secretaria nacional de defesa agropecuÁria...
TRANSCRIPT

SECRETARIA NACIONAL DE DEFESA AGROPECUÁRIA DEPARTAMENTO NACIONAL DE DEFESA ANIMAL.
DIVISÃO DE LABORATÓRIO ANIMAL
MÉTODOS ANALÍTICOS PARA CONTROLE DE ALIMENTOSPARA USO ANIMAL NORMAS GERAIS DE AMOSTRAGEM PARA ANÁLISES DE ROTINA
A principal meta de um laboratório deve ser a produção de dados analíticos de
alta qualidade, através de medidas que sejam exatas, confiáveis e reprodutíveis. Estes resultados somente refletirão a real composição de um produto se a colheita do material for convenientemente efetuada. Para uma boa amostragem, as seguintes normas técnicas gerais devem ser observadas:
1. A quantidade de material deve ser suficiente para realização de toda a parte analítica bem como armazenamento em arquivo, destinada a revisão ou perícia. Adotar no mínimo 1 kg para esta operação. Produtos cuja homogeneidade pode comprometer o resultado analítico (rações e concentrados que contenham uréia, farelo de algodão, etc), a colheita deve ser superior a 2 kg ou, em casos especiais, conforme especificação do laboratório.
2. Fazer o quarteamento, dividindo em 4 sub-amostras e acondicionando-as de maneira a conservar "in natural! os caracteres físicos, químicos e organolépticos. Para produtos sólidos adotar sacos plásticos resistentes e para líquidos frascos de polietileno ou vidro com tampa (melaço, gordura, etc).
3. Identificar e rotular da forma mais completa possível. Exemplo: farelo de soja, local da colheita, fabricante, data de fabricação, data da validade, data da colheita, código da amostra, representatividade da amostra, órgão e responsável pela colheita. Anexar, se necessário, um breve relatório contendo informações sobre anormalidades apresentadas, tais como: condição da embalagem, transporte, contaminação do piso por outros materiais, estado de conservação, etc.
4. O responsável pelas colheitas de amostras deve ter conhecimentos tecnológicos e analiticos. Isto assegurará informações úteis relativas as matérias primas e mais ainda a industrialização de produtos acabados, levando a correção de possíveis erros.
5. Efetuar as colheitas em vários pontos do carregamento, lote, partida, etc., principalmente para produtos que não apresentam uma homogeneidade perfeita ou tenham tendência a separação.
6. Para produtos ensacados utilizar um calador de metal (F1G. 3), retirando a amostra das partes superior, média e inferior do saco (FIG. 4), misturando-as posteriormente.
Proceder a colheita segundo a tabela abaixo: a) Até dez unidades: cinco amostras de unidades diferentes. b) De dez até cem unidades: quinze por cento da partida, com um número
mínimo de dez unidades. . c) Acima de cem unidades: cinco por cento da partida, com um número minimo
de quinze unidades. IMPORTANTE
Nunca retirar a amostra de um único ponto, pois a mesma é casual e não permite conclusões quanto a qualidade do produto.

7. Para acondicionamentos a granel utilizar uma sonda de profundidade com cruzeta móvel (fig. 3), nas medidas aproximadas de 1,60m de comprimento e 0,05 m de diâmetro, introduzindo-a por impulsão forçada. Adotar um mínimo de oito pontos localizados, aplicados a cargas menores e 16 pontos para quantidades maiores (carretas e vagões), sempre intercalando as posições vertical e inclinada da sonda (FIG. 5). IMPORTANTE
O instrumento de colheita deverá necessariamente abranger as partes superior, média e inferior do material em todos os pontos escolhidos.
8. No caso de cargas ensacadas e cobertas com lona, descobri-las para possibilitar melhor retirada das amostras. Em todos os casos, proceder uma amostragem complementar no ato da descrição abrangendo os sacos centrais.
9. Para produtos no estado líquido e pastoso, amostrá-los com sonda específica, abrangendo as partos superior, média e inferior do recipiente.
10. Na chegada da amostra ao laboratório, o responsável pelo preparo deverá processá-la, adotando novamente o sistema de quarteamento (fIg. 1 ou 2).
11. Após esse procedimento, moer a amostra em moinho adequado, passando-a integralmente em peneira com malha pe 1 mm (16 mesh), homogeneizar e acondicionar em frasco de vidro ou plástico com tampa.
Quebra de página
PROCEDIMENTO PARA PREPARAÇÃO DA AMOSTRA DESTINA
A ANALISE LABORATORIAL
Fig 1 - Sistema de Quarteamento Manual
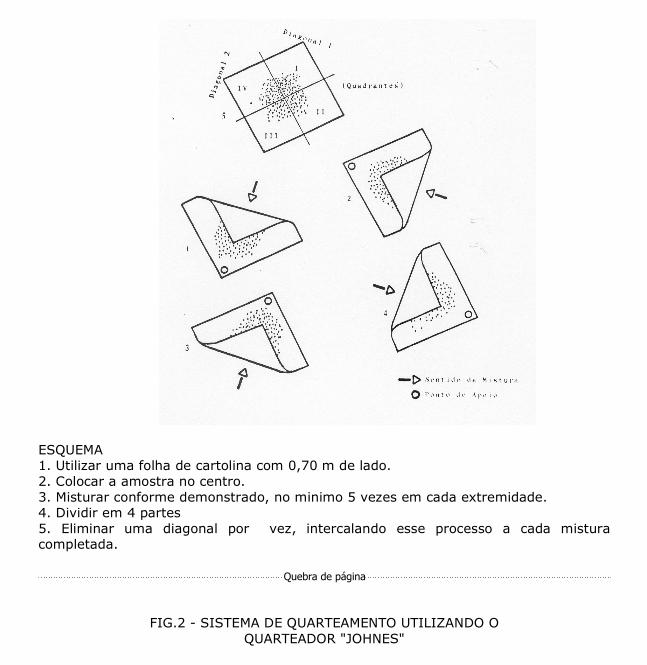
ESQUEMA 1. Utilizar uma folha de cartolina com 0,70 m de lado. 2. Colocar a amostra no centro. 3. Misturar conforme demonstrado, no minimo 5 vezes em cada extremidade. 4. Dividir em 4 partes 5. Eliminar uma diagonal por vez, intercalando esse processo a cada mistura completada.
Quebra de página
FIG.2 - SISTEMA DE QUARTEAMENTO UTILIZANDO O QUARTEADOR "JOHNES"

ESQUEMA 1. Derramar a amostra no interior do quarteador, passando pelas canaletas. 2. Colher as duas porções divididas igualmente em duas bandejas 3. Eliminar uma porção e a outra restante passar novamente pelo mesmo, colhendo as divisões semelhantemente a forma descrita em 2. 4. Proceder a ssim alternadamente até a obtenção da porção de amostra desejada.
Quebra de página
FIG.3 - INSTRUMENTOS UTILIZADOS PARA AMOSTRAGEM
SONDA DE PROFUNDIDADE
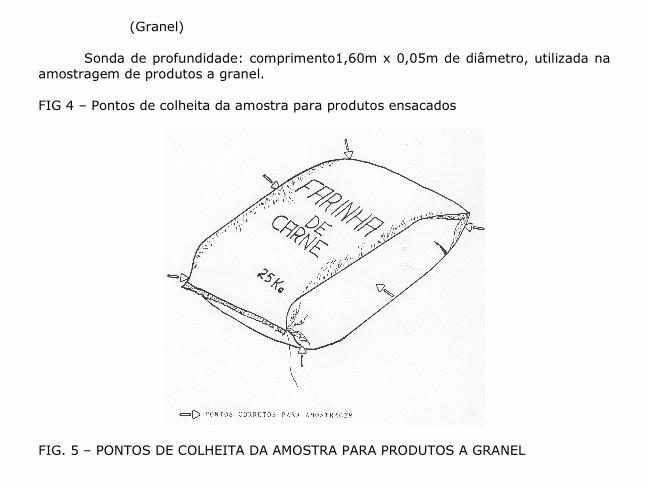
(Granel) Sonda de profundidade: comprimento1,60m x 0,05m de diâmetro, utilizada na amostragem de produtos a granel. FIG 4 – Pontos de colheita da amostra para produtos ensacados
FIG. 5 – PONTOS DE COLHEITA DA AMOSTRA PARA PRODUTOS A GRANEL

MÉTODOS FÍSICO-QUÍMICOS MÉTODO N.º 01- GRANULOMETRIA APLICACÃO: Produtos ou sub-produtos de origem animal, vegetal ou mineral. PRINCÍPIO: Baseia-se na determinação das proporções com que as partículas de diferentes dimensões entram na composição da amostra. MATERIAL E EQUIPAMENTO: - Aparelho vibrador para peneiras - Balança técnica (Precisão +/- 0,1 g) - Conjunto padrão de peneiràs metálicas - diâmetro de 203 mm - Bolas de borracha maciça com 2 a 3 cm de diâmetro. PROCEDIMENTO: 1. Colocar 2 Bolas de borracha em cada peneira e montar o conjunto. 2. Pesar 100g de amostra e transferir para a peneira superior do conjunto. Tampar e fixar no vibrador. 3. Ligar e deixar por um período de 10 minutos em reostato Nº 10.

4. Pesar as frações retidas em cada peneira. CÁLCULO:
RETIDO = Peso retido x 100
Peso total amostra OBSERVAÇAO: O número de peneiras e o tamanho das malhas deverão ser estabelecidos conforme o tipo de produto. MÉTODO N.º 02 - UMIDADE (INDIRETO) APLICAÇÃO: Produtos ou sub-ptodutos de origem vegetal, animal, mineral, rações e concentrados. PRINCÍPIO: Fundamenta-se na perda de umidade e substâncias voláteis a temperatura de 105º C. MATERIAL E EQUIPAMENTO: - Balança analítica (Precisão +/- 0,0001 g) - Estufa de secagem - Pesa filtro ou cápsula de alumínio, com tampa - Dessecador com cloreto de cálcio ou silica-gel anidros PROCEDIMENTO: 1. Pesar a cápsula ou pesa filtro, limpo e previamente seco em estufa a 105ºC. por uma hora, resfriado em dessecador até temperatura ambiente. 2. Pesar de 3 a 5g da amostra, dependendo da capacidade do recipiente. 3. Colocar em estufa pré-aquecida a 105ºC (.+,/- 1ºC) até peso constante (4 a 6 horas). 4. Retirar da estufa e deixar em dessecador até temperatura ambiente. 5. Pesar CÁLCULO:
Umidade % (A - B) = x 100
C ONDE: A = Peso do recipiente + amostra B = Peso do recipiente + amostra após secagem C = Peso da amostra OBSERVAÇAO: Ao utilizar estufa sem renovação de ar recomenda-se não processar grande número de amostras simultaneamente. MÉTODO N.º 03 - UMIDADE (DIRETO) APLICAÇÃO: Amostras líquidas, forragens, silagens ou outras passíveis de alterações quando analisadas pelo método indireto. PRINCÍPIO: Baseia-se na determinação da umidade por destilação da amostra, utilizando-se um líquido não miscível com água e de ponto de ebulição superior a esta.

A água e o líquido volatilizam-se, e após condensação são depositados em um coletar graduado. MATERIAL E EQUIPAMENTO: - Balança semi analítica. (precisão +/- 0,01 g). - Manta ou chapa elétrica com controle de temperatura. - Condensador Liebig com junta esmerilhada. - Balo de fundo redondo ou chato, capacidade 500 ml com junta esmerilhada. - Sistema receptor graduado de "Dean and Stark" ou "Bidwell Stirling" com capacidade 10 ou 20 ml, divisão 1/10 com juntas superior e inferior esmerilhadas. REAGENTE: - Toluol P.A. PROCEDIMENTOS: 1. Pesar 20 a 30 g da amostra "in natura" (ou conforme a quantidade de água estimada), diretamente no balão. 2. Adicionar +/- 200 ml de toluol P.A., conectar ao sistema e este ao condensador. 3. Aquecer a mistura lentamente, controlando a temperatura para manter o líquido em constante ebulição. 4. Destilar até volume constante da fase aquosa no tubo graduado. 5. Desligar e proceder a leitura do volume final à temperatura ambiente. CÁLCULO:
V x.100 Umidade % = P ONDE: V = Volume da fase aquosa no tubo graduado P = Peso da amostra em grama MÉTODO N.º 04 - PROTEÍNA BRUTA (MACRO) APLICAÇÃO: Amostras nitrogenadas de origem orgânica e inorgânica, com exceção de nitratos e nitritos. PRINCÍPIO: Baseia-se na transformação do nitrogênio da amostra em sulfato de amônio por digestão ácida, e em nitrogênio amoniacal por destilação em meio alcalino. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/-0,0001 g). - Conjunto Kjeldahl para digestão e destilação macro. - Bureta de 50 ml. div. 1/10. - Frasco Kjeldahl 500 ou 800 Mi ou tubo de digestão macro. - Erlenmeyer de 300 ou 500 ml. - Pérolas de vidro 4 a 6 mm. - Provetas de 100 ml. REAGENTES: - ÁcIdo sulfúrico P.A. (d = 1.84 g/l). - Hidróxido de sódio P.A. - Sul fato de cobre pentahidratado P. A. - Sulfato de sódio anidro P.A. ou sulfato de potássio anidro P.A. - Vermelho de metiIa P. A.

- Zinco metálico granulado P.A. - Biftalato de potássio P.A. - Ácldo bórico P.A. - Verde de bromocresol P.A. - Ácido clorídrico P.A. - Carbonato de sódio P.A. PROCEDIMENTOS: 1. Pesar 1,0 g da amostra e transferir para frasco Kjeldhal ou tubo de digestão. 2. Adicionar aproximadamente 15 g de mistura catalítica, 5 a 7 pérolas de vidro e 20 ml de ácido sulfúrico P.A. pela parede do frasco. 3. Iniciar a digestão.com temperatura baixa. Prosseguir, elevando até o máximo de 38º C, evitando deixar pontos aderidos a parede do frasco. Continuar por mais 30 minutos após clareamento completo da mistura. 4. Esfriar, juntar de 250 a 350 ml de água destilada- (dependendo da capacidade do frasco). Agitar até dissolução e esfriar. 5. Levar ao destilador, adicionar cinco grânulos de zinco para controlar a ebulição e cuidadosamente, 50 ml de hidróxido de sódio 50%. Arrolhar rapidamente, agitar e proceder a destilação. 6. Recolher cerca de 200 ml de destilado em erlenmeyer de 300 ou 500 ml contendo 50 ou 100 ml de água destilada mais um volume de ácido sulfúrico 0,2 N a ser determinado de acordo com o percentual de proteína bruta estimada e 2 gotas de indicador vermelho de metila 0,1%. OBSERVAÇÃO: Alternativamente poderá ser empregado 50 ml de ácido do bórico 4% com 8 a 10 gotas do Indicador misto para receber a destilado. 7. Retirar e titular o excesso de ácido com solução de hidróxido de sódio até a viragem do vermelho para amarelo. OBSERVAÇAO: Ao utilizar o ácido bórico 4% empregar como titulante ácido clorídrico até viragem do ind'1cador (verde para rosa). 8. Proceder uma prova em branco, testando a qualidade dos reagentes. CÁLCULOS: Proteína = (Va - Vb) x F1 - (Vs x F2) x N x 6,25 x 0.014 x 100 Bruta % P ONDE: Va = Volume de H2S04 0.2 N utilizado Vb = Volume de H2S04 0.2 N consumido pela prova em branco F1 = Fator de correção do H2S04 0.2 N Vs = Volume de NaOH 0.2 N gasto na titulação F2 = Fator de correção do NaOH 0.2 N N = Normalidade 6,25 = Fator de transformação do nitrogênio em proteína, considerando' 16% nitrogênio (100/16=6,25) 0,014 = Miliequivalente grama do nitrogênio P = 'Peso da amostra em g ou no caso de utilização do ácido bórico 4% Proteína (Va - Vb) x F x N x 6,25 x 0,014 x 100 Bruta % = P ONDE:

Va = Volume de HCl 0,1 N gasto na titulação Vb = Volume de HCl 0,1 N gasto na prova em branco F = Fator de correção do HCl 0,1 N N = Normalidade 6,25 = Fator de transformação do nitrogênio em proteína, considerando 16% nitrogênio (100/16=6,25). 0,014 = Miliequivalente grama do nitrogênio P = Peso da amostra em g OBSERVAÇÕES: 1. Destilar de modo que o condensado seja obtido a temperatura ambiente. 2. O terminal do condensador deve ficar mergulhado na solução durante a fase de destilação. 3. Quando houver viragem da cor vermelha para amarela durante a fase de destilação, desconsidera a análise em processo. 4. Orientação para volumes de H2S04 0.2 N a ser utilizado, de acordo com o percentual de proteína bruta esperado. %. PB ml H2S04 até 10 % 10 20 % 15 30 % 25 40 % 30 60 % 50 80 % 60 5. Proceder sempre, uma prova de recuperação utilizando caseína P.A., sulfato de amônio anidro P.A. ou cloreto de amônio P.A. 6. Em amostras com alto teor de gordura ou que apresentem formação de espuma durante a fase de digestão ou destilação, utilizar algumas gotas de solução antiespumante. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução volumétrica padronizada de ácido sulfúrico Medir 5,5 ml de H2S04 P.A. e transferir para balão volumétrico de 1000 ml contendo aproximadamente 500 ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar exatamente 0,4 g de carbonato de sódio (Na2C03) P.A. previamente seco em mufla a 270ºC por uma hora. Dissolver em erlenmeyer, adicionando aproximadamente 30 ml de H2S04 0,2 N medidos e bureta de 4 gotas de vermelho de metila. Prosseguir a titulação gotejando a solução de H2S04 0.2 N até leve coloração rósea. Aquecer até ebulição e resfriar. Havendo permanência da coloração rósea, considerar terminada a titulação. Caso contrário, prosseguir, repetindo a operação até coloração permanente. CÁLCULO DO FATOR:
P NR NR = V x E F= NE ONDE: P = Peso do Na2C03 em miligrama NR ~ Normalidade real da solução V = Volume gasto na titulação em ml

E = Equivalente grama de Na2C03 NE = Normalidade esperada b. Solução volumétrica padronizada de hidróxido de sódio 0,2N Pesar cerca de 8,2 g de NaOH P.A. e dissolver em água destilada. Transferir quantitativamente para balão volumétrico de 1000 ml e completar o volume. PADRONIZAÇÃO: Pesar, com exatidão, cerca de 1,5 9 de biftalato de potássio P.A., previamente seco em estufa a 120ºC por duas horas. Dissolver em erlenmeyer com aproximadamente 75 ml de água destilada. Adicionar 4 gotas de solução alcóolica de fenolftaleína e titular gotejando a solução de NaOH 0.2 N até coloração levemente rósea. CÁLCULO DO FATOR:
P NR NR = V x E F= NE ONDE: E = Equivalente grama do biftalato de potássio P = Peso do Biftalato em miligrama V = Volume de NaOH 0.2 N consumido. NR = Normalidade real . NE = Normalidade esperada c. Solução indicadora de vermelho de metila 0,1% Dissolver 0,1 9 de vermelho de metila P.A. em aproximadamente 60 ml de álcool etílico. Transferir para balão volumétrico de 100 ml e completar o volume. d. Solução indicadora de fenolftaleína 1% Dissolver 1,0 g de .fenolftaleína P.A. em aproximadamente 60 ml de álcool etílico P.A. Transferir para balão volumétrico de 100 ml e completar o volume. e. Solução reagente de hidróxido de sódio 50% Dissolver 500 9 de NaOH P.A. ou grau técnico de boa qualidade em água destilada. Adicionar 30 9 de tiossulfato de sódio, quando o período de armazenamento for longo, para evitar a carbonatação e completar para 1000 ml. f. Mistura catalítica Peneirar separadamente o sulfato de sódio ou potássio anidros e o sulfato de cobre pentahidratado em peneira com malha de 1 mm. Juntar 10 partes de Na2S04 ou K2S04 e uma parte de CuS04. 5H20. Homogeneizar e guardar em frasco com tampa. g. Solução volumétrica padronizada de ácido clorídrico 0,1 N Medir 8,5 ml de HC.l P.A. e transferir para balão volumétrico de 1000ml, contendo aproximadamente 500 ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar exatamente 5,299g de carbonato de sódio (Na2CO3) P.A., previamente seco a 270ºC; por uma hora. Dissolver em água destilada, transferir para balão volumétrico de 1000 ml e completar o volume. Pipetar 20 ml da solução de Na2C03 e transferir para erlenmeyer de 250ml. Adicionar 3 gotas de solução de vermelho de metila 0,1%. Titular até viragem para coloração rósea. Aquecer a ebulição, para eliminar o gás carbônico. Esfriar e prosseguir a titulação. Repetir esta operação até coloração ;rósea permanente.

CÁLCULO DO FATOR:
P NR NR = V x E F= NE ONDE: P = Peso em mg do Na2C03 na alíquota V = Volume gasto de HCl 0,1N E = Equivalente grama do Na2C03 NR = Normalidade real NE = Normalidade esperada h. Solução reagente de ácido bórico 4% Pesar 40g de H3BO3 P.A. e transferir para bequer de 1.000 ml. Dissolver em aproximadamente 500 ml de água destilada, utilizando agitação. Transferir quantitativamente, através de papel filtro qualitativo, para balão volumétrico de 1.000 ml e completar o volume. i. Solução indicadora mista Dissolver 0,132 g de vermelho de metila P.A. em 70ml de álcool etílico P.A. Transferir quantitativamente para balão volumétrico de 200ml. Dissolver 0,066g de verde de bromocresol P.A. em 70ml de álcool etílico P.A. Juntar ao balão contendo vermelho de metila e completar o volume com álcool etilico P.A. OBSERVAÇÃO: O indicador misto poderá ser incorporado à solução de ácido bórico 4% (8ml por litro). MÉTODO N.º 05 - PROTEÍNA BRUTA (SEMI-MICRO) APLICAÇÃO: Amostras nitrogenadas de origem orgânica e inorgânica, com exceção de nitratos e nitritos. PRINCÍPIO: Baseia-se na transformação do nitrogênio da amostra em sulfato de amônio, por digestão ácida, e em nitrogênio amoniacal por destilação em meio alcalino. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g). - Bloco digestor e destilador por arraste de vapor. - Bureta de 25 ml (1/20). - Frasco Kjeldahl de 300 mI ou tubo de digestão (semi micro). - Erlenmeyer de 125 ml - Proveta de 25 ml REAGENTES: - Ácido sulfúrico P.A. (d = 1,84 g/l) - Hidróxido de sódio P.A. - Sulfato de cobre pentahidratado P.A. - Sulfato de sódio anidro P.A. ou sulfato de potássio anidro P.A. - Vermelho de metila P.A. - Zinco metálico granulado. - Biftalato de potássio P.A. - Ácido bórico P.A. - Verde bromocresol P.A. - Ácido clorídrico P.A.

- Carborloto dé sódio P.A. PROCEDIMENTOS: 1. Pesar de 300 a 500 mg da amostra e 'transferir para frasco Kjeldahl ou tubo. 2. Adicionar aproximadamente, 2g de mistura catalítica e 5 a 10ml de ácido sulfúrico P.A. pela parede do frasco. 3. Proceder a digestão até que a solução fique límpida, cuidando para não deixar pontos pretos aderidos a parede do frasco. Continuar por mais 30 minutos após o clareamento. Esfriar e dissolver com aproximadamente 10 a 20ml de água destilada. 4. Colocar em um erlenmeyer aproximadamente 50ml de água destilada, volume de H2S04; 0,2 N de acordo com a porcentagem de proteína estImada e 2 gotas de vermelho de metila 0,1%. OBSERVAÇÃO: Alternativamente poderá ser empregado de 20 a 50ml de ácido bórico 4% para receber o destilado, com 8 a 10 gotas de indicador misto. 5. Transferir quantitativamente a amostra digerida para o destilador, lavando o frasco três vezes com aproximadamente 10ml de água destilada. OBSERVAÇÃO: No caso de destiladores automáticos, conectar o tubo no Suporte e bocal apropriados. 6. Adicionar aproximadamente 20ml de NaOH 50%. Destilar por arraste mantendo o terminal do condensador mergulhado na solução receptora até que toda amônia seja liberada. 7. Retirar o erlenmeyer, lavar o terminal do condensador e titular o excesso de ácido, com solução de NaOH 0,2 até viragem do indicador (vermelho para amarelo). OBSERVAÇAO: Ao utilizar ácido bórico 4% empregar como titulante HCl 0,1N até viragem do indicador misto (verde para rosa). 8. Proceder paralelamente prova em banco dos reagentes utilizados. CÁLCULOS: Proteína = (Va - Vb) x F1 - (Vs x F2) x N x 6,25 x 0.014 x 100 Bruta % P ONDE: VA = Volume de H2S04 0,2 N utilizado V8 = Volume de H2S04 0,2 N consumido na prova em branco F1 = Fator de correção do H2S04 0,2 N VS = Volume de NaOH 0,2 N Gasto na titulação F2 = Fator de correção do NaOH 0,2 N N = Normalidade 6,25 = Fator de transformação do nitrogênio em proteína, considerando 16% de nitroganio (100/16=, 6,25) 0;014 = Miliequivalente grama do nitrogênio P = Peso da amostra em grama - No caso de utilização do Ácido Bórico 4% Proteína (Va - Vb) x F x N x 6,25 x 0,014 x 100 Bruta % = P ONDE: Va = Volume de HCl 0,1 N gasto na titulação Vb = Volume de HCl 0,1 N gasto na prova em branco F = Fator do HCI 0,1 N N = Normalidade

6,25 = Fator de transformação do nitrogênio em proteína, considerando 16% de Nitrogênio (100/16 = 6,25) 0,014=, Miliequivalente grama do nitrogênio P = Peso da amostra em grama OBSERVAÇOES: 1. Destilar de modo que o condensado seja obtido a temperatura ambiente. 2. O terminal do condensador deve ficar mergulhado na solução durante a fase de destilação. 3. Quando houver viragem da cor vermelha para amarela durante a fase de destilação, desconsiderar a análise em processo. 4. Orientação para volumes de H2S04 0,2N a ser utilizado, de acordo com o percentual de proteína bruta esperado. % PB ml H2S04 até 10% 10 20% 15 30% 25 40% 30 60% 50 80% 60 5. Proceder sempre uma prova de recuperação utilizando caserna P.A., sulfato de amônio anidro P.A. ou cloreto de amônio P.A. 6. Em amostras com alto teor de gordura ou que apresentem formação de espuma durante a fase de digestão ou destilação, utilizar algumas gotas de solução antiespumante. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução volumétrica padronizada de ácido sulfúrico 0,2 N. Medir 5,5 ml de H2S04 P.A. e transferir para balão volumétrico de 1.000ml contendo aproximadamente 500 ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar exatamente 0,4 g de carbonato de sódio (Na2C03) P.A. previamente seco em mufla a 270ºC por uma hora. Dissolver em erlenmeyer, adicionando aproximadamente 30 ml de H2S04 0,2N medidos em bureta e 4 gotas de vermelho de metila 0.1%. Prosseguir a titulação gotejando a solução de H2S04 0.2 N até leve coloração rósea. Aquecer até ebulição e resfriar. Havendo permanência da coloração rósea, considerar terminada a titulação. Caso contrário, prosseguir, repetindo a operação até coloração permanente. CÁLCULO DO FATOR:
P NR NR = V x E F= NE ONDE: P = Peso do Na2C03 em miligrama NR = Normalidade real da solução V = Volume gasto na titulação em ml. E = Equivalente grama do Na2C03 NE = Normalidade esperada b. Solução volumétrica padronizada de hidróxido de sódio 0,2N

Pesar 8,2g de NaOH P.A., dissolver em água destilada. Transferir quantitativamente para balão volumétrico de 1.000ml e completar o volume. PADRONIZAÇÃO: Pesar, com exatidão, cerca de 1,a g de biftalato de potássio P.A., previamente seco em estufa a 120ºC por duas horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcoólica de fenolftaleína 1% e titular gotejando a solução de NaOH 0,2N até coloração levemente rósea. CÁLCULO DO FATOR:
P NR NR = V x E F= NE ONDE: E = Equivalente grama do biftalato de potássio P = Peso do biftalato em miligrama V = Volume de NaOH 0, N consumido NR = Normalidade real INE = Normalidade esperada c. Solução indicadora de vermelho de metila 0,1% Dissolver 0,1g de vermelho de metila P.A. em aproximadamente 60 ml de álcool etílico. Transferir para balão volumétrico de 100 ml e completar o volume. d. Solução indicadora de fenolftaleína 1% Dissolver 1,0g de fenolftaleína PA.. em aproximadamente 60 ml de álcool etílico P.A.. Transferir para balão volumétrico de 100ml e completar o volume. e. Solução reagente de hidróxido de sódio 50% Dissolver 500g de NaOH P.A. ou grau técnico de boa qualidade em água destilada. Adicionar 30 g de tiossulfato de sódio para evitar a carbonatação e completar para 1.000ml em béquer. f. Mistura catalítica Peneirar separadamente o sulfato de sódio ou potássio anidros e o sulfato de cobre pentahidratado em peneira com malha de 1mm. Juntar 10 partes de Na2S04 ou K2S04 e uma parte de CuS04. 5H20. Homogeneizar e guardar em frasco com tampa. g. Solução volumétrica padronizada de ácido clorídrico 0,1 N. Medir 8,5ml de HCl P.A., transferir para balão volumétrico de 1.000ml, contendo aproximadamente 500 ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar exatamente 5,299g de carbonato de sódio (Na2C03) P.A. previamente seco a 270ºC por uma hora. Dissolver em água destilada, transferir para balão volumétrico de 1.000ml e completar o volume. Pipetar 20 ml da solução de Na2C03 e transferir para erlenmeyer de 250ml. Adicionar 3 gotas de solução de vermelho de metila 0,1%. Titular até viragem para coloração rósea. Aquecer a ebulição, para eliminar o gás carbônico. Esfriar e prosseguir a titulação. Repetir esta operação até coloração rósea permanente. CÁLCULO 00 FATOR:

P NR
NR = V x E F= NE ONDE: P = Peso em mg do Na2C03 na alíquota V = Volume gasto de HCl E = Equivalente grama do Na2C03 NR = Normalidade real NE = Normalidade esperada. h. Solução reagente de ácido bórico 4% . Pesar 40g de H3B03 P.A. e transferir para bequer de 1.000ml. Dissolver em aproximadamente 500ml de água destilada, utIlizando agitação. Transferir quantitativamente, através de papel filtro qualitativo, para balão volumétrico de 1.000ml e completar o volume. 1. Solução IndIcadora mIsta Dissolver 0,132 g de vermelho de metiIa P.A. em 70mI. de álcool etílico P.A. Transferir quantitativamente para balão volumétrico de 200ml. Dissolver 0,066g de verde de bromocresol P.A. em 70 ml de álcool etílico P.A. Acrescentar ao balão contendo vermelho de metila e completar o volume com álcool etílico P.A. OBSERVAÇAD: O indicador misto poderá ser incorporado a solução de ácido bórico 4% (8ml por litro) MÉTODO N.º 06 - DIGESTIBILIDADE EM PEPSINA 0,2% APLICAÇÃO: Produtos ou sub-produtos protéicos de origem animal. PRINCÍPIO: Fundamenta-se na digestão da amostra por ação da pepsina. MATERIAL E EQUIPAMENTO: - Balança analítica (Precisão +1- 0,0001g) - Estufa bacteriológica - Estufa de secagem - Agitador tipo Wagner (Modelo AOAC para 15 RPM) ou similar - Cadinho de vidro sinterizado NQ 01 ou funil de Buchner - Papel de filtro quantitativo filtração média. - Proveta de 250 ml. - Frascos com tampa ou erlenmeyer capacidade 500 ml REAGENTES: 1. Ácido clorídrico P.A. (d = 1,19g/ml) 2. Pepsina c/ atividade 1:10.000 3. Acetona P.A. PROCEDIMENTOS: 1. Pesar 1g da amostra, desengordurar e transferir quantitativamente para frasco com tampa ou erlenmeyer com rolha esmerilhada. 2. Adicionar 150ml de solução de pepsina 0.2% em HCl 0,075 N, previamente aquecida a 45ºC. Fixar no agitador e incubar por 16h a 45ºC com agitação constante. 3. Filtrar sob sucção em cadinho de vidro sinterizado ou papel filtro previamente tarados. Usando-se a segunda opção, ajustar o papel ao funil Buchner. Lavar o resíduo com 2 a 3 porções de água a 45 – 50ºC e após, com 2 porções de acetona P.A.

4. Colocar o cadinho ou papel de filtro com o resíduo em estufa a 105ºC por 1 hora. Retirar e esfriar em dessecador até temperatura ambiente. Pesar. CÁLCULOS:
A - B Digestibilidade % = 100 - (______) x 100
P ONDE: A = Peso do cadinho ou papel de filtro + resíduo B = Peso do cadinho ou papel de filtro C = Peso da amostra em grama OBSERVAÇOES: 1. A solução de pepsina 0,2% em HCl 0,075N deverá ser preparada momentos antes de sua utilização. 2. Em substituição ao agitador tipo Wagner, poderá ser utilizado banho maria com agitação (tipo Kline) ou similar. A velocidade de agitação deverá ser ajustada de maneira que o resultado obtido seja semelhante aquele do método indicado. SOLUÇOES REAGENTES E VOLUMÉTRICAS a. Solução volumétrica padronizada de ácido clorídrico 0,075N Transferir 12,8ml de HCI P.A. para balão volumétrico de 2.000ml, contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar exatamente 0,12g de Na2C03 P.A. previamente seco em mufla e 270ºC por 1 hora. Dissolver em erlenemeyer,. adicionando aproximadamente 20ml de HCl 0,75N, medidos em bureta e adicionar 4 gotas de vermelho de metila 0,1%. Prosseguir a titulação, gotejando a solução de HCl 0,075N até leve coloração rósea. Aquecer até ebulição e esperar. Havendo permanência de coloração rósea, considerar terminada a titulação. Caso contrário, prosseguir até coloração rósea constante. CÁLCULO DA NORMALIDADE:
P NR = ______
V X E ONDE: P = Peso de Na2C03 em miligrama V = Volume gasto de HCI 0,075N na titulação E = Equivalente grama do Na2C03 NR = Normalidade real b. Solução indicadora de vermelho de metila 0,1% Dissolver 0,1g de vermelho de metila P.A. em aproximadamente 60 ml de álcool etílico. Transferir para balão volumétrico de 100 ml e completar o volume. c. Solução reagente de pepsina 0,2% em ácido clorídrico 0.075N Pesar em bequer exatamente 2,0 g de pepsina - 1:10.000. Adicionar aproximadamente 100 ml de solução HCI 0,075N, dissolver, transferir

quantitativamente para balão volumétrico de 1000 ml e completar o volume com HCI 0,075N. OBSERVAÇAO: Manter a pepsina 1:10.000 estocada em refrigerador. MÉTODO N.º 07 - PROTEíNA DIGESTÍVEL EM PEPSINA 0,2% APLICAÇÃO: Produtos e sub produtos protéicos de origem animal. PRINCÍPIO: Fundamenta-se na determinação indireta da fração nitrogenada da amostra digerida por ação da pepsina. MATERIAL E EQUIPAMENTO: - Balança analítica (Precisão +/- 0,0001g) - Estufa bacteriológica - Estufa de secagem - Agitador tipo Wagner (Modelo AOAC para 15 RPM) ou similar - Funil de Buchner - Papel de filtro quantitativo, filtração média - Proveta de 250ml - Frascos ou erlenmeyer capacidade 500 ml com tampa - Material e equipamento usados para determinação de proteína bruta (macro ou semi-micro) REAGENTES: - Ácido clorídrico P.A. (d = 1,19g/ml) - Pepsina com atividade 1:10.000 - Acetona P.A. - Reagentes utilizados na determinação de proteína bruta (macro ou semi-micro) PROCEDIMENTO: 1. Pesar 19 da amostra, desengordurar e transferir quantitativamente para frasco ou erlenmeyer com tampa. 2. Adicionar 150ml de solução de pepsina 0,2% em HCI 0,075 N, previamente aqueclda a 45ºC. Flxar no agitador e incubar por 16 horas a 45ºC com agitação constante. 3. Filtrar com sucção sobre papel de filtro ajustado a um funil de Buchner lavar o resíduo com 2 a 3 porções de água a 45 – 50ºC e após, com 2 porções de acetona. 4. Transferir o papel com o resíduo para balão Kjeldahl ou tubo de digestão. Proceder conforme descrito nos métodos para determinação de Proteína Bruta (macro ou semi-micro). CALCULO:
Prot. Bruta - Prot. do resíduo Proteína digestível % = x 100
Prot. Bruta OBSERVAÇOES: 1. A solução de pepsina 0,2 em HCI 0,075 N deverá ser preparada momentos antes de sua utilização. 2. Em substituição ao agitador tipo Wagner. poderá ser utilizado banho-maria com agitação (tipo Kline) ou similar. A velocidade de agitação deverá ser ajustada de maneira que o resultado obtido seja semelhante aquele do método indicado. 3. Considerar o peso da amostra original no cálculo da proteína do resíduo.

SOLUÇOES REAGENTES: a. Solução voIumétrica padronizada de HCI 0.075N. Medir 12,8 ml de HCI P.A. e transferir para balão volumétrico de 2.000 ml contendo aproximadamente 500 ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar 0,12 gramas de Na2C03 P.A. previamente seco em mufla a 270ºC por 1 hora. Dissolver em erlenmeyer, adicionando aproximadamente 20ml de HCI 0,075N, medidos em bureta, e adicionar 4 gotas de vermelho de metila 0,1%. Prosseguir a titulação, gotejando a solução de HCl 0,075N até leve coloração rósea. Aquecer até ebulição e esperar. Havendo permanência de coloração rósea, considerar terminada a titulação. Caso contrário, prosseguir até coloração rósea constante. CALCULO DA NORMALIDADE:
P NR = ______
V X E ONDE: P = Peso de Na2C03 em mg V = Volume gasto de HCl 0,075N na titulação E = Equivalente grama do Na2C03 NR = Normalidade real b. Solução reagente de pepsina 0,2% em ácido clorídrico 0,075N Pesar em bequer exatamente 2,0 q de pepsina - 1:10.000. Adicionar aproximadamente 100ml de solução HCl 0,075N, dissolver, transferir quantitatIvamente para balão volumétrico de 1000ml e completar o volume com HCl 0,075 N. OBSERVAÇÃO: Manter a pepsina 1:10.000 estocada em refrigerador. c. Soluções reagentes usadas na determinação de proteína bruta (macro ou semi-mIcro). MÉTODO N.º 08 - PROTEÍNA SOLÚVEL EM KOH APLICAÇÃO: Grãos de oleogInosas e seus derivados. PRINCÍPIO: Baseia-se na extração e determinação da fração nitrogenada da amostra, solúvel em solução de KOH de concentração conhecida. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Agitador magnético ou tIpo Kline, com controle de rotação. - Centrífuga - Erlenmeyer de 250ml ou 300ml - Material e equipamento usados para determinação da proteína bruta (macro ou semi-micro). REAGENTES: - Hidróxido de potássio P.A. (Min. 85% KOH)

- Soluções reagentes usadas na determinação de proteína bruta (macro ou semi-micro). PROCEOIMENTO: 1. Pesar 2,0g da amostra. Transferir para erlenmeyer de 250 ou 300ml e adicionar volumétricamente 100 ml de solução de KOH 0,2% (0,036N). 2. Agitar brandamente por 20 minutos, usando agitador magnético ou tipo Kline. 3. Centrifugar o sobrenadante por 5 a 10 minutos a 1.500 rpm. 4. Pipetar volumétricamente 25ml do centrifugado e transferir para frasco Kjeldahl ou 10ml para tubo de digestão. 5. Proceder conforme descrito nos métodos para determinação de proteína bruta (macro ou semi-micro). 6. Proceder paralelamente prova em branco .dos reagentes utilizados. CÁLCULO:
Proteína do sobrenadante x 100 Proteína solúvel % =
Proteína Bruta i Utilizar peso da amostra para cálculo, de acordo com alíquota empregada. OBSERVAÇOES: 1. A agitação da mistura deve ser branda mas suficiente para provocar revolução completa do material. 2. Devido a alta sensibilidade do método, trabalhar com solução de KOH exatamente 0,036N. SOLUÇOES REAGENTES E VOLUMÉTRICAS: 1. Solução volumétrica padronizada de hidróxido de potássio 0,2% (0,036N). Pesar 2,376 g de KOH P.A., solubilizar em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml, completar o volume e homogeneizar. PADRONIZAÇAO Pesar com exatidão 0,2g de Biftalato de Potássio P.A., previamente seco em estufa a 120ºC por 2 horas. Dissolver em erlenmeyer com aproximadamente 7.5ml de água destilada. Adicionar 4 gotas de solução alcóolica de fenolftaleína a 1% e titular gotejando a solução de KOH 0,036N até coloração levemente rósea. CALCULO DA NORMALIDADE:
P N = Y x E
ONDE: E = Equivalente grama do biftalato de potássio P = Peso do biftalato de potássio em miligrama Y = volume KOH 0,036N consumido. NR = Normalidade real b. Soluções reagentes usadas na determinação de proteína bruta (macro ou semi-micro).

MÉTODO Nº 09 - URÉIA APLICAÇÃO: Uréia ou produtos que a contenham. PRINCÍPIO: Consiste na determinação do nitrogênio da uréia, liberado pela ação enzimática da urease MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Conjunto Kjeldahl para destilação macro - Frasco Kjeldahl capac. 500 ou 800ml - Erlenmeyer 300 ou 500ml - Bureta capacidade 50ml div. 1/10 - Provetas 10 e 100ml - Pérolas ,de vidro 4 a 6mm diâmetro REAGENTES: - Uréia P.A. - Urease P.A. - Cloreto de cálcio P.A. . - Óxido de magnésio P.A. - Solução antiespumante - Reagentes utilizados na determinação de proteína bruta - fase de destilação {macro ou semi-micro). PROCEDIMENTO: 1. Pesar de 0,2 a 2g da amostra, conforme o teor esperado e transferir para frasco Kjeldahl de 500 ou 800 ml. 2. Adicionar aproximadamente 300ml de água destilada e 10ml de solução neutra de urease 1%. tampar hermeticamente e deixar a temperatura ambiente por 1 hora ou 20 minutos a 40ºC. 3. Adicionar 2g de óxido de magnésio, 5ml de solução de cloreto de cálcio 25%, 3ml de solução antiespumante e 7-8 pérolas de vidro. 4. Levar ao destilador, arrolhando rapidamente o frasco. Agitar e proceder a destilação. 5. Recolher cerca de 150-200ml do destilado em erlenmeyer de 300 ou 500ml, contendo 20 e 50ml de solução de ácido bórico 4% ou volume de ácido sulfúrico 0,2N a ser determinado, de acordo com o percentual de uréia esperado (Neste caso, utilizar indicador vermelho de metila 0,1%). 6. Retirar e titular utilizando ácido clorídrico 0,1N ou hidróxido de sódio 0,2N, procedendo a titulação conforme descrito na metodologia ,para determinação de proteína bruta. 7. Proceder paralelamente prova em branco dos reagentes utilizados. CÁLCULOS:
(VA - VB) x F1 - (Vs x F2) x N x 2,143 x.0,014 x 100 Uréia % = P ONDE: VA = Volume H2S04 0,2N utilizado VB = Volume H2S04 0,2N consumido pela prove branco F1= Fator de correção do H2S04 0,2N Vs = Volume de NaOH 0,2N gasto na titulação F2 = Fator de correção do NaOH 0,2N N = Normalidade

2,143 = Fator de transformação do nitrogênio em uréia 0,014 = Miliequivalente grama do nitrogênio P = Peso da amostra em grama ou no caso de utilização de Ácido Bórico 4%
(VA - VB) x F x N x 2,143 x 0,014 X 100 Uréia % = P ONDE: VA = Volume HCl 0,1N gasto na titulação VB = Volume HCl 0,1N consumido na prova branco. F1 = Fator de correção do HCl 0,1N N = Normalidade 2,143 = Fator de transformação do nitrogênio em uréia 0,014 = Miliequivalente grama do nitrogênio P = Peso da amostra em grama OBSERVAÇÕES: 1. Volume de H2S04 0,2N a ser utilizado, de acordo com o percentual de uréia esperado.
% Volume 1 5 2 10 5 25
2. Proceder prova de recuperação utilizando uréia P.A. (100 a 200 mg). 3. O terminal do condensador deve ficar mergulhado na solução durante a fase de destilação. 4. Quando houver viragem da coloração vermelha para amarela durante a fase de destilação, desconsiderar a análise em processo (no caso de usar H2S04 0,2 N) 5. Destilar de modo que o condensado seja obtido a temperatura ambiente. 6. Em substituição a solução neutra de urease 1%, pode-se utilizar (Jackbean Meal) em excesso (500 a 600mg). Conservar em geladeira. SOLUÇÕES REAGENTES E YOLUMÉTRICAS: a. Solução reagente neutra de urease 1% - Determinação da alcalinidade - dissolver exatamente 0,1g de urease P.A. em 50ml de água destilada e titular com HCI 0,1N, utilizando vermelho de metila 0,1% como indicador, até viragem para rosa. Anotar a quantidade de HCI 0,1N necessário para neutralizar 0,1g de urease. - Preparação da solução neutra de urease 1%; pesar exatamente, 1g de urease P.A. em béquer de 100ml e dissolver com aproximadamente 50ml de água destilada. Neutralizar esta solução utilizando a relaçao gramas de urease P.P.:.ml de HCI 0,1N obtida na terminação da alcalinidade. Transferir para balão volumétrico de 100ml e completar o volume com água destilada. - Determinação da atividade enzimática - adicionar quantidades crescentes de solução neutra de urease 1% a várias amostras de 0,1g de uréia P.A. e prosseguir a partir do item 2. "procedimento". De acordo com os resultados obtidos, determina-se a quantidade mínima de solução necessária para hidrolizar 0,1g de uréia. b. Solução reagente de cloreto de cálcio 25% Pesar exatamente 25g de CaCl2.P.A. em béquer e dissolver com água destilada. Transferir quantitativamente para balão volumétrico de 100ml e completar o volume.

c. Soluções reagentes utilizadas a partir da fase de destilação na determinação de Proteína Bruta (macro ou semi-micro). MÉTODO Nº 10:- EXTRATO EtÉREO APLICAÇÃO: Produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. PRINCÍPIO: Baseia-se na extração da fração gordurosa e demais substâncias solúveis através de arraste por solvente. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g). - Estufa de secagem. - Dessecador com cIoreto de cálcio ou silica gel anidros. - Balão de fundo chato ou copo Goldfisch - Apatelho extrator tipó Soxhlet ou Goldfisch - Papel de filtro qualitativo ou cartucho extrator de cerâmica ou celulose - Algodão hidrófilo desengordurado. REAGENTES: - Éter de petróleo P.A. PROCEDIMENTO: 1. Pesar de 2 a 5g da amostra, transferir para cartucho extrator ou cartucho preparado com papel filtro. 2. Secar a 105ºC + / - 1ºC durante 2 horas. 3. Secar o balão ou copo em estufa a 105ºC por uma hora, esfriar em dessecador até a temperatura ambiente e pesar. 4. Introduzir o cartucho no extrator. 5. Adicionar quantidade suficiente de solvente ao balão ou copo, conectando-o ao extrator, Ajustar o conjunto ao condensador. 6. Extrair por um período de, no mínimo, 6 horas a velocidade de condensação de 2 a 4 gotas por segundo. 7. Recuperar o solvente e completar a secagem do balão ou copo em estufa a 105ºC por 30 minutos. 8. Esfriar em dessecador até temperatura ambiente e pesar. 9. Repetir a operação de secagem até que a diferença entre duas pesagens sucessivas não seja superior a 0,1% do peso da amostra. CÁLCULO:
(A – B) Extrato Etéreo % = C x 100 ONDE: A = peso do balão ou copo + resíduo B = peso do balão ou copo C = peso da amostra em grama OBSERVAÇÃO: Conduzir sempre uma prova em branco, testando a qualidade do solvente utilizado. MÉTODO Nº 11 - FIBRA BRUTA APLICAÇÃO: Produtos ou sub-produtos de origem vegetal, rações e concentrados.

PRINCÍPIO: Baseia-se na determinação do resíduo orgânico insolúvel da amostra, após uma digestão ácida e outra alcalina. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Estufa de secagem. - Forno mufla. - Conjunto para determinação de fibra bruta. - Sistema de vácuo. - Dessecador com cloreto de cálcio ou silica gel anidros. - Copo Berzelius de 600mI ou erlenmeyer de 500ml com junta esmerilhada. - Frasco kitassato - Funil de Buchner +/- 10cm de diâmetro. - Cadinho de Gooch capacidade de 40/50mI ou cadinho de vidro sinterizado porosidade 1 (90 a 150 micras). - Fibras de óxido de alumínio ou amianto purificado. - Tela de nailon ou poliester ou aço inox (200 mesh) ou papel de filtro qualitativo. REAGENTES: - Ácido sulfúrico P.A. (d = 1,84 g/ml) - Hidróxido de sódio P.A. - Álcool etílico ou acetona P.A. - Solução anti-espumante PROCEDIMENTO: 1. Pesar de 1 a 3g da amostra, conforme o teor de fibra bruta estimado. Desengordurar.e secar em estufa a 105ºC por uma hora. 2. Transferir o resíduo para bequer, erlenmeyer ou cadinho de vidro sinterizado (sistema automático). Adicionar 200ml de H2S04 1,25% (0,225N) em ebulição (sistema automático - injetar 150ml), e algumas gotas de solução anti-espumante. Digerir com refluxo por exatamente 30 minutos. 3. Filtrar quantitativamente a quente, sob vácuo em funil de Buchner provido de tela de nailon, aço inox ou cadinho de vidro sinterizado (sistema automático). Proceder lavagens sucessivas do resíduo com água fervente até completa neutralização. 4. Retornar o resíduo ao bequer ou erlenmeyer, lavando a tela com 200ml de NaOH 1,25% (0,313N) em ebulição (sistema automático -, injetar 150ml de NaOH 1,25% - 0,313N). Adicionar algumas gotas de solução anti-espumante. Digerir com refluxo por exatamente 30 minutos. 5. Filtrar quantitativamente a quente sob vácuo em funil Buchner provido de tela de nailon ou poliester, aço inox ou diretamente em cadinho de vidro sinterizado. 6.. Transferir quantitativamente o resíduo com auxílio de água quente para cadinho de vidro sinterizado ou cadinho de Gooch contendo camada densa de fibras de óxido de alumínio ou amianto purificado. Lavar com aproximadamente 20ml de álcool etílico P.A. ou acetona P.A. e 20ml de acetona P.A. ou éter etílico ou petróleo P..A.o 7. Colocar em estufa a 105 C até peso constante (4 a 6 horas). Retirar, deixar em dessecador até temperatura ambiente e pesar. 8. Incinerar em mufla a 550ºC por 2 horas ou a 600°C por 1 hora. Retirar a temperatura de 250ºC / 300ºC. Resfriar em dessecador até temperatura ambiente e pesar. CÁLCULO:

(A – B) Extrato Etéreo % = C x 100 ONDE: A = Peso do cadinho + resíduo B = Peso do cadinho + cinzas C = Peso da amostra OBSERVAÇÕES: 1. Em substituição a tela de nailon ou aço inox, poderá ser utilizado um tecido de linho com textura equivalente. 2. Poderão ser eregados como anti-espumantes: a. álcool n-amílico b. solução de silicone (1g de silicone + 50ml de éter etílico + 50ml de éter de petróleo) estocar em refrigerador. 3. A camada de fibras de óxido de alumínio ou amianto no cadinho de Gooch deverá ter, após boa compactação, espessura suficiente para não permitir a passagem de partículas de resíduo. 4. Ao Utilizar cadinho de vidro sinterizado, adotar temperatura máxima de 500ºC e tempo mínimo de 3 horas. 5. O amianto deverá ser previamente calcinado, tratado com solução ácida e alcalina, conforme o ensaio e lavado cuidadosamente de maneira a eliminar todo o resíduo. Calcinar novamente. SOLUÇÕES REAGENTES E VOLUMÉTR1CAS: a. Solução reagente de ácido sulfúrico 1,25º (0;255N) Medir 7.0 ml de H2S04 P.A. (d=1.84g/ml) e transferir para balão volumétrico de 1000ml, contenDo aproximadamente 500ml de água destilada. Esfriar e completar o volume. Padronizar esta solução com NaOH 0,2N e considerar adequada se estiver entre 0,252N e 0,258N. b. Solução reagente de hidróxido de sódio 1,25% (0,313N) Pesar exatamente 12,5g de NaOH P.A. em bequer, solubilizar e transferir quantitativamente para balão volumétrico de 1000ml, esfriar e completar o volume. Padronizar esta solução com H2S04 0,2N e considerar adequada se a normalidade estiver entre 0,310N e 0,316N. MÉTODO Nº 12 - CINZAS OU MATÉRIA MINERAL APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: Fundamenta-se na eliminação da matéria orgânica e inorgânica volátil a temperatura de 550ºC a 600ºC. O resíduo obtido é denominado cinzas. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Forno mufla - Dessecador com cloreto de cálcio ou silica gel anidros - Cadinho ou cápsula de porcelana PROCEDIMENTO: 1. Pesar o cadinho ou cápsula de porcelana, limpo e previamente calcinado em mufla a 550ºC/600ºC por 30 minutos, e resfriado em dessecador até temperatura ambiente. 2. Pesar de 2 a 3g da amostra no cadinho ou cápsula.

3. Levar à mufla e gradualmente aumenta temperatura (550/600ºC) até obtenção de cinzas claras (3 horas no mínimo). 4. Retirar a 250/300ºC, resfriar em dessecador até temperatura ambiente e pesar. CÁLCULO:
A - B Cinzas ou Matéria Mineral % = (___________) x 100
C ONDE: A = Peso do cadinho ou cápsula + resíduo B = Peso dó cadinho ou cápsula C = Peso da amostra em grama OBSERVAÇOES: 1. Nem sempre o resíduo obtido representa toda a substância inorgânica presente na amostra, pois alguns sais podem sofrer redução ou volatilização nessa faixa de temperatura. 2. Não se obtendo cinzas claras após decorrido o período mínimo de calcinação, recomenda-se a adição de 2 a 3 gotas de HN03 concentrado ou água oxigenada 30 volumes, e retorno a mufla entre 550/600ºC por mais uma hora. Essa adição facilitará a oxidação do carbono. 3. Opcionalmente ao item 3 (procedimento), poderá ser efetuada uma pré-queima em placa aquecedora e/ou bico de Buzen, transferindo em seguida para mufla (55Q/600ºC). MÉTODO Nº 13 - RESÍDUO INSOLÚVEL EM HCL APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal e mineral. PRINCÍPIO: Baseia-se na determinação da fração mineral insolúvel em ácido clorídrico. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/:. 0,0001g) - Forno mufla - Dessecador com cloreto de cálcio ou silica gel anidros - Cadinho ou cápsula de porcelana - Papel filtro quantitativo, filtração média - Bequer de 250ml REAGENTES: - Ácido clorídrico P.A. PROCEDIMENTO: 1. Pesar o cadinho ou cápsula de porcelana, limpo e previamente calcinado em mufla a 550/600ºC por 30 minutos, e resfriado em dessecador até temperatura ambiente. 2. Pesar de 2 a 3g da amostra no cadinho ou cápsula. 3. levar à mufla e gradualmente aumentar a temperatura até (550/600ºC) até obtenção de cinzas claras (3 horas no mínimo). 4. Retirar a 250/300ºC, resfriar até a temperatura ambiente. 5. Transferir quantitativamente as cinzas contidas no cadinho para bequer de 250ml, lavando com três porções de aproximadamente 20ml de solução de ácido clorídrico 1:1. Ferver por 5 minutos.

6. Filtrar quantitativamente sobre papel de filtro, lavando o resíduo com H20 destilada quente até eliminação de todo o ácido. 7. Transferir o papel de filtro com resíduo para cápsula ou cadinho de porcelana, previamente tarado. levar à mufla e gradualmente aumentar a temperatura. Manter por duas horas. (no mínimo) à temperatura de 550/600ºC. 8. Retirar a 250/300ºC. Resfriar em dessecador até temperatura ambiente e pesar. CÁLCULO:
A - B Resíduo insolúvel % = (__________) x 100
C ONDE: A = Peso da cápsula ou cadinho + resíduo B = Peso de cápsula ou cadinho C = Peso original da amostra em grama OBSERVAÇÃO: Opcionalmente aos itens 3 e 7, poderá ser efetuada uma pré-queima em placa aquecedora, transferindo em seguida para a mufla (500/600ºC). SOLUCÕES REAGENTES E VOLUMÉTRICAS: a. Solução reagente de ácido clorídrico 1:1 Diluir uma parte de HCI em uma parte de água destilada. MÉTODO Nº 14 - CÁLCIO (OXIDIMÉTRICO) APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: Consiste em precipitar o cálcio de uma solução obtida das cinzas da amostra, por adição de oxalato de amônio. O precipitado formado (oxalato de cálcio) é dissolvido em meio ácido formando ácido oxálico, que é determinado por oxidimetria (permanganatometria). MATERIAL E EQUIPAMENTO: - Placa aquecedora. ou banho maria - Bequer de 250 ou 300ºC - Funil analítico - Cadinho de Gooch - Fibras de Óxido de Alumínio ou Amianto - Vidro de relógio - Pipetas volumétricas - Pipetas graduadas (ou repipetador automático) - Proveta de 50ml - Balões volumétricos - Papel de filtro qualitativo - Bastão de vidro - Bureta ambar de 50ml (1/20) REAGENTES: - Ácido clorídrico P.A. . - Ácido sulfúrico P.A.

- Oxalato de amônio P.A. '" - Hidróxido de amônio P.A.. . - Permanganato de potássio P.A. - Vermelho de metila P.A. PROCEDIMENTO: 1. Com o auxílio de bastão de vidro, transferir quantitativamente para béquer as cinzas obtidas na determinação de matéria mineral. Lavar o cadinho com pequenas porções de solução de HCl 1/1 totalizando 40 ml, e em seguida com água destilada. Cobrir com vidro de relógio e levar a placa aquecedora até que haja redução de 1/3 do volume. Esfriar. 2. Filtrar sobre papel de filtro qualitativo para balão volumétrico adequado. Completar o volume com água destilada e homogeneizar (solução estoque). Reservar esta solução também para a determinação de fósforo. 3. Pipetar volumétricamente uma alíquota adequada para bequer de 250 ou 300ml. Adicionar 2 a 3 gotas de vermelho de metila 0,1% e diluir com água destilada até aproximadamente 50 ml. 4. Aquecer brandamente em placa aquecedora ou banho maria, (70/80ºC) e acrescentar sob agitação constante 25ml de oxalato de amônio 4,2% quente. Adicionar NH40 1/1, gota a gota sob agitação constante, até que a coloração mude do vermelho para rosa. Se houver mudança da coloração para amarelo, gotejar HCI 1:3 até retorno para rosa. Deixar em repouso no mínimo 1 hora e máximo de 3 horas. 5. Filtrar sob vácuo em cadinho de Gooch com camada de fibra de óxido de alumínio ou amianto. Lavar o bequer e o precipitado 3 vezes com NH40H 1/50 ou até que algumas gotas do filtrado não reajam com solução de nitrato de prata acidulada com ácido nítrico. 6. Transferir o cadinho de Gooch com o precipitado para o bequer original. Adicionar água destilada até cobrir o cadinho e 5ml de H2S04 P.A. 7. Aquecer até completa dissolução do precipitado (próximo a ebulição), e titular com KMn04 0,05 N sob constante agitação- até coloração rósea clara persistente por 30 segundos. Cuidar para que durante a titulação a temperatura não fique abaixo de 60ºC. CÁLCULO:
V x N x F x 0,02004 x S x 100 Cálcio % = P x A ONDE: V = Volume de KMn04 0,05 N gasto na titulação. N = Normalidade. F = Fator de correção da solução de KMn04 0,05 N. S = Volume da solução estoque. P = Peso da amostra em grama. A = Volume da alíquota utilizado 0,02034 = miliequivalente grama do cálcio. OBSERVAÇOES: 1. Não é necessário calcinar amostras isentas de matéria orgânica. 2. O Manganês é reduzido pelo oxalato em meio ácido, de + 7.para + 2, tornando a solução de permanganato incolor. A primeira gota que não for reduzida, dará a mesma, uma tonalidade rosa. 3. Orientação para volumes estoques e alíquotas de acordo com o percentual de cálcio esperado.

cálcio esperado % volume estoque alíquota 0 - 1 % 200ml 100 ml 1 - 4 % 200ml 50ml 4 - 10 % 250ml 20ml 10 - 20 % 250ml 10ml 20 - 40 % 500ml 10ml SOLUÇÕES REAGENTES E VOLUMÉTRICAS: a. Solução reagente de ácido clorídrico 1:3 , Diluir uma parte de HCl P.A. em três partes de água destilada. b. SoluçAo reagente de ácido clorídrico 1:1 Diluir uma parte de HCl em uma parte ::1e á~ya destilada. c. SoluçAo de vermelho de metiIa 0,1% Dissolver 0,1g de vermelho de metila P.A. em aproximadamente 60ml de álcool etilico. Transferir para balão volumétrico de 100 ml e completar o volume. d. Solução reagente de hidróxido de amônio 1:1 Diluir uma parte de NH40HP.A. em uma parte de água destilada. e. Solução reagente de oxalató de amônio 4,2% Pesar 42g de oxalato de amônia P.A. em bequer, solubilizar e transferir para balão volumétrico de 1000 ml. Adicionar aproximadamente 700ml de água destilada, agitar manualmente ou mecanicamente até completa dissolução e completar o volume. f. Solução reagente de hidróxido de amônio 1:50 Diluir uma parte de NH40H P.A. em 50 partes de água destilada. g. Solução volumétrica padronizada de permanganato de potássio 0,05 N Dissolver 1,6g de KMn04 P..A. em bequer contendo aproximadamente 300/400ml de água destilada e aquecer a 60/70ºC por duas horas. Esfriar, transferir quantitativamente para balão volumétrico de 1000ml e completar o volume. Deixar em repouso por 72 horas ao abrigo da luz. Filtrar sobre cadinho de vidro sinterizado, amianto ou lã de vidro e estocar em frasco ambar. PADRONIZACÃO: Pesar exatamente 3,35g de oxalato de sódio Na2C204 P.A. previamente seco em estufa a 105ºC por duas horas. Dissolver em bequer com aproximadamente 100ml de água destilada, transferir para balão volumétrico de 1000ml e completar o volume. Pipetar 20ml da solução de Na2C204 para erlenmeyer de 250ml, adicionar 10ml de H2S04 1:1. Aquecer a 70/80ºC e titular nessa temperatura gotejando a solução de KMn04 0,05N numa velocidade de 2 a 3 gotas/segundo, até coloração levemente rósea persistente por 30 segundos. CALCULO DO FATOR
P NR N = V x E NE ONDE: E = Equivalente grama do oxalato de sódio

P = Peso do oxalato de sódio em miligrama na alíquota V = Volume de KMn04 gasto na titulação NR = Normalidade real NE = Normalidade esperada MÉTODO Nº 15 - CALCIO (COMPLEXOMÉTRICO) APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: Fundamenta-se na titulação çomplexométrica de sais de cálcio por uma solução de sal sódico do EDTA em presença de indicador. MATERIAL E EQUIPAMENTo - Balão vólumétrico de 100 ou 250ml - Pipeta volumétrica de 10 ml e 25ml - Erlenmeyer de 250 e 500ml - Placa aquecedora - Papel de filtro - Pipeta graduada - Buretas de 25ml - Pérolas de vidro REAGENTES: - Ácido ClorIdrico P.A. - Molibdato de amônia P.Á. - Hidróxido de sódio P. A. - Trietanolamina P.A. - Cianeto de potássio P.A. - Calcelna P.A. - Timolftaleína P.A. - Nitrato de potássio P.A. - EDTA P.A. PROCEOIMENTO: 1. Com o auxílio de bastão de vidro, transferir quantitativamente as cinzas obtidas na determinação de matéria mineral para bequer, lavar o cadinho, com pequenas porções de solução de HCI 1:1, totalizando 40ml e em seguida, com água destilada, cobrir com vidro de relógio e levar à placa aquecedora ate que haja redução de 1/3 do volume. Esfriar. 2. Filtrar sobre papel de filtro qualitativo para balão volumétrico adequado. Completar o volume com água destilada (solução estoque). 3. Pipetar volumetricamente uma alíquota adequada da solução estoque e transferir para bequer de 250 m,i. 4, Adicionar 25ml de solução de molibdato de amônio 5%, 2,5ml de ácido clorídrico e levar à placa aquecedora a 70ºC durante uma hora. 5. Deixar esfriar. 6. Filtrar em papel de filtro, recolhendo o filtrado em erlenmeyer de 500ml. Fazer lavagens sucessivas com água destilada até extração completa. 7: Adicionar 20mI de hidróxido de sódio 4N, água destilada, 5ml de cianeto de potássio 0,1N e 5ml de trietanolamina 20% (realizar esta operação em capela de exaustão).

8. Elevar o volume a 300ml com água destilada e adicionar pequena quantidade de calceina mista, sob agitação. 9. Titular com EDTA 0,02M. O ponto final da titulação a coloração passa de uma fluorescência verde amarelada para uma não fluorescência violácea. CÁLCULO;
V x M x F x 40,08 x 100 Cálcio % = P x 1000 ONDE: V = Volume gasto de EDTA M = Molaridade da solução de EDTA F = Fator de correção dá solução de EDTA 40,08= Peso molecular do cálcio SOLUÇÕES REAGENTES VOLUMÉTRICAS: a. Calceina mista Misturar 0,2g de calceína., 0,12g de timolftaleina e 20g de nitrato de potássio. Manter em frasco ambar. b.Solução reagente de molibdato de amônia 5%. Dissolver 50g de molibdato de amônio em água destilada, completando a seguir para 1000ml em balão volumétrico. c. Solução reagente de hidróxido de sódio 4N Pesar 160g de NaOH P.A., dissolver e completar o volume com água destilada para 1.000ml. d. Solução reagente de cianeto de potás$io 0,1N Dissolver 6,512g de KCN P.A. e completar o volume com água destilada para 1000ml. e. Solução reagente de trietanolamina 20% Medir 200ml de trietanolamina P.A. e completar o volume com água destilada para 1000ml. f. Solução reagente de ácido clorídrico 10% Medir 100ml de ácido clorídrico e transferir para balão volumétrico de 1.000ml, contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. g. Solução volumétrica padrozinada de EDTA 0,02 M Pesar 7,4449g de EDTA dissódico, dissolver, transferir quantitativamente para balão volumétrico de 1000ml e completar o volume. PADRONIZAÇÃO: Pesar, em erlenmeyer de 250ml 0,5055g de carbonato de cálcio P.A. previamente seco em estufa a 105ºC por três horas. Adicionar quantidade de HCI 10% suficiente para dissolver o sal e transferir quantitativamente para balão volumétrico de 250ml com água destilada, completando o volume. Depois de bem homogeneizado, pipetar alíquota de 25ml e proceder de maneira idêntica a determinação de cálcio.
25 ml FC = V

ONDE: V = volume gasto de EDTA MÉTODO Nº 16 - FÓSFORO (COLORIMÉTRICO I) APLICAÇÃO: Produtos ou sub-produtos de origem animal,' vegetal, mineral, rações e concentrados. PRINCÍPIO: Fundamenta-se na reação de Misson. A partir de uma reação em meio ácido do ortofosfato presente com solüção de vanadato mais molibdato de amônio, forma-se um complexo de coloração amarela, que é medido colorimetricamente. MATERIAL E EQUIPAMENTO: - Colorímetro fotoelétrlco - Pipetas volumétricas - Balões volumétricos de 100ml - Pipeta graduada REAGENTES: - Metavanadato de amônio P.A. - Ácido nítrico P.A. - Molibdato de amônio P.A. - Fosfato de potássio monobásico P.A. - Ácido sulfúrico P.A. PROCEDIMENTO: 1. Pipetar volumetricamente para balão de 100ml, alIquota da solução estoque de maneira que a leitura esteja em uma faixa de 15% a 50% transmitância ou 0,824 a 0,222 de absorbância. 2. Adicionar 4ml de H2S04 10N e 20ml do reagente misto. Completar o volume com água destilada, homogeneizar e aguardar no mínimo por 10 minutos antes de fazer a leitura. 3. Paralelamente, preparar um branco contendo os mesmos volumes de H2S04 10N, reagente misto e água destilada. 4. Determinar a absorbâncla ou transmitância em fotocolorImetro a 420nm, zerando anteriormente com o branco. 5. Comparar a absorbância ou transmitância com a curva padrão previamente elaborada, ou usar equação de regressão. CÁLCULO:
L x S x D x 100 Fósforo % = P X A ONDE: L = Leitura do gráfico em mg D = Fator de diluição S = Volume da solução estoque P = Peso da,amostra em ,miligrama A = Volume da alíquota utilizado OBSERVAÇOES: A coloração amarela do complexo formado possui estabilidade limitada, devendo ser determinada de acordo com a qualidade dos reagentes empregados na solução mista.

SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução padrão de fósforo Pesar exatemente O,4394g de KH2P04 (mono) P.A. previamente seco em estufa a 105ºC por duas horas. Solubilizar e transferir quantitativamente para balão volumétrico de 1000ml. Adicionar aproximadamente 500ml de água destilada e 10ml de H2S04 10N. Completar o volume. Cada ml desta solução contém 100 microgramas de fósforo. Fazer diluições de maneira a obter concentrações adequadas a confecção da curva. b. Solução reagente de ácido sulfúrico 10N , Transferir cuidadosamente 266ml de H2S04 P.A. para balão volumétrico de 1000ml, contendo aproximadamente 700ml de água destilada. Esfriar e completar o volume. c. Solução reagente de metavanadato de amônio O,25% Pesar 2,5g de metavanadato de amônio P..A. e solubilizar em 500ml de água destilada fervente. Esfriar e adicionar lentamente com agitação, 350ml de HN03 P.A. Transferir para balão volumétrico de 1000ml, esfriar e completar o volume. Estocar em frasco ambar. d Solução reagente de molibdato de amônio 5% Pesar 50g de molibdato de amônio e solubilizar em 500ml de água destilada quente (60/70ºC). Esfriar e transferir para balão volumétrico de 1000ml, completar o volume. Estocar em frasco ambar. e. Reagente misto Juntar 1 parte da solução de metavanadato com 1 parte da solução de molibdato de amônio e homogeneizar. Preparar momentos antes da utilização. MÉTODO Nº 17 - FÓSFORO (COLORIMÉTRICO II) APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: A ação do molibdenio sobre o ion fósforo, resulta em ácido fosfomólibdico, que a seguir é reduzido pelo ácido 1,2,4-aminonaftolsulfônico a óxido de molibdenio, produzindo uma coloração azul que é medida colorimetricamente. MATERIAL E EQUIPAMENTO: - Colorímetro fotoelétrico - Pipetas volumétricas - Pipetas graduadas, - Balões voiumétricos capacidade 200, 100 e 50ml REAGENTES: - Molibdato de amônio P.A. - Ácido sulfúrico P.A. - Fosfato de potássio monobásico P.A. - Bissulfito de sódio P.A. - Sulfito de s6dio P.A. - Ácido - 1 amino- 2 naftol-4 sulfônico ou sulfato de p-aminofenol P.A. PROCEDIMENTO:

1. Pipetar volumetricamente para balão de 50ml, alíquota da "solução estoque" de maneira que a leitura a ser obtida esteja em uma faixa de 15 a 60% de transmitância ou 0,824 a 0,222 de absorbância. 2. Adicionar cerca de 25ml de água destilada e 5ml de molibdato de amônio. Misturar girando o balão. 3. Adicionar 2ml de ácido 1-2-4 aminonaftolsulfônico ou sulfato de p-aminofenol e marcar o tempo. Completar o volume, tampar e agitar. 4. Paralelamente preparar um branco contendo os mesmos volume de molibdato de amônio, ácido 1,2,4 aminonaftolsulfônico ou sulfato de p.aminofenol e água destilada. 5. Após 20 minutos exatamente, fazer a leitura a 720 nm, zerando anteriormente com o branco. 6. Comparar a absorbância ,ou transmitância com a curva padrão previamente preparada, ou usar a equação de regressão: CALCULO:
L x :S x D x 100 Fósforo % =
P x A ONDE: L= Leitura do gráfico em miligrama D= Fator de diluição S= Volume da solução estoque P= Peso da amostra em miligrama A= Volume da alíquota utilizado SOLUCÕES REAGENTES E VOLUMÉTRICAS: a. Solução padrão de fósforo Pesar exatemente 0,4394 9 de KH2P04 (nono) P.A. previamente seco em estufa a 105ºC por duas horas. Solubilizar e transferir quantitativamente para balão volumétrico de 1000ml. Adicionar aproximadamente 500ml de água destilada e 10ml de H2S04 10 N. Completar o volume. Cada ml desta solução contém 100 microgramas de fósforo. Fazer diluições de maneira a obter concentrações adequadas a confecção da curva. b. Solução reagente de ácido sulfúrico 10N Transferir cuidadosamente 266ml de H2S04 P.A. para balão volumétrico de 1000ml, contendo aproximadamente 700ml de água destilada. Esfriar e completar o volume. c. Solução reagente de molibdato de amônio 2,5% Pesar 12,5g de molibdato de amônio P.A. e solubilizar em 100ml de água destilada. Colocar 150ml de H2S04 10N em balão volumétrico de 500ml. Adicionar a solução de molibdato de amônio sobre a de ácido sulfúrico. Completar o volume com água destilada e agitar vigorosamente. Estocar em frasco ambar sob refrigeração. d. Solução reagente de bissulfito de sódio 15% Pesar 15g de bissulfito de sódio P.A., solubilizar em água destilada, transferir para balão volumétrico de 100ml, e completar o volume. e. Solução reagente sulfito de sódio 20% Pesar 10 g de sulfito de sódio P.A., solubilizar em água destilada, transferir para balão volumétrico de 50ml e completar o volume.

f. Solução reagente de ácido 1-amino 2-naftol 4-sulfOnico ou sulfato de p-aminfenol Pesar 250mg do reagente em bequer. Adicionar uma ou duas gotas de solução de bissulfito de södio 15% para formar uma pasta, Adicionar, transferindo para um balão de 100ml, 97ml de solução de bissulfito de sódio 15% e 2,5ml de solução de sulfito de sádio 20%. Não havendo completa dissolução, adicionar mais solução de sulfito de sódio 20%, evitando-se o excesso. Descansar por uma noite, filtrar através de papel filtro qualitativo filtração rápida e estocar em frasco ambar. MÉTODO Nº 18 - FÓSFORO (VOLUMÉTRICO) APLICAÇÃO: Produtos ou sub-produtos de origem anirl!al, vegetal, mineral, rações e concentrados. PRINCÍPIO: Baseia-se na determinação do ion fósforo, que é precipitado na forma de fosfomolibdato em presença de molibdato de amônio, seguido de dissolução por alcali e titulação do excesso coM solução ácida. MATERIAL E EQUIPAMENTO: - Bequer 300 ou 400ml - Proveta graduada - Funil analítico - Pipetas volumétricas - Placa aquecedora ou banho-maria com controle de temperatura - Pipetas graduadas, - Buretas de 50ml (1/10) - Papel de filtro quantitativo filtração lenta - Bastão de vidro REAGENTES: - Ácido clorídrico P.A. - Âcido nítrico P.A. - Hidróxido de amônio P.A. - Hidroxido de sódio P.A. - Nitrato de potássio P.A. - Nitrato de amônio P.A. - Fenolftaleina P.A. - Molibdato de amônio P.A. PROCEDIMENTO: 1. Pipetar volumetricamente uma aliquota adequada da solução estoque, e transferir para bequer. 2. Utilizando uma tira de papel tornassol, alcalinizar com NH40H 1/1 e adicionar aproximadamente 10g de NH4NO3 P.A., agitando até dissoluçao. 3. Acldular com HNO3 1/1. elevar a 100ml com água destilada e adicionar 2 gotas de HN03 1/1 em excesso. 4. Levar à placa aquecedora ou banho-maria a 40/50ºC. Gotejar com agitação constante, volume adequado de solução de molibdato de amônio até completa precipitação. Manter nessa faixa de temperatura por uma hora com agitaçao ocasional. Esfriar. 5. Filtrar quantitativamente sobre papel de filtro, lavando o bequer 2 vezes com HN03 2% e 4 vezes com KN03 1% com o auxílio de bastao de vidro de ponta de latex. Transferir o papel contendo o precipitado para o bequer original.

6. Adicionar NaOH 0,2N até completa dissoluçao do precipitado e registrar o volume gasto utilizando 3 a 5 gotas de fenolftale!na 1%, titular com HCI 0,2N até viragem do indicadbr (violeta para incolor). CÁLCULO:
(VS x F1 - VA x F2) x N x S x 0,0269) P % = P x A ONDE: VS = Volume de NaOH 0,2 N VA = Volume de HCI 0,2 N gasto na tltulaçao F1 = Fatór de correção do NaOH 0,2 N F2 = Fator de correçao de HCI 0,2 N N = Normalidade S = Volume da soluçao estoque P = Peso original da amostra em grama A = Volume da slíquota utilizado 0,0269 = Fator de conversao de P205 em P OBSERVAÇÃO: Orientaçao para alíquotas de acordo com o percentual de fósforo estimado. FÓSFORO ESPERADO ALÍQUOTA VOL. MOLIB. AMÔMIO 0-2% 40ml 20ml 2-5% 25ml 25ml 5-10% 10ml 30ml 10-20ml 10ml 25ml 20-25% 10ml 50ml SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução reagente de hidr6xido de amônio 1:1 Diluir uma parte de NH40H P.A. em uma parte de água destilada. b. Solução reagente de ácido nítrico 1:1 Diluir uma parte de HNO3 P:A. em uma parte de água destilada. c. Solução reagente de molibdato de amônio Pesar 10g de moibdato de amônio P. A., adicionar 14ml de NH40H P. A:; e dlssolver sob agitação. Adicionar 6ml de HN03 P.A., e em seguida, com constante agitação, 136ml de HN03 42%. Deixar em repouso por 24 horas mantendo em frasco ambar. OBSERVAÇAO: Caso haja formação de um precipitado branco, preparar nova solução. d. Solução reagente de HNO) 42% Transfetir cuidadosamente 105ml deHN03 P.A. para balão volumétrico de 250ml contendo aproximadamente 100ml de água destilada. Esfriar e completar o volume. e. Solução reagente de ácido nítrico 2% Transferir 2ml de HNO3 P.A. para balão volumétrico de 100ml, completando o volume com água destilada. f. Solução reagente de nitrato de potássio 1%

Pesar 10g de KNO3 P.A solubilizar.em água destilada, e transferir para balão volumétrico de 1000ml, completar o volume. g. Solução volumétrica padronizada de NaOH O,2N Dissolver cerca de 8,20g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml e completar o volume. PADRONIzAÇÃO: Pesar com exatidão 1,5g de biftalato de potássio P.A. previamente seco em estufa 105ºC por duas horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcóolica de fenolftaleína 1% e titular gotejando a solução de NaOH O,2N até coloração levemente rósea. CALCULO DO FATOR:
P NR NR = V x E NE ONDE: E = Equivalente grame de biftalàto de potássio, P = Peso do biftaLato de potassio em miligrama V = Volume de NaOH 0,2N consumidq NR = NormalIdade real NE = Normalidade esperada h. Solução indicadora de fenolftaleína 1% Dissolver 1,0g de feholftalelna P.A. em 100ml de álcool P.A. i. Solução volumétrica padronizada de HCl 0,2 N Medir 17,9ml de HCl P.A. e transferir para balão volumétrico de 1000ml contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar com exatidão 0,26g de Na2CO3 P..A. previamente seco, em mufla 270ºC por 1 hora. Dissolver em erlenmeyer, adicionando aproximadamente 20ml de HCl 0,2N medidos em bureta, e 4 gotas de alaranjado de metila 0,2%.Prosseguir a titulação gotejando a solução de HCl 0,2N até leve coloração rósea. Aquecer até ebulição e esfriar. Havendo permanência da coloração rósea, considerar terminada a titulação. Caso contrário, prosseguir até rósea constante. CALCULO DO FATOR:
P NR NR = V x E NE ONDE: E = Equivalente grama de Na2C03 P = Peso de Na2C03 em miligrama V = Volume gasto na titulação em ml j. Solução indlcadora de alaranjado de metila 0,2% Pesar 0,2g,de alaranjado de metila P.A., solubilizar em água destilada e transferir para balão volumétrico de 100ml, Completar o volume.

MÉTODO Nº 19 - FÓSFORO SOLÚVEL EM ÁCIDO CÍTRICO 2% APLICAÇÃO: Fosfatos naturais, industria+izados e misturas que os contenham. PRINCÍPIO: Fundamenta-se na determinação da fração de fósforo solúvel em ácido cítrico 2%. MATERIAL E EQUIPAMENTO: - Colorímetro fotoelétrico - Pipetas volumétricas - Pipetas graduadas - Balões volumétricos ou garrafas Stohlman . - Agitador tipo Wagher ou similar ou agitador magnético - Papel de filtro quantitativo, filtração média. REAGENTES: - Ácido cítrico P.A, ' - Reagentes usados na determinação de fósforo colorimétrico(molibdato vanadato) PROCEDIMENTO: 1..Pesar1,0g de amostra e transferlr para um balão volumétrico de 250ml ou garrafa stohlman. 2.Adicionar 100ml de solução de ácido cítrico 2%. 3. Agitar durante 30 minutos a velocidade de 30-40 RPM (agitador tipo Wagner). Empregando-se outro sistema, a velocldade deve ser branda mas suficiente para provocar revolução completa do material. 4. Completar o volume homógeneizar e filtrar em papel filtro. 5. Transferir para balão volumetrico de 100ml, uma alíquota da amostra conforme o teor esperado. 6. Adicionar 20ml de água destilada, 4ml de H2S04 10N e 20ml.de solução vanadomolíbdica. Agitar, completar e homogeneizar. 7. Paralelamente preparar um branco. 8. Esperar 10 minutos e determinar absorbância ou transmitância a 420nm. Comparar com uma curva padrão previamente preparada ou usar equação de regressão. CÁLCULO: .
L x S x 100 Fósforo solúvel % = P x A ONDE: L = Leitura do gráfico em miligrama S = Volume da solução P = Peso da amostra em rniligiama A = Volume da alíquota utilizado
Ps Solubilidade Cítrica % =_____________x 1.00
Pt ONDE: Ps = Fósforo solúvel Pt = Fósforo total OBSERVAÇOES:

1. A amostra deve apresentar granulometria correspondente a no máximo 0,425mm (ABNT). 2. A alíquota a ser pipetada da solução estoque deverá adequar-se de tal forma que a leitura encontrada no aparelho esteja sempre na faixa entre 15 a 60% de transmitância ou 0,624 a 0,222 de absorbância. 3. A coloração amarela do complexo formado possui estabilidade limitada, devendo ser determinada de acordo com a qualidade dos reagentes empregados na solução mista. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução reagente ácido cítrico 2% Pesar 20g de ácido cítrico P.A.,soblubilizar em água destilada e transferir para balão volumétrico de 1000ml. Completar o volume e homogeneizar. b. Soluções reagentes usadas para determinação de fósforo colorimétrÍco 1 (vanadato - molibdato). MÉTODO Nº 20 - ATIVIDADE UREÁTICA APLICAÇÃO: Grãos de soja, outros feijões e seus sub-produtos. PRINCÍPIO: Baseia-se na variação de pH em- função da amônia que é liberada pela ação enzimática da urease. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Tubos de ensaio 25 x 150mm com tampa - Pipetas volumétricas - Banho termostático sorológlco - Bequer de 10ml - Potenciômetro (sensibilidade 0,01 de pH) REAGENTES: - Fosfato monobásico de potássio P.A. - Fosfato dibásico de potássio P.A. - Uréia P.A; - Ácido fosfórico P.A. - Hidróxido de potássio P.A. PREPARO DA AMOSTRA: Moer a amostra, evitando: aquecimento, de tal forma que pelo menos 60% passe em peneira de 0,425 mm. PROCEDIMENTO: 1. Pesar exatamente duas porções de 0,2g de amostra e transferir quantitativamente para dois tubos de ensaio. 2. Adicionar volumetricamente 10ml de solução tampão de fósforo pH 7,0 ao tubo A. Agitar sem invertê-Io. Tampar, colocar em banho termostático a 30 +/- 0,5ºC anotando o tempo. Esta será a prova em branco. 3. Adicionar volumetricamente 10ml de solução tamponada de uréia pH 7,0 ao tubo B. Proceder conforme o item anterior. 4. Para facilidade operacional, manter entre os mesmos um intervalo de 5 minutos. 5. Retirar os tubos do banho, após transcorridos exatamente 30 minutos. 6. Transferir o líquido sobrenadante para bequer de 10ml e em seguida medir o pH de A e B.

CÁLCULO: Atividade ureática = pH amostra B - pH amostra A OBSERVAÇOES: 1. A não estabilização do potenciômetro pode ser atribuida a obstrução do elétrodo de calomelano por uma película da fração solúvel de proteina de soja. Deve-se proceder uma limpeza apropriada, empregando-se uma solução de pepsina 1%, onde o eletrodo deverá ficar imerso por tempo suficiente. 2. Os fatores: pH das soluções A e B, tempo de reação enzimática e temperatura da prova, devem ser observados rigorosamente pois são críticos na precisão da análise SOLUÇÕES REAGENTES E VOLUMÉTRICAS: a. Solução tampão de fosfato pH 7,0 / 0,05M. Pesar exatamente 3,4022g de fosfato monobásico de Potássio P.A. e 4,3545g de fosfato dibásico de potássio P.A., e dissolver em quantidade suficiente de água recentemente destilada. Elevar a 1000ml em balão volumétrico e homogeneizar. Conferir o pH da mesma e se necessário e ajustar o pH 7,0 com uma solução concentrada de ácido fosfórico ou hidróxido de potássio. b. Solução tamponada de uréia a pH 7,0 Dissolver 1,5g de uréia P.A. em 50ml da solução tampão de fosfato. Conferir o pH e, se necessário, ajustá-Io a 7,0 com uma solução concentrada de ácido fosfórico ou hidróxido de potássio. c. Observações, 1) Eliminar a solução tampão de fosfato após 30 dias do seu preparo. 2) Preparar a solução tamponada de uréia imediatamente antes do uso. 3) Soluções de ácido fosfórico e hidróxido de potássio poderão ser usadas na concentração 0,2N. MÉTODO Nº 21 - ÍNDICE DE ACIDEZ I , APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, rações e concentrados. PRINCÍPIO: Fundamenta-se na neutralização com solução alcalina padrão dos ácidos graxos livres, extraídos por um solvente. MATERIAL E EQUIPAMENTO: - Balança semi-analítica (precisão +/- 0,01g) - Erlenmeyer ou bequer de 250ml - Funil analítico - Papel de filtro Qualitativo - Bureta de 25ml div. 1/10 - Proveta - Erlenmeyer de 300ml - Bastão de vidro REAGENTES: - Álcool, etílico P.A. - Hidróxido de sódio P.A. - Fenolftaleína P.A.

PROCEDIMENTO: 1. Pesar 5g de amostra e transferir para erlenmeyer ou bequer de 250ml. 2. Adicionar 150ml de álcool etílico previamente neutralizado, deixar em repouso por 30 minutos, fazendo agitações ocasionais (5 em 5 minutos). 3. Filtrar o sobrenadante sobre papel de filtro para erlenmeyer de 300ml. 4. Adicionar ao resíduo 100ml de álcool etílico previamente neutralizado e deixar em repouso por 15 minutos com agitações ocasionais. 5. Fllfrar sobre o mesmo papel de filtro, juntando ao filtrado obtido no item 3. 6. Titular com solução de NaOH 0,1N, utilizando 4 a 5 gotas de fenolftaleína 1%, até atingir coloração rósea persistente por 30 segundos. CÁLCULOS: Índice Acidez em meq. V x N x f x 100 NaOH 0,1N/100g de amostra = P Índice Acidez em mg V x N x f x 40 NaOH/g. de amostra = P Índice Acidez em mg V x N x f x 56,1 KOH/g. de amostra = P Acidez em ácido oléico % = V x N x f x 100 x 0,282 P ONDE: V = Volume gasto de NaOH 0,1N na titulação. N = Normalidade da solução de NaOH. f = Fator de correção do NaOH 0,1N. P = Peso da amostra em grama OBSERVAÇOES: 1. Utilizar ácool etílico neutralizado, preparado imediatamento antes do uso. 2. Recomenda-se conduzir prova em branco dos reagentes. 3. Manter o frasco de álcool etílico fechado, para evitar formação de ácido carbônico. SOLUÇÕES REAGENTES E VOLUMÉTRICAS: a. Solução indicadora de fenolftaleína 1% Dissolver 1,0g de fenolftaleína P.A. em 100ml de álcool etílico P.A. b. Solução volumétrica padronizada de hidróxido de sódio 0,1N Dissolver cerca de 4,20g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume e homegeneizar. PADRONIZAÇÃO: Pesar com exatidão, cerca de 0,40 g de biftalato de potássio P.A. previamente seco em estufa a 120ºC por 2 horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcóolica de fenolftaleíria 1% e titular gotejando a solução de NaOH 0,1N até coloração levemente rósea CÁLCULO DO FATOR: NR = P F = NR

V x E NE ONDE: E = Equivalente grama de biftalato de potássio P = Peso do biftalato de potássio em miligrama V = Volume gasto na titulação em ml NR = Normalidade real NE = Normalidade esperada c. Alcool etílico neutralizado Adicionar 4 gotas de fenolftaleína 1% e com agitação constante, algumas gotas de NaOH 0,1 N até atingir coloração levemente rósea. MÉTODO Nº 22 - ÍNDICE DE ACIDEZ II APLICAÇÃO: Sebos, óleos e gorduras de origem animal ou vegetal. PRINCÍPIO: Fundamenta-se na neutralização com solução alcalina padrão dos ácidos graxos livres, extraídos por solvente. MATERIAL E EQUIPAMENTO: - Balança semi-analítica (precisão +/ - 0,01g) - Erlenmeyer de 250ml - Bureta de 25ml (1/10) - Proveta REAGENTES: - Éter etílico P.A. - Álcool etílico P.A. - Hidróxido de Sódio P.A. - Fenolftaleína P.A PROCEDIMENTO: 1. Pesar 5g da amostra em erlenmeyer. 2. Adicionar 100ml da mistura éter-álcool (2/1) previamente neutralizada e agitar até completa.dissolução. 3. Titular com solução de NaOH 0,1N, utilizando 4 a 5 gotas de fenolftaleína 1%, até atingir coloração rósea persistente por 30 segundos. CÁLCULO: Acidez em ácido olélco % = V x N x f x 0,282 x 100
P ONDE: V - Volume gasto de NaOH 0,1N na titulação N = Normalidade f = Fator de correção do NaOH 0,1N P = Peso da amostra em grama OBSERVAÇOES: 1. Amostras de difícil dissolução, levar após pesagem ao Banho-maria e aquecer até dissolver completamente. O excesso de aquecimento pode provocar elevação do valor de ácidos graxos livres. 2. Utilizar mistura éter.álcool neutralizada, preparada imediatamente antes do uso.

3. Recomenda-se conduzir prova em branco dos reagentes. 4. Manter o frasco da mistura éter-álcool fechado para evitar formação de ácido carbônico. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução reagente éter-álcool (2/1) neutralizada Juntar duas partes de éter etilico P.A., com uma parte de álcool etílico P.A. Adicionar 4 gotas de feriolftaleína 1% e com agitação constante, algumas gotas de NaOH 0,1N até atingir coloração levemente rósea. b. Solução indicadora de fenolftalelna II Dissolver 1,0g de fenolftaleína P.A. em 100ml de álcool etílico P.A. c. Solução volumétrica padronizada de hidróxido de sódio 0,1N Dissolver cerca de 4,20g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume e homogeneizar. PADRONIZAÇÃO: Pesar com exatidão, cerca de 0,40 gramas de Biftalato de potássio P.A. previamente seco em estufa a 120ºC por 2 horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcóolica de fenolftaleína 1% e titular gotejando a solução de NaOH 0,1N até coloração levemente rósea. CÁLCULO DO FATOR: NR = P F = NR
V x E NE ONDE: E – Equivalente grama de biftalato de potássio P - Peso do biftalato de potássio em miligrama. V = Volume gasto na titulação em ml NR = Normalidade real NE = Normalidade esperada METODO Nº 23 - ÍNDICE DE ACIDEZ III APLICAÇÃO: Melaço de cana PRINCÍPIO: Fundamenta-se na neutralização com solução alcalina padrão dos ácidos extraídos por solvente. MATERIAL E EQUIPAMENTO: - Balança semi-analítica (precisão +/ - 0,01g) - Erlenmeyer ou bequer de 250ml - Proveta - Bureta de 25ml divisão 1/10 REAGENTES: - Álcool etílito P.A. - Hidróxido de sodio P.A. - Fenolftaleína P.A. PROCEDIMENTO: 1. Pesar 2g da amostra em erlenmeyer ou bequer de 250ml.

2. Adicionar 100 a 150ml mistura água-álcool (1/1) previamente neutralizada e agitatar até completa dissolução. 3.. Titular com solução de NaOH 0,1N utilizando, 4 a 5 gotas de fenolftaleína 1% até atingir a coloração rósea persistente por 30 segundos. CÁLCULO:
Índice de acidez em V x f x N x 0,06 x 100 Acido Acético % = P
ONDE: V -= Volume gasto de NaOH 0,1 N na titulação f = Fator de correção do NaOH 0,1N. N = Normalidade N = Peso da amostra em grama 0,06 = Miliequivalente grama do ácido acético OBSERVAÇOES: 1. Utilizar água-álcool neutralizada preparada imediatamente ante do uso. 2. Recomenda-se conduzir prova em branco dos reagentes. 3. Manter o frasco da mistura água-álcool tampado para evitar formação de ácido carbonico. SOLUÇÕES REAGENTES E VOLUMÉTRICAS: a. Solução reagente água-álcool (1/1) neutralizada Juntar uma parte de água destilada com uma parte de álcool etílico. Adicionar 4gotas de fenolftaleína 1% e com agitação constante algumas gotas de NaOH 0,1N até atingir coloração levemente rósea. b. Solução indicadora de fenolftaleínia 1% Dissolver 1,0g de fenolftaleína P.A. em aproximadamente 60ml de álcool etílico P.A. Transferir para balão volumétrico de 100ml e completar o volume. c. Ttilução volumétrica padronizada de hidróxido de sódio 0,1N Dissolver cerca de 4,20g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume homogeneizar. PADRONIZAÇÃO: Pesar, com exatidão, cerca de 0,40g de Biftalato de potássio P.A. previamente seco em estufa a 120ºC por 2 horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcoolica de fenolftaleína 1% e titular gotejando a soluçao de NaOH 0,1N até coloração levemente rósea CÁLCULO DO FATOR: NR = P F = NR
V x E NE ONDE: E = Equivalente grama de biftalato de potássio P = Peso do biftalato de potássio em miligrama V = Volume de NaOH 0,1N gasto na titulação em ml NR = Normalidade real

NE = Normalidade esperada MÉTODO Nº 24 - CLORETOS SOLÚVEIS I APLICAÇÃO: Produto ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: o nitrato mercúrio em presença de ion cloreto forma o cloreto mercúrico pouco ionizável. O excesso de ions mercúrio II produz com o indicador difenilcarbazona, um complexo de coloração azul vloláceo. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Bureta 25 ml divisão 1/10 - Erlenmeyer de 250ml - Forno mufla - Funil analítico - Papel de filtro qualitativo - Pipeta graduada 10 ml - Cadinho ou cápsula de porcelana REAGENTES: - Ácido nítrico P.A. - Difenilcarbazona P.A. - Nitrato mercúrico P.A. PROCEDIMENTO: 1. Pesar 5 g da amostra em cápsula de porcelana ou cadinho e levar à mufla a 500ºC por 2 horas. Retirar e esfriar até a temperatura ambiente. 2. Transferlr quantitativamente o resíduo para erlenmeyer de 250ml atraves de papel filtro qualitativo lavando várias vezes com água destilada quente. 3. Utilizando solução de ácido nítrico 1:4, ajustar o pH do filtrado entre 2,3 e 2,8 e adicionar 3 a 4 gotas de solução alcóolica de difenilcarbazona 0,5%. 4. Titular com nitrato mercúrico 0,1N até viragem de incolor para azul violáceo. CÁLCULOS: CLORETO % = V x F x N x 0,0:355 x 100
P NaCI % = V x F x: N x 0,0585 x 100
P ONDE: V = Volume de nitrato de mercúrio 0,1N gasto na titulação F = Fator de correção de nitrato de mercúrio 0,1N N = Normalidade do nitrato mercúrico P = Peso amostra em grama OBSERVAÇÕES: 1. O cálcuio do NaCI % considera que todo o cloreto seja proveniente do NaCI. 2. O método é aplicável somente para cloretos solúveis em água. - ,

3. Amostras com_alto teor de cloretos (sais e mist~ras minerais), proceder diluições adequadas. SOLUÇÕES REAGENTES E.VOLUMÉTRICAS: a. Solução reagente de ácido nítrico 1:4 Diluir uma parte de HNO3 P.A. em 4 partes de água destilada. b. Solução indicadora de difenilcarbazona 0,5% Dissolver 0,5g de difenilcarbazona P.A. em aproximadamente 60ml de álcool etílico P.A.. Transferir para balão volumétrico de 100ml e completar o volume. Estocar em refrigerador. c. Solução volumétrica padronizada de nitrato mercúrico 0,1N Pesar exatamente 16,70g de nitrato mercúrico.Dissolver em quantidade sufuciente de HNO3 conc. P.A. Transferir para balão volumétrico de 1000ml e completar o volume com água destilada. Filtrar e guardar em frasco escuro. PADRONIZAÇÃO: Pesar com exatidão, cerca de 0,117g de NaCl P.A. previamente seco em mufla a 300ºC por uma hora. Transferir para erlenmeyer 125ml e dissolver com aproximadamente 20ml de água destilada. Acidular com HN03 1:4 até pH 2,3 - 2,8. Titular, gotejando a solução de Nitrato Mercúrico 0,1N até viragem para cor azul violácea. CÁLCULO DO FATOR: NR = P F = NR
V x E NE ONDE: P = Pesodo cloreto de sodio E = Equivalente grama do cloreto de sódio V = Volume gasto na titulação NR = Normalidade real NE = Normalidade esperada MÉTODO Nº 25 - CLORETOS SOLÚVEIS II APLICAÇÃO: Produtos ou sub-produtos de origem animal, vegetal, mineral, rações e concentrados. PRINCÍPIO: Fundamenta-se na precipitação de cloretos sob a forma de cloreto de prata, que entre pH 7 e 9 na presença de cromato de potássio como indicador, produz um precipitado vermelho tijolo. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Forno mufla - Chapa aquecedora - Erlenmeyer de 250 ou 300ml - Papel de filtro quafltativo - Balão volumétrico de 100ml - Pipeta vo.lumétrica de 50ml - Bureta de 25ml divisão 1/10 - Cadinho ou cápsula de porcelana

REAGENTES: - Nitrato de prata P.A. - Cromato de potássio P.A. - Ácido nítrico P.A. - Carbonato de cálcio P.A. - Cloresto de sódio P.A. PROCEDIMENTO. 1. Pesar 5g da amostra em cápsula de porcelana ou cadinho e levar à mufla a 500ºC por 2 horas. Retirar e esfriar a temperatura ambiente. 2. Solubilizar o resíduo com HN03 2% quente. Transferir quantitatlvamente para balão volumétrlco a de 100ml através de papel de filtro qualitativo, lavando 2 a 3 vezes com HN03 2% quente. Completar o volume com água destilada e homogeneIzar. 3. Retirar alíquota de 50ml para erlenmeyr de 250 ou 300ml e neutralIzar com carbonato de cálcio P.A. utilizando papel, tornassol como indicador. AdIcionar um excesso de +/- 0,5g de carbonato de cálcio. 4. Aquecer em chapa aquecedora até ebulição, mantendo por 2 ou 3 minutos até desprendimento de todo C02 formado. Esfriar. 5. Adjcionar 2 a 3 gotas de cromato de potássio 5% e titular com ni'trato de prata 0,05 N até vlragem para cor vermelho tijolo clara. CALCULOS: CLORETOS % = V x N x Fx S x 0,0355 x 100
P x A NaCI % = V x N x F x S x 0,0585 x 100
P x A ONDE: V = Volume de nitrato de prata 0,05 N gasto na titulação N = Normalidade F = Fator de correção do Nitrato de prata 0,05 N S=,Volume da solução estoque P = Peso original da amostra em grama A = Volume da alíquota utilizada OBSERVAÇOES: 1. Para produtos de origem mineral, adotar o seguinte procedimento: a. Pesar 5g da alllostra, pre,vIamente triturada e homogeneizada em gral. Transferir para erlenmeyer de 500ml adicionar 100 a 150 ml água destilada e levar a fervura por 5 min. Esfriar. b. Filtrar sobre papel de filtro em balão volumétrico de 500ml, lavando o erlenmeyer e o papel com sucessivas porções de água destilada. Completar o volume e homogeneizar. c. Retirar alíquota conforme tabela, para erlenmeyer de 250 ou 300ml e continuar a partir do ítem 4 do procedimento: NÍVEL SAL ESPERADO % ALIQUOTA (ml)
0 a 2% 100 5% 50 15% 20 30% 10 50% 10

70% 7 98% 5
2. O cálculo do NaCI% considera que todo o cloreto seja proveniente do NaCl. 3. Este método é aplicável somente para clotetos solúveis em água. 4. Amostras com alto teor de cloretos (sais e misturas minerais) proceder diluições adequadas. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução reagente de ácido nítrico 2% Transferir 2ml de HN03 P.A. para balão volumétrico de 100ml, contendo aproximadamente 50ml de água destilada. Esfriar e completar o volume. b. Solução reagente de cromato de p~tássio 5% Pesar exatamente 5g deK2CrO4 P.A. em bequer e adicionar aproximadamente 50ml de água destilada. Dissolver e transferir quantitativamente para balão volumétrico de 100ml, completando o volume com água destilada. Armazenar em frasco escuro ao abrigo de luz, no máximo por 30 dias. c. Solução vo!umétrica padronizada de nitrato de prata 0,05N Pesar exatamente 4,2469g de AgN03 e transferir para balão volumétrico 500ml ambar ou envolto com folha alumínio. Dissolver e completar o volume com água destilada, e agitar. PADRONIZAÇAo: Pesar com exatidão, cerca de 0,07g de NaCl P.A. previamente seco em mufla a 300ºC por .1 hora. Transferir para erlenmeyer de 125ml e adicionar cerca de 30 a 40ml de água destilada. Dissolver, acrescentar 1ml de K2CrO4 5% e titular com AgN03 0,05N até aparecimento da cor vermelho tijolo clara. CÁLCULO DO FATOR: F = ___________P___________
0,00585 x N x V ONDE: F = Fator de correção do nitrato de prata 0,05N P = Peso em grama do cloreto de sódio N = Normalidade V = Volume de AgNO3 gasto na titulação MÉTODO Nº 26 - CROMO (QUALITATIVO) APLICAÇÃO: Produtos ou sub-produtos suspeitos da edição de aparas de couro tratadas com cromo. PRINÍPIO: A difenilcarbazida, na presença de cromatos em meio ácido, produz um composto solúvel de cor violeta. MATERIAL E EQUIPAMENTO: - Cadinho ou cápsula de porcelana - Placas de tock - Balança técnica (precisão + / - 0,1g). - Chapa aquecedora - Conta gotas

- Forno mufla - Pipeta graduada REAGENTES: - Ácido nítrico P.A. - Nitrato de prata P.A. - Persulfato de potássio P.A. - Difenilcarbazida P.A. - Álcool etílico P.A. PROCEDIMENTO: 1. Pesar cerca de 2g P.A. amostra em cadinho ou cápsula de porcelana. 2. Incinerar a 600ºC por 20 minutos. Retirar a 250/300ºC e resfriar. 3. Adicionar 5ml de HN03 P.A. e aquecer até iniciar ebulição. Esfriar. 4. Aplicar na placa de tock:
1 gota da qmostra digerida 1 gota de persulfato de potássio saturado 1 gota de nitrato de prata 1 N '
Esperar 3 minutos e aplicar 1 gota de solução dedifebilcarbazida 1% 5. O aparecimento de coloração violeta no momento da aplicação da solução de difenilcarbazida, indica presença de cromo na amostra. OBSERVAÇOES: 1. A presença de cromo na amostra pode ser um indicativo de adição de raspas de couro. 2: Testar os reagentes com uma solução 5 ppm de cromo (dicromato de potássio, óxido crômico, etc.) antes da análise. 3. A estocagem das soluções de persulfato de potássio e difenilcarbazida por longo perIodo, podem comprometer a sensibilidade do método. SOLUÇÕES REAGENTES: a. Solução reagente de persulfato de potássio Pesar 3g de K2S208 em bequer e dissolver em 100ml de água destilada. Estocar em geladeira. b. Solução reagente de nitrato de prata 1 N Pesar 1,7g de AgNO3. Transferir para balão volumétrico.de 10ml ambar ou, envolto com folha de alumínio. Completar o volume com água destilada. c. Solução reagente de difenilcarbazida 1% Pesar 0,1g de difenilcarbazida, transferir para balão Volumétrico de 10ml ambar ou envolto em folha de alumínio. Completar o volume com álcool etílico P.A. Estocar em geladeira. MÉTODO Nº 27 - FLUOR (VOLUMÉTRICO) APLICAÇÃO: Fosfatos naturais, industrializados e misturas que os contenham. PRINCÍPIO: Baseia-se na separação por destilação do ion fluoreto de outros constituintes, como ácido fluor silícico, de uma solução ácida com ponto de ebulição mais elevado que o da água e posterior titulação. MATERIAL E EQUIPAMENTO: - Frasco de destilação com 3 bocas ou Claysen ou Barret, capacidade 500 ou 1000ml

- Condensador de Allihn ou Liebig - Termômetro - escála até 150ºC - Juntas e conexões - Duas fontes de aquecimento - Frasco gerador de vapor capac. 1000 ou 2000ml - Balança analítica - precisão +/- 0,0001g - Pérolas de vidro - Agitador magnético e barra - Funil de separação - capac. 50 ou 100ml - Balões volumétricos 100, 250 e 500ml - Pipetas volumétricas 50ml - Micro bureta capac. 2 ou 5ml divisão 1/100 - Cadinho de níquel REAGENTES: - Fluoreto de sódio P.A. - Alizarina sulfonato de sódio P.A. - Hidróxido de sódio P.A. - Permanganato de potário P.A. - Ácido perclórico ou sulfúrico P.A. - Nitrato de tório P.A. - Ácido clorídrico P.A. - Ácido monocloroacético P.A. - Fenoftaleína P.A. PROCEDIMENTO: 1. Triturar e homogeneizar uma porção da amostra em gral. Pesar de 0,5 a 5,0g dependendo do teor de flúor esperado e transferir quantitativamente para frasco de destilação. ATENÇÃO: Produtos contendo matéria orgânica deverão ser previamente submetidos a fusão, antes da transferência para o frasco de destilação. Quando se trabalha com ácido perclórico, a presença de matéria orgânica pode provocar explosão violenta. (vide observações). 2. Adicionar certa de 30ml de água destilada, 5 gotas de solução saturada de permanganato de potássio e 6 a 8 pérolas de vidro ou lã de vidro. 3. Fechar o frasco de destilação com rolhas de borracha, através das quais passam: a. Um tubo de vidro concectado ao frasco gerador de vapor, cuja extremidade deve estar mergulhada no líquido. b. Um funil de separação e um termômetro (mergulhado no líquido). c. Um tubo de vidro conectado ao condensador. 4. Aquecer a ebulição o frasco gerador de vapor (usar água destilada), deixando a saída de segurança aberta e a saída de vapor para o frasco de destilação fechadas; 5. Colocar 35ml de ácido perclórico ou sulfúrico no funil de separação. Abrir cuidadosamente a torneira e deixar o ácido escorrer lenta e totalmente fechando-a em seguida. 6. Iniciar o aquecimento do frasco de destilação. Quando a temperatura atIngir 135ºC, abrIr a saída do gerador de vapor para o frasco, fechando a saída de segurança em seguida. 7. Regular o aquecimento dos frascos, de maneira a manter constante o volume da solução e a temperatura entre 132 e 135ºC. 8. Prosseguir com a destilação (mínimo de 30 minutos), até que cerca de 200ml do destilado tenha sido recolhido em balão volumétrico de 250ml ou 500ml, contendo 30ml de NaOH 1N com algumas gotas de fenolftaleína.

9. Abrir a saída de segurança do gerador de vapor e desligar as fontes de aquecimento. 10. Completar o volume do balão volumétrico com água destilada e homogeneizar. Pipetar volumétricamente, uma aliquota.de 100ml para erlenmeyer de 250ml e adicionar de 8 a 10 gotas de solução indicadora de alizarina sulfonato de sódio 0,05%. 11. Adicionar HCl 0,5% até pH 3 - 3,5 ou coloração amarela. Adicionar 1ml de solução tampão. 12. Titular lentamente com solução de nitrato de tório 0,05N até coloração levemente rósea (mesma coloração considerada na fatoração do nitrato de tório). CÁLCULO: Fluor % = V x N x F x 0,019 x 100 x 5
P x A ONDE: V = Volume de nitrato de tório gasto na titulação N = Normalidade F = Fator de correção do nitrato de tório P = Peso inicial da amostra em grama S = Volume da solução estoque A = Volume da alíquota utilizado. 0,019 = Miliequivalente grama de Fluor 0BSERVAÇOES: 1. O destilado deverá ser obtido a temperatura ambiente. Caso contrário, desconsiderar a prova e reavaliar o sistema de condensação. 2. Proceder prova de recuperação em fluoretos de cálcio e de sódio, utilizando água destilada deionizada, 3. Em substituição ao ácido perclórico, poderá ser utilizado ácido sulfúrico P.A. (mínimo 30ml) e máximo de 60ml para 5g de amostra) 4. Quando for necessário a fusão da amostra, usar cadinho de níquel e aproximadamente 2,5g do fundente (hidróxido de sódio, carbonato de cálcio ou óxido de cálcio). A fusão deve ser efetuada em capela, usando bico de Bunsen, até formação de uma pasta homogênea. Tomar os cuidados necessários, para evitar a projeção da amostra, esfriar à temperatura ambiente e transferir a amostra fundida para balão de destilação. 5. A fusão da amostra da ração poderá ser substituída por calcinação a 600ºC, em mistura com oxido de cálcio. 6. Na destilação com ácido perclórico, a temperatura não deve ultrapassar 135ºC. 7. Durante a titulação é recomendável a permanência de um eletrodo na solução, objetivando assegurar a não variação do pH, que é crítico nessa prova.
FIG. I - FLUOR - MÉTODO VOlUMÉTRICO
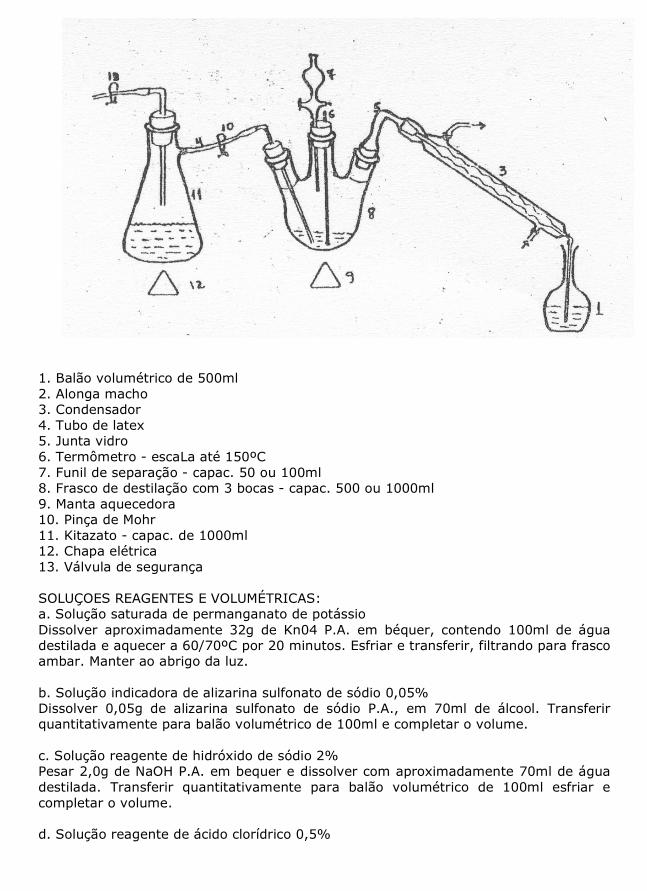
1. Balão volumétrico de 500ml 2. Alonga macho 3. Condensador 4. Tubo de latex 5. Junta vidro 6. Termômetro - escaLa até 150ºC 7. Funil de separação - capac. 50 ou 100ml 8. Frasco de destilação com 3 bocas - capac. 500 ou 1000ml 9. Manta aquecedora 10. Pinça de Mohr 11. Kitazato - capac. de 1000ml 12. Chapa elétrica 13. Válvula de segurança SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução saturada de permanganato de potássio Dissolver aproximadamente 32g de Kn04 P.A. em béquer, contendo 100ml de água destilada e aquecer a 60/70ºC por 20 minutos. Esfriar e transferir, filtrando para frasco ambar. Manter ao abrigo da luz. b. Solução indicadora de alizarina sulfonato de sódio 0,05% Dissolver 0,05g de alizarina sulfonato de sódio P.A., em 70ml de álcool. Transferir quantitativamente para balão volumétrico de 100ml e completar o volume. c. Solução reagente de hidróxido de sódio 2% Pesar 2,0g de NaOH P.A. em bequer e dissolver com aproximadamente 70ml de água destilada. Transferir quantitativamente para balão volumétrico de 100ml esfriar e completar o volume. d. Solução reagente de ácido clorídrico 0,5%

Transferir 5ml de HCI P.A. para balão volumétrico de 1000ml contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. e. Solução tampão de ácido monocloroacético pH 3,0 Pesar exatamente 9,4480g de ácído monocloroacético P.A.em bequer e dissolver com aproximadamente 70ml de NaOH 0,5N. Transferir quantitativamente para balão volumétrico de 100ml, lavando o bequer e completando o volume do balão com NaOH 0,5N. f. Solução volumétrica de hidróxido de sódio 0,5N Dissolver exatamente 2,05g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 100ml, completar o volume e homogeneizar. g. Solução volumétrica padronizada de nitrato de tório 0,05N Pesar exatamente 7,1266g de Th(N03)4. 5H2O em bequer e dissolver com água destilada. Transferir quantitativamente para balão volumétrico de 1000ml, completar o volume e homogeneizar. Cada ml desta solução reage com 0,95mg de flúor. PADRONIZAÇÃO: Dissolver exatamente 0,525g de NaF.P.A. previamente seco em estufa a 105ºC durante 2 horas, em água destilada e transferir quantitativamente para balão volumétrico de 250ml. Completar o volume e homogeneizar. Pipetar volumétricamente uma alíquota de 10ml para erlenmeyer de 250ml, adicionar 50ml de água destilada e 3 gotas de solução indicadora de alizarina sulfonato de sódio 0,05%. Continuar como descrito nos itens 11 e 12 do procedimento. Concentração dessa solução = 0,95mg de flúor/ml ou 2,1mg de NaF/ml. CÁLCULO DO FATOR:
P NR N = V x E F = NE ONDE: E = Equivalente grama de fluoreto de sódio P = Peso de fluoreto de sódio em miligrama na alíquota V = Volume de Th (N03)4 gasto na titulação emml NR = Normalidade real NE = Normalidade esperada MÉTODO Nº 28 - FLUOR (POTENCIOMÉTRICO I) APLICAÇÃO: Fosfatos naturais, industrializados e misturas que os contenham . PRINCÍPIO: A atividade iônica do fluoreto em solução é medida através de eletrodo ion específico cuja resposta Corresponde a variação do potencial elétrico proporcional a concentração. MATERIAL E EQUIPAMENTO: - Potenciômetro com escala em milivolts - Agitador magnético - Eletrodo ion específico para Flúor - Eletrodo de referência - Balança analítica precisão +/- 0,0001g - Cadinho de platina ou níquel

- Bico de Bunsen - Bequer de 150 e 600ml - Balões volumétricos de 100, 250, 500 e 1000ml - Funis analíticos - Pipetas volumétricas - Placa aquecedora REAGENTES: - Fluoreto de sódio P.A. - Cloreto de sódio P.A. - Ácido acético P.A. - CDTA (Ácido 1,2 - ciclohexylenediamino tetra acético) P.A. - Hidróxido de sódio P.A. - Verde de bromocresol P:A. - Álcool etílico P.A. PROCEDIMENTO: 1. Triturar e homogeneizar uma porção da amostra em gral. Pesar em Cadinho de platina ou níquel de 0,25 a 0,5g da amostra dependendo do teor de fluor esperado. 2. Adicionar aproximadamente 215g de NaOH. Fundir a amostra (brandamente no início) em capela usando bico de Bunsen, até formação de uma pasta homogênea. Tomar os cuidados necessários para evitar a projeção da amostra. Esfriar a temperatura ambiente. 3. Transferir o cadinho (com a amostra fundida para bequer de 600ml, adicionar água destilada e deionizada de modo a cobri-lo e, aproximadamente 20ml de HCl 1:1. Deixar em repouso até completa solubilização do resíduo. 4. Transferir quantitativamente para balão de 250 ou 500ml, sem filtrar. Acertar o pH no próprio balão, com NaOH 6N e/ou HCI 10%, utili'zandor verde de bromocresol como indicador, de maneira que o pH esteja entre 5,3 e 5,4. Completar com água destilada e deionizada. 5. Pipetar volumetricamente, alíquota de 50ml para balão volumétrico de 100ml. Adicionar 10ml de solução tisab IV. Completar o volume com água destilada e deionizada: O pH desta solução (sol de leitura) deve estar entre 5,3 e 5,4. 6. Transferir a solução para bequer de 150ml e proceder a determinação potenciométrica em escala mV., com agitação constante e, após estabilização (mínima 3 minutos), fazer a leitura. CÁLCULO:
Ppm x 5 Fluor % = P x As X 100 ONDE: ppm = ppm de fluor obtido na curva de mV x conc. S = volume da solução estoque (250 ou 500ml) P = peso original da amostra As = volume da aliquota utilizado da solução estoque OBSERVAÇOES: 1. As condições de leitura protenciométrica da amostra (tempo de estabilização, temperatura da solução de leitura; agitação constante) deverão ser as mesmas aplicadas na preparação da curva padrão. 2. PREPARAÇÃO DA CURVA PADRÃO:

a. Em balão volumétrico de 100ml; adicionar 10ml de solução tisab IV e completar o volume com água destilada e deionizada. b. Transferir para bequer de 100ml e mergulhar os eletrodos de ion específico e de referência. Manter velocidade de agitação constante (utilizar a mesma velocidade para leitura da amostra), c. Utilizando micro-pipeta de 100 microlitros, acrescentar sucessivamente os volumes indicados na tabela abaixo, anotando os potenciais desenvolvidos para cada volume adicionado. Vol. sol. padrão (1000 ppm) Concentração após adição 100 microlitros 1 ppm 100 microlitros 2 ppm 100 microlltros 3 ppm 100 microlitros 4 ppm 100 microlitros 5 ppm 100 microlitros 6 ppm 100 microlitros 7 ppm 100 microlitros 8 ppm 100 microlitros 9 ppm 100 mlcrolitros 10 ppm d. Traçar a curva, anotando os potenciais obtidos em ordenada e a concentação em abcissa (papel semi-logarítmico), ou então, uma equação de regressão, na qual o coeficiente de correlação deverá ser próximo a 1,0000. 3. Proceder prova de recuperação de fluoretos de cálcio ou sódio, utilizando água destilada é deionizada. SOLUCOES REAGENTES E VOLUMÉTRICAS: a. Solução reagente de hidróxido de sódio.6N
Pesar exatamente 24,6g de NaOH P.A. em bequer. Dissolver com água destilada e deionizada e transferir quantitativamente para balão volumétrico de 100ml. Esfriar, completar o volume e homogeneizar. b. Solução resgente de ácIdo clorídrico 1:1 Diluir uma parte de HCl em uma parte de água destilada. c. Solução reagente de ácido clorídrico 10% . Transferir 8,4ml de HCl P.A. (d = 1,19 g/ml) para balão volumétrico de 100ml contendo aproximadamente 70 ml de água destilada. Esfriar e completar o volume. d. Solução indicadora de verde de bromocresol 0,1% Dissolver 0,1g de verde de bromocresol P.A. em álcool etílico P.A. Transferir quantitativamente para balão volumétrico de 100ml e completar o volume (pH = 3,8 - cor amarela; pH = 5,4; - cor azul). e. Solução tampão TISAB IV pH 5,3 - 5,4 Em bequer de 500ml dissolver em água destilada e deionizada 145g de NaCl P.A., 142,5ml de ácido acético P.A., 10g de CDTA. Ajustar o pH em 5,3 - 5,4 com NaOH 6N, transferir quantitativamente para balão volumétrico de 1000ml e completar o volume com água destilada e deionizada. O pH final deverá ser 5,3 a.5,4.

OBSERVAÇÃO: No preparo da solução tampão, a temperatura deve ser conrolada, pois o CDTA decompõe-se em temperaturas superiores a 55ºC. f. Solução padrão de Flúor 1.000 ppm. Dissolver em água destilada deionizada exatamente 2,2105g de NaF P.A. previamente seco em estufa 105ºC, durante 2 horas e transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume e homogeneizar. MÉTODO Nº 29 - FLÚOR (POTENCIOMÉTRICO II) APLICAÇÃO: Fosfatos naturais, industrializados e misturas que os contenham. PRINCÍPIO: A atividade iônica do fluoreto em solução é medida através de eletrodo ion especifico cuja resposta corresponde a variação do potencial elétrico proporcional a concentração. MATERIAL E EQUIPAMENTO: - Potenciômetro com escala em milivolts - Eletrodo ion especifico para flúor - Eletrodo de referência - Agitador magnético. - Balança analítica precisão +/- 0,0001g - Bequer de 50 e 30ml. - Balões de .1000, 500 e 250ml - Funis analiticos - Pipetas volumétricas de 20, 25 e 50ml - PIaca aquecedora REAGENTES: - Fluoreto de sódio P.A. - Hidróxido de amônio P.A. - Ácido cítrico P.A. - Tampão citrato neutro de amônio pH 7,0 - Azul de bromotimol P.A. - Ácido clorídrico P.A. PROCEDIMENTO: 1. Pesar 0,5000g de amostra, transferir para bequer de 300ml. 2. Adicionar 5ml de HCI concentrado, aquecer brandamente e adicionar água destilada até 100ml. 3. Adicionar solução de hidróxido de amônio 1:1 para precipitação dos metais (até a turvação da solução). 4. Adicionar algumas gotas de azul de bromotimol e ácido cítrico 7% para complexação dos metais, até a viragem de azul para amarelo. 5. Transferir quantitativamente para balão 250ml sem filtrar. Avolumar e agitar. 6. Pipetar volumétricamente uma alíquota de 20ml para bequer de 50ml e adicionar 20ml de citrato neutro de amônio pH 7,0. 7. Proceder a determinação potenciométrica em milivolts com agitação constante, e após estabilização (mínimo 3 minutos), fazer a leitura. CÁLCULO: As leituras são realizadas em milivolts e transformadas em ppm. Através de um gráfico ou de uma equação de regressão. Flúor % = L x 100 x S x 10-6

P ONDE: P = Peso amostra em grama S = Volume da solução estoque L = Leitura de flúor obtida na curva em ppm PREPARO DA CURVA DE CALIBRAÇÃO: 1. A partir de uma solução de 1000ppm de flúor, preparar padrões de 1; 10 e 100ppm. 2. Em bequeres de 50ml identificados como: 1ppm, 10ppm e 100ppm, adicionar 20ml de citrato neutro de amônio e 20ml dos respectivos padrões. 3. Leituras - Acertar o padrão de 10ppm em zero relação milivolts. Deixar estabilizar, em seguida ler os padrões de 1 e 100ppm, com agitação constante. 4. Plotar as leituras em gráfico (papel monolog) ou construir uma equação de regressão (logarítmo da concentração x milivolts), na qual o coeficiente de correlação deverá ser próximo de 1,0000. OBSERVAÇOES: 1. As condições de leitura potenciométrica da amostra (tempo de estabilização, temperatura da solução, agitação) deverão ser as mesmas aplicadas na preparação da curva padrão. 2. No cálculo da concentração de fluoreto contido na amostra, o volume da alíquota utilizada não é considerado, como também não é considerado na realização da curva. O gráfico fornece diretamente, o teor de flúor no balão de diluição. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução padrão de flúor 1000ppm Secar o NaF a 105ºC por duas horas. Pesar exatamente 2,2105g e transferir para balão volumétrico de 1000ml. Completar o volume com água destilada deionizada. b. Solução padrão de flúor 100 ppm Pipetar 50ml da solução padrão 1000ppm, transferir para o balão volumétrico 500ml. Completar o volume com água destilada deionizada. c. Solução padrão de flúor 10 ppm Pipetar 50ml da solução de 100ppm, transferir para o balão 500ml. Completar o volume com água destilada deionizada. d. Solução padrão de flúor 1ppm Pipetar 50ml da solução, de 10ppm, transferir para balão volumétrico de 500ml. Completar o volume com água destilada deionizada. e. Acldo cítrico 7% Pesar 7,0g de ácido cítrico P.A., solubilizar em água destilada. Transferir para o balão 100ml. Completar o volume com água destilada deionizada. f. Hidróxido de amônia 1:1 Adicionar 50ml de NH4OH em 50ml de água destilada e homogeneizar. g. Citrato neutro de amônia - pH 7,0 Dissolver 370g de ácido cítrico em 1,5 I de água destilada. Neutralizar com 345ml de hidróxido de amônio. Esfriar e acertar o pH em 7,0 com hidróxido de amônio e/ou ácido cítrico. Guardar em frasco fechado. Conferir o pH periodicamente. Aceita-se uma variação de 6,97 a 7,03.

h. Indicador azul de bromotimol 0,1% Dissolver 0,1g de azul bromotimol em 20ml de álcool, transferir para balão de 100ml e completar o volume com água destilada. MÉTODO Nº 30-AFLATOXINAS I - CROMATOGRAFIA EM CAMADA DELGADA APLICAÇÃO: Produtos ou sub-produtos de origem vegetal, rações e concentrados. PRINCÍPIO: O método baseia-se na separação cromatográfica e a detecção é feita pela fluorescência das aflatoxinas quando expostas à luz ultravioleta (366nm). Obtém-se o resultado pela comparação com padrões de concentração conhecida igualmente aplicados na placa cromatográfica; MATERIAL E EQUIPAMENTO: - Cromatoplacas silicagel 60 para TLC-0,2mm espessura, tamanho 20 x 20cm sem indicador de flúorescência - Microseringa graduada de 25 microlitros - Balões volumétricos de 25, 50 e 100ml - Agitador magnético - Funil de Buchner de 12cm de diâmetro - Funil de separação de 125ml - Pipetas volumétricas - Erlenmeyer - Papel de filtro - Cromato visor munido de lâmpada com emissão a 3650 A e 15 watts - Cadinho de vidro com placa filtrante - Tubo cromatográfico (22 x 300mm). - Silicagel,60 (0,063 - 0,2mm) ativada para cromatografia em coluna - LS de vidro - Câmara cromatográfica com tampa (22 x 23 x 10cm) REAGENTES: - Éter etílico anidro P.A. - Clorofórmio P.A. - Acetona P.A. - Alcool metílico P.A. - Hexano P.A. - Sulfato de sódio anidro P.A. - Carbonato básico de cobre II P.A. - Padrões de aflatoxinas B1 - B2 - G1 - G2 PROCEDIMENTO: a. PREPARAÇÃO DAS SOLUÇOES PADRÕES: Diluir os padrões de aflatoxinas B1 - B2 - G1 - G2 na forma cristalina, com solução de benzeno + acetonitrila (98 + 2) de modo a se obter concentrações de 8 a 10ug/ml.Manter sob refrigeração constante a 0ºC e em frasco ambar herméticamente fechado. Diluir novamente obtendo-se soluções padrões de trabalho com concentração 0,1 a 0,5ug/ml e manter sob as mesmas condições de estocagem. Observar se não está havendo perda de solvente por evaporação, pois neste caso ocorrerá alteração na concentração da solução padrão. b. EXTRAÇÃO:

Pesar 50g de amostra moída em erlenmeyer de 500ml. Umedecer completamente a amostra e adicionar 250ml de cloroformio. Tampar a boca do frasco com papel alumínio e deixar sob agitação magnética por 30 minutos. Filtrar em papel de filtro. Se a filtração for muito lenta, usar vácuo fraco. c. PURIFICAÇÃO DO EXTRATO: Preparar tubo cromatográfico (22 x 300mm) colocando bolas de lã de vidro no fundo, seguindo-se 5g de Na2SO4 anidro (base para a silica gel). A seguir juntar 10g de silicagel 60 (0,063 - 0,2mm) ativada por secagem durante uma hora em estufa. Adicionar 5g de sulfato de sódio anidro, e lavar a coluna com 30ml de clorofórmio. Transferir 50ml de extrato da amostra para a coluna e diluir em fluxo máximo com 100ml de hexano seguido por 100ml de éter etílico P.A. e descartar. Diluir aflatoxinas com 150ml de metanol + clorofórmio (5 + 145) coletando totalmente esta fração desde o seu início até o final. Adicionar pérolas de vidro e evaporar até próximo de 5ml em banho-maria. Acrescentar 4g de sulfato de sódio anidro e 2g de carbonato básico de cobre II. Agitar vigorosamente e filtrar através de cadinho de vidro com placa filtrante para bequer de 50ml e evaporar até a secura. Diluir o extrato purificado com cloroformio a volume: apropriado e aplicar em CCD. d. CROMATOGRAFIA EM CAMADA DELGADA: Aplicar a 2cm da base da cromatoplaca com auxílio de uma microseringa, alíquotas de extrato purificado e do padrão de aflatoxinas. A aplicação deve ser feita de modo que o diâmetro do spot não seja superior a 5mm. Desenvolver em câmara cromatográfica contendo 200ml da mistura cloroformio + acetona + água (72 + 25 + 3) recem preparada, até 10cm acima do local das alíquotas. Para auxiliar a saturação da câmara, colocar papel de filtro no seu interior encostado nas paredes. Remover a placa da câmara, evaporar o solvente a temperatura ambiente e examinar sob luz ultravioleta de comprimento de onda longo em cabine Cromato visor. Observar a fluorescência no mesmo patamar em relação aos padrões das aflatoxinas. Em ordem decrescente de Rf teremos B1 - B2 - G1 - G2. e. INTERPRETAÇÃO DO CROMATOGRAMA: Comparar as alturas de Rf entre a amostra e o padrão spots fluorescentes na amostra atribuidos a aflatoxinas deverão ter valor idêntico de Rf e ser similar na coloração. NOTA: Desde que spots da amostra e padrão são comparados diretamente na mesma placa, o valor do Rf não é importante, pois este pode variar de placa para placa. Para quantificar a toxina deverão ser considerados os spots do padrão daamostra contendo a mesma intensidade de fluorescência. CÁLCULO:
S. Y. V ug/kg = X. W ONDE: S = ul aplicado do padrão Y = Concentração do padrão em ug/ml V = ul final do extrato da amostra X = Volume aplicado da amostra em ul W = Peso da amostra no extrato em gramas

CONFIRMAÇAO QUÍMICA DA IDENTIDADE DAS AFLATOXINAS: Desde que a detecção de aflatoxinas por CCD não é evidência conclusiva da sua identidade, uma confirmação mais eficiente deve ser feita. Desta forma, testes confirmativos envolvendo a formação de derivados químicos tem sido desenvolvidos. Estes métodos confirmativos envolvem o isolamento da aflatoxina por coluna e CCD, são morosos e difíceis para alguns analistas. Um método simples e rápido para confirmação da aflatoxina B1 foi desenvolvido por Przbylski no qual a aflatoxina B1 extraída da amostra é convertida em aflatoxina B2a diretamente na placa de CCD. O teste é baseado na ação catalítica do ácido trifluoracético com a adição de água através da dupla ligação do anel terminal de furano da aflatoxina B1. Aflatoxina G1 é convertida da mesma maneira em G2a; B2 e G2 não são afetadas. Essa reação tem sido utilizada como base em vários artigos publicados como método de confirmação de aflatoxina B1 e G1. Um microlitro de ácido trifluoracético é aplicado diretamente sobre o spot contendo o extrato de aflatoxina assim como no spot padrão na linha de origem da placa CCD. Após reação, o ácido trifluoracético catalítico é evaporado por corrente de vapor; a placa é desenvolvida com acetona-clorofórmio (15 + 85). O derivado B2a aparece como uma fluorescencia azul a um Rf cerca 1/4 do valor normal ao da aflatoxina B1. Em adição com a condição de evidenciar a confirmação, o teste poderia revelar a presença de spots estranhos no Rf da aflatoxina B1 e G1, os quais contribuiriam na fluorescência total dos spots, resultando em superestimação do conteúdo de aflatoxina na amostra. A técnica de Przybylki também inclui um teste no qual a aflatoxina, após desenvolvimento CCD, e feito spray com H2S04 25%, e por esse meio mudança na fluorescencia de azul para amarelo. Este teste oferece confirmação adicional das aflatoxinas, e é especialmente muito usado quando presentes a pequenos níveis em extratos de amostras com altos background, e particularmente útil na detecção de uma análise falsamente positiva para aflatoxina. DETERMINAÇÃO: Dividir a placa de silica gel em duas metades idênticas. Cobrir uma delas com um vidro limpo. Na outra colocar duas alíquotas (spots) de 1-10ul da amostra contendo 0,5 - 5,0ng de aflatoxina. Colocar também dois spots de padrão com a mesma concentração das amostras. Colocar 1ul de ácido trifluoracético em cada um dos três spots, e deixar a reação ocorrer por 5 minutos. Manter corrente de vapor sobre a placa por 10 minutos. Na outra metade coberta da placa aplicar alíquotas idênticas eliminando o tratamento com ácido trifluoracético. Desenvolver a placa em clorofórmio + acetona (85 + a15) ou acetado de etila saturado com água. Colocar 200 ml do solvente diretamente na cuba de eluição. Após desenvolvimento examinar a placa de CCD sob luz UV de comprimento de onda longo. Aflatoxinas sem tratamento com ácido trifluoracético devem aparecer próximas ao topo da placa. Derivados fluorescentes azuis (B2a e G2a) deverão aparecer a Rf aproximadamente 1/4 aos da Bl e G1. Borrifar a placa com solução de H2SO4 (1 + 3) para mudar a fluorescência da aflatoxina de azul ou azul-esverdeado para amarelo como uma confirmação adicional de aflatoxinas. Usar este teste somente para confirmar a ausência de aflatoxinas, isto é, spots os quais não se tornaram amarelos são positivamente ausentes de aflatoxinas, visto que muitos outros materiais, além da aflatoxina, podem dar reação com H2S04 desenvolvendo coloraçó amarela. OBSERVAÇOES:

A seguir são notificadas algumas precauções que servem como advertência, envolvendo o uso de compostos tóxicos e inflamáveis. a. Destilações, extrações e evaporações com líquidos inflamáveis. Executar estas operações atraves de equipamentos apropriados como banno-maria, vapor ou manta aquecedora. Utilizar sistema eficaz de eliminação de gases para remover vapores inflamáveis que possam ser produzidos. Deixar abertura ampla no frasco e adicionar pérolas de ebulição antes de iniciar o aquecimento. b. Acetonitrila. Tóxico. Evitar contato com a pele e os olhos. Usar sistema eficaz para eliminação de vapores. HCN é liberado quando em contato com ácido. c. Benzeno.Tóxico. Altamente inflamável. Evitar contato com a pele e inalação. Utilizar sistema eficiente para eliminação de vapores. Considerado carcinogênico. d. Acetona. Inflamável. Forma peróxidos explosivos com agentes oxidantes. Utilizar sistema eficaz para eliminação de vapores. e. Hexano. Inflamável. Utilizar sistema eficaz para eliminação de vapores. f. Éter Etílico. Armazenar sob proteção da luz. Inflamável. Utilizar sistema eficaz para eliminação de vapores. Evitar eletricidade estática. Peróxidos instáveis podem se formar após armazenamento prolongado ou exposição dos frascos a luz solar. g. Alcool Metílico. Inflamável. Tóxico. Evitar contatos com a pele, olhos e inalação. Utilizar sistema eficaz para eliminação dos vapores. h. Clorofórmio. Evitar inalação. Utilizar sistema eficaz para elimina os vapores. Considerado carcinogênico. i. Micotoxinas são substâncias de toxicidade elevada e tidas como carcinogênicas. Devem ser manipuladas com muito cuidado e a máxima proteção possível. j. Usar luvas quando manipular toxinas em sua forma cristalizada. k. Descontaminar a vidraria com hipoclorito de sódio 1%. MÉTODO Nº 31-AFLATOXINAS II-CROMATOGRAFIA EM CAMADA DELGADA APLICAÇÃO: Produtos e sub-produtos de origem vegetal, raçoes e concentrados . PRINCÍPIO: O método baseia-se na separação cromatográfica e a detecção é feita pela fluorescência das aflatoxinas quando expostas a luz ultravioleta (366nm). Obtém-se o resultado pela comparação com padrões de concentração conhecida, igualmente aplicados na placa cromatográfica. MATERIAL E EQUIPAMENTO: - Balança semi-analítica - Banho-maria - Agitador magnético ou liquidificador - Câmara escura com lâmpada ultra-vloleta - Secador - Pipetas - Provetas de 50ml, 100ml - Funil de vidro 100mm

- Bequer de 100ml, 250ml - Balões volumétricos 5ml, 10ml - Funil de separação de 500ml tipo pera - Cápsulas de porcelana capacidade de 200ml - Micro seringas 10ul, 50ul - Cuba de vidro com tampa esmerilhada - 20 x 20 x 10 - Erlenmeyer com tampa esmerilhada de 250ml, 500ml - Cromatoplacas silicagel 60/0,2mm de espessura 20 x 20cm - Micropipetas REAGENTES: - Clorofórmio P.A. - Metanol P.A. - Cloreto de sódioP.A: - Hexano P.A. - Benzeno P.A. - Acetato de etila P.A. - Etanol P.A. - Ácido fórmico P.A. - Acetona P.A. - Padrões de aflatoxinas B1, B2, G1 e G2 - Ácido sulfúrico P.A. PROCEDIMENTO: 1. Pesar 25g de amostra previamente moída e homogeneizada (desengordurar se necessário) em um erlenmeyer de 500ml. 2. Adicionar água destilada quente (40 a 60ml), suficiente para formar uma pasta; esfriar. 3. Adicionar 100ml de clorofórmio, tampar e agitar vigorosamente por 30 segundos. Retirar a tampa cuidadosamente. 4. Tampar e deixar em agitação branda e constante por 30 minutos. 5. Filtrar o extrato em papel de filtro 24cm (80g), recebendo em erlenmeyer ou balão de fundo chato com junta 24/40, ou compatível ao condensador. 6. Evaporar no soxhlet, em banho-maria. 7. Dissolver o resíduo em 50ml de metanol e transferir para o funil de separação de 500ml. 8. Adicionar 50ml de solução de NaCl 4% e agitar cautelosamente, eliminando a pressão pela torneira. 9. Adicionar mais 50ml de hexano, agitar vigorosamente eliminando a pressão. 10. Deixar em repouso até a separação das fases. OBSERVAÇÃO: Se a fase hexano não estiver límpida, fazer uma nova lavagem com hexano. 11. Recolher a parte inferior em um funil de separação contendo 50ml de hexano, agitar. 12. Receber a camada inferior em outro funil de separação, contendo 50ml de clorofórmio, agitar, deixar em repouso até a separação das fases. 13. Recolher a parte inferior em um frasco evaporado e reservar. 14. Fazer mais uma extração com 50ml de clorofórmio, recolhendo o extrato no mesmo frasco evaporador. 15. Evaporar em banho-maria até quase secura. Completar a evaporação usando corrente de nitrogênio. 16. Dissolver o extrato com clorofórmio de acordo com os teores esperados (1ml a 10ml).

17. Na cromatofolha, marca linha de origem 1,5cm da margem. Marcar linha de chegada 8,5cm da linha de origem. Dividir a linha de origem de 2 em 2cm. Fazer as aplicações com auxílio de micro seringas na linha de origem. 18. Saturar a cuba com uma das misturas abaixo: Benzeno + acetato dé etila + etanol(60 + 38 + 2) ou, Cloroformio + acetona (90 + 10) ou, Tolueno + acetato de etila + acido fórmico (60 +30 + 10) ou, Tolueno + acetato de etila + ácido fórmico (60 + 40 + 0,5). 19. Aplicar 2 ou 3 pontos de padrões para 3 pontos de amostra. 20. Colocar a placa cuidadosamente na cuba já saturada e aguardar até que a fase móvel atinja a marca de 8,5cm. 21: Retirar a placa da cuba e secar ao ambiente ou com auxílio de secador. 22. Fazer a leitura da placa, em câmara escura, sob luz ultravioleta, comparando a intensidade da fluorescência da amostra com os padrões. Se a intensidade de fluorescência da mancha de amostra for maior que a intensidade de fluorescência da mancha de maior concentração do padrão, a amostra deverá ser diluída e recromatografada. OBSERVAÇAO: Para confirmação, adicionar uma gota de H2S04 50%, sobre a mancha que se supõe ser Aflatoxina B1 e/ou B2. Se a mancha permanecer com fluorescência azul, indica que não se trata de aflatoxina. Se a mancha mudar para amarelo; pode indicar aflatoxina; mas o teste não é conclusivo. Neste caso, proceder ao teste com ácido trifluoracético. CÁLCULO:
5 x V x V = ug Aflatoxina/kg = ppb W x Z
ONDE: S = ul de aflatoxina do padrão com igual intensidade de fluorescência da amostra. Y = Concentração aflatoxina do padrão em ug/ml. V = ul do solvente requerido para diluir o extrato final. W = Peso da amostra em grama. Z = ul de aflatoxina da amostra com igual intensidade de fluorescência do padrão. SOLUÇÕES REAGENTES: 1. Soluções de saturação: a. Homogeneizar 60 partes de benzeno; 38 partes de acetato de etila e 2 partes de etanol. b. Homogeneizar 90 partes de clorofórmio e 10 partes de acetona. c. Homogeneizar 60 partes de tolueno, 30 partes de acetato de etila e 10 partes de ácido fórmico. d. Homogeneizar 60 partes de tolueno, 40 partes de acetato de etila e 0,5 parte de ácido fórmico. 2. Solução de Cloreto de Sódio a 4% Diluir 4 gramas de NaCl em água destilada e completar o volume para 100ml. MÉTOOO Nº 32 - AFLATOXINAS - MINI-COLUNAS APLICAÇÃO: Produtos ou sub-produtos de origem vegetal, rações e concentrados. PRINCÍPIO: O método baseia-se na separação através de colunas e a detecção é feita pelo aparecimento de uma banda azul fluorescente na camada de florisil.

MATERIAL E EQUIPAMENTO: - Balança semi-analítica - Liquidificador - Bastão de vidro - Banho-maria - Suporte para minicoluna - Cromatoplacas silicagel 60, sem indicador de fluorescência, 20 x 20cm espessura 0,2mm - Minicoluna - Papel filtro qualitativo, filtragem rápida, 24cm de diâmetro. - Microseringa de 5 a 10 microlitros - Funil +/- 15cm de diâmetro - Lã de vidro - Funil de separação 125, 250 e 500ml - Vidro de relógio 15cm de diâmetro - Proveta 250, 100, 50 e 10ml - Cromatovisor com lâmpada UV, emissão 3.650 A e 15 watts - Bequer 400ml, 600 ml - Cuba de desenvolvimento 20 x 20 x 5cm, com tampa REAGENTES: - Clorofórmio P.A. - Acetato de etila P.A. - Acetona P.A. - Ácido fórmico 90% P.A. - Hidróxido de sódio P.A. - Carbonato básico de cobre P.A. - Cloreto férrico anidro P.A. - Cloreto de potássio P.A. - Hidróxido de potássio P.A. - Ácido sulfúrico P.A. - Hyflo super-cel ou equivalente - Florisil (100-200 mesh) - Padrões de aflatoxina B1, B2, G1 e G2 - Benzeno P.A. - Acetonitrila P.A. - Tolueno P.A. - Silica gel 60 (0,063 - 0,2mm) - Alumina neutra P.A. - Sulfato de sódio anidro P.A. - Sulfato de cálcio anidro P.A. PROCEDIMENTO: a. EXTRAÇAO: 1. Pesar 50g da amostra previamente homogeneizada (deve passar em peneira malha 0,85mm). 2. Transferir a amostra para um liquidificador (copo de vidro) ou erlenmeyer de 500ml. 3. Adicionar 250ml de acetona + água (85+15) e agitar em alta velocidade por 3 minutos ou agitar em erlenmeyer de 500ml por 30minutos a 1 hora. 4. Filtrar em papel de filtro qualitativo pregueado, usando um funil de 15cm de diâmetro. Tampar com vidro de relógio 5. Recolher em proveta de 250ml os primeiros 150ml.

b. PURIFICAÇAO: 1. Transferir os 150ml do filtrado para um bequer de 400ml. 2. Adicionar 3g de carbonato básico de cobre e homogeneizar. 3. Adicionar 170ml de NAOH 0,2N (8g/l H20) e 30ml de cloreto férrico gel (20g + 300ml H20) em bequer de 600ml. Homogeneizar. 4. Adicionar (2.) em (3). Homogeneizar. 5. Medir 150ml de Hyflo Super-Cel e adicionar ao bequer de 600ml. Homogeneizar 6. Filtrar, usando papel de filtro qualitativo pregueado e funil analítico de 15cm de diâmetro. Tampar com vidro de relógio. 7. Recolher os primeiros 150ml em proveta de 250ml. c. PARTlÇÃO: 1. Transferir os 150ml obtidos na purificação para um funil de separação de 500ml. 2. Adicionar 150ml de H2SO4 0,03% (0,3ml H2SO4/l H2O) e 10ml de clorofórmio. 3. Agitar vigorosamente por 2 minutos e deixar separar as camadas. 4. Transferir a camada inferior para um funil de separação de 125ml (+/- 13 - 14ml). 5. Adicionar 100ml da solução de KOH-KCI (1,12g + 10g + 11). Agitar suavemente por 30 segundos e deixar separar as fases. 6. Se formar emulsão, drenar a emulsão para proveta com tampa de 50, 25 ou 10ml e adicionar 1g de sulfato de sódio anidro. Agitar fortemente. 7. Rotular: extrato (1). Retirar 2ml para a pesquisa na coluna. 8. Se a pesquisa da coluna der positiva seguir o seguinte esquema: a. Adicionar 10ml de clorofórmio ao funil de separação de 500ml do item C-1. Desprezar qualquer resíduo de clorofórmio presente nos funis. b. RepetIr o processo de extração, transferindo a camada inferior de clorofórmio para o mesmo funil de 125ml que contém a solução de KOH-KCl (Trocar a solução de KOH-KCl colocando outros 100ml. Desprezar pela tampa superior, não pela torneira). Seguir a sequência usada nos itens 5 e 6. d. AVALIAÇÃO: 1. Transferir.4ml do extrato (1) e 4ml do extrato (2) para um funil de separação de 125ml. 2. Adicionar 50ml de ácido sulfúrico 0,03% e agitar suavemente por 30 segundos. 3. Coletar 6ml da camada inferior de clorofórmio (medir em proveta de 15ml). 4. Concentrar em banho-maria sob atmosfera de nitrogênio. 5. Diluir para volume conhecido em benzeno acetonitrila (98+2) e realizar a cromatografia em camada delgada usando a seguinte fase móvel: tolueno: acetato de etila: cloroformio: ácido fórmico 90% (50 : 70 : 50 : 20). 6. PESQUISA POR CROMATOGRAFIA EM COLUNA: a) Preparo da Coluna: I. A minicoluna deverá ter 6mm de diâmetro interno e comprimento de 190mm. No final e afilada para 2mm de diâmetro. Todo material deve ser secado a 110ºC por 1-2 horas. II. Colocar lã de vidro na parte final da coluna. III. Adicionar 8-10mm de Drierite (CaSO4 anidro). Bater suavemente após cada adição para facilitar o assentamento. IV. Adicionar 8-10mm de Florisil 100-200 mesh. V. Adicionar 16-20mm de silica gel. VI. AdicIonar 8-10mm de alumina neutra. VII. Adicionar 8-10mm de Drierite. VIII. Colocar lã de vidro no topo da coluna.

b) Cromatografia: Transferir 2ml para coluna (drenar por gravidade 15-30min). Quando o solvente atingir o topo do adsorvente adicionar 3ml do eluente clorofórmio + acetona (9+1). Drenar por gravidade. c) Detecção qualitativa: Será positivo quando aparecer banda azul fluorescente no topo da camada de fiorisil, formando um anel bem nítido. 7. QUANTIFICAÇÃO: Caso o resultado for positivo, fazer quantificação: a) Diluir o extrato para 500 ou 1000ul com benzeno acetonitrila (98:2). b) Aplicar alíquota da amostra é dos padrões em placa de sílica gel 60. c) Correr usando a fase móvel tolueno : acetato de etila : clorofórmio : ácido fórmico 90%(70 : 50 : 50 : 20). d) Calcular a concentração de aflatoxina usando a seguinte fórmula:
A.C.V Aflatoxina em ug/ml = P.S. ONDE: A = Volulme em ul da alíquota de aflatoxina padrão com fluorescência igual a da amostra. C = Concentração em ug/ml da aflatoxina padrão. V = Volume do solvente em ul requeridos para diluir o extrato final. P = Quantidade da amostra em g contida no extrato final (3,9g). S = Volume em ul da alíquota do extrato da amostra com fluorescência igual a Amostra. MÉTODO Nº 33 -AFLATOXINAS-ELISA (EnzIme Linked Imuno Sorbent Assay) APLICAÇÃO: Produtos ou sub-produtos de origem vegetal, rações concentrados e leite. PRINCÍPIO: Ensaio lmunoenzimático competitivo para quantificação e/ou avaliação qualitativa das aflatoxinas B1, B2 e G1. As placas são sensibilizadas com anticorpos (monoclonais) anti-B1. Em um ensaio competitivo a amostra e o conjugado são adicionados simultaneamente e competem por um número limitado de sítios de ligação. Após um determinado período de incubação, durante o qual a ligação competitiva chega a um equilíbrio adiciona-se o substrato para haver desenvolvimento de cor, que será inversamente proporcional a quantidade de aflatoxina contida na amostra (quanto mais cor, menos aflatoxina na amostra, e vice-versa). MATERIAL E EQUIPAMENTO: - Liquidificador ou homogeneizador equivalente, - Leitora de placas - manual ou automática (espectrofotômetro 490nm) - Lavadora de placas - Manual ou automática - Micropipeta monocanal - 25 ul - Micropipeta multicanal (8) - 50 a 200ul (volume variável) - Ponteiras para micropipetas (TIPS) - Pipeta graduada - 10ml - Papel filtro qualitativo ou lã de vidro - Erlenmeyer - 250ml - Balança analítica

REAGENTES: - Metanol P.A. (MEOH) - N, N-dimetilformamida P.A. (DMF) - Ácido clorídrico P.A. concentrado d = 1,19 g/l - Hipoclorito de sódio 5% - Acetona P.A. - Kit para detecção aflatoxina: �Placas sensibilizadas com anticorpos Anti~B1 �B1 conjugado com peroxidase �5 padrões de aflatoxina em diferentes concentrações �diluente de amostra �tabletes de OPD (O - fenilenediamina-2) �diluente de OPD. PROCEDIMENTO: a. AMOSTRAGEM: Colher a amostra segundo instruções e moer de maneira que passe integralmente por uma peneira de 0,85mm. Fazer uma segunda homogenização e armazenar para o procedimento analítico. b. EXTRACÃO: 1. Pesar 50g da amostra 2. Adicionar 250ml de solvente de extração (70% MEOH 29% H2O destilada e 1% DMF). 3. Homogeneizar em liquidificador a velocidade média por 3 minutos (alta- velocidade 1 a 2 minutos), ou em um agitador mecânico por 45 minutos a alta velocidade. 4. Deixar sedimentar. 5. Filtrar o sobrenadante através de papel de filtro ou lã de vidro. 6. Diluir o extrato filtrado 1:20 com diluente da amostra (incluso no kit) - 25ul do extrato em 475ul de diluente de amostra. c. TÉCNICA: Os melhores resultados são obtidos sob rígida observação destas instruções. A precisão e a exatidão serão mantidas se a pipetagem e a lavagem forem cuidadosas. As instruções a seguir são de caráter técnico e permitem a manutenção da precisão do ensaio: 1. Utilizar a técnica de pipetagem reversa para agilizar a adição do reagente (isto é, aspire mais líquido que o necessário e liberar quantidade exata). 2. Adotar uma organização na adição do reagente sempre obedecendo a mesma sequência que a da adição do reagente anterior. 3. Calcular o tempo de incubação após a adição de reagentes a primeira fileira de poços. A cronometragem do ensaio é muito sensível. Padronizar os tempos em todos os passos do ensaio, tanto na adição dos reagentes como na determinação dos tempos de incubação. 4. Depositar os reagentes diretamente no fundo da cavidade, tomando preocuções para que não permaneçam resíduos nas bordas do poço de titulação ou bolhas de ar no fundo. Após a adição da amostra às cavidades, fazer uma checagem rápida para se certificar que não existem bolhas de ar antes da adição do conjugado. 5. Para que o tempo de incubação seja preciso, recomenda-se que não se faça o ensaio de mais de quatro tiras ao mesmo tempo. Para outras amostras, iniciar um novo ensaio com novos padrões e marque novamente o tempo de incubação. 6. Lavar completamente uma fileira de poços antes de passar a seguinte. Aspirar o conteúdo líquido da tira. Completar com solução de lavagem até o eminente

transbordamento. Aspirar o líquido da tira, repetir a lavagem mais três/quatro vezes, então, proceder a lavagem da tira seguinte. 7. Para maior precisão dos resultados quantitativos, recomenda-se que cada amostra seja corrida em duplicata, a variação da densidade óptica deve ser avaliada. É natural que exista uma variação da +/- 10% na densidade óptica. Variações maiores podem ser indicativas de erros técnicos. d. ENSAIO: 1. Permitir que os reagentes do kit atinjam um equilíbrio com a temperatura ambiente. 2. Completar a identificação poço/amostra na tabela de cálculos. 3. Adicionar 50ul de Padrão A, não diluído, aos poços A1 e A2; os Padrões de B a E em duplicata, aos poços de A3 a A10. 4. Adicionar 50ul das amostras diluídas a poços em duplicata. 5. Adicionar imediatamente 50ul de conjugado a cada poço. 6. Incubar 30 minutos para teste quantitativo, 10 minutos para avaliaçao qualitativa de presença ou não de aflatoxina na amostra. 7. Preparar o substrato (OPD) -10ml diluente para 1 tablete de OPD. 8. Ao final da incubação, lavar cada fileira de poços 3 a 5 vezes (água destilada e deionizada). 9. Adicionar 100ul do substrato a cada poço. 10. Incubar por 10 minutos. 11. Adicionar 100ul de HCl 1N a cada poço (para paralisar a reação). 12. Zerar a leitura com ar utilizando uma absorbância de 490nm. 13. Proceder a leitura da placa e registrar as absorbâncias, a 490nm, na tabela de cálculos coluna A. e. RESULTADOS: A concentração das amostras pode ser determinada graficamente (Curva Padrão) ou por regressão do logit vs concentração. CÁLCULOS: 1. Cálculo das médias de absorbância de cada Padrão (A a E) e amostras desconhecidas, coluna B. 2. Cálculo da razão B/B0 para cada Padrão e amostra (coluna C). 3. Cálculo do logit B/B0:
logit. B/B0 =Ln (Coluna C) Coluna D
ONDE: Ln = Logarítmo natural 4. Representar a curva padrão em papel gráfico semi-logarítmico, utilizando os valores dos Padrões (encontrado no rótulo) no eixo X e os valores correspondentes do logit B/B0 no eixo Y. 5. Extrapolar a concentração de aflatoxinas das amostras diretamente da curva padrão pela determinação do logit B/B0, seguindo sobre a linha padrão e então perpendicularmente ao eixo X. A interseção com o eixo X é a concentração de aflatoxina na amostra desconhecida. CURVA PADRÃO: 1. A curva padrão deve ser uma ligeira curva que une os valores dos padrões. A execução cuidadosa do ensaio observando-se a técnica discutida em "Procedimentos

irá produzir uma curva precisa assim como irá possibilitar uma quantificação precisa das amostras. 2. A área mais sensível da curva padrão para quantificação é a área mais inclinada da curva, ou de cerca de 3 a 40ppb. 3. Portanto quando uma amostra équantificada acima de 50ppb, a amostra não estará na porção mais sensível da curva. Se houver necessidade de quantificação de uma amostra em exatamente 0,1ppb, utiliza-se maiores diluições desta amostra para que possa ser enquadrada na porção mais sensível da curva. CURVA PÃDRAO: ZONA ÓTIMA DE SENSITIVIDADE
OBSERVAÇÕES: a. As micotoxinas devem ser manipuladas como substâncias tóxicas. b. Limpar os resíduos com hipoclorito de sódio 1% e deixar em repouso. Adicionar solução de acetona a 5%. c. Os recipientes de vidro devem ser descontaminados com alvejante e acetona antes do descarte. SOLUÇÕES E REAGENTES: a. Solução de Extração: Homogeneizar 70 partes de metanol P.A. 29 partes de água destilada e 1 parte de N, N dimetilformamida P.A. b. Solução para Paralisação - Ácido Clorldrico 1N Transferir 42ml de HCI concentra de (12M) para balão volumétrico de 500ml, contendo aproximadamente 250ml de destilada. Esfriar e completar o volume. OBS: Adicionar sempre os ácidos concentrados a água. c. Solução de Lavagem e Descontaminação: Diluir 100ml de água sanitária (hipoclorito de sódio a 5%) em 2000ml de água. TABELA DE CÁLCULOS

Identificação da Amostra
Poço Amostra
A Padrão Conc.
B Média
A (490)
C B /B0
D -1B/B0
E B/B0
1-B/B0
F Logit. B/B0 N 1-B/B0
G Resultados
ppb
MÉTODO Nº 34 - GLlCÍDIOS REDUTORES EM GLICOSE – GLICÍDIOS NÃO REDUTORES EM SACAROSE - DETERMINAÇÃO DE AMIDO – GLICÍDIOS REDUTORES EM LACTOSE. APLICAÇÃO: Produtos ou sub-produtos de origem vegetal, rações, concentrados e lácteos. PRINCÍPIO: O método de Lane-Eynon, baseia-se na redução de um volume conhecido do reagente de cobre alcalino (Fehling) a óxido cuproso. O ponto final é indicado pelo azul de metileno, que é reduzido a sua forma leuco, por um pequeno excesso de açúcar redutor. MATERIAL E EQUIPAMENTO: - Balança analítica (precisão +/- 0,0001g) - Erlenmeyer de 250ml - Bureta de 25mI ou 50ml - Pipetas volumétricas de 5ml - Pipeta graduada de 1ml - Bico de Bunsen ou placa aquecedora - Banho-maria - Papel de tornassol - Papel de filtro qualitativo REAGENTES: - Tartarato duplo de sódio e potássio P.A. - Sulfato de cobre pentahidratado P.A. - Hidróxido de sódio P.A. - Glicose P.A. - Ferrocianeto de potássio P.A. - Acetato ou sulfato de zinco P.A. - Ácido clorídrico P.A. - Azul de metileno P.A. PROCEDIMENTO PARA GLICÍDEOS REDUTORES EM GLICOSE: a. Preparo da amostra: Pesar em balança analítica 5g de amostra em bequer de 150ml e dissolver com cerca de 50ml de H2O. Transferir para balão volumétrico de 250ml, lavando bem o bequer.

Adicionar 5ml da solução de ferro-cianeto de potássio 15% e 5ml de acetato ou sulfato de zinco 30%. Agitar e completar o volume com água destilada e deixar sedimentar por 15 minutos. Filtrar em papel de filtro seco e receber o filtrado em erlenmeyer seco. b. Titulação: Em erlenmeyer de 500ml, pipetar volumétricamente 10ml das soluções de Fehling A e B e adicionar 40ml de H2O destilada e aquecer a ebulição. Colocar a solução de amostra na bureta e gotejar no erlenmeyer sem agitação, até o desaparecimento da coloração azul. Mantendo a ebulição, adicionar de 1 a 2 gotas de azul de metileno 0,5% e continuar a titulação até que a coloração azul desapareça. A titulação não deve ultrapassar a três minutos. CÁLCULO: Glicídeos redutores em 100 x 250 x T glicose % = V x P ONDE: T = Título da solução de Fehling em açúcar redutor. V = Volume da amostra gasto na titulação em ml P = Peso da amostra em grama 100 = Porcentagem 250 = Volume da solução PROCEDIMENTO PARA GLICÍDEOS NÃO REDUTORES EM GLICOSE: a. Preparo da amostra: Transferir 50ml do filtrado (da amostra) para balão volumétrico de 100ml. Adicionar 2ml de ácido clorídrico concentrado e levar a banho-maria a 60ºC por 60 minutos. Esfriar e neutralizar com solução de hidróxido de sódio 10%, usando papel de tornassol como indicador. Esfriar e completar o volume a 100ml. Filtrar em papel filtro qualitativo para erlenmeyer seco. b. Titulação: Proceder como para glicídeos redutores em glicose. CÁLCULO: Glicídeos totais em 100 x 500 x T glicose % = (açúcar invertido) V x P Sacarose % = (% glicídeos totais - % glicose) x 0,95 0,95 = fator de transformação de héxoses para sacaroses. PROCEDIMENTO PARA DETERMINAÇÃO DE AMIDO: a. Preparo da amostra: Pesar 5g de amostra, adicionar 100ml de água destilada e misturar. Adicionar 10ml de ácido clorídrico concentrado. Aquecer a ebulição por 30 minutos. Esfriar e neutralizar com hidróxido de sódio 40%, usando papel de tornassol como indicador, Transferir para balão volumétrico de 250ml e adicionar 10ml de ferrocianeto de potássio 15% e 10ml de acetato ou sulfato de zinco 30%, Agitar esfriar e completar o volume do balão volumétrico com água destilada. Filtrar em filtro qualitativo para erlenmeyer seco. b. Titulação:

Proceder como para glicídeos redutores em glicose. CÁLCULOS:
100 x 500 x T % açúcares totais = V x P a. Quando a amostra só contiver amido, multiplicar o resultado por 0,90, que é o fator de transformação da glicose em amido.
% amido = % açúcares totais X 0,90 b. Se a amostra contém sacarose:
% amido = ( % açúcares totais - % sacarose) x 0,90
c. Se a amostra contém sacar os e e glicose: % amido = % açúcares totais - (sacarose + glicose) x 0,90
OBSERVAÇOES: 1. A hidrólise ácida pode ser feita em autoclave a 120ºC por 20 minutos para obter glicídios redutores em lactose, faz-se o procedimento como em glicídios redutores em glicose e multiplica-se o resultado pela constante da lactose 1,39.
100 x 250 x T Glicídios redutores em lactose % = V x P x 1,39 SOLUÇOES: a. SOLUÇÃO DE FEHLING A: Dissolver exatamente 34,639G de sulfato de cobre pentahidratado em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume e homogeneizar. b. SOLUCÃO DE FEHLING B: Dissolver exatamente 173g de tartarato duplo de sódio e potássio (sal de Rochelle) e 125g de hidróxido de sódio em água destilada. Transferir quantitativamente para balão volumétrico de 1000ml. Completar o volume e homogeneizar. c. SOLUÇÃO PADRÃO DE GLICOSE: Pesar exatamenbe 500 miligramas de glicose, previamente seca em estufa a vácuo a 70ºC durante 1 hora. Transferir quantitativamente para balão volumétrico de 100ml com auxílio de água destilada. Dissolver, completar o volume e homogeneizar. A solução de glicose deve ser recentemente preparada. d. PADRONIZAÇÃO DA SOLUÇAO DE FEHLING: Transferir volumetricamente 10ml de cada uma das soluções de FEHLING para erlenmeyer de 500ml, adicionar 40ml de água destilada até a ebulição. Titular com a solução padrão de glicose, sem agitação, até o desaparecimento da coloração azul. Manter a ebulição adicionando uma gota de azul de metileno 0,5% e completar a titulação até descoloração do indicador. O consumo estimado da glicose é aproximadamente de 10ml.
Volume de Glicose x 0,5 Título da solução de Fehling = 100

OBSERVAÇAO: O tempo de titulação não deve ultrapassar a três minutos. e. SOLUÇAO DE HIDRÓXIDO DE SÓDIO 40% Dissolver 40G de NaOH P.A. em água destilada. Esfriar e transferir quantitativamente para um balão volumétrico de 100ml. Completar o volume e homogeneizar. f. SOLUÇÃO fERROCIANETO DE POTÁSSIO 15% Dissolver 15g de ferrocianeto de potássio em água destilada. Transferir quantitativamente para um balão volumétrico de 100ml, completar o volume e homogeneizar. g. SOLUÇÃO DE ACETATO DE ZINCO 30% Dissolver 30g de acetato de zinco em água destilada. Transferir quantitativamente para um balão volumétrico de 100ml, completar o volume e homogeneizar. h. SOLUÇÃO INDICADORA DE AZUL DE METILENO 0,5% Dissolver 0,5g de azul de metileno em 10ml de álcool etílico P.A. Transferir com água destilada para um balão volumétrico de 100ml, completar o volume e homogeneizar. MÉTODO Nº 35 - CÁLCIO - ABSORÇÃO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas - Pipetas volumétricas - Proveta - Funis de vidro - Papel de filtro qualitativo REAGENTES E SOLUÇÕES: a. Ácido clorídrico P:A, (d= 1,19 g/l) b. Solução de ácido clorídrico (1 + 1) c. Solução de lantânio 5%: Dissolver 58,65g de trióxido de lantânio em 100ml de água deionizada. Lenta e cuidadosamente, adicionar 250ml de ácido clorídrico concentrado. Deixar esfriar e completar o volume com água deionizada para 1000ml. d. Solução padrão estoque de cálcio 1000ug/ml. Dissolver 1g de cálcio (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. e. Solução padrão de cálcio 100ug/ml.

Pipetar 10,0ml da solução do cálcio 1000ug/ml, transferir para balão volumétrico de 100ml. Adicionar 10ml da solução de lantânio e completar o volume com água deionizada. f. Solução padrão de trabalho de cálcio 2,0ug/ml. Pipetar 2,0 ml de solução de cálcio 100ug/ml, transferir para balão volumétrico de 10ml. Adicionar 10ml da solução de lantânio (item c) e completar o volume com água deionizada. g. Solução padrão de trabalho de cálcio 4,0ug/ml. Pipetar 4,0ml de solução de cálcio 100ug/ml, transferir para balão volumétrico de 10ml. Adicionar 10ml de solução de lantânio (item c) e completar o volume com água deionizada. h. Branco. Transferir 10ml da solução de lantânio para balão volumétrico de 100ml e completar o volume com água deionizada. PROCEDIMENTO: 1. Pesar a amostra em cadinho de porcelana previamente seco e calcinar por 4 horas a 560ºC. A quantidade deverá ser proporcional ao teor de cálcio esperado para o produto: Farinha de carne, peixe ossos: 1,0g de amostra. Rações e concentrados: 2.0g de amostra. Matérias-primas com baixos teores de cálcio: 5,0g de amostra. 2. Adicionar 20ml de ácido clorídrico (1 + 1) e levar à chapa aquecedora. 3. Filtrar em balão volumétrico de 250ml e completar o volume com água deionizada. 4. Fazer as outras diluições dependendo da quantidade de cálcio na amostra. 5. Na última diluição, adicionar solução de lantânio (item c) na quantidade de 1/10 da capacidade do balão volumétrico. 6. Fazer leitura em espectrofotômetro de absorção atômica a 422,7n usando o branco para zerar o aparelho e as soluções padrões para calibração. OBSERVAÇÃO: Fosfato bicálcico, calcáreo e misturas minerais, pesar e bequer e solubilizar com 20ml de ácido clorídrico concentrado. CÁLCULO: Cálcio (ug/g ou ppm) = (C)x(V)xFD
P ONDE: C = Concentração do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml FD = Fator de diluição P = Peso da amostra em gramas FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) %Cálcio na amostra = concentração (ppm ou ug/g)
10.000

MÉTODO Nº 36 - MAGNÉSIO - ABSORÇAO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduaqas - Pipetas volumétricas - Proveta - Funis de vidro - Papel de filtro qualitativo REAGENTES E SOLUÇÕES: a. Ácido clorídrico P.A. (d = 1,19 g/l) b. Solução de ácido clorídrico (1 + 1) c. Solução de lantânio 5%: Dissolver 58,65g de trióxido de lantânio em 100ml de água deionizada. Lenta e cuidadosamente, adicionar 250ml de ácido clorídrico concentrado. Deixar esfriar e completar o volume com água deionizada para 1000ml. d. Solução padrão estoque de magnésio 1000 ug/ml. Dissolver 1g de magnésio (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. e. Solução padrão de magnésio 50ug/ml. Pipetar 5,0ml da solução de magnésio 1000ug/ml, transferir para balão volumétrico de 100ml adicionar 10ml da solução de lantânio e completar o volume com água deionizada. f. Solução padrão de trabalho de magnésio 0,25ug/ml. Pipetar 0,5ml da solução de magnésio 50ug/ml, transferir para balão volumétrico de 100ml adicionar 10ml da solução de lantânio e completar o volume com água deionizada. g. Solução padrão de trabalho de magnésio 0,50ug/ml. Pipetar 1,0ml da solução de magnésio 50ug/ml, transferir para balão volumétrico de 100ml adicionar 10ml da solução de lantânio e completar o volume com água deionizada. h. Branco. Transferir 10ml, da solução de lantânio para balão volumétrico de 100ml e completar o volume com água deionizada. PROCEDIMENTO: 1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,0g de amostra em bequer de 100ml.

Adicionar 20ml de ácido clorídrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PROOUTOS E SUB-PRODUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇOES E CONCENTRADOS: Pesar 5,0g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir con.forme o item 3. 3. PARA TODOS OS TIPOS DE AMOSTRAS, APÓS A DISGESTÃO COM ÁCIDO CLORÍDRICO: Filtrar em balão volumétrico de 250ml. Completar o volume com água deionizada. Fazer outra diluição, dependendo do teor de magnásio na amostra. Adicionar na última diluição, solução de latânio na quantidade de 1/10 da capacidade do balão volumétrico. Fazer a leitura em espectrofotômetro de absorção atômicac a 285,2nm, usando o branco para zerarao aparelho e as soluções padrões para calibração CÁLCULO: Magnésio (ug/g ou ppm = (C) x (V) x FD
P ONDE: C = ConcentraçÃo do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml FD = Fator de diluição FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) % Magnésio na amostra = concentração (ppm ou ug/g)
10.000 MÉTODO Nº 37 - COBALTO - ABSORÇÃO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas - Pipetas volumétricas - Proveta - Funil de vidro - Papel de filtro qualitativo REAGENTES E SOLUÇÕES: a. Ácido clorídrico P.A. (d = 1,19g/l)

b. Solução de ácido clorídrico (1 + 1) c. Solução padrão estoque de cobalto 1000ug/ml: Dissolver 1g de cobalto (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. d. Solução padrão de cobalto 100 ug/ml: Pipetar 10,0ml da solução de cobalto 1000ug/ml, transferir para balão volumétrico de 100ml; adicionar 1ml de ácido clorídrico concentrado e completar o volume com água deionizada. e. Solução padrão de trabalho 2,0ug/ml: Pipetar 2,0ml da solução de cobalto 100ug/ml, transferir para balão volumétrico de 100 ml; adicionar a mesma quantidade de ácido clorídrico utilizada na solução amostra e completar o volume com água deionizada. f. Solução padrão de trabalho de 4,0ug/ml. Pipetar. 4,0ml da solução de cobalto 100ug/ml, transferir para balão volumétrico de 100ml, adicionar a mesma quantidade de ácido clorídrido utilizada na solução amostra e completar o volume com água deionizada. g. Branco. Diluir em balão volumétrico de 100ml, a mesma quantidade de ácido clorídrico utilizada na solução amostra. Completar o volume com água delonizada. PROCEDIMENTO: 1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,0g de amostra em bequer de 100ml. Adicionar 20ml de ácido clorídrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PRODUTOS E SUB-PROOUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇOES E CONCENTRADOS: Pesar 1,0 a 5,8g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC, as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir conforme o item 3. 3. PARA TODOS OS TIPOS DE AMOSTRAS, APÓS A DIGESTAO COM ÁCIDO CLORÍDRICO: Filtrar em balão volumétrico de 250ml. Completar o volume com água deionizada. Fazer outras diluições, dependendo do teor do cobalto da amostra. Fazer leitura em espectrofotômetro de absorção atômica a 240,7nm usando o branco para zerar o aparelho e as soluções padrões para calibração. CÁLCULO: Cobalto (ug/g ou ppm) = (C) x (V) x FD
P ONDE: C = Concentração do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml FD = Fator de diluição P = Peso da amostra em grama

FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) % Cobalto na amostra = concentração (ppm ou ug/g)
10.000 MÉTODO Nº 38 - COBRE - ABSORÇÃO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas - Pipetas volumétricas - Proveta - Funis de vidro - Papel de filtro qualitativo REAGENTES E SOLUÇÕES: a. Ácido clorídrico P.A. (d = 1,19 g/l) b. Sblução de ácido clorídrico (1 + 1) c. Solução padrão estoque de cobre 1000 ug/ml: Dissolver 1g de cobre (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. d. Solução padrão de cobre 100 ug/ml: Pipetar 10,0ml da solução de cobre 1000ug/ml, transferir para balão volumétrico de 100 ml adicionar 1ml de ácido clorídrico concentrado e completar o volume com água deionizada. e. Solução padrão de trabalho de cobre 2,0ug/ml: Pipetar 2,0ml da solução de cobre 100ug/ml, transferir para balão volumétrico de 100ml adicionar a mesma quantidade de ácido clorídrico utilizada na solução amostra e compl~tar o volume com água deionizada. f. Solução padrão de trabalho de cobre 4,0 ug/ml. Pipetar 4,0ml da solução de cobre 100ug/ml, transferir para balão volumétrico de 100 ml, adicionar a mesma quantidade de ácido clorídrido utilizada na solução amostra e completar o volume com água deionizada. g. Branco. Diluir em balão volumétrico de 100ml, a mesma quantidade de acido clorídrico utilizada na solução amostra. Completar o volume com água deionizada. PROCEDIMENTO:

1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,0g de amostra em bequer de 100ml. Adicionar 20ml de ácido clorídrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PRODUTOS E SUB-PRODUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇOES E CONCENTRADOS: Pesar 1,0 a 5,0g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir conforme o item 3. 3. PARA TOOOS OS TIPOS DE AMOSTRAS, APÓS A DIGESTÃO COM ÁCIDO CLORÍORICO: Filtrar em balão volumétrico de 250ml. Completar o volume com água deionizada. Fazer outras diluições, dependendo do teor do cobre da amostra. Fazer leitura em espectrofotômetro de absorção atômica a 324,8nm usando o branco para zerar o aparelho e.as soluções padrões para calibração. CÁLCULO: Cobre (ug/g ou ppm) = (C) x (V) x FD
P ONDE: C = Concentração do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml FD = Fator de diluição P = Peso da amostra em gramas FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) % Cobre na amostra = concentração (ppm ou ug/g)
10.000 MÉTODO Nº 39 - ZINCO - ABSORÇAO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas - Pipetas volumétricas - Proveta - Funil de vidro - Papel de filtro qualitativo

REAGENTES E SOLUtOES: a. Ácido clorídrico P.A. (d = 1,19g/l) b. Solução de ácido clorídrico (1 + 1) c. Solução padrão estoque de zinco 1000 ug/ml: Dissolver 1g de zinco (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. d. Solução padrão de zinco 50 ug/ml: Pipetar 5,0ml da solução de zinco 1000ug/ml, transferir para balão volumétrico de 100ml; adicionar 1ml de ácido clorídrico concentrado e completar o volume com água deionizada. e. Solução padrão de trabalho de zinco 0,5 ug/ml: Pipetar 1,0ml da solução de zinco 50ug/ml, transferir para balão volumétrico de 100ml; adicionar a mesma quantidade de ácido clorídrico utilizada na solução amostra e completar o volume com água deionizada. f. Solução padrão de trabalho de zinco 1,0 ug/ml. Pipetar 2,0ml da solução de zinco 50ug/ml, transferir para balão volumétrico de 100ml, adicionar a mesma quantidade de ácido clorídrido utilizada na amostra e completar o volume com água deionizada. g. Branco. Diluir em balão volumétrico de 100ml, a mesma quantidade de ácido clorídrico utilizada na solução amostra. Completar o volume com água deionizada. PROCEDIMENTO: 1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,9g de amostra em bequer de 100 ml. Adicionar 20ml de ácido cloridrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PRODUTOS E SUB-PRODUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇOES E CONCENTRADOS: Pesar 1,0 a 5,8g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC, as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir conforme o item 3. 3. PARA TODOS OS TIPOS DE AMOSTRAS, APÓS A DIGESTÃO COM ÁCIDO CLORÍDRICO: Filtrar em balão volumétrico de 250mI. Completar o volume com água deionizada. Fazer outras diluições, dependendo do teor do zinco da amostra. Fazer leitura em espectrofotômetro de absorção atômica a 213,9nm usando o branco para zerar o aparelho e as soluções padrões para calibração. CÁLCULO: Zinco (ug/g ou ppm) = (C) x (V) x FD
P

ONDE: C = Contentração do elemento na solução da amostra (ug/mI) V = Volume inicial da solução da amostra em ml FD = Fator de diluição P = Peso da amostra em grama FD = volume inicial da solução da amostra diluída (ml)
volume da alíquota tomada (ml) % de Zinco na amostra = concentração {ppm ou ug/g)
10.000 MÉTODO Nº 40 - FERRO - ABSORÇÃO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas - Pipetas volumétricas - Proveta - Funil de vidro - Papel de filtro qualitativo REAGENTES E SOLUÇÕES: a. Ácido clorídricoP.A. (d = 1,19g/l) b. Solução de ácido clor!drico (1 + 1) c. Solução padrão estoque de ferro 1000ug/ml: Dissolver 1g de ferro (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. d. Solução padrão de ferro 100 ug/ml: Pipetar 10,0ml da solução de ferro 1000ug/ml, transferir para balão volumétrico de 100ml adicionar 1ml de ácido clorídrico e completar o volume com água deionizada. e. Solução padrão de trabalho de ferro 2,0ug/ml: Pipetar 2,0ml da solução de ferro 100ug/ml, transferir para balão volumétrico de 100 ml adicionar a mesma quantidade de ácido clorídrido utilizado na solução amostra e completar o volume com água deionizada. f. Solução padrão de trabalho de ferro 4,0ug/ml: Pipetar 4,0ml da solução de ferro 100ug/ml, transferir para balão volumétrico de 100 ml adicionar a mesma quantidade de ácido clorídrido utilizada na solução amostra e completar o volume com água deionizada.

g. Branco: Diluir em balão volumétrico de 100mI, a mesma quantidade de ácido clorídrico utilizada na solução amostra. Completar o volume com água deionizada. PROCEDIMENTO: 1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,0g de amostra em bequer de 100 ml. Adicionar 20ml de ácido clorídrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PRODUTOS E SUB-PRODUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇÕES E CONCENTRADOS: Pesar 1,0 a 5,8g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC, as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir conforme o item 3. 3. PARA TODOS TIPOS DE AMOSTRAS, APÓS A DIGESTÃO COM ÁCIDO CLORÍDRICO: Filtrar em balão volumétrico de 250ml. Completar o volume com água deionizada. Fazer outras diluições, dependendo do teor do ferro da amostra. Fazer leitura em espectrofotômetro de absorção atômica a 248,3nm usando o branco para zerar o aparelho e as soluções padrões para calibração. CÁLCULO: Ferro (ug/g ou ppm) = (C) x (V) x FD
P ONDE: C = Concentração do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml
FD = Fator de diluição . P = Peso da amostra em grama
FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) % Ferro na amostra = concentração (ppm ou ug/g)
10.000 MÉTODO Nº 41 - MANGANÊS - ABSORÇÃO ATÔMICA APLICAÇÃO: Ingredientes minerais e suas misturas, produtos ou sub-produtos de origem animal e vegetal, rações e concentrados. MATERIAL E EQUIPAMENTO: - Espectrofotômetro de absorção atômica - Forno mufla - Balança analítica - Cadinho de porcelana - Chapa aquecedora - Balões volumétricos - Pipetas graduadas

- Pipetas volumétricas - Proveta - Funil de vidro - Papel de filtro qualitativo REAGENTES E SOLUCÕES: a. Ácido clorídrico P.A. (d = 1,19g/l) b. Solução de ácido clorídrico (1 + 1) c. Solução padrão estoque de manganês 1000ug/ml: Dissolver 1g de manganês (ampola) em balão volumétrico de 1000ml e completar o volume com água deionizada. d. Solução padrão de manganês 50ug/ml: Pipetar 5,0ml da solução de manganês 1000ug/ml. Transferir para balão volumétrico de 100ml adicionar 1ml de ácido clorídrico e completar o volume com água deionizada. e. Solução padrão de trabalho de manganês 0,5ug/ml: Pipetar 1,0 ml da solução de manganês 50ug/ml, transferir para balão volumétrico de 100ml adicionar a mesma quantidade de ácido clorídrico utilizada na solução amostra e completar o volume com água deionizada. f. Solução padrão de trabalho de manganês 1,0ug/ml. Pipetar 2,0ml da solução de manganês 50ug/ml, transferir para balão volumétrico de 100ml, adicionar a mesma quantidade de ácido clorídrido utilizada na solução amostra e completar o volume com água deionizada. g. Branco. Diluir em balão volumétrico de 100ml, a mesma quantidade de ácido clorídrico utilizada na solução amostra. Completar o volume com água deionizada. PROCEDIMENTO: 1. PARA INGREDIENTES MINERAIS: Pesar 0,1 a 1,0g de amostra em bequer de 100ml. Adicionar 20ml de ácido clorídrico concentrado e levar a chapa aquecedora. Seguir conforme o item 3. 2. PARA MISTURAS MINERAIS, PRODUTOS E SUB-PRODUTOS DE ORIGEM ANIMAL E VEGETAL, RAÇÕES E CONCENTRADOS: Pesar 1,0 a 5,9g de amostra em cadinho de porcelana. Calcinar por 4 horas a 560ºC as amostras que contenham matéria orgânica. Adicionar 20ml de ácido clorídrico (1 + 1) e levar a chapa aquecedora. Seguir conforme o item 3. 3. PARA TODOS OS TIPOS DE AMOSTRAS, APÓS A DIGESTAO COM ÁCIDO CLORÍDRICO: Filtrar em balão volumétrico de 250ml. Completar o volume com água deionizada. Fazer outras diluições, dependendo do teor do manganês da amostra. Fazer leitura em espectrofotômetro de absorção atômica a 279,5nm usando o branco para zerar o aparelho e as soluções padrões para calibração. CÁLCULO:

Ferro (ug/g ou ppm) = (C) x (V) x FD P
ONDE: C = Concentração do elemento na solução da amostra (ug/ml) V = Volume inicial da solução da amostra em ml FD = Fator de diluição P = Peso da amostra em grama FD = volume inicial da solução da amostra diluida (ml)
volume da alíquota tomada (ml) % de manganês na amostra = concentração (ppm ou ug/g)
10.000 MÉTODO Nº 42 - MANGANÊS VIA ÚMIDA APLICAÇÃO: Sais de manganês. PRINCÍPIO: Fundamenta-se na oxidação do manganês em meio ácido. O periodato oxida lentamente os sais de manganês a permanganato que é medido colorimetricamente. MATERIAL E EQUIPAMENTO: - Fotocolorímetro com capacidade de 530nm - Chapa aquecedora - Bureta 25 ml divisão 1/10 - Pipeta volumétrica de 5ml - Balões volumétricos 100ml e 500ml - Bequer 100ml, 250ml e 600ml - Papel filtro qualitativo - Almofariz com pistilo REAGENTES: - Ácido clorídrico P.A. (d = 1,19g/l) - Ácido nítrico P.A. (d = 1,42g/l) - Metaperiodato de Potássjo P.A. - Ácido fosfórico P.A. - Sulfato de manganês monohidratado P.A. PROCEDIMENTO: a. Curva padrão: 1) Pipetar volumetricamente alíquotas de 0,5/0,1/2,0/ 3,0ml de MnSO4. H2O (solução padrão 1ml = 1mg Mn). 2) Adicionar em cada alíquota:
- 100ml água destilada - 10ml HNO3 P.A. - 10ml H3PO4 P.A. - 0,5g KIO4 P.A.
3) Ferver por 10 minutos até que a solução adquira coloração semelhante a do permanganato. 4) Esfriar e completar a 200ml em balão volumétrico. 5) Efetuar leitura em absorbância ou transmitância (530nm) usando como branco os reagentes mencionados no item 2, elevando a 200ml com água destilada.

b. Preparaçào da amostra: 1) Homogeneizar a amostra em almofariz 2) Pesar 0,5g em bequer de 250ml 3) Adicionar 8ml HCl P.A. aquecer em chapa aquecedora. Evaporar até quase secura. 4) Dissolver em água destilada e transferir quantitativamente para balão volumétrico de 500ml completando o volume. Filtrar desprezando os 10 primeiros ml. 5) Retirar do filtrado uma alíquota de 5ml, transferir para bequer de 600ml. Prosseguir como descrito nos itens a-2, a-3 e a-4. 6) Determinar a absorbância ou transmitância, comparando a leitura obtida com a de um padrão mais próximo a mesma. CÁLCULOS: Manganês % = Absorbância amostra x 100
Absorbância padrão x Peso da amostra em g na alíquota OBSERVAÇÃO: A alíquota a ser tomada da solução estoque, deverá adequar-se de tal forma que a leitura esteja sempre na faixa entre 15 a 60% T ou 0,824 a 0,222 de absorbância. SOLUÇÃO: a. Solução - Padrão de Manganês 1,0 mg/ml Dissolver 0,3076g MnSO4. H2O em balão volumétrico de 100ml. Completar o volume e homogeneizar. MÉTODO Nº 43 - FERRO VIA ÚMIDA APLICAÇÃO: Sais de ferro. PRINCÍPIO: Fundamenta-se na redução do Fe+++ a Fe++ pelo cloreto estanoso e posterior titulação. MATERIAL E EQUIPAMENTO: - Chapa aquecedora - Erlenmeyer de 500ml - Bureta ambar capacidade 50ml divisão 1/10 REAGENTES: - Ácido clorídrico P.A. (d =1,19g/l) - Cloreto estanoso P.A. - Cloreto mercuroso P.A. - Sulfato de manganês P.A. - Ácido fosfórico P.A. (d = 1,7g/l) - Permanganato de Potássio P.A. - Oxalato de sódio P.A. - Ácido sulfúrico P.A. (d = 1,84g/l) PROCEDIMENTO: 1. Homogeneizar a amostra em almofariz. 2. Pesar 0,3g e transferir para erlenemeyer de 500ml. Adicionar 50ml de HCl 20% e digerir até quase secura. 3. Gotejar solução de cloreto estanoso quente até tornar-se incolor. 4. Esfriar em água corrente.

5. Adicionar 10ml de solução cloreto mercuroso, 100ml de água destilada e 25ml de solução Zimmermann-Reinhart. 6. Deixar em repouso por 5 minutos. Adicionar 25ml de água destilada. 7. Titular com KMnO4 0,1N até a coloração levemente rósea persistente. CÁLCULO: Fe % = V x F x 0,5585
P ONDE: V = Volume gasto de KMnO4 0,1N F = Fator de correção do KMnO4 0,1N P = Peso da amostra em grama SOLUÇOES: a. Solução de ácido clorídrico 1 + 2 Diluir uma parte de ácido clorídrico P.A. em duas partes de água destilada. b. Solução cloreto estanoso 15% (P/V) Pesar 7,5g de cloreto estanoso P.A, e dissolver em bequer com +/- 20ml de solução de ácido clorídrico 1 + 2. Transferir para balão volumétrico de 50ml, e completar o volume com ácido clorídrico 1+2. NOTA: Preparar esta solução momentos antes do uso. c. Ácido clorídrico 20% (P/V) Medir 47,5ml de ácido clorídrico P.A. e transferir para balão volumétrico de 100ml, contendo aproximadamente 40ml de água destilada. Esfriar e completar o volume. d. Solução de Zimmermann-Reinhardt Dissolver 70g de MnSO4. H2O P.A. em 500ml de água destilada. Adicionar 125ml de H2SO4 P.A. e 125ml de H3PO4. Agitar e completar o volume para 1000ml com água destilada. e. Solução permanganato de potássio 0,1N Dissolver 3,3g de KMnO4 P.A. em bequer contendo aproximadamente 300/400ml de água destilada a 60/70ºC por duas horas. Esfriar, transferir quantitativamente para balão volumétrico de 1000ml e completar o volume. Deixar em repouso por 72 horas ao abrigo da luz. Filtrar em cadinho de vidro sinterizado ou amianto e estocar em frasco ambar. PADRONIZAÇÃO: Pesar exatamente 3,35g de oxalato de sódio (Na2C2O4) P.A. previamente seco em estufa de 105ºC por 2 horas. Dissolver em bequer contendo aproximadamente 100ml de água destilada. Transferir para balão volumétrico de 1000ml e completar o volume. Transferir 10ml de solução Na2C2O4 para erlenmeyer de 250ml e adicionar 10ml H2SO4 1 + 1. Aquecer a 70/80ºC e titular até coloração levemente rósea persistente por 30 segundos. CÁLCULO DO FATOR:
P NR N = V x E F = NE

ONDE: E = Equivalente grama de oxalato de sódio P = Peso do oxalato de sódio em mg na alíquota V = Volume de KMnO4 gasto na titulação NR = Normalidade real NE = Normalidade esperada F = Fator de correção MÉTODO Nº 44 - ZINCO VIA ÚMIDA APLICAÇÃO: Sais de zinco, PRINCÍPIO: Baseia-se na reação do zinco com cloreto de amônia em meio ácido. O excesso do ácido presente é titulado com solução alcalina de título conhecido. MATERIAL E EQUIPAMENTO: - Forno mufla - Balança analítica - Chapa aquecedora - Cápsula .ou cadinho de porcelana - Erlenmeyer de 500ml - Bureta capacidade 50ml divisão 1/10 - Almofariz com pistilo REAGENTES: - Ácido sulfúrico P.A. (d = 1,84g/l) - Hidróxido de sódio P.A. - Cloreto de amônio P.A. - Vermelho de metila P.A. PROCEOIMENTO: 1. Homogeneizar a amostra em almofariz. 2. Pesar 1,5g da amostra em cadinho ou cápsula de porcelana. 3. Levar à mufla e aumentar gradualmente a temperatura até 550/600ºC. Manter durante 2 horas. 4. Retirar a 250ºC e resfriar em dessecador até temperatura ambiente. 5. Transferir quantitativamente com o auxílio de bastão de vidro e água destilada, para erlenmeyer de 500ml, contendo 2,5g de cloreto de amônia. 6. Adicionar 50ml de H2SO4 1N. Aquecer até dissolução. 7. Adicionar 5 a 10 gotas de vermelho de metila a 0,1 % e titular com NaOH 1N, até viragem de vermelho para amarelo. CÁLCULO: Zinco % = (V1 )( F1) - (V2 x F2) x 0,032685 x 100
P ONDE: V1 = Volume de H2SO4 1N empregado F1 = Fator de correção de H2SO4 1N V2 = Volume de NaOH 1N gasto na titulação F2 = Fator de correção de NaOH 1N P = Peso da amostra em grama

SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução volumétrica de ácido sulfúrico 1N Medir 27,5ml de H2SO4 P.A. e transferir para balão volumétrico de 1000ml, contendo aproximadamente 700ml de água destilada. Esfriar e completar o volume. PADRONIZAÇÃO: Pesar com exatidão 1,0g de Carbonato de sódio anidro (Na2CO3) previamente seco. Adicionar 20ml da solução H2SO4 1N a ser padronizada e 4 gotas de vermelho metila. Titular gotejando a solução H2SO4 até o aparecimento da coloração rósea. Aquecer até a ebulição. Esfriar. Havendo permanência da cor rósea, considerar terminada. CÁLCULO DO FATOR:
P NR N = V x E F = NE ONDE: P = Peso do Na2CO3 em mg NR = Normalidade real da solução V = Volume gasto na titulação em ml E = Equivalente grama de Na2CO3 NE = Normalidade esperada b. Solução volumétrica de hidróxido de sódio 1N Dissolver aproximadamente 45g de NaOH P.A. em água destilada. Transferir quantitativamente para balão volumétrico de 1.000ml e completar o volume. PADRONIZAÇÃO: Pesar com exatidão 1,5g de Biftalato de Potássio previamente seco em estufa por duas horas. Dissolver em erlenmeyer com aproximadamente 75ml de água destilada. Adicionar 4 gotas de solução alcoólica de fenolftaleína 1%; titular com a solução de NaOH até coloração rósea. CÁLCULO DO FATOR:
P NR N = V x E F = NE ONDE: P = Peso do biftalato de potássio em mg NR = Normalidade real da solução V = Volume gasto na titulação em ml E = Equivalente grama de biftalato de potássio NE = Normalidade esperada c. Solução indicadora de vermelho de metila 0,1N Dissolver 0,1g de vermelho de metila em 60ml álcool etílico. Transferir para balão volumétrico de 100ml e completar o volume. d. Solução indicadora de fenolftaleína 1% Dissolver 1,0g fenolftaleína em aproximadamente 60ml.de álcool etílico P.A. Transferir para balão volumétrico de 100ml e completar o volume.

MÉTODO Nº 45- COBRE - VIA ÚMIDA APLICAÇÃO: Sais de çobre. PRINCÍPIO: Os sais cúpricos, quando tratados çom Iodeto de Potássio liberam iodo. O iodo cuproso formado é insolúvel em ácido acético e solúvel em excesso de Iodeto de Potássio. O iodo liberado pela ação do Acetato Cúprico sobre o Iodeto de Potássio é determinado por titulação com Tiossulfato de Sódio 0,1N. MATERIAL E EQUIPAMENTO: - Balança analítica - Erlenmeyer de 250 ml com tampa esmerilhada - Provetas graduadas de 50 e 100ml - Bureta ambar 50ml çom divisão 1/10 - Almofariz com pistilo - Chapa aquecedora REAGENTES: - Ácido nítrico P.A. (d = 1,42g/l) - Hidróxido de amônio P.A. - Ácido acético glacial P.A. - Amido P.A. - Tiossulfato de sódio P.A. - Bicromato de potássIo P.A. - Iodeto de potássio P.A. PROCEDIMENTO: 1. Homogeneizar a amostra em almofariz. 2. Pesar de 0,1 a 0,3g da amostra, dependendo do teor de cobre esperado. Transferir para erlenmeyer de 250ml. 3. Adicionar 12,5ml de HNO3 1 + 3 e dissolver. 4. Adicionar 12,5ml de água destilada e NHOH 1 + 1 gota a gota, até coloração azul escuro, formando um precipitado de CU(OH)2. 5. Ferver em chapa aquecedora por 3 minutos. 6. Acrescentar 3 a 4ml de ácido acético glacial, para dissolver o precipitado. Esfriar. 7. Adicionar 1,0g de Iodeto de Potássio e titular com solução padronizada de Na2S2O3 0,1N, até que a cor mude de marrom para amarelo. OBSERVAÇÃO: Titular rapidamente para que não ocorra perda por oxidação. 8. Adicionar 1ml de solução de amido 1% e prosseguir a titulação cautelosamente até que a cor azul desapareça. CÁLCULO: Cobre % = 0,636 x V x F
P ONDE: V = Volume de Na2S203 0,1N gasto na titulação F = Fator de correção do Na2S2O3 0,1N P = Peso da amostra em grama SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Ácido Nítrico 1 + 3

Transferir 250ml de HNO3 P.A. para balão volumétrico de 1000ml, contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. b. Hidróxido de Amônio 1 + 1 Transferir 500ml de hidróxido de amônio P.A. para balão volumétrico de 1000ml e completar o volume com água destilada. c. Solução de Amido 1% Pesar 1 grama de amido em bequer de 100ml. Adicionar gotas de água destilada fria até formar uma pasta lisa e fina. Acrescentar 80ml de água fervente, agitando constantemente com um bastão de vidro. Ferver por um minuto. Esfriar e transferir para balão volumétrico de 100ml completando o volume. OBSERVAÇÃO: Preparar esta solução momentos antes do uso. d. Solução de Ácido Clorídrico 1N Transferir 83,5ml HCI P.A. para balão volumétrico de 1000ml, contendo aproximadamente 500ml de água destilada. Esfriar e completar o volume. e. Solução Volumétrica de Tiossulfato de Sódio 0,1N Pesar 24,9g de Na2S2O3.5H20 e diluir em 1000ml de água destilada. PADRONIZAÇAO: Pesar 0,20g de K2Cr2O7 seco a 100ºC / 2 horas em erlenmeyer de 250ml, com tampa esmerilhada. Dissolver com 80ml de água destilada contendo 2 gramas de iodeto de potássio. Adicionar 20ml de HCI 1N e guardar imediatamente no escuro por exatamente 10minutos. Titular imediatamente com Na2S2O3.5H2 0,1N até viragem de cor marrom para amarelo. Adicionar 1 ml de solução de amido 1% e prosseguir a titulação até o desaparecimento da coloração azul. CÁLCULO DO FATOR:
P NR N = V x E F = NE ONDE: NR = Normalidade real da solução P = Peso de K2Cr2O7 em miligrama V = Volume gasto na titulação em ml E = Equivalente grama de K2Cr2O7 NE = Normalidade esperada F = Fator de Correção MÉTODO Nº 46 - COBALTO - VIA ÚMIDA APLICAÇÃO: Sulfato de cobalto. PRINCÍPIO: Fundamenta-se na titulação complexométrica do cobalto por uma solução de sal sódico do EDTA em presença do indicador. MATERIAL E EQUIPAMENTO: - Balança analítica - Balão volumétrico 500ml

- Bequer 400ml - Pipeta volumétrica 25ml - Bureta50 ml divisão 1/10 REAGENTES: - EDTA P.A. sal sódico - Hidróxido de amônio P.A. - Acetato de amônio P.A. - Murexida P.A. PROCEDIMENTO: 1. Pesar 5,0g da amostra e dissolver em água destilada com leve aquecimento. Esfriar e filtrar se necessário com papel de filtro qualitativo, recolhendo em balão volumétrica de 500mI. Completar o volume com água destilada. 2. Transferir uma alíquota de 25ml para bequer de 400ml. Adicionar 5,0g de acetato de amônio e ajustar o pH com hidróxido de amônia para um valor próximo de 12. 3. Adicionar uma pitada de murexida e titular com EDTA 0,1M padronizado, até viragem de amarelo para lilás, passando pela coloração vinho intermediária. CÁLCULO: Cobaldo % = V x F x 0,5893
P ONDE: V = Volume de EDTA 0,1M gasto na titulação F = Fator de correção de EDTA 0,1M P = Peso da amostra em grama OBSERVAÇAO: Se alguma amostra for insolúvel em água, recomeçar a análise, procedendo a uma digestão com +/- 30ml de HCI 1 + 1. Ferver em bequer de 250ml, provido de vidro de relógio, até perto da secura. SOLUÇOES REAGENTES E VOLUMÉTRICAS: a. Solução volumétrica de EDTA 0,1 M Pesar com exatidão 37,22g de EDTA, dissolver e transferir para balão volumétrico de 1000ml. Completar o volume com água destilada. NOTA: Guardar esta solução em frasco de polietileno PADRONIZAÇÃO: Pesar 2,5275g de Carbonato de cálcio P.A. previamente seco e transferir para erlenmeyer de 250ml. Adicionar ácido clorídrico 10% em quantidade suficiente para dissolver o sal. Transferir quantitativamente para balão volumétrico de 250ml com água destilada, completando o volume. Pipetar uma alíquota de 25ml e adicionar 5ml de trietanolamina 20% e 3 ml NaOH4N. Adicionar uma pitada de calcon e titular com EDTA 0 1M. CALCULO DOFATOR:
P NR N = V x E F = NE ONDE:

P = Peso do CaCO3 em miligramas NR = Normalidade real da solução V = Volume gasto na titulação em ml E = Equivalente grama de CaCO3 N~ = Normalidade esperada MÉTODO Nº 47 - IODO - VIA ÚMIDA APLICAÇÃO: Iodatos de cálcio e potássio. PRINCÍPIO: Os sais iodatados, quando tratados com Iodeto de potássio em meio ácido, liberam o iodo. O iodo liberado pela ação do iodeto de potássio e ácido sulfúrico é determinado por titulação com tiossulfato de sódio 0,1N. MATERIAL E EQUIPAMENTO: - Erlenmeyer 300 ml com tampa esmerilhada - Proveta 100ml - pipeta de 10ml - Bureta ambar de 50ml, divisão 1/10 REAGENTES: - Iodeto de potássio P.A. - Acido sulfúrico P.A. (d = 1,84g/l) - Tiossulfato de sódio P.A.. - Amido P.A. - Bicromato de potássio P.A. PROCEDIMENTO: 1. Pesar de 0,1g da amostra em erlenmeyer de 300ml com tampa esmerilhada. 2. Adicionar 100ml de KI 10%. 3. Adicionar 10ml H2SO4 10%. 4. Agitar até dissolução e deixar em repouso por 10 minutos ao abrigo da luz. 5. Titular com Na2S203 0,1 N até cDIDral;ãD amarelo pálido. 6. Adicionar 1ml de solução de amido e titular até descoloração total do azuI. CÁLCULO: Iodo = V x F x 0,2116
P ONDE: V = Volume de Na2S2O3 0,1N gasto F = Fator de correção do Na252O3 0,1N P = Peso da amostra em grama SOLUÇÕES REAGENTES E VOLUMÉTRICAS: a. Solução de iodeto de potássio 10% Pesar 50g de KI P.A. e dissolver em bequer com +/-200 ml de água destilada. Transferir para balão volumétrico de 500ml e completar o volume. NOTA: Preparar esta solução, momentos antes do uso. b. Solução de ácido sulfúrico 10% Medir 5,6ml de H25O4 P.A. (d = 1,84g/l) e transferir para balão volumétrico de 100ml, contendo +/- 70ml de água destilada. Esfriar e completar o volume.

c. Solução de Amido 1% Pesar 1 grama de amido P.A. e transferir para bequer de 250ml. Adicionar +/- 50ml de água destilada e aquecer até ebulição. Esfriar. Transferir para balão volumétrico de 100ml e completar o volume. d. Solução volumétrica de Na2S2O3 0,1N Pesar 24,9g de Na2S2O3. 5H2O P.A. e 0,2g de Na2CO3 P.A. anidro. Transferir para balão volumétrico de 1.000ml e dissolver com água destilada completando o volume. PADRONIZAÇÃO: Pesar 0,2g de K2Cr2O7 seco em erlenmeyer de boca esmerilhada. Dissolver com 80ml de água destilada. Adicionar 2g de KI P.A. e 20ml de HCl 1N. Tampar e guardar no escuro por 10 minutos. Efetuar uma pré-titulação com solução de Tiossulfato de Sódio 0,1N até coloração amarela. Adicionar 1ml de solução de amido 1% e prosseguir a titulação até a descoloração total do azul. CÁLCULO DO FATOR:
P NR N = V x E F = NE ONDE: NR = Normalidade real da solução P = Peso de K2Cr2O7 em miligrama V = Volume gasto na titulação E = Equivalente grama de K2Cr2O7 NE = Normalidade esperada F = Fator de Correção
MÉTODOS MICROBIOLÓGICOS MÉTODO Nº 01 - CONTAGEM DE BOLORES E LEVEDURAS APLICAÇÃO: Produtos ou sub-produtos de origem animal, rações e concentrados. MEIOS DE CULTURA: - Agar oxitetraciclina glicose extrato de levedura - Água peptonada a 1%, tamponada. PROCEDIMENTO: 1. Pesar assepticamente 25g da amostra homogeneizada, adicionar 225ml de água peptonada a 1%, tamponada. Homogeneizar. Preparar as diluições necessárias. 2. Transferir, de cada diluição 1ml para placas de Petri, em duplicata. 3. Adicionar em cada placa 15ml de agar oxitetraciclina glicose extrato de levedura, previamente fundida e mantida a 45ºC. Homogeneizar e deixar solidificar. 4. Incubar a 22/25ºC por 5 a 7 dias. 5. Selecionar placas com 10 - 100 colônias. 6. Contar as colônias e expressar o resultados como número de bolores e leveduras viáveis por grama do produto.

PREPARO DO MEIO DE CULTURA A. Agar Oxitetraciclina Glicose Extrato de Levedura Extrato de Levedura 5,0g D (+) glicose 10,0g Ágar 15,0g Suspender os componentes em um litro de água destilada ou deionizada, ferver até a completa dissolução. Distribuir em frascos e esterilizara 121ºC por 15 minutos. Antes da utilização adicionar 0,1g de oxitetraciclina por 1 litro do meio. Preparar solução aquosa do antibiótico. pH final: 6,5 +/-0,2 B. Agua Peptonada a 1% Tamponada Peptona de carne 10,0g Cloreto de sódio 5,0g Hidrogeno fostato de sódio (NaHPO4) 9,0g Dihidrogenofosfato de potássio (KH2P)4) 5,0g Dissolver os componentes em 1 litro de água destilada ou deionizada. Distribuir em frascos volumes de 225ml. Autoclavar a 121ºC por 15 minutos. pH final: 7,2 +/- 0,2 MÉTODO Nº 02 - CONTAGEM DE E. COLI I APLICAÇÃO: Produtos ou sub-produtos de origem animal, rações e concentrados. MEIOS DE CULTURA: - Água peptonada a 1%, tamponada. - Ágar cristal violeta vermelho neutro bile - Caldo triptona - Caldo VM - VP - Ágar citrato de Simmons - Ágar nutritivo PROCEDIMENTO: 1. Pesar assepticamente 25g da amostra homogeneizada e adicionar 225ml de água peptonada a 1%. Homogeneizar. Manter à temperatura ambiente P9r 2 horas para revitalizar as células debilitadas. Preparar as diluições necessárias. 2. Pipetar alíquotas de 1ml de cada diluição e transferir para placas de Petri, em duplicata, acrescentar 15ml de agar cristal violeta vermelho neutro bile, previamente fundido e mantido a 45ºC. Homogeneizar e deixar solidificar. Acrescentar uma segunda camada do mesmo agar (aproximadamente 10ml) e deixar solidificar. 3. Incubar a 44ºC por 24 horas. 4. Selecionar placas que apresentem entre 15 - 150 colônias de cor roxo avermelhada (fucsia) rodeadas por um halo de precipitação da mesma cor. 5. Contar as colônias típicas. Confirmar realizando as provas de vermelho de metila, Voges-Proskauer, indol e citrato. 6. Repicar em agar nutritivo inclinado, 5 colônias ou um numero igual à raiz quadrada das colônias contadas na placa. Incubar a 35ºC por 24 horas. 7. Transferir a cultura do agar inclinado para os seguintes meios: caldo triptona, caldo VM-VP e agar citrato de SIMMONS. 8. Incubação: Caldo Triptona e Caldo VM-VP - 48 horas a 35ºC.
Agar Citrato de SIMMONS - 96 horas a 35ºC. 9. Considerar como E. coli as culturas que apresentarem o resultado do IMVIC como: + + - - ou - + - -

CÁLCULOS: Calcular o número de E.coli por grama do produto, utilizando a seguinte fórmula: R = C x c x d
r ONDE: R = resultado C = Colônias contadas c = Colônias confirmadas r = Colônias repicadas d = diluição da amostra PREPARO DO MEIO DE CULTURA A. Água Peptonada a 1% tamponada Peptona de carne 10,0g Cloreto de sódio 5,0g Hidrogeno fosfato de sódio (NaHPO4) 9,0g Dihidrogeno fosfato de potássio(KH2PO4) 5,0g Dissolver os componentes em um litro de água destilada ou deionizada. Distribuir em frascos volumes de 225ml. Autoclavar a 121ºC por 15 minutos.
pH final: 7,2 +/- 0,2 B. Ágar Cristal Violeta Vermelho Neutro Bile Peptona de carne 7,09g Extrato de levedura 3,0g Lactose 10,0g Cloreto de sódio 5,0g Sais biliares 1,5g Vermelho neutro 0,03g Cristal .violeta 0,002g Ágar ágar 15,0g Suspender os componentes em 1 litro de água destilada ou deionizada. Deixar em repouso por 15 minutos. Ferver até dissolução completa. Distribuir em frascos e esterilizar em vapor fluente por 30 minutos.
pH final: 7,4 +/- 0,1 c. Caldo Triptona Triptona 10,0g Cloreto de sódio 5,0g Dissolver os componentes em um litro de água destilada ou deionizada, distribuir em tubos e esterilizar a 121ºC por 15 minutos.
pH final: 7,3 +/- 0,3 d. Caldo VM-VP Prciteose-peptona 7,0g Glicose 5,0g Fosfato dipotássico 5,0g Dissolver os compohentes em um litro de água destilada ou deionizada, distribuir 10ml por tubos e esterilizar a 121ºC por 15 minutos.
pH final: 7,5 +/- 0,1 e. Ágar Citrato de Simmons Sulfato de magnésio 0,2g

Dihidrogeno fosfato de amônio 1,0g Dihidrogeno fostato de potássio 1,0g Citrato de sódio tribásico 2,0g Cloreto de sódio 5,0g Azul de bromotimol 0,08g Ágar ágar 12,0g Suspender os componentes em 1 litro de água destilada ou deionizada, deixar em repouso por 15 minutos. Ferver até dissolução completa, distribuir em tubos e esterilizar a 121ºC por 15 minutos. Inclinar os tubos antes da solidificação.
pH final: 6,6 +/- 0,1 F. Ágar Nutritivo Extrato de levedura 2,0g Extrato de carne 1,0g Peptona de carne 5,0g Cl9reto de sódio 5,0g Ágar ágar 15,0g Suspender os componentes em 1 litro de água destilada ou deionizada, deixar em repouso por 15 minutos. Ferver até dissolução completa. Distribuir em frascos e esterilizar a 121ºC por 15 minutos.
pH final: 7,0 +/- 0,2 MÉTODO Nº 03 - CONTAGEM DE E. COLI II APLICAÇÃO: Produtos sub-produtos de origem animal, rações e concentrados. MEIOS DE CULTURA: - Água peptonada a 1%, tamponada. - Agar cristal violeta vermelho neutro bile - Caldo lauril triptose-MUG (4 - metilumbelliferil Beta - D - glicuronídeo) PROCEDIMENTO: 1. Pesar assepticamente 25g da amostra, adicionar 225ml de água peptonada a 1%. Homogeneizar. Manter à temperatura ambiente por 2 horas, para revitalizar as células debilitadas. Preparar as diluições necessárias. 2. Transferir de cada diluição 1ml para placas de Petri, em duplicata. 3. Adicionar em cada placa 15ml de agar cristal violeta vermelho neutro bile, previamente fundido e mantido a 45ºC. Homogeneizar e deixar solidificar. Acrescentar uma segunda camada menos espessa de aproximadamente 10 ml. 4. Incubar a 44ºC por 24 horas. 5. Características das colônias típicas: cor roxo - avermelhada (fucsia) e 0,5 a 2mm de diâmetro. 6. Selecionar as placas com 15 - 150 colônias típicas, efetuar a contagem e anotar. CONFIRMAÇÃO: 1. Repicar 3 a 5 colônias típicas em caldo lauril triptose - MUG. 2. Incubar a 35ºC por 20 horas. 3. Fazer a leitura em câmara com lâmpada ultravioleta a 336nm. A Beta-Glicuronidase da E.Coli hidrolisa o MUG, incolor, liberando o composto 4-metilumbelliferona fortemente fosforescente. CÁLCULOS: O número de E.coli é calculado aplicando a seguinte fórmula: R = C x c x d

r ONDE: R = resultado C = Colônias contadas c = colônias confirmadas r = colônias repicadas d = diluição da amostra PREPARO DO MEIO DE CULTURA. a. Água Peptonada a 1% Tamponada Peptona de carne 10,0g Cloreto de sódio 5,0g Hidrogeno fosfato de sódio (NaHPO4) 9,0g Dihidrogeno fosfato de potássio(KH2PO4) 5,0g Dissolver os componentes em um litro de água destilada ou deionizada. Distribuir em frascos volumes de 225ml. Autoclavar a 121ºC por 15 minutos.
pH final: 7,2 +/-0,2 b. Ágar Cristal Violeta Vermelho Neutro Bile Peptona 7,0g Extrato de levedura 3,0g Lactose 10,0g Cloreto de sódio 5,0g Sais biliares (nº. 3) 1,5g Vermelho neutro 0,03g Cristal violeta 0,002g Ágar ágar 15,0g Suspender os componentes em 1 litro de água destilada e/ou deionizada. Ferver até dissolução completa. Distribuir em frascos e esterilizar em vapor fluente por 30 minutos.
pH final: 7,4 +/- 0,1 c. Caldo Lauril Triptose - MUG Triptose 20,0g Hidrogenofosfato dipotássico (K2HPO4) 2,75g Dinidrogenofosfato de potássio (KH2PO4) 2,75g Cloreto de sódio 5,0g Lactose 5,0g Lauril sulfato de sódio 0,1g 4-metilumbelliferil - Beta - D – glucoronídeo 100,0mg Dissolver os componentes em um litro de água destilada e/ou deionizada. Distribuir em tubos de ensaio com tubos de Durhan, esterilizar a 121ºC por 15 minutos.
pH final: 6,8 +/- 0,1 MÉTODO Nº 04 - PESQUISA DE SALMONELLA. APLICAÇÃO: Produtos e sub-produtos de origem animal, rações e concentrados. MEIOS DE CULTURA: - Caldo Lactosado ou Água Peptona a 1%, tamponada - Caldo Rappaport Modificado - Caldo Selenito Cistina - Ágar Verde Brilhante Vermelho de Fenol Lactose Sacarose

- Ágar para Enterobactérias (Hektoen) - Ágar Xilose Lisina Desoxicolato (XLD) - Ágar Tríplice Açúcar Ferro (TSI) - Ágar Lisina Ferro (LIA) - Ágar Citrato de Simmons - Ágar Nutritivo - Meio SIM - Caldo Uréia - Caldo KCN PROCEDIMENTO: 1. Pré-Enriqueclmento Pesar assepticamente 25g da amostra homogeneizada, adicionar 225m de caldo lactosado ou água peptonada a 1% tamponada. Incubar a 35 C por 18 - 24 horas. 2. Enriquecimento Seletivo Pipetar duas porções de 1ml da cultura pré-enriquecida, uma para tubo contendo 10 ml de caldo Rappaport modificado e outra para tubo contendo 10ml de caldo selenito cistina. Incubar a 42 – 43ºC por 24 - 36 horas. 3. Isolamento e Seleção A partir dos caldos de enriquecimento seletivo semear em placas de ágar verde brilhante ou ágar Hektoen e ágar XLD. Incubar a 35 - 37oC por 24 horas. As colônias típicas de SALMONELLA em ágar verde brilhante apresentam-se incolores ou de coloração rosa avermelhada, entre translúcidas ou ligeiramente opacas. Algumas colônias poderão apresentar coloração verde amarelada, quando estiverem rodeadas do microorganismos fermentadores de lactose. Em ágar Hektoen apresentam-se azuladas com ou sem centro escuro. Em ágar XLD, apresentam-se transparentes da mesma cor do meio, às vezes com centro escuro. De cada placa repicar 3 a 5 colônias típicas em tubos com caldo uréia. Incubar a 37ºC por 24 horas. Repicar em ágar TSI, LIA, agar citrato SIMMONS os tubos que apresentarem resultados negativos na prova da uréia. lncubar a 35 – 37ºC por 24 - 48 horas o TSI e LIA e até 96 horas o ágar citrato. Para leitura e interpretação verificar os quadro a seguir: 1 Ágar TSI.
MICROORGANISMO COLUNA VERTICAL BISEL FORMAÇÃO H2S Salmonella typhy Amarelo Sem alteração
ou vermelho Sem alteração + só na parte ou vermelho superior da coluna vertical
S.paratyphi A e S.choleraesuis
Amarelo com produção de gás
Sem alteração ou vermelho
S. pullorum Amarelo com produção de gás
Sem alteração ou vermelho
S.paratyphi B, S.typhimurium e S.enteritidis
Amarelo com produção de gás
Sem alteração ou vermelho
+(coluna vertical preta)
S.gallinarum Amarelo Sem alteração ou vermelho
+(coluna vertical preta)
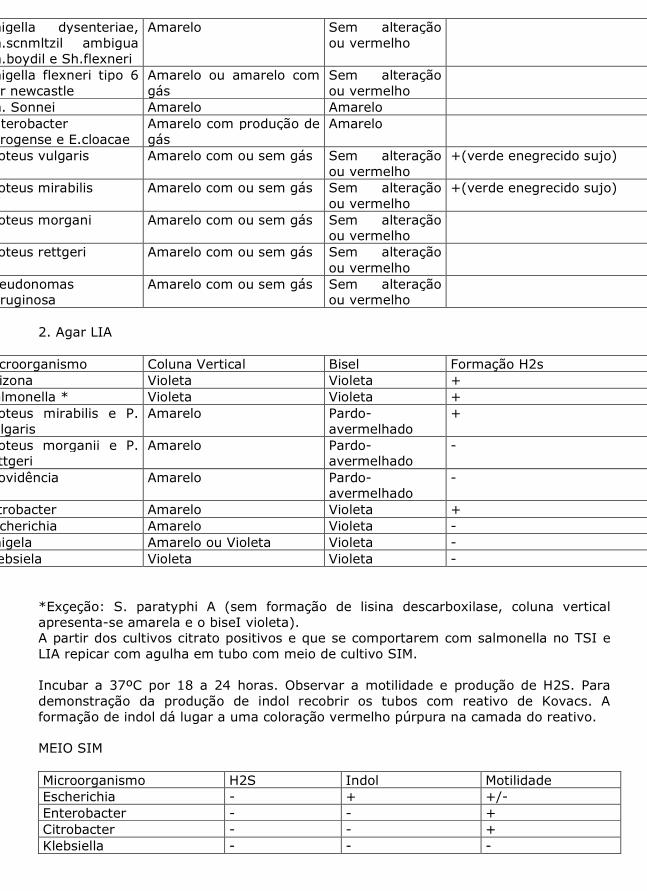
Shigella dysenteriae, Sh.scnmltzil ambigua Sh.boydil e Sh.flexneri
Amarelo Sem alteração ou vermelho
Shigella flexneri tipo 6 var newcastle
Amarelo ou amarelo com gás
Sem alteração ou vermelho
Sh. Sonnei Amarelo Amarelo Enterobacter aerogense e E.cloacae
Amarelo com produção de gás
Amarelo
Proteus vulgaris Amarelo com ou sem gás Sem alteração ou vermelho
+(verde enegrecido sujo)
Proteus mirabilis Amarelo com ou sem gás Sem alteração ou vermelho
+(verde enegrecido sujo)
Proteus morgani Amarelo com ou sem gás Sem alteração ou vermelho
Proteus rettgeri Amarelo com ou sem gás Sem alteração ou vermelho
Pseudonomas aeruginosa
Amarelo com ou sem gás Sem alteração ou vermelho
2. Agar LIA
Microorganismo Coluna Vertical Bisel Formação H2s Arizona Violeta Violeta + Salmonella * Violeta Violeta + Proteus mirabilis e P. vulgaris
Amarelo Pardo-avermelhado
+
Proteus morganii e P. rettgeri
Amarelo Pardo-avermelhado
-
Providência Amarelo Pardo-avermelhado
-
Citrobacter Amarelo Violeta + Escherichia Amarelo Violeta - Shigela Amarelo ou Violeta Violeta - Klebsiela Violeta Violeta -
*Exçeção: S. paratyphi A (sem formação de lisina descarboxilase, coluna vertical apresenta-se amarela e o biseI violeta). A partir dos cultivos citrato positivos e que se comportarem com salmonella no TSI e LIA repicar com agulha em tubo com meio de cultivo SIM. Incubar a 37ºC por 18 a 24 horas. Observar a motilidade e produção de H2S. Para demonstração da produção de indol recobrir os tubos com reativo de Kovacs. A formação de indol dá lugar a uma coloração vermelho púrpura na camada do reativo. MEIO SIM Microorganismo H2S Indol Motilidade Escherichia - + +/- Enterobacter - - + Citrobacter - - + Klebsiella - - -

Salmonella + - +* Shigella - +/- - Proteus vulgaris + + - Proteus Mirabilis + - + Morganella - + + Rettgeralla - + + Arizona + - + Hafnia - - + Serratia - - + Providência - + + Edwardsiella + + + Y-enterocolítica - -(+) -
(*) S.pullorum e S.gallinarum são imóveis. Dos cultivos que mostrarem os resultados a seguir; realizar prova sorológica.
- Uréia - negativa - Produção H2S - positiva ou negativa - Descarboxilação lisina - positiva - Utilização de citrato como única fonte de carbono - positiva - Produção de indol - negativa - Motilidade - positiva - Crescimento em KCN - negativa
Teste sorológico Teste de aglutinação: em uma lâmina fazer uma suspensão espessa de colônias suspeitas em solução salina a 0,85%. Adicionar 1 - 2 gotas do anti-soro. Homogeneizar imprimindo movimento de rotação. Leitura: Fazer a leitura no máximo em um minuto. Aglutinação - positivo Sem aglutinação = negativo OBSERVAÇÕES: - A aglutinação na suspensão de bactérias em solução salina a 0,85% significa uma reação inespecífica, portanto, negativa para salmonela. - Deve-se realizar o teste com anti-soros polivalentes somáticos "O" e flagelares "H". - Se o teste de aglutinação com anti-soros for positivo, proceder a tipificação final. PREPARO DO MEIO DE CULTURA a. Caldo Lactosado Extrato de levedura 3,0g Peptona 5,0g Lactose 5,0g Dissolver os componentes em um litro de água destilada ou deionizada, dlstribuir em frascos com 225ml e esterllizar a 121ºC por 15 minutos.
pH final: 6,9 +/- 0,1 b. Água Peptonada a 1% Tamponada Peptona de carne 10,0g Cloreto de sódio 5,0g Hidrogeno fosfato de sódio (NaHPO4) 9,0g

Dihidrogeno fosfato de potássio(KH2PO4) 5,0g Água destilada 1000 ml Dissolver os componentes em um litro de água destilada ou deionizada. Distribuir em frascos com de 225ml, esterilizar a 121ºC por 15 minutos.
pH final: 7,2 +/-0,1 c. Caldo Rappaport Modificado Triptona 10,0g Cloreto de sódio 8,0g KH2PO4 1,5g Dissolver os componentes em um litro de água destilada ou deionizada. Autoclavar a 121ºC por 15 minutos. Após o resfriamento, acrescentar: 100ml de cloreto de magnésio a 40%, 100ml de solução de tiossulfato de sódio a 2% e 40ml de solução de amarelo de metacromo a 2%. Homogeneizar e distribuir 10ml em cada tubo esteril e adicionar 0,2ml da solução iodo-iodeto. Em lugar da solução de amarelo de metacromo, poderá ser utilizada 30ml de solução de verde malachita a 0,4%. - Solução de Cloreto de Magnésio Cloreto de magnésio hexahidratado 40g Água destilada q.s.p. 100ml Esterilizar em vapor fluente por 30 minutos. - Solução de Tiossulfato de Sódio a 2% Tiossulfato de sódio 2g Água destilada q.s.p. 100ml Esterilizar a 121ºC por 15 minutos., - Solução de Amarelo de Metacromo a 2% Amarelo de metacromo 2g Água destilada q.s.p. 100ml Esterilizar a 121ºC por 15 minutos. - Solução de Iodo-lodeto Iodo 5g Iodeto de potássio 8g Água destilada 40ml d. Caldo Selenito-cistina Triptona 7,0g L .(-) cistina 0,01g Lactose 4,0g Fosfato dissódico dihidratado 10,0g Bi-selenito de sódio 4,0g Dissolver os componentes em um litro de água destilada ou deionizada. Se necessário, pode-se aquecer em banho-maria fervente por 10 minutos. Distribuir em frascos estereis.
pH final: 7,0 +/- 0,2 Usar o meio no mesmo dia da preparação. e.Ágar Verde Brilhante - Vermelho de Fenol Lactose-Sacarose (BPLS) Proteose peptona 10,0g Extrato de levedura 3,0g

Lactose 10,0g Sacarose 10,0g Cloreto de sódio 5,0g Fosfato dissódico 2,0g Vermelho defenol 0,08g Verde brilhante 0,0125g Ágar agar 12,0g Dissolver os componentes em 1 litro de água destilada, deixar em repouso 15 minutos. Ferver até completa dissolução. Esterilizar a 121ºC por 15 minutos.
pH final: 6,9 +/- 0,1 f. Agar para Enttrobacterias de HeKtoen Proteose-peptona 12,0g Cloreto de sódio 5,9g Extrato de levedura 3,0g Sacarose 12,0g Lactose 12,0g Salicina 2,0g Tiossulfato de sódio 5,0g Citrato de ferro e amônia 1,5g Sais biliares 9,0g Azul de bromotimol 0,064g Fucsina ácida 0,04g Ágar agar 13,5g Dissolver em 1 litro de água destilada deixar em repouso, Aquecer em banho-maria ou vapor fluente até completa dissolução. Não esterilizar.
pH final: 7,5 +/- 0,1 g. Agar Xilose-Lisina - Desoxicolato (Ágar XLD) Extrato de levedura 3,0g Cloreto de sódio 5,0g D(+) Xilose 3,5g Lactose 7,5g Sacarose 7,5g L(+) lisina 5,0g Desoxicolato de sódio 2,5g Tiossulfato de sódio 6,8g Citrato de amônia e ferro 0,8g Vermelho de fenol 0,08g Ágar agar 13,5g Dissolver em 1 litro de água destilada aquecendo brevemente para completa dissolução. Verter em placas. Usar imediatamente. Não esterilizar.
pH final: 7,4; +/- 0,2 h. Caldo Uréia Extrato de levedura 0,1g Dihldrogenofosfato de potássio 9,1g Hidrogenofosfato dissódico 9,5g Uréia 20,0g Vermelho fenol 0,01g Dissolver os componentes em 1 litro de água destilada e esterilizar por filtração, distribuindo em tubo (cerca de 3 ml/tubo) Não esterelizar em autoclave.

pH final: 6,8 +/- 0,1 i. Ágar Lisina Ferro (LIA) Peptona 5,0g Extrato de levedura 3,0g Glicose 1,0g Citrato férrico amoniacal 0,5 g Tiossulfato de sódio 0,04g Púrpura de bromocresol 0,02 g Agar Agar 13,5g Suspender os componentes em 1 litro de água destilada e/ou deionizada. Deixar em repouso por 15 minutos. Ferver até dissolução completa. Distribulr em tubos e esterelizar a 121ºC, por 15 minutos. Deixar o meio solidificar com os tubos em posição levemente inclinada (bico de flauta). pH final: 6,7 +/- 0,1 j. Ágar Tríplice Açúcar Ferro (TSI) Extrato de carne 3,0g Extrato de levedura 3,0g Peptona de caseína 15,0g Peptona de carne 5,0g Lactose 10,0g Sacarose 10,0g D(+) glicose 1,0g Citrato de amônia e ferro 0,5g Cloreto de Sódio 5,0g Tiossulfato de sódio 0,3g Vermelho fenol 0,024g Agar agar 12,0g Suspender os componentes em 1 litro de água destilada e/ou deionizada. Deixar em repouso por 15 minutos. Ferver até dissolução completa. Distribuir em tubos e esterilizar a 121ºC, por 15 minutos. Solidificar o ágar com os tubos em posição inclinada, de modo a obter uma coluna de +/- 3cm e sobre ela uma superfície inclinada de igual comprimento.
pH final: 7,4 +/- 0,1 k. Ágar Citrato de Simmons Dihidrogenofosfato de amônio 1,0g Oihidrogenosfosfato de potássio 1,0g Cloreto de sódio 5,0g Citrato de sódio 2,0g Sulfato de magnésio 0,2g Azul de bromotimol 0,08g Ágar agar 12,0g Dissolver em 1 litro de água destilada e distribuir em tubos. Esterilizar por 15 minutos a 121ºC. Inclinar os tubos, antes da solidificação.
pH final: 6,6 +/- 0,1 l. Meio Sim Peptona de caseína 25,0g Peptona de carne 6,6g Citrato amônio e ferro III 0,2g

Tiossulfato de sódio 0,2g Ágar agar 3,0g Dissolver os componentes em 1 litro de água destilada e distribuir em tubos até cerca de 4cm de altura e esterilizar a 121ºC por 15 minutos.
pH final: 7,3 +/- 0,1 m. Caldo KCN Proteose-peptona 25,0g Dihidrogenosfosfato de potássio 0,225g Hidrogenofosfato dissódico 5,64g Cloreto de sódio 5,0g Aditivo-cianeto de potássio 0,075g Dissolver em 1 litro de água destilada e deionizada e esterilizar em autoclave por 15 minutos. Deixar atingir a temperatura ambiente, adicionar 15 ml/l de uma solução estéril a 0,5% de cianeto de potássio e distribuir em tubos com tampa de rosca (4ml por tubo). Não aquecer.
pH final: 7,6 +/- 0,1 n. Ágar Nutritivo Extrato de levedura2,0g Extrato de carne1,0g Peptona5,0g Ágar ágar15,0g Cloreto de sódio5,0g Suspender os componentes em 1 litro de água destilada e/ou deionizada. Deixar em repouso por 15 minutos. Ferver até dissolução completa. Distribuir em tubos e esterilizar a 121ºC, por 15 minutos.
pH final: 7,0 +/- 0,2 Após a esterilização, colocar os tubos em posição inclinada, até solidificar o ágar. MÉTODO Nº 05 - CONTAGEM DE ENTEROBACTÉRIAS TOTAIS, VIÁVEIS APLICAÇÃO: Produtos e sub-produtos de origem animal, rações e concentrados. MEIO DE CULTURA: - Água peptonada a 1%, tamponada. - Ágar cristal violeta vermelho neutro bile glicose. PROCEDIMENTO: 1. Pesar assepticamente 25 gramas da amostra homogeneizada, adicionar 225ml de água peptonada a 1%. Homogeneizar. Manter a temperatura ambiente por 2 horas, para revitalizar as células debilitadas. Preparar as diluições necessárias. 2. Transferir alíquotas de 1ml de cada diluição e transferir para placas de Petri em duplicata. 3. Acrescentar em cada placa +/- 15ml de ágar cristal violeta vermelho neutro glicose, mantido a 45ºC. Homogeneizar e deixar solidificar. Cobrir com uma segunda camada do msâmo ágar (aproximadamente 10ml) deixar solidificar. Incubar a 35 - 3.7ºC por 24 horas. 4. Selecionar placas que apresentem entre 20 - 20 colônias roxo-avermelhada (fucsia) rodeadas por halo de precipitação da mesma cor. Contar as colônias. 5. Confirmar efetuando o teste de oxidase em 5 colônias ou em número igual a raiz quadrada das colônias contadas na placa. As enterobactérias são oxidase negativa.

CÁLCULO: Calcular o número de enterobactérias totais viáveis por grama do produto aplicando a seguinte fórmula: R = C x c x d
r ONDE: R = Resultado C = Colônias contadas c = Colônias confirmadas d = Diluiçgo da amostra na placa r = Colônias repicadas PREPARO DOS MEIOS DE CULTURA: a. Água Peptonada a 1% Tamponada Peptona de carne 10,0g Cloreto de sódio (NaCl) 5,0g Hidrogenofosfato de sódio (NaHPO4) 9,0g Dihidrogeno fosfato de potássio(KH2PO4) 5,0g Dissolver os componentes em 1 litro de água destilada. Distribuir 225ml em frascos. Autoclavar a 121ºC por, 15 minutos.
pH final: 7,2 +/- 0,2 b. Ágar Cristal Violeta Vermelho Neutro Bile Glicose Extrato de levedura 3,0g Peptona7,0g Cloreto de sódio 5,0g Saisbiliares 1,5g Glicose 10,0g Vermelho neutro 0,03g Cristal violeta 0,002g Agar ágar 15,0g Suspender os componentes em 1 litro de água destilada e/ou deionizada. Deixar em repouso por 15 minutos. Ferver até dissolução completa; Esfriar na água da torneira até- +/-50ºC, manter assim até sua utilização. Não esterilizar em autoclave. (Of. nº 168/91)