divisão de biologia celular instituto nacional de câncer...
TRANSCRIPT

FERNANDA LEVE
PARTICIPAÇÃO DA GTPASE RHO NA DINÂMICA DO COMPLEXO
JUNCIONAL APICAL E DO CITOESQUELETO DE ACTINA NA
PROGRESSÃO DO CÂNCER COLORRETAL
RIO DE JANEIRO 2011
Divisão de Biologia Celular Grupo de Biologia Estrutural
Instituto Nacional de Câncer Pós-graduação em Oncologia

ii
INSTITUTO NACIONAL DE CÂNCER - INCA
Pós-Graduação em Oncologia
FERNANDA LEVE
PARTICIPAÇÃO DA GTPASE RHO NA DINÂMICA DO COMPLEXO
JUNCIONAL APICAL E DO CITOESQUELETO DE ACTINA NA
PROGRESSÃO DO CÂNCER COLORRETAL
Dissertação apresentada ao Instituto Nacional de Câncer para obtenção do título de
Doutora em Oncologia
Orientador: Prof. Dr. José Andrés Morgado Díaz
Examinadores:
Dra Eliana Saul Furquim Werneck Abdelhay (INCA)
Dra Raquel Ciuvalschi Maia (INCA)
Dr. Fernando Costa e Silva Filho (UFRJ)
Dra. Thereza Christina Barja Fidalgo (UERJ)
Dr. Luís Felipe Severiano Ribeiro Pinto (INCA)
Dr. João Paulo de Biaso Viola (INCA)
RIO DE JANEIRO 2011

iii
Esta tese de Doutorado foi desenvolvida no Laboratório de Biologia Estrutural da
Divisão de Biologia Celular no Centro de Pesquisas do Instituto Nacional de Câncer (INCA),
sobre a orientação do Dr. José Andrés Morgado Díaz.

iv
ÓRGÃOS FINANCIADORES:
Instituto Nacional de Câncer – Ministério da saúde
(INCA – MS)
Fundação do Câncer
(FAF)
Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq)
Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ)

v
DEDICATÓRIA
Dedico esta Tese ao meu marido,
melhor amigo e maior incentivador,
Alex Morais Ponte.

vi
AGRADECIMENTOS
Ao orientador e amigo, José Morgado, pela dedicação, paciência, confiança, incentivo e
ensinamentos que permitiram o meu desenvolvimento profissional e pessoal;
A minha aluna de iniciação científica Taline Marcondes, sempre muito prestativa e centrada;
Aos integrantes antigos do laboratório, que contribuíram imensamente na minha carreira e
muitos continuam participando da minha vida como grandes amigos: Andréia Deiró, Patrícia
Redondo, Silvia Oliveira, Ana Cristina Xavier, Leandro Augusto, Livia Goto, Rodrigo
Madeiro, Paloma Souza;
Aos integrantes atuais do laboratório que, além do auxílio técnico e excelentes discussões
profissionais, tornaram a rotina de trabalho mais divertida: Simone Fernandes, Lílian Bastos,
Sarah Rabello, Waldemir Fernandes, Júlio César Freitas, Wallace Martins, Flávia Vidal,
Marcelo Tanaka, Pedro Shumman, Pedro Daniel, Natalia Fortunato, Priscila Marcondes e
Carlos Alberto;
Aos amigos da ciência que, embora de outros laboratórios, contribuíram de alguma forma
nesses quatro anos: Cláudia Maia, Gabriel Wajnberg, Giuliana Mognol;
Aos colaboradores: Marcelo Pelajo, Pedro Paulo de Abreu Manso e Bernardo Pascarelli, do
laboratório de Patologia do Instituto Oswaldo Cruz, pela ajuda na microscopia confocal; e
Tatiana Tilli, da divisão de Medicina Experimental do INCA, pelo auxílio com a PCR e
excelentes discussões;
Aos familiares e amigos que, mesmo sem entender o que eu faço, sempre me incentivaram!

vii
RESUMO
A desmontagem do complexo juncional apical (CJA), que compreende as junções
aderentes (JAs) e junções tight (JTs), e a reorganização do citoesqueleto de actina são etapas iniciais da progressão do câncer colorretal (CCR). Subsequentemente, as células adquirem um fenótipo migratório e invasivo, no entanto, os mecanismos moleculares que modulam esses eventos iniciais permanecem desconhecidos. O objetivo do presente estudo foi identificar as vias de sinalização envolvidas na regulação da dinâmica do CJA e do citoesqueleto de actina durante a progressão do CCR. Células de adenocarcinoma de cólon humano Caco-2, utilizadas como modelo de CCR, foram submetidas ao ensaio de retirada do cálcio do meio extracelular e ao tratamento com ácido lisofosfatídico (LPA), um ativador da GTPase Rho. Pré-tratamentos com inibidores específicos de vias de sinalização foram também utilizados nos diferentes ensaios. Na primeira etapa do estudo, verificamos que a retirada do cálcio extracelular ocasionou redistribuição das proteínas do CJA, conforme mostrado por imunofluorescência e immunoblotting usando frações solúveis e insolúveis em Triton X-100; perda da funcionalidade das JTs, como visto pelo decréscimo na resistência elétrica transepitelial (RET) e microscopia eletrônica de transmissão utilizando o traçador vermelho de rutênio; alterações no citoesqueleto de actina na região perijuncional e na distribuição das fibras de estresse, observados por microscopia confocal; e ativação localizada das GTPases Rho e Rac. Todos esses efeitos foram mediados por uma via de sinalização envolvendo a ativação de PKA e Rho-ROCK. Na segunda parte do estudo, observamos que o tratamento com LPA ocasionou redistribuição das proteínas do CJA e redução da expressão de E-caderina bem como reorganização do citoesqueleto de actina perijuncional, e aumento na formação de fibras de estresse. Além disso, o LPA causou: aumento da migração, mas não da invasão celular, como mostrado pelas técnicas do wound healing e de invasão em matrigel, respectivamente; formação de adesões focais, analisada pelo aumento da fosforilação da quinase de adesão focal (FAK) e imunofluorescência; ativação da GTPase Rho, mas não de Rac, conforme o ensaio de pull-down; e aumento do crescimento independente de ancoragem. Esses resultados mostram que o LPA modula as JAs, o citoesqueleto de actina e a migração celular através de uma via de sinalização que integra Rho-ROCK e Src-FAK. Em conclusão, juntos esses resultados mostram um papel central da via de sinalização Rho-ROCK na modulação do CJA e do citoesqueleto de actina durante a progressão do CCR, via que pode constituir um novo e alternativo alvo terapêutico no tratamento deste tipo de câncer.

viii
ABSTRACT
The disassembly of the apical junctional complex (AJC), which encompasses adherent junctions (AJs) and tight junctions (TJs), and the actin cytoskeleton reorganization are initial steps of colorectal cancer (CRC) progression. Subsequently, the cells acquire a migratory and invasive phenotype, but the molecular mechanisms that modulate these initial events remain unknown. The aim of the present study was to identify the signaling pathways that regulate the AJC and actin cytoskeleton dynamics during the CRC progression. Caco-2 cells, obtained from human adenocarcinoma, were used as a CRC model and submitted to the low calcium medium assay and treatment with lysophosphatidic acid (LPA), a Rho GTPase activator. Pre-treatments with specific signaling pathways inhibitors were also used in different assays. In the first part of this study, we verified that the low calcium medium caused redistribution of AJC proteins, as showed by immunofluorescence and immunoblotting using TritonX-100 soluble and insoluble fractions; loss of TJ function, as seen by the transepithelial electrical resistence (TER) decrease and ruthenium red marker permeation; actin cytoskeleton alteration at perijuncional region and stress fibers distribution, observed using confocal microscopy; and spatial Rho and Rac GTPases activation. We demonstrated that these effects were mediated by a signaling pathway involving PKA and Rho-ROCK activation. In the second part of the study, we observed that LPA treatment caused AJC proteins redistribution and E-cadherin downregulation as well as perijunctional actin cytoskeleton reorganization, and increased stress fibers formation. Moreover, LPA induced: increased cell migration, but not cell invasion, as showed by the wound healing technique and matrigel invasion assay, respectively; focal adhesion formation, as analyzed by the increased focal adhesion kinase (FAK) phosphorylation and immunofluorescence; activation of Rho GTPase, but not of Rac, showed by the pull-down assay; and increase in anchorage independent growth. These results show that LPA modulates AJs and actin cytoskeleton organization to induce a migratory phenotype through a cross-talk between Rho-ROCK and Src-FAK signaling pathways. Taken together, our results indicate that Rho-ROCK signaling plays a central role in AJC and actin cytoskeleton modulation during CRC progression, and indicate that it may constitute a new and alternate therapeutic target for the treatment of this cancer type.

ix
LISTA DE ILUSTRAÇÕES
Figura 1 Topologia do intestino grosso 1
Figura 2 Sequência adenoma-carcinoma 2
Figura 3 Tipos de câncer mais incidentes estimados para 2010, exceto
pele não melanoma, na população brasileira 4
Figura 4 Desenvolvimento da polaridade epitelial 5
Figura 5 Complexos envolvidos na polaridade epitelial 7
Figura 6 Organização e composição molecular do complexo juncional 8
Figura 7 Composição molecular das JTs 12
Figura 8 Composição molecular das JAs e participação da β-catenina na
via de sinalização Wnt 15
Figura 9 Proteínas adaptadoras do CJA que se ligam em actina 18
Figura 10 Configurações de Src inativa e ativa 20
Figura 11 Regulação da ativação de PKA 21
Figura 12 Via de sinalização do EGFR 22
Figura 13 Mecanismos de ativação da PI3K de classe I 23
Figura 14 Modelo mostrando o ciclo de ativação de GTPases da família Rho 25
Figura 15 Estrutura molecular e mecanismo de ativação de ROCK 28
Figura 16 Síntese do LPA 30
Figura 17 Progressão tumoral e processo metastático 33
Figura 18 Principais moduladores da TEM 34
Figura 19 mecanismo de sinalização da FAK na migração celular 41
Figura 20 Dinâmica do citoesqueleto de actina durante a formação de
projeções de membrana na migração celular 43
Figura 21 Inibição farmacológica de PKA previne a redistribuição de proteínas
do CJA 58
Figura 22 Associação da E-caderina e claudina-1 ao citoesqueleto de actina é
modulada por PKA 60
Figura 23 A funcionalidade do complexo juncional é regulada por PKA 62
Figura 24 PKA modula a organização do citoesqueleto de actina de forma
diferencial através do volume celular 64
Figura 25 A inibição farmacológica de ROCK não previne a desmontagem
das JTs mas impede a internalização de E-caderina 66

x
Figura 26 A inibição de ROCK não modula a funcionalidade das JTs em
células Caco-2 depletadas de cálcio 69
Figura 27 ROCK não regula a desorganização do citoesqueleto de actina
em células depletadas de cálcio 72
Figura 28 A depleção de cálcio induz translocação de Rho, Rac e RhoGDI
para a membrana em células Caco-2 74
Figura 29 O LPA restaura a formação de fibras de estresse rompidas pela
retirada do cálcio do meio extracelular 77-78
Figura 30 O LPA causa redistribuição das proteínas das JAs em células Caco-2 80-81
Figura 31 A redistribuição das proteínas das JAs induzida por LPA é acompanhada
da redução da expressão de E-caderina 82
Figura 32 O LPA causa redistibuição das proteínas das JTs de forma reversível 84-85
Figura 33 O LPA induz queda da RET em tempos iniciais de tratamento e este
efeito é reversível ao longo do tempo 86
Figura 34 O LPA modula F-actina perijuncional 88-89
Figura 35 O LPA induz formação de adesões focais 91
Figura 36 A ativação de Src, Rho e ROCK medeia a desmontagem das JAs
induzida por LPA 93
Figura 37 O LPA ativa Rho e induz a translocação de RhoA e ROCKII para os
contatos célula-célula 96
Figura 38 A desorganização de F-actina mediada por LPA ocorre através da
ativação Src e da via Rho-ROCK 98
Figura 39 A desorganização do citosesqueleto de actina mediada por
LPA é independente de PKA, PI3K e EGFR 100
Figura 40 O LPA ocasiona redistribuição das proteínas das JTs de forma
dependente de Rho 102
Figura 41 A migração celular induzida por LPA depende da ativação de Src e da
via de sinalização Rho-ROCK 104
Figura 42 A migração celular induzida por LPA é independente de PKA, PI3K
e EGFR 99
Figura 43 Src e ROCK regulam a formação de projeções de membrana induzidas
por LPA em células migratórias 105
Figura 44 O LPA aumenta o crescimento independente de ancoragem em
células Caco-2 107

xi
Figura 45 O LPA não aumenta o potencial invasivo de células Caco-2
em matrigel 110
Figura 46 Modelo para a regulação da desmontagem do CJA e organização
do citoesqueleto de actina em células depletadas de cálcio 116
Figura 47 Modelo para a indução do fenótipo migratório induzido por LPA 125

xii
LISTA DE TABELAS
Tabela 1 Expressão das GTPases Rho e suas proteínas regulatórias/efetoras em CCR 26

xiii
LISTA DE ABREVIATURAS E SIGLAS
ADAM: desintegrina-metaloprotease
ADF: fator despolimerizante de actina
AF: adesão focal
AKAP: proteína de ancoragem em quinase-A
AMPc: Adenosina monofosfato cíclica
AP-1: proteína ativadora 1
APC: Adenomatous poliposis coli
Arp : proteína relacionada à actina
ATX: autaxina
bJT: junção tight bicelular
BSA: Soro albumina bovina
CAR: cocksakie e adenovírus
CBD: domínio de ligação em catenina
CCR: câncer colorretal
Cdc42: proteína homóloga de controle de divisão celular 42
EC: domínio extracelular de caderina
CF: contato focal
Chk: homóloga de Src quinase C-terminal
CIA: crescimento independente de ancoragem
CJA: complexo juncional apical
CK1: caseína quinase 1
Cox-2: ciclooxigenase 2
CNF-1: Fator necrotizante citotóxico-1 de E. coli
CRB: crumb
CRD: domínio rico em cisteína
CREB: elemento responsivo de ligação em AMP cíclico (cAMP response element-binding)
Csk: Src quinase C-terminal
C-terminal: carboxi-terminal
DIL: domínio dilute
DLC-1:deletado em câncer de fígado-1
DLG: disc large
DRF: diaphanous homóloga a formina
Dsv: dishevelled

xiv
EDG: gene de diferenciação endotelial
EGF: fator de crescimento epidérmico
EGFR: receptor de fator de crescimento epidérmico
EGTA: Ácido tetracético etileno glicol
Ena/Vasp : fosfoproteína estimulada por vasodilatador
EPLIN: proteína epitelial perdida em neoplasias
ERK: quinase regulada por sinal extracelular
ERM: complexo ezrina-radixina-moesina
ESAM: molécula de adesão seletiva de célula endotelial
F-actina: actina filamentar
FAK: quinase de adesão focal
FAP: polipose adenomatosa familiar
FAT: alvo de adesão focal
FERM : 4.1 (four.one), ezrina, radixina, e moesina
FHA: domínio associado a forkhead
FI: filamento intermediário
FTase: farnesiltransferase
FU: fluorouracil
Fz: frizzled
G-actina: actina globular
GAP: proteína ativadora de GTPases
GDF: fator de desacoplamento de GDI
GDI: inibidores de dissociação de nucleotídeo de guanina
GEF: fator de troca de nucleotídeo de guanina
GGTase: geranilgeraniltransferase
GPCR: receptor acoplado em proteína G
GSK3β: glicogênio sintase quinase 3 β
GUK: guanilato quinase
HNPCC: câncer colorretal hereditário não polipóide
IQGAP: proteína ativada por GTPase com motivo IQ
ISM: Instabilidade em microssatélite
JAM: molécula de adesão juncional
JMD: domínio justamembranar
JNK: Jun N-terminal quinases

xv
JT: junção tight
LC: baixa concentração de calico (low calcium)
LGL: lethal giant larvae
LIMK: LIM quinase
LMW-PTP: proteína tirosina fosfatase de baixo peso molecular (low molecular weight)
LPA: ácido lisofosfatídico
LPC: lisofosfatidilcolina
LPD: lisofosfolipase D
LPE: lisofosfatidiletanolamina
LPS: lisofosfatidilserina
MAGI: proteína guanilato quinase invertido associada à membrana
MAGUK: guanilato quinases associadas à membrana
MAPK: proteína quinase ativada por mitógeno
MBS: subunidade ligadora de miosina da miosina fosfatase
MDCK: células de rim canino
mDia: formina diaphanous
MEC: matriz extracelular
MET: Microscopia Eletrônica de Transmissão
MLC: cadeia leve da miosina
MLH: homóloga de mutL
MMP: metaloprotease
MSH: homóloga de mutS
MT-MMPs: porções transmembranares de metaloprotease
MT: microtúbulo
MUPP: proteína multi-PDZ-1
NC: concentração normal de cálcio (normal calcium)
NFkB - fator nuclear Kappa b
N-terminal: amino-terminal
N-WASP: WASP neural
PA: ácido fosfatídico
PAK: quinase P21-ativada
PALS1: proteína associada com Lin sete
PAR: proteína de partição defeituosa
PATJ: proteína associada em junção tight com domínio PDZ

xvi
PBS: Solução salina tamponada com fosfato
PCP: polaridade celular planar
PDE: fosfodiesterases
PDGF: fator de crescimento derivado de plaquetas
PDK: quinase dependente de fosfoinositídeo
PDZ: PSD95 (densidade-95 pós-sináptica humana), DLGA (Drosophila disc large) e ZO-1
PGE2: prostaglandina E2
PH: região homóloga a plecstrina
PI: fosfatidil-inositol
PI3K: fosfatidil-inositol-3-quinase
PIP2: fosfatidil-inositol-4,5-bifosfato
PIP3: fosfatidil-inositol-3,4,5-trifosfato
PKA: proteína quinase A
PKC: proteína quinase C
PKCa: proteína quinase C atípica
PLA1: fosfolipase A1
PLA2: fosfosfolipase A2 solúvel
PPARγ: receptor gama de peroxissomo ativador de proliferação
PR: domínio rico em prolina
PTEN: fosfatase homóloga a tensina
PTP-PEST: proteína tirosina fosfatase que contém a sequência peptídica PEST, rica em
prolina (P), ácido glutâmico (E), serina (S) e treonina (T)
RA: domínio associado a Ras
RBD: região de ligação em Rho
RET: resistência elétrica transepitelial
Rho: homóloga a Ras
ROCK: Rho-quinase
SAF-B: scaffold attachment factor B
SDS-PAGE: eletroforese em gel de poliacrilamida usando sódio dodecil sulfato
SFB: soro fetal bovino
SH3: homólogo a Src
Src: proteína quinase derivada de sarcoma
SRF: fator de resposta ao soro
STAT: transdutor de sinal e ativador de transcrição

xvii
TCF: fator de célula T
TEM: transição epitélio-mesenquimal
TGF-β: fator de crescimento transformante beta
TGFβR: receptor de fator de crescimento transformante beta
TGI: trato gastrintestinal
Tiam: proteína de linfoma de célula T de invasão e metástase
TIMP: inibidores teciduais de metaloprotease
tJT: junção tight tricelular
TME: transição mesênquima-epitelial
TNF-α: fator alfa de necrose tumoral
Trio: proteína domínio triplo funcional
TX-100: triton X-100
VEGF: fator de crescimento vascular endotelial
VH1-3: domínio homólogo a vinculina1-3;
WASP: proteína da síndrome Wiskott Aldrich
WAVE: proteína homóloga a verprolina da família WASP
Wnt: Wingless
ZEB: Zinc finger E-box-binding
ZO: zonula ocludens
ZONAB: proteína ligadora de ácido nucleico associada a ZO

xviii
SUMÁRIO
RESUMO ..............................................................................................................................vii
ABSTRACT .........................................................................................................................viii
LISTA DE ILUSTRAÇÕES..................................................................................................ix
LISTA DE TABELAS ..........................................................................................................xii
LISTA DE ABREVIATURAS E SIGLAS ........................................................................xiii
I INTRODUÇÃO 1
I.1 Intestino grosso, desenvolvimento e incidência do câncer colorretal .............................1
I.2 Polaridade Celular .............................................................................................................4
I.3 Complexo juncional Apical (CJA) ....................................................................................7
I.3.1 Junções tight (JTs) .......................................................................................................9
I.3.2 Junções aderentes (JAs) .............................................................................................12
I.4 Citoesqueleto de actina ...................................................................................................16
I.4.1 Inter-relação entre o CJA e o citoesqueleto de actina .................................................17
I.5 Componentes de vias de sinalização envolvidas na modulação do CJA e do
citoesqueleto de actina .......................................................................................................19
I.5.1 Proteínas quinases ......................................................................................................19
I.5.2 Pequenas GTPases Rho .............................................................................................23
I.5.2.1 Proteínas efetoras de Rho ...................................................................................26
I.6 Modulação do complexo juncional apical (CJA) pelas GTPases Rho ...........................28
I.7 Ácido lisofosfatídico (LPA) ............................................................................................29
I.8 Progressão tumoral e metástase .......................................................................................32
I.8.1 Transição epitélio-mesenquimal (TEM) e migração celular ....................................34
I.8.2 Projeções de membrana reguladas por GTPases Rho na migração celular ...............36
I.8.3 Matriz extracelular (MEC) e migração celular .........................................................38
I.8.4 Regulação da organização do citoesqueleto de actina na migração celular ..............41
I.9 Justificativa do estudo ......................................................................................................44
II OBJETIVOS 45
II.1 Geral ...............................................................................................................................45
II.2 Específicos ......................................................................................................................45

xix
III. MATERIAL E MÉTODOS 47
III.1 Anticorpos e Reagentes .................................................................................................47
III.2 Cultura de células ..........................................................................................................48
III.3 Modelo de depleção do cálcio extracelular ...................................................................48
III.4 Tratamento com LPA ....................................................................................................48
III.5 Tratamentos com inibidores de vias de sinalização ......................................................49
III.6 Ensaios de Imunofluorescência .....................................................................................49
III.7 Detecção dos filamentos de actina ................................................................................50
III.8 Obtenção de frações solúveis e insolúveis em Triton X-100 e de lisados
celulares totais ..................................................................................................................50
III.9 Distribuição subcelular de Rho, Rac e RhoGDI ...........................................................50
III.10 Eletroforese em gel de poliacrilamida usando sódio dodecil sulfato
(SDS-PAGE) e Immunoblotting........................................................................................51
III.11 Ensaio de pull-down ....................................................................................................51
III.12 Resistência Elétrica Transepitelial (RET)....................................................................52
III.13 Microscopia Eletrônica de Transmissão (MET)..........................................................52
III.14 Ensaios de migração e invasividade celular ................................................................52
III.15 Proliferação celular .....................................................................................................53
III.16 Crescimento independente de ancoragem (CIA) ........................................................54
III.17 Análise da expressão e atividade de metaloproteases .................................................54
III.18 Análise estatística ........................................................................................................55
IV RESULTADOS 56
PARTE 1: Análise da inter-relação entre o CJA e o citoesqueleto de actina
durante a perda da adesão célula-célula ...........................................................................56
IV.1 PKA modula a desmontagem do CJA e a funcionalidade das JTs ...............................56
IV.2 PKA modula a organização do citoesqueleto de actina de forma diferencial através
do volume celular .......................................................................................................63
IV.3 A inibição farmacológica de ROCK não previne a desorganização das JTs mas
impede a internalização de E-caderina ...........................................................................65
IV.4 ROCK não regula a organização do citoesqueleto de actina em células crescidas
em meio com baixa concentração de cálcio ....................................................................71
IV.5 A depleção de cálcio ocasiona translocação de Rho, Rac e RhoGDI para a
membrana, de forma dependente de PKA .........................................................................73

xx
IV.6 A ativação de Rho restaura a formação de fibras de estresse, mas não a
organização da actina perijuncional ..................................................................................76
PARTE II: Papel do LPA na aquisição do fenótipo migratório ........................................79
IV. 7 O tratamento com LPA causa redistribuição das proteínas das JAs ............................79
IV. 8 O LPA ocasiona desorganização das JTs de forma reversível ...................................83
IV.9 O LPA modula a organização do citoesqueleto de actina perijuncional
concomitante à formação de fibras de estresse e adesões focais ..................................87
IV.10 O LPA induz desorganização das JAs através da ativação de Src, Rho e ROCK .....92
IV.11 O LPA induz desorganização do citoesqueleto de actina através da ativação de
Src, Rho e ROCK........................................................................................................97
IV.12 O LPA modula a desorganização das JTs através da ativação de Rho .....................101
IV.13 O LPA aumenta a migração celular de forma dependente de Src, Rho e ROCK .....103
IV.14 O fenótipo migratório induzido por LPA depende da ativação de Src e ROCK ......106
IV.15 O LPA aumenta o crescimento independente de ancoragem de células Caco-2 ......108
IV.16 O LPA não induz aumento de invasividade e atividade de MMP-9 e 2
das células Caco-2 .........................................................................................................109
V DISCUSSÃO 111
V.1 PARTE I: Análise da inter-relação entre o CJA e o citoesqueleto de actina
durante a perda da adesão célula-célula .......................................................................111
V.2 PARTE II: Papel do LPA na aquisição do fenótipo migratório ...................................117
V.3 Considerações finais: via Rho-ROCK, um possível alvo terapêutico em CCR? .........127
VI CONCLUSÕES 131
VI.1 PARTE I: Análise da inter-relação entre o CJA e o citoesqueleto de actina
durante a perda da adesão célula-célula ........................................................................131
VI.2 PARTE II: Papel do LPA na aquisição do fenótipo migratório .................................131
VII REFERÊNCIAS ............................................................................................................132

xxi
Essa Tese gerou os seguintes trabalhos:
1. Leve, F., de Souza, W. & Morgado-Díaz, J.A. (2008). A cross-link between protein kinase
A and Rho-family GTPases signaling mediates cell-cell adhesion and actin cytoskeleton
organization in epithelial cancer cells.Pharmacol Exp Ther. 327(3), 777-788.
2. Leve, F., Marcondes, T.G.C., Bastos, L.G.R., Rabello, S.V., Tanaka, M.N. & Morgado-Díaz,
J.A. (2011). Lysophosphatidic acid induces a migratory phenotype through a crosstalk
between RhoA-Rock and Src-FAK signalling in colon cancer cells.Submetido.
3. Leve, F., de Souza, W. & Morgado-Díaz, J.A. Rho GTPases and their role in colorectal
cancer. A ser submetido.

1
I INTRODUÇÃO
I.1 Intestino grosso, desenvolvimento e incidência do câncer colorretal
A função primária do trato intestinal é a digestão e absorção de nutrientes. O lúmen
intestinal dos mamíferos é revestido por uma camada simples e especializada de células
epiteliais colunares, e é dividido em intestino delgado e grosso. O intestino grosso é
subdividido em ceco, cólon, sigmoide, reto e ânus. O cólon é coberto pelo peritônio e
apresenta numerosas criptas, estruturas da camada mucosa com abertura para a superfície, não
apresentando vilosidades. As criptas apresentam um revestimento epitelial composto por
enterócitos, células que absorvem nutrientes e secretam enzimas; células caliciformes, que
secretam mucina; células enteroendócrinas, que secretam hormônios e outras substâncias; e
raras células de paneth no cólon ascendente, que secretam defensinas e lisosima. A formação
de um eixo cripta-vilosidade é um processo dinâmico no qual os enterócitos iniciam seu
desenvolvimento na base das criptas a partir de células-tronco e migram para a porção apical,
onde perdem a capacidade de se dividir e se diferenciam em enterócitos maduros (Figura 1).
Dessa forma, ocorre uma constante renovação do epitélio colônico a cada cinco dias, com
uma regulação ótima para manter um equilíbrio entre proliferação e morte celular (Flier &
Clevers, 2009).
Figura 1. Topologia do intestino grosso. A) Células-tronco localizadas na base das criptas originam células progenitoras proliferativas, que migram para o ápice das criptas e se diferenciam. B) Células progenitoras proliferativas se diferenciam em células absortivas, caliciformes e enteroendócrinas. As células de paneth não estão representadas. Adaptado de Reya & Clevers, 2005.

2
A exposição da mucosa intestinal aos agentes cancerígenos inicialmente se manifesta
por lesões inflamatórias que podem se desenvolver e ocasionar uma displasia, alterações
citológicas em um epitélio que predispõe um órgão ao desenvolvimento de câncer. As criptas
aberrantes são alterações histológicas precoces caracterizadas por aumento no tamanho dessas
estruturas, abertura luminal alterada e espessamento do epitélio (Tanaka, 2009). Acredita-se
que essas lesões displásicas podem predizer a formação de câncer colorretal (CCR), pois
podem progredir a pólipos epiteliais denominados adenomas. Um pólipo é uma massa bem
delimitada com divisão celular descontrolada nas criptas, que se projeta na luz do intestino.
Tem sido mostrado que a remoção de pólipos adenomatosos reduz o risco de desenvolvimento
de CCR embora possam levar anos para progredirem a adenocarcinomas (Tanaka, 2009).
O surgimento de um carcinoma a partir de lesões adenomatosas recebe a designação
de sequência adenoma-carcinoma (Figura 2). Nessa sequência, denominada via da
instabilidade cromossômica, as células tumorais passam por modificações gênicas sequenciais
em oncogenes e genes supressores de tumor, e perdas alélicas (Takayama et al., 2006). Os
principais oncogenes mutados em CCR são o K-Ras, em 50% dos casos (Wu et al., 2008), e o
da β-catenina de 10-50% (Akisik et al., 2010); e dentre os genes supressores de tumor, o APC
está mutado em 80% dos casos de CCR (Tanaka, 2009), e o TP53, em 20% (Oki et al., 2009).
A formação de carcinoma também pode ser desencadeada por instabilidade em
microssatélites (ISM) ocasionada por mutações em enzimas de reparo do DNA ou
hipermetilação dos seus promotores. O reparo inadequado do DNA implica na perda de
mecanismos de manutenção da fidelidade genômica durante a divisão celular, permitindo a
proliferação clonal de células mutantes e propagação da instabilidade genômica (Takayama et
al., 2006). Os principais genes de reparo mutados em CCR são o MSH2, MLH1, MSH6, o que
ocorre em 15-20% em pacientes com CCR esporádico (Poulogiannis et al., 2010).
Figura 2. Sequência adenoma-carcinoma. Modelo multipasso da carcinogênese colorretal onde mutações no gene APC ocorrem durante o desenvovimento de pólipos, seguidas por mutações em K-Ras durante o estágio adenomatoso, e mutações em TP53 na transição para a malignidade. Adaptado de Fodde et al., 2001.

3
O câncer representa uma das principais causas de morte no mundo. Aproximadamente
90% das malignidades são de origem epitelial, e o CCR apresenta-se como o terceiro mais
incidente em ambos os sexos na população brasileira (Figura 3), e o segundo em países
desenvolvidos. Além disso, o CCR representa o segundo tipo de câncer com maior índice de
mortalidade, embora a sobrevida média global dos pacientes com essa neoplasia em cinco
anos seja considerada boa, em torno de 40% para países em desenvolvimento (Instituto
Nacional de Câncer & Ministério da Saúde, 2009). Os principais fatores de risco para o CCR
são a história familiar; a predisposição genética ao desenvolvimento de doenças crônicas do
intestino, por exemplo, colite de Crohn; bem como a alta ingestão de gorduras animais e baixa
de frutas, vegetais e cereais, e o consumo excessivo de álcool e tabagismo; sedentarismo;
obesidade; e idade (Instituto Nacional de Câncer & Ministério da Saúde, 2009; Van den
Brandt & Goldbohm, 2006).
Estimativas recentes mostram que em torno de 75% dos casos de CCR são de origem
esporádica, resultantes da ação cumulativa de agentes carcinógenos ambientais ou
alimentares, sendo o restante de origem familiar. Acredita-se que a perda de heterozigosidade
do gene APC seguida por uma segunda mutação neste mesmo gene seja o evento inicial para a
formação de adenomas colônicos esporádicos (Tanaka, 2009). Dentre as desordens
hereditárias mais importantes na susceptibilidade ao CCR, destacam-se: a) a polipose
adenomatosa familial (FAP), uma síndrome autossômica dominante causada por mutação
germinativa no gene APC que ocasiona a formação de centenas de pólipos adenomatosos,
com incidência menor que 1% dos CCR; e b) o câncer de cólon hereditário não poliposo
(HNPCC), outra desordem autossômica dominante causada por mutações em genes de reparo
do DNA, gerando ISM, e com incidência de 2 a 5% dos CCR. Indivíduos acometidos por essa
síndrome desenvolvem poucos pólipos, mas altamente favoráveis à progressão maligna
(Rowley, 2005).
Em suma, a homeostase intestinal é mantida por um balanço entre auto-renovação e
diferenciação celular, e mutações genéticas de origem esporádica ou familiar podem ocasionar
a desregulação desse processo. Atualmente, duas teorias tentam justificar a perda de
diferenciação e polaridade celular na iniciação do câncer, incluindo o CCR: um modelo
estocástico, onde se postula que todas as células apresentam mesmo potencial de sofrer
mutações iniciais que levem à perda de diferenciação e polaridade celular para ocorrer a
formação do tumor; e um modelo de célula-tronco tumoral, onde subpopulações de células
indiferenciadas não polarizadas são capazes de se auto-renovar e originar células tumorais
(Flier & Clevers, 2009).

4
Figura 3. Tipos de câncer mais incidentes estimados para 2010, exceto pele não melanoma, na população brasileira. Instituto Nacional de Câncer & Ministério da Saúde, 2009.
I.2 Polaridade Celular
Entende-se por polaridade celular o padrão de organização assimétrica que permite a
compartimentalização especializada na célula, sendo fundamental para a morfogênese,
diferenciação e proliferação celular (Assémat et al., 2008). A polaridade de células epiteliais
resulta na diferenciação entre os domínios de membrana apical, em contato com o meio
externo, e basolateral, em contato com as células adjacentes e ao tecido conectivo. A
composição proteica e lipídica desses domínios difere, refletindo suas funções específicas na
célula. A porção apical intestinal mantém funções primárias, como absorção e secreção,
contendo canais iônicos e transportadores; a porção basolateral contém proteínas envolvidas
na adesão e comunicação célula-célula; e a porção basal contém sítios de ligação para
constituintes da lâmina basal, receptores de hormônios e outras moléculas de sinalização
(Nelson, 2003; Hartsock & Nelson, 2008). Portanto, a polaridade celular é essencial para as
funções de barreira e de transporte intracelular, e para manter a integridade dos epitélios.
Múltiplos processos são requeridos para o estabelecimento da polaridade em células
epiteliais, incluindo a entrega de vesículas contendo proteínas de diferentes domínios de
membrana (transcitose), a polarização do citoesqueleto, e a adesão celular (Figura 4). Em
células epiteliais polarizadas, o citoesqueleto de actina se localiza no córtex dos domínios de
membrana apical, lateral, basal, e nas microvilosidades, fornecendo principalmente um
suporte estrutural para as células; e os microtúbulos se organizam em feixes alinhados
formando um eixo apico-basal, com a terminação negativa orientada apicalmente e a positiva
basalmente, e participando da transcitose. Nesse contexto, a adesão intercelular é importante

5
A B
Membrana basal
Filamentos de actina
Feixe de actina
Microtúbulos
Microvilos
Adesão intercelular
Endossomo apical
Complexo de Golgi
Membrana lateral
Transcitose
Anel de actina
Endossomo basal
Fibras de estresse
porque previne a difusão de solutos pela via paracelular, restringindo a livre difusão de
lipídios e algumas proteínas associadas à membrana entre o eixo apico-basal (Nelson, 2003).
Figura 4. Desenvolvimento da polaridade epitelial. A polarização celular depende da organização do citoesqueleto nas regiões apical, medial e basal (A); da adesão intercelular; e da transcitose, que entrega vesículas em domínios específicos de membrana, com participação do Complexo Golgi (B). Adaptado de Nelson, 2003.
Em epitélios, o desenvolvimento da polaridade celular é regulado por quatro
complexos proteicos: o complexo Scribble, o complexo de polaridade celular planar (PCP), o
complexo das proteínas de partição defeituosa (PAR), e o complexo das proteínas Crumb
(CRB). O complexo Scribble é formado por três proteínas: Scrib, Discs large (DLG), e Lethal
giant larvae (LGL), que se localizam na região basolateral de células epiteliais do cólon, com
expressão aumentada no ápice das criptas. A redução da expressão de Scrib e DLG durante a
progressão de tumores de cólon indica o complexo Scribble como supressor tumoral. Além
disso, esse complexo pode interagir com o complexo PCP que, por sua vez, modula a
polaridade e a migração celular, e compreende um conjunto de produtos gênicos modulados
pelas proteínas Dishevelled (Dsv) e Frizzled (Fz). Ambas são componentes da denominada
via Wnt não canônica, e a desregulação dessas proteínas leva a um fenótipo mais maligno
(Katoh et al., 2005). Vale mencionar que o complexo Scribble, Dsv e Fz modulam APC,
importante no desenvolvimento de CCR (Humbert et al., 2006).
O complexo PAR foi descrito em Caenorhabditis elegans como modulador do
posicionamento assimétrico do fuso mitótico, resultando na divisão celular desigual.
Posteriormente, foi demonstrado em outras células animais, adotando padrões de localização
assimétrica no desenvolvimento de polaridade. São conhecidos seis membros da família PAR:

6
PAR-1 e -4 são serina treonina quinases; PAR-5 é uma proteína adaptadora; PAR-3 e -6
ancoram proteínas transmembranares ao citoesqueleto através do domínio PDZ, cujo nome foi
dado utilizando as iniciais das três proteínas onde foi descoberto (densidade-95 pós-sináptica
humana ou PSD95, drosophila disc large ou DLGA e zonula occludens 1 ou ZO-1); e PAR-2
modula processos de ubiquitinação (Goldstein & Macara, 2007). Dentre elas, a PAR-4 é
destacada por polarizar completamente células individuais. Essa proteína é transportada do
núcleo para o citosol pela proteína Strad, e a expressão de ambas ocasiona a formação de
citoesqueleto polarizado e de microvilosidades em células intestinais, mesmo na ausência de
contatos célula-célula (Baas et al., 2004). Um dos substratos de PAR-4 é a PAR-1, que se
localiza na membrana lateral e exclui PAR-3 desse domínio. PAR-3 forma um complexo com
PAR-6 e a proteína quinase C atípica (PKCa), que é mantida inativa no complexo PAR-3-
PKCa-PAR-6. Esse complexo é recrutado para a região de contato célula-célula, onde induz
sua formação através da interação com a molécula de adesão juncional (JAM) (Balda &
Matter, 2008; Miyoshi & Takai, 2008). Foi mostrado que as GTPases Cdc42 e Rac ativadas
podem se ligar em PAR-6 para organizar o citoesqueleto de actina (Georgeou et al., 2008), e
ainda, ativadores de Rac, denominados proteína de linfoma de célula T de invasão e metástase
(Tiam), podem se ligar em PAR-3 e organizar a formação de junções tight (JTs) (Chen &
Macara, 2005). Assim, o complexo PAR-3-PKCa-PAR-6 coordena a organização simétrica de
células epiteliais modulando os contatos célula-célula e o citoesqueleto polarizado (Georgeou
et al., 2008).
O complexo PAR pode atuar com o complexo CRB para regular a formação das
junções célula-célula. As proteínas CRB são exclusivas de membranas apicais e contatos
intercelulares, e são transmembranares com domínios de ligação em domínios PDZ e
domínios denominados FERM devido às iniciais das proteínas onde foram descobertos: 4.1
(four.one), ezrina, radixina, e moesina. Três isoformas de CRB foram descritas em
mamíferos, sendo CRB-3 a mais estudada em células epiteliais. CRB-3 estabiliza PAR-6 nas
junções nascentes modulando a formação de JTs (Iden & Collard, 2008). Posteriormente, foi
mostrado que a CRB-3 se associa com a proteína associada em JT com domínio PDZ (PATJ)
através da proteína associada com Lin sete (PALS1), formando o complexo CRB-
3/PALS1/PATJ, e interage com ZOs (Assémat et al., 2008). No entanto, o papel
desempenhado pelo complexo CRB e outros complexos proteicos na perda da polaridade
celular em CCR permanece por ser estabelecido, e cada vez mais estudos mostram a
importância da redistribuição das proteínas desses complexos durante a migração de células
epiteliais (Dow et al., 2007; Du et al., 2010). A Figura 5 ilustra de forma simplificada o

7
modelo proposto de exclusão mútua entre os complexos Scribble/PCP e PAR/CRB para a
manutenção da polaridade apico-basolateral.
Figura 5. Complexos envolvidos na polaridade epitelial. O complexo PAR é constituído pelas proteínas PAR3-PKCa-PAR6 e o Crumb compreende CRB, PALSJ e PATJ. Ambos se localizam nas JTs, e por isso são denominados complexos de polaridade apicais. No complexo Scribble, as proteínas LGL e DLG apresentam localização basolateral. A exclusão mútua entre os complexos Scribble e os complexos apicais controlam a polaridade apico-basal, e a fosforilação de LGL, PAR3 e CRB por PKCa mantém essa distribuição assimétrica. Adaptado de Iden & Collard, 2008.
A perda da polaridade celular e dos contatos célula-célula constitui um fenótipo
comumente observado em diferentes tipos de câncer. Estes eventos não representam somente
uma consequência da transformação celular, mas contribuem diretamente na tumorigênese,
visto que as proteínas que integram os contatos intercelulares atuam como supressoras
tumorais. Ainda, alterações na localização, fosforilação, glicosilação e expressão de proteínas
dos contatos célula-célula e dos complexos de polaridade medeiam a ativação de vias de
sinalização que regulam a proliferação e a migração celular.
I.3 Complexo juncional Apical (CJA)
O complexo juncional, representado na Figura 6, é a estrutura responsável pela
manutenção dos contatos entre células vizinhas epiteliais, sendo constituído por junções
comunicantes (GAP), desmossomos, JTs e JAs (Thiery & Sleman, 2006). Resumidamente, as
junções GAP são canais que intercomunicam o citoplasma de células adjacentes realizando a
troca direta de pequenos componentes citoplasmáticos. Em humanos, essas junções são

8
formadas por vinte e uma isoformas de conexinas, proteínas que se agregam em torno de um
poro central formando um hexâmero chamado hemicanal ou conexon (Maeda & Tsukihara,
2010). Os desmossomos são junções de adesão estáveis encontradas em tecidos expostos a
altos níveis de estresse mecânico, incluindo o trato gastrintestinal (TGI), relacionadas com a
integridade tecidual. São morfologicamente bem definidos como duas placas eletrondensas
idênticas entre células justapostas, associadas pela ancoragem aos filamentos intermediários
(FIs). Além de mediarem a adesão celular, tem-se postulado um papel para os desmossomos
na diferenciação celular e transdução de sinal (Thomanson et al., 2010).
Atualmente, tem sido utilizada a terminologia complexo juncional apical para designar
apenas as JTs e JAs (Ivanov et al., 2004a), que se ancoram ao citoesqueleto de actina. As JTs
são as principais estruturas responsáveis pelo controle da permeabilidade, bloqueando a livre
difusão de moléculas e íons através da via paracelular, assim como a difusão de moléculas
entre os domínios apical e basolateral, participando da manutenção da polaridade celular
(Miyoshi & Takai, 2008). Já as JAs são as principais responsáveis pela adesão física entre
células adjacentes, participando também na manutenção da polaridade celular e controle da
proliferação celular (Nelson, 2003; Niessen, 2007).
Figura 6. Organização e composição molecular do complexo juncional. O complexo juncional é formado por JTs, na região mais apical da célula, e JAs, logo abaixo, formando o CJA; desmossomos, que formam placas densas ancoradas em filamentos intermediários; e junções comunicantes, que formam canais de conexinas (não representadas). O esquema mostra ainda que as JTs são responsáveis pela manutenção da via paracelular (difusão de íons e solutos), indicada pela seta amarela. Adaptado de Steed et al., 2010.

9
I.3.1 Junções tight (JTs)
As JTs são essenciais para a funcionalidade da barreira semipermeável do epitélio e a
polaridade celular. Seus constituintes incluem proteínas integrais com um domínio
transmembranar, JAM; com quatro domínios transmembranares, ocludina, claudinas e
tricelulina; proteínas citoplasmáticas que associam as JTs ao citoesqueleto de actina, ZOs e
cingulina; e os complexos de polaridade celular PAR e CRB (Figura 7).
As JAMs apresentam duas imunoglobulinas no domínio extracelular, e medeiam
adesão célula-célula homotípica de forma independente de cálcio, sendo localizadas em JTs e
JAs. Compreendem os membros JAM-A, -B, -C, a proteína receptora associada a CAR
(cocksakie e adenovírus), molécula de adesão seletiva de célula endotelial (ESAM) e JAM4.
As JAMs interagem com as proteínas das JTs ocludina, cingulina, ZO-1, entre outras, sendo
importantes na correta localização das mesmas. Conforme já mencionado, as JAMs modulam
a polaridade celular através da interação com proteínas do complexo PAR (Balda & Matter,
2008), mas também são importantes na manutenção da permeabilidade paracelular e da
interação célula-matriz extracelular (MEC) (Mandell et al., 2005). Posteriormente, foi
mostrado que JAM-A inibe a migração de células de câncer de mama (Naik et al., 2008).
A ocludina, primeira proteína transmembranar da JT identificada, possui seus
domínios amino- e carboxi-terminais (N- e C-terminal, respectivamente) no citoplasma. Os C-
terminais são sítios de ligação para as proteínas ZOs, quinases, fosfatases e GTPases. Além
disso, a ocludina interage com o citoesqueleto de actina diretamente, ou através da cingulina e
as três isoformas de ZO (Paris et al., 2008); e com a E3 ubiquitina ligase denominada Itch
pelo domínio N-terminal, que ocasiona a degradação da ocludina por proteassomo (Traweger
et al., 2002). Em células epiteliais em repouso, a ocludina encontra-se predominantemente
hiperfosforilada em resíduos serina e treonina, e em complexo com ZO-1. Em células
epiteliais de cólon a ocludina pode ser fosforilada em resíduo serina e treonina por proteína
quinase C (PKC), modulando positivamente a função de barreira das JTs (Suzuki et al.,
2009). Já a fosforilação em resíduos de tirosina reduz sua interação com ZO e está relacionada
com o aumento da permeabilidade em células epiteliais (Kale et al., 2003). No entanto, um
estudo utilizando camundongos deficientes em ocludina, mostrou que a permeabilidade
paracelular intestinal nesses animais não era alterada, e foi sugerido o envolvimento dessa
proteína na diferenciação epitelial (Schulzke et al., 2005). Um estudo recente mostrou que a
localização de ocludina fosforilada em tirosina no fronte de monocamadas celulares é
necessária para o recrutamento das proteínas do complexo de polaridade PAR-3 e PATJ, e na

10
formação de estruturas necessárias para a migração celular polarizada (Du et al., 2010),
indicando um importante papel para ocludina em câncer.
A família das claudinas compreende aproximadamente vinte e quatro membros em
humanos, possuem seus domínios N- e C-terminal no citoplasma, e o C-terminal contém um
domínio PDZ para interação com proteínas que também contêm esse domínio, por exemplo,
as proteínas de polaridade PAR-3 e -6, PALS, multi-PDZ (MUPP-1) e ZO-1, -2 e -3. A
interação das claudinas é homotípica e permite a passagem seletiva de íons, indicando sua
principal função de regular a permeabilidade paracelular (Paris et al., 2008). Foi mostrado que
a claudina-1 possui um papel crucial na função de barreira da epiderme, já que os
camundongos deficientes para essa proteína exibem desidratação severa por consequência de
excessiva perda transepidérmica de água (Furuse et al., 2002). As diferenças entre as
propriedades de barreira das JTs podem ser justificadas por uma composição heterogênea de
claudinas em diferentes tecidos epiteliais. Por exemplo, nas criptas intestinais, que são
fracamente resistentes à permeabilidade paracelular, a claudina-2 reduz a resistência elétrica
transepitelial (RET); já em tecidos com alta resistência, como a porção distal dos néfrons, as
claudinas-7 e -8 aumentam a RET (Oliveira & Morgado-Díaz, 2007). É importante ressaltar
que as claudinas podem ser reguladas por fosforilação, por exemplo, a claudina-5 pode ser
fosforilada por proteína quinase dependente de AMP cíclico (PKA) (Soma et al., 2004);
transcrição, através do repressor transcricional Snail (Ikenouchi et al., 2003) e do fator de
transcrição sox-9 (Darido et al., 2008); por fatores de crescimento; e ainda podem interagir
com as metaloproteases de MEC (MMPs) -2 e -14 (Paris et al., 2008). Além disso, as
claudinas-1, -3 e -4 encontram-se aumentadas em CCR humano, embora não funcionais
(Oliveira et al., 2005) e o aumento de claudina-4 em linhagens de CCR humano induziu perda
da funcionalidade das JTs e aumento da migração celular e expressão de MMP-2 (Takehara et
al., 2009). A função e importância das claudinas em epitélios e em diversos carcinomas foram
revisadas por Oliveira & Morgado-Díaz, 2007.
As quatro isoformas de tricelulinas descritas em humanos apresentam seus domínios
C- e N-terminais citoplasmáticos (Ikenouchi et al., 2005) e localização predominante nos
pontos de contato entre três células, denominados tricelulares (tJTs), mas também em contatos
bicelulares (bJTs) (Figura 7B). Foi mostrado que a tricelulina modula a permeabilidade
paracelular de células epiteliais nesses dois tipos de JTs, mas as tJTs são de fundamental
importância na permeabilidade a macromoléculas (Krug et al., 2009). No entanto,
camundongos nocautes para tricelulina apresentaram apenas deficiências auditivas sem afetar
outros tecidos onde as JTs desempenham funções essenciais, indicando que outras proteínas

11
das JTs poderiam compensar a ausência de tricelulina, bem como de ocludina. Dessa forma,
foi identificada a proteína MarvelD3, que interage com tricelulina nas tJTs e com ocludina nas
bJTs (Raleigh et al., 2010).
As ZOs (-1, -2 e -3) são proteínas citoplasmáticas pertencentes à família de guanilato
quinases associadas à membrana (MAGUKs), e interagem com proteínas transmembranares
das JTs através do seu domínio PDZ na região N-terminal e com o citoesqueleto de actina
através do domínio C-terminal. As ZOs também podem interagir com proteínas das JAs em
células que não formam JTs e são importantes na formação correta do eixo apico-basal, pois a
ausência das suas três isoformas em células epiteliais resulta na perda da formação de JTs
(Umeda et al., 2006). Além disso, foi relatado que a perda da interação entre PALS1 e ZOs
causa defeitos na biogênese do CJA (Wang et al., 2007). Os domínios guanilato quinase
(GUK), pelo qual ocorre a interação com ocludina, e o homólogo a Src (SH3) de ZO-1, são
importantes na interação com a proteína ligadora de ácido nucleico associada a ZO (ZONAB),
proteínas quinase e proteínas G (Matter & Balda, 2007). Embora existam controvérsias sobre
a localização das ZOs no núcleo, essas proteínas regulam a localização de fatores de
transcrição, inclusive de ZONAB. A interação entre ZO-1 e ZONAB resulta na localização
citosólica desta última, inibindo genes do ciclo celular. É interessante notar que o alvo desse
fator de transcrição é o gene de E2F, comumente superexpresso em carcinomas (Balda et al.,
2003). A ZO-2 também regula o ciclo celular, através da repressão transcricional da ciclina
D1 (Huerta et al, 2007).
Outras proteínas recrutadas para as junções célula-célula podem apresentar localização
nuclear e regular a transcrição gênica, como simplequina, proteína ativadora 1 (AP-1) e SAF-
B, embora com funções ainda não esclarecidas (Matter & Balda, 2007). Recentemente foi
mostrado um aumento de simplequina nuclear em amostras de pacientes com CCR. Esse
estudo mostrou que a inibição dessa proteína foi suficiente para reduzir a proliferação celular
e a tumorigenicidade em camundongos, bem como de reduzir os níveis de ZONAB nuclear e
induzir a polarização celular. Esses eventos foram mediados através da redução da expressão
de claudina-2, e indicaram uma importante função para a simplequina na tumorigênese
(Buchert et al., 2010). Outras proteínas adaptadoras identificadas, mas com funções pouco
compreendidas, incluem cingulina, MUPP-1, proteína guanilato quinase invertido associada à
membrana (MAGI), entre outras (Balda & Matter, 2008).

12
Figura 7: Composição molecular das JTs. A) As JTs são compostas pelas proteínas JAM, ocludina e claudinas, que se ancoram ao citoesqueleto de actina através das ZOs e proteínas dos complexos de polaridade celular. B) Distribuição das proteínas das JTs nas tJTs e bJTs. Adaptado de Chiba et al, 2008 (A); Raleigh et al., 2010 (B).
I.3.2 Junções aderentes (JAs)
As JAs fornecem uma adesão celular flexível e dinâmica, e regulam a inibição por
contato do crescimento celular. Em nível molecular, as JAs podem ser subdivididas em dois
tipos de complexos de adesão, ilustrados na Figura 8A: o complexo nectina/afadina e o
complexo E-caderina/catenina (Niessen & Gottardi, 2008).
As caderinas são glicoproteínas transmembranares que medeiam a adesão celular
através de interação homofílica dependente de cálcio, e interagem com proteínas da família
das cateninas através do seu domínio citoplasmático (Niessen, 2007). São subdivididas em
seis subfamílias: caderinas clássicas do tipo I e do tipo II, caderinas desmossomais, caderinas
transmembranares de sete passagens, grandes caderinas e protocaderinas (Stemmler, 2008). A
E-caderina, integrante da subfamília de caderinas clássicas do tipo I, é a principal proteína
transmembranar de células epiteliais. Essa proteína possui cinco domínios extracelulares de
caderina repetitivos (EC), um domínio justamembranar (JMD), e um domínio de ligação em
catenina (CBD) (Hartsock & Nelson, 2008). As E-caderinas se associam lateralmente
formando homodímeros através dos domínios EC, de forma dependente de cálcio, atuando na
manutenção da adesão intercelular.
A perda dos contatos célula-célula e concomitante perda de E-caderina aumenta a
motilidade celular, invasividade e metástase (Onder et al., 2008). Por exemplo, foi relatado
que, a proteína tirosina fosfatase (PTP) que contém a sequência peptídica rica em prolina (P),
ácido glutâmico (E), serina (S) e treonina (T) (PEST), localizada nas JAs, suprime a migração
de células de câncer de cólon através da formação de contatos mediados por E-caderina
A B

13
(Espejo et al., 2010). A expressão da E-caderina é negativamente regulada pelos fatores
transcricionais Snail, Slug, Twist, E12 (E47, TCFE2A), SIP1 (ZEB2) e δEF1 (ZEB1). Esses
fatores se ligam na sequência E-box do promotor da E-caderina, reprimindo sua transcrição, o
que é comumente observado em tumores (Wheelock et al., 2007). Ainda, na progressão de
alguns carcinomas pode ser observada a clivagem do domínio extracelular da E-caderina
pelas MMP-2, -9 e -14, resultando na E-caderina solúvel (sE-cad) e na liberação do seu
domínio intracelular para o citoplasma. Subsequentemente, a sE-cad pode se associar ao
domínio extracelular da E-caderina de células adjacentes e desorganizar as JAs. Já o domínio
intracelular pode ser translocado para o núcleo onde ativa genes relacionados com o processo
tumorigênico, como ciclina D1, AP-1 e transdutor de sinal e ativador de trascrição (STAT)
(Symowicz et al., 2007). Eventos epigenéticos também estão envolvidos na redução da
expressão de E-caderina durante a progressão tumoral, como a hipermetilação de um sítio
próximo a sua região promotora em carcinomas de mama, próstata e colorretal, e a
deacetilação da histona H3 em CCR (Liu et al., 2008). Mais recentemente, foi descrito o fator
de transcrição específico de intestino Cdx2, que aumenta o tráfego de E-caderina para a
membrana, regula positivamente a adesão célula-célula e reduz a migração celular (Funakoshi
et al., 2010).
Conforme mencionado, a E-caderina interage com proteínas da família das cateninas
por seu domínio CBD. Essa interação, com β-catenina e γ-catenina, ocorre de forma
mutuamente exclusiva, e com a α-catenina de forma dinâmica através da β-catenina (Hartsock
& Nelson, 2008). A α-catenina se liga direta ou indiretamente ao citoesqueleto de actina
através de α-actinina, vinculina, ZO-1, afadina e formina (Drees et al., 2005). A α-catenina
também é importante para o recrutamento de uma proteína denominada EPLIN, impedindo
sua função de despolimerizar actina (Abe & Takeichi, 2008). Vale ressaltar que a perda de α-
catenina ocorre em linhagens celulares tumorais, incluindo de CCR. No entanto, mutações no
gene que codifica esta proteína não têm sido observadas em tumores humanos, indicando que
a redução da sua expressão ocorre por inativação epigenética ou modificações pós-
traducionais (Benjamim & Nelson, 2008). Ainda, células epiteliais onde foi induzida a perda
de α-catenina apresentaram-se hiperproliferativas, multinucleadas e com aumento na
migração celular, e com adesões intercelulares desorganizadas (Vasioukhin et al., 2001). O
acúmulo citosólico de α -catenina também tem sido associado ao potencial tumorigênico, o
que ocorre pela redução da interação entre α- e β-catenina por fosforilação mediada por fator
de crescimento epidérmico (EGFR) e proteína quinase derivada de sarcoma (Src), ou

14
competição para ligação em proteína ativada por GTPase com motivo IQ (IQGAP), uma
proteína recrutada pela GTPase Rac para os contatos célula-célula, que será descrita adiante
(Benjamim & Nelson, 2008). Ainda, na ausência de contatos célula-célula, a α -catenina foi
observada no núcleo de linhagens de câncer de cólon (El-Bahrawy et al., 2002). A vinculina,
por sua vez, se localiza nas JAs ligando-se em α-catenina e actina filamentar (F-actina) e nos
contatos de adesão com a MEC (Niessen & Gottardi, 2008).
É bem estabelecido que a ligação de β-catenina em E-caderina mantém uma adesão
completa, e ainda previne a degradação de E-caderina por proteólise (Maher et al., 2009). A
β-catenina interage com uma proteína tirosina fosfatase de baixo peso molecular (LMW-PTP,
low molecular weight) que impede a fosforilação de E-caderina, prevenindo assim o
recrutamento da E3 ubiquitina ligase Hakai, que promove sua ubiquitilação (Fujita et al.,
2002). Além disso, a β-catenina é regulada pelo gene APC, comumente mutado em CCR. A
mutação em APC ou em β-catenina regula a via de sinalização Wnt, na qual proteínas
solúveis Wingless (Wnt) secretadas por células do estroma se associam aos receptores Fz e
LRP5/6. Essa associação (Figura 7B) desencadeia uma cascata de sinalização que impede que
o complexo formado pelas proteínas APC/Axina/caseína quinase 1α (CK1α)/ glicogênio
sintase quinase 3β (GSK3β) direcione a β-catenina para degradação por proteossomo,
ocasionando seu acúmulo no citoplasma e posterior translocação para o núcleo. No núcleo, a
β-catenina atua como cofator de transcrição de genes que participam da proliferação celular e
da apoptose, como o fator de célula T (TCF) (Flier & Clevers, 2009).
Portanto, o complexo E-caderina-β-catenina estável modula a localização e função da
β-catenina, pois sua localização nuclear culminaria na ativação de fatores de transcrição
(Maher et al., 2009). No entanto, a regulação de β -catenina é complexa e essa proteína pode
ser fosforilada em vários resíduos: na tirosina 654 resulta em dissociação de E-caderina; na
tirosina 142 impede sua ligação em α -catenina e resulta no aumento da transcrição de genes
alvos da via Wnt; e na região N-terminal de β-catenina, a fosforilação nos resíduos serina
33/37/41 leva a sua degradação, e no resíduo serina 45 parece modular a sua localização
nuclear. Ainda, a fosforilação de β-catenina na região N-terminal é necessária para sua
localização no fronte invasivo de células migratórias de CCR (Faux et al., 2010). Além disso,
a β-catenina se liga diretamente em EGFR, indicando seu papel na sinalização intracelular, e
na inibição do crescimento por contato (Niessen, 2007). A p120-catenina é outro membro das
JAs e se associa ao domínio JMD da E-caderina, sendo responsável pela regulação local de F-

15
actina através da modulação da atividade de GTPases homólogas a Ras (Rho) e estabilidade
do complexo caderina-catenina (Anastasiadis, 2007).
As nectinas pertencem à superfamília das imunoglobulinas, que compreende as
nectinas-1,-2,-3 e -4. A maioria das suas isoformas contém um motivo na região C-terminal
que se liga ao domínio PDZ da afadina que, por sua vez, possui um domínio de ligação às
proteínas ligadoras de F-actina (α-catenina e ZO-1), e também pode se ligar em Ras. Nectinas
e afadinas participam da formação de JAs e JTs (Niessen & Gottardi, 2008). Embora tenha
sido mostrado que as nectinas podem interagir com α- e p120-catenina, não se conhece como
pode ocorrer a inter-relação entre o complexo E-caderina e nectina (Niessen, 2007). Estudos
têm mostrado que a redução da expressão de nectina-1α está associada ao aumento do
potencial maligno em queratinócitos (Matsushima et al., 2003) e que sua superexpressão está
associada com a formação da JAs em células de câncer gástrico (Peng et al., 2002). Outra
proteína pouco descrita que também pode ser encontrada nas JAs é a MAGI, que possui seis
domínios PDZ ancorando as JAs em F-actina, e ativa vias de sinalização importantes para a
biogênese do CJA (Niessen, 2007; Zhang et al., 2007a).
Figura 8: Composição molecular das JAs e participação da β -catenina na via de sinalização Wnt. A) E-caderina e nectina associam-se ao citoesqueleto de actina através de α-catenina ou afadina, respectivamente. A α-catenina mantém a polimerização de actina no contato célula-célula, mas não se liga nas JAs e actina simultaneamente. A ligação de β -catenina impede a proteólise da E-caderina, e a p120-catenina estabiliza as JAs. B) Na ausência de estimulação (i), baixos níveis de β -catenina são mantidos através da sua fosforilação pelo complexo APC/CKI/GSK3/axina que induz sua degradação via proteossomo. Com a ativação da via Wnt (ii), os ligantes dos receptores Fz/LRP induzem a inibição do complexo proteico, impedindo a degradação da β -catenina, que acumula no citosol e pode ser direcionada ao núcleo, onde atua como fator transcricional em complexo com TCF. Adaptado de Niessen & Gottardi, 2008 (A); Flier & Clevers, 2008 (B).
E-caderina nectina
i ii
Degradação via proteossomo
a f a d i n a
B A

16
I.4 Citoesqueleto de actina
O citoesqueleto organiza o conteúdo celular, conecta física e bioquimicamente a célula
com o ambiente externo e gera forças coordenadas capazes de mover a célula ou modificar
sua forma. Essa estrutura pode ser definida como uma rede dinâmica de proteínas, composta
por três tipos de filamentos proteicos: microtúbulos (MTs), filamentos intermediários (FIs), e
F-actina ou microfilamentos. Resumidamente, os MTs são polímeros compostos por
heterodímeros de tubulina α e β. Em células epiteliais, a maioria dos MTs não está ancorada
ao centrossomo como em outros tipos celulares, pois a formação dos contatos célula-célula e a
polarização celular envolvem a migração dos centríolos para a superfície apical, permitindo o
arranjo apico-basal (Harris & Tepass, 2010). Recentemente, tem sido mostrado que os MTs
regulam a biogênese das JAs e interagem diretamente com p120-catenina (Meng et al., 2008).
Os FIs constituem um sistema citoplasmático que conecta os desmossomos, proporcionando
resistência tensional às células e tecidos, e também formam a lâmina nuclear. Estruturalmente,
os FIs citoplasmáticos são tetrâmeros formados a partir de uma ligação anti-paralela entre dois
dímeros formados por uma haste central em α-hélice flanqueada pelos domínios N- e C-
terminais. Os FIs são agrupados pela homologia de suas sequências, e se expressam de acordo
com a diferenciação celular; por exemplo, células musculares diferenciadas expressam
predominantemente desmina, mas suas precursoras expressam vimentina (Herrmann et al.,
2007). Células epiteliais são constituídas por diferentes filamentos de queratina, mas células
com alto potencial invasivo e baixa expressão de E-caderina podem expressar vimentina,
conforme descrito adiante (Zhao et al., 2008b).
A actina determina a morfologia celular, fornecendo a base estrutural para a
motilidade, contratilidade, e transporte intracelular. Em mamíferos, há seis isoformas de
actina, sendo as quatro α musculares, e as β e γ ubiquitosamente expressas. Postula-se que as
isoformas β e γ executem funções celulares distintas em virtude das suas diferentes
especificidades com proteínas ligadoras de actina e diferente localização subcelular (Perrin &
Ervasti, 2010). Ainda, tem sido mostrada a localização nuclear de β -actina em resposta ao
estresse, assim como de proteínas ligadoras de actina, por exemplo, zixina, cofilina e
profilina. Acredita-se que a actina nuclear desempenhe funções estruturais e regulatórias na
remodelagem de cromatina e na transcrição (Bettinger et al., 2004). Em monocamadas de
células epiteliais polarizadas, a F-actina citoplasmática é distribuída na porção basal, como
fibras de estresse que mantêm a ancoragem da célula ao substrato; apical, como feixes de
actina nos sítios de adesão célula-célula conhecidos como anéis de actina e onde estão
ancoradas proteínas do CJA; e na porção central das vilosidades. Em adição, a F-actina forma

17
o córtex celular logo abaixo da membrana plasmática e controla os movimentos da superfície
celular através da interação com moléculas de miosina (Nelson, 2003).
Os microfilamentos formam-se a partir da actina globular (G-actina), que se liga em
ATP para ligar-se em outra G-actina e formar a F-actina, uma fita polarizada de dupla hélice
que apresenta uma região de crescimento rápido ou terminal+ e uma de crescimento lento ou
terminal-. A polimerização ocorre quando a G-actina-ATP se liga em F-actina, havendo a
hidrólise do ATP em ADP, e a liberação de um fosfato inorgânico. A adição de G-actina-ATP
ocorre preferencialmente no terminal+, e a dissociação de monômeros-ADP no terminal-. Pode
também ocorrer um evento denominado treadmilling, onde há um balanço entre o número de
moléculas que são adicionadas aos terminais+ e - (Disanza et al., 2005).
Mais de cento e cinquenta proteínas contêm domínio de ligação em actina, e aquelas
que alteram sua organização podem ser agrupadas de acordo com sua função: 1) proteínas que
se ligam em monômeros, sequestram G-actina e previnem sua polimerização, como a
profilina; 2) proteínas que despolimerizam filamentos no terminal- e induzem a conversão de
F-actina para G-actina, fornecendo monômeros para a montagem de filamentos no terminal+,
como a despolimerizante de actina (ADF)/cofilina; 3) proteínas que bloqueiam o terminal+
dos filamentos bloqueando sua elongação e permitindo que a polimerização ocorra de forma
tempo e espaço específicos, como a tropomodulina e a gelsolina; 4) proteínas que cortam a F-
actina em duas partes, como a gelsolina; 5) proteínas nucleadoras que se ligam em actina e
iniciam a formação de nova F-actina a partir de filamentos pré-existentes originando feixes
altamente ramificados, como o complexo de proteínas relacionadas à actina (Arp) 2/3 e
forminas; 6) proteínas estabilizadoras, que previnem a despolimerização da actina, como a
tropomiosina; e 7) proteínas motoras constituídas por dois pares de cadeias leves e duas
cadeias pesadas que se ligam em actina e hidrolizam ATP, como a miosina 2 em epitélios.
Vale ressaltar que essas proteínas não se restringem a apenas um desses grupos, podendo
executar mais de uma dessas funções, por exemplo, a profilina não só previne a polimerização
de F-actina, mas também facilita a troca de nucleotídios (de ADP para ATP) dos monômeros
de actina (Dos Remedios et al., 2003).
I.4.1 Inter-relação entre o CJA e o citoesqueleto de actina
Dentre as evidências que sugerem a participação da F-actina na biogênese e função do
CJA, além da interação direta com seus componentes (ZO-1, α-catenina e afadina), como
ilustrado na Figura 9, pode-se mencionar: a) a remodelagem do CJA é concomitante à
reorganização da actina perijuncional (Ivanov et al., 2004a); b) a desorganização da F-actina

18
por drogas previne a formação do CJA (Yamada et al., 2004); c) o rompimento do CJA pode
ser desencadeado por GTPases Rho, moduladores da F-actina (Hopkins et al., 2003; Ivanov,
2008); d) a proteína de JA p120-catenina modula a ativação de Rho e inibição de Rac, que
modulam F-actina (Anastasiadis, 2007); e) A mutação de E-caderina em seu domínio de
ligação nas proteínas adaptadoras, que ancoram as JAs em F-actina, inibe a adesão celular
(Miyoshi & Takai, 2008); e f) proteínas moduladoras de actina localizam-se nas JAs, por
exemplo α-actinina, cortactina, Arp2/3, fosfoproteína estimulada por vasodilatador
(Ena/Vasp), proteína homóloga a verprolina da família WASP (WAVE), proteína da
síndrome Wiskott Aldrich (WASP) (Niessen & Gottardi., 2008) e miosina2 (Smutny et al.,
2010). No entanto, ainda não está bem esclarecido como o CJA e F-actina se inter-relacionam
para regular a atividade migratória de células epiteliais tumorais e quais vias de sinalização
regulam este evento.
Figura 9. Proteínas adaptadoras do CJA que se ligam em actina. A Figura mostra a estrutura das proteínas ZO-1, α-catenina e afadina. Essas proteínas contêm domínios de ligação em actina na região C-terminal, podem interagir com diferentes proteínas (em azul) e também podem interagir entre si. Abreviações: SH3, domínio homólogo a Src3; GUK, domínio guanilato quinase; VH1-3, domínio homólogo a vinculina1-3; RA, domínio associado a Ras; FHA, domínio associado a forkhead (de sinalização nuclear e interação com proteínas fosforiladas); DIL, domínio dilute (de interação proteína-proteína); e PR, domínio rico em prolina. Adaptado de Miyoshi & Takai, 2008.

19
I.5 Componentes de vias de sinalização envolvidas na modulação do CJA e do
citoesqueleto de actina
Dentre as vias de sinalização celular relacionadas ao processo tumorigênico e
aquisição do fenótipo maligno, serão abordados os componentes relacionados à modulação do
CJA e do citoesqueleto de actina, particularmente as proteínas quinases, tais como Src,
proteína quinase ativada por mitógeno (MAPK), EGFR, fosfatidil-inositol-3-quinase (PI3K),
PKA e, finalmente as GTPases Rho.
I.5.1 Proteínas quinases
A Src pertence a uma família de proteínas tirosinas quinase dividida nas subfamílias
Src (Src, Yes, Fyn e Fgr) e Lyn (Lyn, Hck, Lck e Blk). A Src possui quatro domínios SH, um
N-terminal miristoilado que a ancora à membrana, e um C-terminal contendo o resíduo
tirosina 530, cuja fosforilação pela Src quinase C-terminal (Csk) ou sua homóloga (Chk)
resulta numa conformação fechada e inativa da proteína. A desfosforilação dessa tirosina
permite que Src adote uma conformação aberta e ativa, mas sua completa ativação requer a
autofosforilação no resíduo tirosina 416/419 no domínio catalítico (Figura 10). A perda do
domínio C-terminal implica no aumento da atividade de Src e do potencial de transformação
celular. Alternativamente, Src também pode ser ativada pela interação direta com proteínas
quinase como EGFR e quinase de adesão focal (FAK) através do seu domínio SH2,
impedindo as associações intramoleculares inibitórias de Src (Wozniak et al., 2004).
A Src é mantida no seu estado inativo por supressores, e pode ser ativada de forma
transiente na mitose, ou de forma constitutiva por mutação. No entanto, mutações que ativam
Src são raras e não justificam a frequente superexpressão e/ou ativação de Src em CCR. Src
fosforila FAK para regular os contatos célula-MEC, modula a formação de lamelipódios, a
proteólise da MEC e a expressão de fator de crescimento vascular endotelial (VEGF),
relacionados à migração, invasão e angiogênese (Mitra & Schlaepfer, 2006). Ainda, linhagens
tumorais com alta atividade de Src são mais móveis e invasivas in vitro (Frame, 2004), e a
superexpressão de Src induz fenótipos de transformação celular, incluindo crescimento
independente de ancoragem e tumorigenicidade in vivo (Oneyama et al., 2008).
Além disso, a proteína p120-catenina foi identificada como substrato de Src, indicando
que essa quinase pode regular o CJA. Foi então mostrado que a inibição de Src previne a
desorganização de JTs em células epiteliais (Basuroy et al., 2003) e a translocação de β-
catenina para o núcleo em células de câncer de cólon, reduzindo o crescimento e a migração
celular, e aumentando a adesão célula-célula (Coluccia et al., 2006). Também em células de

20
câncer de cólon, a ativação de Src restaurou a localização de E-caderina nas JAs de forma
dependente de integrina, apontando Src como mediadora entre os contatos célula-célula e
célula-MEC (Avizienyte et al., 2002). Portanto, Src desempenha um papel dual nas JAs,
podendo modular positiva ou negativamente os contatos mediados por E-caderina,
dependendo do estímulo celular (Shindo et al., 2008).
Figura 10: Configurações de Src inativa e ativa. Na ativação basal ou inativação de Src, o domínio SH2 interage com o resíduo tirosina 530 fosforilado na região C-terminal. Nessa condição, o domínio quinase SH1 e o SH3 também interagem, resultando numa estrutura molecular fechada. Já na sua ativação, mediada por integrinas ou por receptores de tirosina quinase, a interação intramolecular entre SH1 e SH3 é desfeita resultando na configuração molecular aberta, onde o resíduo tirosina 530 é desfosforilado e o 416/419 fosforilado. Adaptado de Aleshin & Finn, 2010.
A PKA é uma serina treonina quinase heterotrimérica ativada pela interação com o
AMP cíclico (AMPc), um segundo mensageiro gerado através de receptor acoplado em
proteína G (GPCR). PKA compreende dois domínios regulatórios (R) e dois catalíticos (C).
Os domínios R apresentam três funções: a) mantêm a enzima inativa; b) liberam o domínio C
quando duas moléculas de AMPc se ligam em cada R; e c) são responsáveis pela dimerização
e ligação em proteínas de ancoragem em quinase-A (AKAP), que integram múltiplos sinais de
fosforilação. É sugerido que a inibição de PKA seja requerida para aumentar a adesão célula-
célula e célula-MEC, e para reduzir a migração celular (Pidoux & Taskén, 2010). A ativação
de PKA regula as GTPases: inibe RhoA por fosforilação direta ou de uma proteína regulatória
de Rho (Qiao et al., 2008) e, apesar de Rac não ter sítio para ser fosforilada por essa quinase,
suas proteínas ativadoras Tiam-1 e a proteína domínio triplo funcional (Trio), podem ser
fosforiladas por PKA. PKA tem múltiplos substratos, mas sua ativação é compartimentalizada
através de gradientes de AMPc produzidos localmente e pelo recrutamento por AKAPs

21
específicas para cada função celular (Figura 11). Por exemplo, foi observado que células de
câncer de cólon migratórias sobre laminina apresentam gradientes de PKA ativa a partir do
fronte de migração, restrita à superfície basal das células (Paulucci-Holthauzen et al., 2009).
Figura 11: Regulação da ativação de PKA. GPCR ativa localmente a adenilato ciclase, que gera AMPc (círculos vermelhos), evento limitado pelas fosfodiesterases (PDEs). O GPCR é confinado em domínios específicos da membrana celular e, através de AKAPs, as estruturas subcelulares podem recrutar PKA. Adaptado de Pidoux & Taskén 2010.
A cascata das MAPKs é a transdução de sinal mediada pela ativação sequencial das
proteínas Raf-MEK-ERK. Essa via pode ser ativada por Ras ou EGFR (Figura 12). As Raf
quinases (ou MAPKKK) catalisam a fosforilação e ativação de MEK1 e 2 (MAPKK) que, por
sua vez, medeiam fosforilação em resíduo tirosina e treonina nos seus únicos substratos, as
quinases reguladas por sinal extracelular 1 ou 2 (ERKs ou MAPKs). As MAPKs são serina
treonina quinases inicialmente caracterizadas como transdutoras de sinais proliferativos, pois
sua ativação pela via EGFR-Ras-Raf-MEK induz a expressão de ciclina D. São divididas em
três subgrupos: as c-Jun N-terminal quinases (JNKs), as p38 e as ERKs (Roskoski Jr, 2010).
A perda da expressão de JNK1 diminuiu a proliferação e a migração em células de
hepatocarcinoma (Zhang et al., 2010); porém, trabalhos também relataram a participação de
JNK em processos pró-apoptóticos induzidos por quimioterápicos, mostrando que sua
ativação depende do contexto celular (Johnson & Nakamura, 2007). Em camundongos, a
perda de expressão de JNK1 aumentou a atividade trancricional de β-catenina, regulando a
formação de tumores intestinais (Hu et al., 2008). Foi também mostrado que JNK é ativada
pela GTPase Rho e sua efetora Rho-quinase (ROCK) na perda de adesão intercelular de
células de câncer de cólon (Naydenov et al., 2009). Nesse mesmo tipo celular, a inibição de
p38 suprimiu a proliferação celular e aumentou a apoptose (Tsuchiya et al., 2008) e em
carcinomas, foi mostrado que as ERKs aumentam a motilidade e invasão celular (Wu et al.,
2008), formação de CCR in vivo e metástase (Voisin et al., 2008). Vale destacar que a

22
redução da proliferação celular ocasionada pela inibição de ERK em células de câncer de
cólon com Ras ativa, pode ser compensada pela ativação de p38 (van Houdt et al., 2010),
indicando a interação entre os membros da família ERK.
Em humanos, a família de receptores ErbB ou EGFR é dividida nas subfamílias
EGFR, ErbB2, ErbB3 e ErbB4 . O EGFR é uma tirosina quinase transmembranar unipasso
ativada por ligação do fator de crescimento epidérmico (EGF) e do fator alfa de necrose
tumoral (TNF-α). Sob ativação, o EGFR homodimeriza ou forma heterodímeros com outros
membros da família ErbB. Isso estimula a fosforilação nos seus resíduos de tirosina no
domínio regulatório e subsequente ligação de moléculas, propagando a sinalização iniciada
por EGF extracelular até o núcleo, induzindo proliferação celular (Figura 12). Vários tipos de
carcinomas humanos apresentam aumento de expressão de EGFR, e a ativação de Src
potencializa seu efeito mitogênico (Tice et al., 1999). O EGFR também pode se associar com
E-caderina nas JAs, prevenindo a sua dimerização e subsequente aumento da migração,
proliferação e sobrevivência celular (Bremm et al., 2008), e com F-actina, o que reduz sua
autofosforilação (Tang & Gross, 2003).
Figura 12: Via de sinalização do EGFR. O EGF se liga no EGFR induzindo sua dimerização e autofosforilação em resíduos de tirosina. O receptor fosforilado recruta proteínas adaptadoras (Grb2/SOS) para ativar a via Ras-Raf-MEK-ERK e fatores de transcrição. Adaptado de Kolch et al., 2002.
As PI3Ks são tirosinas quinases lipídicas agrupadas em três classes, dependendo das
suas afinidades por substratos e homologia de sequência. As PI3Ks de classe I fosforilam
fosfatidil-inositol(PI)-4,5-bifosfato (PIP2) para gerar o segundo mensageiro PI-3,4,5-trifosfato
(PIP3). A PTEN reverte esse processo, sendo considerada uma supressora tumoral (Figura
13). A classe I é ainda subdividida de acordo com sua ativação por GPCRs ou receptores
tirosina quinases, como o EGFR. A geração de PIP3 causa o recrutamento da serina treonina
Transcrição gênica
Substrato no citosol e citoesqueleto
Substratos no núcleo
EGFR
Membrana celular
núcleo
Quinares

23
quinase Akt para a membrana, e sua subsequente ativação pela quinase dependente de
fosfoinositídeo-3 (PDK1), entre outras. A atividade de Akt está relacionada à migração,
invasão e proliferação celular, via ativação de vários substratos, por exemplo, inibindo
GSK3β (de Araújo et al., 2010). A PI3K pode atuar de forma independente de Akt, por
exemplo, fosforilando Rac para modular F-actina e a formação de lamelipódios. As classes II
e III de PI3K geram PIP3 a partir de PI, com funções relacionadas ao tráfego de membrana e
crescimento celular, e autofagia, respectivamente (Chalhoub & Baker, 2008).
Em CCR, 15% de tumores humanos apresentam mutações que levam a ativação de
PI3K e 9% a inibição de PTEN (Chalhoub & Baker, 2008). A superexpressão de PI3K de
classe I conferiu um fenótipo maligno em células epiteliais mamárias humanas (Zhao et al.,
2003), e a sua ativação aumentou a proliferação de células epiteliais intestinais (Sheng et al.,
2003). Além disso, é conhecido que a formação de JAs recruta PI3K e Akt para a membrana
plasmática, e nosso Grupo recentemente mostrou que a inibição farmacológica da via PI3K-
Akt induziu diferenciação celular, formação de contatos célula-célula funcionais e redução da
migração celular em linhagens invasivas de câncer de cólon (de Araújo et al., 2010).
Figura 13: Mecanismos de ativação da PI3K de classe I. A ativação dos receptores tirosina quinase ocasiona o recrutamento da subunidade regulatória p85 da PI3K e ativação da subunidade catalítica (p110 αβδ). Alternativamente, GPCRs podem se ligar na subunidade regulatória p101 de PI3K através da subunidade Gβγ, ativando a sua subunidade catalítica p110γ. Ambos os mecanismos levam a adição de fosfato ao PIP2 na posição 3 do anel inositol e geração de PIP3. PTEN defosforila PIP3 agindo como regulador negativo dessa via. Adaptado de Chalhoub & Baker, 2008.
I.5.2 Pequenas GTPases Rho
As pequenas GTPases pertencem à superfamília Ras, subdividida em 7 famílias: Ras,
que possui as isoformas H-, N-, e K-Ras; Ran; Rab, com aproximadamente 70 membros;
Rho, cujos membros mais descritos são RhoA, B e C, Rac 1 e 2 e Cdc42; Arf, dividida nos

24
grupos Arf, Sar e Arl; RAP1 e 2; e RAL1A e 1B (Vega & Ridley 2008). Estudos iniciais
mostraram que a inibição de Rho com a exoenzima C3 de Clostridium botulinum ocasionava
perda de fibras de estresse, arredondamento celular, e perda da adesão célula-MEC (Ridley &
Hall, 1992). Investigações subsequentes mostraram que as GTPases Rho são ativadas por
ligantes extracelulares, ocasionando modificações de membrana e do citoesqueleto em
fibroblastos: O tratamento com ácido lisosfatídico (LPA), que ativa Rho, induziu a montagem
de fibras de estresse e adesões focais (AFs), estruturas de contato célula-MEC. Já o tratamento
com o fator de crescimento derivado de plaquetas (PDGF) e insulina, que ativam Rac e Cdc42,
induziu a formação de lamelipódios e filopódios, respectivamente (Hall, 1998). A partir daí, foi
mostrado o envolvimento dessas GTPases na regulação do crescimento, polaridade e adesão
celular, apoptose, sobrevivência e tráfego de vesículas (Benitah et al., 2004). Embora as
GTPases Rho regulem funções importantes na progressão do câncer e estejam superexpressas
em diferentes tipos de tumor, pouco se sabe sobre o papel delas em CCR.
As GTPases Rho são proteínas monoméricas com a capacidade intrínseca de clivar
guanosina trifosfato (GTP), e ciclam entre um estado ativo ligado em GTP, predominantemente
associado em membranas, e um inativo ligado em guanosina difosfato (GDP),
predominatemente no citoplasma (Figura 14). As proteínas que regulam esse ciclo são: a) o
fator de troca de nucleotídeo de guanina (GEF), com 82 membros, na qual apenas Tiam1 está
super-regulada em CCR. As GEFs catalisam a troca entre GDP por GTP para gerar a forma
ativada de Rho, capaz de reconhecer seus efetores downstream (Vega & Ridley, 2008; Malliri
et al., 2006), b) as proteínas ativadoras de GTPases (GAPs) compreendem 67 membros, cuja
função é acelerar a capacidade intrínseca de Rho para hidrolisar GTP, restaurando o estado
ligado em GDP, ocasionando então a inativação da GTPase. Apenas duas GAPs foram
relacionados ao CCR, a proteína denominada deletado em câncer de fígado-1 (DLC-1) e a
RhoGAP 8 (ARHGAP8) (Johnstone et al. 2004; Kim et al., 2009); e c) os inibidores de
dissociação de nucleotídeo guanina (GDIs), uma família com 3 membros (α, -β e -γ RhoGDI),
na qual RhoGDI α e β estão alterados em CCR (Polley et al., 2006; Saito et al., 2007; Zhao et
al., 2008a). Convencionalmente, RhoGDIs são inibidores de Rho porque mantêm as GTPases
no citosol. No entanto, estudos mostraram que RhoGDIs exibem afinidades similares por Rac e
Cdc42 ativas e inativas, indicando seu possível papel no transporte e compartimentalização das
GTPases ativas (Nomanbhoy & Cerione, 1996). A dissociação do complexo Rho-RhoGDI é
mediada pelos fatores de desacoplamento de GDI (GDFs), como o complexo ezrina-radixina-
moesina (ERM) e neutrotrofina R (Moissoglu et al., 2006). Além disso, as GTPases Rho ativas
adquirem uma mudança conformacional que as permitem interagir com aproximadamente 100

25
proteínas efetoras, que desempenham a função biológica de Rho propriamente dita (Vega &
Ridley, 2008). A Tabela 1 ilustra as GTPases Rho e suas proteínas regulatórias/efetoras com
expressão alterada em CCR. Além da expressão, a atividade de RhoA está aumentada em
linhagens de câncer de cólon invasivas (Rapier et al., 2010).
Algumas quinases podem regular tanto as GTPases Rho quanto suas proteínas
regulatórias. Por exemplo, PKA inibe RhoA por fosforilação do resíduo serina 188 (Qiao
Figura 14. Modelo mostrando o ciclo de ativação de GTPases da família Rho. As GTPases Rho ativas (RhoGTP) são estimuladas pela GAP a clivarem GTP, e são mantidas no seu estado inativo (RhoGDP) através da ligação em GDI até que a GEF seja estimulada e catalise a troca de GDP por GTP, permitindo a interação entre a Rho GTPase ativa e sua proteína efetora, e reiniciando o ciclo. Adaptado de Vega & Ridley, 2008.
et
al., 2008), e Src fosforila RhoGDI na tirosina 156, impedindo sua interação com Rho, Rac e
Cdc42 (DerMardirossian et al., 2006). As GTPases Rho podem ser reguladas por
ubiquitilação, por exemplo Smurf1 induz degradação de RhoA ativa por proteassomo, de
forma local específica. Em adição, foi mostrado que células tumorais podem apresentar
redução no mecanismo de ubiquitilação de RhoA, Rac 1 e Cdc42 ocasionando sua ativação
sustentada (Boyer et al., 2006). Ainda, as GTPases Rho podem modular fatores de transcrição
envolvidos na tumorigênese, tais como fator de resposta ao soro (SRF), fator nuclear κB
(NFκB), STAT e CREB (Benitah et al., 2004). Recentemente, foi identificado um complexo
proteico que regula a transcrição de RhoA, modulando a migração e invasão de células de
câncer de mama (Chan et al., 2010).

26
Tabela 1. Expressão das GTPases Rho e suas proteínas regulatórias/efetoras em CCR.
Gene/proteína Expressão Referências
GTPase
RhoA
aumentada
Joshi et al., 2008; Takami et al., 2008; Carothers et al., 2006
RhoB aumentada Saito et al., 2007
RhoC aumentada Bellovin et al., 2006; Saito et al., 2007
Rac1 aumentada Takami et al., 2008
Rac1b aumentada Matos et al., 2003
Cdc42 aumentada Gomes Del Pulgar, 2008
Efetora
IQGAP1 aumentada Nabeshima et al., 2002; Hayashi et al., 2010
N-WASP aumentada Yanagawa et al., 2001
WAVE2 aumentada Iwaya et al., 2007
PAK1 aumentada Carter et al., 2004
PAK4 aumentada Liu et al., 2008
PAK5 aumentada Gong et al., 2009
ROCKI * aumentada Croft et al., 2004
ROCKII * aumentada Croft et al., 2004
Regulatórias
GDI
RhoGDIα aumentada Zhao et al., 2008a
RhoGDIβ/Ly diminuída Saito et al. 2007; Polley et al., 2006
GAP
DLC1 diminuída Kim et al., 2009
ARHGAP8 aumentada Johnstone et al., 2004
GEF
Tiam1 aumentada Liu et al., 2005; Minard et al., 2005; Malliri et al., 2006
*Estudo realizado em células de carcinoma de cólon expressando ROCK ativa e não em amostras de tumores colorretais
humanos, no entanto, foi mostrado que ROCK promove a invasão de tumores sólidos em camundongos nude.
I.5.2.1 Proteínas efetoras de Rho
As primeiras efetoras de Rho identificadas foram a ROCK I (ROKβ ou p160ROCK) e
II (ROKα), que são ubiquitosamente expressas. Essas proteínas compreendem um domínio
serina/treonina quinase na região N-terminal, seguido pela região de ligação em Rho (RBD),
região homóloga a plecstrina (PH), e um domínio rico em cisteína (CRD) na região C-
terminal. Na forma inativa, os domínios PH e RBD interagem com o domínio quinase
formando uma alça auto-inibitória. Estímulos exógenos capazes de ocasionar a ligação entre
Rho ativa e a região RBD de ROCK ocasionam a liberação do domínio quinase, ativando

27
ROCK (Figura 15A). Além disso, a ativação de Rho ocasiona transolcação das ROCKs do
citosol para a membrana (Mueller et al., 2005). As ROCKs foram caracterizadas por
induzirem fibras de estresse e AFs através de proteínas que regulam o citoesqueleto de actina
e contratilidade celular tais como miosina (Figura 15B), LIM quinase (LIMK) e FAK
(Kimura et al., 1996), o que motivou pesquisas sobre a função de ROCK na migração celular.
Considerando que o citoesqueleto de actina se associa ao CJA, foi mostrado que a inibição
farmacológica de ROCK aumenta a permeabilidade paracelular em células intestinais (Walsh
et al., 2001) e reduz a marcação de E-caderina nas JAs (Anderson et al., 2002). Esse efeito
também foi observado utilizando a forma dominante negativa de ROCK em células MDCK
(Takaishi et al., 2000), e o silenciamento gênico dessa quinase mostrou que ROCK II, mas
não ROCK I, desorganiza os contatos célula-célula (Samarin et al., 2007).
Outras efetoras da família Rho que modulam o CJA incluem IQGAP, quinase P21-
ativada (PAK) e formina diaphanous (mDia). IQGAP é efetora de Rac e Cdc42 localizada
nos contatos célula-célula. Baixos níveis de ativação de Rac permitem a interação entre
IQGAP e β-catenina, induzindo a dissociação de α-catenina do complexo E-caderina-
catenina. A interação entre Rac ativa e IQGAP previne a desorganização das JAs (Noritake et
al., 2005). É interessante notar que o aumento de expressão de IQGAP em pacientes com
CCR sugere sua localização no fronte invasivo celular, uma vez que a sua inibição não
alterou a localização de α-catenina em linhagens de câncer de cólon mas impediu a invasão
celular induzida por fator de crescimento de hepatócito (Hayashi et al., 2010). Ao contrário, o
recrutamento de PAK1 por Rac promoveu a desmontagem das JAs. Vale destacar que PAK1
é necessária para a formação de lamelipódios, indicando seu papel na interface entre a perda
das JAs e aumento de motilidade (Lozano et al., 2008). Além disso, mDia1, outra efetora de
Rac, restaurou a localização de E-caderina e α-catenina nas JAs, desorganizadas pela
ativação de ROCK em CCR (Sahai & Marshall, 2002), sugerindo que Rho e Rac exercem
funções opostas em epitélios. Esses dados também justificam os achados conflitantes na
literatura de que Rac pode ser pró- ou anti-tumoral, dependendo da proteína efetora recrutada.

28
Contração Miosina
fosfatase
ativo
inativo
Rho quinase
B A
Figura 15: Estrutura molecular e mecanismo de ativação de ROCK. A) O domínio catalítico de ROCK está na região N-terminal, seguida pelas regiões RBD, PH e CRD na região C-terminal. Os domínios PH e RBD se dobram sobre o domínio quinase formando uma alça auto-inibitória, desfeita pela ativação de Rho, que se liga em RBD e ativa ROCK. B) ROCK ativa fosforila a cadeia leve da miosina (MLC), ou a subunidade ligadora de miosina da miosina fosfatase (MBS), inibindo-a e impedindo a desfosforilação da MLC. Isso ocasiona o deslizamento entre F-actina e miosina para a contração celular. Adaptado de Mueller et al., 2005 (A); Kimura et al., 1996 (B).
I.6 Modulação do complexo juncional apical (CJA) pelas GTPases Rho
As GTPases da família Rho foram inicialmente descritas como reguladoras da dinâmica
do citoesqueleto de actina, na formação de projeções de membrana relacionadas com a
migração celular. Posteriormente, o interesse na inter-relação entre o CJA e Rho se originou
através da observação da localização dessas GTPases nos contatos célula-célula (Akhtar &
Hotchin, 2001). Em células de carcinoma, a função dessas GTPases é contraditória pois elas
são requeridas tanto para a montagem quanto para a desmontagem da adesão célula-célula. Por
exemplo: a) ambos, inativação e ativação constitutiva de Rho ocasionaram desorganização das
JTs e aumento da permeabilidade paracelular (Hopkins et al., 2003; Bruewer et al., 2004); b) a
ativação constitutiva de Rac aumentou os níveis de E-caderina e β-catenina nas JAs, no
entanto, a ativação sustentada de Rac frequentemente desmonta JAs (Braga et al., 2000),
indicando que a ativação de Rac pode estabilizar ou desorganizar o CJA; c) a inibição de Rac
por manipulação genética e a inibição farmacológica de Rho impediram o acúmulo de E-
caderina nas JAs em queratinócitos (Braga et al., 1997), indicando que tanto Rho quanto Rac
regulam essas estruturas. Já o engajamento da E-caderina nos contatos célula-célula, reduz a
atividade de RhoA enquanto ativa Rac e Cdc42 (Noren et al., 2001). A p120 catenina também
modula a atividade das GTPases, pois a depleção dessa proteína em células epiteliais aumentou
a atividade de RhoA e diminuiu a atividade de Rac (Anastasiadis, 2007). Estudos posteriores
mostraram que as ativações de Rho e Rac ocorrem em tempo e espaço específicos, e de forma

29
dependente do estímulo celular (Wozniak et al., 2005; Pertz et al., 2006), justificando os dados
conflitantes da literatura.
Além da modulação do citoesqueleto de actina e adesão celular, as GTPases Rho
também são importantes no controle da polaridade celular. Foi mostrado que células
dominantes negativas para Rac perderam a orientação da superfície apical de forma dependente
da ativação da via Rho-ROCKI (Yu et al., 2008). As GTPases Rho também modulam proteínas
dos complexos de polaridade em vários tipos celulares, por exemplo, Rac1 e Cdc42 ativas se
ligam em PAR6 para ativar PKCa e induzir polaridade celular epitelial
Fisiologicamente, o LPA é uma mistura de ácidos graxos saturados e insaturados com
diferentes atividades biológicas e especificidades por receptores, atuando como precursores de
fosfolipídios complexos (Aoki et al., 2008). No entanto, o LPA está presente em altos níveis
no plasma e fluido de ascite de pacientes com câncer ovariano (Erickson et al., 2001) e
(Georgeou et al., 2008);
PAR3 interage com Tiam, GEF de Rac e Cdc42; e PATJ e PALS1 ativam Cdc42 para modular
a formação de JTs (Iden & Collard, 2008). Ainda, o fator de crescimento transformante beta
(TGF-β) induz perda de polaridade de células epiteliais mediando degradação da GTPase
RhoA através da ubiquitina ligase Smurf1 (Sahai et al., 2007; Xu et al., 2009).
I.7 Ácido lisofosfatídico (LPA)
O LPA é um agente comumente utilizado na ativação da GTPase Rho. Trata-se de um
componente do soro que está relacionado a diferentes respostas celulares, como proliferação,
resistência a apoptose, quimiotaxia, secreção de fatores angiogêncios, ativação de MMPs,
entre outras (Mills & Moolenaar, 2003). É um fosfolipídio produzido localmente em resposta
a injúrias, gerado e secretado por plaquetas, macrófagos, células epiteliais e tumorais, mas
principalmente gerado extracelularmente por ecto-enzimas num processo ainda não elucidado
(Mills & Moolenaar, 2003). A Figura 16 ilustra as duas vias de síntese do LPA. A primeira
ocorre pela remoção de uma cadeia de ácido graxo do ácido fosfatídico (PA, gerado a partir
de fosfolipídios e diacilglicerol) através da ação da fosfosfolipase A2 solúvel (PLA2) ou da
fosfolipase A1 (PLA1). A segunda ocorre a partir de lisofosfatidilserina (LPS),
lisofosfatidiletanolamina (LPE) e lisofosfatidilcolina (LPC), pela ação da lisofosfolipase D ou
autaxina, uma ecto-enzima inicialmente denominada fator de motilidade autócrino ao ser
descoberta no sobrenadante de cultutras de melanócitos (Mills & Moolenaar, 2003; Aoki et
al., 2008). Os níveis de LPA são regulados pelo balanço entre essas vias de síntese e pela
reversão do LPA a monoacilglicerol por lipídiofosfatases ou a PA através da adição de uma
cadeia de ácido graxo pela endofilina (Mills & Moolenaar, 2003).

30
modula o desenvolvimento de carcinomas, incluindo o CCR (Zhao et al., 2007; Sun et al.,
2009; Müller et al., 2010). Vale ressaltar que a isoforma de LPA gerada por PLA2 tem uma
habilidade limitada para hidrolisar lipídios de membranas celulares intactas e é responsável
por manter baixos níveis de LPA no plasma. Ainda, porque PLA2 hidrolisa membranas
danificadas e microvesículas produzidas por células tumorais e em fluido de ascite, acredita-
se que essa enzima contribua para a produção aberrante de LPA em pacientes com câncer.
Vale mencionar que os níveis de LPC, um dos precursores do LPA, estão reduzidos no plasma
de pacientes com CCR. Os autores desse trabalho relatam ainda um aumento dos níveis da
enzima autaxina no plasma desses pacientes, o que sugere a conversão de LPC em LPA no
plasma de indivíduos com esse tipo de neoplasia (Zhao et al., 2007).
Figura 16: Síntese do LPA. O LPA pode ser produzido por: A) hidrólise do ácido fosfatídico (PA) pela fosfolipase A1 (PLA1) ou A2 solúvel (PLA2), que clivam a cadeia do ácido graxo do glicerol; e B) hidrólise de lisofosfolipídios, ilustrados pela lisofosfatidilcolina (LPC), por autaxina (ATX) ou lisofosfolipase D (LPD), que libera a colina. Adaptado de Mills & Moolenaar, 2003.
Os efeitos celulares mediados por LPA ocorrem em resposta à ativação de seus
receptores (LPA1-6), que são GPCRs (Noguchi et al., 2009; Yanagida et al., 2009). Os três
membros de receptores do LPA da família do gene de diferenciação endotelial (EDG-1,-4 e -
7, ou LPA1, 2 e 3, respectivamente), têm sua expressão alterada em diversos tipos de tumores,
estando o LPA2 superexpresso em CCR (Shida et al., 2004; Rusovici et al., 2007; Müller et
al., 2010). Foi mostrado que o LPA1 medeia o espalhamento celular em carcinomas do TGI
(Shin et al., 2009) e aumenta o potencial metastático de células de câncer de cólon (Shida et

31
al., 2003). Além disso, a ausência do LPA2 atenuou a formação de tumor em camundongos
com APC mutado (Lin et al., 2009) e ambos, LPA2 e 3, participaram da proliferação celular
em células de câncer de cólon (Yang et al., 2005). Menos estudados, os receptores não
pertencentes à família EDG (LPA4-6), podem ativar Rho e Rac, assim como LPA1 e 2, e
induzem a formação de projeções de membrana (Yamada et al., 2005; Valentine et al., 2008;
Harper et al., 2010). Foi mostrado que o LPA4 medeia a formação de invadopódios
aumentando o potencial invasivo de células de fibrossarcoma através da ativação de Rac1
(Harper et al., 2010). O LPA ainda se liga ao receptor gama de peroxissomo ativador de
proliferação (PPARγ), um membro da superfamília de receptores de hormônios nucleares
relacionados ao metabolismo energético, diferenciação celular, apoptose e inflamação (Mills
& Moolenaar, 2003). E, recentemente, foi mostrado que o tratamento com LPA regula a
proliferação de células de câncer de cólon através desse receptor (Tsukahara et al., 2010).
Muitos estudos utilizam LPA como ativador de Rho, pois a função inicial atribuída ao
LPA foi a formação de fibras de estresse e AFs (Hall, 1998). No entanto, em decorrência do
grande número de receptores desse agente, e sabendo que suas expressões variam com o tipo
celular, a via de sinalização ativada por LPA é bastante complexa. Essa via inclui não só a
ativação de Rho, mas também de Ras, geração de segundos-mensageiros que ativam proteínas
quinases e transcrição gênica de fatores de crescimento, pró-angiogênicos e MMPs (Mills &
Moolenaar, 2003). Por exemplo, o LPA ativou EGFR e ERK em células de câncer de cólon
(Mori et al., 2006; Zhang et al., 2007a); Fyn quinase, um membro da família Src, em células
de câncer de ovário (Huang et al., 2008); e PKA em células endoteliais (Nguyen et al., 2004).
Alguns estudos mostraram que o LPA modula o CJA e outros eventos importantes em
câncer. Em células de câncer de ovário, o LPA causou dispersão celular e perda de contatos
célula-célula através da fosforilação de p120-catenina mediada por Src (Huang et al., 2008).
Em células invasivas de câncer de cólon, o LPA induziu dissociação de E-caderina e β-
catenina e aumentou a proliferação (Yang et al., 2005, Kam & Quaranta, 2009), migração
celular e a secreção dos fatores pró-angiogênicos VEGF e interleucina-8 (Shida et al., 2003).
Em células epiteliais colônicas, o LPA previniu apoptose induzida por quimioterápicos ou
irradiação (Deng et al., 2002; Rusovici et al., 2007). Mais recentemente, foi mostrado que a
proteína do CJA MAGI-3 regula negativamente a invasão celular induzida por LPA,
aumentando a interação entre o LPA2 e a subunidade α12 de proteína G (Lee et al., 2010b).
Embora esses estudos tenham começado a investigar o efeito de LPA em CCR, pouco se sabe
sobre as vias de sinalização que medeiam os efeitos desse agente na organização do CJA
concomitante às alterações do citoesqueleto de actina nesse tipo de câncer.

32
I.8 Progressão tumoral e metástase
Durante a progressão tumoral as células perdem seus contatos célula-célula e a
polaridade celular, proliferam de forma desordenada formando multicamadas, adquirem
motilidade e se destacam do seu local de origem para migrar em direção a vasos sanguíneos
e/ou linfáticos a fim de atingir e colonizar sítios secundários, caracterizando o processo
metastático, a principal causa de morte por câncer. No entanto, ainda é necessária uma
detalhada compreensão dos mecanismos celulares e moleculares das etapas compreendidas
entre a perda da adesão célula-célula até o estabelecimento de novos tumores.
A análise de células móveis in vivo utilizando microscopia multifóton permitiu a
divisão do processo metastático nas seguintes etapas (Figura 18): a) perda da adesão célula-
célula e aquisição de fenótipo móvel, b) intravasamento, caracterizado pela entrada das
células tumorais na vasculatura; c) parada das células tumorais nos vasos do órgão alvo, por
adesão à parede dos vasos ou obstrução de pequenos capilares; d) extravasamento, que é a
saída das células dos vasos; e e) crescimento nos órgãos alvo (Sahai, 2007). Em todas essas
etapas, a resistência à morte celular permite que as células ultrapassem diversos tipos de
estresse como fragmentação celular, perda de adesão com o substrato (que induz morte por
anoikis), desorganização do citoesqueleto (induz morte por amorfose), depleção de nutrientes
(induz morte por autofagia) e hipóxia (Mehlen & Puisieux, 2006).
Além disso, alterações morfológicas finamente reguladas permitem que as células
comportem a aquisição de motilidade e propriedades invasivas. Esta motilidade é dirigida por
ciclos de polimerização de actina e de adesão ao substrato e por contração actina-miosina, que
medeiam a formação de projeções de membrana necessárias ao direcionamento do movimento
em resposta a um estímulo, por exemplo, para a célula atravessar a MEC (Thiery & Sleeman,
2006).

33
Figura 17: Progressão tumoral e processo metastático. O tumor primário interage com células não tumorais, como fibroblastos e macrófagos, e com proteínas da MEC, por exemplo, fibras de colágeno. As células perdem os contatos célula-célula e a polaridade celular; adquirem capacidade migratória e invasiva; e intravasam para vasos sanguíneos ou linfáticos ativamente ou por mimetismo de células endoteliais. Dentro dos vasos, as células podem causar embolia ou aderir às paredes dos vasos e extravasar. As células devem resistir à apoptose, entrando em dormência ou proliferando dentro ou fora dos vasos. Células tumorais em azul, não tumorais em bege, e endoteliais em vermelho. Adaptado de Sahai, 2007.
Conforme já mencionado as GTPases Rho modulam o citoesqueleto de actina e a
formação de projeções de membrana, indicando que essas proteínas desempenham um
importante papel na migração celular. A esse respeito, um primeiro estudo foi realizado
utilizando o ensaio de wound healing em monocamadas confluentes de células intestinais. Os
resultados indicaram que a inibição de Rho com as toxinas A e B de Clostridium difficile,
microinjeção de RhoGDI, ou microinjeção da toxina C3, inibiam a migração celular (Santos
et al., 1997). Posteriormente, foi mostrado que a ativação de Rho aumenta a migração de
células de câncer de cólon em câmaras Transwell (Wilkinson et al., 2005). Em adição, a
ativação de Tiam1, uma GEF de Rac e Cdc42 superexpressa em CCR, foi necessária para o
aumento da migração celular induzido por ativação de Rac (Minard et al., 2006), e também
foi mostrado que a inibição de ROCKII reduziu a invasividade de células de câncer de cólon
(Vishnubhotla et al., 2007). Estudos in vivo mostraram que a inoculação de células de câncer
de cólon que superexpressam Rac1 em camundongos aumenta a formação de tumores e
metástases em vasos linfáticos e peritônio (Espina et al., 2008), e ainda, que o LPA ativa
RhoA e aumenta a incidência de metástase peritoneal em um modelo de indução de
adenocarcinomas intestinais em ratos utilizando azoximetano (Tatsuta et al., 2005). Embora
esses estudos de fato indiquem que as GTPases Rho modulam a migração celular e a
invasividade, as vias de sinalização que regulam esses eventos permanecem desconhecidas.

34
I.8.1 Transição epitélio-mesenquimal (TEM) e migração celular
A TEM vem sendo extensivamente estudada em câncer, e trabalhos recentes originaram
sua classificação conforme a seguir: Tipo 1, ocorre na embriogênese, independente de
inflamação; tipo 2, presente no reparo tecidual e fibrose, e cessam quando o estímulo
inflamatório é retirado; e tipo 3, em células tumorais, com alterações genéticas e epigenéticas
(Kalluri, 2009). Nos três tipos de TEM, as células epiteliais perdem a polaridade, desregulam
a adesão célula-célula com concomitante contração actina-miosina, perdem marcadores
epiteliais específicos, como E-caderina e cateninas, e moléculas mesenquimais como
vimentina e N-caderina são expressas (Figura 18).
Figura 18. Principais moduladores da TEM. A) As células epiteliais desenvolvem TEM, através da dissociação do CJA e desorganização do citoesqueleto de actina. De forma reversível, as células mesenquimais adquirem um fenótipo epitelial na transição mesênquima-epitelial (TME). Adaptado de Thiery & Sleman, 2006.
Em linhagens de câncer de cólon, foi realizado um estudo da assinatura genética da
TEM induzida pela ativação de Ras, e foi mostrada a redução da expressão de E-caderina,
indução da expressão de vimentina e alteração dos níveis de claudinas (Joyce et al., 2009). A
p120-catenina também é essencial na TEM, visto que níveis endógenos dessa proteína foram
requeridos para invasividade em células deficientes em E-caderina, através da ligação em
caderinas mesenquimais via ativação de Rac e inibição de Rho (Anastasiadis, 2007). E ainda,
alguns trabalhos têm indicado que a sinalização mediada por integrinas pode contribuir para a
desorganização das adesões célula-célula na TEM (Bates et al., 2005). Vale ressaltar que na
TEM ocorre o aumento da expressão de integrinas αvβ6, receptores para fibronectina e
Células mesenquimais
TME
Dissociação das JTs
Dissociação das JAs e desmossomos
Células epiteliais
Efetores TME Adesão
Actina cortical
Marcadores Epiteliais E-caderina
Claudina, ocludina Desmoplaquina
Citoqueratina -8, -9, -18 Mucina-1
Marcadores Mesenquimais Fibronectina Vitronectina Vimentina
Actina de músculo liso FGFR
Formação das JTs
Associação dos
desmossomos
Ativação de GTPases Rho Reorganização do citoesqueleto de actina
Montagem de JAs
TME
TEM
Início do contato aderente
por E-caderina
Efetores TEM Fatores de crescimento
Citocinas
TEM
TME
TEM
TME
Ativação de GTPases Rho

35
tenascina expressos na embriogênese (Bates et al., 2005), bem como a ativação da via de
sinalização Wnt e desorganização do complexo PAR (Iden & Collard, 2008). Esses dados
refletem a perda de diferenciação do epitélio para fenótipos mais embriogênicos, e
demonstram que alterações na adesão ao substrato facilitam o escape e a disseminação de
células de carcinoma, ocasionando a metástase. Além disso, essas integrinas ativam a via do
TGF-β (Yue & Mulder, 2001), sugerindo sua utilização como um novo indicador de
prognóstico para CCR invasivo (Bates et al., 2005).
O principal indutor da TEM é o TGF-β, cujos receptores são do tipo I (TGFβR-I) ou II
(TGFβR-II). A ativação de ambos desencadeia uma cascata de sinalização que pode ser a)
dependente das proteínas Smad-3 e -4, ativando fatores transcricionais como TCF/LEF-1, ou b)
independente de Smad, mimetizando o sinal de receptores tirosina quinase capazes de ativar
vias de sinalização como a Ras-MAPK, PI3K, Rho e Rac. Ainda, a ativação da via Ras-MAPK
por TGF-β acentuou a expressão de Snail, um repressor transcricional da E-caderina em
células de câncer de cólon (Medici et al, 2006; Wang et al., 2010), assim como a sinalização
Wnt/β-catenina, que potencializou a TEM induzindo a expressão de Slug, outro repressor
transcricional de E-caderina (Baum et al., 2008). Além disso, foi mostrado que o TGF-β pode
ativar a via Rho-ROCK para modular o citoesqueleto de actina e induzir formação de fibras de
estresse, enquanto induz a degradação de Rho nas JTs através da fosforilação de PAR6, que
recruta Smurf, uma E3 ubiquitina ligase de RhoA, causando perda dos contatos célula-célula.
Em células de câncer de cólon, a ativação de Rho em resposta ao tratamento com TGF-β
também foi responsável pela expressão de actina não muscular, um marcador de TEM, e pela
liberação das MMP-2 e -9, mediando a invasividade (Paduch et al., 2010). Assim, TGF-β pode
mediar a ativação local de Rho para aquisição de um fenótipo mesenquimal e capacidade
migratória aumentada, mas se outros ativadores de Rho podem induzir TEM permanece
desconhecido. A associação entre TEM e invasão celular foi recentemente confirmada num
estudo mostrando que a expressão de vimentina modula a elongação de invadopódios
(Schoumacher et al., 2010).
No entanto, a aquisição de um fenótipo mesenquimal durante a TEM não é o único
mecanismo de aumento na capacidade migratória. Nesse contexto, a migração celular pode ser
divida em três mecanismos: 1) a migração coletiva, onde os contatos célula-célula são
mantidos e as células do fronte migram de forma mesenquimal; 2) a migração mesenquimal,
onde a célula adquire uma morfologia alongada através da transição epitélio-mesenquimal, de
forma dependente da ação de MMPs; e 3) a migração ameboide, onde a célula adquire uma

36
morfologia arredondada de forma dependente da ativação da via Rho-ROCK (Sanz-Moreno et
al., 2008; Parri & Chiarugi, 2010). Essas formas de migração dependem do estímulo e tipo
celular e, embora a migração coletiva tenha sido relatada em alguns tipos de câncer como
mama, próstata e pulmão, células de câncer de cólon costumam migrar individualmente.
Inicialmente, estudos in vivo mostraram que células tumorais epiteliais migravam
preferencialmente de forma ameboide (Sahai, 2007). No entanto, tem sido mostrado que esses
fenótipos são interconversíveis, por exemplo, a migração coletiva de explantes de melanomas
foi revertida em ameboide pela inibição de integrina β1 (Hegerfeldt et al., 2002); e células de
fibrossarcoma e de câncer de cólon optaram pela migração mesenquimal na presença de
inibidores de ROCK e ameboide na presença de inibidores de MMPs (Torka et al., 2006). Por
outro lado, a ativação de Rac reprimiu o movimento ameboide através da inibição de MLC em
células epiteliais (Sanz-Moreno et al., 2008); e a expressão de efrina A em câncer de próstata,
e redução de p53 e p27 em melanoma levaram a transição mesênquima-ameboide (Parri &
Chiarugi, 2010). Esses dados indicam que o tipo de migração celular é um mecanismo
adaptativo da célula tumoral e justifica as dificuldades no desenvolvimento de terapias que se
baseiam em moléculas individuais.
I.8.2 Projeções de membrana reguladas por GTPases Rho na migração celular
O citoesqueleto celular pode ser modificado na aquisição de um fenótipo
morfologicamente transformado com habilidade para migrar e invadir. Nessa condição, o
citoesqueleto de actina se reorganiza em resposta à ativação das GTPases Rho, Rac e Cdc42,
formando fibras de estresse que mantêm sua ancoragem a MEC, e projeções de membrana
denominadas lamelipódios e filopódios, respectivamente.
Os lamelipódios são grandes projeções de membrana achatadas com formato de folha,
e que contêm F-actina altamente ramificada, formados pela polimerização de actina localizada
com a geração de terminais+ livres, conforme descrito adiante. Acredita-se que sua função
seja dirigir a migração celular através de adesão ao substrato e geração de força capaz de
puxar o corpo celular para frente. Já os filopódios são estruturas longas e finas com feixes
paralelos de actina que se interconectam, e que frequentemente se projetam além da borda do
lamelipódio. São abundantes em estágios iniciais do espalhamento celular e sua função está
correlacionada às respostas da célula com relação ao ambiente externo (Yamaguchi &
Condeelis, 2006). Tem sido sugerido que a principal proteína formadora dessas estruturas é a
fascina, cuja alta expressão em células tumorais induz a formação de filopódios e maior
invasividade (Machesky & Li, 2010). Os blebs são outro tipo de projeção de membrana, com

37
formato esférico e inicialmente descritos como marcadores de apoptose. Essas projeções
ocorrem quando a pressão hidrostática do córtex dirige uma expansão externa, isto é, ocorre a
dissociação da membrana do córtex ou a ruptura da actina cortical através da ativação da via
Rho-ROCK, seguida pelo fluxo de citosol para dentro dessa expansão. O processo termina
com a repolimerização de actina por um mecanismo ainda desconhecido causando a retração
do bleb. Embora sua função ainda não esteja completamente elucidada, sabe-se que essas
estruturas participam da citocinese e da migração ameboide (Charras & Paluch, 2008).
Mais recentemente foram descritas em sistemas tridimensionais estruturas
denominadas invadopódios, projeções de membrana ventrais formadas por células cancerosas
altamente invasivas em substratos fisiológicos e com atividade proteolítica da MEC. São
compostos por um centro de actina e uma variedade de proteínas que modulam o
citoesqueleto, moléculas de adesão, de sinalização e de degradação de MEC. Essas estruturas
são consideradas análogas às AFs em sistemas bidimensionais, que serão descritas a seguir, e
requerem ambas, as maquinarias responsáveis pela formação de filopódios e de lamelipódios
(Schoumacher et al., 2010). Os invadopódios são utilizados por células de carcinoma para
invadir o estroma e vasos sanguíneos no processo de metástase (Linder et al., 2007). Alguns
autores diferenciam invadopódios de podossomos: os invadopódios são menores, estão em
menor quantidade nas células, mas apresentam maior capacidade proteolítica de MEC.
Também foi relatado que células mais invasivas de carcinoma oral formam invadopódios,
enquanto menos invasivas formam podossomos (Takkunen et al., 2010). No entanto, ambas
as estruturas possuem função biológica, morfologia e composição moleculares semelhantes.
De maneira geral, o que as distingue são os tipos celulares onde são formadas: Podossomos
são formados por células de origem monocítica; estruturas denominadas similares a
podossomos formam-se em células endoteliais e de músculo liso; e invadopódios em células
epiteliais e células transormadas por ativação de Src (Linder et al., 2007). Embora os estudos
sobre invadopódios sejam recentes e a sua regulação pouco conhecida, foi mostrado que
ROCKII se localiza em invadopódios em células de câncer de cólon, modulando a invasão em
colágeno I e a atividade das MMPs -2 e -13 (Vishnubhotla et al., 2007). Ainda nesse tipo
celular, a atividade de Src foi requerida para a formação e maturação de invadopódios e para a
atividade de degradação da MEC (Kelley et al., 2010).

38
I.8.3 Matriz extracelular (MEC) e migração celular
Além da necessidade da formação de projeções de membrana durante a transformação
neoplásica, a interação da célula com a MEC também apresenta alterações. As moléculas da
MEC, como fibronectinas, lamininas, colágenos, proteoglicanos e vitronectinas não apenas
criam a arquitetura tecidual, mas também desempenham funções celulares através da ligação
em receptores de superfície, principalmente as integrinas. Em mamíferos, são conhecidas
dezoito subunidades α e oito β que se associam em diferentes combinações para formar pelo
menos vinte e quatro tipos de integrinas. As integrinas são receptores proteicos
transmembranares que possuem um domínio extracelular de ligação na MEC e outro
citoplasmático de ligação a uma placa submembranar constituída por múltiplas proteínas
adaptadoras, ligadoras do citoesqueleto, ou de vias de sinalização (Mitra et al., 2005). É
interessante notar que diferentes respostas celulares são desencadeadas dependendo do
substrato celular. Por exemplo, a ativação de Rac inibiu a migração de células epiteliais em
substratos revestidos com fibronectina e laminina-1 e, ao contrário, promoveu motilidade em
substratos de colágeno (Sander et al., 1998). Então, a composição da MEC controla o
recrutamento de integrinas específicas nas AFs.
Células móveis montam adesão ao substrato, fraca e dinâmica, através de contatos
focais (CFs). Essas estruturas se formam principalmente na periferia celular e funcionam
como âncoras capazes de exercer uma força contrátil que permite a locomoção da célula. Foi
mostrado que, durante a formação de lamelipódios, ocorre um acúmulo de proteínas de
maneira hierárquica até que se formem AFs estáveis ao redor de um heterodímero central de
integrina α-β. Por exemplo, os CFs contêm talina, vinculina, paxilina e FAK, enquanto zixina
e tensina são recrutadas após a maturação dessas estruturas em AFs (Zaidel-Bar et al., 2003).
Vale destacar que pequenos CFs na periferia de células migratórias são regulados por Rac e
Cdc42 (Hall, 1998), precedendo a formação de grandes AFs reguladas por Rho. As AFs são
localizadas tanto na periferia celular como centralmente, associadas às terminações das fibras
de estresse (Chrzanowska-Wodnicka & Burridge, 1996). As fibras de estresse são compostas
por ramos de aproximadamente 10-30 filamentos de actina, miosina2 e proteínas acessórias
do citoesqueleto de actina, como α-actinina, fascina, espina e filamina, formadas pela
ativação de Rho (Hall, 1998). A organização dessas estruturas sugere uma função contrátil, e
acredita-se que seu encurtamento é oposto ao estado de adesão forte mediado por AFs. Nesse
contexto, é importante mencionar que a ligação de integrina à MEC por si só é insuficiente
para induzir a formação de CFs, o que originou a hipótese de que eventos de sinalização

39
vindos de dentro da célula são requeridos para a formação de AFs, mais especificamente, a
ativação de Rho (Wozniak et al., 2004).
Além disso, as AFs são fosforiladas em resíduos de tirosina, o que cria um sítio de
recrutamento de proteínas que contêm o domínio SH2, regulando a subsequente ativação de
quinases e fosfatases (Wozniak et al., 2004). As duas principais quinases localizadas nas AFs
são a FAK e a Src, que se ligam em diferentes parceiros para regularem a dinâmica das AFs e
o comportamento celular. A FAK é uma tirosina quinase constituída por um domínio FAT (do
inglês, focal adhesion targeting) na região C-terminal, que a mantém nas AFs, um domínio
FERM na região N-terminal e dois motivos ricos em prolina, que a permite interagir com
diferentes alvos. O domínio FERM tem um papel autoinibitório, e é liberado quando a FAK
interage com a cauda citoplasmática de integrina β1. Foi mostrado que a inibição da
expressão de integrina β1 em linhagens de CCR inibiu a migração celular e a metástase
hepática em camundongos (Zhang et al., 2010), sugerindo um papel para FAK nesses eventos.
Vários resíduos de tirosina são fosforilados quando a FAK é ativada, o que ocorre por
autofosforilação na tirosina 397 iniciada por engajamento de integrina em seu ligante. Essa
fosforilação ocasiona ativação e recrutamento de Src para as AFs, que remove a interação
autoinibitória de FAK e causa o acúmulo de FAK nas AFs, aumentando sua autofosforilação.
Src também fosforila FAK nas tirosinas 576 e 577 permitindo sua ativação, e nas tirosinas
862 e 925, criando um sítio de ligação para outras proteínas, permitindo a ativação de Ras e
MAPK (Wozniak et al., 2004). A FAK também pode ativar as GTPases Rho ligando-se
diretamente e fosforilando suas GEFs, o que é importante já que Rho regula a formação de
AFs. FAK ativa Rho e Rac através da fosforilação de p190RhoGEF e Trio, respectivamente
(Zhai et al., 2003;
Em resumo, para a migração ocorrer, as células devem extender uma protrusão para
formar um contato inicial com a MEC. Alguns desses contatos mediados por Rac se
desenvolvem em AFs dependentes de Rho, que estabilizam as células durante a migração. No
entanto, para migrarem, as células devem ser capazes de remodelar continuamente os CFs em
AFs e vice-versa. Então, a formação de novos CFs é localizada no fronte migratório das
células conforme são emitidas as projeções de membrana, ao passo que a desmontagem
dessas estruturas deve ocorrer na região posterior da célula permitindo a sua retração. Ainda,
essa dinâmica é mantida por ciclos de polimerização de actina que permitem a contração das
fibras de estresse e da ativação de AFs através da fosforilação de FAK no resíduo tirosina
Medley et al., 2003). Além disso, foi mostrado que FAK medeia a
migração celular em resposta ao LPA de forma dependente da ativação de Rho e ROCK em
fibroblastos (Iwanicki et al., 2008).

40
397, seguindo o recrutamento e ativação de Src, ambos eventos importantes na ciclagem de
AFs. As vias de sinalização desencadeadas pela ativação de AFs durante a migração celular
estão ilustradas na Figura 19. Vale destacar que FAK foi encontrada em altos níveis em uma
variedade de tumores, incluindo o CCR (Ayaki et al., 2001); foi mostrado um aumento na
fosforilação do resíduo de tirosina 397 em um modelo de formação de adenomas em
camundongos (Weyant
As MMPs e as desintegrinas-metaloproteases (ADAMs) cumprem função importante
na remodelagem da MEC durante a invasão celular, e estão presentes em podossomos e
invadopódios. A família de MMPs humanas compreende vinte e quatro membros que
degradam diversas proteínas. Além disso, as MMPs também podem estar localizadas na
superfície celular através de porções transmembranares (MT-MMPs). As MMPs são
sintetizadas como pró-enzimas e são ativadas por clivagem proteolítica, e bloqueadas por
inibidores teciduais (TIMPs). As ADAMs interagem especialmente com as subunidades β das
integrinas e funcionam clivando proteínas da superfície celular, liberando seu ectodomínio.
Existem também outros moduladores da MEC, como serina proteases denominadas seprases,
e invadolisina, ainda com pouco conhecimento no processo de invasão celular (Linder, 2007).
As MMPs -2 e -9 degradam a membrana basal, estando frequentemente associadas à invasão
celular e metástase. No entanto, essas enzimas também participam de outras etapas do
desenvolvimento do tumor, inclusive da clivagem de outros substratos, como E-caderina
et al., 2001), e a inibição de FAK reduziu o tamanho do tumor e a
angiogênese em um um modelo murino de CCR (Lei et al., 2010).
Algumas evidências indicam que a desregulaçao da adesão célula-MEC contribui para
a desmontagem das adesões célula-célula. Por exemplo, foi mostrado que a inibição de
integrinas β1 em células de câncer de mama induz a montagem de JAs e a diferenciação
celular (Weaver et al., 1997), e a superexpressão dessas integrinas em células normais
desorganiza as JAs (Gimond et al., 1999). Em linhagens de CCR, foi mostrado que o
colágeno I reduz a expressão de E-caderina e β-catenina, levando a um fenótipo mais
indiferenciado dependente de integrina α2β1 (Kirklank, 2009). Nesse sentido, Src tem sido
apontada como a proteína que integra a sinalização entre esses dois tipos de adesão: Foi
mostrado que a ativação de Src em células de câncer de cólon ocasionou endocitose de E-
caderina de forma dependente de FAK (Avizyenite et al., 2002). É interessante notar que Src
é ativada por FAK e é capaz de fosforilar E-caderina, levando a sua degradação (Fujita et al.,
2002), conforme anteriormente mencionado. Além disso, a sinalização mediada por integrina
ativa Snail/Slug para suprimir a expressão de E-caderina (Tan et al., 2001).

41
(Symovicz et al., 2007). Deve-se destacar que MMP-2, -9 e -12 estão superexpressas em
pólipos intestinais de camundongos com APC mutado. Em contraste, MMP-19 é produzida
pelo epitélio intestinal normal, mas sua expressão é perdida durante a transformação celular
(Sinnamon et al., 2008).
Figura 19: mecanismo de sinalização da FAK na migração celular. FAK é ativada por receptores de fatores de crescimento e integrinas durante a migração. A sinalização de FAK modula os contatos célula-célula mediados por E-caderina e os contatos célula-MEC, recrutando proteínas dos CFs e permitindo a ciclagem (montagem e desmontagem) dessas estruturas. A atividade de FAK promove mudanças na F-actina e MTs, através de GTPases da família Rho. Adaptado de Mitra et al., 2005.
I.8.4 Regulação da organização do citoesqueleto de actina na migração celular
Foi mostrado que drogas despolimerizantes de actina reduziram a migração de células
de carcinoma (Hayot et al., 2006) e que o aumento da proporção entre F- e G-actina aumenta a
capacidade metastática de CCR (Rapier et al., 2010). Durante a migração, a polimerização de
F-actina ocorre através da exposição do terminal+ por três mecanismos: a) a nucleação, pela
qual nova F-actina é gerada; b) a entrega de monômeros de actina de filamentos pré-existentes
pela cofilina; e c) o descapeamento de filamentos pré-existentes (Figura 20).
A nucleação ocorre a partir da atividade de WASP, proteína relacionada a Diaphanous
homóloga a formina (DRF), e Ena/VASP. Em mamíferos, a família WASP compreende os
membros WASP, WASP neural (N-WASP), WAVE-1,-2 e -3, relacionados à formação de
MEC
Integrina Receptor tirosina quinase
Membrana plasmática
Montagem
Desmontagem
CFs E-caderina
Migração celular
Fibras de estresse
lamelipódio filopódio microtúbulos

42
lamelipódios e invadopódios em conjunto com Arp 2/3 (Miyoshi & Takai, 2008; Albiges-Rizo
et al., 2009). As WASPs são efetoras de Rac e Cdc42 que, quando ativas, interagem com o
complexo Arp 2/3. Esse complexo contém sete subunidades, onde duas são relacionadas à
actina (Arp 2 e Arp 3), e criam junções em forma de Y em filamentos existentes, resultando em
ramificações estabilizadas por cortactina (Weaver et al., 2002). Foram descritas quinze
isoformas de proteínas DRF em mamíferos, onde a mDia1, efetora de Rho, inicia a formação
de feixes de actina não ramificados, acelerando a incorporação de monômeros e impedindo o
capeamento de F-actina. Além disso, a geração de força contrátil entre actina e miosina
aumenta a polimerização de actina dirigida por forminas (Spiering & Hodgson, 2011). Já as
proteínas Ena/VASP atuam impedindo o bloqueio do terminal+ por proteínas como a gelsolina,
e podem ser fosforiladas por PKA, o que inibe a sua atividade de nucleação (Gertler, 1996).
Algumas dessas proteínas estão alteradas em CCR, por exemplo, a redução da
expressão e alteração da localização de gelsolina leva a um fenótipo invasivo (Litwin et al.,
2009). Já a co-localização de Arp2 e WAVE aumenta o risco de metástase hepática (Iwaka et
al., 2007) e de WASP com Vasp de metástase para os linfonodos (Zhang et al., 2007b). Além
disso, a profilina, que promove a polimerização regional de actina por altas concentrações
locais de complexos profilina-actina através da troca de ADP por ATP, exerce um papel
importante na aquisição de um fenótipo tumoral. Foi mostrado que uma mutação no seu sítio
de ligação em actina é altamente tumorigênica in vivo, indicando seu papel supressor de tumor
(Wittenmayer et al., 2004). Vale destacar que os níveis de profilina-2 estão aumentados em
pacientes com doença inflamatória associada a CCR (Knaan et al., 2010).
A migração celular necessita de acelerada renovação de actina, ocasionada pela
(ADF)/cofilina. Inicialmente, acreditava-se que a cofilina atuasse apenas aumentando a
dissociação de ADP-actina no terminal-, induzindo a conversão de actina-F em actina-G
(Miyoshi & Takai, 2008). No entanto, mostrou-se que sua atividade está relacionada à invasão
e metástase através de polimerização de actina e formação de projeções de membrana
(Yamaguchi & Condeelis, 2006). Portanto, a cofilina coordena eventos de polimerização e
despolimerização de actina na migração celular. Sua atividade é negativamente afetada por
fosforilação em resíduo serina pela LIMK, ativada pela via Rho-ROCK ou Rac-PAK1
(Spiering & Hodgson, 2011). Ainda, utilizando RNA de interferência ou uma forma
constitutivamente ativa de LIMK, foi mostrado que a inibição de cofilina inibiu a migração em
células de carcinoma (Wang et al., 2006).

43
Figura 20: Dinâmica do citoesqueleto de actina durante a formação de projeções de membrana na migração celular. Os terminais+ da actina são bloqueados por proteínas capeadoras (CP, quadrados azuis), como a gelsolina (retângulos rosa-claro), que também cliva F-actina. Os terminais- perdem monômeros de actina através das proteínas despolimerizantes ADF/cofilina (retângulos rosa-claro). Para haver polimerização, esses eventos são desfeitos e a cofilina fornece G-actina ao termial+ para a nucleação por Arp 2/3 e formina, que geram actina ramificada. A G-actina, antes sequestrada por profilina, é convertida na sua forma ligada em ATP e recrutada aos terminais+. Os lamelipódios contêm actina ramificada formada por Arp 2/3, cortactina e N-WASP e os filopódios contêm F-actina elongada disposta em paralelo, formada por forminas e Ena/Vasp. Adaptado de Miyoshi & Takai, 2008.

44
I.9 Justificativa do estudo
O CCR representa a terceira causa mais incidente de câncer no mundo e, apesar do
crescente conhecimento da base genética e molecular deste tipo de câncer, o desenvolvimento
de um tratamento efetivo tem sido limitado. Particularmente, doenças metastáticas são fatais
porque malignidades em estágio tardio respondem pobremente a regimes terapêuticos comuns,
e o prognóstico dos pacientes é frequentemente grave. Tratamentos utilizando como estratégia
alvos moleculares para o câncer têm sido relatados. No entanto, esses agentes estão baseados
nas propriedades de tumores primários e, considerando que a transformação do fenótipo
benigno para o maligno é crítica em CCR, é de fundamental importância elucidar a biologia
que medeia a evolução metastática dessa doença. A progressão para carcinomas invasivos e
metastáticos envolve profundas alterações na estrutura e função do epitélio, como perda da
polaridade e diferenciação celular, desorganização do sistema de adesão celular e do
citoesqueleto de actina. Apesar do aumento dos conhecimentos relacionados com esses
eventos, os mecanismos celulares e moleculares que os desencadeiam ainda não estão
completamente elucidados, particularmente em CCR. Dessa forma, um estudo detalhado dos
processos iniciais da aquisição do fenótipo maligno, como a perda da adesão célula-célula e
alterações na dinâmica do citoesqueleto de actina, bem como das vias de sinalização que os
governam, pode contribuir no entendimento da carcinogênese colorretal. Além disso, os
resultados obtidos podem contribuir também para propor novos candidatos terapêuticos para
esta doença.

45
II OBJETIVOS
II.1 Geral
Analisar eventos celulares e moleculares na dinâmica do citoesqueleto de actina e do
CJA na progressão tumoral em células de adenocarcinoma de cólon humano, Caco-2.
Para melhor compreensão, a apresentação desta tese foi dividida em duas partes:
Parte I – Análise da inter-relação entre o CJA e o citoesqueleto de actina durante a perda da
adesão célula-célula;
Parte II – Papel do LPA na adquisição do fenótipo migratório.
II.2 Específicos
Parte I: Análise da inter-relação entre o CJA e o citoesqueleto de actina durante a perda
da adesão célula-célula
- Avaliar a redistribuição de proteínas do CJA (E-caderina, ocludina, claudina-1 e ZO-
1), sua funcionalidade e ultraestrutura após a retirada do cálcio do meio extracelular;
- Verificar se a perda dos contatos célula-célula induzida pela retirada do cálcio do
meio extracelular ocasiona mudanças do citoesqueleto de actina e formação de projeções de
membrana;
- Analisar a inter-relação entre as vias de sinalização envolvidas nas mudanças do
citoesqueleto de actina e desorganização do CJA durante a formação do fenótipo maligno,
enfatizando a participação das proteínas quinases PKA, MAPK, PI3K e EGFR, e dos
membros da família Rho de GTPases Rho e Rac.
Parte II: Papel do LPA na aquisição do fenótipo migratório
- Avaliar a localização de proteínas do CJA (E-caderina, β-catenina, p120 catenina,
ocludina, claudina-1 e ZO-1), sua funcionalidade, e concomitantes mudanças do citoesqueleto
de actina em resposta ao tratamento com LPA;
- Investigar a participação do LPA nas mudanças do citoesqueleto de actina na
formação de adesões focais (verificando a ativação de Src e FAK), fibras de estresse e
projeções de membrana;

46
- Analisar a inter-relação entre as vias de sinalização ativadas pelo LPA envolvidas na
desorganização do CJA e do citoesqueleto de actina e na migração celular, enfatizando a
participação das proteínas quinases Src, PKA, PI3K e EGFR, e da família Rho de GTPases
Rho e Rac, e suas efetoras ROCK e IQGAP;
- Verificar se o LPA aumenta o potencial tumorigênico celular, avaliando o potencial
de invasividade, atividade das MMP-2 e -9, e crescimento independente de ancoragem.

47
III. MATERIAL E MÉTODOS
III.1 Anticorpos e Reagentes
O anticorpo monoclonal produzido em camundongo anti-E-caderina (clone 36) foi
obtido da BD Biosciences (San Diego, CA, EUA). Os anticorpos policlonais produzidos em
coelho anti-claudina-1 (clone JAY.8), ocludina, ZO-1 (clone Z-R1), Rho-GDI-α (clone NGA-
25), e os monoclonais produzidos em camundongo anti-p120-catenina (6H11), anti-IQGAP1
e anti-α-tubulina (Z022) foram obtidos da Invitrogen Corporation (Carlsbad, CA, EUA). Os
anticorpos produzidos em coelho anti-ROCK II, anti-fosfo-FAK (Tyr 397), anti-p125 FAK e
anti-β-catenina, os anticorpos monoclonais produzidos em camundongo anti-Rac (23A8) e
anti-vimentina (V9), e a faloidina conjugada à rodamina foram obtidos da Sigma-Aldrich
(Saint Louis, MO, EUA). O anti-fosfo-Src (Tyr 416) produzido em coelho (100F9) foi obtido
da Cell Signaling Technology, Inc (Danvers, MA, EUA). Os kits de ativação de Rho e Rac
EZ-Detect TM para os ensaios de pull-down, incluindo os anticorpos anti-Rho e anti-Rac,
foram obtidos da Pierce (Rockford, IL, EUA). O anticorpo secundário Alexa 488 e 546 foram
obtidos da Invitrogen Corporation (Carlsbad, CA, EUA). Os anticorpos secundários
conjugados à peroxidase anti-coelho e anti-camundongo foram obtidos da Sigma-Aldrich
(Saint Louis, MO, EUA).
Os inibidores foram diluídos em dimetilsulfóxido e mantidos nas respectivas
concentrações estoques: N-[2-((p- Bromocinamil) amino) etil]- S –isoquinolinesulfonamida_
(H-89), de PKA, 10 mM; 2’– amino – 3´- metoxiflavona (PD98059), de MAPK, 25 mM; e
LY294002 (2-(4-morpholinil)-8-phenil-4H-1-benzopiran-4-ona), de PI3K, 10 mM, foram
obtidos da Biomol Research Laboratories Inc. (Plymouth Meeting, PA, EUA). Os inibidores 4
- [(3-Bromofenil) amino] - 6, 7 – dimetoxiquimazolina (PD153035), de EGFR, 1 mM; [(R) –
(+) – trans – N– (4-piridil) – 4- (1- aminoetil) - ciclohexanecarboxamida] (Y-27632), de
ROCK, 10 mM e toxina A de Clostridium difficile, de GTPase Rho, 665 mM, e PP2 (4-
amino-5-(4-clorophenil)-7-(t-butil) pirazolo[3,4-d] pirimidina, de Src, 170 nM foram obtidos
da Calbiochem (EMD Biosciences, Inc, San Diego, CA, EUA); o L-α-ácido lisofosfatídico
(LPA), o ativador de PKA forskolina, na concentração estoque de 10 mM, e o coquetel de
inibidores de protease foram obtidos da Sigma Chemical Co. (St Louis, MO, EUA). A resina
epon foi obtida da Electron Microscopy Sciences (Washington, PA, EUA); o kit de
quimioluminescência da Amersham Biosciences (GE healthcare, UK); as câmaras de

48
policarbonato da Costar (Corning, NY, EUA); e o matrigel da BD Biosciences (San Diego,
CA, EUA).
III.2 Cultura de células
Foram utilizadas as linhagens celulares de adenocarcinoma de cólon humano Caco-2
(ATCC, # HTB-37) e HT-29 (ATCC, # HTB-38). As células Caco-2 se diferenciam quando
atingem confluência e desenvolvem complexo juncional funcional, enquanto células HT-29
apresentam fenótipo indiferenciado, crescem em multicamada, e possuem maior potencial
invasivo e metastático. Ambas foram cultivadas em meio DMEM suplementado com 10% de
soro fetal bovino (SFB), penicilina G (60 mg/L) e estreptomicina (100 mg/L) e mantidas a
37°C em 5% CO2. Para ensaios bioquímicos, as células foram cultivadas em garrafas de 25
cm2 ou placas de 8,8 cm2
Para avaliar o papel do LPA na indução do fenótipo migratório (Parte II da Tese),
soluções de LPA na concentração de 20 mM em etanol 20% foram preparadas e estocadas a -
80°C. Inicialmente, foram realizados testes nas concentrações finais de 1, 10 e 20 µM de LPA
em meio sem SFB e em meio LC. Foi estabelecida a concentração de 10 µM para os
experimentos subsequentes, considerando que esta concentração não afeta a viabilidade
, para técnicas de imunocitoquímica em lamínulas de vidro, e para
medida de RET e microscopia eletrônica de transmissão, em membranas de policarbonato
Transwell com 6,5 mm de diâmetro e poro de 0,4 µm. Foram também utilizadas as células
OVCAR-3, epiteliais de ovário, cultivadas em meio RPMI suplementado com 20% de SFB,
penicilina G (60 mg/L) e estreptomicina (100 mg/L) e mantidas a 37°C em 5% CO2. Essa
linhagem é conhecidamente responsiva ao LPA (Sawada et al., 2002) e foi gentilmente cedida
pela Dra. Etel Gimba.
III.3 Modelo de depleção do cálcio extracelular
Na Parte I deste trabalho, meio de cultura em baixa concentração de cálcio (LC) foi
preparado suplementando o meio DMEM sem SFB com uma solução estoque de EGTA na
concentração de 200 mM, atingido uma concentração final de 4 mM. As células em
confluência foram pré-incubadas por 1 h a 37°C em meio sem SFB, e então incubadas por 2,5
h em meio contendo baixa concentração de cálcio. O efeito da retirada do cálcio extracelular
sobre a monocamada celular foi monitorado por microscopia de contraste de fase.
III.4 Tratamento com LPA

49
celular e é suficiente para induzir a formação de fibras de estresse. As células foram
depletadas de SFB por 24 h e o tratamento com LPA nesta concentração foi feito nos tempos
de 5, 15, 30 min, 1, 6, 24 e 48 h.
III.5 Tratamentos com inibidores de vias de sinalização
Após atingirem confluência, e antes de serem depletadas de cálcio, as monocamadas
celulares foram incubadas por 1 h em meio DMEM sem SFB e por 2,5 h com os respectivos
inibidores em meio com baixa concentração de cálcio. Para os estudos utilizando LPA, as
monocamadas foram depletadas de SFB por 24 h, pré-tratadas por 1 h com os respectivos
inibidores em meio DMEM sem SFB e em seguida incubadas com meio depletado de SFB
contendo LPA e os inibidores. As concentrações finais dos inibidores utilizadas foram:
inibidor de PKA H-89, 20 μM; inibidor de MAPK PD98059, 50 µM; inibidor de Src PP2, 100
nM; inibidor de EGFR PD153035, 100 nM; inibidor de PI3K LY294002, 10 µM; inibidor de
ROCK Y-27632, 10µM; inibidor de GTPases da família Rho toxina A de Clostridium
difficile, 10 µM. Foi também utilizada a forskolina como ativadora de PKA, na concentração
final de 10 µM.
III.6 Ensaios de Imunofluorescência
Células crescidas sobre lamínulas de vidro, após atingirem confluência, foram
submetidas a diferentes tratamentos, lavadas com salina tamponada com fosfato (PBS), fixadas
com paraformaldeído 4% durante 30 min a 37°C, e incubadas com NH4Cl por 10 min. Logo
permeabilizadas com Triton X-100 (TX-100) 0,5% por 5 min e o bloqueio foi feito com BSA
3% em PBS por 2 h. Posteriormente foram incubadas com os anticorpos primários contra
proteínas de JA (E-caderina, na diluição de 1:100; β-catenina, 1:220; e p120-catenina 1:200),
GTPases e suas efetoras (RhoA, 1:200; Rac, 1:60; IQGAP, 1:50; e ROCKII, 1:500), e FAK
(1:1000). Alternativamente, a fixação foi realizada com metanol 100% por 20 min a -20°C para
utilização dos anticorpos primários contra proteínas das JTs (ocludina, 1:20; claudina-1, 1:25; e
ZO-1, 1:25). Nova lavagem foi realizada, e as células foram incubadas com os respectivos
anticorpos secundários conjugados a Alexa 488 ou 546 na diluição de 1:500. Para marcação do
núcleo, foi utilizado 4',6-diamidino-2-phenylindole (DAPI, 1:500) por 1 min. Posteriormente
as lâminas foram montadas com N-propil-galacto, e observadas em microscópio invertido
(AXIOVERT S 100) acoplado a um programa de processamento de imagens KS300 (Zeiss), e
alternativamente em microscópio confocal LSM Meta 510 (Zeiss).

50
III.7 Detecção dos filamentos de actina
Para marcação dos filamentos de actina, as células foram fixadas e permeabilizadas
com TX-100 0,5%, como acima indicado, e incubadas com faloidina conjugada a rodamina
(500 ng/mL) por 40 min no escuro. As lâminas foram rigorosamente lavadas, montadas com
N-propil-galacto e observadas em microscópio confocal LSM510 Meta, utilizando o laser de
excitação de 543 nm. Foram realizadas seções ópticas próximas ao plano apical, medial (5 µm)
e basal (9 µm) da monocamada (plano X-Y) e no plano perpendicular (plano X-Z).
III.8 Obtenção de frações solúveis e insolúveis em Triton X-100 e de lisados celulares
totais
A fim de avaliar a distribuição das proteínas do CJA após os respectivos tratamentos, as
monocamadas celulares foram lavadas em PBS e homogeneizadas em tampão CSK, pH 6,8
(NaCl 50 mM, Tris-HCl 10 mM, MgCl2 3 mM, TX-100 0,1%, sacarose 300 mM), contendo
inibidores de proteases (1:100, Sigma), fluoreto de sódio (NaF) 20mM e ortovanadato 1mM
por 10 min a 4°C. Após centrifugação a 10.000 g por 10 min a 4°C, o sobrenadante obtido
corresponde à fração solúvel em TX-100 (proteínas solúveis). O pellet foi homogeneizado em
tampão SDS pH 7,5 (Tris-HCl 20 mM, EDTA 5 mM, EGTA 2,5 mM e SDS 1%) e fervido por
10 min. Após centrifugação a 10.000 g por 10 min, o sobrenadante corresponde à fração
insolúvel em TX-100 (proteínas ligadas ao citoesqueleto). Ambas as frações foram estocadas
em –200
Para obtenção de lisados totais, as amostras foram lavadas e incubadas por 20 min a
4°C em tampão de extração (NaCl 150 mM, Hepes 10 mM, pH 7.3, SDS 0,2%, TX-100 1%,
deoxicolato 0.5%, e EDTA 2 mM) contendo ortovanadato 2 mM, NaF 20 mM, e coquetel de
inibidores de proteases (1:100). As células foram raspadas das places, homogeneizadas e
centrifugadas a 10.000 g por 10 min a 4°C. O sobrenadante foi coletado e estocado a -20°C.
até seu uso correspondente.
III.9 Distribuição subcelular de Rho, Rac e RhoGDI
Para determinar a localização das GTPases após os respectivos tratamentos, frações de
membrana e citosol foram obtidas como se segue: As células foram coletadas e
homogeneizadas usando um homogenizador tipo Potter em tampão Tris-HCl 10 mM, pH 7.5,
contendo sacarose 250 mM, MgCl2 1 mM, ortovanadato 1 mM, NaF 20 mM e inibidores de
protease (1:100) Os lisados celulares foram centrifugados a 3.000 g por 10 min a 4°C. O
sobrenadante foi coletado e centrifugado a 15.000 g por 1 h a 4°C. O sobrenadante resultante

51
foi coletado como fração citosólica e o pellet como fração de membrana. A fração de
membrana foi ressuspensa e homogeneizada em tampão Hepes 10 mM, pH 7.3, contendo
EDTA 2 mM, SDS 0.2%, deoxicolato de sódio 0.5%, TX-100 0.1%, NaCl 150 mM,
ortovanadato 2 mM, NaF 20 mM e inibidores de protease (1:100).
III.10 Eletroforese em gel de poliacrilamida usando sódio dodecil sulfato (SDS-PAGE) e
Immunoblotting
Para analisar o padrão de distribuição das proteínas por efeito dos respectivos
tratamentos, quantidades iguais de proteínas (30-60 μg) foram separadas por SDS-PAGE
usando géis de 7,5, 10 e 12%. As proteínas foram transferidas para membranas de nitrocelulose
utilizando-se o aparelho de transferência semi-seco (Bio Rad Lab. Hercules, CA, EUA) a 10 V
por 60 min. As membranas foram mantidas em tampão de bloqueio: TBS-T (Tris-HCl 20 mM,
pH 7,6, NaCl 137 mM e Tween 20 0,1%) contendo leite desnatado 5% ou BSA 1-3%, durante
60 min, e incubadas com os anticorpos primários contra proteínas de adesão célula-célula (E-
caderina, na diluição de 1:5000; β-catenina, 1:4000; p120-catenina 1:1000; ocludina, 1:250;
claudina-1, 1:200; e ZO-1, 1:250), GTPases e reguladoras (Rho na diluição de 1:2000; Rac,
1:500; RhoGDI, 1:250), FAK fosforilada no resíduo tirosina 397, na diluição 1:1000, Src
fosforilada no resíduo tirosina 416, na diluição 1:500, vimentina 1:500, e a proteína
constitutiva como controle (β-tubulina, na diluição de 1:500) por 4 h ou overnight. Após
sucessivas lavagens com TBS-T, as membranas foram incubadas com os anticorpos
secundários produzidos em coelho ou camundongo conjugados a peroxidase por 1 h na
diluição de 1: 50.000. As membranas foram lavadas e a reatividade para as proteínas em estudo
foi feita usando um kit de quimioluminescência (ECL). A intensidade das bandas foi
quantificada por densitometria utilizando o software LabWorks 4.6 (Bio Rad Lab. Hercules,
CA, EUA).
III.11 Ensaio de pull-down
A atividade das proteínas Rho e Rac foi determinada usando ensaios específicos de
pull-down, seguindo as instruções do fabricante. Resumidamente, Rotekina RBD e PAK-1
PBD ligadas a agarose-glutationa, foram usadas para precipitar Rho e Rac dos lisados
celulares, respectivamente. As formas ativas e totais de Rho e Rac foram separadas por SDS-
PAGE e visualizadas por immunoblotting.

52
III.12 Resistência Elétrica Transepitelial (RET)
Para avaliar a funcionalidade das JTs em relação à permeabilidade a íons, as células
foram crescidas em membranas Transwell até atingirem confluência. Após os respectivos
tratamentos, a RET foi mensurada, usando um voltímetro com eletrodo de prata/cloreto
(Ag/AgCl) Millicel-ERS (Millipore). Um eletrodo foi inserido na câmara superior, acessando a
monocamada celular, e o outro na câmara inferior. A passagem de íons através da via
paracelular é quantificada através da medição da diferença de potencial elétrico entre as duas
câmaras, utilizando uma corrente constante de 20 µA. A resistência das membranas sem
células foi subtraída dos valores encontrados nos tratamentos: (Ramostra – Rbranco = Rmonocamada ),
e posteriormente multiplicada pela área das membranas (0,33 cm2
A migração celular foi avaliada através da técnica de wound healing. Células Caco-2
foram plaqueadas na densidade de 4 X 10
).
III.13 Microscopia Eletrônica de Transmissão (MET)
Para avaliar a funcionalidade das JTs em relação à permeabilidade paracelular a
moléculas, foi realizada a citoquímica utilizando o traçador vermelho de rutênio, seguida pela
análise por MET. As células crescidas em filtros de policarbonato Transwell com 6,5 mm de
diâmetro e poros de 0,4 µm foram fixadas numa solução contendo glutaraldeído 2,5%,
paraformaldeído 1%, sacarose 8% e CaCl2 2 mM preparado em tampão cacodilato de sódio 0,1
M, pH 7,2 por 1 h. Posteriormente foram lavadas em tampão cacodilato por 3 vezes durante 10
min e a pós-fixação realizada com tetróxido de ósmio 1%, ferrocianeto de potássio 0,8% e
CaCl2 2,5 mM em tampão cacodilato. Nas etapas de fixação, lavagem e pós-fixação, 6 µg/mL
de vermelho de rutênio foram aplicados na câmara superior do Transwell (região apical das
células). As amostras foram lavadas em tampão cacodilato por 3 vezes durante 10 min,
desidratadas nas concentrações crescentes de acetona (30%, 50%, 70%, 90%, 2 vezes em
100% e outras 2 vezes em 100% super seca) por 10 min cada, e infiltradas em resina Epóxi
(Electron microscopy Science, Washington, PA, EUA). Cortes ultrafinos de 60 nm foram
obtidos em ultramicrótomo Reichert Ultracute da Leica, e contrastados com citrato de chumbo
por 3 min para observação no microscópio eletrônico de transmissão Zeiss 900.
III.14 Ensaios de migração e invasividade celular
4 células/ cm2 e, após atingirem confluência,
depletadas de SFB por 24 h e pré-tratadas por 1 h com os inibidores. Em seguida, um risco foi
feito manualmente sobre a monocamada utilizando uma ponteira de micropipeta estéril. Para

53
cada tratamento, foram feitos três riscos e três campos de cada risco foram selecionados e
marcados. Após lavagem em PBS, meio de cultura contendo os inibidores e LPA foi
adicionado às células, que foram incubadas a 37°C. Células tratadas e não tratadas migraram
para as áreas riscadas e foram fotografadas imediatamente (0 h), após 6 h, e no final do
experimento (24 h), em microscópio invertido (AXIOVERT S 100) acoplado a um programa
de processamento de imagens KS300 (Zeiss). A distância entre as duas margens da
monocamada celular riscada foi quantificada utilizando o Programa Adobe Photoshop 6.0 em
três experimentos independentes. Os valores foram representados em porcentagens e plotados
em um gráfico.
A invasão celular foi avaliada utilizando membranas Transwell com poros de 8,0 µm
revestidos com matrigel (1:10), que contém os componentes de membrana basal (colágeno,
laminina, proteoglicanos, enzimas e fatores de crescimento). As células Caco-2 e OVCAR-3
(2,5 X 104 células) foram cultivadas na câmara superior sobre o matrigel em meio depletado
de SFB com ou sem tratamento com LPA. Na câmara inferior, foi utilizado meio contendo
20% de SFB como quimioatraente. Após 48 h em 37°C, as células que invadiram o matrigel
foram fixadas com paraformaldeído 3,7%, coradas com azul de toluidina 0,5% e fotogradas
em microscópio invertido (AXIOVERT S 100) acoplado a um programa de processamento de
imagens KS300 (Zeiss).
III.15 Proliferação celular
O número relativo de células viáveis foi determinado pelo ensaio do cristal violeta.
Resumidamente, a mesma densidade de células utilizada no ensaio de migração celular (1.5 x
104 células, ou 4 X 104 células/ cm2) foram plaqueadas por 4 h em placas de 96 poços,
incubadas com meio de cultura contendo ou não SFB e tratadas com LPA por 24 e 48 h. O
meio de cultura foi removido, as células foram lavadas com PBS, fixadas com etanol absoluto
por 10 min, e coradas com cristal violeta por 10 min na concentração de 0,05% em etanol
20% à temperatura ambiente. Após lavagem em água e secagem ao ar, o corante foi
solubilizado com metanol, e as densidades ópticas foram mensuradas em espectrofotômetro
em 595 nm (Spectra Max 190, Molecular Devices, Sunnyvale, CA, EUA) e avaliadas
utilizando o software Soft Max pro 4.3 LS.

54
III.16 Crescimento independente de ancoragem (CIA)
Placas de 12 poços foram revestidas com ágar 0,6% em meio DMEM contendo 10% de
SFB. Após a solidificação a 4°C por 30 min, 250 células por poço foram suspensas em ágar
0,3% em DMEM contendo 10% de SFB na presença ou ausência de 10 µM de LPA, e
plaqueadas sobre a camada de ágar inferior. Após solidificação, meio de cultura contendo 10%
de SFB na presença ou ausência de 10 µM de LPA foi adicionado. As células cresceram por
duas semanas, trocando o meio de cultura a cada três dias. As colônias foram observadas em
microscópio invertido (AXIOVERT S 100), contadas e plotadas num gráfico utilizando o
programa GraphPad Prisma 4.0.
III.17 Análise da expressão e atividade de metaloproteases
A expressão do RNAm das MMP-2 e -9 foi analisada por qRT-PCR. Para isso, o RNA
total de células Caco-2 sem tratamento e após os respectivos tratamentos foi extraído
utilizando o Kit RNeasy (Qiagen), seguindo o protocolo descrito pelo fabricante. Em seguida,
o RNA foi dosado no espectrofotômetro NanoDRop, e um micrograma de cada amostra de
RNA total foram reversamente transcritos para cDNA, segundo o protocolo do kit ImProm-II
Reverse Transcriptase for RT-PCR (Promega). Foi feito qRT-PCR utilizando o sistema de
detecção de SYBR Green (SYBR™ Green PCR Master Mix, Applied Biosystems). Os
oligonucleotídeos utilizados foram os seguintes: MMP-2 forward AAA ATG GAT CCT GGC
TTC CC, MMP-2 reverse TAG CCA GTC GGA TTT GAT GC, MMP-9 forward TGA CAG
CGA CAA GAA GTG, MMP-9 reverse CAG TGA AGC GGT ACA TAG G, actina forward
TGA CCC AGA TCA TGT TTG AGA, actina reverse ACT CCA TGC CCA GGA AGG A.
O volume final da PCR foi de 20 µL, contendo 10 µL de SYBR Green, 5 µL da solução 800
nM de mistura dos oligonucleotídeos senso e antisenso e 5 µL de cDNA (sintetizado a partir
de 1 µg de RNA total). O programa de ciclagem consiste de uma incubação inicial a 50°C por
2 min e 94°C por 5 minutos, seguido de 40 ciclos de 94°C por 30 s (desnaturação), 58°C
por 30 s (anelamento) e 72°C por 90 s (extensão). Essa etapa foi seguida de uma incubação
final a 72°C for 15 min. Ao término da ciclagem, foi realizada uma curva de dissociação com
o objetivo de confirmar a especificidade de amplificação. A curva de dissociação foi realizada
com um aumento de temperatura que se inicia em 60°C e estende-se até 90°C, com leituras de
fluorescência efetuadas a cada 0,2°C. Todas as amostras foram amplificadas em duplicatas,
inclusive os controles negativos. As reações de qRT-PCR foram realizadas e analisadas no

55
aparelho CFX 96 Real Time System (Bio Rad). O nível de expressão relativa das MMP-2 e -9
foram calculados pelo método de quantificação relativa, utilizando a fórmula 2-∆∆CT
A atividade das MMP-2 e -9 foi analisada por zimografia utilizando sobrenadantes de
meio de cultura de células Caco-2 confluentes correspondentes a diferentes condições
experimentais. Proteínas (30 µg) foram separadas por SDS-PAGE contendo gelatina 0,2%.
Após separação eletroforética, as proteínas foram renaturadas em tampão contendo TX-100
2,5% por 1 h, e incubadas em tampão contendo CaCl2 5mM por 20 h a fim de verificar a
capacidade enzimática de degradação da gelatina. Em seguida, os geis foram corados
utilizando Coomassie Brilliant Blue R-250 0,1% em solução com metanol 40% e ácido
acético 10% por 30 min e descorados em solução contendo metanol 50% com ácido acético
10%. Os géis foram mantidos em solução secante por 24 h e escaneados. Os sobrenadantes da
linhagem HT-1080, de fibrossarcoma, usados como controle da atividade metaloprotease,
foram gentilmente cedidos pela Dra. Silvya Stuchi Maria-Engler, da Universidade de São
Paulo.
.
III.18 Análise estatística
A análise estatística foi realizada usando o programa GraphPad Prism version 5.0
(GraphPad Software, San Diego, CA, EUA). Para cada experimento, os dados das triplicatas
foram calculados e foi feita a análise de significância utilizando os seguintes testes: one-way
ANOVA com pós-teste de Bonferroni para análise da RET; teste T-student para análise para
análise densitométrica das frações de membrana e citosólica e ensaio de crescimento
independente de ancoragem; e two-way ANOVA com pós-teste de Dunnett para análise
densitométrica das frações solúveis e insolúveis em TX-100 e ensaio de migração celular. As
diferenças foram consideradas significativas para p < 0,05.

56
IV. RESULTADOS
PARTE 1: Análise da inter-relação entre o CJA e o citoesqueleto de actina durante a
perda da adesão célula-célula
Um modelo frequentemente utilizado para estudar a dinâmica do CJA é a retirada do
cálcio do meio extracelular. Em condições fisiológicas, a concentração de cálcio no meio
extracelular (Cae) é próxima de 1,8 mM, e no meio intracelular (Cai) próxima de 0,1 mM. A
utilização de quelante ocasiona queda nas concentrações de Cae (entre 1-5 μM), e Cai (para
aproximadamente 15 nM) (Gonzalez-Mariscal et al., 1990). Estudos preliminares avaliaram a
biogênese do CJA após submeterem células epiteliais a baixas concentrações de Cae e
subsequente restauração da concentração fisiológica de Cae. Nessas condições, ocorre um
súbito aumento na concentração de Cai (300 nM), não requerido para a formação do CJA pois
a concentração de 100 nM foi suficiente para que 96% dos contatos célula-célula fossem
selados (Tobey et al., 1994). Além disso, a presença de drogas que impedem a penetração do
cálcio na célula não interferiu na formação das junções célula-célula, confirmando que o
aumento no Cai não é necessário para a formação do CJA. A redução ou o aumento do Cai
utilizando um quelante intracelular ou um ionóforo, respectivamente, não alteraram a
permeabilidade paracelular ao se utilizar o modelo de retirada do Cae, ou após restaurá-lo.
Esses resultados indicam que o aumento da permeabilidade paracelular ocorrido em resposta à
retirada do Cae ocorre via efeito extracelular (Tobey et al., 1994). Além disso, esse mesmo
estudo mostrou que, após submeter células a baixas concentrações de Cae, e ao se colocar um
anticorpo contra a porção extracelular (DECMA ou HECD) ou intracelular (4A2C7) da E-
caderina e restaurar os níveis de cálcio no meio de cultura, apenas a RET do segundo se
restabelece, mostrando que o cálcio atua através da porção extracelular da E-caderina. Foi
então sugerido que as caderinas ligam-se ao cálcio através da sua porção extracelular e
interagem com proteínas quinases através de sua cauda citoplasmática. A partir daí, o modelo
de retirada do cálcio extracelular passou a ser amplamente utilizado no estudo da dinâmica
dos contatos célula-célula.
IV.1 PKA modula a desmontagem do CJA e a funcionalidade das JTs
Para determinar as vias de sinalização envolvidas na desmontagem do CJA causadas pela
retirada do cálcio do meio extracelular, células Caco-2 foram cultivadas em meio normal
(NC) ou em baixa concentração de cálcio (LC), pré-tratadas com inibidores específicos de
quinases antes da incubação em meio LC, e a distribuição das proteínas juncionais foi

57
inicialmente analisada por imunofluorescência. A Figura 21A mostra que nas células
cultivadas em meio NC, E-caderina, ocludina, claudina-1 e ZO-1 localizam-se
predominantemente nos contatos célula-célula. No entanto, em meio LC, as células
apresentaram uma morfologia arredondada e perda dos contatos entre células vizinhas, com
internalização das proteínas do CJA, mas o contato com o substrato foi mantido. A
desmontagem do CJA e a internalizaçaõ das proteínas causadas pela retirada do cálcio do
meio extracelular foram prevenidas pelo pré-tratamento com o inibidor de PKA, H-89. Os
outros inibidores utilizados como PD153035, PD98059 e LY294002, que inibem
respectivamente EGFR, MAPK e PI3K foram incapazes de reverter o efeito do meio LC.
Além disso, as células foram tratadas com forskolina, um agente conhecido por aumentar os
níveis intracelulares de AMPc e ativar PKA. Este tratamento ocasionou significativa
internalização das proteínas do CJA, um efeito similar ao causado pelo meio LC, embora
menos intenso (Figura 21B).
É bem estabelecido que as proteínas juncionais são funcionais quando associadas ao
citoesqueleto. Então, em seguida nós avaliamos a distribuição de E-caderina e claudina-1,
uma proteína de JT que desempenha uma importante função na permeabilidade paracelular,
usando frações solúveis e insolúveis em TX-100 após a incubação em meio NC e LC, e pré-
tratamento com o inibidor de PKA, H-89. O padrão de distribuição e a análise densitométrica
de E-caderina e claudina-1 mostraram uma significativa translocação da fração insolúvel para
a solúvel em células incubadas em meio LC, e esse efeito foi prevenido por H-89 (Fig. 22).
Vale destacar que, embora o IC50 desse inibidor de PKA esteja entre 48 e 135 nM (Davies et
al., 2000), nós utilizamos a concetração de 20µM baseados em estudos prévios mostrando que
essa concentração também inibe PKA (Klingler et al., 2000).
Juntos, esses resultados mostram que a desmontagem do CJA causada pela retirada do
cálcio do meio extracelular é mediada por ativação de PKA.

58
E-caderina ocludina claudina-1 ZO-1
B
A

59
Figura 21: Inibição farmacológica de PKA previne a redistribuição de proteínas do CJA. A) Células Caco-2 foram incubadas em meio normal (NC) e em baixa concentração de cálcio (LC) por 2,5 h ou pré-tratadas com os inibidores: H-89 (PKA), PD98059 (MAPK), PD153035 (EGFR) e LY294002 (PI3K) antes da incubação com o meio LC. B) As células também foram incubadas em meio NC contendo forskolina, um ativador de PKA. As células foram fixadas e marcadas para E-caderina, ocludina, claudina-1 e ZO-1. Note que as proteínas do CJA foram redistribuídas em meio LC e esse efeito foi prevenido apenas pelo H-89. A forskolina causou internalização significativa das proteínas do CJA. Barra, 12 µm.

60
Figura 22. Associação da E-caderina e claudina-1 ao citoesqueleto de actina é modulada por PKA. Immunoblottings representativos e análise densitométrica de E-caderina e claudina-1 nas frações insolúveis (i) e solúveis (s) em TX-100 em células que foram incubadas em meio NC e LC e pré-tratadas com H-89. Observe que o H-89 preveniu a redistribuição das proteínas das frações insolúveis para as solúveis causada pelo meio LC. O valor densitométrico foi calculado usando a seguinte equação: valor arbitrário = (quantidade de proteína na fração insolúvel)/quantidade de proteína na fração solúvel). O valor obtido para células do grupo NC foi normalizado para 1 em cada caso. O desvio padrão de três experimentos independentes está representado. Foram considerados significativos valores para *, p < 0,05 em relação ao grupo NC, e #, p < 0,05 em relação ao grupo LC.

61
Após os diferentes tratamentos, nós verificamos a funcionalidade das JTs monitorando
a resistência elétrica transepitelial (RET) nas monocamadas celulares e nas junções celulares
individuais usando a técnica do vermelho de rutênio associado à análise por microscopia
eletrônica. Como mostrado na Figura 23A, a análise da permeabilidade a íons em
monocamadas confluentes de células Caco-2, acessadas por quantificação da RET, indica que
as células cultivadas em meio NC apresentaram valores próximos a 400 Ω.cm2 (100%). No
entanto, em meio LC, houve uma acentuada queda na RET após 1 h e foram observados
valores próximos a zero após 150 min. Esse efeito foi prevenido de forma significativa por H-
89, que restaurou em torno de 80% da RET, enquanto os inibidores de EGFR, MAPK e PI3K
não preveniram a queda na RET, mostrando que essas quinases não estão envolvidas na
desorganização dos contatos célula-célula causada pelo meio LC. Esse resultado foi
confirmado através da técnica do vermelho de rutênio. Foi observado que o traçador não
permeou as JTs das células em meio NC, permanecendo restrito à região apical das
monocamadas (Fig. 23B), mas permeou em células incubadas com meio LC (Fig. 23C). Dos
inibidores de quinase utilizados, apenas o H-89 preveniu a permeabilidade do corante
eletrondenso causada pelo meio LC (Fig. 23D). Os outros inibidores de quinase, como o
PD98059, LY294002 e PD15035 não reverteram o efeito do meio LC sobre a funcionalidade
das JTs.
Juntos, esses resultados indicam que a ativação de PKA é requerida para regular a
desmontagem de proteínas do CJA causada pelo meio LC e para regular a função de barreira
das JTs.
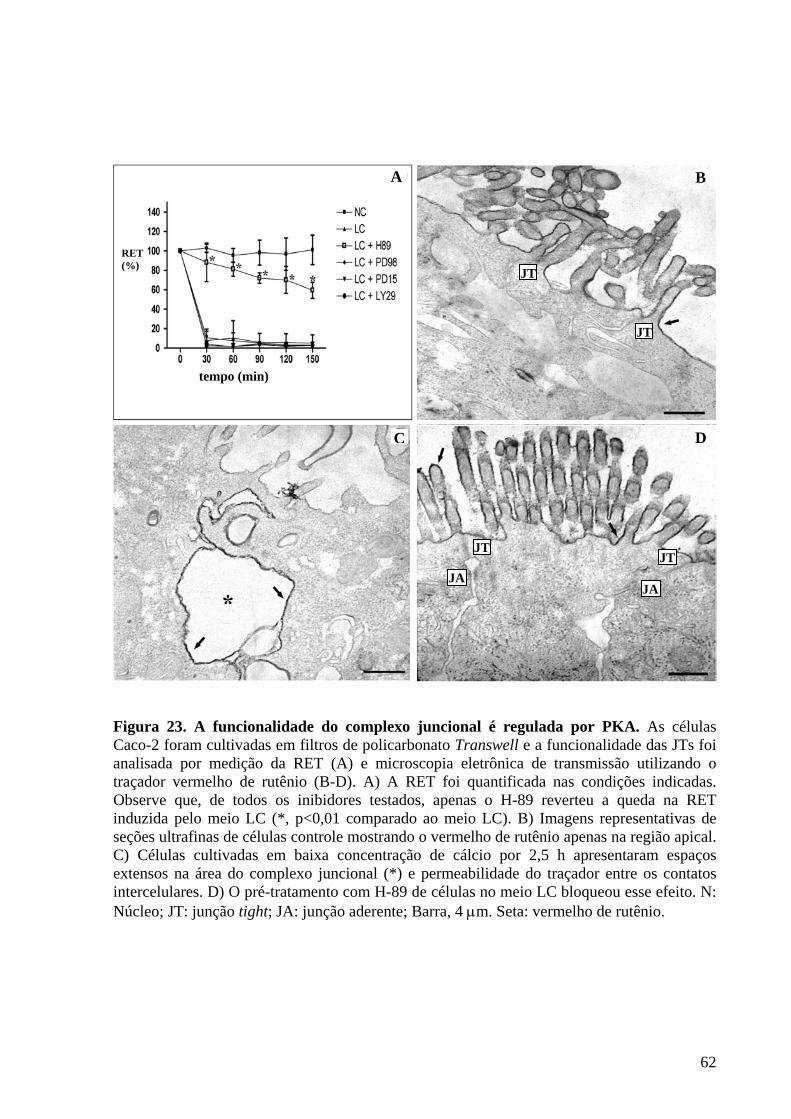
62
Figura 23. A funcionalidade do complexo juncional é regulada por PKA. As células Caco-2 foram cultivadas em filtros de policarbonato Transwell e a funcionalidade das JTs foi analisada por medição da RET (A) e microscopia eletrônica de transmissão utilizando o traçador vermelho de rutênio (B-D). A) A RET foi quantificada nas condições indicadas. Observe que, de todos os inibidores testados, apenas o H-89 reverteu a queda na RET induzida pelo meio LC (*, p<0,01 comparado ao meio LC). B) Imagens representativas de seções ultrafinas de células controle mostrando o vermelho de rutênio apenas na região apical. C) Células cultivadas em baixa concentração de cálcio por 2,5 h apresentaram espaços extensos na área do complexo juncional (*) e permeabilidade do traçador entre os contatos intercelulares. D) O pré-tratamento com H-89 de células no meio LC bloqueou esse efeito. N: Núcleo; JT: junção tight; JA: junção aderente; Barra, 4 µm. Seta: vermelho de rutênio.
JT JT
JA JA
JT
JT
B A
C D
RET (%)
tempo (min)

63
IV.2 PKA modula a organização do citoesqueleto de actina de forma diferencial
através do volume celular
Para explorar a relação entre a reorganização do citoesqueleto de actina e
desmontagem do CJA, nós marcamos o citoesqueleto de actina com faloidina conjugada a
rodamina e realizamos análise por microscopia confocal. A Figura 24 mostra a distribuição
dos filamentos de actina nas regiões apical, medial e basal no plano X-Y (Fig. 24A) e no
plano X-Z (Fig. 24B) das monocamadas celulares cultivadas em meio NC com ou sem
forskolina e meio LC na presença ou ausência de H-89. No meio NC, as células apresentaram
marcação pontual na região apical, caracterizando actina nas microvilosidades e nos contatos
célula-célula. Na região medial, a actina foi predominante nas junções intercelulares; e na
região basal, formando as fibras de estresse. O tratamento com forskolina em meio NC
desorganizou o citoesqueleto de actina principalmentre nas regiões apical e medial, sem
interferir nas fibras de estresse. Em meio LC, as células adquiriram morfologia arredondada
com microfilamentos corticais na periferia celular e desmontagem das fibras de estresse na
região basal. O H-89 preveniu o efeito nas regiões apical e medial, mas as fibras de estresse
não foram restabelecidas; ao contrário, foi possível observar estruturas semelhantes a
lamelipódios na região basal.
Esses resultados indicam que a atividade de PKA modula a desmontagem do CJA em
paralelo à desorganização do citoesqueleto de actina nas regiões apical e medial, mas não a
formação de fibras de estresse.

64
Figura 24. PKA modula a organização do citoesqueleto de actina de forma diferencial através do volume celular. As monocamadas celulares foram incubadas em meio NC, NC com forskolina, LC ou LC com H-89, e marcadas com faloidina conjugada a rodamina. (A) Imagens representativas no plano X-Y em seções próximas a região apical, medial (5 μm) ou basal (9 μm). (B) Imagens do plano X-Z. Note que o meio LC desorganizou o citoesqueleto de actina e esse efeito foi prevenido nas regiões apical e medial pelo H-89, mas não na região basal. Setas: fibras de estresse; cabeça de seta: lamelipódio. Barra: 20 µm.
Apical
Medial
Basal
X-Z
A NC NC+FK LC LC+H89
B

65
IV.3 A inibição farmacológica de ROCK não previne a desorganização das JTs mas
impede a internalização de E-caderina
Considerando que prévios estudos mostraram que PKA modula a função de RhoA
(Qiao et al., 2008), e que ROCK é uma proteína alvo dessa GTPase (Sahai & Marshall, 2002),
nós decidimos investigar se a sinalização Rho-ROCK poderia estar envolvida na
desmontagem do CJA de células Caco-2. Usamos toxina A, um inibidor de GTPases da
família Rho e Y-27632, um inibidor seletivo de ROCK I e II, e inicialmente analisamos a
distribuição das proteínas do CJA por imunofluorescência. A Fig. 25A mostra que, em meio
NC, a toxina A causou arredondamento celular e marcação difusa de E-caderina, ocludina,
claudina-1 e ZO-1 em relação à distribuição normal dessas proteínas nos contatos célula-
célula. Além disso, essa droga causou aumento no tamanho celular e intensa invaginação de
membrana, como visualizado na marcação para ZO-1. O inibidor Y-27632 parece preservar a
distribuição de E-caderina, ocludina e ZO-1, como observado em NC, mas causou aparente
desorganização da distribuição da claudina-1. Além disso, o inibidor de ROCK não preveniu a
internalização das proteínas de JTs ocludina, claudina-1 e ZO-1, mas reverteu a internalização
de E-caderina causada pelo meio LC.
Nós confirmamos esse resultado por immunoblotting usando frações solúveis e
insolúveis em TX-100 e observamos que o Y-27632 preveniu a translocação de E-caderina
das frações insolúveis para solúveis, mas não de claudina-1 (Fig. 25B). O inibidor de ROCK
não teve efeito sobre a solubilidade de ocludina e ZO-1 em TX-100.

66

67
Figura 25. A inibição farmacológica de ROCK não previne a desmontagem das JTs mas impede a internalização de E-caderina. A) As monocamadas celulares foram tratadas com toxina A ou Y-27632 em meio NC ou LC, fixadas e marcadas para E-caderina, ocludina, claudina-1 e ZO-1. Observe a redistribuição das proteínas do CJA e aumento do tamanho das células causado pela inibição de Rho com toxina A (TXA). O inibidor de ROCK (Y27) em meio LC preservou a localização de E-caderina nas JAs, mas não a internalização das proteínas das JTs. Barra, 12 µm. B) Immunoblottings representativos e análise densitométrica de E-caderina e claudina-1 das frações insolúveis (i) e solúveis (s) em TX-100 de células incubadas em meio NC e LC e pré-tratadas com Y-27632. Note que o inibidor de ROCK preveniu a redistribuição de E-caderina da fração insolúvel para a solúvel causada pelo meio LC, mas não de claudina-1. Em cada caso, os valores da densitometria foram calculados como na Fig. 22. O valor obtido para células do grupo NC foi normalizado para 1 em cada caso. O desvio padrão de três experimentos independentes está representado. Foram considerados significativos valores para *, p < 0,05 em relação ao grupo NC e #, p < 0,05 em relação ao grupo LC.

68
Em seguida, analisamos o efeito do inibidor de ROCK na funcionalidade das JTs
usando a quantificação da RET e a técnica do vermelho de rutênio (Fig. 26). A inibição de
ROCK não preveniu a queda da RET causada pelo meio LC e, em meio NC não ocasionou
redução significativa da RET (Fig. 26A). Por microscopia eletrônica, verificamos que o
traçador vermelho de rutênio adicionado à região apical não permeou nas JTs das células em
meio NC (Fig. 26B), mas permeou em células incubadas com meio LC (Fig. 26C). Nós
observamos que a presença do inibidor de ROCK em meio NC não causou permeabilidade do
traçador (Fig 26D), mostrando um padrão similar às células controle, e ocasionou alterações
morfológicas (microvilosidades elongadas) em algumas áreas apicais próximas às JTs. Além
disso, o inibidor de ROCK não impediu a permeabilidade do vermelho de rutênio causada
pelo meio LC (Fig. 26E). Esses dados sugerem fortemente que as proteínas ROCKs modulam
as JAs e as JTs de foma independente em células depletadas de cálcio.

69
Tempo (min)
RET %
JT
JA
A
B C
E D

70
Figura 26: A inibição de ROCK não modula a funcionalidade das JTs em células Caco-2 depletadas de cálcio. Monocamadas celulares cresceram em filtros de policarbonato Transwell e a funcionalidade das JTs foi analisada pela medição da RET (A) e pela técnica do vermelho de rutênio seguida por microscopia eletrônica de transmissão (B-E). A) A redução da RET ocasionada pelo inibidor de ROCK não foi significativa (*, p > 0,05 comparada ao meio NC), e a queda na RET em meio LC não foi prevenida pelo Y-27632. Os dados são representativos de três experimentos independentes. B) Células Caco-2 mantidas em meio NC; C) meio LC após 2,5 h; D) meio NC com o inibidor Y-27632; e E) meio LC com Y-27632. Observe grandes espaços na área do complexo juncional (*) em meio LC, mostrando a permeabilidade do traçador, não prevenida pelo inibidor de ROCK. N: Núcleo; JT: Junção tight; JA: Junção aderente; D: Desmossomos. Barra, A, C e D: 0,8 μm; B: 1,2 μm. Setas, vermelho de rutênio.

71
IV.4 ROCK não regula a organização do citoesqueleto de actina em células crescidas
em meio com baixa concentração de cálcio
Visto que ROCK é uma proteína efetora que interage com Rho ativa causando
contração actina-miosina, investigamos o efeito dos seus respectivos inibidores, Y-27632 e
toxina A, sobre a organização dos filamentos de actina por microscopia confocal. As seções
ópticas das monocamadas cultivadas em meio NC com toxina A mostraram aumento no
tamanho das células e alteraçãos na distribuição de F-actina nos contatos célula-célula nas
regiões apical e medial, e reorientação das fibras de estresse quando comparadas às células
controle. O inibidor de ROCK não preveniu o efeito do meio LC sobre a organização do
citoesqueleto de actina por todo o volume celular, mas ocasionou um intenso rompimento das
fibras de estresse e formação de lamelipódios na região basal (Fig. 27). Além disso, o
tratamento com toxina A ocasionou desorganização das fibras de estresse. É importante
ressaltar que linhagens celulares tumorais, como a Caco-2, apresentam alta atividade de Rho
em relação a linhagen não tumorais, justificando o fato do tratamento com toxina A não ter
causado rompimento das fibras de estresse. Coletivamente, esses dados se contrapõem ao
envolvimento de ROCK na desorganização do citoesqueleto de actina durante a depleção de
cálcio em células Caco-2.
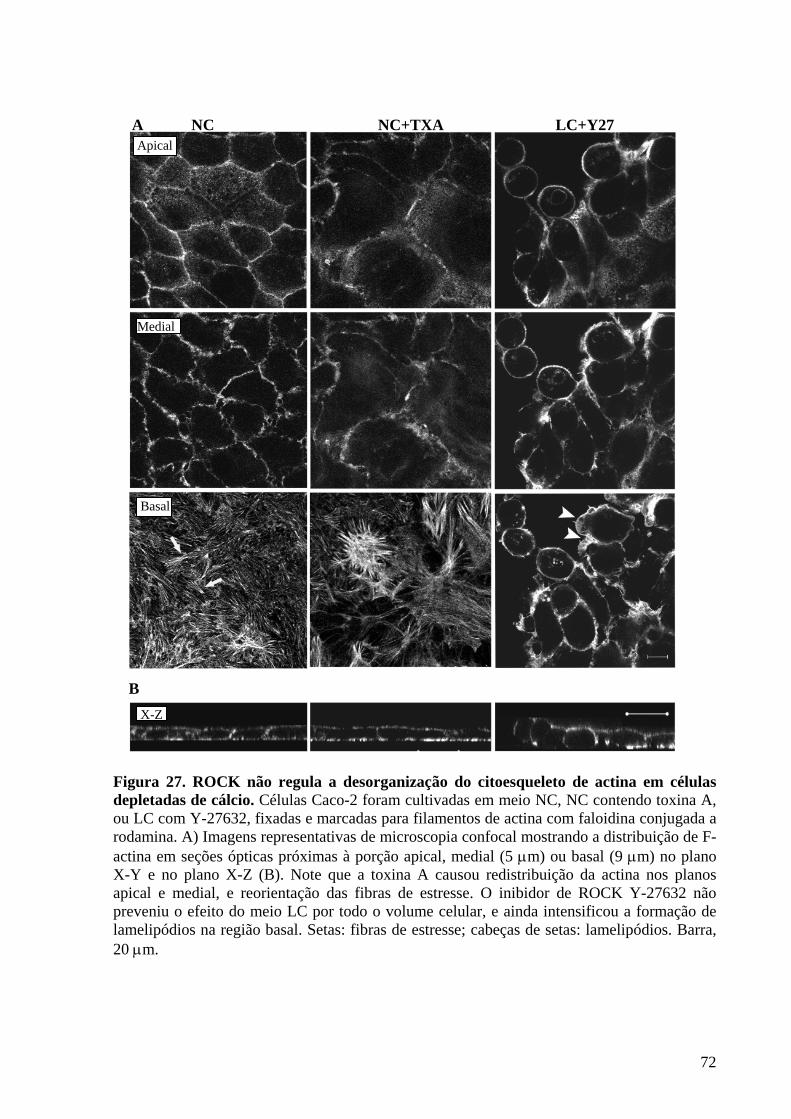
72
Figura 27. ROCK não regula a desorganização do citoesqueleto de actina em células depletadas de cálcio. Células Caco-2 foram cultivadas em meio NC, NC contendo toxina A, ou LC com Y-27632, fixadas e marcadas para filamentos de actina com faloidina conjugada a rodamina. A) Imagens representativas de microscopia confocal mostrando a distribuição de F-actina em seções ópticas próximas à porção apical, medial (5 µm) ou basal (9 µm) no plano X-Y e no plano X-Z (B). Note que a toxina A causou redistribuição da actina nos planos apical e medial, e reorientação das fibras de estresse. O inibidor de ROCK Y-27632 não preveniu o efeito do meio LC por todo o volume celular, e ainda intensificou a formação de lamelipódios na região basal. Setas: fibras de estresse; cabeças de setas: lamelipódios. Barra, 20 µm.
A NC NC+TXA LC+Y27 Apical
Medial
Basal
X-Z
B

73
IV.5 A depleção de cálcio ocasiona translocação de Rho, Rac e RhoGDI para a
membrana, de forma dependente de PKA
Finalmente, nós decidimos avaliar os efeitos da depleção de cálcio e a participação de
PKA sobre a translocação de Rho para a membrana, que tem sido reportada como um reflexo
da ativação de RhoA e Rac (Hirakawa et al., 2007). Para isso, frações de membrana e citosol
foram obtidas e a localização de RhoA, Rac1 e RhoGDI foi avaliada por immunoblotting. A
Fig. 28A mostra que ambos, retirada do cálcio (LC) e tratamento com forskolina, aumentaram
significativamente os níveis de RhoA na fração de membrana, e esse efeito foi prevenido pela
inibição de PKA com H-89. O aumento de Rho na fração de membrana foi concomitante à
redução na fração citosólica. Esse resultado reforça a ideia de que a desorganização do CJA
causada pelo meio LC induz ativação de Rho através de um mecanismo que requer PKA.
Visto que o meio LC induziu perda das fibras de estresse e formação de lamelipódio (Fig. 24),
e esses efeitos estão relacionados à ativação de Rac (Hall, 1998), decidimos verificar em
paralelo se PKA também modula a atividade de Rac. Conforme visto na Fig. 28B, Rac foi
predominantemente localizada na fração de membrana em células mantidas em meio LC, mas
não forskolina. Surpreendentemente, esse efeito foi inibido pelo tratamento com H-89,
indicando que PKA também pode modular a atividade de Rac. Uma banda de menor peso
molecular e logo acima de Rac1 observada na fração de membrana possivelmente
corresponde à isoforma Rac1b, descrita em linhagens de câncer de cólon (Matos et al., 2003).
É conhecido que a toxina A de Clostridium difficile causa mono-glicosilação de Rho e
Rac, fazendo-as acumular nas membranas biológicas, mas prevenindo a interação entre essas
GTPases e suas efetoras e, portanto, inibindo-as (Genth et al., 1999). Então, utilizamos essa
abordagem como controle de translocação para a membrana de Rho e Rac e, conforme
mostrado nas Fig. 28A e B, ambas as GTPases localizaram-se nas membranas após o
tratamento com toxina A. Embora esses resultados pareçam conflitantes, foi reportado que a
fosforilação in vivo de RhoA aumenta sua interação com RhoGDI, previne sua ativação,
impede a interação de RhoA com suas proteínas efetoras, e a dissocia da membrana
plasmática (Dong et al., 1998; Forget et al., 2002; Dransart et al., 2005). É importante
mencionar que embora RhoGDIs sejam consideradas inibidoras de Rho porque mantêm as
GTPases no citosol, estudos mostraram que essas proteínas exibem afinidades similares por
Rac e Cdc42 ativas e inativas, sugerindo seu possível papel no transporte e
compartimentalização das GTPases ativas (Nomanbhoy & Cerione, 1996). Então, para
verificar se a ativação de Rho induzida pela ativação de PKA é mediada por RhoGDI, nós
analisamos a localização dessa proteína por immunoblotting em frações de membrana.

74
Conforme mostrado na Fig. 28C, em meio LC e em células tratadas com forskolina, Rho-GDI
foi predominantemente localizada na fração de membrana, acompanhando a localização de
Rho e Rac; e ainda, o pré-tratamento com H-89 preveniu esse efeito. Juntos, esses resultados
sugerem que a ativação de Rho e Rac mediadas por PKA ocorre através da associação dessas
GTPases com RhoGDI, que poderia translocá-las para a membrana. Vale ressaltar que
RhoGDI não é mono-glicosilada por toxina A e, portanto, permanece no citosol após esse
tratamento.

75
Figura 28. A depleção de cálcio induz translocação de Rho, Rac e RhoGDI para a membrana em células Caco-2. As células foram cultivadas em meio NC (controle) e em NC contendo forskolina ou toxina A, e em meio LC, na presença ou ausência de H-89. Após os respectivos tratamentos, os lisados totais e frações de membrana foram obtidos. A expressão de RhoA (A), Rac (B) e RhoGDI (C) foi verificada por immunoblotting e bandas representativas dos ensaios são mostrados à esquerda no topo dos painéis. A análise densitométrica das respetivas bandas está mostrada à direita da Figura. Cada barra representa valores significativos ± desvio padrão, obtidos de três experimentos independentes. A marcação com Ponceau S foi utilizada como controle da aplicação de proteínas das frações de membrana. *, p < 0,05, em relação ao grupo controle.

76
IV.6 A ativação de Rho restaura a formação de fibras de estresse, mas não a
organização da actina perijuncional
Nós mostramos que a perda dos contatos célula-célula induzida por meio LC
ocasionou ativação de Rho (Fig. 28) e ruptura das fibras de estresse (Fig. 24). Como esses
resultados são aparentemente contraditórios visto que a ativação de Rho ocasiona a formação
de fibras de estresse (Hall, 1998), levantamos a hipótese de que a perda dos contatos célula-
célula poderia ocasionar uma ativação local de Rho, isto é, o meio LC induziria ativação de
Rho na região apical, levando à desorganização do CJA, e inibição na região basal,
permitindo a ruptura das fibras de estresse. Então, sabendo que o LPA é um conhecido
ativador de Rho, decidimos verificar se a ativação de Rho com este agente é capaz de modular
o citoesqueleto de actina nas regiões apical, medial e basal de células Caco-2. Para isso, as
células foram tratadas com LPA e a organização do citoesqueleto de actina em seções ópticas
nos planos X-Y e X-Z da monocamada celular foi analisada por microscopia confocal
utilizando faloidina conjugada a rodamina.
Inicialmente, foram utilizadas as doses de 1, 10 e 20 µM de LPA em células mantidas
em meio LC por 2,5 h. A Fig. 29A mostra que as monocamadas de Caco-2 perderam os
contatos célula-célula e as fibras de estresse em meio LC, e os tratamentos das células
mantidas neste meio com 10 e 20 µM de LPA restauram as fibras de estresse na região basal
dessas células, no entanto, os contatos célula-célula permaneceram desorganizados. A Fig.
29B mostra que esse efeito também ocorreu em tempos menores de tratamento com 10 µM de
LPA. Esses resultados confirmaram nossa hipótese de que a perda dos contatos célula-célula
induzida pelo meio LC ocasiona ativação de Rho na região apical e inibição de Rho na região
basal, necessária para romper fibras de estresse.
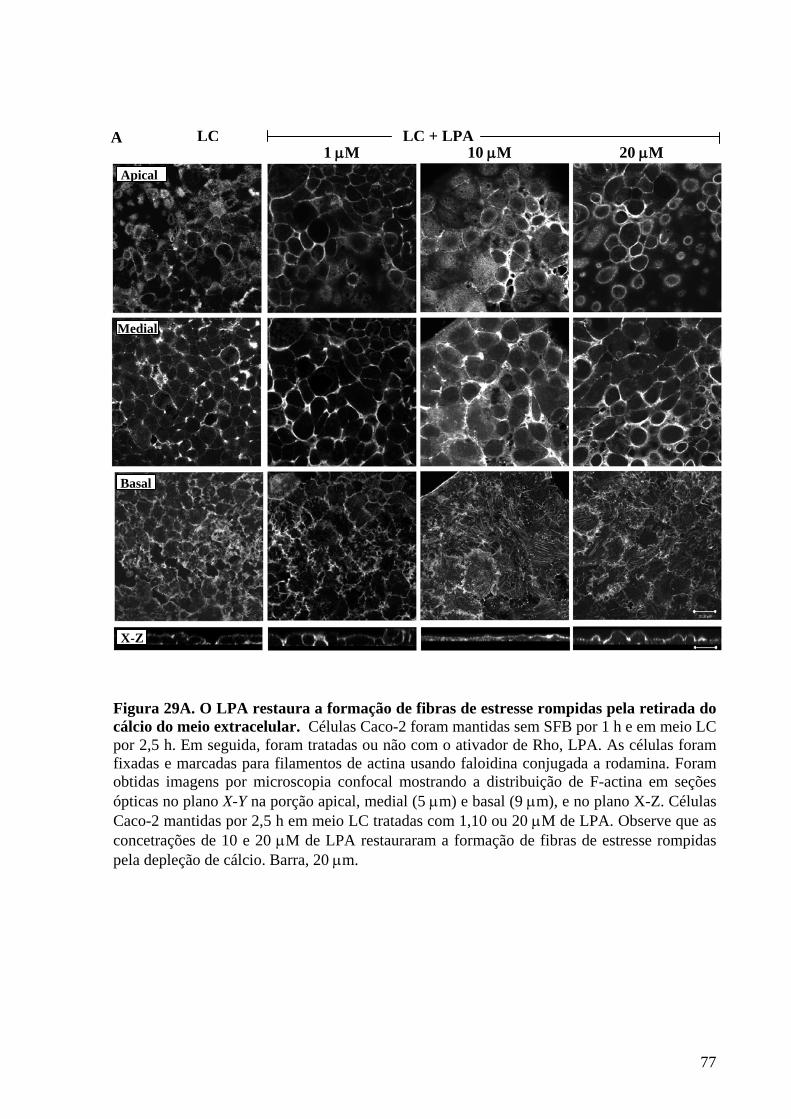
77
Figura 29A. O LPA restaura a formação de fibras de estresse rompidas pela retirada do cálcio do meio extracelular. Células Caco-2 foram mantidas sem SFB por 1 h e em meio LC por 2,5 h. Em seguida, foram tratadas ou não com o ativador de Rho, LPA. As células foram fixadas e marcadas para filamentos de actina usando faloidina conjugada a rodamina. Foram obtidas imagens por microscopia confocal mostrando a distribuição de F-actina em seções ópticas no plano X-Y na porção apical, medial (5 µm) e basal (9 µm), e no plano X-Z. Células Caco-2 mantidas por 2,5 h em meio LC tratadas com 1,10 ou 20 µM de LPA. Observe que as concetrações de 10 e 20 µM de LPA restauraram a formação de fibras de estresse rompidas pela depleção de cálcio. Barra, 20 µm.
A
Apical
Medial
Basal
X-Z
LC LC + LPA 1 µM 10 µM 20 µM

78
Figura 29B. O LPA restaura a formação de fibras de estresse rompidas pela retirada do cálcio do meio extracelular. Células Caco-2 foram mantidas sem SFB por 1 h e em meio LC por 2,5 h. Em seguida, foram tratadas ou não com o ativador de Rho, LPA. As células foram fixadas e marcadas para filamentos de actina usando faloidina conjugada a rodamina. Foram obtidas imagens por microscopia confocal mostrando a distribuição de F-actina em seções ópticas no plano X-Y na porção apical, medial (5 µm) e basal (9 µm), e no plano X-Z. O tratamento com LPA por 20, 60 e 150 min não restaurou a desorganização de actina perijuncional (região medial), mas preveniu a ruptura das fibras de estresse (cabeças de seta). Barra, 20 µm.

79
PARTE II: Papel do LPA na aquisição do fenótipo migratório.
Na segunda etapa da Tese objetivamos analisar os efeitos do LPA na indução do fenótipo
migratório enfatizando as vias de sinalização envolvidas na desorganização do CJA em
paralelo às mudanças do citoesqueleto de actina.
IV. 7 O tratamento com LPA causa redistribuição das proteínas das JAs
Nossos resultados prévios mostraram que a perda de contatos célula-célula ocasiona
ativação de Rho (Fig. 28A), e o tratamento com LPA não restaurou a organização do
citoesqueleto de actina na região próxima aos contatos célula-célula (Fig. 29). Assim,
resolvemos verificar se o LPA ocasiona desorganização do CJA. Inicialmente, realizamos
uma imunofluorescência para analisar a localização subcelular das proteínas de JA E-
caderina, β-catenina e p120-catenina após o tratamento com LPA. A Fig. 30 mostra que essas
proteínas são localizadas preferencialmente nos contatos célula-célula em células não tratadas.
Após o tratamento com LPA por 15, 30 e 60 min, e 6, 24 e 48 h as células das monocamadas
se dissociaram umas das outras e exibiram um padrão de marcação descontínuo, com
internalização das proteínas das JAs, no entanto mantendo a adesão ao substrato.
Então, nós investigamos a funcionalidade das JAs através da sua ligação ao
citoesqueleto de actina avaliando a distribuição subcelular de E-caderina, β-catenina e p120-
catenina por immunoblotting usando frações celulares solúveis e insolúveis em TX-100, após
o tratamento com LPA. A análise densitométrica revelou uma translocação significativa das
proteínas das JAs da fração insolúvel para solúvel quando as células foram incubadas com
LPA (Fig. 31). Nós também verificamos a expressão das proteínas das JAs após 24 h de
tratamento com LPA e observamos que esse agente induziu redução da expressão de E-
caderina, mas não de β-catenina ou p120-catenina em células Caco-2. O agente não induziu a
expressão de vimentina, como visto nas células HT-29 utilizadas como controle de expressão
dessa proteína (Fig. 32). Esses resultados indicam que o LPA medeia a desorganização das
JAs concomitante à perda de expressão de E-caderina.

80
Figura 30A. O LPA causa redistribuição das proteínas das JAs em células Caco-2. Monocamdas celulares depletadas de SFB por 24 h (controle) foram incubadas em meio contendo 10 µM de LPA por 15, 30 e 60 min, fixadas, marcadas para E-caderina, β-catenina e p120-catenina e analisadas por imunofluorescência. A perda de marcação contínua das proteínas das JAs indica desmontagem dos contatos célula-célula (cabeças de setas). Barra, 10 µm.
LPA 15’
LPA 30’
LPA 1 h
Controle
E-caderina β-catenina p120-catenina A

81
Figura 30B. O LPA causa redistribuição das proteínas das JAs em células Caco-2. Monocamdas celulares depletadas de SFB por 24 h (controle) foram incubadas em meio contendo 10 µM de LPA por por 6, 24 e 48 h, fixadas, marcadas para E-caderina, β-catenina e p120-catenina e analisadas por imunofluorescência. O LPA causou reditribuição das proteínas das JAs indicando desmontagem dos contatos célula-célula (cabeças de setas). Barra, 10 µm.
LPA 6 h
LPA 24 h
LPA 48 h
Controle
E-caderina β-catenina p120-catenina B
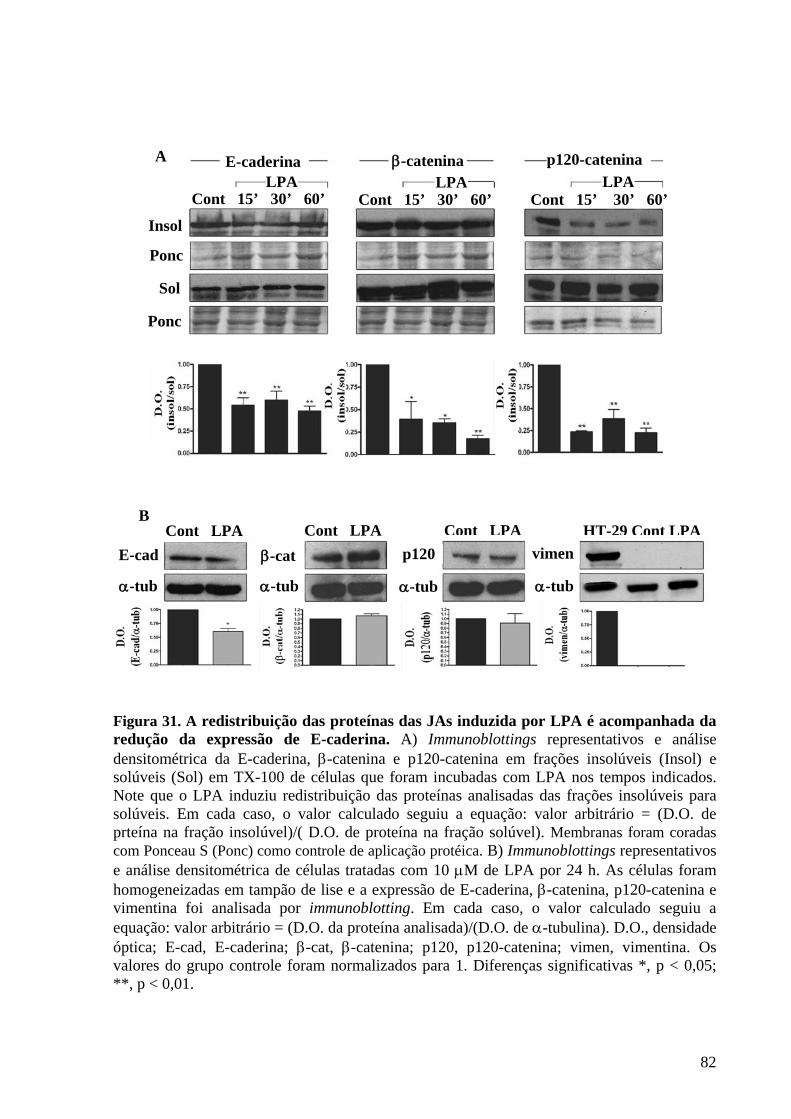
82
Figura 31. A redistribuição das proteínas das JAs induzida por LPA é acompanhada da redução da expressão de E-caderina. A) Immunoblottings representativos e análise densitométrica da E-caderina, β-catenina e p120-catenina em frações insolúveis (Insol) e solúveis (Sol) em TX-100 de células que foram incubadas com LPA nos tempos indicados. Note que o LPA induziu redistribuição das proteínas analisadas das frações insolúveis para solúveis. Em cada caso, o valor calculado seguiu a equação: valor arbitrário = (D.O. de prteína na fração insolúvel)/( D.O. de proteína na fração solúvel). Membranas foram coradas com Ponceau S (Ponc) como controle de aplicação protéica. B) Immunoblottings representativos e análise densitométrica de células tratadas com 10 µM de LPA por 24 h. As células foram homogeneizadas em tampão de lise e a expressão de E-caderina, β-catenina, p120-catenina e vimentina foi analisada por immunoblotting. Em cada caso, o valor calculado seguiu a equação: valor arbitrário = (D.O. da proteína analisada)/(D.O. de α-tubulina). D.O., densidade óptica; E-cad, E-caderina; β-cat, β-catenina; p120, p120-catenina; vimen, vimentina. Os valores do grupo controle foram normalizados para 1. Diferenças significativas *, p < 0,05; **, p < 0,01.
p120
α-tub α-tub α-tub
E-cad β-cat
α-tub
vimen
B
E-caderina β-catenina p120-catenina LPA LPA
Cont 15’ 30’ 60’ Cont 15’ 30’ 60’ Cont 15’ 30’ 60’ Insol
Ponc
Ponc
Sol
LPA A
Cont LPA Cont LPA HT-29 Cont LPA Cont LPA

83
IV. 8 O LPA ocasiona desorganização das JTs de forma reversível
Para verificar se o LPA modula as JTs, realizamos uma análise por
imunofluorescência marcando as proteínas ocludina, claudina-1 and ZO-1 (Fig. 32). As
células Caco-2 foram depletadas de SFB por 24 h e tratadas com LPA nos tempos indicados.
As três proteínas analisadas localizaram-se preferencialmente nos contatos célula-célula no
grupo controle. Embora seja possível observar uma discreta maracação descontínua das
proteínas de JT após a incubação com LPA por 15 min, esse efeito foi revertido ao longo do
tempo. Surpreendentemente, foi observado um agregado perinuclear de ZO-1 no tempo de 48
h. Nós também verificamos a funcionalidade das JTs, mensurando a RET de células Caco-2
confluentes. A Fig. 33 mostra que as monocamadas apresentam valores de RET de
aproximadamente 500 Ω.cm2 após a depleção do SFB por 24 h (controle), e que o LPA
causou uma queda de aproximadamente 65% em 5 e 15 min de tratamento. Esta queda da
RET foi restaurada após 30 min de tratamento, confirmando os dados obtidos no ensaio de
imunofuorescência.

84
Figura 32A. O LPA causa redistibuição das proteínas das JTs de forma reversível. Células Caco-2 depletadas de SFB foram tratadas com 10 µM de LPA por 15, 30 e 60 min e processadas para imunofluorescência usando anticorpos contra ocludina, claudina-1 e ZO-1. O LPA causou uma discreta marcação descontínua dessas proteínas nos contatos célula-célula no tempo de 15 min (cabeça de seta), efeito revertido ao longo do tempo. Barra, 10 µm.

85
Figura 32B. O LPA causa redistibuição das proteínas das JTs de forma reversível. Células Caco-2 depletadas de SFB foram tratadas com 10 µM de LPA por 6, 24 e 48 h e processadas para imunofluorescência usando anticorpos contra ocludina, claudina-1 e ZO-1. Note que em 48 h de tratamento podem ser observados agreados perinucleares de ZO-1 (seta). Barra, 10 µm.
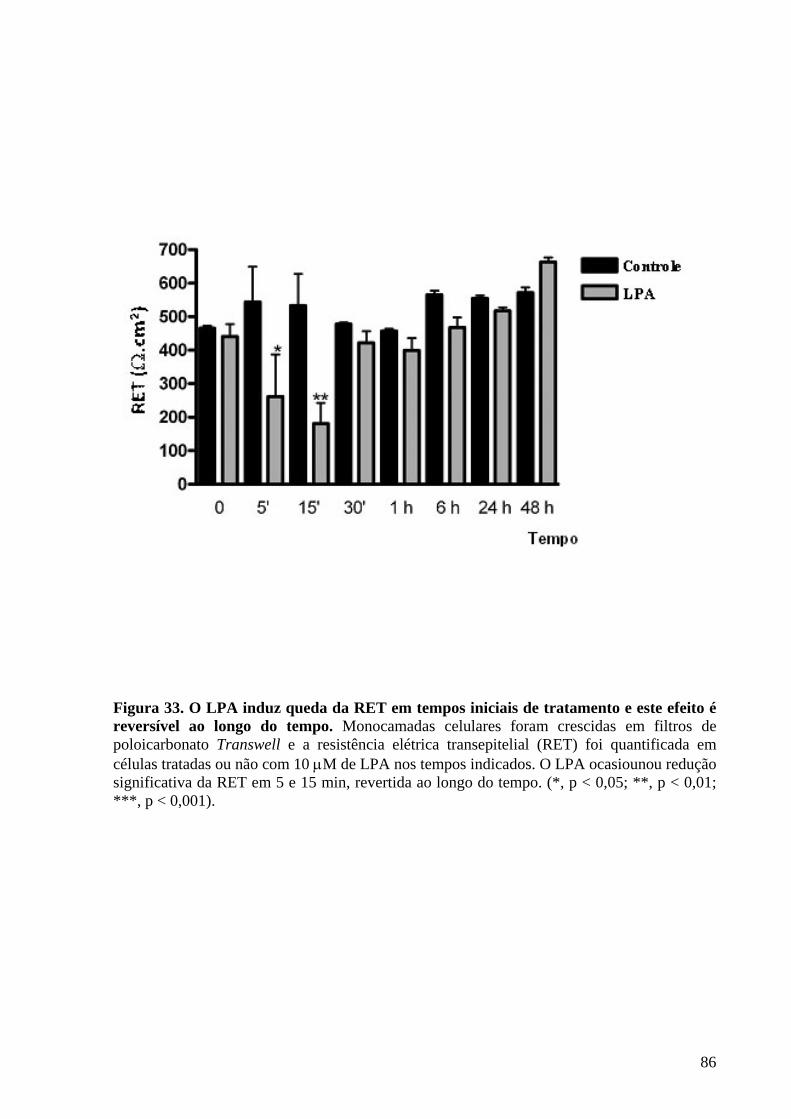
86
Figura 33. O LPA induz queda da RET em tempos iniciais de tratamento e este efeito é reversível ao longo do tempo. Monocamadas celulares foram crescidas em filtros de poloicarbonato Transwell e a resistência elétrica transepitelial (RET) foi quantificada em células tratadas ou não com 10 µM de LPA nos tempos indicados. O LPA ocasiounou redução significativa da RET em 5 e 15 min, revertida ao longo do tempo. (*, p < 0,05; **, p < 0,01; ***, p < 0,001).

87
IV.9 O LPA modula a organização do citoesqueleto de actina perijuncional
concomitante à formação de fibras de estresse e adesões focais
Para explorar a relação entre a desmontagem do CJA e a organização do citoesqueleto
de actina mediada pelo LPA, monocamdas de células Caco-2 foram depletadas de SFB por 24
h (controle) ou tratadas com a droga na concentração de 10 µM nos tempos indicados. Então,
a organização do citoesqueleto de actina foi analisada utilizando faloidina conjugada à
rodamina e microscopia confocal nos planos X-Y e X-Z. A Fig. 34 mostra que, no grupo
controle, as células apresentam marcação pontual na região apical, refletindo a presença de
actina nas microvilosidades, e nos contatos célula-célula. Na região medial, a actina é
predominante na região perijuncional, e poucas fibras de estresse podem ser observadas na
região basal. Já o tratamento com LPA alterou a morfologia celular nos tempos de 15, 30 min
e 1 h, aumentando o tamanho das células. Aparentemente, o LPA não alterou a distribuição de
F-actina na região apical, mas desorganizou a distribuição de F-actina na região medial e,
conforme esperado, induziu a formação de longas fibras de estresse na região basal (Fig.
34A). Nos tempos de 6, 24 e 48 h de tratamento com LPA observamos uma evidente
dispersão celular e redistribuição do citoesqueleto de actina na região medial, ainda com
numerosas fibras de estresse na região basal após 6 h de tratamento. No entanto, essas
estruturas são formadas em menor número e com localização periférica nos tempos de 24 e 48
h, além de ser possível observar a redução do tamanho das células em relação a períodos
curtos de tratamento (Fig. 34B).

88
Figura 34A. O LPA modula F-actina perijuncional. Imagens representativas obtidas por microscopia confocal de células Caco-2 depletadas de SFB (controle), incubadas com 10 µM de LPA por 15, 30 min e 1 h e marcadas com faloidina-rodamina. As imagens mostram seções ópticas do plano X-Y regiões celulares apical, medial (5 µm), ou basal (9 µm) e plano X-Z. Note que o LPA induziu desorganização de F-actina na região medial a partir de 15 min de tratamento. Cabeças de setas: fibras de estresse. Barra, 20 µm.
Controle
LPA 15’
LPA 30’
LPA 1 h
Apical Medial Basal X-Z
A

89
Figura 34B. O LPA modula F-actina perijuncional. Imagens representativas obtidas por microscopia confocal de células Caco-2 depletadas de SFB (controle), incubadas com 10 µM de LPA por 6,24 e 48 h e marcadas com faloidina-rodamina. As imagens mostram seções ópticas do plano X-Y regiões celulares apical, medial (5 µm), ou basal (9 µm) e plano X-Z. Note que o LPA induziu desorganização de F-actina na região medial a em todos os tempos de tratamento. Cabeças de setas: fibras de estresse. Barra, 20 µm.

90
A formação das fibras de estresse por vários fatores intra e extracelulares é
acompanhada pela formação, estimulação e montagem das AFs (Ridley & Hall, 1992), que são
essenciais para a migração celular. Uma vez que nossos resultados prévios mostraram que o
LPA aumentou a formação de fibras de estresse (Fig. 34), decidimos investigar se há formação
de AFs em resposta ao LPA. É bem estabelecido que a autofosforilação de FAK, uma proteína
constituinte de AFs, no resíduo de tirosina 397 induz o recrutamento de Src para essas
estruturas. Src, por sua vez, fosforila outros sítios de FAK, necessários para sua ativação
máxima (Schlaepfer et al., 2004). Esse complexo FAK-Src promove a ativação de cascatas de
sinalização intracelular envolvidas na desmontagem dos contatos célula-célula e migração
celular (Avizienyte et al., 2002; Jiang et al., 2006). Examinamos os níveis de fosforilação
dessas quinases, utilizando um anticorpo que detecta o resíduo de tirosina 397 fosforilado de
FAK e um que detecta o resíduo de tirosina 416 fosforilado de Src. Immunoblottings
representativos são mostrados nas Figs. 35A-B indicando que o LPA estimula a fosforilação de
FAK e Src, predominatemente nos tempos de 15 e 30 min de tratamento.
Para confirmar que nossos dados obtidos por análise bioquímica do complexo FAK-Src
se correlacionam com a análise da distribuição de FAK, utilizamos a microscopia confocal. As
monocamdas confluentes de células Caco-2 foram depletadas de SFB por 24 h e incubadas
com LPA por 15, 30 e 60 min. A Fig. 35C mostra que FAK localiza-se nas monocamadas
celulares nos complexos focais como marcações pontuais nas células controle. Após 15 min de
tratamento com LPA, além de um drástico aumento dos feixes de actina, a extensão da
marcação de FAK aumentou e se tornou elongada, e FAK foi localizada na terminação das
fibras de estresse, indicando formação de AFs. Esse mesmo efeito foi observado no tempo de
30 min e foi reduzido após 1 h de tratamento.

91
Figura 35. O LPA induz formação de adesões focais. (A-B) Análise da fosforilação de Src e FAK. As monocamadas celulares foram tratadas com 10 µM de LPA conforme indicado, e os lisados totais foram obtidos e preparados para o western blotting usando anticorpos específicos contra as formas fosforiladas de Src na tirosina 416 (p-Src) (A) e FAK na tirosina 397 (p-FAK) (B). As imagens das bandas foram quantificadas por densitometria óptica em três experimentos independentes. Os valores foram calculados usando a razão entre a proteína analisada e α-tubulina como controle. Os valores do grupo controle foram normalizados para 1 em cada caso, e foi realizada a análise estatística. *, p < 0,05 e ***, p < 0,001. D.O., densidade óptica. C) Monocamadas de células Caco-2 sem SFB (controle) foram incubadas com meio contendo LPA por 15, 30 e 60 min. As células foram fixadas, marcadas para F-actina (vermelho) e FAK (verde), e analisadas por microscopia confocal. Após o tratamento com LPA, a marcação de FAK tornou-se elongada e localizada nas terminaçãoes das fibras de estresse, indicando a formação de AFs (cabeças de setas). Insertos na parte superior à direita de cada painel das imagens sobrepostas indicam um maior aumento da área marcada com um asterisco. Barra, 20 µm.

92
IV.10 O LPA induz desorganização das JAs através da ativação de Src, Rho e ROCK
Baseados em estudos anteriores mostrando que o LPA ativa diferentes vias de
sinalização (Noguchi et al., 2009; Yanagida et al., 2009), nós decidimos investigar por qual
via o LPA medeia a desmontagem das JAs em células Caco-2. As monocamadas celulares
foram pré-incubadas por 1 h com os diferentes inibidores de sinalização celular, incluindo
H89 (inibidor de PKA), LY294002 (inibidor de PI3K), PD153035 (EGFR inhibitor), PP2 (Src
inhibitor), toxina A de Clostridium difficile (inibidor de Rho), e Y-27632 (inibidor de ROCK).
As células foram então tratadas por 1 h com meio sem SFB contendo 10 µM de LPA
suplementado com os respectivos inibidores químicos. Dentre os inibidores utilizados, o
inibidor de Src PP2 preveniu a redistribuição da p120-catenina, mas não impediu a
redistribuição de E-caderina e β-catenina. Além disso, os inibidores de Rho e ROCK
preveniram a desmonategm das JAs, conforme evidenciado pela localização de E-caderina, β-
catenina e p120-catenina nos contatos célula-célula (Fig. 36A e B).
Para confirmar esses resultados, após os respectivos tratamentos, foram obtidas
frações insolúveis e solúveis em TX-100 seguida por immunoblotting. A Fig. 36C mostra que
o tratamento com LPA por 1 h reduziu os níveis de p120-catenina na fração insolúvel em TX-
100, e esse efeito foi prevenido pela inibição de Src com PP2. Os inibidores de Rho e ROCK
também reduziram os níveis de p120-catenina na fração solúvel em TX-100, em relação ao
grupo tratado com LPA. Esses resultados, junto com o achado de que Src é requerida para a
formação de AFs, indicam que a ativação de Src mediada por LPA modula a adesão célula-
célula e célula-substrato.
A P I C A L
M É D I A
B A S A L

93

94
Figura 36. A ativação de Src, Rho e ROCK medeia a desmontagem das JAs induzida
por LPA. Células Caco-2 depletadas de SFB por 24 h (controle) foram tratadas com LPA
e/ou pré-tratadas com os inibidores conforme indicado, antes da incubação com LPA. A)
Imunofluorescência mostrando que a inibição de Src com PP2 preveniu a redistribuição de
p120-catenina, mas não de E-caderina ou β-catenina. A inibição de Rho com toxina A e
ROCK com Y27632, impediu a redistribuição das três proteínas analisadas. B) Imagens
mostrando que os inibidores de PKA (H-89), PI3K (LY294002) e EGFR (PD153035) não
participam da desmontagem das JAs induzida por LPA. As cabeças de setas em (A) indicam a
redistribuição das proteínas das JAs induzida por LPA. Barra, 10 µm. C) Immunoblotting de
um único experimento mostrando que o LPA causa translocação de p120-catenina para a
fração solúvel e que o tratamento com os inibidores de Src, Rho e ROCK restauram sua
localização na fração insolúvel em TX-100.

95
Considerando que a formação das fibras de estresse é uma resposta celular bem
conhecida da ativação de Rho (Ridley & Hall, 1992), e como nós observamos que a inibição
de Rho previniu a desmontagem das JAs induzida por LPA, decidimos investigar a ativação
das GTPases Rho e Rac após o tratamento com LPA usando o ensaio de pull-down. A Fig.
37A mostra que monocamadas confluentes de células Caco-2 apresentam níveis basais de
Rho ativa no grupo controle, que foram aumentados após 1 h de tratamento com LPA. No
entanto, o tratamento com LPA não aumentou os níveis de Rac ativa. Então nós avaliamos a
distribuição celular de ROCK II e IQGAP, proteínas efetoras de Rho e Rac, respectivamente
por ensaio de imunofluorescência. Foi possível observer que o tratamento com LPA aumentou
RhoA e ROCK II nos contatos célula-célula, mas nem Rac1 ou IQGAP foram redistribuídas
(Fig. 37B). Como GTPases Rho ligadas à membrana estão predominantemente no seu estado
ativo, a marcação de Rac1 nos contatos célula-célula visualizada no grupo controle
provavelmente corresponde ao nível de atividade basal visto no ensaio de pull-down.
Portanto, esses resultados mostram que o LPA modula o citoesqueleto de actina através da
ativação da via RhoA-ROCKII, e sugerem que a desmontagem do CJA seja regulada por
contração de F-actina.
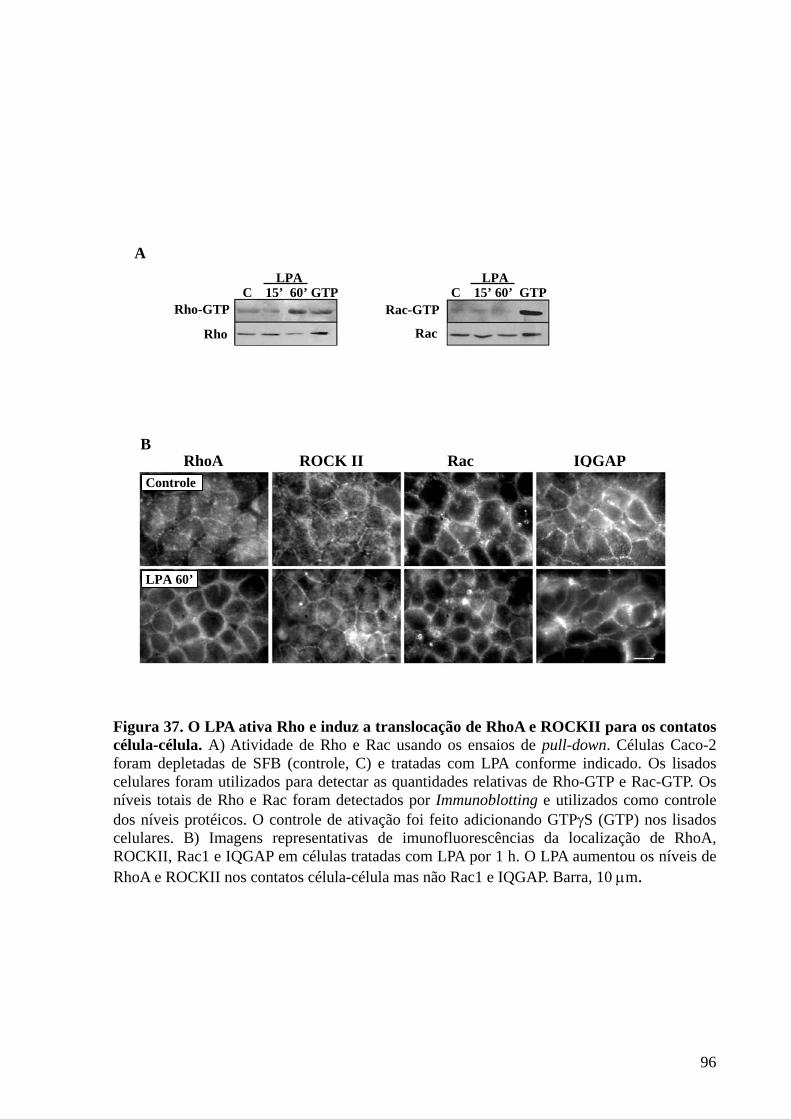
96
Figura 37. O LPA ativa Rho e induz a translocação de RhoA e ROCKII para os contatos célula-célula. A) Atividade de Rho e Rac usando os ensaios de pull-down. Células Caco-2 foram depletadas de SFB (controle, C) e tratadas com LPA conforme indicado. Os lisados celulares foram utilizados para detectar as quantidades relativas de Rho-GTP e Rac-GTP. Os níveis totais de Rho e Rac foram detectados por Immunoblotting e utilizados como controle dos níveis protéicos. O controle de ativação foi feito adicionando GTPγS (GTP) nos lisados celulares. B) Imagens representativas de imunofluorescências da localização de RhoA, ROCKII, Rac1 e IQGAP em células tratadas com LPA por 1 h. O LPA aumentou os níveis de RhoA e ROCKII nos contatos célula-célula mas não Rac1 e IQGAP. Barra, 10 µm.
C 15’ 60’ GTP
C 15’ 60’ GTP Rho-GTP
Rho
Rac-GTP Rac
LPA
LPA
A
B
Controle
LPA 60’
RhoA ROCK II Rac IQGAP

97
IV.11 O LPA induz desorganização do citoesqueleto de actina através da ativação de
Src, Rho e ROCK
Em seguida, examinamos as vias de sinalização envolvidas na desorganização do
citoesqueleto de actina induzidas pelo tratamento com LPA. As monocamadas celulares
foram depletadas de SFB por 24 h, pré-tratadas com inibidores seletivos antes da incubação
com LPA, e as imagens de células marcadas com faloidina conjugada a rodamina foram
obtidas por microscopia confocal nas regiões apical, medial e basal nos planos X-Y e X-Z.
Conforme mostrado na Fig. 38A, o inibidor de Src PP2 e o inibidor de Rho toxina A
claramente restauraram a redistribuição dos filamentos de actina na região medial induzida
por LPA, e a formação de fibras de estresse na região basal. A inibição de ROCK com Y-
27632 parcialmente preveniu os efeitos na região perijuncional e, conforme esperado, impediu
a formação de fibras de estresse induzida por LPA. Nós também verificamos se Src é
requerida para ativação de Rho induzida or LPA. As monocamdas celulares foram depletadas
de SFB por 24 h, pré-tratadas com o inibidor de Src PP2 por 1 h e então tratadas com 10 µM
de LPA por 1 h, e os níveis de RhoA foram analisados pelo ensaio de pull-down. A Figura
38B mostra que a inibição de Src preveniu a ativação de RhoA induzida por LPA, indicando
que Src atua upstream à via Rho-ROCK em nosso modelo de estudo. Além disso, também
mostramos que os inibidores de PKA, PI3K e EGFR não preveniram os efeitos do LPA sobre
a desorganização do citosesqueleto de actina (Fig. 39).

98
LPA LPA GTP
Rho-GTP
Rho
B +PP2
Controle
LPA
LPA+PP2
LPA+TXA
Apical Medial Basal X-Z
LPA+Y27632
A

99
Figura 38. A desorganização de F-actina mediada por LPA ocorre através da ativação de Src e da via Rho-ROCK. A) Células Caco-2 sem SFB por 24 h (controle) e incubadas com 10 µM de LPA por 1 h ou pré-tratadas por 1 h com os inibidores como indicado, foram processadas para análise de F-actina por microscopia confocal. Observe que os inibidores de Src e Rho (PP2 e toxina A) restauraram a distribuição de F-actina na região perijuncional e a organização das fibras de estresse (cabeça de seta) induzida pelo LPA, e o inibidor de ROCK (Y-27632) reverteu parcialmente os efeitos do LPA na região medial. Barra, 10 µm. B) Ensaio de pull-down mostrando que o aumento da atividade de RhoA induzido por LPA foi prevenido pela inibição de Src com PP2.

100
Figura 39. A desorganização do citosesqueleto de actina mediada por LPA é independente de PKA, PI3K e EGFR. Monocamadas de células Caco-2 foram depletadas de SFB por 24 h e incubadas com 10 µM de LPA por 1 h ou pré-tratadas por 1 h com os inibidores químicos de PKA (H-89), PI3K (LY29) e EGFR (PD15). As células foram fixadas, marcadas com faloidina-rodamina e analisadas por microscopia confocal. As seções ópticas obtidas mostram que os três inibidores não preveniram a reorganização do citoesqueleto de actina induzida por LPA. As cabeças de setas indicam longas fibras de estresse. Barra, 10 µm.
LPA
LPA+H89
LPA+LY29
Apical Medial Basal X-Z
LPA+PD15

101
IV.12 O LPA modula a desorganização das JTs através da ativação de Rho
A fim de verificar se a mesma via de sinalização que modula ambos, JAs e
citoesqueleto de actina também modula as JTs em resposta ao LPA, avaliamos a distribuição
da proteína claudina-1 e a funcionalidade das JT por medição da RET. Células Caco-2 foram
depletadas de SFB por 24 h, pré-tratadas por 1 h com os inibidores de Src, Rho, ROCK, PKA,
PI3K e EGFR , incubadas com 10 µM de LPA contendo os respectivos inibidores por 15 min,
e processadas para imunofluorescência. A Figura 40A mostra que apenas os inibidores de Rho
e ROCK restauraram a morfologia celular, restaurando também a localização das proteínas
das JTs nos contatos célula-célula. Surpreendentemente, através da análise funcional das JTs
quantificada por medição da RET, observamos que o aumento da permeabilidade paracelular
de monocamadas celulares tratadas com LPA por 15 min foi prevenido apenas pelo inibidor
de Rho (Fig. 40B).

102
Figura 40. O LPA ocasiona redistribuição das proteínas das JTs de forma dependente de Rho. As células foram mantidas por 24 h sem SFB (Controle), pré-tratadas por 1 hora com os inibidores: de Src (PP2), Rho (toxina A), ROCK (Y27632), PKA (H89), PI3K (LY294992) e EGFR (PD153035), e tratadas com LPA contendo os respectivos inibidores por 15 min para análise da distribuição de claudina-1 por imunofluorescência (A) e análise da funcionalidade das JTs através da mensuração da permeabilidade a íons (B). Embora a inibição de Rho e ROCK tenham restaurado morfologia celular e a distribuição das proteínas analisadas, a funcionalidade das JTs foi restabelecida apenas com a inibição de Rho. Cada barra representa valores significativos obtidos de três experimentos independentes. * , p < 0,05, em relação ao tratamento com LPA. Barra, 10 µm.
Controle LPA LPA+PP2 LPA+TXA
LPA+Y27 LPA+H89 LPA+LY29 LPA+PD15
LPA LPA+PP2 LPA+TXA LPA+Y27 LPA+H89 LPA+LY29 LPA+PD15
Tempo (min) 0 15 0 15 0 15 0 15 0 15 0 15 0 15
A Claudina-1
B

103
IV.13 O LPA aumenta a migração celular de forma dependente de Src, Rho e ROCK
Estudos anteriores mostraram que o LPA aumenta a migração celular em diferentes
tipos celulares (Xu et al., 2007; Shida et al., 2008). Aqui, utilizando a técnica do wound
healing, nós mostramos que o LPA promove a migração em células Caco-2. Nas células
controle depletadas de SFB, observamos que os riscos permaneceram abertos no tempo de 6 h
e apenas começaram a fechar após 24 h em cultura. Contudo, células tratadas com LPA
migraram mais rapidamente que as células do grupo controle, e iniciaram o fechamento do
risco no tempo de 6 h de tratamento, encontrando-se quase inteiramente fechados em 24 h
(Fig. 41A e B).
Para investigar as vias de sinalização envolvidas na migração celular induzida por
LPA, as monocamadas celulares foram pré-tratadas com os inibidores de quinases específicos,
antes do ensaio de wound healing. Nós observamos que os inibidores que o PP2, toxina A e
Y-27632, que inibem Src, Rho e ROCK, respectivamente, preveniram a migração celular
induzida por LPA no tempo de 6 h de tratamento. Além disso, o inibidor de ROCK impediu
completamente a migração celular mesmo após 24 h em cultura. Os outros inibidores (H-89
de PKA, LY294002 de PI3K e PD153035 de EGFR) não preveniram o aumento da migração
celular induzido pelo LPA (Fig. 42). O número relativo de células, determinado pela técnica
do cristal violeta nos tempos da análise de migração celular, confirmou que o efeito observado
após o tratamento com LPA ocorreu em decorrência a um aumento de migração mas não de
proliferação celular (Fig. 41C). Esse resultado indica que a mesma via de sinalização que
modula a desmontagem dos contatos célula-célula é também responsável pelo controle da
migração celular.

104
Figura 41. A migração celular induzida por LPA depende da ativação de Src e da via de sinalização Rho-ROCK. A-B) A migração celular foi analisada usando a técnica de wound healing. Células Caco-2 foram depletadas de SFB (controle), incubadas com 10 µM de LPA por 1 h ou pré-tratadas por 1 h com os inibidores farmacológicos antes da incubação com LPA. As monocamdas celulares foram rompidas manualmente e a migração celular foi monitorada nos tempos de 6 e 24 h. A) Os inibidores de Src (PP2), Rho (toxina A) e ROCK (Y-27632), preveniram o aumento da migração celular induzida por LPA no tempo de 6 h, e a inibição de ROCK preveniu completamente o fechamento do risco, mesmo após 24 h. Barra, 100 µm. B) A quantificação do ensaio foi realizada medindo a distância entre as duas margens da monocamada celular em três experimentos independentes. C) O ensaio de proliferação foi realizado em 6, 24 e 48 h de tratamento com LPA, em paralelo ao ensaio de migração celular. A análise estatística foi feita utilizando ANOVA com pós-teste Dunnett. ***, p < 0,001 comparado ao grupo controle; ###, p < 0,001 e ##, p < 0,01 comparado às células tratadas com LPA.
0 6 24 48 0 6 24 48
Controle LPA LPA+PP2 LPA+TXA LPA+Y27632
LPA
+ SFB - SFB
Tempo (h)
Tempo (h)
0 6 24 0 6 24 0 6 24 0 6 24 0 6 24
B C
Controle LPA LPA+PP2 LPA+TXA LPA+Y27632
A 0 h
6 h
24 h

105
Figura 42. A migração celular induzida por LPA é independente de PKA, PI3K e EGFR. Células Caco-2 foram depletadas de SFB (controle), incubadas com 10 µM de LPA por 1 h ou tratadas por 1 h com os inibidores farmacológicos antes e durante a incubação com LPA. As monocamdas celulares foram rompidas manualmente e a migração celular foi monitorada pela técnica de wound healing nos tempos de 6 e 24 h. A) Os inibidores de PKA (H89), PI3K (LY294992) e EGFR (PD153035) não preveniram o aumento da migração celular induzido pelo tratamento com LPA. Barra, 100 µm. B) A quantificação do ensaio foi realizada medindo a distância entre as duas margens da monocamada celular em três experimentos independentes.
LPA LPA+H89 LPA+LY29 LPA+PD15
0 6 24 0 6 24 0 6 24 0 6 24
Tempo (h)
0 h
6 h
24 h
LPA LPA+H89 LPA+LY29 LPA+PD15
A
B

106
IV.14 O fenótipo migratório induzido por LPA depende da ativação de Src e ROCK
Uma vez que o LPA induziu a formação de fibras de estresse e AFs em monocamadas
de células confluentes (Figs. 34-35) e o aumento da migração celular (Figs. 41-42), decidimos
investigar se esse agente lipídico também induz a formação dessas estruturas no fronte de
células migratórias. Para isso, foram utilizadas monocamadas subconfluentes depletadas de
SFB por 24 h e incubadas com LPA em diferentes tempos, e a distribuição de FAK e F-actina
foi analisada por microscopia confocal. A Fig. 43 mostra que as células do grupo controle não
emitem projeções de membrana e apresentam marcação pontual de FAK por todo o volume
celular. O tratamento das células por 60 min com LPA ocasionou a formação de grandes
protrusões de membrana, longas fibras de estresse, e AFs elongadas. Observamos que estas
estruturas foram formadas também com 15 e 30 min.
Logo verificamos a participação de Src e da via Rho-ROCK na indução desse
fenótipo migratório por LPA. Monocamadas celulares subconfluentes foram pré-tratadas por
1 h com os respectivos inibidores, mantidos por mais 1 h em meio de cultura sem SFB
contendo LPA. Foi possível observar que a inibição de Src com PP2 e de ROCK com Y-
27632 preveniram a formação de projeções de membrana e o fenótipo migratório induzido
pelo LPA, corroborando nossos resultados obtidos pela análise da migração celular. No
entanto, somente a inibição de ROCK impediu a formação de AFs elongadas.

107
Figura 43. Src e ROCK regulam a formação de projeções de membrana induzidas por LPA em células migratórias. Monocamadas de células Caco-2 subconfluentes depletadas de SFB (controle) foram incubadas com LPA por 1 h ou pré-tratadas por 1 h com os inibidores químicos de Src (PP2) e ROCK (Y-27632) antes do tratamento com LPA. As células foram fixadas e marcadas para F-actina (vermelho) e FAK (verde) e analisadas por microscopia confocal. O tratamento com LPA induziu a formação de grandes projeções de membrana (cabeças de setas), prevenidas pela inibição de Src e ROCK, e elongadas adesões focais (setas), prevenidas pela inibição de ROCK. Os insertos mostrados na região superior à direita de cada painel indicam um maior aumento das regiões das imagens marcadas com um asterisco. Núcleos foram marcados com DAPI (azul). Barra, 20 µm.

108
IV.15 O LPA aumenta o crescimento independente de ancoragem de células Caco-2
É conhecido que a tumorigenicidade celular pode ser avaliada in vitro através do ensaio
de crescimento independente de ancoragem (CIA), e que a ativação de oncogenes, como Src,
aumenta essa característica (Dohn et al., 2009). Sabe-se também que a ativação do compelxo
FAK-Src é capaz de sustentar a sobrevivência celular na ausência de substrato (Bouchard et al.,
2007; Zouq et al., 2009). Então, resolvemos verificar se o LPA induz o CIA em células Caco-2, e
se a inibição da via Rho-ROCK participa desse processo. Os resultados da Fig. 45A mostram que
as células Caco-2 são capazes de crescer na ausência de adesão ao substrato formando colônias.
Embora o tratamento com 10 µM de LPA tenha ocasionado aumento do número de colônias, o
tamanho das mesmas não teve alteração. O tratamento das células com o inibidor Y-27632 de
ROCK não foi capaz de prevenir o aumento da formação de colônias induzido pelo tratamento
com LPA.
Figura 44. O LPA aumenta o crescimento independente de ancoragem em células Caco-2. Células plaqueadas em um substrato de agarose cresceram por 14 dias na presença ou ausência de LPA, com ou sem o inibidor químico de ROCK (Y-27632). O número de colônias formadas em cada condição experimental foi contado, os valores obtidos em dois experimentos independentes foram normalizados para 1 e plotados no gráfico. *, p < 0,05 em relação ao grupo controle.

109
IV.16 O LPA não induz aumento de invasividade e atividade de MMP-9 e 2 das células
Caco-2
Tem sido mostrado que o LPA aumenta a capacidade invasiva de diversos tipos
celulares, incluindo da linhagem de câncer de ovário OVCAR-3 (Sawada et al., 2002), bem
como a expressão e ativide de MMPs (Wu et al., 2005). Para verificar se o LPA aumenta o
potencial invasivo em nosso modelo de estudo, utilizamos insertos Transwell revestidos com
matrigel e avaliamos a capacidade de invasão celular das células Caco-2. Conforme
apresentado na Fig. 45A, o tratamento com LPA não aumentou a capacidade invasiva das
células Caco-2, embora esse agente tenha induzido invasão das células OVCAR-3 usadas
como controle de invasividade. Além disso, o ensaio de zimografia (Fig. 45B) mostrou que o
LPA ocasiona um discreto aumento na atividade de MMP-2 mas não de MMP-9, mesmo
ocasionando aumento de expressão do RNAm de MMP-2 e MMP-9. (Fig. 45C).

110
Figura 45. O LPA não aumenta o potencial invasivo de células Caco-2 em matrigel. A) As células OVCAR-3 e Caco-2 foram plaqueadas na câmara superior dos insertos Transwell revestidos com matrigel em meio sem SFB (controle) ou contendo LPA. Na câmara inferior foi adicionado meio com 20% de SFB como quimioatraente. Após 48 h, os insertos foram corados e as células que invadiram foram contadas. Imagens representativas de três experimentos independentes. B) O sobrenadante de monocamadas de células Caco-2 foi coletado e o ensaio de zimografia foi realizado. O sobrenadante de células de fibrossarcoma HT-1080 foi utilizado como controle, assim como o sobrenadante de monocamadas de Caco-2 mantidas em meio com SFB. Após depletar as células de SFB por 24 h, não são observadas bandas correspondentes à atividade das MMP-2 e -9, observadas nos controles com SFB. O tratamento por 24 h com LPA ocasionou um discreto aumento da atividade de MMP-2, mas não de MMP-9. Imagem representativa de dois experimentos independentes. C) qRT-PCR mostrando que a expressão das MMP-2 e -9 foram aumentadas pelo tratamento com LPA, em um único experimento.
tratamento
A
B
C

111
V. DISCUSSÃO
V.1 PARTE I: Análise da inter-relação entre o CJA e o citoesqueleto de actina durante
a perda da adesão célula-célula
De forma geral, a participação de GTPases e de PKA tem sido implicada na
desmontagem do CJA bem como na organização do citoesqueleto de actina em células
epiteliais. No entanto, esses mecanismos celulares têm sido estudados separadamente, e a
existência de uma interação entre essas duas moléculas de sinalização em células tumorais,
particularmente em CCR, permanece indefinida. Nessa primeira parte da Tese, usamos as
células Caco-2 como modelo de CCR, depleção do cálcio extracelular como agente de
desmontagem das junções celulares, e tratamento com inibidores de vias de sinalização para
identificar as vias que medeiam esse evento. Nós identificamos uma cascata de sinalização
que integra PKA, Rho-ROCK e Rac para regular a desmontagem dos contatos célula-célula e
a organização de F-actina. Essas conclusões são suportadas pelas seguintes observações: a) A
inibição de PKA preveniu a redistribuição de proteínas do CJA, a perda de funcionalidade das
JTs, e a desorganização de F-actina nas regiões apical e medial da monocamada celular; b) A
inibição de Rho causou desorganização do CJA e reorganização do citoesqueleto de actina por
todo o volume celular; c) A inibição de ROCK impediu a redistribuição de E-caderina, mas
não de proteínas das JTs e desorganização de F-actina; e d) o tratamento com forskolina e
meio LC, que ativa PKA, causaram a translocação de RhoA, Rac e RhoGDI do citosol para a
membrana plasmática, indicando a ativação dessas GTPases.
A participação de PKA na desmontagem do CJA tem sido relatada com relação às JTs.
Nossos resultados, mostrando que PKA está envolvida no aumento da permeabilidade
paracelular e redistribuição das proteínas do complexo juncional, estão em concordância com
observações prévias (Klingler et al., 2000). Adicionalmente, reportamos que outras vias de
sinalização celular como MAPK, EGFR e PI3K não estão envolvidas na perda de
funcionalidade das JTs. A queda da RET (Fig. 23A) observada nas células tratadas com a
baixa concentração de cálcio pode ser justificada pela alteração na distribuição de claudina-1,
visto que a família das claudinas é a principal responsável pela formação de JTs e função de
barreira em epitélios (Furuse et al., 2002). Em relação às JAs, estudos recentes vêm
mostrando que a ativação de PKA aumenta a atividade do repressor transcricional de E-
caderina, Snail (McPherson et al., 2010), e ativa a via Wnt através da inibição de
GSK3β, permitindo assim a translocação de β-catenina para o núcleo (Park et al., 2010). Nós

112
mostramos que ativação de PKA em resposta à depleção do cálcio extracelular causou a
internalização de E-caderina para o citosol, mas não avaliamos a distribuição de β-catenina.
Portanto, mais experimentos são necessários para determinar se a retirada do cálcio do meio
extracelular é capaz de ativar Snail e a via Wnt.
É importante mencionar que muitos estudos têm demonstrado que a internalização das
proteínas do CJA pode ocorrer pelos três mecanismos clássicos de endocitose, dependendo do
estímulo. Isso inclui endocitose mediada por clatrina das JTs e JAs utilizando estímulos não
fisiológicos, como o meio LC (Ivanov et al., 2004b); macropinocitose em resposta a citocinas
pró-inflamatórias como o interferon-γ (Bruewer et al., 2005), resultando na internalização de
proteínas das JTs mas não de JAs, e dessa forma aumentando a permeabilidade paracelular
sem alterar a adesão célula-célula; e endocitose mediada por caveolina em resposta à toxina
de E. coli CNF-1, que ativa RhoA (Hopkins et al., 2003), levando a internalização da ocludina
e concomitante redistribuição de outras proteínas das JTs ou JAs. Ainda, acredita-se que o
meio LC medeie a endocitose similar à fisiológica, necessária para manter o turnover das
proteínas do CJA, que são degradadas via endossomos tardios ou recicladas de volta à
membrana. No entanto, se células tumorais apresentam maiores níveis de endocitose das
proteínas do CJA e se esse processo é mediado por PKA permanece por ser investigado.
Adicionalmente, o principal mecanismo descrito por modular a perda de proteínas dos
contatos intercelulares em células tumorais é a repressão transcricional (Wheelock et al.,
2007; McPherson et al., 2010). Nesse contexto, um estudo mostrou que a redução dos níveis
de cálcio na dieta de camundongos com mutação no gene APC ocasiona redução da expressão
de E-caderina nos tumores de cólon desses animais (Carothers et al., 2006), mas se PKA e/ou
os repressores de E-caderina participam desse evento é completamente desconhecido.
Considerando que as proteínas do CJA são ligadas ao citoesqueleto de actina, também
verificamos se PKA participa da organização de citoesqueleto de actina, em paralelo à
modulação deste complexo. Mostramos que o meio LC causou reorganização de F-actina em
todos os níveis do volume celular, e a ativação de PKA por forskolina nos níveis apical e
medial, contudo, causou apenas reorientação e não dissolução das fibras de estresse (Fig. 24).
Vale menacionar que a forskolina induziu perda de fibras de estresse em células musculares
(Hirakawa et al., 2007) e também observamos esse efeito em células MDCK (não mostrado).
Então, essa menor susceptibilidade das fibras de estresse de células Caco-2 à ativação de PKA
pode ser justificada pelos maiores níveis de expressão ou atividade de GTPases Rho em
linhagens tumorais (Tabela 1). Ainda assim, a reorientação das fibras de estresse observada
em células Caco-2 aponta para a possibilidade de PKA mediar a aquisição de um fenótipo

113
móvel alterando a ancoragem das células ao substrato, conforme sugerido por Thiery &
Sleeman (2006) e mostrado por Hirakawa et al. (2007) e Lim et al. (2008). É válido
mencionar que PKA regula proteínas acessórias do citoesqueleto de actina. Por exemplo, foi
mostrado que PKA pode se ligar em WAVE-1 para ocasionar reorganização de F-actina
(Westphal et al., 2000) e que a proteína VASP é um substrato de PKA em resposta à
formação de contatos célula-célula (Lawrence et al., 2002). PKA também fosforila LIMK
diretamente, permitindo a fosforilação de cofilina e subsequente despolimerização de actina
(Nadella et al., 2009). Ainda, PKA regula a polaridade celular durante a migração, tendo sua
ativação compartimentalizada na região anterior das células (Lim et al., 2008).
No presente estudo, mostramos que o meio LC causou arredondamento celular e
formação de actina em forma de anel nas regiões apical e medial, características típicas de
aquisição de motilidade dependente de Rho (Sanz-Moreno et al., 2008). Aqui, exploramos
essa possibilidade utilizando toxina A, um inibidor de Rho conhecido por perturbar a função
de barreira epitelial (Nusrat et al., 2001). Verificamos que essa droga causou redistribuição
das proteínas do CJA (Fig. 25 e 26) de forma concomitante à desorganização de F-actina nas
regiões apical e medial e formação de fibras de estresse (Fig. 27). Posteriormente,
hipotetizamos que o envolvimento de RhoA na desmontagem do CJA induzida por depleção
de cálcio poderia ser dependente de ROCK, uma vez que essa proteína representa uma efetora
clássica de Rho (Sahai & Marshall, 2002). Nossos experimentos mostraram que o inibidor
dessa proteína, Y-27632, em meio depletado de cálcio, não impediu a translocação das
proteínas das JTs das áreas do contato célula-célula para o citosol e não preveniu a perda da
função de barreira paracelular, mas preveniu a internalização de E-caderina, e ainda, a
presença de Y-27632 em meio NC não afetou a RET de forma significativa (Figs. 25 e 26).
Esses dados sugerem que a ativação de ROCK desorganiza as JAs, mas não JTs em nosso
modelo de estudo. Esses resultados são condizentes com alguns estudos usando células de
CCR, por exemplo, foi mostrado que a utilização de Y-27632, que inibe as duas isoformas de
ROCK (ROCK I e II), aumentou os níveis de E-caderina na fração insolúvel em TX-100
(Sahai & Marshall, 2002), e preveniu a localização de proteínas das JAs e JTs nos contatos
intercelulares após depleção seguida de restauração de cálcio no meio de cultura (Walsh et al.,
2001). Ao contrário, outros estudos mostraram que a indução de adesão célula-célula mediada
por um metabólito de vitaminaD3, que inclusive regula os níveis de cálcio intracelular, é
dependente da ativação de Rho-ROCK, que aumenta os níveis de E-caderina e cateninas na
membrana (Ordóñez-Morán et al., 2008). Em células de cólon, foi reportado que a inibição de
ROCK com Y-27632 bloqueou completamente a reorganização de F-actina e a translocação

114
de proteínas do CJA em meio depletado de cálcio (Samarin et al., 2007). Esses autores
usaram o silenciamento gênico dos membros homólogos ROCKI e ROCKII e reportaram o
envolvimento apenas de ROCKII na desmontagem das JTs. Portanto, é possível sugerir que as
ROCKs participam da regulação de JAs e JTs de forma independente, isto é, ROCKI seria
responsáveis pela regulação das JAs e ROCKII pelas JTs e organização do citoesqueleto de
actina.
Considerando que a ativação de Rho ocorre quando ela é translocada para a membrana
celular (Hirakawa et al., 2007), analisamos esse evento usando frações de membrana após a
depleção de cálcio e pré-tratamentos com forskolina e H-89. Observamos que o meio LC e a
forskolina induziram translocação de Rho para a membrana, e esse efeito foi revertido pelo H-
89, indicando que PKA medeia ativação de RhoA induzida pela depleção do cálcio
extracelular. Alguns estudos mostraram que a ativação de Rho-ROCK ocorre como resposta à
perda de contatos célula-célula através de contração actina-miosina. Por exemplo, a depleção
de cálcio induziu a fosforilação da cadeia leve da miosina de forma dependente da via Rho-
ROCK em células de câncer de mama (Fan et al., 2007), e a ativação de Rho resultou em
contração actina-miosina na desmontagem do CJA em células de CCR (Samarin et al., 2007).
Então, sugerimos que a ativação de PKA cause desmontagem do complexo juncional e,
subsequentemente, esse evento ocasione ativação de Rho na região celular apical.
Aparentemente, existe uma contradição nesses resultados uma vez que o meio LC aumentou
os níveis de RhoA na membrana enquanto desorganiza as fibras de estresse, o que deveria
ocorrer na inibição de RhoA (Hall, 1998). Contudo, a ativação local de GTPases Rho durante
a migração celular e formação de projeções de membrana tem sido sugerida (Wozniak et al.,
2005; Pertz et al., 2006). Então, a partir desses resultados, hipotetizamos que a ativação de
PKA subsequente à depleção de cálcio é capaz de inibir RhoA na região basal das células,
possivelmente por fosforilação direta (Dong et al., 1998; Vega & Ridley, 2008), causando
perda das fibras de estresse, enquanto ativa Rho na região apical para recrutar ROCKI ou II e
regular a desmontagem da JAs e JTs, respectivamente. Isso poderia ser explicado como
consequência da formação de gradientes de atividade de PKA gerados através de uma
proteína AKAP específica em células com fenótipo migratório. Essa quinase apresentou alta
atividade restrita à região basal em células de CCR (Paulucci-holthauzen et al., 2009). Assim,
o resultado da Figura 29 confirma essa hipótese, uma vez que o tratamento com LPA, um
ativador de Rho, restaurou as fibras de estresse, mas não a organização da actina perijuncional
e a formação de contatos célula-célula.

115
Nós mostramos que o pré-tratamento com Y-27632 em células mantidas em meio LC
(Fig. 27) não reverteu a formação de lamelipódios, um indício de ativação de Rac na região
basal, e até acentuou a formação dessas estruturas. Esse resultado está de acordo com outros
mostrando que a ativação de Rho-ROCK no fronte de células migratórias de carcinoma de
mama inibe a formação de projeções de membrana induzidas por ativação de Rac (El Sibai et
al., 2008). Em se sabendo que: a) Rho e Rac atuam de forma oposta em células epiteliais, b)
ambas foram translocadas e, portanto, ativadas utilizando o meio LC, c) a depleção de cálcio
induziu a formação de lamelipódios, e d) A perda de contatos célula-célula pode ocorrer por
inibição de Rac (Nakagawa et al., 2001), é possível especular que Rac é ativada por PKA na
região basal para haver a formação de lamelipódios, e inibida na região apical, para ocasionar
desmontagem de JAs. No entanto, se essa ativação de Rac é dependente da inibição de Rho e
vice-versa, permanece por ser demonstrado. De qualquer maneira, o fato do H-89 prevenir a
reorganização de actina nas regiões apical e medial (Fig. 24), sugere que PKA induz uma
modulação diferencial da ativação de Rac. A existência de uma distribuição espacial de PKA
durante a migração celular quimiotática em fibroblastos e a ativação de Rac por PKA durante
a formação de pseudópodos (Howe et al., 2005) suportam essa possibilidade. Além disso,
embora Rac não apresente uma região diretamente fosforilada por PKA, sua ativação pode ser
ocasionada de forma indireta através de uma proteína GAP (O’Connor & Mercurio, 2001).
Prévios estudos mostraram que GTPases Rho ativas estão ligadas à membrana,
enquanto no estado inativo são localizadas predominantemente no citoplasma devido à
associação GTPase-GDI-GDP (Forget et al., 2002). Em adição, foi proposto que RhoGDI
poderia servir como uma proteína transportadora de GTPases ativas (DerMardirossian et al.,
2005) e um modelo sugere que RhoGDI pode se ligar em membranas por três mecanismos: a)
atração eletrostática, b) recrutamento do complexo Rho-RhoGDI por receptores, e c)
desestabilização do complexo Rho-RhoGDI por fatores de dissociação (GDFs) ou
fosforilação (Dransart et al., 2005). Aqui, observamos que o meio LC e a forskolina
induziram translocação de RhoGDI para a membrana de forma concomitante com RhoA e
Rac, indicando que a ativação de PKA poderia modular a associação de GTPases Rho com
RhoGDI para translocá-las para a membrana. Adicionalmente, vale ressaltar que os sítios de
ligação de RhoGDI e RhoGEF se sobrepõem, o que indica que o complexo Rho-RhoGDI na
membrana deve se dissociar antes de ocorrer ativação pela RhoGEF. Uma possível RhoGEF
candidata a mediar a ativação de RhoA após translocação do complexo Rho-RhoGDI para a
membrana é a GEF-H1, ativada por depleção de cálcio (Samarin et al., 2007). No entanto,
mais estudos são necessários para confirmar essa possibilidade.

116
Em resumo, esses resultados mostram a existência de uma inter-relação entre PKA e
GTPases da família Rho que medeiam a desmontagem do CJA e a desorganização do
citoesqueleto de actina em células Caco-2 depletadas de cálcio extracelular. Finalmente,
sugerimos um mecanismo novo envolvendo ativação espacial de uma cascata regulatória
consistindo de PKA, Rho/ROCKs, RhoGDI e Rac (Fig. 46) e nos leva a propor que um
mecanismo molecular similar pode regular passos iniciais da progressão tumoral em células
de câncer de cólon.
Figura 46: Modelo para a regulação da desmontagem do CJA e organização do citoesqueleto de actina em células depletadas de cálcio. A depleção do cálcio extracelular causa a desorganização de interações mediadas por E-caderina entre células vizinhas de forma direta, induzindo a perda da polaridade celular; e em paralelo PKA é ativada por um mecanismo upstream desconhecido, independente de E-caderina. A) PKA ativada poderia modular a desmontagem do CJA e causar ativação de Rho através de sua translocação para a membrana e subsequente recrutamento de proteínas efetoras. A desmontagem de JA poderia modular a sinalização Rho-ROCK (I ou II), e a desmontagem da JT o recrutamento de ROCKII. Na região basal, PKA inibe a sinalização RhoA-ROCK através do sequestro citoplasmático mediado por RhoGDI, para desorganizar as fibras de estresse. B) No domínio apical, a internalização da E-caderina poderia inibir Rac através do sequestro citoplasmático mediado por RhoGDI ou outro mecanismo desconhecido. No domínio basal, a ativação de PKA ou a inibição de Rho poderiam ativar Rac através da sua translocação para a membrana, induzindo a formação de lamelipódios.

117
V.2 PARTE II: Papel do LPA na aquisição do fenótipo migratório
Tem sido proposto que o LPA causa diferentes respostas celulares em vários modelos
de câncer, como aumento da proliferação celular, inibição da apoptose e indução da migração
celular (Mills & Moolenaar, 2003). No entanto, poucos estudos investigaram os efeitos do
LPA nos contatos célula-célula e na organização do citoesqueleto de actina, particularmente
na progressão do CCR. Então, decidimos verificar o papel desempenhado pelo LPA durante
as etapas de aquisição de características migratórias e invasivas em CCR. Nessa segunda parte
da Tese nosso objetivo foi explorar os mecanismos pelo qual o LPA promove perda dos
contatos célula-célula, desorganização de F-actina e, consequentemente, aumento da migração
celular. Nossos resultados mostram pela primeira vez que o LPA modula ambas as estruturas
para induzir um fenótipo migratório em células de CCR. Além disso, mostramos que uma
cascata regulatória envolvendo a inter-relação entre Rho-ROCK e Src-FAK pode
desempenhar uma importante função na regulação desses eventos.
Aqui, nós mostramos que o LPA provoca a redistribuição das proteínas das JAs E-
caderina, β-catenina e p120-catenina em células de CCR humanas Caco-2. Curiosamente, a E-
caderina também teve sua expressão reduzida após 24 h de tratamento com LPA. Um estudo
recente usando células de câncer de ovário tratadas com LPA mostrou que a ocorrência de
estruturas alongadas em contatos célula-célula, chamadas de hemi-junções (half-junctions),
podem representar uma fase de transição antes da perda de adesão célula-célula (Huang et al.,
2008). Nossos achados mostrando que o LPA induz o rompimento das JAs concordam com
esses resultados (Fig. 30 e 31), e a concomitante desorganização do citoesqueleto de actina na
região medial (Fig. 34) sugere que o LPA induz desmontagem das JAs por consequência da
contração de actina. Um estudo anterior, utilizando células de CCR HCT-116 analisou os
efeitos a longo prazo do LPA sobre proteínas juncionais e mostrou que este biolipídio
ocasiona acúmulo nuclear de β-catenina, o que promoveu a proliferação celular (Yang et al.,
2005). No presente estudo, utilizando células Caco-2, não observamos aumento da
proliferação celular (Fig. 41C), em concordância com resultados in vivo de Tatsuta et al.,
2005. De qualquer forma, estes resultados contraditórios podem ser explicados pelas
diferenças nas linhagens celulares utilizadas, uma vez que as células Caco-2 são bem
diferenciadas, exibem contatos célula-célula funcionais, e são menos invasivas que as células
HCT-116 (de Freitas Júnior et al., 2010). Na maioria dos carcinomas, a E-caderina se
comporta como supressora de migração, e a expressão ou função comprometida dessa
proteína em estágio tardio do câncer aumenta a transição para a metástase (Bellovin et al.,
2006). Além disso, a E-caderina é parcialmente regulada pelos seus parceiros citoplasmáticos

118
α-, β- e p120-catenina. α- e β-cateninas estão associados com o citoesqueleto de actina (Drees
et al., 2005), enquanto a p120-catenina modula a estabilidade de E-caderina na superfície da
célula (Davis et al., 2003). Assim, o fato do LPA induzir a redistribuição de β-catenina e
p120-catenina dos contatos célula-célula para o citosol poderia explicar a baixa expressão de
E-caderina (Fig. 31B) e o aumento da migração celular aqui observados (Fig. 41A e B). Nesse
contexto, foi mostrado que mutações no domínio extracelular de E-caderina aumentaram a
migração de células de ovário (Mateus et al., 2009), e que a redução da expressão de E-
caderina aumentou a proliferação, migração e invasão em células de câncer de próstata, de
forma dependente de β-catenina (Syed et al., 2008). Vale destacar que, embora muitos
estudos indiquem a inativação da via Wnt e subsequente desfosorilação na região N-terminal
e localização de β-catenina nuclear na promoção de um fenótipo tumoral, recentemente foi
mostrado que a β-catenina fosforilada também está localizada na borda principal de células
migratórias de CCR (Faux et al., 2010). Assim, seria interessante investigar se o LPA induz
fosforilação de β-catenina, e se esse evento estaria modulando a migração celular em nosso
modelo de estudo.
Poucos trabalhos avaliaram a modulação das JTs por LPA, no entanto, alguns estudos
que investigaram o papel de Rho na regulação das JTs utilizaram o LPA como ativador dessa
GTPase. Os resultados da literatura são muito controversos, pois tanto a ativação de Rho com
LPA ou sua forma constitutivamente ativa, assim como a inibição de Rho com toxina C3,
toxina A e sua forma dominante negativa aumentaram a permeabilidade paracelular (Nusrat et
al., 1995; Hopkins et al., 2003; Bruewer et al., 2004). Esses resultados indicam que a
regulação das JTs por Rho é complexa, e ainda que a resposta celular desencadeada em
resposta à ativação dessa GTPase deve depender da proteína efetora recrutada. Essa
controvérsia também reforça a ideia de que a ativação de Rho ocorre localmente, e que sua
desregulação por todo o volume celular gera resultados difíceis de interpretar. Nossos
resultados mostraram uma perda de funcionalidade das JTs em tempos iniciais de tratamento
com LPA (5-15 min) e de forma reversível (Fig. 32 e 33). Embora a ativação de Rho induzida
por LPA tenha ocorrido no tempo de 1 h, o inibidor de Rho toxina A preveniu esse efeito,
indicando que a ativação de Rho pode também ocorrer em tempos anteriores aos utilizados
por nós no ensaio de pull-down. Por exemplo, o LPA ocasionou ativação de Rho nos tempos
de 1 a 3 min de forma reversível ao longo do tempo em células de câncer pancreático (Stähle
et al., 2003). De acordo com o que observamos anteriormente utilizando o meio LC, a

119
inibição de ROCK não foi capaz de prevenir a redução da RET induzida por LPA (Fig. 40B),
reforçando a hipótese de que essa efetora modula preferencialmente as JAs.
Vale ressaltar que o efeito reversível do LPA sobre as JTs não parece ser uma
consequência da saturação dos receptores ou degradação desse biolipídio no meio de cultura
pois o efeitos do LPA sobre as JAs continuaram sendo observados nessas mesmas condições.
No entanto, é possível especular que ocorra um papel compensatório mediado por outra
proteína de JT não avaliada em nosso estudo. Em outras palavras, o aumento da
permeabilidade paracelular induzido por LPA seria revertido pelo aumento de função de outra
proteína. Uma possibilidade seria a tricelulina ou a MarvelD3, proteínas que têm sido
mostradas por exercerem papéis compensatórios na perda de função de JTs (Ikenouchi et al.,
2005; Raleigh et al., 2010). Além disso, foi mostrado que o LPA aumenta a expressão de
claudina-1 em células de câncer de ovário. Todavia, o aumento de expressão de claudina-1
nesse tipo de câncer aumenta seu potencial migratório (Murph et al., 2009). Se o LPA modula
a expressão das proteínas das JTs em CCR permanece desconhecido. Surpreendentemente,
encontramos a localização de ZO-1 na região perinuclear após tratarmos as células com LPA
por 48 h (Fig. 32). Esse é o primeiro relato da modulação de ZO-1 por LPA na literatura.
Conforme mencionado, outras proteínas podem translocar ao núcleo em reposta ao LPA para
modular a proliferação celular, como a β-catenina. Assim, seria interessante investigar se
tratamentos mais longos com LPA induziriam o acúmulo de ZO-1 no núcleo ou do fator de
transcrição que essa proteína regula, denominado ZONAB, de forma concomitante ao
aumento de proliferação, pois avaliamos esse evento apenas até 48 h. Nesse contexto, foi
mostrado que a superexpressão da Rho GEF-H1 ocasionou translocação de ZONAB para o
núcleo de células MDCK, de forma dependente da ativação de Rho mas não de ROCK (Nie et
al., 2009). Outro possível mecanismo pelo qual o LPA poderia modular o CJA seria através
da enzima Ciclooxigensase-2 (Cox-2). Foi mostrado que o LPA aumenta a expressão dessa
proteína em linhagens de epitélio ovariano e CCR (Symowicz et al., 2005; Shida et al., 2005),
e nosso grupo mostrou que o tratamento de células Caco-2 com prostaglandina E2 (PGE2),
um lipídio bioativo produzido por Cox-2, ocasiona desorganização do CJA (Tanaka et al.,
2008). Ainda, um estudo mostrou que a inibição ou o silenciamento de Cox-2 em diferentes
células epiteliais colônicas resultou em redução da atividade de RhoA, revertida pelo
tratamento com PGE2. Ainda, esse estudo também mostrou que a ativação de RhoA por Cox-
2 desorganizou as JAs e aumentou a motilidade celular (Chang et al., 2006).
Encontramos que a desmontagem das JAs induzida por LPA e a desorganização do
citoesqueleto de actina são dependentes de Src e Rho-ROCK (Figs. 36A e 38A). Além disso,

120
a proteína p120-catenina foi identificada como substrato para a quinase oncogênica Src. Um
estudo recente mostrou que o LPA aumenta a interação entre Src ativa e p120-catenina
levando a diminuição da interação entre α- e β-cateninas em células de câncer de ovário
(Huang et al., 2008). Nossos resultados mostraram que Src participa principalmente da
redistribuição de p120-catenina (Fig. 36A), mas se essa redistribuição de p120-catenina
mediada por Src em resposta o LPA também pode ocasionar a redução da expressão de E-
caderina observada na Fig. 31B permanece por ser determinado. É importante ressaltar que
um estudo recente mostrou que o LPA aumenta o crescimento de células de câncer de cólon e
a proteção de apoptose após três dias de tratamento, de forma dependente de MAPK e PI3K
(Sun et al., 2009). Considerando que PI3K e EGFR não foram capazes de reverter os efeitos
do LPA em nosso estudo, é possível que suas ativações sejam importantes em eventos tardios
que modulam o crescimento celular, enquanto a atividade Src atuaria como um sinal inicial
necessário para ocasionar desmontagem das JAs e desorganização do citoesqueleto de actina.
Em adição, observamos que, ao contrário do modelo de retirada do cálcio do meio
extracelular, a inibição de PKA não preveniu a desorganização do CJA em células tratadas
com LPA. De fato, PKA ativa é capaz de inibir Rho em diferentes modelos de estudo (Qiao et
al., 2008); no entanto e conforme descrito a seguir, a ativação de Rho pode ser ocasionada
diretamente por receptores de LPA, indicando a ativação sustentada dessa GTPase. Em
concordância, foi mostrado que o tratamento com forskolina, que ativa PKA, não reduziu os
níveis de Rho ativa em células de câncer de ovário tratadas com LPA (Nguyen et al., 2004).
Sabe-se que as GTPases Rho modulam a estrutura e função das JAs (Bruewer et al.,
2004; Samarin et al., 2007). No entanto, se por um lado a atividade de Rho é necessária para a
formação e manutenção de junções célula-célula, sua contínua atividade ou hiperativação
pode levar a desmontagem das JAs e a promoção da migração celular (Bruewer et al., 2004).
Aqui, nós mostramos que o LPA ativa RhoA (mas não Rac1) e Rho recruta ROCKII para os
contatos célula-célula (Fig. 34B), indicando que essa via poderia ocasionar a contração actina-
miosina que, por sua vez, levaria a desorganização das JAs (Ivanov, 2008). Sabe-se que os
GPCRs, tais como os de LPA, ativam diferentes vias de sinalização através de uma variedade
de subunidades α. Além disso, já foi demonstrado que a via de sinalização dependente de Rho
pode ser ativada através da família Gα12 e Gα13 (Bian et al., 2006). Curiosamente, tem sido
mostrado que, em células MDCK, a sinalização de Gα12 desorganiza o CJA via Src (Meyer
et al., 2003), e a sinalização de Gα13 desorganiza o CJA via Rho-ROCK (Donato et al.,
2009). Portanto, nossos resultados sugerem que o LPA pode ativar RhoA diretamente através

121
de GPCR ou através de Src para perturbar o CJA. Já foi mostrado que linhagens celulares
intestinais tumorais, incluindo a Caco-2, apresentam aumento de expressão de LPA2, e
redução de LPA1 e 3 (Shida et al., 2004). Além disso, sabe-se que os receptores LPA1 e
LPA2 interagem diretamente com uma GEF de Rho, levando a sua ativação (Yamada et al.,
2005). Ainda, MAGI-3, uma proteína de JAs, interage diretamente com LPA2 para ativar
RhoA em células de CCR (Zhang et al., 2007). Juntos, esses dados sugerem que o LPA2
poderia ativar Rho diretamente em nosso modelo de estudo. Assim, pode-se concluir que a via
de sinalização ativada em resposta ao LPA depende dos receptores expressos em cada tipo
celular bem como da subunidade α por eles recrutada. No entanto, não podemos descartar a
possibilidade da participação dos receptores de LPA não pertencentes à família EDG (LPA4-
6), que também podem ativar Rho (Valentine et al., 2008) e ainda não foram investigados em
células de CCR.
As principais etapas da migração celular são a perda de adesão célula-célula, a
montagem e desmontagem dinâmicas das AFs na região anterior da célula, e o concomitante
movimento e liberação da adesão na região posterior da célula (Mills & Moolenaar, 2003).
Uma vez que o ensaio de wound healing, aqui utilizado para analisar a migração celular
induzida por LPA, depende da remodelagem coordenada de adesões célula-célula e célula-
MEC, decidimos investigar se a mesma via de sinalização que modula a perda de contatos
célula-célula e a reorganização do citoesqueleto de actina também pode modular a formação
das AFs. Sabe-se que Src se localiza nas adesões periféricas com o substrato celular, onde
regula o turnover de AFs através da fosforilação da FAK (Webb et al., 2004; Schlaepfer et
al., 2004; Lee et al., 2010a). Nossos resultados mostram que Src participa dos efeitos do LPA
sobre a redistribuição de p120-catenina, formação das fibras de estresse e de AFs, ativação de
FAK e migração celular, e sugerem que a regulação recíproca entre os dois tipos de adesão
pode ocorrer durante a migração celular mediada por Src em células Caco-2. Além disso, o
fato de que: a) o aumento da atividade de Src em uma ampla variedade de tipos de tumor se
correlaciona com o estágio da doença (Summy & Gallick, 2003), b) linhagens celulares de
tumores com níveis elevados de Src são mais móveis e invasivos in vitro através de alterações
tanto nos contatos célula-célula e célula-substrato (Frame et al., 2004), e c) a superexpressão
de Src induz fenótipos de transformação característicos, incluindo o crescimento independente
de ancoragem e a malignidade in vivo (Oneyama et al., 2008), corroboram com o
envolvimento de Src na indução de migração induzida por LPA em nosso modelo.
Vale ressaltar que, o fato do LPA ter induzido a perda de JAs mas não de JTs e
aumento da migração celular não significa que a migração celular ocorra com JTs íntegras,

122
dadas as diferenças nas condições experimentais de cada ensaio. O ensaio de wound healing
por si só induz a migração celular já que a lesão da monocamada celular funciona como
quimioatraente. Para esclarecer a importância das proteínas das JTs e também das JAs no
processo de migração, seria necessário depletar as proteínas do CJA nessas células e verificar
se ainda assim ocorre aumento na migração induzido por LPA. Nesse sentido, foi relatado que
a redução da expressão de E-caderina, α−, β− ou p120-catenina aumentaram a migração de
células de câncer de próstata (Syed et al., 2008; Kümper & Ridely, 2010), e a redução da
expressão ou fosforilação de ocludina ocasionou defeitos na montagem de projeções de
membrana requeridas para a migração de células MDCK (Du et al., 2010).
FAK é uma tirosina quinase não-receptora localizada em sítios de adesão de contato
entre a célula e a MEC, sendo crucial para o movimento celular e retração da região posterior
de células migratórias. FAK é ativada em resposta ao engajamento de integrina, fatores de
crescimento e GPCRs, incluindo aqueles para LPA (Frame et al., 2004), e sua fosforilação
leva ao recrutamento e ativação de Src (Schlaepfer et al., 2004). Nossos resultados concordam
com estudos anteriores que mostraram que a estimulação com LPA induz a fosforilação e
ativação de FAK em diversos tipos celulares (Stähle et al., 2003; Bian et al., 2006; Jiang et
al., 2006). Tem sido sugerido que a FAK recruta proteínas que contribuem para a
remodelagem dinâmica de adesões através de uma cascata dependente de Src e Rho (Webb et
al., 2004). Nossos resultados mostrando que a) o LPA ativa Rho de forma dependente de Src
(Fig. 37B); b) o LPA induz a ativação de FAK através da fosforilação do resíduo de tirosina
específico para Src (Fig. 35B); e c) a atividade de Src, Rho e ROCK são necessárias para a
migração induzida por LPA (Fig. 41) suportam essa hipótese.
Além disso, mostramos que as longas projeções de membrana, cruciais para a
migração celular, não são formadas em monocamadas de células subconfluentes tratadas com
LPA contendo os inibidores de Src e ROCK (Figura 43). Curiosamente, AFs maduras foram
prevenidas pela inibição ROCK, mas não de Src. Isso é consistente com estudos anteriores
mostrando que células sem expressão de membros da família Src ainda são capazes de montar
AFs elongadas, mas com menor turnover (Webb et al., 2004; Lee et al., 2010a), e com um
estudo recente mostrando que as AFs maduras requerem a contração actina-miosina induzida
por ativação de ROCK (Pattla et al., 2010). Juntos, estes dados sugerem que a inibição de Src
e ROCK previnem a migração celular impedindo a dinâmica correta das AFs. No entanto, o
mecanismo pelo qual LPA ativa FAK in vivo permanece por ser elucidado. É importante
mencionar que altos níveis de FAK têm sido encontrados em uma variedade de tumores,
incluindo CCR (Ayaki et al., 2001) e a autofosforilação de FAK na tirosina 397 foi

123
encontrada aumentada nos adenomas intestinais em um modelo de camundongo (Weyant et
al., 2001). Também tem sido relatado que tumores de cólon com mutações na proteína p53
apresentam aumento na expressão de FAK (Golubovskaya et al., 2008), e que o LPA pode
suprimir a atividade de p53 permitindo a progressão do ciclo celular em fibroblastos, de forma
dependente da ativação de Rho via LPA2 (Kortlever et al., 2008). Devido a essas importantes
funções desempenhadas por FAK na progressão do câncer, essa quinase tem sido postulada
como um alvo terapêutico promissor nessa doença. Dentre os diferentes compostos químicos
testados contra FAK, o inibidor PF-562,271 foi o único que, além de ser eficiente em estudos
in vitro e modelos animais, se mostrou bem tolerado em testes clínicos, permanencedo ainda
em estudo (Hao et al., 2009).
Tem sido mostrado que a ativação de tirosinas quinases por GPCRs é necessária para a
estimulação eficiente da sinalização mediada por Rho (Sah et al., 2000). De fato, muitos
GPCRs são potentes ativadores de FAK, o que indica uma estreita relação entre ativação de
FAK e sinalização de Rho (Bian et al., 2006) e sugere que os receptores de LPA poderiam
ativar FAK. Em um estudo utilizando fibroblastos estimulados com LPA, a ativação de RhoA
levou a reorganização de F-actina acompanhada pela localização de Src em sítios de adesão
periféricos nas terminações das fibras de estresse (Timpson et al., 2001). Além disso, foi
mostrado que o recrutamento de actina para fibras de estresse não ocorre em células
deficientes em FAK ou que não são fosforiladas em tirosina 397 (Serrels et al., 2007). Da
mesma forma, mostramos que o LPA induz o recrutamento de FAK para as terminações das
fibras de estresse, mesmo na presença de adesões célula-célula (Fig. 35C), sugerindo que o
LPA recruta o complexo FAK-Src para as AFs permitindo que as células adquiram um
fenótipo migratório.
Alguns estudos sugerem que Src é uma molécula upstream a RhoA porque Src inibe
RhoA por uma via dependente de p190RhoGAP (Haskell et al., 2001). Src também fosforila
RhoGDI, diminuindo sua capacidade de ligar e inibir RhoA, Rac e Cdc42 (DerMardirossian et
al., 2006). Consistente com essa última possibilidade, e de acordo com resultados que
demonstram que RhoA está ativa na região anterior de células migratórias (Pertz et al., 2006),
nossos resultados indicam que Src ativa modula a ativação de RhoA, pois a inibição de Src
com PP2 diminuiu a ativação de Rho (Fig. 38B). Também é possível que Src fosforile ROCK
diretamente para reduzir a força de contração mediada pela via Rho-ROCK, o turnover de
AFs e a migração direcional, como mostrado recentemente (Lee et al., 2010a). No entanto, se
a ativação de Src ocorre diretamente por qualquer receptor específico de LPA, pela
internalização de E-caderina, ou diretamente por FAK permanece por ser determinado.

124
Alguns estudos têm mostrado que o LPA induz ativação de Rac e formação de
lamelipódios (Jung et al., 2004; Jourquin et al., 2006). Além disso, foi demonstrado que Src
fosforila Tiam, uma GEF que ativa Rac levando a sua degradação, desorganização das JAs e
migração de células epiteliais (Woodcock et al., 2009). Nós não observamos a formação de
lamelipódios em monocamadas confluentes de células Caco-2, mas observamos projeções de
membrana em células subconfluentes após tratamento com LPA por 1 h (Fig. 43), em
concordância com o aumento da migração de células tratadas com LPA. Também não
detectamos Rac ativa em monocamadas confluentes e subconluentes, indicando que a
atividade de Rac pode ser tipo celular específico. Além disso, embora não seja frequente, já
foi demonstrado que a formação de lamelipódio pode induzir a migração celular de maneira
independente de Rac (Spaargaren et al., 1999; Yip et al., 2007). Vale ressaltar que a inibição
de ROCK com Y-27632 pode induzir formação de lamelipódios via ativação de Rac, o que
ocorreu utilizando meio LC (Parte I, Fig. 27 e 28) e no fronte de células individuais
migratórias de carcinoma de mama (El Sibai et al., 2008), mas não em células tratadas com
LPA, indicando que esse agente modula a formação dessas estruturas por outro mecanismo.
Nesse contexto, foi mostrado que FAK interage diretamente com Arp2/3 em lamelipódios, e a
autofosforilação de FAK foi necessária para a formação dessas estruturas. Ainda, o tratamento
de fibroblastos com LPA aumentou os níveis de Arp2/3 nos lamelipódios de forma
dependente da ativação da via Rho-ROCK (Mingle et al., 2009). Nossos resultados mostrando
que o LPA ocasiona ativação de Rho e autofosforilação de FAK permitem sugerir a
possibilidade de o LPA ocasionar a formação de lamelipódios através da modulação de
Arp2/3. Ainda, esses dados também poderiam justificar a ausência de lamelipódios nas
células tratadas com LPA e o inibidor de ROCK. No entanto, seriam necessários mais
experimentos para esclarecer essa questão.
Juntos estes resultados mostram que o LPA modula a desmontagem de adesão célula-
célula, a remodelagem do citoesqueleto de actina e turnover de AFs, três eventos importantes
para a migração celular. Sugerimos um novo mecanismo envolvendo a ativação de Src no
compartimento celular apical (onde causa internalização de p120-catenina, diretamente ou
através da ativação da via Rho-ROCK para modular a desmontagem das JAs) e no
compartimento celular basal (onde a atividade de Src ocasiona a formação do complexo Src-
FAK para recrutar Rho-ROCK e induzir a formação fibras de estresse e AFs) (Fig. 47). Além
disso, propomos que um evento semelhante molecular pode regular os passos iniciais da
progressão do CCR.

125
Figura 47. Modelo para a indução do fenótipo migratório induzido por LPA. (A) O LPA, através dos receptors acoplados em protein G (GPCRs), induz desmontagem das JAs por um mecanismo dependente ou independente de Src. A ativação de Src pode induzir a redistribuição de p120-catenina e ativar a via Rho-ROCK. Alternativamente, o LPA pode ativar a via Rho-ROCK diretamente e induzir desmongtagem das JAs. (B) O LPA ativa GPCR para ativar o complex FAK-Src que, por sua vez, recruta Rho-ROCK para mediar a formação de fibras de estresse e adesões focais (FAs). PP2: inibidore de Src; toxina A: inibidor de Rho; Y27632: inibidor de ROCK. MEC, matriz extracelular; FAK, quinase de adesão focal.
Uma vez que: a) o LPA ocasionou ativação de FAK e Src (Fig 35A-B), e essas
quinases são ativadas em resposta à ativação de integrinas, o que tem sido relacionado ao
aumento da proliferação, sobrevivência celular e resitência à morte celular por anoikis
(Bouchard et al., 2007; Zouq et al., 2009), b) estudos prévios sugerem que proteínas das AFs,
na ausência de substrato, modulam o CIA (Bouchard et al., 2007), e c) nossos resultados
mostraram que o LPA ocasiona a formação de AFs em monocamadas confluentes (Fig. 35C)
e subconfluentes de forma dependente de ROCK (Fig. 43), decidimos verificar se esse
biolipídio também modula o crescimento de forma independente de ancoragem (CIA) de
células Caco-2.
Nós observamos que o LPA aumentou o número de colônias formadas (Fig. 44) e,
surprendentemente, a inibição de ROCK não preveniu esse efeito, indicando que o CIA
induzido por LPA é independente de ROCK. Vale destacar que, durante o crescimento

126
celular, o papel da via Rho-ROCK é controverso: Se por um lado a ativação de Rho é
associada com a sinalização pró-mitogênica, transformação celular e geração de tensão para
adesão celular ao substrato (Dohn et al., 2009), por outro, alguns estudos têm mostrado que a
tensão dependente de RhoA promove morte celular (Ma et al., 2007). Além disso, Dohn et al.
(2009) mostraram que a depleção de p120-catenina ocasiona ativação de RhoA e inibição de
CIA em células MDCK, efeito revertido pela inibição farmacológica de ROCK. Nesse estudo
foi mostrada uma complexa regulação do CIA por proteínas do CJA, onde a perda de
proteínas das JAs reduziu o CIA de forma estímulo dependente: a depleção de p120-catenina
suprimiu o CIA induzido pela ativação de Src e Rac mas não por Ras, e a depleção de E-
caderina suprimiu o CIA induzido pela ativação de Rac mas não por Src. Se por um lado
nossos resultados são contraditórios em relação à modulação do CIA por ROCK, por outro,
aparentemente concordam com essa última possibilidade, pois nós mostramos que a) o LPA
ocasionou ativação de Src, mas não Rac (Figs. 35A e 37A; b) o LPA aumentou o CIA (Fig.
44); e c) o LPA reduziu a expressão de E-caderina (Fig. 31B). Além disso, é possível que a
redistribuição das proteínas do CJA também poderia modular o CIA, já que a formação de
colônias requer adesão célula-célula. Assim, é possível especular que o inibidor de ROCK não
reduziu a formação de colônias porque aumentou os níveis das proteínas das JAs nos contatos
célula-célula, como observado nas Figuras 25 e 36A e como previamente relatado (Sahai &
Masrhall, 2002; Chang et al., 2006). Portanto, mais experimentos são necessários pra
esclarecer como a via Rho-ROCK modula as proteínas do CJA no CIA.
Embora o LPA tenha aumentado a migração celular, nossos resultados mostraram que
esse agente não aumentou a invasividade de células de Caco-2, ao contrário do ocorrido na
linhagem de câncer de ovário OVCAR-3. Permanece necessário esclarecer que outros fatores
seriam requeridos para que o LPA seja capaz de aumentar a invasividade de células Caco-2,
uma vez que ambas as linhagens são bem diferenciadas e apresentam contatos célula-célula
íntegros (Huang et al., 2008; de Freitas et al., 2010), características que podem atuar como
supressoras de invasão celular. Foi mostrado que o LPA ocasiona aumento de expressão e
atividade de MMP-2, levando ao aumento da invasão de células de ovário (Fishman et al.,
2001). Nossos resultados preliminares mostram que o LPA aumenta a expressão do RNAm
mas ocasiona somente um discreto aumento na atividade de MMP-2 em células Caco-2, o que
poderia justificar a falta de capacidade dessas células de invadirem o matrigel; no entanto,
seria necessário verificar se essas células são capazes de invadir colágeno IV, principal
componente de MEC de células epiteliais (Borghi et al., 2010). Nesse contexto, foi mostrado
que células de CCR invadiram colágeno I de forma dependente da ativação de ROCK em

127
resposta ao LPA (Sun et al., 2009). Ainda, é interessante notar que, embora o tratamento com
LPA tenha reduzido a expressão de E-caderina, não ocorreu transição epitélio-mesenquimal
uma vez que não foi observada a indução da expressão de vimentina (Fig. 31B). Isso sugere
que o LPA poderia induzir migração do tipo ameboide em nosso modelo de estudo, onde as
principais características são a ausência de atividade proteolítica de componentes da MEC,
ativação de Rho-ROCK e perda de contatos célula-célula (Parri & Chiarugi, 2010
Estudos recentes têm sugerido altos níveis de LPA (maiores que 1µM) no plasma ou
fluido de ascite como biomarcador para câncer de ovário (Erickson et al., 2001). Assim como
). Também é
possível especular que a ausência de atividade de MMP e de invasão celular em matrigel
poderiam ser consequências da ausência de vimentina em células Caco-2, uma vez que a
expressão dessa proteína modulou a invasão em células de câncer de próstata (Zhao et al.,
2008b) e a maturação de invadopódios em células invasivas de câncer de cólon HCT-116
(Schoumacher et al., 2010). De qualquer maneira, esses resultados sobre a invasividade
mediada pelo LPA em células de CCR ainda são preliminares e requerem mais experimentos
para obtermos conclusões mais contundentes.
V.3 Considerações finais: via Rho-ROCK, um possível alvo terapêutico em CCR?
Embora tenhamos utilizado somente dois estímulos que desencadeiam a perda de
contatos célula-célula, e existam muitos outros (por exemplo, éster de forbol, PGE2, radiação,
etc.), ambos os modelos apresentaram como evento comum a ativação da via Rho-ROCK. Na
primeira parte da Tese mostramos que a perda de contatos célula-célula induzida pela retirada
do cálcio do meio extracelular causa reorganização de F-actina e aumento da atividade das
GTPases Rho em células de adenocarcinoma de cólon humano. Usando essa mesma linhagem
celular, na segunda parte da Tese, observamos que o LPA induz a desorganização do CJA e
do citoesqueleto de actina, bem como aumento do potencial migratório de forma dependente
da ativação de Rho e ROCK. É interessante notar que vários estudos têm mostrado que a
perda de contato célula-célula induzida por diferentes estímulos requer reorganização de F-
actina (Bruewer et al., 2004, Samarin et al., 2007; Ivanov, 2008; Miyoshi & Takai, 2008).
Dessa forma, é cada vez mais aceita a hipótese de que a ativação de Rho-ROCK ocasione
contração actina-miosina para mediar a desmontagem do CJA (Ivanov et al., 2004a). No
entanto, não se pode descartar a possibilidade dessa GTPase regular o CJA diretamente,
conforme já mencionado. Ainda, a inibição de ROCK foi importante para a manutenção das
JAs nos dois modelos utilizados e também impediu a migração das células tratadas com LPA,
indicando um possível papel terapêutico para a via de sinalização Rho-ROCK.

128
em câncer de ovário, a presença de ascite reduz a sobrevivência de pacientes com CCR (Chu
et al., 1989). E ainda, foram relatados baixos níveis de lisofosfatidilcolina (LPC), um dos
precursores do LPA, com concomitante aumento dos níveis da enzima autaxina, que converte
LPC em LPA, no plasma de pacientes com CCR (Zhao et al., 2007), sugerindo elevados
níveis de LPA no plasma de indivíduos com esse tipo de neoplasia. No entanto, essas
pesquisas limitam-se a apontar o LPA como biomarcador e não como fator capaz de predizer
a terapia adequada a determinado paciente. Vale ressaltar que a administração de LPA ou a
utilização do modelo de retirada do cálcio do meio extracelular em camundongos com
mutação no gene APC ocasionaram ativação de Rho e redução da expressão de E-caderina
nos tumores de cólon desses animais (Carothers et al., 2006). Nesse mesmo modelo, foi
mostrado que o LPA aumentou a incidência de tumor via receptor LPA2 e ainda aumentou a
proliferação celular e os níveis de β-catenina nuclear (Lin et al., 2009). No entanto, esses
eventos ainda não foram avaliados em tumores de pacientes com CCR.
Embora estudos mostrem o aumento de expressão de Rho e suas proteínas reguladoras
em CCR (tabela 1), mutações no gene que codifica as GTPases Rho não foram encontradas, e
o mecanismo que modula o aumento de expressão dessas proteínas em câncer permanece
desconhecido. Esses estudos indicam um possível papel para LPA na modulação da expressão
e ativação de Rho que, por sua vez, poderia modular a expressão de proteínas do CJA e
desencadear perda de contatos célula-célula. Isso considerando que alterações na expressão e
localização de proteínas das JAs, por exemplo, a redução da expressão de E-caderina e a
localização de β-catenina no núcleo (Carothers et al., 2006; Lin et al., 2009), e das JTs, como
o aumento da expressão das claudinas-1, -3 e -4 (Oliveira et al., 2005), são comumente
observadas em CCR. ROCK é a principal proteína responsiva à ativação de Rho, nossos
resultados mostraram que sua inibição induziu efeitos antitumorais in vitro, como
reorganização dos contatos célula-célula e redução na migração celular, e experimentos in
vivo mostraram reduzida metástase em modelos animais de câncer hepático, de pulmão e
mama (Croft et al., 2004; Ying et al., 2006). Esses dados sugerem que a inibição
farmacológica de ROCK apresenta grande potencial como terapia de alguns tipos de câncer, o
que já vem sendo postulado (Olson, 2008), no entanto, a expressão dessa proteína em tumores
de pacientes com CCR ainda não foi investigada. Assim, seria relevante correlacionar os
níveis plasmáticos de LPA em pacientes com CCR com a expressão de seus receptores em
tumores e com a atividade das GTPases Rho e sua efetora ROCK e concomitante alterações
na expressão de proteínas do CJA. Se a inibição de ROCK poderia trazer benefícios a

129
pacientes com CCR com níveis aumentados de LPA no plasma ou fluido de ascite e/ou seus
receptores nos tumores, permanece por ser investigado.
O tratamento de CCR requer uma abordagem multidisciplinar cirúrgica, quimioterápica
e radioterápica. Até recentemente, o único tratamento quimiotarápico disponível para CCR era
a utilização de 5-fluorouracil (5-FU) sozinho ou combinado com oxaliplatina e irinotecan
(Waldner & Neurath, 2010). Embora novos alvos terapêuticos, como a inibição de VEGF e
EGFR utilizando bevacizumab e cetuximab respectivamente, tenham contribuído no
tratamento de CCR (Cohen & Hochster, 2008), apenas subgrupos de pacientes se beneficiam
dessa abordagem devido à resistência dos tumores a essas drogas (Waldner & Neurath, 2010).
Vale destacar que VEGF ativa Rac1 em células endoteliais e de câncer gástrico (Garret
Conforme mencionado, a via Rho-ROCK parece ser um alvo terapêutico promissor em
câncer. A inibição de ROCK com fasudil tem sido utilizada sem qualquer reação adversa grave
no tratamento de vasoespasmo no Japão, e muitos estudos clínicos de inibição de ROCK estão
et al.,
2007; Xue et al., 2004) e EGFR ativa Rac para promover migração em células de CCR (Dise et
al., 2008), indicando que a inibição de Rac também pode ser efetiva no tratamento de CCR.
Alguns estudos utilizando linhagens de CCR têm proposto o uso de inibidores de
enzimas farnesiltransferases (FTases) e geranilgeraniltransferases (GGTases) para bloquear a
adição de lipídio isoprenoide nas GTPases, inibindo assim sua ancoragem à membrana e
subsequente ativação. Contudo, inibidores de FTases individualmente não apresentaram
eficácia significativa em vários tipos de câncer, incluindo o CCR (Sebti & Adjei, 2004) e
inibidores de GGTases ocasionaram toxicidade (Lobell et al., 2001). Inibidores de ambas,
GGTases e FTases, foram desenvolvidos mas não apresentam eficácia satisfatória (Reid et al.,
2004). Outra estratégia é o uso de inibidores contra a enzima HMG-CoA redutase, também
conhecidos como estatinas, que reduzem os níveis de colesterol prevenindo a síntese de seus
precursores, farnesil pirofosfato e geranil geranil pirofosfato. Os inibidores dessa enzima
reduzem a isoprenilação de Ras e Rho, prevenindo a ancoragem delas na membrana necessária
para suas ativações. A inibição de HMG-CoA foi reportada por inibir o desenvolvimento de
CCR em camundongos (Cho et al., 2008), mas estudos clínicos ainda são necessários para
estabelecer sua eficácia como alvo terapêutico em CCR. Além disso, a inibição de RhoA em
células de CCR foi associada ao aumento da sensibilidade a doxorrubicina, uma droga
citotóxica intercalante de DNA, indicando que poderia ser utilizada para melhorar sua eficácia
nesse tipo de câncer (Doublier et al., 2008). E ainda, considerando que o LPA está envolvido
na ativação de Rho, a inibição de seus receptores também constitui uma interessante estratégia
terapêutica no controle da progressão do CCR (Lin et al., 2009).

130
em desenvolvimento, em sua maioria para doenças cardiovasculares (Olson, 2008). No
entanto, não estão sendo realizados estudos clínicos que avaliem se o fasudil pode ser utilizado
na terapia de CCR humano. Estudos in vitro mostraram que o fasudil reduziu a migração
celular e a formação de colônia em células de mama e fibrossarcoma, e in vivo, a disseminação
do tumor e metástase peritoneal, reduzindo a ascite em modelos animais de hepatoma (Ying et
al., 2006), indicando assim que essa droga poderia representar uma importante ferramenta no
tratamento de CCR. Embora mais estudos sejam necessários para entender as respostas
celulares mediadas por GTPases da família Rho, todos esses estudos apontam essas GTPases
como potenciais alvos terapêuticos em pacientes com CCR.

131
VI CONCLUSÕES
VI. 1 PARTE I: Análise da inter-relação entre o CJA e o citoesqueleto de actina durante
a perda da adesão célula-célula
- A retirada de cálcio do meio extracelular ocasionou desorganização e perda da
funcionalidade do CJA concomitante à desorganização do citoesqueleto de actina de forma
diferencial através da ativação de PKA;
- A perda de contatos célula-célula e concomitante reorganização de F-actina, induzidas pela
depleção do cálcio do meio de cultura, foram dependentes da via de sinalização Rho-ROCK;
- A retirada do cálcio do meio extracelular ocasionou ativação das proteínas RhoA e Rac1
como visto pela translocação dessas proteínas do citosol para a membrana, e esse evento
também foi modulado por PKA;
- A retirada de cálcio do meio extracelular ocasionou ativação local de Rho nos contatos
célula-célula, uma vez que a ativação dessa GTPase com LPA em meio depletado de cálcio
restaurou a formação das fibras de estresse na região basal mas não restabeleceu os contatos
intercelulares.
VI. 2 PARTE II: Papel do LPA na aquisição do fenótipo migratório
- O tratamento com LPA induziu a desmontagem das JAs, desorganização reversível das JTs e
reorganização do citoesqueleto de actina de forma dependente de Src e da via de sinalização
Rho-ROCK;
- O LPA induziu a formação de fibras de estresse e adesões focais, e causou aumento da
migração celular através da ativação de Src, e da via Rho-ROCK;
- O LPA não induziu transição epitélio-mesenquimal, visto que esse agente não ocasionou
morfologia celular alongada e pela ausência de expressão de vimentina;
- O LPA aumentou o crescimento independente de ancoragem de forma independente de
ROCK;
- O tratamento com LPA não induziu invasividade de células Caco-2, embora tenha
ocasionado aumento de expressão de MMP-2 e MMP-9.

132
VII REFERÊNCIAS
Abe, K. & Takeichi, M. (2008). EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl Acad. Sci. 105, 13-19.
Akhtar, N. & Hotchin, N.A. (2001). Rac1 regulates adherens junctions through endocytosis of E-cadherin. Mol Biol Cell. 12, 847-862.
Akisik, E., Bugra, D., Yamaner, S., & Dalay, N. (2010). Analysis of beta-catenin alterations in colon tumors: a novel exon 3 mutation. Tumor Biol. In Press.
Albiges-Rizo, C., Destaing, O., Fourcade, B., Planus, E., Block, M.R. (2009). Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci. 122, 3037-3049.
Aleshin, A. & Finn, R.S. (2010). SRC: A Century of Science Brought to the Clinic. Neoplasia. 12, 599-607.
Anastasiadis, P.Z. (2007). p120-ctn: A nexus for contextual signaling via Rho GTPases.
Anderson, S.C., Stone, C., Trach, L. & SundarRaj, N. (2002). Rho and Rho-kinase (ROCK) signaling in adherens and gap junction assembly in corneal epithelium. Investigative Ophthal Vis Sci. 43, 978-986.
Aoki, J., Inoue, A. & Okudaira, S. (2008). Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 1781, 513–518.
Assémat, E., Bazellières, E., Pallesi-Pocachard, E., Le Bivic, A., and Massey-Harroche, D. (2008). Polarity complex proteins. Biochim Biphys Acta. 1778, 614-630.
Avizienyte, E., Wyke, A.W., Jones, R.J., McLean, G.W., Westhoff, M.A., Brunton, V.G. & Frame, M.C. (2002). Src-induced de-regulation of e-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 4, 632-638.
Ayaki, M., Komatsu, K., Mukai, M., Murata, K., Kameyama, M., Ishiguro, S., Miyoshi, J., Tatsuta, M. & Nakamura, H. (2001). Reduced expression of focal adhesion kinase in liver metastases compared with matched primary human colorectal adenocarcinomas. Clin Cancer Res. 7, 3106-3112.
Basuroy, S., Sheth, P., Kuppuswamy, D., Balasubramanian, S., Ray, R.M., Rao, R.K. (2003). Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and
Baas, A.F., Kuipers, J., van der Wel, N.N., Batle, E., Koerten, H.K., Peters, P.J. & Clevers, H.C. (2004). Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 116, 457-466.
Balda, M.S. & Matter, K. (2008). Tight junctions at a glance. J Cell Sci. 121, 3677-82.
Balda, M.S., Garret, M.D. & Matter, K. (2003). The ZO-1 associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol. 160, 423-432.

133
accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. J Biol Chem. 278, 11916–11924.
Bates, R.C., Bellovin, D.I., Brown, C., Maynard, E., Wu, B., Kawakatsu, H., Sheppard, D., Oettgen, P. & Mercurio, A.M. (2005). Transcriptional activation of integrin β6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 115, 339-347.
Baum, B., Settleman, J. & Quinlan, M.P. (2008). Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 19, 294-308.
Bellovin, D.I., Simpson, K.J., Danilov, T., Maynard, E., Rimm, D.L., Oettgen, P. & Mercurio, A.M. (2006). Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 25, 6959-6967.
Benitah, S.A., Valerón, P.F., van Aelst, L., Marshall, C.J. & Lacal, J.C. (2004). Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta. 1705, 121-132.
Benjamin, J.M., & Nelson, W.J. (2008). Bench to Bedside and Back Again: Molecular Mechanisms of α- Catenin Function and Roles in Tumorigenesis. Semin Cancer Biol. 18, 53–64.
Bettinger, B.T., Gilbert, D.M. & Amberg, D.C. (2004). Actin up in nucleus. Nat Rev Mol Cell Biol. 5, 410-415.
Bian, D., Mahanivong, C., Yu, J., Frisch, S.M., Pan, Z.K., Ye, R.D. & Huang, S. (2006). The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene. 25, 2234-2244.
Borghi, N., Lowndes, M., Maruthamuthu, V., Gardel, M.L. & Nelson, J. (2010). Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proc Natl Acad Sci. 107, 13324-13329.
Bouchard, V., Demers, M.J., Thibodeau, S., Laquerre, V., Fujita, N., Tsuruo, T., Beaulieu, J.F., Gauthier, R., Vézina, A., Villeneuve, L., Vachon, P.H. (2007). Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. J Cell Physiol. 212, 717-28.
Boyer, L., Turchi, L., Desnues, B., Doye, A., Ponzio, G., Mege, J.L., Yamashita, M., Zhang, Y.E., Bertoglio, J., Flatau, G., Boquet, P. & Lemichez, E. (2006). CNF1-induced ubiquitylation and proteasome destruction of activated RhoA is impaired in smurf-/- cells. Mol Cel Biol. 17, 2489-2497.
Braga, V.M, Betson, M., Li, X. & Lamarche-Vane, N. (2000). Activation of the small GTPases Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Mol Biol Cell. 11, 3703-3721.
Braga, V.M., Machesky, L.M., Hall, A. & Hotchin, N.A. (1997). The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol. 137, 1421-1431.

134
Bremm, A., Walch, A., Fuchs, M., Mages, J., Duyster, J., Keller, G., Hermannstädter, C., Becker, F.F., Rauser, S., Langer, R., von Weyhern, C.H., Höfler, H. & Luber, B. (2008). Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res. 68, 707-14.
Bruewer, M. Utech, M., Ivanov, A.I. Hopkins, A.M., Parkos, C.A., Nusrat, A. (2005). Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 19, 923–933.
Bruewer, M., Hopkins, A.M., Hobert, M.E., Nusrat, A., Madara, J.L. (2004), RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am J Physiol Cell Physiol. 287, C327-C335.
Buchert, M., Papin, M., Bonnans, C., Darido, C., Raye, W.S., Garambois, V., Pélegrin, A., Bourgaux, J.F., Pannequin, J., Joubert, D. & Hollande, F. (2010). Symplekin promotes tumorigenicity by up-regulating claudin-2 expression. Proc Natl Acad Sci. 107, 2628–2633.
Carothers, A.M., Javid, S.H., Moran, A.E., Hunt, D.H., Redston, M., Bertagnolli, M.M. (2006). Deficient E-cadherin adhesion in C57BL/6j-Min/+ mice is associated with increased tyrosine kinase activity and RhoA-dependent actomyosin contractility. Exp Cell Res. 312, 387-400.
Carter, J.H., Douglass, L.E., Deddens, J.A., Colligan, B.M., Bhatt, T.R., Pemberton, J.O., Konicek, S., Hom, J., Marshall, M. & Graff, J.R. (2004). Pak-1 expression increases with progression of colorectal carcinomas to metastasis. Clin Cancer Res. 10, 3448-3456.
Chalhoub, N. & Baker, S.J. (2008). PTEN and the PI3-Kinase Pathway in Cancer. Annu Rev Pathol.
Chang, Y.E., Marlin, J.W., Chance, T.W. & Jakobi, R. (2006). RhoA mediates cyclooxygenase-2 signaling to disrupt the formation of adherens junctions and increase cell motility. Cancer Res. 66, 11700-11708.
4, 199-227.
Chan, C.H., Lee, S.W., Li, C.F., Wang, J., Yang, W.L., Wu, C.Y., Wu, J., Nakayama, K.I., Kang, H.Y., Huang, H.Y., Hung, M.C., Pandolfi, P.P. & Lin, H.K. (2010). Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 12, 457-469.
Charras, G. & Paluch, E. (2008). Blebs lead the way: how to migarte without lamellipodia. Nat Rev Mol Cel Biol. 9, 730-736.
Chen, X. & Macara, I.G. (2005) Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 7, 262–269.
Chiba, H., Osanai, M., Murata, M., Kojima, T., Sawada, N. (2008). Transmembrane proteins of tight junctions. Biochim Biphys Acta. 1778, 588-600.
Cho, S.J., Kim, J.S., Kim, J.M., Lee, J.Y., Jung, H.C. & Son, I.S. (2008). Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer. 123, 951-957.

135
Chrzanowska-Wodnicka, M. & Burridge, K. (1996) Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 133, 1403-1415.
Chu, D.J., Lang, N.P.,Thompson, C., Osteen, P.K. & Westbrook, K.C. (1989) Peritoneal Carcinomatosis in Nongynecolagic Malignancy A Prospective Study of Prognostic Factors. Cancer. 63, 364-367.
Cohen, D.J. & Hochster, H.S. (2008). Rationale for Combining Biotherapy in the Treatment of Advanced Colon Cancer. Gastrointest Cancer Res.
Croft, D.R., Sahai, E., Mavria, G., Li, S., Tsai, J., Lee, W.M., Marshall, C.J. & Olson, M.F. (2004).
2, 145-151.
Coluccia, A.M., Benati, D., Dekhil, H., De Filippo, A., Lan, C., Gambacorti-Passerini, C. (2006). SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 66, 2279-2286.
Conditional ROCK activation in vivo induces tumor cell dissemination and angiogenesis. Cancer Res. 64, 8994-9001.
Darido, C., Buchert, M., Pannequin, J., Bastide, P., Zalzali, H., Mantamadiotis, T., Bourgaux, J.F., Garambois, V. & Jay, P. (2008). Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells.Cancer Res. 68, 4258-4268.
Davies, S.P., Reddy, H., Caivano, M. & Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 351, 95-105.
Davis, M.A., Ireton, R.C. & Reynolds, A.B. (2003). A core function for p120-catenin in cadherin turnover. J Cell Biol. 163, 525-34.
de Araújo WM, Vidal FC, de Souza WF, de Freitas JC Jr, de Souza W, Morgado-Diaz JA. (2010). PI3K/Akt and GSK-3β prevents in a differential fashion the malignant phenotype of colorectal cancer cells. J Cancer Res Clin Oncol.136, 1773-1782.
de Freitas Junior, J.C.M., D´aguilar Silva B.D.R., de Souza, W.F., de Araújo W.M., Abdelhay, E.S.F.W. & Morgado-Díaz, J.A. (2010). Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell-cell adhesion and inhibitrs cell proliferation in undifferentiated human colon cancer cells. Cancer chemother Pharmacol. In Press.
Deng, W., Balazs, L., Wang, D.A., Middlesworth, L.V., Tigyl, G. & Johnson, L.R. (2002). Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 123, 206-216.
DerMardirossian, C., Rocklin, G., Seo, J.Y. & Bokoch, G.M. (2006). Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol Biol Cel. 17, 4760-4768.
Disanza, A., Steffen, A., Hertzog, M., Frittoli, E., Rottner, K. & Scita, G. (2005). Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cel Mol Life Sci. 62, 955-970.

136
Dise, R.S., Frey, M.R., Whitehead, R.H. & Polk, D.B. (2008). Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 294, G276-285.
Dohn, M.R., Brown, M.V. & Reynolds, A.B. (2009). An essential role for p120-catenin in Src-and Rac1-mediated anchorage-independent cell growth. J Cell Biol. 184, 437-450.
Donato, R., Wood, S.A., Saunders, I., Gundsambuu, B., Yan, Mak. K., Abbott, C.A., Powell, B.C. (2009). Regulation of epithelial apical junctions and barrier function by Galpha13. Biochim Biophys Acta. 1793, 1228-1235.
Dong, J.M., Leung, T., Manser, E. & Lim, L. (1998). cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 273, 22554-22562.
Dos Remedios, C.G., Chhabra, D., Kekic, M., Dedova, I.V., Tsubakihara, M., Berry, D.A & Nosworthy, N.J. (2003). Actin Binding Proteins: Regulation of cytoskeletal microfilaments. Physiol Rev. 83, 433-473.
Doublier, S., Riganti, C., Voena, C., Costamagna, C., Aldieri, E., Pescarmona, G., Ghigo, D. & Bosia, A. (2008). RhoA silencing reverts the resistance to doxorubicin in human colon cancer cells. Mol Cancer Res. 6, 1607-1620.
Dow, L.E., Kauffman, J.S., Caddy, J., Zarbalis, K., Peterson, A.S., Jane, S.M., Russell, S.M. & Humbert, P.O. (2007). The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 26, 2272–2282.
Dransart, E., Olofsson, B. & Cherfils, J. (2005). RhoGDIs revisited: Novel roles in Rho regulation. Traffic. 6, 957–966.
Drees, F., Pokutta, S., Yamada, S., Nelson, W.J. & Weis, W.I. (2005). α-catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell. 123, 903-915.
Du, D., Xu, F., Yu, L., Zhang, C., Lu, X., Yuan, H., Huang, Q., Zhang, F., Bao, H., Jia, L., Wu, X., Zhu, X., Zhang, X., Zhang, Z., Chen, Z. (2010). The Tight Junction Protein, Occludin, Regulates the Directional Migration of Epithelial Cells. Develop Cell. 18, 52–63
El Sibai, M., Pertz, O., Pang, H., Yip, S.C., Lorenz, M., Symons, M., Condeelis, J.S., Hahn, K.M. & Backer, J.M. (2008). RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp Cell Res. 314, 1540-1552.
El-Bahrawy M, Talbot I, Poulsom R, Alison M. (2002). Variable nuclear localization of alpha-catenin in colorectal carcinoma. Lab Invest. 82, 1167–74.
Erickson, A., Hasegawa, Y., Fang, X., Eder, A., Mao, M., Furui, T., Aoki, J., Morris, A. & Mills, G.B. (2001). Lysophosphatidic acid and ovarian cancer: a paradigm for tumourigenesis and patient management. Prostaglandins. 64, 63-81.

137
Espejo, R., Rengifo-Cam, W., Schaller, M.D., Evers, B.M., Sastry, S.K. (2010). PTP-PEST controls motility, adherens junction assembly, and Rho GTPase activity in colon cancer cells. Am J Physiol Cell Physiol. 299, C454-63.
Espina, C., Céspedes, M.V., García-Cabezas, M.A., Gomes Del Pulgar, M.T., Boluda, A., Oroz, L.G., Cejas, P., Nistal, M., Mangues, R., Lacal, C. (2008). A critical role for Rac1 in tumor progression of human colorectal adenocarcinoma cells. Am J Pathol. 172, 156-166.
Fan, L., Sebe, A., Péterfi, Z., Masszi, A., Thirone, A.C., Rotstein, O.D., Nakano, H., McCulloch, C.A., Szászi, K., Mucsi, I. & Kapus, A. (2007). Cell contact-dependent regulation of epithelialmyofibroblast transition via the Rho-Rho kinase-phospho-myosin pathway. Mol Biol Cell. 18, 1083–1097.
Faux, M.C., Coates, J.L., Kershaw, N.J., Layton, M.J., Burgess, A.W. (2010). Independent interactions of phosphorylated β-catenin with E-cadherin at cell-cell contacts and APC at cell protrusions. PLoS One. 5, e14127.
Fishman, D.A., Liu, Y., Ellerbroek, S.M. & Stack, M.S. (2001). Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 61, 3194-3199.
Flier, L.G. van der & Clevers, H. (2009). Stem Cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 71, 5.1-5.20.
Fodde, R., Smits, R. & Clevers, H. (2001). APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 1, 55-67.
Forget, M.A., Desrosiers, R.R., Gingras, D. & Béliveau, R. (2002). Phosphorylation states of cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 361, 243-254.
Frame, M.C. (2004). Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 117, 989-998.
Fujita, Y., Krause, G., Scheffner, M., Zechner, D., Leddy, H.E.M., Behrens, J., Sommer, T. & Birchmeier, W. (2002). Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 4, 222–231.
Funakoshi, S., Kong, J., Crissey, M.A., Dang, L., Dang, D., Lynch, J.P. (2010). Intestine-specific transcription factor Cdx2 induces E-cadherin function by enhancing the trafficking of E-cadherin to the cell membrane. Am J Physiol Gastrointest Liver Physiol. 299, G1054-G1067.
Furuse, M., Hata, M., Furuse, K., Yoshida, Y., Haratake, A., Sugitani, Y., Noda, T., Kubo, A. & Tsukita, S. (2002). Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 156, 1099-1111.
Garret, T.A., Van, Buul. J.D. & Burridge, K. (2007). VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp Cell Res. 313, 3285-3297.

138
Genth, H., Aktories, K. & Just, I. (1999). Monoglycosylation of RhoA at threonine 37 block cytosol-membrane cycling. J Biol Chem. 274, 29050–29056.
Georgeou, M., Marinari, E., Burden, J. & Baum, B. (2008). Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr Biol. 18, 1631-1638.
Gertler, F.B., Niebuhr, K., Reinhard, M., Wehland, J. & Soriano, P. (1996). Mena, a relative of Vasp and Drosophila enabled, is implicated in the control of microfilaments dynamics. Cell. 87, 227-239.
Gimond, C. van Der Flier, A., van Delft, S., Brakebusch, C., Kuikman, I., Collard, J.G., Fässler, R. & Sonnenberg, A. (1999). Induction of cell scattering by expression of β1 integrins in β1-deficient epithelial cells requires activation of members of the rho family of GTPases and downregulation of cadherin and catenin function. J Cell Biol. 147, 1325–1340.
Goldstein, B. & Macara, I.G. (2007). The PAR Proteins: Fundamental Players in Animal Cell Polarization. Develop Cell. 13, 609-622.
Golubovskaya,V.M., Finch, R., Kweh, F., Massoll, N.A., Campbell-Thompson,M., Wallace, M.R. & Cance, W.G. (2008). p53 Regulates FAK Expression in Human Tumor Cells Mol carcinogenesis. 47, 373–382.
Gomes Del Pulgar, T., Valdés-Mora, F., Bandrés, E., Pérez-Palacios, R., Espina, C., Cejas, P., García-Cabezas, M.A., Nistal, M., Casado, E., González-Barón, M., García-Foncillas, J. & Lacal, J.C. (2008). Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int J Oncol. 33, 185-193.
Gong, W., An, Z., Wang, Y., Pan, X., Fang, W., Jiang, B. & Zhang, H. (2009). P21-activated kinase 5 is overexpressed during colorectal cancer progression and regulates colorectal carcinoma cell adhesion and migration. Int J Cancer. 125, 548-55.
Gonzalez-Mariscal, L., Contreras, R.G., Bolívar, J.J., Ponce, A., De Ramirez, B.C & Cereijido, M. (1990). Role of calcium in tight junction formation between epithelial cells. Am J Physiol. 259, C978-C986.
Hall, A. (1998). Rho GTPases and the actin cytoskeleton. Science. 279, 509-514.
Hao, H., Naomoto, Y., Bao, X., Watanabe, N., Sakurama, K., Noma, K., Motoki, T., Tomono, Y., Fukazawa, T., Shirakawa, Y., Yamatsuji, T., Matsuoka, J., Wang, Z.G. & Takaoka, M. (2009). Focal adhesion kinase as potential target for cancer therapy. Oncology Rep. 22, 973-979.
Harper K., Arsenault, D., Boulay-Jean, S., Lauzier, A., Lucien, F & Dubois, C.M. (2010). Autotaxin Promotes Cancer Invasion via the Lysophosphatidic Acid Receptor 4: Participation of the Cyclic AMP/EPAC/Rac1 Signaling Pathway in Invadopodia Formation. Cancer Res. 70, 4634-4643.
Harris, T.J.C. & Tepass, U. (2010). Adherens junctions: from molecules to morphogenesis. Nat Rev. 11, 502-514.

139
Hartsock, A. & Nelson, W.J. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biphys Acta. 1778, 660- 669.
Haskell, M.D., Nickles, A.L., Agati, J.M., Su, L., Dukes, B.D. & Parsons, S.J. (2001). Phosphorylation of p190 on Tyr1105 by c-Src is necessary but not sufficient for EGF-induced actin disassembly in C3H10T1/2 fibroblasts. Cell Sci. 114, 1699-1708.
Hayashi, H., Nabeshima, K., Aoki, M., Hamasaki, M., Enatsu, S., Yamauchi, Y., Yamashita, Y. & Iwasaki, H. (2010). Overexpression of IQGAP1 in advanced colorectal cancer correlates with poor prognosis-critical role in tumor invasion. Int J Cancer. 126, 2563-2574.
Hayot, C., Debeir, O., Vam Ham, P., Vam Damme, M., Kiss, R. & Decaestecker, C. (2006). Characterization of the activities of actin-affecting drugs on tumor cell migration. Toxicol Appl Pharmacol. 211, 30-40.
Hegerfeldt, Y., Tusch, M., Brocker, E. B. & Friedl, P. (2002). Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, 1 -integrin function and migration strategies. Cancer Res. 62, 2125-2130.
Herrmann, H., Bär, H., Kreplak, L., Strelkov, S.V. & Aebi, U. (2007). Intermediate filaments: from cell architecture to nanomechanics. Nat Rev. 8, 562-573.
Hirakawa, M., Karashima, Y., Watanabe, M., Kimura, C., Ito, Y. & Oike, M. (2007). Protein kinase A inhibits lysophosphatidic acid-induced migration of airway smooth muscle cells. J Pharmacol Exp Ther. 321, 1102–1108.
Hopkins, A.M., Walsh, S.V., Verkade, P., Boquet, P & Nusrat, A. (2003). Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci. 116, 725-742.
Howe, A.K., Baldor, L.C. & Hogan, B.P. (2005). Spatial regulation of the cAMPdependent protein kinase during chemotactic cell migration. Proc Natl Acad Sci. 102, 4320-14325.
Hu, D., Fang, W., Han, A., Gallagher, L., Davis, R.J., Xiong, B. & Yang, W. (2008). c-Jun N-terminal kinase 1 interacts with and negatively regulates Wnt/beta-catenin signaling through GSK3beta pathway. Carcinogenesis. 29, 2317-2324.
Huang, R.Y.J., Wang, S.M., Hsieh, C.Y. & Wu, J.C. (2008). Lysophosphatidic acid induces ovarian cancer cell dispersal by activating Fyn kinase associated with p120-catenin. Int J Cancer. 123, 801-809.
Huerta, M., Muñoz, R., Tapia, R., Soto-Reyes, E., Ramírez, L., Recillas-Targa, F., González-Mariscal, L., López-Bayghen, E. (2007). Cyclin D1 is transcriptionally down-regulated by ZO-2 via an E Box and the transcription factor c-Myc. Mol Biol Cell. 18, 4826-36.
Humbert, P.O., Dow, L.E. & Russell, S.M. (2006). The Scribble and Par complexes in polarity and cell migration: friends or foes? Trends Cel Biol. 16, 622-630.
Iden, S. & Collard, J.G. (2008). Crosstalk between small GTPases and polarity proteins in cell polarization. Nature. 9, 846-859.

140
Ikenouchi, J., Furuse, M., Furuse, K., Sasaki, H., Tsukita, S. (2005). Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 171, 939–945.
Ikenouchi, J., Matsuda, M., Furuse, M. & Tsukita, S. (2003). Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by snail. J Cell Sci. 116, 1959-1967.
Instituto Nacional de Câncer. (2009). Estimativa 2010: incidência de câncer no Brasil. Ministério da Saúde. Instituto Nacional de Câncer, INCA, Rio de Janeiro.
Ivanov, A.I, McCall, I.C., Parkos, C.A. & Nusrat, A. (2004a). Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Molecular Biology of the Cell. 15, 2639-51.
Ivanov, A.I. (2008). Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci. 13, 6662-6681.
Ivanov, A.I., Nusrat, A. & Parkos, C.A. (2004b). Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 15, 176-188.
Iwanicki, M.P., Vomastek, T., Tilghman, R.W., Martin, K.H., Banerjee, J., Wedegaertner, P.B. & Parsons, J.T. (2008). FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J Cell Sci. 121, 895-905.
Iwaya, K., Oikawa, K., Semba, S., Tsuchiya, B., Mukai, Y., Otsubo, T., Nagao, T., Izumi, M., Kuroda, M., Domoto, H. & Mukai, K. (2007). Correlation between liver metastasis of the colocalization of actin-related protein 2 and 3 complex and WAVE2 in colorectal carcinoma. Cancer Sci. 98, 992-999.
Jiang, X., Jacamo, R., Zhukova, E., Sinnett-Smith, J. & Rozengurt, E. (2006). RNA interference reveals a differential role of FAK and Pyk2 in cell migration, leading edge formation and increase in focal adhesions induced by LPA in intestinal epithelial cells. J Cell Physiol. 207, 816-828.
Johnson, G.L. & Nakamura, K. (2007). The c-Jun Kinase/Stress-activated Pathway: Regulation, Function and Role in Human Disease. Biochim Biophys Acta. 1773, 1341–48.
Johnstone, C.N., Castellví-Bel, S., Chang, L.M., Bessa, X., Nakagawa, H., Harada, H., Sung, R.K., Piqué, J.M, Castells, A. & Rustgi, A.K. (2004). ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/ CDC42GAP/ p50RHOGAP: mutation and expression analyses in colorectal and breast cancers. Gene. 336, 59-71.
Joshi, B., Strugnell, S.S., Goetz, J.G., Kojic, L.D., Cox, M.E., Griffith, O.L., Chan, S.K., Jones, S.J., Leung, S.P., Masoudi, H., Leung, S., Wiseman, S.M. & Nabi, I.R. (2008). Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res. 68, 8210- 8220.
Jourquin, J., Yang, N., Kam, Y., Guess, C. & Quaranta, V. (2006). Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA). J Cell Physiol. 206, 337-346.

141
Joyce, T., Cantarella, D., Isella, C., Medico, E. & Pintzas, A. (2009). A molecular signature for Epithelial to Mesenchymal transition in a human colon cancer cell system is revealed by large-scale microarray analysis. Clin Exp Metastasis. 26, 569-87.
Jung, I.D., Lee, J., Lee, K.B., Park, C.G., Kim, Y.K., Seo, D.W., Park, D., Lee, H.W., Han, J.W.& Lee, H.Y. (2004). Activation of p21-activated kinase 1 is required for lysophosphatidic acid-induced focal adhesion kinase phosphorylation and cell motility in human melanoma A2058 cells. Eur J Biochem. 271, 1557-1565.
Kale, G., Naren, P., Sheth, P. & Rao, R.K. (2003). Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2 and ZO-3. Biochem Biophys Res Comm. 302, 324-329.
Kalluri, R. (2009). EMT: When epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 119, 1417–1419.
Kam, Y. & Quaranta, V. (2009). Cadherin-Bound β-Catenin Feeds into the Wnt Pathway upon Adherens Junctions Dissociation: Evidence for an Intersection between β-Catenin Pools. PLoS One. 4, e4580.
Kanaan, Z., Qadan, M., Eichenberger, M.R. & Galandiuk, S. (2010). The actin-cytoskeleton pathway and its potential role in inflammatory bowel disease-associated human colorectal cancer. Genet Test Mol Biomarkers. 14, 347-353.
Katoh, M. (2005). WNT/PCP signaling pathway and human cancer. Oncol Rep. 14(6), 1583-1588.
Kelley, L.C., Ammer, A.G., Hayes, K.E., Martin, K.H., Machida, K., Jia, L., Mayer, B.J., Weed, S.A. (2010). Oncogenic Src requires a wild-type counterpart to regulate invadopodia maturation. J Cell Sci. 123, 3923-32.
Kim, T.Y., Vigil, D., Der, C.J. & Juliano, R.L. (2009). Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer metastasis Rev. 28, 77-83.
Kimura, K., Ito, M., Amano, M., Chihara, K., Fukata, Y., Nakafuku, M., Yamamori, B., Feng, J., Nakano, T., Okawa, K., Iwamatsu, A. & Kaibuchi, K. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 273, 245-248.
Kirklank, SC. (2009). Type I collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br J Cancer. 101, 320-326.
Klingler, C., Kniesel, U., Bamforth, S.D., Wolburg, H., Engelhardt, B. & risau, W. (2000). Disruption of epithelial tight junctions is prevented by cyclic nucleotide-dependent protein kinase inhibitors. Histochem Cell Biol. 113, 349-361.
Kolch, W., Kotwaliwale, A., Vass, K. & Janosch, P. (2002). The role of Raf kinases in malignant transformation. Expert Rev Mol Med
Kortlever, R.M, Brummelkamp, T.R., van Meeteren, L.A., Moolenaar, W.H. & Bernards, R. (2008). Suppression of the p53-Dependent Replicative Senescence Response by Lysophosphatidic Acid Signaling Mol Cancer Res. 6, 1452-1460.
. 4, 1-18.

142
Krug, S.M., Amasheh, S., Richter, J.F., Milatz, S., Günzel, D., Westphal, J.K., Huber, O., Schulzke, J.D. & Fromm, M. (2009). Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell. 20, 3713–3724.
Kümper S & Ridley AJ. (2010). p120ctn and P-cadherin but not E-cadherin regulate cell motility and invasion of DU145 prostate cancer cells. PLoS One. 5, e11801.
Lawrence, D.W., Comeford, K.M & Colgan, S.P. (2002). Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Cell Physiol. 282, C1235-45.
Lobell, R.B., Omer, C.A., Abrams, M.T., Bhimnathwala, H.G., Brucker, M.J., Buser, et al. (2001). Evaluation of Farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61, 8758-8768.
Lee, H.H., Tien, S.C., Jou, T.S., Chang, Y.C., Jhong, J.G., Chang, Z.F. (2010). Src-dependent phosphorylation of Rock participates of focal adhesion dynamics. J Cell Sci. 123, 3368-3377.
Lee, S.J., Ritter, S.L., Zhang, H., Shim, H., Hall, R.A. & Yun, C.C. (2010). MAGI-3 competes with NHERF-2 to negatively regulate LPA2 receptor signaling in colon cancer cells. Gastroenterol. In Press.
Lei, K., Ye, L., Yang, Y., Wang, G.J., Jiang, Q.Y., Jiang, Y., Wei, Y.Q., Deng, H.X. (2010). RNA interference-mediated silencing of focal adhesion kinase inhibits growth of human colon carcinoma xenograft in nude mice. J Biomed Nanotechnol. 6, 272-278.
Lim, C.J., Kain, K.H., Tkachenko, E., Goldfinger, L.E., Gutierrez, E., Allen, M.D., Groisman, A., Zhang, J. & Ginsberg, M.H. (2008). Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol Biol Cell. 19, 4930-4941.
Lin, S., Wang, D., Iyer, S., Ghaleb, A.M., Shim, H., Yang, V.W., Chun, J. & Yun, C.C. (2009). The absence of LPA2 attenuates tumour formation in an experimental model of colitis-associated cancer. Gastroenterol. 136, 1711-1720.
Linder, S. (2007). The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trend in Cell Biol. 17, 107-117.
Litwin, M., Mazur, A.J., Nowak, D., Mannherz, H.G. & Malicka-Błaszkiewicz, M. (2009). Gelsolin in human colon adenocarcinoma cells with different metastatic potential. Acta Biochim Pol. 56, 739-743.
Liu, L., Wu, D.H. & Ding, Y.Q. (2005). Tiam1 gene expression and its significance in colorectal carcinoma. World J Gastroenterol. 11, 705-707.
Liu, Y., Hong, Y., Zhao, Y., Ismail, T.M., Wong, Y., Eu, K.W. (2008). Histone H3 (lys-9) deacetylation is associated with transcriptional silencing of E-cadherin in colorectal cancer cell lines. Cancer Invest. 26, 575-82.
Liu, Y., Xiao, H., Tian, Y., Nekrasova, T., Hao, X., Lee, H.J., Suh, N., Yang, C.S. & Minden, A. (2008). The pak4 protein kinase plays a key role in cell survival and tumorigenesis in athymic mice. Mol Cancer Res. 6, 1215-1224.

143
Lozano, E., Frasa, M.A., Smolarczyk, K., Knaus, U.G. & Braga, V.M. (2008). PAK is required for the disruption of E-cadherin adhesion by the small GTPase Rac. J Cell Sci
Machesky LM & Li A. (2010). Fascin: Invasive filopodia promoting metastasis.
121, 933-938.
Ma, Z., Myers, D.P., Wu, R.F., Nwariaku, F.E. & Terada, L.S. (2007). P66Shc mediates anoikis through RhoA. J Cell Bio. 179, 23-31.
Commun Integr Biol. 3, 263-70.
MacPherson, M.R., Molina, P., Souchelnytskyi, S., Wernstedt, C., Martin-Pe´rez, J., Portillo, F. & Cano, A. (2010). Phosphorylation of Serine 11 and Serine 92 as New Positive Regulators of Human Snail1 Function: Potential Involvement of Casein Kinase-2 and the cAMP-activated Kinase Protein Kinase A. Mol Biol Cell. 21, 244–253.
Maeda, S. & Tsukihara, T. (2010). Structure of the gap junction channel and its implications for its biological functions. Cell. Mol. Life Sci. In press.
Maher, M.T., Flozak, A.S., Stocker, A.M., Chenn, A., Gottardi, C.J. (2009). Activity of the beta-catenin phosphodestruction complex at cell-cell contacts is enhanced by cadherin-based adhesion. J Cell Biol. 186, 219-28.
Malliri, A., Rygiel, T.P., van der Kammen, R.A., Song, J.Y., Engers, R., Hurlstone, A.F., Clevers, H.& Collard, J.G. (2006). The rac activator Tiam1 is a Wnt-responsive gene that modifies intestinal tumor development. J Biol Chem. 281, 543-548.
Mandell, K.J., Babbin, B.A., Nusrat, A. & Parkos, C.A. (2005). Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J Biol Chem. 280, 11665-11674.
Mateus, A.R., Simões-Correia, J., Figueiredo, J., Heindl, S., Alves, C.C., Suriano, G., Luber, B. & Seruca, R. (2009). E-cadherin mutations and cell motility: a genotype-phenotype correlation. Exp Cell Res. 315, 1393-1402.
Matos, P., Collard, J.G. & Jordan, P. (2003). Tumor-related Alternatively Spliced Rac1b Is Not Regulated by Rho-GDP Dissociation Inhibitors and Exhibits Selective Downstream Signaling. J Biol Chem. 278, 50442–50448.
Matsushima, H., Utani, A., Endo, H., Matsuura, H., Kakuta, M,, Nakamura, Y., Matsuyoshi, N., Matsui, C., Nakanishi, H., Takai, Y., Shinkai, H. (2003). The expression of nectin-1alpha in normal human skin and various skin tumours. Br J Dermatol. 148, 755-62.
Matter, K. & Balda, M.S. (2007). Epithelial tight junctions, gene expression and nucleojunctional interplay. J Cell Sci. 120, 1505-1511.
Medici, D., Hay, E.D. & Goodenough, D.A. (2006). Cooperation between Snail and LEF-1 transcription factors is essential for TGF-β1–induced epithelial–mesenchymal transition. Mol Biol Cell. 17, 1871-1879.
Medley, Q.G., Buchbinder, E.G., Tachibana, K., Ngo, H., Serra-Pagès, C., Streuli, M. (2003). Signaling between focal adhesion kinase and trio. J Biol Chem. 278, 13265-13270.

144
Mehlen, P & Puisieux, A. (2006). Metastasis: A question of life or death. Nat Rev Cancer. 6, 449-458.
Meng, W., Mushika, Y., Ichii, T. & Takeichi, M. (2008). Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell–cell contacts. Cell. 135, 948–959
Meyer, T.N., Hunt, J., Schwesinger, C. & Denker, B.M. (2003). Galpha12 regulates epithelial cell junctions through Src tyrosine kinases. Am J Physiol Cell Physiol. 285, C1281-1293.
Mills, G.B. & Moolenaar, W.H. (2003). The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 3, 582-591.
Minard, M.E., Ellis, L.M., Gallick, G.E. (2006). Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin Exp Metastasis. 23, 301-313.
Minard, M.E., Herynk, M.H., Collard, J.G. & Gallick, G.E. (2005). The guanine nucleotide exchange factor Tiam1 increases colon carcinoma growth at metastatic sites in an orthotopic nude mouse model. Oncogene. 24, 2568-2573.
Mingle, L.A., Bonamy, G., Barroso, M., Liao, G. & Liu, G. (2009). LPA-induced mutually exclusive subcellular localization of active RhoA and Arp2 mRNA revealed by sequential FRET and FISH. Histochem Cell Biol. 132, 47-58.
Mitra, S.K. & Schlaepfer, D.D. (2006). Integrin-regulated FAK–Src signaling in normal and cancer cells.
Mueller, B.K., Mack, H. & Teusch, N. (2005). Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Disc. 4, 387-398.
Curr Opin Cell Biol. 18, 516-23.
Mitra, S.K., Hanson, D.A. & Schlaepfer, D.D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat Rev Cell Biol. 6, 56-68.
Miyoshi, J. & Takai, Y. (2008). Structural and functional associations of apical junctions with cytoskeleton. Biochim Biphys Acta. 1778, 670-691.
Moissoglu, K., Slepchenko, B.M., Meller, N., Horwitz, A.F. & Schwartz, M.A. (2006). In vivo dynamics of Rac-membrane interactions. Mol Biol Cell. 17, 2770-2779.
Mori, K.M.D., Kitayama, J., Shida, D., Yamashita, H., Watanabe, T., Nagawa, H., (2006). Lysophosphatidic acid-induced effects in human colon carcinoma DLD1 cells are partially dependent on transactivation of epifermal growth factor receptor. J. Surg Res. 132, 56-61.
Müller, R., Berliner, C., Leptin, J., Pörtner, D., Bialecki, W., Kleuser, B., Schumacher, U. & Milićević, N.M. (2010). Expression of sphingosine-1-phosphate receptors and lysophosphatidic acid receptors on cultured and xenografted human colon, breast, melanoma, and lung tumour cells. Tumour Biol.
Murph, M.M., Liu, W., Yu, S., Lu, Y., Hall, H., Hennessy, B.T., Lahad, J., Schaner, M., Helland, A., Kristensen, G., Børresen-Dale, A.L. & Mills B.G. (2009). Lysophosphatidic Acid-Induced Transcriptional Profile Represents Serous Epithelial Ovarian Carcinoma and Worsened Prognosis. PLoS One. 4, e5583.
31, 341-349.

145
Nabeshima, R., Shimao, Y., Inoue, T., Koono, M. (2002). Immunohistochemical analysis of IQGAP1 expression in human colorectal carcinomas: its overexpression in carcinomas and association with invasion fronts. Cancer Lett. 176, 101-109.
Nadella, K.S., Saji, M., Jacob, N.K., Pavel, E., Ringel, M.D. & Kirschner, L.S. (2009). Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 10, 599-605.
Naik, M.U., Naik, T.U., Suckow, A.T., Duncan M.K. & Naik, U.P. (2008). Attenuation of Junctional Adhesion Molecule-A Is a Contributing Factor for Breast Cancer Cell Invasion. Cancer Res.
Nakagawa, M., Fukata, M., Yamaga, M., Itoh, N. & Kaibuchi, K. (2001). Recruitment and activation of Rac 1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J Cell Sci. 114, 1829-1838.
Naydenov, N.G., Hopkins, A.M. & Ivanov, A.I. (2009). c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle. 8, 2110-2121.
Nelson, W.J. (2003). Adaptation of core mechanisms to generate cell polarity. Nature. 422, 766-774.
Nguyen, G.H., French, R. & Radhakrishna, A. (2004). Protein kinase A inhibits lysophosphatidic acid induction of serum response factor via alterations in the actin cytoskeleton. Cel Signal. 16, 1141-1151.
Nie, M., Aijaz, S., San, I.V.L.C., Balda, M.S. & Matter, K. (2009). The Y-box factor ZONAB/DbpA associates with GEF-H1/Lfc and mediates Rho-stimulated transcription. EMBO Rep. 10, 1125–1131.
Niessen, C.M. & Gottardi, C.J. (2008). Molecular components of the adherens junction. Biochim Biphys Acta. 1778, 562-71.
Niessen, C.M. (2007). Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 127, 2525-32.
Noguchi, K., Herr, D., Mutoh, T. & Jerold, C. (2009). Lysophosphatidic acid (LPA) and its receptors. Curr Op Pharmacol. 9, 15-23.
Nomanbhoy, T.K. & Cerione, R. (1996). Characterization of the interaction between RhoGDI and Cdc42Hs using fluorescence spectroscopy. J Biol Chem. 271, 10004-9.
Noren, N.K., Niessen, C.M., Gumbiner, B.M. & Burridge, K. (2001). Cadherin engagement regulates Rho family GTPases. J Biol Chem. 276, 33305-33308.
Noritake, J., Watanabe, T., Sato, K., Wang, S. & Kaibuchi, K. (
68, 2194
2005). IQGAP1: a key regulator of adhesion and migration. J Cell Sci. 118, 2085–2092.
Nusrat, A., von Eichel-Streiber, C., Turner, J.R., Verkade, P., Madara, J.L. & Parkos, C.A. (2001). Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. 69, 1329-1336.

146
O´Connor, K.L. & Mercurio, A.M. (2001). Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 276, 47895-47900.
Oki, E., Zhao, Y., Yoshida, R., Egashira, A., Ohgaki, K., Morita, M., Kakeji, Y. & Maehara, Y. (2009). The difference in p53 mutations between cancers of the upper and lower gastrointestinal tract. Digestion. 79, 33-39.
Oliveira, S.S. & Morgado-Díaz, J.A. (2007). Claudins: multifunctional players in epithelial tight junctions and their role in cancer. Cell Mol Life Sci. 64, 17-28.
Oliveira, S.S., Oliveira, I.M., Souza, W., Morgado-Díaz, J.A. (2005). Claudins upregulation in human colorectal cancer. FEBS Letters. 579, 6179-6185.
Olson, M.F. (2008). Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 20, 242-248. Biochim Biophys Acta. 1773, 34-46.
Onder, T.T., Gupta, P, B., Mani, S.A., Yang, J., Lander, E.S. & Weinberg, R.A. (2008). Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 68, 3645-3654.
Oneyama, C., Hikita, T., Nada, S. & Okada, M. (2008). Functional dissection of transformation by c-Src and v-Src. Genes Cells. 13, 1-12.
Ordóñez-Morán, P., Larriba, M.J., Pálmer, H.G., Valero, R.A., Barbáchano, A., Duñach, M., de Herreros, A.G., Villalobos, C., Berciano, M.T., Lafarga, M. & Muñoz, A. (2008). RhoA–ROCK and p38MAPK-MSK1 mediate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J Cell Biol. 183, 697-710.
Paduch, R., Kandefer-Szerszeń, M., Szuster-Ciesielska, A. & Plewka, K. (2010). Transforming growth factor-beta1 modulates metalloproteinase-2 and -9, nitric oxide, RhoA and alpha-smooth muscle actin expression in colon adenocarcinoma cells. Cell Biol Int. 34, 213-223.
Paris, L., Tonutti, L., Vannini, C., Bazzoni, G. (2008). Structural organization of the tight junctions. Biochim et Biophys Acta 1778, 646–659.
Park, C.H., Moon, Y., Shin, C.M. & Chung, J.H. (2010). Cyclic AMP suppresses matrix metalloproteinase-1 expression through inhibition of MAPK and GSK-3beta. J Invest Dermatol. 130, 2049-2056.
Parri, M & Chiarugi, P. (2010). Rac and Rho GTPases in cancer cell motility control Cell Comm Signal. 8, 23-37.
Pattla, I., Volgberg, T., Elad, N., Hirschfels-Warneken, V., Grashoff, C., Fässler, R., Spatz, J.P., Geiger, B. & Medalia, O. (2010). Dissecting the molecular architecture of integrin adhesion sites by cryo-electron tomography. Nat Cell Biol. 12, 909-915.
Paulucci-Holthauzen, A.A., Vergara, L.A., Bellot, L.J., Canton, D., Scott, J.D., & O'Connor, K.L. (2009). Spatial Distribution of Protein Kinase A Activity during Cell Migration Is Mediated by A-kinase Anchoring Protein AKAP Lbc. J Biol Chem. 284, 5956–5967.

147
Peng, Y.F., Mandai, K., Nakanishi, H., Ikeda, W., Asada, M., Momose, Y., Shibamoto, S., Yanagihara, K., Shiozaki, H., Monden, M., Takeichi, M. & Takai, Y. (2002). Restoration of E-cadherin-based cell-cell adhesion by overexpression of nectin in HSC-39 cells, a human signet ring cell gastric cancer cell line. Oncogene. 21, 4108-19.
Perrin, B.J. & Ervasti, J.M. (2010). The Actin Gene Family: Function Follows Isoform. Cytoskeleton. 67,630–634.
Pertz, O., Hodgson, L., Klemke, R.L. & Hahn, K.M. (2006). Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 440, 1069-1072.
Pidoux, G. & Taskén, A. (2010). Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. Journal of Molecular Endocrinology . 44, 271–284
Polley, A.C.J., Mulholland, F., Pin, C., Williams, E.A., Bradburn, D.M., Mills, S.J., Mathers, J.C. & Johnson, I.T. (2006). Proteomic analysis reveals field-wide changes in protein expression in the morphologically normal mucosa of patients with colorectal neoplasia. Cancer Res. 66, 6553-6562.
Poulogiannis, G., Frayling, I.M., Arends, M.J. (2010). DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 56, 167–179.
Qiao, J., Holian, O., Lee, B.S., Huang, F., Zhang, J. & Lum, H. (2008). Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am J Physiol Cell Physiol. 295, C1161-1168.
Raleigh, D.R., Marchiando, A.M., Zhang, Y., Shen, L., Sasaki, H., Wang, Y., Long, M. & Turner, J.R. (2010). Tight junction–associated MARVEL Proteins marvelD3,tricellulin, and occludin have distinct but overlapping functions. Molecular Biology of the Cell. 21, 1200–1213.
Rapier, R., Huq, J., Vishnubhotla, R., Bulic, M., Perrault, C.M., Metlushko, V., Cho, M., Tay, R.T. & Glover, S.C. (2010). The extracellular matrix microtopography drives critical changes in cellular motility and Rho A activity in colon cancer cells. Cancer Cell Int. 10, 24-34.
Reid, T.S., Long, S.B. & Beese, L.S. (2004). Crystallographic analysis that anticancer clinical candidate L-778,123 inhibits protein farnesyltransferase and geranylgeranyltransferase-I by different binding modes. Biochem. 43, 9000-9008.
Reya, T. & Clevers, H. (2005). Wnt signalling in stem cells and cancer. Nature. 434, 843-850.
Ridley, A.J. & Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibres in response to growth factors. Cell. 70, 389-399.
Roskoski Jr, R. (2010). RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Comm. 399, 313–317.
Rowley, P.T. (2005). Inherited susceptibility to colorectal cancer. Annu Rev Med. 56, 539-554.

148
Rusovici, R., Ghaleb, A., Shim, H., Yang, V.W. & Yun, C.C. (2007). Lysophosphatidic acid prevents apoptosis of Caco-2 colon cancer cells via activation of mitogen-activated protein kinase and phosphorylation of Bad. Biochim Biophys Acta. 1770, 1194-203.
Sah, V.P., Seasholtz, T.M., Sagi, S.A. & Brown, J.H. (2000). The role of Rho in G protein-coupled receptor signal transduction. Annu. Rev. Pharmacol. Toxicol. 40, 459-489.
Sahai, E. & Marshall, C.J. (2002). Rho GTPases and cancer. Nat Rev Cancer. 2, 133-142.
Sahai, E. (2007). Illuminating the metastatic process. Nat Rev Cancer. 7, 737-749.
Saito, N., Kameoka, S. & Furukawa, R. (2007). Gene profile analysis of colorectal cancer cell lines by cDNA macroarray. Oncology Reports. 17, 1061-1065.
Samarin, S.N., Ivanov, A.I., flatau, G., Parkos, C.A. & Nusrat, A (2007). Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol Biol Cel.
Serrels, B., Serrels, A., Brunton, V.G., Holt, M., McLean, G.W., Gray, C.H., Jones, G.E. & Frame, M.C. (2007). Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol. 9, 1046-1056.
18, 3429-3439.
Sander, E.E., vanDelft, S., ten Klooster, J.P., Reid., T., van der Kammen, R.A., Michiels, F. & Collard, J.D. (1998). Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 143, 1385-1398.
Santos, M.F., McCormack, S.A., Guo, Z., Okolicany, J., Zheng. Y. & Johnson, L.R., Tigyl, G. (1997). Rho proteins play a critical role in cell migration during the early phase of mucosal restitution. J Clin Invest. 100, 216-225.
Sanz-Moreno, V., Gdea, G., Ahn, J., Paterson, H., Marra, P., Pinner, E.S. & Marshall. (2008). Rac activation and inactivation control plasticityt of tumor cell movement. Cell. 135, 510-523.
Sawada, K., Morishige, K-I., Tahara, M., Ikebuchi, Y., Kawagishi, R., Tasaka, K & Murata, Y. (2002). Lysophosphatidic Acid Induces Focal Adhesion Assembly through Rho/Rho-Associated Kinase Pathway in Human Ovarian Cancer Cells. Gynecologic Oncology 87, 252–259.
Schlaepfer, D.D., Satyajit, K.M. & Ilic, D. (2004). Control of motile and invasive cell phenotypes by focal adhesion kinse. Biochim Biophys Acta. 1692, 77-102.
Schoumacher, M., Goldman, R.D., Louvard, D. & Vignjevic, D.M. (2010). Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 189, 541-556.
Schulzke, J.D., Gitter, A.H., Mankertz, J., Spiegel, S., Seidler, U., Amasheh, S., Saitou, M., Tsukita, S. & Fromm, M. (2005). Epithelial transport and barrier function in occludin-deficient mice Biochim Biophys Acta 1669 (1), 34-42.
Sebti, S.M. & Adjei, A.A. (2004). Farnesyltransferase inhibitors. Sem Oncol 31, 28-39.

149
Sheng, H., Shao, J., Townsend jr, C. M. & Evers, B. M. (2003). Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut. 52, 1472–1478.
Shida, D., Fang, X., Kordula, T., Takabe, K., Lépine, S., Alvarez, S.E., Milstien, S. & Spiegel, S. (2008). Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 68, 6569-6577.
Shida, D., Kitayama, J., Yamaguchi, H., Yamashita, H., Mori, K., Watanabe, T., Nagawa, H. (2005). Lysophosphatidic acid transactivates both c-Met and epidermal growth factor receptor, and induces cyclooxygenase-2 expression in human colon cancer LoVo cells. World J Gastroenterol.11, 5638-5643.
Shida, D., Kitayama, H., Yamaguchi, H., Okaji, Y., Tsuno, N.H., Watanabe, T, Takuwa, Y. & Nagawa, H. (2003). Lisophopsphatidic acid enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 63, 1706-1711.
Shida, D., Watanabe, T., Aoki, J., Hama., K., Kitayama, J., Sonoda, H., Kishi, Y., Yamaguchi, H., Sasaki, S., Sako, A., Konishi, T., Arai, H. & Nagawa, H. (2004). Aberrant expression of lysophodphatidic acid (LPA) receptors in human colorectal cancer. Lab. Invest. 84, 1352-1362.
Shin, K.J., Kim, Y.L., Lee, S., Kim, D.K., Ahn, C., Chung, J., Seong, J.Y. & Hwang, J.I. (2009). Lysophosphatidic acid signaling through LPA receptor subtype 1 induces colony scattering of gastrointestinal cancer cells. J. Cancer Res. Clin. Oncol
Soma, T., Chiba, H., Kato-Mori, Y., Wada, T., Yamashita, T., Kojima, T., Sawada, N. (2004). Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP.
. 135, 45-52.
Shindo, M., Wada, H., Kaido, M., Tateno, M., Aigaki, T., Tsuda, L. & Hayashi, S. (2008). Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Develop. 135, 1355-1364.
Sinnamon, M.J., Carter, K.J., Fingleton, B. & Matrisian, L.M. (2008). Matrix metalloproteinase-9 contributes to intestinal tumourigenesis in the adenomatous polyposis coli multiple intestinal neoplasia mouse. Int J Exp Path. 89, 466–475.
Smutny, M., Cox, H.L., Leerberg, J.M., Kovacs, E.M., Conti, M.A., Ferguson, C., Hamilton, N.A., Parton, R.G., Adelstein, R.S. & Yap, A.S. (2010). Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 12, 696-702.
Exp Cell Res
Spaargaren, M. & Bos, J.L. (1999). Rab5 Induces Rac- independent lamellipodia formation and cell migration. Mol Biol. Cell. 10, 3239–3250.
Spiering, D. & Hodgson, L. (2011). Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adhesion & Migration 5:2, 1-11.
. 300, 202-12.
Stähle, M., Veit, C., Bachfischer, U., Scierling, K., Skripczynsky, B., Hall, A., Gierschik, P. & Giehl, K. (2003). Mechanisms in LPA-induced tumor cell migration: critical role of phosphorylated ERK. J Cell Sci. 116, 3835-3846.

150
Steed, E., Balda, M.S. & Matter, J. (2010). Dynamics and functions of tight Junctions. Trends in Cell Biology Vol.20, 143-149.
Stemmler, M.P. (2008). Cadherins in development and cancer. Mol Bio Syst. 4, 835-50.
Summy, J.M. & Gallick, G.E. (2003). Src family kinases in tumour progression and metastasis. Cancer Metast Rev. 22, 337-358.
Sun, H., Ren, J., Zhu, Q., Kong, F.Z., Wu, L. & Pan, B.R., (2009). Effects of lysophosphatidic acid on human colon cancer cells and its mechanisms of action. W J Gastroenterol. 15, 4547-4555
Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, Desiderio D, Guntaka R, Rao R. (2009). PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci. 106, 61-66.
Syed V., Mak, Cheng, D. & Balaji, K.C. (2008). B-catenin mediates alteration in cell proliferation, motility and invasion of prostate cancer cells by differential expression of E-cadherin and protein kinase D1. J Cell Biochem. 104, 82-95.
Symowicz, J., Adley, B.P., Gleason, K.J., Johnson, J.J., Ghosh, S., Fishman, D.A., Hudson, L.G. & Stack, M.S. (2007).
Tan, C., Costello, P., Sanghera, J., Dominguez, D., Baulida, J., de Herreros, A.G. & Dedhar, S. (2001). Inhibition of integrin linked kinase (ILK) suppresses β-catenin-Lef/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC–/– human colon carcinoma cells. Oncogene. 20, 133–140.
Engagement of Collagen-Binding Integrins Promotes Matrix Metalloproteinase-9–Dependent E-Cadherin Ectodomain Shedding in Ovarian Carcinoma Cells. Cancer Res. 67, 2030-2039.
Symowicz, J., Adley, B.P., Woo, M.M., Auersperg, N., Hudson, L.G. & Stack, M.S. (2005). Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 65, 2234-42.
Takaishi, K., Matozaki, T., Nakano, K. & Takai, Y. (2000). Multiple downstream signalling pathways from ROCK, a target moplecule of Rho small G protein, in reorganization of the actin cytoskeleton in Madin-Darby canine kidney cells. Genes to Cells. 5, 929-936.
Takami, Y., Higashi, M., Kumagai, S., Kuo, P.C., Kawana, H., Koda, K., Miyazaki, M. & Harigawa, K. (2008). The activity of RhoA is correlated with lymph node metastasis in human colorectal cancer. Dig Dis Sci. 53, 467-473.
Takayama, T., Miyanishi, K., Hayashi, T., Sato, Y. & Niitsu, Y. (2006). Colorectal cancer: genetics of development and metastasis. J Gastroenterol. 41, 185–192.
Takehara, M., Nishimura, T., Mima, S., Hoshino, T. & Mizushima, T. (2009). Effect of Claudin Expression on Paracellular Permeability, Migration and Invasion of Colonic Cancer Cells. Biol. Pharm. Bull. 32, 825-831.
Takkunen, M., Hukkanen, M., Liljeström, M., Grenman, R. & Virtanen, I. (2010). Podosome-like structures of non-invasive carcinoma cells are replaced in epithelial-mesenchymal transition by actin comet-embedded invadopodia. J Cell Mol Med. 14, 1569-1593.

151
Tanaka, M.N., Diaz, B.L., de Souza, W., Morgado-Diaz, J.A. (2008). Prostaglandin E2-EP1 and EP2 receptor signaling promotes apical junctional complex disassembly of Caco-2 human colorectal cancer cells. BMC Cell Biol. 9, 63.
Tanaka, T. (2009). Colorectal carcinogenesis: Review of human and experimental animal studies. J carcinog 8, 1-19.
Tang, J. & Gross, D.J. (2003). Regulated EGF receptor binding to F-actin modulates receptor phosphorylation.Biochem Biophys Res Commun. 312, 930-936.
Tatsuta, M., Iishi, H., Baba, M., Uedo, N., Ishihara, R., Higashino, K., Mukai, M. & Ishiguro, S. (2005) Induction by lysophosphatidic acid of peritoneal and pleural metastases of intestinal cancers induced by azoxymethane in Wistar rats. Cancer letters. 219, 137-145.
Thiery, J.P. & Sleeman, J.P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 7, 131-142.
Thomason, H.A., Scothern, A., Mc Harg, S. & Garrode, D,R. (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem. J. 429, 419–433
Tice, D.A., Biscardi, J.S., Nickles, A.L., & Parsons S.J. (1999). Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. PNAS. 96,
Tsukahara T, Hanazawa S, Kobayashi T, Iwamoto Y & Murakami-Murofushi K. (2010). Cyclic phosphatidic acid decreases proliferation and survival of colon cancer cells by inhibiting peroxisome proliferator-activated receptor γ.
415-1420.
Timpson, P., Jones, G.E., Frame, M.C. & Brunton, V.G. (2001). Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr Biol. 11, 1836-1846.
Tobey, N.A., Argote, C.M., Hosseini, S.S. & Orlando, R.C. (1994). Calcium-switch technique and junctional permeability in native rabbit esophageal epithelium. American Journal of Physiology. 286, 1042-1049.
Torka, R., Thuman, F., Herzog, V. & Kirfel, G. (2006). ROCK signaling mediates the adoption of different modes of migration and invasion in human mammary epithelial tumor cells. Exp Cell Res. 312, 3857-3871.
Traweger, A., Fang, D., Liu, Y.C., Stelzhammer, W., Krizbai, I.A., Fresser, F., Bauer, H.C. & Bauer, H. (2002). The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch, J. Biol. Chem. 277, 10201–10208.
Tsuchiya, T., Tsuno, N.H., Asakage, M., Yamada, J., Yoneyama, S., Okaji, Y., Sasaki, S., Kitayama, J., Osada, T., Takahashi, K. & Nagawa, H. (2008). Apoptosis induction by p38 MAPK inhibitor in human colon cancer cells. Hepatogastroenterology. 55, 930-935.
Prostaglandins Other Lipid Mediat. 93, 126-33.
Umeda, J., Ikenouchi, K., Katahira-Tayama, S., Furuse, K., Sasaki, H., Nakayama, M., Matsui, T., Tsukita, S., Furuse, M., Tsukita, S. (2006). ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 126, 741–754.

152
Valentine, W.J., Fujiwara, Y., Tsukahara, R. & Tigyi, G. (2008). Lysophospholipid signaling: Beyond the EDGs. Biochim Biophy Acta. 1780, 597-605.
van den Brandt, P. &, Goldbohm, R.A. (2006). Nutrition in the prevention of gastrointestinal cancer. Best Pract Res Clin Gastroenterol. 20, 589-603.
van Houdt, W.J., de Bruijn, M.T., Emmink, B.L., Raats, D., Hoogwater, F.J., Borel Rinkes, I.H. & Kranenburg, O. (2010). Oncogenic K-ras activates p38 to maintain colorectal cancer cell proliferation during MEK inhibition. Cell Oncol. 32, 245-57.
Vasioukhin, V., Bauer, C., Degenstein, L., Wise, B., Fuchs, E. (2001). Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell. 104, 605–17
Vega, F.M. & Ridley, A.J. (2008). Rho GTPases in cancer cell biology. FEBS Letters. 582, 2093-2101.
Vishnubhotla, R., Sun, S., Huq, J., Bulic, M., Ramesh, A., Guzman, G., Cho, M. & Glover, S.C. (2007). ROCK-II mediates colon cancer invasion via regulation of MMP-2 and MMP-13 at the site of invadopodia as revealed by multiphoton imaging. Lab invest. 87, 1149-1158.
Voisin, L., Julien, C., Duhamel, S., Gopalbhai, K., Claveau, I., Saba-El-Leil, M.C., Rodrigue-Gervais, I.G., Gaboury, L., Lamarre, D., Basik, M. & Meloche, S. (2008). Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer, 8, 337-354.
Waldner, M.J. & Neurath, M.F. (2010). The molecular therapy of colorectal cancer. Mol Aspects Med. 31, 171-178.
Walsh, S.V., Hopkins, A.M., Chen, J., Narumiya, S., Parkos, C.A. & Nusrat, A. (2001). Rho kinase regulate tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology. 121, 566-579.
Wang, Q., Chen, X.W. & Margolis, B. (2007). PALS1 Regulates E-Cadherin Trafficking in Mammalian Epithelial Cells. Mol Biol Cell. 18, 874-885.
Wang, W., Mouneimne, G., Sidani, M., Wyckoff, J., Chen, X., Makris, A., Goswami, S., Bresnick, A.R. & Condeelis, J.S. (2006). The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 173, 395-404.
Wang, Y., Ngo, V.N., Marani, M., Yang, Y., Wright, G., Staudt, L.M. & Downward, J. (2010). Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 29, 4658-4670.
Weaver, A.M., Heuser, J.E., Karginov, A.V., Lee, W-L., Parsons, J.T & Cooper, J.A. (2002). Interaction of cortactin and N-Wasp with Arp2/3 complex. Curr Biol. 12, 1270-1278.
Webb, D.J., Donais, K., Whitmore, L.A., Thomas, S.M., Turner, C.E., Parsons, J.T. & Horwitz, A.F. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 6, 154-161.

153
Westphal, R.S., Soderling, S.H., Alto, N.M., Langeberg, L.K. & Scott, J.D. (2000). Scar/Wave-1, a Wiskott-Aldrich syndrom protein, assembles an actin-associated multi-kinase scaffold. EMBO J. 19, 4589-4600.
Weyant, M.J., Carothers, A.M., Dannenberg, A.J., Bertagnolli, M.M. (2001). (+)-Catechin inhibits intestinal tumor formation and suppresses focal adhesion kinase activation in the min/+ mouse. Cancer Res. 61, 118-125.
Wheelock, M.J., Shintani, Y., Maeda, M., Fukumoto, Y., & Johnson, K.R. (2007). Cadherin switching. J. Cell Sci. 121, 727-735.
Wilkinson, S., Paterson, H.F. & Marshall, C.J. (2005), Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol.
Wittenmayer, N., Jandrig, B., Rothkegel, M., Schlüter, K., Arnold, W., Haensch, W., Scherneck, S. & Jockusch, B.M. (2004). Tumor suppressor activity of profiling requires a functional actin binding site. Molecular Biology of the Cell. 15, 1600-1608.
7, 255-261.
Woodcock, S.A., Rooney, C., Liontos, M., Connolly, Y., Zoumpourlis, V., Whetton, A.D., Gorgoulis, V.G. & Malliri, A. (2009). SRC-induced disassembly of adherens junctions requires localized phosphorylation and degradation of the rac activator tiam1. Mol Cell.
Wu, W.T., Chen, C.N., Lin, C.I., Chen, J.H. & Lee, H. (2005). Lysophospholipids enhance matrix metalloproteinase-2 expression in human endothelial cells. E
33, 639-653.
Wozniak, M.A., Kwong, L., Chodniewics, D., Klemke, R.L. & Keely, P.J. (2005). R-Ras controls membrane protrusion and cell migration through the spatial regulation of Rac and Rho. Mol Biol Cell. 16, 84-96.
Wozniak, M.A., Modzelewska, K., Kwong, L. & Keely, P.J. (2004). Focal adhesion regulation of cell behavior. Biochim et Biophys Acta. 1692, 103–119.
Wu, W.S., Wu, J.R. & Hu, C.T. (2008). Signal cross talks for sustained MAPK activation and cell migration: the potential role of reactive oxygen species. Cancer Metastasis Rev. 27, 303-14.
ndocrinology
Xu, K.P., Yin, J. & Yu, FS. (2007). Lysophosphatidic acid promoting corneal epithelial wound healing by transactivation of epidermal growth factor receptor. Invest. Ophtalmol. & visual sci. 48, 636-643.
Xu, M.Y., Porte, J., Knox, A.J., Weinreb, P.H., Maher, T.M., Violette, S.,M., McAnulty, R.J., Sheppard, D. & Jenkins, R.G. (2009). Lysophosphatidic acid induces αvβ6 integrin-mediated TGF-β activation via the LPA2 receptor and the small G protein Gαq. Am J Pathol.
. 146, 3387-3400.
174, 1264-1279.
Xue, Y., Bi, F., Zhang, X., Pan, Y., Liu, N., Zheng, Y. & Fan, D. (2004). Inhibition of endothelial cell proliferation by targeting Rac1 GTPase with small interference RNA in tumor cells. Biochem Biophys Res Commun. 320, 1309-1315.

154
Yamada, A., Irie, K., Fukuhara, A., Ooshio, T. & Takai, Y. (2004). Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in MDCK cells. Genes Cells. 9, 843-855.
Yamada, T., Yoshiharu, O., Kogo, M. & Inagaki, S. (2005). Physical and functional interactions of the lysophosphatidic acid receptors with PDZ domain-containing Rho guanine nucleotide exchange factors (RhoGEFs). J. Biol. Chem. 19, 19358-19363.
Yamaguchi, H. & Condeelis, J. (2006). Regulation of actin cytoskeleton in cancer cell migration and invasion. Biochim Byoph Acta. 1773, 642-652.
Yanagawa, R., Furukawa, Y., Tsunoda, T., Kitahara, O., Kameyama, M., Murata, K., Ishikawa, O. & Nakamura, Y. (2001). Genome wide-screeningof genes showing altered expression in liver metastases of human colorectal cancers by cDNA microarray. Neoplasia. 3, 395-401.
Yanagida, K., Masago, K., Nakanishi, H., Kihara, Y., Hamano, F., Tajima, Y., Taguchi, R., Shimizu, T. & Ishii, S. (2009). Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 284, 17731-17741.
Yang, M., Zhong, W.W., Srivastava, N., Slavin, A., Yang, J., Hoey, T. & An, S. (2005). G protein-coupled lisophopsphatidic acid receptors stimulate proliferation of colon cancer cells through the β-catenin pathway. Proc Natl Acad Sci. 102, 6027-6032.
Ying, H., Biroc, S.L., Li, W.W., Alicke, B., Xuan, J.A., Pagila, R., Ohashi, Y., Okada, T., Kamata, Y. & Dinter, H. (2006). The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 5, 2158-2164.
Yip, SC., El Sibai, M., Coniglio, SJ., Mouneimne, G., Eddy, R.J., Drees, B.E., Neilsen, P.O., Goswami, S., Symons, M., Condeelis, J.S. & Backer, J.M. (2007). The distinct roles of Ras and Rac in PI 3-kinasedependent protrusion during EGF-stimulated cell migration. J Cell Sci. 120, 3138-3146.
Yu, W., Shewan, A.M., Brakeman, P., Eastburn, D.J., Datta, A., Bryant, D.M., Fan, Q.W., Weiss, W.A., Zegers, M.M., Mostov, K.E. (2008). Involvement of RhoA, ROCK I and myosin II in inverted orientation of epithelial polarity. EMBO Rep. 9, 923-929.
Yue, J. & Mulder, K.M. (2001). Tranforming growth factor β signal transduction in epithelial cells. Pharmacol Therap. 91, 1-34.
Zaidel-Bar, R., Ballestrem, C., Kam, Z. & Geiger, B. (2003). Early molecular events in the assembly of matriz adhesions at the leading edge of migrating cells. Journal of Cell Science. 116, 4605-4613.
Zhai, J., Lin, H., Nie, Z., Wu, J., Cañete-Soler, R., Schlaepfer, W.W. & Schlaepfer, D.D. (2003). Direct interaction of focal adhesion kinase with p190RhoGEF. J Biol Chem. 278, 24865-24873.
Zhang J, Gao J, Tan X, Wang M, Qin R. (2010). Effects of down-regulation of integrin-beta1 expression on migration and hepatic metastasis of human colon carcinoma. J Huazhong Univ Sci Technolog Med Sci. 30, 464-9.

155
Zhang, H., Wang, D., Sun, H., Hall, R.A. & Yun, C.C. (2007a). MAGI-3 regulates LPA-induced activation of Erk and RhoA. Cell Signal. 19, 261-268.
Zhang, J., Gao, J., Tan, X., Wang, M., Qin, R. (2010). Effects of down-regulation of integrin-beta1 expression on migration and hepatic metastasis of human colon carcinoma. J Huazhong Univ Sci Technolog Med Sci. 30, 464-9.
Zhang, J., Peng, X & Wei, L. (2007b). Coexpression of WAVE2 and VASP in colorectal cancer. FASEB J. 21,708.2.
Zhao, J.J., Gjoerup, O.V, Subramanian, R.R., Cheng,Y. Chen, W., Roberts, T.M. & Hahn, W.C. (2003). Human mammary epithelial cells transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 3, 483-495.
Zhao, L., Wang, H., Li, J., Liu, Y. & Ding, Y. (2008a). Overexpression of Rho GDP-dissociation inhibitor alpha is associated with tumor progression and poor prognosis of colorectal cancer. J Proteome Res. 7, 3994-4003.
Zhao, Y., Yan, Q., Long, X., chen, X. & Wang, Y. (2008b). Vimentin affects the motility and invasiveness of prostate cancer cell. J Biochem Funct. 26, 571-577.
Zhao, Z., Xiao, Y., Elson, Tan, P.H., Plummer, S.J., Berk, M., Aung, P.P., Lavery, I.C., Achkar, J.P., Li, L., Casey, G. & Xu, Y. (2007). Plasma lysophosphatidylcholine levels: potential biomarkers for colorectal cancer. J Clin Oncol. 25, 2696-2701
Zouq, N.K., Keeble, J.A., Lindsay, J., Valentijn, A.J., Zhang, L., Mills, D., Turner, C.E.,Streuli, C.E. & Gilmore, A.P. (2009). FAK engages multiple pathways to maintain survival of fibroblasts and epithelia – differential roles for paxillin and p130Cas. J Cell Sci.
122, 357-367.