procedimento tÉcnico de avaliaÇÃo e otimizaÇÃo...
TRANSCRIPT

Comissão de Laboratório Clínico e Genética
Humana de Bioquímicos Clínicos
Associação Nacional de Bioquímicos
PROCEDIMENTO TÉCNICO DE
AVALIAÇÃO E OTIMIZAÇÃO DE
CARTAS DE CONTROLO DE
QUALIDADE INTERNO NO
LABORATÓRIO CLÍNICO
Colégio de Biologia Humana e Saúde
Ordem dos Biólogos

14-05-2016 2
O presente procedimento técnico de avaliação e otimização de cartas de controlo de qualidade
interno no laboratório clínico é resultante do trabalho conjunto entre a Comissão de Laboratório Clínico
e Genética Humana de Bioquímicos Clínicos da Associação Nacional de Bioquímicos (LabGen-ANBIOQ) e
o Colégio de Biologia Humana e Saúde da Ordem dos Biólogos (CBHS). Foi elaborado com base na visão
comum de equipa plural e de consciência integradora de diferentes competências, experiências e
conhecimentos, e que se pretende alargada a toda a comunidade de profissionais de saúde.
É competência da Comissão de Qualidade da LabGen-ANBIOQ, sistematizar, concentrar e
divulgar a informação atualizada sobre a Gestão Total da Qualidade nos Laboratórios Clínicos e de
Genética Humana, contribuindo para a melhoria contínua e constante atualização de todos os
profissionais, nesta área.
Do mesmo modo o Colégio de Biologia Humana e Saúde da Ordem dos Biólogos, criado como
consequência da forte ligação profissional do Biólogo à Saúde Humana, tem como objetivos prioritários
a valorização do conhecimento e a promoção da formação e do bom exercício da atividade profissional
dos seus especialistas, nas várias vertentes da saúde humana. A sua ação incide especialmente nas áreas
de análises clínicas, genética humana e embriologia/reprodução humana, abrangendo todas as
vertentes de laboratório, investigação, desenvolvimento de produtos de ação terapêutica, assessoria
técnico-científica e implementação de sistemas de gestão da qualidade.
O documento agora proposto, foi revisto e atualizado, apresentando as seguintes alterações em
relação à revisão de 2012, para além de correções de gralhas nos cálculos referentes à HBA1C nas pags
17 e 42 em relação à revisão de 2015:
• Substituição da Métrica do “Erro Total”, pela “Incerteza da Medição”;
• Introdução do processo de “Avaliação do Risco Clínico” associado aos doseamentos
efetuados num laboratório clínico;
• Introdução do procedimento de ajuste primário da média-alvo para vários equipamentos
com o mesmo método de doseamento;
• Introdução do procedimento de avaliação e adaptação das Normas de Orientação Clínica
da DGS (NOCs), ao Laboratório Clínico.
• Apresentação das fórmulas de cálculo: a) deteção de alterações fisiopatológicas
relevantes; b) dispersão máxima de doseamento; c) número de amostras necessárias
para detetar o ponto homeostático real do paciente;
• Introdução de exemplo associado a parâmetros de cálculo dependentes de outras
magnitudes biológicas (Fórmula de Friedewald para LDL colesterol);
• Apresentação de 2 exemplos concretos de procedimento de otimização segundo as
Normas de Orientação Clínica em vigor, para o Colesterol Total e HbA1c, nos apêndices 1
e 2.

14-05-2016 3
Com este procedimento, a LabGen-ANBIOQ e o CBHS/OBIO pretendem contribuir para uma
melhoria significativa da competência técnica dos laboratórios, prestando apoio diretamente não só aos
profissionais do laboratório clínico, responsáveis de setores, unidades, equipamentos ou métodos, ou
ainda com responsabilidades na gestão da qualidade, mas também todos aqueles que desejam adicionar
valor aos laboratórios onde exercem funções.
A informação contida neste procedimento orienta o profissional de laboratório no processo da
implementação de melhorias, com impacto clinico quantificável, diretamente nas cartas de controlo de
qualidade interno, que já se encontrem em vigor nos seus laboratórios ou propostas pelos fabricantes.
Convidamos todos os interessados a partilharem ideias e sugestões que possam melhorar este
procedimento, certos de que a experiência acumulada pelos laboratórios permite uma crescente
competência técnica, objetivo de todos os profissionais com interesse em produzir resultados válidos.
Para pedidos de esclarecimentos, ou qualquer outro assunto, contate-nos p.f. pelo correio
eletrónico:
[email protected] (LabGen-ANBIOQ)
[email protected] (Colégio Biologia Humana e Saúde, Ordem dos Biólogos)
(Revisão de maio de 2016)
Jorge Pinheiro
Coordenador da Comissão de Laboratório
Clínico e Genética Humana de Bioquímicos
Clínicos – Associação Nacional de
Bioquímicos
Ana Sousa
Presidente do Colégio de Biologia Humana e Saúde –
Ordem dos Biólogos

14-05-2016 4
Índice
I – INTRODUÇÃO ....................................................................................................................... 6
II - INCERTEZA DE MEDIÇÃO VS ERRO TOTAL ...................................................................... 8
III - CÁLCULO DA INCERTEZA MÁXIMA DE MEDIÇÃO DO LABORATORIO A
PARTIR DOS SEUS INDICADORES TÉCNICOS OPERACIONAIS DE QUALIDADE .............. 11
1. - Caso particular de parâmetros de análise dependentes de outras magnitudes biológicas doseadas no laboratório (parâmetros de cálculo).
Exemplo Fórmula de Friedewald LDL. ............................................................................................. 12
1.1.- Incerteza Máxima de Medição calculada através do SD máximo
(2SD) da Fórmula de LDL Friedewald num dado laboratório ........................................... 12
IV - AVALIAÇÃO DO RISCO CLÍNICO DOS DOSEAMENTOS EFETUADOS NUM
LABORATÓRIO CLÍNICO ......................................................................................................... 14
1. – Avaliação do Risco Clínico no Laboratório ............................................................................. 14
2. - Cálculo da capacidade de deteção de alterações fisiopatológicas
relevantes entre dois doseamentos consecutivos. ..................................................................... 15
3. - Cálculo do número de amostras necessárias para determinar o ponto
homeostático real de uma magnitude biológica. ........................................................................ 16
4. - Cálculo da dispersão máxima de doseamento de uma magnitude
biológica doseada no laboratório clínico ........................................................................................ 17
V - DEFINIÇÃO DA MÉDIA-ALVO DO MÉTODO DE DOSEAMENTO PARA
UMA MAGNITUDE BIOLÓGICA ............................................................................................. 19
1. - Definição da média-alvo de vários métodos de doseamento para uma
mesma magnitude biológica .............................................................................................................. 19

14-05-2016 5
VI - DEFINIÇÃO DO DESVIO PADRÃO DO MÉTODO DE DOSEAMENTO
/EQUIPAMENTO: A PEDRA BASILAR DO PROCESSO DE OTIMIZAÇÃO PARA O
MÉTODO DE DOSEAMENTO ........................................................................................ 21
1. - Definição do Desvio Padrão obtido em vários equipamentos com o mesmo
método de doseamento ....................................................................................................................... 21
VII - PROCEDIMENTO DE OTIMIZAÇÃO DE CARTAS DE CONTROLO INTERNO DO
LABORATÓRIO CLÍNICO ......................................................................................................... 26
1- Capacidade de detetar alterações fisiopatológicas relevantes: ......................................... 27
2- Dispersão máxima de um doseamento: .................................................................................... 27
3- Número de amostras para determinar o ponto homeostático real do paciente: ........ 28
VIII – CONSIDERAÇÕES FINAIS .............................................................................................. 32
BIBLIOGRAFIA .......................................................................................................................... 34
APÊNDICE 1 .............................................................................................................................. 36
APÊNDICE 2 .............................................................................................................................. 40

14-05-2016 6
I – INTRODUÇÃO
O Laboratório clínico deve criar mecanismos de avaliação da qualidade independentes,
mensuráveis e contínuos, e assegurar a sua correta implementação, monitorização e aplicação de ações
corretivas. Deve simultaneamente aperfeiçoar o sistema de controlo adotado, contribuindo assim para a
melhoria da segurança do paciente.
Cada vez mais se assume o cálculo da incerteza de medição, associado a qualquer valor
quantitativo no laboratório clínico, sendo um requisito de qualidade da ISO 15189: 2014 (1). Não
obstante, os resultados de um inquérito internacional realizado em janeiro de 2015 pela Fundação
Westgard a laboratórios clínicos (2), mostram que menos de 40% dos laboratórios usam os requisitos de
qualidade para monitorizar a sua incerteza de medição e apenas cerca de 32% definem requisitos de
qualidade de uso clínico nos seus programas de controlo interno. Cerca de 52% dos laboratórios referem
usar ainda os requisitos de qualidade segundo as especificações dos fabricantes de reagentes e
amostras de controlo. Expressivos foram também os cerca de 51% de laboratórios que declaram sentir
necessidade de apoio na definição dos seus requisitos de qualidade.
O presente documento serve de orientação à implementação de cartas de controlo interno, de
acordo com a métrica da Incerteza de Medição, que substitui agora a métrica do Erro Total.
Resultante do uso da métrica da incerteza de medição, outra melhoria introduzida neste
procedimento é a integração dos requisitos de qualidade clínicos para o laboratório, diretamente nas
cartas de controlo de qualidade interna (CQI), que permitem assim assegurar a monitorização
personalizada e a maior segurança dos seus pacientes (3). O Documento de Consenso de Estocolmo de
1999 (4) e o Documento de Consenso de Milão de 2014 (5), que atualiza o de Estocolmo, definem a
hierarquia dos requisitos de qualidade no laboratório clínico. De acordo com estes documentos, os
requisitos de qualidade clínicos são considerados prioritários, seguidos dos requisitos constantes nas
bases de dados de variabilidade biológica. Assim, através do método exposto no atual procedimento, é
conferida “autoridade clínica”, com base no conhecimento científico atual, ao processo de otimização
das cartas de CQI de qualquer laboratório médico.
As Normas de Orientação Clínica da Direção Geral da Saúde são emitidas de acordo com os Guias
de Prática Clínica (GPC) Internacionais. Os GPC são sistematicamente avaliados pela “AGREE” (6), abaixo
dos 50% nos indicadores de qualidade como a “Aplicabilidade” e “Rigor do desenvolvimento”, com o
associado risco para o paciente, como o demonstram vários estudos internacionais (7,8,9).

14-05-2016 7
Os Laboratórios devem garantir a observância dos requisitos clínicos e/ou analíticos, referidos
nas Normas de Orientação Clínica da DGS, que se encontrem direta ou indiretamente ligadas ao
Laboratório, de forma a garantir o cumprimento do princípio base das Normas da DGS de maior
segurança ao paciente com a melhor eficácia produtiva.
Este procedimento apresenta também 3 ferramentas essenciais para avaliar o Risco Clínico no
cumprimento dos requisitos operacionais de qualidade clínica (na forma de incerteza máxima de
medição), dos Guias de Pratica Clinica Internacionais ou das Normas de Orientação Clínica da DGS
(NOC). O processo de avaliação do Risco Clínico, permite também adaptar os GPC ou as NOC às
condições particulares de cada laboratório clínico, caso este não consiga cumprir com o requisito clínico
inicial, para garantir a mesma qualidade clínica da resposta desses laboratórios. Normalmente os GPC
ou as NOC não fornecem diretamente os dados operacionais de qualidade a cumprir pelo laboratório
clínico. Por esse motivo, é necessário extrair esses dados através: a) do cálculo da capacidade de
deteção de alterações fisiopatológicas relevantes (dados da monitorização clinica ou tratamento); b) do
número de amostras necessárias para obter o ponto homeostático real do paciente (dados de cut-offs
de decisão clinica para diagnóstico ou tratamento); c) da dispersão máxima esperada (cut-offs de
decisão clinica para diagnóstico ou terapêutica), para cada magnitude biológica.
Nos Apêndices 1 e 2, encontramos 2 exemplos práticos da otimização das cartas de controlo
interno. Para o colesterol total (Apêndice 1), a otimização das cartas de controlo é efetuada para duas
situações práticas possíveis: no caso do laboratório clínico que consegue otimizar as suas cartas de
controlo interno e cumprir com os requisitos clínicos patentes nas NOC 066/2011 “Prescrição de Exames
Laboratoriais para Avaliação de Dislipidémias no Adulto” (10) e NOC 005/2013 “Avaliação do Risco
Cardiovascular SCORE (Systematic Coronary Risk Evaluation)” (11); e no caso do laboratório que não
otimiza as suas cartas de controlo ou não consegue cumprir diretamente com os requisitos clínicos das
referidas NOCs, mas que sabe quantificar o risco clínico que incorre.
O segundo exemplo prático, Apêndice 2, refere-se à otimização das cartas de controlo interno de
um laboratório para a HbA1c, de forma a respeitar o requisito de qualidade analítico diretamente
referido na forma de CV% máximo a cumprir na NOC 033/2011 “Prescrição e Determinação da
Hemoglobina Glicada A1c” (12), comparando-o com o laboratório que não otimiza a suas cartas de
controlo interno.

14-05-2016 8
II – INCERTEZA DE MEDIÇÃO VS ERRO TOTAL
O sistema métrico do Erro Total nasceu com o Dr James Westgard nos anos 70 do século
passado, numa altura em que os laboratórios efetuavam, por norma, os doseamentos dos seus
pacientes em duplicado (3). Nesse período o laboratório centrava-se mais sobre si próprio, concentrado
em avaliar e comparar a exatidão e a precisão das suas medições, com os outros laboratórios. Desse
modo confiava-se que quanto mais preciso e exato fosse o método, mais útil seria para o paciente (3).
Nestes anos, os laboratórios não possuíam um segundo método/equipamento para confirmar os
resultados, e assumiam que os seus pacientes eram sempre avaliados (monitorizados e diagnosticados)
no mesmo local, pelo mesmo médico, e pelo mesmo equipamento/método de doseamento. A métrica
do Erro Total também é indicada para efetuar comparações de desempenho entre métodos de
doseamento, sendo usada como ferramenta de atribuição de rankings/posições de comparação analítica
entre os diferentes laboratórios ou de comparação com os requisitos de qualidade analítica expressos
na forma de Erro Total (3, 13, 14, 15).
No laboratório do século XXI, os doseamentos são feitos de forma única (mesmo quando
repetidos para confirmação de resultados críticos, são efetuados geralmente num segundo
equipamento, deixando de ser um duplicado de cada equipamento como era comum no laboratório do
século passado). Por esse motivo o laboratório moderno necessita saber qual a incerteza associada a
cada medição/doseamento que efetua (1, 3). Esta análise implica conhecer a contribuição do erro
sistemático (bias) e do erro aleatório, numa única métrica que quantifique a incerteza de medição de
cada doseamento efetuado.
A expressão dos doseamentos em química clinica, hematologia e imunologia, na forma de
incerteza, pretende ajudar o utilizador dos resultados do laboratório a tomar uma decisão informada (e
personalizada para cada paciente) do impacto de cada doseamento, podendo inferir se um tratamento
teve, ou está a ter, um efeito quantitativo relevante (3, 16, 17).
Atualmente o laboratório clínico assume cada vez mais a responsabilidade de se concentrar na
perspetiva e necessidades do utilizador final dos seus dados. A métrica da incerteza de medição permite
descrever tanto a performance do método ou do sistema de doseamento, como o impacto clínico ou
contribuição de cada doseamento individual, quer na forma de comparação entre dois resultados do
mesmo paciente (monitorização personalizada) quer comparando um resultado com um valor de
decisão clínica (cut-off clinico ou de tratamento) que por definição é sem incerteza (3, 16, 17, 18).
O laboratório hospitalar atual possui também vários métodos/equipamentos no mesmo espaço
físico, ou distribuídos por unidades laboratoriais em vários hospitais que supostamente podem servir os
mesmos utentes em situações, tempos e locais diferentes (episódio de urgência, consulta de rotina,
internamento hospitalar no mesmo hospital ou entre hospitais do mesmo grupo/centro hospitalar).
Esses pacientes necessitam de ser tratados como se estivessem a ser monitorizados pelo mesmo

14-05-2016 9
equipamento/método para a mesma magnitude biológica. Também no caso do laboratório privado,
quer tenha mais do que um equipamento para a mesma magnitude biológica, quer esteja integrado
num grupo empresarial, existe a preocupação da manutenção de um registo único do paciente.
Para além disso, projetos como a Plataforma de Dados de Saúde - PDS, desenvolvido pela CIC -
Comissão para Informatização Clínica e pela SPMS - Serviços Partilhados do Ministério da Saúde, EPE,
concentram registos personalizados de saúde de cada paciente em Portugal, incluindo os diversos
resultados de análises dos diferentes laboratórios clínicos utilizados pelo paciente (hospitais, centros de
saúde, laboratórios privados), pressionam à harmonização da resposta laboratorial.
Assim, através da aplicação da métrica da incerteza de medição no laboratório clínico e
independentemente dos laboratórios do mesmo grupo empresarial, centro hospitalar ou diversos
laboratórios independentes aos quais recorre um paciente ao longo do tempo, torna-se tecnicamente
possível tratar os dados obtidos nos diversos analisadores como se o paciente estivesse a ser
monitorizado pelo mesmo equipamento ou método para a mesma magnitude biológica e no mesmo
local, permitindo uma monitorização personalizada do estado do paciente.
A diferença entre a métrica do Erro Total e a da Incerteza de Medição, reside na forma como
tratam o erro sistemático (bias). A figura 1 mostra como o erro sistemático e o erro aleatório são
integrados na métrica da incerteza de medição.
Figura 1 - Componentes de incerteza combinados: A- Conceito do Erro Total, com adição linear dos erros
sistemáticos e aleatórios. B- Conceito da incerteza, integra as duas componentes como variantes do teorema de
Pitágoras (In “Bias in clinical chemistry”, Bioanalysis (2014) 6(21), 2855–2875, Elvar Theodorsson, Bertil Magnusson & Ivo
Leito).
14-05-2016 10
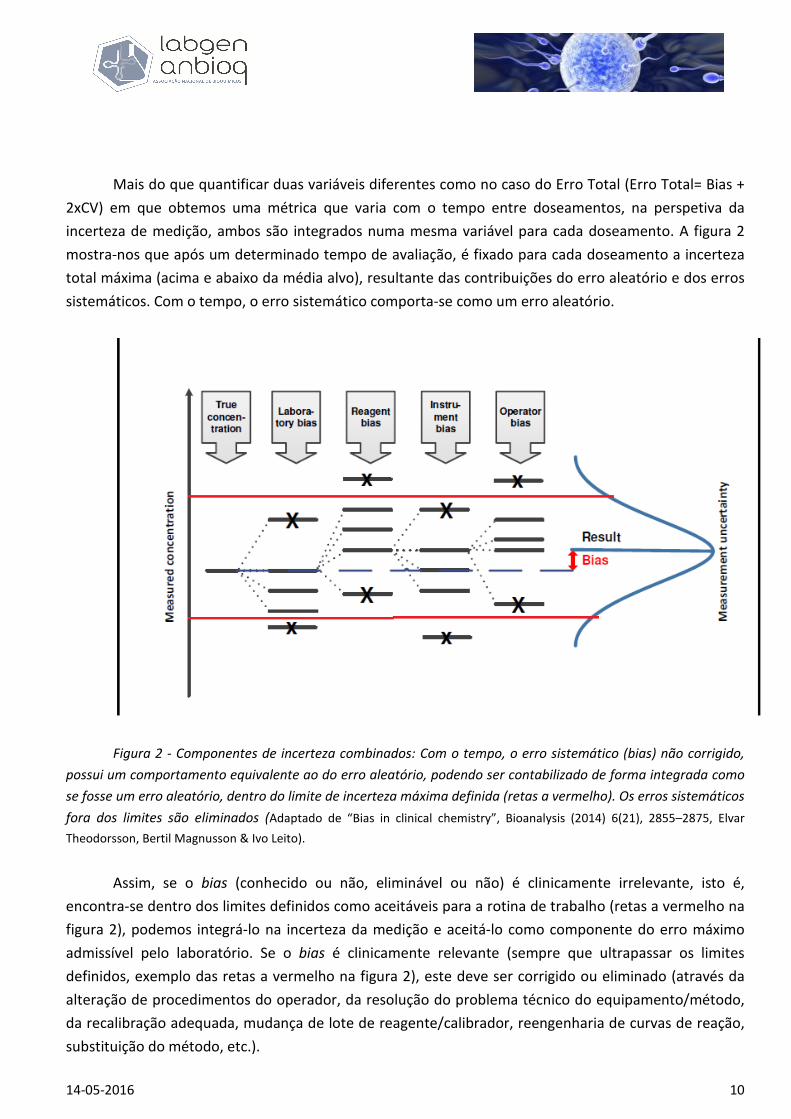
Mais do que quantificar duas variáveis diferentes como no caso do Erro Total (Erro Total= Bias +
2xCV) em que obtemos uma métrica que varia com o tempo entre doseamentos, na perspetiva da
incerteza de medição, ambos são integrados numa mesma variável para cada doseamento. A figura 2
mostra-nos que após um determinado tempo de avaliação, é fixado para cada doseamento a incerteza
total máxima (acima e abaixo da média alvo), resultante das contribuições do erro aleatório e dos erros
sistemáticos. Com o tempo, o erro sistemático comporta-se como um erro aleatório.
Figura 2 - Componentes de incerteza combinados: Com o tempo, o erro sistemático (bias) não corrigido,
possui um comportamento equivalente ao do erro aleatório, podendo ser contabilizado de forma integrada como
se fosse um erro aleatório, dentro do limite de incerteza máxima definida (retas a vermelho). Os erros sistemáticos
fora dos limites são eliminados (Adaptado de “Bias in clinical chemistry”, Bioanalysis (2014) 6(21), 2855–2875, Elvar
Theodorsson, Bertil Magnusson & Ivo Leito).
Assim, se o bias (conhecido ou não, eliminável ou não) é clinicamente irrelevante, isto é,
encontra-se dentro dos limites definidos como aceitáveis para a rotina de trabalho (retas a vermelho na
figura 2), podemos integrá-lo na incerteza da medição e aceitá-lo como componente do erro máximo
admissível pelo laboratório. Se o bias é clinicamente relevante (sempre que ultrapassar os limites
definidos, exemplo das retas a vermelho na figura 2), este deve ser corrigido ou eliminado (através da
alteração de procedimentos do operador, da resolução do problema técnico do equipamento/método,
da recalibração adequada, mudança de lote de reagente/calibrador, reengenharia de curvas de reação,
substituição do método, etc.).

14-05-2016 11
III - CÁLCULO DA INCERTEZA MÁXIMA DE MEDIÇÃO DO LABORATORIO A
PARTIR DOS SEUS INDICADORES TÉCNICOS OPERACIONAIS DE QUALIDADE
Qualquer Programa de CQI tem como base as cartas de controlo de qualidade implementadas no
laboratório clínico.
A Média-Alvo (M), o Desvio Padrão (SD) e o Coeficiente de Variação (%CV= (SD/M) X 100) (14,15),
são facilmente obtidos a partir dos gráficos de Levey-Jennings. Quando conhecemos a regra de exclusão
em uso nesse laboratório, a média-alvo e o desvio padrão traduzem a qualidade com que o laboratório
está a admitir/aceitar trabalhar, sendo por isso denominados neste procedimento de “indicadores
operacionais técnicos de qualidade”.
Figura 3: Gráficos de controlos de qualidade interna para o doseamento da HbA1c do equipamento HA-8160 da A.
Menarini diagnostics, das amostras de controlo interna da qualidade de nível 1 e nível 2 (ambas do lote de
controlos da mesma casa comercial número 201201). Os tracejados a vermelho definem o limite máximo de
incerteza analítica (erro aleatório e erro sistemático) permitida para cada nível de controlo e que correspondem a
um CV máximo <2%, quando usada a regra de controlo Westgard 1/2s. Estes gráficos mostram um programa de
controlos internos que respeita o requisito de qualidade da NOC DGS 033/2011, como o descrito no exemplo
prático do Apêndice 2.

14-05-2016 12
No exemplo da figura 3, o gráfico de controlo nível 1 tem definido a média-alvo para 5,9%
HbA1C, com um desvio padrão de 0,059% o que perfaz um
CV = (0,059/5,9) X100 = 1% (CV de 1 SD)
Como a regra de exclusão está definida para 1/2s (exclusão perante um desvio de 2SD ou mais),
então o CV% máximo definido para esta carta de controlo representa a incerteza máxima de medição
permitida no laboratório e é tal que CVmáx <2% (note-se que quando CV=2%, a regra de exclusão do
controlo é aplicada e as ações corretivas ativadas), o que nos indica que esta carta de controlo interno
respeita o requisito de qualidade intralaboratorial definido na NOC da Hemoglobina Glicada, ou seja,
CVmáximo intralaboratorial < 2% (6).
1. - Caso particular de parâmetros de análise dependentes de outras magnitudes
biológicas doseadas no laboratório (parâmetros de cálculo). Exemplo Formula de
Friedewald LDL
No caso particular de parâmetros de cálculo dependentes de outros parâmetros doseados pelo
laboratório, como a Fórmula de Friedwald para o cálculo da LDL, usa-se o cálculo combinado da
incerteza de medição dos vários componentes da Fórmula de Friedewald, segundo o método referido
no "Appendix C: Combining Uncertainty Components” da referência bibliográfica 18 (“Uncertainty of
Measurement in Quantitative Medical Testing. A Laboratory Implementation Guide”).
Sendo assim, o valor obtido de CV máximo para o LDL colesterol é feito através da fórmula
tradicional de cálculo do CV = (SD/M) x 100, com determinação direta do desvio padrão total
equivalente ao máximo permitido pela Regra de exclusão implementada (neste exemplo assumimos a
regra de exclusão 1/2s, exclusão para lá de 2 SD), para ambos os níveis de controlo de qualidade de cada
uma das magnitudes biológicas, combinadas na fórmula de Friedewald:
1.1.- Incerteza Máxima de Medição calculada através do SD máximo (2SD) da Fórmula de LDL
Friedewald num dado laboratório
Fórmula Friedwald em mmol/L:
LDL = Col – HDL - (Trig/2,22) (10)

14-05-2016 13
O valor da média-alvo equivalente (MLDL) é dado pela equação:
MLDL = MCol – MHDL - M (Trig/2,22)
Fórmula de cálculo combinado da imprecisão:
(SD max LDL)2 = (SD max Col)2+ (SD max HDL)2 +(SD max Trigl/2,22)2
(referência bibliográfica 18 - Appendix C)
Exemplo de um Controlo Nivel 1
(SD max LDL)2 = (0,09875)2+ (0,0451)2 +(0,033/2.22)2=> SD max LDL= 0,118 para MLDL= MCol-
MHDL-M (Trig/2,22)=3,95-0,82-(1,65/2,22)=2.387 LOGO: CV max = 4,94%
Exemplo de um Controlo Nível 2
(SD max LDL)2 = (0,1905)2+ (0,0891)2 +(0,079 /2,22)2 => SD max LDL= 0,2246 para MLDL= MCol-
MHDL-M (Trig/2,22)= 7,62-1,62-(3,94/2,22)= 4.22 LOGO: CV max = 5,3%
Conclusão: O CV máximo que corresponde a incerteza máxima de medição para o LDL calculado é
CV max=5,3% (o maior CV dos dois níveis de controlo).

14-05-2016 14
IV - AVALIAÇÃO DO RISCO CLÍNICO DOS DOSEAMENTOS EFETUADOS NUM
LABORATÓRIO CLÍNICO
1. – Avaliação do Risco Clínico no Laboratório
A análise do risco é uma componente essencial da implementação dos programas de CQI e é
competência do laboratório clínico quantificar o risco admitido nos seus métodos de doseamento (19).
Estamos perante uma avaliação do risco clinico quando quantificamos o impacto do risco
admitido pelo laboratório, diretamente nas decisões clínicas.
Na medicina laboratorial, a avaliação do Risco Clínico de um doseamento deve compreender
todas as fases envolvidas no processo do laboratório clínico:- Pré-analítica (através do conhecimento da
variabilidade biológica esperada para cada magnitude biológica e assumindo que todos os
procedimentos de colheita, manuseamento e transporte das amostras são cumpridos de igual forma
para todos os pacientes); Analítica (através do conhecimento da incerteza máxima de medição
permitida no laboratório e assumindo que todos os procedimentos de operação são cumpridos para
todas as amostras de todos os pacientes e que são detetados todos os interferentes do doseamento) e
Pós-Analítica (correlacionando os dados de variabilidade biológica, com os dados da incerteza máxima
de medição, confrontando-os com os requisitos clínicos e o seu impacto nos pacientes).
Na base da avaliação pré-analítica do Risco Clínico de um doseamento, está o conhecimento da
variabilidade biológica. Por exemplo, sabendo que a variabilidade biológica intraindividual conhecida
para a Prolactina plasmática é de 39,2% e que diminui para 23,3% quando determinada em amostras
séricas, o laboratório consegue diminuir em quase 50% o risco da decisão clínica do doseamento da
prolactina associada à variabilidade pré-analítica, se eleger as amostras séricas para o doseamento da
prolactina.
A avaliação do Risco Clínico na fase analítica do laboratório, depende diretamente da incerteza
máxima de medição permitida no programa de CQI do laboratório. Por exemplo, um programa de CQI
para a HbA1c que permita uma incerteza máxima de medição que ultrapassa a variabilidade biológica
intraindividual da HbA1c (1,85%), aumenta o peso da fase analítica no cálculo da incerteza total
associada à medição da HbA1c e aumenta proporcionalmente o risco da decisão clínica (no diagnóstico
ou monitorização).
No processo da avaliação do Risco Clínico na fase pós-analítica de um método de doseamento no
laboratório clínico, os indicadores técnicos operacionais de qualidade definidos no laboratório (incerteza
máxima de medição permitida pelo laboratório), são confrontados com o seu impacto clínico direto no

14-05-2016 15
paciente, em conjunto com os dados da variabilidade biológica, na forma de: a- deteção de alterações
fisiopatológicas relevantes; b- dispersão máxima de doseamento; c- número de amostras necessárias
para detetar o ponto homeostático real do paciente.
A avaliação final do Risco Clínico de um método de doseamento no Laboratório Clínico, depende
dos requisitos clínicos de qualidade, derivados das NOC da DGS ou dos GPC. As NOC ou os GPC definem
procedimentos clínicos (por exemplo, o número de observações ou amostras biológicas necessários
antes de definir um diagnóstico ou iniciar um tratamento, como na NOC 066-2011 (10) que define que
devem ser efetuados dois doseamentos de colesterol total, espaçados 4 semanas entre si), ou contêm
indicadores de cut-offs de decisão clínica (por exemplo na definição de um diagnóstico de Diabetes
Mellitus, com o valor de HbA1c=7%, como referido na NOC 033-2011 (12)), ou ainda contêm objetivos de
tratamento e monitorização terapêutica (por exemplo, uma redução ≥ 50% do valor basal do c-LDL,
quando o valor alvo não pode ser atingido em doentes com risco cardiovascular muito elevado – NOC
066-2011). Estes podem ser convertidos em requisitos operacionais de qualidade clínica para o
laboratório, quando conhecemos as respetivas fórmulas.
Os requisitos de qualidade clínica que se obtêm através das NOC ou GPC, são necessários para
comparar a eficácia clínica entre o programa de controlo interno da qualidade existente no laboratório e
o programa de CQI proposto através da otimização das cartas de CQI.
2. - Cálculo da capacidade de deteção de alterações fisiopatológicas relevantes
entre dois doseamentos consecutivos
A fórmula do cálculo da capacidade de deteção de alterações fisiopatológicas relevantes entre dois
doseamentos consecutivos, para alterações unilaterais significativas (só aumento ou só diminuição, com
95% de confiança) é (18, 20, 21, 22):
Δ-Check = 2,33 x √(CVa2 + CVb
2)
Sendo CVa a incerteza máxima permitida pelo laboratório e CVb a variabilidade biológica intraindividual
conhecida para a magnitude biológica em estudo.

14-05-2016 16
Exemplo:
Cálculo da capacidade de deteção de alterações fisiopatológicas relevantes entre dois doseamentos
consecutivos de HbA1c, garantidos pela NOC 033/2011 (12):
Como o CV máximo permitido é <2% e o CVb (variabilidade biológica intraindividual HbA1c) = 1,85% (22),
então obtemos:
Δ-Check = 2,33 x √(22+1,852) = 6,35%
Assim, a NOC 033/2011 garante que é detetada uma alteração fisiopatológica relevante entre dois
doseamentos consecutivos de um paciente, quando a diferença entre os dois doseamentos ultrapassa a
amplitude de 6,35% (por exemplo, quando um paciente passa de 7% de HBA1C para 7,45%, o
laboratório garante estarmos perante uma subida fisiopatológica relevante da HbA1c para este
paciente, que é diretamente proporcional ao aumento do risco da condição clínica neste paciente).
3. - Cálculo do número de amostras necessárias para determinar o ponto
homeostático real de uma magnitude biológica.
A fórmula de cálculo do número de amostras necessárias para obter um valor cerca de D% perto do
valor homeostático real de qualquer paciente é (18, 20, 21, 22):
onde n é o número de amostras necessárias para obter um valor D% perto do ponto homeostático real
do paciente, com uma probabilidade dependente da confiança Z (normalmente Z=1,96, para 95% de
confiança), CVa é a incerteza máxima de medição permitida no laboratório e CVb é a variabilidade
biológica intraindividual conhecida para a magnitude biológica em estudo.

14-05-2016 17
4. - Cálculo da dispersão máxima de doseamento de uma magnitude biológica
doseada no laboratório clínico
Também a dispersão máxima de um doseamento depende da contribuição não só da incerteza de
medição máxima permitida pelo laboratório, como também da variabilidade biológica intraindividual
conhecida para a magnitude biológica em estudo.
Assim, da fórmula anterior obtemos a seguinte fórmula de dispersão de cada doseamento (fórmula
anterior para n=1):
D = 1,96 x √(CVa2 + CVb
2)
Em que D é a dispersão resultante de um único doseamento de um método com a incerteza máxima de
medição CVa e a variabilidade biológica CVb da magnitude biológica em estudo, com 95% probabilidade.
Exemplo:
Cálculo do número de amostras necessárias para determinar o ponto homeostático real da HbA1C, e
respetiva dispersão máxima de cada doseamento, segundo os requisitos patentes na NOC 033/2011.
Sabendo que a variabilidade biológica intraindividual para a HbA1C é CVb=1,85% (23), temos que:
n = 2 ���� 2 = [1,96 x √(22 + 1,852)/D]2 ���� D = 3,78%
onde n = 2 é o número de amostras necessárias para obter um valor D = 3,78% perto do ponto
homeostático real, com uma probabilidade dependente da confiança Z = 1,96 para 95% de confiança.
Assim, quando é feito um segundo doseamento da HbA1C no laboratório médico, para obter a
confirmação do diagnóstico de diabetes Mellitus (como requerido pela Norma 033/2011), obtemos uma
média do valor de HbA1c dos dois doseamentos, que se encontra cerca de 3,78% perto do valor
homeostático real do paciente, e esse facto é decisivo para reduzir ao mínimo o risco do diagnóstico
clínico. Por exemplo, se com dois doseamentos obtemos uma média de 7%, o valor homeostático real
do paciente encontra-se entre 6,74% - 7,26% para a HbA1C.
A dispersão máxima de um doseamento de HbA1c efetuado num laboratório clínico que cumpra os
requisitos da Norma (CV HbA1c máximo = 2%) é:

14-05-2016 18
Para uma probabilidade de 95% temos a
% dispersão = ± 1,96 √[CVa2 + CVb2] = ± 1,96 √[22 + 1.852]= ±5,34%
(ou seja, com 1 doseamento ficamos a ± 5,34% do valor homeostático real do paciente: Se 1 único
doseamento obtiver HbA1C= 7%, o valor real da HbA1C no paciente pode estar entre 6,63%-7,37%).
Assim, se confrontarmos com a dispersão da média de dois doseamentos de HbA1C (3,78%),
observamos que com duas amostras de doseamento de HbA1c, conseguimos reduzir o risco clínico do
diagnóstico da diabetes em cerca de 30%, quando comparado com apenas um doseamento de HbA1C.

14-05-2016 19
V - DEFINIÇÃO DA MÉDIA-ALVO DO MÉTODO DE DOSEAMENTO PARA UMA
MAGNITUDE BIOLÓGICA
Todas as recomendações técnicas apontam uniformemente para a necessidade de ajuste das
cartas de Levey-Jennings propostas pelos fabricantes dos controlos de qualidade interna, à qualidade
real observada na rotina de trabalho dos métodos analíticos que controlam (14,15,18). Nesse ajuste, a
média-alvo (M) é adaptada à média observada para o método de doseamento na rotina diária do
laboratório ao fim de pelo menos 20 pontos obtidos ao longo de 20 dias (14,15). Esta média é reavaliada e
torna-se consolidada com os dados referentes de 3 a 6 meses da rotina do laboratório, a partir dos quais
é definitivamente ajustada. Esse valor apenas volta a ser ajustado quando se verifica a alteração da
estabilidade do lote de controlo para a magnitude biológica em causa.
1. - Definição da média-alvo de vários métodos de doseamento para uma mesma
magnitude biológica
No caso particular de vários equipamentos, de um único laboratório ou vários laboratórios de um
mesmo centro hospitalar ou grupo laboratorial privado, podem ser efetuados dois tipos de ajustes com
os diferentes equipamentos que doseiam a mesma magnitude biológica, de forma a garantir uma
resposta harmonizada entre todos os equipamentos:
a) Ajuste primário (apenas possível para equipamentos supostamente idênticos da mesma casa
comercial e desde que o erro sistemático entre os equipamentos/métodos permita cumprir com os
requisitos de incerteza máxima de doseamento definidos pelo laboratório);
b) Ajuste secundário (3). É usado primariamente entre equipamentos e métodos diferentes e de
diferentes modelos/fabricantes. Apesar de cientificamente validado e usado nos países escandinavos
na avaliação dos métodos POCTs (por imposição legal), esta metodologia levanta no entanto algumas
questões à luz da legislação IVD da União Europeia e da FDA Americana, quanto à responsabilidade
na rastreabilidade dos diferentes métodos de doseamento (fabricantes de reagentes), pelo que não é
abordada nesta versão do procedimento de otimização de cartas de controlo interno.
Decorrem ações junto das autoridades competentes europeias e americanas no sentido de admitir
este tipo de procedimento de ajuste entre métodos analíticos diferentes. Ocorrendo alteração da
moldura legal, este tipo de ajuste será incluído neste procedimento.
No procedimento de ajustamento primário, para cada magnitude biológica doseada nos vários
equipamentos do mesmo laboratório ou de vários laboratórios do centro hospitalar ou grupo

14-05-2016 20
laboratorial privado, é calculada uma média da média dos valores–alvo, obtidos nos vários
equipamentos/métodos idênticos do mesmo laboratório e/ou das várias unidades laboratoriais. Por
exemplo a média-alvo para o método de doseamento de glicose em 3 equipamentos diferentes
(equipamentos A, B e C) pode ser expressa como M total = [(MA+MB+MC)/3]. Desta forma, é definida
uma mesma carta de controlo interno da qualidade (mesma média e CV máximo admissível) para todos
os equipamentos/métodos de doseamento, supostamente idênticos do mesmo fabricante.
Depois de determinada a média-alvo total dos vários equipamentos e consolidada para o mesmo lote de
controlos (após reavaliação ao fim de 3 a 6 meses de trabalho), não se deve voltar a alterar essa média
(exceto se devido a alteração da estabilidade do lote de controlo). Desta forma o laboratório pode
detetar, além dos desvios aleatórios inaceitáveis, os desvios sistemáticos inaceitáveis provocados, por
exemplo, pelo operador, pelo equipamento ou pela matriz de produção dos diferentes lotes de
reagentes ou calibradores, nos diferentes equipamentos. O laboratório garante assim a mesma resposta
harmonizada com a mesma incerteza máxima associada (CV% máximo) para os mesmos valores
absolutos de doseamento, entre doseamentos diferentes no tempo, para qualquer paciente, em
qualquer dos equipamentos, e a mesma dispersão máxima para cada doseamento em todos os valores
absolutos obtidos. Este procedimento permite ao laboratório (isolado ou, inserido no mesmo centro
hospitalar ou grupo laboratorial), conhecer a dispersão máxima esperada em cada doseamento ao
mesmo tempo em todos os seus equipamentos, e concluir se o resultado obtido é significativamente
diferente dos valores de decisão clínica (cut-offs), assim como permite a monitorização personalizada e
eficiente dos pacientes. De facto, o laboratório conhece a sua capacidade em detetar alterações
fisiopatológicas relevantes em todos os seus equipamentos como se de um equipamento único de
tratasse. Isto é, o laboratório, centro hospitalar ou grupo laboratorial sabe, no momento da deteção do
erro, se este é clinicamente relevante, de forma idêntica em todos os seus equipamentos.

14-05-2016 21
VI – DEFINIÇÃO DO DESVIO PADRÃO DO MÉTODO DE DOSEAMENTO
/EQUIPAMENTO: A PEDRA BASILAR DO PROCESSO DE OTIMIZAÇÃO PARA O
MÉTODO DE DOSEAMENTO
Depois de determinada a média-alvo, a otimização das cartas de controlo começa com o ajuste
do Desvio Padrão (SD), com base no SD observado pelo método/equipamento. Quando estamos
perante um método de doseamento novo no laboratório, apenas ao fim de 3 a 6 meses de dados da
rotina de trabalho, podemos avaliar qual o desvio padrão real que engloba todos os erros aleatórios e
sistemáticos associados à rotina de trabalho. Com esse indicador operacional de qualidade,
determinamos o coeficiente de variação médio do método (CV%, para 1 SD). Depois de determinarmos
o CV% médio real de trabalho do método, estamos em condições de atribuir um máximo de coeficiente
de variação.
O erro máximo admissível atribuído normalmente nos processos de definição de requisitos de
qualidade analítica é o dobro do erro médio da rotina de trabalho.
Assim:
CV% máx = 2 x CV% médio real
A partir do CV% máx, podemos calcular e definir o menor Delta-Check possível que revela a
melhor capacidade de deteção de alterações fisiopatológicas relevantes (ver exemplo do capitulo IV.2.).
Com a definição do CV%máximo permitido no laboratório, podemos também calcular qual o número de
amostras necessárias para determinar o ponto homeostático real do paciente para a magnitude
biológica em estudo, bem como qual a dispersão máxima de doseamento esperada segundo o CV% máx
permitido pelo laboratório e a variação biológica intraindividual conhecida para a magnitude biológica
em estudo (ver exemplos do capítulo IV.4.).
1. - Definição do Desvio Padrão obtido em vários equipamentos com o mesmo
método de doseamento
Quando definimos o desvio padrão real com os dados da rotina de vários equipamentos para o
mesmo método de doseamento para uma dada magnitude biológica, de um laboratório, centro
hospitalar ou grupo laboratorial privado, devemos ter em consideração os erros aleatórios de todos os
equipamentos, integrado com os erros sistemáticos que existem em cada equipamento, como se de um
equipamento único se tratasse.

14-05-2016 22
Por exemplo: num dado laboratório, centro hospitalar ou grupo laboratorial, o equipamento A
do laboratório A possui média-alvo de 5,5 mmol/L para a glicose, o equipamento B do laboratório B
possui média-alvo de 5,6 mmol/L e o equipamento C do laboratório C possui média-alvo de 5,4 mmol/L,
e todos possuem um CV médio real de trabalho de 2,10%, 2,05% e 2,13%, respetivamente. Podemos
definir um CV médio real de trabalho dos 3 equipamentos de forma integrada, que inclua os erros
sistemáticos e aleatórios resultantes da rotina laboratorial nos 3 equipamentos como se de um único
equipamento se tratasse. Dessa forma, o valor absoluto dos extremos a definir numa única carta de
controlo interno, para os 3 equipamentos, é atribuído ao extremo mais baixo do valor absoluto do
equipamento C (pois possui a média-alvo mais baixa, para um CV muito idêntico aos dos outros 3
equipamentos) para o CV% máximo = 2 x 2,13% = 4,26%, o que nos dá SD máx = 0,0426 x 5,4 = 0,23 =>
valor absoluto mais baixo = 5,17 mmol/L. Ao valor absoluto mais elevado atribuímos o extremo obtido
com o equipamento B (pois possui a média-alvo mais elevada, para um CV muito idêntico aos dos outros
3 equipamentos), CV% máx = 2 x 2,0535%, o que nos dá SD máx = 0,04107 x 5.6= 0,23 => valor absoluto
mais elevado = 5,83 mmol/L. Como vimos no capitulo VI, a média-alvo a atribuir a uma carta de controlo
que integre os erros aleatórios e sistemáticos dos 3 equipamentos é dada pela média aritmética das 3
médias alvo determinadas para cada equipamento:
M total = [(MA + MB + MC)/3] e neste exemplo é M Total=[(5,5 + 5,6 + 5,4)/3] = 5,5 mmol/L
Logo, neste caso temos os seguintes indicadores operacionais de qualidade integrados dos 3
equipamentos:
M Total = 5,5 mmol/L
extremo superior máximo admissível (limite superior LS) LS=5,83 mmol/L
extremo inferior máximo admissível (limite inferior LI) LI= 5,17 mmol/L.
O que nos dá um CV máximo admissível de 6% para a carta de controlo nível 1, comum que
integre todos os erros sistemáticos e aleatórios admissíveis na rotina laboratorial dos 3 equipamentos
referidos. Neste exemplo, assumimos que o erro cometido na rotina laboratorial dos 3 equipamentos
para o nível 1 é maior do que para o nível 2 de controlo (aqui não são apresentados os cálculos para o
nível 2) e por isso é o erro máximo (CV máximo) definido para o nível 1 de controlo que define o erro
máximo admissível para o método de doseamento de todos os 3 equipamentos. É a partir desse CV
máximo integrado que calculamos a capacidade de deteção de alterações fisiopatológicas relevantes e a
dispersão máxima real de trabalho, dos 3 equipamentos com o mesmo método de doseamento da
glicose, para qualquer paciente que se dirija ao longo do tempo a locais diferentes do mesmo centro
hospitalar ou grupo laboratorial.

14-05-2016 23
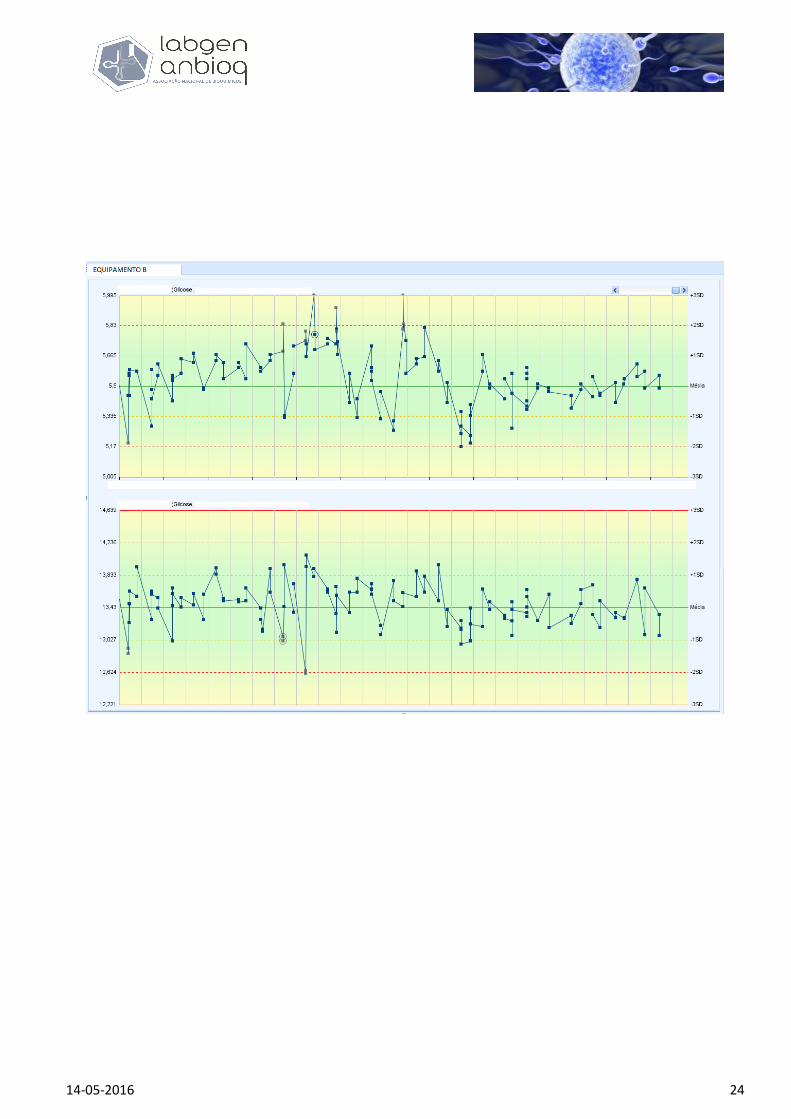
A figura 4 mostra-nos os gráficos de dois níveis de controlos para a glicose, referentes aos 3
equipamentos diferentes do laboratório, centro hospitalar ou grupo laboratorial, do exemplo anterior e
cuja incerteza máxima de medição está definida para CV% máximo = 6%.
14-05-2016 24

14-05-2016 25
Figura 4: Gráficos de controlo de qualidade interna para o método de determinação da glicémia nos
equipamentos A, B e C, da mesma casa comercial, em 3 equipamentos idênticos de um mesmo laboratório ou
unidades laboratoriais diferentes de um mesmo grupo laboratorial ou centro hospitalar. Os tracejados a vermelho
definem o limite máximo de incerteza analítica permitida para cada nível de controlo e que correspondem a um
CV máximo < 6%, quando usada a regra de exclusão Westgard 1/2s. A média-alvo e desvio padrão de cada nível
de controlo são os mesmos para todos os equipamentos. Apesar das diferenças de comportamento entre eles (o
equipamento C parece ser o mais estável), a todos são impostos os mesmos limites e desta forma, o grupo
laboratorial ou centro hospitalar define a mesma incerteza máxima de medição para os mesmos valores absolutos
de doseamento da glicose para os seus 3 laboratórios/equipamentos.

14-05-2016 26
VII – PROCEDIMENTO DE OTIMIZAÇÃO DE CARTAS DE CONTROLO INTERNO
DO LABORATÓRIO CLÍNICO
O procedimento de implementação de cartas de controlo interno otimizadas, começa com uma
avaliação às especificações técnicas operacionais de qualidade da carta de controlo interno em vigor no
laboratório (ou do fabricante), na forma de incerteza máxima de medição permitida. Conforme se refere
na introdução deste procedimento, um inquérito internacional promovido pela Fundação Westgard
revelou que mais de 50% dos laboratórios ainda usam como requisitos operacionais de qualidade os
dados fornecidos pelo fabricante do seu controlo interno de qualidade (2). Por outro lado alguns dos
fabricantes de amostras de controlo, mais recentemente, já não definem os limites a implementar nas
cartas de controlo, assumindo que o laboratório clínico já definiu o seu requisito de qualidade e
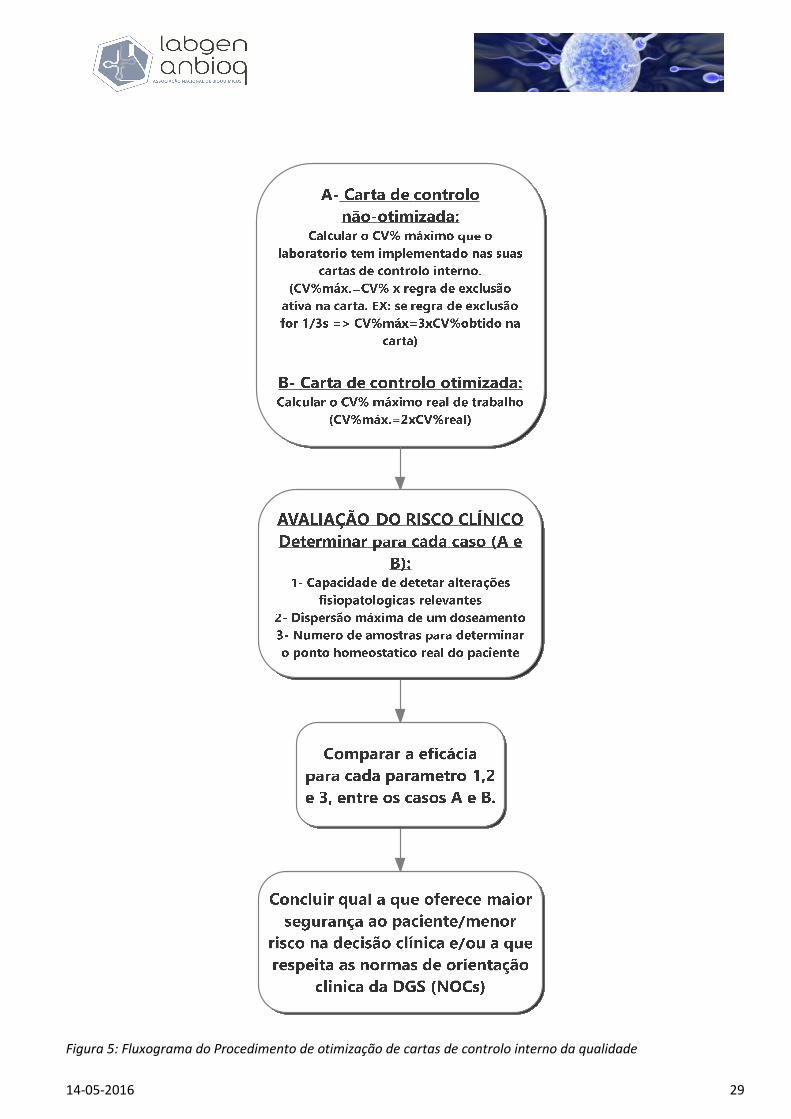
portanto, os limites com que aceita trabalhar. A figura 5 esquematiza o procedimento de otimização das
cartas de controlo interno em vigor num laboratório clínico, cujo procedimento se descreve a seguir
para os casos A (cartas não otimizadas) e B (cartas otimizadas):
O procedimento de otimização começa por calcular qual o CV% máximo do laboratório com base
nas suas cartas de controlo aplicando a regra de exclusão em uso. Assim, por exemplo, se um
laboratório nas suas cartas de controlo da glicose possui CV% = 10% e usa a regra de exclusão Westgard
1/3s, quer dizer que esse laboratório permite um erro máximo de incerteza de CV% máximo = 3 x 10 =
30% (laboratório caso A com cartas não otimizadas).
Para os laboratórios otimizarem as suas cartas de controlo interno (laboratórios caso B), calcula-
se qual o CV% real associado ao método de doseamento (com dados de pelo menos 3 a 6 meses da
rotina do laboratório). Normalmente o CV real é muito inferior ao obtido nas cartas de controlo interno
definidas pelos fabricantes.
Por exemplo no mesmo laboratório em estudo, podemos assumir o CV real como sendo CV% real
= 4%. Esse revela a incerteza média real no laboratório para o mesmo método de doseamento. Como
referido no capítulo VII, a incerteza máxima permitida nesse método normalmente é 2x a incerteza
média real (desta forma só se executam ações corretivas quando o método passa a ter o dobro do erro
resultante na rotina normal do laboratório trabalho).
Assim, neste exemplo assumimos um novo erro máximo de
CV% max = 2 x 4 = 8% (laboratório caso B com cartas otimizadas)

14-05-2016 27
Para cada caso (laboratórios caso A com cartas não-otimizadas e caso B com cartas otimizadas)
avaliamos o risco clínico do doseamento da glicose calculando (sabendo que a CVi glicose=5,6% (23), para
amostras de soro):
1 - Capacidade de detetar alterações fisiopatológicas relevantes
Δ-Check = 2,33 x √(CVa2+CVb
2)
A- Δ-Check = 2,33 x √(CVa2+CVb
2) = 2,33 x √(302+5,62) = 71,11%
B- Δ-Check = 2,33 x √(CVa2+CVb
2) = 2,33 x √(82+5,62) = 22,75%
Conclusão: A carta otimizada (caso B), permite ao laboratório ser 3x mais eficiente na deteção de
alterações fisiopatológicas da glicose relevantes, comparativamente com as cartas que tem em uso (não
otimizadas do caso A). Isto é, garante detetar alterações fisiopatológicas relevantes para diferenças a
partir de 22,8% entre dois doseamentos consecutivos, para cada paciente.
A carta não otimizada, só garante detetar alterações fisiopatológicas relevantes a partir de uma
diferença de 71,11% entre dois doseamentos consecutivos para cada paciente. Logo, o laboratório
consegue reduzir em mais de 300% o risco clínico durante a monitorização dum paciente, apenas com as
cartas de controlo interno otimizadas (caso B).
2 - Dispersão máxima de um doseamento
D = 1,96 x √(CVa2 + CVb
2)
A- D = 1,96 x √(CVa2 + CVb
2) = 1,96 x √(302 + 5,62) = 59.82%
B- D = 1,96 x √(CVa2 + CVb
2) = 1,96 x √(82 + 5,62) = 19.14%
Conclusão: A carta otimizada (caso B), permite ao laboratório calcular uma dispersão para cada
doseamento de glicose, 3x inferior da que possui com a carta em uso (não otimizada, caso A). Por
exemplo, se nesse laboratório se obtiver um valor de glicose de 100mg/dL, o valor real do paciente varia
entre 80,86 e 119,14 mg/dL com a carta otimizada. Com a carta em uso (não-otimizada, caso A), a
dispersão obtida situa-se entre 40,18 e 159,82 mg/dL para a glicose.

14-05-2016 28
3 - Número de amostras para determinar o ponto homeostático real do paciente
n = [Z x √(CVa2 + CVb
2)/D]2
Por exemplo se dosearmos 2 amostras do mesmo paciente para a glicose (com intervalo entre si
de 4 semanas), obtemos para cada situação o valor de dispersão perto do ponto homeostático
real do paciente de:
A- n = [1,96 x √(CVa2 + CVb
2)/D]2 <=> 2 = [1,96 x √(302 + 5,62)/D]2 <=> D= 42,30 %
B- n = [1,96 x √(CVa2 + CVb
2)/D]2 <=> 2 = [1,96 x √(82 + 5,62)/D]2 <=> D= 13,53 %
Conclusão: Mais uma vez comprovamos que com as cartas otimizadas, o laboratório obtém uma
eficiência 3 vezes superior na dispersão do valor médio de dois doseamentos, para as duas amostras
consecutivas em cada paciente. Isto é, estamos 3 vezes mais próximos do ponto hemostático real do
paciente, quando comparado com o valor obtido nas cartas de controlo interno não otimizadas,
reduzindo desta forma proporcionalmente, acima de 300%, o risco da decisão clínica.
14-05-2016 29
Figura 5: Fluxograma do Procedimento de otimização de cartas de controlo interno da qualidade

14-05-2016 30
Este procedimento de otimização das cartas de controlo interno da qualidade serve vários
propósitos:
- Harmonização da resposta laboratorial e fundamentação do Programa de CQI no processo de
monitorização da qualidade clínica definida para os diferentes métodos/equipamentos do(s)
laboratório(s) do mesmo grupo laboratorial ou centro hospitalar.
- Garantia de deteção precoce de erros, ou falhas, associados ao método em monitorização (o ajuste do
CV e o uso de regras simples de Westgard, permitem detetar o erro antes de ser clinicamente relevante,
no início, ou dentro do próprio “Run” analítico, e não alguns “Runs” depois, como sucede no caso das
multirregras Westgard, ou dos limites calculados pelos fabricantes que são demasiado permissivos
relativamente à imprecisão do método);
- Garantia de controlo e correta aplicação do delta-check ao laboratório (as cartas de controlo devem
estar preparadas para conter as variações na precisão e exatidão do método, ao definirem qual o CV
máximo permitido ou admitido pelo laboratório, por isso definem o delta-check/capacidade de deteção
de alterações fisiopatológicas relevantes, de trabalho do laboratório);
- Permitir efetuar o cálculo do risco clínico associado a cada doseamento e avaliar se o laboratório
consegue cumprir com os requisitos clínicos das Normas de Orientação Clínica da DGS, ou Guias de
Prática Clínica; no caso de não conseguir cumprir com as NOCs, o laboratório sabe como adaptar as
NOCs de forma a garantir a mesma segurança ao paciente comparativamente ao laboratório que
consegue cumprir com as NOCs.
As figuras 6 e 7 mostram duas cartas de controlo para os mesmos pontos, do mesmo nível de
controlo interno, para um doseamento de Ureia. A figura 6 representa uma carta de controlo otimizada.
A figura 7 representa uma carta de controlo elaborada, com os típicos 10% de variabilidade (CV) pré-
definida pelos fabricantes de amostras de controlo interno, para 1SD.
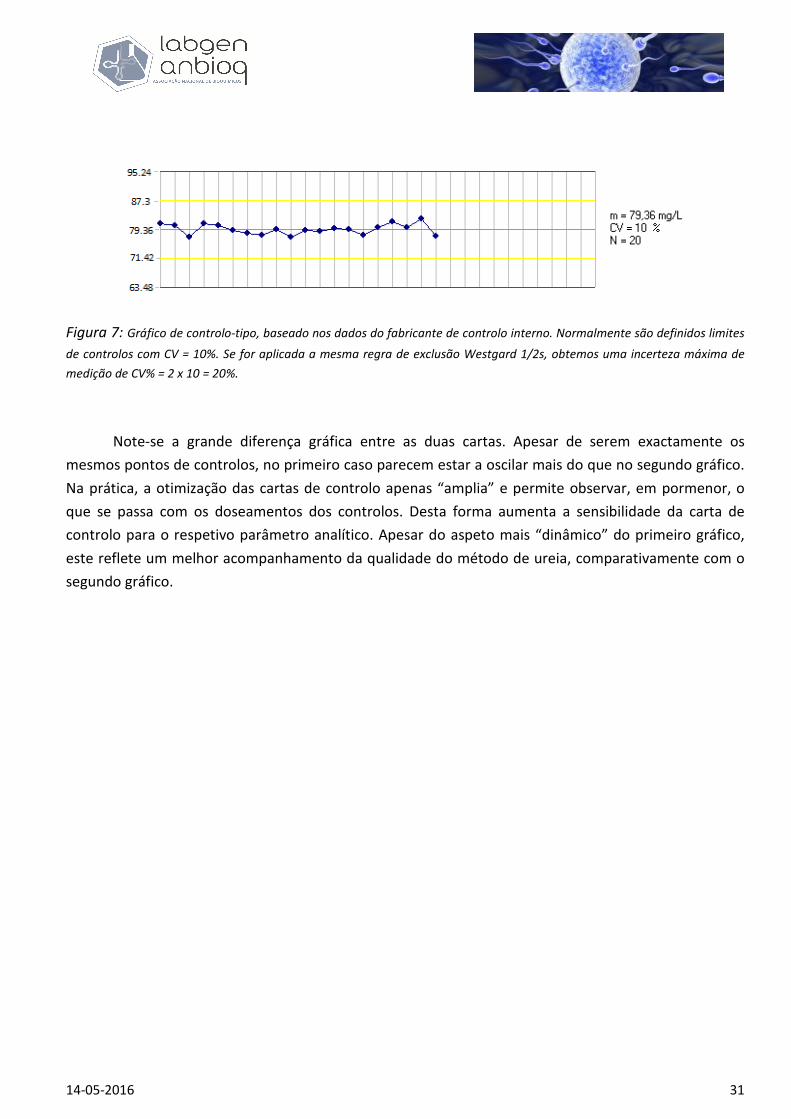
Figura 6: Gráfico de controlo otimizado, ajustado ao desvio padrão real do método de doseamento de ureia. Ao lado do
gráfico podemos observar a média e o Coeficiente de variação observados para o referido método e usados para elaborar
esta carta. Com o CV de 1.8% para 1 desvio padrão, podemos ver que se estipularmos a regra de exclusão Westgard 1/2s, o
máximo de incerteza de medição permitido passa a ser CV% máx = 2 x 1,8 = 3,6%.
14-05-2016 31
Figura 7: Gráfico de controlo-tipo, baseado nos dados do fabricante de controlo interno. Normalmente são definidos limites
de controlos com CV = 10%. Se for aplicada a mesma regra de exclusão Westgard 1/2s, obtemos uma incerteza máxima de
medição de CV% = 2 x 10 = 20%.
Note-se a grande diferença gráfica entre as duas cartas. Apesar de serem exactamente os
mesmos pontos de controlos, no primeiro caso parecem estar a oscilar mais do que no segundo gráfico.
Na prática, a otimização das cartas de controlo apenas “amplia” e permite observar, em pormenor, o
que se passa com os doseamentos dos controlos. Desta forma aumenta a sensibilidade da carta de
controlo para o respetivo parâmetro analítico. Apesar do aspeto mais “dinâmico” do primeiro gráfico,
este reflete um melhor acompanhamento da qualidade do método de ureia, comparativamente com o
segundo gráfico.

14-05-2016 32
VIII – CONSIDERAÇÕES FINAIS
Estima-se que 60 a 70 % das decisões clínicas (diagnóstico, tratamento, internamento e alta
hospitalar) são baseadas em resultados laboratoriais (25). O papel do Laboratório é essencial, sendo o
rigor e controlo analítico pontos fulcrais pela importância informativa de que se revestem. Cabe cada
vez mais ao Laboratório, não só produzir bons resultados assegurando a sua qualidade analítica, mas
sobretudo aprimorar e otimizar o controlo de qualidade Interna, fornecendo elementos de decisão
clinica relevantes, contribuindo para a redução do risco clínico.
O procedimento de otimização de controlos de qualidade interna, aliado à métrica da incerteza
de medição, permite ao laboratório conhecer qual o impacto clínico de cada doseamento nos seus
pacientes, ao mesmo tempo permite a adaptação às NOCs, ou então permite ao laboratório adaptar as
NOCs à sua realidade de trabalho e assegurar a mesma segurança/risco clínico, aos seus pacientes.
As cartas de controlo fornecidas nas bulas pelos fabricantes, são normalmente muito permissivas
quanto ao desvio padrão (SD) definido, e muitas vezes não apresentando a média-alvo real associada ao
método. Cientes dessa realidade, os fabricantes dos materiais de controlo de qualidade interno, tendem
a disponibilizar a todos os laboratórios que usam os seus produtos, a possibilidade de poderem colocar
a informação das suas médias e desvio padrão, numa página online preparada para o efeito. Nessas
condições, cada laboratório pode aferir a média-alvo obtida por consenso (peer-group), bem como um
desvio-padrão observado no conjunto dos laboratórios, para o método em estudo. O uso deste serviço
oferecido pelos fabricantes do material de controlo interno da qualidade é vital para planear,
implementar e monitorizar não só as cartas de controlo interno otimizadas, bem como todo o seu
programa de CQI.
Da nossa experiência, sabemos que as especificações operacionais técnicas observadas no
equipamento/método em uso no laboratório são, na maioria das vezes, muito mais precisas e de maior
qualidade. O procedimento de Avaliação e Otimização das Cartas de Controlo Interno aqui apresentado,
permite garantir o delta-check respetivo, que melhor se adequa a cada laboratório, garantindo a
deteção precoce das alterações fisiopatológicas relevantes para os pacientes. Ao mesmo tempo,
permite o uso das regras simples de Westgard, que detetam os erros no início ou no próprio “Run”
analítico.
A otimização das cartas de controlo deve ser efetuada sempre que a qualidade observada na
rotina laboratorial do método avaliado o permita. Assim, devemos certificar-nos que os requisitos
operacionais técnicos a aplicar às cartas de controlo otimizadas, são alcançáveis pelo
método/equipamento em estudo e para isso deve ser feita uma avaliação com um número mínimo de
pontos de controlos de qualidade internos (no caso das provas controladas diariamente, são
recomendados cerca de 3 a 6 meses de dados).
Se não houver alteração da estabilidade do método ao longo do tempo, a incerteza máxima de
medição definida mantem-se, mesmo quando se inicia um novo lote de controlo (onde muda
normalmente a média-alvo), revelando qual o requisito de qualidade que queremos cumprir (CV% máx).

14-05-2016 33
O único cuidado a ter, é a possível contribuição adicional associada à diferente estabilidade dos
diferentes tipos de amostras de controlos entre os diferentes fabricantes.
Uma das consequências visíveis na introdução de cartas de controlo interno otimizados é a
alteração do aspeto geral nos gráficos. A adoção de SD mais apertados do que os do fabricante, leva a
que as cartas de controlo apresentem um aspeto mais flutuante das suas curvas, aparentemente com
fortes oscilações entre os seus pontos (embora sempre dentro dos 2s ou 3s, conforme fique definido no
Programa de CQI). Trata-se de uma situação normal que deve ser devidamente explicada aos
profissionais do laboratório, sensibilizando-os para este facto.
Todas as alterações/otimizações efetuadas às cartas de controlo interno da qualidade devem
constituir um registado do Sistema de Gestão da Qualidade do Laboratório clínico, por exemplo através
de relatórios internos com a descrição do procedimento que é aqui exposto.

14-05-2016 34
BIBLIOGRAFIA
1 - “NP EN ISO 15189: 2014– Laboratórios clínicos, Requisitos para a qualidade e competência (ISO
15189:2012).” Instituto Português da Qualidade (IPQ).
2 - http://www.westgard.com/global-goal-results.htm
3 - “Bias in clinical chemistry”, Elvar Theodorsson, Bertil Magnusson & Ivo Leito, Bioanalysis (2014)
6(21), 2855–2875
4 - “Consensus agreement Conference on Strategies to set global quality specifications in laboratory
medicine.”, Kenny D, Fraser CG, Hyltoft Petersen P, KallnerA. ,Stockholm, April 24-26, 1999.
Scand J Clin Lab Invest 1999; 559:585.
5 - “Defining analytical performance specifications: Consensus Statement from the 1st Strategic,
Conference of the European Federation of Clinical Chemistry and Laboratory Medicine”, Sverre
Sandberg * , Callum G. Fraser , Andrea Rita Horvath , Rob Jansen , Graham Jones , Wytze
Oosterhuis , Per Hyltoft Petersen , Heinz Schimmel , Ken Sikaris and Mauro Panteghini, Clin
Chem Lab Med 2015, (http://www.efcclm.eu/files/efcc/2%20CCLM-
Consensus%20Statement.pdf , datado de 19/02/2015)
6 - AGREE “Appraisal of Guidelines and Research and Evaluation Instrument”:
http://www.agreetrust.org/
7 - “International Assessment of the Quality of Clinical Practice Guidelines in Oncology Using the
Appraisal of Guidelines and Research and Evaluation Instrument” J.S. Burger et al , JOURNAL OF
CLINICAL ONCOLOGY , VOLUME 22 NUMBER 10 MAY 15 2004
8 - “Endocrine clinical practice guidelines in North America. A systematic assessment of quality.”,
Bancos et al, Journal of Clinical Epidemiology 65 (2012) 520-525.
9 - “Critical appraisal of clinical practice guidelines of hepatocelular carcinoma”, Schmidt et al, J
Gastroenterol. Hepatol. 2011;26(12):1779-1786
10 - “Prescrição de Exames Laboratoriais para Avaliação de Dislipidemias”, NORMA DGS 066/2011.
11 - “Avaliação do Risco Cardiovascular SCORE”, Norma 005/2013, de 19/03/2013
12 - “Prescrição e Determinação da Hemoglobina Glicada A1c”, NORMA DGS 033/2011.
13 - “TIETZ Fundamentals of Clinical Chemistry”, Carl A. Burtis, et al, Saunders Elsevier, Capítulo 13,
pagina 211; Edição 2008.
14 - “Six Sigma Quality Design & Control”, 2d Edition, James O. Westgard, Westgard QC Inc.
15 - “Basic QC Practices”, 3rd Edition, James O. Westgard, Westgard QC Inc.
16 - “Requirements for the Estimation of Measurement Uncertainty”, National Pathology
Accreditation Advisory Council (NPAAC), Australian Government Department of Health and
Ageing, Online ISBN: 1 74186 165 9, Edition 2007
17 - “Basics of Estimating Measurement Uncertainty”, Graham H White, Clin Biochem Rev Vol 29
Suppl (i) August 2008

14-05-2016 35
18 - “Uncertainty of Measurement in Quantitative Medical Testing. A Laboratory Implementation
Guide”, G.H. White, I. Farrance, AACB Uncertainty of Measurement Working Group, Clin
Biochem Rev. 2004 November; 25(4): S1–S24
19 - “Six Sigma Risk Analysis”, 2011 Edition, James O. Westgard, Westgard QC Inc.
20 - “Test result variation and the quality of evidence-based clinical guidelines”, Callum G. Fraser,
Clinica ChimicaActa 346 (2004) 19–24
21 - “Improved Monitoring of Differences in Serial Laboratory Results” Callum G. Fraser, Clinical
Chemistry 57:12 1635–1637 (2011)
22 - ”Variación; biológica. Revisión desde una perspectiva práctica” Carmen Ricós et al, Rev. Lab.
Clin. 2010;3(4):192–200
(http://www.elsevier.es/sites/default/files/elsevier/pdf/282/282v03n04a13187891pdf001.pdf)
23 - Base de dados de Variabilidade Biológica desejável (SEQC)
(http://www.seqc.es/dl.asp?188.130.194.227.4.20.10.7.116.159.3.109.230.192.43.7.166.148.6
7.154.139.7.59.166.255.16.254.106.111.25.121.156.10.216.202.23.73.131.117.112.234.91.232.
193.203.180.35.124.252.201.72.134.190.225.237.105.54.30.163.73.103.151.247.249.163.113.2
52.156.249.166.72.176.199.226.107.90.96.162.97.139 /, consultado a 9/07/2015).
24 - Liquid Assayed Multiqual lote 45680, BioRad,
(http://myeinserts.qcnet.com/EI_customselection.aspx)
25- “Why is the laboratory an afterthought for managed care organizations?”, Forsman W. , Clinical
Chemistry 1996; 42 :813-816

14-05-2016 36
APÊNDICE 1
EXEMPLO DE OTIMIZAÇÃO DE CARTAS DE CONTROLO PARA O COLESTEROL TOTAL
O procedimento de implementação de cartas de controlo interno otimizadas começa com uma
avaliação às especificações técnicas operacionais de qualidade da carta de controlo interno em vigor no
laboratório, na forma de incerteza máxima de medição permitida.
Para além disso, deve ser feita uma avaliação dos requisitos clínicos presentes nas NOCs e no
caso do colesterol total temos:
A NOC 066/2011 define que se devem dosear 2 amostras do paciente para determinarmos o
colesterol total (espaçadas 4 semanas entre si), antes da definição do risco cardiovascular ou início de
terapêutica.
A NOC 005/2013 define como alvos clínicos da estratificação do risco cardiovascular os valores
de 4, 5, 6, 7 e 8 mmol/L de ColT, que devem ser significativamente diferentes com a menor
sobreposição possível entre si.
Tendo esses dados, avançamos com o processo de otimização usando a metodologia referida no
capítulo VII deste procedimento.
A - Carta de controlo em uso no laboratório (não-otimizada):
O coeficiente de variação intralaboratorial (CV %) máximo definido nas cartas de controlo de
qualidade interna de um dado laboratório são as definidas para 2 desvios padrões (2s), quando
aplicada a regra Westgard de 1/2s, aos limites definidos no lote 45680 das amostras de controlo
Assayed Multiqual da BioRad (24).
Assim, o denominador máximo dos CV máximo dos níveis 1, 2 e 3: CV max = 13,18% (13,18%
para o nível 1; 12,35% para o nível 2 e 12,13% para o nível 3, para os equipamentos AUs
Beckman) (24)

14-05-2016 37
B - Carta de controlo otimizada:
A seguir calcula-se qual o CV% real de trabalho que o método de doseamento possui (com dados
de pelo menos 3 a 6 meses da rotina de trabalho).
CV médio real de trabalho do laboratório em estudo: Neste exemplo assumimos CV% média Col
= 2%, para os 3 níveis de controlo.
Logo o CV% máx a aplicar na carta de controlo usando a regra Westgard 1/2s é de CV%
máx=2xCV%médio=2x2=4%.
Avaliação comparativa do Risco Clínico
Para cada caso (A e B) calculamos a seguir (sabendo que a CVi colesterol total=5,95%, para
amostras de soro (23)):
1 - Capacidade de detetar alterações fisiopatológicas relevantes (Δ-Check=2,33 x
√(CVa2+CVb
2) ):
A- Δ-Check=2,33 x √(CVa2+CVb
2) = 2,33 x √(13,182+5,952) = 33,7% B- Δ-Check=2,33 x √(CVa
2+CVb2) = 2,33 x √(42+5,952) = 16,7%
Conclusão: A carta otimizada permite ao laboratório ser 2x mais eficiente na redução do risco da
decisão clínica na deteção das alterações fisiopatológicas relevantes para o colesterol total do que com
as cartas que tem em uso (não otimizadas). Isto é, garante detetar alterações fisiopatológicas relevantes
para diferenças a partir de 16,7% entre dois doseamentos consecutivos para cada paciente. Com a carta
não otimizada, apenas garante detetar alterações fisiopatológicas relevantes a partir de 33,7% de
diferença entre dois doseamentos consecutivos de cada paciente.
2 - Dispersão máxima de um doseamento (D = 1,96 x √(CVa2 + CVb
2)):
A- D = 1,96 x √(CVa2 + CVb
2) = 1,96 x √(13,182+5,952) = 28,34 %
B- D = 1,96 x √(CVa2 + CVb
2) = 1,96 x √(42+5,952) = 14,05 %

14-05-2016 38
Conclusão: A carta otimizada permite ao laboratório possuir uma dispersão para cada doseamento de
colesterol total, 2x inferior (torna-se 2x mais eficiente na menor dispersão) à que possui com a carta em
uso (não otimizada) e reduz proporcionalmente o risco da decisão em cut-off clínicos do Colesterol
Total.
3- Número de amostras para determinar o ponto homeostático real do paciente (n = [Z
x √(CVa2 + CVb
2)/D]2):
Assim, cumprindo com a NOC 066/2011, para 2 amostras de doseamento de colesterol total,
obtemos para cada situação o valor de dispersão perto do ponto homeostático real do paciente de:
A- n = [1,96 x √(CVa2 + CVb
2)/D]2 <=> 2 = [1,96 x √(13,182+5,952)/D]2 <=> D= 20,0 % B- n = [1,96 x √(CVa
2 + CVb2)/D]2 <=> 2 = [1,96 x √(42+5,952)/D]2 <=> D= 9,9 %
Conclusão: Mais uma vez comprovamos que com as cartas otimizadas, o laboratório obtém 2x
mais eficiência na redução do risco da decisão clínica associada à dispersão da média de dois
doseamentos de duas amostras consecutivas de cada paciente. Isto é, encontra-se 2x mais perto do
ponto homeostático real do paciente, do que com as cartas de controlo interno não otimizadas.
Desta forma e se formos avaliar os alvos de decisão clinica da NOC 005/2013, para a avaliação do
risco cardiovascular nas tabelas SCORE, verificamos que nas cartas otimizadas apenas possuímos
sobreposição dos alvos 5-6, 6-7 e 7-8 mmol/L de colesterol total e apenas nesses sentidos. Com as cartas
de controlo original da empresa produtora de amostras de controlo, o laboratório obtém doseamentos
que sobrepõem os alvos em todos os sentidos para 4-5, 4-5-6, 4-5-6-7-8 e 5-6-7-8 mmol/L de colesterol
total.

14-05-2016 39
4 5 6 7 8 Sobreposições
9,9% dispersão de 2 amostras
(CV máx ColT= 4%) 3,60-4,4 4,51-5,5 5,41-6,59 6,31-7,69 7,21-8,79 5-6; 6-7; 7-8
20% dispersão de 2 amostras
(CV máx ColT= 13,18%) 3,2-4,8 4,0-6,0 4,8-7,2 5,6-8,4 6,4-9,6 4-5; 4-5-6; 4-5-6-7-8; 5-6-7-8
Figura 1 – Apêndice 1: Dados das dispersões esperadas para cada valor de decisão clinica de
colesterol total definido na NOC 005/2013 da avaliação do risco cardiovascular SCORE, para as duas
situações de incerteza máxima de medição deste exemplo (ColT CV máx = 4% => dispersão de 2 amostras
= 9,9% para a carta de controlo otimizada; e ColT CV máx = 13,18% => dispersão de 2 amostras = 20,0%
para a carta de controlo não otimizada).
No caso particular de um laboratório que não consegue garantir um ColT CV máx = 4% =>
dispersão de 2 amostras = 9,9%, para a carta de controlo otimizada (laboratório C), sabendo qual o CV
máximo que consegue definir no seu programa de CQI, esse laboratório está em condições de definir o
número de amostras que necessita para garantir a mesma segurança ao paciente que o laboratório do
caso B.
Para isso, basta efetuar o cálculo do número de amostras necessárias para determinar o valor
médio de colesterol total perto de 9,9% do valor real de ColT dos seus pacientes.
Assim, se por exemplo o laboratório C apenas consegue garantir um ColT CV máx = 7%, então
obtemos:
C- n = [1,96 x √(CVa2 + CVb
2)/D]2 <=> n = [1,96 x √(72+5,952)/9,9]2 <=> n= 3 amostras (arredondado
de 3,3)
Conclusão: Desta forma o laboratório C demonstra que, no seu caso, com 3 doseamentos de
ColT consegue garantir a mesma segurança na estratificação do risco cardiovascular pela SCORE (NOC
005/2013) aos seus pacientes, equivalente à conseguida pelo laboratório B.

14-05-2016 40
APÊNDICE 2
EXEMPLO DE OTIMIZAÇÃO DE CARTAS DE CONTROLO PARA A HBA1C
O procedimento de implementação de cartas de controlo interno otimizadas começa com uma
avaliação às especificações técnicas operacionais de qualidade da carta de controlo interno em vigor no
laboratório, na forma de incerteza máxima de medição permitida.
Para além disso deve ser feita uma avaliação dos requisitos clínicos presentes nas NOCs e no
caso da HbA1C temos:
A NOC 033/2011 (12) define diretamente o CV máximo intralaboratorial admissível para a HBA1C
com CV máx < 2%.
Tendo esses dados, avançamos com o processo de otimização usando a metodologia referida no
capítulo VII deste procedimento.
A - Carta de controlo em uso no laboratório (não-otimizada):
O coeficiente de variação intralaboratorial (CV %) máximo definido no Programa de controlo de
qualidade interna para o doseamento da HbA1c do equipamento HA-8160 da A.Menarini diagnostics
usando os controlos da A. Menarini lote 201201, observamos que definiam um erro máximo que
permite uma incerteza máxima de medição de CV nível 1 = 16.9% e CV nível 2 = 9.17% (usando a mesma
regra de controlo Westgard 1/2s como limite).
Assim, o denominador máximo dos CVmáximo dos níveis 1 e 2, é: CVmax = 16,9%
B - Carta de controlo otimizada:
A seguir calcula-se qual o CV% real de trabalho que o método de doseamento possui (com dados
de pelo menos 3 a 6 meses da rotina de trabalho).
O coeficiente de variação analítica médio observado no Sistema de Informação do Laboratório
para os dois níveis do lote de controlo número 201201, durante o período de 6 meses, foi: CVnível 1 de
controlo= 0,90%; CV nível 2 de controlo=0,78%.
Logo o CV médio (maior observado) = 0,90% => CV máx = 1,8%.

14-05-2016 41
Como a NOC 033/2011 define claramente que a incerteza máxima de medição intralaboratorial
deve ser <2%, podemos definir CVmáx=2% com a regra de exclusão Westgard 1/2s, pois garante o
cumprimento do requisito de qualidade clinica definida na NOC. Para além disso a rotina de trabalho
desse laboratório demonstra que consegue cumprir com esse requisito, dado que o dobro do CV médio
de trabalho é 1,8% (<2%).
Para isso a carta de controlo interno da qualidade é montada com um CV=2% sob a regra de
controlo Westgard 1/2s (regra de alerta para a resolução de problema no método de doseamento da
HbA1c, no caso do valor de um controlo interno igualar ou ultrapassar os 2 desvios padrões da média-
alvo). Note-se que quando o erro iguala os dois desvios padrão (acima ou abaixo do valor alvo), estamos
na situação limite CV=2% que ultrapassa a incerteza máxima permitida pela Norma, pelo que a regra
ativa o alerta relativo ao erro a ser corrigido de modo a garantir CV máximo < 2%.
Avaliação comparativa do Risco Clínico
Para cada caso calculamos (sabendo que a CVi HbA1c=1,85%, para amostras de sangue total (23)):
1 - Capacidade de detetar alterações fisiopatológicas relevantes (Δ-Check=2,33 x
√(CVa2+CVb
2) ):
A- Δ-Check=2,33 x √(CVa2+CVb
2) = 2,33 x √(16,92+1,852) = 39,61%
B- Δ-Check=2,33 x √(CVa2+CVb
2) = 2,33 x √(22+1,852) = 6,35%
Conclusão: A carta otimizada permite ao laboratório cumprir com o requisito de qualidade
intralaboratorial (CVmáx<2%) requerido pela NOC 033/2011 e ao mesmo tempo passa a ser 6x mais
eficiente na redução do risco da decisão clínica na deteção de alterações fisiopatológicas relevantes
para a HbA1C, do que com as cartas que tem em uso neste exemplo (não otimizadas). Isto é, o
laboratório que cumpre com a NOC 033/2011 garante detetar alterações fisiopatológicas relevantes
para diferenças a partir de 6,35% entre dois doseamentos consecutivos para cada paciente. Com a carta
não otimizada, só garante detetar alterações fisiopatológicas relevantes a partir de 39,61% de diferença
entre dois doseamentos consecutivos de HbA1c de cada paciente.

14-05-2016 42
2 - Dispersão máxima de um doseamento (D = 1,96 x √(CVA2 + CVb
2)):
A- D = 1,96 x √(CVa2+CVb
2) = 1,96 x √(16,92+1,852) = 33,32%
B- D = 1,96 x √(CVa2+CVb
2) = 1,96 x √(22+1,852) = 5,34%
Conclusão: A carta otimizada de modo a cumprir com a NOC 033/2011, permite ao laboratório
possuir uma dispersão para cada doseamento de HbA1C, 6x inferior à que possui com a carta em uso
(não otimizada) e reduz proporcionalmente o risco da decisão em cut-off clínicos da HbA1C.
3- Número de amostras para determinar o ponto homeostático real do paciente (n = [Z
x √(CVa2 + CVb
2)/D]2):
Assim, cumprindo com a NOC 066/2011, para 2 amostras de doseamento de HbA1C, obtemos para cada
situação o valor de dispersão perto do ponto homeostático real do paciente de:
A- n = [1,96 x √(CVa2+CVb
2)/D]2 <=> 2 = [1,96 x √(16,92+1,852)/D]2 <=> D= 23,56 % B- n = [1,96 x √(CVa
2+CVb2) /D]2 <=> 2 = [1,96 x √(22+1,852)/D]2 <=> D= 3,78%
Em conclusão, mais uma vez comprovamos que com as cartas otimizadas, o laboratório obtém 6x
mais eficiência na redução do risco da decisão clínica associada à dispersão da média de dois
doseamentos de HbA1C referentes a duas amostras consecutivas de cada paciente. Isto é, com as cartas
de controlo otimizadas, em harmonia com a NOC 033/2011, com duas amostras consecutivas dos
pacientes, o Laboratório encontra-se 6x mais perto do ponto homeostático real do paciente, do que
com as cartas de controlo interno não otimizadas.
Assim, quando é feito um segundo doseamento da HbA1C no laboratório com as cartas de
controlo otimizadas com CVmáx=2% (com regra de exclusão 1/2s), para obter a confirmação do
diagnóstico de Diabetes Mellitus (como requerido pela Norma 033/2011), obtemos uma média do valor
de HbA1c entre os dois doseamentos, que se encontra cerca de 3,78% perto do valor homeostático real
do paciente, e esse facto é decisivo para o correto diagnóstico.
Desta forma e se formos avaliar o alvo de decisão clinica HbA1C =7%, verificamos que com as
cartas de controlo interno otimizadas, qualquer valor obtido no intervalo 6,74% - 7,26% de HBA1C,
representa o cut-off de decisão clínica.

14-05-2016 43
Coordenação técnico-científica
Jorge Pinheiro
Bioquímico Clínico – LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Membro da Direção do Colégio de Biologia Humana e Saúde da Ordem dos Biólogos (CBHS/OBIO)
Coordenador da Comissão da Qualidade de Laboratório Clínico e Genética Humana de Bioquímicos Clínicos da
Associação Nacional de Bioquímicos (LabGen-ANBIOQ)
Revisão
António Dinis
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Luisa Ponte
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Olga Rebelo
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Paulo Vieira
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Rita Ribeiro
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Ana Sousa
Especialista em Genética Humana – CBHS/OBIO
Presidente do Colégio de Biologia Humana e Saúde –
Ordem dos Biólogos
Sílvia Lopo
Especialista em Análise Clínicas – CBHS/OBIO
Membro da Direção do Colégio de Biologia Humana e
Saúde da Ordem dos Biólogos
Claudia Julio
Especialista em Análise Clínicas – CBHS/OBIO Vogal Direção da OBIO para a Saúde
Bárbara Marques
Especialista em Genética Humana – CBHS/OBIO Vogal Direção da OBIO para a Saúde
Nuno Cunha
Bioquímico Clínico - LabGen-ANBIOQ
Especialista em Análises Clínicas – CBHS/OBIO
Membro da Direção do Colégio de Biologia Humana e
Saúde da Ordem dos Biólogos
Comissão de Laboratório Clínico e Genética
Humana de Bioquímicos Clínicos
Associação Nacional de Bioquímicos
Colégio de Biologia Humana e Saúde
Ordem dos Biólogos