particularmente para o Domínio Archaea, Esta estação havia
chamado atenção previamente devido a sua baixa concentração de DNA purificado e ao aspecto
compactado do sedimento.
As análises de agrupamento com coeficiente de similaridade de Jaccard permitiram
observar que o Porto do Pecém possui um ambiente mais homogêneo que o Porto do Mucuripe,
pois a similaridade total entre as estações de coleta, tanto para Bacteria como para Archaea, foi
mais elevada, acima de 55%. Já no Porto do Mucuripe, essa similaridade ficou em torno de
45%. Além disso, com exceção da estação de coleta P7, de maior granulometria, a qual se
separou das demais na análise da assembleia de Archaea, as estações foram separadas de acordo
com a localização geográfica, o que permitiu diferenciar as amostras obtidas entre os píeres 1
e 2 das obtidas entre píeres 2 e 3. Já no Porto do Mucuripe, uma avaliação visual preliminar da
localização dos canais de acesso interno e externo, da Bacia de evolução e dos principais berços
de atracação sugere que há influência dessa arquitetura na organização espacial da assembleia
de Bacteria. As estações M1 e M2 ficam localizadas na área de atracação destinada a granéis
líquidos, que transportam principalmente derivados do petróleo; as estações M12, M13, M14 e
M15 já ficam próximas ao canal de acesso externo, enquanto as estações M4 e M5 ficam na zona
de atracação de navios de passageiros.
Outros dados que apontam para a maior heterogeneidade das estações de coleta do Porto
do Mucuripe são as medidas de beta diversidade, apresentadas na Tabela 11. A beta diversidade
pode ser definida como a extensão da reposição de espécies ao longo de um gradiente ambiental,
podendo indicar o grau de particionamento dos hábitats pelas espécies, o que a Ecologia trata

103
de “diversidade diferenciada”. As medidas utilizadas, beta diversidade de Whitakker, w
(Whittaker, 1960), e “beta turnover”, t (Wilson e Shmida, 1984), medem a razão entre o
número total de espécies encontradas e o número médio de espécies descrito para cada micro-
hábitat estudado. Particularmente a última medida, t, é mais complexa por acrescentar a
quantificação da perda e do ganho de espécies à proposição original de reposição de espécies.
A distribuição das UTOs das comunidades analisadas, principalmente no Porto do Mucuripe,
parece seguir a “Teoria do Nicho”, a qual prevê que mudanças na composição das espécies são
relacionadas a modificações nas variáveis ambientais locais (MENDES et al., 2014), pois tais
comunidades teriam seus membros reunidos de forma determinística, e não aleatória conforme
prevê a “Teoria Neutra”. Entretanto, uma caracterização físico-química mais detalhada de todas
as estações de coleta assim como o conhecimento da identidade dos membros da comunidade
seriam necessários para testar tal hipótese. Van der Gast e colaboradores, em 2008, estudando
a dinâmica de comunidades em reatores contendo uma concentração crescente de efluentes
industriais, encontraram que, conforme aumentava-se a concentração de efluentes, havia uma
troca de processos estocásticos para processos determinísticos (com base no nicho) devido ao
aumento da pressão seletiva sobre os membros das comunidades.
Os coeficientes de similaridade encontrados para o Domínio Archaea foram mais
baixos nos dois portos, indicando que, não apenas o Mucuripe parece ser um ambiente de maior
heterogeneidade, mas também que as assembleias de Archaea são mais variáveis do que as de
Bacteria em ambos os portos, sugerindo a existência de mais nichos para arqueias e menor
competição entre elas. Para avaliar se o padrão de diversidade encontrado nos portos, ou seja,
com elevada riqueza de bandas de DGGE para o Domínio Archaea, representa também uma
alta diversidade taxonômica, faz-se necessária a utilização de ferramentas que permitam
conhecer a identidade das UTOs encontradas, como excisão das bandas do gel de DGGE para
sequenciamento, utilização de biblioteca de clones ou sequenciamento direto dos metagenomas.
Quando as amostras do Porto do Pecém e do Porto do Mucuripe foram submetidas
à análise de agrupamento em conjunto, tanto para o Domínio Bacteria como para Archaea, os
padrões de agrupamento resultantes mostraram dois grandes grupos referentes às amostras do
Porto do Pecém e as do Porto do Mucuripe (Figuras 17 e 18). Dentro de cada um dos grupos,
os padrões observados previamente para cada um dos portos foram mantidos, com pequenas
modificações (Figuras 15 e 16). Os padrões de agrupamento para o Domínio Bacteria foram
idênticos; entretanto, no Domínio Archaea, foram observadas algumas divergências apenas no

104
Porto do Mucuripe, com a estação M6 participando de um agrupamento com as estações M4 e
M5. A análise reforça a maior heterogeneidade do Porto do Mucuripe.
A análise dos géis de DGGE dos pools de DNA de cada porto em comparação com
uma região isenta da influência de atividades portuárias foi capaz de distingui-las, tanto para
Bacteria como para Archaea. As diferenças entre os portos e a área sem atividade portuária
foram maiores no Domínio Archaea, pois observou-se um menor número de UTOs para ICA,
onde encontrou-se apenas 10 UTOs, assim como uma quantidade mais baixa de UTOs
compartilhadas pelas três áreas, somente cinco UTOs. Além disso, a quantidade de bandas
exclusivas de cada porto e compartilhadas apenas pelos portos foi maior para Archaea. A baixa
quantidade de UTOs de Archaea na região de referência vai ao encontro de relatos prévios de
baixa diversidade de Archaea em sedimentos marinhos (BOWMAN et al., 2000; MUSAT et
al., 2006). Por usa vez, a maior riqueza de Archaea nas amostras dos portos pode ser um
indicativo de ambientes cronicamente contaminados (ALONSO-GUTIÉRREZ et al., 2009).
É possível fazer algumas especulações a respeito da natureza das UTOs
compartilhadas e exclusivas de cada área em estudo, expostas pelos diagramas de Venn da
Figura 19. As bandas compartilhadas referem-se, possivelmente, a organismos comumente
presentes em sedimentos marinhos, o “core microbiome” ou microbioma fundamental, uma vez
que a ocorrência dos mesmos microrganismos em todas as amostras sugere que eles
desempenham um papel crucial na manutenção dessa comunidade, enquanto as UTOs
exclusivas podem corresponder a organismos adaptados a condições ou tipos de impactos
particulares a cada área, como o tempo de operação dos mesmos, superior a 50 anos no Porto
do Mucuripe e menor que 15 anos no Porto do Pecém. Ademais, a características estruturais e
geomorfológicas locais podem estar atuando na diferenciação dessa microbiota. As UTOs
compartilhadas apenas pelos portos são, provavelmente, relacionadas a organismos resistentes
ou organismos oportunistas, adaptados a impactos ambientais comuns a zonas portuárias
(JOHNSTON; ROBERTS, 2009). Contudo, os dados apresentados neste capítulo não permitem
avaliar tais hipóteses. Uma análise das sequências presentes em cada um dos portos pode
esclarecer se há realmente a presença de um microbioma fundamental característico dos portos
em estudo.
É importante destacar que os estimadores de riqueza avaliados nesse trabalho, Chao
2, Jackknife 1 e bootstrap, mostraram que as riquezas de UTOs de Bacteria e de Archaea
encontradas nas diferentes estações de coleta cobriram bem a diversidade presente nos
sedimentos dos portos analisados (Tabela 10). Entretanto, quando estes estimadores foram
calculados para os perfis obtidos pelos pools de DNA dos portos, os valores resultantes foram

105
mais elevados que a riqueza de UTOs revelada pelas DGGEs (Tabela 13). Esses estimadores
de riqueza são utilizados para estimar o número real da riqueza de espécies com base em
espécies raras compartilhadas entre as amostras utilizando a quantidade de uniques (espécies
que ocorrem em apenas uma amostra) e em duplicates (espécies que ocorrem em somente duas
amostras). Dessa forma, os estimadores indicam que a riqueza de UTOs presentes no ambiente
pode ser maior que a revelada pelos pools. Isso fica bem mais evidente quando comparamos o
número total de UTOs reveladas pelas estações de coleta, entre 42 e 56, e pelos pools, entre 29-
32, mostrando que houve diminuição no total de UTOs encontradas. Os totais de UTOs
encontrados pelos pools são semelhantes aos números médios de UTOs encontradas em cada
estação de coleta, o que sugere que essa análise deixou de lado uma parcela de grupos menos
abundantes. Apenas as UTOs de Archaea do Porto do Pecém apresentaram números
compatíveis com os estimadores de riqueza calculados para os pools, indicando que esse
domínio teve uma boa cobertura nesse porto.
A DGGE possui limiares de detecção de abundâncias abaixo dos quais táxons raros
não são detectados. Outras limitações incluem o limitado valor para a identificação das
populações presentes, a indefinição sobre o número de cópias de rRNA presentes em uma banda
e as polêmicas estimativas de abundância de bandas (GOBET, BOETIUS, RAMETTE, 2013).
Mesmo assim, juntamente com outras técnicas de ribotipagem, representa uma excelente opção
para o processamento rápido, reprodutível e economicamente viável de muitas amostras
ambientais concomitantes, tendo contribuído para o avanço na ecologia microbiana ao
possibilitar valiosos insights a respeito dos organismos dominantes nas amostras ambientais
(OLIVER, LILLEY, VAN DER GAST, 2012).
Nesse estudo, a técnica permitiu comparar um grande número de amostras em uma
malha amostral bem detalhada ao longo das áreas portuárias estudadas. Considerando que,
muitas vezes, perturbações induzem mudanças na composição das assembleias microbianas, tal
conhecimento é chave para melhor entender os processos de resistência e resiliência, que
contribuem para a estabilidade dos ambientes, assim como de redundância funcional. Para
complementar tais dados, a obtenção de informações sobre a composição taxonômica e
funcional das assembleias microbianas se faz necessária. Para tanto, outras abordagens são
sugeridas, como o sequenciamento de clones de genes codificadores do rRNA 16S,
sequenciamento de nova geração de amplicons ou WGS (NOGALES et al., 2011).
Particularmente para o Porto do Pecém, localizado na região onde está sendo
instalado o Complexo Industrial e Portuário do Pecém, este trabalho representa o ponto de
partida para acompanhar os efeitos das mudanças ambientais que possivelmente ocorrerão em

106
decorrência da instalação de indústrias e seus respectivos sistemas de escoamento de produção
e de efluentes. Além disso, para também distinguir modificações decorrentes de perturbações
antropogênicas das relacionadas à dinâmica temporal própria do ecossistema, sugere-se um
monitoramento periódico dessas áreas.
4.6. Conclusões
Os dados mostraram que os sedimentos dos portos do Mucuripe e do Pecém
possuem características ambientais distintas e que cada estação de coleta em cada porto
representa um micro-hábitat, abrigando assembleias de Bacteria e Archaea também distintas. O
Porto do Mucuripe destacou-se como um ambiente de maior riqueza e heterogeneidade que o
Porto do Pecém, possivelmente decorrentes da contaminação crônica desse porto. Algumas
variáveis analisadas, como a concentração de carbonato, matéria orgânica, COT e
benzofluorantenos, parecem ter papel relevante nos padrões observados nas estruturas das
assembleias procarióticas nesses portos. A comparação dos portos entre si e com uma região
sem atividade portuária sugere a existência de unidades taxonômicas operacionais selecionados
pelas condições ambientais operantes em cada porto.

107
5. Capítulo 2 - COMPOSIÇÃO TAXONÔMICA E FUNCIONAL DO PATRIMÔNIO
MICROBIANO REGIONAL DE SEDIMENTOS MARINHOS SOB INFLUÊNCIA
DE REGIÕES PORTUÁRIAS
O espectro completo de biodiversidade reúne níveis múltiplos de organização
biológica, desde a diversidade genética em populações à diversidade de espécies em
comunidades ou à diversidade de comunidades ao longo de paisagens e ecossistemas. Portanto,
a biodiversidade em um determinado ecossistema pode ser conceituada por três elementos
principais: a composição, a estrutura e a função (ELLIS et al., 2011). Contudo, estudar esses
três elementos não é tarefa fácil. Para tanto, é necessário o uso de várias ferramentas ou de uma
ferramenta mais moderna e abrangente.
Uma aplicação mais avançada do sequenciamento de nucleotídeos para a análise da
biodiversidade de comunidades microbianas é a metagenômica, a qual permite uma avaliação
detalhada da composição, da estrutura e do funcionamento de comunidades constituintes de um
metagenoma. Em um análise simples, pode-se identificar mais de 1.2 milhões de novos genes
e 1700 novas famílias de proteínas (KARL, 2007). Nas últimas décadas, o uso de sequências
genômicas e abordagens relacionadas superaram a necessidade de cultivo para caracterizar e
identificar micro-organismos na natureza. Parece impossível a tarefa de identificar e inter-
relacionar os trilhões de micro-organismos presentes em um metro cúbico de água do mar,
quanto mais em alguns hectares de oceano. Felizmente, o advento das tecnologias de
sequenciamento de nova geração permitiram aos cientistas alcançar consideráveis progressos
(FUHRMAN, 2009).
Neste capítulo, utilizou-se uma abordagem metagenômica em combinação com
NGS para caracterizar o perfil taxonômico e metabólico de assembleias microbianas presentes
em dois sedimentos derivados de diferentes sistemas portuários localizados no Estado do Ceará,
o Porto do Pecém e o Porto do Mucuripe. Considerando-se as diferenças estruturais e
operacionais apresentadas por tais portos, essa pesquisa investigou se tais divergências
poderiam refletir na riqueza e na composição das comunidades microbianas que habitam tais
ecossistemas, assim como também em seu funcionamento metabólico. Sugere-se que tal
conhecimento poderá ser aplicado como uma ferramenta adicional para a análise dos impactos
da poluição marinha e na prospecção de novas ferramentas de biorremediação.
Atualmente, tem-se dado bastante atenção à pesquisa de novas estratégias e de
tecnologias sustentáveis aplicáveis à remediação de sedimentos contaminados. Esse inventário
representa portanto, o primeiro passo para o possível uso desse tipo de tecnologia para

108
monitorar e/ou diagnosticar ambientes alterados pelo homem e que demandem estratégias de
biorremediação.
5.1. Hipótese
A metagenômica é capaz de detalhar a composição taxonômica e funcional de uma
comunidade, podendo ser utilizada para diferenciá-la de outras. A hipótese de trabalho deste
estudo é de que regiões portuárias com diferenças estruturais e operacionais, assim como em
seus parâmetros físico-químicos, apresentam composição taxonômica e funcional distintas das
assembleias de Bacteria e Archaea associadas aos seus sedimentos, podendo essas informações
ser utilizadas para apontar grupos taxonômicos indicativos do status desses ecossistemas.
5.2. Materiais e Métodos
5.2.1. Locais de Estudo
O estudo foi realizado em duas áreas portuárias, o Porto do Pecém (PEC)
(03°32’52.03’’ S; 38°48’46.14’’ O) e o Porto do Mucuripe (MUC) (03°42’45.55’’ S;
38°28’26.80’’ O), ambos localizados no Estado do Ceará, Brasil. O Porto do Pecém está
posicionado na região destinada à instalação do Complexo Industrial do Pecém, distante 60 Km
da região, localizado na região metropolitana. Tais portos diferem em relação ao seu tempo de
operação, com PEC estando ativo há 12 anos e MUC há mais de 50 anos. Outra diferença refere-
se a suas infraestruturas, pois PEC é um porto moderno do tipo offshore, construído a 2000 m
da linha de costa e conectado à terra através de uma ponte sobre pilastras. O objetivo de tal
ponte foi preservar a linha de costa e minimizar o impacto negativo associado com as
arquiteturas portuárias tradicionais, que afetam as correntes costeiras e o transporte de
sedimentos (BURUAEM et al., 2012). Por sua vez, MUC é um porto marítimo artificial
construído entre dois espigões tradicionais. Nesse porto, ao longo dos anos de funcionamento,
a estrutura portuária gerou uma forte alteração no balanço sedimentar regional com interrupção
do transporte de sedimentos e consequente deposição de sedimentos finos na área portuária.
Por essa razão, atividades de dragagem são efetuadas periodicamente no porto, objetivando a
manutenção de seu calado e do canal de navegação (MAIA et al., 1998).

109
5.2.2. Coleta das amostras e processamento
No Porto do Pecém, a amostragem foi realizada em Novembro do ano de 2011 em dez
diferentes estações (P1-P10) enquanto no Porto do Mucuripe as amostras foram coletadas em
Dezembro do mesmo ano em 15 diferentes estações (M1-M15) (Seção 4.3.1; Figura 9). As
distâncias entre as estações de coleta em ambos os portos foram de cerca de 500 m, de modo a
cobrir toda a área de operação dos portos estudados.
A amostragem foi conduzida com o pegador de fundo de aço inoxidável do tipo Van Veen
em profundidades medindo entre 10 e 30 m. Após a coleta, as amostras obtidas em cada estação
foram transferidas para recipientes plásticos esterilizados e transportados sob refrigeração para
o Laboratório de Ecologia Microbiana e Biotecnologia (Lembiotech), localizado no Campus do
Pici da Universidade Federal do Ceará. A profundidade e a transparência da água, medida com
disco de Sechii, foram mensuradas in situ, enquanto a salinidade e o pH foram mensurados após
a chegada ao laboratório em refratômetro e potenciômetro, respectivamente. A profundidade
das coletas variou de 15 a 20 m, com pH de 7.5-7.6, salinidade de 40 e transparência da água
de 1.9 a 2.9 m.
5.2.3. Extração do DNA ambiental
Para cada porto, alíquotas de sedimento de cada estação de amostragem foram reunidas,
homogeneizadas e submetidas à extração de DNA utilizando o protocolo proposto por Zhou,
Bruns e Tiedje (1996). Após a extração, as amostras de DNA foram ressuspendidas em água
MilliQ estéril e purificadas por adição de uma mistura de clorofórmio:álcool isoamílico (24:1),
sendo em seguida precipitadas pela adição de isopropanol p.a. gelado e incubadas a -20 °C por
10-30 min. Então, foram lavadas com etanol 70%, deixadas evaporar e ressuspendidas em
tampão Tris-EDTA 10 mM:1 mM pH 8,0 com RNAse 20g/mL. Após incubação por 1 h a
37°C para ação da RNAse, as amostras de DNA extraídas foram então quantificadas por
espectrofotometria (NanoDrop Technologies, EUA) para estimar os parâmetros de qualidade
do DNA, indicativos da contaminação com proteínas, fenóis, ácidos húmicos e outros
compostos, razão de absorbância 260/280 nm e 260/230 nm. As amostras foram também
avaliadas através de eletroforese em gel de agarose a 1% para checar-se a integridade do

110
material extraído. Em seguida, as amostras foram enviadas ao Laboratório Nacional de
Computação Científica (LNCC, Petrópolis – RJ) para sequenciamento.
5.2.4. Pirosequenciamento para análise metagenômica
Os pools das amostras de DNA ambiental extraídas de cada porto foram submetidos
a pirosequenciamento utilizando a tecnologia 454 GS FLX Titanium, da Roche Applied
Sciences (EUA). Para criação das bibliotecas metagenômicas, 5 mg de DNA de cada porto
foram utilizados. Os procedimentos de preparação e sequenciamento do material foram
realizados no Laboratório Nacional de Computação Científica (LNCC, Petrópolis – RJ, Brasil)
de acordo com o protocolo padrão do sequenciador GS FLX Titanium. Agrupamentos de
sequências replicadas artificialmente gerados pelo sequenciamento do tipo 454 foram
identificados e removidos utilizando o programa Replicates (Gomez-Alvarez, Teal e Schmidt,
2009). As demais sequências foram, então, filtradas para remover as sequências de pequeno
tamanho (menores de 150 pb) ou que ocorrem a intervalos menores que 150 pb e com qualidade
phred menor que 20 utilizando o programa Lucy (CHOU E HOLMES, 2001). As sequências
limpas foram adicionadas ao MG-RAST sob os números de acesso 4488272.3 e 4488273.3 para
os metagenomas do Porto do Mucuripe e do Porto do Pecém, respectivamente.
5.2.5. Classificação taxonômica das sequências ambientais
A classificação taxonômica das sequêncais foi feita no servidor MG-RAST versão
3.0 (http://metagenomics.nmpdr.org) (MEYER et al., 2008) com um cutoff máximo de E-value
de 1e-10, cutoff de tamanho mínimo de sequência de 50 pb e um cutoff mínimo de identidade
de 75%. As comparações entre as comunidades microbianas foram feitas com base nas
abundâncias relativas dos componentes de cada comunidade. Para classificação taxonômica, as
sequências foram alinhadas contra as sequências dos bancos de dados M5NR (WILKE et al.,
2012), GenBank (NCBI-nr; BENSON et al., 2013) e RefSeq (PRUITT et al., 2009).

111
5.2.6. Análise funcional utilizando os bancos de dados KEGG e SEED
Para a análise funcional, foram utilizados os bancos de dados KEGG (Kanehisa et
al., 2004) e o SEED (OVERBEEK et al., 2005) com um cutoff máximo de E-value de 1e-05,
cutoff de tamanho mínimo de alinhamento de 15 pb e um cutoff mínimo de identidade de 60%.
5.2.7. Comparação estatística dos perfis metagenômicos
Para investigar se as diferenças observadas entre os metagenomas dos portos eram
estatisticamente significantes, os resultados de anotação do MG-RAST foram visualizados
utilizando o programa STAMP de comparação estatística de perfis metagenômicos na versão
2.0.0 (PARKS, BEIKO, 2010). O programa permite detectar diferenças biologicamente
relevantes nas proporções relativas das sequências classificadas. Tais análises foram realizadas
utilizando comparação par a par dos metagenomas, sendo as diferenças estatisticamente
significantes entre os mesmos avaliadas pelo Teste Exato de Fisher para duas amostras
independentes com o método FDR de Benjamini-Hochberg utilizado para correção múltipla do
teste, conforme recomendado pelos desenvolvedores do programa. Os táxons mais importantes
foram selecionados e filtrados por q-value <0,05 e utilizando somente categorias que tivessem
efeitos de tamanho maior que dois, tanto para diferença como para razão entre as proporções.
5.2.8. Classificação taxonômica dos principais grupos procarióticos envolvidos em rotas
metabólicas do KEGG
As sequências classificadas pelo MG-RAST em catogorias funcionais do KEGG
relacionadas aos metabolismos do nitrogênio, enxofre e metano, assim como relacionadas à
biodegradação de xenobióticos e degradação de HPAs foram extraídas e classificadas
taxonomicamente utilizando BLASTX contra o banco de dados M5NR no servidor MG-RAST
v3.0., com os mesmos parâmetros utilizados na seção 5.2.5.

112
5.2.9. Comparação dos metagenomas de sedimentos dos portos com outros metagenomas
de ecossistemas marinhos
As sequências obtidas dos metagenomas dos sedimentos portuários estudados
foram comparadas com conjuntos de dados oriundos de outros ecossistemas marinhos. A
comparação foi feita utilizando os resultados gerados pelo MG-RAST com o banco de dados
M5NR. A frequência de diferentes grupos taxonômicos em cada metagenoma foi usada para
gerar uma árvore enraizada através de uma análise de agrupamento baseada na média aritmética
entre os membros do grupo (UPGMA; do inglês Unweighted Pair-Group using Arithmetic
Average) no programa PAST v2.17c (HAMMER, HARPER, RYAN, 2001).
5.3. Resultados
5.3.1. Pirosequenciamento dos metagenomas dos portos
As diferenças entre os portos do Pecém e Mucuripe, já indicadas pelos dados
ambientais e pelo estudo dos perfis de DGGE, foram examinadas em maior detalhe utilizando
uma abordagem metagenômica baseada no sequenciamento direto das amostras de DNA
extraídas de pools dos sedimentos obtidos nas diferentes estações de coleta em cada um dos
portos estudados (P1-P10 para o PEC e M1-M15 para o MUC).
Foi gerado um total de 470.000 sequências de ambas as amostras, com um tamanho
médio de 320 pb. Previamente ao processamento das sequências, os dados brutos foram
submetidos a um filtro de controle de qualidade (QC) para remover sequências de baixa
qualidade e artificialmente duplicadas (ADR; do inglês, artificial duplicate reads), um
fenômeno frequentemente observado durante o pirosequenciamento, removendo ~0,5% das
sequências de cada metagenoma. Cerca de metade das sequências que passaram pelo filtro QC
continham proteínas previstas com funções conhecidas e foram, subsequentemente, submetidas
a outras análises focando nas anotações taxonômicas e funcionais. O restante das sequências
continha proteínas com função desconhecida. Somente 0,6% das sequências pertencia a genes
do RNA ribossomal 16S. As curvas de rarefação para os metagenomas são mostradas na Figura
19, podendo-se observar que ainda não atingiram o platô, podendo ainda haver incremento no
número de espécies nos dois portos.

113
Figura 19 - Curvas de rarefação dos metagenomas sequenciados descrevendo o crescimento do número de
espécies encontradas como função do número de sequências sequenciadas. PEC representa a curva de rarefação
para o metagenoma do Porto do Pecém (total de sequências = 330.470) e MUC representa a curva de rarefação
para o metagenoma do Porto do Mucuripe (total de sequências = 146.724).

114
5.3.2. Perfil taxonômico dos metagenomas dos portos
As distribuições em domínios das duas amostras demonstraram a dominância de
Bacteria (~94%). Esta foi seguida de Archaea (~3%) e de pequenas frações de Eucarya, vírus e
outros organismos não classificados (~3%). Em razão das pequenas quantidades de sequências
classificadas para o domínio Eukarya, a análise foi concentrada em Bacteria e Archaea.
Com base no banco M5NR, o Domínio Archaea foi caracterizado pela presença dos
filos Thaumarchaeota, Euryarchaeota e Crenarchaeota. No Porto do Pecém, o filo
Thaumarchaeota foi dominante, com frequência de 96%, enquanto Euryarchaeota e
Crenarchaeota apresentaram abundâncias relativas de 1,7% e 0,9%. No Porto do Mucuripe,
67% das sequências não foram classificadas e o restante foi classificado como Euryarchaeota e
Thaumarchaeota, ambas com 1,7%. As sequências de Thaumarchaeota do Pecém pertenceram
todas à espécie Nitrosopumilus maritimus, uma arqueia oxidadora de amônia de grande
importância para o ciclo global do nitrogênio realizando a transformação de amônia em nitrito
(nitrificação) (Figura 20).
Em PEC, também segundo o banco M5NR, Domínio Bacteria foi dominado pelo
filo Proteobacteria (57,6%), seguido de Actinobacteria (15.1%). Cyanobacteria e Firmicutes
constituíram cada um 3.8% e 2.4%, respectivamente. Todos os demais filos presentes
corresponderam a ~8% das sequências classificadas. Diferentemente, a classificação obtida
para as sequências no MUC pertencentes ao Domínio Bacteria foi constituída por
Cyanobacteria (32.1%), seguida por apenas 21% de Proteobacteria. Os filos Bacteroidetes,
Chloroflexi e Actinobacteria corresponderam a 15.2%, 13.7%, 10% das sequências,
respectivamente (Figura 21).
No nível de Classe, em PEC houve uma predominância de Deltaproteobacteria,
representando 42.1% das sequências. Outras classes abundantes foram Actinobacteira (15%) e
Gammaproteobacteria (6.9%). Já no MUC houve dominância de classes não classificadas
derivadas de Cyanobacteria (32%), seguida de Bacteroidetes (14.8%), Thermomicrobia
(13.6%), Gammaproteobacteria (11.9%) e Actinobacteria (~10%). Em ambos os portos,
Alphaproteobacteria e Betaproteobacteria representaram, cada, ~3% e 6%, respectivamente
(Figura 20).
A classificação em nível de ordem reforçou as diferenças taxonômicas observadas
para classes e revelou grupos particulares relacionados a cada um dos portos. As ordens
Myxococcales e Solirubrobacterales foram exclusivas do PEC, enquanto Bacteroidetes order II
Incertae sedis, Sphaerobacterales, Acidimicrobiales, Pasteurellales, Legionellales e

115
Thiotrichales foram limitadas ao MUC. Chroococcales, Actinomycetales e Alteromonadales
também apresentaram números expressivos em MUC, porém não foram exclusivas (Figura 22).
Algumas ordens mostraram-se presentes em ambos os portos em números semelhantes, como
Vibrionales e Burkholderiales.
Quando comparados no Programa STAMP, os portos exibiram baixos valores de
correlação em todos os níveis taxonômicos, evidenciando que possuem uma composição
taxonômica bem diversa. As dissimilaridades nas abundâncias dos grupos dominantes são
responsáveis por grande parte das variâncias observadas, com Proteobacteria e Cyanobacteria
alternando-se entre PEC e MUC e com muito pouca sobreposição entre as distribuições dos
membros da microbiota. Chroococcales apareceu como uma ordem importante para MUC,
enquanto para PEC tal papel é representado por Myxococcales (Figura 23A). No nível de
espécies, Haliangium ochraceum (Deltaproteobacteria), Conexibacter woesei (Actinobacteria)
e bactérias redutoras de sulfato não cultiváveis (SRB, Deltaproteobacteria) apresentaram-se
com quantidades significativamente aumentadas no PEC, enquanto espécies do gênero
Synechococcus (Cyanobacteria), Rhodothermus marinus (Bacteroidetes) e Sphaerobacter
thermophilus (Thermomicrobia) foram dominantes no MUC (Figura 23B).
As distribuições de abundância dos gêneros identificados nos dois portos
mostraram a predominância de poucos grupos muito abundantes e muitos grupos de baixa
frequência, correspondendo ao padrão de long tail de táxons raros frequentemente observado
em comunidades microbianas. No Porto do Pecém, o gênero Haliangium (Deltaproteobacteria)
destacou-se como mais abundante, enquanto no Porto do Mucuripe, Synechococcus
(Cyanobacteria) apresentou-se como gênero dominante (Figuras 24 e 25).
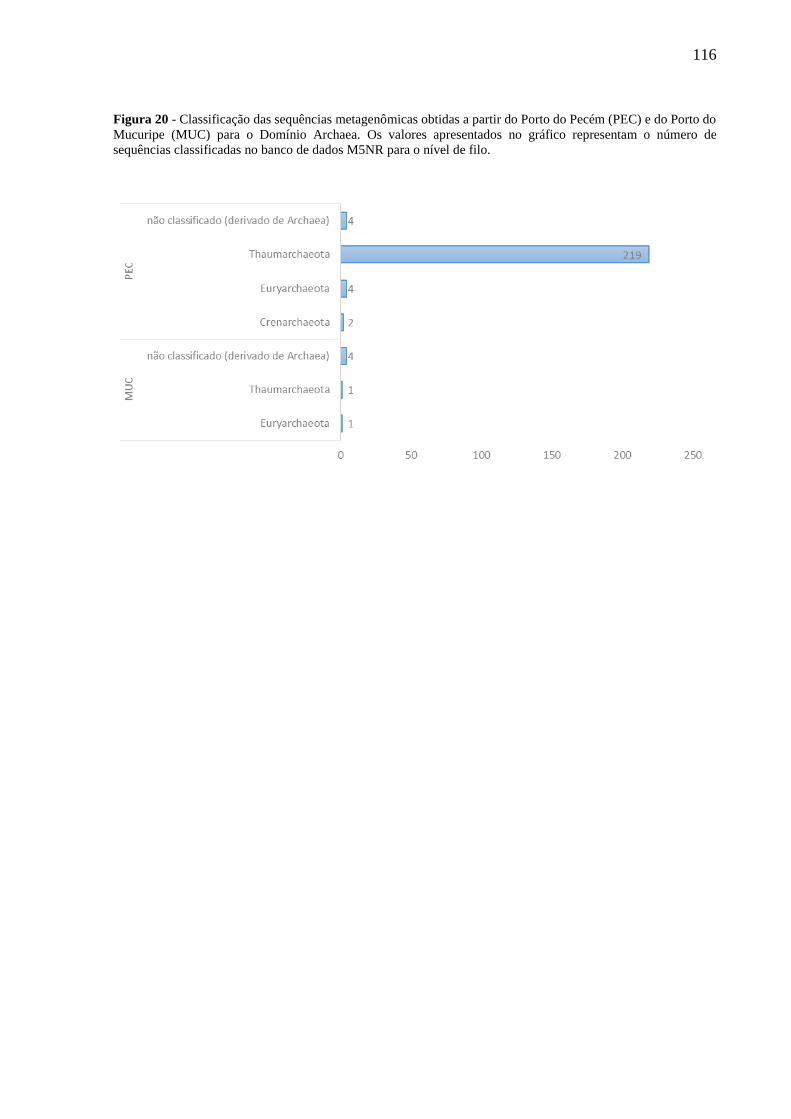
116
Figura 20 - Classificação das sequências metagenômicas obtidas a partir do Porto do Pecém (PEC) e do Porto do
Mucuripe (MUC) para o Domínio Archaea. Os valores apresentados no gráfico representam o número de
sequências classificadas no banco de dados M5NR para o nível de filo.

117
Figura 21 - Classificação das sequências metagenômicas obtidas a partir do Porto do Pecém (PEC) e do Porto do
Mucuripe (MUC). À esquerda é mostrada a afiliação taxonômica baseada nos bancos de dados RefSeq, GenBank
e M5NR para o nível de filo. Os valores apresentados acima do gráfico representam o número de sequências
classificadas em cada um dos bancos de dados. Posicionada à direita, é mostrada a classificação taxonômica
baseada no banco de dados M5NR para o nível de classe.

118
Figura 22 - Esquema comparativo dos perfis taxonômicos em nível de ordem para o Porto do Pecém (PEC) e
Porto do Mucuripe (MUC). Os perfis taxonômicos para PEC (colorido em verde) e MUC (colorido em laranja)
foram computados usando a plataforma MG-RAST v3.0 e o programa STAMP v2.0.0. O diâmetro das esferas
mostradas na figura foram calculados com base nas abundâncias relativas de cada táxon. O símbolo * designa
ordens que apresentaram baixas abundâncias relativas, porém ocorreram em apenas um dos portos. A distribuição
das ordens apresentada não está correlacioada a sua posição nos portos, mas representa apenas uma organização
das mesmas em ordem de maior para menor abundância relativa.

119
Figura 23 - Comparação dos perfis taxonômicos em nível de ordem (A) e em nível de espécie (B) para o Porto do
Pecém (PEC) e Porto do Mucuripe (MUC). Os perfis taxonômicos para PEC (colorido em verde) e para MUC
(colorido em laranja) foram computados usando a plataforma MG-RAST v3.0 e o programa STAMP v2.0.0. Os
pontos mostrados em cada lado a linha de tendência tracejada encontram-se enriquecidos em uma das amostras
analisadas. Os pontos mais distantes dessa linha indicam que tais táxons possuem maior diferença proporcional
entre os dois metagenomas e foram identificados na figura. Um filtro foi aplicado para remover pontos com p >
0,05.

120
Figura 24 - Curva de distribuição das abundâncias dos gêneros associados aos sedimentos do Porto do Pecém.

121
Figura 25 - Curva de distribuição das abundâncias dos gêneros associados aos sedimentos do Porto do
Mucuripe.

122
5.3.3. Perfil funcional dos metagenomas dos portos
Os perfis funcionais dos portos foram estudados utilizando os bancos de dados
funcionais SEED e KEGG para observar detalhadamente quaisquer diferenças de
funcionamento existentes entre os ambientes estudados.
Apesar das diferenças observadas entre os perfis taxonômicos, houve pouca
variação funcional entre PEC e MUC, mostrando que os portos são bastante similares neste
aspecto, um reflexo da redundância funcional frequentemente exibida por comunidades
microbianas (Figura 26).
Analisando as categorias do KEGG, foram observadas diferenças estatísticas
apenas para o metabolismo do metano e para a degradação de HPAs. Na subcategoria de
metabolismo do metano, sequências relacionadas a genes para o transporte de sódio e prótons
(id. K03314) foram super-representadas no MUC, enquanto genes relacionados à degradação
de HPAs foram detectados apenas no PEC (Figura 26B).
Em ambos os portos, dentro da categoria de metabolismo energético, as
subcategorias de metabolismo do metano, do enxofre e do nitrogênio representaram cerca de
30%, 7.5% e 7% das sequências em ambos os portos (Figura 26A). Já a subcategoria de
transporte por membrana foi dominada por sequências de transportadores ABC (~78%). Para a
subcategoria de biodegradação de xenobióticos e metabolismo, a via de degradação de
nitrotolueno correspondeu a 25% nos dois metagenomas, seguida da degradação de benzoato e
de clorociclohexano/clorobenzeno, com 20% e 15%, respectivamente (Figura 26C).
Quando as sequências foram analisadas com base no banco de dados do SEED, os
portos se mostraram estatisticamente similares em todas as subcategorias. Na categoria de
carboidratos, o metabolismo de compostos com um carbono representou entre 16 e 18% das
sequências. As subcategorias deste último foram dominadas por assimilação de formaldeído
(81%), mas apresentaram também sequências relacionadas à metanogênese (10% em PEC e 7%
em MUC). O subsistema de transporte através da membrana apresentou 35% das sequências
associadas a transportadores ABC. Já quando se analisou o metabolismo de compostos
aromáticos, notou-se que mais de 60% das sequências foram relacionadas a vias periféricas para
o catabolismo de compostos aromáticos, seguido do metabolismo de intermediários aromáticos
centrais (~25%) e degradação anaeróbica de compostos aromáticos (~16%) em ambos os
portos. O metabolismo do nitrogênio foi dominado pelo processo de assimilação de amônia
(~55%), seguido de amonificação de nitrato e nitrito (~12%). A fixação de nitrogênio e a
denitrificação, cada qual, correspondeu a somente 5% das sequências. No metabolismo do

123
enxofre, encontrou-se somente 8% das sequências relacionas a complexos associados à redução
de sulfato e 5% de oxidação do enxofre. Já a assimilação de enxofre inorgânico correspondeu
a 21% das sequências dessa subcategoria. Estudando as sequências relacionadas à Virulência,
pode-se observar que uma quantidade significante delas pertenceu ao subsistema de Bombas de
Efluxo multirresistentes a drogas (15%), o que pode estar relacionado à liberação de efluentes
contaminados no ambiente. Ademais, sistemas relacionados à resistência a metais se mostraram
presentes, como a resistência a cobalto, zinco e cádmium (10%), homeostase do cobre (~7%),
resistência ao zinco (~4%), resistência ao arsênio (3%), tolerância ao cobre (2%) e redutase do
mercúrio (1%) (Figura 26D).
Figura 26 - Comparação dos perfis metagenômicos funcionais obtidos para o Porto do Pecém (PEC) e para o
Porto do Mucuripe (MUC) para as categorias do KEGG de metabolismo energético (A), metabolismo do metano
(B) e metabolismo e degradação de xenobióticos (C), e para a categoria do SEED de Virulência (D). Os perfis
funcionais do PEC (colorido em verde) e do MUC (colorido em laranja) foram computados usando a plataforma
MG-RAST v3.0 e o programa STAMP v2.0.0. Os pontos mostrados em cada lado da linha de tendência tracejada
encontram-se enriquecidos em uma das amostras analisadas. Os pontos mais distantes dessa linha indicam que tais

124
táxons possuem maior diferença proporcional entre os dois metagenomas e foram identificados na figura. Um
filtro foi aplicado para remover pontos com p > 0.05.

125
5.3.4. Classificação taxonômica dos principais grupos procarióticos envolvidos em rotas
metabólicas do KEGG
Após a extração das sequências relacionadas a algumas categorias funcionais do
banco de dados KEGG, estas foram classificadas contra o banco de dados M5NR de modo a
revelar os principais grupos taxonômicos envolvidos nesses metabolismos. Como resultado
observou que, apesar das similaridades funcionais compartilhadas entre os portos, os táxons
relacionados a tais metabolismos são diferentes. A Tabela 14 sumariza algumas importantes
diferenças.
Todas as sequências do Porto do Mucuripe foram classificadas como
Gammaproteobacteria, Thermomicrobia ou Cyanobacteria, enquanto as sequências do Porto do
Pecém foram mais diversas, pertencentes a Deltaproteobacteria, Gammaproteobacteria,
Actinobacteria ou Alphaproteobacteria. Nenhuma das funções analisadas parece ser
desempenhada pelos mesmos grupos taxonômicos. Particularmente para a degradação de
HPAs, subcategoria encontrada apenas no Porto do Pecém, os gêneros Mycobacterium,
Acinetobacter e Coraliomargarita foram encontrados.
Tabela 14 - Classificação taxonômica das sequências metagenômicas obtidas obtidas no Porto
do Pecém (PEC) e no Porto do Mucuripe (MUC) relacionadas a algumas categorias da
classificação funcional pelo banco de dados KEGG.
Categoria funcional PEC MUC
Metabolismo do Enxofre Desulfovibrionales Chroococcales
Metabolismo do Nitrogênio Rhodobacterales Sphaerobacterales
Enterobacterales
Metabolismo do Metano Myxococcales
Enterobacterales
Sphaerobacterales
Degradação e metabolismo de
Xenobióticos
Myxococcales
Solirubrobacterales
Chromatiales
Degradação de HPAs Actinomycetales
Puniceicoccales
Pseudomonadales
-

126
5.3.5. Comparação com perfis metagenômicos de outros hábitats marinhos
Os perfis taxonômicos e funcionais dos dois metagenomas estudados foram
comparados com outros metagenomas derivados de ecossistemas marinhos, obtidos por WGS
e disponíveis publicamente no servidor do MG-RAST. Estes foram: dois metagenomas de
sedimentos da Bacia Brazos-Trinity no Golfo do México (MG RAST id. 4441214.3 e
4441215.3), um metagenoma derivado de amostras de água costeira nas Ilhas Seychelles no
Oceano Índico (id. 4441613.3), um metagenoma derivado de amostras de água de zonas de
oceano aberto do Oceano Índico (id. 4441610.3) e três metagenomas derivados de zonas
estuarinas da Austrália, um de Porto Flinders (id. 4446342.3), um de Ponto Turton (id.
4446341.3) e outro da lagoa hipersalina Coorong (id. 4440984.3, Jeffries et al., 2011). Além
desses, também foram utilizados quatro metagenomas de sedimentos de manguezais da costa
cearense – Timonha (id. 4450297.3), Jaguaribe (id. 4450116.3), Pacoti (id. 4471561.3) e Cocó
(4471562.3) - igualmente obtidos por WGS.
Todos os metagenomas comparados exibiram somente cerca de 6% de similaridade
em nível de classe. Após a separação dos metagenomas pelágicos do Oceano Índico, todos os
demais apresentaram similaridade de 20%, como pode ser observado no agrupamento
apresentado na Figura 27. O agrupamento, além de mostrar as diferenças entre os metagenomas
comparados, também revelou padrões biogeográficos. Um dos grupos foi formado pelos
metagenomas derivados da costa brasileira, incluindo ecossistemas costeiros (PEC e MUC) e
ecossistemas estuarinos (os quatro manguezais; Timonha, Jaguaribe, Pacoti e Cocó), com 35%
de similaridade. Outro agrupamento foi formado pelos metagenomas originados na costa
australiana (Porto Flinders, Ponto Turton e Lagoa Coorong), com 50% de similaridade. Os
metagenomas do Golfo do México afluíram em outro grupo, sendo quase 100% idênticos. Já
os metagenomas de Oceano Índico, de região costeira e de mar aberto, formaram um grupo
externo, com 45% de similaridade.
Em geral, o filo Proteobacteria foi dominante em todos os ecossistemas marinhos
analisados, enquanto outros filos foram mais abundantes em alguns ecossistemas particulares.
Actinobacteria mostrou-se mais característico de MUC e PEC. Além desse táxon, quantidades
elevadas de Cyanobacteria e Chloroflexi também caracterizaram MUC. No nível de classes,
Deltaproteobacteria dominou as sequências de PEC, enquanto Cyanobacteria e Bacteroidetes
dominaram em MUC. Já Alphaproteobacteria foi caracteristicamente menos abundante em
ambos os portos.

127
Outros ecossistemas apresentaram relação com outras classes, como
Betaproteobacteria nos sedimentos do Golfo do México, Fibrobacteria no manguezal do
Jaguaribe, Sphingobacteria nos manguezais do Jaguaribe e do Pacoti, e Epsilonproteobacteria
no manguezal do Pacoti.
As ordens dominantes foram ainda mais diversas. PEC foi representado por
Myxococcales e Solirubrobacterales, enquanto MUC o foi por Chroococcales, Bacteroidetes
order II Incertae sedis, Sphaerobacterales, Acidimicrobiales, Actinomycetales e
Alteromonadales. Por sua vez, Pseudomonadales, Pasteurellales e Burkholderiales foram
características dos sedimentos do Golfo do México, Rhodobacteriales dos sedimentos
estuarinos da Austrália e Prochlorales dos metagenomas aquáticos do Oceano Índico.
Figura 27 - Análise de agrupamento dos perfis taxonômicos em nível de classe para os 13 metagenomas de
ecossistemas marinhos analisados, incluindo o Porto do Pecém (PEC) e Porto do Mucuripe (MUC). A frequência
dos grupos taxonômicos em cada metagenoma foi extraída da plataforma MG-RAST v3.0 e comparada no
programa PAST v2.17c usando o Índice de Similaridade de Bray-Curtis.
%
Similaridade

128
Apesar das diferenças observadas na classificação taxonômica, os ecossistemas
comparados se mostraram muito similares funcionalmente. Os únicos metagenomas marinhos
que diferiram estatisticamente dos portos foram os derivados dos sedimentos do Golfo do
México. Eles exibiram menores quantidades de sequências relacionadas a processos celulares
e metabolismo lipídico. Por outro lado, apresentaram quantidades mais expressivas das
seguintes funções: cbiO (K02006; ligado ao sistema de transporte de níquel e cobalto), ppa
(K01507; ligado à fosforilação oxidativa), rho (K03628; ligado a degradação de RNA), tbp
(K03120; uma proteína de ligação ao TATA-box relacionada à transcrição), fen1 (K04799;
ligada à replicação do DNA) e E2.6.1.83 (K10206; ligada à biossíntese de lisina).
5.4. Discussão
Os sistemas portuários do Pecém e do Mucuripe têm estruturas bem diferentes
assim como tempo de operação. Baseado em tais diferenças estruturais e operacionais, pode-se
hipotetizar que os ambientes que abrigam tais portos apresentam-se diferentes um do outro;
diferenças essas que podem ser percebidas através de análises físico-químicas ou biológicas.
Além disso, pode-se especular que a história de cada porto pode influenciar na sua
biodiversidade e, como consequência, na resistência e na resiliência das comunidades
microbianas presentes nesse sistema antropizado. Alguns trabalhos anteriores mostraram que
tais diferenças podem ser investigadas e utilizadas para indicar modificações ambientais ligadas
a atividades antrópicas.
Com base em tais ideias, o trabalho em questão teve por objetivo ligar as
divergências estruturais e operacionais, e possíveis diferenças físico-químicas advindas das
mesmas, entre o Porto do Pecém e o Porto do Mucuripe aos perfis taxonômicos e funcionais
obtidos por pirosequenciamento dos metagenomas associados aos sedimentos desses portos.
Este trabalho constitui o primeiro inventário sobre a diversidade microbiana para a região
tropical do Oceano Atlântico, passando a compor o conjunto dos poucos trabalhos existentes
atualmente dedicados ao estudo das comunidades microbianas de sedimentos sob influência de
atividades portuárias. Tais trabalhos são bastante recentes e tiveram por objetivo o estudo de
sedimentos poluídos e a biodegradação dos poluentes envolvidos (WANG; TAM, 2011;
IANNELLI et al., 2012; DELL’ANNO et al., 2012; CHIELLINI et al., 2013; GOMES et al.,
2013; ISAAC et al., 2013).

129
A composição taxonômica do Domínio Archaea teve como destaque a dominância
no Porto do Pecém do filo Thaumarchaeota. Em camadas superficiais de sedimentos, esse filo
pode contribuir significativamente com a reserva de óxidos de nitrogênio e, portanto, para a
produtividade. No Pecém, todas as sequências de Thaumarchaeota pertenceram à espécie
Nitrosopumilus maritimus. Essa espécie é uma importante arqueia oxidadora de amônia que faz
parte do ciclo do nitrogênio na reação de nitrificação.
Considerando-se a composição taxonômica do Domínio Bacteria, é possível
observar primeiramente que Proteobacteria, geralmente dominante em muitos ecossistemas, foi
substituída pelo filo Cyanobacteria em MUC. Analisando-se as classes encontradas em PEC
com maior detalhe, nota-se que Deltaproteobacteria se destaca como a classe principal,
enquanto que em MUC, Thermomicrobia, Gammaproteobacteria e Actinobacteria destacaram-
se, juntamente com sequências de Cyanobacteria e Bacteroidetes não classificadas em nível de
classe. Números tão expressivos quanto esses de Deltaproteobacteria, Gammaproteobacteria e
Cyanobacteria foram encontrados por Acosta-González, Rosseló-Moóra e Márquez (2013) em
sedimentos do infralitoral contaminados com petróleo após o derramamento do navio Prestige
na costa da Espanha em 2002. A comunidade microbiana foi avaliada cinco anos após o
acidente e exibiu quantidades menores de Cyanobacteria que as observadas apenas dois anos
após o evento de contaminação.
Os procariotos que vivem em sedimentos marinhos participam em uma variedade
de rotas bioquímicas envolvendo compostos orgânicos e inorgânicos. Análises de composição
da comunidade microbiana em vários sedimentos de diferentes partes do mundo mostraram que
os grupos filogenéticos predominantes são geralmente similares. Sedimentos do Mar do Norte
e do Mar Ártico, assim como do Mar Antártico, revelaram a presença de Gammaproteobacteria,
Deltaproteobacteria, Flavobacteria e Planctomycetes. Em sedimentos da Baía de Tóquio
(Japão), a predominância foi de grupos de Deltaproteobacteria, Gammaproteobacteria,
Epsilonproteobacteria e Verrucomicrobia. Um dos primeiros estudos moleculares de
sedimentos marinhos, na Baía de Puget Sound (Seattle, EUA) mostrou os mesmos grupos
taxonômicos. Outros estudos recentes no Mar Mediterrâneo descreveram a abundância de
membros de Deltaproteobacteria e Gammaproteobacteria, assim como Betaproteobacteria,
Plantomycetes, Acidobacteria e Firmicutes (KÖCHLING et al., 2011).
Zinger et al. (2011) publicaram a primeira síntese global contendo sequências
microbianas obtidas ao longo do projeto de Censo Internacional de Micróbios Marinhos
(ICoMM; do inglês, International Census of Marine Microbes). Os autores compararam a
diversidade beta para ambientes bentônicos e pelágicos em alguns de seus ecossistemas chave,

130
como água de superfície e de fundo, zona costeira e oceano aberto. Eles encontraram que
Gammaproteobacteria e Deltaproteobacteria, seguidas de Planctomycetes, Actinobacteria e
Acidobacteria, dominaram as comunidades bentônicas, especialmente em ecossistemas
bentônicos costeiros. Além desses, Clostridia e Bacilli, do filo Firmicutes, também apareceram
em maiores proporções. Esses táxons contêm muitos representantes microaerófilos ou
heterotróficos anaeróbicos e quimioautotróficos, com Deltaproteobacteria sendo fortemente
representada por grupos redutores de sulfato. Isso ocorre porque, em ambientes costeiros
bentônicos, a depleção de oxigênio e a disponibilidade de compostos químicos reduzidos pode
ser capaz de suportar algumas populações bacterianas com adaptações genéticas e fenotípicas
à vida em quimioclinas ou em condições de anoxia. Os autores também mostraram que os
sedimentos costeiros são extremamente variáveis espacial e temporalmente em termos de
fatores físico-químicos. Essa heterogeneidade ambiental pode aumentar a resiliência do
ecossistema, o que é essencial para sua sobrevivência quando se considera que tais ambientes
são constantemente submetidos a eventos de poluição e perda de hábitats. Diferentemente do
observado neste trabalho, Cyanobacteria, o filo mais abundante no Porto de Mucuripe, foi
mostrado como táxon de importância apenas em áreas pelágicas de oceano aberto.
Contudo, já foram relatadas quantidades significativas de Cyanobacteria em
sedimentos. Esses relatos foram observados após a Guerra do Golfo, em 1991, quando toneladas
de petróleo foram lançadas nas costas do Golfo Arábico (também conhecido como Golfo
Pérsico) em decorrência do conflito. Posteriormente, a colonização dessas costas por
cianobactérias (AL-THUKAIR, 2002; COHEN, 2002; BARTH, 2003; ABED, AL-THUKAIR,
DE BEER, 2006; AL-THUKAIR, ABED, MOHAMED, 2007) assim como sua habilidade de
formar consórcios microbianos capazes de biodegradação (ABED, KÖSTER, 2005;
SÁNCHEZ et al., 2006) foram relatadas. Dois anos após os derramamentos, grandes áreas da
região entre-marés foram cobertas com tapetes de cianobactérias. Esse táxon é conhecido por
crescer extensivamente construindo lâminas espessas na superfície do sedimento contendo óleo,
sendo capaz de selar tal superfície. Consequentemente, o óleo derramado pode ser aprisionado
em um ambiente anaeróbico no qual a transformação deste pode ocorrer, mesmo que em baixas
taxas (BARTH, 2003). Na verdade, polissacarídeos produzidos pelas cianobactérias podem ter
papéis essenciais na emulsificação do óleo, quebrando-o em pequenas gotas que são
subsequentemente atacadas por heterótrofos capazes de degradar o óleo que vivem associados
às cianobactérias. Adicionalmente, as cianobactérias previnem que tais heterótrofos sejam
levados pelas correntes ao os imobilizarem em seus polissacarídeos extracelulares. Podem
ainda atuar de forma importante ao suprir tais micro-organismos com oxigênio e/ou nitrogênio

131
fixado, necessários para o sucesso da degradação (COHEN, 2002). É importante destacar que
as cianobactérias não degradam os hidrocarbonetos do petróleo, mas parecem desempenhar um
papel indireto, porém significativo, na biodegradação ao suportarem o crescimento e atividade
dos verdadeiros degradadores – bactérias heterotróficas anaeróbicas associadas (ABED,
KÖSTER, 2005; SANCHÉZ et al., 2006).
Acidentes envolvendo derramamento de óleo já foram registrados no Porto do
Mucuripe, o último em 2012, quando 70 toneladas de óleo pesado foram derramadas, mas não
no Porto do Pecém. Houve ainda outros acidentes menores registrados, um março de 2008 e
outro em janeiro de 2010. Dessa forma, é possível que a ocorrência significativa de
Cyanobacteria somente no Mucuripe esteja relacionada a tais derramamentos. Além disso,
Chroococcales, a ordem de Cyanobacteria mais abundante no Mucuripe, já foi relatada em
sedimentos contaminados com óleo na costa da Arábia Saudita (AL-THUKAIR, ABED,
MOHAMED, 2007). Interessantemente, o táxon marinho Synechococcus, o gênero dominante
no Mucuripe, mostrou possuir duas estratégias de vida principais: especialistas de oceano
aberto, que dominam em ambientes quentes e oligotróficos ou temperados/polares e
mesotróficos, e oportunistas de zonas costeiras, que podem ser encontrados em quantidades
relativamente baixas tanto em áreas costeiras como ao longo de uma grande faixa de
ecossistemas, mas ocasionalmente atingindo números mais expressivos após perturbações
ambientais (ZWIRGLMAIER et al., 2008). Possivelmente, esse táxon, juntamente com outros
gêneros prevalentes no Porto do Mucuripe, como Rhodothermus e Sphaerobacter, podem ser
alvo de estudos futuros para avaliar seu potencial como indicadores de contaminação em
ambientes atingidos por derramamentos de óleo ou, de modo genérico, em ambientes portuários
cronicamente contaminados, como o Porto do Mucuripe. O gênero Sphaerobacter foi
previamente relatado estar presente em amostras de sedimentos marinhos anóxidos da Baía de
Cadíz, na Espanha (KÖCHLING et al., 2011).
Acosta-González, Rosselló-Moóra e Márques (2013) relataram a dominância das
ordens de Deltaproteobacteria, Myxococcales e Desulfobacterales, em sedimentos
contaminados após o derrame de óleo do navio Prestige. Tais ordens prevaleceram nos
sedimentos mesmo após a remoção mecânica do óleo. Particularmente Myxococcales destacou-
se como ordem dominante no Porto do Pecém, não sendo nesse caso relacionada ao registro de
derramamentos de óleo. As mixobactérias têm sido genericamente associadas a ambientes
terrestres, apesar de números cada vez mais altos de sequências de origem marinha estarem
sendo descritos recentemente (BRINKHOFF et al., 2012). Algumas mixobactérias cultiváveis
são únicas dentre os procariotos por seu estilo de vida singular e complexo, sendo capazes de

132
degradar polímeros complexos através da excreção de enzimas hidrolíticas (ACOSTA-
GONZÁLEZ, ROSSELLÓ-MOÓRA e MÁRQUES, 2013). A abundância de mixobactéria em
sedimentos marinhos vem sendo ligada ao tamanho dos grãos, havendo uma tendência a
maiores quantidades em areias bem finas (BRINKHOFF et al., 2012), como é o caso dos
sedimentos de ambos os portos estudados. Entretanto, Myxococcales apareceu como ordem
dominante somente no Pecém provavelmente devido ao carácter crônico da contaminação e/ou
aos derramamentos de óleo que ocorreram no Mucuripe ao longo dos anos, que levaram à
ocorrência de ordens de Cyanobacteria em seu lugar.
Baseado nos padrões observados até agora em sedimentos marinhos, pode-se
propor que as assembleias microbianas do Mucuripe vêm sofrendo os efeitos dos impactos
acumulados ao longo de mais de 50 anos de funcionamento, sendo bem peculiares. Ambos os
portos podem ser caracterizados como contaminados com base em sua caracterização físico-
química apresentada no Capítulo 1, mas o Porto do Mucuripe apresenta uma contaminação
crônica. Alteromonadales, presente em maiores quantidades no Mucuripe, foi mostrada
aumentar após a contaminação de manguezais com óleo (DOS SANTOS et al., 2011).
Thiotrichales e Legionellales, que ocorreram somente no Mucuripe, foram previamente
relacionadas à degradação de hidrocarbonetos (GOMES et al., 2008) ou a ambientes
antropizados enriquecidos com fosfatos orgânicos e Cyanobacteria (CARVALHO et al., 2007),
respectivamente.
No nível de espécies, o número de sequências de bactérias redutoras de sulfato
(SRB) não cultiváveis é capaz de diferenciar ambos os portos estatisticamente. Procariotos
redutores de sulfato são componentes diversos das comunidades bacterianas de sedimentos
(MIYATAKE, MACGREGOR E BOSCHKER, 2009). Nogales et al. (2011) relataram a
ocorrência de potenciais SRB capazes de degradar hidrocarbonetos em várias áreas costeiras
dos EUA, Itália, Venezuela, Porto Rico e Coreia do Sul. Outro estudo recente a respeito da
composição de assembleias de redução de sulfato em sedimentos portuários revelou sequências
relacionadas a redutores de sulfato cultiváveis capazes de catabolizar compostos aromáticos,
como o benzoato e o naftaleno (CHIN et al., 2008). Os dados obtidos neste trabalho mostram
que a microbiota associada aos sedimentos do Porto do Pecém possui sequências de SRB, o que
é importante dada à capacidade catabólica desse táxon, o que deve contribuir fortemente para a
resistência dessa comunidade.
As curvas de abundância de gêneros de ambos os portos mostrou a dominância de
poucos táxons muito abundantes e a presença da biosfera rara, caracterizada pela presença de
muitos táxons de baixa frequência. Esse padrão é característico de comunidades microbianas,

133
onde há uma microbiota fundamental, correspondente aos membros dominantes, e um banco
de sementes constituído de vários táxons pouco abundantes, constituindo uma long tail.
característica da distribuição. Os organismos que fazem parte do microbioma fundamental são
bem adaptados ao ecossistema onde residem. Dessa forma, o microbioma fundamental não é
estático, pois pode haver seleção de membros do banco de sementes em decorrência de
alterações ambientais (PEDRÓS-ALIÓ, 2006). É bem aceito em Ecologia que a distribuição de
abundância de espécies de uma comunidade de animais ou plantas, após uma perturbação,
mudará de uma estrutura de alta equabilidade para uma de dominância aumentada. Dessa forma,
tais mudanças na equabilidade têm sido utilizadas como indicadores da integridade biológica
de uma comunidade (AGER et al., 2010).
No ambiente marinho, em geral os hidrocarbonetos são degradados sob condições
aeróbicas (MCGRODDY, FARRINGTON E GSCHWEND, 1995). Entretanto, grande parte do
metabolismo da matéria orgânica em sedimentos marinhos é, na verdade, anaeróbico
(CANFIELD et al., 1993). Estudos recentes mostraram que muitos processos de degradação
anaeróbica de xenobióticos ocorreram sob condições de redução do sulfato (COATES et al.,
1997; TOWNSEND, PRINCE e SUFLITA, 2003; WIDDEL, RABUS, 2001). Coates et al.
(1997) relataram a degradação de naftaleno, fenantreno, fluoranteno e metilnaftaleno em
sedimentos marinhos com o sulfato agindo como aceptor final de elétrons. Esses dados são
muito importantes, pois sugerem que alguns contaminantes persistentes podem ser degradados
em condições de redução de sulfato (HAYES, NEVIN e LOVLEY, 1999), contribuindo para
ressaltar o potencial de purificação in situ dos portos estudados.
Gomes et al. (2013) estudaram assembleias bacterianas associadas aos sedimentos
do Oceano Atlântico na costa portuguesa sob influência de atividades portuárias. A
classificação relatada pelos autores foi comparada com os filos bacterianos encontrados no
Pecém e no Mucuripe. Similarmente ao Mucuripe, observou-se uma super-representação de
Bacteroidetes nas sequências obtidas por Gomes. Entretanto, a presença massiva de
Cyanobacteria no Porto do Mucuripe permanece um achado único.
As atividades antropogênicas têm um importante impacto sobre os ecossistemas
marinhos. As pressões podem ser oriundas de diferentes ações, porém todas têm efeitos sobre
os vários componentes das cadeias tróficas marinhas, de micro-organismos a predadores de
topo. Tais pressões podem causar profundas mudanças na composição e na função da
comunidade. As perturbações capazes de causar mudanças no funcionamento da comunidade,
contudo, são consideradas mais sérias, pois conseguem ir além da redundância funcional com

134
consequências sobre os serviços ecossistêmicos desempenhados pelas comunidades
microbianas (NOGALES et al., 2011).
Os portos do Pecém e do Mucuripe não apresentaram diferenças funcionais
significativas apesar das divergências taxonômicas encontradas. Apesar de quase todos os
fatores físico-químicos analisados neste trabalho serem estatisticamente diferentes entre os
portos estudados, poucas diferenças funcionais foram encontradas nos bancos de dados KEGG
e SEED. Portanto, é possível afirmar que a redundância funcional não foi afetada pelos estresses
ambientais que ocorrem nesses portos. Porém, embora a manutenção da diversidade genética
dos sedimentos portuários analisados confirmem a redundância funcional das assembleias
procarióticas, é possível hipotetizar que a persistência dos impactos atuais ou seu agravamento
possam levar a mudanças na biodiversidade microbiana que comprometam o funcionamento do
ecossistema. E, uma vez que tenham sofrido modificações funcionais, as mudanças podem se
tornar irreversíveis. Logo, deve-se considerar algumas precauções para melhorar ou manter a
qualidade dos sedimentos. Além disso, o monitoramento regular de tais ambientes é fortemente
recomendado.
Algumas categorias funcionais relacionadas ao acúmulo de contaminantes, como
resistência ao cobalto, zinco e cádmio, homeostase do cobre, resistência ao zinco, resistência
ao arsênio, tolerância ao cobre e redutase do mercúrio, foram detectadas em ambos os portos.
Contudo, apesar dos dois portos exibirem sintomas de enriquecimento orgânico, apresentaram
elevadas proporções de transportadores ABC dentro do subsistema de Transporte por
Membrana. Esse tipo de transporte é típico de ambientes oligotróficos, onde os nutrientes são
escassos, mas retirados do ambiente eficientemente (KISAND et al., 2012). Normalmente essa
é a condição dos sedimentos encontrados sob águas equatoriais oligotróficas, considerando que
os sedimentos marinhos superficiais são formados a partir do afundamento de partículas
orgânicas e inorgânicas das zonas pelágicas acima. Entretanto, esse fato pode ser considerado
uma evidência de que, como discutido anteriormente, a funcionalidade dos sedimentos dos
portos estudados parece estar preservada.
Apesar dos portos estudados terem divergido muito pouco em termos de
transformações metabólicas, a afiliação taxonômica das sequências metagenômicas
classificadas em diferentes categorias funcionais segundo o banco de dados KEGG reforçou as
diferenças filogenéticas encontradas entre os portos. Em geral, a maioria dos organismos
envolvidos nos metabolismos analisados foram do filo Proteobacteria e Actinobacteria no Porto
do Pecém, e Chloroflexi e Cyanobacteria no Porto do Mucuripe.

135
Desulfovibrionales e Chroococcales, principais responsáveis pelo metabolismo do
enxofre no Porto do Pecém e no Porto do Mucuripe, respectivamente, foram anteriormente
descritas como contribuintes para o ciclo biogeoquímico do enxofre, mas em diferentes rotas.
Chroococcales pode participar da redução assimilatória do sulfato, enquanto
Desulfovibrionales participa na redução dissimilatória do sulfato. Os redutores assimilatórios
do sulfato reduzem o sulfato a sulfito para cobrir suas necessidades pelo elemento e para
incorporar em novas moléculas sintetizadas. Por outro lado, os redutores dissimilatórios do
sulfato são ubíquos em ambientes anaeróbicos e contribuem de forma marcante para os ciclos
globais do enxofre e do carbono, principalmente em ambientes marinhos, onde a concentração
de sulfato é elevada. Estima-se que a redução dissimilatória do sulfato seja responsável por
cerca de 50% da remineralização de carbono em ambientes marinhos (CASPI et al., 2010).
As sequências relacionadas ao metabolismo do nitrogênio foram classificadas em
sua maioria como Sphaerobacterales (filo Chloroflexi), associada ao processo de denitrificação.
A ordem Enterobacteriales, também encontrada no Mucuripe, mas em menores frequências,
vem sendo associada ao processo de redução dissimilatória do nitrato. A redutase respiratória
do nitrato A, de Enterobacteriales, utiliza o nitrato como o aceptor de elétrons de uma cadeia
de respiração anaeróbica e é expressa em resposta a altos níveis de nitrato no ambiente (CASPI
et al., 2010). No Pecém, somente um táxon foi relacionado ao metabolismo do nitrogênio, a
ordem Rhodobacterales, a qual é composta de diazotróficos de vida livre possuidores de
enzimas fixadoras do nitrogênio, como a NifX. Esta enzima foi prevista a partir da anotação do
genoma completo de Rhodobacter sphaeroides (LIM et al., 2009).
Muitas informações podem ser obtidas a partir da comparação entre diferentes
ecossistemas, auxiliando a responder algumas questões relacionadas a aspectos importantes da
ecologia microbiana (DELMONT et al., 2011). Para inferir-se sobre a robustez dos resultados
encontrados neste estudo, comparou-se nossos conjuntos de dados com uma coleção de
metagenomas disponíveis publicamente e pertencentes a outros ecossistemas marinhos. A
comparação envolvendo somente os metagenomas dos portos do Pecém e do Mucuripe não
revelou diferenças funcionais significativas, mas somente taxonômicas. Por sua vez, as
divergências taxonômicas observadas entre os ecossistemas marinhos comparados sugeriram
um padrão biogeográfico, capaz de diferenciar ecossistemas da costa brasileira daqueles das
costas australianas, do Oceano Índico e do Golfo do México.
As bactérias dos gêneros Synechococcus, Rhodothermus e Sphaerobacter,
encontradas abundantemente no Porto do Mucuripe, constituem potenciais bioindicadores.
Porém, uma avaliação mais detalhada de suas características de crescimento na presença de

136
contaminantes é necessária para comprovar tal utilidade. Como nesse estudo os micro-
organismos foram detectados por um método independente de cultivo, reforça-se a necessidade
de isolar tais organismos ou de estudá-los através de um método como do DNA-SIP (DNA
stable-isotope probing), capaz de relacionar a detecção de um organismos a sua habilidade
metabólica.
Felizmente, o cenário do monitoramento ambiental pode mudar bastante com a
implementação das modernas técnicas de sequenciamento e abordagens metagenômicas.
Consequentemente, é possível que se tenha uma visão ampla e global da diversidade de
comunidades microbianas, as quais podem ser utilizadas como indicadores de condições
ambientais, com tais técnicas representando opções razoáveis para o monitoramento ambiental
futuro.
5.5. Conclusões
A análise taxonômica e funcional dos metagenomas mostrou que os portos
estudados possuem composições taxonômicas diferenciadas caracterizadas pela predominância
de Deltaproteobacteria e Actinobacteria no Porto do Pecém e por Cyanobacteria, Bacteroidetes
e Chloroflexi no Porto do Mucuripe. Entretanto, os portos não diferiram funcionalmente. As
diferenças encontradas podem ser atribuídas às características ambientais de cada porto,
decorrentes das diferenças de arquitetura e tempo de operação existentes entre ambos. A
composição taxonômica do Porto do Mucuripe destacou-se pela presença de táxons não
encontrados no Porto do Pecém ou em outros sedimentos costeiros, sugerindo a forte influência
dos impactos ambientais acumulados durante os mais de 50 anos de funcionamento. Também
foi possível selecionar três táxons, Synechococcus, Rhodothermus e Sphaerobacter, dominantes
no Porto do Mucuripe, e indicá-los como promissores candidatos a bioindicadores de
sedimentos portuários cronicamente contaminados.

137
6. CONSIDERAÇÕES FINAIS E PERSPECTIVAS FUTURAS
Os micro-organismos são essenciais para todos os ecossistemas em termos de
biomassa, diversidade e funcionamento. Portanto, entender seus padrões de distribuição é
crucial para poder antecipar eventos resultantes de futuras mudanças ambientais nos
ecossistemas marinhos. Contudo, por muitos anos, estudar micro-organismos foi considerada
uma tarefa desafiadora devido à falta de métodos apropriados que permitissem que os estudos
fossem realizados em cronogramas razoáveis e com resolução adequada, considerando-se os
elevados números de micro-organismos não cultiváveis. Felizmente, as técnicas independentes
de cultivo e, mais recentemente, as metodologias de sequenciamento de nova geração passaram
a permitir novas perspectivas através de uma abordagem metagenômica aplicável a qualquer
tipo de ambiente.
Concretizou-se por meio de tais tecnologias descrever profundamente os membros
microbianos e seu funcionamento em duas áreas portuárias, representando uma opção razoável
para o monitoramento ambiental no futuro. As conclusões obtidas no estudo foram limitadas
pelo pequeno número de amostras e aspecto pontual da pesquisa, porém, devido à profundidade
das análises e à metodologia de amostragem, foi possível conhecer de forma muito abrangente
as assembleias microbianas presentes nos locais de estudo. Ao fim, pôde-se ainda sugerir táxons
de bactérias a serem utilizados para o monitoramento de tais ecossistemas.
Considerando a importância desse tema, sugere-se que os táxons indicados sejam
avaliados quanto a suas características de crescimento, habilidade de responder a contaminantes
típicos de áreas portuárias ou capacidade de metabolização dos mesmos com o intuito de revelar
futuramente ferramentas biológicas que possam auxiliar na detecção de perigos ambientais em
ecossistemas costeiros ou mesmo em sua remediação.

138
7. REFERÊNCIAS
ABED, R. M. M.; AL-THUKAIR, A.; DE BEER, D. Bacterial diversity of cyanobacterial mat
degrading petroleum compounds at elevated salinities and temperatures. FEMS
Microbiology Ecology, v. 57, p. 290-301, 2006.
ABED, R. M. M.; KÖSTER, J. The direct role of aerobic heterotrophic bacteria associated
with cyanobacteria in the degradation of oil compounds. International Biodeterioration &
Biodegradation, v. 55, p. 29-37, 2005.
ACOSTA-GONZÁLEZ, A. Comunidades microbianas en sedimentos marinos
contaminados por el vertido de Prestige. 2013. 221 f. Tese (Doutorado em Bioquímica e
Biologia Molecular) - Universidad de Granada, Granada, 2013.
ACOSTA-GONZÁLEZ, A.; ROSSELLÓ-MÓRA, R.; MARQUÉS, S. Characterization of the
anaerobic microbial community in oil-polluted subtidal sediments: aromatic biodegradation
potential after the Prestige oil spill. Environmental Microbiology, v. 15, n. 1, p. 77–92,
2013.
AGER, D.; EVANS, S.; LI, H.; LILLEY, A. K.; GAST, C. J. VAN DER. Anthropogenic
disturbance affects the structure of bacterial communities. Environmental Microbiology , v.
12, n. 3, p. 670–8, 2010.
ALONSO-GUTIÉRREZ, J.; FIGUERAS, A.; ALBAIGÉS, J.; et al. Bacterial communities
from shoreline environments (costa da morte, northwestern Spain) affected by the prestige oil
spill. Applied and Environmental Microbiology, v. 75, n. 11, p. 3407–18, 2009.
AL-THUKAIR, A. A.; ABED, R. M. M.; MOHAMED, L. Microbial community of
cyanobacteria mats in the intertidal zone of oil-polluted coast of Saudi Arabia. Marine
Pollution Bulletin , v. 54, p. 173-179, 2007.
AL-THUKAIR, A. Effect of oil pollution on euendolithic cyanobacteria of the Arabian
Gulf. Environmental Microbiology, v. 4, p. 125-129, 2002.
ANGELIM, A. L. Seleção e imobilização de consórcios de bactérias
hidrocarbonoclásticas e produtoras de compostos ativos de superfície para aplicações em
processos de biorremediação. 2012. 150 f. Tese (Doutorado em Biotecnologia) - Rede
Nordeste de Biotecnologia, Universidade Federal do Ceará, Fortaleza, 2012.
ANSORGE, W. J. Next-generation DNA sequencing techniques. New Biotechnology, v. 25,
n. 4, p. 195–203, 2009.
ARNDT, D.; XIA, J.; LIU, Y.; ZHOU, Y.; GUO, A. C.; CRUZ, J. A.; SINELNIKOV, I.;
BUDWILL, K.; NESBO, C. L.; WISHART, D. S. METAGENassist: a comprehensive web
server for comparative metagenomics. Nucleic Acids Research, v. 40, n. Web Server issue, p.
W88–95, 2012.
ATLAS, R. M.; BARTHA, R. Microbial Ecology, Fundamentals and Applications. Redwood
City, CA: Benjamin/Cummings, 1998.

139
ATLAS, R. M.; HOROWITZ, A; KRICHEVSKY, M.; BEJ, A K. Response of microbial
populations to environmental disturbance. Microbial Ecology, v. 22, n. 1, p. 249–56, 1991.
BALL, I. Port waste reception facilities in UK ports Iwan Ball. Marine Policy, v. 23, n. 4-5,
p. 307–327, 1999.
BARTH, H-J. The influence of cyanobacteria on oil polluted intertidal soils at the Saudi
Arabian Gulf shores. Marine Pollution Bulletin, v. 46, p. 1245-1252, 2003.
BAZINET, A. L.; CUMMINGS, M. P. A comparative evaluation of sequence classification
programs. BMC Bioinformatics, v. 92, p. 1-13, 2012.
BEGON, M.; TOWNSEND, C.R.; HARPER, J.L. Ecologia: de indivíduos a ecossistemas.
4ª ed. Porto Alegre: Artmed, 2007. 759 p.
BENSON, D. A.; CAVANAUGH, M.; CLARK, K.; KARSCH-MIZRACHI, I.; LIPMAN. D.
J.; OSTELL, J.; SAYERS, E. W. GenBank. Nucleic Acids Research, v. 41, D36-42, 2013.
BEOLCHINI, F.; ROCCHETTI, L.; REGOLI, F.; DELL’ANNO, A. Bioremediation of
marine sediments contaminated by hydrocarbons: experimental analysis and kinetic
modeling. Journal of Hazardous Materials, v. 182, n. 1-3, p. 403–7, 2010.
BÖER, S. I.; HEDTKAMP, S. I. C.; BEUSEKOM, J. E. E. VAN; et al. Time- and sediment
depth-related variations in bacterial diversity and community structure in subtidal sands. The
ISME Journal, v. 3, n. 7, p. 780–91, 2009.
BOLHUIS, H.; FILLINGER, L.; STAL, L. J. Coastal microbial mat diversity along a natural
salinity gradient. PloS ONE, v. 8, n. 5, p. e63166, 2013.
BOWMAN, J. P.; REA, S. M.; MCCAMMON, S. A.; MCMEEKIN, T. A. Diversity and
community structure within anoxic sediment from marine salinity meromictic lakes and a
coastal meromictic marine basin, Vestfold Hills, Eastern Antarctica. Environmental
Microbiology , v. 2, n. 2, p. 227–237, 2000.
BREITBART, M. Marine viruses: truth or dare. Annual Review of Marine Science, v. 4, p.
425–48, 2012.
BRINKHOFF, T.; FISCHER, D.; VOLLMERS, J.; VOGET, S.; BEARDSLEY, C.; THOLE,
S.; MUSSMANN, M.; KUNZE, B.; WAGNER-DÖBLER, I.; DANIEL, R.; SIMON, M.
Biogeography and phylogenetic diversity of a cluster of exclusively marine
myxobacteria. The ISME Journal, v. 6, n. 6, p. 1260-1272, 2012.
BURUAEM, L. M.; HORTELLANI, M. A; SARKIS, J. E.; COSTA-LOTUFO, L. V;
ABESSA, D. M. S. Contamination of port zone sediments by metals from Large Marine
Ecosystems of Brazil. Marine Pollution Bulletin, v. 64, n. 3, p. 479–88, 2012.
CANFIELD, D. E.; JORGENSEN, B. B.; FOSSING, H.; GLUD, R.; GUNDERSEN, J.;
RAMSING, N. B.; THAMDRUP, B.; HANSEN, J. W.; NIELSEN, L. P.; HALL, P. O. J.
Pathways of organic carbon oxidation in three continental margin sediments. Marine
Geology , v. 113, p. 27-40, 1993.

140
CARDENAS, E.; TIEDJE, J. M. New tools for discovering and characterizing microbial
diversity. Current Opinion in Biotechnology, v. 19, n. 6, p. 544–9, 2008.
CARVALHO, F. R. S.; VAZOLLER, R. F.; FORONDA, A. S.; PELLIZARI, V. H.
Phylogenetic Study of Legionella Species in Pristine and Polluted Aquatic Samples from a
Tropical Atlantic Forest Ecosystem. Current Microbiology, v. 55, p. 288-293, 2007.
CASADO-MARTÍNEZ, M. C.; BUCETA, J. L.; BELZUNCE, M. J.; DELVALLS, T. A.
Using sediment quality guidelines for dredged material management in commercial ports
from Spain. Environment International, v. 32, n. 3, p. 388–96, 2006.
CASAMAYOR, E. O.; SCHÄFER, H.; BAÑERAS, L.; PEDRÓS-ALIÓ, C.; MUYZER, G.
Identification of and Spatio-Temporal Differences between Microbial Assemblages from Two
Neighboring Sulfurous Lakes: Comparison by Microscopy and Denaturing Gradient Gel
Electrophoresis. Applied and Environmental Microbiology, v. 66, n. 2, p. 499 - 508, 2000.
CASE, R. J.; BOUCHER, Y.; DAHLLÖF, I.; et al. Use of 16S rRNA and rpoB genes as
molecular markers for microbial ecology studies. Applied and Environmental
Microbiology , v. 73, n. 1, p. 278–88, 2007.
CASPI, R.; ALTMAN, T.; DALE, J. M.; DREHER, K.; FULCHER, C. A.; GILHAM, F.;
KAIPA, P.; KARTHIKEYAN, A. S.; KOTHARI, A.; KRUMMENACKER, M.;
LATENDRESSE, M.; MUELLER, L. A.; PALEY, S.; POPESCU, L.; PUJAR, A.; SHEARER,
A. G.; ZHANG, P.; KARP. P. D. The MetaCyc database of metabolic pathways and enzymes
and the BioCyc collection of pathway/genome databases. Nucleic Acids Research, v. 38, p.
D473 - D479, 2010.
CHEN, K.; PACHTER, L. Bioinformatics for whole-genome shotgun sequencing of microbial
communities. PLoS Computational Biology, v. 1, n. 2, p. 106–12, 2005.
CHIELLINI, C.; IANNELLI, R.; VERNI, F.; PETRONI, G. Bacterial communities in
polluted seabed sediments: a molecular biology assay in Leghorn Harbor. The Scientific
World Journal , v. 2013, p. 165706, 2013.
CHIN, K.-J.; SHARMA, M. L.; RUSSELL, L. A; O’NEILL, K. R.; LOVLEY, D. R.
Quantifying expression of a dissimilatory (bi)sulfite reductase gene in petroleum-
contaminated marine harbor sediments. Microbial Ecology, v. 55, n. 3, p. 489–99, 2008.
CHISTOSERDOVA, L. Recent progress and new challenges in metagenomics for
biotechnology. Biotechnology Letters, v. 32, n. 10, p. 1351–9, 2010.
CHOU, H-H.; HOLMES, M. H. DNA sequence quality trimming and vector
removal. Bioinformatics, v. 17, n. 12, p. 1093-1104, 2001.
CLEARY, D. F. R.; SMALLA, K.; MENDONÇA-HAGLER, L. C. S.; GOMES, N. C. M.
Assessment of variation in bacterial composition among microhabitats in a mangrove
environment using DGGE fingerprints and barcoded pyrosequencing. PloS ONE, v. 7, n. 1, p.
e29380, 2012.
COATES, J. D.; WOODWARD, J.; ALLEN, J.; PHILP, P.; LOVLEY, D. R. Anaerobic
degradation of polycyclic aromatic hydrocarbons and alkanes in petroleum-contaminated

141
marine harbor sediments. Applied and Environmental Microbiology, v. 63, n. 9, p. 3589–
93, 1997.
COHEN Y. Bioremediation of oil by marine microbial mats. International Microbiology, v.
5, p. 189-193, 2002.
COMMENDATORE, M. G.; ESTEVES, J. L. An assessment of oil pollution in the coastal
zone of patagonia, Argentina. Environmental Management, v. 40, n. 5, p. 814–21, 2007.
DELL’ANNO, A.; BEOLCHINI, F.; ROCCHETTI, L.; LUNA, G. M.; DANOVARO, R. High
bacterial biodiversity increases degradation performance of hydrocarbons during
bioremediation of contaminated harbor marine sediments. Environmental Pollution, v. 167,
p. 85–92, 2012.
DELMONT, T. O.; MALANDAIM, C.; PRESTAT, E.; LAROSE, C.; MONIER, J-M.;
SIMONET, P.; VOGEL, T. M. Metagenomic mining for microbiologists. The ISME
Journal , v. 5, n. 12, p. 1837-1843, 2011.
DELONG, E. F. Archaea in coastal marine environments. Proceedings of the National
Academy of Sciences of the United States of America, v. 89, n. 12, p. 5685–9, 1992.
DIAS, J. C. DE S. Gestão e operação portuária: experiência em cingapura. Brasília, 2013.
DOS SANTOS, H. F.; CURY, J. C.; DO CARMO, F. L.; DOS SANTOS, A. L.; TIEDJE, J.;
VAN ELSAS, J. D.; ROSADO, A. S.; PEIXOTO, R. S. Mangrove Bacterial Diversity and the
Impact of Oil Contamination Revaeled by Pyrosequencing: Bacterial Proxies for Oil
Pollution. PLoS ONE, v. 6, n. 3, p. e16943, 2011.
DUMONT, M.; MURRELL, J. Stable isotope probing—linking microbial identity to
function. Nature Reviews Microbiology, v. 3, n. June, p. 499–504, 2005.
DUNBAR, J.; TICKNOR, L. O.; KUSKE, C. R. Assessment of microbial diversity in four
southwestern United States soils by 16S rRNA gene terminal restriction fragment
analysis. Applied and Environmental Microbiology, v. 66, n. 7, p. 2943–50, 2000.
EDLUND, A.; JANSSON, J. K. Changes in active bacterial communities before and after
dredging of highly polluted Baltic Sea sediments. Applied and Environmental
Microbiology, v. 72, n. 10, p. 6800–7, 2006.
EEA. Priority issues in the Mediterranean environment. EEA Report, n. 4, 2006. ISBN: 92-
9167-812-0.
ELLIS, S. L.; INCZE, L. S.; LAWTON, P.; et al. Four regional marine biodiversity studies:
approaches and contributions to ecosystem-based management. PloS ONE, v. 6, n. 4, p.
e18997, 2011.
ESPO – European Sea Ports Organisation. Annual Report 2012-2013. Disponível em:
<http://www.espo.be/images/stories/Publications/annual_reports/espo_annual%20report_2013
.pdf >. Acesso em 16 jul. 2014.
FALKOWSKI, P. G.; FENCHEL, T.; DELONG, E. F. The microbial engines that drive
Earth’s biogeochemical cycles. Science, v. 320, n. 5879, p. 1034–9, 2008.

142
FREIRE, G. S. S.; LIMA, S.; MAIA, L. P.; LACERDA, L. D. Geochemistry of continental
shelf sediments of the Ceara coast, NE Brazil. In: LACERDA, L.D.; SANTELLI, R.E.;
DUURSMA, E.K.; ABRÃO, J.J. (Ed.). Facets of environmental Geochemistry in tropical
and subtropical environments. Berlin: Springer Verlag, 2004, p. 365-378.
FUHRMAN, J. A. Microbial community structure and its functional implications. Nature, v.
459, n. 7244, p. 193–199, 2009.
GARRISON, T. S. Essentials of Oceanography. Belmont/CA, Cengage Learning, 2011.
GIACOBO, Fabiano. Relatório Técnico. Plano Mestre do Porto de Pecém. 2012.
GLASS, E. M.; WILKENING, J.; WILKE, A.; ANTONOPOULOS, D.; MEYER, F. Using
the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold
Spring Harbor Protocols, v. 2010, pdb.prot5368, 2010.
GOBET, A.; BÖER, S. I.; HUSE, S. M.; et al. Diversity and dynamics of rare and of resident
bacterial populations in coastal sands. The ISME Journal, v. 6, n. 3, p. 542–53, 2012.
GOBET, A.; BOETIUS, A.; RAMETTE, A. Ecological coherence of diversity patterns
derived from classical fingerprinting and Next Generation Sequencing
techniques. Environmental Microbiology, 2013.
GOMES, N. C. M.; BORGES, L. R.; PARANHOS ,R.; PINTO, F. N.; MENDONÇA-
HAGLER, L. C. S.; SMALLA, K. Exploring the diversity of bacteria communities in
sediments of urban mangrove forests. FEMS Microbiology Ecology, v. 66, n. 1, p. 96-109,
2008.
GOMES, N. C. M.; MANCO, S. C.; PIRES, A. C. C.; et al. Richness and composition of
sediment bacterial assemblages in an Atlantic port environment. The Science of the Total
Environment, v. 452-453, p. 172–80, 2013.
GOMEZ-ALVAREZ, V.; TEAL, T. K.; SCHMIDT, T. M. Systematic artifacts in metagenomes
from complex microbial communities. The ISME Journal, v. 3, n. 11, p. 1314–1317, 2009.
GRECH, A; BOS, M.; BRODIE, J.; et al. Guiding principles for the improved governance of
port and shipping impacts in the Great Barrier Reef. Marine Pollution Bulletin, v. 75, n. 1-2,
p. 8–20, 2013.
GROSS, M. G. Carbon determination. In: Carver RE, editors. Procedures in Sedimentary
Petrology. New York: Wiley-Interscience; 1971. p. 573–596.
GUILBEAU, E. T. The effects of sediment size and oil exploration on microbial ATP
biomass. 2003. 28 p. Thesis (Master of Science) – The Department of Oceanography and
Coastal Sciences, Agricultural and Mechanical College, Lousiana State University, 2003.
HALPERN, B. S.; SELKOE, K. A; MICHELI, F.; KAPPEL, C. V. Evaluating and ranking the
vulnerability of global marine ecosystems to anthropogenic threats. The Journal of the
Society for Conservation Biology, v. 21, n. 5, p. 1301–15, 2007.
HAMMER, O.; HARPER, D. A. T.; RYAN, P. D. Past: Paleontological Statistics Software
Package for Education and Data Analysis. Paleontologia Electronic, v. 4, n. 1, 2001.

143
HANDELSMAN, J. Metagenomics : Application of Genomics to Uncultured Microorganisms
Metagenomics. Microbiology and Molecular Biology Reviews, v. 68, n. 4, 2004.
HANDELSMAN, J.; RONDON, M. R.; BRADY, S. F.; CLARDY, J.; GOODMAN, R. M.
Molecular biological access to the chemistry of unknown soil microbes: a new frontier for
natural products. Chemistry & Biology, v. 5, n. 10, p. R245–9, 1998.
HAYES, L. A.; NEVIN, K. P.; LOVLEY, D. R. Role of prior exposure on anaerobic
degradation of naphthalene and phenanthrene in marine harbor sediments. Organic
Geochemistry , v. 30, n. 8, p. 937–945, 1999.
HAYES, L. A; LOVLEY, D. R. Specific 16S rDNA sequences associated with naphthalene
degradation under sulfate-reducing conditions in harbor sediments. Microbial Ecology , v.
43, n. 1, p. 134–45, 2002.
HEUER H, KRSEK, M.; BAKER, P.; SMALLA, K.; WELLINGTON, E. M. H. Analysis of
actinomycete communities by specific amplification of genes encoding 16S Rrna and gel-
electrophoretic separation in denaturing gradients. Applied and Environmental
Microbiology, v. 63, p. 3233-3241, 1997.
HOWELL, E. A; BOGRAD, S. J.; MORISHIGE, C.; SEKI, M. P.; POLOVINA, J. J. On
North Pacific circulation and associated marine debris concentration. Marine Pollution
Bulletin , v. 65, n. 1-3, p. 16–22, 2012.
HUSON, D. H.; AUCH, A. F.; QI, J.; SCHUSTER, S. C. MEGAN analysis of metagenomic
data. Genome Research, v. 17, p. 377-386, 2007.
IANNELLI, R.; BIANCHI, V.; MACCI, C.; PERUZZI, E.; CHIELLINI, C.; PETRONI, G.;
MASCIANDARO, G. Assessment of pollution impact on biological activity and structure of
seabed bacterial communities in the Port of Livorno (Italy). The Science of the Total
Environment , v. 426, p. 56–64, 2012.
INGOLE, B.; SIVADAS, S.; NANAJKAR, M.; SAUTYA, S.; NAG, A. A comparative study
of macrobenthic community from harbours along the central west coast of
India. Environmental monitoring and assessment, v. 154, n. 1-4, p. 135–46, 2009.
IRWIN, R. J.; VANMOUWERIK, M.; STEVENS, L.; SEESE, M. D.; BASHAM,
W. Environmental Contaminants Encyclopedia. National Park Service, Water Resources
Division, Fort Collins, Colorado. 1997. Distributed within the Federal Government as an
Electronic Document.
ISAAC, P.; SÁNCHEZ, L. A.; BOURGUIGNON, N.; CABRAL, M. E.; FERRERO, M. A.
Indigenous PAH-degrading bacteria from oil-polluted sediments in Caleta Cordova, Patagonia
Argentina. International Biodeterioration & Biodegradation, v. 82, p. 207–214, 2013.
JACKSON, C. R.; WEEKS, A. Q. Influence of particle size on bacterial community structure
in aquatic sediments as revealed by 16S rRNA gene sequence analysis. Applied and
Environmental Microbiology, v. 74, n. 16, p. 5237–40, 2008.

144
JACOB, M.; SOLTWEDEL, T.; BOETIUS, A.; RAMETTE, A. Biogeography of Deep-sea
benthic bacteria at regional scale (LTER HAUSGARTEN, Fram Strait, Arctic). PloS ONE , v.
8, n. 9, p. e72779, 2013.
JEFFRIES T. C.; SEYMOUR, J. R.; GILBERT, J. A.; DINSDALE, E. A.; NEWTON, K.;
LETERME, S. S. C.; ROUDNEW, B.; SMITH, R. J.; SEURONT, L.; MITCHELL, J. G.
Substrate Type Determines Metagenomic Profiles from Diverse Chemical Habitats. PLoS
ONE , v. 6, n. 9, p. e25173, 2011.
JOHNSEN, A. R.; WICK, L. Y.; HARMS, H. Principles of microbial PAH-degradation in
soil. Environmental Pollution, v. 133, n. 1, p. 71–84, 2005.
JOHNSTON, E. L.; ROBERTS, D. A. Contaminants reduce the richness and evenness of
marine communities: a review and meta-analysis. Environmental Pollution, v. 157, n. 6, p.
1745–52, 2009.
KAKIRDE, K. S.; PARSLEY, L. C.; LILES, M. R. Size Does Matter: Application-driven
Approaches for Soil Metagenomics. Soil Biology & Biochemistry, v. 42, n. 11, p. 1911–
1923, 2010.
KANEHISA, M.; GOTO, S.; KAWASHIMA, S.; OKUNO, Y.; HATTORI, M. The KEGG
resource for deciphering the genome. Nucleic Acids Research, n. 32, p. D277–D280, 2004.
KARL, D. M. Microbial oceanography: paradigms, processes and promise. Nature Reviews
Microbiology, v. 5, n. 10, p. 759–69, 2007.
KENNEDY, J.; FLEMER, B.; JACKSON, S. A; LEJON, D. P. H.; MORRISSEY, J. P.;
O’GARA, F.; DOBSON, A. D. W. Marine metagenomics: new tools for the study and
exploitation of marine microbial metabolism. Marine Drugs, v. 8, n. 3, p. 608–28, 2010.
KENT, W. J. BLAT---The BLAST-Like Alignment Tool. Genome Research, v. 12, n. 4, p.
656–664, 2002.
KIRCHMAN, D.L. Processes in Microbial Ecology. Oxford University Press, 2012, 312 p.
KISAND, V.; VALENTE, A.; LAHM, A.; TANET, G.; LETTIERI, T. Phylogenetic and
functional metagenomic profiling for assessing microbial biodiversity in environmental
monitoring. PloS ONE, v. 7, n. 8, p. e43630, 2012.
KNOPPERS, B.; EKAU, W.; FIGUEIREDO, A. The coast and shelf of east and northeast
Brazil and material transport. Geo-Marine Letters, v. 19, p. 171–178, 1999.
KÖCHLING, T.; LARA-MARTÍN, P.; GONZÁLEZ-MAZO, E.; AMILS, R.; SANZ, J. L.
Microbial community composition of anoxic marine sediments in the Bay of Cádiz
(Spain). International Microbiology, v. 14, p. 143–154, 2011.
KRISTIANSSON, E.; HUGENHOLTZ, P.; DALEVI, D. ShotgunFunctionalizeR: an R-
package for functional comparison of metagenomes. Bioinformatics, v. 25, n. 20, p. 2737-
2738., 2009.

145
LACERDA, L. D.; MARINS, R. V. Geoquímica de sedimentos e o monitoramento de metais
na plataforma continental nordeste oriental do Brasil. Geochimica Brasiliensis, v. 20, n. 1, p.
123-135, 2006.
LAGUERRE, G.; ALLARD, M. R.; REVOY, F.; AMARGER, N. Rapid Identification of
Rhizobia by Restriction Fragment Length Polymorphism Analysis of PCR-Amplified 16S
rRNA Genes. Applied and Environmental Microbiology, v. 60, n. 1, p. 56–63, 1994.
LATIMER, J. S.; ZHENG, J. The sources, Transport, and Fate of PAHs in the Marine
Environment. In: DOUBEN, P.E.T. PAHs: an ecotoxicological perspective. John Wiley &
Sons Ltd, West Sussex, England. 2003.
LIM, S. K.; KIM, S. J.; CHA, S. H.; OH, Y. K.; RHEE, H. J.; KIM, M. S.; LEE, J. K.
Complete genome sequence of Rhodobacter sphaeroides KD131.Journal of Bacteriology, v.
191, n. 3, p. 1118-1119, 2009.
LIU, L; LI, Y; LI, S. Comparison of Next-Generation Sequencing Systems. Journal of
Biomedicine and Biotechnology, p. 1–11, 2012.
LLOBET-BROSSA, E.; RABUS, R.; BÖTTCHER, M. E.; KÖNNEKE, M.; FINKE, N.;
SCHRAMM, A.; MEYER, R. L.; GRÖTZSCHEL, S.; ROSSELLÓ-MORA, R.; AMANN, R.
Community structure and activity of sulfate-reducing bacteria in an intertidal surface
sediment : a multi-method approach. Aquatic Microbial Ecology, v. 29, n. 3, p. 211–226,
2002.
LUO, C.; TSEMENTZI, D.; KYRPIDES, N.; READ, T.; KONSTANTINIDIS, K. T. Direct
comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial
community DNA sample. PloS ONE, v. 7, n. 2, p. e30087, 2012.
MA, Z. S.; GENG, J.; ABDO, Z.; FORNEY, L. J. A Bird’s Eye View of Microbial
Community Dynamics. In: Microbial Ecological Theory. Eds: OGILVIE, L. A.; HIRSCH, P.
R. Caister Academic Press, Norfolk, UK, 2012.
MAIA, L. P.; JIMENEZ, J.; SERRA, J.; MORAIS, J. O. The Coastline of Fortaleza City. A
product of environmental impacts caused by the Mucuripe Harbor. Arquivo de Ciências do
Mar , v. 31, n. 1-2, p. 93-100, 1998.
MALIK, S.; BEER, M.; MEGHARAJ, M.; NAIDU, R. The use of molecular techniques to
characterize the microbial communities in contaminated soil and water. Environment
International , v. 34, n. 2, p. 265–76, 2008.
MARCHESI, J. R.; SATO, T.; WEIGHTMAN, A. J.; MARTIN, T. A.; FRY, J. C.; HIAM, S.
J.; DYMNOCK, D.; WADE, W. G. Design and evaluation of useful bacterium-specific PCR
primers that amplify genes coding for bacterial 16S rRNA. Applied and Environmental
Microbiology, v.64, p.795-799, 1998.
MARKOWITZ, V. M.; IVANOVA, N. N.; SZETO, E.; PALANIAPPAN, K.; CHU, K.;
DALEVI, D.; CHEN, I. A.; GRECHKIN, Y.; DUBCHAK, I.; ANDERSON, I.; LYKIDIS, A.;
MAVROMATIS, K.; HUGENHOLTZ, P.; KYRPIDES, N. C. IMG/M: a data management
and analysis system for metagenomes. Nucleic Acids Research, v. 36, n. Database issue, p.
D534–8, 2008.

146
MARTINY, J. B. H.; EISEN, J. A.; PENN, K.; ALLISON, S. D.; HORNER-DEVINE, M. C.
Drivers of bacterial β -diversity depend on spatial scale. Proceedings of the National
Academy of Science, v. 108, n. 19, p. 7850 - 7854, 2011.
MAXAM, A. M.; GILBERT, W. A new method for sequencing DNA. Proceedings of the
National Academy of Science, v. 74, p. 560-564, 1977.
MCGRODDY, S. E.; FARRINGTON, J. W.; GSCHWEND, P. M. Comparison of the in situ
and desorption sediment-water partitioning of polycyclic aromatic hydrocarbons and
polychlorinated biphenyls. Environmental Science & Technology, v. 30, n. 1, p. 172-177,
1995.
MCLEOD, E.; CHMURA, G. L.; BOUILLON, S.; SALM, R.; BJÖRK, M.; DUARTE, C. M.;
LOVELOCK, C. E.; SCHLESINGER, W. H.; SILLIMAN, B. R. A blueprint for blue carbon :
toward an improved understanding of the role of vegetated coastal habitats in sequestering
CO2 . Frontiers in Ecology and Environment, 2011.
MENDES, L. W.; KURAMAE, E. E.; NAVARRETE, A. A; VEEN, J. A VAN; TSAI, S. M.
Taxonomical and functional microbial community selection in soybean rhizosphere. The
ISME Journal, p. 1–11, 2014.
METZKER, M. L. Sequencing technologies - the next generation. Nature Reviews Genetics,
v. 11, n. 1, p. 31–46, 2010.
MEYER, F.; PAARMANN, D.; D’SOUZA, M.; OLSON, R.; GLASS, E. M.; KUBAL, M.;
PACZIAN, T.; RODRIGUEZ, A.; STEVENS, R.; WILKE, A.; WILKENING, J.; EDWARDS,
R. A. The metagenomics RAST server - a public resource for the automatic phylogenetic and
functional analysis of metagenomes. BMC Bioinformatics, v. 9, p. 386, 2008.
MILOSLAVICH, P.; KLEIN, E.; DÍAZ, J. M.; et al. Marine biodiversity in the Atlantic and
Pacific coasts of South America: knowledge and gaps. PloS ONE, v. 6, n. 1, p. e14631, 2011.
MITRA, S.; KLAR, B.; HUSON, D. H. Visual and statistical comparison of
metagenomes. Bioinformatics, v. 25, n. 15, p. 1849–55, 2009.
MIYATAKE, T.; MACGREGOR, B. J.; BOSCHKER, H. T. S. Linking microbial community
function to phylogeny of sulfate-reducing Deltaproteobacteria in marine sediments by
combining stable isotope probing with magnetic-bead capture hybridization of 16S
rRNA. Applied and Environmental Microbiology, v. 75, n. 15, p. 4927–35, 2009.
MOXLEY, K.; SCHMIDT, S. Isolation of a phenol-utilizing marine bacterium from Durban
Harbour (South Africa) and its preliminary characterization as Marinobacter sp. KM2. Water
Science and Technology, v. 65, n. 5, p. 932-939, 2012.
MUSAT, N.; WERNER, U.; KNITTEL, K.; et al. Microbial community structure of sandy
intertidal sediments in the North Sea, Sylt-Rømø Basin, Wadden Sea. Systematic and
Applied Microbiology, v. 29, n. 4, p. 333–48, 2006.
MUYZER, G.; DE WAAL, E. C.; UITTERLINDEN, A. G. Profiling of Complex Microbial
Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain
Reaction-Amplified Genes Coding 16S rRNA. Applied and Environmental Microbiology,
v. 59, n. 3, p. 695-700, 1993.

147
MUYZER, G.; SMALLA, K. Application of denaturing gradient gel electrophoresis (DGGE)
and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van
Leeuwenhoek, v. 73, n. 1, p. 127–41, 1998.
NAKATSU, C. H. Soil microbial community analysis using denaturing gradient gel
electrophoresis. Soil Science Society of America Journal, v. 71, p. 562-571, 2007.
NASCIMENTO, F. S.; FREIRE, G. S. S; MIOLA, B. Geochemistry of Marine Sediments of
the Brazilian Northeastern Continental Shelf. Brazilian Journal of Oceanography, v. 58, p.
1-11, 2010.
NIPPER, M. Current approaches and future directions for contaminant-related impact
assessments in coastal environments: Brazilian perspective. Aquatic Ecosystem Health &
Management , v. 3, n. 4, p. 433–447, 2000.
NOGALES, B.; LANFRANCONI, M. P.; PIÑA-VILLALONGA, J. M.; BOSCH, R.
Anthropogenic perturbations in marine microbial communities. FEMS Microbiology
Reviews , v. 35, n. 2, p. 275–98, 2011.
OECD - The Organisation for Economic Co-operation and Development. Environmental
Impacts of International Shipping: The Role of Ports, OECD Publishing, 2011.
OLIVEIRA, M. M. N. Aspectos morfológicos e sedimentares associados à dinâmica do
litoral oeste de Icapuí, Ceará. 2012. 188 f. Dissertação (Mestrado em Ciências Marinhas
Tropicais) – Instituto de Ciências do Mar, Universidade Federal do Ceará, Fortaleza, 2012.
OLIVER, A.; LILLEY, A. K.; VAN DER GAST, C. J. Species-Time Relationships for
Bacteria. In: Microbial Ecological Theory. Eds: OGILVIE, L. A.; HIRSCH, P. R. Caister
Academic Press, Norfolk, UK, 2012.
ORCUTT, B. N.; SYLVAN, J. B.; KNAB, N. J.; EDWARDS, K. J. Microbial ecology of the
dark ocean above, at, and below the seafloor. Microbiology and Molecular Biology
Reviews , v. 75, n. 2, p. 361–422, 2011.
OVERBEEK, R.; BEGLEY, T.; BUTLER, R. M.; CHOUDHURI, J. V.; CHUANG, H-Y.;
COHOON, M.; CRÉCY-LAGARD, V.; DIAZ, N.; DISZ, T.; EDWARS, R.; FONSTEIN, M.;
FRANK, E. D.; GERDES, S.; GLASS, E. M.; GOESMANN, A.; HANSON, A.; REUYL,
D.I.; JENSEN, R.; JAMSHIDI, N.; KRAUSE, L.; KUBAL, M.; LARSEN, N.; LINKE, B.;
MCHARDY. A.C.; MEYER, F.; NEUWEGER, H.; OLSEN, G.; OLSON, R.; OSTERMAN,
A.; PORTNOY, V.; PUSCH, G. D.; RODIONOV, D. A.; RÜCKERT, C.; STEINER, J.;
STEVENS, R.; THIELE, I.; VASSIEVA, O.; YE, Y.; ZAGNITKO, O.; VONSTEIN, V. The
subsystems approach to genome annotation and its use in the project to annotate 1000
genomes. Nucleic Acids Research, v. 33, n. 17, p. 5691–702, 2005.
OVREÅS, L.; FORNEY, L.; DAAE, F. L.; TORSVIK, V. Distribution of bacterioplankton in
meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of
PCR-amplified gene fragments coding for 16S rRNA. Applied and Environmental
Microbiology, v. 63, n. 9, p. 3367–73, 1997.
PACE, N. R.; STAHL, D. A.; LANE, D. J.; OLSEN, G. J. Analyzing natural microbial
populations by rRNA sequences. ASM News, v. 51, p. 4-12, 1985.

148
PAISSÉ, S.; COULON, F.; GOÑI-URRIZA, M.; et al. Structure of bacterial communities
along a hydrocarbon contamination gradient in a coastal sediment. FEMS Microbiology
Ecology , v. 66, n. 2, p. 295–305, 2008.
PAN, L.; REN, J.; LIU, J. Effects of benzo(k)fluoranthene exposure on the biomarkers of
scallop Chlamys farreri. Comparative Biochemistry and Physiology. Toxicology &
Pharmacology , v. 141, n. 3, p. 248–56, 2005.
PARKS, D. H.; BEIKO, R. G. Identifying biologically relevant differences between
metagenomic communities. Bioinformatics, v. 26, n. 6, p. 715–21, 2010.
PEDRÓS-ALIÓ, C. Marine microbial diversity: can it be determined? Trends in
Microbiology, v. 14, n. 6, p. 257–63, 2006.
PETROSINO, J. F.; HIGHLANDER, S.; LUNA, R. A.; GIBBS, R. A.; VERSALOVIC, J.
Metagenomic Pyrosequencing and Microbial Identification. Clinical Chemistry, v. 55, n. 5,
p. 856–866, 2009.
POMMIER, T.; NEAL, P.; GASOL, J.; MONTSERRAT, C.; ACINAS, S. G.; PEDRÓS-
ALIÓ, C. Spatial patterns of bacterial richness and evenness in the NW Mediterranean Sea
explored by pyrosequencing of the 16S rRNA. Aquatic Microbial Ecology, v. 61, n. 3, p.
221–233, 2010.
PORT, J. A; WALLACE, J. C.; GRIFFITH, W. C.; FAUSTMAN, E. M. Metagenomic
profiling of microbial composition and antibiotic resistance determinants in Puget
Sound. PloS ONE , v. 7, n. 10, p. e48000, 2012.
PROSSER, J. I. Replicate or lie. Environmental Microbiology, v. 12, n. 7, p. 1806-1810,
2010.
PRUITT, K. D.; TATUSOVA, T.; KLIMKE, W.; MAGLOTT, D. R. NCBI Reference
Sequences: current status, policy and new iniatiaves. Nucleic Acids Research, v. 37, n. 1, p.
D32-D36, 2009.
QUAIL, M A.; SMITH, M.; COUPLAND, P.; OTTO, T. D.; HARRIS, S. R.; CONNOR, T.
R.; BERTONI, A.; SWERDLOW, H. P.; GU, Y. A tale of three next generation sequencing
platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq
sequencers. BMC Genomics , v. 341, p. 1-13, 2012.
QUINCE, C.; LANZÉN, A.; CURTIS, T. P.; DAVENPORT, R. J.; HALL, N.; HEAD, I. M.;
READ, L.F., SLOAN, W. T. Accurate determination of microbial diversity from 454
pyrosequencing data. Nature Methods, v. 6, n. 9, p. 639–41, 2009.
ROH, S. W.; KIM, K.-H.; NAM, Y.-D.; et al. Investigation of archaeal and bacterial diversity
in fermented seafood using barcoded pyrosequencing. The ISME Journal, v. 4, n. 1, p. 1–16,
2010.
SALEH-LAKHA, S.; MILLER, M.; CAMPBELL, R. G.; SCHNEIDER, K.;
ELAHIMANESH, P.; HART, M. M.; TREVORS, J. T. Microbial gene expression in soil:
methods, applications and challenges. Journal of Microbiological Methods, v. 63, n. 1, p. 1–
19, 2005.

149
SÁNCHEZ, O.; FERRERA, I.; VIGUÉS, N.; DE OTEYZA, T. G.; GRIMALT, J.; MAS, J.
Role of cyanobacteria in oil degradation by microbial mats. International Biodeterioration &
Biodegradation, v. 58, p. 186-195, 2006.
SANGER, F.; NICKLEN, S.; COULSON, A. R. DNA sequencing with chain-terminating
inhibitors. Proceedings of the National Academy of Science, v. 74, p. 5463-5467, 1977.
SCHAUER, R.; BIENHOLD, C.; RAMETTE, A.; HARDER, J. Bacterial diversity and
biogeography in deep-sea surface sediments of the South Atlantic Ocean. The ISME
Journal , v. 4, n. 2, p. 159–70, 2010.
SCHNEIDER, T.; RIEDEL, K. Environmental proteomics: analysis of structure and function
of microbial communities. Proteomics, v. 10, p. 785-798, 2010.
SCHULTE, E. E.; HOPKINS, B. G. Estimation of sediment organic matter by weight loss-on
ignition. In: Magdoff FR, Tabatabai MA, Hanlon EA, editors. Sediment Organic Matter:
Analysis and Interpretation. Sediment. Madison: Science Society of America; 1995. p. 21–
31.
SCHWIEGER, F.; TEBBE, C. C. A new approach to utilize PCR-single-strand-conformation
polymorphism for 16S rRNA gene-based microbial community analysis. Applied and
Environmental Microbiology, v. 64, n. 12, p. 4870–6, 1998.
SHADE, A.; HANDELSMAN, J. Beyond the Venn diagram: the hunt for a core
microbiome. Environmental Microbiology, v. 14, n. 1, p. 4–12, 2012.
SHYU, C.; SOULE, T.; BENT, S. J.; FOSTER, J. A; FORNEY, L. J. MiCA: a web-based tool
for the analysis of microbial communities based on terminal-restriction fragment length
polymorphisms of 16S and 18S rRNA genes. Microbial Ecology, v. 53, n. 4, p. 562–70,
2007.
WILSON, M. V.; SHMIDA, A. Measuring beta diversity with presence-absence data. Journal
of Ecology, n. 72, v. 3, p. 1055-1062, 1984.
SIMON, C.; DANIEL, R. Metagenomic analyses: past and future trends. Applied and
Environmental Microbiology, v. 77, n. 4, p. 1153–61, 2011.
SLATER, G. F.; COWIE, B. R.; HARPER, N.; DROPPO, I. G. Variation in PAH inputs and
microbial community in surface sediments of Hamilton Harbour: implications to remediation
and monitoring. Environmental Pollution, v. 153, n. 1, p. 60–70, 2008.
SOREK, R.; COSSART, P. Prokaryotic transcriptomics: a new view on regulation, physiology
and pathogenicity. Nature Reviews Genetics, v. 11, p. 9-16, 2010.
SPALDING, M. D.; FOX, H. E.; ALLEN, G. R.; DAVIDSON, N.; FERDAÑA, Z. A.;
FINLAYSON, M.; HALPERN, B. S.; JORGE, M. A.; LOMBANA, A.; LOURIE, S. .A;
MARTIN, K. D.; EDMUND, M.; MOLNAR, J.; RECCHIA, C. A.; ROBERTSON, J.. Marine
Ecoregions of the World : A Bioregionalization of Coastal and Shelf Areas. BioScience , v. 57,
n. 7, p. 573–583, 2007.

150
SPIEGELMAN, D.; WHISSEL, G.; GREER, C. W. A survey of the methods for the
characterization of microbial consortia and communities. Canadian Journal of
Microbiology , v. 51, p. 355-386, 2005.
STALEY, J. T.; KONOPKA, A. Measurement of in situ activities of nonphotosynthetic
microorganisms in aquatic and terrestrial habitats. Annual Review of Microbiology , v. 39, p.
321-346, 1985.
SU, C.; LEI, L.; DUAN, Y.; ZHANG, K.-Q.; YANG, J. Culture-independent methods for
studying environmental microorganisms: methods, application, and perspective. Applied
Microbiology and Biotechnology, v. 93, n. 3, p. 993–1003, 2012.
SUGUIO K. Introdução à sedimentologia. São Paulo: Edgard Blücher; 1973.
SUN, S.; CHEN, J.; LI, W.; ALTINTAS, I.; LIN, A.; PELTIER, S.; STOCKS, K.; ALLEN E.
E.; ELLISMAN, M.; GRETHE, J.; WOOLEY, J. Community cyberinfrastructure for
Advanced Microbial Ecology Research and Analysis: the CAMERA resource. Nucleic Acids
Research , v. 39, n. Database issue, p. D546–51, 2011.
TESKE, A.; SØRENSEN, K. B. Uncultured archaea in deep marine subsurface sediments:
have we caught them all? The ISME Journal, v. 2, n. 1, p. 3–18, 2008.
THIES, J. E. Molecular Mechanisms of Plant and Microbe Coexistence. (C. S. Nautiyal & P.
Dion, Eds.), Soil Biology, v. 15, p. 411–436, 2008. Berlin, Heidelberg: Springer Berlin
Heidelberg.
THOMAS, T.; GILBERT, J.; MEYER, F. Metagenomics - a guide from sampling to data
analysis. Microbial Informatics and Experimentation, v. 2, n. 1, p. 3, 2012.
TOMARU, A.; KAWACHI, M.; DEMURA, M.; FUKUYO, Y. Denaturing gradient gel
electrophoresis shows that bacterial communities change with mid-ocean ballast water
exchange. Marine Pollution Bulletin, v. 60, n. 2, p. 299–302, 2010.
TORSVIK, V.; ØVREÅS, L. Microbial diversity and function in soil: from genes to
ecosystems. Current Opinion in Microbiology, v. 5, n. 3, p. 240–5, 2002.
TOWNSEND, G. T.; PRINCE, R. C.; SUFLITA, J. M. Anaerobic oxidation of crude oil
hydrocarbons by the resident microorganisms of a contaminated anoxic
aquifer. Environmental Science & Technology, v. 37, p. 5213–5218, 2003.
TROZZI, C.; VACCARO, R. Environmental impact of port activities. Maritime Engineering
and Ports II, p 151-161. Editors Brebbia, C. A.; Olivella, J., 2000. ISBN 1-85312-829-5.
TYSON, G. W.; CHAPMAN, J.; HUGENHOLTZ, P.; ALLEN, E. E.; RAML, R. J.;
RICHARDSON, P. M.; SOLOVYEV, V. V.; RUBIN, E. M.; ROKHSAR, D. S.; BANFIELD,
J. F. Community structure and metabolism through reconstruction of microbial genomes from
the environment. Nature, v. 428, n. 6978, p. 37–43, 2004.
VAN DER GAST, C. J.; AGER, D.; LILLEY, A. K. Temporal scaling of bacteria taxa is
influenced by both stochastic and deterministic ecological factors. Environmental
Microbiology, n. 10, p. 1411-1418, 2008.

151
VENTER, J. C.; REMINGTON, K.; HEIDELBERG, J. F.; HALPERN, A.L.; RUSCH, D.;
EISEN, J. A.; WU, D.; PAULSEN, I.; NELSON, K. E., NELSON, W.; FOUTS, D. E.; LEVY,
S.; KNAP, A. H.; LOMAS, M. W.; NEALSON, K.; WHITE, O.; PETERSON, J.;
HOFFMAN, J.; PARSONS, R.; BADEN-TILLSON, H.; PFANNKOCH, C.; ROGER, Y. H.;
SMITH, H. O. Environmental genome shotgun sequencing of the Sargasso Sea. Science, v.
304, n. 5667, p. 66–74, 2004.
WANG, G.; DONG, J.; LI, X.; SUN, H. The bacterial diversity in surface sediment from the
South China Sea. Acta Oceanologica Sinica, v. 29, n. 4, p. 98–105, 2010.
WANG, Y. F.; TAM, N. F. Y. Microbial community dynamics and biodegradation of
polycyclic aromatic hydrocarbons in polluted marine sediments in Hong Kong. Marine
Pollution Bulletin , v. 63, n. 5-12, p. 424–30, 2011.
WHITE, J. R.; NAGARAJAN, N.; POP, M. Statistical methods for detecting differentially
abundant features in clinical metagenomic samples. PLoS Computational Biology, v. 5, n. 4,
p. e1000352, 2009.
WHITTAKER, R. H. Vegetation of the Siskiyou Mountains, Oregon and California.
Ecological Monographs, n. 30, p. 279-338, 1960.
WIDDEL, F.; RABUS, R. Anaerobic biodegradation of saturated and aromatic
hydrocarbons. Current Opinion in Biotechnology, v. 12, n. 3, p. 259–76, 2001.
WILKE, A.; HARRISON, T.; WILKENING, J.; FIELD, D.; GLASS, E. M.; KYRPIDES, N.;
MAVROMMATIS, K.; MEYER, F. The M5nr: a novel non-redundant database containing
protein sequences and annotations from multiple sources and associated tools. BMC
Bioinformatics , v. 13, n. 1, p. 141, 2012.
WILMES, P.; BOND, P. L. The application of two-dimensional polyacrylamide gel
electrophoresis and downstream analysis to a mixed community of prokaryotic
microorganisms. Environmental Microbiology, v. 6, p. 911-920, 2004.
WOESE, C. R. Bacterial Evolution. Microbiological Reviews, v. 51, n. 2, 1987.
WOOLEY, J. C.; GODZIK, A.; FRIEDBERG, I. A primer on metagenomics. PLoS
Computational Biology, v. 6, n. 2, p. e1000667, 2010.
XIONG, J.; YE, X.; WANG, K.; et al. Biogeography of the sediment bacterial community
responds to a nitrogen pollution gradient in the East China Sea. Applied and Environmental
Microbiology, v. 80, n. 6, p. 1919–25, 2014.
YILMAZ, P.; KOTTMANN, R.; FIELD, D.; et al. Minimum information about a marker gene
sequence (MIMARKS) and minimum information about any (x) sequence (MIxS)
specifications. Nature Biotechnology, v. 29, n. 5, p. 415–20, 2011.
ZHOU, J. Z.; THOMPSON, D. K. Challenges in applying microarrays to environmental
studies. Current Opinion in Biotechnology, v. 13, p. 204-207, 2002.
ZHOU, J.; BRUNS, M. A.; TIEDJE, J. M. DNA recovery from soils of diverse
composition. Applied Environmental Microbiology, v. 62, n. 2, p. 316-322, 1996.

152
ZINGER, L.; AMARAL-ZETTLER, L. A; FUHRMAN, J. A; HORNER-DEVINE, M. C.;
HUSE, S. M.; WELCH, D. B. M.; MARTINY, J. B. H.; SOGIN, M.; BOETIUS, A.;
RAMETTE, A. Global patterns of bacterial beta-diversity in seafloor and seawater
ecosystems. PloS ONE, v. 6, n. 9, p. e24570, 2011.
ZINGER, L.; BOETIUS, A; RAMETTE, A. Bacterial taxa-area and distance-decay
relationships in marine environments. Molecular Ecology, v. 23, n. 4, p. 954–64, 2014.
ZWIRGLMAIER K.; JARDILLIER, L.; OSTROWSKI, M.; MAZARD, S.; GARCZAREK,
L.; VAULOT, D.; NOT, F.; MASSANA, R.; ULLOA, O.; SCANLAN, D. J. Global
phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning
of lineages among oceanic biomes. Environmental Microbiology, v. 10, n. 1, p. 147-161,
2008.