sÍntese e reatividade de novos calcogenofenos via reaÇÕes de...
TRANSCRIPT

1
UNIVERSIDADE FEDERAL DE SANTA MARIA CENTRO DE CIÊNCIAS NATURAIS E EXATAS
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
SÍNTESE E REATIVIDADE DE NOVOS CALCOGENOFENOS VIA REAÇÕES DE
CICLIZAÇÃO ___________________
Dissertação de Mestrado
ANDRÉ LUIZ AGNES STEIN
PPGQ
Santa Maria, RS, Brasil
2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
SÍNTESE E REATIVIDADE DE NOVOS CALCOGENOFENOS VIA REAÇÕES DE CICLIZAÇÃO
Por
ANDRÉ LUIZ AGNES STEIN
Dissertação apresentada no Programa de Pós-Graduação em Química,
Área de Concentração em Química Orgânica, da Universidade Federal
de Santa Maria (RS), como requisito parcial para a obtenção do grau de
Mestre em Química
PPGQ
Santa Maria, RS, Brasil
2010

3
UNIVERSIDADE FEDERAL DE SANTA MARIA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
A COMISSÃO EXAMINADORA ABAIXO ASSINADA, APROVA A DISSERTAÇÃO
SÍNTESE E REATIVIDADE DE NOVOS CALCOGENOFENOS VIA REAÇÕES DE
CICLIZAÇÃO
ELABORADA POR:
André Luiz Agnes Stein
COM REQUISITO PARCIAL PARA A OBTENÇÃO DO GRAU DE MESTRE EM
QUÍMICA
COMISSÃO EXAMINADORA
Prof. Dr. Gilson Zeni – Orientador – UFSM
Prof. Dr. Paulo Henrique Schneider – UFRGS
Prof. Dr. Luciano Dornelles – UFSM
Santa Maria, 23 de Fevereiro de 2010.

4
A minha família, pessoas fundamentais em minha vida
que tiveram paciência e jamais mediram esforços para
me proporcionar uma educação de qualidade. Este
trabalho é dedicado a vocês.
iv

5
Ao Prof. Gilson, meus sinceros agradecimentos
pela orientação desde a Iniciação Científica. Fica aqui expressado
o meu reconhecimento pelos conhecimentos transmitidos e pelo
companheirismo durante esses anos.
v

6
AGRADECIMENTOS
Aos antigos: Rodrigo Panatieri, Jesus, Angélica, Olga, Patrícia, Joel,
Elvis, Twana, Diego e atuais: Alisson, Anderson, Zé Neto, Juliano, Helton,
Ricardo Schumacher, Benhur, Flávia, Caroline, Daniela, Adriane, colegas,
amigos e irmãos de Laboratório, deixo o meu muito obrigado pela amizade,
paciência, parceria, conversa e apoio em todos os momentos.
Aos colegas do laboratório da Prof. Cristina, desde os primeiros tempos
de Iniciação Científica, pela amizade, companheirismo, cervejadas,
coloborações e convivência diária como “vizinhos de porta”.
Aos amigos do Laboratório do Prof. Braga, antigos e novos, pela
amizade e companheirismo. Aos Colegas e ex-colegas dos laboratórios dos
professores Cláudio, João Batista, Oscar e Luciano pela amizade e
convivência.
A Prof. Cristina Nogueira, um agradecimento especial, pela amizade,
paciência com todas as brincadeiras, caronas, e colaboração em todas as
etapas de realização deste trabalho.
A Juliana, minha namorada, amiga, professora de português,
companheira em todas as horas. Um agradecimento especial pelo carinho,
compreensão nos momentos difíceis e paciência. Essa conquista também é
sua.
Aos professores e funcionários do curso de Pós-graduação em Química
pela colaboração e atenção prontamente dispensadas durante a realização
deste trabalho.
Aos Cirilo e Cabelo, pessoas com quem morei durante a graduação,
pela amizade, ajuda, façanhas e consideração demonstradas durantes todos
estes anos de convivência.
vi

7
A todos meus demais amigos, pessoas fundamentais, pela amizade,
humildade, risadas, festas, conversas e incríveis composições nas “álvores”.
Papa Capim, Gordo, Marlão, Bóris, Cirilome, Cabelo, Gandus, Cléo, Willian,
Tiagão, Boliviano, Boss, Galetto, Faoro, Carminaire, Sika, Carlota, Adri, Cechin,
Tatu, Ander...a todos só digo uma coisa: “oooooohhhhhhhh manecoo, o bolivia
ta mal cara”.
Aos funcionários, Ademir e Valéria pela amizade e trabalho eficiente
frente à Coordenação do PPGQ.
Às agencias financiadoras FAPERGS, CNPq e CAPES, pelas bolsas e
auxílios concedidos.
vii

8
RESUMO
Título: Síntese e Reatividade de Novos Calcogenofenos via Reações de Ciclização
Autor: André Luiz Agnes Stein
Orientador: Prof. Dr. Gilson Zeni
Na primeira etapa desse trabalho, desenvolveu-se uma metodologia de
obtenção de derivados de calcogenofenos funcionalizados na posição 3 do
anel heteroaromático 2, via reações de ciclização de (Z)-calcogenoeninos 1,
catalisadas por cobre. Utilizando-se este protocolo, uma série de (Z)-seleno- e
teluroeninos 1 foi preparada através de reações de hidroselenação e
hidroteluração, sendo estes posteriormente ciclizados com dicalcogenetos de
diorganoíla, catalisados por CuI, fornecendo os respectivos produtos em
rendimentos de moderados a excelentes.
Em seguida, a reatividade dos calcogenofenos obtidos foi testada frente
as reações de acoplamento cruzado do tipo Suzuki, catalisadas por paládio,
com ácidos arilborônicos e ariltrifluoroboratos fornecendo os 3-
arilcalcogenofenos em bons rendimentos.
Na última etapa desse trabalho, são apresentados os resultados das
reações de ciclização eletrofílica dos 2-organocalcogeno-3-alquiniltiofenos 6
com diferentes eletrófilos, tais como I2, ICl, e PhSeBr. As reações de ciclização
procederam-se em condições brandas, resultando nos respectivos heterocíclos
fundidos 4-iodo-calcogenofeno[2,3-b]tiofenos 7 em bons rendimentos.
viii

9
.
Uma vez obtidos os calcogenofenos 7, a reatividade dos mesmos foi
testada frente as reações de acoplamento cruzado do tipo Suzuki e Negishi,
catalisadas por paládio, e acoplamento cruzado com tióis, catalisadas por
cobre.
.
UNIVERSIDADE FEDERAL DE SANTA MARIA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
Dissertação de Mestrado
Santa Maria, Fevereiro de 2010
ix

10
ABSTRACT
Title: Synthesis and Reactivity of New Chalcogenophenes by Cyclization Reactions
Author: André Luiz Agnes Stein
Academic Advisor: Prof. Dr. Gilson Zeni
In the first step of this study, we present our results on the efficient
copper-catalyzed cyclization reactions of chalcogenoenynes 1 and established
a route to obtain 3-substituted chalcogenophenes 2. A range of (Z)-seleno- and
telluroenines 1 were synthesized by hydroselenation and hydrotelluration
reactions, which in a second step were cyclized by reaction with diorganoyl
dichalcogenides catalyzed by CuI furnishing the corresponding products in
moderate to excellent yields. In addition, the obtained chalcogenophenes were
readily transformed to more complex products using the palladium-catalyzed
cross-coupling reactions with boronic acids and aryltrifluoroborates to give
Suzuki-type products in good yields.
In the last stage of this study, we present our results on the electrophilic
cyclization reaction of 2-organochalcogen-3-alkynylthiophenes 6 with different
electrophiles such as I2, ICl, and PhSeBr. The cyclization reaction proceeded
cleanly under mild reaction conditions giving the fused heterocycles 4-iodo-
selenophene[2,3-b]thiophenes 7 in good yields. The application of these
compounds in the palladium- or copper-catalyzed cross-coupling reactions with
thiols, boronic acids, and organozinc reagents was also presented.
x

11
S YR1E+, CH2Cl2, r.t.
R, R1 = alkyl, aryl; Y = Se, Te; E+ = I2, ICl, PhSeBr
YS
E
R
6 7
R
38-86%
UNIVERSIDADE FEDERAL DE SANTA MARIA
PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA
Master degree dissertation in Chemistry
Santa Maria, February 2010
xi

12
ÍNDICE
Agradecimentos......................................................................................
Resumo..................................................................................................
Abstract..................................................................................................
Lista de Tabelas......................................................................................
vi
viii
x
xv
Lista de Figuras....................................................................................... xvi
Lista de Siglas, Abreviaturas e Símbolos................................................ xvii
Introdução e Objetivos............................................................................. 2
Capítulo 1: Revisão da Literatura........................................................
1.1 Compostos Heterocíclicos e heteroaromáticos.................................
1.2 Reações de Ciclização Eletrofílica....................................................
1.3 Síntese e Reatividade de selenofenos e telurofenos........................
1.4 Reações de Ciclização Catalisadas por Cobre.................................
Capítulo 2: Apresentação e Discussão dos Resultados...................
8
8
10
18
25
32
2.1 Reações de ciclização de (Z)-calcogenoeninos catalisadas por
Cobre.......................................................................................................
32
2.1.1 Síntese de 3-ariltelurofenos via reações de acoplamento
cruzado do tipo Suzuki............................................................................
43
2.2 Síntese de 4-iodoselenofenos[2,3-b]tiofenos, via reações de
ciclização eletrofílica de 2-organocalcogeno-3- alquiniltiofenos..............
45
2.2.1 Aplicações do composto 7a em reações de acoplamento
cruzado catalisadas por sais de paládio e cobre.....................................
55
2.2.1.1 Reação de acoplamento cruzado do composto 7a com p-
cloro-benzenotiol catalisada por CuI.......................................................
55
2.2.1.2 Reação de acoplamento cruzado do tipo Suzuki do
composto 7a com o ácido p-bromofenilborônico.....................................
56
2.2.1.3 Procedimento geral para a reação de acoplamento
cruzado do tipo Negishi do composto 7a com cloreto de p-toluilzinco...
57
Considerações Finais e Conclusões................................................... 60
xii

13
Capítulo 3: Parte Experimental............................................................ 63
3.1. Materiais e Métodos......................................................................... 63
3.1.1. Espectroscopia de Ressonância Magnética Nuclear.......... 63
3.1.2. Espectrometria de Massas.................................................. 63
3.1.3. Análise Elementar............................................................... 63
3.1.4 Rota-evaporadores............................................................. 64
3.1.5 Solventes e Reagentes...................................................... 64
3.2 Procedimentos Experimentais........................................................... 65
3.2.1 Preparação do Pd(PPh3)4................................................... 65
3.2.2 Preparação do PdCl2(PPh3)2.............................................. 65
3.2.3 Procedimento geral para a preparação de disselenetos e
de diorganoila..........................................................................................
66
3.2.4 Procedimento geral para a preparação de disseleneto de
dipiridina..................................................................................................
66
3.2.5 Procedimento geral para a preparação de disseleneto e
ditelureto de dibutila................................................................................
67
3.2.6 Procedimento geral para preparação dos (Z)-teluroeninos
1a-d.........................................................................................................
67
3.2.7 Procedimento geral para preparação dos (Z)-
selenoeninos 3a e 3c..............................................................................
68
3.2.8 Obtenção do (Z)-1-(n-Butilseleno)-4-fenil-but-1-en-3-ino
(3b)..........................................................................................................
68
3.2.9 Procedimento geral para preparação dos telurofenos....... 69
3.2.10 Procedimento geral para preparação dos selenofenos..... 73
3.2.11 Procedimento geral para a reação de acoplamento
cruzado do telurofeno 2o com ácidos borônicos.....................................
78
3.2.12 Procedimento geral para a reação de acoplamento
cruzado do telurofeno 2o com ariltrifluoroboratos...................................
78
3.2.13 Procedimento geral para preparação dos 2-alquilselanil-
3-alquiniltiofenos 6a-l, 6p-q.....................................................................
79
3.2.14 Procedimento geral para preparação do 3-(feniletinil)-2-
(fenilselenil)tiofeno 6r..............................................................................
84
3.2.15 Procedimento geral para preparação do 2-(butiltelanil)-
3-(feniletinil)tiofeno 6o.............................................................................
85
xiii

14
3.2.16 Procedimento geral para as reações de ciclização
eletrofílica................................................................................................
85
3.2.17 Procedimento geral para a reação de acoplamento
cruzado do composto 7a com p-cloro-benzenotiol via catálise de CuI...
89
3.2.18 Procedimento geral para a reação de acoplamento
cruzado do tipo Suzuki do composto 7a com o ácido p-
bromofenilborônico..................................................................................
90
3.2.19 Procedimento geral para a reação de acoplamento
cruzado do tipo Negishi do composto 7a com cloreto de p-toluilzinco...
Referências Bibliográficas...................................................................
91
93
Capítulo 4: Espectros Selecionados................................................... 102
Anexos.................................................................................................... 154
xiv

15
LISTA DE TABELAS
Tabela 1 - Influência do grupamento R ligado ao telúrio nas
reações de ciclização.....................................................
36
Tabela 2 - Reação de ciclização de (Z)-teluroeninos catalisadas
por CuI com diferentes dicalcogenetos de diorganoíla..
37
Tabela 3 - Reações de ciclização de (Z)-selenoeninos com
diferentes dicalcogentetos de diorganoíla......................
39
Tabela 4 - Estudo de solventes nas reações de ciclização do
composto 6a com I2.......................................................
48
Tabela 5 - Influência do grupamento R1 ligado as selênio nas
reações de ciclização.....................................................
49
Tabela 6 - Reações de ciclização com diferentes eletrófilos ou 2-
organocalcogeno-3-alquiniltiofenos...............................
51
xv

16
LISTA DE FIGURAS
Figura 1 - Figura 2 - Figura 3 -
Fármacos contendo unidade heterocíclica.................. 8
Exemplos de calcogenofenos.................................... 9
Estrutura geral do calcogenofeno 2............................ 33
Figura 4 - Estrutura geral do calcogenofeno fundido 7................... 46
xvi

17
LISTA DE SIGLAS, ABREVIATURAS E SÍMBOLOS
acac – acetilacetonato
Ar – Arila
DMF – N,N-Dimetilformamida
NBS – N-bromosuccinimida
NIS – N-iodosuccinimida
LHMDS – Hexametildisililazida de lítio
m-CPBA – Ácido metacloroperbenzóico
TBAF – Fluoreto de tetrabutil amônio
DMAP – Dimetilaminopiridina
dba – Dibenzilideno acetona
DBN – Diazabiciclo[3.4.0]-noneno
TMEDA – N,N,N,N-Tetrametiletilenodiamina
RMN 13C –Ressonância Magnética Nuclear de Carbono Treze
RMN 1H – Ressonância Magnética Nuclear de Hidrogênio
t. a. –Temperatura Ambiente
TMS – Tetrametilsilano
DBN – Diazabiciclo[3.4.0]-noneno
LHMDS – Hexametildisililazida de lítio
DME - 1,2-Dimetoxietano
xvii

18
Introdução e Objetivos

19
A síntese de compostos heterocíclicos altamente funcionalizados é um
importante alvo em Química Orgânica, sendo altamente explorada pelo fato de
muitos compostos biologicamente ativos que imitam produtos naturais serem
heterocíclicos. Esses compostos são de grande interesse em diversas áreas,
tais como biologia, farmacologia, óptica e eletrônica. De maneira geral, a
importância dos compostos heterocíclicos sintéticos vêm crescendo
exponencialmente, apresentando enormes aplicações farmacêuticas,
agroquímicas, entre outras. Um dado interessante é que 85% dos fármacos
disponíveis na medicina moderna são de origem sintética. Destes, 62% são
heterocíclicos, sendo que 91% contém nitrogênio, 24% enxofre e 16,5%
oxigênio em seu núcleo base.1
Assim, por ser uma fonte inesgotável de importantes compostos, fica
claro entender a necessidade do desenvolvimento de novas e eficientes
metodologias, bem como melhorias nos métodos já conhecidos para síntese de
compostos heterocíclicos.
Dentre essas, as reações de ciclização eletrofílica de compostos
insaturados tornaram-se uma metodologia versátil para a síntese de unidades
heterocíclicas.2 Importantes compostos heterocíclicos, tais como, indóis,2a-b
benzo[b]furanos,2c-d benzo[b]tiofenos,2e-f benzo[b]selenofenos,2g tiofenos,2h
furanos,2i pirróis,2j entre outros,2k-v podem ser sintetizados utilizando-se este
protocolo. 1 Barreiro, E. J.; Fraga, C. A. F. Química Medicinal: As Bases Moleculares de ação de Fármacos, Artemed Editora, Porto Alegre, RS, 2001, 53. 2 (a) Barluenga, J.; Trincado, M.; Rubio, E.; Gonzalez, J. M. Angew. Chem., Int. Ed. 2003, 42, 2406. (b) Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2006, 71, 62. (c) Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 10292. (d) Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L. Synlett 1999, 1432. (e) Yue, D.; Larock, R. C. J. Org. Chem. 2002, 67, 1905. (f) Hessian, K. O.; Flynn, B. L. Org. Lett. 2003, 5, 4377. (g) Kesharwani, T.; Worlikar, S. A.; Larock, R. C. J. Org. Chem. 2006, 71, 2307. (h) Flynn, B. L.; Flynn, G. P.; Hamel, E.; Jung, M. K. Bioorg. Med. Chem. Lett. 2001, 11, 2341. (i) Sniady, A.; Wheeler, K. A.; Dembinski, R. Org. Lett. 2005, 7, 1769. (j) Knight, D. W.; Redfern, A. L.; Gilmore, J. J. Chem. Soc., Perkin Trans. 1 2002, 622. (k) Huang, Q.; Hunter, J. A.; Larock, R. C. J. Org. Chem. 2002, 67, 3437. (l) Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68, 5936. (m) Yue, D.; Della Ca, N.; Larock, R. C. Org. Lett. 2004, 6, 1581. (n) Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 1432. (o) Yao, T.; Campo, M. A.; Larock, R. C. J. Org. Chem. 2005, 70, 3511. (p) Zhou, C.; Dubrovsky, A. V.; Larock, R. C. J. Org. Chem. 2006, 71, 1626. (q) Waldo, J. P.; Larock, R. C. Org. Lett. 2005, 7, 5203. (r) Arcadi, A.; Cacchi, S.; Giuseppe, S. D.; Fabrizi, G.; Marinelli, F. Org. Lett. 2002, 4, 2409. (s) Dabdoub, M. J.; Dabdoub, V. B.; Pereira, M. A. J. Org. Chem. 1996, 61, 9503. (t) Bellina, F.; Biagetti, M.; Carpita, A.; Rossi, R. Tetrahedron 2001, 57, 2857. (u) Peng, A.; Ding, Y. J. Am. Chem. Soc. 2003, 125, 15006. (v) Djuardi, E.; McNelis, E. Tetrahedron Lett. 1999, 40, 7193. (x) Alves, D.; Luchese, C.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2007, 72, 6726.

20
Dentre estas inúmeras classes de compostos heterocíclicos que vêm
sendo preparadas, os compostos contendo enxofre, selênio e telúrio surgem
como uma importante alternativa, o que estimula testes bioquímicos ou
farmacológicos. A incorporação do átomo de selênio em moléculas orgânicas
permite a preparação de inúmeros compostos com propriedades já
reconhecidas.3
Adicionalmente, compostos orgânicos de selênio têm atraído
considerável atenção em síntese orgânica devido a sua utilidade em um
extraordinário número de reações,4 incluindo a formação de novas ligações
carbono-carbono,5 bem como por apresentarem propriedades toxicológicas e
farmacológicas.3
Ainda sobre compostos orgânicos de selênio, os derivados de
selenofenos são de grande importância em química orgânica devido às suas
excelentes propriedades elétricas.6 Uma classe que vem sendo bastante
estudada para essas propriedades são os heterociclos fundidos fazendo com
que a síntese destes se tornem bastante atrativa.7,8
3 (a) Parnham, M. J.; Graf, E. Prog. Drug. Res. 1991, 36, 9. (b) Mugesh, G.; du Mont, W. W.; Sies, H. Chem. Rev. 2001, 101, 2125. (c) Nogueira, C. W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255. 4 (a) Organoselenium Chemistry. Em Topics in Current Chemistry 208; Wirth, T., Ed.; Springer-Verlag: Heidelberg, 2000. (b) Krief, A. Em Comprehensive Organometallic Chemistry II; Abel, E. V.; Stone, F. G. A.; Wilkinson, G., Eds.; Pergamon Press: New York, 1995; Vol. 11, Chapter 13. (c) Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Em Organic Chemistry Series 4; Baldwin, J. E., Ed.; Pergamon Press: Oxford, 1986. (d) Sharpless, K. B.; Young, M. W.; Lauer, R. F. Tetrahedron Lett. 1973, 22, 1979. (e) Reich, H. J. J. Org. Chem. 1975, 40, 2570. (f) Sharpless, K. B.; Lauer, R. F. J. Am. Chem. Soc. 1972, 94, 7154. (g) Sevrin, M.; Vanende, D.; Krief, A. Tetrahedron Lett. 1976, 30, 2643. (h) Sevrin, M.; Dumont, W.; Hevesi, L. D.; Krief, A. Tetrahedron Lett. 1976, 30, 2647. (i) Seebach, D.; Peleties, N. Chem. Ber. 1972, 105, 511. (j) Seebach, D.; Beck, A. K. Angew. Chem., Int. Ed. Engl. 1974, 13, 806. (k) Reich, H. J.; Shah, S. K. J. Am. Chem. Soc. 1975, 97, 3250. (l) Perin, G.; Lenardão, E. J.; Jacob, R. G.; Panatiere, R. B. Chem. Rev. 2009, 109, 1277. (m) Freudendahl, D. M.; Shahzad, S. A.; Writh, T. Eur. J. Org. Chem. 2009, 1649. 5 Silveira, C. C.; Braga, A. L.; Vieira, A. S.; Zeni, G. J. Org. Chem. 2003, 68, 662. 6 (a) Nakayama, J.; Konishi, T. Heterocycles 1988, 27, 1731. (b) Kuroda, M.; Nakayama, J.; Hoshino, M.; Furusho, N.; Kawata, T.; Ohba,S. Tetrahedron 1993, 49, 3735. (b) Emiko, F.; Zhou, B.; Akiko, K.; Hayao, K.; Yuichi, F.; Eiji, N.; Makoto, S.; Hhoji, I.; Kiyoyuki, T. Eur. J. Inorg. Chem. 2009, 12, 1585. 7 (a) Skotheim T. A.; Elsenbaumer R. L.; Reynolds J. R. Handbook of Conducting Polymers, second ed., Dekker, New York, 1998; (b) Nalwa H. S. Handbook of Conductive Materials and Polymers, Wiley, New York, 1997; (c) Kraft A.; Grimsdale A.; Holmes A. B. Angew. Chem. Int. Ed. Engl. 1998, 37, 403.

21
Sendo assim, devido ao grande interesse de nosso grupo de pesquisa
na síntese, reatividade,9 avaliação toxicológica e farmacológica de
organocalcogênios,5c,10 planejou-se duas metodologias para síntese de
derivados calcogenofenos.
No primeiro trabalho, foi explorada uma nova metodologia para a síntese
de seleno- e telurofenos, funcionalizados na posição 3 do anel heteroaromático
2, via reação de (Z)-seleno- e teluroeninos 1 com dicalcogenetos de
diorganoíla, catalisadas por sais de cobre (Esquema 1).
Esquema 1
Estes derivados de calcogenofenos, uma vez obtidos, do ponto de vista
sintético, tornam-se compostos potenciais para a síntese de calcogenofenos
com outros substituintes.
8 (a) Arbizzani C.; Catellani M.; Mastragostino M.; Cerroni M. G. J. Electroanal. Chem. 1997, 423, 23. (b) Henssler, J. T.; Matzger, A. Org. Lett. 2009, 11, 3144. 9 (a) Zeni, G.; Nogueira, C. W.; Panatieri, R. B.; Silva, D. O.; Menezes, P. H.; Braga, A. L.; Silveira, C. C.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron Lett. 2001, 42, 7921. (b) Zeni, G.; Ludtke, D. S.; Nogueira, C. W.; Panatieri, R. B.; Braga, A. L.; Silveira, C. C.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron Lett. 2001, 42, 8927. (c) Zeni, G.; Nogueira, C. W.; Silva, D. O.; Menezes, P. H.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron Lett. 2003, 44, 1387. (d) Zeni, G.; Silva, D. O.; Menezes, P. H.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron Lett. 2003, 44, 685. (e) Barros, O. S. R.; Favero, A.; Nogueira, C. W.; Menezes, P. H.; Zeni, G. Tetrahedron Lett. 2006, 47, 2179. (f) Panatieri, R. B.; Reis, J. S.; Borges, L. P.; Nogueira, C. W.; Zeni, G. Synlett 2006, 3161. (g) Prediger, P.; Moro, A. V.; Nogueira, C. W.; Savegnago, L.; Rocha, J. B. T.; Zeni, G. J. Org. Chem. 2006, 71, 3786. (h) Barros, O. S. R.; Nogueira, C. W.; Stangherlin, E. C.; Menezes, P. H.; Zeni, G. J. Org. Chem. 2006, 71, 1552. (i) Zeni, G. Tetrahedron Lett. 2005, 46, 2647 10 (a) Savegnago, L.; Jesse, C. R.; Pinto, L. G.; Rocha, J. B. T.; Nogueira, C. W. Brain Res. 2007, 1175, 54. (b) Luchese, C.; Brandão, R.; Oliveira, R.; Nogueira, C. W.; Santos, F. W. Toxicology Lett. 2007, 173, 181. (c) Savegnago, L.; Pinto, L. G.; Jesse, C. R.; Rocha, J. B. T.; Nogueira, C. W.; Zeni, G. Brain Res. 2007, 1162, 32. (d) Borges, V. C.; Dadalt, G.; Savegnago, L.; Moro, A. V.; Rocha, J. B. T.; Nogueira, C. W. Toxicology in Vitro 2007, 21, 387. (e) Luchese, C.; Stangherlin, E. C.; Ardais, A. P.; Nogueira, C. W.; Santos, F. W. Toxicology 2007, 230, 189. (f) Savegnago, L.; Pinto, L. G.; Jesse, C. R.; Alves, D.; Rocha, J. B. T.; Nogueira, C. W.; Zeni, G. Eur. J. Pharmacol 2007, 555, 129. (g) Savegnago, L.; Borges, V. C.; Alves, D.; Jesse, C. R.; Rocha, J. B. T.; Nogueira, C. W. Life Sci. 2006, 79, 1546. (h) Stangherlin, E. C.; Favero, A. M.; Zeni, G.; Rocha, J. B. T.; Nogueira, C. W. Brain Res. Bull. 2006, 69, 311.
22
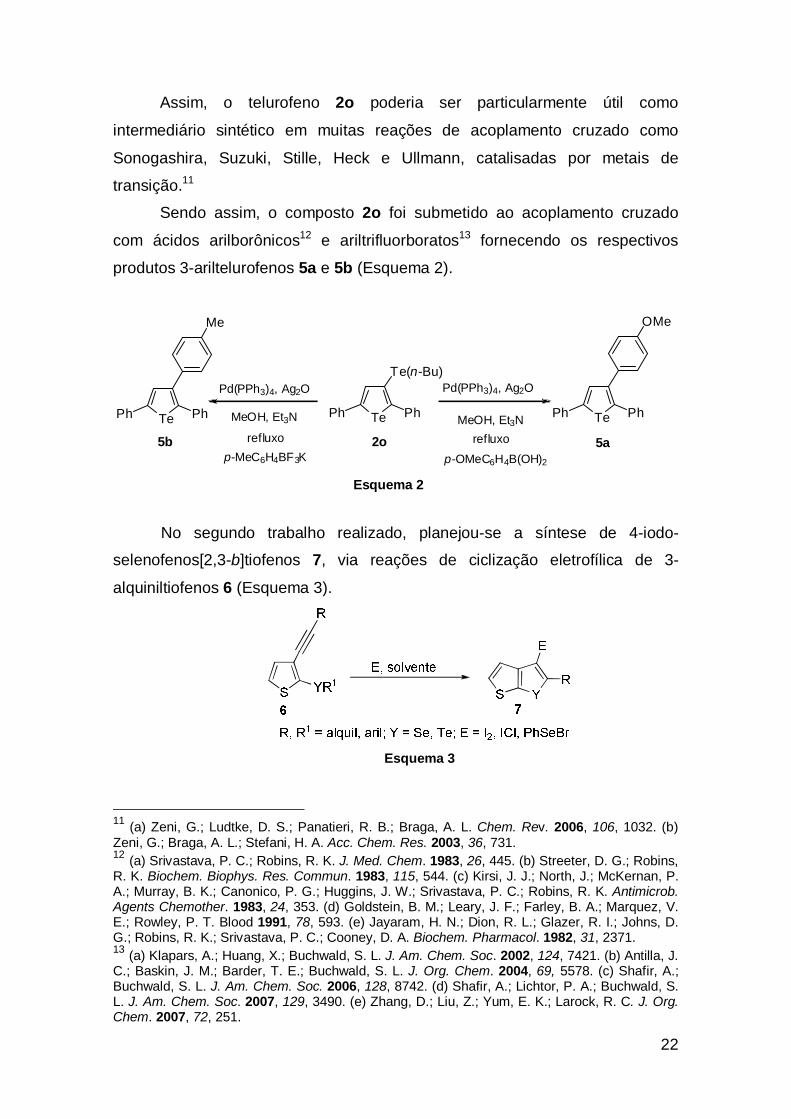
Assim, o telurofeno 2o poderia ser particularmente útil como
intermediário sintético em muitas reações de acoplamento cruzado como
Sonogashira, Suzuki, Stille, Heck e Ullmann, catalisadas por metais de
transição.11
Sendo assim, o composto 2o foi submetido ao acoplamento cruzado
com ácidos arilborônicos12 e ariltrifluorboratos13 fornecendo os respectivos
produtos 3-ariltelurofenos 5a e 5b (Esquema 2).
Te PhPh
Te(n-Bu)
Te PhPhTe PhPh
Me OMe
Pd(PPh3)4, Ag2O
MeOH, Et3N
refluxop-MeC6H4BF3K
Pd(PPh3)4, Ag2O
MeOH, Et3Nrefluxo
p-OMeC6H4B(OH)25b 2o 5a
Esquema 2
No segundo trabalho realizado, planejou-se a síntese de 4-iodo-
selenofenos[2,3-b]tiofenos 7, via reações de ciclização eletrofílica de 3-
alquiniltiofenos 6 (Esquema 3).
Esquema 3
11 (a) Zeni, G.; Ludtke, D. S.; Panatieri, R. B.; Braga, A. L. Chem. Rev. 2006, 106, 1032. (b) Zeni, G.; Braga, A. L.; Stefani, H. A. Acc. Chem. Res. 2003, 36, 731. 12 (a) Srivastava, P. C.; Robins, R. K. J. Med. Chem. 1983, 26, 445. (b) Streeter, D. G.; Robins, R. K. Biochem. Biophys. Res. Commun. 1983, 115, 544. (c) Kirsi, J. J.; North, J.; McKernan, P. A.; Murray, B. K.; Canonico, P. G.; Huggins, J. W.; Srivastava, P. C.; Robins, R. K. Antimicrob. Agents Chemother. 1983, 24, 353. (d) Goldstein, B. M.; Leary, J. F.; Farley, B. A.; Marquez, V. E.; Rowley, P. T. Blood 1991, 78, 593. (e) Jayaram, H. N.; Dion, R. L.; Glazer, R. I.; Johns, D. G.; Robins, R. K.; Srivastava, P. C.; Cooney, D. A. Biochem. Pharmacol. 1982, 31, 2371. 13 (a) Klapars, A.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 7421. (b) Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org. Chem. 2004, 69, 5578. (c) Shafir, A.; Buchwald, S. L. J. Am. Chem. Soc. 2006, 128, 8742. (d) Shafir, A.; Lichtor, P. A.; Buchwald, S. L. J. Am. Chem. Soc. 2007, 129, 3490. (e) Zhang, D.; Liu, Z.; Yum, E. K.; Larock, R. C. J. Org. Chem. 2007, 72, 251.
23
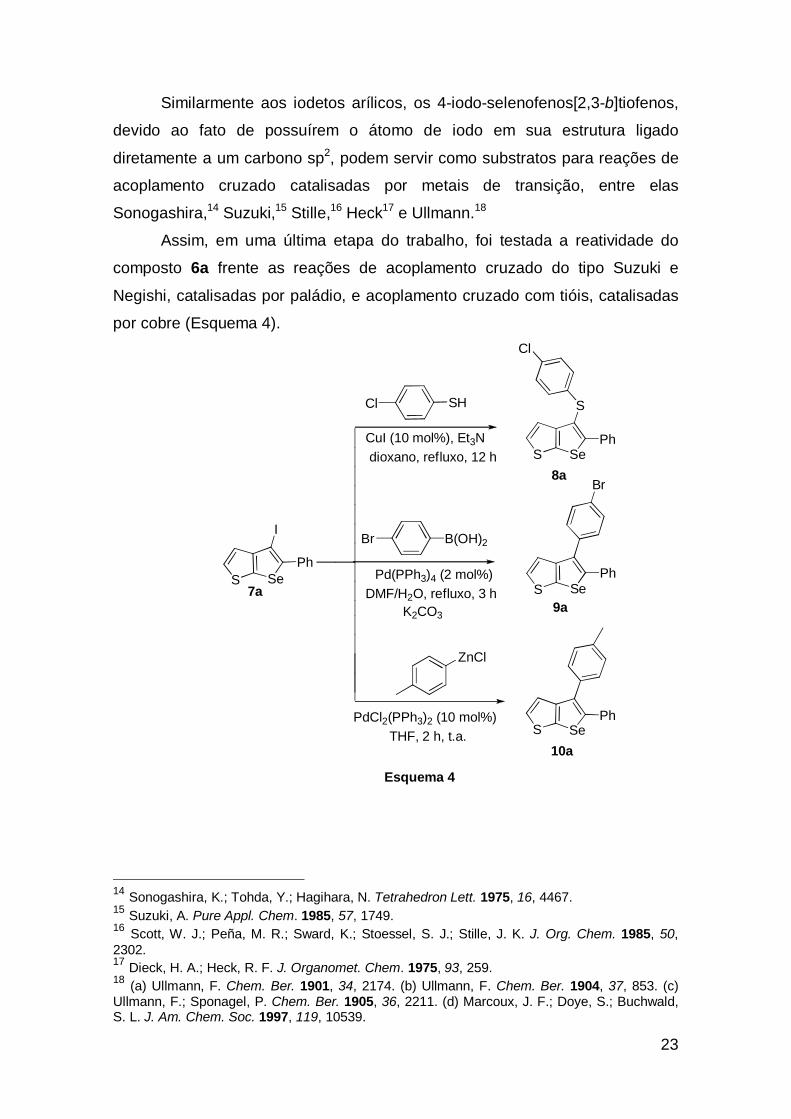
Similarmente aos iodetos arílicos, os 4-iodo-selenofenos[2,3-b]tiofenos,
devido ao fato de possuírem o átomo de iodo em sua estrutura ligado
diretamente a um carbono sp2, podem servir como substratos para reações de
acoplamento cruzado catalisadas por metais de transição, entre elas
Sonogashira,14 Suzuki,15 Stille,16 Heck17 e Ullmann.18
Assim, em uma última etapa do trabalho, foi testada a reatividade do
composto 6a frente as reações de acoplamento cruzado do tipo Suzuki e
Negishi, catalisadas por paládio, e acoplamento cruzado com tióis, catalisadas
por cobre (Esquema 4).
SeS
I
Ph
CuI (10 mol%), Et3Ndioxano, refluxo, 12 h
Pd(PPh3)4 (2 mol%)DMF/H2O, refluxo, 3 h
PdCl2(PPh3)2 (10 mol%)THF, 2 h, t.a.
SeS
S
Ph
SeSPh
SeSPh
7a9a
10a
Cl
Br
Cl SH
B(OH)2Br
ZnCl
K2CO3
8a
Esquema 4
14 Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467. 15 Suzuki, A. Pure Appl. Chem. 1985, 57, 1749. 16 Scott, W. J.; Peña, M. R.; Sward, K.; Stoessel, S. J.; Stille, J. K. J. Org. Chem. 1985, 50, 2302. 17 Dieck, H. A.; Heck, R. F. J. Organomet. Chem. 1975, 93, 259. 18 (a) Ullmann, F. Chem. Ber. 1901, 34, 2174. (b) Ullmann, F. Chem. Ber. 1904, 37, 853. (c) Ullmann, F.; Sponagel, P. Chem. Ber. 1905, 36, 2211. (d) Marcoux, J. F.; Doye, S.; Buchwald, S. L. J. Am. Chem. Soc. 1997, 119, 10539.

24
Capítulo 1
Revisão da Literatura

25
REVISÃO DE LITERATURA
1.1 COMPOSTOS HETEROCÍCLICOS E HETEROAROMÁTICOS
Vários compostos heterocíclicos são fármacos mundialmente
consumidos que apresentam atividades farmacológicas diversificadas, tais
como, inibidor do HIV (AZT 11),1 antitumoral (D-501036 12);19 antifúngica (5-(3-
buten-1-inil)-2,2’-bitienila 13);20 antinflamatória e analgésica (dipirona 14);1
antiprotozoária (metronidazol 15)1 e antiviral (ribavirina 16)1 (Figura 3).
Figura 1. Fármacos contendo unidade heterocíclica
Os compostos heterocíclicos apresentam uma nomenclatura bastante
complexa, devido a diversidade de características inerentes as suas estruturas.
A nomenclatura recomendada por Hantzsch-Widman, estabelece hierarquias
de prioridades: tais como tipo de heteroátomos, tamanho do anel, tipos de
19 (a) Shiah, H. S.; Lee, W. S.; Juang, S. H.; Hong, P. C.; Lung, C. C.; Chang, C. J.; Chou, K. M.; Chang, J. Y. Biochemical Pharmacology 2007, 73, 610. (b) Juang, S. H.; Lung, C. C.; Hsu, P. C.; Hsu, K. S.; Li, Y. C.; Hong, P. C.; Shiah, H. S.; Kuo, C. C.; Huang, C. W.; Wang, Y. C.; Huang, L.; Chen, T. S.; Chen, S. F.; Fu, K. C.; Hsu, C. L.; Lin, M. J.; Chang, C. J.; Ashendel, C. L.; Chan, T. C. K.; Chou, K. M. Chang, J. Y. Molecular Câncer Therapeutics 2007, 6, 193. (c) Shiah, H. S.; Lee, W. S.; Juang, S. H.; Chang, C. J.; Chang, J. Y. Clinical Cancer Research 2005, 11, 9101. 20 Chan, G. F. Q.; Towers, G. H. N.; Mitchell, J. C. Phytochemistry 1975, 14, 2295.

26
moléculas: monocíclica com um único tipo de heteroátomo, monocíclicas com
dois tipos de heteroátomos, bicíclicas com dois anéis, moléculas policíclicas,
entre outras peculiaridades estruturais.21
Sistemas heterocíclicos estão divididos em quatro grupos distintos,
sendo eles, heterocicloalcanos, heterocicloalquenos, heteroanulenos e
heteroaromáticos. Compostos heteroaromáticos seguem a regra de Hückel,
onde possuem (4n + 2) elétrons π deslocalizados ao longo do anel
heterocíclico. O mais importante grupo destes compostos possui aromaticidade
semelhante à do benzeno, sendo esta classe também chamada de
heteroarenos. Os compostos mais importantes desta classe são o furano,
tiofeno, pirrol, piridina e íons pirílio, sendo a reatividade e estabilidade desta
classe de compostos comparada à do benzeno.
Heterociclos aromáticos de cinco membros contendo átomos de
calcogênio pertencem à classe de substâncias denominada genericamente de
calcogenofenos, sendo que o mais simples deles é o furano. Também estão
inclusos nesta classe o tiofeno, selenofeno e telurofeno (Figura 4). Ainda estão
nesta classe os benzo derivados de calcogenofenos, sendo mais comumente
encontrados os benzo[b]furanos e benzo[b]tiofenos.
Figura 2. Exemplos de calcogenofenos
21 (a) Katritzky, A. R.; Pozharskii, A. F. Em Handbook of Heterocyclic Chemistry, Second Edition; Pergamon: Oxford 2000. (b) Eicher, T.; Hauptmann, S. Em The Chemistry of Heterocycles, Second Edition; Wiley-VCH 2003.

27
Furanos, tiofenos e seus derivados, têm despertado o interesse de
pesquisadores na química orgânica sintética, pois suas ocorrências em
produtos naturais que apresentam alguma atividade biológica é relativamente
frequente, incentivando a procura de metodologias para a síntese destes
compostos. Selenofenos, telurofenos e seus derivados vêm recebendo menos
atenção da comunidade científica por estes não apresentarem relatos de
ocorrência natural. Por outro lado, a síntese deste tipo de compostos
heteroaromáticos vem crescendo nos últimos anos devido ao fato dos
polímeros destes compostos apresentarem propriedades ópticas e
eletroquímicas.22
1.2 REAÇÕES DE CICLIZAÇÃO ELETROFÍLICA
Reações de ciclização eletrofílica de compostos insaturados, tornaram-
se uma metodologia versátil para a síntese de unidades heterocíclicas.
Importantes compostos heterocíclicos, tais como, indóis, benzofuranos,
benzotiofenos, benzo[b]selenofenos, benzopiranos, isocumarinas,
isoquinolinas, tiofenos, furanos, pirróis, telurofenos, entre outros, podem ser
sintetizados utilizando-se este protocolo.
Dentre as reações de ciclização eletrofílicas, ciclizações
intramoleculares de sistemas π-alquinílicos catalisadas por metais de transição,
são descritas como uma das principais metodologias de síntese de compostos
heterocíclicos.23 Estes processos normalmente envolvem complexação rápida
e reversível do alquino com um complexo de Pd (II). Os complexos π-
alquinílicos resultantes são relativamente estáveis e suscetíveis a ataques
nucleofílicos. Diferentes nucleófilos podem ser utilizados nestes tipos de
reações, destacando-se contendo átomos de oxigênio, nitrogênio e enxofre.
Após o ataque nucleofílico aos sistemas π-alquinílicos, ocorre uma etapa de
eliminação redutiva, acarretando na formação do composto ciclizado e também
em uma espécie de Pd (0), onde esta retorna ao ciclo catalítico da reação.
22 (a) Pu, S.; Hou, J.; Xu, J.; Nie, G.; Zhang, S.; Shen, L.; Xiao, Q. Materials Lett. 2005, 59, 1061. (b) Salzner, U.; Lagowski, J. B.; Pickup, P. G.; Poirier, R. A. Synthetic Metals 1998, 96, 177. (c) Otsubo, T.; Inoue, S.; Nozoe, H.; Jigami, T.; Ogura, F. Synthetic Metals 1995, 69, 537. 23 (a) Zeni, G.; Larock, R. C. Chem. Rev. 2004, 104, 2285. (b) Zeni, G.; Larock, R. C. Chem. Rev. 2006, 106, 4644.

28
Um exemplo desta metodologia de ciclização eletrofílica mediada por
metais de transição foi descrita por Cacchi e colaboradores.24 Este trabalho
relatou a síntese de benzo[b]furanos através da reação entre haletos de 2-
hidroxi-arila com alquinos terminais catalisadas por sais de paládio. Quando os
autores utilizaram quantidades catalíticas de Pd(OAc)2(PPh3)2 e CuI, as
reações entre os haletos de 2-hidroxi-arila 17 com os alquinos terminais
forneceram como produtos de ciclização os benzo[b]furanos 18 em bons
rendimentos (Esquema 4).
Esquema 5
Uma metodologia alternativa para as reações de ciclização eletrofílica de
alquinos contendo um nucleófilo (átomos de O, N, S, Se, Te) em proximidade a
ligação tripla é quando se utiliza uma fonte eletrofílica, sendo que as mais
utilizadas são de iodo (I2, ICl, NIS), bromo (Br2, NBS) e selênio (PhSeCl,
PhSeBr). Diversas metodologias de síntese de compostos heterocíclicos foram
descritas utilizando-se este método.
O mecanismo destas reações envolvendo espécies eletrofílicas de
halogênios e selênio segue as seguintes etapas, em concordância com
experimentos descritos na literatura. 2b, 2g, 2r, 2x
1 – Coordenação da molécula do eletrófilo a ligação tripla do alquino
formando o intermediário A;
2 – Um ataque nucleofílico anti do heteroátomo em proximidade ao
intermediário A, fornecendo o intermediário heterocíclico B;
3 – O ânion remanescente derivado do eletrófilo reage nucleofilicamente
com o grupamento R ligado ao heteroátomo, levando a formação do produto
desejado (Esquema 6). 24 (a) Arcadi, A.; Marinelli, F.; Cacchi, S. Synthesis 1986, 749. (b) Arcadi, A.; Cacchi, S.; Del Rosario, M.; Fabrizi, G.; Marinelli, F. J. Org. Chem. 1996, 61, 9280.
29
Y
ER
A B
E
YR
R1
YR
R1+E
E-
E = Eletrófilo de I, Br, SeY = O, N, S, Se, Te
E
R1
Y
E
R1
R
Esquema 6
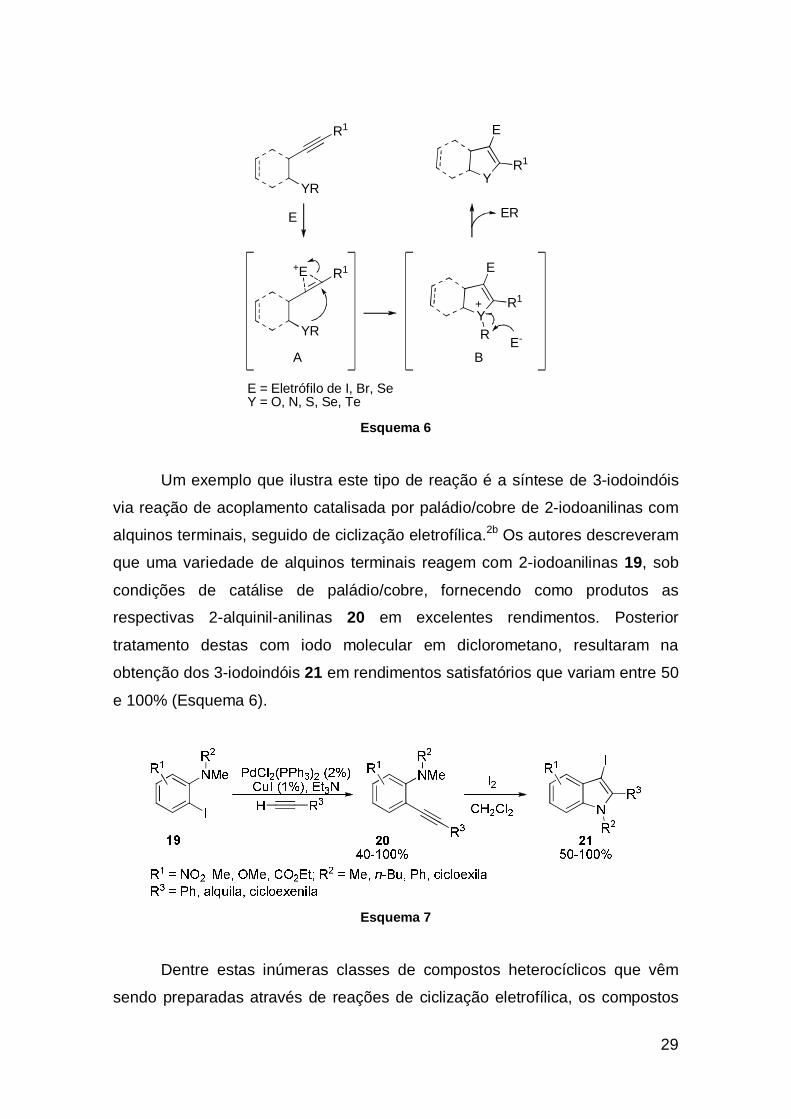
Um exemplo que ilustra este tipo de reação é a síntese de 3-iodoindóis
via reação de acoplamento catalisada por paládio/cobre de 2-iodoanilinas com
alquinos terminais, seguido de ciclização eletrofílica.2b Os autores descreveram
que uma variedade de alquinos terminais reagem com 2-iodoanilinas 19, sob
condições de catálise de paládio/cobre, fornecendo como produtos as
respectivas 2-alquinil-anilinas 20 em excelentes rendimentos. Posterior
tratamento destas com iodo molecular em diclorometano, resultaram na
obtenção dos 3-iodoindóis 21 em rendimentos satisfatórios que variam entre 50
e 100% (Esquema 6).
Esquema 7
Dentre estas inúmeras classes de compostos heterocíclicos que vêm
sendo preparadas através de reações de ciclização eletrofílica, os compostos

30
contendo átomos de calcogênio em seu núcleo base surgem como uma
importante alternativa para síntese de compostos, que estimulam testes
bioquímicos ou farmacológicos.
Tendo isto em vista, furanos substituídos puderam ser sintetizados
através de ciclizações eletrofílicas de butinonas 1,4-dissubstituídas 22 com
eletrófilos de bromo e iodo. Eletrófilos como NBS, NIS e ICl foram usados,
sendo os 3-iodo ou 3-bromofuranos 23 obtidos em bons rendimentos tanto em
reações em acetona quanto em CH2Cl2 (Esquema 8).2i
Esquema 8
O mecanismo proposto pelos autores passa em um primeiro momento
pela formação de um intermediário catiônico na ligação tripla. A etapa posterior
pode ser tanto por abstração do hidrogênio alfa seguida de ciclização A, quanto
por ciclização seguida de desprotonação B, conforme equilíbrio mostrado no
esquema 9.2i
Esquema 9
Alternativamente a esta metodologia de síntese de furanos, Liu e
colaboradores descreveram a ciclização de cetonas α,β-insaturadas
alquinílicas 24, na presença de um nucleófilo, utilizando-se para isto iodo,
K3PO4 e CH2Cl2.25 Sob estas condições de reação foi possível a obtenção de 3-
25 Liu, Y.; Zhou, S. Org. Lett. 2005, 7, 4609.

31
iodofuranos 25 altamente substituídos, com a incorporação do nucleófilo na
molécula, onde os rendimentos variaram entre 47-99% (Esquema 10). Os
autores relataram que a reação se processa primeiramente com formação de
um intermediário iodônio. Após uma etapa de ciclização com o oxigênio da
carbonila, ocorre uma reação de adição 1,4 do nucleófilo a ligação dupla C-C,
fornecendo como produto os iodofuranos 25 desejados.
O
Nu
I
R1
I2, K3PO4
NuH, CH2Cl2t. a., 30 min.47-99%
R1 = Ph, 4-MeC6H4, TMSNu = MeO, PhO, HCCCH2O, H2C=CHCH2O
R1O
R1O
O+
I
R1
I+
NuH
I2
24 25
Esquema 10
Ainda sobre síntese de compostos heterocíclicos contendo o átomo de
oxigênio, benzo[b]furanos foram sintetizados através de ciclização de 2-
alquinilanisóis com diferentes fontes eletrofílicas.2c Partindo-se de 2-iodoanisóis
26, com acoplamento posterior do tipo Sonogashira catalisado por sais de
paládio e cobre com alquinos terminais, forneceram os respectivos 2-
alquinilanisóis 27 em excelentes rendimentos. Numa etapa subsequente,
diferentes eletrófilos foram utilizados para as reações de ciclização, fornecendo
os correspondentes benzo[b]furanos dissubstituídos 28 em excelentes
rendimentos. Através desta metodologia, furopiridinas biologicamente
importantes também puderam ser sintetizadas (Esquema 11).

32
Esquema 11
Devido ao grande interesse na síntese desses compostos, nosso grupo
de pesquisa relatou, recentemente, duas metodologias para síntese de
heterocíclos, contendo o átomo de oxigênio, altamente substituídos via reações
de ciclização eletrofílica com diferentes eletrófilos.
Assim, foi relatada uma eficiente metodologia para obtenção de
benzopiranos devido ao fato desses produtos serem encontrados em diversos
produtos naturais com importantes propriedades farmacêuticas e biológicas.26
Para isso, foi sintetizado uma grande variedade de éteres arílicos
organocalcogeno propargílicos 30, via um intermediário acetileto de lítio,
gerado pela reação de n-BuLi com os éteres arílicos 29, e com a subsequente
captura de espécies eletrofílicas de enxofre, selênio e telúrio. Tendo esses
éteres arílicos organocalcogeno propargílicos 30 em mãos, uma grande
variedade de 3-halo-4-calcogeno-2H-benzopiranos 31 foram preparados via
reações de ciclização eletrofílica usando-se espécies eletrofílicas de iodo e
telúrio. (Esquema 12).
Esquema 12
26 Godoi, B.; Sperança, A.; Back, D. F.; Brandão, R.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 3469.
33
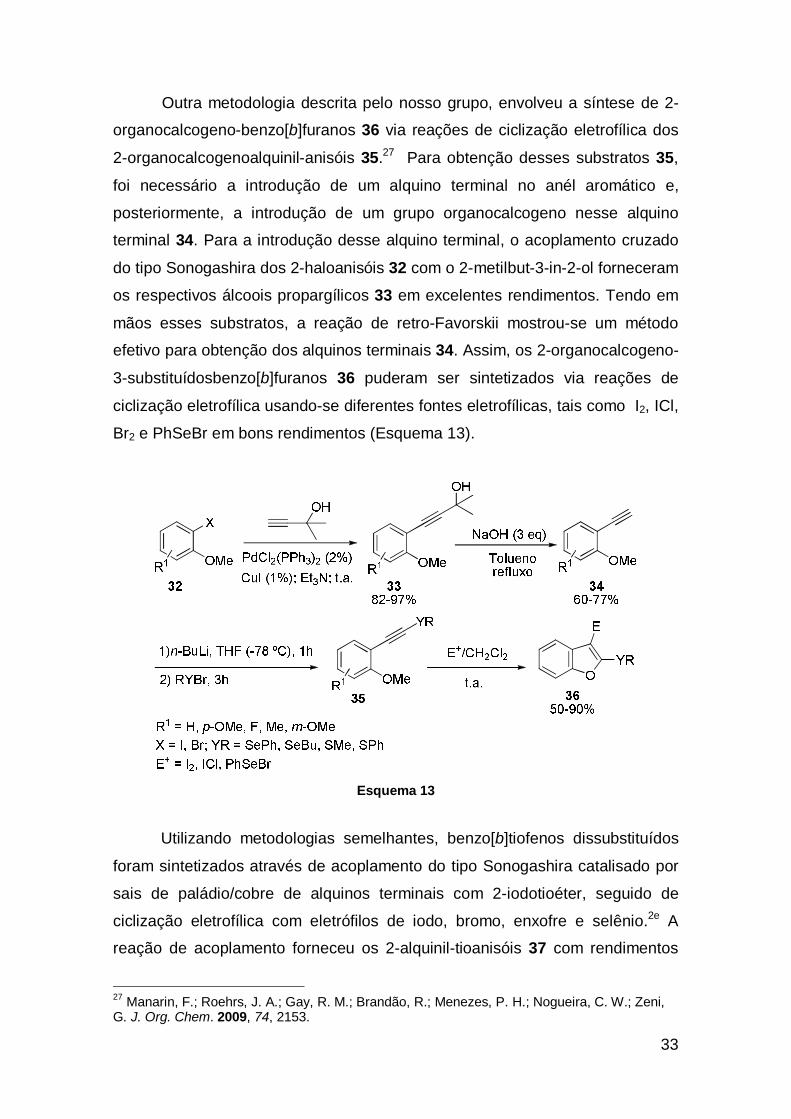
Outra metodologia descrita pelo nosso grupo, envolveu a síntese de 2-
organocalcogeno-benzo[b]furanos 36 via reações de ciclização eletrofílica dos
2-organocalcogenoalquinil-anisóis 35.27 Para obtenção desses substratos 35,
foi necessário a introdução de um alquino terminal no anél aromático e,
posteriormente, a introdução de um grupo organocalcogeno nesse alquino
terminal 34. Para a introdução desse alquino terminal, o acoplamento cruzado
do tipo Sonogashira dos 2-haloanisóis 32 com o 2-metilbut-3-in-2-ol forneceram
os respectivos álcoois propargílicos 33 em excelentes rendimentos. Tendo em
mãos esses substratos, a reação de retro-Favorskii mostrou-se um método
efetivo para obtenção dos alquinos terminais 34. Assim, os 2-organocalcogeno-
3-substituídosbenzo[b]furanos 36 puderam ser sintetizados via reações de
ciclização eletrofílica usando-se diferentes fontes eletrofílicas, tais como I2, ICl,
Br2 e PhSeBr em bons rendimentos (Esquema 13).
Esquema 13
Utilizando metodologias semelhantes, benzo[b]tiofenos dissubstituídos
foram sintetizados através de acoplamento do tipo Sonogashira catalisado por
sais de paládio/cobre de alquinos terminais com 2-iodotioéter, seguido de
ciclização eletrofílica com eletrófilos de iodo, bromo, enxofre e selênio.2e A
reação de acoplamento forneceu os 2-alquinil-tioanisóis 37 com rendimentos
27 Manarin, F.; Roehrs, J. A.; Gay, R. M.; Brandão, R.; Menezes, P. H.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 2153.

34
elevados. Os bons rendimentos foram atribuídos pela complexação do átomo
de enxofre com o intermediário σ-arilpaládio (II) formado na reação após a
etapa de adição oxidativa. Os correspondentes 2-alquinil-tioanisóis 37 obtidos
reagiram com diferentes fontes eletrofílicas, utilizando-se CH2Cl2 como solvente
a temperatura ambiente. Sob estas condições benzo[b]tiofenos 38 foram
obtidos em rendimentos satisfatórios de 52-100% (Esquema 11).
Esquema 14
Recentemente, Larock e colaboradores descreveram a síntese de
benzo[b]selenofenos 40 substituídos nas posições 2 e 3 do anel heterocíclico
através de reação de ciclização eletrofílica de 1-alquinil-2-metilseleno-arenos
39. Estas reações de ciclização toleraram uma grande variedade de grupos
funcionais, incluindo, álcool, éster, nitrila, nitro e grupos silanos, fornecendo os
produtos ciclizados em bons rendimentos e em condições de reação suaves
(Esquema 15).2g
Esquema 15
Telurofenos, também puderam ser sintetizados através da metodologia
de iodociclização. Teluroenínos de estereoquímica Z 41 diferentemente
substituídos, quando tratados com 2 equivalentes de iodo em éter de petróleo,

35
forneceram como produtos de reação 3-iodo-telurofenos 42 substituídos nas
posições 2 e 5 do anel heterocíclico em altos rendimentos. Experimentos de
raios-X ajudaram a elucidar a estrutura do produto de iodociclização 2,5-difenil-
3-iodotelurofeno (Esquema 16).2s
Esquema 16
Similarmente a essa metodologia, nosso grupo de pesquisa relatou a
eficiente síntese de 3-iodoselenofenos 44, substituídos nas posições 2 e 5 do
anel heterocíclico, a partir dos selenoeninos de estereoquímica Z 43
diferentemente substituídos.2x A reação procedeu-se em condições brandas
fornecendo os respectivos selenofenos 44 em bons rendimentos com
diferentes eletrófilos, tais como I2, ICl, PhSeBr e PhSeCl (Esquema 17).
Esquema 17
1.3 SÍNTESE E REATIVIDADE DE SELENOFENOS E TELUROFENOS
Quando comparado com seus análogos tiofenos e furanos, os
telurofenos e selenofenos são escassamente relatados na literatura tanto na
área biológica, quanto na área de síntese e reatividade destes compostos. Este
fato incentiva estudos que busquem demonstrar possibilidades de síntese e
uso dos mesmos em diferentes metodologias de síntese orgânica, bem como a
busca de compostos com possíveis atividades biológicas. Curiosamente,

36
apesar do selenofeno e telurofeno serem conhecidos a mais de cinquenta
anos, poucos são os estudos que os utilizam como matérias primas para a
preparação de compostos de interesse biológico.
Sendo assim, uma das primeiras metodologias de síntese de
selenofenos foi desenvolvida por Gronowits e colaboradores. O selenofeno 46
foi obtido em rendimentos de 40-60%, passando-se um fluxo de acetileno em
um tubo de vidro contendo sílica gel e selênio elementar, sendo este sistema
aquecido a 450 °C. Os autores acreditam que a altas temperaturas ocorreu a
formação e posterior reação entre H2Se e butadiíno, formando o intermediário
selenoenínico 45. Subsequente etapa de ciclização intramolecular forneceu
como produto uma molécula de selenofeno 46 (Esquema 18).28
Esquema 18
Por sua vez, telurofenos 47 podem ser preparados através da reação
entre uma espécie bivalente negativa de telúrio e compostos diacetilênicos.
Através destas metodologias, diínos, diinóis e também sililbutadiínos reagiram
com Na2Te em MeOH fornecendo como produto de ciclização os telurofenos
47 em rendimentos entre 25-99% (Esquema 19).29
Esquema 19
28 Gronowits, S.; Fredj, T.; Moberg-Ogard, A.; Trege, L. J. Heterocycl. Chem. 1976, 13, 1319. 29 (a) Mack, W. Angew. Chem. 1966, 78, 940; (b) Angew. Chem., Int. Ed. Engl. 1966, 5, 896. (c) Barton, T. J.; Roth, R. W. J. Organomet. Chem. 1972, 39, 66.

37
Recentemente foi descrita a síntese de selenofenos altamente
funcionalizados com grupamentos organofosfonatos em suas moléculas.
Reagindo-se hidrosselenolato de sódio com duas moléculas do bis-fosfonato
de alquinila 48, seguido de ciclização intramolecular, resultou no composto 2,3-
dihidro-selenofeno 49 contendo quatro grupamentos fosfonato em 36% de
rendimento. A oxidação do composto 49 com m-CPBA forneceu como produto
o selenofeno 50 em rendimento satisfatório. A formação do selenofeno 50 pôde
ser explicada devido à oxidação do composto 49 ao seu correspondente
selenóxido, seguido de desidratação (Esquema 20).30
Esquema 20
Benzo[b]dicalcogenofenos foram sintetizados através da metodologia
desenvolvida por Takimiya. Nesta metodologia de síntese, o composto
dibromado 51 sofreu reação de de-halometalação com t-BuLi. O intermediário
dilitiado formado, reagiu com selênio ou telúrio elementar, com posterior reação
de ciclização em etanol, fornecendo como produtos os respectivos
benzo[b]disselenofenos 52 e benzo[b]ditelurofenos 53 em 65 e 55% de
rendimento. Uma etapa posterior de desproteção do alquino com TBAF,
forneceu os produtos desejados 54 e 55 em excelentes rendimentos (Esquema
21).31
30 Sasaki, S.; Adachi, K.; Yoshifuji, M. Org Lett. 2007, 9, 1729. 31 Takimiya, K.; Konda, Y.; Ebata, H.; Niihara, N.; Otsubo, T. J. Org. Chem. 2005, 70, 10569.

38
Esquema 21
Uma metodologia de síntese de benzo[c]selenofenos foi descrita por
Cava e colaboradores. Partindo-se do 1,3-dihidrobenzo[c]selenofeno 56, sendo
este submetido a etapas de bromação e de-hidrobromação, seguida por uma
reação de oxidação, obtiveram-se os benzo[c]selenofenos 58 em rendimentos
satisfatórios. Entretanto, estes compostos eram extremamente instáveis em
solução aquosa e decompunham com o passar do tempo (Esquema 22).32
Esquema 22
Anos mais tarde o mesmo grupo de pesquisa modificou esta
metodologia de síntese. Tratando-se o composto dibromado 57 com bases em
soluções não aquosas, tais como, DBN e LHMDS, utilizando-se THF como
solvente, obtêve-se como produto benzo[c]selenofenos 58 em bons
rendimentos. Através desta metodologia, foi possível a obtenção de
benzo[c]selenofenos em solução de THF, onde este permaneceu relativamente
estável nesta solução a frio. Os autores também relataram a síntese de
diésteres derivados de benzo[c]selenofenos 59 através da reação do
32 Saris, L. E.; Cava, M. P. J. Am. Chem. Soc. 1976, 78, 867.

39
benzo[c]selenofeno 58 com excesso de n-BuLi e posterior captura do
intermediário dilitiado com cloroformiato de etila (Esquema 23).33
Esquema 23
Derivados halogenados de selenofenos e telurofenos são de grande
interesse em síntese orgânica, sendo esses aplicados em diferentes reações,
mais precisamente na formação de novas ligações carbono-carbono e carbono-
heteroátomo.
Um exemplo de síntese de derivados halogenados de selenofenos foi
descrito em 1996 por Takahashi, onde 2-Iodo-selenofenos 60 foram obtidos
através da reação de metalação do selenofeno com n-BuLi em éter etílico a 0
°C. Após esta reação, o intermediário litiado reagiu com iodo molecular,
fornecendo o produto em 70% de rendimento.34 Esta metodologia foi estendida
também para a síntese de 2,5-diiodo-selenofenos 61, onde para esta síntese
utilizou-se TMEDA juntamente com n-BuLi para gerar o ânion dilitiado, hexano
como solvente, obtendo-se o produto desejado em 50% de rendimento
(Esquema 24).
Esquema 24
Analogamente a iodo-selenofenos, iodotelurofenos foram sintetizados
por nosso grupo de pesquisa, através de reação de metalação de telurofenos
substituídos na posição 2 do anel heterocíclico, onde o intermediário litiado
33 Aqad, E.; Lakshmikantham, M. V.; Cava, M. P.; Broker, G. A.; Rogers, R. D. Org. Lett. 2003, 5, 2519. 34 Takahashi, K.; Tarutani, S. Heterocycles 1996, 43, 1927.

40
formado reagiu com I2 resultando nos 2-iodo-5-organoiltelurofenos 63 em bons
rendimentos (Esquema 25).7f
Esquema 25
A estabilidade de telurofenos frente a reagentes organometálicos foi
estudada por Müller e colaboradores. Neste estudo, os autores realizaram a
troca Te/Li do composto 2,5-difeniltelurofeno 64 utilizando n-BuLi em TMEDA,
levando ao intermediário 1,4-dilítio-1,4-difenilbuta-1,3-dieno 65. Este foi
capturado in situ com diferentes eletrófilos, levando aos dienos conjugados 66
substituídos com retenção de configuração (Esquema 26).35
Esquema 26
Recentemente, 2-iodo-selenofenos foram utilizados por nosso grupo de
pesquisa em reações de formação de ligação carbono-nitrogênio com amidas,
catalisada por sais de cobre.7h Verificou-se que esta reação apresenta os
melhores rendimentos das N-selenofenoamidas 67 utilizando-se K3PO4 como
base, na presença de etilenodiamina como ligante, sob refluxo de dioxano por
24 horas, suportando como fonte de nitrogênio uma série de amidas, tais como,
oxazolidinonas, lactamas, amidas alifáticas e aromáticas (Esquema 27).
Esquema 27
35 Luppold, E.; Müller, E.; Winter, W. J. Chem. Sci. 1976, 31, 1654.

41
Outro método descrito para a formação de uma nova ligação carbono-
heteroátomo utilizando-se 2-halocalcogenofenos foi publicado em 2005.9i Neste
artigo foi descrito o uso de 2-halo-selenofenos ou telurofenos como substratos
no acoplamento com organotióis sob catálise de CuI. Esta rota mostrou-se
pouco sensível ao efeito eletrônico, pois tolera organotióis substituídos tanto
por grupamentos retiradores como por doadores de elétrons, onde os produtos
de acoplamento 68 foram obtidos em bons rendimentos (Esquema 28).
Esquema 28
Um outro exemplo da utilização de selenofenos em reações de
acoplamento pôde ser observado quando se promoveu a reação de 2-halo-
selenofenos com ácidos borônicos sob catálise de sais de paládio.9g A reação
de obtenção dos selenofenos mono 69 ou di-arilados 70, partindo-se de 2-iodo-
selenofenos, ocorreu na presença de Pd(OAc)2, K2CO3/H2O em DME, podendo
ser realizada com ácidos borônicos arílicos contendo grupamentos elétrons
retiradores, doadores ou neutros. Adicionalmente, este procedimento permitiu a
obtenção de cetonas não simétricas contendo selenofeno 71 pelo tratamento
de 2-iodoselenofeno e ácido borônico em um processo de carbonilação
catalisado por Pd(PPh3)4 (Esquema 29).
Esquema 29

42
Compostos 2-halo-selenofenos também se mostraram úteis em reações
de acoplamento do tipo Sonogashira. Neste trabalho foi mostrado os resultados
do acoplamento entre 2-iodo e 2-bromo-selenofenos com alquinos terminais
em reação catalisada por Pd(PPh3)2Cl2 na presença de Et3N como base, DMF
como solvente e na ausência de sal de cobre, estabelecendo um novo
procedimento para a preparação de 2- e 2,5-alquinil-selenofenos 72 e 73
respectivamente, em bons rendimentos. Este procedimento mostrou-se
tolerante a uma série de alquinos terminais, incluindo álcoois, aminas e éteres
propargílicos, bem como alquinos alquílicos e arílicos (Esquema 30).9e
Esquema 30
1.4 REAÇÕES DE CICLIZAÇÃO CATALISADAS POR COBRE
A obtenção de diferentes heterocíclos vem sendo estudada a mais de
um século e uma grande variedade de metodologias para sua síntese são
encontradas na literatura. O desenvolvimento de novas metodologias,
eficientes e com economia de átomos, são atraentes e vem sendo bastante
exploradas. Dentre as inúmeras metodologias descritas, as reações catalisadas
por metais de transição vem se destacando, uma vez que essas reações
podem fornecer moléculas complexas e em condições brandas Reações de
ciclização catalisadas por metais de transição de alcinos contendo um átomo

43
nucleofílico em suas proximidades é um dos processos mais importantes na
química orgânica para obtenção de heterociclos.36
Um exemplo que ilustra esse tipo de reação, é a síntese de furanos
substituídos 76 via um sistema catalítico de Cu(I) em DMF de 2-(1-alquinil)-2-
alquen-1-onas 74 na presenção de um álcool 75 (Esquema 31).37 O uso de 1,5
equivalentes do álcool 75 e 10 mol% do catalisador mostrou-se a
estequiometria ideal para obtenção dos furanos substituídos 75.
Esquema 31
Os autores relataram que a reação se processa por dois caminhos
possíveis: (i) o ataque nucleofílico da carbonila no alcino complexado com o
cobre, resultando na formação do íon oxônium I estabilizado por ressonância
(parte A), seguido do ataque do álcool com subsequente protonação
regenerando a espécie de Cu(I) e formando o respectivos furanos 76. (ii) a
ativação do grupo carbonila com a coordenação do cobre com o oxigênio da
carbonila e a ligação π do alcino, o que aumenta a eletrofilicidade da posição β
facilitando a adição de Michael do álcool (parte B) resultando no intermediário
II. O intermediário cetona alquinílica II formada reage então in situ na presença
de Cu(I) para formar os furanos 76 (Esquema 32).
36 Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395. 37 Patil, N. T.; Wu, H.; Yamamoto, Y. J. Org. Chem. 2005, 70, 4531.
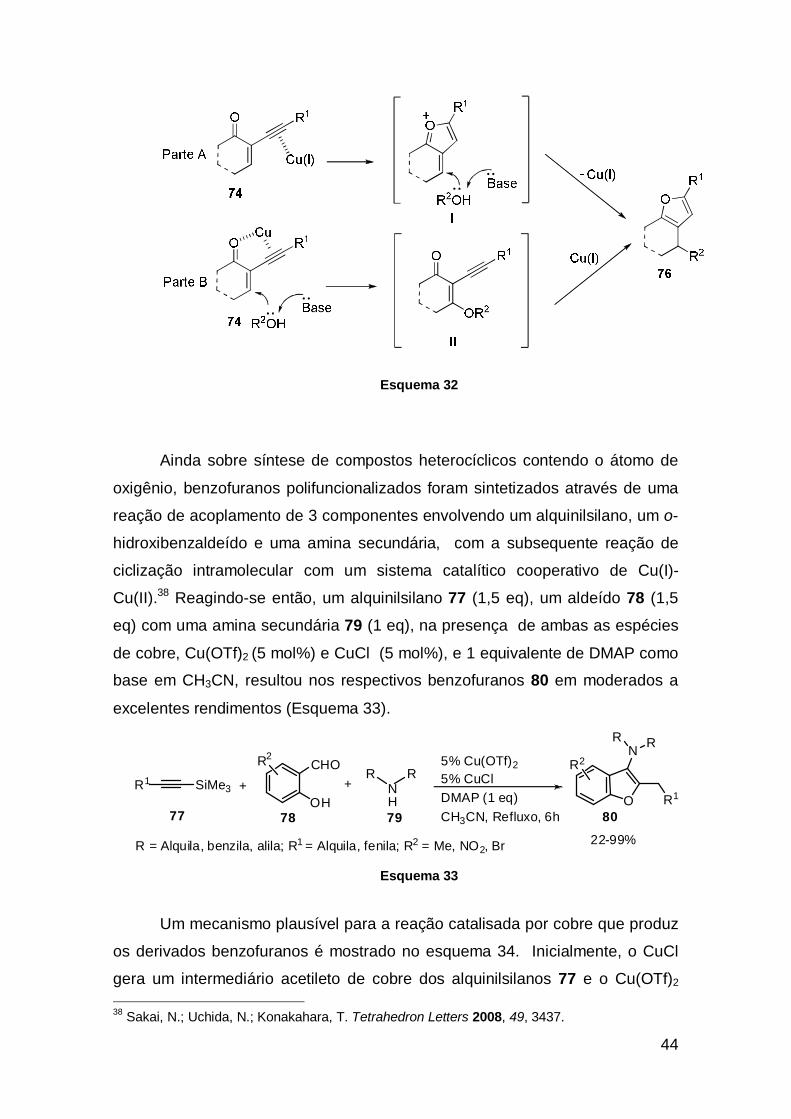
44
Esquema 32
Ainda sobre síntese de compostos heterocíclicos contendo o átomo de
oxigênio, benzofuranos polifuncionalizados foram sintetizados através de uma
reação de acoplamento de 3 componentes envolvendo um alquinilsilano, um o-
hidroxibenzaldeído e uma amina secundária, com a subsequente reação de
ciclização intramolecular com um sistema catalítico cooperativo de Cu(I)-
Cu(II).38 Reagindo-se então, um alquinilsilano 77 (1,5 eq), um aldeído 78 (1,5
eq) com uma amina secundária 79 (1 eq), na presença de ambas as espécies
de cobre, Cu(OTf)2 (5 mol%) e CuCl (5 mol%), e 1 equivalente de DMAP como
base em CH3CN, resultou nos respectivos benzofuranos 80 em moderados a
excelentes rendimentos (Esquema 33).
Esquema 33
Um mecanismo plausível para a reação catalisada por cobre que produz
os derivados benzofuranos é mostrado no esquema 34. Inicialmente, o CuCl
gera um intermediário acetileto de cobre dos alquinilsilanos 77 e o Cu(OTf)2 38 Sakai, N.; Uchida, N.; Konakahara, T. Tetrahedron Letters 2008, 49, 3437.
R1 SiMe3 +
R2CHO
OH+ N
H
RR5% Cu(OTf)25% CuClDMAP (1 eq)CH3CN, Refluxo, 6h
O
N
R1
R2
RR
77 78 79 80
22-99%R = Alquila, benzila, alila; R1 = Alquila, fenila; R2 = Me, NO2, Br

45
assume duas funções: (i) primeiro, ele se comporta como um ácido de Lewis na
geração in situ do intermediário imínio, que é proveniente dos substratos
aldeídos 78 e das aminas 79, e (ii) ativa a molécula do alcino para facilitar o
ataque nucleofílico intramolecular do grupo hidroxila na 5-exo-dig ciclização
(Esquema 34).
Esquema 34
Uma metodologia para a síntese de fosfaisocumarinas foi relatado por
Ding e colaboradores. Nesse trabalho, relataram a eficiente ciclização
intramolecular, catalisadas por sais cobre, dos ácidos o-etinilfenilfosfônicos
monoésteres 84, levando à formação dos respectivos fosfaisocumarinas 85. Os
materiais de partida foram preparados a partir da hidrólise básica dos
compostos 83, que foram sintetizados pelo acoplamento cruzado catalisados
por paládio dos correspondentes fenilperfluoralcanesulfonatos 82 com alquinos
terminais. Tendo-se os substratos 84 preparados, a ciclização intramolecular
usando-se CuI (10 mol%) em DMF resultou nos respectivos fosfaisocumarinas
85 em bons rendimentos (Esquema 35).39
39 Peng, A-Y.; Ding, Y-X. J. Am. Chem. Soc. 2003, 125, 15006.
N
R1O
R2RR
H
Cu(OTf)2
X
X = DMAP ou OH
OH
NR
RCu(OTf)2
R1
R2
O
NR
RR2
R1
Ar
NR
R
OH
Cu(OTf)2
R2
OHAr = Ar
NRR
OH
Cu(OTf)2
78 + 79
Cu-Cl
Cu R1 Me3Si-Cl
Me3 Si R1
77
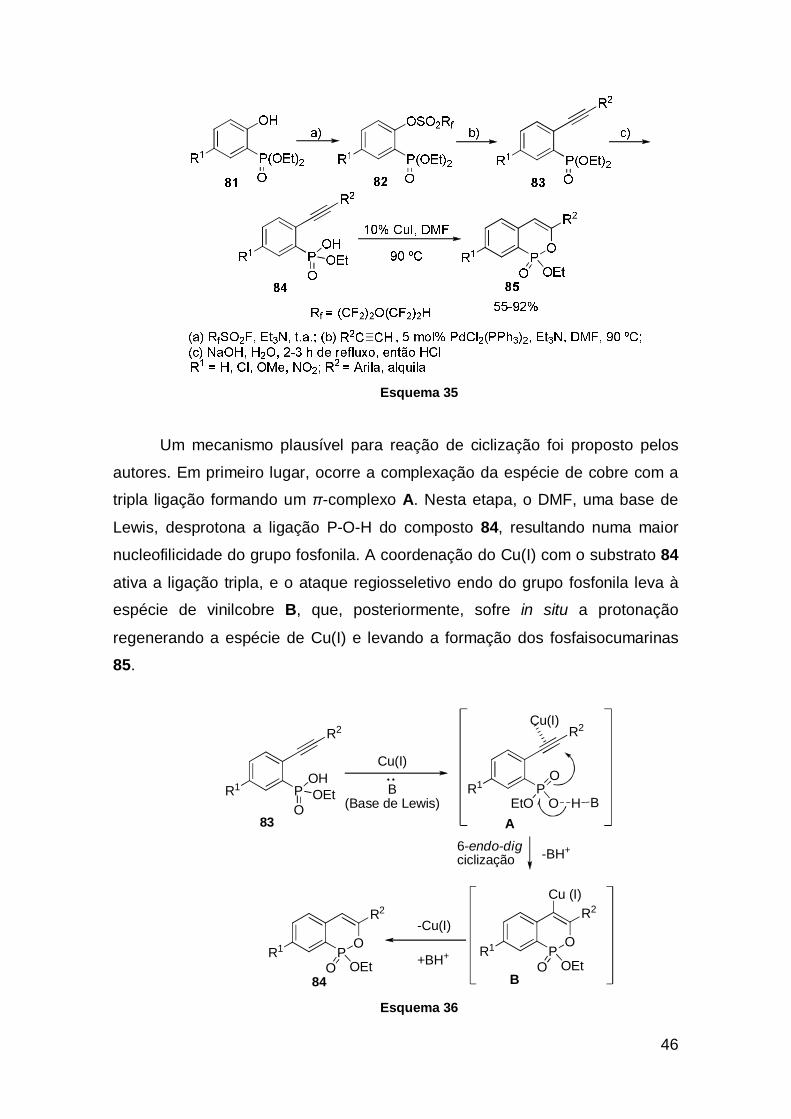
46
Esquema 35
Um mecanismo plausível para reação de ciclização foi proposto pelos
autores. Em primeiro lugar, ocorre a complexação da espécie de cobre com a
tripla ligação formando um π-complexo A. Nesta etapa, o DMF, uma base de
Lewis, desprotona a ligação P-O-H do composto 84, resultando numa maior
nucleofilicidade do grupo fosfonila. A coordenação do Cu(I) com o substrato 84
ativa a ligação tripla, e o ataque regiosseletivo endo do grupo fosfonila leva à
espécie de vinilcobre B, que, posteriormente, sofre in situ a protonação
regenerando a espécie de Cu(I) e levando a formação dos fosfaisocumarinas
85.
PR1
R2
R1 PO
O OEt
R2
PR1
O
R2
OEtOH
83
Cu(I)
O
EtO O H B
Cu(I)
B(Base de Lewis)
R1 PO
O OEt
R2
84
Cu (I)
-Cu(I)
+BH+
-BH+6-endo-digciclização
A
B Esquema 36
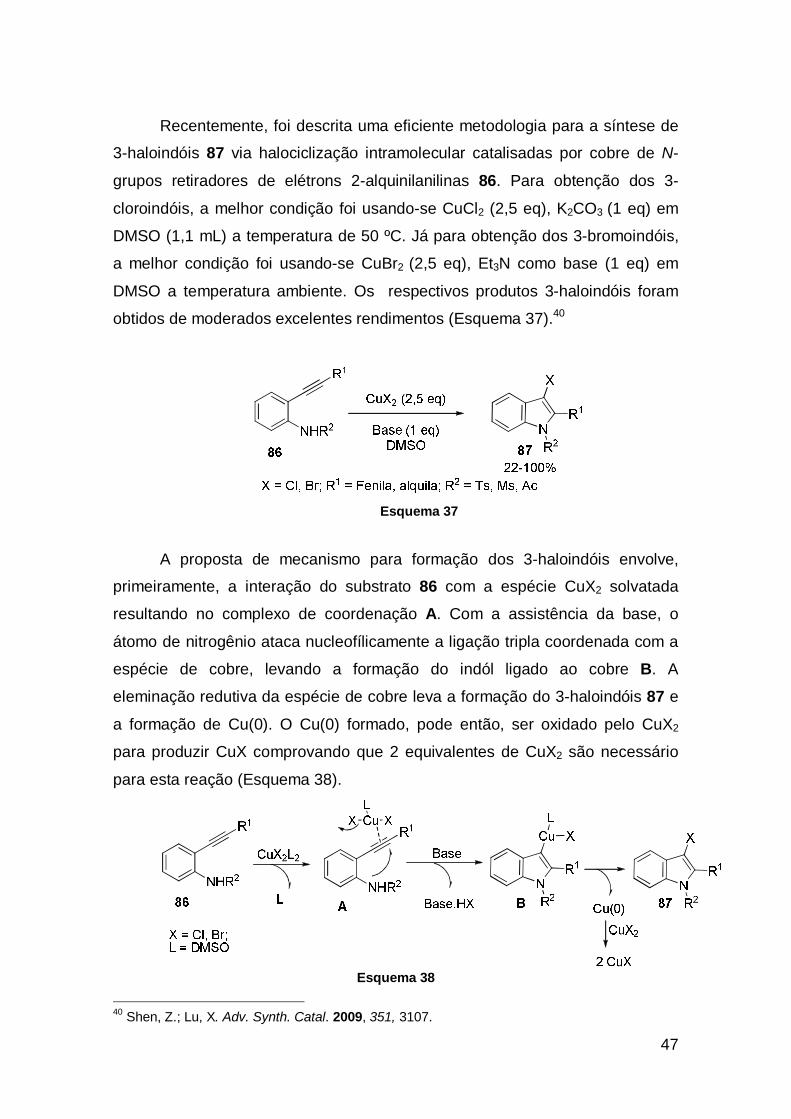
47
Recentemente, foi descrita uma eficiente metodologia para a síntese de
3-haloindóis 87 via halociclização intramolecular catalisadas por cobre de N-
grupos retiradores de elétrons 2-alquinilanilinas 86. Para obtenção dos 3-
cloroindóis, a melhor condição foi usando-se CuCl2 (2,5 eq), K2CO3 (1 eq) em
DMSO (1,1 mL) a temperatura de 50 ºC. Já para obtenção dos 3-bromoindóis,
a melhor condição foi usando-se CuBr2 (2,5 eq), Et3N como base (1 eq) em
DMSO a temperatura ambiente. Os respectivos produtos 3-haloindóis foram
obtidos de moderados excelentes rendimentos (Esquema 37).40
Esquema 37
A proposta de mecanismo para formação dos 3-haloindóis envolve,
primeiramente, a interação do substrato 86 com a espécie CuX2 solvatada
resultando no complexo de coordenação A. Com a assistência da base, o
átomo de nitrogênio ataca nucleofílicamente a ligação tripla coordenada com a
espécie de cobre, levando a formação do indól ligado ao cobre B. A
eleminação redutiva da espécie de cobre leva a formação do 3-haloindóis 87 e
a formação de Cu(0). O Cu(0) formado, pode então, ser oxidado pelo CuX2
para produzir CuX comprovando que 2 equivalentes de CuX2 são necessário
para esta reação (Esquema 38).
Esquema 38
40 Shen, Z.; Lu, X. Adv. Synth. Catal. 2009, 351, 3107.

48
Capítulo 2
Apresentação e Discussão dos Resultados

49
2 APRESENTAÇÃO E DISCUSSÃO DOS RESULTADOS
A seguir, serão apresentados e discutidos os resultados obtidos durante
a realização do presente trabalho. Na primeira parte do nosso trabalho, será
discutida a síntese de derivados de calcogenofenos, via reação de ciclização
de (Z)-seleno- e teluroeninos com dicalcogenetos de diorganoíla, catalisadas
por CuI. Em seguida, discutir-se-á a aplicação dos mesmos, em reações de
acoplamento cruzado do tipo Suzuki, catalisada por sais de paládio. Na
segunda parte, será discutida a síntese de 4-iodo-selenofenos[2,3-b]tiofenos,
via reações de ciclização eletrofílica de 2-butilselenil-3-alquiniltiofenos e o
estudo da reatividade destes compostos frente as reações de acoplamento
cruzado do tipo Suzuki, Negishi e com tióis catalisadas por sais de paládio e
cobre.
2.1 REAÇÃO DE CICLIZAÇÃO DE (Z)-CALCOGENOENINOS CATALISADAS POR COBRE
Nos últimos anos, nosso grupo de pesquisa vem atuando
primordialmente na área de síntese e reatividade de compostos
organocalcogenos. O foco dos trabalhos que vem sendo desenvolvidos está
centrado, principalmente, na síntese de novas moléculas contendo átomos de
selênio e telúrio suscetíveis a reações de acoplamento catalisadas por metais
de transição. Estes compostos desenvolvidos vêm sendo aplicados com
sucesso em diferentes classes de reações de acoplamento cruzado catalisadas
por sais de paládio, destacando-se, Sonogashira,9a-f, 41Suzuki,9g Negishi42 e,
mais recentemente, reações de acoplamento catalisadas por sais de cobre.9h-i
41 (a) Zeni, G.; Alves, D.; Pena, J. M.; Braga, A. L.; Stefani, H. A.; Nogueira, C. W. Org. Biom. Chem. 2004, 2, 803. (b) Zeni, G.; Menezes, P. H.; Moro, A. V.; Braga, A. L.; Silveira, C. C.; Stefani, H. A. Synlett 2001, 1473. (c) Braga, A. L.; Andrade, L. H.; Silveira, C. C.; Moro, A. V.; Zeni, G. Tetrahedron Lett. 2001, 42, 8563. (d) Zeni, G.; Perin, G.; Cella, R.; Jacob, R. G.; Braga, A. L.; Silveira, C. C.; Stefani, H. A. Synlett 2002, 975. (e) Zeni, G.; Nogueira, C. W.; Pena, J. M.; Pilissão, C.; Menezes, P. H.; Braga, A. L.; Rocha, J. B. T. Synlett 2003, 579. (f) Braga, A. L.; Vargas, F.; Zeni, G.; Silveira, C. C.; Andrade, L. H. Tetrahedron Lett. 2002, 43, 4399. 42 (a) Zeni, G.; Alves, D.; Braga, A. L.; Stefani, H. A.; Nogueira, C. W. Tetrahedron Lett. 2004, 45, 4823. (b) Alves, D.; Schumacher, R. F.; Brandão, R.; Nogueira, C. W.; Zeni, G. Synlett 2006, 7, 1035.

50
Neste contexto, grandes avanços foram feitos na formação de novas
ligações carbono-heteroátomo em reações de acoplamento cruzado, usando-
se sais de cobre como catalisador.13 Além disso, reações de compostos
orgânicos catalisadas por metais de transição foram ganhando destaque, e
transformações altamente seletivas de organocalcogênios têm sido
desenvolvidas usando-se catalisadores de paládio ou cobre.43
De acordo com nosso interesse no desenvolvimento de novos
compostos contendo átomos de selênio e telúrio em sua estrutura e em
concordância com os objetivos traçados, propôs-se a preparação de derivados
de calcogenofenos substituídos na posição 3 do anel heterocíclico 2, com a
estrutura geral mostrada na Figura 1.
Figura 3
Dessa forma, através de uma análise retrossintética do calcogenofeno 2
ilustrado na figura 3 (Esquema 39), acreditou-se que o anel heteroaromático
poderia ser formado através de uma reação de ciclização, utilizando-se para
isso, como substrato, um calcogenoenino de estereoquímica Z 1. Esta seria a
etapa chave do processo de obtenção dos selenofenos e dos telurofenos
desejados. Estes selenoeninos e teluroeninos de estereoquímica Z 1, poderiam
ser obtidos através de uma reação de hidrosselenação e hidroteluração de
diínos conjugados, onde estes, por sua vez seriam obtidos através de reações
de dimerização de alquinos (diínos simétricos)44 ou reações de acoplamento de
espécies alquinílicas catalisadas por sais de cobre (diínos não-simétricos)45. Os
alquinos utilizados para a obtenção dos correspondentes diínos, foram obtidos
através de fontes comerciais e disponíveis em nosso laboratório.
43 (a) Beletskaya, I. P.; Sigeev, A. S.; Peregudov, A. S.; Petrovskii, P. V. J. Organomet. Chem. 2000, 605, 96. (b) Beletskaya, I. P.; Ananikov, V. P. Pure Appl. Chem. 2007, 79, 1041. 44 Hay, A. S. J. Org. Chem. 1962, 27, 3320. 45 Alami, M.; Ferri, F. Tetrahedron Lett. 1996, 37, 2763.

51
Esquema 39
Inicialmente, planejou-se a síntese dos calcogenoeninos46 de
estereoquímica Z, estes considerados substratos chave para a síntese dos
derivados de calcogenofenos desejados. Reagindo-se o composto 1,4-
difenilbutadiíno com o ânion butiltelurolato, gerado da reação entre ditelureto de
dibutila com NaBH4 em etanol, sob refluxo por 8 horas, obteve-se o
correspondente (Z)-teluroenino 1a como único estereoisômero, em 75% de
rendimento. Da mesma maneira, o (Z)-selenoenino 3a foi obtido em 70% de
rendimento, sob refluxo por 5h (Esquema 40).
Esquema 40
A partir da síntese do composto 1a, estudamos as melhores condições
para as reações de ciclização, para a síntese dos derivados de telurofenos.
Para isto, o (Z)-teluroenino 1a e disseleneto de difenila foram escolhidos como
substratos padrões para estas reações. Reagindo-se o composto 1a (0,25
mmol) com disseleneto de difenila (1,1 eq), utilizando-se DMSO como solvente
(3mL), temperatura de 110 ºC e usando-se CuI como catalisador (10 mol%),
obteve-se o produto de ciclização 2,5-difenil-3-(fenilseleno)-telurofeno 2a em
79% de rendimento após 10 horas de reação (Esquema 41).
46 (a) Dabdoub, M. J.; Baroni, A. C. M.; Lenardão, E. J.; Gianeti, T. R.; Hurtado, G. R. Tetrahedron 2001, 57, 4271. (b) Zeni, G.; Stracke, M. P.; Nogueira, C. W.; Braga, A. L.; Menezes, P. H.; Stefani, H. A. Org. Lett. 2004, 6, 1135. (c) Zeni,G.; Formiga, H. B.; Comasseto, J.V. Tetrahedron Lett. 1999, 41, 1311.

52
Esquema 41
Outros sais de cobre, como CuCl, CuBr, CuCN, Cu(OTf)2, Cu(OAc)2,
CuCl2 e CuBr2, foram menos eficazes nestas condições. Em relação a
quantidade do catalisador, 5 mol % foi insuficiente para o consumo total do
substrato 1a e o produto foi isolado em 62% após 36 horas de reação.
Aumentando-se a quantidade de CuI para 15 e 20 mol%, não houve nenhuma
melhora quanto ao rendimento do produto 2a. Quando a reação foi realizada na
ausência do catalisador, apenas os materiais de partidas foram recuperados.
Em comparação, quando CuI foi usado, na ausência de disseleneto de difenila,
apenas os substratos foram recuperandos. Estas observações sugerem a
necessidade de um complexo PhSe/CuI47 para a conversão catalítica do
substrato 1a ao 2a.
A quantidade do disseleneto de difenila também foi estudada.
Observamos que, aumentando-se a quantidade para 2 equivalentes, o
rendimento e o tempo da reação não foi alterado. No entando, quando
diminuída para 0.5 equivalentes, o rendimento da obtenção do 2a diminuiu para
40% de rendimento.
Quanto a influência do solvente nessa reação, os melhores resultados
obtidos foram usando-se DMSO. Outros solventes como THF, CH2Cl2 e MeCN
apresentaram resultados insatisfatórios, com rendimentos abaixo de 20%.
Solventes, como tolueno, dioxano e DMF exigiram um tempo de reação
superior (48 h) ao DMSO além de apresentarem rendimentos inferiores. Assim,
concluímos que manter a temperaturada reação entre 110 a 120 ºC é um fator
determinante para o bom desenvolvimento da mesma. Para comprovar esse
fato, a reação foi realizada em DMSO a 100 ºC e a reação exigiu 48 horas para
conversão do 1a em 2a.
Sendo assim, em uma etapa posterior a determinação da melhor
condição da reação, analisou-se a influência do grupamento R ligado ao átomo 47 (a) Taniguchi, N. J. Org. Chem. 2007, 72, 1241. (b) Taniguchi, N.; Onami, T. J. Org. Chem. 2004, 69, 915.

53
de telúrio nas reações de ciclização. Acreditando que esses grupos estejam
diretamente ligados ao desenvolvimento da reação, decidimos explorar essa
influência com diferentes grupos alquila e arila, e os resultados são mostrados
na tabela 1.
Tabela 1. Influência do grupamento R ligado ao telúrio nas reações de
ciclização.a
# (Z)-Teluroenino 1a-d Tempo (h) Rendimento 2a (%)
1 1a (R = Bu) 24 79
2 1b (R = Et) 24 44
3 1c (R = Me) 24 47
4 1d (R = Ph) 48b - a Reações realizadas com (Z)-Teluroeninos (0,25 mmol), PhSeSePh (1,1 eq), CuI (10 mol%) em DMSO (3 mL). b (Z)-teluroenino 1d foi recuperado quantitativamente. Uma análise mais detalhada desses resultados reveleram que a reação
com teluroeninos contendo um grupo alquila ligado ao átomo calcogênio
resultou no telurofeno 2a em rendimentos satisfatórios, apesar de ter sido
menor para teluroeninos contendo um grupo metílico ou etílico (Tabela 1;
exemplos 2-3). No entanto, a reação com o teluroenino 1d, o qual tem um
grupo fenila ligado ao átomo de telúrio, o produto desejado não foi obtido
mesmo em 48 horas de reação (Tabela 1; exemplo 4). Estes resultados
demonstram que a formação do produto 2a depende significantemente de
teluroeninos que contenham um C-sp3 ligado diretamente ao átomo de telúrio.
Após uma análise detalhada dos experimentos realizados até então,
considerou-se como condição ideal para as reações de ciclização a utilização
do (Z)-teluroenino 1a (0,25 mmol), CuI como catalisador (10 mol%), 1,1
equivalentes do dicalcogeneto de diorganoíla em DMSO (3 mL) a temperatura
de 110 ºC. Assim, estendeu-se essa condição para diferentes dicalcogenetos
de diorganoíla e os resultados são mostrados na tabela 2.
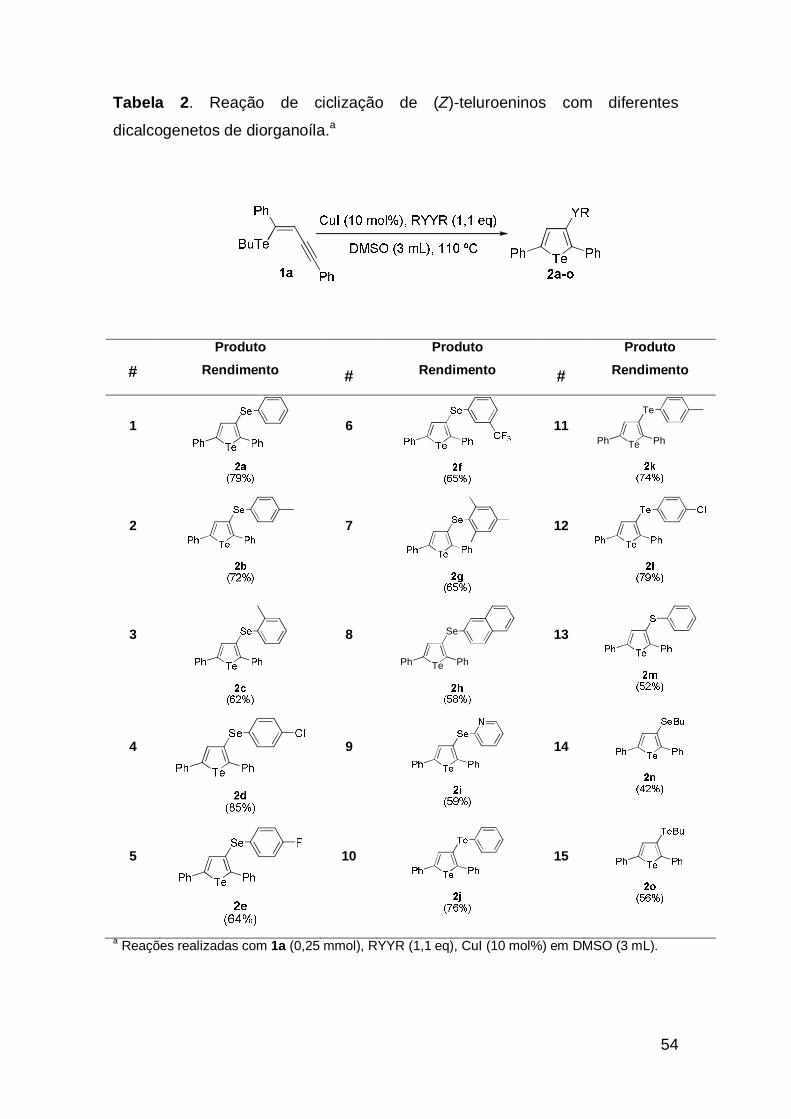
54
Tabela 2. Reação de ciclização de (Z)-teluroeninos com diferentes
dicalcogenetos de diorganoíla.a
# Produto
Rendimento
#
Produto Rendimento
#
Produto Rendimento
1
6
11
Te PhPh
Te
2
7
12
3
8
Te PhPh
Se
13
4
9
14
5
10
15
a Reações realizadas com 1a (0,25 mmol), RYYR (1,1 eq), CuI (10 mol%) em DMSO (3 mL).

55
Analisando-se a Tabela 2, pode-se perceber que estas condições de
reação promoveram com eficiência a reação de ciclização do (Z)-teluroenino 1a
com diferentes dicalcogenetos de diorganoíla. Ambos disselenetos impedidos e
desimpedidos estericamente levaram aos derivados telurofenos em bons
rendimentos (Tabela 2, exemplos 1, 7-8). Uma análise mais detalhada dos
resultados revelou que a reação também não é sensível aos efeitos eletrônicos
dos substituíntes do anel aromático. Por exemplo, disselenetos de diarila
contendo substituíntes retiradores de elétrons forneceram os produtos
desejados em rendimentos semelhantes aos substituíntes doadores de elétrons
(Tabela 2, exemplos 2-6).
Através dessa metodologia, foi possível preparar telurofenos altamente
funcionalizados, não só usando disselenetos de diarila como substratos
(Tabela 2, exemplos 1-9), mas também diteluretos de diarila (Tabela 2,
exemplos 10-12). Percebe-se que para todos os dicalcogenetos de diorganoíla,
os telurofenos foram obtidos em bons rendimentos. Na prepararação do
telurofeno 2m, o dissulfeto de difenila não comportou-se como os demais
dicalcogenetos e o telurofeno 2m foi obtido em apenas 32% de rendimento.
Então, quando foi usado 2 equivalentes de benzenotiól no lugar do dissulfeto
de difenila, o produto foi obtido em 52% de rendimento (Tabela 2, exemplo 13).
Observa-se na tabela que o disseleneto de dialquila e o ditelureto de dialquila
resultaram nos respectivos produtos 2n e 2o em rendimentos moderados
(Tabela 2, exemplos 14-15).
É válido ressaltar que na preparação do telurofeno 2a, o produto
BuSePh foi quantificado em 65% de rendimento. Devido a dificuldade de
separação dos demais dicalcogenetos de diorganoíla, apenas foi observado
por cromatografia em plaquinha delgada a formação dos produtos BuYAr.
No intuito de aumentar o escopo da reação, aplicamos essa metodologia
de preparação dos telurofenos na preparação de derivados de selenofenos a
partir dos (Z)-selenoeninos 3. Os resultados são mostrados na tabela 3.
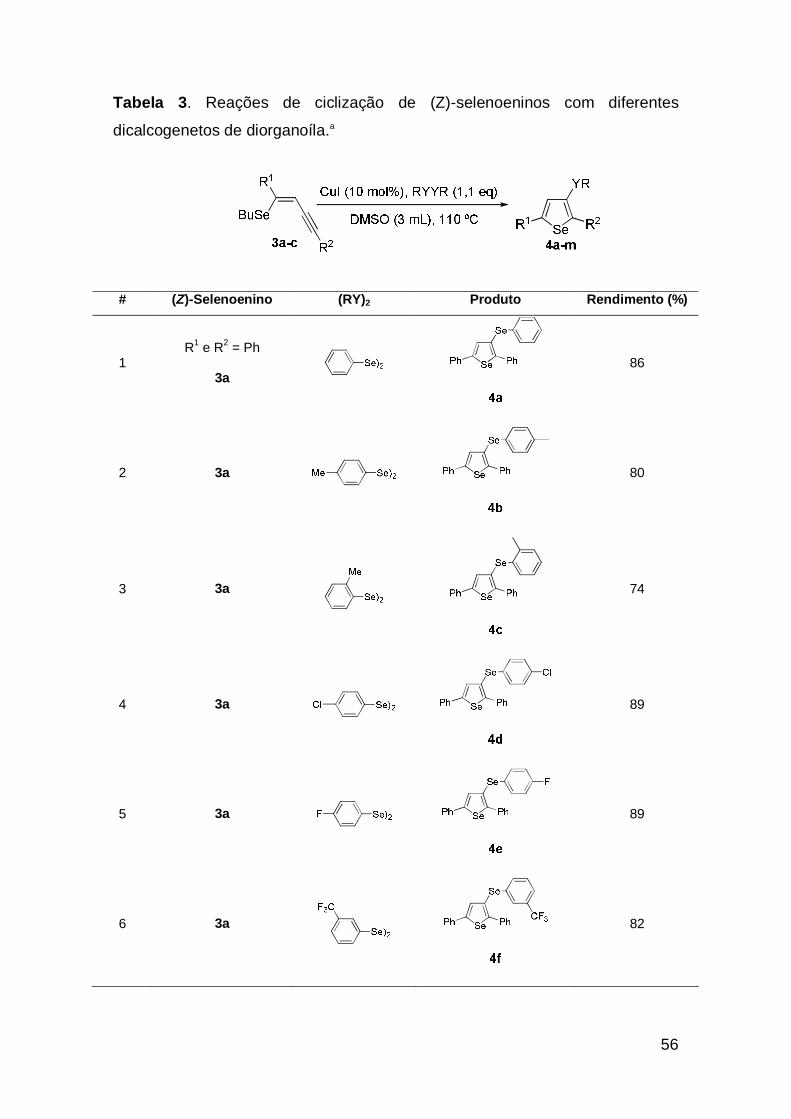
56
Tabela 3. Reações de ciclização de (Z)-selenoeninos com diferentes
dicalcogenetos de diorganoíla.a
# (Z)-Selenoenino (RY)2 Produto Rendimento (%)
1 R1 e R2 = Ph
3a
86
2
3a
80
3
3a
74
4 3a
89
5 3a
89
6 3a
82
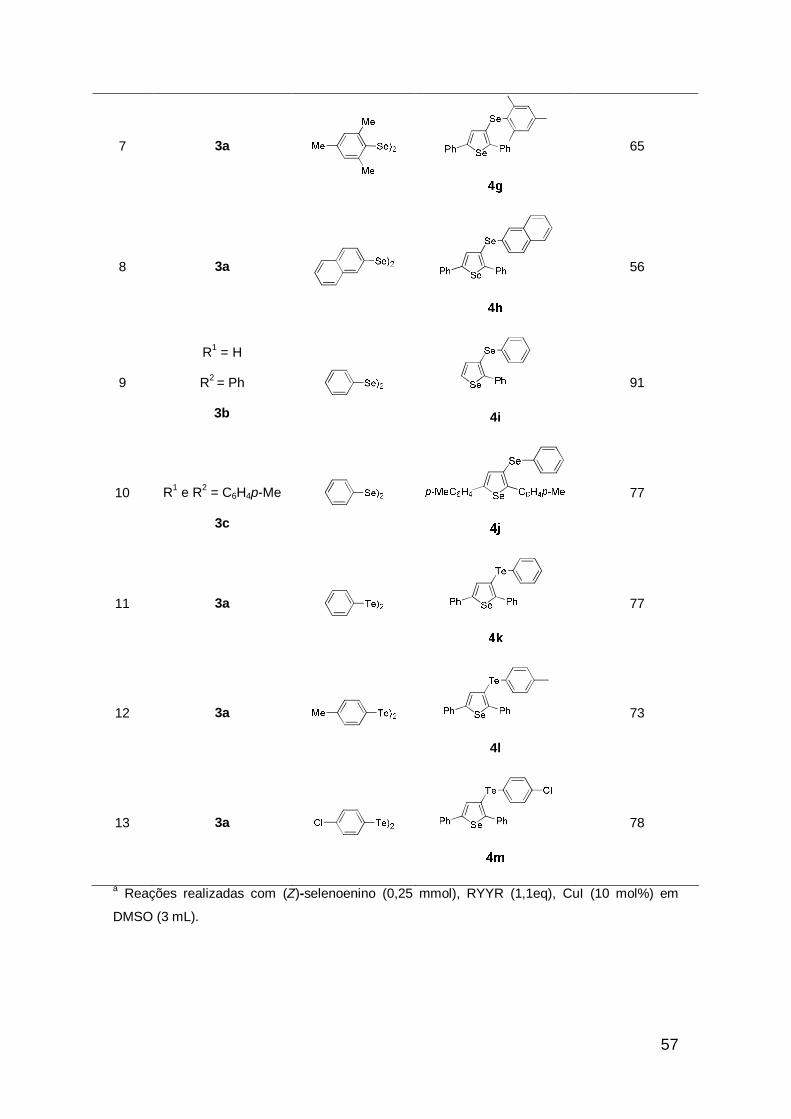
57
7 3a
65
8 3a
56
9
R1 = H
R2 = Ph
3b
4i
91
10
R1 e R2 = C6H4p-Me
3c
77
11 3a
77
12 3a
4l
73
13 3a
78
a Reações realizadas com (Z)-selenoenino (0,25 mmol), RYYR (1,1eq), CuI (10 mol%) em
DMSO (3 mL).

58
Analisando-se a tabela 3, percebe-se que estas condições puderam ser
estendidas à síntese de selenofenos com uma grande variedade de
substituíntes na posição 3 do anel heterocíclico. Tanto grupos doadores quanto
retiradores de elétrons no anel aromático dos disselenetos de diarila, levaram
aos derivados selenofenos em bons rendimentos, mostrando que a reação não
é sensível aos efeitos eletrônicos (Tabela 3, exemplos 2-6). Quando usados
disselenetos impedidos, as reações também não mostraram-se sensíveis aos
efeitos estéricos (Tabela 3, exemplos 7 e 8). Excelente rendimento foi obtido
usando o selenoenino 3b que resultou no selenofeno 4i em 91%, com
hidrogênio como substituinte na posição 5 do anel heterocíclico (Tabela 3,
exemplo 9). O uso de grupos doadores nos anéis aromáticos do selenoenino
não levou a melhores resultados para obtenção do produto (Tabela 3, exemplo
10). Vale mencionar que na tentativa de aumentarmos a classe de selenofenos
funcionalizados, estendemos a metodologia para uso de diteluretos de diarila e
assim, bons resultados também puderam ser obtidos (Tabela 3, exemplos 11-
13).
Para elucidar o mecanismo da reação de ciclização dos
calcogenoeninos nos derivados de calcogenofenos, baseamo-nos em alguns
aspectos experimentais obtidos: (1) em todos os casos foi observado a
formação de BuYAr como subproduto das reações; (2) não foi obtido o produto
de ciclização quando havia um carbono sp2 ligado ao átomo de calcogênio; (3)
nenhum produto foi obtido na ausência do sal de cobre ou do dicalcogeneto de
diorganoila. Estes aspectos nos levam acreditar que:
(a) O CuI pode estar se inserindo na ligação Y-Y do dicalcogeneto
formando uma espécie de selenolato quadrado planar tetracoordenado de
Cu(III);48
(b) Ocorre, então, a coordenação do metal central com a ligação tripla
levando a uma espécie organocatiônica de Cu(III);
(c) Após, o ataque anti do átomo calcogênio a ligação tripla ativada
produz um sal de calcogênio;
48 Vicente, J.; Herrero, P. G.; Sánchez, Y. G.; Jones, P. G.; Bautista, D. Eur. J. Inorg. Chem. 2006, 115.

59
(d) Por fim, um ataque nucleofílico do calcogenolato remanescente ao
grupo alquila ligado ao calcogênio leva a formação do produto BuYAr
(Esquema 42).
n-BuY
Ar
Ar
ArYYAr
Y ArAr
Cu
[CuI]
n-BuY
Ar
Ar
[Cu]
n-Bu
YAr
I
Y ArAr
YAr
n-Bu
+
+
+
Y ArAr
YAr
n-BuYAr+
CuArY
ArY
I
ICu
YAr
YArIII III
ArY
[CuI(YAr)]¯
III
(a)
(b)
(c)
(d)
Esquema 42

60
2.1.1 SÍNTESE DE 3-ARILTELUROFENOS VIA REAÇÕES DE ACOPLAMENTO CRUZADO DO TIPO SUZUKI.
Os derivados calcogenofenos, uma vez obtidos, do ponto de vista
sintético, tornam-se compostos potenciais para a síntese de calcogenofenos
altamente substituídos. Acreditamos que as reações de acoplamento cruzado
catalisadas por metais de transição seriam úteis para transformar a
funcionalidade da posição 3 do anel calcogenofeno em outros substituintes.
Nesse contexto, as reações de acoplamento cruzado do tipo Suzuki
catalisadas por paládio, de haletos de arila com ácidos ou ésteres borônicos,
tornou-se um método comum e conveniente na química orgânica sintética.49
Recentemente, avanços significativos têm sido realizados na utilização de
reagentes organoboro na formação de novas ligações carbono-carbono em um
grande número de reações catalisadas por sais de paládio. Entre eles, o uso de
organotrifluoroboratos de potássio apresentam algumas vantagens em
comparação aos ácidos e ésteres borônicos, como por exemplo, ser mais
nucleofílico, estáveis ao ar e por serem sólidos cristalinos.50
Na última década, a química de compostos organotelúrio foi
extensivamente explorada e muitos métodos que empregam compostos de
telúrio foram reportados.51 Dentre esses métodos, reagentes de organotelúrio
foram utilizadas com sucesso como espécie eletrofílica11b em muitas reações
de acoplamento cruzado catalisadas por metais de transição como,
Sonogashira,19b,19d Negishi,20a Heck,52 e Suzuki-Miyaura.53
49 (a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. (b) Suzuki, A. Pure Appl. Chem. 1994, 66, 213. (c) Suzuki, A. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F., Stang, P. J., Eds.; Wiley-VHC: Weinheim, 1998. (d) Lloyd-Williams, P.; Giralt, E. Chem. Soc. Rev. 2001, 30, 145. 50 (a) Molander, G. A.; Ellis, N. Acc. Chem. Res. 2007, 40, 275. (b) Molander, G. A.; Petrillo, D. E.; Landzberg, N. R.; Rohanna, J. C.; Biolatto, B. Synlett 2005, 1763. (c) Molander, G. A.; Figueroa, R. Aldrichimica Acta 2005, 38, 49. (d) Molander, G. A.; Felix, L. A. J. Org. Chem. 2005, 70, 3950. (e) Molander, G. A.; Yun, C. S.; Ribagorda, M.; Biolatto, B. J. Org. Chem. 2003, 68, 5534. (f) Molander, G. A.; Biolatto, B. J. Org. Chem. 2003, 68, 4302. (g) Molander, G. A.; Katona, B. W.; Machrouhi, F. J. Org. Chem. 2002, 67, 8416. (h) Molander, G. A.; Reviro, M. R. Org. Lett. 2002, 4, 107. 51 Para revisão: (a) Petragnani, N.; Stefani, H. A. Tetrahedron 2005, 61, 1613. (b) Petragnani, N.; Comasseto, J. V. Synthesis 1986, 1. (c) Petragnani, N.; Comasseto, J. V. Synthesis 1991, 793, 897. (d) Comasseto, J. V.; Ling, L. W.; Petragnani, N.; Stefani, H. A. Synthesis 1997, 373. 52 (a) Braga, A. L.; Rhoden, C. R. B.; Zeni, G.; Silveira, C. C.; Andrade, L. H. J. Organomet. Chem. 2003, 682, 35. (b) Nishibayashi, Y.; Cho, C.-S.; Ohe, K.; Uemura, S. J. Organomet. Chem. 1996, 507, 197. (c) Nishibayashi, Y.; Cho, C.-S.; Ohe, K.; Uemura, S. J. Organomet. Chem. 1996, 526, 335.

61
Desde que a reação de Suzuki tem se mostrado bem sucedida para o
arilação seletiva de heterociclos,54 decidimos explorar o potencial sintético do
telurofeno 2o frente as reações de acoplamento cruzado do tipo Suzuki usando
ácidos borônicos e organotrifluorboratos de potássio como reagentes. Assim, o
telurofeno 2o (0,25 mmol), MeOH (2 mL), Pd(PPh3)4 (5 mol%), a respectiva
espécie de boro (1,2 eq), Ag2O (2 eq) e Et3N (3 eq) reagiram por 90 minutos
para levar aos correspondentes 3-ariltelurofenos 5a e 5b em bons rendimentos
(Esquema 43).
Esquema 43
53 (a) Stefani, H. A.; Cella, R.; Dorr, F. A.; Pereira, C. M. P.; Zeni, G., Jr.; Gomes, M. Tetrahedron Lett. 2005, 46, 563. (b) Kang, S. K.; Hong, Y. T.; Kim, D. H.; Lee, S. H. J. Chem. Res. 2001, 283. 54 (a) Tung, D. T.; Tuan, D. T.; Rasool, N.; Villinger, A.; Reinke, H.; Fischer, C.; Langer, P. Adv. Synth. Cat. 2009, 351, 1595. (b) Ullah, I.; Khera, R. A.; Hussain, M.; Villinger, A.; Langer, P. Tetrahedron Lett. 2009, 50, 4651. (c) Dang, T. T.; Rasool, N.; Dang, T. T.; Reinke, H.; Langer, P. Tetrahedron Lett. 2007, 48, 845. (d) Tuan, D. T.; Tung, D. T.; Langer, P. Synlett 2006, 2812. (e) Bellur, E.; Langer, P. Eur. J. Org. Chem. 2005, 22, 4815.

62
2.2 SÍNTESE DE 4-IODOSELENOS[2,3-b]TIOFENOS VIA REAÇÕES DE CICLIZAÇÃO ELETROFÍLICA DE 2-ORGANOCALCOGENO-
3-ALQUINILTIOFENOS
Os derivados de heterocíclos fundidos são de grande importância devido
a grande aplicação em química orgânica e, pricipalmente, pelas suas
importantes propriedades biológicas.55 A síntese de heterocíclos fundidos
também têm atraído considerável atenção devido suas aplicações na síntese
de materiais com propriedades eletrônicas.7,8 Entretanto, pouco se sabe sobre
heterocíclos fundidos de selênio e enxofre com diferentes características e
aplicações na literatura. Desta forma, o desenvolvimento de novas
metodologias para a síntese desses compostos, vem a ser útil devido ao
interesse de explorar seu potencial biológico ou farmacêutico.56
Neste contexto, reações de ciclização eletrofílica de alquinos, contendo
um nucleófilo em proximidade a ligação tripla é uma eficiente via de preparação
de uma grande variedade de carbociclos e heterociclos, destacando-se,
benzofuranos, benzotiofenos, indóis, tiofenos, furanos, entre outros, como
também compostos heterocíclicos contendo selênio, por exemplo,
benzo[b]selenofenos.2
De acordo com nosso interesse no desenvolvimento de novos
compostos contendo átomos de selênio em sua estrutura e em concordância
com os objetivos traçados, foi proposto a preparação de derivados de
selenofenos[2,3-b]tiofenos fundidos funcionalizados na posição 4 do anel
heterocíclico 7, com a estrutura geral mostrada na Figura 4.
55 (a) Manetti, F.; Santucci, A.; Locatelli, G. A.; Maga, G.; Spreafico, A.; Serchi, T.; Orlandini, M.; Bernardini, G.; Caradonna, N. P.; Spallarossa, A.; Brullo, C.; Schenone, S.; Bruno, O.; Ranise, A.; Bondavalli, F.; Hoffmann, O.; Bologna, M.; Angelucci, A.; Botta, M. J. Med. Chem. 2007, 50, 5579. (b) Naya, S.; Ohtoshi, H.; Nitta, M. J. Org. Chem. 2006, 71, 176. (c) Wendt, J. A.; Deeter, S. D.; Bove, S. E.; Knauer, C. S.; Brooker, R. M.; Augelli-Szafran, C. E.; Schwarz, R. E.; Kinsora, J. J.; Kilgore, K. S. Bioorg. Med. Chem. Lett. 2007, 17, 5396. 56 (a) Sommen, G. Comel, A.; Kirsch, G. Phosphorus, Sulfur and Silicon and the Related Elements. 2005, 180, 939. (b) Sommen, G.; Comel, A.; Kirsch, G. Synthesis. 2004, 451. (c) Yasuike, S.; Kurita, J.; Tsuchiya, T. Heterocycles 1997, 45, 1891. (d) Kazuo Takimiya, K.; Konda, Y.; Ebata, H.; Niihara, N.; Otsubo, T. J. Org. Chem. 2005, 70, 10569.

63
Figura 4
Dessa forma, através de uma análise retrossintética do selenofeno 7,
pode-se inferir que o anel heteroaromático poderia ser formado através de uma
reação de ciclização eletrofílica, utilizando-se para isso, como substratos, os 2-
organoseleno-3-alquinilltiofenos 6 (Esquema 44). Estes, poderiam ser obtidos a
partir dos 3-alquiniltiofenos através da introdução do grupo organocalcogeno.
Esta seria a etapa chave do processo de obtenção dos produtos desejados.
Esquema 44
Para a preparação desses 2-organoseleno-3-alquiniltiofenos, utilizamos
o acoplamento do tipo Sonogashira de 3-bromotiofenos, obtido por fontes
comerciais, com diferentes alquinos terminais catalisados por sais de paládio.57
Depois disso, para a introdução do grupo organoseleno, gerou-se o
intermediário 2-lítio-3-alquiniltiofeno via reação do 3-alquiniltiofeno com n-BuLi
(1 eq), em THF a -78 ºC por 1 hora, seguido da adição do selênio elementar e
da alquilação usando-se o adequado brometo de alquila (Esquema 45).11
Na obtenção dos substratos 6, os rendimentos variaram entre 60 e 80%.
Em todos os casos foram observados a litiação da posição 5 do anel tiofeno
resultando nos 5-organoseleno-3-alquiniltiofenos como subprodutos da reação.
57 Alves, D.; Reis, J. S.; Luchese, C.; Nogueira, C. W.; Zeni,G. Eur. J. Org. Chem. 2008, 377

64
Esquema 45
A partir da síntese do composto 6a (R = Ph e R1 = n-Bu), estudamos as
melhores condições para as reações de ciclização eletrofílica, para a tentativa
de síntese dos derivados heterocíclos fundidos. Para isto, o 2-(butilselenil)-3-
(feniletinil)-tiofeno 6a e iodo molecular (I2) foram escolhidos como substratos
padrões para estas reações. Adicionando-se lentamente uma solução de I2 (1,1
eq em 3 mL de CH2Cl2) numa solução do composto 6a (0,25 mmol em 2 mL de
CH2Cl2), obteve-se o produto de ciclização eletrofílica 4-iodo-5-
fenilselenofeno[2,3-b]tiofeno 7a em 81% de rendimento após 5 minutos de
reação (Esquema 46).
Esquema 46
Após este resultado preliminar, um estudo relativo à influência do
solvente nesta reação de ciclização foi realizado. Diferentes solventes foram
testados em reações do substrato 6a com I2, sendo os resultados obtidos
mostrados na Tabela 4.

65
Tabela 4. Estudo de solventes nas reações de ciclização do composto 6a com
I2.a
# Solvente Tempo (min.) Rendimento (%)
1 CH2Cl2 5 81 2 THF 120 87 3 Et2O 90 66 4 MeOH 45 80 5 Hexano 45 77
6 MeCN 10 76 a Reações utilizando-se o composto 6a (0,25 mmol), I2 (1,1 eq) em 5 mL de solvente.
Analisando-se a Tabela 4, bons rendimentos do produto de ciclização 7a
foram obtidos em todos solventes testados (Tabela 4, exemplos 1-6), enquanto
o CH2Cl2 resultou no bom rendimento de 81% num curto espaço de tempo
(Tabela 4, exemplo 1). Quando THF, Et2O, MeOH, MeCN e hexano foram
empregados como solventes, bons rendimentos do produto 7a foram obtidos,
porém requerendo maiores tempos de reação.
Adicionalmente, a quantidade do eletrófilo também foi estudada.
Observamos que, aumentando-se a quantidade de iodo para 1,5 ou 2
equivalentes, o rendimento da reação não foi alterado.
Numa etapa posterior do estudo, analisou-se a influência do grupamento
R ligado ao átomo de selênio, tais como, alquila, arila e benzila nas reações de
ciclização. Assim, os 2-organoseleno-3-alquiniltiofeno 6a, 6p-r sintetizados
foram submetidos a reações de ciclização com iodo utilizando-se CH2Cl2 como
solvente, e os resultados destas reações estão expressos na Tabela 5.

66
Tabela 5. Influência do grupamento R1 ligado as selênio nas reações de
ciclização.a
# Substrato 6a-f Tempo (min) Rendimento 7a (%)
1 6a - nBu 5 81
2 6p - nEt 15 84
3 6q - Bz 120 46
4 6r - Ph 120 - a Reações realizadas com os substratos 6 (0,25 mmol), I2 (1,1 eq) em CH2Cl2 (5 mL).
Uma análise dos resultados demonstrados na Tabela 5 mostram que os
substratos contendo grupos R n-Butila e etila ligados diretamente ao átomo de
selênio, fornecem o respectivo produto de ciclização 7a rapidamente em bons
rendimentos (Tabela 5; exemplos 1-2). O 2-benzilseleno-3-feniletiniltiofeno,
também levou a obtenção do produto 7a, porém, o rendimento foi moderado e
com maior tempo de reação (Tabela 5, exemplo 3). Nota-se que quando o
grupamento R ligado ao átomo de selênio é fenila a reação não ocorre e,
consequentemente, o substrato 6r foi recuperado. Estes resultados
demonstram que a eficiência na formação do produto 7a depende
significantemente de efeitos estéricos e esta reação de ciclização ocorre
apenas em compostos que contenham um C-sp3 ligado diretamente ao átomo
de selênio (Tabela 5, exemplo 4).
De acordo com os trabalhos descritos na literatura para moléculas
análogas,2g acredita-se que esta reação de ciclização eletrofílica de 2-
organoseleno-3-alquiniltiofenos, segue as seguintes etapas de reação:
1 – Coordenação da molécula de I2 na ligação tripla do substrato 6
formando o intermediário iodônio A;
2 – Um ataque nucleofílico anti do átomo de selênio ao intermediário
iodônio A, fornecendo o intermediário heterocíclico B;

67
3 – Por fim, o ânion iodeto remanescente reage nucleofilicamente com o
grupamento R ligado ao átomo de selênio, levando a formação do produto
desejado e a formação de um iodeto orgânico. É relevante destacar que para a
reação acontecer, o grupamento R necessita ter um carbono sp3 ligado
diretamente ao átomo de selênio (Esquema 47).
Esquema 47
Após uma análise detalhada dos experimentos realizados até então,
considerou-se como condição ideal para as reações de ciclização eletrofílica a
utilização de 2-butilseleno-3-alquiniltiofenos (0,25 mmol), I2 (1,1 eq), CH2Cl2
como solvente a temperatura ambiente.
Tendo-se determinado esta condição e considerando-a satisfatória,
estendeu-se a mesma para a reação de ciclização eletrofílica com diferentes
eletrófilos, bem como, a série de 2-butilseleno-3-alquiniltiofenos previamente
preparados, e os resultados são mostrados na Tabela 6.
68
Tabela 6. Reações de ciclização com diferentes eletrófilos ou 2-butlicalcogeno-
3-alquiniltiofenos.a
# Substrato E tempo (min)
Produto rendimento
1
I2 5
2 ICl 5
3 PhSeBr 10
4
I2
5
5 ICl 5
6
I2
7
7
I2
5
69
8
I2
7
9 ICl 7
10
I2
5 SeS
I
C5H11
11 ICl 5
12
I2
30
13 ICl 7
14
I2 15
15 ICl 60
16
I2 15
17
I2
30

70
18
I2
15
19
I2
20
20
S
C5H11
SenBu
I2
120
_
21
I2 120
_
22 ICl 120 _
23
I2
15
a Reações realizadas com substrato (0,25 mmol), E (1.1 eq) em CH2Cl2 (5 mL).
Analisando-se a Tabela 6, pode-se perceber que estas condições de
reação promoveram com eficiência a reação de ciclização do substrato 6a com
diferentes fontes eletrofílicas. Rendimentos satisfatórios foram obtidos
utilizando-se tanto eletrófilos de iodo (Tabela 6; exemplos 1 e 2), quanto um
eletrófilo de selênio (Tabela 6; exemplo 3).
Uma análise detalhada da tabela 6 revelou que o anel aromático ligado
ao alquino sem qualquer substituínte (Tabela 6, exemplos 1-3), com grupos

71
doadores de elétrons (Tabela 6, exemplos 4-6) e grupos retiradores de elétrons
(Tabela 6, exemplos 8-9) resultaram nos respectivos produtos em rendimentos
semelhantes. Estes resultados mostram que a reação não depende de efeitos
eletrônicos dos substituíntes ligados ao anel aromático da ligação tripla. A
utilização de um grupo volumoso, como naftaleno (Tabela 6, exemplo 7), levou
ao produto desejado em apenas 5 minutos de reação, com o rendimento de
78%.
Além dos anéis aromáticos, a reação procedeu-se bem com grupos
alquila (Tabela 6, exemplos 10-11) e alquila volumoso (tabela 6, exemplos 12-
13) diretamente ligados ao grupamento alquino, tanto com I2 quanto com ICl
como fonte eletrofílica. Bons rendimentos foram obtidos quando foram
utilizados derivados de benzotiofenos 6h-6l como substratos, porém um maior
tempo de reação foi exigido (Tabela 6, exemplos 14-19). Em relação a
seletividade da ciclização quanto a competição do átomo de selênio e oxigênio,
é importante ressaltar que não foi obtido qualquer derivado de benzofurano e o
único produto obtido foram os derivados de selenofenos (Tabela 6, exemplos
18 e 19). Esta alta seletividade pode ser atribuída ao efeito eletrônico:
nucleofilicidade relativa do átomo de selênio, a natureza catiônica do
intermediário formado e ao fato de que o grupo metóxila parece ser mais
resistente a desmetilação e fechamento do anel.58
Quando usados benzotiofenos contendo um grupo alquila ou um álcool
propargila, encontramos algumas limitações dessa metodologia. Por exemplo,
não foram observadas a formação dos respectivos produtos de ciclização
usando-se os substratos 6m-6n e apenas foi identificado a presença do
produto de redução da ligação tripla (Tabela 6, exemplos 20-22).
A fim de aumentar o escopo da presente metodologia, foram usadas as
mesmas condições para teluretos. Assim, o 2-butilteluro-3-feniletiniltiofeno 6o
pôde reagir também para formar 4-iodotelurofeno[2,3-b]tiofeno 7n, em
rendimento moderado (Tabela 6, exemplo 23).
58 Para outras reatividades relativas de vários grupos funcionais nas reações de ciclização eletrofílica de alcinos ver: (a) Mehta, S.; Waldo, J. P.; Larock, R. C. J. Org. Chem. 2009, 74, 1141. (b) Arcadi, A.; Marinelli, F.; Cacchi, S. Synthesis 1986, 749. (c) G. Kirsch, S. Hesse, A. Comel, Curr. Org. Chem. 2004, 1, 47.

72
2.2.1 APLICAÇÃO DO COMPOSTO 7a EM REAÇÕES DE ACOPLAMENTO CATALISADAS POR SAIS DE PALÁDIO E COBRE
No intuito de demonstrar o potencial sintético dos derivados selenofenos
como precursores para o aumento da complexidade molecular, foi testada a
reatividade do 4-iodoselenofeno[2,3-b]tiofeno 7a em reações de acoplamento
cruzado, como Negishi, Suzuki e com tióis, catalisadas por paládio e cobre.
2.2.1.1 Reações de Acoplamento Cruzado do Composto 7a com p-cloro-benzenotiól Catalisada por CuI
O interesse na síntese de compostos organosulfurados aumentou
significativamente na química orgânica sintética devido suas importantes
propriedades de auxiliar sequências sintéticas.59 Ao longo dos últimos anos,
grandes progressos foram feitos na formação de novas ligações carbono-
heteroátomo através do acoplamento cruzado de espécies nucleofílicas com
haletos utilizando-se sistemas catalisados por cobre.21 No entanto, há poucos
relatos sobre selenofenos ou telurofenos halogenados, como substratos, para a
formação de novas ligações carbono-enxofre, via reações de acoplamento
cruzado catalisados por paládio ou cobre.60 Considerando-se as vantagens do
uso de catalisadores de cobre, menos caros e tóxicos do que sais de paládio, o
uso de uma metodologia para formação de uma nova ligação carbono-enxofre,
sem o uso de ligante ou co-catalisador, seria de grande interesse na química
sintética.9i Assim, o intermediário 7a (0,5 mmol) reagiu de modo eficaz frente ao
acoplamento com 4-cloro-benzenotiol (0,6 mmol) via catálise de CuI (10 mol%),
Et3N (1 mmol) como base em dioxano (3 mL). Nessas condições, o produto 4-
(p-clorofeniltio)-5-fenilselenofeno[2,3-b]tiofeno 8a foi obtido em 66% de
rendimento em 12 horas de reação (Esquema 48).
59 a) Takeda, T.; Furukawa, H.; Fujimori, M.; Suzuki, K.; Fujiwara, T. Bull. Chem. Soc. Jpn. 1984, 57, 1863. b) Trost, B. M.; Lavoie, A. C. J. Am. Chem. Soc. 1983, 105, 5075. 60 Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2004, 248, 2337.

73
SeS
I
Ph
CuI (10 mol %), Et3N
dioxano, refluxo, 12 h
SeS
S
Ph7a
8a(66%)
Cl
Cl SH
Esquema 48
2.2.1.2 Reações de Acoplamento Cruzado do tipo Suzuki do Composto 7a com o Ácido p-bromofenilborônico
Os derivados calcogenofenos, uma vez obtidos, do ponto de vista
sintético, tornam-se compostos potenciais para a síntese de calcogenofenos
com outros substituintes. Nesse contexto, as reações de acoplamento cruzado
do tipo Suzuki catalisadas por paládio, de haletos de arila com ácidos ou
ésteres borônicos, tornou-se um método comum e conveniente na química
orgânica sintética para formação de novas ligações carbono-carbono.28 Desde
que a reação de Suzuki tem se mostrado bem sucedida para a arilação seletiva
de heterociclos36, decidimos explorar o intermediário 4-iodoselenofeno[2,3-
b]tiofeno 7a frente a reação de Suzuki afim de demonstrar seu potencial como
intermediário sintético. Assim, quando o substrato 7a (0.5 mmol) reagiu com o
ácido p-bromo-fenilborônico (1,5 eq) usando-se Pd(PPh3)4 (2 mol%) como
catalisador, em 5 mL de DMF/H2O (5:1) na presença de base (2 eq), o produto
4-(4-bromofenil)-5-fenilselenofeno[2,3-b]tiofeno 9a pôde ser obtido em 63% de
rendimento (Esquema 49).
Esquema 49

74
2.2.1.3 Reações de Acoplamento Cruzado do tipo Negishi do Composto 7a com Cloreto de p-toluilzinco
Reações de acoplamento cruzado catalisadas por metais de transição
entre centros de Csp2 têm sido amplamente utilizadas na preparação de
produtos farmacêuticos e agroquímicos.61 Nesse contexto, têm-se desenvolvido
novos sistemas de acoplamento catalisados por paládio, os quais são mais
econômicos e mais facilmente acessíveis e reativos, mesmo sob condições
brandas. Neste sentido, o acoplamento cruzado de eletrófilos de organozinco
com compostos orgânicos, catalisados por paládio, conhecida como reação de
Negishi, tornou-se uma eficiente ferramenta para formação de novas ligações
carbono-carbono.62 Os compostos de calcogênios encontraram uma ampla
utilidade em um grande número de reações diferentes para formação de novas
ligações carbono-carbono sob condições muito suaves,11a e para a síntese de
compostos com atividades biológicas.5c Entre esses compostos de calcogênio,
os calcogenofenos desempenham um papel importante em síntese orgânica
pois muitos deles mostram atividade biológica.63 Assim, halocalcogenofenos
são derivados importantes e tendem a fornecer oportunidades para
tranformações posteriores.47 Portanto, decidimos explorar a reatividade do 4-
iodoselenofeno[2,3-b]tiofeno 7a frente a reação de Negishi catalisada por
paládio (Esquema 50).
61 (a) Miyaura, N. Cross-Coupling Reactions. A Practical Guide; Springer: Berlin, 2002; (b) Diederich, F.; Stang, P. J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, 1998. 62 (a) Negishi, E.; Anastasia, L. Chem. Rev. 2003, 103, 1979; (b) Negishi, E. In Handbook of Organopalladium Chemistry for Organic Synthesis; Wiley and Sons: New York, 2002; Vols. 1 and 2; (c) Li, J. J.; Gribble, G. W. Palladium in Heterocyclic Chemistry. In Tetrahedron Organic Chemistry Series; Pergamon: Amsterdam, 2000; Vol. 20; (d) Negishi, E.; Hu, Q.; Huang, Z.; Qian, M.; Wang, G. Aldrichim. Acta 2005, 38, 71. 63 Chemistry and Biology of Naturally-occurring Acetylenes and Related Compounds; Lain, J., Breteler, H., Arnason, T., Hansen, L., Eds.; Elsevier: Amsterdam, 1988.

75
SeS
I
Ph PdCl2(PPh3)2 (10 mol %)THF, 2 h, t. a. SeS
Ph7a
10a(69%)
ZnCl
Esquema 50
Uma análise mais detalhada do esquema 15 revela que quando o
substrato 7a (0,5 mmol) reage com 3 equivalentes do cloreto de p-toluilzinco
(gerado pela adição de n-BuLi ao p-bromotolueno em THF a -78ºC, seguido da
adição ZnCl2) por 2 horas, na presença de PdCl2(PPh3)2 (10 mol%) em THF, a
temperatura ambiente, formou o 5-fenil-4-p-toluilselenofeno[2,3-b]tiofeno 10a,
com 69% de rendimento.

76
Considerações Finais e Conclusões

77
Considerando-se os objetivos propostos para o presente trabalho e
analisando-se os resultados obtidos, é possível fazer algumas considerações
pertinentes frente às reações estudadas.
Na primeira parte do nosso trabalho, desenvolveu-se uma metodologia
de obtenção de derivados de selenofenos 2, funcionalizados na posição 3 do
anel heteroaromático, via reações de ciclização de (Z)-seleno- e teluroeninos 1
com dicalcogenetos de diorganoíla, catalisadas por sais de cobre. A natureza
do solvente (DMSO), bem como dos grupamentos ligados diretamente ao
átomo de calcogênio (Csp3-calcogênio), foram fatores primordiais para a
formação dos produtos desejados em bons rendimentos. Através deste
protocolo, uma série de telurofenos e selenofenos, contendo diferentes
substituíntes na posição 3 do anel heterocícliclo, puderam ser preparados em
rendimentos de moderados a excelentes.
Após o êxito das reações de ciclização, decidimos explorar o potencial
sintético do telurofeno 2o frente as reações de acoplamento cruzado do tipo
Suzuki. Observou-se que tanto usando ácidos borônicos, quanto
organotrifluoroboratos de potássio como reagentes, os correspondentes 3-
ariltelurofenos 5a e 5b foram obtidos em bons rendimentos.
Na segunda parte do nosso trabalho, planejou-se a síntese de 4-iodo-
selenofenos[2,3-b]tiofenos 7, via reações de ciclização eletrofílica de 3-
alquiniltiofenos 6. A natureza do solvente (CH2Cl2), bem como dos
grupamentos ligados diretamente ao átomo de selênio (Csp3-Se), foram fatores
determinantes para a formação dos produtos desejados em bons rendimentos
e em tempos de reação curtos. Através deste protocolo, uma série de
derivados de selenofenos fundidos puderam ser preparados usando-se tanto
eletrófilos de iodo quanto de selênio em rendimentos de moderados a
excelentes.
Assim, em uma última etapa do trabalho, foi testada a reatividade do
composto 7a frente as reações de acoplamento cruzado do tipo Negishi e
Suzuki, catalisadas por paládio, e acoplamento cruzado com tióis, catalisadas
por cobre. Os respectivos produtos 8a, 9a e 10a puderam ser obtidos em bons
rendimentos.

78
Por fim, convém destacar que de um modo geral, as metodologias
descritas mostraram-se satisfatórias para a síntese de uma variedades de
derivados de calcogenofenos funcionalizados em bons rendimentos e sob
condições reacionais relativamente brandas.
Cabe salientar que os resultados aqui apresentados para a defesa desta
dissertação resultaram na produção de dois artigos, publicados em periódicos
de nível internacional (ver Anexos).

79
Capítulo 3
Parte Experimental

80
PARTE EXPERIMENTAL
2.1 MATERIAIS E MÉTODOS 2.1.1 Espectroscopia de Ressonância Magnética Nuclear.
Os espectros de RMN 1H, RMN 13C, foram obtidos em espectrômetros
Bruker DPX, que operam na freqüência de 200 MHz e 400 MHz,
(Departamento de Química – UFSM). Os deslocamentos químicos (δ) estão
relacionados em parte por milhão (ppm) em relação ao tetrametilsilano (TMS,
utilizado como padrão interno para os espectros de RMN 1H) e CDCl3 (para os
espectros de RMN 13C), colocando-se entre parênteses a multiplicidade (s =
singleto, d = dupleto, t = tripleto, quart = quarteto, quint = quinteto, sex =
sexteto, m = multipleto, dd = duplo dupleto, ddd = duplo duplo dubleto dt =
duplo tripleto, td = triplo dupleto), o número de hidrogênios deduzidos da
integral relativa e a constante de acoplamento (J) expressa em Hertz (Hz).
2.1.2 Espectrometria de Massas.
Os espectros de massas de baixa resolução (MS) foram obtidos a partir
de um aparelho Shimatzu QP2010PLUS 70 eV (Universidade Federal de Santa
Maria).
Os espectros de massas de alta resolução (HRMS) foram obtidos a
partir de um aparelho Kratos-MS50TC-70 eV da Iowa State University – Ames,
EUA. Os valores calculados foram baseados no isótopo de 80Se.
2.1.3 Análise Elementar
As análises elementares foram realizadas através do analisador
elementar Carlo Erba EA 1110 (Departamento de Química Fundamental da
Universidade Federal de Pernambuco).

81
2.1.4 Rota-evaporadores.
Para remoção dos solventes das soluções orgânicas, foram utilizados:
- Rota-evaporador Heidolph VV 60;
- Rota-evaporador Heidolph 4011 - Digital;
- Rota-evaporador Fisatom – Modelo 558;
- Linha de vácuo equipada com uma bomba de alto-vácuo Boc Edwards
modelo RV8 Rotary Vane.
2.1.5 Solventes e Reagentes.
Os solventes foram purificados e secos antes de serem utilizados,
conforme técnicas usuais.64 Os reagentes restantes foram obtidos de fontes
comerciais e utilizados sem prévia purificação.
O THF foi refluxado sobre sódio metálico, utilizando como indicador a
benzofenona e destilado imediatamente antes do uso. Diclorometano e
acetonitrila foram destilados sobre pentóxido de fósforo e armazenados sob
peneira molecular. O metanol foi destilado de magnésio metálico. Pirrolidina e
trietilamina foram destiladas sobre KOH; o tolueno foi destilado sobre sódio
metálico e o etanol foi seco com óxido de cálcio.
A concentração do reagente de alquil-lítio foi determinada através de
titulação do mesmo com isopropanol, utilizando-se 1,10-fenantrolina como
indicador.65 O selênio e telúrio elementar utilizados (~200 mesh – ALDRICH)
foram secos em estufa a 80 °C durante 12 horas.
As placas de cromatografia em camada delgada foram obtidas de fontes
comerciais; Sílica G/UV254 (0,20 mm). Utilizou-se, como método de revelação,
cuba de iodo, luz ultravioleta e solução ácida de vanilina.
Para os produtos purificados utilizando cromatografia em coluna, o
material usado foi uma coluna de vidro, gel de sílica 60 (230-400 mesh –
MERCK) e, como eluente, um solvente ou mistura de solventes adequados.
64 Perrin, D. D.; Armarego, W. L. Em Purification of Laboratory Chemicals, 4th ed. Pergamon Press, New York, 1997. 65 Watson, S. C.; Eastham, J. F. J. Organomet. Chem. 1967, 9, 165.

82
3.2 PROCEDIMENTOS EXPERIMENTAIS
3.2.1 Preparação do Pd(PPh3)4
66
A uma suspensão de PdCl2 (0,301g; 1,7 mmol) em água (2,5 mL),
adicionou-se NaCl (0,198g; 3,4 mmol). A mistura foi aquecida de forma lenta e
cuidadosa, em chapa de aquecimento, sob agitação, até quase a secura.
Resfriou-se a reação e adicionou-se água (2,5 mL) e repetiu-se a evaporação
até a secura total da reação. Em seguida, adicionou-se etanol (50 mL),
aqueceu-se a 60 °C e adicionou-se PPh3 (2,67g; 10,2 mmol). Retirou-se o
aquecimento e adicionou-se N2H4.H2O (0,25 mL). Após 2-3 minutos de
agitação, o sólido levemente esverdeado foi separado por filtração em funil de
Büchner, lavado com éter etílico (2x 10 mL), e seco sob pressão reduzida, em
bomba de alto vácuo. Rendimento: 1,61g (80%).
3.2.2 Preparação do PdCl2(PPh3)2 67
A uma suspensão de PdCl2 (0,301g; 1,7 mmol) em água (2,5 mL),
adicionou-se NaCl (0,198g; 3,4 mmol). A mistura foi aquecida de forma lenta e
cuidadosa, em chapa de aquecimento, sob agitação, até quase a secura.
Resfriou-se a reação e adicionou-se água (2,5 mL) e repetiu-se a evaporação
até a secura total da reação. Em seguida, adicionou-se etanol (50 mL),
aqueceu-se a 60 °C e adicionou-se PPh3 (1,78g; 6,8 mmol). Depois de 1-2
minutos, formou-se um precipitado amarelo. Retirou-se o aquecimento e
manteve-se a agitação por mais 2-3 minutos. Filtrou-se a suspensão em funil
de Büchner, lavou-se o sólido com éter etílico (2x 10 mL) e secou-se em
bomba de alto vácuo. Rendimento: 1,15g (97%).
66 Coulson, R. D. Inorg. Synth. 1972, 13, 121. 67 Hartely, F. R. Organometal. Chem. Rev. A. 1970, 6, 119.

83
3.2.3 Procedimento geral para a preparação de disselenetos de diorganoila68
Em um balão equipado com condensador de refluxo e agitação
magnética, sob argônio, contendo magnésio elementar (50 mmol; 1,2 g) e
flambou-se o sistema. Alguns cristais de iodo e THF (30 mL) foram adicionados
ao balão. Em seguida, iniciou-se lentamente a adição do haleto desejado (50
mmol) diluido em THF. Após todo o magnésio ser consumido, adicionou-se
selênio elementar (50 mmol; 6,7 g), em pequenas porções utilizando-se funil de
adição de sólidos. A reação foi colocada em refluxo por 1 hora e depois a
temperatura ambiente por 8 horas. O balão foi envolto em um banho de gelo e
adicionou-se, cuidadosamente, solução saturada de cloreto de amônio. Deixou-
se a mistura reacional sob oxidação ao ar por 4 horas. Extraiu-se 5 vezes com
acetato, secou-se com MgSO4, filtrou-se e evaporou-se o solvente sob pressão
reduzida. O produto foi purificado por recristalização em etanol. Rend.: 65-80%
3.2.4 Procedimento geral para a preparação de disseleneto de dipiridina69
Em um balão equipado com condensador de refluxo e agitação
magnética, sob argônio, contendo magnésio elementar (55 mmol; 1,27 g)
flambou-se o sistema. Alguns cristais de iodo e THF (50 mL) foram adicionados
ao balão. Em seguida, iniciou-se lentamente a adição do cloreto de isopropila
(55 mmol) diluido em THF (20 mL). Após todo o magnésio ser consumido,
adicionou-se gota a gota a 2-bromopiridina (50 mmol; 1,3 g) diluído em THF (10
mL), a 0 ºC, utilizando-se funil de adição de líquidos. Após, remove-se o banho
de gelo e deixa-se reagir a temperatura ambiente por 2 horas. Em seguida,
baixou-se novamente a temperatura para 0ºC e adicionou-se selênio elementar
(50 mmol; 6,7 g), em pequenas porções utilizando-se funil de adição de sólidos.
Após 15 minutos, remove-se o banho de gelo e deixa-se consumir todo o Seº.
O balão foi envolto em um banho de gelo e adicionou-se, cuidadosamente,
solução saturada de cloreto de amônio. Deixou-se a mistura reacional sob
oxidação ao ar por 4 horas. Extraiu-se 5 vezes com acetato de etila, secou-se
68 Reich, H. J.; Renga, J. M.; Reich, I. L. J. Am. Chem. Soc. 1975, 97, 5435. 69 Bhasin, K. K.; Arora, V. Appl. Organometal. Chem. 2004. 18. 359.

84
com MgSO4, filtrou-se e evaporou-se o solvente sob pressão reduzida. O
produto foi purificado por cromatografia em coluna de gel de sílica utilizando-se
um gradiente de hexano e acetato de etila (5:1) como eluentes. Rend.: 80%
3.2.5 Procedimento geral para a preparação de disseleneto e ditelureto de dibutila70
Em uma suspensão do calcogênio elementar requerido (50 mmol)
[previamente seco em estufa à 85 ºC] e THF (50 mL), foi adicionado
lentamente a 0ºC n-BuLi (1.1 eq). A mistura foi agitada durante 1 hora a
temperatura ambiente. Após este período, foi adicionado lentamente uma
solução saturada de NH4Cl (25 mL) e a mistura foi agitada por 2 horas
em contato com o ar. Passado esse tempo, dilui-se com acetato de etila
(30 mL) e a fase orgânica foi separada e lavada sucessivamente com
água (3 x 20 mL) e solução saturada de NaCl. A fase orgânica foi seca
com MgSO4 e o solvente evaporado. Rend.: 90%
3.2.6 Procedimento geral para preparação dos (Z)-teluroeninos 1a-d.
Em um balão de 2 bocas, munido de condensador de refluxo e agitação
magnética, à temperatura ambiente, sob atmosfera de argônio, adicionou-se
NaBH4 (0,472g; 12,5 mmol) a uma solução do ditelureto de diorganoíla
apropriado (2,5 mmol) em etanol (50 mL). Em seguida, adicionou-se 1,4-difenil-
1,3-butadiíno (1,010g; 5 mmol). A reação foi mantida sob refluxo, por 5 horas.
Após este tempo, a reação foi diluída em acetato de etila (60 mL) e lavada com
solução saturada de NaCl (3x 30 mL). A fase orgânica foi seca com MgSO4 e
concentrada sob vácuo. Os (Z)-selenoeninos obtidos foram purificados por
cromatografia em coluna de gel de sílica utilizando-se hexano como eluente.
70 Engman, L.; Cava, M. P. Synth. Commun. 1982, 12, 163.

85
3.2.7 Procedimento geral para preparação dos (Z)-selenoeninos 3a e 3c.25b
A uma suspensão de selênio elementar (0,395 g; 5 mmol) em THF seco
(25 mL) sob atmosfera de argônio e provido de agitação magnética, foi
adicionado n-BuLi (2,0 mL de uma solução 2,5 M; 5 mmol). Uma solução
amarela foi formada. A esta solução foi adicionado o diíno apropriado (5 mmol)
juntamente com etanol desoxigenado (25 mL). A reação foi mantida sob refluxo
por 24 h. Após este tempo, a reação foi diluída em acetato de etila (60 mL) e
lavada com solução saturada de NH4Cl (30 mL) e solução saturada de NaCl
(2x 30 mL). A fase orgânica foi seca com MgSO4 e concentrada sob vácuo. Os
(Z)-selenoeninos obtidos foram purificados por cromatografia em coluna de gel
de sílica utilizando-se um gradiente de hexano e acetato de etila como
eluentes.
3.2.8 Obtenção do (Z)-1-(n-Butilseleno)-4-fenil-but-1-en-3-ino (3b).
Em um balão de duas bocas, sob atmosfera de argônio e munido de
condensador de refluxo, contendo uma solução de 2-hidroxi-2-metill-6-fenil-3,5-
hexadiíno (0,830g; 5,0 mmol) em tolueno seco (10 mL), adicionou-se NaOH
(0,220g; 5,5 mmol) e refluxou-se por 2 horas. A solução do 1-fenil-1,3-butadiíno
obtida foi resfriada a temperatura ambiente, onde se adicionou solução de
disseleneto de dibutila (0,680g; 2,5 mmol) em etanol 95% (50 mL). NaBH4
(0,472g; 12,5 mmol) foi adicionado sob forte agitação. A reação foi refluxada
por 4 horas. Após este tempo, a reação foi diluída em acetato de etila (60 mL) e
lavada com solução saturada de NaCl (3x 30 mL) e H2O (3x 30 mL). A fase
orgânica foi seca com MgSO4 e concentrada sob vácuo. Os (Z)-selenoenino
obtido foi purificado por cromatografia em coluna de gel de sílica utilizando-se
hexano como eluente. Rend.: 0,841g (64%).

86
3.2.9 Procedimento geral para preparação dos telurofenos.
A uma solução de DMSO (3 mL), CuI (10 mol%), sob atmosfera de
argônio, foi adicionado a temperatura ambiente o teluroenino (0,25 mmol) e o
apropriado dicalcogeneto de diorganoíla (1,1 eq). A mistura da reação foi
agitada sob temperatura de 110 ºC durante 10 horas. Após, resfriou-se o meio
reacional a temperatura ambiente, diluiu-se com acetato de etila (30 mL) e
lavou-se com NH4Cl (3 x 20 mL). Em seguida separou-se a fase orgânica e
secou-se com Mg2SO4 e retirou-se o solvente no rota-evaporador a pressão
reduzida. O resíduo foi purificado por cromatografia em coluna, usando-se
hexano como eluente.
2,5-difenil-3-(fenilseleno)-telurofeno (2a). Rendimento:
0,192 g (79%). RMN 1H (CDCl3, 200 MHz): δ 7,82 (s, 1H); 7,49-7,18 (m, 15H);
RMN 13C (CDCl3, 50 MHz): δ 147,86; 146,22; 139,82; 139,69; 139,11; 132,91;
131,26; 129,19; 129,16; 128,92; 128,26; 127,88; 127,74; 126,72; 126,64;
126,02. MS (intensidade relativa) m/z: 488 (24); 360 (13); 279 (10); 202 (100);
77 (10); 51 (6). Anal. Calculado para C22H16SeTe: C: 54,27; H: 3,88.
Encontrado: C: 54,58; H: 3,72.
2,5-difenil-3-(p-toluilseleno)-telurofeno (2b).
Rendimento: 0,180 g (72%). RMN 1H (CDCl3, 200 MHz): δ 7,65-7,57 (m, 3H);
7,42-7,16 (m, 10H); 7,00 (d, J = 7,82 Hz, 2H); 2,30 (s, 3H). RMN 13C (CDCl3,
100 MHz): δ 148,36; 146,99; 140,36; 140,23; 139,62; 138,81; 134,28; 131,92;
130,56; 129,61; 129,43; 129,09; 128,76; 128,23; 127,32; 127,22; 22,45; MS
(intensidade relativa) m/z: 502 (23); 293 (17); 202 (100); 91 (16); 77 (6). HRMS
calculado para C23H18SeTe: 503,9636. Encontrado: 503,9651.
2,5-difenil-3-(o-toluillseleno)-telurofeno (2c). Rendimento:
0,155 g (62%). RMN 1H (CDCl3, 400 MHz): δ 7,75 (s, 1H); 7,47-7,45 (m, 2H);

87
7,34-7,22 (m, 10H); 7,13-7,08 (m, 2H); 2,30 (s, 3H). RMN 13C (CDCl3, 50 MHz):
δ 147,87; 146,55; 139,83; 139,67; 139,04; 138,31; 133,78; 131,29; 130,03;
129,08; 128,90; 128,49; 128,23; 127,85; 127,71; 126,76; 126,70; 126,68; 21,93;
MS (intensidade relativa) m/z: 502 (21); 293 (17); 202 (100); 91 (21); 77 (9); 51
(6). Anal. Calculado para C23H18SeTe: C: 55,14; H: 3,62. Encontrado: C: 55,39;
H: 3,82.
2,5-difenil-3-(p-clorofenilseleno)-telurofeno (2d).
Rendimento: 0,222 g (85%). RMN 1H (CDCl3, 200 MHz): δ 7,76 (s, 1H); 7,48-
7,00 (m, 14H); 2,29 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ 148,36; 147,02;
139,46; 138,88; 132,64; 132,32; 131,16; 129,20; 129,10; 128,92; 128,31;
128,30; 128,24; 127,94; 127,79; 126,65. MS (intensidade relativa) m/z: 522
(19); 394 (10); 202 (100); 77 (12); 51 (6). Anal. Calculado para C22H15ClSeTe:
C: 50,68; H: 2,90. Encontrado: C: 50,81; H: 3,17.
2,5-difenil-3-(p-fluorofenilseleno)-telurofeno (2e).
Rendimento: 0,162 g (64%). RMN 1H (CDCl3, 200 MHz): δ 7,75 (s, 1H); 7,47-
7,21 (m, 12H); 6,96-6,88 (d, J = 8,80 Hz, 2H). RMN 13C (CDCl3, 50 MHz): δ
150,85; 147,90; 142,66; 141,60; 139,09; 134,20; 129,57; 128,97; 128,86;
128,76; 128,40; 127,93; 127,88; 126,89; 115,29; 113,48. MS (intensidade
relativa) m/z: 506 (23); 378 (12); 297 (7); 202 (100); 77 (6). Anal. Calculado
para C22H15FSeTe: C: 52,33; H: 2,99. Encontrado: C: 52,55; H: 3,21.
2,5-difenil-3-(m-trifluorometilfenilseleno)-telurofeno
(2f). Rendimento: 0,180 g (65%). RMN 1H (CDCl3, 400 MHz): δ 7,86 (s, 1H);
7,56 (s, 1H); 7,46-7,22 (m, 13H). RMN 13C (CDCl3, 100 MHz): δ 148,78; 148,47;
139,61; 139,39; 138,89; 134,40; 133,81; 131,22 (q, J = 32,20 Hz); 129,36;
129,11; 128,98; 128,27; 128,05; 127,91; 127,54; 127,15 (q, J = 3,66 Hz);
126,71; 123,66 (q, J = 272,97 Hz); 123,13 (q, J = 3,66 Hz). MS (intensidade

88
relativa) m/z: 554 (22); 479 (5); 407 (5); 347 (7); 202 (100); 134 (8); 77 (10).
Anal. Calculado para C23H15F3SeTe: C: 49,78; H: 2,72. Encontrado: C: 50,02;
H: 2,91.
2,5-difenil-3-(2,4,6-trimetilfenilseleno)-telurofeno (2g).
Rendimento: 0,172 g (65%). RMN 1H (CDCl3, 400 MHz): δ 7,53-7,51 (m, 2H);
7,41-7,38 (m, 2H); 7,26-7,21 (m, 6H); 7,25 (s, 1H); 6,92 (s, 2H); 2,40 (s, 6H);
2,28 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ 146,80; 142,59; 140,00; 139,33;
138,56; 137,70; 136,47; 131,39; 129,03; 128,86; 128,79; 128,69; 128,35;
127,44; 126,83; 126,67; 24,31; 20,95. MS (intensidade relativa) m/z: 530 (25);
321 (24); 202 (100); 119 (22); 105 (12); 91 (16); 77 (16); 51 (7). Anal. Calculado
para C25H22SeTe: C: 56,76; H: 4,19. Encontrado: C: 56,42; H: 3,81.
2,5-difenil-3-naftilseleno-telurofeno (2h). Rendimento:
0,156 g (58%). 1H RMN (CDCl3, 400 MHz): δ 7,85-7,84 (m, 2H); 7,75-7,73 (m,
1H); 7,68-7,65 (m, 2H); 7,50-7,48 (m, 2H); 7,43-7,39 (m, 3H); 7,35-7,19 (m,
8H). RMN 13C (CDCl3, 100 MHz): δ 146,25; 139,72; 139,67; 139,03; 133,92;
132,05; 130,36; 129,80; 129,19; 129,04; 128,90; 128,87; 128,55; 128,29;
127,87; 127,77; 127,70; 127,28; 126,83; 126,70; 126,39; 125,83. MS
(intensidade relativa) m/z: 538 (23); 410 (17); 329 (21); 202 (100); 128 (14); 77
(8). Anal. Calculado para C26H18SeTe: C: 58,15; H: 3,38. Encontrado: C: 57,89;
H: 3,01.
2,5-difenil-3-(2-piridilseleno)-telurofeno (2i). Rendimento:
0,144 g (59%). RMN 1H (CDCl3, 200 MHz): δ 8,40 (ddd, J = 4,8, 1,8, 0,9 Hz,
1H); 8,06 (s, 1H); 7,49-7,27 (m, 11H); 7,08 (td, J = 7,6, 1,6 Hz, 1H); 6,99 (ddd, J
= 7,6, 4,8, 1,1 Hz, 1H). RMN 13C (CDCl3, 100 MHz): δ 159,37; 150,50; 149,79;
148,60; 140,97; 139,47; 138,98; 136,69; 129,19; 129,01; 128,25; 128,02;
127,92; 127,19; 126,73; 124,09; 120,23. MS (intensidade relativa) m/z: 489

89
(28); 360 (51); 280 (20); 202 (100); 181 (17); 77 (23); 51 (18). HRMS calculado
para C21H15NSeTe: 490,9432. Encontrado: 490,9445.
2,5-difenil-3-(fenilteluro)-telurofeno (2j). Rendimento: 0,204
g (76%). RMN 1H (CDCl3, 400 MHz): δ 7,70-7,67 (m, 3H); 7,42-7,18 (m, 13H).
RMN 13C (CDCl3, 100 MHz): δ 150,31; 146,93; 142,80; 141,77; 139,31; 138,04;
129,39; 128,92; 128,79; 128,38; 127,84; 127,82; 127,75; 126,90; 115,76;
115,61. MS (intensidade relativa) m/z: 536 (17); 330 (5); 202 (100); 176 (6); 77
(22); 51 (11). Anal. calculado C22H16Te2: C: 49,34; H: 3,01. Encontrado: C:
49,62; H: 3.31.
2,5-difenil-3-(p-toluilteluro)-telurofeno (2k). Rendimento:
0,203 g (74%). RMN 1H (CDCl3, 200 MHz): δ 7,65-7,57 (m, 3H); 7,42-7,16 (m,
10H); 7,00 (d, J = 7,82 Hz, 2H); 2,30 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ
150,03; 145,75; 142,35; 141,71; 139,27; 138,57; 137,87; 130,25; 128,80;
128,68; 128,31; 127,70; 127,60; 126,83; 115,91; 111,38; 21,17. MS
((intensidade relativa) m/z: 457 (52); 330 (30); 266 (12); 202 (100); 150 (11); 77
(10). Anal. Calculado para C23H18Te2: C: 50,26; H: 3,30. Encontrado: C: 50,58;
H: 3,61.
2,5-difenil-3-(p-clorofenilteluro)-telurofeno (2l).
Rendimento: 0,225 g (79%). RMN 1H (CDCl3, 200 MHz): δ 7,70 (s, 1H); 7,51 (d,
J = 8,38 Hz, 2H); 7,36-7,23 (m, 10H); 7,11 (d, J = 8,38 Hz, 2H). RMN 13C
(CDCl3, 50 MHz): δ 150,85; 147,90; 142,66; 141,60; 139,09; 134,20; 129,57;
128,97, 128,86; 128,76; 128,40; 127,93; 127,88; 126,89; 115,29; 113,48. MS
(intensidade relativa) m/z: 572 (10); 333 (12); 202 (100); 77 (22); 51 (9). Anal.
Calculado para C22H15ClTe2: C: 46,36; H: 2,65. Encontrado: C: 46,59; H: 2,87.

90
2,5-difenil-3-(fenilteluro)-telurofeno (2m). Rendimento:
0,114 g (52%). RMN 1H (CDCl3, 200 MHz): δ 7,89 (s, 1H); 7,42-7,19 (m, 15H).
RMN 13C (CDCl3, 100 MHz): δ 150,31; 146,93; 142,80; 141,77; 139,31; 138,04;
129,39; 128,92; 128,79; 128,38; 127,84; 127,82; 127,75; 126,90; 115,76;
115,61. MS (intensidade relativa) m/z: 442 (43); 312 (26); 279 (9); 234 (17); 202
(100); 77 (11); 51 (7). HRMS calculado para C22H16STe: 442,0035. Encontrado:
442,0061.
2,5-difenil-3-(butilseleno)-telurofeno (2n). Rendimento: 0,098 g
(42%). RMN 1H (CDCl3, 400 MHz): δ 7,96 (s, 1H); 7,52-7,27 (m, 10H); 2,79 (t, J
= 7,50 Hz, 2H); 1,56 (quint, J = 7,50 Hz, 2H); 1,29 (sex, J = 7,50 Hz, 2H); 0,82
(t, J = 7,50 Hz, 3H). RMN 13C (CDCl3, 100 MHz): δ 147,44; 143,86; 140,02;
139,35; 139,06; 129,31; 129,15; 128,94; 128,17; 127,79; 127,45; 126,76; 32,20;
28,66; 22,71; 13,49. MS (intensidade relativa) m/z: 468 (24); 412 (9); 284 (23);
202 (100); 126 (3); 77 (6); 57 (3); 51 (4). Anal. Calculado para C20H20SeTe: C:
51,45; H: 4,32. Encontrado: C: 51,03; H: 3,92.
2,5-difenil-3-(butilteluro)-telurofeno (2o).2s Rendimento: 0,144 g
(56%). RMN 1H (CDCl3, 200 MHz): δ 7,88 (s, 1H); 7,20-7,60 (m, 10H); 2,78 (t, J
= 7,00 Hz, 2H); 1,64 (quint, J = 7,00 Hz, 2H); 1,26 (sex, J = 7,00 Hz, 2H); 0,81
(t, J = 7,00 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 150,10; 142,70; 141,70;
139,30; 128,80; 128,10; 127,60; 126,80; 112,50; 33,60; 24,80; 13,30; 8,90. MS
(intensidade relativa) m/z: 518 (8); 460 (5); 332 (13); 202 (100). Anal. Calculado
para C20H20Te2: C: 46,16; H: 3,88. Encontrado: C: 45,95; H: 3,56.
3.2.10 Procedimento geral para preparação dos selenofenos.
A uma solução de DMSO (3 mL), CuI (10 mol%), sob atmosfera de
argônio, foi adicionado a temperatura ambiente o apropriado selenoenino (0,25
mmol) e o apropriado dicalcogeneto de diorganoíla(1,1 eq). A mistura da

91
reação foi agitada sob temperatura de 110 ºC durante 10 horas. Após, resfriou-
se o meio reacional a temperatura ambiente, diluiu-se com acetato de etila (30
mL) e lavou-se com NH4Cl (3 x 20 mL). Em seguida separou-se a fase orgânica
e secou-se com Mg2SO4 e retirou-se o solvente no rota-evaporador a pressão
reduzida. O resíduo foi purificado por cromatografia em coluna, usando-se
hexano como eluente.
2,5-difenil-3-(fenilseleno)-selenofeno (4a). Rendimento:
0,188 g (86%). RMN 1H (CDCl3, 400 MHz): δ 7,56-7,54 (m, 2H); 7,49-7,47 (m,
2H); 7,40-7,19 (m, 12H). RMN 13C (CDCl3, 100 MHz): δ 149,44; 135,90; 135,52;
132,43; 131,03; 129,32; 129,24; 128,95; 128,33; 128,24; 128,13; 127,94;
127,86; 126,67; 126,05; 122,16. MS (intensidade relativa) m/z: 439 (100); 284
(46); 207 (58); 156 (75); 129 (37); 77 (21). HRMS calculado para C22H16Se2:
439,9582. Encontrado: 439,9585.
2,5-difenil-3-(p-metilfenilseleno)-selenofeno (4b).
Rendimento: 0,181 g (80%). RMN 1H (CDCl3, 400 MHz): δ 7,55-7,46 (m, 4H);
7,38-7,22 (m, 9H); 7,15-7,09 (m, 2H); 2,33 (s, 3H). RMN 13C (CDCl3, 100 MHz):
δ 149,46; 138,07; 135,91; 135,50; 133,49; 132,49; 131,44; 130,98; 130,08;
129,21; 128,91; 128,31; 127,91; 126,74; 126,01; 121,73; 21.83. MS
(intensidade relativa) m/z: 454 (56); 284 (79); 202 (100); 169 (6); 126 (7); 91
(18); 77 (7). HRMS calculado para C23H18Se2: 453,9739. Encontrado: 453,9743.
2,5-difenyil-3-(m-metilfenilseleno)-selenofeno (4c).
Rendimento: 0,167 g (74%). RMN 1H (CDCl3, 400 MHz): δ 7,54-7,08 (m, 15H;
2,32 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ 149,66; 149,45; 138,02; 135,88;
135,47; 133,49; 132,47; 130,94; 130,06; 129,18; 128,92; 128,90; 128,29;
128,07; 127,87; 126,73; 125,99; 121,70; 21,82. MS (intensidade relativa) m/z:

92
454 (54); 282 (56); 202 (100); 126 (7); 91 (22); 77 (8). HRMS calculado para
C23H18Se2: 453,9739. Encontrado: 453,9741.
2,5-difenil-3-(p-clorofenilseleno)-selenofeno (4d).
Rendimento: 0,210 g (89%). RMN 1H (CDCl3, 400 MHz): δ 7,52-7,46 (m, 4H);
7,38-7,24 (m, 9H); 7,16 (d, J = 8,53 Hz, 2H). RMN 13C (CDCl3, 100 MHz): δ
149,88; 149,78; 135,68; 135,34; 132,74; 132,17; 132,15; 130,86; 129,30;
129,26; 128,95; 128,34; 128,21; 128,03; 126,01; 121,65. MS (intensidade
relativa) m/z: 474 (47); 394 (51); 358 (15); 282 (20); 202 (100); 77 (8). HRMS
calculado para C22H15ClSe2: 473,9193. Encontrado: 473,9196
2,5-difenil-3-(p-fluorofenilseleno)-selenofeno (4e).
Rendimento: 0,203 g (89%). RMN 1H (CDCl3, 200 MHz): δ 7,55-7,27 (m, 13H);
6,93 (t, J = 8,80 Hz, 2H). RMN 13C (CDCl3, 100 MHz): δ 162,16 (d, J = 246,63
Hz), 149,56; 148,47; 135,62 (d, J = 38,05 Hz); 133,70 (d, J = 7,32 Hz); 131,75;
131,39; 129,26; 128,95; 128,36; 128,14; 127,99; 126,57; 126,02; 122,72;
116,39 (d, J = 21,95). MS (intensidade relativa) m/z: 458 (48); 378 (50); 282
(26); 220 (15); 202 (100); 176 (5); 96 (6); 77 (7). HRMS calculado para
C22H15FSe2: 457,9488. Encontrado: 457,9490.
2,5-difenil-3-(m-trifluorometilfenilseleno)-selenofeno
(4f). Rendimento: 0,207 g (82%). RMN 1H (CDCl3, 400 MHz): δ 7,57 (s, 1H);
7,52-7,25 (m, 14H). RMN 13C (CDCl3, 100 MHz): δ 151,06; 150,11; 135,58;
135,32; 134,10; 133,60; 132,27; 131,33 (q, J = 32,20 Hz); 129,45; 129,30;
128,99; 128,35; 128,32; 128,11; 126,97 (q, J = 3,66 Hz); 126,05; 123,66 (q, J =
272,97 Hz); 123,20 (q, J = 3,66 Hz); 120,76. MS (intensidade relativa) m/z: 508
(51); 428 (43); 347 (8); 282 (23); 202 (100); 126 (7); 89 (11); 77 (6). HRMS
calculado para C23H15F3Se2: 507,9456. Encontrado: 507,9459.

93
2,5-difenil-3-(2,4,6-trimetilfenilseleno)-selenofeno (4g).
Rendimento: 0,156 g (65%). RMN 1H (CDCl3, 400 MHz): δ 7,42-7,19 (m, 11H);
6,91 (s, 2H); 2,40 (s, 6H); 2,56 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ 148,69;
142,78; 141,85; 138,70; 136,37; 135,72; 129,38; 128,83; 128,81; 128,78;
128,43; 128,28; 128,26; 127,69; 126,03; 125,26; 24,31; 20,98. MS (intensidade
relativa) m/z: 482 (45); 401 (6); 284 (100); 202 (71); 119 (19); 105 (10); 91 (21);
77 (12). HRMS calculado para C25H22Se2: 482,0052. Encontrado: 482,0057.
2,5-difenil-3-(2-naftilseleno)-selenofeno (4h).
Rendimento: 0,137 g (56%). RMN 1H (CDCl3, 400 MHz): δ 7,85 (s, 1H); 7,68-
7,66 (m, 2H); 7,58-7,56 (m, 2H); 7,46-7,23 (m, 13H). RMN 13C (CDCl3, 100
MHz): δ 149,53; 149,27; 135,87; 135,44; 133,96; 132,32; 132,08; 130,02;
129,60; 129,29; 128,90; 128,82; 128,63; 128,34; 128,12; 127,91; 127,72;
127,25; 126,43; 126,01; 125,87; 122,18. MS (intensidade relativa) m/z: 488
(49); 410 (55); 329 (36); 282 (18); 202 (100); 126 (20); 77 (9); 51 (9). HRMS
calculado para C26H18Se2: 489,9739. Encontrado: 489,9743.
2-fenil-3-(fenilseleno)-selenofeno (4i). Rendimento: 0,165 g
(91%). RMN 1H (CDCl3, 400 MHz): δ 7,88 (d, J = 5,87 Hz, 1H); 7,51-7,49 (m,
2H), 7,36-7,16 (m, 9H). RMN 13C (CDCl3, 100 MHz): δ 149,85; 136,89; 135,94;
132,52; 131,19; 130,02; 129,38; 129,13; 128,25; 127,98; 126,63; 121,40. MS
(intensidade relativa) m/z: 364 (65); 284 (62); 202 (47); 126 (59); 115 (100); 77
(33); 51 (18). HRMS calculado para C16H12Se2: 363,9269. Encontrado:
363,9271.
2,5-bis(p-metilfenil)-3-(fenilseleno)-selenofeno (4j).
Rendimento: 0,179 g (77%). RMN 1H (CDCl3, 400 MHz): δ 7,44-7,42 (m, 2H);

94
7,37-7,34 (m, 3H); 7,25-7,08 (m, 9H); 2,35-2,32 (m, 6H). RMN 13C (CDCl3, 100
MHz): δ 149,18; 149,16; 138,01; 137,82; 133,09; 132,82; 131,88; 130,83;
129,56; 129,35; 129,17; 129,01; 128,79; 126,50; 125,88; 121,56; 21,23; 21,13.
MS (intensidade relativa) m/z: 468 (100); 388 (87); 310 (8); 293 (24); 229 (26);
215 (92); 91 (14); 77 (18); 51 (6). HRMS calculado para C24H20Se2: 467,9895.
Encontrado: 467,9898.
2,5-difenil-3-(fenilteluro)-selenofeno (4k). Rendimento:
0,187 g (77%). 1H RMN (CDCl3, 400 MHz): δ 7,65-7,63 (m, 2H); 7,48-7,17 (m,
14H). RMN 13C (CDCl3, 50 MHz): δ 151,22; 150,87; 137,48; 135,56; 135,34;
129,43; 129,12; 129,01; 128,89; 128,36; 128,16; 127,79; 127,74; 126,15;
115,44; 107,58. MS (intensidade relativa) m/z: 488 (29); 360 (31); 282 (15); 202
(100); 77 (22); 51 (9). HRMS calculado para C22H16SeTe: 489,9479.
Encontrado: 489,9481.
2,5-difenil-3-(p-metilfenilteluro)-selenofeno (4l).
Rendimento: 0,183 g (73%). RMN 1H (CDCl3, 200 MHz): δ 7,58 (d, J = 7,94 Hz,
2H); 7,50-7,24 (m, 11H); 7,02 (d, J = 7,94 Hz, 2H); 2,32 (s, 3H). RMN 13C
(CDCl3, 100 MHz): δ 151,02; 149,88; 138,22; 137,95; 137,61; 135,68; 134,97;
130,38; 129,09; 128,86; 128,38; 128,10; 127,74; 126,20; 110,98; 107,98; 21,20.
MS (intensidade relativa) m/z: 502 (30); 374 (40), 282 (14), 202 (100), 91 (24),
77 (5). HRMS calculado para C23H18SeTe: 503,9636. Encontrado: 503,9639.
2,5-difenil-3-(p-clorofenilteluro)-selenofeno (4m).
Rendimento: 0,203 g (78%). RMN 1H (CDCl3, 200 MHz): δ 7,53-7,26 (m, 13H);
7,14 (d, J = 8,23 Hz, 2H). RMN 13C (CDCl3, 100 MHz): δ 151,61; 151,59;
138,64; 137,38; 135,47; 135,25; 134,17; 129,62; 129,15; 128,95; 128,38;
128,27; 127,92; 126,19; 113,11; 107,27. MS (intensidade relativa) m/z: 522

95
(30); 394 (37); 282 (17); 202 (100); 126 (6); 77 (8). HRMS calculado para
C22H15ClSeTe: 523,9090. Encontrado: 523,9094.
3.2.11 Procedimento geral para a reação de acoplamento cruzado do telurofeno 2o com ácidos borônicos.
Em uma solução do telurofeno 2o (0,25 mmol) em MeOH (2 mL) foi
adicionado Pd(PPh3)4 (5 mol%), o ácido borônico (1,2 eq), Ag2O (2 eq) e Et3N
(3 eq). A solução resultante foi agitada por 90 minutos sob refluxo, resfriado a
temperatura ambiente, diluído com acetato de etila (30 mL) e lavado com
NH4Cl (3 x 30 mL). Após, secou-se a fase orgânica com MgSO4 anidro, o
solvente foi removido sob pressão reduzida e o resíduo purificado por
cromatografia em coluna, usando-se hexano como eluente. 5a: Rendimento:
0,088 g (81 %). RMN 1H: CDCl3, 200 MHz, δ(ppm): 8,08 (s, 1H); 7,50-7,46 (d,
J = 8,8 Hz, 2H); 7,40-7,18 (m, 10H); 6,82-6,78 (d, J = 8,8 Hz, 2H); 3,79 (s, 3H).
RMN 13C: CDCl3, 100 MHz, δ(ppm): 158, 45; 137,95; 131,74; 130,56; 129,27;
128,94; 128,37; 127,63; 126,74; 113,63; 55,19.
3.2.12 Procedimento geral para a reação de acoplamento cruzado do telurofeno 2o com ariltrifluoroboratos.
Em uma solução do telurofeno 2o (0,25 mmol) em MeOH (2 mL) foi
adicionado Pd(PPh3)4 (5 mol%), o ariltrifluoroborato (1,2 eq), Ag2O (2 eq) e
Et3N (3 eq). A solução resultante foi agitada por 90 minutos em refluxo,
resfriado a temperatura ambiente, diluído com acetato de etila (30 mL) e lavado
com NH4Cl (3 x 30 mL). Após, secou-se a fase orgânica com MgSO4 anidro, o
solvente foi removido sob pressão reduzida e o resíduo purificado por
cromatografia em coluna, usando-se hexano como eluente. 5b: Rend: 0,09 g
(86%). RMN 1H: CDCl3, 400 MHz, δ(ppm): 7,59-7,57 (m, 3H); 7,39-7,35 (m,
2H); 7,30-7,19 (m, 8H); 7,10-7,08 (m, 2H); 2,33 (s, 3H); RMN 13C: CDCl3, 100
MHz, δ(ppm): 148,25; 140,84; 136,57; 136,32; 136,17; 134,71; 129,58; 129,25;
129,21; 129,05; 129,01; 128,92; 128,38; 127,64; 127,19; 126,02; 21,19; MS m/z

96
(%) 373 (100); 281 (65); 296 (45); 220 (42); 128 (72); 91 (65). HRMS
Calculado. C23H18Se: 374,0574. Encontrado: 374,0580.
3.2.13 Procedimento geral para preparação dos 2-alquilselanil-3-alquiniltiofenos 6a-l, 6p-q.
Em um balão de duas bocas, sob atmosfera de argônio e agitação
magnética, foi adicionado n-BuLi (20 mmol, 1,5 M em hexano) gota a gota a
uma solução do adequado 3-alquiniltiofeno (20 mmol) em THF seco (100 mL) a
-78 ºC. A mistura da reação foi agitada por 30 minutos nesta temperatura e
então, elevou-se a -40 ºC e adicionou-se numa só porção o selênio elementar
(20 mmol, 1.58 g) e deixou-se reagir por uma hora nessa temperatura. Após, o
adequado brometo de alquila (25 mmol) foi adicionado a -10 ºC e a mistura foi
agitada por duas horas a temperatura ambiente. Após este tempo, foi
adicionado uma solução concentrada de NH4Cl (50 mL) e extraída com acetato
de etila (3 x 50 mL). A fase orgânica foi seca com MgSO4 e concentrada sob
vácuo. Os produtos obtidos foram purificados por cromatografia em coluna de
gel de sílica utilizando-se hexano como eluente.
2-(butilselenil)-3-(feniletinil)-tiofeno (6a). RMN 1H (CDCl3, 400
MHz): δ 7,55-7,53 (m, 2H); 7,37-7,32 (m, 3H); 7,29 (d, J = 5,4 Hz, 1H); 7,12 (d,
J = 5,1 Hz, 1H); 2,98 (t, J = 7,3 Hz, 2H); 1,71 (quint, J = 7,3 Hz, 2H), 1,44(sex, J
= 7,3 Hz, 2H), 0,89 (t, J = 7,3 Hz, 3H). MS (intensidade relativa) m/z: = 316
(100); 261 (91); 182 (29); 137 (31); 76 (1).
2-(etilselenil)-3-(feniletinil)tiofeno (6p). RMN 1H (CDCl3, 200
MHz): δ 7,57-7,52 (m, 2H); 7,38-7,34 (m, 3H); 7,32 (d, J = 5,3 Hz, 1H); 7,14 (d,
J = 5,3 Hz, 1H); 2,98 (q, J = 7,3 Hz, 2H); 1,46 (t, J = 7,3 Hz, 3H). RMN 13C

97
(CDCl3, 100 MHz): δ 131,5; 130,6; 128,5; 128,3; 128,2; 99,9; 92,5; 84,6; 24,3;
15,6. MS (intensidade relativa) m/z: = 289 (100); 274 (34); 259 (59); 216 (21);
182 (16); 137 (41); 112 (15); 87 (8).
2-(benzilselenil)-3-(feniletinil)tiofeno (6q). RMN 1H (CDCl3, 200
MHz): δ 7,57-7,47 (m, 2H); 7,37-7,11(m, 10H); 1,56 (s, 2H). RMN 13C (CDCl3,
100 MHz): δ 138,3; 137,9; 134,1; 131,6; 131,5; 130,6; 129,2; 128,9; 128,8;
128,5; 128,4; 128,3; 127,1; 127,0; 123,2; 35,5. MS (intensidade relativa) m/z:
349 (14); 270 (21); 259 (7); 137 (11); 90 (100); 64 (11); 50 (2).
2-(butilselenil)-3-(hept-1-inil)tiofeno (6f). RMN 1H (CDCl3, 200
MHz): δ 7,23 (d, J = 5,3 Hz, 1H); 6,99 (d, J = 5,3 Hz, 1H); 2,92 (t, J = 7,3 Hz,
2H); 2,44 (t, J = 6,9 Hz, 2H); 1,75-1,27 (m, 10H); 0,96-0,86 (m, 6H). RMN 13C
(CDCl3, 50 MHz): δ MS (intensidade relativa) m/z: 139,9; 124,6; 122,6; 121,5;
97,1; 74,2; 32,4; 31,1; 29,6; 28,5; 22,8; 22,2; 19,7; 14,1; 13.5.
2-(butilselenil)-3-(3,3-dimetilbut-1-inil)tiofeno (6g). RMN 1H
(CDCl3, 400 MHz): δ 7,21 (d, J = 5,3 Hz, 1H); 6,98 (d, J = 5,3 Hz, 1H); 2,93 (t, J
= 7;3 Hz, 2H); 1,67 (quint, J = 7,3 Hz, 2H); 1,43 (sex, J = 7.3 Hz, 2H); 1,33 (s,
9H); 0,90 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 100 MHz): δ 130,6; 128,5; 127,8;
127,4; 101,9; 99,9; 74,2; 32,2; 31,0; 29,9; 28,2; 22,7; 13,5. MS (intensidade
relativa) m/z: 297 (43); 226 (74); 161 (31); 146 (92); 56 (29); 50 (3).

98
2-(butilselenil)-3-(p-toliletinil)tiofeno (6b). RMN 1H (CDCl3, 400
MHz): δ 7,43 (d, J = 8,1 Hz, 2H); 7,28 (d, J = 5,3 Hz, 1H); 7,15 (d, J = 8,1 Hz,
2H); 7,10 (d, J = 5,3 Hz, 1H); 2,97 (t, J = 7,3 Hz, 2H); 2,36 (s, 3H); 1,7 (quint, J
= 7,3 Hz, 2H); 1,44 (sex, J = 7,3 Hz, 2H); 0,89 (t, J = 7,3 Hz, 3H). RMN 13C
(CDCl3, 50 MHz): δ 134,2; 132,7; 130,5; 128,7; 128,5; 127,5; 121,8; 91,3; 82,0;
49,0; 32,2; 30,7; 22,6; 13,5. MS (intensidade relativa) m/z: 330 (100); 275 (33);
196 (10); 151 (37); 76 (3).
2-(butilselenil)-3-[(4-metoxifenil)etinil]tiofeno (6c). RMN 1H
(CDCl3, 200 MHz): δ 7,48 (d, J = 8,1 Hz, 2H); 7,29 (d, J = 5,3 Hz, 1H); 7,1 (d, J
= 5,3 Hz, 1H); 6,88 (d, J = 8,1 Hz, 2H); 3,83 (s, 3H); 2,97 (t, J = 7,3 Hz, 2H); 1,7
(quint, J = 7,3 Hz, 2H); 1,44 (sex, J = 7.3 Hz, 2H); 0,89 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 159,5; 137,4; 132,9; 132,8; 130,5; 128,3; 123,7; 113,9;
92,5; 78,2; 55,3; 32,1; 30,5; 22,6; 13,5. MS (intensidade relativa) m/z: 346
(100); 303 (34); 290 (28); 276 (76); 169 (25); 125 (14); 73 (5); 56 (4).
2-(butilselenil)3-(naftaleno-2-iletinil)tiofeno (6d). RMN 1H
(CDCl3, 200 MHz): δ 8,1 (s, 1H); 7,75-7,70 (m, 3H); 7,62-7,47 (m, 3H); 7,32 (d,
J = 5,3 Hz, 1H); 7,17 (d, J = 5,3 Hz, 1H); 3,01 (t, J = 7,3 Hz, 2H); 1,73 (quint, J
= 7,3 Hz, 2H); 1,45 (sex, J = 7,3 Hz, 2H); 0,90 (t, J = 7,3 Hz, 3H). RMN 13C
(CDCl3, 50 MHz): δ 133,4; 133,0; 132,8; 131,3; 130,6; 129,2; 128,3; 127,9;
127,9; 127,7; 126,6; 126,5; 120,6; 92,9; 84,9; 32,2; 30,7; 22,7; 13,5. MS
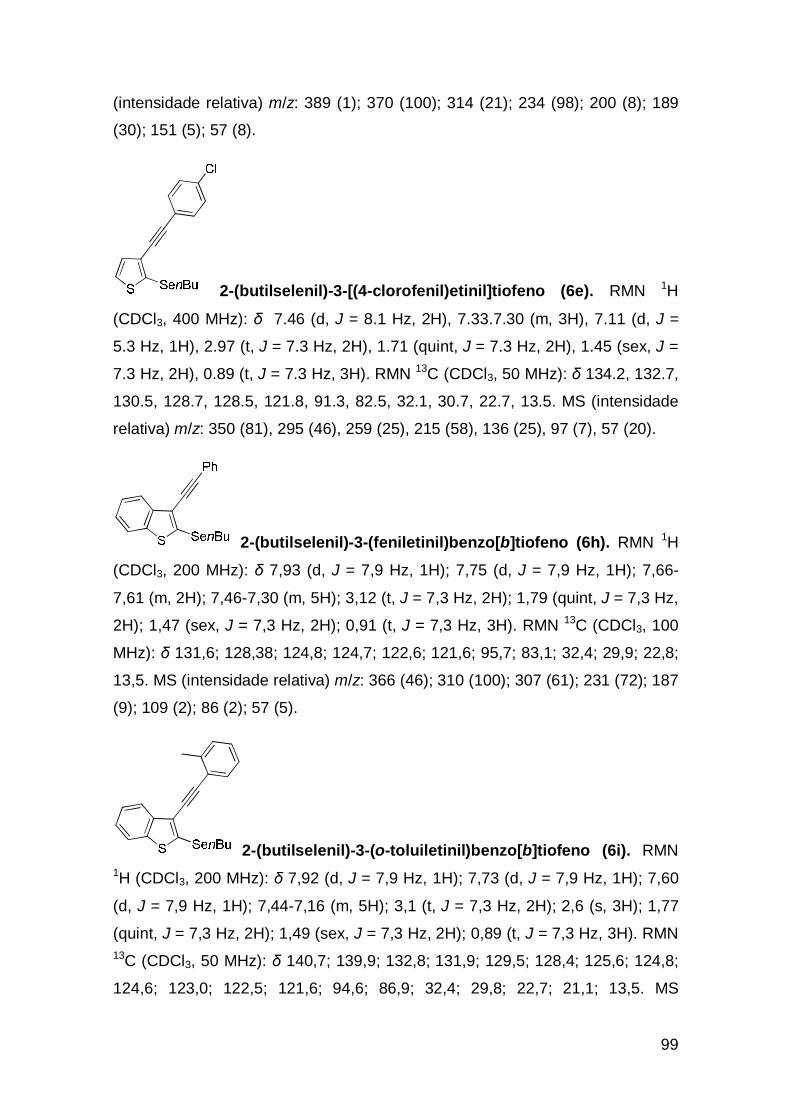
99
(intensidade relativa) m/z: 389 (1); 370 (100); 314 (21); 234 (98); 200 (8); 189
(30); 151 (5); 57 (8).
2-(butilselenil)-3-[(4-clorofenil)etinil]tiofeno (6e). RMN 1H
(CDCl3, 400 MHz): δ 7.46 (d, J = 8.1 Hz, 2H), 7.33.7.30 (m, 3H), 7.11 (d, J =
5.3 Hz, 1H), 2.97 (t, J = 7.3 Hz, 2H), 1.71 (quint, J = 7.3 Hz, 2H), 1.45 (sex, J =
7.3 Hz, 2H), 0.89 (t, J = 7.3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 134.2, 132.7,
130.5, 128.7, 128.5, 121.8, 91.3, 82.5, 32.1, 30.7, 22.7, 13.5. MS (intensidade
relativa) m/z: 350 (81), 295 (46), 259 (25), 215 (58), 136 (25), 97 (7), 57 (20).
2-(butilselenil)-3-(feniletinil)benzo[b]tiofeno (6h). RMN 1H
(CDCl3, 200 MHz): δ 7,93 (d, J = 7,9 Hz, 1H); 7,75 (d, J = 7,9 Hz, 1H); 7,66-
7,61 (m, 2H); 7,46-7,30 (m, 5H); 3,12 (t, J = 7,3 Hz, 2H); 1,79 (quint, J = 7,3 Hz,
2H); 1,47 (sex, J = 7,3 Hz, 2H); 0,91 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 100
MHz): δ 131,6; 128,38; 124,8; 124,7; 122,6; 121,6; 95,7; 83,1; 32,4; 29,9; 22,8;
13,5. MS (intensidade relativa) m/z: 366 (46); 310 (100); 307 (61); 231 (72); 187
(9); 109 (2); 86 (2); 57 (5).
2-(butilselenil)-3-(o-toluiletinil)benzo[b]tiofeno (6i). RMN 1H (CDCl3, 200 MHz): δ 7,92 (d, J = 7,9 Hz, 1H); 7,73 (d, J = 7,9 Hz, 1H); 7,60
(d, J = 7,9 Hz, 1H); 7,44-7,16 (m, 5H); 3,1 (t, J = 7,3 Hz, 2H); 2,6 (s, 3H); 1,77
(quint, J = 7,3 Hz, 2H); 1,49 (sex, J = 7,3 Hz, 2H); 0,89 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 140,7; 139,9; 132,8; 131,9; 129,5; 128,4; 125,6; 124,8;
124,6; 123,0; 122,5; 121,6; 94,6; 86,9; 32,4; 29,8; 22,7; 21,1; 13,5. MS

100
(intensidade relativa) m/z: 384 (78); 327 (30); 247 (100); 215 (31); 115 (9); 77
(1).
4-[(2-butilselenil)benzo[b]tiofen-3-il)etinil]-N,N-dimetilanilina (6j). RMN 1H (CDCl3, 200 MHz): δ 7,93 (d, J = 7,9 Hz, 1H);
7,74 (d, J = 7,9 Hz, 1H); 7,52-7,31 (m, 4H); 6,69 (d, J = 8,5 Hz, 2H); 3,2 (t, J =
7,3 Hz, 2H); 2,9 (s, 6H); 1,78 (quint, J = 7,3 Hz, 2H); 1,49 (sex, J = 7,3 Hz, 2H);
0,91 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 100 MHz): δ 150,2; 140,5; 132,8;
124,9; 124,6; 124,5; 122,7; 121,6; 111,8; 109,9; 99,9; 81,1; 40,2; 40,2; 32,4;
29,8; 22,7; 13,5. MS (intensidade relativa) m/z: 413 (100); 370 (37); 356 (66);
341 (13); 312 (17); 232 (19); 156 (5); 41 (13).
2-(butilselenil)-3-[(2-metoxifenil)etinil]benzo[b]tiofen (6k).
RMN 1H (CDCl3, 400 MHz): δ 7,99 (d, J = 7,9 Hz, 1H); 7,36 (d, J = 7,9 Hz, 1H);
7,59-7,57 (m, 1H); 7,43- 7,39 (m, 1H); 7,35-7,31 (m, 2H); 6,99-6,33 (m, 2H);
3,96 (s, 3H); 3,15 (t, J = 7,3 Hz, 2H); 1,78 (quint, J = 7,3 Hz, 2H); 1,48 (sex, J =
7,3 Hz, 2H); 0,91 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 160,1; 140,7;
140,3; 133,3; 132,2; 129,8; 124,8; 124,7; 122,8; 122,7; 121,5; 120,5; 112,6;
110,8; 92,3; 87,2; 55,8; 32,4; 29,7; 22,7; 13,5. MS (intensidade relativa) m/z:
470 (15); 328 (19); 262 (17); 164 (10); 131 (26); 73 (12); 44 (8).
2-(butilselenil)-3-[(2-metoxi-5-metilfenil)etinil]benzo[b]tiofeno (6l). RMN 1H (CDCl3, 400 MHz): δ 7,99 (d, J

101
= 7,9 Hz, 1H); 7,74 (d, J = 7,9 Hz, 1H); 7,43-7,49 (m, 2H); 7,36-7,31 (m, 1H);
7,12 (d, J = 7,9 Hz, 1H); 6,83 (d, J = 8,5 Hz, 1H); 3,93 (s, 3H); 3,15 (t, J = 7,3
Hz, 2H); 2,31 (s, 3H); 1,78 (quint, J = 7,3 Hz, 2H); 1,48 (sex, J = 7,3 Hz, 2H);
0,91 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 100 MHz): δ 158,1; 140,3; 133,7;
132,0; 130,3; 129,7; 124,7; 124,6; 122,8; 122,8; 121,5; 112,3; 110,8; 99,9; 92,5;
86,8; 56,0; 32,4; 29,7; 22,8; 20,3; 13,5. MS (intensidade relativa) m/z: 478 (84);
475 (44); 338 (100); 335 (43); 273 (15); 169 (6); 137 (17); 72 (1).
3.2.14 Procedimento geral para preparação do 3-(feniletinil)-2-
(fenilselenil)tiofeno 6r.
Em um balão de duas bocas, sob atmosfera de argônio e agitação
magnética, foi adicionado n-BuLi (20 mmol, 1,5 M em hexano) gota a gota a
uma solução do 3-feniletiniltiofeno (20 mmol, 3,68 g) em THF seco (100 mL) a
-78 ºC. A mistura da reação foi agitada por 30 minutos nesta temperatura e
então, elevou-se a -40 ºC e adicionou-se numa só porção o PhSeBr (25 mmol,
5,9 g) e deixou-se reagir por duas horas a temperatura ambiente. Após este
tempo, foi adicionado uma solução concentrada de NH4Cl (50 mL) e extraída
com acetato de etila (3 x 50 mL). A fase orgânica foi seca com MgSO4 e
concentrada sob vácuo. Os produtos obtidos foram purificados por
cromatografia em coluna de gel de sílica utilizando-se hexano como eluente.
3-(feniletinil)-2-(fenilselenil)tiofeno (6r). RMN 1H (CDCl3, 200
MHz): δ 7,52-7,43 (m, 4H); 7,36-7,22 (m, 7H); 7,17 (d, J = 5,3 Hz, 1H). RMN 13C (CDCl3, 100 MHz): δ 131,9; 131,6; 130,9; 130,6; 129,7; 129,4; 129,3; 129,2;
128,9; 128,8; 128,3; 128,2; 127,8; 127,3; 123,0; 92,9; 84,2. MS (intensidade
relativa) m/z: 340 (17); 260 (100); 215 (31); 129 (26); 77 (1); 51 (9).

102
3.2.15 Procedimento geral para preparação do 2-(butiltelanil)-3-(feniletinil)tiofeno 6o.
Em um balão de duas bocas, sob atmosfera de argônio e agitação
magnética, foi adicionado n-BuLi (20 mmol, 1,5 M em hexano) gota a gota a
uma solução do adequado 3-feniletiniltiofeno (20 mmol, 3,68 g) em THF seco
(100 mL) a -78 ºC. A mistura da reação foi agitada por 30 minutos nesta
temperatura e então, elevou-se a -40 ºC e adicionou-se numa só porção o
telúrio elementar (20 mmol, 2,56 g) e deixou-se reagir por uma hora nessa
temperatura. Após, o bromobutano (25 mmol, 3,43 g) foi adicionado a -10 ºC e
a mistura foi agitada por duas horas a temperatura ambiente. Após este tempo,
foi adicionado uma solução concentrada de NH4Cl (50 mL) e extraída com
acetato de etila (3 x 50 mL). A fase orgânica foi seca com MgSO4 e
concentrada sob vácuo. Os produtos obtidos foram purificados por
cromatografia em coluna de gel de sílica utilizando-se hexano como eluente.
2-(butiltelanil)-3-(feniletinil)tiofeno (6o). RMN 1H (CDCl3, 200
MHz): δ 7,58-7,52 (m, 2H); 7,42 (d, J = 5,3 Hz, 1H); 7,38- 7,32 (m, 3H), 7,12 (d,
J = 5,3 Hz, 1H); 2,98 (t, J = 7,3 Hz, 2H); 1,79 (quint, J = 7,3 Hz, 2H); 1,39 (sex,
J = 7,3 Hz, 2H); 0,89 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 143,5;
132,5; 131,5; 130,4; 128,4; 128,3; 128,2; 123,2; 104,2; 91,9; 86,2; 33,6; 25,9;
13,3; 10,9. MS (intensidade relativa) m/z: 370 (35); 312 (10); 184 (100); 139
(49); 57 (9).
3.2.16 Procedimento geral para as reações de ciclização eletrofílica.
A uma solução do 2-alquilcalcogênio-3-alquiniltiofeno apropriado (0,25
mmol) em CH2Cl2 (2 mL) foram adicionados gradativamente 1,1 equivalentes
do eletrófilo dissolvido em 3 mL de CH2Cl2. Manteve-se a reação a temperatura
ambiente sob agitação pelos tempos indicados na Tabela 3. O excesso do
eletrófilo foi removido lavando-se a reação com solução saturada de Na2S2O3
(20 mL). Extraiu-se a reação com CH2Cl2 (3x 20 mL). A fase orgânica foi seca

103
com MgSO4 e concentrada sob vácuo. Os produtos obtidos foram purificados
por cromatografia em coluna de gel de sílica utilizando-se um gradiente de
hexano e acetato de etila como eluentes.
4-iodo-5-fenilselenofeno[2,3-b]tiofeno (7a). Rendimento: 0,074
g; 76%. RMN 1H (CDCl3, 200 MHz): δ 7,61-7,57 (m, 2H); 7,48-7,41 (m, 4H);
7,33 (d, J = 5,3 Hz, 1H). RMN 13C (CDCl3, 50 MHz): δ 151,8; 136,5; 129,7;
128,5; 128,5; 127,9; 124,2; 73,6. MS (intensidade relativa) m/z: 385 (100); 260
(69); 216 (20); 169 (31); 137 (36); 86 (19); 50 (6). Anal. (%) Calculado para
C12H7ISSe: C: 37,04, H: 1,81. Encontrado: C: 37,29, H: 2,12.
5-fenil-4-(fenilselenil)selenofeno[2,3-b]tiofeno (7b).
Rendimento: 0,071 g; 68%. RMN 1H (CDCl3, 400 MHz): δ 7,54-7,52 (m, 4H);
7,40-7,36 (m, 6H); 7,32 (d, J = 5,3 Hz, 1H); 7,13 (d, J = 5,3 Hz, 1H). MS
(intensidade relativa) m/z: 415 (100); 336 (84); 256 (71); 169 (56); 137 (55); 76
(24); 50 (11). ). Anal. (%) Calculado para C18H12SSe2: C: 61,59, H: 2,89.
Encontrado: C: 61,72, H: 3,11.
4-iodo-5-pentilselenofeno[2,3-b]tiofeno (7g). Rendimento:
0,06798 g; 71%. RMN 1H (CDCl3, 400 MHz): δ 7,37 (d, J = 5,3 Hz, 1H); 7,21 (d,
J = 5,3 Hz, 1H); 2,90 (t, J = 7,3 Hz, 2H); 1,70 (quint, J = 7,3 Hz, 2H); 1,45-1,34
(m, 4H); 0,91 (t, J = 7,3 Hz, 3H). RMN 13C (CDCl3, 50 MHz): δ 150,7; 149,9;
130,8; 127,6; 123,3; 102,3; 78,2; 74,4; 35,4; 31,2; 31,2; 22,4; 13,9. MS
(intensidade relativa) m/z: 380 (22); 378 (33); 323 (100); 321 (56); 254 (13); 198
(19); 120 (20); 74 (9); 50 (3). Anal. (%) Calculado para C11H13ISSe: C: 34,48, H:
3,42. Encontrado: C: 34,62, H: 3,70.
5-tert-butil-4-iodoselenofeno[2,3-b]tiofeno (7h). Rendimento:
0,0618 g; 67%. RMN 1H (CDCl3, 400 MHz): δ 7,36 (d, J = 5,3 Hz, 1H); 7,28 (d,
J = 5,3 Hz, 1H); 1,58 (s, 9H). RMN 13C (CDCl3, 100 MHz): δ 158,1; 153,3;

104
129,2; 127,1; 124,3; 68,5; 37,8; 30,6. MS (intensidade relativa) m/z: 366 (24);
351 (100); 225 (15); 146 (63); 120 (18); 50 (4). Anal. (%) Calculado para
C10H11ISSe: C; 32,54, H: 3,00. Encontrado: C: 32,71, H: 3,27.
4-iodo-5-p-toluilselenofeno[2,3-b]tiofeno (7c).
Rendimento: 0,0866 g; 86%. RMN 1H (CDCl3, 400 MHz): δ 7,47 (d, J = 8,1 Hz,
2H); 7,42 (d, J = 5,3 Hz, 1H); 7,31 (d, J = 5,3 Hz, 1 H); 7,23 (d, J = 8,1 Hz, 2H);
2,39 (s, 3H). RMN 13C (CDCl3, 50 MHz): δ 151,7; 146,8; 138,5; 133,5; 132,9;
129,5; 128,2; 127,8; 124,1; 73,3; 21,3. MS (intensidade relativa) m/z: 399 (100);
274 (22); 195 (31); 151 (28); 137 (32); 51 (4). Anal. (%) Calculado para
C13H9ISSe: C: 38,73, H: 2,25. Encontrado: C: 39,01, H: 2,47.
4-iodo-5-(4-metoxifenil)selenofeno[2,3-b]tiofeno (7d).
Rendimento: 0,0649 g; 62%. RMN 1H (CDCl3, 200 MHz): δ 7,50 (d, J = 8,1 Hz,
2H); 7,44 (d, J = 5,3 Hz, 1H); 7,32 (d, J = 5,3 Hz, 1H); 6,96 (d, J = 8,1 Hz, 2H);
3,86 (s, 3H). RMN 13C (CDCl3, 100 MHz): δ 159,9; 151,7; 146,7; 132,7; 131,0;
128,9; 127,8; 124,1; 113,9; 99,9; 73,4; 55,3. MS (intensidade relativa) m/z: 415
(100); 290 (19); 275 (25); 167 (17); 124 (14); 74 (8). Anal. (%) Calculado para
C13H9IOSSe: C: 37,25, H: 2,16. Encontrado: C: 37,50, H: 2,35.
4-iodo-5-(naftalen-2-il)selenofeno[2,3-b]tiofeno (7e).
Rendimento: 0,09 g; 78%. RMN 1H (CDCl3, 400 MHz): δ 8,03 (s, 1H); 7,88-7,83
(m, 3H); 7,69 (dd, J = 8,8 Hz, 1H); 7,52-7,49 (m, 2H), 7,44 (d, J = 5,3 Hz, 1H);
7,34 (d, J = 5,3 Hz, 1H). Anal. (%) Calculado para C20H11ISSe: C: 49,10, H:
2,27. Encontrado: C: 49,25, H: 2,41.
5-(p-clorofenil)-4-iodoselenofeno[2,3-b]tiofeno (7f).
Rendimento: 0,0773 g; 73%. RMN 1H (CDCl3, 200 MHz): δ 7,54- 7,39 (m, 5H);
7,33 (d, J = 5,3 Hz, 1H). RMN 13C (CDCl3, 50 MHz): δ 151,8; 134,9; 134,7;
133,4; 131,0; 128,8; 128,2; 124,2; 74,2. MS (intensidade relativa) m/z: 420 (57);

105
295 (44); 259 (52); 136 (31); 86 (4); 50 (3). Anal. (%) Calculado para
C12H6ClISSe: C: 34,03, H: 1,43. Encontrado: C: 34,33, H: 1,71.
3-iodo-2-fenilbenzo[d]selenofeno[2,3-b]tiofeno (7i).
Rendimento: 0,0659g; 60%. RMN 1H (CDCl3, 200 MHz): δ 9,05 (d, J = 7,9 Hz,
1H); 7,85 (d, J = 7,9 Hz, 1H); 7,61-7,56 (m, 2H); 7,52-7,36 (m, 5H). MS
(intensidade relativa) m/z: 435 (48); 309 (76); 229 (21); 187 (4); 154 (46); 115
(18); 50 (30). Anal. (%) Calculado para C16H9ISSe: C: 43,76, H: 2,07.
Encontrado: C: 43,59, H: 1,88.
3-iodo-2-o-toluilbenzo[d]selenofeno[2,3-b]tiofeno (7j).
Rendimento: 0,043 g; 38%. RMN 1H (CDCl3, 400 MHz): δ 8,99 (d, J = 7,9 Hz,
1H); 7,85 (d, J = 7,9 Hz, 1H); 7,49-7,36 (m, 5H); 7,32 (d, J = 7,9 Hz, 1H); 2,28
(s, 3H). RMN 13C (CDCl3, 100 MHz): δ 146,5; 144,6; 137,8; 136,5; 134,3; 131,0;
130,3; 129,5; 129,3; 125,7; 124,4; 123,8; 122,7; 121,6; 68,7; 20,4. MS
(intensidade relativa) m/z: 454 (16); 327 (11); 247 (100); 202 (20); 163 (14); 123
(47); 51 (2). Anal. (%) Calculado para C17H11ISSe: C: 45,05, H: 2,45.
Encontrado: C: 45,29, H: 2,61.
4-(3-iodobenzo[d]selenofeno[2,3-b]tiofen-2-il)-
N,N-dimetilanilina (7k). Rendimento: 0,0819 g; 68%. RMN 1H (CDCl3, 200
MHz): δ 9,06 (d, J = 7,9 Hz, 1H); 8,04 (d, J = 7,9 Hz, 1H); 7,84 (d, J = 8,1 Hz,
2H); 7,46 (d, J = 8,1 Hz, 2H); 6,79-6,64 (m, 2H); 3,06 (s, 6H). RMN 13C (CDCl3,
100 MHz): δ 155,4; 149,8; 132,8; 131,0; 128,2; 124,9; 124,5; 124,2; 123,6;
122,6; 122,5; 121,9; 11,6; 72,3; 40,3. MS (intensidade relativa) m/z: 355 (7);
327 (17); 277 (100); 207 (73); 138 (28); 73 (36); 40 (5).
3-iodo-2-(2-metoxifenil)benzo[b]selenofeno[2,3-d]tiofeno (7l). Rendimento: 0,076 g; 65%. RMN 1H (CDCl3, 200 MHz): δ 9,02
(d, J = 7,9 Hz, 1H); 7,83 (d, J = 7,9 Hz, 1H); 7,49-7,35 (m, 4H); 7,09-6,98 (m,
2H); 3,84 (s, 3H). RMN 13C (CDCl3, 50 MHz): δ 156,9; 144,6; 134,4; 132,6;

106
130,6; 125,7; 124,2; 123,6; 122,6; 121,7; 120,4; 111,4; 99,4; 60,3; 55,6. MS
((intensidade relativa) m/z: 470 (79); 328 (100); 262 (20); 234 (20); 164 (29);
131 (26); 44 (8). Anal. (%) Calculado para C17H13IOSSe: C: 43,52, H: 2,36.
Encontrado: C: 43,71, H: 2,52.
3-iodo-2-(metoxi-5-metilfenil)benzo[d]selenofeno[2,3-b]tiofene (7m). Rendimento: 0,0749 g; 62%. RMN 1H (CDCl3, 400 MHz): δ
9,02 (d, J = 7,9 Hz, 1H); 7,82 (d, J = 7,9 Hz, 1H); 7,47-7,35 (m, 2H); 7,22-7,19
(m, 2H); 6,89 (d, J = 7,9 Hz, 1H); 3,81 (s, 3H); 2,35 (s, 3H). RMN 13C (CDCl3,
50 MHz): δ 154,8; 144,6; 134,4; 132,9; 130,9; 129,6; 125,3; 124,2; 123,6;
122,6; 121,7; 111,4; 78,7; 55,7; 20,4. MS (intensidade relativa) m/z: 478 (20);
338 (100); 273 (15); 169 (6); 137 (17); 116 (3); 72 (1).
4-iodo-5-feniltelurofeno[2,3-b]tiofeno (7n). Rendimento: 0,053-
25 g; 48%. RMN 1H (CDCl3, 400 MHz): δ 7,54 (d, J = 5,3 Hz, 1H); 7,51-7,49 (m,
3H); 7,40-7,38 (m, 3H). RMN 13C (CDCl3, 50 MHz): δ 140,3; 137,6; 131,6;
129,7; 129,5; 128,5; 128,4; 128,1; 127,9; 80,4. MS (intensidade relativa) m/z:
440 (36); 341 (54); 281 (65); 207 (100); 139 (62); 73 (73); 41 (23).
3.2.17 Procedimento geral para a reação de acoplamento cruzado do composto 7a com p-cloro-benzenotiol via catálise de CuI.
Em um tudo de Schlenck, sob atmosfera de argônio, contendo o 4-iodo-
5-fenilselenofeno[2,3-b]tiofeno 7a (0,5 mmol) em dioxano seco (3 mL) foi
adicionado o p-cloro-benzenotiol (0,6 mmol). Após, a Et3N (1 mmol) é
adicionado gota a gota, seguido da adição do CuI (0,0095 g, 10 mol %). A
mistura da reação foi refluxada por 12 horas. Após, a solução foi diluída em
diclorometano (20 mL) e lavada com uma solução concentrada de NH4Cl (3 ×
20 mL). A fase orgânica foi seca com MgSO4 e concentrada sob vácuo. Os
produtos obtidos foram purificados por cromatografia em coluna de gel de sílica
utilizando-se hexano como eluente.

107
SeS
S
Ph
Cl
4-(p-clorofeniltio)-5-fenilselenofeno[2,3-b]tiofeno (8a).
Rendimento: 0,134 g; 66%. RMN 1H (CDCl3, 200 MHz): δ 7,57-7,52 (m, 3H);
7,41-7,32 (m, 4H); 7,14 (d, J = 8,1 Hz, 2H); 7,00 (d, J = 8,1 Hz, 2H). RMN 13C
(CDCl3, 50 MHz): δ 135,8; 135,1; 133,4; 131,2; 129,4; 129,0; 128,8; 128,6;
128,5; 127,9; 122,4. MS (relative intensity) m/z: 408 (11); 406 (100); 326 (57);
290 (58); 171 (43); 145 (44); 89 (8); 45 (3).
3.2.18 Procedimento geral para a reação de acoplamento cruzado do tipo Suzuki do 7a com o ácido p-bromofenilborônico.
A uma solução de 4-iodo-5-fenilselenofeno[2,3-b]tiofeno 7a (0,5 mmol)
em DMF/ H2O (5:1; 5mL) foi adicionado o Pd(PPh3)4 (2 mol %) e K2CO3 (2 eq).
Após, o ácido borônico (1,5 eq em DMF 0,5 mL) foi adicionado gota a gota, e a
mistura da reação foi refluxada por 8 horas. Após, a solução foi diluída em
acetato de etila (20 mL) e lavada com uma solução concentrada de NH4Cl (3 ×
20 mL). A fase orgânica foi seca com MgSO4 e concentrada sob vácuo. Os
produtos obtidos foram purificados por cromatografia em coluna de gel de sílica
utilizando-se hexano como eluente.
SeSPh
Br
4-(p-bromofenil)-5-fenilselenofeno[2,3-b]tiofeno (9a).
Rendimento: 0,131 g; 63%. RMN 1H (CDCl3, 200 MHz): δ 7,46 (d, J = 8,1 Hz,
2H); 7,39 (d, J = 5,3 Hz, 1 H); 7,27-7,17 (m, 7H); 7,08 (d, J = 5,3 Hz, 1H). RMN 13C (CDCl3, 100 MHz): δ 149,7; 146,4; 135,9; 131,7; 131,3; 129,4; 128,5; 128,5;
127,6; 122,1; 121,3. MS (relative intensity) m/z: 421 (15); 418 (100); 338 (24);
258 (70); 169 (30); 129 (58); 51 (2).

108
3.2.19 Procedimento geral para a reação de acoplamento cruzado do tipo Negishi do 7a com cloreto de p-toluilzinco.
Em um tubo de Schlenk, munido de agitação magnética e atmosfera de
argônio, contendo o cloreto de p-toluilzinco previamente preparado (1,5 mmol),
foi adicionado 4-iodo-5-fenilselenofeno[2,3-b]tiofeno 7a (0,5 mmol) e Pd(PPh3)4
(10 mol %, 0,057 g). Deixou-se reagir a mistura amarela por 3 horas a
temperatura ambiente. Após, a solução foi diluída em diclorometano (20 mL) e
lavada com uma solução concentrada de NH4Cl (3 × 20 mL). A fase orgânica
foi seca com MgSO4 e concentrada sob vácuo. Os produtos obtidos foram
purificados por cromatografia em coluna de gel de sílica utilizando-se hexano
como eluente.
5-fenil-4-p-toluilselenofeno[2,3-b]tiofeno (10a). Rendimento:
0,121 g; 69%. RMN 1H (CDCl3, 200 MHz): δ 7,62-7,11 (m, 11H); 2,39 (s, 3H).
RMN 13C (CDCl3, 100 MHz): δ 149,1; 138,3; 136,6; 129,4; 128,9; 128,6; 127,8;
126,8; 126,1; 121,7; 118,3; 21,1; Anal. (%) Calculado para C19H14SSe: C:
64,58; H: 3,99. Encontrado: C: 64.71, H: 4.20.

109
Referências Bibliográficas

110
REFERÊNCIAS BIBLIOGRÁFICAS
1. Barreiro, E. J.; Fraga, C. A. F. Química Medicinal: As Bases Moleculares
de ação de Fármacos, Artemed Editora, Porto Alegre, RS, 2001, 53.
2. (a) Barluenga, J.; Trincado, M.; Rubio, E.; Gonzalez, J. M. Angew. Chem.,
Int. Ed. 2003, 42, 2406. (b) Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem.
2006, 71, 62. (c) Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70,
10292. (d) Arcadi, A.; Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L. Synlett
1999, 1432. (e) Yue, D.; Larock, R. C. J. Org. Chem. 2002, 67, 1905. (f)
Hessian, K. O.; Flynn, B. L. Org. Lett. 2003, 5, 4377. (g) Kesharwani, T.;
Worlikar, S. A.; Larock, R. C. J. Org. Chem. 2006, 71, 2307. (h) Flynn, B.
L.; Flynn, G. P.; Hamel, E.; Jung, M. K. Bioorg. Med. Chem. Lett. 2001, 11,
2341. (i) Sniady, A.; Wheeler, K. A.; Dembinski, R. Org. Lett. 2005, 7,
1769. (j) Knight, D. W.; Redfern, A. L.; Gilmore, J. J. Chem. Soc., Perkin
Trans. 1 2002, 622. (k) Huang, Q.; Hunter, J. A.; Larock, R. C. J. Org.
Chem. 2002, 67, 3437. (l) Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68,
5936. (m) Yue, D.; Della Ca, N.; Larock, R. C. Org. Lett. 2004, 6, 1581. (n)
Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 1432. (o) Yao, T.; Campo,
M. A.; Larock, R. C. J. Org. Chem. 2005, 70, 3511. (p) Zhou, C.;
Dubrovsky, A. V.; Larock, R. C. J. Org. Chem. 2006, 71, 1626. (q) Waldo,
J. P.; Larock, R. C. Org. Lett. 2005, 7, 5203. (r) Arcadi, A.; Cacchi, S.;
Giuseppe, S. D.; Fabrizi, G.; Marinelli, F. Org. Lett. 2002, 4, 2409. (s)
Dabdoub, M. J.; Dabdoub, V. B.; Pereira, M. A. J. Org. Chem. 1996, 61,
9503. (t) Bellina, F.; Biagetti, M.; Carpita, A.; Rossi, R. Tetrahedron 2001,
57, 2857. (u) Peng, A.; Ding, Y. J. Am. Chem. Soc. 2003, 125, 15006. (v)
Djuardi, E.; McNelis, E. Tetrahedron Lett. 1999, 40, 7193. (x) Alves, D.;
Luchese, C.; Nogueira, C. W.; Zeni, G. J. Org. Chem. 2007, 72, 6726.
3. (a) Parnham, M. J.; Graf, E. Prog. Drug. Res. 1991, 36, 9. (b) Mugesh, G.;
du Mont, W. W.; Sies, H. Chem. Rev. 2001, 101, 2125. (c) Nogueira, C.
W.; Zeni, G.; Rocha, J. B. T. Chem. Rev. 2004, 104, 6255.
4. (a) Organoselenium Chemistry. Em Topics in Current Chemistry 208;
Wirth, T., Ed.; Springer-Verlag: Heidelberg, 2000. (b) Krief, A. Em
Comprehensive Organometallic Chemistry II; Abel, E. V.; Stone, F. G. A.;
Wilkinson, G., Eds.; Pergamon Press: New York, 1995; Vol. 11, Chapter
(c) Paulmier, C. Selenium Reagents and Intermediates in Organic

111
Synthesis; Em Organic Chemistry Series 4; Baldwin, J. E., Ed.; Pergamon
Press: Oxford, 1986. (d) Sharpless, K. B.; Young, M. W.; Lauer, R. F.
Tetrahedron Lett. 1973, 22, 1979. (e) Reich, H. J. J. Org. Chem. 1975, 40,
2570. (f) Sharpless, K. B.; Lauer, R. F. J. Am. Chem. Soc. 1972, 94, 7154.
(g) Sevrin, M.; Vanende, D.; Krief, A. Tetrahedron Lett. 1976, 30, 2643. (h)
Sevrin, M.; Dumont, W.; Hevesi, L. D.; Krief, A. Tetrahedron Lett. 1976, 30,
2647. (i) Seebach, D.; Peleties, N. Chem. Ber. 1972, 105, 511. (j)
Seebach, D.; Beck, A. K. Angew. Chem., Int. Ed. Engl. 1974, 13, 806. (k)
Reich, H. J.; Shah, S. K. J. Am. Chem. Soc. 1975, 97, 3250. (l) Perin, G.;
Lenardão, E. J.; Jacob, R. G.; Panatiere, R. B. Chem. Rev. 2009, 109,
1277. (m) Freudendahl, D. M.; Shahzad, S. A.; Writh, T. Eur. J. Org. Chem.
2009, 1649.
5. Silveira, C. C.; Braga, A. L.; Vieira, A. S.; Zeni, G. J. Org. Chem. 2003, 68,
662.
6. Nakayama, J.; Konishi, T. Heterocycles 1988, 27, 1731. (b) Kuroda, M.;
Nakayama, J.; Hoshino, M.; Furusho, N.; Kawata, T.; Ohba,S. Tetrahedron
1993, 49, 3735.
7. (a) Skotheim T. A.; Elsenbaumer R. L.; Reynolds J. R. Handbook of
Conducting Polymers, second ed., Dekker, New York, 1998; (b) Nalwa H.
S. Handbook of Conductive Materials and Polymers, Wiley, New York,
1997; (c) Kraft A.; Grimsdale A.; Holmes A. B. Angew. Chem. Int. Ed.
Engl. 1998, 37, 403.
8. Arbizzani C.; Catellani M.; Mastragostino M.; Cerroni M. G. J. Electroanal.
Chem. 1997, 423, 23.
9. (a) Zeni, G.; Nogueira, C. W.; Panatieri, R. B.; Silva, D. O.; Menezes, P.
H.; Braga, A. L.; Silveira, C. C.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron
Lett. 2001, 42, 7921. (b) Zeni, G.; Ludtke, D. S.; Nogueira, C. W.; Panatieri,
R. B.; Braga, A. L.; Silveira, C. C.; Stefani, H. A.; Rocha, J. B. T.
Tetrahedron Lett. 2001, 42, 8927. (c) Zeni, G.; Nogueira, C. W.; Silva, D.
O.; Menezes, P. H.; Stefani, H. A.; Rocha, J. B. T. Tetrahedron Lett. 2003,
44, 1387. (d) Zeni, G.; Silva, D. O.; Menezes, P. H.; Stefani, H. A.; Rocha,
J. B. T. Tetrahedron Lett. 2003, 44, 685. (e) Barros, O. S. R.; Favero, A.;
Nogueira, C. W.; Menezes, P. H.; Zeni, G. Tetrahedron Lett. 2006, 47,
2179. (f) Panatieri, R. B.; Reis, J. S.; Borges, L. P.; Nogueira, C. W.; Zeni,

112
G. Synlett 2006, 3161. (g) Prediger, P.; Moro, A. V.; Nogueira, C. W.;
Savegnago, L.; Rocha, J. B. T.; Zeni, G. J. Org. Chem. 2006, 71, 3786. (h)
Barros, O. S. R.; Nogueira, C. W.; Stangherlin, E. C.; Menezes, P. H.; Zeni,
G. J. Org. Chem. 2006, 71, 1552. (i) Zeni, G. Tetrahedron Lett. 2005, 46,
2647.
10. (a) Savegnago, L.; Jesse, C. R.; Pinto, L. G.; Rocha, J. B. T.; Nogueira,
C. W. Brain Res. 2007, 1175, 54. (b) Luchese, C.; Brandão, R.; Oliveira,
R.; Nogueira, C. W.; Santos, F. W. Toxicology Lett. 2007, 173, 181. (c)
Savegnago, L.; Pinto, L. G.; Jesse, C. R.; Rocha, J. B. T.; Nogueira, C. W.;
Zeni, G. Brain Res. 2007, 1162, 32. (d) Borges, V. C.; Dadalt, G.;
Savegnago, L.; Moro, A. V.; Rocha, J. B. T.; Nogueira, C. W. Toxicology in
Vitro 2007, 21, 387. (e) Luchese, C.; Stangherlin, E. C.; Ardais, A. P.;
Nogueira, C. W.; Santos, F. W. Toxicology 2007, 230, 189. (f) Savegnago,
L.; Pinto, L. G.; Jesse, C. R.; Alves, D.; Rocha, J. B. T.; Nogueira, C. W.;
Zeni, G. Eur. J. Pharmacol 2007, 555, 129. (g) Savegnago, L.; Borges, V.
C.; Alves, D.; Jesse, C. R.; Rocha, J. B. T.; Nogueira, C. W. Life Sci. 2006,
79, 1546. (h) Stangherlin, E. C.; Favero, A. M.; Zeni, G.; Rocha, J. B. T.;
Nogueira, C. W. Brain Res. Bull. 2006, 69, 311.
11. (a) Zeni, G.; Ludtke, D. S.; Panatieri, R. B.; Braga, A. L. Chem. Rev.
2006, 106, 1032. (b) Zeni, G.; Braga, A. L.; Stefani, H. A. Acc. Chem. Res.
2003, 36, 731.
12. (a) Srivastava, P. C.; Robins, R. K. J. Med. Chem. 1983, 26, 445. (b)
Streeter, D. G.; Robins, R. K. Biochem. Biophys. Res. Commun. 1983,
115, 544. (c) Kirsi, J. J.; North, J.; McKernan, P. A.; Murray, B. K.;
Canonico, P. G.; Huggins, J. W.; Srivastava, P. C.; Robins, R. K.
Antimicrob. Agents Chemother. 1983, 24, 353. (d) Goldstein, B. M.; Leary,
J. F.; Farley, B. A.; Marquez, V. E.; Rowley, P. T. Blood 1991, 78, 593. (e)
Jayaram, H. N.; Dion, R. L.; Glazer, R. I.; Johns, D. G.; Robins, R. K.;
Srivastava, P. C.; Cooney, D. A. Biochem. Pharmacol. 1982, 31, 2371.
13. (a) Klapars, A.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124,
7421. (b) Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org.
Chem. 2004, 69, 5578. (c) Shafir, A.; Buchwald, S. L. J. Am. Chem. Soc.
2006, 128, 8742. (d) Shafir, A.; Lichtor, P. A.; Buchwald, S. L. J. Am.

113
Chem. Soc. 2007, 129, 3490. (e) Zhang, D.; Liu, Z.; Yum, E. K.; Larock, R.
C. J. Org. Chem. 2007, 72, 251.
14. Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16,
4467.
15. Suzuki, A. Pure Appl. Chem. 1985, 57, 1749.
16. Scott, W. J.; Peña, M. R.; Sward, K.; Stoessel, S. J.; Stille, J. K. J. Org.
Chem. 1985, 50, 2302.
17. Dieck, H. A.; Heck, R. F. J. Organomet. Chem. 1975, 93, 259.
18. (a) Ullmann, F. Chem. Ber. 1901, 34, 2174. (b) Ullmann, F. Chem. Ber.
1904, 37, 853. (c) Ullmann, F.; Sponagel, P. Chem. Ber. 1905, 36, 2211.
(d) Marcoux, J. F.; Doye, S.; Buchwald, S. L. J. Am. Chem. Soc. 1997,
119, 10539.
19. (a) Shiah, H. S.; Lee, W. S.; Juang, S. H.; Hong, P. C.; Lung, C. C.;
Chang, C. J.; Chou, K. M.; Chang, J. Y. Biochemical Pharmacology 2007,
73, 610. (b) Juang, S. H.; Lung, C. C.; Hsu, P. C.; Hsu, K. S.; Li, Y. C.;
Hong, P. C.; Shiah, H. S.; Kuo, C. C.; Huang, C. W.; Wang, Y. C.; Huang,
L.; Chen, T. S.; Chen, S. F.; Fu, K. C.; Hsu, C. L.; Lin, M. J.; Chang, C. J.;
Ashendel, C. L.; Chan, T. C. K.; Chou, K. M. Chang, J. Y. Molecular
Câncer Therapeutics 2007, 6, 193. (c) Shiah, H. S.; Lee, W. S.; Juang, S.
H.; Chang, C. J.; Chang, J. Y. Clinical Cancer Research 2005, 11, 9101.
20. Chan, G. F. Q.; Towers, G. H. N.; Mitchell, J. C. Phytochemistry 1975, 14, 2295.
21. (a) Katritzky, A. R.; Pozharskii, A. F. Em Handbook of Heterocyclic
Chemistry, Second Edition; Pergamon: Oxford 2000. (b) Eicher, T.;
Hauptmann, S. Em The Chemistry of Heterocycles, Second Edition; Wiley-
VCH 2003.
22. (a) Pu, S.; Hou, J.; Xu, J.; Nie, G.; Zhang, S.; Shen, L.; Xiao, Q. Materials
Lett. 2005, 59, 1061. (b) Salzner, U.; Lagowski, J. B.; Pickup, P. G.; Poirier,
R. A. Synthetic Metals 1998, 96, 177. (c) Otsubo, T.; Inoue, S.; Nozoe, H.;
Jigami, T.; Ogura, F. Synthetic Metals 1995, 69, 537.
23. (a) Zeni, G.; Larock, R. C. Chem. Rev. 2004, 104, 2285. (b) Zeni, G.;
Larock, R. C. Chem. Rev. 2006, 106, 4644.

114
24. (a) Arcadi, A.; Marinelli, F.; Cacchi, S. Synthesis 1986, 749. (b) Arcadi, A.;
Cacchi, S.; Del Rosario, M.; Fabrizi, G.; Marinelli, F. J. Org. Chem. 1996,
61, 9280.
25. Liu, Y.; Zhou, S. Org. Lett. 2005, 7, 4609.
26. Godoi, B.; Sperança, A.; Back, D. F.; Brandão, R.; Nogueira, C. W.; Zeni,
G. J. Org. Chem. 2009, 74, 3469.
27. Manarin, F.; Roehrs, J. A.; Gay, R. M.; Brandão, R.; Menezes, P. H.;
Nogueira, C. W.; Zeni, G. J. Org. Chem. 2009, 74, 2153.
28. Gronowits, S.; Fredj, T.; Moberg-Ogard, A.; Trege, L. J. Heterocycl.
Chem. 1976, 13, 1319.
29. (a) Mack, W. Angew. Chem. 1966, 78, 940; Angew. Chem., Int. Ed. Engl.
1966, 5, 896. (b) Barton, T. J.; Roth, R. W. J. Organomet. Chem. 1972, 39,
66.
30. Sasaki, S.; Adachi, K.; Yoshifuji, M. Org Lett. 2007, 9, 1729.
31. Takimiya, K.; Konda, Y.; Ebata, H.; Niihara, N.; Otsubo, T. J. Org. Chem.
2005, 70, 10569.
32. Saris, L. E.; Cava, M. P. J. Am. Chem. Soc. 1976, 78, 867.
33. Aqad, E.; Lakshmikantham, M. V.; Cava, M. P.; Broker, G. A.; Rogers, R.
D. Org. Lett. 2003, 5, 2519.
34. Takahashi, K.; Tarutani, S. Heterocycles 1996, 43, 1927.
35. Luppold, E.; Müller, E.; Winter, W. J. Chem. Sci. 1976, 31, 1654.
36. Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395.
37. Patil, N. T.; Wu, H.; Yamamoto, Y. J. Org. Chem. 2005, 70, 4531.
38. Sakai, N.; Uchida, N.; Konakahara, T. Tetrahedron Letters 2008, 49,
3437.
39. Peng, A-Y.; Ding, Y-X. J. Am. Chem. Soc. 2003, 125, 15006.
40. Shen, Z.; Lu, X. Adv. Synth. Catal. 2009, 351, 3107.
41. (a) Zeni, G.; Alves, D.; Pena, J. M.; Braga, A. L.; Stefani, H. A.; Nogueira,
C. W. Org. Biom. Chem. 2004, 2, 803. (b) Zeni, G.; Menezes, P. H.; Moro,
A. V.; Braga, A. L.; Silveira, C. C.; Stefani, H. A. Synlett 2001, 1473. (c)
Braga, A. L.; Andrade, L. H.; Silveira, C. C.; Moro, A. V.; Zeni, G.
Tetrahedron Lett. 2001, 42, 8563. (d) Zeni, G.; Perin, G.; Cella, R.; Jacob,
R. G.; Braga, A. L.; Silveira, C. C.; Stefani, H. A. Synlett 2002, 975. (e)
Zeni, G.; Nogueira, C. W.; Pena, J. M.; Pilissão, C.; Menezes, P. H.; Braga,

115
A. L.; Rocha, J. B. T. Synlett 2003, 579. (f) Braga, A. L.; Vargas, F.; Zeni,
G.; Silveira, C. C.; Andrade, L. H. Tetrahedron Lett. 2002, 43, 4399.
42. (a) Zeni, G.; Alves, D.; Braga, A. L.; Stefani, H. A.; Nogueira, C. W.
Tetrahedron Lett. 2004, 45, 4823. (b) Alves, D.; Schumacher, R. F.;
Brandão, R.; Nogueira, C. W.; Zeni, G. Synlett 2006, 7, 1035.
43. (a) Beletskaya, I. P.; Sigeev, A. S.; Peregudov, A. S.; Petrovskii, P. V. J.
Organomet. Chem. 2000, 605, 96. (b) Beletskaya, I. P.; Ananikov, V. P.
Pure Appl. Chem. 2007, 79, 1041.
44. Hay, A. S. J. Org. Chem. 1962, 27, 3320.
45. Alami, M.; Ferri, F. Tetrahedron Lett. 1996, 37, 2763.
46. (a) Dabdoub, M. J.; Baroni, A. C. M.; Lenardão, E. J.; Gianeti, T. R.;
Hurtado, G. R. Tetrahedron 2001, 57, 4271. (b) Zeni, G.; Stracke, M. P.;
Nogueira, C. W.; Braga, A. L.; Menezes, P. H.; Stefani, H. A. Org. Lett. 2004, 6, 1135. (c) Zeni,G.; Formiga, H. B.; Comasseto, J.V. Tetrahedron
Lett. 1999, 41, 1311.
47. (a) Taniguchi, N. J. Org. Chem. 2007, 72, 1241. (b) Taniguchi, N.; Onami,
T. J. Org. Chem. 2004, 69, 915.
48. Vicente, J.; Herrero, P. G.; Sánchez, Y. G.; Jones, P. G.; Bautista, D. Eur.
J. Inorg. Chem. 2006, 115.
49. (a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. (b) Suzuki, A.
Pure Appl. Chem. 1994, 66, 213. (c) Suzuki, A. In Metal-Catalyzed Cross-
Coupling Reactions; Diederich, F., Stang, P. J., Eds.; Wiley-VHC:
Weinheim, 1998. (d) Lloyd-Williams, P.; Giralt, E. Chem. Soc. Rev. 2001,
30, 145.
50. (a) Molander, G. A.; Ellis, N. Acc. Chem. Res. 2007, 40, 275. (b)
Molander, G. A.; Petrillo, D. E.; Landzberg, N. R.; Rohanna, J. C.; Biolatto,
B. Synlett 2005, 1763. (c) Molander, G. A.; Figueroa, R. Aldrichimica Acta
2005, 38, 49. (d) Molander, G. A.; Felix, L. A. J. Org. Chem. 2005, 70,
3950. (e) Molander, G. A.; Yun, C. S.; Ribagorda, M.; Biolatto, B. J. Org.
Chem. 2003, 68, 5534. (f) Molander, G. A.; Biolatto, B. J. Org. Chem.
2003, 68, 4302. (g) Molander, G. A.; Katona, B. W.; Machrouhi, F. J. Org.
Chem. 2002, 67, 8416. (h) Molander, G. A.; Reviro, M. R. Org. Lett. 2002,
4, 107.

116
51. Para revisão: (a) Petragnani, N.; Stefani, H. A. Tetrahedron 2005, 61,
1613. (b) Petragnani, N.; Comasseto, J. V. Synthesis 1986, 1. (c)
Petragnani, N.; Comasseto, J. V. Synthesis 1991, 793, 897. (d)
Comasseto, J. V.; Ling, L. W.; Petragnani, N.; Stefani, H. A. Synthesis
1997, 373.
52. (a) Braga, A. L.; Rhoden, C. R. B.; Zeni, G.; Silveira, C. C.; Andrade, L. H.
J. Organomet. Chem. 2003, 682, 35. (b) Nishibayashi, Y.; Cho, C.-S.; Ohe,
K.; Uemura, S. J. Organomet. Chem. 1996, 507, 197. (c) Nishibayashi, Y.;
Cho, C.-S.; Ohe, K.; Uemura, S. J. Organomet. Chem. 1996, 526, 335.
53. (a) Stefani, H. A.; Cella, R.; Dorr, F. A.; Pereira, C. M. P.; Zeni, G., Jr.;
Gomes, M. Tetrahedron Lett. 2005, 46, 563. (b) Kang, S.-K.; Hong, Y.-T.;
Kim, D.-H.; Lee, S.-H. J. Chem. Res. 2001, 283.
54. (a) Tung, D. T.; Tuan, D. T.; Rasool, N.; Villinger, A.; Reinke, H.; Fischer,
C.; Langer, P. Adv. Synth. Cat. 2009, 351, 1595. (b) Ullah, I.; Khera, R. A.;
Hussain, M.; Villinger, A.; Langer, P. Tetrahedron Lett. 2009, 50, 4651. (c)
Dang, T. T.; Rasool, N.; Dang, T. T.; Reinke, H.; Langer, P. Tetrahedron
Lett. 2007, 48, 845. (d) Tuan, D. T.; Tung, D. T.; Langer, P. Synlett 2006,
2812. (e) Bellur, E.; Langer, P. Eur. J. Org. Chem. 2005, 22, 4815.
55. (a) Manetti, F.; Santucci, A.; Locatelli, G. A.; Maga, G.; Spreafico, A.;
Serchi, T.; Orlandini, M.; Bernardini, G.; Caradonna, N. P.; Spallarossa,
A.; Brullo, C.; Schenone, S.; Bruno, O.; Ranise, A.; Bondavalli, F.;
Hoffmann, O.; Bologna, M.; Angelucci, A.; Botta, M. J. Med. Chem. 2007,
50, 5579; (b) Naya, S.; Ohtoshi, H.; Nitta, M. J. Org. Chem. 2006, 71,
176; (c) Wendt, J. A.; Deeter, S. D.; Bove, S. E.; Knauer, C. S.; Brooker,
R. M.; Augelli-Szafran, C. E.; Schwarz, R. e.; Kinsora, J. J.; Kilgore, K.
S. Bioorg. Med. Chem. Lett. 2007, 17, 5396.
56. (a) Sommen, G. Comel, A.; Kirsch, G. Phosphorus, Sulfur and Silicon
and the Related Elements, 2005, 180, 939. (b) Sommen, G.; Comel, A.;
Kirsch, G. Synthesis, 2004, 451. (c) Yasuike, S.; Kurita, J.; Tsuchiya, T.
Heterocycles 1997, 45, 1891. (d) Kazuo Takimiya, K.; Konda, Y.; Ebata,
H.; Niihara, N.; Otsubo, T. J. Org. Chem. 2005, 70, 10569.
57. Alves, D.; Reis, J. S.; Luchese, C.; Nogueira, C. W.; Zeni, G. Eur. J. Org.
Chem. 2008, 377

117
58. Para outras reatividades relativas de vários grupos funcionais nas
reações de ciclização eletrofílica de alcinos ver: (a) Mehta, S.; Waldo, J.
P.; Larock, R. C. J. Org. Chem. 2009, 74, 1141. (b) Arcadi, A.; Marinelli, F.;
Cacchi, S. Synthesis 1986, 749. (c) G. Kirsch, S. Hesse, A. Comel, Curr.
Org. Chem. 2004, 1, 47.
59. (a) Takeda, T.; Furukawa, H.; Fujimori, M.; Suzuki, K.; Fujiwara, T. Bull.
Chem. Soc. Jpn. 1984, 57, 1863. b) Trost, B. M.; Lavoie, A. C. J. Am.
Chem. Soc. 1983, 105, 5075.
60. Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2004, 248, 2337.
61. (a) Miyaura, N. Cross-Coupling Reactions. A Practical Guide; Springer:
Berlin, 2002; (b) Diederich, F.; Stang, P. J. Metal-Catalyzed Cross-
Coupling Reactions; Wiley-VCH: Weinheim, 1998.
62. (a) Negishi, E.; Anastasia, L. Chem. Rev. 2003, 103, 1979; (b) Negishi, E.
In Handbook of Organopalladium Chemistry for Organic Synthesis; Wiley
and Sons: New York, 2002; Vols. 1 and 2; (c) Li, J. J.; Gribble, G. W.
Palladium in Heterocyclic Chemistry. In Tetrahedron Organic Chemistry
Series; Pergamon: Amsterdam, 2000; Vol. 20; (d) Negishi, E.; Hu, Q.;
Huang, Z.; Qian, M.; Wang, G. Aldrichim. Acta 2005, 38, 71.
63. Chemistry and Biology of Naturally-occurring Acetylenes and Related
Compounds; Lain, J., Breteler, H., Arnason, T., Hansen, L., Eds.; Elsevier:
Amsterdam, 1988.
64. Perrin, D. D.; Armarego, W. L. Em Purification of Laboratory Chemicals,
4th ed. Pergamon Press, New York, 1997.
65. Watson, S. C.; Eastham, J. F. J. Organomet. Chem. 1967, 9, 165.
66. Coulson, R. D. Inorg. Synth. 1972, 13, 121.
67. Hartely, F. R. Organometal. Chem. Rev. A. 1970, 6, 119.
68. Reich, H. J.; Renga, J. M.; Reich, I. L. J. Am. Chem. Soc. 1975, 97, 5435.
69. Bhasin, K. K.; Arora, V. Appl. Organometal. Chem. 2004. 18. 359.
70. Engman, L.; Cava, M. P. Synth. Commun. 1982, 1.

118
Capítulo 4
Espectros Selecionados
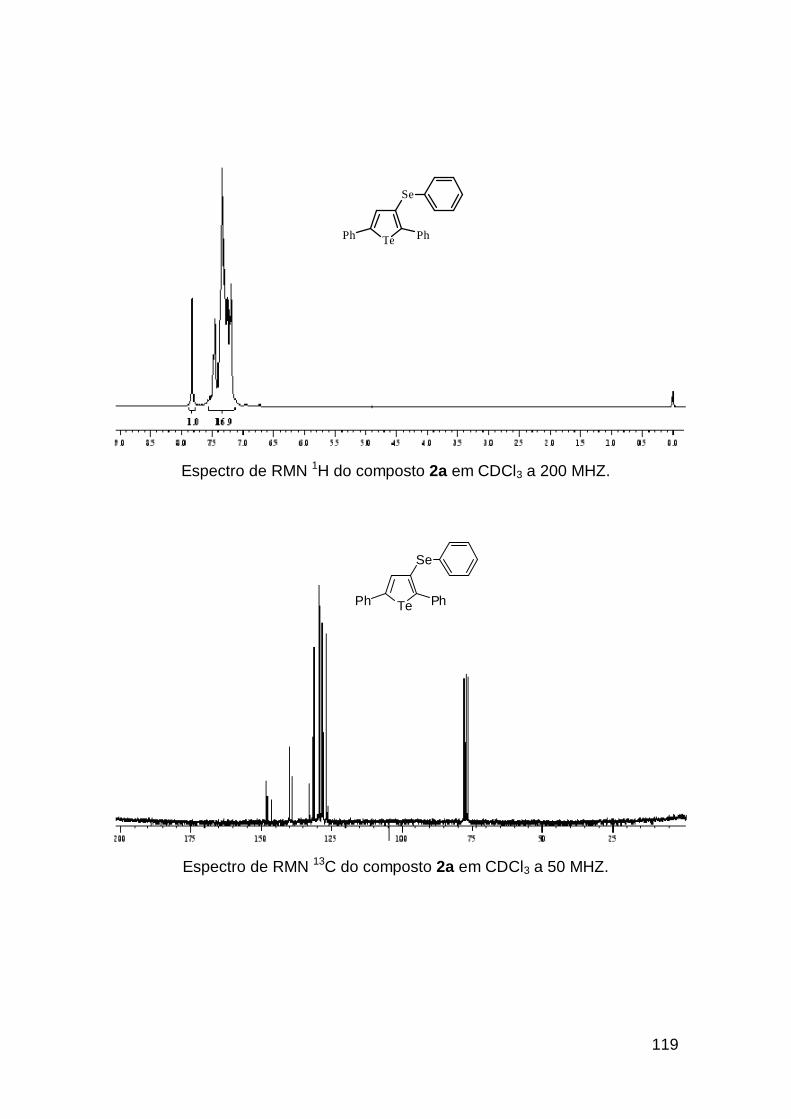
119
Espectro de RMN 1H do composto 2a em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2a em CDCl3 a 50 MHZ.
Te PhPh
Se
Te PhPh
Se
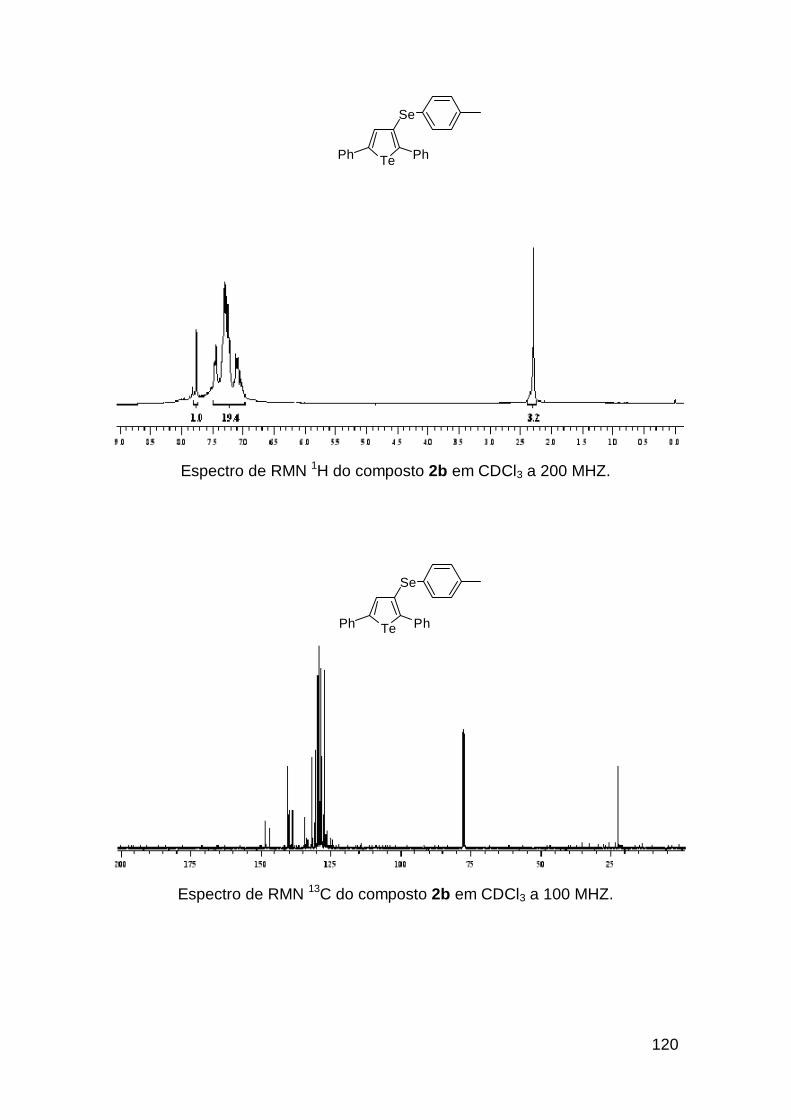
120
Espectro de RMN 1H do composto 2b em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2b em CDCl3 a 100 MHZ.
Te PhPh
Se
Te PhPh
Se

121
Espectro de RMN 1H do composto 2c em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2c em CDCl3 a 50 MHZ.
Te PhPh
Se
Te PhPh
Se
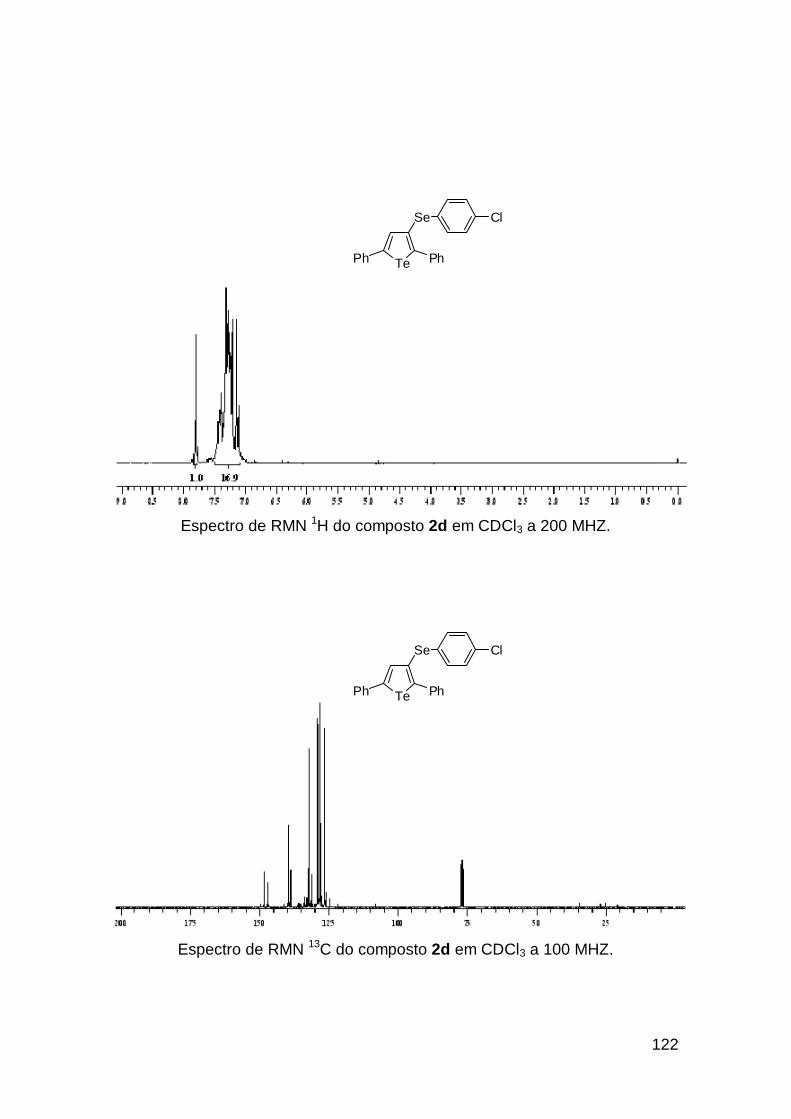
122
Espectro de RMN 1H do composto 2d em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2d em CDCl3 a 100 MHZ.
Te PhPh
Se Cl
Te PhPh
Se Cl
123
Espectro de RMN 1H do composto 2e em CDCl3 a 200 MHZ.
Te PhPh
Se F
124
Espectro de RMN 1H do composto 2f em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2f em CDCl3 a 100 MHZ.
Te PhPh
Se
CF3
Te PhPh
Se
CF3
125
Espectro de RMN 1H do composto 2g em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2g em CDCl3 a 100 MHZ.
TePh
Se
Ph
TePh
Se
Ph
126
Espectro de RMN 1H do composto 2h em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2h em CDCl3 a 100 MHZ.
Te PhPh
Se
Te PhPh
Se
127
Espectro de RMN 1H do composto 2i em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2i em CDCl3 a 100 MHZ.
Te PhPh
SeN
Te PhPh
SeN
128
Espectro de RMN 1H do composto 2j em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2j em CDCl3 a 100 MHZ.
Te PhPh
Te
Te PhPh
Te
129
Espectro de RMN 1H do composto 2k em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2k em CDCl3 a 100 MHZ
Te PhPh
Te
Te PhPh
Te

130
Espectro de RMN 1H do composto 2l em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 2l em CDCl3 a 50 MHZ.
Te PhPh
Te Cl
Te PhPh
Te Cl
131
Espectro de RMN 1H do composto 2m em CDCl3 a 200 MHZ.
Te PhPh
S
132
Espectro de RMN 1H do composto 2n em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 2n em CDCl3 a 100 MHZ.
Te PhPh
SeBu
Te PhPh
SeBu
133
Espectro de RMN 1H do composto 4a em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4a em CDCl3 a 100 MHZ.
Se PhPh
Se
Se PhPh
Se
134
Espectro de RMN 1H do composto 4b em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4b em CDCl3 a 100 MHZ.
Se PhPh
Se
Se PhPh
Se
135
Espectro de RMN 1H do composto 4c em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4c em CDCl3 a 100 MHZ.
Se PhPh
Se
Se PhPh
Se
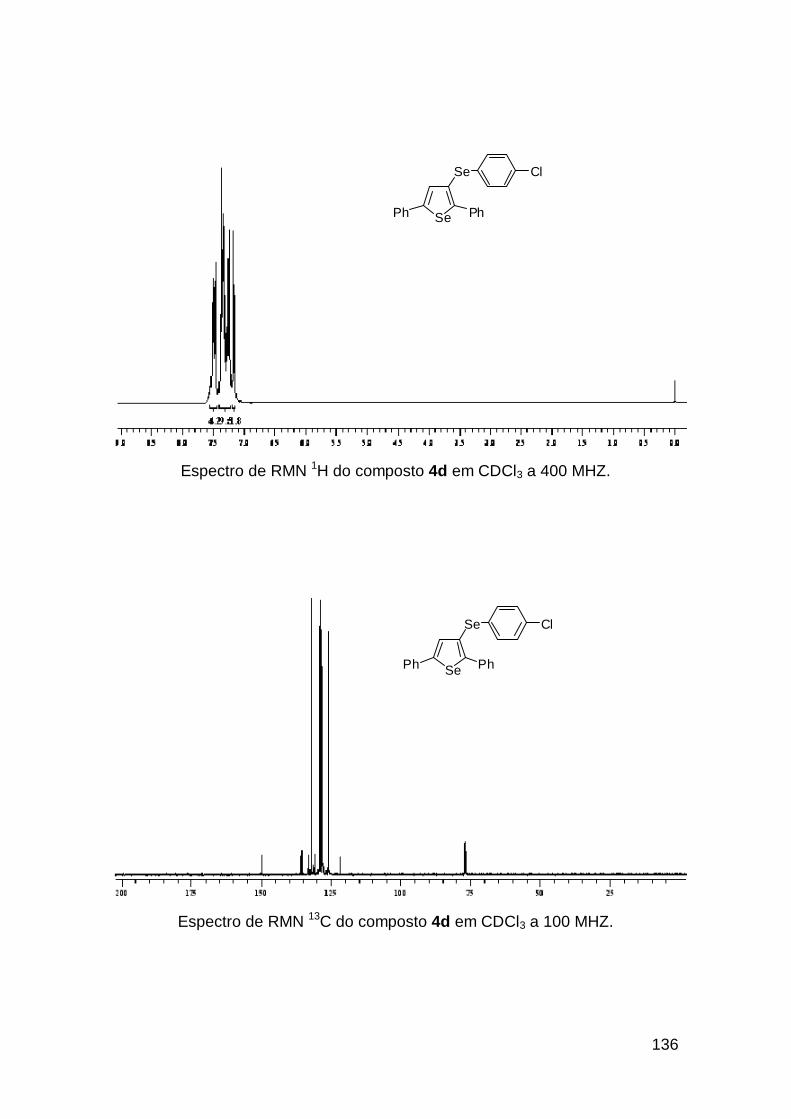
136
Espectro de RMN 1H do composto 4d em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4d em CDCl3 a 100 MHZ.
Se PhPh
Se Cl
Se PhPh
Se Cl
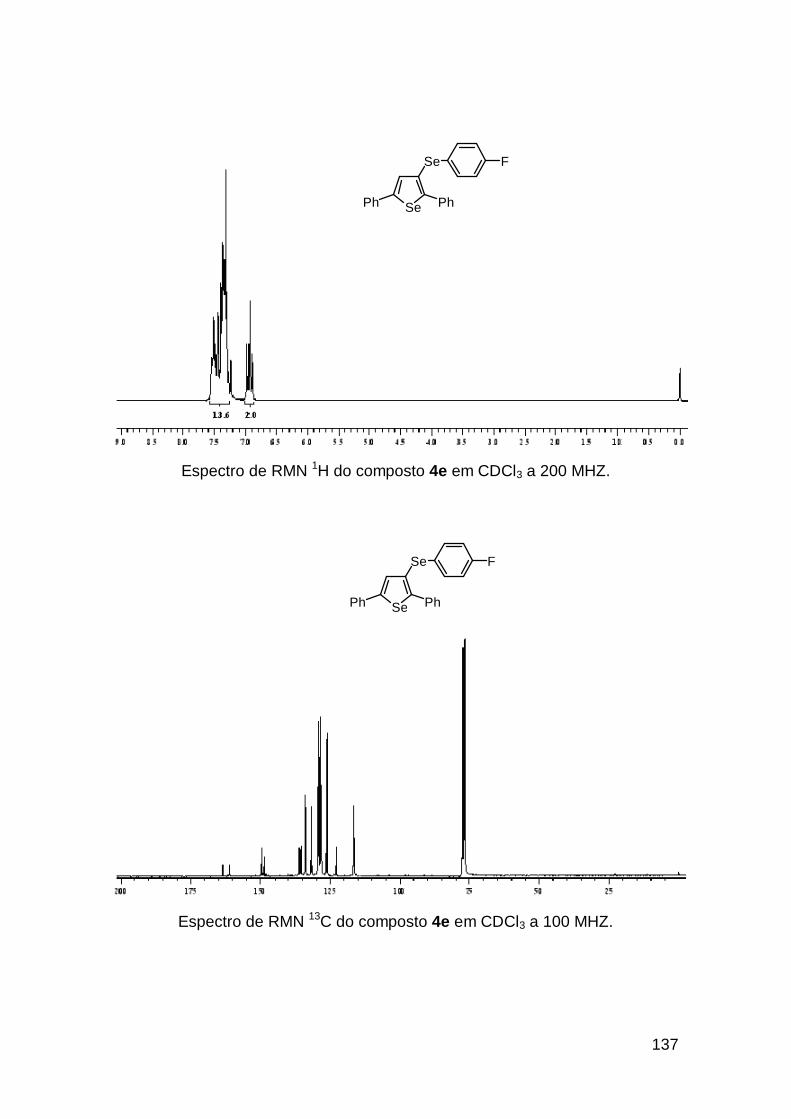
137
Espectro de RMN 1H do composto 4e em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 4e em CDCl3 a 100 MHZ.
Se PhPh
Se F
Se PhPh
Se F

138
Espectro de RMN 1H do composto 4f em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4f em CDCl3 a 100 MHZ.
Se PhPh
Se
CF3
Se PhPh
Se
CF3

139
Espectro de RMN 1H do composto 4g em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4g em CDCl3 a 100 MHZ.
SePh
Se
Ph
SePh
Se
Ph

140
Espectro de RMN 1H do composto 4h em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4h em CDCl3 a 100 MHZ.
Se PhPh
Se
Se PhPh
Se
141
Espectro de RMN 1H do composto 4i em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4i em CDCl3 a 100 MHZ.
Se Ph
Se
Se Ph
Se
142
Espectro de RMN 1H do composto 4k em CDCl3 a 400 MHZ.
Espectro de RMN 13C do composto 4k em CDCl3 a 50 MHZ.
Se PhPh
Te
Se PhPh
Te
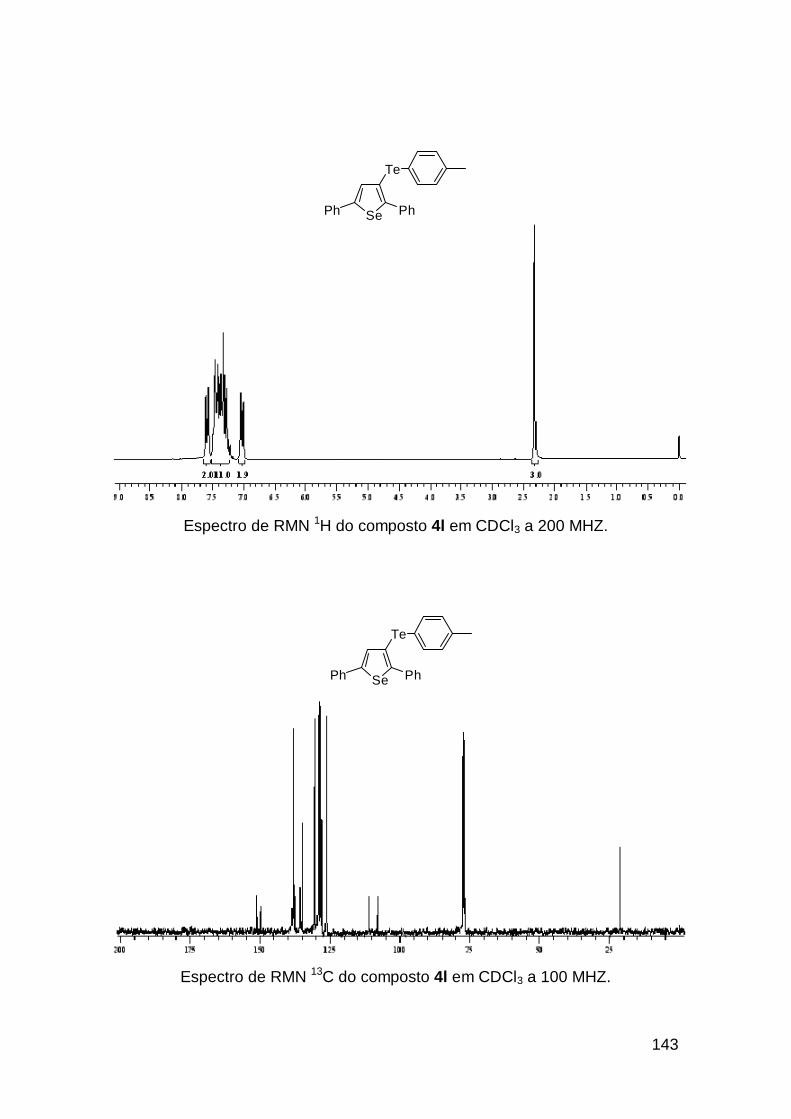
143
Espectro de RMN 1H do composto 4l em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 4l em CDCl3 a 100 MHZ.
Se PhPh
Te
Se PhPh
Te

144
Espectro de RMN 1H do composto 4m em CDCl3 a 200 MHZ.
Espectro de RMN 13C do composto 4m em CDCl3 a 100 MHZ
Se PhPh
Te Cl
Se PhPh
Te Cl
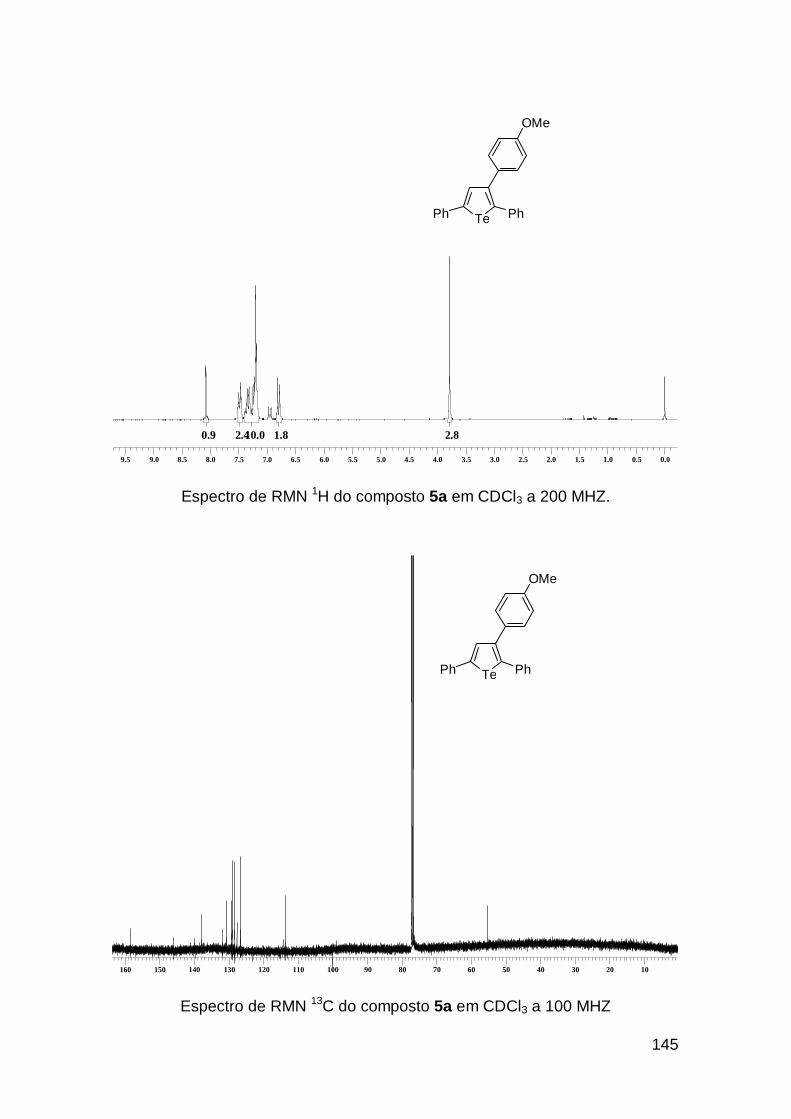
145
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.08.58.59.09.09.59.5
2.8 1.8 0.9 2.4 10.0
Espectro de RMN 1H do composto 5a em CDCl3 a 200 MHZ.
101020203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 5a em CDCl3 a 100 MHZ
Te PhPh
OMe
Te PhPh
OMe

146
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
1.9 2.1 2.3 3.2
7.107.207.307.407.507.60
2.1 3.6 1.1 1.0
Espectro de RMN 1H do composto 6a em CDCl3 a 400 MHZ.
S
Ph
SenBu
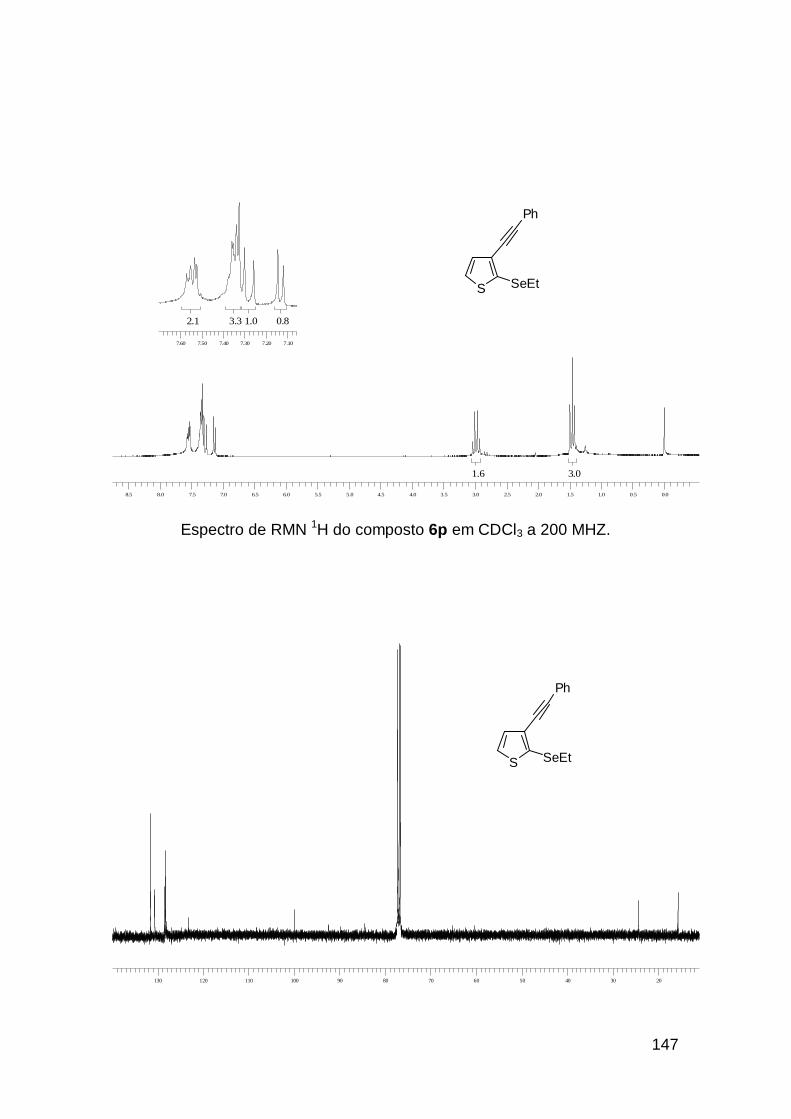
147
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
1.6 3.0
7.107.207.307.407.507.60
0.8 2.1 3.3 1.0
Espectro de RMN 1H do composto 6p em CDCl3 a 200 MHZ.
20203030404050506060707080809090100100110110120120130130
S
Ph
SeEt
S
Ph
SeEt
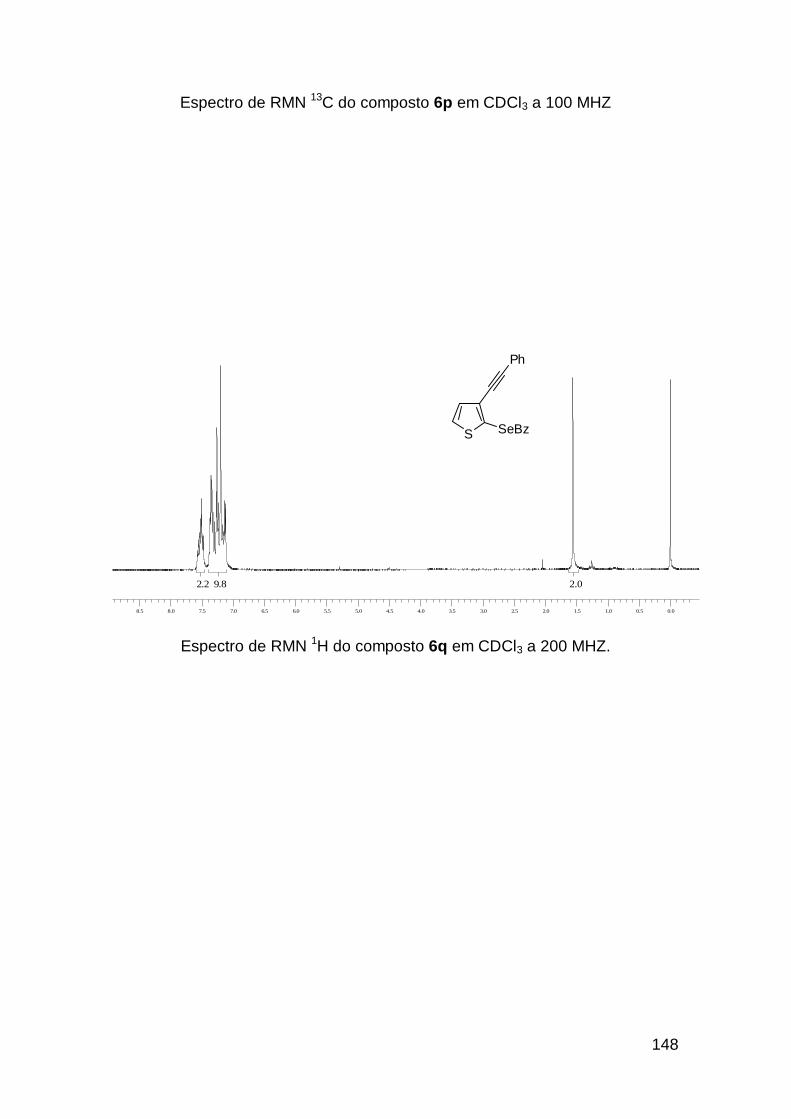
148
Espectro de RMN 13C do composto 6p em CDCl3 a 100 MHZ
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.08.58.5
2.0 2.2 9.8
Espectro de RMN 1H do composto 6q em CDCl3 a 200 MHZ.
S
Ph
SeBz
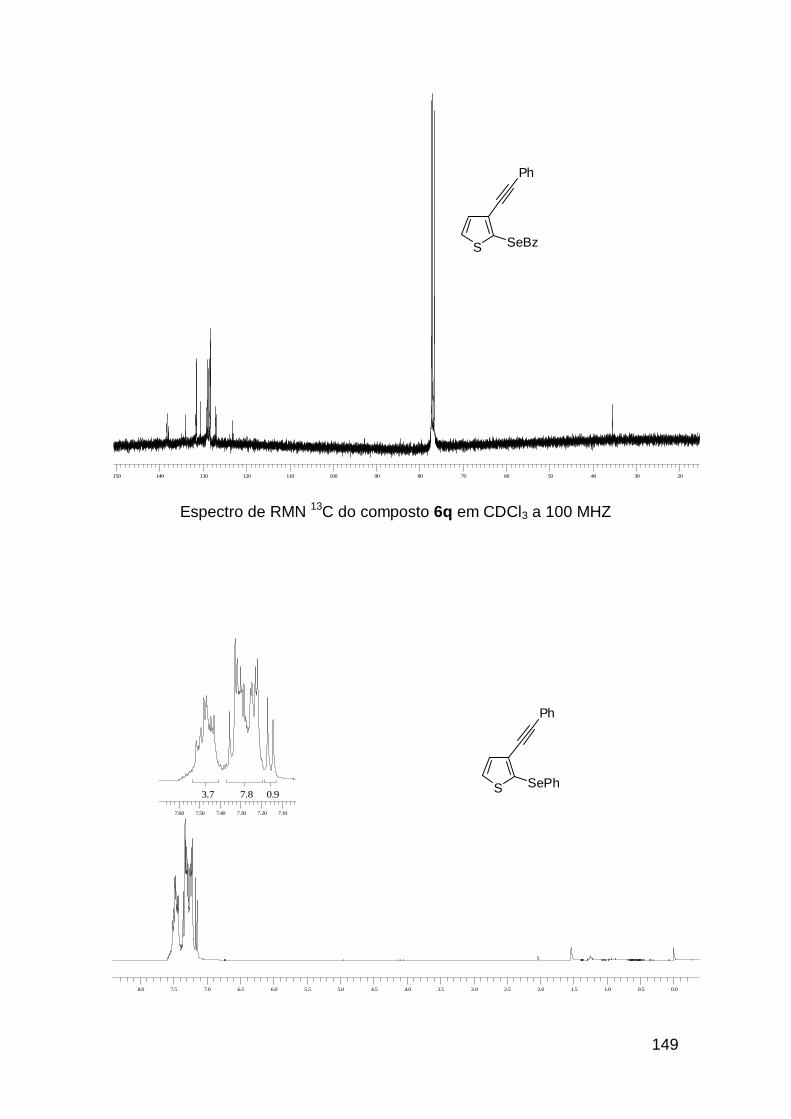
149
20203030404050506060707080809090100100110110120120130130140140150150
Espectro de RMN 13C do composto 6q em CDCl3 a 100 MHZ
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
7.107.207.307.407.507.60
0.9 3.7 7.8
S
Ph
SeBz
S
Ph
SePh
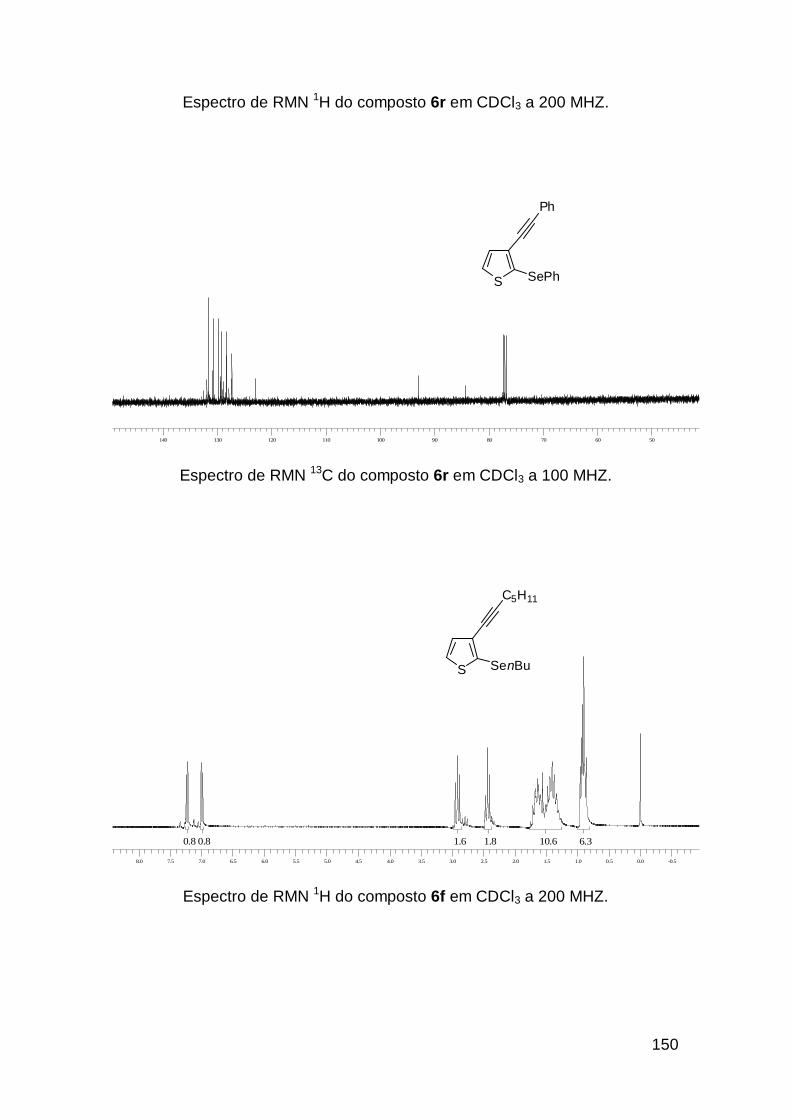
150
Espectro de RMN 1H do composto 6r em CDCl3 a 200 MHZ.
50506060707080809090100100110110120120130130140140
Espectro de RMN 13C do composto 6r em CDCl3 a 100 MHZ.
-0.5-0.50.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.0
1.6 1.8 10.6 0.8 0.8 6.3
Espectro de RMN 1H do composto 6f em CDCl3 a 200 MHZ.
S
Ph
SePh
S
C5H11
SenBu
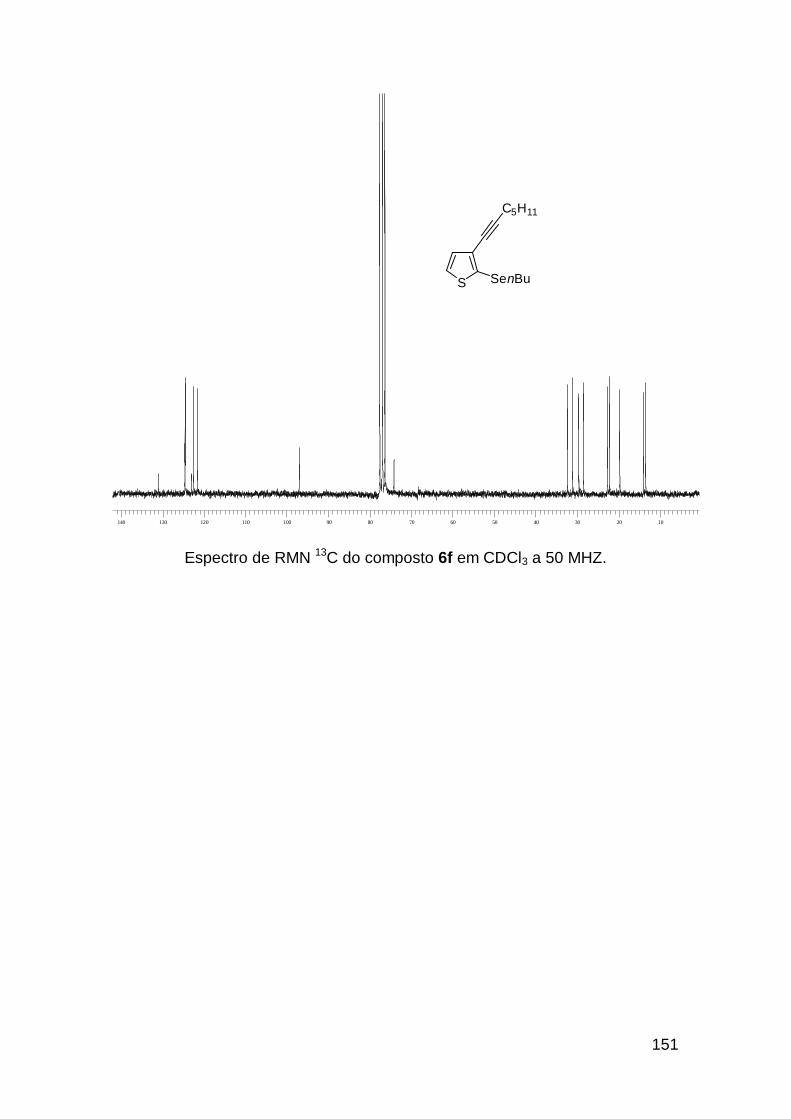
151
101020203030404050506060707080809090100100110110120120130130140140
Espectro de RMN 13C do composto 6f em CDCl3 a 50 MHZ.
S
C5H11
SenBu
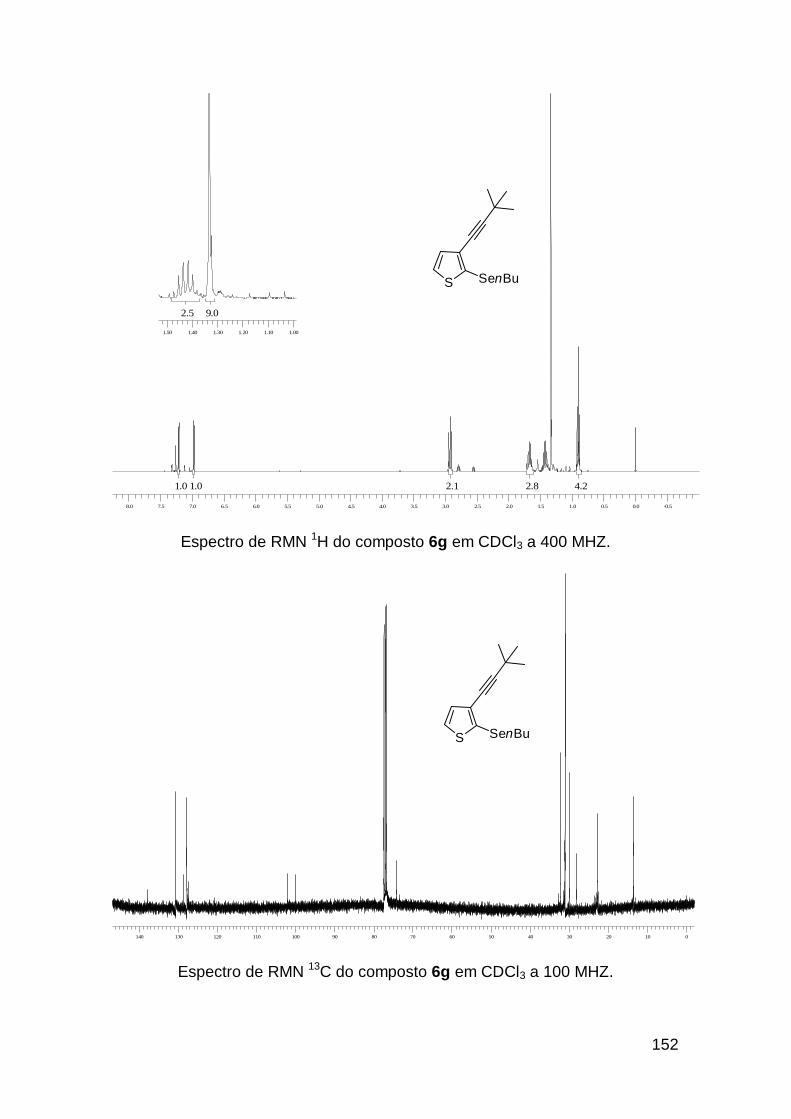
152
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
1.0 1.0 2.8 2.1 4.2
1.001.101.201.301.401.50
9.0 2.5
Espectro de RMN 1H do composto 6g em CDCl3 a 400 MHZ.
00101020203030404050506060707080809090100100110110120120130130140140
Espectro de RMN 13C do composto 6g em CDCl3 a 100 MHZ.
S SenBu
S SenBu
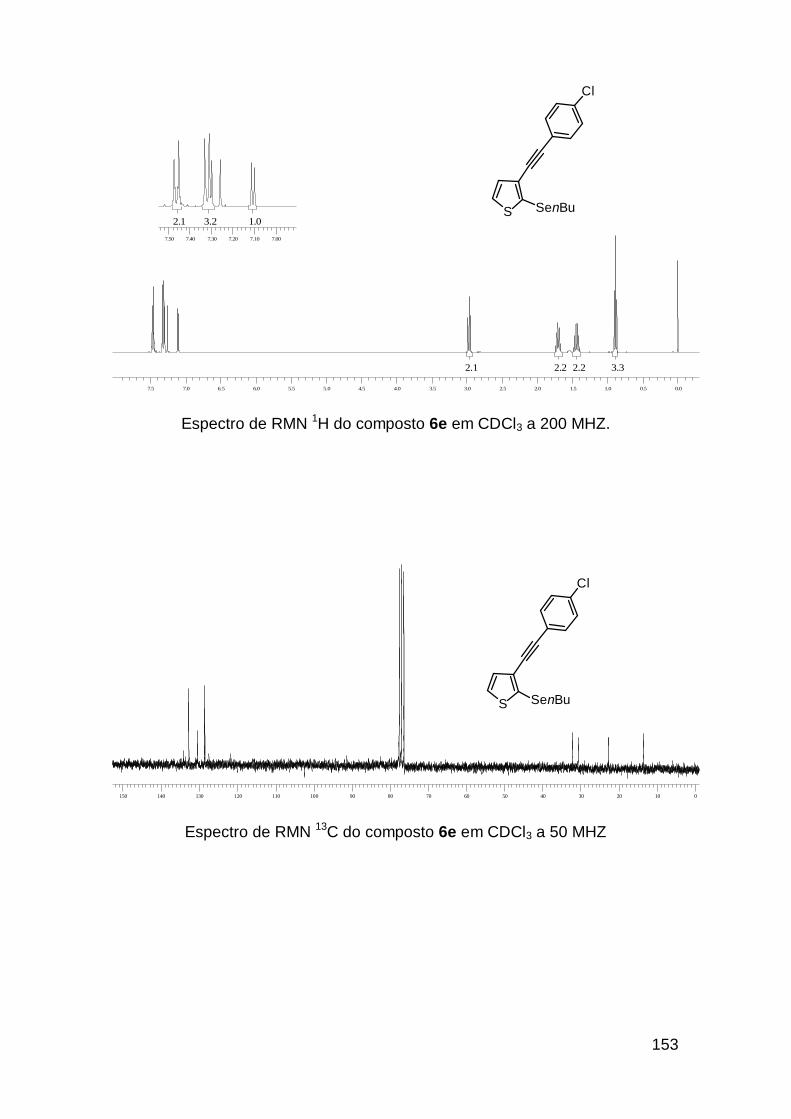
153
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
2.1 2.2 2.2 3.3
7.007.107.207.307.407.50
1.0 3.2 2.1
Espectro de RMN 1H do composto 6e em CDCl3 a 200 MHZ.
00101020203030404050506060707080809090100100110110120120130130140140150150
Espectro de RMN 13C do composto 6e em CDCl3 a 50 MHZ
S SenBu
Cl
S SenBu
Cl
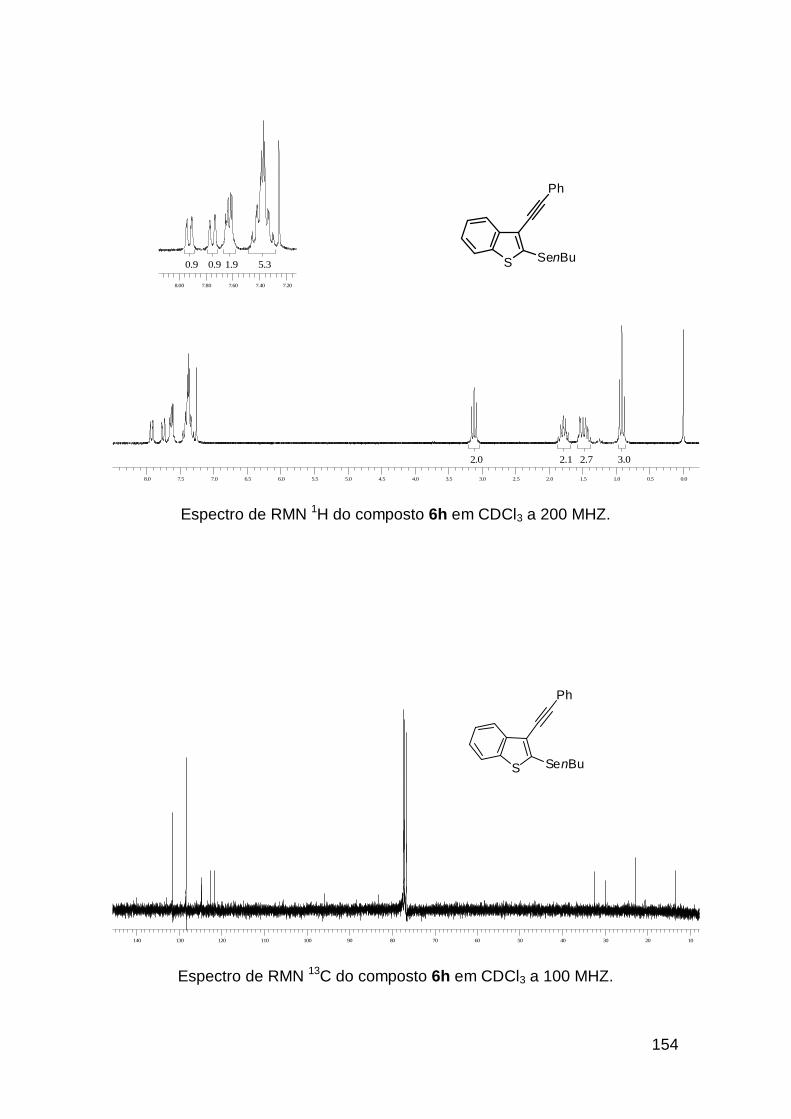
154
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
2.0 2.1 3.0 2.7
7.207.407.607.808.00
0.9 0.9 1.9 5.3
Espectro de RMN 1H do composto 6h em CDCl3 a 200 MHZ.
101020203030404050506060707080809090100100110110120120130130140140
Espectro de RMN 13C do composto 6h em CDCl3 a 100 MHZ.
S
Ph
SenBu
S
Ph
SenBu
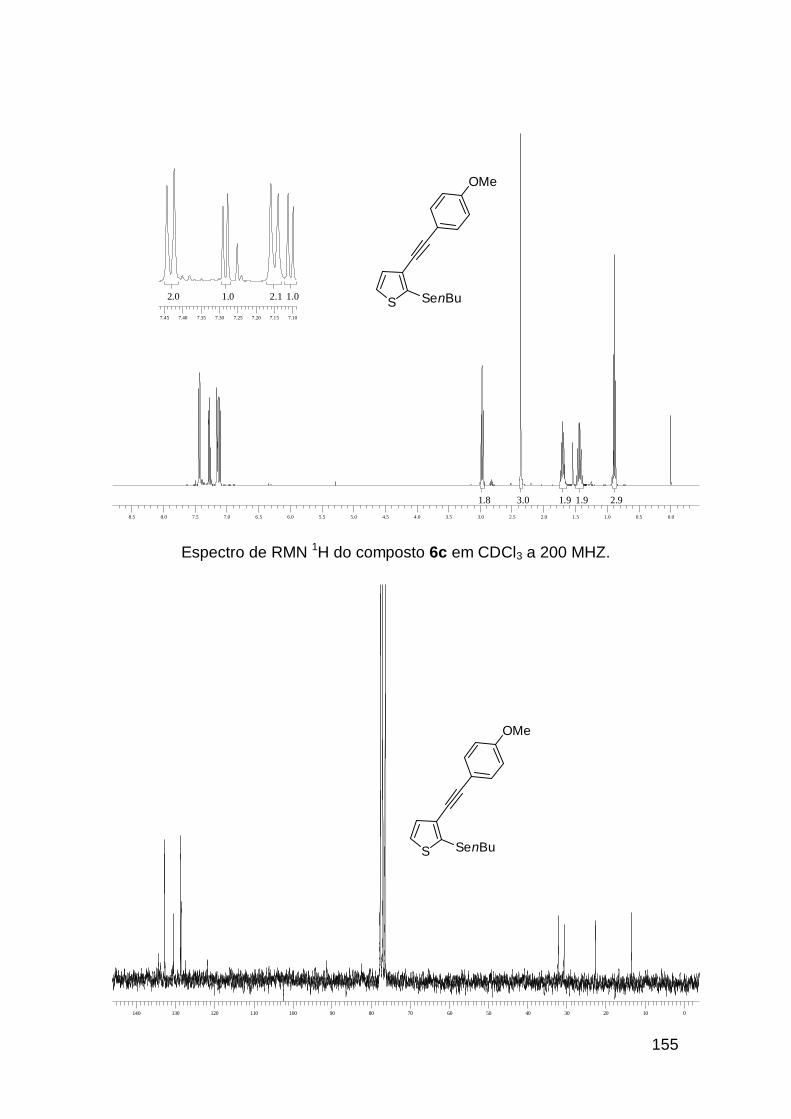
155
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
3.0 1.8 1.9 1.9 2.9
7.107.157.207.257.307.357.407.45
1.0 2.1 1.0 2.0
Espectro de RMN 1H do composto 6c em CDCl3 a 200 MHZ.
00101020203030404050506060707080809090100100110110120120130130140140
S SenBu
OMe
S SenBu
OMe

156
Espectro de RMN 13C do composto 6c em CDCl3 a 50 MHZ.
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
3.0 1.7 3.0 1.7 1.8 2.6
6.807.007.207.407.607.808.00
1.0 0.9 1.0 2.1 1.2
Espectro de RMN 1H do composto 6l em CDCl3 a 400 MHZ.
101020203030404050506060707080809090100100110110120120130130140140150150160160170170
S SenBu
MeO
S SenBu
MeO
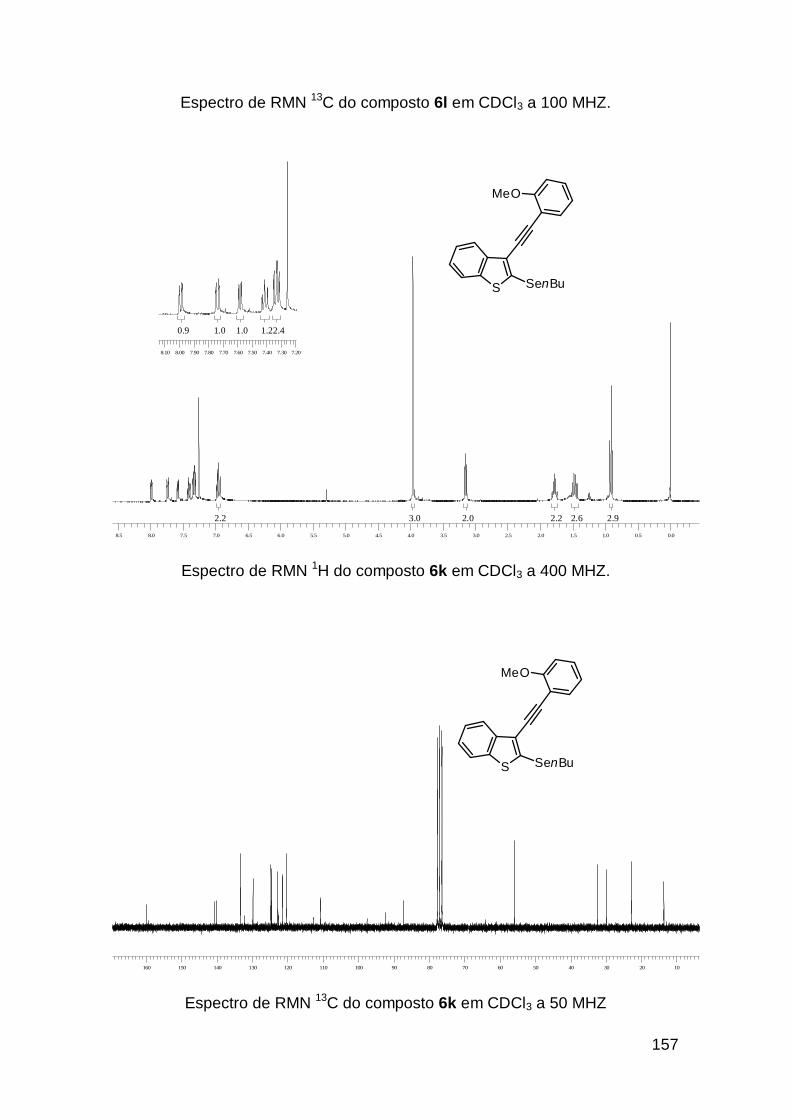
157
Espectro de RMN 13C do composto 6l em CDCl3 a 100 MHZ.
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
3.0 2.0 2.2 2.6 2.9 2.2
7.207.307.407.507.607.707.807.908.008.10
0.9 1.0 1.0 1.2 2.4
Espectro de RMN 1H do composto 6k em CDCl3 a 400 MHZ.
101020203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 6k em CDCl3 a 50 MHZ
S SenBu
MeO
S SenBu
MeO
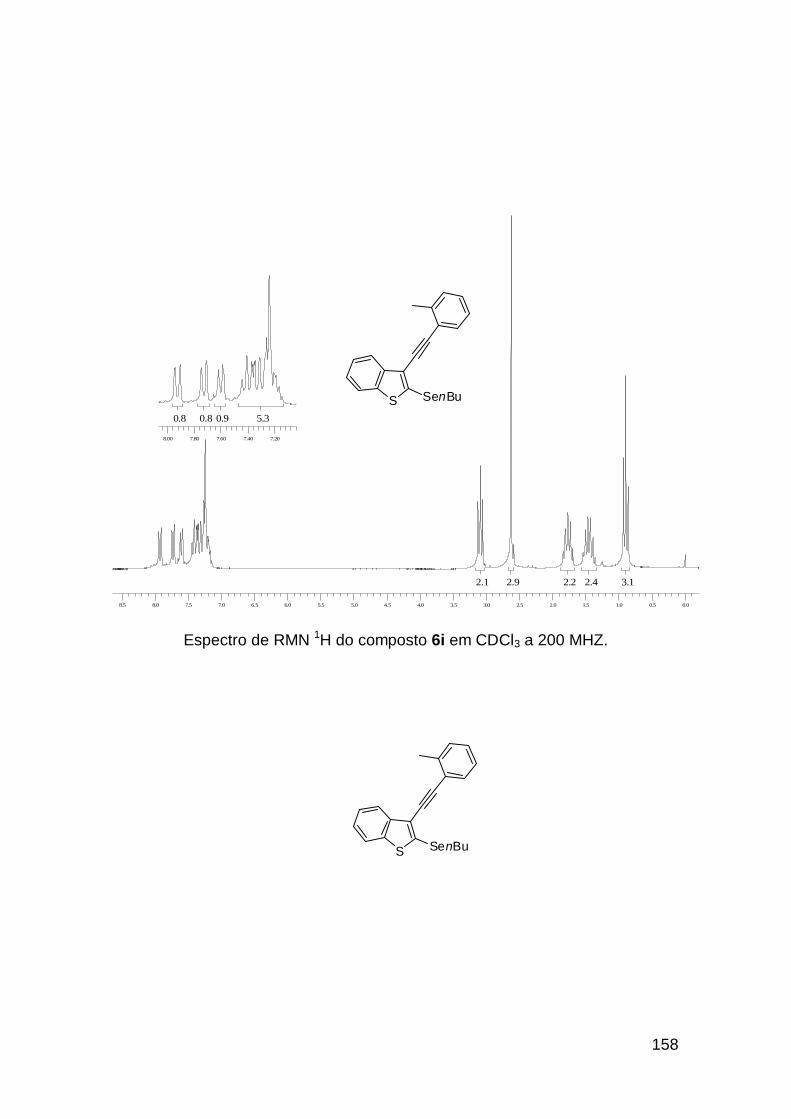
158
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
2.1 2.9 2.2 2.4 3.1
7.207.407.607.808.00
0.8 0.8 0.9 5.3
Espectro de RMN 1H do composto 6i em CDCl3 a 200 MHZ.
S SenBu
S SenBu

159
101020203030404050506060707080809090100100110110120120130130140140150150
Espectro de RMN 13C do composto 6i em CDCl3 a 50 MHZ
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.08.58.5
1.7 2.4 2.7 3.3 0.7 0.9 1.0 3.2 3.3
Espectro de RMN 1H do composto 6d em CDCl3 a 200 MHZ.
S SenBu
S SenBu
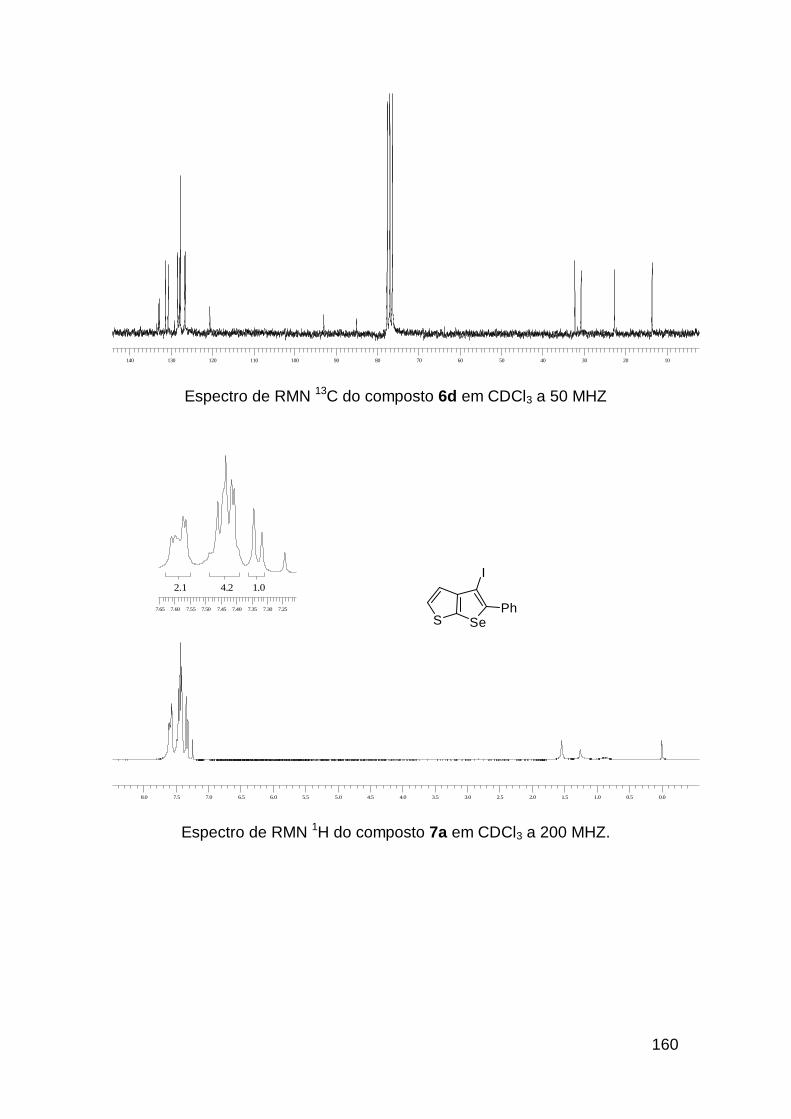
160
101020203030404050506060707080809090100100110110120120130130140140
Espectro de RMN 13C do composto 6d em CDCl3 a 50 MHZ
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
7.257.307.357.407.457.507.557.607.65
1.0 2.1 4.2
Espectro de RMN 1H do composto 7a em CDCl3 a 200 MHZ.
SeS
I
Ph
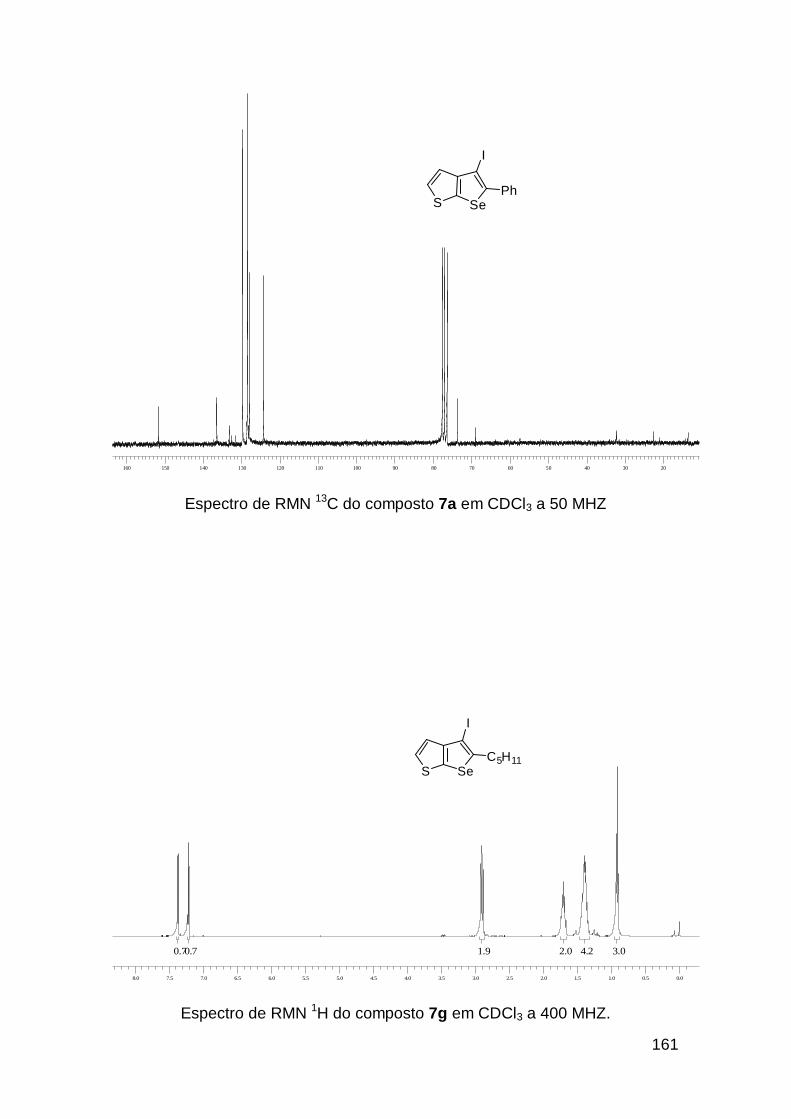
161
20203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 7a em CDCl3 a 50 MHZ
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.0
0.7 0.7 1.9 2.0 4.2 3.0
Espectro de RMN 1H do composto 7g em CDCl3 a 400 MHZ.
SeS
I
Ph
SeS
I
C5H11
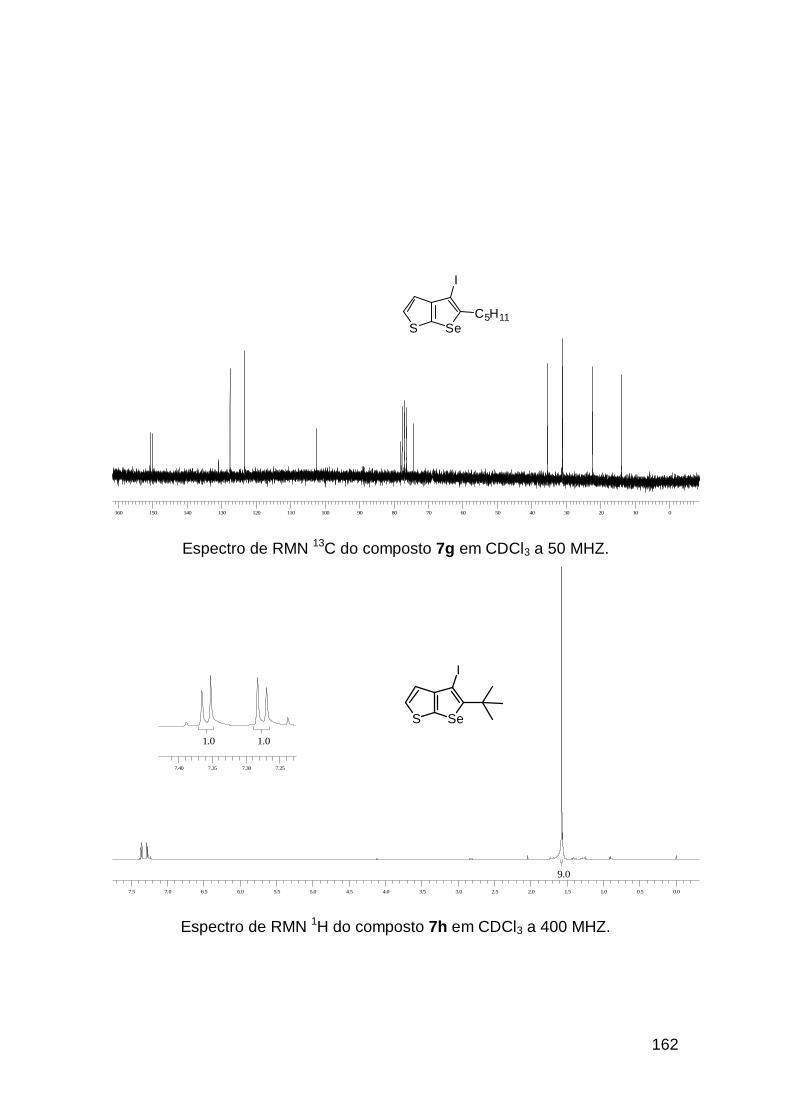
162
00101020203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 7g em CDCl3 a 50 MHZ.
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
9.0
7.257.307.357.40
1.0 1.0
Espectro de RMN 1H do composto 7h em CDCl3 a 400 MHZ.
SeS
I
C5H11
SeS
I

163
20203030404050506060707080809090100100110110120120130130140140150150160160170170
Espectro de RMN 13C do composto 7h em CDCl3 a 100 MHZ
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.08.58.59.09.0
0.7 1.0 2.1 4.8
Espectro de RMN 1H do composto 7l em CDCl3 a 200 MHZ.
SeS
I
SeS
I
Ph

164
0123456789
3.0
7.157.207.257.307.357.407.457.50
2.0 0.9 0.9 1.9
Espectro de RMN 1H do composto 7c em CDCl3 a 400 MHZ.
SeS
I

165
101020203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 7c em CDCl3 a 50 MHZ
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
3.0
6.806.907.007.107.207.307.407.507.60
1.0 1.1 2.2 2.2
Espectro de RMN 1H do composto 7d em CDCl3 a 200 MHZ.
SeS
I
SeS
I
OMe
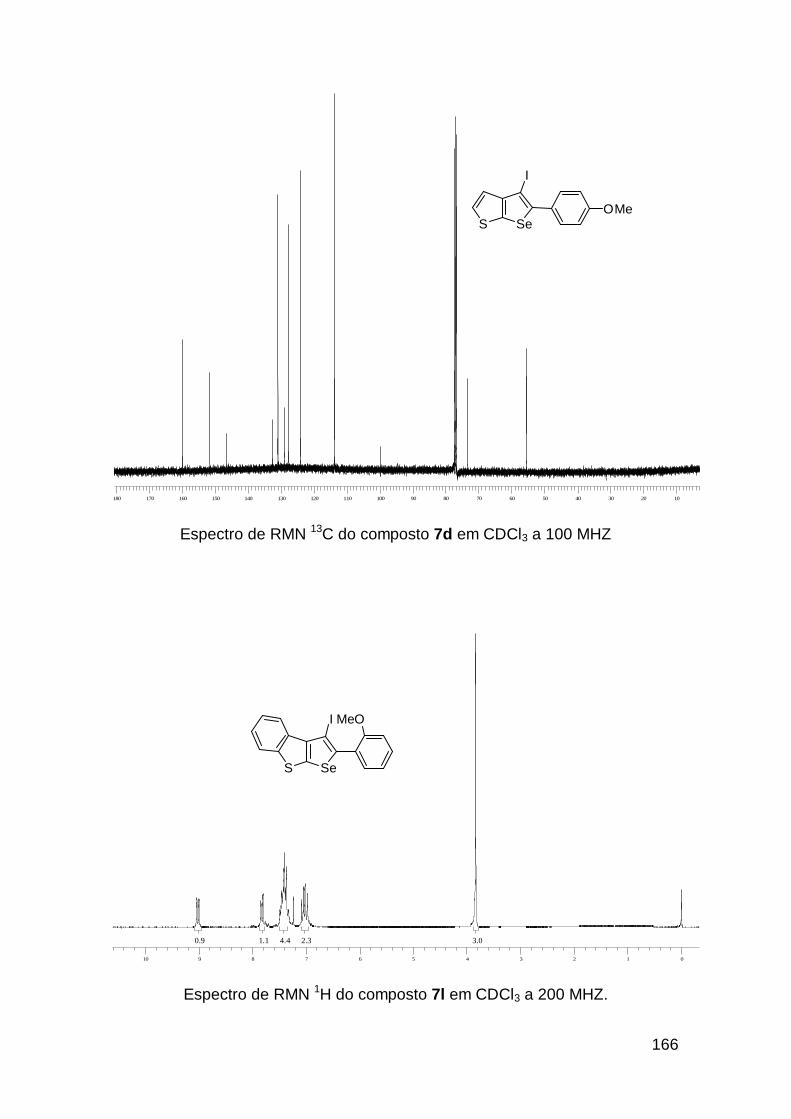
166
101020203030404050506060707080809090100100110110120120130130140140150150160160170170180180
Espectro de RMN 13C do composto 7d em CDCl3 a 100 MHZ
001122334455667788991010
3.0 0.9 1.1 2.3 4.4
Espectro de RMN 1H do composto 7l em CDCl3 a 200 MHZ.
SeS
I
OMe
SeS
I MeO
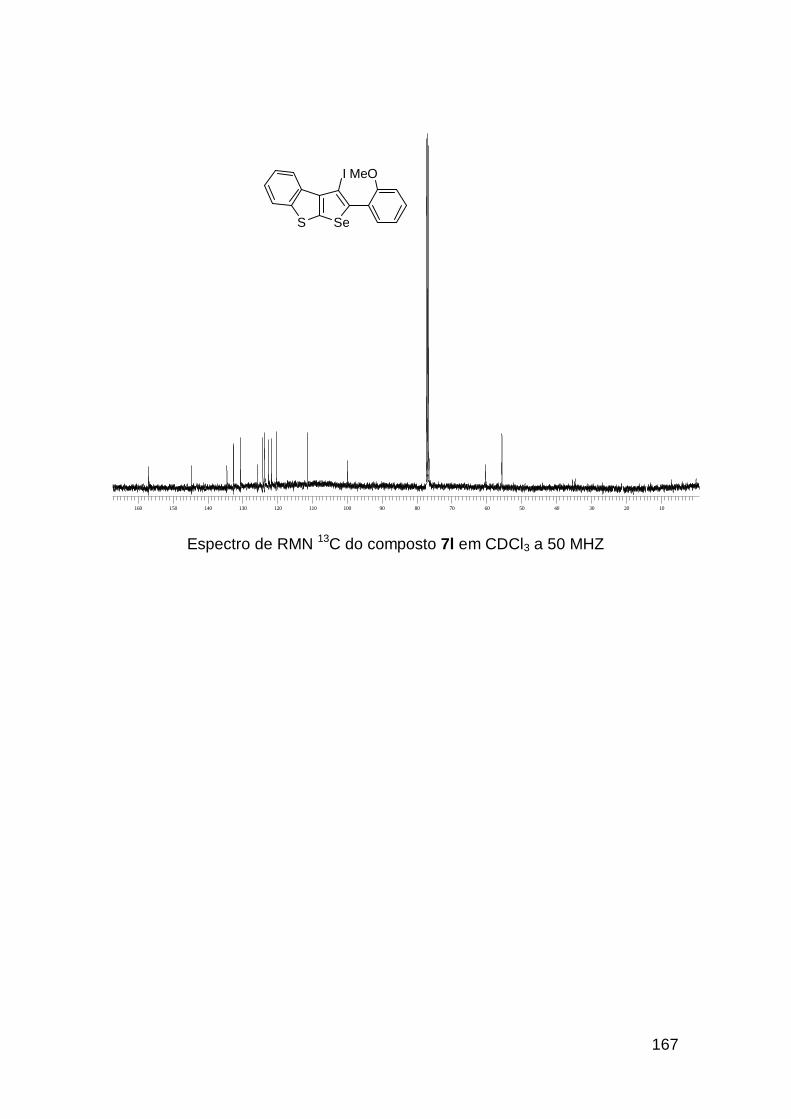
167
101020203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 7l em CDCl3 a 50 MHZ
SeS
I MeO

168
00112233445566778899
2.7 0.9 1.0 1.0 2.3 1.9 2.6
Espectro de RMN 1H do composto 7m em CDCl3 a 400 MHZ.
00101020203030404050506060707080809090100100110110120120130130140140150150160160170170
Espectro de RMN 13C do composto 7m em CDCl3 a 50 MHZ
SeS
I MeO
SeS
I MeO
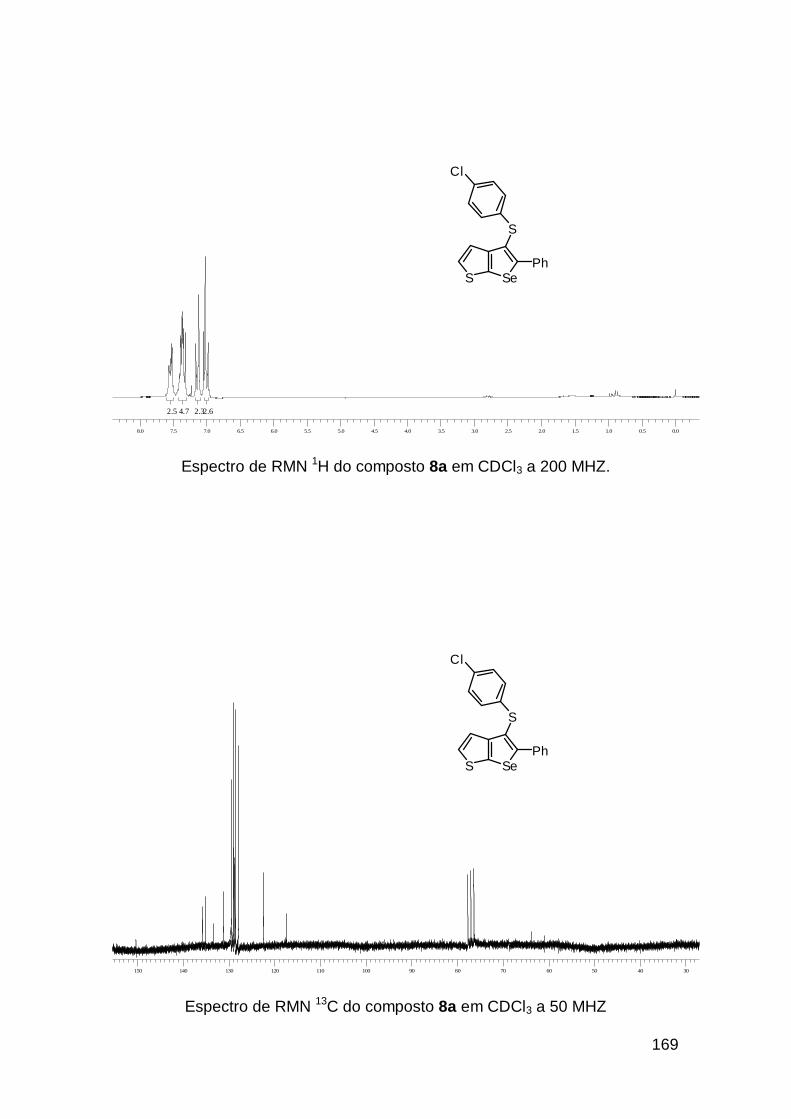
169
0.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.0
2.3 2.5 2.6 4.7
Espectro de RMN 1H do composto 8a em CDCl3 a 200 MHZ.
3030404050506060707080809090100100110110120120130130140140150150
Espectro de RMN 13C do composto 8a em CDCl3 a 50 MHZ
SeS
S
Ph
Cl
SeS
S
Ph
Cl
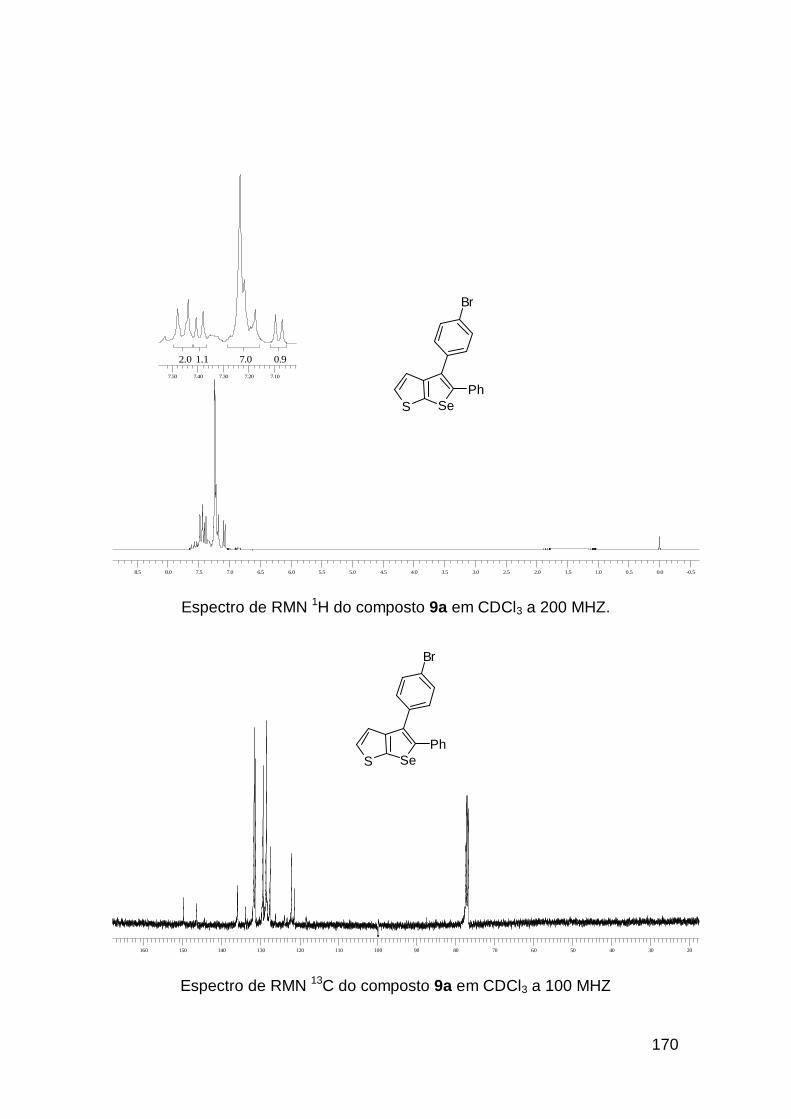
170
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
7.107.207.307.407.50
2.0 1.1 7.0 0.9
Espectro de RMN 1H do composto 9a em CDCl3 a 200 MHZ.
20203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 9a em CDCl3 a 100 MHZ
SeSPh
Br
SeSPh
Br

171
-0.5-0.50.00.00.50.51.01.01.51.52.02.02.52.53.03.03.53.54.04.04.54.55.05.05.55.56.06.06.56.57.07.07.57.58.08.0
11.5 3.1
Espectro de RMN 1H do composto 10a em CDCl3 a 200 MHZ.
20203030404050506060707080809090100100110110120120130130140140150150160160
Espectro de RMN 13C do composto 10a em CDCl3 a 100 MHZ.
SeSPh
SeSPh

172
Anexos

173

174

175

176

177

178
179

180

181

182
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo