efeito da n-acetilcisteína sobre a inflamação, estresse ...... ferreira nc. efeito da...
TRANSCRIPT

NATALIE CHAVES FERREIRA
Efeito da N-acetilcisteína sobre a inflamação, estresse
oxidativo e morte celular em camundongos com esteato-
hepatite submetidos à isquemia e reperfusão hepática
Tese apresentada à Faculdade de Medicina
da Universidade de São Paulo para a
obtenção do título de Doutor em Ciências
Programa de Clínica Cirúrgica
Orientadora: Profa. Dra. Edna Frasson de
Souza Montero
(Versão corrigida, Resolução CoPGr 6018/11, de 1 de novembro de 2011. A versão
original está disponível na Biblioteca da FMUSP)
SÃO PAULO
2018


Ferreira NC. Efeito da N-acetilcisteína sobre a inflamação, estresse oxidativo e
morte celular em camundongos com esteato-hepatite submetidos à isquemia e
reperfusão hepática [doutorado]. São Paulo: Universidade de São Paulo, Faculdade
de Medicina; 2018.
Banca examinadora
Presidente da Banca: Profa. Dra. Edna Frasson de Souza Montero
Instituição: Faculdade de Medicina da Universidade de São Paulo
Membro Titular: Prof. Dr. Edivaldo Massazo Utiyama
Instituição: Faculdade de Medicina da Universidade de São Paulo
Membro Titular: Prof. Dr. Francisco de Salles Collet e Silva
Instituição: Instituto Central do Hospital das Clínicas
Membro Titular: Prof. Dr. Marcelo Moura Linhares
Instituição: Escola Paulista de Medicina-Universidade Federal de São Paulo
Membro Suplente: Prof. Dr. Alfredo Luiz Jacomo
Instituição: Faculdade de Medicina da Universidade de São Paulo
Membro Suplente: Prof. Dr. Franz Robert Apodaca Torrez
Instituição: Escola Paulista de Medicina-Universidade Federal de São Paulo
Membro Suplente: Profa. Dra. Margareth Pauli Lallée
Instituição: Instituto Central do Hospital das Clínicas

Dedico aos meus pais, Diva e Cecílio, pelo apoio e dedicação
durante toda a minha caminhada até aqui.
Aos meus avós maternos, Teodolino (in memoriam) e Tertulina
(in memoriam), pelos ensinamentos e conselhos, e aos meus
avós paternos, Gerolino (in memoriam) e Maria (in memoriam),
pelo carinho em família.
Aos meus familiares, tios, tias, primos, primas e a Federico
Martín Cayuela por todo incentivo em cada momento de minha
vida.

AGRADECIMENTOS
À minha orientadora, Profª Drª Edna Frasson de Souza Montero, pela
oportunidade de desenvolver esta tese, pela disponibilidade e dedicação em
transmitir seus conhecimentos científicos, e pela confiança em meu trabalho.
Ao Prof. Dr. Edivaldo Massazo Utiyama, Professor Titular da Divisão de
Clínica Cirúrgica III, pelo apoio no desenvolvimento dos projetos no Laboratório de
Fisiopatologia Cirúrgica (LIM-62).
Aos membros do Laboratório de Fisiopatologia Cirúrgica (LIM-62), Mário
Matsuo Itinoshe, Marco de Luna, Luci Yukiko Takasaka, Elisabete Hiroe Minami e
Ana Maria de Lima Cattani, pelo suporte técnico no desenvolvimento desse trabalho,
e à estagiária Gabrielly Pascoa Negreti por sua participação voluntária nesse
projeto.
À secretária do Programa de Pós-Graduação em Clínica Cirúrgica, Eliane
Falconi Monico Gazetto, por todo auxílio prestado com as questões burocráticas da
Pós-Graduação.
À Drª Marcia Kiyomi Koike, pelas orientações e pelo suporte profissional
durante a execução do projeto de doutorado.
Ao Prof. Dr. José Antonio Picanço Diniz Júnior por todo apoio para a
conclusão desse trabalho, e aos funcionários do Laboratório de Microscopia
Eletrônica do Instituto Evandro Chagas pelo suporte técnico.
À Faculdade de Medicina da Universidade de São Paulo, onde pude ampliar
minha qualificação profissional e consolidar meus conhecimentos.
Agradeço especialmente a Federico Martín Cayuela, aos meus pais,
familiares e amigos, que são pessoas importantes na minha vida e, de alguma
forma, contribuíram para a realização desse trabalho e para o meu crescimento
profissional e pessoal.

“De tudo ficarão três coisas: a certeza de que estamos
começando, de que é preciso continuar e de que podemos ser
interrompidos antes de terminar. Fazer da interrupção um
caminho novo. Da queda, um passo de dança. Do medo, uma
escada. Do sonho, uma ponte. Da procura, um encontro”
(Fernando Sabino, 1956).

RESUMO
Ferreira NC. Efeito da N-acetilcisteína sobre a inflamação, estresse oxidativo e
morte celular em camundongos com esteato-hepatite submetidos à isquemia e
reperfusão hepática [tese]. São Paulo: Faculdade de Medicina, Universidade de São
Paulo; 2018.
A manipulação cirúrgica do fígado com esteato-hepatite pode levar a complicações
pós-operatórias importantes. A N-acetilcisteína (NAC) apresenta efeito protetor em
modelos de I/R hepática, bem como na doença hepática gordurosa não alcoólica.
Porém, há carência de estudos relacionados a seu efeito em lesões de I/R em fígado
com esteato-hepatite. Portanto, esse estudo teve como objetivo avaliar o efeito da
NAC sobre a reação inflamatória, estresse oxidativo e morte celular induzidos pela
I/R hepática associada à esteato-hepatite. Foram utilizados camundongos
C57BL/6J, machos, com oito semanas e peso entre 25 e 30 gramas, que receberam
ração normal AIN-93M ou deficiente em Metionina e Colina (DMC) de acordo com o
grupo de estudo. A NAC (150mg/kg) foi administrada 15 minutos antes da isquemia
(DMC-I/R+NAC). Nos grupos DMC-I/R e DMC-I/R+NAC, os animais foram
submetidos a 30 minutos de isquemia e 24 horas de reperfusão. Após esse período,
o sangue foi coletado, para análise bioquímica das transaminases, e o fígado,
removido cirurgicamente para avaliação histopatológica, dosagem das citocinas IL-
1β, TGF-β1 e IFN-, nitrito e Substâncias Reativas ao Ácido Tiobarbitúrico (TBARS),
análise da expressão celular de Bcl-2 e apoptose (TUNEL). A histopatologia mostrou
redução de focos inflamatórios no grupo DMC-I/R+NAC. Os níveis AST foram
elevados no grupo DMC-I/R enquanto que os de ALT diminuíram em relação ao
controle. No grupo DMC-I/R+NAC houve redução em ambas as transaminases,
sendo significante apenas para a avaliação de AST. A produção das citocinas IL-1β
e TGF-β1 foi elevada no fígado esteatótico de camundongos submetidos à I/R em
comparação aos animais com fígado normal; porém, o tratamento com NAC
promoveu uma redução significante dessas citocinas. A análise do estresse
oxidativo mostrou que os animais com esteato-hepatite submetido à I/R tiveram as
concentrações de TBARS e de nitrito elevadas em relação aos camundongos com
fígado normal. No entanto, ao receberem o tratamento com NAC, esses animais

apresentaram uma redução de ambos. A análise da expressão de Bcl-2 mostrou que
essa proteína estava elevada em camundongos com fígado normal, e reduzida na
presença de esteato-hepatite ou quando esta afecção está associada à I/R. A NAC,
nesse caso, promoveu o aumento da expressão de Bcl-2 em camundongos com
esteato-hepatite submetido à I/R. Esse resultado estava associado aos dados de
apoptose, nos quais foi observado aumento do número de células apoptóticas no
fígado esteatótico de camundongos em que a I/R foi induzida e redução nos animais
tratados com NAC. Desta forma, conclui-se que o uso da NAC promove efeitos
benéficos sobre o fígado de camundongos com esteato-hepatite associada à lesão
de I/R por meio da redução do infiltrado inflamatório, das concentrações de IL-1β e
TGF-β1, do estresse oxidativo e da morte celular.
Descritores: fígado gorduroso; citocinas; estresse oxidativo; morte celular; fígado;
camundongo; acetilcisteína.

ABSTRACT
Ferreira NC. Effect of N-acetylcysteine on inflammation, oxidative stress, and cell
death in mice with steatohepatitis submitted to hepatic ischemia and reperfusion
[thesis]. São Paulo: “Faculdade de Medicina, Universidade de São Paulo”; 2018.
The surgical manipulation of the liver with steatohepatitis may lead to important
postoperative complications. The N-acetylcysteine (NAC) has a protective effect in
hepatic I/R models, as well as in non-alcoholic fatty liver disease. However, there is a
lack of studies related to its effect inI/R injury to liver with steatohepatitis. Therefore,
the purpose of this study was to assess the effect of NAC over inflammatory,
oxidative stress and cell death induced by hepatic I/R associated to steatohepatitis.
The mice used were C57BL/6J, male, eight weeks old and weighing between 25 and
30 grams, which received normal diet AIN-93M or Methionine and Choline-deficient
diet (DMC) according to their study group. The NAC (150mg/kg) was administered 15
minutes before the ischemia (DMC-I/R+NAC). In groups DMC-I/R and DMC-
I/R+NAC, the animals were submitted to 30 minutes of ischemia and 24 hours of
reperfusion. After this period, blood was collected, for biochemical analysisof
transaminases, and the liver, surgically removed for histopathological assessment,
dosage of cytokines levels IL-1β, TGF-β1 and IFN-, nitrite and Reactive Substances
to Thiobarbituric Acid (TBARS), analysis of cell expression of Bcl-2 and apoptosis
(TUNEL). The histopathology showed a reduction of inflammatory foci in DMC-
I/R+NAC group. The AST levels were elevated in DMC-I/R group while the ALT
reduced in comparing to control. In the DMC-I/R+NAC group there was a reduction in
both transaminases, being significant only for AST assessment. The production of
cytokines IL-1β and TGF-β1 was elevated in the liver with steatosis of mice
submitted to I/R when compared to the animals with normal liver; however, the
treatment with NAC provided a significant reduction of these cytokines. The oxidative
stress analysis showed that the animals with steatohepatitis submitted to I/R had the
concentrations of TBARS and nitrite elevated when compared to the mice with
normal liver. However, when they received treatment with NAC, these animals
presented a reduction in both. The analysis of the Bcl-2 expression showed that this
protein was high in mice with normal liver, and reduced in the presence of

steatohepatitis or when this affection is associated to I/R. A NAC, in this case, lead to
an increase of Bcl-2 expression in mice with steatohepatitis submitted to I/R. This
result was associated to data of apoptosis, where increase in the number of apoptotic
cells in liver with steatosis of mice in which theI/R was induced and reduction in
animals treated with NAC was observed. Therefore, the conclusion is that the use of
NAC promotes beneficial effects on the liver of mice with steatohepatitis associated
to I/R injury through the reduction of inflammatory infiltrate, of concentrations of IL-1β
and TGF-β1, of oxidative stress and cell death.
Descriptors: fatty liver; cytokines; oxidative stress; cell death; liver; mice;
acetylcysteine.

LISTA DE FIGURAS
Figura 1 – Distribuição dos grupos experimentais........................................... 36
Figura 2 – Fotografias da cavidade abdominal e da oclusão do pedículo
hepático clampeado dos camundongos.......................................... 38
Figura 3 – Fotomicrografia do fígado de camundongos do grupo Controle..... 46
Figura 4 – Fotomicrografia do fígado de camundongos do grupo DMC.......... 46
Figura 5 – Fotomicrografias do fígado de camundongos do grupo DMC-I/R... 47
Figura 6 – Fotomicrografia do fígado de camundongos do grupo DMC-
I/R+NAC.......................................................................................... 48
Figura 7 – Fotomicrografia do fígado de camundongos do grupo Controle
mostrando a expressão de Bcl-2.................................................... 56
Figura 8 – Fotomicrografia do fígado de camundongos do grupo DMC
mostrando a expressão de Bcl-2.................................................... 56
Figura 9 – Fotomicrografia do fígado de camundongos do grupo DMC-I/R
mostrando a expressão de Bcl-2.................................................... 57
Figura 10 – Fotomicrografia do fígado de camundongos do grupo DMC-
I/R+NAC mostrando a expressão de Bcl-2..................................... 57
Figura 11 – Fotomicrografia do fígado de camundongos do grupo Controle
apresentando apoptose.................................................................. 59
Figura 12 – Fotomicrografia do fígado de camundongos do grupo DMC
apresentando apoptose.................................................................. 60
Figura 13 – Fotomicrografia do fígado de camundongos do grupo DMC-I/R
apresentando apoptose.................................................................. 60
Figura 14 – Fotomicrografia do fígado de camundongos do grupo DMC-
I/R+NAC apresentando apoptose............................................................. 61

LISTA DE GRÁFICOS
Gráfico 1 – Níveis de AST no soro dos camundongos dos grupos
experimentais, expressos em média ± desvio padrão.................. 49
Gráfico 2 – Níveis de ALT no soro dos camundongos dos grupos
experimentais, expressos em média ± desvio padrão.................. 50
Gráfico 3 – Quantificação da citocina IL-1β no fígado de camundongo dos
grupos experimentais, expressos em média ± desvio padrão...... 51
Gráfico 4 – Quantificação da citocina TGF-β1 no fígado dos camundongos
dos grupos experimentais, expressos em média ± desvio padrão 52
Gráfico 5 – Quantificação de TBARS no fígado dos camundongos dos
grupos experimentais, expressos em média ± desvio padrão...... 53
Gráfico 6 – Quantificação de nitrito no fígado dos camundongos dos grupos
experimentais, expressos em média ± desvio padrão.................. 54
Gráfico 7 – Contagem de células apoptóticas no fígado dos camundongos
dos grupos experimentais, expressos em média ± desvio padrão 58

LISTA DE TABELAS
Tabela 1 – Valores de AST e ALT no soro dos camundongos, expressos
em média ± desvio padrão........................................................... 50
Tabela 2 – Valores das citocinas IL-1β e TGF-β1 no fígado dos
camundongos, expressos em média ± desvio padrão................. 52
Tabela 3 – Valores de TBARS e nitrito no fígado dos camundongos,
expressos em média ± desvio padrão......................................... 55
Tabela 4 – Contagem de células apoptóticas no fígado dos camundongos,
expressos em média ± desvio padrão......................................... 59

LISTA DE SIGLAS
AIF: Fator indutor de apoptose
ALT: Alanina aminotransferase
AMP: Adenosina monofosfato
AST: Aspartato aminotransferase
ATF4: Activating transcription factor 4
ATP: Adenosina trifosfato
BAG: Bcl-2 associated athanogenes
Bad: Bcl-2 associated agonist of cell death
Bak: Bcl-2 homologous antagonist/killer
Bax: Bcl-2 associated X protein
Bcl: B-cell Lymphoma (-x, -XL, -XS, -w, -10, -2)
Bid: Bax like BH3 protein
Bik: Bcl-2 interacting killer
Bim: Bcl-2 interacting mediator of cell death
Blk: B lymphoid tyrosine kinase
CHOP: Proteína homóloga à proteína de ligação ao potenciador-CCAAT
DAB: Diaminobenzidina
DAMPs: Padrões moleculares associados a danos
DHGNA: Doença hepática gordurosa não alcoólica
DIABLO: Proteína de ligação direta ao inibidor de proteínas de apoptose com
baixo ponto Isoelétrico
DMC: Deficiente em metionina e colina
DNA: Ácido desoxirribonucléico
EHNA: Esteato-hepatite não alcóolica
ERN: Espécies reativas de nitrogênio
ERO: Espécies reativas de oxigênio
FMUSP: Faculdade de Medicina da Universidade de São Paulo
GRP78: Glucose regulated protein 78
HCl: Ácido clorídrico
H2SO4: Ácido sulfúrico
IEC: Instituto Evandro Chagas

IL-1β: Interleucina-1β
IFN-γ: Interferon-γ
iNOS: Óxido nítrico sintase induzida
I/R: Isquemia e reperfusão
LIM 62: Laboratório de Fisiopatologia Cirúrgica
LLE: Lobo lateral esquerdo
LME: Laboratório de Microscopia Eletrônica
LME: Lobo medial esquerdo
MDA: Malondialdeído
Mcl-1: Myeloid leukemia cell differentiation protein
MnTBAP: Cloreto de porfirina de Mn (III) tetraquis (ácido benzóico-4)
NAC: N-acetilcisteína
NaOH: Hidróxido de sódio
NK: Natural killer
NO: Óxido Nítrico
PBS: Tampão fosfato com salina
pH: Potencial hidrogeniônico
SBF: Soro bovino fetal
SBH: Sociedade Brasileira de Hepatologia
Smac: Segundo ativador mitocondrial de caspase
TBA: Ácido tiobarbitúrico
TBARS: Substâncias reativas ao ácido tiobarbitúrico
TCA: Ácido tricloroacético
TGF-β1: Transforming growth factor-β1
TMB: Tetrametilbenzidina
TNF-α: Fator de necrose tumoral-α
TRL-4: Toll-like receptor-4
TUNEL: Marcação de terminais dUTP em fitas simples de DNA mediadas por
deoxinucleotidil terminal transferase
VLDL: Lipoproteína de muito baixa densidade

SUMÁRIO
1 INTRODUÇÃO........................................................................................... 18
1.1 DOENÇA HEPÁTICA GORDUROSA NÃO ALCOÓLICA (DHGNA)......... 19
1.2 I/R EM FÍGADO ESTEATÓTICO............................................................... 21
1.3 RESPOSTA INFLAMATÓRIA.................................................................... 22
1.4 ESTRESSE OXIDATIVO........................................................................... 24
1.5 MORTE CELULAR.................................................................................... 27
1.6 N-ACETILCISTEÍNA (NAC)....................................................................... 29
2 OBJETIVOS.............................................................................................. 32
3 MÉTODOS................................................................................................. 34
3.1 TIPO DE ESTUDO.................................................................................... 35
3.2 AMOSTRA................................................................................................. 35
3.3 PROCEDIMENTO OPERATÓRIO............................................................ 36
3.4 PROCESSAMENTO HISTOLÓGICO........................................................ 39
3.5 DOSAGEM BIOQUÍMICA DE TRANSAMINASES.................................... 39
3.6 PREPARO DAS AMOSTRAS PARA AS DOSAGENS DE CITOCINAS,
TBARS E NITRITO.................................................................................... 39
3.6.1 Dosagem de citocinas............................................................................. 40
3.6.2 Dosagem de TBARS................................................................................ 40
3.6.3 Dosagem de nitrito.................................................................................. 41
3.7 DETECÇÃO DE Bcl-2 POR IMUNOHISTOQUÍMICA............................... 41
3.8 DETECÇÃO DE APOPTOSE PELA TÉCNICA DE TUNEL..................... . 42
3.9 ANÁLISE ESTATÍSTICA........................................................................... 43
4 RESULTADOS.......................................................................................... 44
4.1 ANÁLISE HISTOPATOLÓGICA................................................................ 45
4.2 ANÁLISE BIOQUÍMICA DAS TRANSAMINASES..................................... 48
4.3 AVALIAÇÃO DA EXPRESSÃO DE CITOCINAS....................................... 51
4.4 ANÁLISE DA QUATIFICAÇÃO DE TBARS E DE NITRITO..................... 53
4.5 ANÁLISE DA EXPRESSÃO DE Bcl-2....................................................... 55
4.6 ANÁLISE DE NECROSE E APOPTOSE NO TECIDO HEPÁTICO....... 58
5 DISCUSSÃO............................................................................................. 62
6 CONCLUSÃO........................................................................................... 72

REFERÊNCIAS......................................................................................... 74
APÊNDICE................................................................................................ 93
APÊNDICE A............................................................................................. 93
ANEXOS.................................................................................................... 94
ANEXO A................................................................................................... 94
ANEXO B................................................................................................... 95
ANEXO C............................................................................... .................... 96

18
1 INTRODUÇÃO

19
1 INTRODUÇÃO
A prevalência de obesidade tem aumentado sensivelmente nos últimos anos,
passando a ser um problema de saúde pública, não somente no Brasil, mas também
em outros países, principalmente nos desenvolvidos. Uma das preocupações com
relação à obesidade é o possível desenvolvimento de esteatose no fígado, e, caso
essa afecção hepática não seja devidamente tratada, pode ser agravada e evoluir
para uma esteato-hepatite1.
Pacientes com esteato-hepatite submetidos a procedimentos cirúrgicos no
fígado podem apresentar complicações importantes que dificultam a sua
recuperação. Dentre os procedimentos, destacam-se operações que promovam
lesão de isquemia e reperfusão (I/R) hepática, tais como ressecção e transplante de
fígado, trauma abdominal e choque hemorrágico2,3,4.
A busca por estratégias de prevenção ou terapêuticas na abordagem cirúrgica
de pacientes acometidos com essa enfermidade é de alta relevância. Os modelos
murinos são bastante adequados para estas avaliações pré-clínicas, pelo baixo
custo e pelo conhecimento que se tem desse modelo experimental em diferentes
áreas5.
1.1 DOENÇA HEPÁTICA GORDUROSA NÃO ALCOÓLICA (DHGNA)
Dentre os diversos tipos de doenças associadas ao acúmulo de lipídeos nas
células, a DHGNA é uma das afecções hepáticas mais presente em países
desenvolvidos e está associada principalmente à obesidade e à síndrome
metabólica6,7. Na população global, a prevalência dessa doença é de 4 a 46%8,9, e
deste valor, 90% ocorre em obesos mórbitos6. No Brasil, a Sociedade Brasileira de
Hepatologia (SBH) estima que cerca de 20% da população do país apresenta o
quadro histológico da DHGNA1.
Nos dias atuais, essa afecção tornou-se a segunda maior causa para a
indicação de transplante hepático nos Estados Unidos10 e chega a estar presente em
até 26% dos potenciais doadores11. Dentre outras causas, o aumento do número de
pacientes com necessidade de transplante hepático em decorrência do acúmulo de
lipídeos no fígado e deve ao estilo de vida ocidental12.

20
A DHGNA é definida como uma condição em que mais de 5-10% dos
hepatócitos apresentam esteatose macroscópica – observada por microscopia de
luz – na ausência de outras etiologias de doenças hepáticas6,13, tais como o
consumo excessivo de álcool, doenças auto-imunes ou induzidas por drogas e por
hepatite viral14. O espectro histológico dessa doença pode variar de uma esteatose
simples a esteatohepatite não alcoólica (EHNA), com fibrose hepática e
cirrose13,14,15.
A esteatose hepática, sendo considerada como uma das fases da DHGNA, é
caracterizada pelo acúmulo de vacúolos de lipídeos nos hepatócitos, principalmente
de triacilgliceróis, sem inflamação importante16,17,18. Mudanças na oxidação das
gorduras ou a diminuição da exportação, no fígado, de lipoproteína de muito baixa
densidade (VLDL) são os principais mecanismos envolvidos na esteatose hepática19.
À medida que o dano provocado pelo acúmulo de lipídeos se mantém no
tecido hepático, a esteatose simples evolui para uma EHNA. Esta, por sua vez, é
caracterizada principalmente pela presença de uma esteatose macro e
microvesicular, infiltrado inflamatório e baloneamento dos hepatócitos14,20,21. Em
alguns indivíduos com o fenótipo de EHNA, o fígado não consegue manter o seu
ritmo de regeneração devido ao aumento da taxa de morte celular, o que resulta na
cicatrização do órgão com fibrose22,23.
Diante dessa condição, a EHNA pode progredir para cirrose – 10 a 29% dos
indivíduos desenvolvem-na no prazo de 10 anos. Na cirrose, os hepatócitos são
substituídos por colágeno tipo 1, o principal componente do tecido cicatricial, que é
produzido pelas células estreladas ou células de Ito ativadas por uma lesão
hepática. Nessa fase, os pacientes podem morrer de complicações de hipertensão
portal, insuficiência hepática e carcinoma hepatocelular, caso o transplante de fígado
não seja realizado24,25.
Essas alterações podem demorar anos para se estabelecer em humanos com
comportamento alimentar hipercalórico e sedentarismo; porém, para melhor
compreender o desenvolvimento da DHGNA, vários modelos animais foram
estabelecidos, como o uso de uma dieta hipercalórica26,27,28 ou de uma dieta
deficiente de metionina e colina29,30, ou ainda o uso de animais geneticamente
modificados para desenvolver obesidade31,32.

21
1.2 I/R EM FÍGADO ESTEATÓTICO
A I/R no fígado caracteriza-se por ser uma interrupção parcial ou completa do
fluxo sanguíneo e do abastecimento de oxigênio nesse órgão, seguido do
restabelecimento do fluxo e consequente reoxigenação do tecido33,34. Nesse
processo, a isquemia provoca várias alterações hepáticas, que estão principalmente
relacionadas à diminuição do metabolismo aeróbico causado pela falta de
disponibilidade de oxigênio; no entanto, é durante a reperfusão, na qual se observa
o recrutamento de leucócitos, a liberação de mediadores inflamatórios e o aumento
da produção de radicais livres, que as lesões no tecido hepático são agravadas35.
A esteatose hepática é um dos fatores que podem aumentar a
susceptibilidade do fígado à lesão causada pelo processo de I/R. Dessa forma, a
esteatose tornou-se o fator de risco clínico mais importante para esse tipo de lesão
hepática36,37,38, aumentando o risco de morbidade e mortalidade pós-operatório,
inclusive em casos de transplante38,39,40. É o fator de maior impacto na disfunção
primária do enxerto e de baixa sobrevida entre os transplantados, assim como de
mau resultado em procedimentos que exijam isquemia transitória41,42,43. Esses
resultados, em relação à isquemia no fíagdo esteatótico, têm sido estudados e sua
causa é atribuída a diversos fatores, tais como alterações microvasculares,
alterações funcionais do hepatócito14, maior sensibilidade às espécies reativas de
oxigênio (ERO), entre outros44,45.
O acúmulo de triacilgliceróis dentro dos hepatócitos, por exemplo, provoca o
aumento do volume dessas células, distorcendo toda a arquitetura sinusoidal e
diminuindo o diâmetro do lúmen dos vasos sanguíneos em até 50%, quando
comparados aos fígados sem esteatose37. A redução do espaço sinusoidal faz com
que a hipóxia persista durante o processo de I/R, causando ainda mais dano ao
tecido hepático. Além disso, em fígados esteatóticos, proteínas da membrana
interna da mitocôndria induzem a diminuição da síntese de adenosina trifosfato
(ATP) e o aumento da produção de ERO46,47.
Quando fígados esteatóticos são submetidos à I/R, apresentam respostas
mais intensas em relação às manifestadas em fígados normais. Selzner e
colaboradores (2000) afirmam que há maior quantidade de necrose do que apoptose
em análises bioquímicas e histológicas48. Em outros estudos, encontram-se dados

22
que reforçam a ideia de uma maior resposta inflamatória em fígados esteatóticos
submetidos a episódios de isquemia transitória, quando comparados com fígados
normais49,50.
1.3 RESPOSTA INFLAMATÓRIA
A resposta imune do fígado está envolvida com a defesa desse órgão, por
meio da apresentação de antígenos para linfócitos e a eliminação de patógenos. No
entanto, as células imunes hepáticas podem também estar relacionadas com a
patogênese metabólica e fibrose do fígado, por meio das interações com outras
células, tais como hepatócitos, células endoteliais sinusoidais ou células
estreladas51,52.
Dentre as células que participam do sistema imune do fígado, estão presentes
os linfócitos hepáticos53,54, células de Kupffer55,56, infiltrado de monócitos57 e
eosinófilos58. Estas interagem com as células adjacentes no fígado e liberam
citocinas, resultando no desenvolvimento de doenças hepáticas51,59.
Citocinas são peptídeos reguladores pleiotrópicos que podem ser sintetizados
por diferentes tipos celulares. Em muitos tecidos, incluindo no fígado, ocorre sua
produção basal; no entanto, a síntese desses mediadores inflamatórios pode ser
aumentada mediante um estímulo, seja ele fisiológico ou patológico, levando a
doenças inflamatórias60,61.
Diferentes processos podem contribuir para o desenvolvimento de doenças
inflamatórias no fígado. Tilg e Moschen (2010) sugerem que mediadores
inflamatórios de vários tecidos, em especial do intestino e do tecido adiposo, podem
ter papel fundamental na ativação da cascata inflamatória, na fibrose e por fim no
desenvolvimento de tumor no fígado62.
A inflamação no fígado pode estar relacionada, por exemplo, ao acúmulo
excessivo de lipídeos nesse órgão e, consequentemente, à doenças metabólicas
hepáticas, tais como a EHNA. Nesses casos, acredita-se que as citocinas pró-
inflamatórias sejam um dos responsáveis pela desregulação metabólica no
fígado63,64.
A lesão hepática causada por I/R também está relacionada ao
desenvolvimento de uma resposta inflamatória exacerbada, caracterizada pela

23
síntese de citocinas inflamatórias e quimiocinas que recrutam leucócitos para o
tecido isquêmico, intensificando ainda mais a lesão no tecido hepático65. Dentre as
citocinas que participam da patogênese da EHNA e da resposta inflamatória
proveniente da I/R no fígado, podem ser destacadas a IL-1β, TGF-β e INF-γ.
AIL-1β é uma citocina pró-inflamatória produzida por macrófagos, monócitos,
células dendríticas, células NK, linfócitos B66, além de células epiteliais67 e células
endoteliais68. Está presente como um precursorinativo que, por meio da clivagem de
sítios específicos realizada pela caspase 166,69 ou por proteases derivadas de
neutrófilos, pode se tornar uma citocina biologicamente ativa70; e quando ativada, se
liga ao seu receptor e desencadeia a resposta inflamatória71.
Na EHNA, a produção de IL-1β é elevada devido ao acúmulo de ácidos
graxos saturados que induz a ativação do inflamossomo nas células hepáticas72, o
que pode aumentar o recrutamento de leucócitos e intensificar o processo
inflamatório, causando ainda mais danos ao tecido hepático73. Durante a I/R, a IL-1β
é uma das principais citocinas responsáveis pelo desenvolvimento do processo
inflamatório. Harada e colaboradores (2002) mostraram que, quando a produção
dessa citocina é reduzida, diversas reações inflamatórias mediadas por ela podem
ser bloqueadas e, consequentemente, a lesão causada pela I/R é atenuada74.
O TGF-β é uma citocina que participa de uma variedade de processos
biológicos, incluindo crescimento celular, diferenciação, adesão celular, migração e
resposta imune, além de ter um papel importante nas mudanças fibroproliferativas
que podem ocorrer em muitos órgãos e tecidos danificados, como fígado, rim,
coração pele, pulmão e parede das artérias75,76. O TGF-β é um dos principais
mediadores da resposta imune responsável pela proliferação e ativação das células
estreladas, e estas, por sua vez, estão relacionadas com a superprodução e
acumulação de colágeno que levam a fibrose no fígado em casos de doenças
hepáticas53,77.
A esteatose no fígado é considerada como um fator de risco para a
progressão da fibrose em casos de doenças hepáticas, já que, durante a injúria
causada pelo processo inflamatório resultante do acúmulo de ácidos graxos, as
células estreladas hepáticas são ativadas e liberam o TGF-β, uma das principais
citocinas fibrogênicas, que por sua vez levam à transdiferenciação das células
estreladas em miofibroblastos para a produção de tecido fibrótico78,79.

24
Na lesão de I/R, também pode ser observado o processo de fibrose mediado
pela citocina TGF-β80. Yu e colaboradores (2012) mostraram que a I/R em lobos
clampeados contribuiu para a regeneração hepática de lobos não clampeados, já
que a produção dessa citocina foi menos estimulada nos lobos não clampeados,
entretanto, a produção dessa citocina foi menos estimulada nos obos não
clampeados, e assim, ocorreu menos fibrose hepática nessa região81.
O IFN-γ é uma citocina pertencente ao grupo de interferon do tipo II e
desempenha um papel muito importante tanto na defesa do organismo quanto na
regulação da resposta imune82. Inicialmente acreditava-se que essa citocina
apresentava apenas função antiviral82, porém, atualmente sabe-se que o IFN-γ está
associado à sinalização de apoptose83, e que também apresenta funções anti ou
pró-tumorais84,85.
De acordo com Inzaugarat e colaboradores (2017), pacientes com DHGNA
apresentaram um aumento de infiltrado inflamatório no fígado, formado por linfócito
T CD4+, e esse aumento está associado à presença de níveis elevados de IFN-γ no
tecido hepático86. Devido o IFN-γ estar associado à sinalização de morte celular por
apoptose mediada pela via JAK2/STAT1, o aumento dessa citocina no fígado
submetido à I/R pode ser responsável pela lesão no tecido hepático87.
Esses mediadores inflamatórios, em geral, podem causar danos ao fígado por
meio da indução do estresse oxidativo e, consequentemente, de morte celular88.
Dessa forma, o controle da síntese e liberação de citocinas em fígado esteatótico
submetidos à I/R é de grande importância para diminuir os efeitos deletéreis do
processo inflamatório no fígado.
1.4 ESTRESSE OXIDATIVO
O estresse oxidativo é caracterizado pelo desequilíbrio entre as
concentrações de substâncias pró e antioxidantes, que pode resultar no aumento da
produção de ERO e espécies reativas de nitrogênio (ERN) e em uma desordem no
estado redox celular89,90. O fígado é um órgão que fisiologicamente produz elevadas
taxas de ERO e ERN, por isso, a presença de afecções hepáticas, tais como
hepatites virais91, DHGNA92, I/R e transplante hepático42, pode intensificar a
produção desses radicais livres e induzir o estresse oxidativo.

25
A patogênese da DHGNA, incluindo a atuação do estresse oxidativo sobre o
tecido hepático, ainda é pouco conhecida. Até então, a melhor hipótese para explicar
o desenvolvimento dessa afecção é o modelo de duas fases93. A primeira fase
consiste em uma alteração metabólica, com a presença de resistência à insulina,
hiperglicemia, hiperlipidemia e o acúmulo de triglicerídeos nos hepatócitos, que
resultam em esteatose; a segunda fase é a progressão para esteatohepatite, fibrose
e cirrose94,95,96.
No fígado, a resistência à insulina, causada pelo bloqueio dos receptores de
insulina por meio do aumento dos níveis de citocina pró-inflamatória derivada do
tecido adiposo97, induz a síntese de ácidos graxos dentro dos hepatócitos, que são
acumulados em forma de triacilgliceróis ou processados pelas vias de catabolização
de ácidos graxos realizados pela mitocôndria e peroxissomos. Também ativa a
enzima Citocromo P450 no retículo endoplasmático98,99. Esses processos resultam
no aumento da síntese de ERO, ERN e outros componentes derivados do estresse
oxidativo100,101.
As ERO e ERN produzidas e liberadas pelas células hepáticas provocam
morte celular no fígado, levando à formação de Padrões Moleculares Associados a
Danos (DAMPs), que se ligam ao receptor semelhante ao Toll-4 (TLR-4) na
superfície das células de Kupffer. Como resultado, essas células induzem a
produção de citocinas e quimiocinas, o recrutamento de células inflamatórias e maior
síntese de ERO e ERN (feedback positivo)102,103.
Dessa forma, a DHGNA acaba evoluindo de uma esteatose simples para uma
esteato-hepatite, e é nesse momento que o estresse oxidativo aparece de forma
mais intensa, causando danos ainda maiores, como peroxidação lipídica,
degeneração e necrose das células do tecido hepático, morte celular por
apoptose104,105,106 e ativação das células estreladas, com consequente indução de
fibrinogênese93,107.
A I/R, na sua fase inicial, provoca a hipóxia celular, o que reduz o ATP a
adenosina monofosfato (AMP) intracelular e hipoxantina. Na fase subsequente de
reperfusão, com aumento da oferta de oxidantes, a hipoxantina é metabolizada pela
enzima xantina oxidase, formando ERO108,109,110, que podem vir a exceder a
capacidade de degradação das enzimas antioxidantes, em especial as superóxido-
desmutases45.

26
Além disso, a isquemia provoca danos nos hepatócitos e também induz a
liberação de DAMPs. Assim como ocorre na DHGNA, esses metabólitos celulares se
ligam aos seus respectivos receptores na superfície das células de Kupffer e as
ativam111, promovendo a liberação de citocinas pró-inflamatórias, quimiocinas,
ciclooxigenases e ERO43,112, e o recrutamento de neutrófilos e monócitos que
potencializam a resposta inflamatória no fígado43,113. O aumento de ERO resulta em
mais ativação das células de Kupffer, que acarretam um ciclo de autoativação10,
causando lesão direta ao DNA e peroxidação lipídica das membranas
plasmáticas108,109.
O óxido nítrico (NO) também é considerado um importante mediador
inflamatório que pode promover ou prevenir danos ao tecido3. Em condições
fisiológicas, o NO induz a vasodilatação e impede o acúmulo de células
polimorfonuclerares e a adesão plaquetária, contribuindo para que não ocorra a
obstrução dos sinusóides hepáticos114. No entanto, quando, em uma reação
inflamatória, citocinas e/ou endotoxinas induzem a síntese exacerbada de NO por
períodos prolongados, esse mediador inflamatório é capaz de reagir com ERO e
produzir peroxinitrito, um compomente reativo, tóxico e altamente oxidativo115,116.
Fuita e colaboradores (2010) mostraram que a presença de NO e nitrotirosina,
produto derivado da nitração de tirosina por ERN, está intimamente associada ao
estresse oxidativo e pode ser um indicativo de progressão da EHNA117. Durante o
processo de I/R, a diminuição de NO pode acarretar na constrição dos vasos
sanguíneos e na intensificação da lesão no fígado118,119. Em contrapartida, o
aumento exagerado na produção de NO na lesão após o procedimento de I/R
hepática, provocam citotoxicidade por meio da ação de componentes secundários,
tais como nitrito e nitrato, formados a partir das reações enzimáticas sobre o NO120.
Varela e colaboradores (2011) afirmam que a produção de NO é um dos principais
responsáveis pelos danos ocorridos no tecido hepático durante a I/R em fígado com
esteatose121.
Essas alterações histopatológicas causadas tanto pela DHGNA quanto pela
lesão de I/R, além de provocar morte celular no fígado, podem resultar na perda
generalizada de células endoteliais dos vasos sanguíneos que nutrem esse órgão, o
que desencadearia uma trombose e, conseqüente, resposta inflamatória sistêmica,
levando à insuficiência de múltiplos órgãos122,123.

27
1.5 MORTE CELULAR
Diversos processos metabólicos, tóxicos ou inflamatórios no fígado, como a
DHGNA e a I/R, podem resultar em lesão hepática por meio da ativação dos
mecanismos de morte celular principalmente por apoptose ou necrose; contudo,
outras formas de morte celular podem acontecer124,125.
A apoptose é um tipo de morte celular programada que está envolvida na
morfogênese durante o desenvolvimento embrionário e também no controle da
homeostase do tecido adulto, eliminando células envelhecidas ou prejudiciais ao
organismo126. No fígado, a apoptose é fundamental para controlar o crescimento e
regeneração desse órgão127; porém, pode intensificar as lesões quando associadas
a doenças hepáticas.
A apoptose predomina na segunda fase da DHGNA, e a ela se deve o
aumento da fragilidade das membranas, tanto externa quanto das organelas128. No
caso das lesões induzidas por I/R, as ERO e a hipóxia são as principais
responsáveis pelos mecanismos de morte celular por apoptose, particularmente os
que estão associados à via intrínseca129.
Os mecanismos de apoptose são considerados como eventos dependentes
de energia, que resultam na condensação e marginalização da cromatina,
fragmentação nuclear, encolhimento celular e formação de corpos apoptóticos,
mediante a ativação de cascatas de eventos moleculares complexos. Esse tipo de
morte celular pode ser desencadeado pela ativação de receptores de morte (via
extrínseca) ou pelo estresse intracelular, com participação da mitocôndria no
processo apoptótico (via intrínseca)129,130. Tanto os eventos de via extrínseca quanto
os de via intrínseca da apoptose são dependentes da ativação de um grupo de
proteínas pertencentes à família das proteases aspartato-específicas dependentes
de cisteína, denominadas de caspases131. Em algumas células, como nos
hepatócitos e colangiócitos, essas duas vias de apoptose podem ocorrer juntas, já
que o envolvimento mitocondrial é necessário para amplificar o sinal apoptótico dos
receptores de morte132.
Na via extrínseca da apoptose, os receptores de morte celular, ao unirem-se
ao seu ligante, recrutam o complexo de sinalização induzido por morte, que promove
a clivagem proteolítica da pró-caspases 8 e/ou 10 em caspases ativadas. Estas, por

28
sua vez, induzem a ativação das caspases efetoras 3, 6 e 7, tanto de forma direta
quanto por mecanismos que envolvem a atuação da mitocôndria125. No que se
refere à via intrínseca da apoptose, diversos estímulos podem ser responsáveis por
desencadeá-la, como estresse oxidativo, radiação gama, dano no DNA, toxinas,
estresse no retículo endoplasmático, dentre outros. As cascatas de sinalização
induzidas por esses estímulos acabam convergindo para os mecanismos de morte
celular que envolvem a mitocôndria125.
A regulação dos eventos mitocondriais da apoptose é feita por um grupo de
proteínas definidas como família Bcl-2. Algumas proteínas dessa família têm ação
pró-apoptótica, como Bcl-10, Bax, Bak, Bid, Bad, Bim, Bik, e Blk, enquanto outras
são caracteristicamente anti-apoptóticas, a exemplo de Bcl-x, Bcl-XL, Bcl-XS, Bcl-w,
BAG e a proteína que dá nome à família, Bcl-2. Todos os membros dessa família
são proteínas que atuam de maneira direta ou indireta na permeabilidade da
membrana mitocondrial, regulando da liberação de citocromo C, SMAC/DIABLO,
Fator Indutor de Apoptose (AIF) e endonuclease G. Enquanto os dois primeiros
contribuem para a cascata apoptótica que induz a ativação de caspases efetoras, os
dois últimos medeiam os mecanismos de degradação do DNA129,133,134.
Citando apenas como exemplos, outras organelas atuam no processo de
morte celular, tais como lisossomo e retículo endoplasmático. Os lisossomos, em
resposta a vários estímulos, podem apresentar aumento da permeabilidade da
membrana e liberar o seu conteúdo para o citosol135. Muitas das enzimas
lisossomais liberadas atuam na clivagem e ativação da proteína Bid e das proteínas
anti-apoptóticas Bcl-2, Bcl-xL e Mcl-1, contribuindo, dessa forma, para a cascata
apoptótica associada à mitocôndria. Este processo é denominado de mecanismo de
apoptose lisossomal136,137.
No retículo endoplasmático, quando o estresse ocorre com grande
intensidade e duração, podem ocorrer erros no mecanismo de restauração da
homeostase, o que resulta em apoptose. Um dos mediadores desse estresse é a
Proteína Homóloga da Proteína de Ligação ao Potenciador-CCAAT (CHOP), que
atua aumentando a concentração das proteínas pró-apoptóticas138 e reduzindo a
quantidade de proteínas Bcl-2139. No fígado, a presença de concentrações levadas
de ácido palmítico, um ácido graxo fisiológico, pode ser tóxico e induzir a ativação da
CHOP e consequentemente levar à apoptose140.

29
A falha em manter todos esses processos, muito dependente do consumo de
ATP, resulta em necrose. Esse tipo de morte celular é caracterizado pela perda da
homeostase iônica, aumento dos níveis de cálcio intracelular, ativação de proteases
e fosfolipases e perda da integridade mitocondrial, que culminam no edema celular e
ruptura da membrana plasmática141,142.
A necrose difere da apoptose por ser um evento com maior liberação de
precursores inflamatórios e maior repercussão inflamatória local e sistêmica. Isto
ocorre devido ao rompimento da membrana plasmática que resulta na liberação dos
componentes do interior da célula para o meio extracelular. Este é um mecanismo
de proteção contra agressões por microrganismos e presente no processo de
cicatrização. Entretanto, quando ocorre em grande intensidade, pode desencadear a
síndrome da resposta inflamatória sistêmica143,144.
Na DHGNA, pode ocorrer lesão hepatocelular na região próxima à veia
centrolobular (zona 3), que inclui baloneamento, apoptose e formação de
corpúsculos de Mallory – acúmulos grumosos ou filamentosos no citoplasma de
parte dos hepatócitos, constituído principalmente de citoqueratina e outras
proteínas145. Além disso, o estresse oxidativo associado ao processo inflamatório
acaba resultando em necrose focal ou zonal e pode provocar destruição dos
hepatócitos e desordem na arquitetura celular146.
Na lesão de I/R, a morte celular hepática é caracterizada principalmente pela
presença de necrose oncótica associada a apoptose. Nesse caso, o dano celular
inicia na fase de isquemia, com a queda na produção de ATP, e se intensifica na
fase de reperfusão, quando ocorre a ativação do sistema imune, com a presença de
células de Kupffer ativadas e infiltrado inflamatório, liberação de citocinas pró-
inflamatórias e indução do estresse oxidativo147,148.
1.6 N-ACETILCISTEÍNA (NAC)
Para combater o estresse oxidativo que pode causar a morte celular, o
organismo estimula a produção de grandes quantidades de componentes celulares
denominados de antioxidantes, que são capazes de evitar ou reduzir a oxidação de
proteínas, DNA e lipídeos no interior das células, e assim contribuir para a proteção
do tecido lesionado149,150.

30
Dentre os antioxidantes presentes nas células, a glutationa é o mais
abundante, e atinge concentrações elevadas no fígado. É um tripeptídeo composto
por glutamato, cisteína e glicina, sendo considerado um importante agente para
manter a homeostase do estado redox celular e, consequentemente, proteger as
células dos efeitos do estresse oxidativo151.
Como principais funções, a glutationa realiza o sequestro direto do radical
hidroxil e superóxido, serve como cofator para a metabolização do peróxido de
hidrogênio e peróxidos de lipídeos, restaura outros antioxidantes – como a vitamina
C e E –, regula a homeostase do óxido nítrico, entre outros152.
Camundongos knockout Gclc (h/h) específico de hepatócitos é usado para
avaliar os efeitos da redução dos níveis de glutationa no fígado. No estudo de Chen
e colaboradores (2007) foi observado que a extensa redução desse antioxidante no
tecido hepático resulta no desenvolvimento de uma esteatose grave, acompanhada
da destruição dos hepatócitos e da presença de infiltrado inflamatório153.
Para tentar reverter essa situação, a terapêutica farmacológica com
administração de antioxidantes exógenos está sendo avaliada em modelos
animais154. Dentre estes antioxidantes, a NAC vem sendo estudada em nosso
laboratório nos diferentes modelos experimentais de isquemia e reperfusão, para
avaliar seu efeito nas lesões morfológicas, bioquímicas e funcionais em diferentes
órgãos155-160.
A NAC é uma substância de baixo custo, alta disponibilidade e de uso clínico
seguro. É utilizada na prática clínica, como substância mucolítica, antídoto para
intoxicação por paracetamol, prevenção da disfunção renal por radiocontrastes,
potencializador dos efeitos hemodinâmicos da nitroglicerina e protetor contra
radiação em investigações militares161. É uma pequena molécula composta do
aminoácido cisteína ligado a um grupo acetil, e é convertida em diferentes
metabólitos, incluindo a cisteína, um precursor da glutationa, que participa
conjuntamente com a enzima glutationa peroxidase na cascata de degradação das
ERO162. Tem sido utilizada em estudos experimentais envolvendo redução de lesão
tecidual hepática em modelos experimentais de transplante de fígado, comprovando
sua eficácia ao prevenir a lesão de isquemia e reperfusão162.
Em modelo de choque hemorrágico, observou-se proteção tecidual em
pulmão, avaliando o comprometimento sistêmico155,156, e em fígado submetido à I/R

31
após choque hemorrágico157, assim como a sobrevida dos animais nesses modelos.
Na I/R hepática associada à hepatectomia foi observado efeito protetor enzimático e
morfológico ao fígado158,159. E, no estudo em que foi realizada a associação da NAC
com o precondicionamento isquêmico, não se observou efeito sinérgico ou
antagônico no tecido hepático ou pulmonar após I/R hepática, uma vez que o efeito
protetor foi similar160.
Apesar das evidências experimentais citadas acima em relação aos efeitos
benéficos da NAC em diferentes afecções, Robinson e colaboradores (2013)
encontraram dificuldades em provar as vantagens do uso da NAC no seu estudo em
pacientes submetidos à ressecção hepática segmentar163. Esse fato evidencia a
necessidade de uma maior compreensão do uso da NAC em lesão hepática
resultante de I/R. Além disso, há carência de estudos na literatura dos efeitos
exercidos pelo uso desse antioxidante exógeno na inflamação, estresse oxidativo e
morte celular em modelos de I/R hepática associada à DHGNA; o que justifica a
busca por fármacos reguladores do potencial inflamatório e oxidativo, como a NAC,
com utilidade clínica para esse tipo de afecção hepática.

32
2 OBJETIVO

33
2 OBJETIVO
Avaliar o efeito da NAC sobre a resposta inflamatória, o estresse oxidativo e a
morte celular em camundongos com esteato-hepatite submetidos à I/R hepática.

34
3 MÉTODOS

35
3 MÉTODOS
3.1 TIPO DE ESTUDO
Esta é uma pesquisa experimental, transversal, controlada e aleatorizada.
Todos os animais foram manipulados de acordo com “Os Princípios Éticos da
Experimentação Animal da União Internacional Protetora dos Animais” e da Lei Nº
11.794, de 8 de outubro de 2008. Este projeto foi submetido à Comissão de Ética em
Pesquisa da FMUSP, e foi aprovado sob o parecer nº 243/13 (Anexo A e B).
3.2 AMOSTRA
A amostra utilizada neste projeto constou de 48 camundongos (Mus
musculus), da linhagem C57BL/6J, machos, com idade de oito semanas e peso
entre 25 e 30 gramas. Os animais provenientes do Centro de Bioterismo da FMUSP
foram operados no Laboratório de Fisiopatologia Cirúrgica (LIM 62) da FMUSP.
Aqueles provenientes do Centro de Bioterismo do Instituto Evandro Chagas (IEC)
foram operados no Laboratório de Microscopia Eletrônica (LME) do IEC. Todos os
animais passaram por esse procedimento operatório após período de adaptação e
controle nutricional no biotério setorial de cada laboratório.
Durante o experimento, os animais foram alojados em gaiolas apropriadas
com as dimensões 40x30x25cm, em grupos de no máximo três animais e foram
alimentados com ração específica para o grupo ao qual pertenciam e água ad
libitum. O ritmo circadiano dos animais foi respeitado, mantendo as condições
sanitárias adequadas. Antes do experimento, os animais tiveram restrição de
alimentos sólidos por quatro horas.
O grupo de animas que foi submetido à alimentação esteatogênica recebeu
uma dieta Deficiente em Metionina e Colina (DMC) (Anexo C). Esta dieta é
constituída majoritariamente de amido de milho, dextrina, sucrose, extrato de
amendoim e óleo vegetal, assim como outros componentes em menor quantidade,
objetivando um total de metionina de 300mg/kg de alimento, e com cloridrato de
colina totalizando 50mg/kg de alimento. É uma dieta elaborada e fornecida

36
comercialmente para o propósito de indução de DHGNA em modelos animais para
estudo29.
Os animais foram separados, por sorteio, em quatro grupos contendo 12
animais cada – seis desses animais foram manipulados no LIM-62 e os outros seis,
no IEC –, sendo esses grupos designados de acordo com a dieta alimentar e o
procedimento operatório realizado: Grupo Controle, composto por animais
submetidos apenas à anestesia e laparotomia; Grupo DMC, composto por animais
alimentados com dieta DMC e submetidos apenas à anestesia e laparotomia; Grupo
DMC-I/R, composto por animais alimentados com dieta DMC e submetidos à I/R; e
Grupo DMC-I/R+NAC, composto por animais alimentados com dieta DMC,
submetidos à I/R e tratados com NAC (Figura 1).
Figura 1– Distribuição dos grupos experimentais
Grupo DMC = Grupo dieta DMC; Grupo DMC-I/R = Grupo dieta DMC-Isquemia/Reperfusão; Grupo DMC-I/R+NAC = Grupo dieta DMC-Isquemia/Reperfusão + N-acetilcisteína. Fonte: desenho esquemático feito pelo próprio autor (2017).
3.3 PROCEDIMENTO OPERATÓRIO
Após pesados, os animais receberam uma solução de atropina intramuscular
de 0,04mg/kg, 10 minutos antes da indução anestésica. Depois, os animais foram

37
anestesiados com uma associação dos fármacos xilazina (10 mg/Kg) e cetamina (70
mg/kg) por via intramuscular, na face lateral da pata traseira direita. O animal foi
considerado em plano anestésico quando houve a perda dos reflexos córneo-
palpebral e de retirada da pata traseira contralateral, ao estímulo doloroso por
preensão digital. Quando necessário, foi utilizado dose complementar de anestésico,
correspondendo à metade da dose inicial.
Em seguida, realizou-se a tricotomia e antissepsia com álcool a 70% da
região abdominal e procedeu-se a hidratação com soro fisiológico a 0,9%, em
volume correspondente a 80mL/Kg do peso corpóreo. Nesse momento, foi feito o
sorteio do procedimento a ser realizado nos animais alimentados com ou sem dieta
DMC: anestesia e laparotomia, I/R ou I/R e tratamento com NAC.
Os animais foram fixados com fita crepe na bancada cirúrgica sobre colchão
térmico a 38oC e, utilizando-se o microscópio cirúrgico nos aumentos 6 a 16 vezes
bem como instrumental microcirúrgico apropriado, foram então submetidos ao
procedimento sorteado.
Para a realização da I/R hepática seletiva, foi ocluído o pedículo hepático
acima da emergência dos ramos dos segmentos lateral direito, lobo caudado e
processo papilar com clampe vascular microcirúrgico durante 30 minutos (Figura 2),
seguido de 24 horas de reperfusão. Nos animais dos grupos apropriados, a NAC foi
administrada lentamente na veia cava abdominal, na dose de 150 mg/kg de peso
corporal, 15 minutos antes da isquemia.
A parede abdominal dos animais foi suturada em dois planos – peritônio-
músculo-aponeurótico e pele –, ambos com fio de polipropileno 7-0. Ao término do
procedimento e recuperação da anestesia, os camundongos foram mantidos em
gaiolas e receberam água e ração ad libitum.
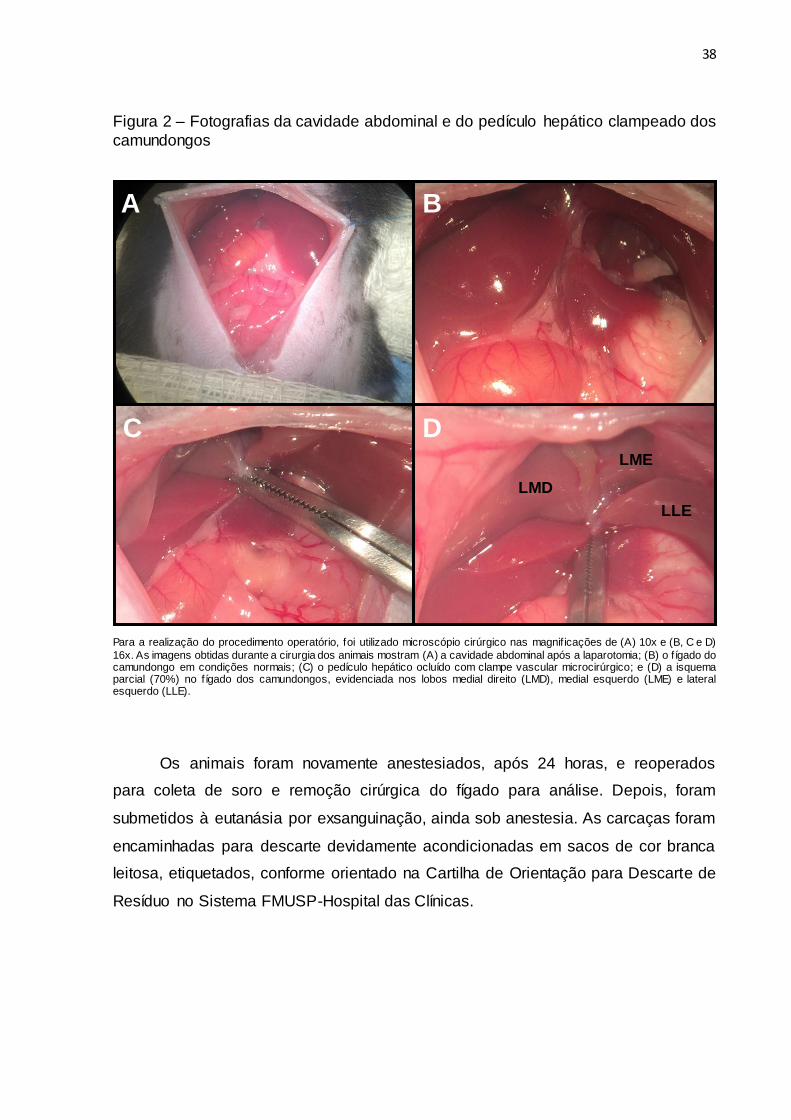
38
Figura 2 – Fotografias da cavidade abdominal e do pedículo hepático clampeado dos
camundongos
Para a realização do procedimento operatório, foi utilizado microscópio cirúrgico nas magnif icações de (A) 10x e (B, C e D)
16x. As imagens obtidas durante a cirurgia dos animais mostram (A) a cavidade abdominal após a laparotomia; (B) o fígado do camundongo em condições normais; (C) o pedículo hepático ocluído com clampe vascular microcirúrgico; e (D) a isquema parcial (70%) no fígado dos camundongos, evidenciada nos lobos medial direito (LMD), medial esquerdo (LME) e lateral esquerdo (LLE).
Os animais foram novamente anestesiados, após 24 horas, e reoperados
para coleta de soro e remoção cirúrgica do fígado para análise. Depois, foram
submetidos à eutanásia por exsanguinação, ainda sob anestesia. As carcaças foram
encaminhadas para descarte devidamente acondicionadas em sacos de cor branca
leitosa, etiquetados, conforme orientado na Cartilha de Orientação para Descarte de
Resíduo no Sistema FMUSP-Hospital das Clínicas.
A B
C D
LMD
LME
LLE

39
3.4 PROCESSAMENTO HISTOLÓGICO
Após a remoção cirúrgica do fígado, uma parte do lobo hepático lateral
esquerdo do camundongo foi fixada em formol 10%, submetido ao processamento
histológico e emblocado em parafina. Os blocos foram posteriormente cortados com
espessura de 4µm em micrótomo Modelo RM2235 (Leica/Alemanha) e corados com
H/E.
Os cortes foram, então, avaliados em microscópio óptico Modelo Imager.A2
(Zeiss/Alemanha) por examinador que desconhecia o grupo ao qual pertencia o
animal, em parâmetros definidos por Kleiner e colaboradores (2005)164, conforme
segue no Apêndice A.
Após essa análise, o material foi fotografado com câmera Modelo AxioCam
MRc (Zeiss/Alemanha) acoplada ao microscópio citado, e as imagens foram
trabalhadas no software AxioVision LE (Zeiss/Alemanha).
3.5 DOSAGEM BIOQUÍMICA DE TRANSAMINASES
O sangue foi coletado por punção cardíaca com seringa hipodérmica de 1mL
e agulha de 25 x 0,60 descartáveis, foi centrifugado a 956g por 15 minutos a 4°C, e
o soro foi coletado, congelado em nitrogênio líquido e mantido em freezer -80°C para
a análise de ALT e AST, realizada no Núcleo Multiusuário do Laboratório de
Análises Especiais da FMUSP.
3.6 PREPARO DAS AMOSTRAS PARA AS DOSAGENS DE CITOCINAS, TBARS E
NITRITO
A porção do tecido hepático dos camundongos que não foi utilizada para o
processamento histológico (item 3.4), foi congelada em nitrogênio líquido e
armazenada em freezer -80°C. Posteriormente, uma parte desse tecido hepático foi
homogeneizada em tampão PHEM 1X, pH 7.2 – para a dosagem de citocinas – e
outra parte em tampão fosfato, pH 7.2 – para as dosagens de TBARS e nitrito –
todos na concentração de 100mg/mL.O sobrenadante coletado foi armazenado em
freezer -80°C até o momento da realização dos testes experimentais citados.

40
3.6.1 Dosagem de citocinas
A dosagem de citocinas no tecido hepático foi feita por meio do Ensaio
Imunoenzimático (ELISA do tipo sandwich). Essa técnica foi realizada conforme
instruções do fabricante do kit BD OptEIATM (BD Biosciences/EUA) para analisar a
expressão das citocinas IL-1β, TGF-β e IFN-γ no tecido hepático.
Placas de 96 poços foram incubadas com anticorpo de captura específico
para cada citocina, overnight a uma temperatura de 4°C. As placas sensibilizadas
foram lavadas com uma solução contendo PBS e Tween 20 a 0,05% e depois
incubadas com 200μL de uma solução de bloqueio composta de PBS e 10% de SBF
por um período de uma hora a temperatura ambiente.
As placas foram novamente lavadas e a elas foram adicionadas os
sobrenadantes do fígado dos camundongos e das diluições seriadas da curva
padrão; seguida da incubação por duas horas à temperatura ambiente – para a
quantificação da citocina TGF-β1, as amostras foram previamente tratadas com
ácido clorídrico (HCl) por uma hora e depois neutralizadas com hidróxido de sódio
(NaOH) a 1N. Após esse intervalo de incubação, as placas foram lavadas cinco
vezes e incubadas por uma hora à temperatura ambiente com uma solução
contendo anticorpo de detecção biotinilado, conjugado de estreptavidina ligada à
peroxidase e solução de trabalho (ver instrução do kit) – para a dosagem da citocina
IL-1β, as amostras e as diluições seriadas da curva padrão devem ser incubadas
primeiramente com o anticorpo de detecção, por uma hora, e depois com o
conjugado de estreptavidina-peroxidase, por 30 minutos. As placas foram lavadas e
incubadas com o cromógeno (TMB - Tetrametilbenzidina) por um período de 15 a 30
minutos, à temperatura ambiente e protegidas da luz. Após incubação, a reação foi
interrompida com ácido sulfúrico (H2SO4) a 2N e lida em espectofotômetro Modelo
ELx800TM (BIO-TEK/EUA), com filtro decomprimento de onda de 450nm.
3.6.2 Dosagem de TBARS
Uma alíquota do sobrenadante do tecido hepático homogeneizado foi utilizada
para a dosagem de proteínas totais pelo método de Bradford. As amostras foram
diluídas na proporção de 1:10, misturadas a 300µL do Reagente de Bradford e lidas

41
em espectrofotômetro Modelo TP-Reader (ThermoPlate/China), com comprimento
de onda de 595nm. Após a dosagem de proteínas totais, foi realizada a dosagem de
TBARS. Às amostras de fígado, também diluídas na proporção de 1:10, foram
adicionadas 250µL de ácido tricloroacético (TCA) a 17,5% e 250µL de ácido
tiobarbitúrico (TBA) a 0,6%, incubando a solução por 15 minutos em banho-maria a
95°C. Depois, 250µL de TCA a 70% foram acrescentados a essa solução,
mantendo-a incubada por 20 minutos a 4°C. A absorbância da solução final foi então
mensurada em espectrofotômetro Modelo TP-Reader (ThermoPlate/China), com
filtro de comprimento de onda de 534nm.
3.6.3 Dosagem de nitrito
Para quantificação de nitrito foi realizada a técnica do Reagente de Griess. O
limite de detecção para esse método é de 1,0 μM de nitrito.
Foram colocados 50 μL de cada amostra em uma placa de 96 poços,
juntamente com as diluições previstas no kit emtampão apropriado para produzir a
curva padrão. Em seguida, nos poços que continham as amostras biológicas e as
diluições seriadas da curva padrão foram adicionados 50μL do reagente de Griess,
anteriormente preparado com volumes iguais do componente A (N-(1-naphthyl)
ethylenediamine dihydrochloride) e do componente B (ácido sulfanílico), incubando a
placa por uma hora à temperatura ambiente. A amostra de referência fotométrica foi
preparada misturando-se 50μL do reagente de Griess e 50μL de água deionizada.
Após incubação, a absorbância das amostras foi medida em espectrofotômetro
Modelo ELx800TM (BIO-TEK/EUA), utilizando filtro de comprimento de onda de
548nm.
3.7 DETECÇÃO DE Bcl-2 POR IMUNOHISTOQUÍMICA
Para a imunomarcação da proteína Bcl-2, foi utilizada a técnica de
imunoperoxidase usando um kit comercial VECTASTAIN® ABC (Vector
Laboratories/EUA), seguindo instruções do fabricante com adaptações. Os cortes
foram incubados com ácido cítrico a 0,1M por 30 minutos em panela de pressão
para fazer a recuperação antigênica. Depois foram permeabilizados com uma

42
solução de Triton X100 a 0,5%, os sítios inespecíficos foram bloqueados em solução
contendo soro de cabra a 10%, soro fetal bovino a 10% e BSA a 3%; foram
incubados overnight em anticorpo primário, depois em anticorpo secundário por três
horas, a peroxidase endógena foi inativada com peróxido de hidrogênio a 0,3%, e
por fimos cortes foram incubados com o reagente ABC por uma hora.
As reações imunohistoquímicas foram reveladas com solução reveladora
DAB/níquel, e os cortes marcados foram montados entre lâmina/lamínula com
Entellan (Merk/Alemanha), analisados em microscópio óptico de campo claro
(Axiophot - Zeiss/Alemanha) e fotografados com câmera digital (AxioCam HRC -
Zeiss/Alemanha). Os anticorpos primário e secundário utilizados nos ensaios de
imunomarcação foram anti-Bcl2 policlonal (Novus Biologicals - EUA) e Reagente IgG
anti-cabra biotinilado (ThermoFisher Scientific - EUA), respectivamente.
3.8 DETECÇÃO DE APOPTOSE PELA TÉCNICA DE TUNEL
A presença de células apoptóticas foi investigada pelo método de marcação
de dUTP terminais em fitas simples de DNA mediadas por deoxinucleotidil terminal
transferase (TUNEL), em cortes histológicos com 4 µm de espessura. Foi utilizado o
In Situ Cell Death Detection Kit, POD (Roche Applied Science/Alemanha) com a
finalidade de identificar morfologicamente as células em processo de apoptose via
marcação de fragmentos terminais de DNA (porção 3'-OH), caracteristicamente
relacionado com a fragmentação do DNA nuclear.
Os cortes foram desparafinizados em xilol, mergulhados em álcool absoluto e
em diluições decrescentes de álcool (95%, 90%, 80%, 70%, 50%) por cinco minutos
em cada solução. Depois, foram lavados em tampão fosfato com salina (PBS), pH
7.2, incubados em solução de proteínas k (20µg/mL) por 20 minutos à temperatura
ambiente, lavados em água destilada, e incubados em peróxido de hidrogênio a 2%
por cinco minutos. Para a recuperação antigênica, os cortes foram incubados em
solução de proteinase k, por 15 minutos à temperatura ambiente, e para bloquear a
peroxidase endógena, foi utilizada uma solução de H2O2 a 3% em metanol puro v/v,
incubando os cortes nessa solução por cinco minutos. Após lavagem com PBS, os
antígenos inespecíficos foram bloqueados com albumina bovina (BSA) a 1% e SBF
a 20% em tampão Tris a 0,1M, pH 7.5, por uma hora à temperatura ambiente. Os

43
cortes foram, então, permeabilizados com Triton X-100 a 0,2% em tampão citrato de
sódio, pH 6,0, durante 30 minutos, depois incubados na solução label (POD kit) por
duas horas, lavados com PBS e incubados com na solução converter (POD kit) por
30 minutos.
Em seguida, os cortes foram novamente lavados em PBS e incubados com o
substrato para peroxidase (3,3’-diaminobenzidine tetrahydrochloride dihydrate -
DAB) (Merck/Alemanha) para a revelação da técnica. Ao final, os cortes foram
montados entre lâmina e lamínula, o material foi analisado e fotografado com
câmera Modelo AxioCam MRc (Zeiss/Alemanha) acoplada ao microscópio Modelo
Imager.A2 (Zeiss/Alemanha), e as imagens foram trabalhadas no software
AxioVision LE (Zeiss/Alemanha).
3.9 ANÁLISE ESTATÍSTICA
Os valores bioquímicos de transaminases, dos níveis de citocinas, TBARS e
nitrito, e o número de células apoptóticas estão expressos em média ± desvio
padrão. Para a análise estatística dos resultados foi aplicado o teste ANOVA one
way, pós-teste de Bonferroni, utilizando o software GraphPad Prism 5. Em todos os
testes foi utilizado o nível de significância em 5% (p<0,05).

44
4 RESULTADOS

45
4 RESULTADOS
4.1 ANÁLISE HISTOPATOLÓGICA
Com a análise histológica, puderam ser observadas as diferenças entre os
grupos experimentais com relação à presença de vesículas de gordura e de focos
inflamatórios e à estrutura morfológica do hepatócito, após a ingestão ou não da
dieta DMC, e a associação dessa dieta com o procedimento de I/R.
Os animais do grupo Controle apresentaram fígado com arquitetura normal do
parênquima (Figura 3). Nos animais do grupo DMC observou-se esteatose hepática
macrovesicular panacinar de nível 3, com presença de inflamação lobular de nível 3
e inflamação portal (Figura 4). Já nos camundongos do grupo DMC-I/R (Figura 5A),
além dessas características mencionadas, foi evidenciada também a presença de
hepatócitos com baloneamento de nível 1 na maioria das lâminas avaliadas (Figura
5B).
No grupo DMC-I/R+NAC, foi observado que a maior parte dos camundongos
apresentou esteatose hepática macrovesicular periportal de nível 3, sendo possível
evidenciar uma diferença na extensão do parênquima comprometido, mas não no
grau de esteatose, quando comparado ao grupo DMC-I/R. Além disso, foi observada
uma redução da quantidade de focos inflamatórios no tecido hepático em quase
todos os animais do grupo DMC-I/R+NAC, em comparação aos animais do grupo
DMC-I/R, apresentando apenas inflamação lobular de nível 2. No entanto, foram
encontrados hepatócitos com baloneamento de nível 1 de forma similar ao grupo
DMC-I/R (Figura 6).
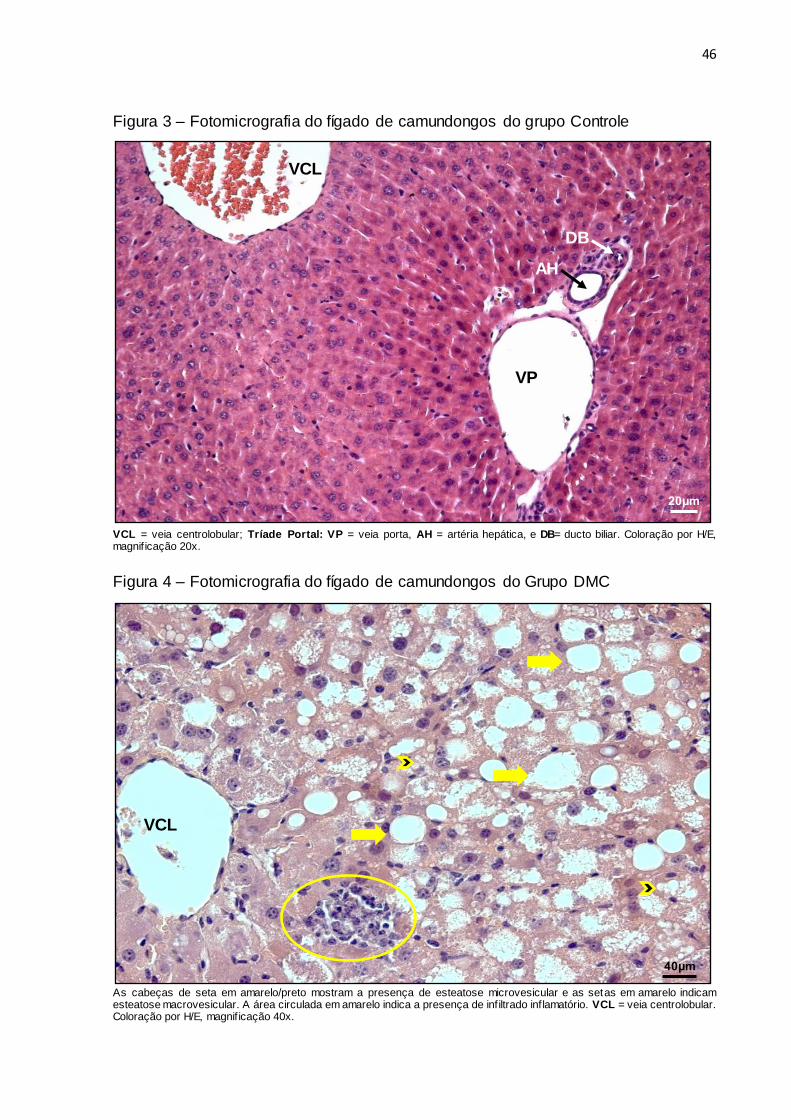
46
Figura 3 – Fotomicrografia do fígado de camundongos do grupo Controle
VCL = veia centrolobular; Tríade Portal: VP = veia porta, AH = artéria hepática, e DB= ducto biliar. Coloração por H/E, magnif icação 20x.
Figura 4 – Fotomicrografia do fígado de camundongos do Grupo DMC
As cabeças de seta em amarelo/preto mostram a presença de esteatose microvesicular e as setas em amarelo indicam esteatose macrovesicular. A área circulada em amarelo indica a presença de infiltrado inflamatório. VCL = veia centrolobular. Coloração por H/E, magnif icação 40x.
20μm20μm
VCL
VP
AH
DB
20μm
40μm
VCL
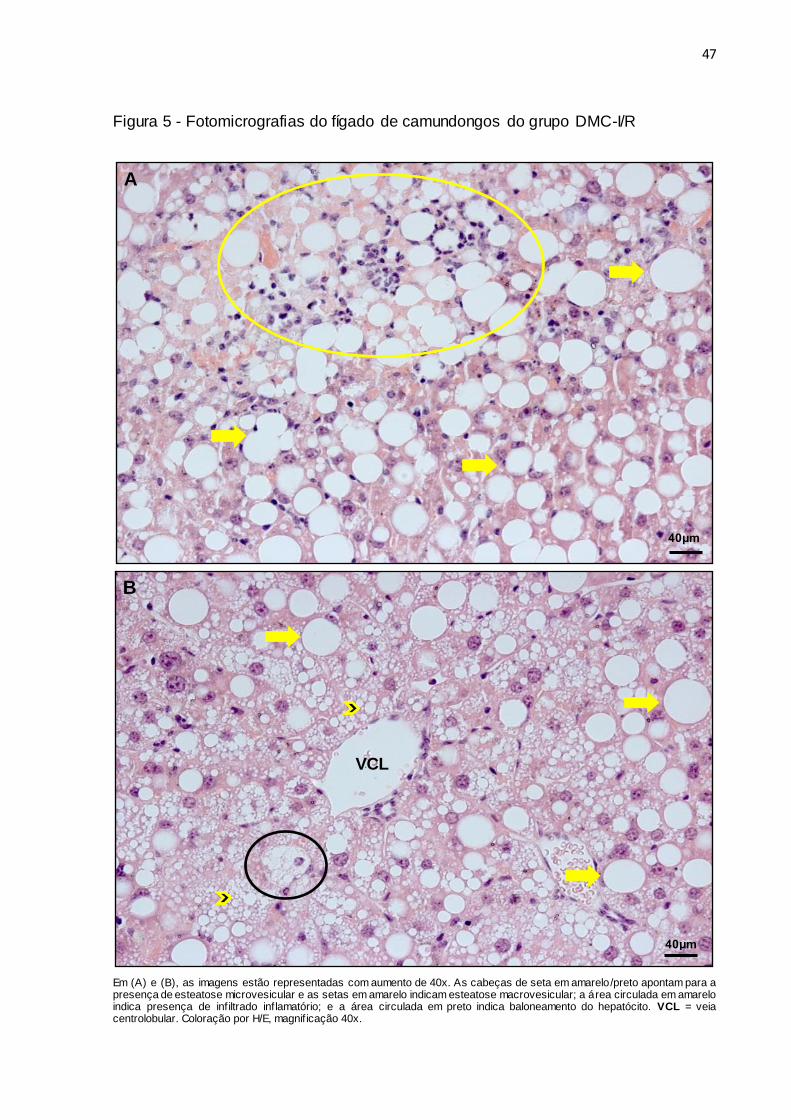
47
Figura 5 - Fotomicrografias do fígado de camundongos do grupo DMC-I/R
Em (A) e (B), as imagens estão representadas com aumento de 40x. As cabeças de seta em amarelo/preto apontam para a presença de esteatose microvesicular e as setas em amarelo indicam esteatose macrovesicular; a área circulada em amarelo indica presença de infiltrado inflamatório; e a área circulada em preto indica baloneamento do hepatócito. VCL = veia centrolobular. Coloração por H/E, magnif icação 40x.
A
40μm
B
40μm
VCL
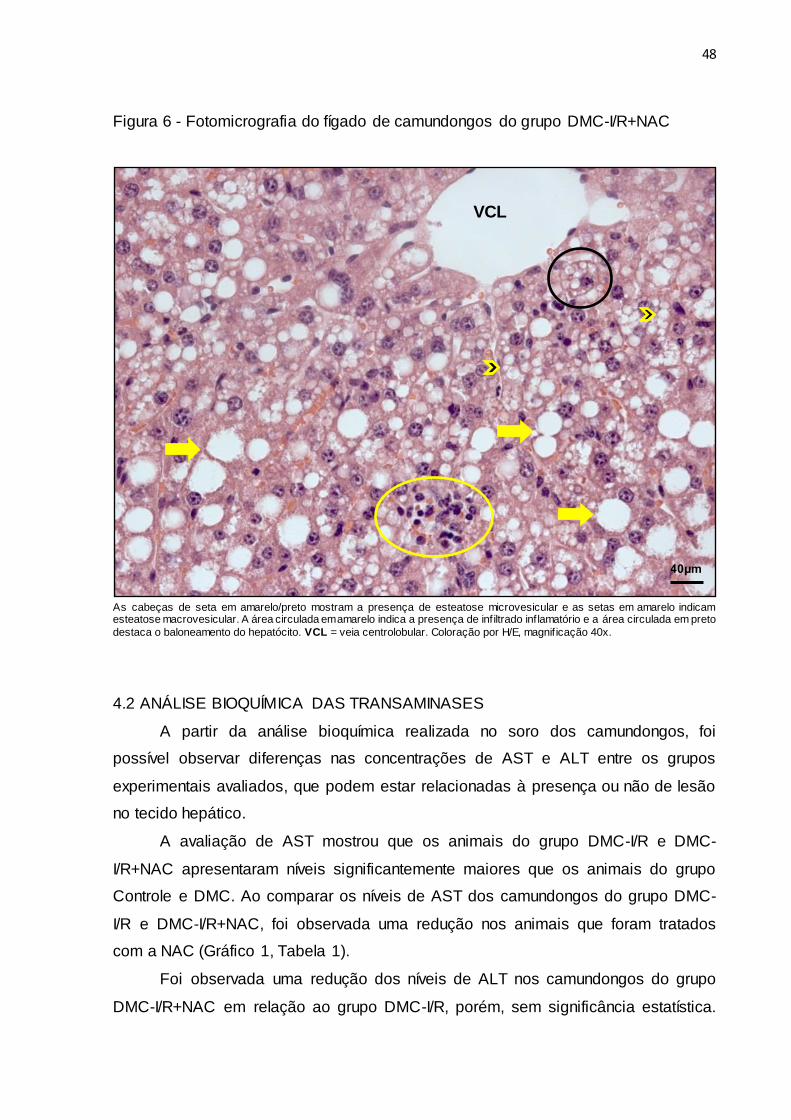
48
Figura 6 - Fotomicrografia do fígado de camundongos do grupo DMC-I/R+NAC
As cabeças de seta em amarelo/preto mostram a presença de esteatose microvesicular e as setas em amarelo indicam esteatose macrovesicular. A área circulada em amarelo indica a presença de infiltrado inflamatório e a área circulada em preto
destaca o baloneamento do hepatócito. VCL = veia centrolobular. Coloração por H/E, magnif icação 40x.
4.2 ANÁLISE BIOQUÍMICA DAS TRANSAMINASES
A partir da análise bioquímica realizada no soro dos camundongos, foi
possível observar diferenças nas concentrações de AST e ALT entre os grupos
experimentais avaliados, que podem estar relacionadas à presença ou não de lesão
no tecido hepático.
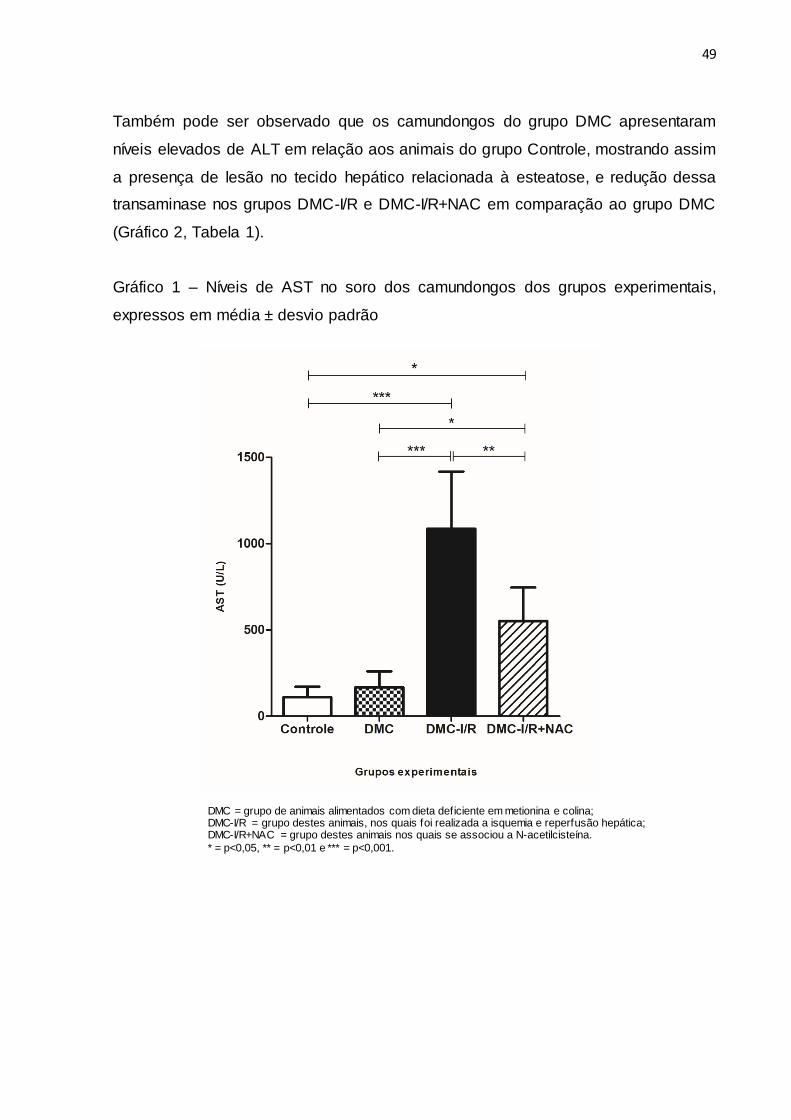
A avaliação de AST mostrou que os animais do grupo DMC-I/R e DMC-
I/R+NAC apresentaram níveis significantemente maiores que os animais do grupo
Controle e DMC. Ao comparar os níveis de AST dos camundongos do grupo DMC-
I/R e DMC-I/R+NAC, foi observada uma redução nos animais que foram tratados
com a NAC (Gráfico 1, Tabela 1).
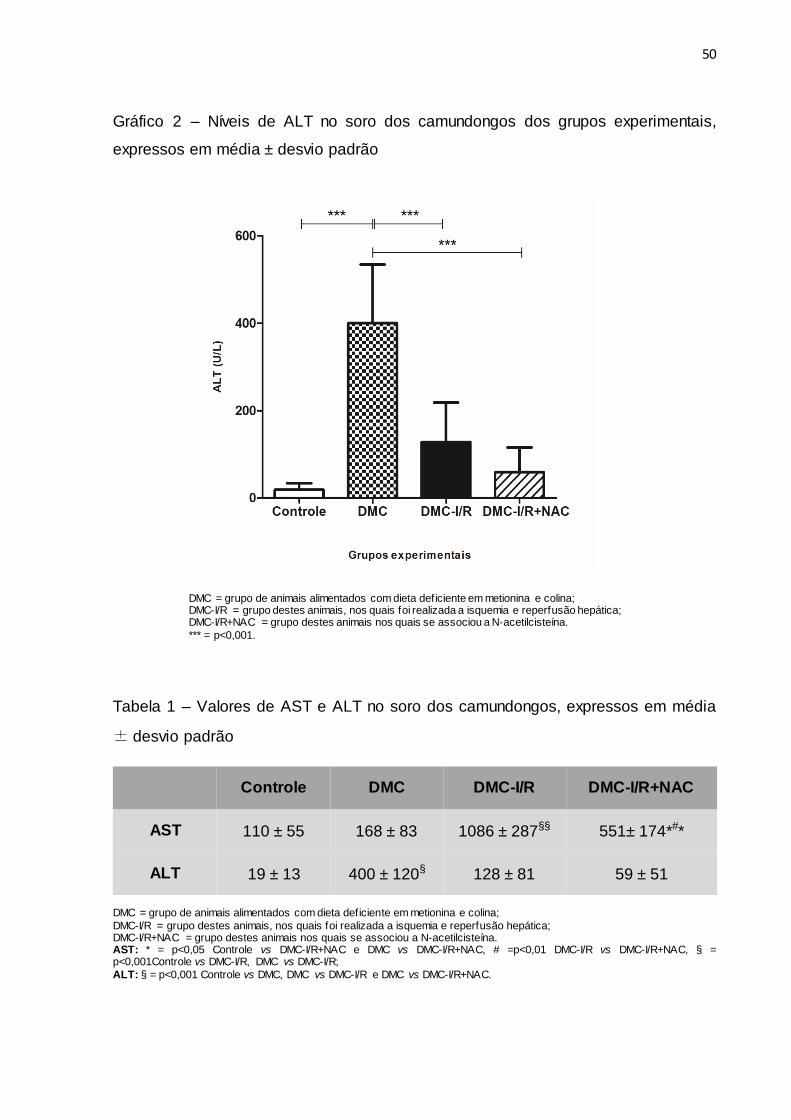
Foi observada uma redução dos níveis de ALT nos camundongos do grupo
DMC-I/R+NAC em relação ao grupo DMC-I/R, porém, sem significância estatística.
VCL
40μm
49
Também pode ser observado que os camundongos do grupo DMC apresentaram
níveis elevados de ALT em relação aos animais do grupo Controle, mostrando assim
a presença de lesão no tecido hepático relacionada à esteatose, e redução dessa
transaminase nos grupos DMC-I/R e DMC-I/R+NAC em comparação ao grupo DMC
(Gráfico 2, Tabela 1).
Gráfico 1 – Níveis de AST no soro dos camundongos dos grupos experimentais,
expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína.
* = p<0,05, ** = p<0,01 e *** = p<0,001.
50
Gráfico 2 – Níveis de ALT no soro dos camundongos dos grupos experimentais,
expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína.
*** = p<0,001.
Tabela 1 – Valores de AST e ALT no soro dos camundongos, expressos em média
± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina;
DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. AST: * = p<0,05 Controle vs DMC-I/R+NAC e DMC vs DMC-I/R+NAC, # =p<0,01 DMC-I/R vs DMC-I/R+NAC, § = p<0,001Controle vs DMC-I/R, DMC vs DMC-I/R;
ALT: § = p<0,001 Controle vs DMC, DMC vs DMC-I/R e DMC vs DMC-I/R+NAC.
Controle DMC DMC-I/R DMC-I/R+NAC
AST 110 ± 55 168 ± 83 1086 ± 287§§ 551± 174*#*
ALT 19 ± 13 400 ± 120§ 128 ± 81 59 ± 51
51
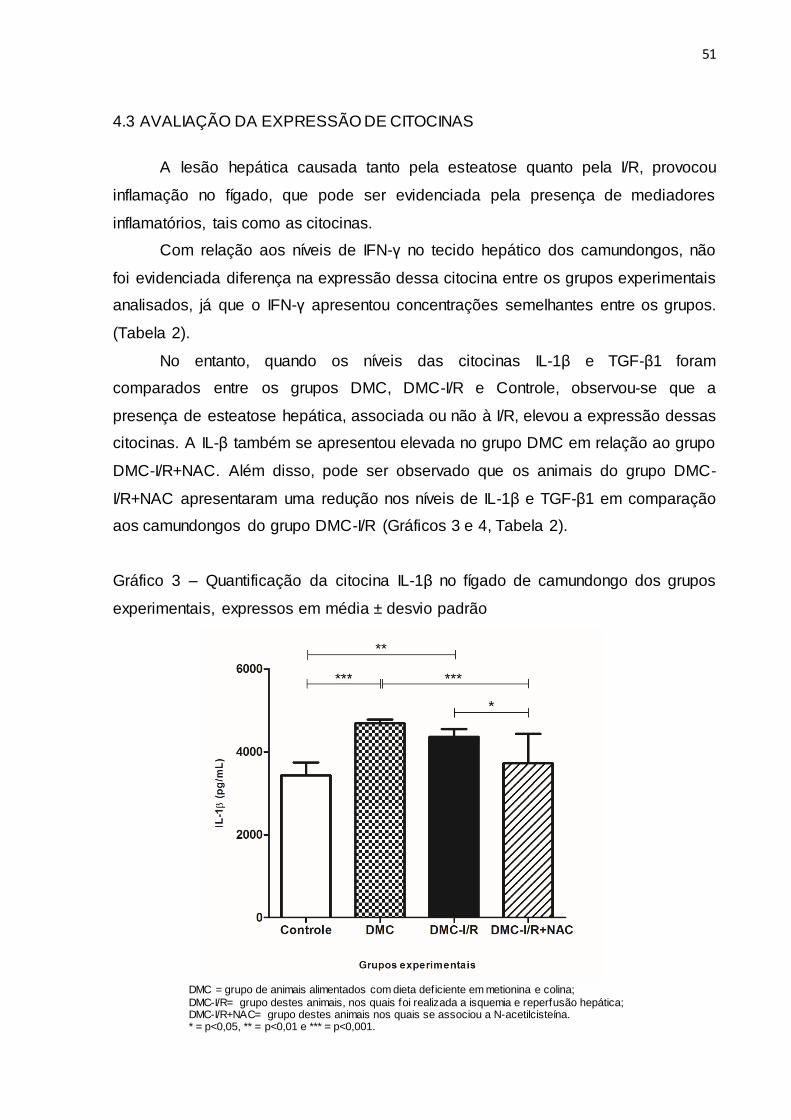
4.3 AVALIAÇÃO DA EXPRESSÃO DE CITOCINAS
A lesão hepática causada tanto pela esteatose quanto pela I/R, provocou
inflamação no fígado, que pode ser evidenciada pela presença de mediadores
inflamatórios, tais como as citocinas.
Com relação aos níveis de IFN-γ no tecido hepático dos camundongos, não
foi evidenciada diferença na expressão dessa citocina entre os grupos experimentais
analisados, já que o IFN-γ apresentou concentrações semelhantes entre os grupos.
(Tabela 2).
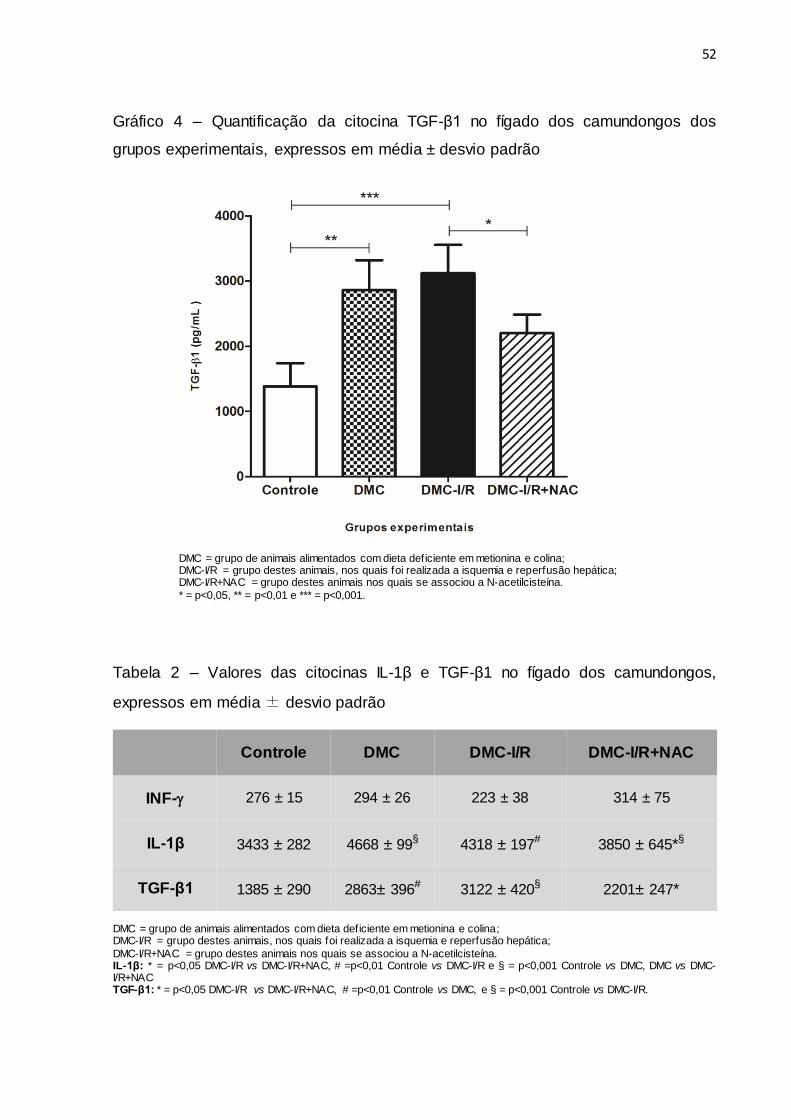
No entanto, quando os níveis das citocinas IL-1β e TGF-β1 foram
comparados entre os grupos DMC, DMC-I/R e Controle, observou-se que a
presença de esteatose hepática, associada ou não à I/R, elevou a expressão dessas
citocinas. A IL-β também se apresentou elevada no grupo DMC em relação ao grupo
DMC-I/R+NAC. Além disso, pode ser observado que os animais do grupo DMC-
I/R+NAC apresentaram uma redução nos níveis de IL-1β e TGF-β1 em comparação
aos camundongos do grupo DMC-I/R (Gráficos 3 e 4, Tabela 2).
Gráfico 3 – Quantificação da citocina IL-1β no fígado de camundongo dos grupos
experimentais, expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina;
DMC-I/R= grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC= grupo destes animais nos quais se associou a N-acetilcisteína. * = p<0,05, ** = p<0,01 e *** = p<0,001.
52
Gráfico 4 – Quantificação da citocina TGF-β1 no fígado dos camundongos dos
grupos experimentais, expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína.
* = p<0,05, ** = p<0,01 e *** = p<0,001.
Tabela 2 – Valores das citocinas IL-1β e TGF-β1 no fígado dos camundongos,
expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática;
DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. IL-1β: * = p<0,05 DMC-I/R vs DMC-I/R+NAC, # =p<0,01 Controle vs DMC-I/R e § = p<0,001 Controle vs DMC, DMC vs DMC-I/R+NAC TGF-β1: * = p<0,05 DMC-I/R vs DMC-I/R+NAC, # =p<0,01 Controle vs DMC, e § = p<0,001 Controle vs DMC-I/R.
Controle DMC DMC-I/R DMC-I/R+NAC
INF- 276 ± 15 294 ± 26 223 ± 38 314 ± 75
IL-1β 3433 ± 282 4668 ± 99§ 4318 ± 197# 3850 ± 645*§
TGF-β1 1385 ± 290 2863± 396# 3122 ± 420§ 2201± 247*
53
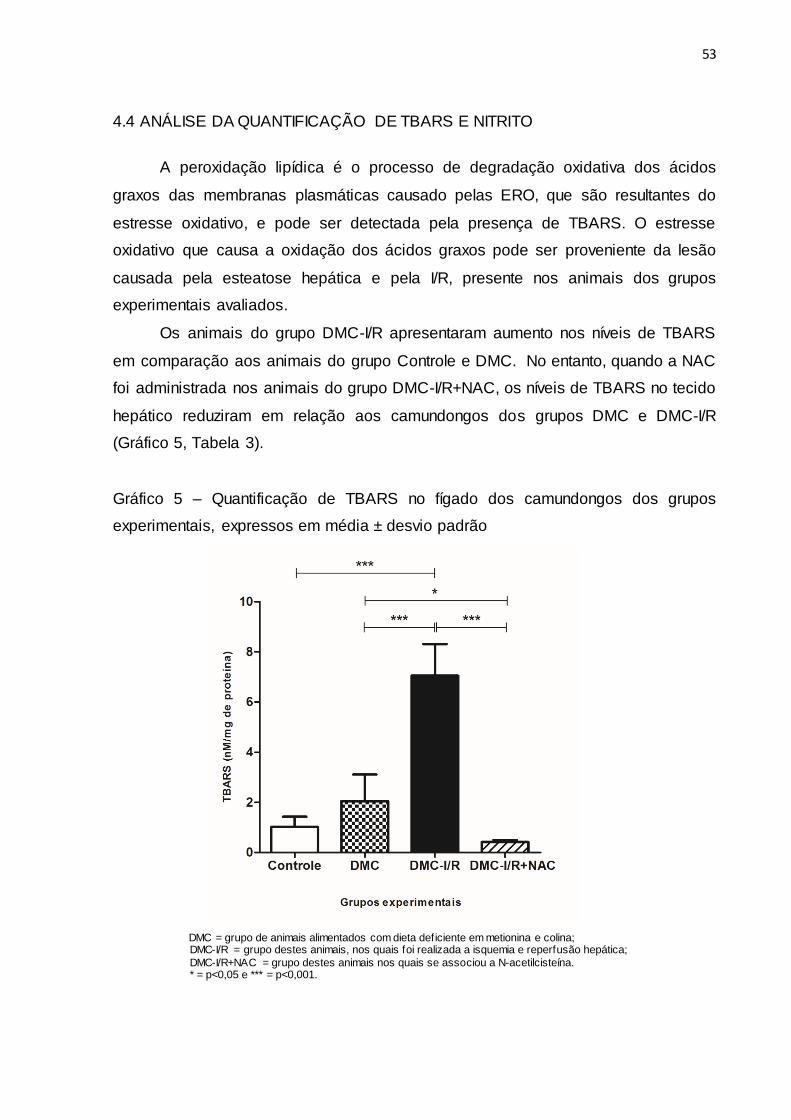
4.4 ANÁLISE DA QUANTIFICAÇÃO DE TBARS E NITRITO
A peroxidação lipídica é o processo de degradação oxidativa dos ácidos
graxos das membranas plasmáticas causado pelas ERO, que são resultantes do
estresse oxidativo, e pode ser detectada pela presença de TBARS. O estresse
oxidativo que causa a oxidação dos ácidos graxos pode ser proveniente da lesão
causada pela esteatose hepática e pela I/R, presente nos animais dos grupos
experimentais avaliados.
Os animais do grupo DMC-I/R apresentaram aumento nos níveis de TBARS
em comparação aos animais do grupo Controle e DMC. No entanto, quando a NAC
foi administrada nos animais do grupo DMC-I/R+NAC, os níveis de TBARS no tecido
hepático reduziram em relação aos camundongos dos grupos DMC e DMC-I/R
(Gráfico 5, Tabela 3).
Gráfico 5 – Quantificação de TBARS no fígado dos camundongos dos grupos
experimentais, expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática;
DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. * = p<0,05 e *** = p<0,001.
54
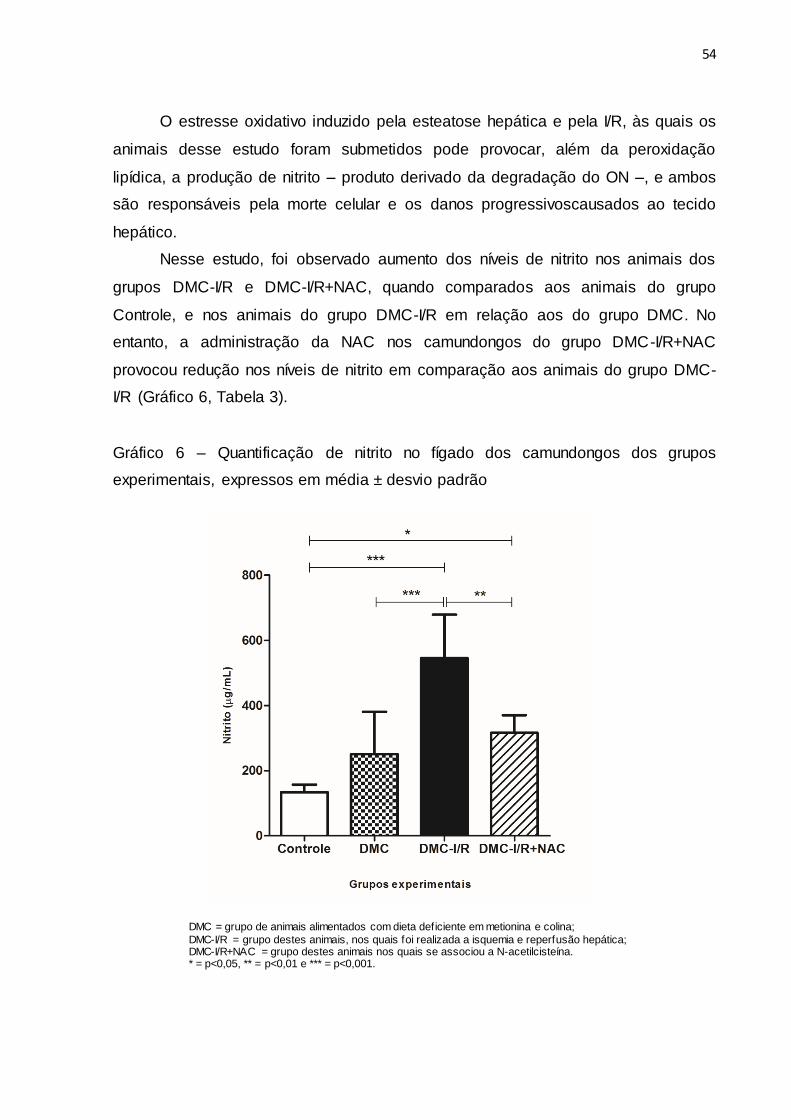
O estresse oxidativo induzido pela esteatose hepática e pela I/R, às quais os
animais desse estudo foram submetidos pode provocar, além da peroxidação
lipídica, a produção de nitrito – produto derivado da degradação do ON –, e ambos
são responsáveis pela morte celular e os danos progressivoscausados ao tecido
hepático.
Nesse estudo, foi observado aumento dos níveis de nitrito nos animais dos
grupos DMC-I/R e DMC-I/R+NAC, quando comparados aos animais do grupo
Controle, e nos animais do grupo DMC-I/R em relação aos do grupo DMC. No
entanto, a administração da NAC nos camundongos do grupo DMC-I/R+NAC
provocou redução nos níveis de nitrito em comparação aos animais do grupo DMC-
I/R (Gráfico 6, Tabela 3).
Gráfico 6 – Quantificação de nitrito no fígado dos camundongos dos grupos
experimentais, expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina;
DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. * = p<0,05, ** = p<0,01 e *** = p<0,001.

55
Tabela 3 – Valores de TBARS e nitrito no fígado dos camundongos, expressos em
média ± desvio padrão
TBARS: * = p<0,05 DMC vs DMC-I/R+NAC, § = p<0,001 Controle vs DMC-I/R, DMC vs DMC-I/R e DMC-I/R vs DMC-I/R+NAC; Nitrito: * = p<0,05 Controle vs DMC-I/R+NAC, # =p<0,01 DMC-I/R vs DMC-I/R+NACe § = p<0,001 Controle vs DMC-I/R, DMC vs DMC-I/R.
4.5 ANÁLISE DA EXPRESSÃO DE Bcl-2
A Bcl-2 é uma proteína que faz parte da família de proteínas que regulam os
eventos mitocondriais relacionados à morte celular por apoptose, atuando direta ou
indiretamente sobre a permeabilidade da membrana das mitocôndrias. A presença
dessa proteína no tecido indica a ação anti-apoptótica das células diante de uma
injúria.
No fígado de camundongos do grupo Controle, foi observada a presença da
proteína Bcl-2 em uma grande área do tecido hepático (Figura 7); enquanto que, nos
animaisdo grupo DMC, a área onde a proteína Bcl-2 foi marcada é menor que a do
grupo Controle (Figura 8). Assim como no grupo DMC, os camundongos do grupo
DMC-I/R apresentaram menor marcação de Bcl-2 no tecido hepático que no fígado
dos animais do grupo Controle (Figura 9). Entretanto, quando a NAC foi
admisnistrada nos camundongos do grupo DMC-I/R+NAC, a presença de Bcl-2 no
tecido hepático foi aumentada (Figura 10), em comparação aos animais dos grupos
DMC e DMC-I/R (Figura 8 e 9).
Controle DMC DMC-I/R DMC-I/R+NAC
TBARS 1,0 ± 0,4 2,0 ± 1,0 7,3 ± 1,3§§ 0,4 ± 0,1§*
Nitrito 134 ± 21 250 ± 117 546 ± 108§§ 316 ± 49*#
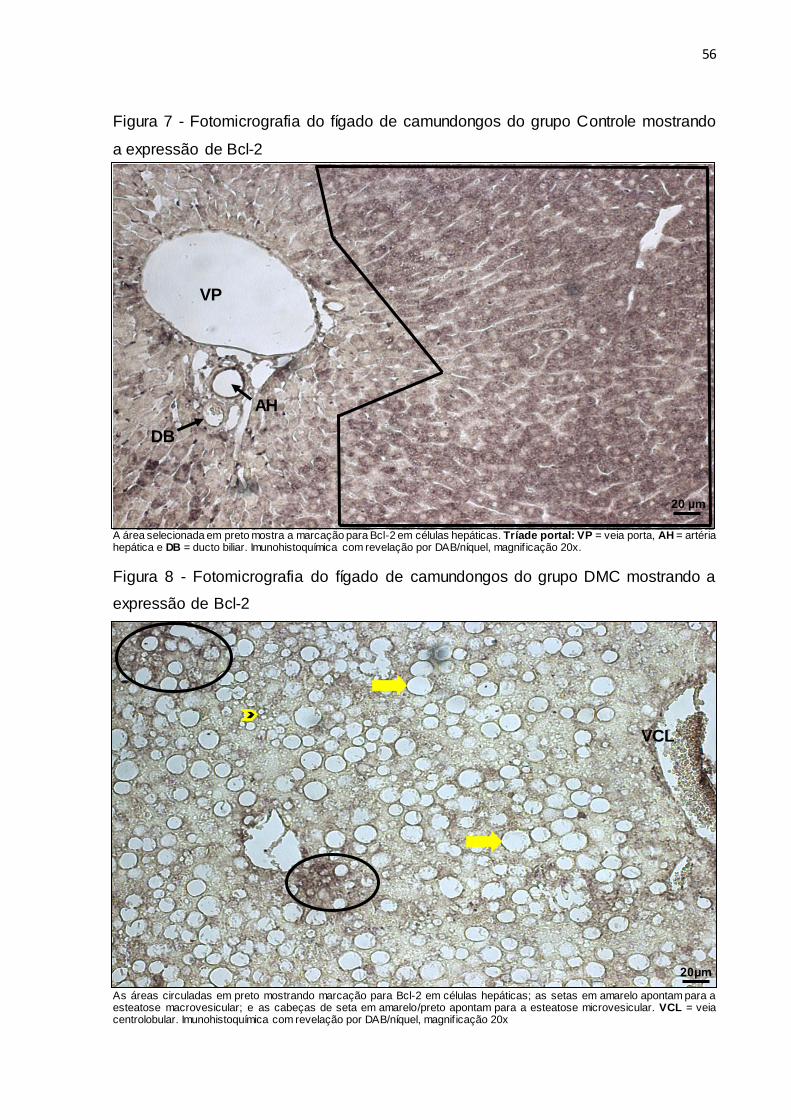
56
Figura 7 - Fotomicrografia do fígado de camundongos do grupo Controle mostrando
a expressão de Bcl-2
A área selecionada em preto mostra a marcação para Bcl-2 em células hepáticas. Tríade portal: VP = veia porta, AH = artéria hepática e DB = ducto biliar. Imunohistoquímica com revelação por DAB/níquel, magnif icação 20x.
Figura 8 - Fotomicrografia do fígado de camundongos do grupo DMC mostrando a
expressão de Bcl-2
As áreas circuladas em preto mostrando marcação para Bcl-2 em células hepáticas; as setas em amarelo apontam para a esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose microvesicular. VCL = veia centrolobular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 20x
VP
20 µm
AH
DB
VCL
20μm
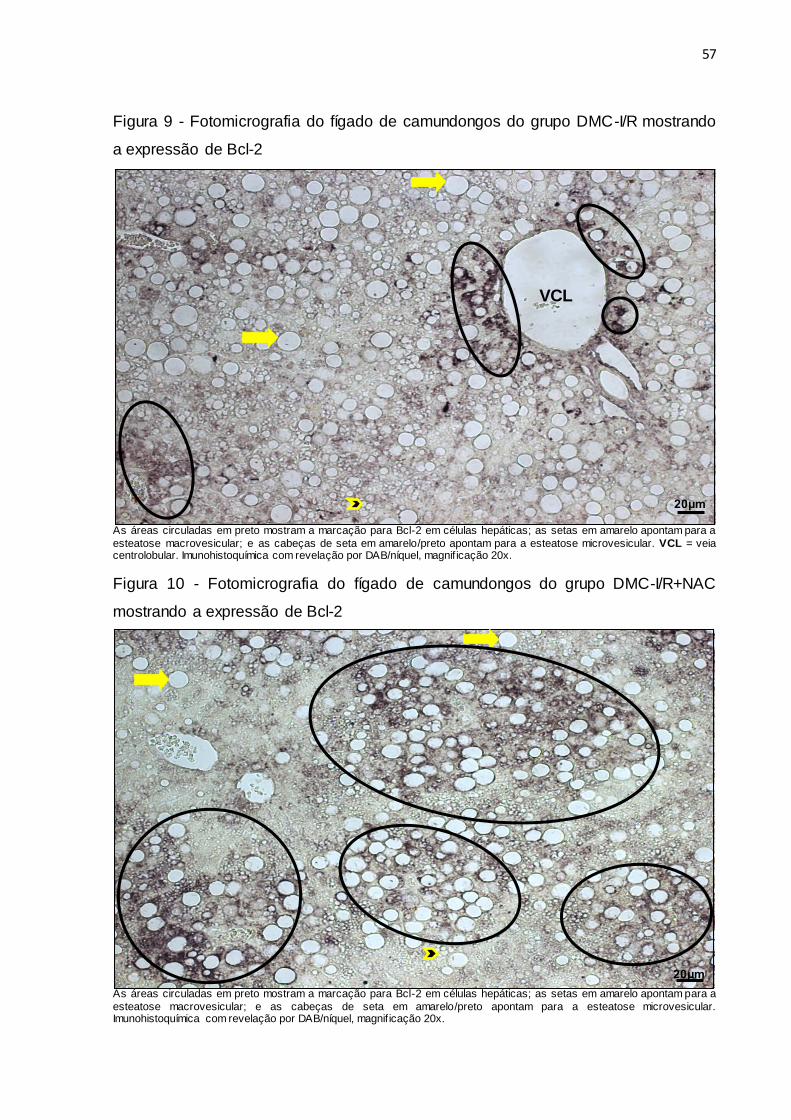
57
Figura 9 - Fotomicrografia do fígado de camundongos do grupo DMC-I/R mostrando
a expressão de Bcl-2
As áreas circuladas em preto mostram a marcação para Bcl-2 em células hepáticas; as setas em amarelo apontam para a
esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose microvesicular. VCL = veia centrolobular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 20x.
Figura 10 - Fotomicrografia do fígado de camundongos do grupo DMC-I/R+NAC
mostrando a expressão de Bcl-2
As áreas circuladas em preto mostram a marcação para Bcl-2 em células hepáticas; as setas em amarelo apontam para a
esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose microvesicular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 20x.
VCL
20μm
20μm
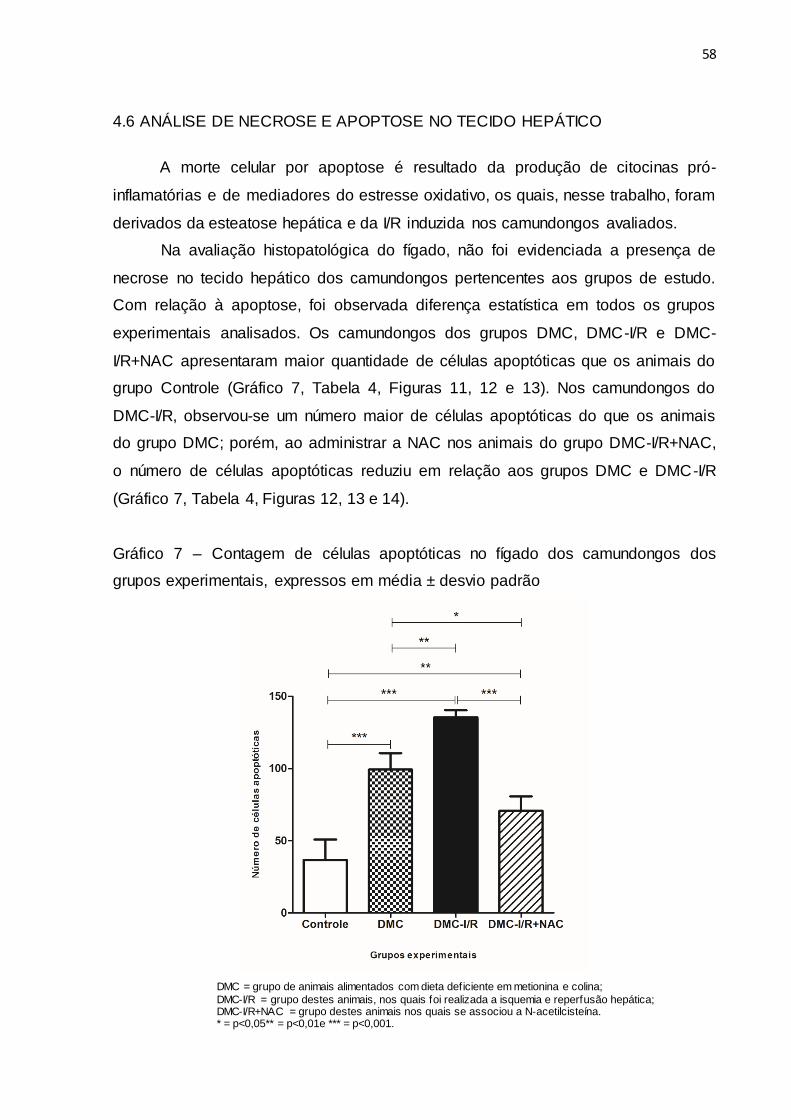
58
4.6 ANÁLISE DE NECROSE E APOPTOSE NO TECIDO HEPÁTICO
A morte celular por apoptose é resultado da produção de citocinas pró-
inflamatórias e de mediadores do estresse oxidativo, os quais, nesse trabalho, foram
derivados da esteatose hepática e da I/R induzida nos camundongos avaliados.
Na avaliação histopatológica do fígado, não foi evidenciada a presença de
necrose no tecido hepático dos camundongos pertencentes aos grupos de estudo.
Com relação à apoptose, foi observada diferença estatística em todos os grupos
experimentais analisados. Os camundongos dos grupos DMC, DMC-I/R e DMC-
I/R+NAC apresentaram maior quantidade de células apoptóticas que os animais do
grupo Controle (Gráfico 7, Tabela 4, Figuras 11, 12 e 13). Nos camundongos do
DMC-I/R, observou-se um número maior de células apoptóticas do que os animais
do grupo DMC; porém, ao administrar a NAC nos animais do grupo DMC-I/R+NAC,
o número de células apoptóticas reduziu em relação aos grupos DMC e DMC-I/R
(Gráfico 7, Tabela 4, Figuras 12, 13 e 14).
Gráfico 7 – Contagem de células apoptóticas no fígado dos camundongos dos
grupos experimentais, expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina;
DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática; DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. * = p<0,05** = p<0,01e *** = p<0,001.
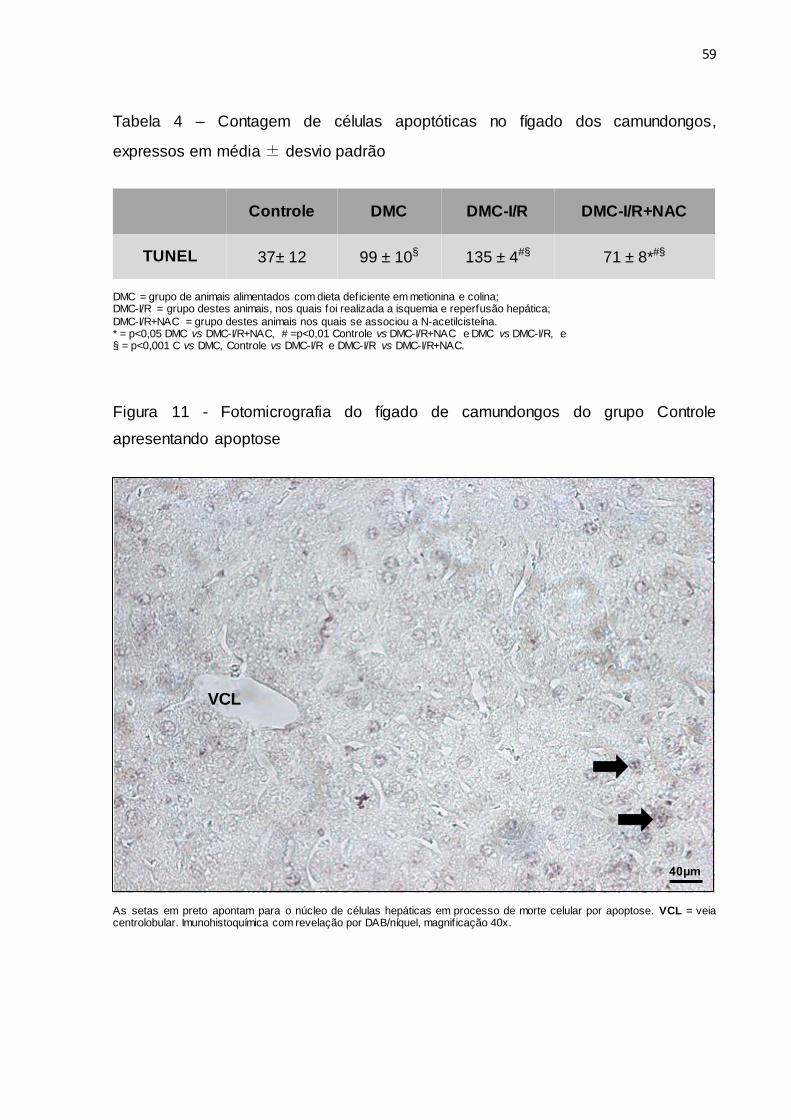
59
Tabela 4 – Contagem de células apoptóticas no fígado dos camundongos,
expressos em média ± desvio padrão
DMC = grupo de animais alimentados com dieta deficiente em metionina e colina; DMC-I/R = grupo destes animais, nos quais foi realizada a isquemia e reperfusão hepática;
DMC-I/R+NAC = grupo destes animais nos quais se associou a N-acetilcisteína. * = p<0,05 DMC vs DMC-I/R+NAC, # =p<0,01 Controle vs DMC-I/R+NAC e DMC vs DMC-I/R, e § = p<0,001 C vs DMC, Controle vs DMC-I/R e DMC-I/R vs DMC-I/R+NAC.
Figura 11 - Fotomicrografia do fígado de camundongos do grupo Controle
apresentando apoptose
As setas em preto apontam para o núcleo de células hepáticas em processo de morte celular por apoptose. VCL = veia centrolobular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 40x.
Controle DMC DMC-I/R DMC-I/R+NAC
TUNEL 37± 12 99 ± 10§ 135 ± 4#§ 71 ± 8*#§
VCL
40μm
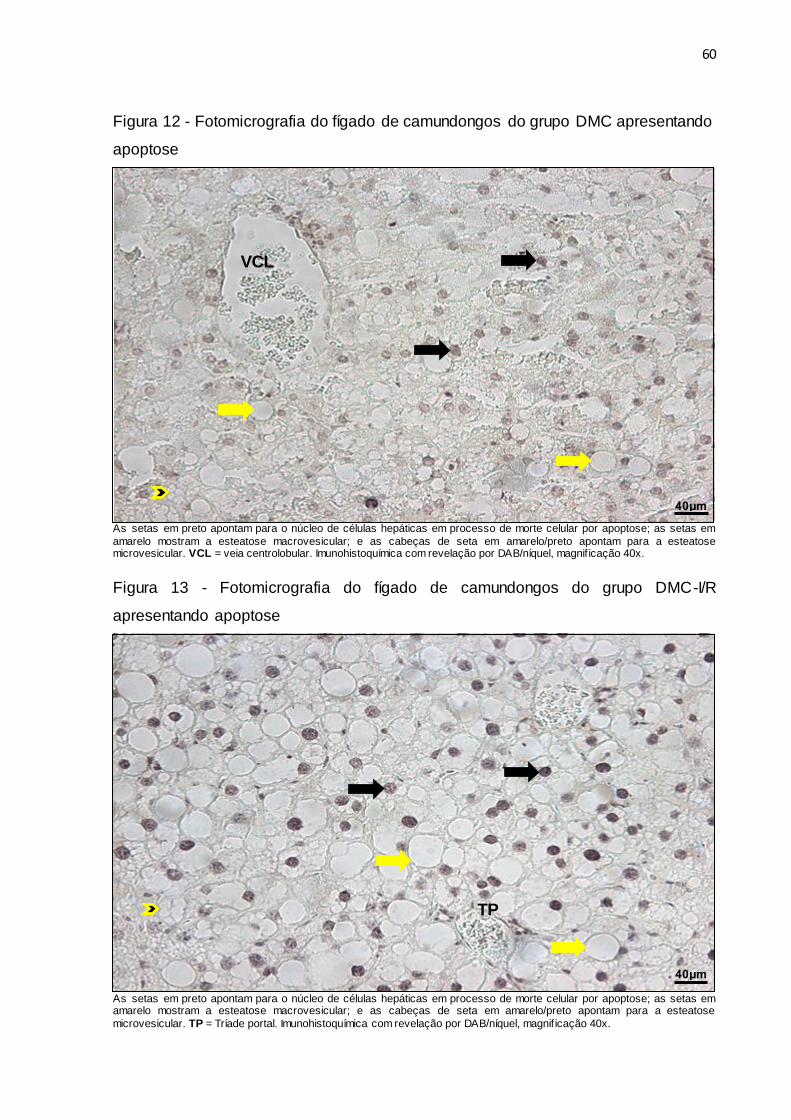
60
Figura 12 - Fotomicrografia do fígado de camundongos do grupo DMC apresentando
apoptose
As setas em preto apontam para o núcleo de células hepáticas em processo de morte celular por apoptose; as setas em
amarelo mostram a esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose microvesicular. VCL = veia centrolobular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 40x.
Figura 13 - Fotomicrografia do fígado de camundongos do grupo DMC-I/R
apresentando apoptose
As setas em preto apontam para o núcleo de células hepáticas em processo de morte celular por apoptose; as setas em amarelo mostram a esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose
microvesicular. TP = Tríade portal. Imunohistoquímica com revelação por DAB/níquel, magnif icação 40x.
VCL
40μm
40μm
TP
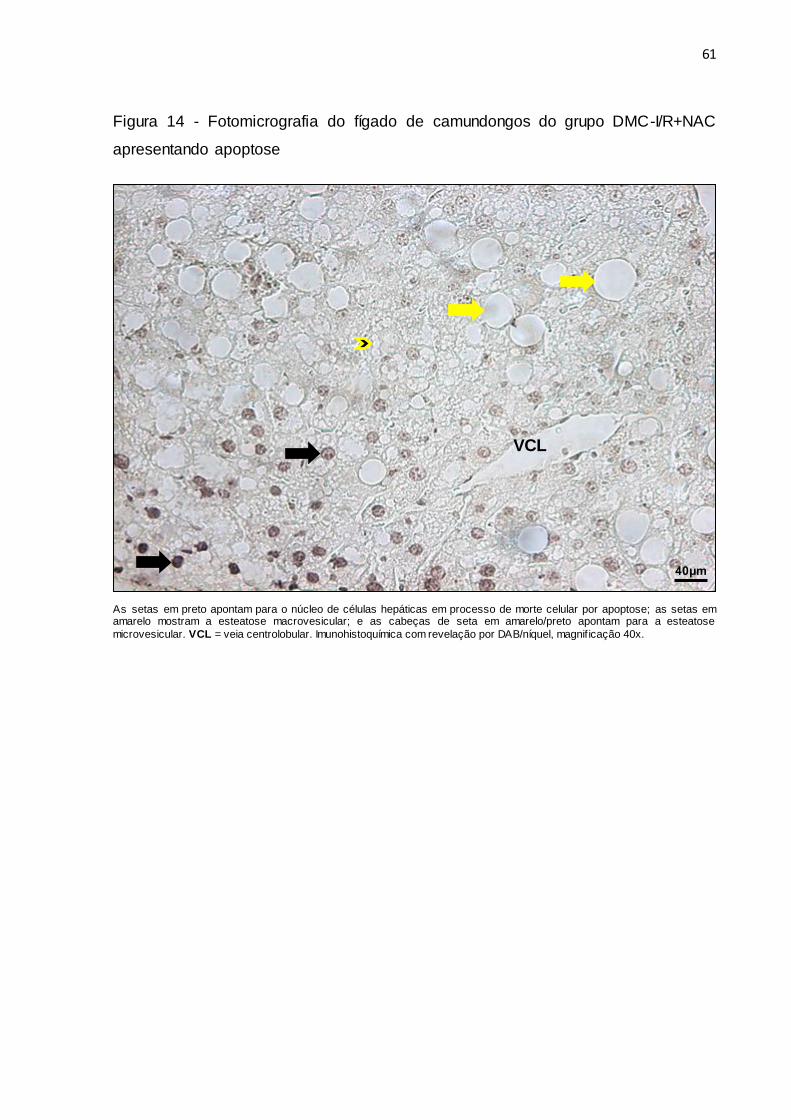
61
Figura 14 - Fotomicrografia do fígado de camundongos do grupo DMC-I/R+NAC
apresentando apoptose
As setas em preto apontam para o núcleo de células hepáticas em processo de morte celular por apoptose; as setas em amarelo mostram a esteatose macrovesicular; e as cabeças de seta em amarelo/preto apontam para a esteatose
microvesicular. VCL = veia centrolobular. Imunohistoquímica com revelação por DAB/níquel, magnif icação 40x.
40μm
VCL

62
5 DISCUSSÃO

63
5 DISCUSSÃO
Em nosso estudo, a NAC foi capaz de reduzir a inflamação, por meio da
diminuição dos focos inflamatórios e dos níveis das citocinas IL-1β e TGF-β1, e o
estresse oxidativo, por meio da redução dos níveis de TBARS e nitrito, no fígado de
camundongos com esteato-hepatite submetidos à I/R hepática. Além disso, a NAC
promoveu a diminuição da quantidade de células apoptóticas no tecido hepático
acometido com essas afecções.
A DHGNA é uma das doenças mais comuns no mundo, principalmente em
países nos quais o comportamento sedentário e a alimentação com alto teor calórico
são bastante evidenciados. Juntamente com a obesidade e a resistência à insulina,
as quais desempenham um papel fundamental na síndrome metabólica, a incidência
da DHGNA vem aumentando ao longo dos anos165,166. Estas afecções podem
agravar situações que requeiram cirurgia, eletiva ou de urgência, traumática ou não.
Em transplantes, tem se intensificado o uso de doadores limítrofes, nos quais
a esteatose é um dos fatores agravantes na lesão de preservação e de isquemia e
reperfusão167,168,169. Desta forma, o presente estudo foi pensado para avaliar uma
estratégia de diminuir as repercussões deletérias na associação da DHGNA com I/R,
baseando-se no controle dos fatores que provocam lesões no tecido hepático, como
a inflamação, o estresse oxidativo e a morte celular, os quais podem prejudicar a
recuperação de um paciente.
Para tanto, foi utilizado nesse trabalho um agente farmacológico de baixa
custo e fácil acesso, a NAC, que tem sido usado como antioxidante no tratamento da
intoxicação por acetaminofeno, bronquite, encefalopatias hepáticas, toxicidade por
metais pesados, entre outros enfermidades170. Quando metabolizada, a NAC torna-
se um precursor da glutationa e atua no sequestro dos radicais livres – responsáveis
pelo processo de morte celular nos diferentes tecidos –, protegendo o fígado do
dano oxidativo171,172. Sagias e colaboradores (2010) mostraram que o uso desse
agente antioxidante é vantajoso durante o isolamento de hepatócitos de doadores de
tecido hepático esteatótico173. A partir desse estudo, a NAC é agora utilizada nos
protocolos de processamento de hepatócitos de fígados com esteatose para
transplante de células. Segundo González e colaboradores (2009), a co-
administração de NAC, coenzima Q10 e MnTBAP, uma substância equivalente à
superóxido dismutase, aumentou a expressão das subunidades do complexo I do

64
canal de transporte de elétrons mitocondrial, e reduziu a produção de ERO, a
disfunção mitocondrial e a morte celular dos hepatócitos174. Wang e colaboradores
(2014) também demonstraram que o uso da NAC pode prevenir os mecanismos de
morte celular por apoptose e autofagia induzidas pela I/R175.
Esses estudos mostram o potencial da NAC na proteção do fígado contra os
danos causados tanto pela DHGNA quanto pela I/R, separadamente. Desssa forma,
consideramos que a NAC também poderia apresentar efeito protetor quando essas
duas afecções fossem associadas. Tal efeito foi confirmado em nosso trabalho, já
que a NAC reduziu a inflamação, o estresse oxidativo e a morte celular em
camundongos com esteato-hepatite submetidos à I/R hepática.
Uma das formas mais empregadas em trabalhos experimentais para a
compreensão das doenças que afetam humanos e a análise de uma possível
estratégia farmacológica para tais doenças é o uso de modelos animais. Em nosso
estudo, foram utilizados camundongos da linhagem C57BL/6J alimentados com
dieta DMC como modelo animal para desenvolver esteato-hepatite, sendo estes
posteriormente submetidos à I/R e tratados com NAC.
A escolha desse modelo foi baseada no fato de que os roedores são os
animais mais rotineiramente utilizados nas pesquisas de DHGNA. Além disso, a
linhagem de camundongos C57BL/6J machos é muito usada para simular algumas
doenças metabólicas ou desordens lipídicas em diferentes modelos experimentais,
tais como os de animais geneticamente modificados para desenvolver obesidade ou
aqueles em que a DHGNA é induzida por meio de dieta específica176.
Os animais geneticamente obesos, como os camundongos ob/ob,
apresentam ocorrência natural de uma mutação de ponto espontânea no gene da
Leptina (Lepob, ob/ob), levando à deficiência absoluta desse hormônio derivado de
adipócitos, o que resulta em hiperfagia e obesidade acompanhada de uma síndrome
semelhante a diabetes do tipo 2, na qual se observa intolerância à glicose e
hiperglicemia177. Além disso, Ohyama e colaboradores (2014) mostraram que esses
camundongos desenvolvem esteatose hepática, com níveis elevados de ALT no
fígado178.
Os camundongos alimentados com dieta DMC, por sua vez, apresentam
perda de peso, redução dos níveis de colesterol, triglicerídeos, glicose, insulina e
leptina, além de apresentarem esteatose hepática, inflamação lobular e

65
baloneamento dos hepatócitos179. Caballero e colaboradores (2010) afirmaram que,
nos animais com DHGNA induzida pela dieta DMC, a falta de metionina pode ser
responsável pela maioria dos efeitos deletéreis no fígado dos camundongos, tais
como, perda de peso, lesão hepatocelular, estresse oxidativo, inflamação e fibrose,
e a deficiência de colina foi a principal responsável pelo desenvolvimento de
esteatose hepática180.
Para definir o melhor modelo animal de DHGNA associada à I/R, realizamos
um estudo prévio, no qual foram avaliados a inflamação e o estresse oxidativo no
tecido hepático de camundongos ob/ob e de camundongos alimentados com dieta
DMC submetidos à I/R. Nesse estudo, observamos que os animais dos dois
modelos experimentais avaliados apresentavam o mesmo nível de inflamação,
porém, nos camundongos alimentados com dieta DMC observou-se níveis mais
elevados de AST, TBARS e nitrito no tecido hepático, após I/R, do que nos animais
ob/ob. Dessa forma, optamos por utilizar o modelo animal de DHGNA induzida pela
dieta DMC em camundongos C57BL/6J devido ter-se mostrado mais adequado para
avaliar a lesão de I/R em fígado com esteato-hepatite e estudar o efeito protetor da
NAC sobre essas afecções associadas.
A partir das avaliações histopatológicas foi observado que a NAC apresentou
efeitos benéficos em relação à reação inflamatória presente no fígado esteatótico
submetido à I/R hepática, já que promoveu a redução dos focos de infiltrado
inflamatório. A redução da inflamação no fígado foi observada por Samuhasaneeto e
colaboradores (2007), no qual foram utilizadas diferentes doses de NAC para avaliar
o efeito desse antioxidante em ratos com EHNA181. Winbladh e colaboradores
(2012), avaliando o efeito da NAC em porcos submetidos à I/R, observaram
diminuição da inflamação, evidenciada pela queda de leucócitos no tecido
hepático154. Esses trabalhos confirmam o efeito benéfico da NAC sobre a inflamação
no tecido hepático tanto na esteato-hepatite quanto na lesão de I/R. Em nosso
estudo, mostramos esse efeito protetor na associação das afecções.
Em nosso trabalho, também foram avaliados os níveis das transaminases
AST e ALT no soro dos camundongos pertencentes aos grupos experimentais
analisados, já que a dosagem de ALT e AST no soro é a técnica mais comum para
avaliar a presença de doença no fígado182.

66
De acordo com os nossos dados, foi observado que a presença de esteato-
hepatite associada à I/R promoveu o aumento do nível de AST no fígado dos
camundongos em relação aos camundongos que não sofreram algum tipo de lesão
no tecido hepático. Considerando o nível de ALT, o aumento significante dessa
transaminase no fígado dos camundongos foi evidenciado apenas na presença de
esteato-hepatite, quando comparado ao fígado dos animais que não sofreram lesão
hepática. No entanto, a NAC foi capaz de reduzir tanto os níveis de AST quanto os
de ALT - apesar desse último não ter apresentado significância estatística - no
fígado dos camundongos que apresentaram esteato-hepatite e sofreram lesão de
I/R, em relação aos animais que se encontravam nas mesmas condições, mas não
receberam a NAC.
Wang e colaboradores (2014) mostraram redução de AST e ALT após o uso
da NAC somente em fígado submetido à I/R175. Níveis de ALT reduzidos, após o
tratamento com NAC e resveratrol associados, foram observados em camundongos
com lesão hepática decorrente apenas da DHGNA induzida pela dieta DMC183. Em
nosso estudo, a diminuição de ALT e AST após o uso da NAC evidencia o efeito
benéfico desse antioxidante exógeno na associação de esteato-hepatite e I/R
hepática.
O fato de a redução de ALT não ter apresentado significância estatística com
o uso da NAC nos animais com fígado esteatótico e lesão de I/R hepática é devido a
intensa diminuição no nível dessa transaminase quando os camundongos com
esteato-hepatite são submetidos à I/R. Nasiri e colaboradores (2018) mostraram
essa redução dos níveis de ALT durante a regeneração do fígado de camundongos
submetidos à I/R184. Nesse trabalho, o nível de ALT aumentou com três horas de
reperfusão e reduziu a praticamente menos da metade com 24 horas, e após 4, 7,
14 e 28 dias, encontrava-se no mesmo nível do Grupo SHAM, mostrando que o
fígado consegue se regenerar rapidamente ao longo do tempo. Em nosso estudo,
entretanto, essa regeneração pode não ter sido o motivo principal da redução de
ALT no fígado. Os camundongos com esteato-hepatite apresentaram níveis
elevados de ALT, porém, a redução de ALT no fígado dos animais em que essa
afecção foi associada à I/R pode ser decorrente da exaustação do hepatócito que já
estava lesionado na ocorrência da esteto-hepatite.

67
Como consequência das injúrias causadas ao fígado, sejam elas provenientes
da presença de gordura ou da indução de I/R, o sistema imune do organismo
responde promovendo a inflamação no tecido hepático por meio da produção de
mediadores inflamatórios, tais como as citocinas185. Em nosso estudo foi observado
um aumento das concentrações das citocinas IL-1β e TGF-β1 tanto nos
camundongos que apresentaram somente esteato-hepatite quanto nos animais que
tinham a presença dessa afecção associada à lesão de I/R. No entanto, a expressão
de IFN-γ não apresentou alteração entre os grupos experimentais analisados.
A elevação de IL-1β no tecido hepático mostra o estado inflamatório do fígado
com DHGNA. Miura e colaboradores (2013) afirmaram em seu estudo que essa
elevação é decorrente da presença de ácido palmítico, um dos ácidos graxos mais
comuns encontrados em animais com esteatose186. Sadatomo e colaboradores
(2017), por sua vez, mostraram que a interação entre neutrófilos e macrófagos
promove a maturação de IL-1β e causa a inflamação derivada dessa citocina na I/R
hepática187.
Com relação ao TGF-β1, Seki e Schwabe (2015) afirmaram que a DHGNA
promove a produção dessa citocina no tecido hepático e esta induz a proliferação de
células estreladas, que são responsáveis pelo processo de fibrose no fígado188.
Yang e colaboradores (2014), por sua vez, afirmaram que a sinalização mediada por
TGF-β1 na DHGNA contribui para a morte de hepatócitos e para a acumulação de
lipídeos por meio da sinalização de Smad e produção de ERO, resultando no
desenvolvimento de EHNA189. Yu e colaboradores (2010) mostraram que em modelo
de transplante de fígado small for size ocorre a produção elevada de TGF-β1, sendo
essa citocina responsável pela fibrose no tecido hepático190.
Esses trabalhos corroboram com os dados obtidos em nosso estudo, pois
evidenciam o aumento IL-1β e TGF-β1 na presença de gordura no fígado ou na
indução de I/R hepática, o que pode intensificar o dano causado ao tecido hepático,
já que essas citocinas inflamatórias são capazes de induzir mediadores de estresse
oxidativo que provocam morte celular. Entretato, nosso estudo mostrou a elevação
de tais citocinas não somente na presença de esteato-hepatite, mas também na
associação dessa afecção à I/R.
Quando os animais com esteato-hepatite submetidos à I/R foram
administrados com NAC, as concentrações dessas citocinas no fígado foram

68
reduzidas em comparação aos animais em que a NAC não foi administrada.
Alexandropoulos e colaboradores (2017) mostraram o efeito benéfico da NAC sobre
a lesão hepática e renal causada por I/R intestinal em ratos ao observar a redução
da concentração de IL-1β e outras citocinas pró-inflamatórias191. El-Lakkany e
colaboradores (2016) mostraram que o uso associado da NAC com metformina no
fígado com DHGNA promoveu a redução da concentração de TGF-β e TNF-α, de
outros mediadores inflamatórios e de estresse oxidativo192. Em nosso estudo,
porém, a NAC isoladamente foi capaz de promover um efeito protetor no fígado com
esteato-hepatite submetido à I/R, por meio da redução das concentrações de ambas
as citocinas, IL-1β e TGF-β1.
Nakamura e colaboradores (2017) mostraram que, na lesão hepática de I/R, a
expressão de INF-γ foi aumentada, e com a ativação da sirtuina 1 pelo resveratrol,
antioxidante exógeno, sua expressão tornou-se reduzida193. Ellet e colaboradores
(2009) demonstraram níveis elevados de INF-γ em fígados esteatóticos submetidos
à isquemia e reperfusão194. Entretanto, em ambos os estudos a isquemia foi
diferente, ou mais prolongada193 ou total194, adicionando-se ainda que a dieta
hiperlipídica induziu DHGNA mais leve, atingindo a fase de esteatose. Estas
variações poderiam justificar os nossos achados, em que a associação da esteato-
hepatite com uma isquemia de curta duração e reperfusão prolongada não alterou a
concentração de INF-γ no fígado dos animais analisados de forma significante.
As citocinas inflamatórias em fígado esteatótico e/ou submetido à I/R são
responsáveis pela liberação celular de diversos fatores oxidantes no tecido hepático,
tais como as ERO e ERN195,196. Em circunstâncias normais, o fígado produz
quantidades fisiológicas de oxidantes, e essa produção é balanceada pela ação dos
antioxidantes naturais do organismo197. No entanto, o desequilíbrio entre as
quantidades de oxidantes e antioxidantes, denominado de estresse oxidativo, está
relacionadoà patogênese de uma série de doenças hepáticas198.
Os produtos aldeídos, que são gerados pela peroxidação lipídica nas
membranas celulares devido à ação das ERO sobre os ácidos graxos
poliinsaturados, têm uma meia-vida longa e são capazes de promover alterações
locais e sistêmicas199. O óxido nítrico, contudo, pode estar relacionado com a
sobrevivência ou a morte de células hepáticas. Por exemplo, o NO pode inibir a ação
das caspases, atuando diretamente nas vias apoptóticas200, ou pode inativar

69
moléculas de sinalização responsáveis por inibir os mecanismos de morte celular e
diminuir a transferência de elétrons na cadeia respiratória da mitocôndria no tecido
hepático201.
O aumento exacerbado do estresse oxidativo é consequência tanto da lesão
causada por I/R202,175 quanto daquela provocada pela esteato-hepatite203,204, sendo
este fato evidenciado em nossos resultados, quando foram observadas elevadas
concentrações de TBARS e de nitrito, nos animais que desenvolveram esteato-
hepatite e que foram submetidos à I/R.
Esse aumento nos níveis de TBARS e nitrito hepáticos pode ser explicado
pelo fato de que o estresse oxidativo na I/R induz a ativação e produção de
citocinas, prostaglandinas e moléculas de adesão, que promovem a inflamação, com
recrutamento de leucócitos, e consequentemente elevam a produção de agentes
oxidantes205. Na DHGNA, por sua vez, o estresse oxidativo está atribuído às
quantidades elevadas de ERO e ERN e peroxidação lipídica, que são gerados
durante o metabolismo dos ácidos graxos e são responsáveis pela morte celular no
fígado180. Outros trabalhos mostraram que o estresse oxidativo tem um papel
fundamental nos danos causados por I/R hepática ou por esteatose no fígado, e as
células de Kupffer são uma das responsáveis pelo aumento na produção de
mediadores inflamatórios nessas doenças206,207.
Em nosso estudo, ao ser administrada em camundongos com fígado
esteatótico submetidos à I/R, a NAC contribuiu para a diminuição acentuada das
concentrações de TBARS e nitrito, o que mostra o efeito benéfico desse agente
antioxidante na redução do estresse oxidativo diante das afecções em questão.
Sun e colaboradores (2014) mostraram que a NAC melhorou a lesão hepática
causada pelo estresse oxidativo do retículo endoplasmático após I/R, reduzindo a
concentração de TBARS e ERO208. Observaram ainda que a NAC foi capaz de inibir
a expressão dos genes GRP78, ATF4 e CHOP, que são característicos do estresse
oxidativo proveniente dessa organela. Hsieh e colaboradores (2014) demonstraram
que a NAC reduziu os níveis de TBARS e das atividades do NO em fígado
submetido à I/R209. Baumgardner e colaboradores (2008) observaram esse efeito da
NAC sobre os níveis de TBARS em ratos que desenvolveram DHGNA por meio de
dieta gordurosa administrada por via entereal total210. Nesses trabalhos, a redução
do estresse oxidativo promovida pela NAC foi verificada na lesão de I/R ou causada

70
por DHGNA, em nosso estudo, entretanto, esse resultado foi obtido na associação
de amabas as afecções hepáticas.
Fusai e colaboradores (2005) observaram que a NAC melhorou a lesão no
tecido hepático de coelhos submetidos à I/R quente, diminuindo as taxas elevadas
de ERO e ERN211. Nakano e colaboradores (1998), por sua vez, mostraram que o
uso da NAC pode evitar as lesões causadas por I/R fria em fígado esteatótico de
ratos, apresentando níveis mais elevados de glutationa nos animais tratados com
NAC212. Esses estudos corroboram com nosso trabalho no que diz respeito à
redução de ERO e ERN, porém, nos mesmos não foi analisado o efeito da NAC
sobre a inflamação e morte celular no tecido hepático com esteato-hepatite e
submetido à I/R.
O aumento dos níveis de ERO e ERN proveniente do processo inflamatório
no tecido hepático esteatótico e/ou submetido à I/R resulta na morte celular por
necrose213 ou apoptose214,215,216. A necrose é caracterizada pela tumefação de
células e organelas, rompimento de membranas e liberação de conteúdo celular que
provoca a ativação de fatores inflamatórios217. A necrose pode ser observada na
DHGNA218 ou na lesão de I/R com tempo prolongado de oclusão do pedículo
hepático, parcial ou total175,219. Em nossas análises histopatológicas não foi
observada a presença de necrose no tecido hepático esteatótico submetido à I/R,
provavelmente devido ao tempo de isquemia, que pode ter sido insuficiente para
causar necrose, e ao fato do clampeamento no fígado ter sido parcial. A morte
celular por apoptose caracteriza-se por retração celular, condensação da cromatina,
fragmentação nuclear e formação de corpos apoptóticos220. A sinalização de
apoptose pode se iniciar tanto pela ativação de receptores de membrana221 quanto
por mecanismos em que há participação da mitocôndria. Nesse último evento, as
proteínas da família Bcl-2 estão intimamente envolvidas222.
A Bcl-2 é uma proteína pertencente à família de proteínas que regulam os
eventos mitocondriais relacionados à morte celular, atuando direta ou indiretamente
sobre a permeabilidade da membrana das mitocôndrias. Quando a expressão de
Bcl-2 está aumentada no fígado, a apoptose pode ser inibida nos hepatócitos223,224.
Em nossos resultados, foi observada a expressão aumentada de Bcl-2 no tecido
hepático de camundongos do grupo controle, que não sofreram lesão no fígado, em
contraste com a baixa expressão dessa mesma proteína no fígado dos animais com

71
esteato-hepatite, inclusive associada à lesão de I/R hepática. Esse dado está em
concordância com os resultados de contagem de células apoptóticas, nos quais
observou-se aumento das células apoptóticas nos camundongos com esteato-
hepatite, bem como nos animais que tinham essa afecção associada aos danos
causados por I/R hepática.
Nos camundongos com esteato-hepatite submetidos à I/R hepática, em que
se administrou NAC, a área marcada com Bcl-2 no fígado mostrou-se aumentada e
o número de células apoptóticas reduzido, em comparação àqueles com as mesmas
afecções, que não receberam a NAC. Resultados semelhantes foram encontrados
no trabalho de Shi e colaboradores (2018), no qual foi evidenciado o aumento da
expressão de Bcl-2 em fígado de ratos com esteatose tratados com microcápsulas
de carbono ativado de NAC (ACNAC), inibindo assim a apoptose dos hepatócitos218.
Wang e colaboradores (2014) demonstraram esse efeito, porém em camundongos
acometidos somente pelas lesões provenientes da I/R hepática e tratados com
NAC175. O efeito protetor da NAC em relação à morte celular tanto na DHGNA
quanto na lesão de I/R evidenciado nesses trabalhos foi mostrado em nosso estudo
quando as duas afecções estavam associadas.
A contribuição desse trabalho está em mostrar os efeitos benéficos da NAC
na presença de duas afecções associadas, esteato-hepatite e I/R. Esta avaliação
refere-se principalmente à atuação desse antioxidante exógeno sobre os danos
causados pela produção de mediadores inflamatórios, de estresse oxidativo e de
morte celular no tecido hepático. A partir desse trabalho, novos estudos podem ser
realizados para melhor avaliar o efeito protetor da NAC, analisando, por exemplo,
sua ação preventiva no desenvolvimento da DHGNA.

72
6 CONCLUSÃO

73
6 CONCLUSÃO
A NAC promoveu redução da resposta inflamatória, do estresse oxidativo e da
apoptose na lesão de isquemia e reperfusão hepática em camundongos com
esteato-hepatite induzida por dieta.

74
*De acordo com o Estilo Vancouver
REFERÊNCIAS*
1. Coelho HSM. Doença hepática gordurosa não alcoólica [editorial]. Sociedade Brasileira de Hepatologia (São Paulo). 2012;1-19.
2. Clarke CN, Kuboki S, Tevar A, Lentsch AB, Edwards M. Cxc chemokines play a critical role in liver injury, recovery, and regeneration. Am J Surg. 2009
Sep;198(3):415-9.
3. Go KL, Lee S, Zendejas I, Behrns KE, Kim J-S. Hindawi Mitochondrial
dysfunction and autophagy in hepatic ischemia/reperfusion injury. Biomed Res Int. 2015 Nov 11;2015(Article ID 183469):14 pages.
4. Miyashita T, Nakanuma S, Ahmed AK, Makino I, Hayashi H, Oyama K,
Nakagawara H, Tajima H, Takamura H, Ninomiya I, Fushida S, Harmon JW,
Ohta T. Ischemia reperfusion-facilitated sinusoidal endothelial cell injury in liver transplantation and the resulting impact of extravasated platelet aggregation. Eur
Surg. 2016 Oct 14;48:92-9.
5. Sanches SCL, Ramalho LNZ, Augusto MJ, Silva DM, Ramalho FS. Nonalcoholic
steatohepatitis: a search for factual animal models. Biomed Res Int.2015 Dec 10; 2015(Article ID 574832):13 pages.
6. Nassir F, Rector RS, Hammoud GM, Ibdah JA. Pathogenesis and prevention of
hepatic steatosis. Gastroenterol Hepatol. 2015 Mac;11(3):167-75.
7. Pham T, Dick TB, Charlton MR. Nonalcoholic fatty liver disease and liver transplantation. Clin Liver Dis. 2016 May;20(2):403-17.
8. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic
steatohepatitis in adults. Aliment Pharmacol Ther. 2011 Aug;34(3):274-85.
9. Sherif ZA, Saeed A, Ghavimi S, Nouraie SM, Laiyemo AO, Brim H, Ashktorab H.
Global epidemiology of nonalcoholic fatty liver disease and perspectives on US minority populations. Dig Dis Sci. 2016 May;61(5):1214-25.
10. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States.
Gastroenterology. 2015 Mar;148(3):547-55.
11. Selzner M, Clavien PA. Fatty liver in liver transplantation and surgery. Semin
Liver Dis. 2001;21(1):105-13.
12. Relatórios anuais da Eurotransplant. Dispõe sobre as informações anuais
relacionadas a transplantes e doadores de órgãos. Annual report (2008 and 2011). [acesso em 16 nov. 2017] Disponível em: http://www.eurotransplant.org.

75
13. Younossi ZM, Koenig AB, Abdellatif D, Fazel Y, Henry L, Wymer M. Global
epidemiology of non-alcoholic fatty liver diseasemeta-analytic assessment of prevalence, incidence and outcomes. Hepatology. 2015 Jul;64(1):73-84.
14. Chalasani N., Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease:
practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012 Jun;55(6):2005-23.
15. Lefere S, Steenkiste CV, Verhelst X, Van Vlierberghe H, Devisscher L, Geerts A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic
fatty liver disease. Cell Mol Life Sci; 2016 Sep;73(18):3419-31.
16. Angulo P. Nonalcoholic fatty liver disease and liver transplantation. Liver Transpl.
2006 Apr;12(4):523-34.
17. Angulo P. The natural history of NAFLD. In: Non-alcoholic fatty liver disease: a
practical guide (edsFarrell GC, McCullough AJ, Day CP), Oxford, UK: Wiley Blackwell Press. 2013 Apr 26;4 chapters:37-45.
18. Mincis M; Mincis, R. Esteatose e esteato-hepatite não alcoólicas. Rev Bras Med. 2007;63(11):564-70.
19. Browning JD, Horto JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004 Jul;114(2):147-52.
20. Brunt EM,Ramrakhiani S, Cordes BG, Neuschwander-Tetri BA, Janney CG,
Bacon BR, Di Bisceglie AM. Concurrence of histologic features of steatohepatitis with other forms of chronic liver disease. Mod Pathol. 2003 Jan;16(1):49-56.
21. Walle P, Takkunena M, Männistö V, Vaittinen M, Lankinen M, Kärjä V, Käkelä P, Âgren J, Tiainen M, Schwab U, Kuusisto J, Laakso M, Pihlajamäki J. Fatty acid
metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism. 2016 May;65(5):655-66.
22. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F. Liver fibrosis, but no other histologic features, is
associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015Aug;149(2):389-97.
23. Boursier J, Mae Diehl A. Nonalcoholic fatty liver disease and the gut microbiome. Clin Liver Dis. 2016 May;20(2):263-75.
24. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011 Jun 24;332(6037):1519-23.

76
25. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and
hepatocellular carcinoma: a weighty connection. Hepatology. 2010 May;51(5):1820-32.
26. Bashiri A, Nesan D, Tavallaee G, Sue-Chue-Lam I, Chien K, Maguire GF, Naples M, Zangh J, Magomedova L, Adeli K, Cummins CL, Ng DS. Cellular cholesterol
accumulation modulates high fat high sucrose (HFHS) diet-induced ER stress and hepatic inflammasome activation in the development of non-alcoholic steatohepatitis. Biochimi Biophys Acta. 2016 Jun;1861(7):594-605.
27. Ham JR, Lee H-I, Choi R-Y, Sim M-O, Seod K-II, Lee MK. Anti-steatotic and anti-inflammatory roles of syringic acid in high-fat diet-induced obese mice. Food
Funct. 2016 Jan 11;7(2):689-97.
28. Yang M, Li X, Zeng X, et al. Rheum palmatum L. Attenuates high fat diet-induced
hepatosteatosis by activating AMP-activated protein kinase. Am J Chin Med. 2016 Apr 25;44(3):551-64.
29. Koteish A, Mae Diehl A. Animal models of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002 Oct;16(5):679-90.
30. Lee SJ, Kang JH, Iqbal W, Kwon O-S. Proteomic analysis of mice fed methionine
and choline deficient diet reveals marker proteins associated with steatohepatitis.
Plos One. 2015 Apr 7;10(4):e0120577.
31. Ohyama T, Sato K, Yamazaki Y, Hashizume H, Horiguchi N, Kakizaki S, Mori M,
Kusano M, Yamada M. MK-0626, a selective DPP-4 inhibitor, attenuates hepatic steatosis in ob/ob mice. World J Gastroenterol. 2014 Nov 21;20(43):16227-35.
32. Sajan MP, Ivey RA, Lee MC, Farese RV. Hepatic insulin resistance in ob/ob mice
involves increases in ceramide, aPKC activity, and selective impairment of Akt-
dependent FoxO1 phosphorylation. J Lipid Res. 2015 Jan;56(1):70-80.
33. Eltzschig HK, Eckle T. Ischemia and reperfusion: from mechanism to translation.
Nat Med. 2011 Nov 7;17(11):1391-401.
34. Cursio, R. Autophagy and liver ischemia-reperfusioniInjury. Biomed Res Int. 2015 Sep 7;2015(Article ID 417590):16 pages.
35. Quesnelle KM, Bystrom PV, Toledo-Pereyra LH. Molecular responses to
ischemia and reperfusion in the liver. Arch Toxicol. 2015 May;89(5):651-7.
36. Ramalho FS, Fernandez-Monteiro I, Rosello-Catafau J, Peralta C. Hepatic
microcirculatory failure. Acta Cir Bras. 2006;21(Suppl 1):48-53.
37. DuBray Jr BJ, Conzen KD, Upadhya GA, Gunter KL, Jia J, Knolhoff BL, Monahakumar T, Chapman WC, Anderson CD. BH3-only proteins contribute to steatotic liver ischemia and reperfusion injury. J Surg Res. 2015 Apr;194(2):653-
8.

77
38. Tashiro H, Kuroda S, Mikuriya Y, Ohdan H. Ischemia-reperfusion injury in
patients with fatty liver and the clinical impact of steatotic liver on hepatic surgery. Surg Today. 2014 Sep;44(9):1611-25.
39. McCormack L, Petrowsky H, Jochum W, Furrer K, Clavien PA. Hepatic steatosis
is a risk factor for postoperative complications after major hepatectomy: a
matched case-control study. Ann Surg. 2007 Jun;245(6):923-30.
40. Reddy SK, Marsh JW, Varley PR, Mock BK, Chopra KB, Geller DA, Tsung A. Underlying steatohepatitis, but not simple hepatic steatosis, increases morbidity after liver resection: a case-control study. Hepatology. 2012 Dec;56(6):2221-30.
41. Trevisani F, Colantoni A, Caraceni P, Van Thiel DH. The use of donor fatty liver for liver transplantation: a challenge or a quagmire? J Hepatol. 1996 Jan
1;24(1):114-21.
42. Marsman WA, Wiesner RH, Rodriguez I, et al. Use of fatty donor liver is associated with diminished early patient and graft survival. Transplantation. 1996 Nov 15;62(9):1246-51.
43. Lee L-Y, Harberg C, Matkowskyj KA, Cook S, Roenneburg D, Werner S,
Johnson DA, Johnson JA, Foley DP. Cell-specific over-activation of Nrf2-mediated gene expression in myeloid cells decreases hepatic ischemia reperfusion injury. Liver Transpl. 2016 Aug;22(8):1115-28.
44. Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, Walajtys-Rode E, Rashid A, Chen CH, Huang CC, Wu TC, Lane MD, Diehl MA. Obesity induces
expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999 Feb 26;274(9):5692-700.
45. Singh S, Hynan LS, Lee WM. The Acute Liver Failure Study Group. Improvements in hepatic serological biomarkers ere associated with clinical
benefit of intravenous n-acetylcysteine in early stage non-acetaminophen acute liver failure. Dig Dis Sci. 2013 May;58(5):1397-402.
46. Caraceni P, Bianchi C, Domenicali M, Maria Pertosa A, Maiolini E, Parenti Castelli G, Nardo B, Trevisani F, Lenaz G, Bernardi M. Impairment of mitochondrial oxidative phosphorylation in rat fatty liver exposed to preservation-
reperfusion injury. J Hepatol. 2004 Jul;41(1):82-8.
47. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004
Dec;114(12):1752-61.
48. Selzner M, Rüdiger HA, Sindram D, Madden J, Clavien PA. Mechanisms of
ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000 Dec;32(6):1280-8.

78
49. Jiang Y, Tang JJ, Wu BQ, Yuan B, Qu Z. The protective effects of different-time-
ischemic preconditioning on the reperfusion injury in fatty livers in rats. Plos One. 2013;8(3):e58086.
50. Veteläinen R, Van Vliet A, Gouma DJ, Van Gulik TM. Steatosis as a risk factor in
liver surgery. Ann Surg. 2007 Jan;245(1):20-30.
51. Byun J-S, Yi H-S. Hepatic Immune Microenvironment in Alcoholic and Nonalcoholic Liver Disease. Biomed Res Int. 2017 Aug;2017:6862439.
52. Naito M, Hasegawa G, Takahashi K. Development, differentiation, and maturation of Kupffer cells. Microsc Res Tech. 1997;39:350-64.
53. Gao B, Jeong W-I, Tian Z. Liver: An Organ with Predominant Innate Immunity. Hepatol. 2008;47(2):729-736.
54. Peng H, Wisse E, Tian Z. Liver natural killer cells: subsets and roles in liver immunity. Cell Mol Immunol.2016;13(3):328-36.
55. Dixon LJ, Barnes M. Tang H, PritchardMT, NagyLE. Kupffer cells in the liver.
Compr Physiol.2013;3(2):785-97.
56. Bieghs V, Trautwein C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013 Sep;34(9):446-52.
57. Lee YS, Eun HS, Kim SY, Jeong JM, Seo W, Byun JS, Jeong WI, Yi HS. Hepatic immunophenotyping for streptozotocin-induced hyperglycemia in mice. Sci Rep. 2016 Jul;28(6):30656.
58. Goh YPS, Henderson NS, Heredia JE, Red Heagle A, Odegaard JI, Lehwald N, Nguyen KD, Sheppard D, Mukunda L, Locksley RM, Chawla A. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci U S A. 2013,
Jun;110(24):9914-9.
59. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu SJ, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct,
opposing roles during liver injury and repair. J Clin Invest. 2005 Jan;115(1):56-65.
60. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996 Mar;87(6):2095-147.
61. Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000 Nov;343(20):1467-76.
62. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease:
the multiple parallel hits hypothesis. Hepatol. 2010 Nov;52(5):1836-46.

79
63. Jin G, Qiu G, Wu D, Hu Y, Qiao P, Fan C, Gao F. Allogeneic bone marrow-
derived mesenchymal stem cells attenuate hepatic ischemia-reperfusion injury by suppressing oxidative stress and inhibiting apoptosis in rats. Int J Mol Med. 2013
Apr;31:1395-401.
64. Wang A-J, Yang Z, Grinchuk V, Smith A, Qin B, Lu N, Wang D, Wang H, Ramalingam TR, Wynn TA, Urban JF, Shea-Donohue T Jr, Zhao A. IL-25 or IL-17E protects against high-fat diet–induced hepatic steatosis in mice dependent
upon IL-13 activation of STAT6. J Immunol. 2015 Nov;195(10):4771-80.
65. Jaeschke H. Mechanisms of liver injury. Mechanisms of neutrophilinduced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory
conditions. Am J Physiol. Gastrointest Liver Physiol. 2006;290:G1083-88.
66. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229-65.
67. Hoffmann E, Thiefes A, Buhrow D, Dittrich-Breiholz O, Schneider H, Resch K, et al. MEK1-dependent delayed expression of Fos-related antigen-1 counteracts c-
Fos and p65 NF-kappaB-mediated interleukin-8 transcription in response to cytokines or growth factors. J Biol Chem. 2005;280(10):9706-18.
68. Bandman O, Coleman RT, Loring JF, Seilhamer JJ, Cocks BG. Complexity of
inflammatory responses in endothelial cells and vascular smooth muscle cells determined by microarray analysis. Ann N Y Acad Sci. 2002;975:77-90.
69. Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57(3):642-
54.
70. Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JWM, Joosten LAB.. IL-1β processing in host defense: beyond the inflammasomes. Plos Pathog. 2010 Feb;6(2):e1000661.
71. O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of
progress. Immunol Rev. 2008;226:10-8.
72. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo J. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger
signals to stimulate immune cells. Hepatol. 2011 Jul;54(1):133-44.
73. Prabhu SD, Chandrasekar B, Murray DR and Freeman GL: beta-adrenergic blockade in developing heart failure: Effects on myocardial inflammatory
cytokines, nitric oxide, and remodeling. Circulation. 2000;101:2103-9.
74. Harada H, Wakabayashi G, Takayanagi A, Shimazu M, Matsumoto K, Obara H, Shimizu N, Kitajima M. Transfer of the interleukin-1 receptor antagonist gene into rat liver abrogates hepatic ischemia-reperfusion injury. Transplantation.
2002;74:1434-41.

80
75. Leask A, Abraham D J. TGF-β signaling and the fibrotic response. FASEB
J. 2004;18:816-27.
76. Achyut BR, Yang L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterol. 2011;141:1167-78.
77. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539-48.
78. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18.
79. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic
disease. J Clin Invest. 2011;121:2111-7.
80. Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg. 2001;181(2):160-6.
81. Yu JH, Zhang WG, Jiang GX, Zhao JY, Li H, Wang ZD, et al. Ischemia/reperfusion in clamped lobes facilitates liver regeneration of
nonclamped lobes after selective portal vein ligation. Dig Dis Sci. 2012;57(12):3178-83.
82. Wheelock EF. Interferon-like vírus-inhibitor induced in human leukocytes by
phytohemagglutinin. Science. 1995;149:310-311.
83. Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95-109.
84. Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer
Res. 2011;17:6118-24.
85. Shen C, Xiao Z, Zhao Q, Li M, Wu X, Zhang L, Hu W, Cho CH. Anti-cancer
therapy with TNFα and IFNγ: A comprehensive review. Cell Prolif. 2018 Jan 4;e12441.
86. Inzaugarat ME, De Matteo E, Baz P, Lucero D, García CC, Gonzalez Ballerga E,
et al. New evidence for the therapeutic potential of curcumin to treat nonalcoholic
fatty liver disease in humans. Plos One. 2017;12(3):e0172900.
87. Lia J, Zhang Q, Lia S, Daia W, Fenga J, Wua L, Liua T, Chena K, Xiaa Y, Lua J, Zhoua Y, Fanc X, Guo C. The natural product fucoidan ameliorates hepatic ischemia-reperfusion injury in mice. Biomed Pharmacother. 2017 Jul 24;94:687-
96.
88. Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver
ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection, Hepatology. 2014 May;59(5):1984-97.

81
89. Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007 Nov;35(Pt5):1147-50.
90. Reiniers MJ, Van Golen RF, Van Gulik TM, Heger M. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid Redox
Signal. 2014 Sep;21(7):1119-42
91. Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C.
Oncogene.2006 Jun 26;25(27):3834-47.
92. Duval F, Moreno-Cuevas JE, González-Garza MT, Rodríguez-Montalvo C, Cruz-
Vega DE. Protective mechanisms of medicinal plants targeting hepatic stellate cell activation and extracellular matrix deposition in liver fibrosis. Chin Med. 2014 Dec 24;9(1):27.
93. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J
Gastroenterol. 2014 Oct 21;20(39):14205-18.
94. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the
pathogenesis of Non-alcoholic fatty liver disease. Free Rad Res. 2015;49(12):1405-18.
95. Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial
adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology.
2013 Oct;58(4):1497-507.
96. García-Ruiz C, Baulies A, Mari M, García-Rovés PM, Fernandez-Checa JC. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: cause or consequence? Free Radic Res. 2013 Nov;47(11):854-68.
97. Li Y, Mouche S, Sajic T, et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond).2012 Dec;36(12):1503-13.
98. Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006
May;290:852-8.
99. Davies MJ. Myeloperoxidase-derived oxidation: mechanisms of biological
damage and its prevention. J Clin Biochem Nutr. 2011 Jun;48(1):8-19.
100. Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and
nonalcoholic fatty liver disease. Antioxid Redox Signal. 2011 Jul 15;15(2):421-4.
101. Li X, Wang Y, Wang H, Huang C, Huang Y, Li J. Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflamm Res. 2015 Jan;64(1):1-7.

82
102. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, and Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011Jul;54(1):133-44.
103. Yang Q, Liu Y, Shi Y, Zheng M, He J, Chen Z. The role of intracellular high-
mobility group box 1 in the early activation of Kupffer cells and the development of Con A-induced acute liver failure. Immunobiology. 2013 Apr 27;218(10):1284-92.
104. Wallace K, Burt AD, Wright MC. Liver fibrosis. Biochem J. 2008 Apr 1;411(1):1-18.
105. Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver
disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol
Hepatol. 2011 Apr;5(2):201-12.
106. Del Ben M, Polimeni L, Carnevale R, Bartimoccia S, Nocella C, Baratta F,
Loffredo L, Pignatelli P, Violi F, Angelico F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-
alcoholic fatty liver disease. BMC Gastroenterol. 2014;14:81-8.
107. Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepato. 2005
Jun;42(6):928-40.
108. Glantzounis GK; Salacinski HJ; Yang W; Davidson BR; Seifalian AM. The
contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: a review. Liver Transpl. 2005 Sep;11(9):1031-47.
109. Cuzzocrea S, Riley DP, Caputi AP, Savemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001 Mar;53(1):135-59.
110. Urakami H, Abe Y, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in partial liver transplantation: lessons learned from reduced-size liver
ischaemia and reperfusion injury. Clin Exp Pharmacol Physiol. 2007 Sep;34(9):912-19.
111. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011 Aug;11(8):1563-69.
112. Wang Y, Shen J, Xiong X, Xu Y. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced
autophagy. PLoS One. 2014 Jun 10;9(6):e98834.
113. Katada K, Bihari A, Mizuguchi S, Yoshida N, Yoshikawa T, Frazer DD, Potter
RF, Cepinskas G. Carbon monoxide liberated from CO-releasing molecule

83
(CORM-2) attenuates ischemia/reperfusion (I/R)-induced inflammation.
Inflammation. 2010 Apr;33(2):92-100.
114. Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhoták S, Austin RC.
Peroxynitrite causes endoplasmic reticulum stress and apoptosis and human vascular endothelium: implications in atherogenesis. Arterioscler Thromb Vasc
Biol. 2005 Dec;25(12):2623-9.
115. Koppenol WH, Kissner R, Beckman JS. Syntheses of peroxynitrite: to go with
the flow or on solid grounds? Methods Enzymol. 1996;269:296-302.
116. G. Ferrer-Sueta, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, Alvarez B, Radi R. The biochemistry of peroxynitrite and protein tyrosine
nitration. Chem Rev. 2018;118(3):1338-408.
117. Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, Inamori M,
Saito S, Iwasaki T, Terauchi Y, Maeyama S, Nakajima A. Nitric oxide play a crucial role in the development-progression of non-alcoholic steatohepatitis in the
choline-deficient, 1-amino acid-defined diet-fed rat model.Alcohol Clin Exp Res. 2010 Feb;34(S1):S18-S24.
118. Duranski MR, Elrod WJ, Calvert JW, Bryan NS, Feelisch M, Lefer DJ. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. Am J PhysiolHeart Circ Physiol. 2006 Jul 28;291(6):2980-6.
119. Diesen DL, Kuo PC. Nitric oxide and redox regulation in the liver: part II. Redox biology in pathologic hepatocytes and implications for intervention. J Surg Res.
2011 May 1;167(1):96-112.
120. Unal B, Ozcan F, Tuzcu H, Kirac E, Elpek GO, Aslan M. Inhibition of neutral
sphingomyelinase decreases elevated levels of nitrative and oxidative stress markers in liver ischemia-reperfusion injury. Redox Rep. 2016 Mar 24;22(4):147-59.
121. Varela AT, Rolo AP, Palmeira CM. Fatty liver and ischemia/reperfusion injury: Are there drugs able to mitigate injury? Curr Med Chem. 2011;18(32):4987-5002.
122. Huang H, Hou M, Xu J, Pang T, Duan J. Roles of heme oxygenase-1 promoting regeneration of peribiliary vascular plexus in bile duct ischemia/reperfusion injury.
Zhonghua Wai Ke ZaZhi. 2014 Mar 1;52(3):193-7.
123. Liu B, Qian J-M. Cytoprotective role of heme oxygenase-1 in liver ischemia
reperfusion injury. Int J Clin Exp Med. 2015 Nov 15;8(11):19867-73.
124. Kaplowitz N. Mechanisms of liver cell injury. J Hepatol. 2000;32(1 Suppl):39-47.
125. Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver.Compr Physiol. 2013 Apr;3(2):977-1010.

84
126. Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014;5(e996):1-10.
127. Benedetti A, Jezequel AM, Orlandi F. A quantitative evaluation of apoptotic bodies in rat liver. Liver. 1988 Jun;8(3):172-7.
128. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF,
Rydzewski R, Bugart LJ, Gore GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004
Jun;40(1):185-94.
129. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol.
2007 Jun;35(4):495-516.
130. Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis.
Nat Rev Cancer. 2002 Apr;2(4):277-88.
131. Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory
signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015 Jan;1(1):17-27.
132. Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver
injury. Gut. 2005 Jul 1;54(7):1024-33
133. Martinez-Caballero S, Dejean LM, Kinnally MS, Oh KJ, Mannella CA, Kinnally
KW. Assembly of the mitochondrial apoptosis-induced channel, MAC. J Biol Chem. 2009 May 1;284(18):12235-45.
134. Chipuk JE, Green DR. How the Bcl-2 proteins induced mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008 Apr;18(4):157-64.
135. Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012 Jan;1824(1):22-33.
136. Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R, Golub TR. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012
Apr 17;21(4):547-62.
137. Repnik U, Turk B. Lysosomal-mitochondrial cross-talk during cell death.
Mitochondrion. 2010 Nov;10(6):662-9
138. Punthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes
PD, Michalak EM, McKimm-Breschki J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim.
Cell. 2007 Jun 29;129(7):1337-49.

85
139. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153
sensitizes cells toendoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001 Feb;21(4):1249-59.
140. Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, Finnberg N, El-Deiry WS, Kaufmann SH, Gores GJ. Death receptor 5 signaling
promotes hepatocyte lipoapoptosis. J Biol Chem. 2011 Nov 11;286(45):39336-48.
141. Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010 Jul;90(3):1165-94.
142. Weigand K, Brost S, Steinebrunner N, Büchler M, Schemmer P, Müller M.
Ischemia/Reperfusion injury in liver surgery and transplantation: pathophysiology. HPB Surg. 2012;2012(Article ID 176723):8 pages.
143. Zong WX, Thompson CB. Necrotic cell death as a fate. Genes Dev. 2006 Jan 1;20(1):1-15.
144. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE.Cell death. N Engl J Med. 2009 Oct 15;361(16):1570-83.
145. Brunt EM, Janney CG, Dia Bisceglie AM, Neuschwander-Tetri A, Bacon BR. Nonalcoholic steatohepatitis: a proposal for granding and staging the histological
lesion. Am J Gastroenterol. 1999;94:2467-74.
146. Chatterjee R, Mitra A. An overview of effective therapies and recent advances in biomarkers for chronicliver diseases and associated liver cancer. Int.
Immunopharmacol. 2015 Feb;24(2):335-45.
147. Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver
ischemia/reperfusion injury: Processes in inflammatory networks-a review. Liver Transplant. 2010 Sep;16(9):1016-32.
148. Qu K, Shen NY, Xu XS,Su HB, Wei JC, Tai MH, Meng FD, Zhou L, Zhang YL, Liu C. Emodin induces human T cell apoptosis in vitro by ROS-mediated
endoplasmic reticulum stress and mitochondrial dysfunction. Acta Pharmacol Sin. 2013 Sep 5;34(9):1217-28.
149. Andrade KQ, Moura FA, dos Santos JM, de Araújo OR, de Farias Santos JC, Goulart MO.Oxidative stress and inflammation in hepatic diseases: therapeutic possibilities of n-acetylcysteine. Int J Mol Sci. 2015 Dec 18;16(12):30269-308.
150. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. 2015 Jun 5;97:55-74.
151. Chen Y, Dong H, Thompson DC, Shertzer HG, Nebert DW, Vasiliou V.
Glutathione defense mechanism in liver injury: insights from animal models. Food
Chem Toxicol. 2013 Oct;60:38-44.

86
152. Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, Gaston B. S-
nitrosothiols signal the ventilatory response to hypoxia. Nature. 2001 Sep 13;413(6852):171-4.
153. Chen Y, Yang Y, Miller ML, Shen D, Shertzer HD, Stringer KF, Wang B, Schneider SN, Nebert DW, Dalton TP. Hepatocyte-specific Gclc deletion leads to
rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology. 2007 May;45(5):1118-28.
154. Winbladh A, Björnsson B, Trulsson L, et al. N-acetylcysteine improves glycogenesis after segmental liver ischemia and reperfusion injury in pigs. Scand J Gastroenterol. 2012 Feb;47(2):225-36.
155. Saad KR, Saad PF, Dantas Filho L, Brito JM, Koike MK, Zanoni FL, Dolhnikoff M, Montero EF. Pulmonary impact of N-acetylcysteine in a controlled
hemorrhagic shock model in rats. J Surg Res. 2013 Jun 1;182(1):108-15.
156. Saad PF, Saad KR, de Oliveira Filho LD, Ferreira SG, Koike MK, Montero EF.
Effect of n-acetylcysteine on pulmonary cell death in a controlled hemorrhagic shock model in rats.Acta Cir Bras. 2012 Aug;27(8):561-5.
157. Portella AOV, Montero EFS, de Figueiredo LFP, Bueno AS, Thurow AA, Rodrigues FG. Effects of n-acetylcysteine in hepatic ischemia-reperfusion injury during hemorrhagic shock. Transplant Proc. 2004 May 1;36(4): 846-8.
158. Lee EJ, Silva SM, Simões Mde J, Montero EF. Effect of n-acetylcysteine in liver ischemia-reperfusion injury after 30% hepatectomy in mice. Acta Cir Bras. 2012
Apr;27(4):346-9.
159. Silva SM, Carbonel AAF, Taha MO, Simões MJ, Montero EFS. Proliferative
activity in ischemia/reperfusion injury in hepatectomized mice: effect of N-acetylcysteine. Transplant Proc. 2012 Oct;44(8):2321-5.
160. Galhardo MA, Quireze Júnior C, Navarro PGR, Morello RJ, Simões MJ, Montero EFS. Liver and lung late alterations following hepatic reperfusion associated to ischemic preconditioning or N-acetylcysteine. Microsurgery. 2007
May 3;27(4):295-9.
161. Takhtfooladi MA, Jahanshahi G, Jahanshahi A, Sotoudeh A, Samiee Amlashi
O, Allahverdi A. Effects of N-acetylcysteine on liver remote injury after skeletal muscle ischemia reperfusion in rats. Turk J Gastroenterol. 2014 Dec;25(Suppl
1):43-7
162. Seguro AC, Poli de Figueiredo LF, Shimizu MH. N-acetylcysteine (NAC)
protects against acute kidney injury (AKI) following prolonged pneumoperitoneum in the rat. J Surg Res. 2012 Jun 15;175(2):312-15.

87
163. Robinson SM, Saif R, Sen G, French JJ, Jaques BC, Charnley RM, Manas DM,
White SA. N-acetylcysteine administration does not improve patient outcome after liver resection. HPB (Oxford). 2013 Jun;15(6):457-62.
164. Kleiner DE, Brunt EM, Van NM, Behling C, Contos MJ, Cummings OW, Ferrel LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, Mc Cullough AJ, Sanyal AJ,
Nonalcoholic Steatohepatitis Clinical Research Network.Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005 Jun;41(6):1313-21.
165. Smith BW, Adams LA. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci. 2011 May-Jun;48(3):97-113.
166. Zhou Y, Wei F, Fan Y. High serum uric acid and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Clin Biochem. 2016
May;49(7-8):636-42.
167. Farrell GC, Teoh NC, and McCuskey RS. Hepatic microcirculation in fatty liver
disease.Anat Rec (Hoboken). 2008 Jun; 291(6):684-92.
168. Bejaoui M, Pantazi E, De Luca V, Panisello A, Folch-Puy E, Serafin A, Capasso
C, Supuran CT, Roselló-Catafau J. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther. 2015 Nov 1;355(2):191-8.
169. Perez-Daga JA, Santoyo J, Suárez MA, Fernández-Aquilar JA, Ramírez C, Rodriguéz-Cañete A, Aranda JM, Sánchez-Pérez B, Montiel C, Palomo D, Ruiz
M, Mate A. Influence of degree of hepatic steatosis on graft function and postoperative complications of liver transplantation. Transplant Proc. 2006
Oct;38(8):2468-70.
170. Gillissen A, Nowak D. Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy. Respir Med. 1998 Apr;92(4):609-23.
171. Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003 Jan;60(1):6-20.
172. Zwingmann C, Bilodeau M. Metabolic insights into the hepatoprotective role of N-acetylcysteine in mouse liver. Hepatology. 2006 Mar;43(3):454-63.
173. Sagias FG, Mitry RR, Hughes RD,Lehec SC, Patel AG, Rela M, Mieli-Vergani G, Heaton ND, Dhawan A. N-acetylcysteine improves the viability of human
hepatocytes isolated from severely steatotic donor liver tissue. Cell Transplant. 2010;19(11):1487-92.
174. González R, Ferrín G, Hidalgo A, Ranchal I, López-Cillero P, Santos-González M, López-Lluch G, Briceño J, Gómez MA, Poyato A, Villalba JM, Navas P, de la Mata M, Muntané J. N-acetylcysteine, coenzyme Q10 and superoxide dismutase
mimetic prevent mitochondrial cell dysfunction and cell death induced by d-

88
galactosamine in primary culture of human hepatocytes. Chem Biol Interact.
2009 Sep 14;181(1):95-106.
175. Wang C, Chen K, Xia Y, Dai W, Wang F, Shen M, Cheng P, Wang J, Lu J,
Zhang Y, Yang J, Zhu R, Zhang H, Li J, Zheng Y, Zhou Y, Guo C. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and
autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. Plos One. 2014 Sep 29;9(9):e108855.
176. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int JExp Pathol. 2006 Feb;87(1):1-16.
177. Church SJ, Begley P, Kureishy N, McHarg S, Bishop PN, Bechtold DA, Unwin RD, Cooper GJ. Deficient copper concentrations in dried-defatted hepatic tissue from ob/ob mice: A potencial model for study of defective copper regulationin
metabolic liver disease. BiochemBiophys Res Commun. 2015 May 8;460(3):549-54.
178. Ohyama T, Sato K, Yamazaki Y, Hashizume H, Horiguchi N, Kakizaki S, Mori
M, Kusano M, Yamada M. MK-0626, a selective DPP-4 inhibitor, attenuates
hepatic steatosis in ob/ob mice. World J Gastroenterol. 2014 Nov 21;20(43):16227-35.
179. Larter CZ, Yeh MM, Williams J, Bell-Anderson KS, Farrel GC. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic
transformation of hepatocytes. J Hepatol. 2008 Sep;49(3):407-16.
180. Caballero F, Fernandéz A, Matías N, Martínez L, Fucho R, Elena M, et al.
Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial s-adenosyl-L-methionine and glutathione. J Biol Chem. 2010 Jun 11;285(24):18528-36.
181. Samuhasaneeto S, Thong-Ngam D, Kulaputaba O, Patumraj S, Klaikeaw N. Effects of N-acetylcysteine on oxidative stress in rats with non-
alcoholic steatohepatitis. J Med Assoc Thai. 2007 Apr 1;90(4):788-97.
182. Amancher DE, Schomaker SJ, Aubrecht J. Development of blood biomarkers
for drug-induced liver injury: an evaluation of their potential for risk assessment and diagnostics. Mol Diagn Ther. 2013 Dec;17(6):343-54.
183. Ali MH, Messiha BA, Abdel-Latif HA. Protective effect of ursodeoxycholic acid, resveratrol, and N-acetylcysteine on nonalcoholic fatty liver disease in rats.
Pharm Biol. 2015Jul;54(7):1198-208.
184. Nasiri M, Karimi MH, Azarpira N, Saadat I. Gene Expression profile of Toll-like
receptor/adaptor/interferon regulatory factor/cytokine axis during liver regeneration after partial ischemia-reperfusion injury. Exp Clin Transplant. 2018
Mar 9; doi: 10.6002/ect.2017.0120.

89
185. Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver
diseases: Therapeutic strategies. World J Hepatol. 2018 Jan;10(1):1-7.
186. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like
receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis throught inflammasome activation in mice.
Hepatology. 2013;57:577-89.
187. Sadatomo A, Inoue Y, Ito H, Karasawa T, Kimura H, Watanabe S, Mizushina Y,
Nakamura J, Kamata R, Kasahara T, Horie H, Sata N, Takahashi M. Interaction of neutrophils with macrophages promotes IL-1β maturation and contributes to hepatic ischemia-reperfusion injury. J Immunol. 2017 Nov;199(9):3306-15.
188. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066-79.
189. Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, Seki E. TGF-β Signaling in Hepatocytes Participates in Steatohepatitis Through Regulation of Cell Death
and Lipid Metabolism. Hepatology. 2014 Feb;59(2):483-95.
190. Yu Y, Lu L, Quian X, Chen N, Yao A, Pu L, Zhang F, Li X, Kong L, Sun B,
Wang X. Antifibrotic effect of hepatocyte growth factor-expressing mesenchymal stem cells in small-for-size liver transplant rats. Stem Cells Dev. 2010 Jun;19(6):903-14.
191. Alexandropoulos D, Bazigos GV, Doulamis IP, Tzani A, Konstantopoulos P, Tragotsalou N, Kondi-Pafiti A, Kotsis T, Arkadopoulos N, Smyrniotis V, Perrea
DN. Protective effects of N-acetylcystein and atorvastatin against renal and hepatic injury in a rat model of intestinal ischemia-reperfusion. Biomed
Pharmacother. 2017 May;89:673-80.
192. El-Lakkany NM, el-Din SHS, Sabra A-NA-L, Hammam OA, Ebeid FA-L. Co-administration of metmorfin and N-acetylcysteine with dietary control improves
the biochemical and histological manifestations in rats with non-alcoholic fatty liver. Res Pharm Sci. 2016 Oct;11(5):374-82
193. Nakamura K, Kageyama S, Ke B, Fujii T, Sosa RA, Reed EF, Datta N, Zarrinpar
A, Busuttil RW, Kupiec-Weglinski JW. Sirtuin 1 attenuates inflammation and
hepatocellular damage in liver transplant ischemia/reperfusion: from mouse to human. Liver Transplant. 2017 Jul 9;23:1282-93.
194. Ellet JD, Evans ZP, Atkinson C, Schmidt MG, Schnellman RG, Chavin KD. Toll-like receptor 4 is a key mediator of murine steatotic liver warm
ischemia/reperfusion injury Liver Transplant. 2009 Sep;15(9):1101-1109. 195. Engin A. Non-alcoholic fatty liver disease. Adv Exp Med Biol. 2017:960:443-67.

90
196. Jaeschke H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced
liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006 Jun;290(6):G1083-8.
197. Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, Varela N, Contreras J, Lazarte R, Csendes A, Rojas J, Maluenda F, Burdiles P, Diaz JC,
Smok G, Thielemann L, Poniachik J. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci. 2004 Mac;106(3):261-8.
198. Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: Hints from the EASL Basic School of Hepatology. J Hepatol. 2013
Feb;58(2):365-74.
199. Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the
pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012 Jan 1;52(1):59-69.
200. Török NJ, Higuchi H, Bronk S, Gores GJ. Nitric oxide inhibits apoptosis downstream Cythocrome C release by nitrosylating caspase 9. Cancer Res. 2002 Mac 15;62(6):1648-53.
201. Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. 2002 Mac;3(3): 214-20.
202. Nakao A, Moore BA, Murase N, Liu F, Zuckerbraun BS, Bach FH, Choi AM, Nalesnik MA, Otterbein LE, Bauer AJ. Immunomodulatory effects of inhaled
carbon monoxide on rat syngeneic small bowel graft motility. Gut. 2003 Sep 1;52(9):1278-85.
203. de Medeiros IC, de Lima JG. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease? - A mechanistic hypothesis. Med Hypotheses. 2015 Aug;85(2):148-52.
204. Santos JC, Valentim IB, de Araujo OR, Ataide Tda R, Goulart MO. Development of nonalcoholic hepatopathy: Contributions of oxidative stress and
advanced glycation end products. Int J Mol Sci. 2013 Oct 1;14(10):19846-66.
205. Zhai Y, Shen XD, Hancock WW, Gao F, Qiao B, Lassman C, Belperio JA,
Strieter RM, Busuttil RW, Kupiec-Weglinski JW. CXCR3+CD4+ T cells mediate innate immune function in the pathophysiology of liver ischemia/reperfusion
injury. J Immunol. 2006;176(10):6313-22.
206. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S,
Prévot S, Makhzami S, Perlemuter G, Cassard-Doulcier AM. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012 Jul;57(1):141-9.

91
207. Yang HJ, Tang LM, Zhou XJ, Qian J, Zhu J, Lu L, Wang XH. Ankaflavin
ameliorates steatotic liver ischemia-reperfusion injury in mice. Hepatobiliary Pancreat Dis Int. 2015 Dec;14(6):619-25.
208. Sun Y, Pu LY, Wang XH, Zhang F, Rao JH. N-acetylcysteine attenuates reactive-oxygen-species-mediated endoplasmic reticulum stress
during liverischemia-reperfusion injury. World J Gastroenterol. 2014 Nov 7;20(41):15289-98.
209. Hsieh CC, Hsieh SC, Chiu JH, Wu YL. Protective Effects of N-acetylcysteine and a Prostaglandin E1 Analog, Alprostadil, Against Hepatic Ischemia: Reperfusion Injury in Rats. J Tradit Complement Med. 2014 Jan;4(1):64-71.
210. Baumgardner JN, Shankar K, Hennings L, Albano E, Badger TM, Ronis MJ. N-acetylcysteine attenuates progression of liver pathology in a rat model of
nonalcoholic steatohepatitis. J Nutr. 2008 Oct;138(10):1872-9.
211. Fusai G, Glantzouniss GK, Hafez T, Yang W, Quaglia A, Sheth H, Kanoria S,
Parkes R, Seifalian A, Davidson BR. N-acetycysteine ameliorates the late phase of liver ischaemia/reperfusion injury in rabbit with hepatic steatosis.Clin Sci. 2005;109(5):465-73.
212. Nakano H, Nagasaki H, Yoshida K, Kigawa G, Fujiwara Y, Kitamura N, Takeuchi S, Sasaki J, Shimura H, Yamaguchi M, Kumada K. N-acetylcysteine
andante-ICAM-1 monoclonal antibody reduce ischemia-reperfusion injury of the steatotic rat liver. Transplant Proc. 1998 Nov;30(7):3763.
213. Weigand K, Brost S, Steinebrunner N, Büchler M, Schemmer P, Müller M. Ischemia/Reperfusion injury in liver surgery and transplantation: pathophysiology.
HPB Surg. 2012;2012:176723.
214. Chang WJ, Toledo-Pereyra LH. Toll-like receptor signaling in liver ischemia and reperfusion. J Invest Surg. 2012;25:271-7.
215. Walsh KB, Toledo AH, Rivera-Chavez FA, Lopez-Neblina F and Toledo-Pereyra LH: Inflammatory mediators of liver ischemiareperfusion injury. Exp Clin
Transplant. 2009;7:78-93.
216. Neuman MG. Apoptosis in diseases of the liver. Crit Rev Clin Lab Sci. 2001;38:109-66.
217. Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci.
2004;19:124-28.
218. Shi T, Yang X, Zhou H, Xil J, Sun J, Kel Y, Zhang J, Shao Y, Jiang X, Pan X,
Liu S, Zhuang R. Activated carbon N-acetylcysteine microcapsule protects against nonalcoholic fatty liver disease in young rats via activating telomerase and inhibiting apoptosis. Plos One. 2018 Jan;13(1):e0189856.

92
219. Jin C, Henao-Mejia J, Flavell RA. Innate immune receptors: key regulators of
metabolic disease progression. Cell Metab. 2013;17:873-82.
220. Grivicich I, Regner A, da Rocha AB. Morte Celular por Apoptose. Br J Cancer Suppl. 2007;53(3):335-43.
221. Kim SJ, Eum HA, Billiar TR, Lee SM. Role of heme oxygenase 1 in TNF/TNF
receptor-mediated apoptosis after hepatic ischemia/reperfusion in rats. Shock. 2013;39:380-8.
222. Lin HC, Lai IR. Isolated mitochondria infusion mitigates ischemiareperfusion injury of the liver in rats: reply. Shock. 2013;39:543.
223. Cao ZG, Yuan SJ. Advances in inhibiting the expression of Bax and
cytoprotection. Clin J Clin Pharmacol Ther. 2003;8(3):245-8.
224. Fu YF, Fan TJ. Bcl-2 family proteins and apoptosis. Acta Biochim. Biophys. Sin.
2002;34(4):389-94.
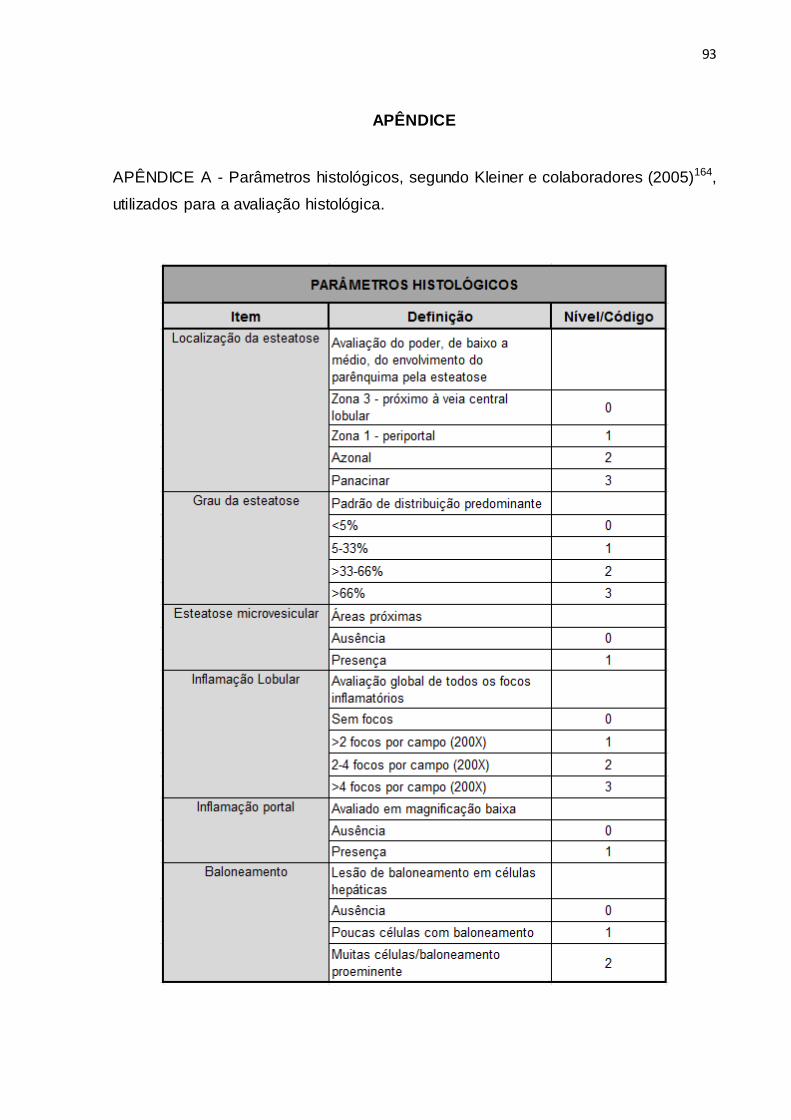
93
APÊNDICE
APÊNDICE A - Parâmetros histológicos, segundo Kleiner e colaboradores (2005)164,
utilizados para a avaliação histológica.

94
ANEXOS
ANEXO A - Parecer de aprovação do projeto pela Comissão de Ética em Pesquisa
da FMUSP

95
ANEXO B - Parecer de aprovação da mudança de pesquisador executante pela
Comissão de Ética em Pesquisa da FMUSP

96
ANEXO C - Composição da Dieta DMC