intemperismo fotoquímico do petróleo colombiano · a composição de petróleo é extremamente...
TRANSCRIPT

. DEPARTAMENTO DE QUÍMICA – CENTRO DE CIÊNCIAS EXATAS
Intemperismo Fotoquímico do Petróleo Colombiano
Gislaine M. Bragagnolo
LONDRINA
2003

RESUMO
Este trabalho tem o intuito de analisar o intemperismo do petróleo
colombiano.
Várias amostras irradiadas e não irradiadas (branco) do óleo e da água foram
irradiadas ao Sol por várias horas e, posteriormente analisadas por espectrofluorimetria para
verificar se ocorreu alguma mudança nas fases.
A fase aquosa além de ter sido irradiada e não irradiada, ainda foi tratada
com solução de peróxido de hidrogênio e suspensão de dióxido de titânio para verificar se
esses oxidantes são capazes de oxidar componentes do petróleo fazendo com que a
intensidade de fluorescência na água diminua.

I. INTRODUÇÃO
I.1 Composição de petróleo
A composição de petróleo é extremamente complexa e varia de acordo com
o tipo de petróleo (NAS, 1985). Essa composição pode mudar com o tempo, mesmo quando a
amostra é retirada de um mesmo poço.
Petróleo é dividido em frações de acordo com a solubilidade, ponto de
ebulição e propriedades cromatográficas em sílica gel. Em termos químicos, o óleo bruto é
dividido em frações alifáticas, frações aromáticas, frações polares e frações asfaltênicas (Gill
et al, 1989,).
As frações alifáticas predominantemente apresentam hidrocarbonetos
saturados de cadeia normal além de muitos ciclos e policiclos (naftenos). As frações
aromáticas apresentam hidrocarbonetos aromáticos alquilados com um a cinco anéis
aromáticos conjugados. A fração polar contém muitos aromáticos heterocíclicos que pode
incluir derivados de porfirina e compostos alifáticos contendo nitrogênio e enxofre, além de
apresentar traços de grupos como álcool, fenol, cetona e ácido carboxílico. A identificação de
ácido carboxílico pode indicar o ano de formação do óleo (Seifert, 1973). A fração asfaltênica
é definida como a fração insolúvel em pentano ou heptano. Essa fração pode variar de
petróleo para petróleo e até mesmo pode estar ausente (Nicodem et al, 2001).
Os aromáticos, nafteno-aromáticos e aromáticos com enxofre (tiofenos) ou
derivados são os principais componentes do óleo bruto responsáveis pelos principais danos ao
meio ambiente e a saúde humana. Esses componentes são usualmente a maior constituição de
frações de hidrocarbonetos com alto ponto de ebulição. Eles incluem um ou vários anéis
aromáticos condensados os quais se fundem com anéis naftênicos e ramificações. As
estruturas mais freqüentes estão representadas na tabela 1.

Tabela 1 – Principais tipos de hidrocarbonetos do óleo bruto ( Tissot et al, 1984)
Fórmula
molecular
Mono-
aromáticos
Di-aromáticos Tri-aromáticos Moléculas
aromáticas com
enxofre
CnH2n-6 R
S
R
CnH2n-10S
CnH2n-8 R
S
R
CnH2n-12S
CnH2n-10 R
S
R
CnH2n-14S
CnH2n-12 R
R
S
R
CnH2n-16S
CnH2n-14 RR
S
R
CnH2n-18S
CnH2n-16 R
S
R
CnH2n-20S
CnH2n-18 RR
S CnH2n-22S
CnH2n-20
S CnH2n-24S

I.2 Intemperismo sofrido pelo óleo
O destino do óleo derramado em ambientes marinhos depende de uma série
de processos físicos, químicos e biológicos, denominados de intemperismo.
Quando petróleo é derramado em águas naturais os processos físicos de
intemperismo iniciam-se rapidamente e a degradação biológica é lenta devido à toxicidade de
seus componentes e, algumas vezes, pela falta de nutrientes. As transformações químicas
acontecem principalmente devido a processos fotoquímicos, os quais são significativos na
oxidação dos componentes recalcitrantes do óleo (nicodem et al, 1997).
O intemperismo fotoquímico ocorre por reações com oxigênio singlete e
radicais livres (Guedes et al, 1998). Produtos da fotooxidação do óleo são foto-reativos em
água, ocorrendo à degradação através de transferência de elétrons e formação de radicais
livres (Nicodem et al, 2001).
Os resultados do intemperismo de petróleo são razoavelmente conhecidos
como resultado de análises ambientais, mas os processos pelos quais ocorrem as alterações
não são bem definidos.

Processos físicos
-evaporação
-lavagem
-dissolução
-“mousse”
-deposição
-sedimentação
⇔
Processos químicos
-destruição de
aromáticos
-formação de
surfactantes
-produtos solúveis em
água
-oxigenação
⇔
Processos biológicos
-mineralização
-oxidação parcial
-metabólitos
Figura 1 Interação entre os processos de intemperismo do petróleo
No início a evaporação aumenta a viscosidade e a densidade do petróleo e
ainda inicia-se a formação de emulsões água em óleo, conhecidas como “mousse" de
chocolate “(Nicodem, 1998).
Processos fotoquímicos modificam as propriedades físicas, a composição do
óleo, e a solubilidade. O aumento na solubilidade afeta a toxicidade e a biodegradação, além
de permitir a fotodegradação na fase aquosa.
A tabela 2 mostra vários hidrocarbonetos aromáticos que são considerados
tóxicos tanto ao meio ambiente quanto ao seres humanos, neste caso eles são considerados
carcinogênicos e eles são encontrados em quantidades variadas no petróleo e são controlados
pela Agência de Proteção Ambiental dos EUA e pela Associação de Saúde (Bedding et al,
1995 e Kumke et al, 1995).
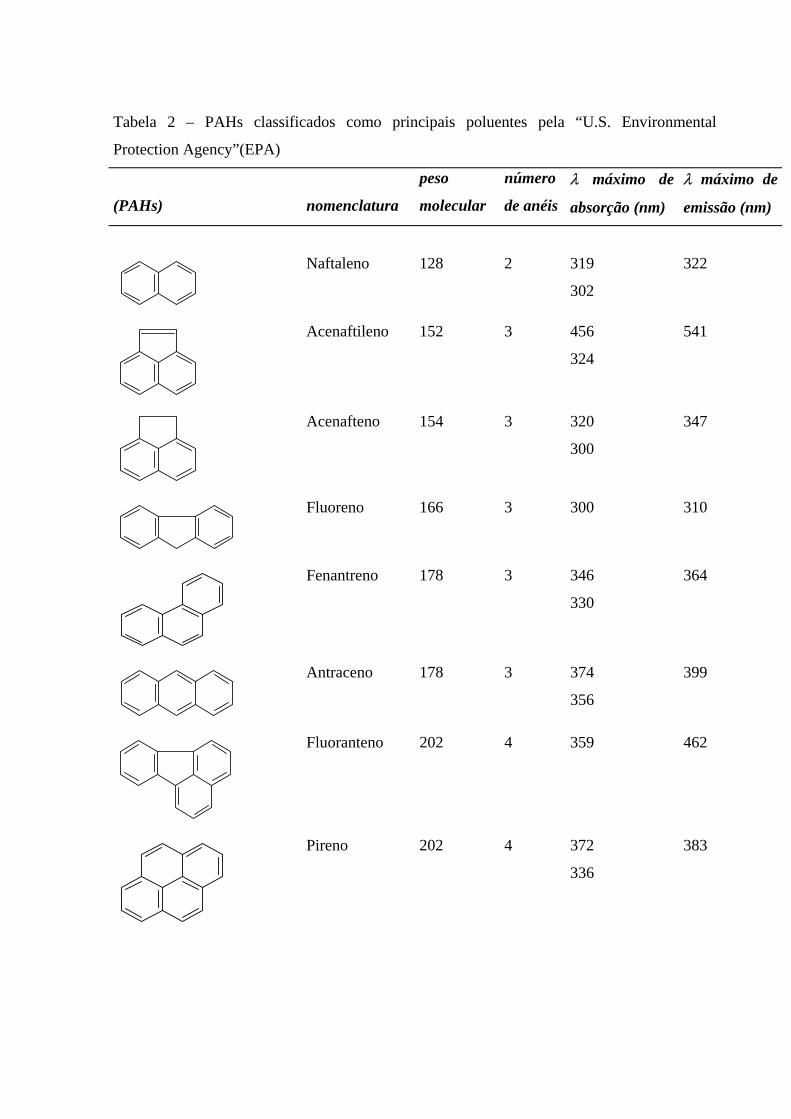
Tabela 2 – PAHs classificados como principais poluentes pela “U.S. Environmental
Protection Agency”(EPA)
(PAHs)
nomenclatura
peso
molecular
número
de anéis
λ máximo de
absorção (nm)
λ máximo de
emissão (nm)
Naftaleno 128 2 319
302
322
Acenaftileno 152 3 456
324
541
Acenafteno 154 3 320
300
347
Fluoreno 166 3 300 310
Fenantreno 178 3 346
330
364
Antraceno 178 3 374
356
399
Fluoranteno 202 4 359 462
Pireno 202 4 372
336
383

(PAHs)
nomenclatura
peso
molecular
número
de anéis
λ máximo de
absorção (nm)
λ máximo de
emissão (nm)
Benz[a]
antraceno
228 4 385
300
385
Criseno 228 4 362
321
381
Benzo[k]
fluoranteno
252 5 402
308
402
Benzo[b]
fluoranteno
252 5 369
302
446
Benzo[a]pireno 252 5 404
385
403
Benzo[g,h,i]
perileno
276 6 406
300
419
Indeno
[1,2,3-cd]
pireno
276 6 460
302
503

(PAHs)
nomenclatura
peso
molecular
número
de anéis
λ máximo de
absorção (nm)
λ máximo de
emissão (nm)
Dibenz[a,h]
antraceno
278 5 394
322
394
Fonte: Guedes, 1998
I.3 Mecanismos de fotodegradação do óleo
Os mecanismos abaixo representados pela figura 2 (Correa, 1997), referem-
se a transferência de energia de estados eletronicamente excitados (triplete) de componentes
aromáticos e polares para oxigênio molecular gerando oxigênio singlete (Gorman, 1992), os
quais podem interagir com compostos aromáticos e heterocíclicos com enxofre por adição.
(a)
singlete (esta-do excitado)
triplete (esta-do excitado)
S0 S1 T1
singlete (esta-do fundamental)

(b)
O OO2
*1
(c)
S S OO*2
1
(d)
Figura 2: Figura (a) representação do estado fundamental e excitado do oxigênio Figuras (b) e
(b) representam o mecanismo da reatividade de oxigênio singlete, figura (d) mecanismo de
fotodimerização de HPA
Muitos trabalhos publicados sobre fotoquímica dos filmes de óleo estão
preocupados com as alterações físicas e químicas do óleo observado. Embora exista
especulação do processo fotoquímico envolvido, poucos trabalhos tem sido realizados.
Os resultados são muito complicados porque diferentes técnicas são
aplicadas, sendo assim, resultando de conclusões freqüentemente divergentes (Nicodem et al,
1998).
I.4 Propriedades físicas do óleo

As mudanças físicas mais importantes que estão relacionadas com o
impacto ambiental são: evaporação, difusão, emulsificação e dissolução. As transformações
químicas afetam todos esses processos físicos.
Quando o óleo é derramado na superfície da água, a ação dos ventos e o
bater das ondas misturam e dispersam o óleo na água. Há emulsões água em óleo as quais
podem conter até 90% de água (Thingstas & Pengerud, 1983) que se tornam completamente
estáveis. Essas emulsões são denominadas “mousse de chocolate” devido a sua aparência.
A formação do “mousse” é um processo importante no intemperismo de
petróleo porque a emulsão água-óleo é altamente viscosa e fica aderida em areias e rochas. É
muito difícil a remoção e o tratamento do “mousse”, por isso ele aumenta o impacto
ambiental. Sua formação esta relacionada com a formação de material polar de alto peso
molecular, e pode ser inibido por β-caroteno que é um bom supressor de oxigênio singlete
(Thingstad & Pengerud, 1983).
A capacidade do petróleo de não sofrer intemperismo para formar emulsões
água-óleo depende da concentração de frações asfaltênicas (Mackay et al, 1973).
I.5 Solubilidade e fotodegradação em água
A irradiação do óleo pode aumentar a solubilidade do material em água a
qual se torna tóxica para muitos organismos aquáticos e seres humanos. O aumento na
toxicidade se deve a formação de derivados polares de todas as frações de petróleo como, por
exemplo, hidroperóxidos, ácidos, fenóis dentre outros.
Produtos de petróleo refinado, tais como gasolina, óleo diesel e outros
combustíveis, são mais solúveis em água (Zürcher & Thuer, 1978). Essa solubilidade esta

relacionada com os componentes de baixo peso molecular, por exemplo, fração aromática
monocíclica do petróleo. Esses compostos tem grande capacidade de serem volatilizados e
rapidamente perdidos à atmosfera.
Processos fotoquímicos são importantes na química de sistemas aquáticos
(Cooper & Herr, 1987), principalmente, quando inclui formação de oxigênio singlete (Zepp et
al, 1977).
A fração solúvel em água de petróleo bruto da Nigéria foi fotodegradado por
luz do Sol com perda preferencial para componentes com maior número de substituintes
“alquil” (Ehrhardt et al, 1992). Os processos fotoquímicos podem ser tão importantes quanto
processos biológicos para degradação de hidrocarbonetos em águas naturais.
Hidrocarbonetos aromáticos policíclicos, diluídos em solução aquosa, foram
destruídos quando irradiados à luz do Sol (Paalme et al, 1990).
I.6 Degradação fotocatalítica
Métodos físico-químicos como troca iônica, a adsorção em carvão ativado e
“air stripping” são empregados para a remoção de contaminantes orgânicos da água. Embora
essas técnicas sejam eficientes no processo de descontaminação, elas apenas envolvem a
transferência de fase do contaminante, sem necessariamente destruí-lo.
Os chamados Processos Oxidativos Avançados (POAs) são capazes de
converter poluentes em espécies químicas inócuas, tais como gás carbônico e água. O termo
POA é usado para definir o processo em que radicais hidroxila (•OH) são gerados para atuar
como agentes oxidantes químicos (Jardim et al, 1995).
O processo é baseado na irradiação de um fotocatalisador, que pode ser um
semicondutor inorgânico como TiO2, ZnO ou CdS, cuja energia do fóton deve ser maior ou

igual à energia do “band gap” do semicondutor para provocar uma transição eletrônica. Com
isso forma-se os sítios oxidantes e redutores capazes de catalisar reações químicas, oxidando
os compostos orgânicos à CO2 e H2O e reduzindo metais dissolvidos ou outras espécies
presentes.
Os POAs podem ser classificados em dois grupos; os que envolvem reações
homogêneas, usando H2O2, O3 e/ou luz, e os que empregam reações heterogêneas, usando
catalisadores. A fotocatálise heterogênea é um processo recente que visa à destruição de
contaminantes orgânicos e inorgânicos presentes em águas e efluentes (Jardim et al, 1995).
I.6.1 Fotocatálise heterogênea
A fotocatálise heterogênea implica no emprego de suspensões de
catalisador, também chamado lama. Nesse caso, após a espécie orgânica sofrer oxidação
segue-se uma série de etapas de separação e recuperação do catalisador.
Em trabalhos que utilizam catalisadores ultrafinos, como o TiO2, essas
etapas podem ser demoradas devido consumir tempo. Um meio alternativo é imobilizar o
catalisador em um suporte estacionário, por onde a água contaminada escoa em contato com
ele. Esse meio torna o método mais barato do que as tecnologias convencionais usadas no
tratamento de águas contaminadas. São usados também suportes como: areia, pequenas
esferas de vidro, placas de vidro, fibra de vidro, teflon, membranas poliméricas e cerâmicas.
A maior desvantagem do uso de fotocatalisadores imobilizados é que eles
limitam a trasferência de massa e a vantagem é de poder fazer uso da energia solar como fonte
de irradiação (Ziolli et al, 1998).
I.6.1.2 Mecanismo geral das reações mediadas por semicondutores

Equações 1 A adsoção na partícula do catalisador
RRsítios
HOOHTiOOHTi
OHTiOHTi
LIV
LIV
IVIV
→+
+→++
→+
−−−
−
−
1
22
22
Equação 2 Excitação do semicondutor
+− +→+ BVBC hehvTiO2
Equações 3 Manutenção das cargas
( )
( )
IIIBC
IV
IIIBC
IV
IVBV
IV
IVBV
IV
TieTi
OHTieOHTi
OHTihOHTi
HOHTihOHTi
→+
→+
→+
+→+
−
−−•
−
•+−−
+•+− 2
Equações 4 Recombinação das cargas

( )
OHTiOHTih
OHTiOHTie
Etérmicahe
IVIIIBV
IVIVBV
BVBc
−−+
−•−
+−
→+
→+
→+
onde:
R1 é substrato;
h+ lacuna fotogerada; −e = elétron fotogerado;
BV= banda de valência do semicondutor;
BC= banda de condução do semicondutor; −2
LO = oxigênio do retículo do TiO2
Para melhor entendimento do mecanismo da reação do TiO2 em fotoquímica
de compostos orgânicos é preciso compreender três mecanismos distintos que podem ser
estudados pela química do estado sólido, química de interface e química de solução. A idéia é
iniciar pelo semicondutor puro e isolado, para chegar aos produtos finais de uma
fotodegradação completa, ou seja, CO2 e H2O (Ziolli et al, 1998).
O TiO2 é o semicondutor mais usado em fotocatálise, e por esta razão,
várias propriedades já foram exaustivamente estudadas (Hoffmann et al, 1995), (Fox et al,
1993) e (Linsebigler et al, 1995).
I.6.1.3 Mecanismos envolvendo h+ e •OH
Vem sendo estudado o envolvimento de espécies como radical hidroxila,
lacuna fotogerada, oxigênio singlete e íon-radical superóxido em transformações
fotocatalíticas com ZnO em meio aquoso (Jardim et al, 1998).

Oxigênio singlete pode formar-se em fotocatálise de acordo com o
mecanismo da equação 5.
Equação 5 Formação de oxigênio singlete
−•
−→+ 22 OTiOTi IVIII
21
2 OhOTi IV →+ +−•−
onde,
21O : oxigênio singlete
A reatividade química do oxigênio singlete tem sido exaustivamente
estudada. Ele pode interagir com substratos para gerar peróxidos, os quais podem iniciar um
processo radicalar em cadeia como mostra a equação 6.
Equação 6 Oxidação envolvendo 1 O2*
produtosROOR
RROOHRHROO
ROOOR
ROHRHOH
RAOHRHAO
OHAOAOOH
AOOHAHO
→
+→+
→+
+→+
+→+
+→
→+
••
••
••
••
••
••
,
23
2
21

I.6.2 Fotocatálise homogênea
Reações homogêneas que usam H2O2, O3 e/ou luz UV como catalisadores.
Ozônio é um gás incolor e se decompõe rapidamente a oxigênio e espécies
radicalares. O ozônio é um agente oxidante poderoso (E0= 2,08V) quando comparado a outros
agentes oxidantes conhecidos como o peróxido de hidrogênio (E0= 1,78V), permitindo que
esta espécie reaja com um grande número de classe de compostos (Masten et al, 1994).
O peróxido de hidrogênio pode se decompor em radical hidroxila que é um
poderoso e não seletivo oxidante (E0= 2,80V) podendo reagir pelo mecanismo da equação 8.
Equação 7 Reações característica de H2O2 em águas naturais
HOOH ou ROOH + λν = 2 •OH ou RO• + •OH
HOOH ou ROOH + Me(red) = Me(ox) + RO• ou •OH + -OH
OHOH •→222 (sob luz e aquecimento)
Equação 8 Reação iniciada pelo radical hidroxila (Kunz, et al, 1999)
+ OH
H
OH

Este trabalho tem como objetivo estudar a degradação de compostos
orgânicos do petróleo em águas naturais utilizando a luz do Sol como fator principal.

II PARTE EXPERIMENTAL
II.1 Reagentes
• Água destilada;
• Diclorometano procedente da Nuclear, grau PA, PM 84,93 e 99,5% de
pureza;
• Petróleo Colombiano fornecido pela REPAR-PETROBRAS e utilizado
sem nenhum tratamento;
• Petróleo mistura (Marlin, Bacia de Campos-70% e Beinngton, Nigéria-
30%), fornecido pela REPAR-PETROBRAS e utilizado sem nenhum
tratamento;
• Peróxido de hidrogênio procedente da Nuclear grau PA, PM 34,01 e
dosagem 32-36,5%;
• Dióxido de titânio, procedente da Degussa AG, pH 3,5-4,5, densidade
aproximadamente 3,8g/cm3, área superficial específica (BET) 50(35-
65)m2/g.
II.2 Materiais e equipamentos
• Balão volumétrico
• Copo de vidro
• Cuba de vidro
• Funil comum

• Funil de separação
• Membrana durapore PUDF 25mm de diâmetro MILLEX GV
• Micropipeta automática
• Papel alumínio
• Papel filtro
• Pipeta Pasteur
• Pipeta volumétrica
• Proveta
• Seringa
• Suporte e garra
• Tubo de ensaio
• Centrífuga FANEM – Excelsa Baby I – modelo 206
• Espectrofluorímetro Shimadzu RF-5301PC
• Geladeira Brastemp
• Infravermelho Shimadzu FTIR-8300
II.3 FILME DE PETRÓLEO SOB LUZ SOLAR
II.3.1 Preparo de amostras
As amostras de petróleo foram preparadas na forma de filmes finos sobre
água destilada. Placas de Petri com diâmetro interno de aproximadamente 9,0cm e com
tampas de vidro “Pyrex” foram utilizadas como recipientes de irradiação. Filmes de petróleo
foram preparados com 5mL do óleo sobre 20mL de água destilada.

II.3.2 Exposição das amostras
Os filmes de petróleo sobre água destilada foram irradiados por exposição à luz
do Sol no pátio do departamento de química da UEL em dias de céu claro no período entre 9 e
15 horas nas estações de primavera e verão de 2000 e 2001. As amostras foram estocadas no
escuro em geladeira a ± 18ºC entre as irradiações, até que o número total de horas fosse
acumulado. Para cada amostra irradiada foi preparada uma outra não irradiada (branco) da
mesma maneira, exceto que as tampas de vidro “Pyrex” foram pintadas com tinta preta para
eliminar a irradiação.
II.3.3 Extração do filme
Após irradiação, as amostras foram transferidas para tubos de ensaio, com
auxílio de um funil de vidro. Em seguida foram centrifugadas durante 6 minutos em
velocidade média. A fase óleo foi coletada com pipeta Pasteur e a fase aquosa filtrada em
papel de filtro. Ambas as fases foram guardadas no escuro em geladeira a ± 18ºC enquanto
aguardavam as análises.
II.3.4 ANÁLISE DO FILME DE PETRÓLEO

II.3.4.1 Emissão por fluorescência
Espectros de fluorescência do óleo com excitação iniciando em 280nm e
emissão na faixa de 300 a 800nm foram, registrados em diclorometano na diluição de 1:1000
v/v em um espectrofluorímetro Shimadzu RF–5301PC. As análises foram registradas na
modalidade “synchronous”, sincronismo de 20nm entre os monocromadores de excitação e
emissão, “sampling interval” de 1,0nm e “scanning speed fast”.
Os espectros de fluorescência foram registrados tanto das amostras
irradiadas quanto das amostras não irradiadas após 0h, 2h, 5h, 15h, 20h, 42h, 60h e 100 horas
de exposição solar.
Dois tipos de células de sílica fundida (quartzo) foram utilizadas para análise:
célula quadrangular e triangular.
II.3.4.2 Absorção no Infravermelho
Espectros de transmitância na região do infravermelho foram obtidos
usando um Shimadzu FTIR-8300 a partir de amostras de petróleo depositadas como filmes
entre placas de NaCl. Espectros na região entre 400 a 4000cm-1 foram registrados para
amostras de petróleo com 0h, 2,0h, 5,0h, 15h, 20h, 42h, 60h e 100 horas de exposição ao Sol.

II.4 ANÁLISE DA FASE AQUOSA
II.4.1 Emissão por fluorescência
Espectros de fluorescência da fase aquosa, com excitação iniciando em
230nm e emissão na faixa de 250 a 650nm foram registrados, sem qualquer diluição ou
concentração, em um espectrofluorímetro Shimadzu RF-5301PC. As análises foram
realizadas na modalidade “synchronous” com sincronismo de 20nm entre os monocromadores
de excitação e emissão, “sampling interval” 1,0nm e “scanning speed fast”.
Todas as amostras de água em contato com petróleo não irradiado e irradiado
por 0h, 2h, 5h, 15h, 20h, 42h, 60h e 100 horas foram analisadas em célula de quartzo
quadrangular.
II.5 ADIÇÃO DE PERÓXIDO DE HIDROGÊNIO EM ÁGUA CONTAMINADA POR PETRÓLEO
II.5.1 Preparo de amostras
As amostras de água contaminada foram preparadas em contato com
petróleo bruto na forma de filme sobre água destilada. Uma cuba de vidro com diâmetro
interno ± 15cm com tampa de vidro “Pyrex” foi utilizada como recipiente de exposição ao

Sol. As amostras ditas “não irradiada” e “irradiadas” foram preparadas com 100mL de óleo
sobre 400mL de água destilada.
II.5.2 Exposição das amostras
Os filmes de petróleo sobre água destilada foram expostos por 40h à luz do
Sol no pátio do departamento de química da UEL, em dias de céu claro no período entre 9 e
15 horas no mês de junho de 2002. As amostras foram estocadas no escuro em geladeira a ±
18ºC até que completasse às 40h de exposição. A cuba e a tampa, contendo a amostra “não
irradiada”, foram pintadas com tinta preta e encapadas com papel alumínio, simulando a
contaminação de água subterrânea. A amostra “irradiada” simula a água superficial,
recebendo à luz do Sol.
II.5.3 Extração de fases
Após 40 horas de exposição as amostras foram transferidas para funis de
separação com auxílio de um funil para sólidos. Esperou-se 1,0h para a separação das fases. A
fase óleo foi retirada e a fase aquosa foi completada ao volume inicial e filtrada em papel
filtro. Ambas as fases foram guardadas no escuro em geladeira a ± 18ºC.
II.5.4 Adição de peróxido de hidrogênio

As amostras foram preparadas utilizando 60mL de água contaminada
(amostra irradiada) e 10µL de peróxido de hidrogênio em um copo de vidro com diâmetro
interno aproximadamente 6,5cm tampado com vidro “Pyrex”. A concentração final de H2O2
em água é 4,9.10-6M.
II.5.5 Exposição da amostra com peróxido de hidrogênio
A água contaminada por petróleo contendo peróxido de hidrogênio foi
exposta ao Sol durante 5 horas no pátio do departamento de química da UEL no dia 03 de
junho de 2002 no período entre 9 e 15 horas.
Alíquotas de 3mL foram retiradas da amostra irradiada e não irradiada com
auxílio de uma seringa nos intervalos de 30, 60, 90, 120, 180, 240 e 300 minutos de
exposição, e logo após analisadas.
II.5.6 Análise por emissão de fluorescência
Espectros de fluorescência foram registrados com excitação iniciando em
230nm e emissão na faixa de 250 a 650nm, sem qualquer diluição ou concentração em um
espectrofluorímetro Shimadzu RF-5301PC. As análises foram feitas na modalidade
“synchronous”, sincronismo de 20nm entre os monocromadores de excitação e emissão,
“sampling interval” de 1,0nm e “scanning speed fast”.

II.6 ADIÇÃO DE DIÓXIDO DE TITÂNIO EM ÁGUA CONTAMINADA POR PETRÓLEO
II.6.1 Preparo de amostras, exposição das amostras e extração de fases
Estes itens seguem a mesma metodologia descrita nos itens II.5.1, II.5.2 e II.5.3.
II.6.2 Adição de dióxido de titânio
Em um balão volumétrico de 100mL foi preparado uma suspensão contendo
aproximadamente 0,20g de TiO2 em água contaminada por petróleo. A amostra foi transferida
para um copo de vidro.
Mais duas amostra foram preparadas com 100mL de água contaminada sem
TiO2: amostra irradiada e não irradiada. Todos os copos foram tampados com vidro “Pyrex”.
A concentração de TiO2 em suspensão é aproximadamente 2,5.10-2M.
II.6.3 Exposição da amostra com TiO2

A água contaminada por petróleo contendo dióxido de titânio foi exposta ao
Sol durante 5 horas no pátio do departamento de química da UEL, no dia 19 de dezembro de
2002 no período entre 10 e 16 horas (horário de verão).
Alíquotas de 3mL foram retiradas da amostra irradiada e não irradiada com
auxílio de uma seringa nos intervalos de 0, 15, 30, 45, 60, 90, 120, 150, 180 e 240 minutos de
exposição, e logo após analisadas.
II.6.4 Análise por emissão de fluorescência
Espectros de fluorescência foram registrados com excitação iniciando em
230nm e emissão na faixa de 250 a 650nm em um espectrofluorímetro Shimadzu RF-5301PC.
As análises foram feitas na modalidade “synchronous”, sincronismo de 20nm entre os
monocromadores de excitação e emissão, “sampling interval”de 1,0nm e “scanning speed
fast”.
As amostras de água contendo TiO2 foram previamente filtradas em um
funil de membrana durapore antes de analisadas no espectrofluorímetro.

III RESULTADOS E DISCUSSÃO
III.1 FILME DE PETRÓLEO SOB LUZ SOLAR NATURAL
III.1.1 CONSIDERAÇÕES GERAIS
O petróleo absorve a luz solar que penetra na atmosfera terrestre, desde o
ultravioleta até o infravermelho próximo, passando por todo o visível (Nicodem et al, 1997).
As tampas dos recipientes de irradiação dos experimentos são de “Pyrex”.
Este tipo de vidro é de uso comum em experimentos utilizando substâncias com considerável
absorção no ultravioleta próximo e visível do espectro solar (Anba-Lurot et al, 1995)
(Lartiges e Garrigues, 1995).
Houve uma preocupação na preparação das amostras de petróleo,
principalmente, para que fosse obtido um filme homogêneo do petróleo. Quando este foi
adicionado sobre a água destilada tomou-se o máximo de cuidado para que o óleo não
entrasse em contato com as paredes do recipiente de irradiação.
As amostras denominadas “não irradiadas” tiveram as tampas dos
recipientes pintadas com tinta preta para evitar o efeito da luz solar, e assim, pudessem
retratar somente o efeito térmico.

III.2 ANÁLISE DA FASE ÓLEO
III.2.1 Emissão por fluorescência
Nas figuras 3 e 4 aparecem espectros de fluorescência do petróleo em
função do tempo de irradiação. Pode-se observar uma notável diminuição da intensidade de
dluorescência, no entanto o decaimento não ocorre de maneira linear por todo o espectro.
0
10
20
30
40
50
60
70
300 350 400 450 500 550 600 650 700 750 800
comprimento de onda (nm)
intensidade
relativa
óleo brutozero hora2h irradiado5h irradiado15h irradiado20h irradiado40h irradiado60h irradiado100h irradiado
Figura 3 – Espectros de fluorescência em célula triangular do petróleo irradiado (1:1000 v/v
óleo/diclorometano)

0
10
20
30
40
50
60
70
300 350 400 450 500 550 600 650 700 750 800
comprimento de onda (nm)
intensidade
relativa
óleo brutozero hora2h irradiado5h irradiado15h irradiado20h irradiado42h irradiado60h irradiado100h irradiado
Figura 4 – Espectros de fluorescência em célula quadrangular do petróleo irradiado (1:1000
v/v óleo/diclorometano)
A diminuição na intensidade de fluorescência do óleo (figura3), logo nas
primeiras 2 horas de irradiação, pode indicar que alguns componentes fluorescentes são muito
reativos e são preferencialmente destruídos. A reação de HPA com oxigênio singlete (figura
2a ) como também, a reação de dimerização de alguns HPA (figura 2c) que reduzem
fluorescência.
A intensa redução da fluorescência (figura3) da fase óleo inicia-se a partir
de 400nm durante a exposição solar e demonstra a degradação fotoquímica da fração polar e
também de asfaltenos do petróleo Colombiano (Nicodem et al, 2001).
0
10
20
30
40
50
60
70
300 350 400 450 500 550 600 650 700 750 800comprimento de onda (nm)
intensidade
relativa
óleo brutozeo hora20h cél. Quadrangular20h cél. Triangular
Figura 5 – Espectros de fluorescência da fase óleo exposta ao sol.
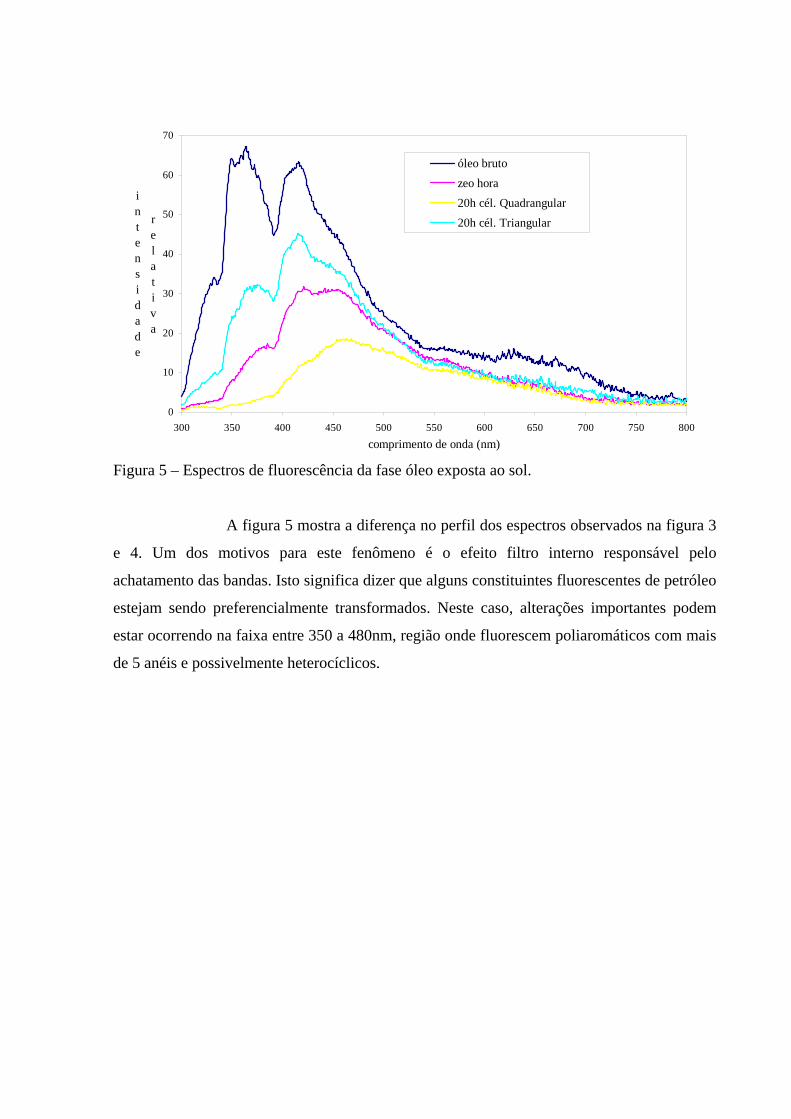
A figura 5 mostra a diferença no perfil dos espectros observados na figura 3
e 4. Um dos motivos para este fenômeno é o efeito filtro interno responsável pelo
achatamento das bandas. Isto significa dizer que alguns constituintes fluorescentes de petróleo
estejam sendo preferencialmente transformados. Neste caso, alterações importantes podem
estar ocorrendo na faixa entre 350 a 480nm, região onde fluorescem poliaromáticos com mais
de 5 anéis e possivelmente heterocíclicos.

0
10
20
30
40
50
60
70
80
90
100
300 350 400 450 500 550 600 650 700 750 800
comprimento de onda (nm)
intensidade relativa
óleo brutozero hora42h irradiado42h não irradiado
Figura - 6 Espectros de fluorescência da fase óleo
Os espectros da figura 6 tem o objetivo de mostrar a diferença de
fluorescência sofrida pelo óleo quando irradiado por 42 horas e óleo não irradiado. Esse
fenômeno se deve ao efeito térmico que pode ser atribuído ao aumento na concentração de
componentes fluorescentes mais pesados do óleo, como conseqüência da volatilização
(processo não fotoquímico) de componentes fluorescentes mais leves, como por exemplo,
mono e di-aromáticos. Nesse caso fica bem evidente que a fotodegradação do óleo é
extremamente dependente das condições as quais lê é submetido.
A figura 7 mostra a degradação de espécies fluorescentes no visível
(416nm), observadas no petróleo correspondendo a emissão das frações polares e asfaltênicas.
A cinética apresentada na figura sugere uma reação de primeira ordem, por apresentar um
decaimento aproximadamente linear com uma constante de velocidade 3 x 10-4min-1.
HPAs ou derivados que fluorescem por volta de 416nm praticamente
desparecem do óleo irradiado pó 60 horas ao Sol.

0 10 20 30 40 50 609,6
9,8
10,0
10,2
10,4
10,6
10,8
11,0
11,2
inte
nsid
ade
rela
tiva
tempo de irradiação
Figura – 7 Cinética de fotodegradação de HPAs do petróleo Colombiano em 416nm

III.2.2 Absorção no infravermelho
400 800 1200 1600 2000 2400 2800 3200 3600 4000
núme ro de onda (1/c m)
400 800 1200 1600 2000 2400 2800 3200 3600 4000
número de onda (1/c m)
C=O OH
Figura 8 – Infravermelho de petróleo não irradiado e irradiado 42 horas respectivamente
A irradiação do filme de petróleo bruto sobre água, em condições naturais e
exclusão de processos biológicos, leva à incorporação de oxigênio ao óleo irradiado na forma
de C=O e OH, detectados por espectroscopia no infravermelho como ilustra a figura 8.
O aumento da banda correspondente aos grupamentos funcionais citados
indicam aparecimentos de fenóis, ácidos carboxílicos e outros, durante a irradiação do óleo.
A banda na faixa de 3400 a 3500cm-1 representa a vibração axial de OH
podendo ser referente a álcool ou fenol. A banda próxima de 1700cm-1 representa a
deformação axial de C=O.

0
10
20
30
40
50
60
70
80
400 1000 1600 2200 2800 3400 4000
número de onda (cm-1)
intensidade
relativa
óleo brutozero horairradiado 2hirradiado 15hirradiado 20hirradiado 42h
Figura 9 – Absorção no infravermelho da fase óleo irradiada A figura 9 mostra os espectros de absorção no infravermelho da fase óleo.
Pode-se observar que a amostra 15 horas irradiada incorporou água próximo de 3400cm-1,
enquanto as amostras 20 e 42 horas irradiadas mostra claramente na mesma região a
incorporação do grupamento OH .
III.3 ANÁLISE DA FASE AQUOSA
III.3.1 EMISSÃO POR FLUORESCÊNCIA
A figura 10 mostra os espectros de fluorescência da fase aquosa irradiada em
função do tempo de exposição do óleo à luz do Sol. Verifica-se que ocorre um aumento na
intensidade de fluorescência nas primeiras horas de exposição devido aos compostos de baixo

peso molecular do óleo estarem migrando para a fase aquosa. Na mesma figura também se
observa deslocamento da emissão para a região visível do espectro, durante o período de
irradiação do óleo.
0
5
10
15
20
25
30
250 300 350 400 450 500
comprimento de onda (nm)
intensidade relativa
irradiado 2hirradiado 5hirradiado 15hirradiado 20hirradiado 42hirradiado 60hirradiado 100hbranco
Figura 10 – Fluorescência em fase aquosa decorrente da irradiação do petróleo à luz do Sol
Tabela 3 – Variação da intensidade de fluorescências na fase aquosa em função do
comprimento de onda e do tempo de irradiação
Comprimento de onda (nm) Tempo de irradiação (h) Intensidade fluorescência
(u.a)
0 8,089
2,0 12,308
5,0 3,279

300 15,0 0,476
20,0 1,293
40,0 0,156
60,0 0,871
100,0 1,147
0 1,049
2,0 3,869
5,0 3,034
350 15,0 5,474
20,0 4,246
40,0 1,979
60,0 3,324
100,0 1,625
0 0,163
2,0 1,353
5,0 1,029
380 15,0 5,887
20,0 2,676
40,0 6,951
60,0 2,602
100,0 0,751

0
2
4
6
8
10
12
14
16
18
20
250 300 350 400 450 500 550 600 650
comprimento de onda (nm)
intensidade
relativa
brancoirradiado 2hirradiado 42h
Figura 11 – Espectros de fluorescência na fase aquosa irradiada 2,0h e 42,0 horas
Observa-se tanto pela tabela 3 como na figura 11 que em 300nm, 350nm e
380nm, é possível notar que nas primeiras horas de irradiação (2 horas e 5 horas ) não ocorreu
ou ocorreu muito discretamente passagem de material fluorescente do petróleo para a fase
aquosa, fotoquimicamente. Com 42 horas de irradiação, nos mesmos picos já citados, surgem
alterações pouco mais significativa.
O pico de 2 horas mostra redução na intensidade de fluorescência devido a
perda de aromáticos fluorescentes com peso molecular relativamente baixo (até 3 anéis),
provavelmente por volatilização ou por degradação fotoquímica em fase aquosa. No pico de
42 horas ocorre aumento na intensidade de fluorescência em água natural marcando,
provavelmente, o ínicio do intemperismo fotoquímico de petróleo; sendo responsáveis os
derivados aromáticos com 4,5 e 6 anéis (Kumke et al, 1995). Os poliaromáticos podem ter
migrado para a água como resultado da fotooxidação de componentes do óleo, após longo
período de irradiação, sendo os produtos fotodegradação bem mais polares.

0
5
10
15
20
25
30
250 300 350 400 450 500 550 600 650
comprimento de onda (nm)
intensidade
relativa
brancoirradiado 42hnão irradiado 42h
Figura 12 – Fluorescência em fase aquosa decorrente do intemperismo químico do óleo
Na figura 12 verifica-se a migração de componentes fluorescentes do óleo bruto para a fase
aquosa sob efeito da luz solar. O aumento de polaridade da fração aromática e/ou
asfaltênica se deve a reações com incorporação de oxigênio (figura 2) Nicodem et al,
2001). Após longos períodos de irradiação (42h) pode ser notada a degradação ou
transferência de fase.

0
5
10
15
20
25
30
250 270 290 310 330 350 370 390 410 430 450
comprimento de onda (nm)
intensidade
relativa
não irradiado 2hnão irradiado 5hnão irradiado 15hnão irradiado 20hnão irradiado 42hnão irradiado 60hnão irradiado 100hóleo bruto
Figura 13 – Fluorescência em fase aquosa decorrente do intemperismo físico do óelo
A figura 13 mostra a fluorescência em fase aquosa como resultado do óleo “não irradiado”.
As amostras não irradiadas apresentam aumento na intensidade de fluorescência no
período inicial de exposição, visto que aumenta a solubilidade de componentes do óleo
devido a elevação de temperatura.
Nesse caso, a passagem de material fluorescente da fase óleo para a fase aquosa ocorreu
simplesmente por extração, não envolvendo qualquer processo fotoquímico, já que essas
amostras foram expostas protegidas a luz do Sol.

A adição de H2O2 foi testada para investigar a eficiência deste oxidante
frente a HPAs de petróleo, contaminantes de águas naturais.
0
1
2
3
4
5
6
7
8
9
10
250 300 350 400 450 500 550 600 650
comprimento de onda (nm)
intensidade
relativa
branco15min30min45min1h1,5min2,0h2,5h3h
Figura 14 – Fluorescência de HPAs em água na presença de luz solar
0
1
2
3
4
5
6
7
8
9
10
250 300 350 400 450 500 550 600 650
comprimento de onda (nm)
intensidade
relativa
15 min30 min45 min1.0 h1.5 h2.0h2.5h3.0hbranco
Figura 15 – Fluorescência de HPAs em água sem presença de luz

Observa-se nitidamente que o perfil dos espectros das figuras 14 e 15 são
completamente diferentes. A figura 14 apresenta água irradiada na presença de peróxido de
hidrogênio. Neste caso a redução de fluorescência é marcada pela degradação fotoquímica
além do alto poder oxidante do peróxido de hidrogênio que, gerando radicais hidroxilas, é
capaz de oxidar uma grande variedade de compostos orgânicos. Já na figura 15, nota-se que
não há uma degradação significativa de fluorescência quando adiciona peróxido de hidrogênio
em água “não irradiada”.
Neste caso o comportamento cinético de ambas as amostras irradiada e “não
irradiada” são bem diferentes. Pelo que mostra a figura 16 a amostra irradiada apresenta
provavelmente uma reação de pseudo-primeira ordem, enquanto que a amostra “não
irradiada” apresenta uma reação de primeira ordem.
-20 0 20 40 60 80 100 120 140 160 180 200
0
1
2
3
4
5
6
7
8
inte
nsid
ade
rela
tiva
tempo de exposição (min)
amostra exposta ao Sol amostra não exposta ao Sol
Figura 16 – Cinética de degradação de HPAs pelo peróxido de hidrogênio

IV. Conclusão As figuras 5 e 6 esclarece a redução na intensidade de fluorescência que
ocorre no petróleo após longos períodos de irradiação ocasionada pelo fato de alguns
componentes fluorescentes serem muito mais reativos e, por isso, serem preferencialmente
destruídos.
Na figura 8 verifica-se que o efeito térmico é responsável pelo aumento na
concentração de componentes fluorescentes mais pesados do óleo, devido a volatilização, que
é um processo não fotoquímico, de componentes fluorescentes mais leves.
Também após longos períodos de irradiação do petróleo, ele incorpora em
sua estrutura oxigênio. A figura 10, mostra o surgimento de bandas características de C=O e
OH no óleo característica da incorporação do oxigênio
A fase aquosa começa a ter fluorescência após ter sido irradiada em curto
período de tempo seja por efeito da luz ou solubilidade devido aos compostos de baixo peso
molecular do óleo migrarem para a fase aquosa (figura 12). Os picos máximos de
fluorescência em curtos períodos de tempo aos poucos desaparecem, e novos picos em
comprimento de onda maiores surgem quando irradiado por mais tempo devido a derivados
aromáticos com 4,5 e 6 anéis. Os poliaromáticos ou derivados podem ter migrado para a água
como resultado da fotodegradação de componentes do óleo após longo período de irradiação,
sendo os produtos de fotodegradação bem mais polares (figura 13).
A fase aquosa não irradiada apresenta o efeito térmico causando aumento na
intensidade de fluorescência no período inicial de exposição devido provavelmente o aumento
da solubilidade de componentes do óleo devido a elevação de temperatura durante a
exposição e, a passagem de material fluorescente ocorre simplesmente por transferência de
fase, não envolvendo nenhum processo fotoquímico.
No caso da utilização de peróxido de hidrogênio em água contaminada, o
processo de fotodegradação de componentes do óleo apresenta uma significativa vantagem
quando compara com a água contaminada somente irradiada. A redução da intensidade de

fluorescência, 71%, é causada pelo poder oxidante que o peróxido de hidrogênio apresenta
sobre os compostos orgânicos presentes nessa água. O decaimento da fluorescência causadoa
pela adição de hidróxido de hidrogênio tem o comportamento de uma reação de pseudo-
primeira ordem.
Pelo tratamento da água irradiada utilizando dióxido de titânio, pode-se
concluir que ele também teve um efeito muito parecido com o do peróxido de hidrogênio, no
entanto, como este possui maior ação oxidante, ele foi mais eficiente para a redução de
fluorescência, 85%. O decaimento apresentado pela adição de dióxido de titânio comporta-se
como se fosse uma reação de pseudo-primeira ordem analisando todos os tempos de
exposição, porque em determinado intervalo de tempo o decaimento apresenta-se como uma
reação de primeira ordem.

V BIBLIOGRAFIA
Alberici, R. M.; Nogueira, R. F. P.; Jardim, W. F. Ciência Hoje, junho 1995. Bedding, N. D.; Taylor, P. N. & Lester, J. N. 1995. Environ. Technology, 16, 801. Cooper, W. J. & Herr, F. L., 1987. Introduction and Overview Chapter 1 in Zilka RG & Cooper WJ (Ed) Photochemistry of Environmental Aquatic Systems ACS Symposium Series #327, Am. Chem., pp 1-8. Ehrhardt, M. G.; Burns, K. A. & Bicego, M. C. 1992 . Mar. Chem. 37, 53. Fox, M. A.; Dulay, M. T.; Chem. Rev. 1993, 93, 341. Gill 1989 Guedes, C. L. B., . Tese de Doutorado Hoffmann, M. R.; Choi, W.; Bahnemann, D. W.; Chem. Rev. 1995, 95, 69. Kumke, M. U.; Löhmannsröben, H.-G. & Roch, Th. 1995. Journal of Fluorescence 5, 139. Kunz, A.; Freire, R. S.; Rowedler, J. J. R.; Mansilla, H.; Rodrigues, J.; Duran, N. Química Nova 1999, 22, 425. Linsebigler, A. L.; Lu, G.; Yates Jr, J. T.; Chem. Rev. 1995, 95, 735. Mackay, G. D. M.; Mclean, A. Y.; Betancourt, O. J. & Johson, B. D. 1973. J. Inst. Pet., 59, 164. Masten 1994 National Academy of Sciences, 1985. Oil in the Sea, Inputs, Fates and Effects. National Academy Press, Washington, DC. Nicodem, D. E.; Fernandes, M. C. Z.; Guedes, C. L. B. & Correa, R. J. 1997. Biogeochem. 39, 121. Nicodem, D. E.; Cunha, F. V. & Guedes, C. L. B.; 1998. A Time Resolved Single Photon Counting Study of the Quenching of Fluorescect Probes by Petroleum: Probing the Energy Distribution of the Aromatic Components. Nicodem, D. E., Guedes, C. L. B. & Correa, R. J., 1998. Photochemistry of Petroleum I: Systematic Study os a Brazilian Intermediate Crude oil. Mar. Chem. Paalme, L.; Irha, N.; Urbas, E.; Tsybsn, A. & Kirso, U. 1990. Mar. Chem. 30, 105.

Seifert, 1973 Tissot , 1984 Thingstad, T. & Pengerud, B., 1983. Mar. Pollut. Bull. 14, 214. Zepp, R. G.; Wolfe, N. L.; Boughman, G. L.; & Hollis, R. C. 1977. Nature 267, 421. Zürcher, F. & Thure, M., 1978. Environ. Sci. Technol. 12, 833.