objectivo i compreender os conceitos básicos … de segurança do fármaco e na validação da dose...
TRANSCRIPT

Farmacocinética – UP1
1
Objectivo I – Compreender os conceitos básicos gerais relativos à farmacocinética e sua
relação com áreas afins, objecto de estudo da farmacocinética, aplicação da farmacocinética
na rotina clínica e na investigação e desenvolvimento de novos fármacos
1. Definir farmacocinética, farmacocinética clínica e toxicocinética. Compreender a sua
relação com a Biofarmácia, Terapêutica e Toxicologia. Conhecer a sua aplicação no
desenvolvimento de novos fármacos e na optimização/individualização de regimes
posológicos. (Shargel 2005 Chap. 1; Tozer 2006 Chap. 1)
Farmacocinética
Depois da libertação do fármaco da sua forma de dosagem, este é absorvido no
tecido envolvente. De seguida, ocorre a disposição do fármaco, que não é mais do que a
descrição da distribuição e eliminação do mesmo -> A caracterização da disposição é um
importante pré-requisito para determinar/modificar regimes de dosagem para os doentes.
O estudo PK envolve estudos experimentais (1) e aproximações teóricas (2):
(1) Envolvem a análise de amostras biológicas, métodos analíticos de medição de fármacos e
metabolitos e procedimentos que facilitem a recolha e manipulação de dados;
(2) Envolve matemática e técnicas computacionais -> Desenvolvimento de modelos
farmacocinéticos que predigam a disposição do fármaco, após a sua administração.
Os métodos estatísticos são parte integral dos estudos PK ->
Usados como um parâmetro de estimação e interpretação de dados,
sendo aplicados para determinar erros de dados e desvios ao modelo
estrutural.
Farmacocinética clássica é o estudo de modelos teóricos
focando-se no desenvolvimento de modelos e parametrização.
Farmacocinética clínica
Durante o processo de desenvolvimento de fármacos, são testados muitos doentes
de forma a determinar os regimes óptimos de dosagem. Neste sentido, Farmacocinética
Clínica é a aplicação dos métodos farmacocinéticos na terapia com fármacos -> Envolve uma
aproximação multidisciplinar a estratégias de doseamento individual optimizado baseadas na
doença, no doente e nas considerações específicas deste -> Idade, sexo, genética, e etnia
podem originar diferenças farmacocinéticas que podem afectar o resultado da farmacoterapia.
O estudo destas diferenças em vários grupos populacionais -> Farmacocinética de população.
Todas as etapas a que um fármaco está sujeito, uma vez
incorporado no organismo têm lugar de forma simultânea e
não sequencial -> Processo dinâmico.

Farmacocinética – UP1
2
Outro aspecto importante da PK é a monitorização da terapia com fármacos, de
forma a optimizar a eficácia e prever qualquer toxicidade adversa. Quando fármacos com
índices terapêuticos estreitos são usados, é necessário monitorizar o doente -> Monitorização
das concentrações plasmáticas do fármaco ou de um parâmetro farmacodinâmico.
Alguns fármacos frequentemente monitorizados são: aminoglicosídeos,
anticonvulsivantes, e fármacos usados em quimioterapia -> Minimizar efeitos adversos.
Toxicocinética
É a aplicação dos princípios farmacocinéticos ao design, conduta, interpretação dos
estudos de segurança do fármaco e na validação da dose experimental usada em animais.
Estudos toxicocinéticos: feitos em animais durante o desenvolvimento pré-clínico do
fármaco e podem continuar depois deste ter sido testado em ensaios clínicos.
Dados toxicocinéticos: ajudam na interpretação de descobertas toxicológicas em
animais e na extrapolação dos resultados para os humanos
Toxicologia clínica: estudo dos efeitos adversos dos fármacos e de substâncias
tóxicas no corpo. A PK do fármaco num doente medicado com altas doses terapêuticas pode
ser bastante diferente da PK quando administrado em baixas doses terapêuticas -> Em doses
altas, a concentração do fármaco pode saturar as enzimas envolvidas na absorção,
biotransformação ou nos mecanismos de secreção renal activa e, por isso, alterar a PK de
linear para não-linear.
Biofarmacêutica
As características físico-químicas do princípio activo, a forma de dosagem e a via de
administração são importantes no que diz respeito à segurança e eficácia do fármaco; podem
afectar em grande margem a sequência de eventos que se segue, pelo que os regimes de
dosagem actuais (dose, forma de dosagem, intervalo de dosagens) são cuidadosamente
determinados com o auxílio de ensaios pré-clínicos e clínicos.
1. É tomado na sua forma de dosagem, por diferentes vias de administração;
2. De seguida, é libertado da sua forma de dosagem de forma previsível e caracterizável;
3. Algumas fracções do fármaco são absorvidas a partir dos seus locais de administração
para os tecidos envolventes, para o corpo (Ex.: via oral), ou para ambos;
4. Finalmente alcança o local de acção -> Concentração de fármaco neste excede a
concentração mínima efectiva (CME) -> Efeito farmacológico.
Biodisponibilidade (medida da disponibilidade sistémica do fármaco) pode diferir de
um fármaco para outro com o mesmo princípio activo, até mesmo pela mesma via de
administração.

Farmacocinética – UP1
3
Biofarmacêutica -> Ciência que examina a relação das propriedades físicas do
fármaco, a sua forma de dosagem e a via de administração com a taxa e extensão da absorção
sistémica do fármaco. Envolve factores que influenciam:
1. A estabilidade do princípio activo;
2. A libertação do fármaco da sua forma de dosagem;
3. A taxa de dissolução/ libertação do fármaco no seu local de absorção;
4. Absorção sistémica do fármaco.
2. Conhecer o objecto de estudo da farmacocinética (curvas concentração-tempo) e a
metodologia geral para a sua obtenção.
As concentrações de fármacos são um elemento importante para determinar a PK.
Métodos invasivos: colheita de amostras de sangue, fluido espinhal, fluido sinuvial, biopsia ou
qualquer tipo de método que necessite de intervenção parenteral ou cirúrgica no doente.
Métodos não invasivos: urina, fezes, saliva, ar expirado ou qualquer tipo de método que possa
ser recolhido sem intervenção parenteral ou cirúrgica no doente.
Concentração do fármaco no sangue, plasma e soro
É a forma mais directa de aceder à farmacocinética de um fármaco no organismo.
O sangue total contém os elementos celulares: glóbulos vermelhos, brancos,
plaquetas e várias proteínas (albumina e globulinas). Mas geralmente, o soro e o plasma são
os mais comummente usados na medição da concentração do fármaco.
- Soro: sangue total coagula e o soro é recolhido do sobrenadante após centrifugação;
- Plasma: obtido do sobrenadante do centrifugado do sangue total ao qual foram adicionados
anticoagulantes (Ex.: heparina) -> Proteínas presentes no soro e no plasma são diferentes.
Significado da medida da concentração de fármacos no plasma
A intensidade dos efeitos farmacológicos ou tóxicos de um fármaco está relacionada
com a concentração deste no alvo (localizado nas células dos tecidos). Neste sentido, como a
maior parte destas células estão em contacto com fluidos tecidulares ou plasma, a medida do
nível plasmático do fármaco é um método sensível para monitorizar o curso da terapêutica.
Concentração de fármacos nos tecidos

Farmacocinética – UP1
4
As biopsias são ocasionalmente efectuadas para diagnóstico (verificar existência de
células malignas) -> Geralmente, é removida apenas uma pequena amostra de tecido, fazendo
com que a medição da concentração do fármaco seja difícil -> Concentrações no tecido
retirado podem não reflectir a concentração do fármaco noutros tecidos ou até mesmo noutra
parte do mesmo tecido.
Concentração de fármaco na urina e nas fezes
Medição do fármaco na urina: método indirecto para verificar a sua
biodisponibilidade -> Taxa e extensão da excreção do fármaco na urina reflecte a taxa da
extensão da absorção sistémica do fármaco.
Medição do fármaco nas fezes: pode reflectir fármacos que não foram absorvidos por
via oral ou que foram expelidos por secreção biliar, após absorção sistémica.
Concentrações de fármacos na saliva (indicador secundário)
Como apenas fármacos livres se dissolvem na saliva -> Níveis detectados nesta
tendem a ser os do fármaco-livre (aprox.), e não a concentração plasmática total do fármaco.
O rácio da concentração de fármacos na saliva/plasma é maioritariamente
influenciado pelo pKa do fármaco e pelo pH da saliva -> Ácidos e bases fracas com pKa muito
diferente de pH 7,4, geralmente, têm uma melhor correlação com os níveis de fármaco no
plasma. As concentrações de fármacos na saliva retiradas após equilíbrio com as
concentrações de fármaco no plasma, geralmente, providenciam uma indicação mais estável
dos níveis de fármaco no sangue.
Medições forenses de fármacos
As medições de fármacos obtidas na autópsia tanto nos tecidos como nos fluidos
corporais (Ex.: Saliva, sangue e urina) podem ser úteis se a vítima/suspeito sofreu uma
overdose, foi envenenada ou usou drogas de abuso (Ex.: heroína, cocaína).
Curva concentração no plasma-tempo
É gerada pela medição da concentração do fármaco a partir de amostras de plasma
recolhidas em vários intervalos de tempo, após a administração do mesmo. Por outro lado, a
PK pode também descrever a curva níveis plasmáticos-tempo considerando o pico plasmático,
tempo para o atingir e área sob a curva.
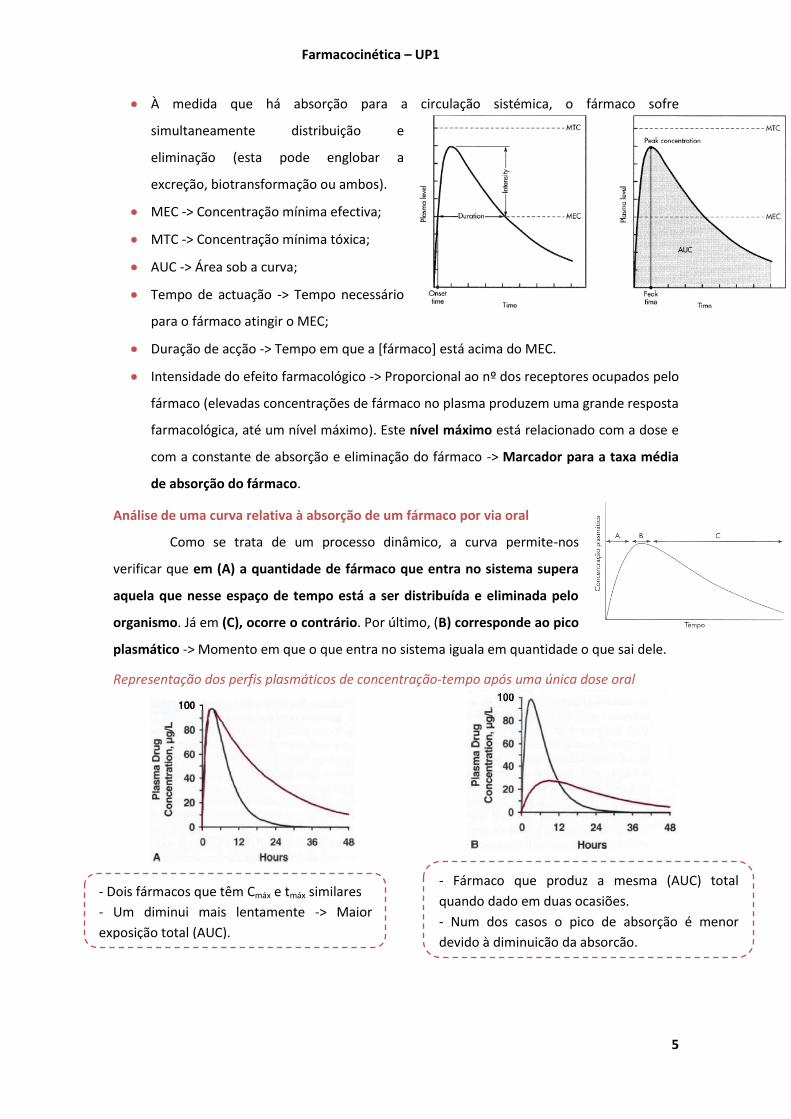
À medida que o fármaco atinge a circulação sistémica, a concentração deste no plasma
vai aumentar até um nível máximo (Cmáx).
Normalmente a absorção do fármaco é mais rápida que a sua eliminação.
Farmacocinética – UP1
5
À medida que há absorção para a circulação sistémica, o fármaco sofre
simultaneamente distribuição e
eliminação (esta pode englobar a
excreção, biotransformação ou ambos).
MEC -> Concentração mínima efectiva;
MTC -> Concentração mínima tóxica;
AUC -> Área sob a curva;
Tempo de actuação -> Tempo necessário
para o fármaco atingir o MEC;
Duração de acção -> Tempo em que a [fármaco] está acima do MEC.
Intensidade do efeito farmacológico -> Proporcional ao nº dos receptores ocupados pelo
fármaco (elevadas concentrações de fármaco no plasma produzem uma grande resposta
farmacológica, até um nível máximo). Este nível máximo está relacionado com a dose e
com a constante de absorção e eliminação do fármaco -> Marcador para a taxa média
de absorção do fármaco.
Análise de uma curva relativa à absorção de um fármaco por via oral
Como se trata de um processo dinâmico, a curva permite-nos
verificar que em (A) a quantidade de fármaco que entra no sistema supera
aquela que nesse espaço de tempo está a ser distribuída e eliminada pelo
organismo. Já em (C), ocorre o contrário. Por último, (B) corresponde ao pico
plasmático -> Momento em que o que entra no sistema iguala em quantidade o que sai dele.
Representação dos perfis plasmáticos de concentração-tempo após uma única dose oral
- Dois fármacos que têm Cmáx e tmáx similares
- Um diminui mais lentamente -> Maior
exposição total (AUC).
- Fármaco que produz a mesma (AUC) total
quando dado em duas ocasiões.
- Num dos casos o pico de absorção é menor
devido à diminuição da absorção.

Farmacocinética – UP1
6
Uma absorção retardada pode ser desvantajosa se a resposta está directamente
relacionada com a concentração (Cmax menor e tmax ocorre mais tarde), com a
possibilidade de não ser alcançada uma resposta clínica adequada.
A velocidade de absorção seria de reduzida importância se o benefício clínico do
fármaco fosse determinado pela exposição total (AUC) e não pela concentração.
3. Conhecer as diferentes fases do ciclo geral dos fármacos no organismo: Biofarmacêutica,
Farmacocinética, Farmacodinâmica e Terapêutica. Perceber o conceito de margem
terapêutica e a sua relação com a resposta farmacológica. Entender a existência de
variabilidade inter-individual na resposta farmacológica e suas implicações terapêuticas.
A fase farmacocinética cobre a
relação existente entre o input dos fármacos,
que depende de factores tais como a dose,
forma farmacêutica, via de administração.
A farmacodinâmica relaciona-se com
a concentração no organismo e os efeitos
adversos produzidos com o tempo.
Margem terapêutica e a sua relação com a resposta farmacológica
1. Intensidade do efeito aumenta com o aumento da exposição ao fármaco (mas só até
certos limites).
2. O efeito máximo produzido por um fármaco pode ser muito diferente do efeito
produzido por outro, embora a resposta medida seja a mesma. Aspirina e morfina -> A
aspirina por mais que se administre em elevadas doses nunca terá o mesmo efeito que a
morfina para o alívio da dor severa.
3. Fármacos produzem uma multiplicidade de efeitos (desejáveis e indesejáveis).
Para que um fármaco possa actuar é necessário que atinja concentrações eficazes
nas estruturas celulares onde a sua acção se exerce (biofase). Assim sendo, a margem ou
janela terapêutica exprime a relação entre a dose máxima tolerada, ou também tóxica
(concentração tóxica), e a dose terapêutica (concentração mínima eficaz) (Dose tóxica/dose
terapêutica).
Ex.: II Ex.: III

Farmacocinética – UP1
7
Exemplo I
Regime A: é conseguido sucesso terapêutico, embora não inicialmente.
Regime B: objectivo terapêutico é alcançado mais rapidamente, originando efeitos adversos.
Exemplo: Quinacrina (antimalárico) -> Em baixas doses, não é eficaz; já em altas doses tem
efeitos adversos. Este fármaco é eliminado muito lentamente quando administrado em
pequenas doses diariamente e acumula-se com a administração continua. Desta forma,
inicialmente deve-se dar elevadas doses deste fármaco para atingir a janela terapêutica e
posteriormente deve reduzir-se a dose para não provocar efeitos adversos.
Exemplo II
A PD também é importante na margem terapêutica -> Efeito da atorvastatina (usada
para combater o colesterol) só tem efeito terapêutico após 6 meses de toma.
Exemplo III
Antes da administração do fármaco, a actividade da protrombina é elevada. A
absorção do fármaco é rápida, mas o seu pico de acção ocorre aproximadamente passados 2
dias da toma.
Variabilidade inter-individual na resposta farmacológica e as suas implicações terapêuticas
A variabilidade inter-individual que está associada à PK (regime posológico
individualizado) toma particular importância em fármacos com margens terapêuticas estreitas
(Ex.: Ciclosporina, fenitoína); já que em casos de ampla margem terapêutica -> Doentes podem
tolerar ampla faixa de exposições para graus similares de benefício (e mesmo assim, o doente
pode não responder à terapia por não possuir o receptor onde os fármacos actuam).
A biotransformação contribui de forma mais significativa para a variabilidade
associada à PK do que, por exemplo, a distribuição.
Legenda:
Curva vermelha – actividade da protrombina
Curva preta – fármaco Varfarina
Ex.: I

Farmacocinética – UP1
8
A concentração de fenitoína em doses
crónicas tende a aumentar com a taxa
da dose – muito variável de indivíduo
para indivíduo.
A concentração mínima alveolar requerida
para anestesia geral (com desflurano) varia
com a idade -> Doentes idosos são mais
sensíveis ao seu efeito anestésico.
Factores genéticos
Malária -> Afecta mais africanos (deficiência na glucose-6-fosfato desidrogenase)
comparativamente aos caucasianos.
Deficiência em enzimas
Deficiência CYP450 -> Debriosoquine -> Não é metabolizada -> Hipotensão
Através da análise do genoma -> Compreender base molecular das diferenças
genéticas, criando-se desta forma a possibilidade de usar a informação genómica para predizer
e individualizar a terapia.
Interacções farmacológicas
Ex.: Doente idoso susceptível ao desenvolvimento de tromboses -> Fenilbutazona
(anti-inflamatório) -> Inibidor das enzimas que metabolizam a varfarina (anticoagulante).
Outros -> Idade, sexo, patologias e ritmos biológicos
Implicações terapêuticas
Ajustar doses;
Estabelecer terapias individuais;
Monitorização dos fármacos.
Importância da compliance (cumprimento da prescrição médica – dose, duração da toma)
Se todos os doentes aderissem ao
regime das doses prescritas tornava-se mais fácil
compreender porque os tratamentos falham ou
as concentrações aumentam para valores tóxicos

Farmacocinética – UP1
9
-> O cumprimento da compliance é fulcral no sucesso da terapia, nomeadamente quando se
utilizam fármacos como antibióticos, fármacos para o HIV, entre outros.
3.1. Entender as diferentes etapas das fases Biofarmacêutica e Farmacocinética e a sua
natureza dinâmica no ciclo geral dos fármacos no organismo (Libertação, Absorção,
Distribuição, Metabolismo e Excreção). Diferenciar incorporação de disposição. Distinguir
eliminação e excreção.
Incorporação Vs. Disposição
Incorporação – Está relacionada com a absorção e libertação do fármaco.
Disposição – Todos os processos cinéticos que ocorrem após a absorção do fármaco. Os
componentes da disposição são a distribuição e eliminação.
Eliminação Vs. Excreção
Eliminação – Perda irreversível do fármaco do local de acção. Ocorre através de 2 processos:
excreção e metabolismo.
Excreção – perda irreversível de fármacos não ionizados.
Fase biofarmacêutica
Comporta as etapas de disponibilização dos princípios activos no organismo.
1. Libertação
Na administração extravascular de uma forma farmacêutica sólida, a libertação pode
fazer-se rapidamente (forma farmacêutica de libertação rápida); ou lentamente (forma de
libertação prolongada). Consiste, geralmente, numa desintegração da forma sólida.
2. Dissolução
Para atravessar as membranas biológicas, o princípio activo deve ser disperso no
estado molecular, em meio aquoso, no local de absorção. A velocidade de dissolução do
princípio activo função das suas características físico-químicas e do pH do meio de absorção.
Fase farmacocinética
Absorção
Passagem desde o local onde o fármaco é depositado até atingir a circulação
sanguínea -> O princípio activo dissolvido, cuja forma não-ionizada é suficientemente
lipossolúvel para atravessar as membranas biológicas do local de absorção e penetrar na
circulação sanguínea.
No caso de uma absorção GI, o princípio activo é primeiro transportado, pela veia
porta, para o fígado, antes de atingir a circulação sistémica, ficando disponível para sofrer
biotransformação pré-sistémica no lúmen intestinal, na parede da membrana GI ou no fígado.

Farmacocinética – UP1
10
Essa transformação explica, em parte, a diminuição da biodisponibilidade de um
fármaco, após a sua administração oral.
Factores susceptíveis de influenciar a absorção
1. Área de absorção
Quanto maior for a superfície através do qual o fármaco possa transpor as barreiras que o
separam da circulação sistémica, maior será a velocidade de absorção.
2. Tempo de contacto
Há zonas (ex: esófago) pelas quais o medicamento passa tão depressa que não é possível
ocorrer absorção significativa. As alterações do trânsito intestinal (diarreia ou osbtipação)
podem modificar quantitativamente o processo de absorção.
3. Intimidade do contacto
Os medicamentos em solução ou em suspensão são mais facilmente absorvidos ao nível
do TGI do que numa preparação sólida, porque, no primeiro caso, não só a área de contacto é
mais extensa, como também o contacto do medicamento com as mucosas é mais íntimo.
4. Intensidade de irrigação
Quanto mais intensa for a irrigação, maior será a diferença de concentração entre o local
de absorção e o sangue para onde o fármaco se dirige.
5. Espessura da estrutura absorvente
A absorção será, em regra, mais rápida através de uma mucosa fina do que através de
uma mucosa espessa.
Distribuição
• O processo de distribuição de um fármaco corresponde à transferência reversível das
moléculas do espaço intravascular para o espaço extravascular.
• O princípio activo pode ligar-se a proteínas plasmáticas e difundir-se em certos órgãos ou
tecidos, contendo ou não receptores farmacológicos.
• O volume de distribuição e a percentagem de ligação às proteínas
plasmáticas são características de um principio activo e determinam a
amplitude da distribuição deste no espaço vascular e extravascular.
• No inicio do processo de distribuição, a maior parte do fármaco vai
aparecer em tecidos muito irrigados, como os rins e o cérebro,
posteriormente em zonas de irrigação intermédia como o musculo
esquelético e vísceras menos irrigadas que as anteriores e finalmente
em zonas escassamente irrigadas como o tecido adiposo.

Farmacocinética – UP1
11
• Quer se encontre no espaço intravascular, quer no espaço
extravascular, o fármaco apresenta-se sempre sob a forma livre e
ligada (em equilíbrio dinâmico e reversível), mas a transferência entre
estes dois compartimentos acontece apenas em consequência do
equilíbrio que se estabelece entre as fracções livres existentes nos
mesmos.
• Assim o equilíbrio do processo de distribuição é atingido quando
todos estes processos reversíveis se encontram estabilizados, sendo
certo que, quando isso acontece, a concentração de fármaco livre e ligado em ambos os
espaços (e nos diferentes órgãos, tecidos e fluidos do espaço extravascular) não tem de ser
igual.
• A forma como um fármaco se distribui no corpo está intimamente relacionada com a
afinidade com que ele se liga às proteínas plasmática e às proteínas tecidulares.
• Maior afinidade para os tecidos que para as proteínas plasmáticas-> maior concentraçao
no espaço extravascular.
Volume de distribuição (Vd)
Parâmetro que permite relacionar doses administradas com concentrações
plasmáticas obtidas-> Se a concentração plasmática é muito baixa, o Vd será muito alto,
indicando que está acumulado nalgum tecido; pelo contrário, se o fármaco está muito unido a
proteínas a concentração plasmática será alta e o Vd será baixo.
• Embora todos os tipos de proteínas plasmáticas possam intervir na ligação de fármacos há
duas que assumem particular importância clínica:
o Albumina ligação de fármacos com características ácidas.
o Glicoproteína-1 ácida ligação de fármacos com características básicas.
• A fracção ligada às proteínas plasmáticas é incapaz de atravessar a parede capilar, não
sofrendo eliminação e apresenta-se farmacologicamente inactiva comportando-se como um
depósito que prolonga a permanência da forma livre de fármaco.
Interacção entre 2 fármacos administrados concomitantemente, ambos com afinidade
significativa para as proteínas plasmáticas:
Como a ligação às proteínas plasmáticas é inespecífica, pode acontecer que a
administração de um fármaco desloque o outro que se encontre previamente unido.
Existem ainda situações patológicas que diminuem de forma importante a
proteinemia nestas condições, um fármaco que se ligue de forma significativa às proteínas
nesta condição terá uma fracção livre maior.

Farmacocinética – UP1
12
Casos especiais de distribuição
Distribuição de fármacos para o SNC A BHE. A barreira placentária é outra barreira
com estrutura morfofuncional especial.
Redistribuição:
A alta concentração que inicialmente se obtém no sangue, a elevada lipofilia das
moléculas e a intensa irrigação do cérebro e de algumas vísceras proporcionam que se
verifique uma acumulação dos fármacos nesses tecidos.
No entanto, o gradiente de concentrações vai-se tornando favorável à passagem lenta
dos fármacos destes locais novamente para o sangue e, porque os processos de
eliminação nem sempre são rápidos, terá lugar uma redistribuição do fármaco para os
tecidos de grande massa e irrigação menos intensa (como os músculos esqueléticos).
Metabolismo ou biotransformação
A biotransformação define-se como o conjunto de reacções bioquímicas que
produzem modificações na estrutura química dos fármacos.
Estas modificações podem produzir metabolitos inactivos, activos ou produtos com uma
actividade farmacológica distinta da do fármaco original-> metabolitos passam a ter mais
grupos funcionais hidrofílicos e de maiores dimensões, aumentando assim a sua excreção e
diminuindo a sua passagem pelas membranas biológicas e a probabilidade de abandonarem o
espaço extravascular (distribuição).
Este ocorre preferencialmente no fígado devido à diversidade enzimática presente
nos microssomas hepáticos e à sua massa.
Reacções de fase I – dependem das enzimas microssómicas
Podem ser reacções de oxidação (hidroxilações, N e O-
desalquilações), redução ou hidrólise.
o Estas reacções podem introduzir um grupo reactivo na
estrutura do fármaco, que aumenta a sua actividade
química.
o As reacções de fase I mais frequentes são as oxidações,
catalisadas por um sistema enzimático complexo conhecido
como sistema de oxigenases de função mista.
O sistema enzimático mais importante é o sistema do
citocromo P450 (CYP), do qual existem cerca de 100
isoenzimas as isoenzimas mais importantes para o
metabolismo de fármacos em humanos são as CYP3A4,

Farmacocinética – UP1
13
2D6, 2C19 e 2C9.
Reacções de fase II
Podem ser reacções de conjugação, que normalmente inactivam o fármaco.
Em termos gerais estas reacções actuam sobre o grupo reactivo introduzido nas reacções
de fase I, adicionando um substituinte maior como um glucuronilo, um sulfato ou um
acetilo, que diminuem a lipossolubilidade. Após a administração conjunta de várias
substâncias pode ocorrer indução (aumento da capacidade metabólica) e inibição
(diminuição da capacidade metabólica) enzimática.
Eliminação
• O princípio activo é eliminado sob a forma inalterada ou sob a forma de vários
metabolitos geralmente inactivos, ou sob ambas as formas, em proporções variáveis.
• O rim é o principal órgão de excreção do principio activo inalterado e dos metabolitos
mais hidrossolúveis, enquanto que o fígado é o principal local da sua biotransformação.
A formação da urina tem lugar nos nefrónios e é o resultado da intervenção de
diferentes mecanismos:
1. Filtração glomerular apenas a fracção livre do fármaco.
2. Reabsorção tubular (transporte passivo maioritariamente)
a. A sua intensidade está intimamente relacionada com o pH do filtrado glomerular
b. A alcalinização da urina aumenta a ionização dos ácidos fracos-> indução da sua
excreção por diminuição da reabsorção tubular (contrário com as bases fracas).
3. Secreção tubular constitui um processo mediante o qual certas substâncias e alguns
fármacos podem ser transportados directamente desde o sangue para o lúmen tubular
por transporte activo.
• Muitos fármacos são também excretados parcialmente por via biliar
o O princípio activo ou os seus metabolitos excretados na bílis podem ser reabsorvidos
(circulação entero-hepática).
• A excreção pulmonar envolve dois aspectos de interesse farmacológico: a excreção pelas
glândulas de secreção brônquica e a excreção através dos alvéolos
A eliminação é avaliada por 3 parâmetros farmacocinéticos:
• Constante de velocidade de eliminação
• Período de semivida de eliminação
• Clearance
Esta fase depende de:
• Características farmacocinéticas do fármaco

Farmacocinética – UP1
14
• Características fisiológicas do doente
Fase Farmacodinâmica
Estudo dos efeitos bioquímicos e fisiológicos dos princípios activos e dos seus
mecanismos de acção -> O princípio activo
difunde-se no local de acção e combina-se
com um receptor, enzima ou qualquer
estrutura celular, provocando uma resposta
farmacodinâmica-> efeito terapêutico.
A sensibilidade do receptor ao
princípio activo pode variar de indivíduo
para indivíduo, em função da idade, sexo,
estado fisiopatológico, da administração de
outros medicamentos, dos ritmos
biológicos, etc.
Fase terapêutica -> efeito terapêutico
3.2 – Conhecer as diferentes vias de administração de medicamentos, suas vantagens e
desvantagens e suas implicações farmacocinéticas.
Entéricas -> Absorção ocorre através da parede do TGI.
Parentéricas -> quando existe absorção, esta ocorre fora do aparelho digestivo.
Aplicação sistémica
o Directamente introduzido no sangue (injecção endovenosa
-> única via em que não se verifica absorção).
o Administrado por outras vias (oral, I.M., inalatória), tendo
que ultrapassar barreiras celulares para chegar ao sangue.
(*) No estômago os ácidos fracos apresentaram uma fracção
não-ionizada favorecendo a sua absorção através da mucosa gástrica, pelo contrário as bases
fracas estarão quase totalmente ionizadas, sendo assim a sua absorção a partir do estômago
pouco significativa.
Mesmo no caso dos ácidos fracos, a absorção tem lugar primordialmente a nível intestinal,
apesar do pH do meio e a proporção relativa das formas ionizada e não ionizada favorecerem
absorção a nível gástrico.
Farmacocinética – UP1
15
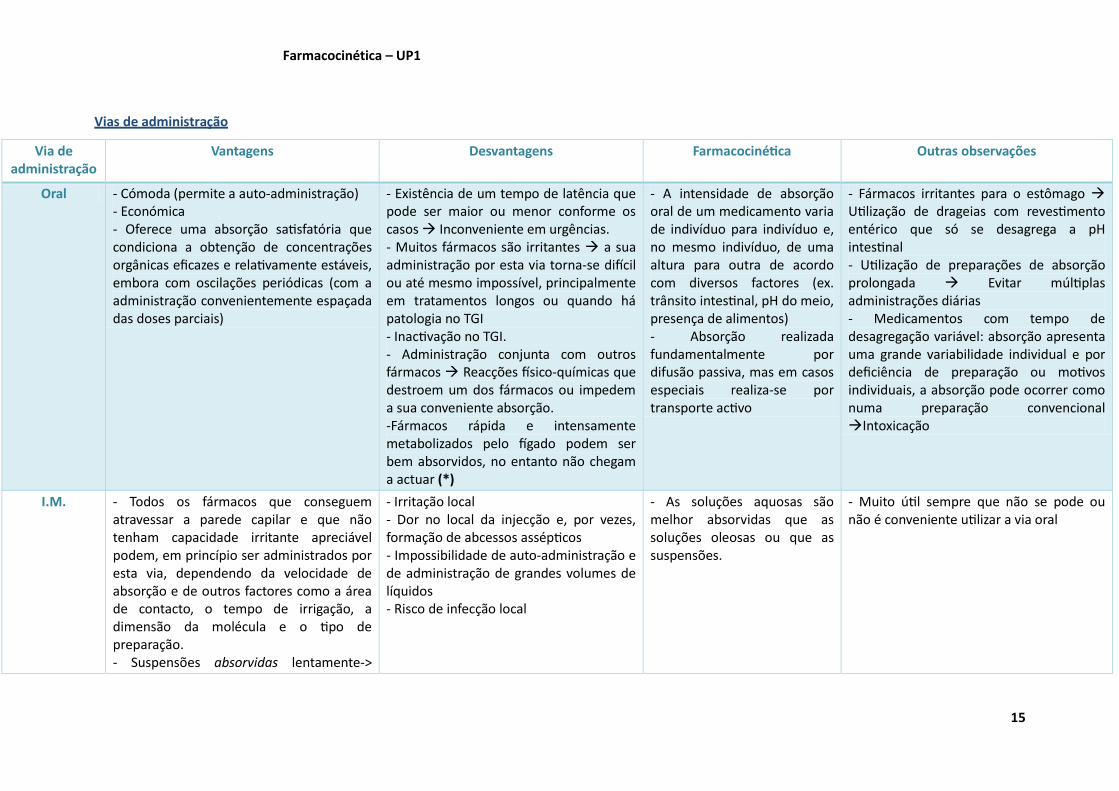
Vias de administração
Via de administração
Vantagens Desvantagens Farmacocinética Outras observações
Oral - Cómoda (permite a auto-administração) - Económica - Oferece uma absorção satisfatória que condiciona a obtenção de concentrações orgânicas eficazes e relativamente estáveis, embora com oscilações periódicas (com a administração convenientemente espaçada das doses parciais)
- Existência de um tempo de latência que pode ser maior ou menor conforme os casos Inconveniente em urgências. - Muitos fármacos são irritantes a sua administração por esta via torna-se difícil ou até mesmo impossível, principalmente em tratamentos longos ou quando há patologia no TGI - Inactivação no TGI. - Administração conjunta com outros fármacos Reacções físico-químicas que destroem um dos fármacos ou impedem a sua conveniente absorção. -Fármacos rápida e intensamente metabolizados pelo fígado podem ser bem absorvidos, no entanto não chegam a actuar (*)
- A intensidade de absorção oral de um medicamento varia de indivíduo para indivíduo e, no mesmo indivíduo, de uma altura para outra de acordo com diversos factores (ex. trânsito intestinal, pH do meio, presença de alimentos) - Absorção realizada fundamentalmente por difusão passiva, mas em casos especiais realiza-se por transporte activo
- Fármacos irritantes para o estômago Utilização de drageias com revestimento entérico que só se desagrega a pH intestinal - Utilização de preparações de absorção prolongada Evitar múltiplas administrações diárias - Medicamentos com tempo de desagregação variável: absorção apresenta uma grande variabilidade individual e por deficiência de preparação ou motivos individuais, a absorção pode ocorrer como numa preparação convencional Intoxicação
I.M. - Todos os fármacos que conseguem atravessar a parede capilar e que não tenham capacidade irritante apreciável podem, em princípio ser administrados por esta via, dependendo da velocidade de absorção e de outros factores como a área de contacto, o tempo de irrigação, a dimensão da molécula e o tipo de preparação. - Suspensões absorvidas lentamente->
- Irritação local - Dor no local da injecção e, por vezes, formação de abcessos assépticos - Impossibilidade de auto-administração e de administração de grandes volumes de líquidos - Risco de infecção local
- As soluções aquosas são melhor absorvidas que as soluções oleosas ou que as suspensões.
- Muito útil sempre que não se pode ou não é conveniente utilizar a via oral
Farmacocinética – UP1
16
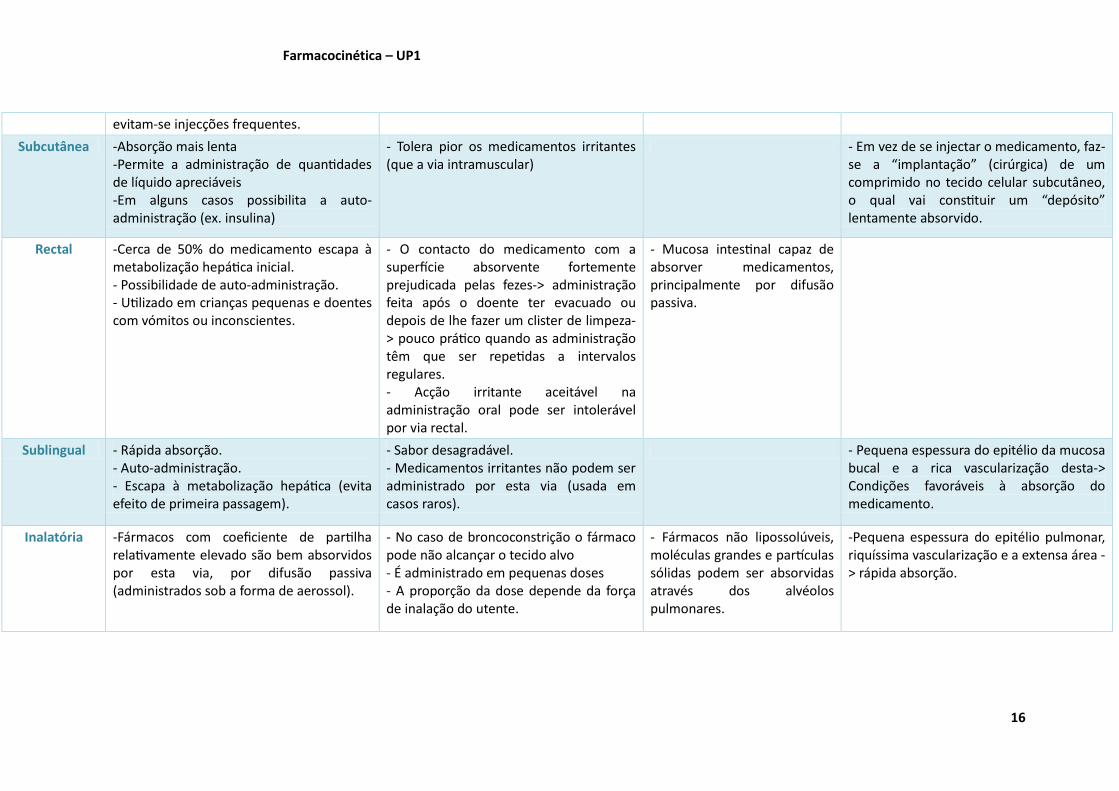
evitam-se injecções frequentes.
Subcutânea -Absorção mais lenta -Permite a administração de quantidades de líquido apreciáveis -Em alguns casos possibilita a auto-administração (ex. insulina)
- Tolera pior os medicamentos irritantes (que a via intramuscular)
- Em vez de se injectar o medicamento, faz-se a “implantação” (cirúrgica) de um comprimido no tecido celular subcutâneo, o qual vai constituir um “depósito” lentamente absorvido.
Rectal -Cerca de 50% do medicamento escapa à metabolização hepática inicial. - Possibilidade de auto-administração. - Utilizado em crianças pequenas e doentes com vómitos ou inconscientes.
- O contacto do medicamento com a superfície absorvente fortemente prejudicada pelas fezes-> administração feita após o doente ter evacuado ou depois de lhe fazer um clister de limpeza-> pouco prático quando as administração têm que ser repetidas a intervalos regulares. - Acção irritante aceitável na administração oral pode ser intolerável por via rectal.
- Mucosa intestinal capaz de absorver medicamentos, principalmente por difusão passiva.
Sublingual - Rápida absorção. - Auto-administração. - Escapa à metabolização hepática (evita efeito de primeira passagem).
- Sabor desagradável. - Medicamentos irritantes não podem ser administrado por esta via (usada em casos raros).
- Pequena espessura do epitélio da mucosa bucal e a rica vascularização desta-> Condições favoráveis à absorção do medicamento.
Inalatória -Fármacos com coeficiente de partilha relativamente elevado são bem absorvidos por esta via, por difusão passiva (administrados sob a forma de aerossol).
- No caso de broncoconstrição o fármaco pode não alcançar o tecido alvo - É administrado em pequenas doses - A proporção da dose depende da força de inalação do utente.
- Fármacos não lipossolúveis, moléculas grandes e partículas sólidas podem ser absorvidas através dos alvéolos pulmonares.
-Pequena espessura do epitélio pulmonar, riquíssima vascularização e a extensa área -> rápida absorção.
Farmacocinética – UP1
17
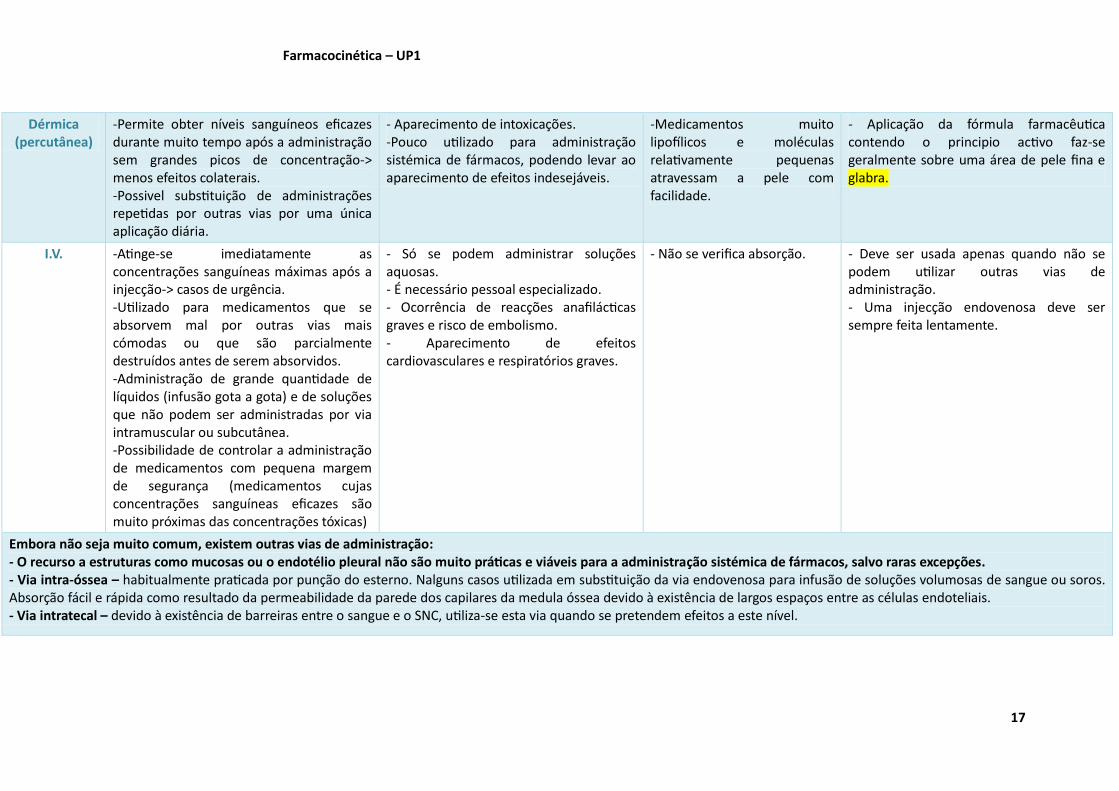
Dérmica (percutânea)
-Permite obter níveis sanguíneos eficazes durante muito tempo após a administração sem grandes picos de concentração-> menos efeitos colaterais. -Possivel substituição de administrações repetidas por outras vias por uma única aplicação diária.
- Aparecimento de intoxicações. -Pouco utilizado para administração sistémica de fármacos, podendo levar ao aparecimento de efeitos indesejáveis.
-Medicamentos muito lipofílicos e moléculas relativamente pequenas atravessam a pele com facilidade.
- Aplicação da fórmula farmacêutica contendo o principio activo faz-se geralmente sobre uma área de pele fina e glabra.
I.V. -Atinge-se imediatamente as concentrações sanguíneas máximas após a injecção-> casos de urgência. -Utilizado para medicamentos que se absorvem mal por outras vias mais cómodas ou que são parcialmente destruídos antes de serem absorvidos. -Administração de grande quantidade de líquidos (infusão gota a gota) e de soluções que não podem ser administradas por via intramuscular ou subcutânea. -Possibilidade de controlar a administração de medicamentos com pequena margem de segurança (medicamentos cujas concentrações sanguíneas eficazes são muito próximas das concentrações tóxicas)
- Só se podem administrar soluções aquosas. - É necessário pessoal especializado. - Ocorrência de reacções anafilácticas graves e risco de embolismo. - Aparecimento de efeitos cardiovasculares e respiratórios graves.
- Não se verifica absorção. - Deve ser usada apenas quando não se podem utilizar outras vias de administração. - Uma injecção endovenosa deve ser sempre feita lentamente.
Embora não seja muito comum, existem outras vias de administração: - O recurso a estruturas como mucosas ou o endotélio pleural não são muito práticas e viáveis para a administração sistémica de fármacos, salvo raras excepções. - Via intra-óssea – habitualmente praticada por punção do esterno. Nalguns casos utilizada em substituição da via endovenosa para infusão de soluções volumosas de sangue ou soros. Absorção fácil e rápida como resultado da permeabilidade da parede dos capilares da medula óssea devido à existência de largos espaços entre as células endoteliais. - Via intratecal – devido à existência de barreiras entre o sangue e o SNC, utiliza-se esta via quando se pretendem efeitos a este nível.

Farmacocinética – UP1
18
3.3 – Perceber o movimento de fármacos através das barreiras biológicas e os factores
condicionantes. Descrever os diferentes compartimentos aquosos do organismo humano.
Existem dois tipos de proteínas associadas às estruturas membranares:
Proteinas Integrais- papel importante na travessia membranar de substâncias.
Proteinas Periféricas- papel praticamente irrelevante na passagem de substâncias de
um lado para o outro da membrana, funcionando basicamente
como enzimas.
Processos de transporte
Transcelular - passagem do fármaco através das células (+ comum)
Paracelular- movimento através de canais estreitos entre as células
pelos fármacos polares que não conseguem atravessar a
membrana lipídica.
Difusão Passiva
• É um processo no qual as moléculas se difundem espontaneamente de uma região de
maior concentração para uma região de menor concentração sem gasto de energia.
• Não é saturavel, ou seja segue uma cinetica de ordem 1
(Lei de Fick)
Nota: duplicando a área de superfície duplica-se a probabilidade de colisão com a membrana e desse modo,
aumenta-se duas vezes mais a velocidade de penetração.
Se os dois lados das membranas têm a mesma concentração, o
movimento para a frente é contra-balançado pelo movimento para trás
das moléculas, não havendo transferências liquida do fármaco.
Quando um dos lados tem maior concentração, num dado momento, o
número de moléculas que se movimentam para a frente é maior que o número de
moléculas que se movimentam para trás.
Como resultado, haverá uma transferência de moléculas para o lado com menor
concentração, como indicado na figura com uma seta maior.
A velocidade de transferência é denominada por fluxo e está representada por um vector
para mostrar a sua direcção no espaço.
Propriedades do fármaco que determinam a permeabilidade
Tamanho e lipofilicidade
Velocidade de penetração = P × AC × (Clado 1 – Clado 2) P: permeabilidade
AC: Área de contacto
Clado 1 – Clado 2: gradiente de concentração

Farmacocinética – UP1
19
O tamanho tem grande impacto no movimento através das membranas -> relativa rigidez
das membranas celulares que estericamente impedem o movimento dos fármacos.
Para algumas membranas, os materiais solúveis em água não podem movimentar-se
através das células, em vez disso, movem-se paracelularmente. Nota: No transporte
paracelular, o tamanho molecular é o principal determinante do transporte.
Carga
Apenas a fracção não polar e não ionizada do fármaco penetra na
membrana atingindo, no equilíbrio, concentrações iguais das
espécies não ionizadas em ambos os lados, embora a
concentração total possa ser diferente devido à diferença do
grau de ionização -> hipótese da partição de pH.
Características da membrana
O movimento do plasma através das membranas (processo convectivo) aumenta o
transporte. Some membranes, such as the renal glomerulus and blood capillaries of most
tissues, are highly permeable to molecules up to 5000 gmol in size with little effect of
charge or lipophilicity. In these cases, drug transfer occurs paracellularly by movement
through large fenestrations (windows) in the membrane. Movement o f plasma water
through the membranes (a convective process) augments the transport
Outro determinante da permeabilidade é a espessura da membrana.
Equilíbrio é rapidamente atingido quando existe uma grande área de superfície de
contacto com a membrana.
A ausência de competição entre moléculas e a inexistência do limite superior para a
velocidade de transporte são características da difusão passiva.
1.Transporte mediado

Farmacocinética – UP1
20
A. Transporte Activo
• É um processo de transporte transmembranar mediado que tem um papel importante na
absorção gastrointestinal e nas secreções renais e biliares de muitos fármacos e
metabolitos.
• É um processo especializado que requer um transportador que se liga ao fármaco para
formar um complexo transportador-fármaco->o fármaco atravessa contra o gradiente de
concentração através da membrana dissociando-se no fim do transportador no outro lado
da membrana.
• O transportador pode ser altamente selectivo para o fármaco sendo que fármacos com
estruturas semelhantes possam competir para o mesmo mecanismo de transporte.
Quando um fármaco é absorvido por difusão passiva (linha A) a
velocidade da absorção aumenta linearmente. No entanto
quando é absorvido por um processo de transporte mediado
(linha B) a velocidade da absorção do fármaco aumenta com a
concentração até os transportadores estarem completamente
saturados. A maiores concentrações, a velocidade da absorção
do fármaco mantém-se constante (ou de ordem zero).
B. Difusão Facilitada
• Também é um sistema de transporte mediado.
• Difere unicamente do transporte activo na medida em que não necessita de energia.
• Parece ter um papel menor na absorção.
Transporte mediado no Intestino
Os sistemas de transporte mediado podem estar presentes na borda em escova do
intestino e na membrana basolateral, para a absorção de iões específicos e nutrientes. Alguns
fármacos são absorvidos devido à semelhança estrutural de substratos naturais.
P-glicoproteína ou proteína de permeabilidade
Transportador dependente de ATP.
Papel importante na secreção hepática para a bílis de alguns fármacos, na secreção renal de
outros e na velocidade de absorção de alguns fármacos no TGI.
Principal transportador de efluxo presente na barreira hemato-encefálica.
3. Transporte Vesicular

Farmacocinética – UP1
21
Envolve as partículas ou dissolve materiais (ex:Pinocitose e Fagocitose)
Pinocitose refere-se à incorporação de solutos ou fluidos.
Fagocitose à incorporação de partículas maiores ou macromoléculas.
Endo e Exocitose: Processos de movimento de macromoléculas especificas para dentro e
fora da célula.
4. Transporte por poros
Pequenas moléculas (uréia, água e açúcar) Capazes de atravessar a membrana
rapidamente se esta tiver canais ou poros.
o Estes poros não são visiveis microscopicamente. O modelo de penetração do fármaco
por poros aquosos explica a excreção renal e a recaptação de fármacos pelo fígado.
Pequenas moléculas (ex: fármacos), movem-se nestes canais por difusão mais rapidamente
do que noutras partes da membrana.
5. Formação de pares de iões
Fármacos ionizados ligam-se a uma carga oposta para formar um par iónico neutro que se
difunde mais facilmente através da membrana.
Corrente sanguínea vs permeabilidade
Limite da perfusão
O movimento do fármaco através das membranas não pode ser
separado das considerações de perfusão (mL/min por massa).
Se o movimento através da membrana for fácil, o passo lento
(limita a velocidade) é a perfusão e não a permeabilidade.
o Velocidade inicial do movimento do fármaco para os tecidos:
Determinada pela velocidade de distribuição que depende da
corrente sanguínea.
Limite da permeabilidade
Permeabilidade: o que está em causa é a penetração na membrana e
não a distribuição do fármaco.
Aumento da espessura da membrana leva ao aumento da resistência
A resistência aumenta com o tamanho e a polaridade da molécula
Estes factores limitam a velocidade de extravasação e o movimento para fora dos vasos.
Natureza reversível do transporte
O transporte pelas membranas é geralmente bidireccional. No entanto, o transporte
unidireccional entre TGI e sangue permite a absorção. Há adsorventes que reduzem a
absorção sistémica e aceleram a remoção de fármacos do corpo.

Farmacocinética – UP1
22
Modelos compartimentais
Corpo: sistema de compartimentos que comunicam reversivelmente uns com os outros.
Compartimento: tecido ou grupo de tecidos com fluxo sanguíneo e afinidade similar para os
fármacos. Dentro de cada compartimento, o fármaco é considerado uniformemente
distribuído.
A mistura de fármaco num compartimento é rápida e homogénea -> [fármaco] representa
uma [] média. Cada molécula tem igual probabilidade de sair do compartimento.
Estes modelos são baseados em hipóteses lineares (equações diferenciais lineares).
O fármaco move-se dinamicamente dentro e fora dos compartimentos.
Taxas constantes são utilizadas para representar a taxa global de processos de entrada e
saída de fármacos.
É um sistema aberto uma vez que os fármacos podem ser eliminadas do sistema.
Maneira simples de agrupar os tecidos num ou mais compartimentos onde os compostos se
deslocam de e para o compartimento central ou plasmático.
Em qualquer momento, a quantidade de fármacos no corpo é igual à soma dos fármacos
presentes no compartimento central mais o fármaco presente no compartimento tecidual.
O compartimento tecidual não representa um tecido específico, mas o balanço de massas
tem em consideração o fármaco presente em todos os tecidos.
Parâmetros do modelo de 2 compartimentos: possível estimar a quantidade de fármaco
presente no lado esquerdo do corpo e a quantidade de fármaco eliminada do corpo, a
qualquer momento.
Modelos compartimentais: Úteis se houver poucas informações sobre os tecidos.
Se as [] de fármaco e a ligação ao tecido forem conhecidos, os modelos PK fisiológicos,
tornam os dados mais realistas.
Os modelos PK fisiológicos são utilizados na descrição da distribuição de fármaco em
animais, pois as amostras são facilmente disponíveis para análise.
Amostras teciduais dos humanos estão frequentemente indisponíveis -> Aproximações.
Ao contrário destes modelos, parâmetros como o T1/2 são determinados a partir de dados.
A maior parte de modelos fisiológicos assume uma média ajustada do fluxo sanguíneo para
objectivos individuais -> Desvantagem para tentar individualizar a dose.
Modelo “Mammillary”
Modelo compartimental mais usado na farmacocinética.
Consiste num ou mais compartimentos periféricos ligados a um compartimento central.

Farmacocinética – UP1
23
Compartimento central: Atribuído para representar plasma e tecidos altamente
perfundidos que rapidamente se equilibram com o fármaco.
Sistema fortemente conectado: possível estimar a quantidade de fármaco em qualquer
compartimento do sistema após introdução do fármaco num compartimento.
Dose IV -> Fármaco entra directamente para o compartimento central.
A eliminação ocorre a partir do compartimento central, pois os órgãos envolvidos na
eliminação, principalmente rins e fígado, são tecidos bem
perfundidos.
Tipos de modelos compartimentais são descritos na
figura. Constantes PK representados por K.
1. Compartimento 1 - Plasma ou compartimento central
2. Compartimento 2 - Compartimento tecidual.
Este modelo permite:
Escrever equações diferenciais que descrevem variações de []
em cada compartimento,
Fornece uma representação visual da taxa de processos, e
Mostra quantas constantes PK são necessárias para descrever o
processo de forma adequada.
Modelo “Catenary”
Compartimentos unidos a um outro como os compartimentos
num comboio.
Modelo “Mammilary”: um ou mais compartimentos em torno de um central como
satélites.
O modelo “Catenary” não se aplica aos órgãos mais funcionais directamente ligados ao
plasma -> Não se aplica tanto como o modelo “Mammillary”.
Modelo Farmacocinético Biológico (Moldelo dos fluidos)
Os modelos fisiológicos PK ou modelos de fluxo sanguíneo ou de perfusão, são modelos
baseados no conhecimentos anatómico e fisiológico que descrevem cineticamente os dados
considerando que o fluxo sanguíneo é responsável pela distribuição do fármaco pelo corpo.
Captação do fármaco pelo órgão -> Depende da ligação do fármaco aos tecidos deste. Como
há muitos tecidos de órgãos no corpo:
Deve ser obtido cada volume de tecidos;
[] do fármaco em cada tecido tem de ser descrito.

Farmacocinética – UP1
24
O modelo pode predizer as [] de fármaco nos tecidos realisticamente, o que falha no
modelo de dois compartimentos.
Informação requerida para descrever o modelo PK fisiológico-> Dificeis de obter.
Contudo, este modelo providencia uma visão melhor de como os factores fisiológicos
podem modificar a distribuição de fármacos, de um animal para outro pois:
Não é preciso montar dados no modelo de perfusão. [] de fármacos nos tecidos, são
previstas pelo tamanho do tecido do órgão, corrente sanguínea, e razões sangue-tecido
determinadas experimentalmente (partição do fármaco entre o tecido e o sangue).
Corrente sanguínea, tamanho do tecido, e razão órgão-tecido do fármaco, podem variar
devido a condições patofisiológicas Efeito dessas variações na distribuição do fármaco,
deve ser tida em conta nos modelos farmacocinéticos fisiológicos.
Modelos FK fisiológicos: Alargados a várias espécies, para alguns fármacos, pode haver
extrapolação dos dados humanos. A extrapolação de dados de animais não é possivel com
o modelo de compartimentos: O volume de distribuição nesses modelos é um conceito
matemático não relacionado só com o volume sanguíneo e a corrente sanguínea.
Modelos farmacocinéticos de perfusão do fármaco.
O ks, representa constante cinéticas:
ke: Constante de 1ª ordem para excreção de fármacos por via urinária.
km: Constante de eliminação hepática
Cada caixa representa um compartimento de tecido.
Órgãos mais importantes na absorção de fármacos: Considerados
separadamente, enquanto que os outros tecidos são agrupados em
RET (Tecidos de equilíbrio rápido) e SET ( Tecidos de equilíbrio lento).
Tamanho ou massa dos compartimento: Determinado fisiologicamente.
[] de fármaco no tecido Determinada pela capacidade do tecido acumular o fármaco,
assim como pela taxa de perfusão sanguínea para o tecido, representada por Q.
O número de compartimentos no modelo de perfusão, varia com o fármaco.
Tecidos ou órgãos que não apresentam penetração ao fármaco, são excluídos.
4. Apreender as etapas gerais do processo de descoberta e desenvolvimento de novos
fármacos e o papel da farmacocinética em cada uma dessas etapas (ensaios in silico, ensaios
in vitro, e estudos in vivo não-clínicos e clínicos).
In silico -> Computadores modelam processo natural ou de laboratório e não os cálculos
computacionais genéricos.

Farmacocinética – UP1
25
In vitro -> Processos biológicos fora dos sistemas vivos, num ambiente controlado e fechado
normalmente feitos em recipientes de vidro.
In vivo -> Processo que ocorre num organismo; experiências feitas dentro ou no tecido vivo de
um organismo vivo.
Benefícios de entender a PK e relação concentração–resposta: Aplicação na indústria
farmacêutica.
Muitos dos processos básicos que controlam a PK e as respostas são semelhantes em
várias espécies de mamíferos -> Extrapolação para prever comportamento em humanos. Em
sistemas in vitro utilizam-se cada vez mais humanos ou materiais humanos.
Isto aumenta as hipóteses de selecção dos compostos e também do intervalo correcto
de doses seguras para o primeiro teste em humanos.
A incorporação de elementos tanto PK como PD na fase I de estudos é realizada em
indivíduos saudáveis, com a avaliação de efeitos secundários -> Ajuda a definir a dosagem e
regimes de avaliação na fase II, realizado num pequeno nº de pacientes para testar eficácia do
fármaco para a indicação pretendida, conhecida como Fase "prova de conceito".
Fases I e II: Definem os regimes de dosagem mais seguro e eficaz do uso nos grandes
ensaios clínicos seguintes -> Fase III (confirmação clínica), envolvendo milhares de doentes.
Modelo hierárquico: Dados ADME recolhidos somente em compostos com potência e
seletividade adequada.
No modelo horizontal dados ADME são recolhidos em todos os compostos.

Farmacocinética – UP1
26
4.1. Compreender a necessidade do estudo precoce das propriedades físico-químicas e
farmacocinéticas de moléculas candidatas a fármacos.
Dissolução e solubilidade
Solubilidade: Parâmetro essencial para a dissolução
dos compostos após a administração oral. É estudada
em fases iniciais no desenvolvimento de fármacos.
Esta depende da área de superfície do sólido a
dissolver e da solubilidade do fármaco na superfície
desse mesmo sólido.
É inversamente proporcional ao nº e tipos de funções lipofílicas da molécula e ao
empacotamento cristalino da molécula.
Foram desenvolvidos métodos (ex: turbidimetria) que medem a solubilidade (grande nº de
compostos).
Aproximações para uma rápida medição dos valores de pKa dos compostos.
Lipofilicidade
Principal parâmetro FQ ligado à permeabilidade da membrana e portanto à absorção e
distribuição de fármacos por via de “clearance” (metabólica ou renal).
A lipofilicidade medida ou calculada de um composto é facilmente sujeita a automação.
o Muitos destes cálculos dependem de valores fragmentados mas métodos simples
baseados no tamanho molecular para calcular valores log P também podem ser uteis.
o A combinação da medição com aproximações fragmentárias permite predizer as
propriedades dos novos compostos.
Para uma real medição, usam-se coeficientes de partição octanol/água alternativos que
incluem a imobilização de membranas artificiais e coeficientes de partição lipossoma/água.
o A ligação do lipossoma pode ser transferida para um sistema “throughput “ e pode
fornecer um “volume de distribuição” quando ligado a outras propriedades.
A capacidade de ligação de H de um soluto é importante no conceito de lipossolubilidade.
Absorção / Permeabilidade
Quando um composto atravessa a membrana por difusão passiva estima-se a
permeabilidade usando propriedades moleculares, como log D ou ligações de hidrogénio.
Métodos in vitro (ex: monocamadas Caco-2) Muito usados em estimativas de absorção.
o Além da contribuição da componente FQ para o transporte na membrana, muitos
compostos são afectados por eventos biológicos (ex: transportadores e metabolismo).

Farmacocinética – UP1
27
Muitos fármacos são substratos de proteinas transportadoras (promovem ou prejudicam a
permeabilidade). Não existe uma teoria de REA que tenha em conta estes efeitos.
O método mais rápido para se estimar a absorção é fazer a estimativa in silico.
Ionização (pKa)
Esta afecta a solubilidade, a lipofilicidade, a permeabilidade, o volume de distribuição,
metabolismo e excreção.
A teoria de partição de pH diz que só as espécies neutras atravessam a membrana (posto
em causa).
Por voltametria ciclica, demonstrou-se que compostos na sua forma ionizada passavam as
membranas nas fases organicas.
Peso molecular, forma e flexibilidade
O peso molecular pode ser um factor limitante relativamente à absorção oral.
o Regra dos 5 de Lipinski: Limite máximo de 500Da para compostos com este tipo de
absorção.
A forma e o tamanho não são medidos, são calculados.
Existe uma propriedade (área de secção cruzada) que é obtida por medições da actividade
de superfície.
o Esta é útil para descriminar compostos que conseguem ou não aceder ao cérebro.
o Nº de ligações rotativas: tomado como uma medida da flexibilidade do composto.
Anfifilicidade
Balanço entre grupos hidrofílicos e hidrofóbicos de uma molécula. Esta pode ser utilizada
para prever efeitos nas membranas que levam a citotoxicidade ou fosfolipidose.
Permeabilidade
Caso se dê o composto por via oral, uma estimativa da absorção precoce é desejada.
A alta capacidade e a alta previsibilidade são preditivos para a permeabilidade e absorção.
4.1.1. Entender o conceito de biodisponibilidade. Conhecer as principais propriedades físico
químicas que podem determinar a biodisponibilidade dos fármacos
Biodisponibilidade: Medição da taxa e extensão do fármaco activo disponível no local de
acção. Porção de princípio activo que passa a circulação sistémica e chega ao local de acção.
Alguma alteração nos processos de ADME pode modificar a acção do fármaco.
Factores farmacêuticos que afectam a biodisponibilidade
1. Tipo de fármaco;
2. Natureza dos excipientes no medicamento;
3. Propriedades físico-quimicas da molécula de fármaco;

Farmacocinética – UP1
28
4. Via de administração do fármaco.
Dissolução e solubilidade
Dissolução - Substância sólida do fármaco dissolve num solvente. Propriedade dinâmica.
Solubilidade - Massa de soluto que se dissolve numa especifica massa ou volume de solvente a
uma dada temperatura. É uma propriedade estática.
4.1.2. Conhecer os principais modelos experimentais usados in vitro para estudar a
Absorção, Distribuição e Eliminação, e para predizer as propriedades farmacocinéticas in
vivo de candidatos a fármacos. Entender o potencial preditivo dos diferentes modelos.
(Shargel 2005 Chap. 13; Smith 2001 Chap. 10; Testa 2006 pgs. 25-41; Artigos Científicos)
Determinação das propriedades metabólicas de
uma nova entidade química (NCE): Passo
importante durante o processos de descoberta e
desenvolvimento de fármacos.
São usados métodos in vitro para se estimar e
prever o metabolismo in vivo dos NCF.
A utilização de métodos in vitro permite prever a
estabilidade metabólica dos NCFs, e o risco de
interacções medicamentosas resultante da
inibição e indução de enzimas metabólicas dos fármacos.
As companhias farmacêuticas realizam estudos do metabolismo do fármaco in vitro, com
software de previsão in silico e screens automáticos de "throughput” elevado (HTS).
Os métodos in vitro são úteis para identificação e eliminação de NCFs com propriedades
metabólicas indesejadas mas o output quantitativo dos métodos tem de ser melhorado.
Modelos in vitro usados para estudar o metabolismo dos fármacos
1. Enzimas recombinantes
Microssomas com enzimas metabólicas expressas em cDNA são importantes modelos
in vitro para estudos do metabolismo dos fármacos. As enzimas recombinantes são usadas
para prever a clearance do fármaco e o risco de interacções entre fármacos relacionado com a
inibição de enzimas.
VantagensModelo in vitro mais simples, contém enzimas individuais, produzidas
no retículo endoplasmático das células eucarióticas. Permite estudar separadamente a
actividade de enzimas especificas.
DesvantagensDiferença na actividade de enzimas metabólicas por unidade de
proteínas microssomais comparada com a dos microssomas hepáticos humanos.

Farmacocinética – UP1
29
SoluçãoDurante a extrapolação in vitro-in vivo, foram propostos 2 parâmetros: factor de
actividade relativa (FAR) e factor de extrapolação intersticial (FEIS).
Os sistemas de expressão mais comumente usados: sistema baculovírus e o sistema
linfoblastóide humano.
2. Microssomas Hepáticos
Podem ser preparados a partir do fígado de animais ou seres humanos por
ultracentrifugação, tendo os microssomas hepáticos humanos (HML) passado a ser largamente
utilizados no modelo in vitro.
Através da suplementação de microssomas hepáticos com cofactores relevantes e
outros componentes é possível investigar e distinguir as actividades de CYPs, de FMO’s e
glucoronosiltransferases modelo muito usado pelas companhias farmacêuticas para
previsão da clearance de NCFs in vivo.
A principal desvantagem deste modelo é o período limitado de incubação pois a
actividade das enzimas diminui após 2h de incubação.
3. Hepatócitos frescos e criopreservados
São importantes in vitro para estimar e prever a clearance e as interacções entre
fármacos (DDI) dos NCFs. Os hepatócitos humanos são também o modelo in vitro mais
importante para estudos em DDIs indutivos e de possíveis efeitos adversos. Este modelo
contém todas as enzimas metabólicas de fase I e fase II, assim como todos os co-factores
necessários.
A principal desvantagem deste modelo é a disponibilidade limitada de hepatócitos
humanos frescostêm que ser colocados em cultura imediatamente após a colheita.
Soluçãotécnicas de criopreservação.
4. “Fatias” de tecido
É o modelo in vitro mais complexo, sendo o que se aproxima mais de uma situação in
vivo e é útil para estudar a formação de metabolitos, mas não para prever a clearance
metabólica e DDI’s.
5. Células Caco-2 monolayers (CCM)
São o ensaio padrão para prever a permeabilidade intestinal ao fármaco e a fracção
da dose oral absorvida em seres humanos, pois para compostos de administração oral, a
permeabilidade através das CCM relaciona-se bem com a absorção nos humanos.
As CCM permitem o estudo do transporte passivo transcelular e paracelular, do
influxo mediado por um transportador e dos mecanismos de efluxo.

Farmacocinética – UP1
30
As células Caco-2 são extraídas do adenocarcinoma de cólon humano.
Durante a sua cultura, em filtros permeáveis e porosos, diferenciam-se
espontaneamente em enterócitos, formando uma membrana composta por células em
monocamada unidas por junções, que se multiplicam rapidamente (vantagem).
As células Caco-2 expressam certos transportadores e enzimas metabólicas, que são
usados para estudar a interacção fármaco-fármaco/fármaco-
excipiente e a estabilidade metabólica de fármacos.
Existem limitações na utilização de células Caco-2 para
avaliar a absorção, entre as quais: o reduzido número de
transportadores (p. ex. péptidos PEPT 1 e PEPT 2); baixa
permeabilidade a compostos hidrofílicos; danos causados à
membrana devido à presença de co-solventes no sistema;
aderência física do fármaco ao filtro de policarbonato,
apresentando reduzida permeabilidade; elevado tempo de crescimento das células Caco-2.
6. Membranas artificiais - PAMPA
Vantagem: oferecem grande reprodutibilidade e um sistema de alto processamento.
Pensou-se em desenvolver uma membrana artificial de Caco-2 paralela ao ensaio de
permeabilidade (PAMPA) para previsão rápida de absorção do potencial transcelular.
Neste sistema, a permeabilidade é avaliada por um membrana formada por uma
mistura de lecitina e um solvente orgânico inerte num suporte
de filtro hidrofóbico.
Embora a absorção oral não seja completamente
previsível para os humanos, dados de PAMPA mostram
tendências definidas na habilidade de moléculas para penetrar
membranas por difusão passiva.
7.MDCK: É um modelo de células de rim canino (“Madin-
Darby”). Estas células quando cultivadas em membranas semi-
permeáveis diferenciam-se em epiteliais colunares, com junções semelhantes às células Caco-

Farmacocinética – UP1
31
2. São largamente utilizadas para estudar a fisiologia tubular distal renal, incluindo o
transporte de iões. E representam um dos poucos modelos que expressam quantidade
adequada de co-transportadores. Estes estudos são relevantes no delineamento da
terapêutica com compostos diuréticos tiazínicos utilizados no tratamento da hipertensão.
Vantagem: células MDCK crescem mais rápido que Caco-2 e 2/4/A1, e têm níveis
muito baixos de transportadores expressos -> Satisfatório para estudar difusão passiva.
4.1.3. Perceber a utilidade da conjugação das propriedades físico-químicas com a informação
de ensaios in vitro na predição da biodisponibilidade de candidatos a fármacos. (Shargel
2005 Chap. 13; Smith 2001 Chap. 10; Testa 2006 pgs. 25-41; Artigos Científicos)
Os testes in vitro -> possibilitam o aumento do conhecimento dos processos de
metabolismo dos fármacos assim como dos seus mecanismos de acção (ex: interacções
fármaco-fármaco, indução do CYP,…). São ferramentas úteis para identificação desde os
primeiros passos à eliminação de NCE’s com propriedades metabólicas não desejáveis.
4.2. Compreender a necessidade de realizar estudos in vivo não-clínicos e conhecer a sua
utilidade. (EMEA Guidelines, Shargel 2005 Chap. 13; Smith 2001 Chap. 10; Testa 2006 pgs.
25-41; Artigos Científicos)
1. Importância dos modelos animais
Podem existir diferenças qualitativas e quantitativas nas respostas biológicas dos
animais, em comparação com os seres humanos. Neste sentido: uma resposta idêntica em
células humanas e animais in vitro não é necessariamente uma garantia de que a resposta in
vivo seja semelhante.
Na prática, estudos em animais com fármacos altamente específicos podem não
reproduzir o efeito farmacológico pretendido em seres humanos; ou dar origem a erros de
interpretação dos resultados de PK e de PD; ou ocultar efeitos tóxicos relevantes.
A demonstração da relevância do modelo animal pode incluir a comparação com seres
humanos, em termos de:
• Expressão do alvo
• Distribuição
• Estrutura primária
• Farmacodinâmica
• Metabolismo e outros aspectos farmacocinéticos
•estudos de reactividade cruzada com tecidos humanos e animais (ex. anticorpos
monoclonais)
No entanto, um alto grau de homologia não implica necessariamente efeitos comparáveis.

Farmacocinética – UP1
32
Quando não existem espécies em causa, o uso de proteínas homólogas ou a utilização
de animais transgénicos que expressam o alvo humano podem ser a única escolha.
1. Farmacodinâmica
Os estudos farmacodinâmicos deverão abordar o modo de acção e devem fornecer o
conhecimento biológico sobre o alvo, por exemplo, ligação ao receptor, duração do efeito e
dose-resposta.
A farmacodinâmica primária e secundária devem ser realizadas em sistemas animais e
humanos in vitro e em modelos animais in vivo.
2. Farmacocinética
Exposição de doses farmacodinâmicas em modelos animais deve ser determinada
especialmente quando os efeitos farmacodinâmicos são suspeitos de contribuir para
potenciais problemas de segurança.
3. Farmacologia de Segurança
Estudos adicionais para investigar os efeitos noutros sistemas devem ser realizados caso
a caso Em particular, para medicamentos de segmentação do sistema imunológico, os
potenciais efeitos indesejados devem ser investigados, p.e., utilizando estudos in vitro,
incluindo material humano.
4. Toxicologia
Deve incluir a toxicocinética.
A utilização de produtos homólogos ou a aproximação de modelos transgénicos ou a
utilização de sistemas humanos in vitro pode fornecer informações adicionais relevantes.
A dosagem em estudos em animais pode não prever os efeitos dessas substâncias em
seres humanos (p.e., a presença de anticorpos neutralizantes).
5. Estimativa da primeira dose em humanos
É um elemento importante para garantir a segurança das primeiras pessoas nos estudos
de fase I.
Os Sem Efeito Adverso Observado (NOAEL) determinados nos estudos não clínicos de
segurança realizados em espécies de animais mais sensíveis e relevantes (ajustados com
factores alométricos ou com base na farmacocinética) dão a informação mais importante.
A abordagem ao “Nível de Efeito Mínimo Biológico Esperado” (MABEL) é recomendada.
• Um factor se segurança pode ser aplicado para o cálculo da primeira dose em humanos de
MABEL.
• O cálculo de MABEL deve recorrer a toda a informação disponível da PK/PD de estudos in
vitro e in vivo, tais como:

Farmacocinética – UP1
33
A informação pré-clinica ajuda a identificar compostos promissores e sugere doses úteis para testar em humanos
Fases I, II e III dos testes em humanos geralmente correspondem, no inicio em determinados pacientes seleccionados e só depois em testes abrangentes a um maior numero de pacientes.
A vigilância após comercialização do fármaco. ajuda a optimizar a relação PK/PD e a segurança
- meta vinculativa e estudos da ligação ao receptor in vitro em células alvo de humanos
ou espécies animais;
- curvas de concentração-resposta in vitro em células alvo ou espécies animais e
dose/exposição-resposta in vivo de espécies animais;
- exposição às doses farmacológicas nas espécies animais.
Quando os métodos de cálculo (p.e., NOAEL, MABEL) dão diferentes estimativas da
primeira dose nos humanos, deve ser usado o valor mais baixo.
4.3. Conhecer as diferentes fases do desenvolvimento clínico de um medicamento. (Tozer
2006 Chap. 1)
A incorporação da farmacocinética e farmacodinâmica nos estudos de fase I ajudam a
definir a forma de dosagem e os regimes para avaliação na fase II conduzida num pequeno
grupo de pacientes para testar se o fármaco é efectivo para a respectiva indicação->“proof of
concept”.
A fase I e II pretendem definir a dose mais eficaz e segura para ser usado em maior
escala na fase III (triagem clinica) em testes clínicos, envolvendo milhares de pacientes.
Objectivo II -
Aprofundar o ciclo geral do
medicamento no organismo: processos cinéticos de libertação, absorção, distribuição e
eliminação de fármacos e factores condicionantes.
1.Apreender os princípios gerais subjacentes à cinética da absorção sistémica de fármacos.
(Tozer, 2006; Chap. 6; Shargel 2005 Chap. 13)

Farmacocinética – UP1
34
4.2. Entender os processos subjacentes à absorção gastrointestinal de fármacos quando
administrados em formas farmacêuticas sólidas ou em solução.
Forma Farmacêutica em solução
Segundo a hipótese de partição de pH, os ácidos fracos são absorvidos mais
rapidamente no estômago a pH = 1 do que a pH = 8 (e o oposto para as bases fracas).
Contudo, a absorção dos ácidos é muito mais rápida ao nível do intestino delgado
(menos acídico – pH=6.6 a 7.5), isto porque, apesar de os fármacos estarem mais ionizados
existem outros factores no intestino como: maior área de superfície, maior permeabilidade da
membrana intestinal aos fármacos e maior perfusão. ->verificar se já tem atrás
A absorção de todos os compostos, quer sejam ácidos, bases ou neutros é mais rápida
a nível do intestino delgado, e, como tal, a velocidade de esvaziamento é o passo limitante da
velocidade de absorção do fármaco.
O esvaziamento gástrico é um dos
passos limitantes da absorção do fármaco.
Quanto mais rápido for o esvaziamento, mais
rapidamente o fármaco chega ao intestino e
mais rápido ele é absorvido.
Um fármaco que seja procinético (ex:
metaclopramida) favorece o esvaziamento e,
consequentemente, a absorção.
Forma Farmacêutica sólida
A forma sólida tem de se desintegrar e desagregar e o fármaco tem de se dissolver. A
dissolução é um factor-chave.
1.2.1. Distinguir formas farmacêuticas de libertação imediata de formas farmacêuticas de
libertação modificada (libertação prolongada e libertação retardada)
Os fármacos são mais frequentemente administrados de
uma forma extravascular (em vez de uma forma intravascular) e
actuam mais sistemicamente do que localmente.
Atrasos ou perdas do fármaco durante a absorção
sistémica, podem contribuir para uma variabilidade na resposta
farmacológica ou até resultar numa falha da terapia. Mesmo para
os medicamentos que são utilizados localmente, a absorção
sistémica pode influenciar o tempo de latência, a intensidade do fármaco e a duração dos
efeitos adversos.->para tirar?

Farmacocinética – UP1
35
Formas orais disponíveis:
Líquidas (xaropes, elixires, tinturas, suspensões e emulsões)
Sólidas (comprimidos e cápsulas) são as mais comuns, geralmente formuladas para
libertar o fármaco, imediatamente após a sua administração e acelerar a absorção ->
Fármacos de libertação imediata:
De libertação modificada -> Libertar os fármacos, numa taxa controlada, com o
propósito de evitar o contacto com fluidos gástricos (meio ácido), ou prolongar a
entrada do fármaco na circulação sistémica.
Estes produtos (libertação modificada) dividem-se em duas categorias:
Libertação prolongada - permite uma redução na frequência das tomas, ou diminui a
flutuação dos níveis do fármaco em administrações repetidas.
Libertação retardada - a libertação do fármaco (parcial ou total) é adiada. São utilizadas
para prevenir a libertação do fármaco no estômago, onde este se iria decompor ou
causar irritação -> assim o fármaco é absorvido só no intestino.->quadro?
Nota: Há outras formas de administração extravascular de libertação prolongada, como é o caso dos
depósitos intramusculares e sub-cutâneos na forma de emulsões, soluções oleosas, suspensões, pensos, etc.
1.2.2. Conhecer a cinética de dissolução (equação de Noyes-Whitney) e compreender a
importância dos ensaios de dissolução no processo de formulação. (Shargel 2005 Chap.14)
Os passos de dissolução inclui a dissolução do fármaco na superfície da
partícula sólida, que forma uma solução saturada à volta da partícula. O
fármaco dissolvido na solução saturada forma a camada estática, difundindo-se
para o seio do solvente de regiões de elevada concentração de fármaco para as
de baixas concentrações.
Equação de Noyes-Whitney:
Onde,
dC/dt é a velocidade de dissolução em função do tempo t
D é a constante de velocidade de difusão
A é a área de superfície da partícula
Cs é a concentração de fármaco (igual à solubilidade do fármaco) na camada estática
C é a concentração de fármaco no seio do solvente
h é a espessura da camada estagnante
A velocidade de dissolução, dC/dt é a velocidade a que o fármaco se dissolve per time
expresso como a mudança de concentração no fluido de dissolução.

Farmacocinética – UP1
36
Entre os factores que afectam a dissolução encontram-se as características físico-
químicas do fármaco, o tipo de formulação, o solvente, a temperatura e a agitação.
A absorção de um fármaco no TGI é afectada pela sua capacidade de se difundir (D) e
pelo coeficiente de partição entre a membrana lipídica Coeficiente de partição favorável
(Kóleo/água) facilitará a absorção do fármaco.
Estudos realizados in vitro permitiram verificar que:->tiro?
- Um aumento na temperatura aumenta a energia cinética das moléculas e aumenta a
constante de difusão, D.
- Um aumento na agitação do solvente reduz a espessura, h, da camada estática,
permitindo uma dissolução mais rápida.
A dissolução é um processo muito importante, pois deste depende a absorção.
Consideremos 2 situações:
A primeira (menos comum->gráfico I) corresponde àquela em
que o processo de dissolução é mais rápido que a absorção.
Consequentemente, muito fármaco é dissolvido rapidamente
antes de uma fracção apreciável de fármaco ser absorvida. Nesta
situação, a permeabilidade limita a velocidade de absorção (em vez da
dissolução). Um fármaco que seja muito polar – sucralfato
(independentemente das formulações) - irá ter uma rápida dissolução
mas fraca absorção.
A segunda (mais comum -> gráfico II) corresponde àquela em
que a dissolução ocorre lentamente e qualquer porção de fármaco
dissolvido rapidamente atravessa as barreiras biológicas (ex. epitelio
intestinal). Nesta situação a absorção não pode ocorrer rapidamente.
A dissolução limita a velocidade de absorção.
Neste caso, mudanças na dissolução afectam profundamente a
velocidade e, às vezes a extensão, da absorção do fármaco.
Nota: o gráfico I pode aplicar-se a fármacos em solução e o gráfico II a fármacos sólidos.
1.2.3. Rever os processos cinéticos de primeira-ordem e de ordem-zero. Resolução exercícios.
(Shargel 2005 Chap. 2 pgs.42-49)
A velocidade de uma reacção química velocidade a que reacção ocorre
Se a quantidade de fármaco A diminuir ao longo do tempo dtdA/
Se a quantidade do fármaco B aumenta ao longo do tempo dtdB /
Apenas o fármaco farmacologicamente activo é medido experimentalmente.
BfármacoAfármaco

Farmacocinética – UP1
37
Reacções de Ordem Zero:
A concentração de fármaco no plasma diminui linearmente com o tempo, eliminando
uma concentração fixa (não percentual) de fármaco por unidade de tempo;
É uma cinética de eliminação de poucos fármacos: etanol, fenitoína e aspirina em
concentrações fortes ou tóxicas;->tiro?
Se a concentração de fármaco A diminui num intervalo de tempo constante, t, a
velocidade de desaparecimento é expressa por: 0kdt
dA , onde k0 é a constante de
velocidade de ordem zero e é expressa em massa/tempo (ex. mg/min).
A integração é dada por: 00 AkA ,
onde A0 é a quantidade de fármaco
em t = 0.
00 CtkC , onde C0 é a concentração do fármaco em t = 0, C é a
concentração do fármaco em t;
Exemplo:
Um farmacêutico pesa exatamente 10g de um fármaco e dissolve em
100mL de água. A solução é conservada na estufa e as amostras são
removidas periodicamente e analisa-se o fármaco. O farmacêutico obtém
os seguintes dados:
A concentração de fármaco declina 5mg/mL por cada intervalo de 2 horas
A construção de um gráfico da concentração do fármaco Vs tempo irá
resultar numa linha linear-> A velocidade de declínio é de ordem zero->A
constante k0 pode ser obtida pelo declive;
Tempo de meia vida
Não é constante. O tempo de meia vida de ordem zero é proporcional à quantidade
inicial ou concentração do fármaco e é inversamente proporcional à constante de velocidade
de ordem zero, k0: 0
0
2/1
5.0
k
At
REACÇÕES DE PRIMEIRA ORDEM
• A velocidade do processo é directamente proporcional à concentração do fármaco;
• A concentração de fármaco no plasma diminui de forma exponencial em função do
tempo, quando é eliminado numa percentagem fixa de fármaco numa unidade de tempo;
• Se a concentração do fármaco A diminui a uma velocidade que é proporcional à
quantidade de fármaco A restante, então a velocidade do desaparecimento do fármaco A é
dada por:
Constante de
velocidade
de ordem
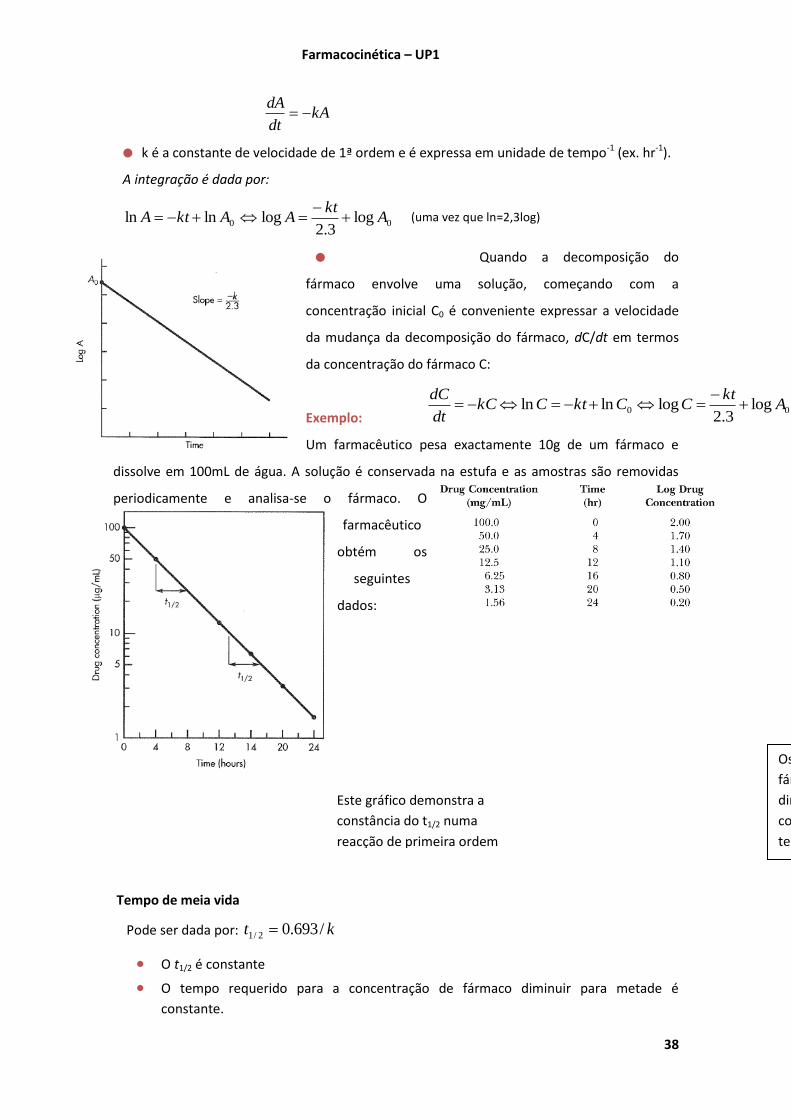
Farmacocinética – UP1
38
kAdt
dA
• k é a constante de velocidade de 1ª ordem e é expressa em unidade de tempo-1 (ex. hr-1).
A integração é dada por:
00 log3.2
loglnln Akt
AAktA
• Quando a decomposição do
fármaco envolve uma solução, começando com a
concentração inicial C0 é conveniente expressar a velocidade
da mudança da decomposição do fármaco, dC/dt em termos
da concentração do fármaco C:
Exemplo:
Um farmacêutico pesa exactamente 10g de um fármaco e
dissolve em 100mL de água. A solução é conservada na estufa e as amostras são removidas
periodicamente e analisa-se o fármaco. O
farmacêutico
obtém os
seguintes
dados:
Tempo de meia vida
Pode ser dada por:
kt /693.02/1
O t1/2 é constante
O tempo requerido para a concentração de fármaco diminuir para metade é
constante.
(uma vez que ln=2,3log)
Este gráfico demonstra a
constância do t1/2 numa
reacção de primeira ordem
Os valores das concentrações do
fármaco podem ser traçados
directamente com o logaritmo da
concentração do fármaco versus o
tempo → cinética de ordem 1
00 log3.2
loglnln Akt
CCktCkCdt
dC
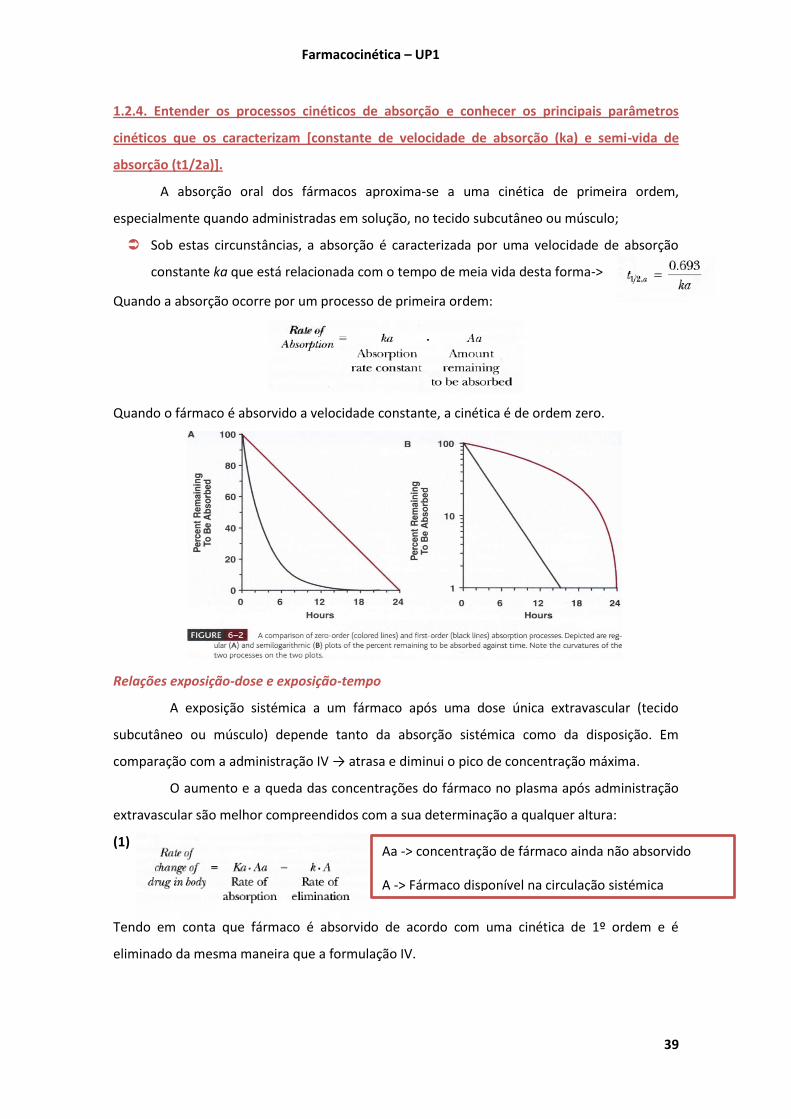
Farmacocinética – UP1
39
1.2.4. Entender os processos cinéticos de absorção e conhecer os principais parâmetros
cinéticos que os caracterizam [constante de velocidade de absorção (ka) e semi-vida de
absorção (t1/2a)].
A absorção oral dos fármacos aproxima-se a uma cinética de primeira ordem,
especialmente quando administradas em solução, no tecido subcutâneo ou músculo;
Sob estas circunstâncias, a absorção é caracterizada por uma velocidade de absorção
constante ka que está relacionada com o tempo de meia vida desta forma->
Quando a absorção ocorre por um processo de primeira ordem:
Quando o fármaco é absorvido a velocidade constante, a cinética é de ordem zero.
Relações exposição-dose e exposição-tempo
A exposição sistémica a um fármaco após uma dose única extravascular (tecido
subcutâneo ou músculo) depende tanto da absorção sistémica como da disposição. Em
comparação com a administração IV → atrasa e diminui o pico de concentração máxima.
O aumento e a queda das concentrações do fármaco no plasma após administração
extravascular são melhor compreendidos com a sua determinação a qualquer altura:
(1)
Tendo em conta que fármaco é absorvido de acordo com uma cinética de 1º ordem e é
eliminado da mesma maneira que a formulação IV.
Aa -> concentração de fármaco ainda não absorvido
A -> Fármaco disponível na circulação sistémica
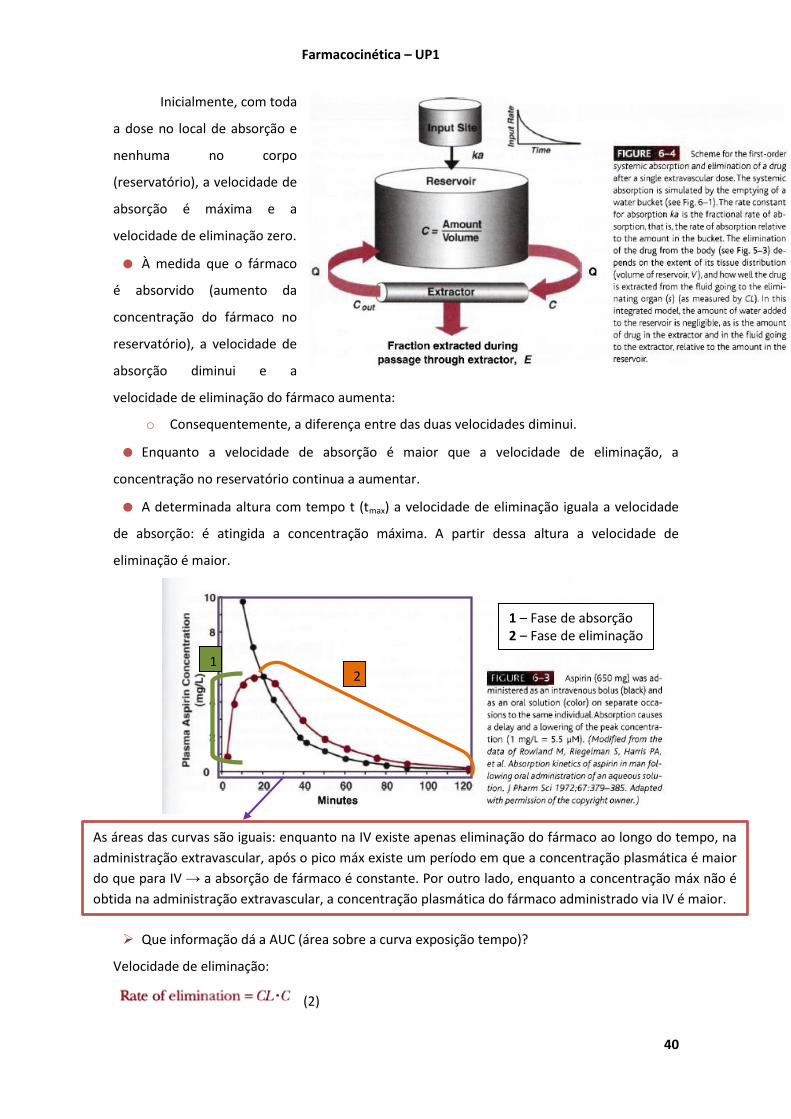
Farmacocinética – UP1
40
Inicialmente, com toda
a dose no local de absorção e
nenhuma no corpo
(reservatório), a velocidade de
absorção é máxima e a
velocidade de eliminação zero.
• À medida que o fármaco
é absorvido (aumento da
concentração do fármaco no
reservatório), a velocidade de
absorção diminui e a
velocidade de eliminação do fármaco aumenta:
o Consequentemente, a diferença entre das duas velocidades diminui.
• Enquanto a velocidade de absorção é maior que a velocidade de eliminação, a
concentração no reservatório continua a aumentar.
• A determinada altura com tempo t (tmax) a velocidade de eliminação iguala a velocidade
de absorção: é atingida a concentração máxima. A partir dessa altura a velocidade de
eliminação é maior.
Que informação dá a AUC (área sobre a curva exposição tempo)?
Velocidade de eliminação:
(2)
As áreas das curvas são iguais: enquanto na IV existe apenas eliminação do fármaco ao longo do tempo, na
administração extravascular, após o pico máx existe um período em que a concentração plasmática é maior
do que para IV → a absorção de fármaco é constante. Por outro lado, enquanto a concentração máx não é
obtida na administração extravascular, a concentração plasmática do fármaco administrado via IV é maior.
1 2
1 – Fase de absorção 2 – Fase de eliminação

Farmacocinética – UP1
41
E a sua integração:
(3)
A quantidade total eliminada após uma dose oral iguala o total absorvido, F.Dose,
onde o parâmetro F (biodisponibilidade) tem em conta que apenas esta fracção da dose oral
atinge a circulação sistémica - isto é: (4)
Biodisponibilidade
Conhecendo a biodisponibilidade (extensão de absorção), é possível assegurar que a
dose oral administrada é a correcta para permitir a exposição terapêutica.
• Apesar da dose ser conhecida, e da área poder ser determinada de acordo com uma dose
extravascular, a partir da equação 4, verifica-se que é necessário a clearance para calcular a
biodisponibilidade.
Para determinar a clearance, o fármaco tem que ser administração intravascularmente onde
F=1. Assim: (5)
Após uma dose extravascular: (6)
A razão entre 6 e 5, leva a: (7)
Biodisponibilidade relativa:
Feita quando não há registo IV devido a falta de preparações administráveis por essa via
(custo de desenvolvimento, fraca solubilidade, potenciais eventos adversos).
É determinada através da comparação de fracções absorvidas em diferentes doses,
diferentes vias de administração ou diferentes condições (dieta ou presença de outro
fármaco).
Exemplo com duas formas de dosagem:
Mudanças na dose ou cinética de absorção
O perfil de concentração - tempo de acordo com a mudança de dose ou das
características de absorção da dosagem podem ser antecipadas.

Farmacocinética – UP1
42
Alteração de dose: Se todos os factores forem constantes, o aumento da dose ou a fracção
da dose absorvida produz um aumento proporcional na concentração plasmática em todos
os tempos.
o O valor de tmáx continua inalterado mas a concentração máxima e AUC aumenta
proporcionalmente com a dose.
Mudanças na cinética de absorção: A mudança na forma de dosagem ou a administração
do produto com comida muda os perfis da concentração plasmática do fármaco.
Esta situação é ilustrada através de 3 situações apresentadas na figura seguinte onde estão
envolvidas apenas alterações na meia-vida da absorção.
A disposição é o passo limitante!

Farmacocinética – UP1
43
No caso A: a meia-vida de absorção é mais pequena que a meia-vida de eliminação: muito do
fármaco foi absorvido e apenas uma pequena parte foi eliminada enquanto o pico é atingido.
o Posteriormente, o declínio do fármaco é determinado principalmente pela disposição do
fármaco. A meia-vida estimada através da fase de declínio é a meia-vida de eliminação.
Caso B: a meia-vida de absorção é maior que em A, mas continua a ser menor que a meia vida
de eliminação.
o O pico ocorre mais tarde (aumento do tmax).
o A concentração máxima é menor porque o fármaco foi sendo absorvido ao longo do
tempo.
o Mesmo assim, a absorção é completa antes que a maioria do fármaco seja eliminado.
o Consequentemente, a fase de declínio determina a meia vida de eliminação e a
disposição é o passo limitante.
A absorção é o passo limitante!
Quando a meia vida de absorção é maior que a de eliminação.
O pico da concentração ocorre mais tarde e mais baixo que os dois primeiros casos, o que
mostra absorção lenta.
Ao contrário de A e B, a absorção é tão lenta que uma quantidade de fármaco fica por
absorver na altura em que o pico máximo é atingido.
Em todos os tempos, o fármaco esta a ser absorvido e eliminado e existe um pouco no
corpo.
De facto, durante a fase de declínio, o fármaco é virtualmente eliminado tão depressa
quanto é absorvido: a absorção é agora o passo limitante da velocidade. Pode, deste modo, ser
feita a seguinte aproximação: Eq. 6-13,ou seja,
Eq.6-14
A concentração plasmática (C=A/V) durante a fase de declínio é directamente proporcional
à quantidade de fármaco que falta absorver.
Exemplo: quando a quantidade que falta absorver diminui para metade, o mesmo acontece
com a concentração plasmática. O tempo necessário para que isto aconteça é a meia-vida de
absorção. Ou seja, a meia vida de declínio do fármaco no corpo corresponde agora a meia-vida
de absorção. Acontece um caso de flip-flop (termo dado para esta situação cinética)-> As fases
de absorção e eliminação na representação gráfica estão misturadas.
Distinção entre limitações da velocidade de absorção e disposição

Farmacocinética – UP1
44
Esta distinção é feita através da alteração da cinética de absorção do fármaco:
administração do fármaco noutra forma de dosagem como solução ou através de outra via.
1.2.5. Perceber a influência do esvaziamento gástrico e do tempo de trânsito intestinal;
entender a influência dos alimentos e de fármacos que condicionam o trânsito
gastrointestinal.
Esvaziamento gástrico e tempo de trânsito intestinal
• Fármacos que influenciam o esvaziamento gástrico afectam também a absorção de outros
fármacos -> Ex.: acetaminofeno (acção analgésica/ antipirética):-> ver atrás
A retenção do acetaminofeno no estômago aumenta a percentagem da dose absorvida
através da mucosa gástrica, mas a maioria da dose
continua a ser absorvida no epitélio intestinal.
O estômago pode assim ser considerado um órgão
de reposição permitindo a ejecção do fármaco, através
de movimentos peristálticos, para os locais de absorção
mo intestino delgado.
Em jejum, o esvaziamento gástrico de sólidos
maiores e mais pequenos é rápido (cerca de 1 hora) no
entanto existe considerável variabilidade interindividual.
• O estômago tem períodos de “descanso” e de
contracção moderada com várias frequências, que move o material para o intestino delgado.
• O tempo exacto de ejecção de uma partícula de sólido depende do seu tamanho, de
quando foi ingerida durante o ciclo de actividade motora e de onde está localizada no
estômago.
• A ejecção é maior quando as partículas de sólido estão próximas do esfíncter pilórico
onde as contracções ocorrem.

Farmacocinética – UP1
45
Quando há comida no estômago o trânsito gástrico dos sólidos é aumentado. Este
aumento é maior depois de uma refeição pesada do que depois de uma refeição ligeira,
sendo também muito maior para uma única grande dose do que para pequenas doses.
INFLUÊNCIA DOS ALIMENTOS E DOS FÁRMACOS
Anatomicamente, o fármaco ingerido atinge rapidamente o estômago, passando
posteriormente para o duodeno.
O duodeno possui a maior capacidade de absorção de fármacos de todo o tracto GI.
Desta forma, um atraso no tempo de esvaziamento gástrico reduz a possibilidade de uma
extensa absorção do fármaco, prolongando o tempo de inicio de acção do mesmo.
Existem diversos factores que afectam o tempo de esvaziamento gástrico, e alguns deles
tendem a retardar o tempo de esvaziamento:
Factor Influência no esvaziamento gástrico
Alim
ento
s
Ácidos gordos
Redução do esvaziamento de uma forma directamente
proporcional com a sua concentração e comprimento da cadeia
de carbonos; uma pequena diferença verifica-se com o ácido
acético e com o ácido octanóico; verifica-se uma maior influência
inibitória em compostos com mais de 10 carbonos.
Triglicerídeos Redução do esvaziamento: triglicerídeos insaturados mais
eficientes que os saturados
Carbohidratos
Redução do esvaziamento, como resultado da pressão osmótica;
a inibição do esvaziamento aumenta quando aumenta a
concentração
Aminoácidos
A redução do esvaziamento depende directamente do aumento
da concentração, provavelmente como resultado do aumento da
pressão osmótica
Fárm
aco
s
Anticolinérgicos Redução do esvaziamento
Analgésicos narcóticos Redução do esvaziamento
Metoclopramida Aumenta o esvaziamento
Etanol Redução do esvaziamento
1.2.6. Conhecer os processos que condicionam a absorção a partir do tracto gastrointestinal
e que podem determinar a biodisponibilidade oral de fármacos. Alguns exemplos de
interacções relevantes no tracto gastrointestinal (fármaco-fármaco e fármaco-alimento) e de
interesse marcado na prática clínica.
BIODISPONIBILIDADE
A biodisponibilidade (F) de fármacos administrados oralmente é normalmente menor que 1,
devido a efeitos de 1ª passagem, tempo de absorção insuficiente e reacções de competição.

Farmacocinética – UP1
46
1. Perda por efeito de primeira passagem
O fármaco passa do lúmen intestinal através da sua parede para o fígado através da
veia porta, antes de entrar na circulação sistémica. ->ver se está atrás
Se a única causa de perda do fármaco é o tempo de absorção incompleto, então a
biodisponibilidade é menor que 1 e o complemento (porção que é excretada nas fezes
inalterada) é uma medida da retenção luminal.
O fármaco pode ser “perdido” por decomposição no lúmen. A fracção que entra nos
tecidos intestinais (FI) é a fracção que nem é perdida nas fezes nem decomposta no lúmen.
Apenas uma fracção escapa à destruição das paredes do tracto gastrointestinal (FG).
Se o fármaco é eliminado no fígado, aparece uma fracção adicional (FH). A biodisponibilidade é
então dada por-> F=FF.FG.FH
Ex.1: Se 50% do fármaco é perdido em cada passo, a biodisponibilidade do fármaco, medida
sistematicamente, será 0,5 x 0,5 x 0,5 = 0,125% ou 12,5%.
• O fármaco pode ser totalmente perdido em qualquer um destes passos.
Ex.2: A aspirina foi um dos primeiros pró-fármacos sintéticos.
• A hidrólise intestinal e hepática é tão rápida que a maioria é convertida em ácido salicilico
numa única passagem
através destes órgãos,
resultando num
substancial metabolismo
de primeira passagem.
Quando o fármaco
perde muito a sua
actividade ou se torna
mesmo inactivo devido ao
efeito de primeira
passagem, a dose oral
deve ser maior do que o equivalente da dose intravenosa ou intramuscular.
2. Tempo de absorção insuficiente
Nos fármacos permeáveis, a absorção é rápida e provavelmente completa no intestino
delgado, ainda que algum fármaco entre no intestino grosso, a permeabilidade ainda é
suficientemente alta de forma a permitir a absorção.
Fármacos com fraca permeabilidade são geralmente mais polares e a sua absorção ocorre
primariamente no intestino delgado, mas a absorção pode não ser completa num período de
trânsito de 2 a 4 horas. ->anh?

Farmacocinética – UP1
47
Os fármacos com baixa permeabilidade demonstraram ter uma biodisponibilidade oral
reduzida não apenas pela baixa permeabilidade mas também porque o tempo de absorção no
tracto gastrointestinal é insuficiente onde o produto da área permeabilidade-superfície (P.SA)
é elevado.
O termo P.SA tende a diminuir drasticamente com o
movimento do fármaco do intestino delgado para o cólon.
Gráfico1- Demonstra a relação entre a fracção de dose absorvida
e a permeabilidade de alguns fármacos.
Fármacos com permeabilidade abaixo de 1.0 são absorvidos de
forma incompleta.
3. Reacções competitivas
• Qualquer reacção que compete com a absorção pode reduzir a biodisponibilidade oral do
fármaco. ->penso que já esteja referido atrás.
• Reacções podem ser: Enzimáticas (causadas por enzimas digestivas, metabólicas e por
enzimas da microflora) e não-enzimáticas (ex. hidrólise ácida que ocorre no estômago)
• Podem ocorrer reacções de complexação com outros fármacos → diminuição da
biodisponibilidade do fármaco.
• Quando o fármaco absorvente e o absorvido são usados frequentemente, a sua
administração deve ser espaçada para evitar que se misturem no tracto gastrointestinal.
• O problema da absorção incompleta pode ser ultrapassado com protecção física do
fármaco ou utilizando derivados mais estáveis que depois são convertidos na molécula activa
no tracto gastrointestinal (pró-fármacos).
• Por vezes, a biodisponibilidade oral de um fármaco é muito baixa mas continua a ser
usada para efeitos sistémicos → utilidade no tratamento de doenças do canal alimentar.
Processos que condicionam a absorção
Aminoácidos, ácidos gordos e outros nutrientes contidos nos alimentos podem afectar
o pH intestinal e a solubilidade dos fármacos.
- Atraso no esvaziamento gástrico
- Estimulação da bílis
- Alterações no pH do tracto GI
- Um aumento no fluxo sanguíneo esplâncnico (“splanchnic blood flow”)
- Alterações no metabolismo do fármaco
- Interacções físico-químicas dos alimentos com o fármaco.

Farmacocinética – UP1
48
O teor calórico, a quantidade de nutrientes, o volume de alimentos ingeridos e a
temperatura da refeição podem causar alterações fisiológicas no tracto GI e
consequentemente afectar a absorção e metabolismo do fármaco.
Fármacos muito lipossolúveis->Absorção mais eficiente quando administrados com alimentos
ricos em gorduras.->penso que já está dito atrás
Alguns fármacos (eritromicina, aspirina, AINEs…) são irritantes para a mucosa gástrica e desta
forma, devem ser administrados com alimentos de forma a reduzir a irritação gástrica.
A quantidade absorvida pode ser reduzida na presença de alimentos, mas a extensão de
absorção pode ser a mesma e a eficácia do fármaco é mantida.
Fármacos que afectam a absorção de outros fármacos:
Os fármacos anticolinérgicos reduzem as secreções ácidas estomacais. O bromido de
propantelina é um fármaco anticolinérgico que pode reduzir o esvaziamento gástrico e a
motilidade no intestino delgado.->penso que isto não seja preciso
Os antidepressivos tricíclicos e as fenotiazinas possuem também um efeito colateral
anticolinérgico que reduz os movimentos peristálticos no tracto GI.
A metoclopramida estimula a contracção do estômago, relaxa o esfíncter pilórico e aumenta
os movimentos peristálticos intestinais, reduzindo o tempo de absorção de determinados
fármacos e reduzindo também o pico de concentração do fármaco e o tempo que este demora
a atingir esse pico de concentração.
Os antiácidos não devem ser administrados com a cimetidina, pois estes reduzem a absorção
do fármaco.
o Os antiácidos contêm alumínio, cálcio ou magnésio que podem complexar fármacos como
as tetraciclinas e ciprofloxacina, resultando na diminuição da absorção do fármaco.
o Para evitar esta interacção os antiácidos devem ser administrados 2h antes das refeições ou
6h após a administração do outro fármaco.
Os inibidores da bomba de protões, como o omeprazol, podem também afectar a absorção de
outros fármacos ao reduzirem o teor ácido do estômago.
A absorção de cálcio no duodeno é um processo activo facilitado pela vitamina D. Acredita-se
que as proteínas que se associam ao cálcio, que aumentam após a administração de vitamina
D, se ligam ao cálcio na célula intestinal e transferem este da célula para a circulação
sanguínea.
1.3. Entender o processo de absorção de fármacos a partir de outros locais para além do
tracto gastrointestinal
A absorção pela via intramuscular e subcutânea está limitada pelo fluxo sanguíneo.

Farmacocinética – UP1
49
A rápida absorção da lidocaína na
administração local depende do fluxo
sanguíneo (ver tabela).
o Administração de adrenalina
provoca vasoconstrição, redução
do fluxo sanguíneo e
prolongamento do efeito
anestésico local.
Quando um fármaco é administrado por via intramuscular ou subcutânea e se necessitar de
uma acção sistémica, o reduzido fluxo sanguíneo pode ser uma desvantagem.
o Em casos extremos, como choque hemorrágico, o fluxo sanguíneo no tecido muscular
encontra-se bastante reduzido (não é apropriado utilizar estas vias).
Esta dependência da absorção do fluxo sanguíneo pode ser explicada pela natureza da barreira
(membrana capilar) entre o sítio de injeção (fluido intersticial) e sangue.
o Esta membrana apresenta uma estrutura mais fina do que a parede epitelial do tracto GI,
oferecendo assim pouco impedimento à passagem dos fármacos para o sangue, mesmo no
caso de fármacos ionizados.
Os pulmões possuem uma extensa superfície de absorção.
o Os seus componentes têm como função remover partículas estranhas das superfícies
periféricas pulmonares com elevada capacidade de absorção, através de clearence mucociliar.
o Se os compostos forem fármacos na forma de aerossóis, podem atingir as regiões
periféricas do pulmão com elevada eficácia, ou seja, sendo muito facilmente absorvidos.
Fármacos de aplicação tópica:
o Apesar do objectivo ser a obtenção de um efeito local, alguns fármacos devem sofrer
absorção sistémica – fármacos de aplicação transdérmica.
1.4. Compreender as implicações no perfil de exposição sistémica (curvas concentração-
tempo) resultantes de alteração na dose ou na cinética de absorção. Perceber outros
parâmetros farmacocinéticos relacionados com a velocidade [concentração máxima (Cmax)
e tempo ao qual é alcançada a concentração máxima (tmax)] e extensão de absorção [área
sob a curva concentração-tempo (AUC)] (ver também: Tozer2006; Chap. 3, pg.38)

Farmacocinética – UP1
50
Em A: Em B:
- Os fármacos possuem AUC diferentes
- Atingem rapidamente a Cmáx (têm Cmáx
iguais)
- Velocidades de eliminação diferentes
- AUC são iguais
- (I) atinge rapidamente a Cmáx
- Absorção rápida
- Velocidade de eliminação rápida
- (II) atinge posteriormente a sua Cmáx com
tmáx mais tardio
- Absorção lenta
- Eliminação lenta
Existem alguns benefícios numa rápida diminuição da concentração plasmática, caso a
exposição prolongada ao fármaco leve a um aumento do risco de efeitos adversos.
ALTERAÇÕES NA DOSE OU NA CINÉTICA DE ABSORÇÃO
Alteração da dose
Se todos os outros factores permanecerem constantes, o aumento da dose ou da fracção
de uma dose absorvida produz um aumento proporcional da concentração plasmática ao
longo do tempo.
O valor tmax permanece inalterado, enquanto que a Cmax e a AUC aumenta proporcionalmente
com a dose.
Alteração da cinética de absorção
Representação esquemática dos perfis de concentração plasmática - tempo após a administração de uma dose oral.
A. Para 2 fármacos que produzem um pico de concentração e de tempo semelhantes, mas um deles decresce mais devagar que o outro, apesar de criar uma maior área sob a curva (AUC) e concentrações mais elevadas nos tempos finais.
B. Para um fármaco que produz a mesma AUC quando administrado em duas ocasiões, atingindo numa delas um pico de concentração plasmática mais baixo atingido mais tarde devido a uma absorção mais lenta.
Iguais perfis de absorção mas diferentes velocidades de excreção.
Diferentes tempos de exposição ao fármaco

Farmacocinética – UP1
51
Alterações na cinética de absorção, como por exemplo a alteração na forma de dosagem
ou a administração do fármaco com alimentos, originam alterações do perfil de tempo das
concentrações plasmáticas. (ver ponto 1.2.4)
2. Apreender a cinética da disposição de fármacos. (Tozer 2006 Chap. 5)
2.1. Compreender os conceitos gerais e os principais parâmetros cinéticos de interesse
subjacentes ao estudo da disposição de fármacos [volume de distribuição e clearance,
constante de velocidade de eliminação de primeira-ordem (k) e semi-vida de eliminação (t
½)].
Por injecções rápidas, podem ser atingidas elevadas concentrações do fármaco
enquanto que por infusão a uma velocidade constante, pode ser mantida uma concentração
constante de fármaco, bem como a resposta a este.
As características de disposição de um fármaco são definidas pela análise do fármaco
no plasma e na urina, tendo em conta que este é administrado por via intravenosa.
Método Cartesiano: é o método mais usado e consiste em traçar os valores de
concentração vs os valores de tempo num papel gráfico regular;
Método Semilogarítmico: consiste em traçar os valores do logaritmo da concentração
vs os valores de tempo. A vantagem deste método é de ser mais fácil prever a
concentração a um terminado tempo.
Volume de Distribuição e Clearance
O fármaco é colocado num reservatório aquoso que é bem homogeneizado (≈ corpo) cujos
componentes são reciclados através de um extractor (≈ fígado/rins) que removem o fármaco.
A concentração do fármaco no reservatório, C, e as concentrações
que conseguem sair do extractor (Cout) podem ser determinadas.
A concentração inicial no reservatório (C0), depende da
quantidade que é introduzida (ou seja da dose) e do volume do
reservatório (V).
Como é possível obter um declive linear quando se traça os dados numa escala semilogarítmica? O que
determina as diferenças observadas nos perfis de diferentes fármacos?
Métodos para dispor
graficamente a informação
concentração-tempo
Esquerda: M. Cartesiano
Direita: M. Semilogarítmico
V
DoseC
""0

Farmacocinética – UP1
52
O fluido passa pelo extractor a uma determinada taxa de fluxo, Q. Admitindo que a
concentração do fármaco que entra no extractor é a mesma do reservatório, C, então a taxa de
entrada no extractor é Q x C.
Do fármaco que entra no extractor, uma fracção, E, é excretada por processos de
eliminação e nunca retorna ao reservatório. Então a taxa correspondente ao fármaco que sai
do extractor e retorna ao reservatório é Q x Cout.
A taxa de eliminação (ou extracção) pode ser dada por:
)(lim outinaçãoE CCQEQCTaxa
Do qual se retira que a taxa de extracção, E, é dada por:
C
CC
QC
CCQE outout )()(
Assim, vemos que a razão de extracção pode ser determinada experimentalmente
medindo as concentrações que entram e saem do extractor, adicionando um factor de
correcção, c.
Conceptualmente, é útil para relatar a taxa de eliminação para a concentração medida.
Este parâmetro é designado por clearance, CL, e obtém-se por:
CCLTaxa inaçãoE lim
As unidades da clearance podem ser (mL/min) ou (L/h), uma vez que a taxa de
eliminação pode ser (µg/min) ou (mg/h) e a concentração pode ser (µg/L) ou (mg/L).
EQCL
Esta equação dá uma interpretação física da clearance: volume de fluído presente no
órgão de eliminação (ou extractor) que é efectivamento purificado do fármaco, por unidade de
tempo.
Q= 1L/min e E=0,5 500mL do fluído que sai do reservatório e entra no extractor por minuto
é efectivamente purificado.
A taxa de fluxo é que limita a quantidade de fluxo que entra no extractor. Assim, a
clearance dá uma estimativa da taxa de eliminação relativamente à concentração inicial, C0 ou
outra concentração.
Eliminação de Primeira Ordem
Considera-se a taxa de eliminação, CL x C, relativa a quantidade que está presente no
reservatório, A, uma taxa comummente referida como taxa fraccional de eliminação, k:
Com que velocidade um fármaco sai do reservatório?
V
CL
CV
CCL
A
CCL
Quantidade
Taxak
servatório
inaçãoE
Re
lim

Farmacocinética – UP1
53
C = C0e-kt
Assim, a taxa fraccional de eliminação depende do volume e da clearance, dois parâmetros
independentes.
Um método mais simples e confiável de calcular
estes vários qualquer seja o tempo, t, é a Fracção de Dose Restante.
kteDose
AFDR
Por vezes é útil o tempo relativo de meia-vida , t ½. A vantagem reside no facto de n
ser o número de semi-vidas decorridas após administração (dose de bolus).
Assim, como k = 0,693 / t ½, obtém-se:
Uma vez que e-0,693 = ½, tem-se que:
nFDR )2
1( n = 1 , permanece 50% ( ½ ) da concentração de fármaco
n = 2, permanece 25% ( ½ . ½ ) da concentração de fármaco
n = 4, permanece 6,25% ( ½ 4) da concentração do fármaco
Existem dois meios de determinar experimentalmente a taxa de eliminação:
1. Medição das taxas de “entrada e saída” do fármaco do organismo.
2. Determinação da taxa de perda de fármaco do reservatório, assumindo que a única forma de
perda a partir do sistema é a sua eliminação no extractor.
Considerando o segundo método, temos que:
kAdt
dATaxa inaçãoE lim
,
sendo –dA a quantidade de fármaco perdido do reservatório no intervalo de tempo dt.
A taxa do processo é directamente proporcional à quantidade present , pelo que
processos como este são denominados de Processos de Primeira-Ordem. Dai, o parâmetro k é
frequentemente chamado de constante de velocidade de primeira-ordem.
Se se substituir A=VC em ambos os membros da equação tem-se:
-dC/dt = kC ->integração-> -> permite uma representação curvilínea.
Como é que a fracção de dose restante no reservatório varia com o tempo?
Equação Mono-Exponencial
CL=1 L/h e V=10 L k= 0,1 h-1
O reservatório elimina 10% a cada hora.
nkt eeFDR 693,0

Farmacocinética – UP1
54
-> permite uma representação linear, com declive –k
O declive, -k, determina a velocidade de diminuição da concentração de fármaco no
organismo, que depende de V e CL.
Tempo de Meia-Vida
A cinética dos fármacos é caracterizada por uma meia-vida/semi-vida (t ½), que é o
tempo necessário para a concentração (e quantidade no reservatório) diminuir para metade
da inicial, tendo em conta que a taxa de eliminação é constante (estes dois parâmetros estão
claramente interligados).
Partindo da equação C = C0e-kt, a uma meia-vida, C = 0.5 x C0, então:
ou
5,021
kt
e
Aplicando logaritmos a ambos os membros da equação, tem-se:
kk
t693,02ln
21
Substituindo k por CL/V, obtém-se:
CL
Vt
693,02
1
Sendo o t ½ directamente proporcional a V e inversamente proporcional a CL.
Considerando a creatinina, produto do catabolismo muscular e marcador da função renal:
Para um paciente de 70 kg, 60 anos de idade, a creatinina tem uma CL = 4,5L/h e está
distribuída pelos 42L de água total do organismo. Pela equação anterior, o seu t ½ é de 6,5
horas.
A inulina, um polissacarídeo também usado como marcador exógeno da função renal,
tem a mesma CL da creatinina nesse paciente, mas t ½ é de apenas 2,5h. Isto deve-se ao facto
da inulina estar restrita a 16L de água extracelular, pelo que o tamanho do seu “reservatório” é
menor que o da creatinina.
2.2. Entender a cinética de distribuição dos fármacos para os tecidos
(velocidade e extensão de distribuição)
O equilíbrio de distribuição dos fármacos no organismo estabelece-se a
diferentes velocidades, segundo os tecidos considerados.
- O mesmo sucede para a quantidade de fármaco que se localizará nos
diferentes tecidos.
- Vários factores podem influir nas características de distribuição dos
ln C = ln C0 – kt
21
005,0kt
eCC

Farmacocinética – UP1
55
medicamentos, nomeadamente, a velocidade de apresentação de um fármaco a um dado
tecido, a facilidade com a qual a substância pode atravessar as membranas biológicas e a
ligação dos fármacos às proteínas plasmáticas e tecidulares.
- Um dos parâmetros a ter em conta é a velocidade e extensão de distribuição, uma vez que
estes são diferentes nos vários tecidos.
O volume aparente de distribuição é um indicador da amplitude da distribuição do
medicamento no organismo.
Similarmente à biodisponibilidade, a distribuição pode ser caracterizada por:
Elemento Cinético: descreve a velocidade de distribuição no organismo
Elemento Quantitativo: descreve a amplitude de distribuição nos tecidos do organismo.
A velocidade de distribuição de um medicamento nos diferentes tecidos ou órgãos é função do
débito sanguíneo e da natureza da sua membrana biológica.
- Essa velocidade pode ser caracterizada pela constante de velocidade de distribuição
ou pelo inverso dessa constante, que exprime o tempo de distribuição médio.
A irrigação sanguínea é, geralmente, o processo que limita a distribuição nos tecidos
para os medicamentos fortemente lipossolúveis, que atravessam as membranas biológicas.
O medicamento não se pode difundir nos tecidos mais rapidamente do que é transportado
pelo sangue.
No caso dos medicamentos menos lipossolúveis e ionizados no pH do plasma, a difusão
através das membranas biológicas é a etapa que limita a distribuição.
A ligação aos tecidos tem um efeito sobre a amplitude da distribuição.
Certos medicamentos têm muita afinidade por certos tecidos, o que contribui para aumentar o
volume aparente de distribuição.
2.2.1. Perceber a influência da perfusão sanguínea e da permeabilidade membranar.
Conhecer os principais parâmetros farmacocinéticos: razão de distribuição no equilíbrio (KP),
constante de velocidade de distribuição (kT), semi-vida de distribuição no tecido (t1/2T),
volume aparente de distribuição, ligação às proteínas plasmáticas e ligação tecidular.
Limitação da Perfusão
As limitações à velocidade de perfusão prevalecem quando as membranas tecidulares
não constituem uma barreira à distribuição.
A velocidade de perfusão varia desde 10 mL/min/g nos pulmões para apenas 0,0025
mL/min/g no tecido adiposo e nos músculos.

Farmacocinética – UP1
56
Existe uma correlação directa entre a
velocidade de perfusão nos tecidos e o tempo
necessário para distribuir o fármaco no tecido, que
pode ser expressa por:
AãoDistribuiç QCExtensão ,
Em que Q é o fluxo sanguíneo e CA é a concentração
do fármaco no fluxo arterial.
Aplicando a equação anterior numa artéria
arterial e venosa e considerando que a artéria se comporta como um caudal de volume
constante, obtemos a equação : )( VAUptake CCQVelocidade (1)
onde CV é a concentração do fármaco na circulação venosa
que deixa o tecido.
Assim sendo, a concentração no tecido, CT, aumenta quando a extensão da distribuição excede
o CV.
Considerando que, em equilíbrio, a quantidade de fármaco nos tecidos é função do
coeficiente de distribuição, KP, da concentração do quociente entre CT/CV:
(2)VpTTTTecido CKVCVQuantidade ,
onde VT é o volume tecidular e Kp é a razão de distribuição no equilíbrio (CT/CV) e mede a
afinidade do tecido para o fármaco.
Quanto maior for o valor de KP, maior é a afinidade do tecido.
A fracção de saída, ou a constante de distribuição, KT, é dada por:
(3)p
T
VpT
V
Tecido
Saída
TK
VQ
CKV
QC
Quantidade
Velocidadek
,
onde Q/VT é a taxa de perfusão no tecido.
Um análogo à constante de distribuição é o tempo de meia vida de distribuição, que se
pode dar por: (4)
O KT permite-nos saber a rapidez com que o fármaco deixa o tecido se a concentração
arterial CA fosse baixando rapidamente para 0. A razão para tal é que quando a afinidade é alta
há muito mais fármaco no tecido e com uma perfusão lenta, demora-se mais tempo a remove-
lo.
A cinética de distribuição pode ser definida a partir do volume de saída (1) e da
quantidade de fármaco no tecido em equilíbrio (3), podendo assim calcular-se a semivida de
distribuição, (t1/2t):
TVQ
p
T
K
kVidaMeia
693,0693,0
TVQ
p
T
t
K
kt
693,0693,02
1

Farmacocinética – UP1
57
A constante KT representa a constante de velocidade de distribuição e indica a
velocidade com que o fármaco sai do tecido.
Exprime-se como h-1 ou min -1. Assim, a semivida de distribuição tecidular permite-nos
prever o tempo que a concentração no tecido levará para diminuir para metade.
O fármaco deixa lentamente um tecido para o qual tem elevada afinidade (KP) e baixa
perfusão.
A explicação para o facto é
que, quando a afinidade é elevada
existe maior concentração de
fármaco no tecido, e com baixa
perfusão é necessário mais tempo
para remover o fármaco do tecido.
A distribuição é mais rápida nos
tecidos em que o tempo de meia vida de distribuição é mais curto.
A distribuição no tempo de meia vida aplica-se não só ao efluxo mas também à
entrada nos tecidos. Esta distribuição é rápida nos tecidos onde a semi-vida de distribuição é
curta.
A influência da velocidade de difusão sobre o equilíbrio de distribuição depende,
portanto, da interacção entre as propriedades físico-quimicas do medicamento e a natureza e
a influência das membranas a atravessar.
O tiopental é uma substância apolar, lipofílica e de características muito pouco ácidas
(pKa=7,6). Só parcialmente será ionizado no pH plasmático. Portanto, difundir-se-á facilmente
em todos os tecidos do organismo, incluindo o sistema nervoso central (SNC) e os músculos. A
distribuição tecidular desta substância será, portanto, limitada pela irrigação tecidular. Como o
débito sanguíneo do cérebro, é muito mais elevado do que o do músculo, o tiopental
penetrará mais rapidamente no cérebro do que no músculo.
Dado o seu carácter muito mais polar, as penicilinas não passam facilmente as membranas
biológicas. A velocidade de entrada mais rápida das penicilinas no tecido muscular,
relativamente ao cérebro, explica-se pela natureza mais porosa dos capilares musculares.
Para a maior parte dos tecidos do organismo, o leito capilar é muito poroso, (por exemplo,
rim, musculo) e tem muito pouco efeito sobre a distribuição de medicamentos de massa
molecular habitual, sejam quais forem as propriedades físico-quimicas destes últimos.

Farmacocinética – UP1
58
O equilíbrio do fármaco no fluido cerebrospinal e no plasma
tem frequentemente uma taxa de permeabilidade-limitada.
O rácio da concentração do fármaco (líquido
cefalorraquidiano / fármaco livre no plasma) é indicado para
vários fármacos em cães.
A concentração plasmática manteve-se relativamente
constante ao longo do estudo.
Quando uma taxa de limitação de permeabilidade ocorre, o
tempo para atingir o equilíbrio é maior do que quando
atinge a taxa limite de perfusão, como ocorre com o
tiopental lipofílico.
Em contrapartida, podem existir limitações de difusão ao nível das membranas celulares.
Para outros tecidos, como o SNC, limitações de difusão capilar de natureza anatómica influem
na entrada de medicamentos (penicilinas não passam para o cérebro).
O início de acção farmacológica depende, muitas vezes, da velocidade à qual o equilíbrio
de distribuição é atingido ao nível tecidular. Isto determina a velocidade a que é atingido o
local de acção pelo medicamento.
O equilíbrio de distribuição é atingido quando as concentrações de medicamento não ligado
são as mesmas no plasma e nos tecidos, desde que a concentração plasmática do
medicamento seja mantida constante, durante um período de tempo suficientemente longo.
Limitação da permeabilidade
Uma limitação da taxa de permeabilidade surge sobretudo para fármacos polares ao
difundir-se pelas membranas lipídicas, tais como o cérebro quando o impedimento está a nível
vascular.
A taxa de limitação da permeabilidade
diminui a velocidade de entrada e,
consequentemente, aumenta o tempo para atingir a distribuição no equilíbrio ao longo de
uma perfusão isolada.
A figura acima mostra o momento certo de equilíbrio de vários compostos entre o
líquido cefalorraquidiano do cérebro e o fármaco livre no plasma.
O anestésico tiopental é muito lipofílico e a sua taxa de entrada por perfusão cerebral
é essencialmente reduzida. Os restantes compostos são mais polares e a absorção é mais
lenta.
Por exemplo, os coeficientes de partição do agente anti-inflamatório, ácido salicílico
(pKa 3) e pentobarbital (pKa 7,8), num sedativo hipnótico, são semelhantes, mas o tempo
necessário para chegar à distribuição de equilíbrio é muito mais curto para o pentobarbital do

Farmacocinética – UP1
59
que para o ácido salicílico, uma vez que, sendo um ácido mais fraco, uma maior percentagem
de pentobarbital é não-ionizado no plasma e nos tecidos com intervalos de pH 7,2-7,4.
No equilíbrio, a concentração de fármaco livre nos tecidos seria de esperar ser a
mesma que no plasma. Porém, essa igualdade não é observada.
Razões para esta falta de igualdade são:
Gradientes de pH através das membranas celulares;
Transporte activo.
Extensão da Distribuição
No sangue, a albumina é a
proteína mais importante para o
transporte dos fármacos. Esta
proteína pode ligar ao mesmo tempo,
os aniões e os catiões. Os fármacos
ácidos ligam-se, em geral, à parte N-terminal da molécula, enquanto os fármacos básicos
também se podem ligar à α1-glicoproteína ácida e às lipoproteínas.
As principais consequências da ligação dos medicamentos às proteínas plasmáticas são:
1) Quando a percentagem ligada ou as forças de ligação são elevadas, a ligação às proteínas
plasmáticas pode servir de armazenamento para um medicamento ou uma substância
endógena.
a. Por exemplo, a bilirrubina é armazenada a nível plasmático, sob a forma ligada às
proteínas;
2) Só o medicamento sob forma livre fica disponível para a difusão nos tecidos, para a
biotransformação ou para a excreção.
a. Uma percentagem elevada de ligação às proteínas poderia, teoricamente, diminuir a
velocidade de eliminação de uma substância.
b. Em contrapartida, a percentagem de ligação não é um parâmetro que permita prever a
semivida de um fármaco.
c. O factor mais importante a considerar é a força de ligação que existe entre o fármaco e a
proteína.
d. Para certos medicamentos, cuja percentagem de ligação é importante, mas o elo de
ligação com as proteínas é fraco, a ligação às proteínas plasmáticas pode aumentar a
velocidade de eliminação, assegurando um melhor transporte da substancia para os órgãos de
eliminação, quando o coeficiente de extracção hepática ou renal é elevado.
e. A dissociação rápida do complexo permite a eliminação do medicamento;

Farmacocinética – UP1
60
3) Quando uma percentagem importante do medicamento está ligada às proteínas plasmáticas,
uma fraca variação da percentagem de ligação pode causar alterações significativas do volume
aparente de distribuição, da depuração e da semivida de eliminação, segundo o volume
aparente de distribuição e o coeficiente de extracção hepática ou renal.
As principais causas de variações da ligação às proteínas plasmáticas situam-se ao nível:
Do medicamento: Características físico-quimicas; Concentração total (saturação dos locais de
ligação)
Da proteína ligante
a) Quantidade diminuída por perda urinaria, difusão no liquido intersticial ou diminuição da
síntese hepática;
b) Qualidade alterada por modificação estrutural
Da afinidade entre o medicamento e a proteína ligante: alteração da afinidade por substâncias
endógenas e pela acidose metabólica
Da deslocação dos locais de ligação por medicamentos ou substâncias endógenas.
Volume Aparente de Distribuição
A concentração no plasma atingida após distribuição por todo o corpo é resultado da
dose administrada e do grau de distribuição nos tecidos, reflectida pelo volume de distribuição
(este parâmetro relaciona a quantidade no corpo e a concentração plasmática).
Recordando a equação:
O parâmetro V varia muito: desde 3 aos 7000 L (um valor muito superior ao tamanho total do
corpo). Os ácidos tendem a ter pequenos volumes de distribuição (<50 L); compostos básicos
muito maiores (> 100 L).
Exemplo: a figura. 5-8 mostra que os valores de V
para fármacos.
- Ácidos (aspirina, a furosemida (diurético) e a
varfarina (anticoagulante oral)) agrupam-se em torno
dos 10 L.
- Básicos (β-bloqueador anti-hipertensivo
propranolol, a morfina (analgésico), e o
antidepressivo nortriptilina) encontram-se >100 L e
podem chegar aos 7000 (ex.:amiodarona
(antiarrítmico)).

Farmacocinética – UP1
61
Sabendo o volume plasmático (Vp) e o volume de distribuição (V), a fracção de fármaco no
corpo que está dentro e fora do plasma pode ser estimada:
A quantidade de plasma é VpC e a quantidade no organismo é VC.
Quanto maior o volume de distribuição menor é a percentagem em plasma.
A fracção restante - Fracção de fármaco no corpo fora do plasma (inclui fármacos em células
do sangue):
Embora esta fracção possa ser facilmente determinada para dados plasmáticos, a distribuição
real de fármaco fora do plasma não pode.
A razão pela qual o volume de distribuição é um volume aparente e por que razão o
seu valor difere entre os fármacos podem verificar-se no modelo simples seguinte:
Neste modelo, o fármaco no corpo é totalmente contabilizado no plasma (Vp) e num
compartimento de tecido (VT). Na distribuição de equilíbrio, a quantidade de fármaco em cada
localização pode ser expressa em termos dos seus respectivos volumes e concentrações (C, CT).
Como A = V C, dividindo-se a equação acima por C e desde que
, segue-se que:
O produto é o volume aparente do tecido para medição do fármaco no plasma.
Assim, através da expansão do modelo para abraçar todos os tecidos do corpo, vê-se
que o volume de distribuição de um fármaco é o volume de plasma mais a soma dos volumes
de distribuição aparente de cada tecido.
Para alguns tecidos, o valor de Kp é grande → explica como o volume de distribuição de
alguns fármacos, tais como de compostos básicos, pode ser muito maior que o tamanho total
do corpo.
Exemplo: a gordura ocupa aproximadamente 20% do volume corporal. Se o Kp na
gordura é 10, então este tecido sozinho tem um volume de distribuição aparente de 140 L (0,2
x 70 x 10), o dobro do volume do corpo.
Quantidade no tecido Quantidade no plasma

Farmacocinética – UP1
62
Mesmo quando há apenas uma limitação da taxa de perfusão, leva muitas horas para o
equilíbrio da distribuição ocorrer na gordura → devido à sua baixa taxa de perfusão
e alta afinidade do fármaco para a gordura.
O volume de distribuição de um fármaco específico também varia muito entre os
doentes/pessoas.
Ligação às proteínas plasmáticas
O grau de ligação é frequentemente expresso como o limite para a relação da concentração
total. Esta relação tem valores limite de 0 e 1,0 (fármacos com valores superiores a 0,90 são
considerados altamente ligados).
A concentração de fármaco não ligado é mais importante na terapêutica que a porção ligada. A
fracção livre no plasma (não ligado), , é:
A ligação é a função da afinidade entre proteína e fármaco.
Como o número de sítios de ligação a uma proteína é limitada, a ligação também depende das
concentrações molares de ambas - as de fármaco e as de proteína.
Quanto maior a concentração de proteína para uma concentração de determinado
fármaco, maior a fracção ligada e o inverso.
Na prática, apenas uma pequena percentagem dos locais de ligação disponíveis em proteínas
está ocupada com as concentrações terapêuticas da maioria dos fármacos. Então:
é relativamente constante para uma dada concentração de proteína e
independente da concentração de fármaco.
Deste modo, a concentração plasmática total é uma boa medida de mudanças na
concentração de fármaco não ligado.
Ocasionalmente, as concentrações terapêuticas são suficientemente elevadas e a maioria dos
sítios disponíveis para ligação são ocupados. Então:
é dependente da concentração.
A ligação às proteínas plasmáticas muitas vezes só é de interesse porque a concentração
plasmática total é comummente medida e por vezes varia, como na doença renal e hepática e
com a concentração de fármaco.
A concentração total é medida devido a problemas na medição da concentração não ligada:
- problemas físicos na separação de fármacos com baixo peso molecular não ligados de
fármacos ligados a proteínas
- problemas da sensibilidade do ensaio, especialmente quando é muito pequena.

Farmacocinética – UP1
63
A concentração plasmática total depende tanto da extensão da ligação às proteínas como da
concentração não ligada:
Ligação Tecidular
O fármaco pode ter uma grande afinidade para proteínas plasmáticas, mas pode estar
localizado/acumulado principalmente no tecido Se os componentes do tecido tiverem uma
maior afinidade com o fármaco do que as proteínas do plasma.
A afinidade de um medicamento por um tecido pode explicar-se pela ligação às
proteínas plasmáticas tecidulares, aos ácidos nucleicos ou, no caso do tecido adiposo, pela
dissolução das substâncias nas gorduras.
A ligação de fármacos ao tecido, ao contrário da ligação a proteínas plasmáticas, não pode ser
medida directamente.
Para separar fármacos ligados de não ligados, o tecido deve ser interrompido → perda
da sua integridade. Mesmo assim, a ligação aos tecidos é importante na distribuição de
fármacos.
A ligação ao tecido pode ser inferida a partir da medição da ligação do fármaco no plasma.
Considere-se, por exemplo, a relação de equilíbrio de massas seguintes:
( )
Em que VT é o volume aquoso fora do plasma nos quais o fármaco se distribui e CT é a
correspondente concentração média total de tecido, que representa a massa de fármaco no
tecido.
Dividindo por C:
Sabendo que
(fracção livre ao nível plasmático).
Analogamente, para o tecido,
(onde fuT é a fracção livre tecidular, CuT é a
concentração tecidular do fármaco livre e CT é a concentração tecidular total).
Dado que o equilíbrio de distribuição é atingido quando a concentração livre no
plasma, Cu, e no tecido, CuT, são iguais, então:
Que substituindo na equação anterior:
Quantidade no plasma Quantidade no tecido Quantidade no corpo
Volume Aparente de Distribuição Volume de plasma
Volume Aparente no tecido
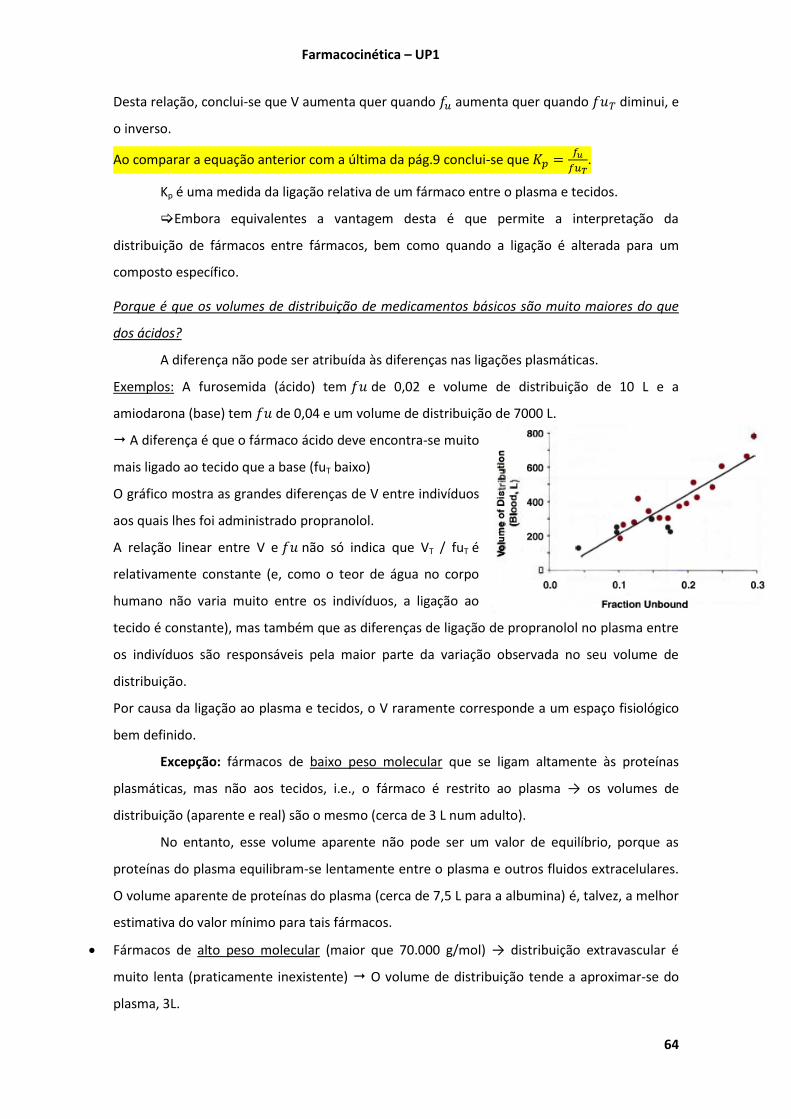
Farmacocinética – UP1
64
Desta relação, conclui-se que V aumenta quer quando aumenta quer quando diminui, e
o inverso.
Ao comparar a equação anterior com a última da pág.9 conclui-se que
.
Kp é uma medida da ligação relativa de um fármaco entre o plasma e tecidos.
Embora equivalentes a vantagem desta é que permite a interpretação da
distribuição de fármacos entre fármacos, bem como quando a ligação é alterada para um
composto específico.
Porque é que os volumes de distribuição de medicamentos básicos são muito maiores do que
dos ácidos?
A diferença não pode ser atribuída às diferenças nas ligações plasmáticas.
Exemplos: A furosemida (ácido) tem de 0,02 e volume de distribuição de 10 L e a
amiodarona (base) tem de 0,04 e um volume de distribuição de 7000 L.
A diferença é que o fármaco ácido deve encontra-se muito
mais ligado ao tecido que a base (fuT baixo)
O gráfico mostra as grandes diferenças de V entre indivíduos
aos quais lhes foi administrado propranolol.
A relação linear entre V e não só indica que VT / fuT é
relativamente constante (e, como o teor de água no corpo
humano não varia muito entre os indivíduos, a ligação ao
tecido é constante), mas também que as diferenças de ligação de propranolol no plasma entre
os indivíduos são responsáveis pela maior parte da variação observada no seu volume de
distribuição.
Por causa da ligação ao plasma e tecidos, o V raramente corresponde a um espaço fisiológico
bem definido.
Excepção: fármacos de baixo peso molecular que se ligam altamente às proteínas
plasmáticas, mas não aos tecidos, i.e., o fármaco é restrito ao plasma → os volumes de
distribuição (aparente e real) são o mesmo (cerca de 3 L num adulto).
No entanto, esse volume aparente não pode ser um valor de equilíbrio, porque as
proteínas do plasma equilibram-se lentamente entre o plasma e outros fluidos extracelulares.
O volume aparente de proteínas do plasma (cerca de 7,5 L para a albumina) é, talvez, a melhor
estimativa do valor mínimo para tais fármacos.
Fármacos de alto peso molecular (maior que 70.000 g/mol) → distribuição extravascular é
muito lenta (praticamente inexistente) O volume de distribuição tende a aproximar-se do
plasma, 3L.

Farmacocinética – UP1
65
Fármacos muito polares → não se ligam a tecidos ou plasma (ex: cafeína e álcool) O volume
de distribuição varia entre o volume do fluido extracelular (16L) e a água total do corpo (42 L),
dependendo do grau em que o fármaco ganha acesso aos fluidos intracelulares.
Quantidade no corpo e concentração livre
Para muitos fármacos o volume de distribuição é superior a 50 L → implica que apenas uma
pequena fracção de fármaco no organismo permanece no plasma.
Portanto, ignorando o primeiro termo da última equação, Vp (3L) e sabendo que
segue-se:
Esta equação indica que a concentração não ligada (Cu) é independente da ligação plasmática
para uma determinada quantidade de fármaco no corpo e que a quantidade no organismo é
melhor reflectida por Cu que C quando a ligação às proteínas plasmáticas é alterada.
2.3. Estudar a cinética de eliminação dos fármacos; 2.3.1 – Identificar os principais processos
de eliminação
Excreção
Para a maioria dos fármacos a excreção é renal. Alguns fármacos são excretados na bílis,
outros através da respiração (substâncias voláteis). Outras vias: fezes, lágrimas, saliva, suor,
leite…
Metabolismo
Frequentemente, um fármaco sofre metabolização por várias vias metabólicas e a
percentagem dos metabolitos resultantes depende das velocidades relativas de cada via.
Ocorrem reacções em série ->reacções
sequenciais.
Geralmente, o fígado é o principal local (e por
vezes único) onde ocorre o metabolismo.
Ocasionalmente, o fármaco pode ser
extensamente metabolizado num ou mais
tecidos como os rins, pulmões, sangue e parede
GI.
As vias metabólicas são classificadas pelas
alterações químicas. A maioria das
transformações ocorre no RE das células do fígado e outros tecidos
Quando se homogeneízam estes tecidos, o RE é destruído levando à formação de pequenas
vesículas chamadas microssomas. Por esta razão, as enzimas metabolizadoras do RE são
chamadas de enzimas microssomais.

Farmacocinética – UP1
66
O metabolismo dos fármacos é então classificado como: Microssomal vs.Não-microssomal
As principais enzimas responsáveis pela oxidação e redução de
muitos fármacos pertencem à superfamília das enzimas do
CYP450. Esta superfamília divide-se em 3 famílias:
o CYP(450) 1
o CYP(450)2
o CYP (450) 3
Estas são posteriormente subdivididas em subfamílias: A-E
Os números referem-se às enzimas individuais da subfamília.
As enzimas de cada família demonstram um alto nível de
especificidade estrutural.
o Um fármaco é bom substrato para uma enzima mas não para
outra
A família mais abundante é a CYP3A4
o Metaboliza muitos fármacos com estrutura e tamanhos diversos
o Encontrada na parede intestinal e fígado
2.3.2 – Aprofundar o conhecimento sobre os parâmetros de eliminação (clearance, clearance
hepática, clearance renal, clearance total). Estudar algumas considerações relativas à
excreção biliar e renal de fármacos.
Clearance e Eliminação
A clearance permite calcular a velocidade de eliminação para qualquer concentração
plasmática. É definida como o factor de propocionalidade que relata a velocidade da
eliminação do fármaco no plasma.
CCLTaxa inaçãoE lim
Alternativamente, a clearance pode ser vista como a perda de
fármaco do órgão por eliminação: QECL ,
onde Q é a velocidade de fluxo e E o rácio de extracção.
Também é possível calcular a quantidade eliminada durante o intervalo de
tempo (dt): CL x C x dt
Multiplicando a área sobre a curva do gráfico num dado tempo [AUC (0,t)] pela Clearance (CL),
dá-nos a quantidade de fármaco que foi eliminada até esse instante.
Quando a área é expressa como uma fracção da AUC total, obtém-se a fracção da dose
eliminada.

Farmacocinética – UP1
67
Excreção Renal como uma Fracção de Eliminação Total
A fracção inalterada excretada de uma dose IV (fe = quantidade inalterada excretada total) é
um importante parâmetro farmacocinético, sendo uma medida quantitativa da contribuição
de excreção renal para a eliminação total do fármaco.
o O conhecimento de fe ajuda a estabelecer modificações apropriadas no regime de dosagem
habitual de um fármaco para pacientes com variados graus de função renal.
O valor de fe para os fármacos varia entre 0 e 1,0.
Fármacos altamente lipofílicos apresentam valores baixos, pois a excreção é uma via
minoritária de eliminação do fármaco.
Uma estimativa de fe é mais facilmente obtida a partir de dados de excreção urinária
cumulativa após administração IV.
Aditividade da Clearance
Velocidade de Eliminação = velocidade de excreção renal + velocidade de metabolismo
hepático-> CLtotal= CLR + CLH
A clearance pode ser determinada experimentalmente a partir de dados de excreção urinária e
da clearance total.
Clearance Renal = fe x CL
A clearance hepática pode ser facilmente estimada como a diferença entre a clearance total
e a clearance renal: CLH = (1-fe) x CL
Velocidade de Eliminação = CL x C
Ou, a clearance perda de fármaco através de eliminação por um órgão, resultando a
relação:
Clearance (CL) = Q x E
Q= velocidade de fluxo; E= rácio de extracção
Esta abordagem fisiológica tem vantagens na previsão e na avaliação dos efeitos de alterações
no fluxo sanguíneo, na ligação a proteínas plasmáticas, na actividade enzimática e secretora e
na eliminação de um fármaco
Quando se calcula o rácio de extracção de um órgão, é importante relacionar a clearance
sanguínea (CLb) com o fluxo sanguíneo e não a clearance plasmática com o fluxo plasmático
CLb = Q x E
CLb= clearance sanguínea; Q= fluxo sanguíneo; E= rácio de extracção
As consequências do metabolismo dos fármacos são variadas. A biotransformação
constitui um mecanismo de eliminação de compostos indesejados e fármacos.

Farmacocinética – UP1
68
Se o rácio de extracção se aproximar de 1,0, a clearance sanguínea aproxima-se de um
máximo
A clearance sanguínea também se pode dar por:
b
bC
CCLCL
C= [fármaco] no plasma; Cb= [fármaco] no sangue
Clearance Hepática
Clearance sanguínea hepática = QH x E
QH= fluxo sanguíneo hepático; EH= rácio de extracção hepática
Rácio de Extracção Elevado: quando o rácio de extracção de um fármaco
se aproxima de 1,0 (EH>0,7). A sua clearance sanguínea hepática
aproxima-se do fluxo sanguíneo hepático.
Rácio de Extracção Baixo: a eliminação de um fármaco com um baixo
rácio de extracção (EH<0,3) tem que ter velocidade limitada.
Quando o EH é baixo espera-se que a clearance seja influenciada por
alterações na ligação a proteínas plasmáticas.
Excreção Biliar e Circulação Entero-Hepática
Fármacos com baixo peso molecular são encontrados na bílis enquanto os
fármacos proteicos são excluídos. Entra no intestino delgado através do
ducto biliar comum após armazenamento na vesícula biliar. Podem ser
reabsorvidos no intestino delgado para completar a circulação entero-
hepática->É um componente da distribuição e não da eliminação.
Se não for reabsorvido, a circulação entero-hepática pertence à
eliminação. Qualquer fármaco que não seja reabsorvido (directa ou
indirectamente como metabolito) é excretado nas fezes.
Para alguns fármacos esta é a maior via de eliminação quando são
administrados IV.
Algumas generalizações podem ser feitas olhando às características do fármaco necessárias
para assegurar a elevada clearance biliar:
1. O fármaco deve ser activamente secretado.
2. Deve ser polar e o PM deve exceder as 350 g/mol.
Estes argumentos não auxiliam na predição da natureza e especificidade dos mecanismos
secretórios mas auxiliam na predição da possibilidade de uma elevada clearance biliar.

Farmacocinética – UP1
69
Exemplo:Conjugados com glucuronidos de fármacos polares, ionizados, pKa ~ 3 com PM > 350
g/mol são muitas vezes altamente eliminados na bílis.
Clearance Renal
Embora os princípios gerais que regulam a clearance renal sejam os mesmos da hepática, os
mecanismos envolvidos e a importância de cada uma delas são diferentes.
A unidade anatómica e funcional básica da função renal é o nefrónio.
Constituição: Glomérulo; túbulo proximal; ansa de Henle; túbulo distal; túbulo colector
O glomérulo recebe e filtra cerca de 120 mL de plasma por minuto, num paciente típico de
20 anos. O filtrado passa através do túbulo, onde a maior parte da água é reabsorvida; apenas
1 - 2 mL/min abandona os rins na forma de urina.
A presença de fármacos na urina resulta dos processos de filtração, secreção e
reabsorção. Os primeiros dois processos adicionam fármaco à parte proximal do lúmen do
nefrónio; o último processo envolve o movimento de retorno do fármaco do lúmen para a
corrente sanguínea. A velocidade de excreção é, portanto:
[ ]
em que FR é a fracção reabsorvida do lúmen.
O fármaco entra no rim pela artéria renal e abandona-o parcialmente através da
veia renal, sendo a restante parte excretada na urina. A excreção urinária (4) é o
efeito de rede da filtração glomerular de fármaco não ligado (1), da secreção
tubular (2), processos estes que adicionam fármaco na parte proximal do lúmen
do túbulo, e da reabsorção de fármaco do lúmen distal para a corrente
sanguínea (3).
Filtração glomerular
Cerca de 20-25% do output cardíaco, ou seja, 1.1 L de sangue/minuto entra nos
rins. Deste volume, cerca de 10% é filtrado no glomérulo por pressão hidráulica
exercida pelo sangue arterial.
Apenas proteínas com peso molecular < 2000 g/mol são filtradas, dependendo a filtração
também da carga e da forma. Geralmente, apenas os fármacos que não se encontram ligados
a proteínas plasmáticas são filtrados.
A velocidade à qual o plasma é filtrado chama-se taxa de filtração glomerular (TFG):
Taxa de filtração = TFG.Cu
Sendo Cu a concentração de fármaco livre (não ligado a proteínas) no plasma.
Tendo em conta que fu é o ratio da concentração de fármaco não ligado/concentração total de
fármaco, vem:

Farmacocinética – UP1
70
Taxa de filtração = fu.TFG.C
Se um fármaco for apenas filtrado e todo o fármaco filtrado for excretado na urina, então
velocidade de excreção = taxa de filtração. Desse modo, a clearance renal é, por definição:
Para o referido fármaco, a clearance renal (por filtração) será fu.TFG
O ratio de extracção de tal fármaco é baixo. Por exemplo, mesmo que um fármaco se
encontre totalmente livre no sangue (Cu = Cb), o ratio de extracção é apenas 0.11. Isto porque:
A creatinina (composto produzido endogenamente) não se liga a proteínas plasmáticas nem é
secretada, sendo todo o conteúdo filtrado excretado na urina. Portanto, a sua clearance renal
é uma medida da TFG. Sob condições normais, a TFG é relativamente estável, não sendo
sensível a alterações no fluxo sanguíneo renal.
Secreção Activa
A secreção (tubo proximal) e o transporte activo do sangue para o lúmen do nefrónio facilitam
a extracção, apesar de não terem um elevado grau de especificidade. Existem mecanismos
para secreção de ácidos (aniões) e de bases (catiões).
A secreção é inferida quando a velocidade de excreção (CLR.C) > taxa de filtração (fu.TFG.C).
A secreção é aparente quando CLR > fu.TFG
Alguns fármacos são tão bons substratos para o sistema secretor que são completamente
removidos do sangue durante o tempo em que estão em contacto com o local de transporte
activo, mesmo quando se encontram ligados a proteínas plasmáticas ou localizados em células
sanguíneas.
O ácido para-aminohipúrico (PAH) é removido deste modo, não sendo reabsorvido.
O ratio de extracção do PAH é, portanto, próximo de 1.0.
Este composto existe apenas no plasma.
Por isso, a sua clearance plasmática renal é uma medida do fluxo plasmático renal, utilizada
clinicamente.
De acordo com estas circunstâncias, a clearance é limitada pela velocidade de perfusão, não
sendo sensível a alterações na ligação às proteínas plasmáticas.
Reabsorção
A reabsorção tubular tem de ocorrer se a clearance renal for menor que a clearance por
filtração calculada (CLR < fu.TFG). Pode ocorrer ainda alguma secreção, mas a sua contribuição
é menor que a da reabsorção.

Farmacocinética – UP1
71
A reabsorção varia de quase ausente a virtualmente completa.
A reabsorção activa ocorre para muitos compostos endógenos, incluindo as vitaminas B e C,
electrólitos, glucose e aminoácidos. Porém, para a maioria dos fármacos a reabsorção ocorre
por difusão passiva.
A força motora para a reabsorção é a reabsorção extensa da água filtrada (120 mL/min no
glomérulo; 1-2 mL/min na urina formada), que tende a concentrar fármaco no lúmen do
nefrónio.
A reabsorção extensa é comumente observada em fármacos lipofílicos, que atravessam a
membrana luminal de retorno ao sangue, perfundindo o nefrónio. Por isso, as suas clearances
renais são muito baixas.
Tratamento renal de fármacos proteicos
Apesar de a insulina ser filtrada no glomérulo, são excretadas pequenas quantidades na urina.
Isto deve-se ao facto de este e muitos outros fármacos protéicos serem completamente
metabolizados virtualmente nos aminoácidos constituintes, quer por enzimas localizadas na
fronteira do lúmen ou, após endocitose, dentro das células luminais.
Tudo indica que o rim é o principal órgão de eliminação dos fármacos proteicos.