disturbi del movimento in età evolutiva. inquadramento e
TRANSCRIPT

DISTURBI DEL MOVIMENTO IN
ETÀ EVOLUTIVA.
CHRONIC PEDIATRIC HYPERKINETIC
MOVEMENT DISORDERS
Dott. Carlo Fusco Dott.ssa Margherita Baga Dott. Carlo Alberto Cesaroni

Chronic pediatric
hyperkinetic
movement disorders
Tremore essenziale
in età evolutiva
Transient Movement disorders of infancy and childhood

Epidemiologia dei disturbi del movimento (PMD)
Tic 43%
Distonia/Sindrome rigido-ipocinetica 24%
Tremore 16%
Mioclono 6%
Coreatetosi 3%
Stereotipie 2%
Misti 4%

Epidemiologia dei disturbi del movimento (PMD)
Primario: d.movimento unico
sintomo; RM normale;
verosimilmente genetico: 46%
Secondario: cause ambientali
(infezioni/stroke): 20%.
Sindromico:
metabolico/degenerativo; 29%
Non classificabile: 2%

Impatto
clinico/epidemiologico
• PMD cronici prevalgono sui
transient PMP.
• Se ad esordio entro i l
primo anno di vita: solo i l
4% è transient!

CHRONIC PEDIATRIC
HYPERKINETIC
MOVEMENT
DISORDERS

DEFINIZIONE DI
MOVIMENTO
IPERCINETICO:
MOVIMENTO IN
ECCESSO
INVOLONTARIO.

Classificazione delle ipercinesie
• Corea - Ballismo
• Distonia
• Mioclono
• Atetosi
• Stereotipie
• Tic

TIC
• Movimenti involontari, aritmici, af inal ist ici che generalmente non
alterano l ’att ività volontaria a di f ferenza del la corea e del
mioclono e pertanto solo raramente generano disabi l ità.
• Disturbo del movimento più frequente in età pediatrica: motorio
o vocale /fonico, transitorio o cronico, semplice o complesso.
• Associazione più frequente con: OCD, def icit di
attenzione /disturbi di apprendimento

TIC
• Predisposizione genetica: combinazione con
eventi ambientali.
• Terapia farmacologica: se invalidanti /se
debilitanti
• Scelta farmacologica spesso condizionata da
comorbidità, possibili effetti collaterali,
conoscenza del farmaco.

PANDAS: pediatric
autoimmune
neuropsychiatric
disorders with
streptococcal
infections. Swedo et a l ,
1998
• Sintomi OC oppure t ic o ambedue secondo DSM IV
temporalmente associat i con infezione da SBEGA.
• Esordio prepuberale dei s intomi.
• Andamento episodico del la sever ità dei s intomi: onset
improvviso o esacerbaz ione improvvisa, con r iduz ione del la
s intomatolog ia o scomparsa dei s intomi tra gl i episodi .
• Associaz ione con infezione SBEGA: tampone o TAS.
• Contemporanea presenza di d isturbi neurologic i come:
alteraz ione dei movimenti f in i del le mani, movimenti
coreic i isolat i (non corea.. .)

PANDAS:
criteri di
Swedo
• Associazione temporale?
Esplosione “neurologica” in contemporanea
con infezione da 1 a 6 mesi.
• Sintomi psichiatrici: irritabilità, disturbo
dell’umore, disattenzione, iperattività,
comportamento oppositivo, ansia.

PANDAS:
criticità
• Diagnosi dif ferenziale con tic /OCD e PANDAS in
bambini con “infezione cronica”? NON stabil ita. TIC
e OCD stesso esordio esplosivo.
• Riscontro TAS elevato in bambini asintomatici!
• E’ stato dimostrato: aumento rischio di Tourette
(TS) in bambini con multiple infezioni da
streptococco
• E’ stato dimostrato: rischio non signif icativo di TS in
bambini con infezioni multiple da streptococco
• Anticorpi verso la superf icie neuronale NON
aumentano in PANDAS e TS

Criteria for
PANDAS,
PANS, CANS
CANS: Childhood acute neuropsychiatric Syndromes - Federica Zibordi, Giovanna Zorzi, Miryam Carecchio, Nardo Nardocci

MIOCLONO

Mioclono
Movimento improvviso,
rapido, involontario. Di
solito di breve durata
(10-50ms), non ritmico,
non identif icabile un
pattern preciso.

MIOCLONO
CLASSIFICAZIONE

SYT-1

Cos è la sinaptotagmina?
Synaptotagmin 1 (SYT1) ha un
ruolo critico nel rilascio rapido,
sincrono e calcio-dipendente
dei neurotrasmettitori e
modula anche l'endocitosi della
vescicola sinaptica.

Alterazioni neurologiche in alterazioni di SYT-1
Ritardo mentale
globale
Linguaggio povero
o assente
Disabilità
intellettiva
moderato-severa
Impossibilità alla
deambulazione
Movimenti
ipercinetici
Movimenti
involontari Distonie Disinesie Coreoatetosi Atassia

EEG in alterazioni
di SYT-1
Disturbo EEG dell’attività
di fondo, con sequenza
intermittenti di onde
lente di grande ampiezza.
Brain. 2018 Sep; 141(9): 2576–2591. Published online 2018 Aug 13. doi: 10.1093/brain/awy209 SYT1-associated neurodevelopmental disorder: a case series Kate Baker, Sarah L Gordon et al.

COREE

Corea -
Semeiologia
Appare come movimento improvviso continuo,
casuale, non prevedibile, come risultante di uno o
più movimenti più”discreti” o più movimenti
(contrazioni muscolari) frammentati.

Corea -
Semeiologia
• Ogn i s ingo lo mov imento può avere un in iz io e un end-po int d is t in to ,
sebbene mol to d i f f i c i l i da ident i f i care perché i s ingo l i mov iment i spesso
s i sovrappongono.
• Non ha come t r igger l ’ avv io d i un movimento vo lontar io e non
scompare con i l manten imento d i una pos tura r i ch ies ta .
• “Motor impers is tence ” : imposs ib i l i tà a mantenere determinate posture
come la prot rus ione l igua le , l ’ es tens ione deg l i ar t i o la prens ione .
Possono co invo lgere tut t i i d is t ret t i musco lar i d i t ronco, ar t i , f acc ia e
l ingua .
• Quando i mov iment i core i c i sono p iù sever i , genera lmente a i c ingo l i e
p iù v io lent i , vengono ch iamat i “mov iment i ba l l i c i ” o ba l l i smo.

Corea - diagnosi
differenziale
• Corea vs tremore: movimento r i tmico e prevedibi le .
• Corea vs distonia: elemento distintivo del la distonia è la
presenza di contrazioni muscolari sostenute che
determinano posture abnormi o movimenti di torsione.
• Corea vs mioclono: movimento stereotipato (se gruppi
muscolari selezionati sono coinvolt i), spesso rapido, ( jerk-
l ike) Anche se possono apparire simil i , i l mioclono è
frequentemente multifocale e asincrono

Corea - diagnosi
differenziale
• Corea vs t ic: Parz ia lmente o completamente (per brev i moment i )
soppress i da l la volontà, stereot ipat i / r ipet i t iv i con “urgenza”
ne l l ’avv iare i l mov imento.
• Corea vs stereotipie : mimano atteggiament i motor i compless i ,
par te de l reper to i re umano f i s io log ico.
• Corea vs atetosi : Mov iment i lent i , cont inui , involontar i “d i
scr i t tura” che ant ic ipano i l mantenimento d i una postura stab i le .
Non s i ind iv iduano moviment i f rammentat i . A d i f fe renza de l la
corea le s tesse reg ioni de l corpo sono costantemente in teressate.
• Corea vs bal l ismo: mov imento core ico ampio e “v io lento”.

Corea - diagnosi
differenziale

COREE
EZIOLOGIA
Primarie: genetiche e neurodegenerative
Neurometaboliche
Disimmuni
Infettive
Metaboliche - Tossiche

Coree
primarie
Huntington’s disease
Huntington’s disease-like 2 ed altre HD like-syndrome
Dentatorubropallidoluysian atrophy (DRPLA)
Neuroacantocitosi
Atassia-teleangectasia- Atassie spinocerebellari (tipo 2,3,17)
Coreatetosi kinesigeniche parossistiche
Forme GNAO1
Corea benigna ereditaria: NKX2-TITF1, ADCY5
Pantothenate kinase-associated neurodegeneration (PKAN) with brain iron accumulation
Ipoplasia ponto-cerebellare tipo 2
FOXG1
Metaemoglobinemia autosomica recessiva ereditaria tipo II
GLUT-1 deficiency syndrome

COREA
BENIGNA
EREDITARIA

Corea benigna
ereditaria
Disordine autosomico dominante (OMIM: 118700),
caratterizzato da disturbo del movimento di t ipo
coreico generalmente non progressivo, associato a
normale svi luppo cognitivo, con ipotiroidismo ed
episodi di distress respiratorio alla nascita.
Mutazioni a carico del gene NKX2 che codif ica per
TITF-1 (thyroid transcription factor 1) che mappa sul
cromosoma 14q13.
Nuovo locus, adult-onset mappa su cromosoma
8q21.3-23.

Corea benigna
ereditaria
Definizione:
Brain-lung-
thyroid
syndrome.
• TITF-1 knock-out mice: nato morto; assenza di
tiroide, polmone e ghiandola pituitaria, assenza
strutture ventral i del prosencefalo, malformazioni dei
pall idi .
• Cervello: durante embriogenesi media la migrazione
degli interneuroni striatal i e del prosenecefalo.
• Unico report di neuropatologia: reperti aspecif ici
(l ieve astrocitosi ed iperplasia, senza perdita
neuronale). PETFDG nello stesso paziente: normale.

Corea benigna
ereditaria
Clinica
• In epoca neonatale: distress respiratorio, apnee del
neonato, frequenti infezioni respiratorie.
• Nel primo anno di vita: ipotonia di origine centrale.
• Ipotiroidismo: ma valori ormoni tiroidei possono
essere nei l imiti di norma con TSH elevato. Ritardo
motorio: frequente, anche se non rappresenta un
criterio necessario.
Corea benigna
ereditaria
Clinica classica
• Distress respiratorio con disfunzione tiroidea
• Ipotonia e ritardo motorio
• Corea non progressiva: movimento ipercinetico
(pertanto non voluto in eccesso), aritmico,
af inalistico, diffuso a volte associato ad atetosi.

Corea benigna
ereditaria
Clinica:
Variabili
fenotipiche
• Nessun problema respiratorio /modesta disfunzione
tiroidea
• Ipotonia e ritardo motorio non necessari /disturbo
cognitivo
• Corea associata a distonia e movimento ipercinetico

Corea benigna
ereditaria
• NEUROFISIOLOGIA:
EEG/EEG sonno: nella norma.
PES/BAEPs: nella norma.
ENG-EMG: nella norma
• NEURORADIOLOGIA:
RMN encefalo-midollo /RMN spettroscopica: nella
norma.
SPECT cerebrale: nella norma.
PET-FDG (F-f luorodesossiglucosio): nella norma.

Corea benigna
ereditaria
Conclusioni
• Patologia spesso non riconosciuta per la presenza di
sintomatologia NON direttamente correlabi le al disturbo
ipercinetico
• Ipotonia centrale isolata
• Distress respiratorio /apnee del neonato “at ipiche”
• Ipotiroidismo
• Diagnosi molecolare su leucocit i e biochimica su f ibroblasti
• Possibi l i tà di buona risposta a L-Dopa soprattutto sul la
marcia /meno ef f icace su ipercinesie .
• Prognosi favorevole
• Malattia autosomica dominante: consigl io genetico

COREA BENIGNA
EREDITARIA:
GENE ADCY5

Corea benigna
ereditaria:
gene ADCY5
• Movimenti distonici, coreici (“piano playing
movements”), mioclonici che interessano
prevalentemente viso collo e arti superiori
• Presenti sin dall’ infanzia, età adolescenziale
(range 0-19 aa)
• Peggiorano in caso di stress, stanchezza o ansia
• Carattere episodico /f luttuante
• Discinesie parossistiche in sonno ed al risveglio

Corea benigna
ereditaria:
gene ADCY5

Discinesie
correlate ad
ADCY5
Ampio fenotipo clinico
Caratteristiche ricorrenti: miochimia facciale, discinesie
motorie all’addormentamento o al risveglio, episodica
postura distonica dolorosa che aumenta con lo stress o la
malattia e l'ipotonia assiale con ritardo psicomotorio.
I fenotipi non comuni includono la corea ad esordio
infantile, la distonia mioclonica, isolata distonia focale ed
emiplegia alternata.

SINDROME
DI LESCH-
NYHAN

Sindrome di
Lesch-Nyhan
• Malatt ia ereditar ia causata dal
def icit del l ’enzima ipoxantina-
guanina-fosfo-r ibosi l-transferasi
(HPRT), or iginato in seguito a
mutazioni nel gene HPRT1,
local izzato sul cromosoma X.

Sindrome di
Lesch-Nyhan
Clinica -
Disturbi del
movimento
Anamnesi prenatale e perinatale muta
Esordio con ipotonia e ritardo di acquisizione TSPM
Distonie (nei primi anni di vita), opistotono, coreoatetosi e ballismo
1/3 ca dei pazienti presenta segni piramidali (spasticità, iperreflessia,
clono)

Sindrome
di Lesch-
Nyhan -
Clinica
Iperuricemia
Agiti autolesivi: Compaiono dai 2-4 aa o in tarda adolescenza.
Disturbo del comportamento: impulsività, aggressività, disturbo oppositivo, coprolalia.
Atrofia testicolare
Anemia macrocitica non responsiva all’integrazione
Atrofia cerebrale
Disfagia
Apnee
Insufficienza respiratoria

ATASSIA
TELEANGECTASIA

Caratteristiche
• Dovuta a mutazione del gene ATM, localizzato
sul cromosoma 11q22-23
• Maschi e femmine colpiti in egual misura
• Seconda forma più comune di atassia dell’età
evolutiva, dopo l’atassia di Friedreich

Clinica
• Atassia sol i tamente come primo segno di manifestazione.
• I pazienti necessitano di sedia a rotel le entro i 10 anni di età
• Teleangectasie evidenti dai 6 anni di età, soprattutto
congiuntival i .
• Possibi le microcefal ia
• Disartria, aprassia oculomotoria, nistagmo
• Distonie, tremori, movimenti coreici e mioclono da
coinvolgimento dei gangl i del la base.
• Dal punto di vista internistico, possibi le associazione con DM,
ipercolesterolemia, steatosi, carenza di ormone del la crescita,
insuf f icienza gonadica, ster i l i tà .

GLUT-1
DEFICIENCY
SYNDROME

GLUT-1
deficiency
syndrome
• Crisi epilettiche precoci refrattarie alla
terapia AED
• Microcefalia evolutiva
• Ritardo mentale e cognitivo
• Spasticità, atassia, disartria, opsoclono,
discinesia parossistica.

GLUT-1 deficiency
syndrome
Fenotipo tipico
Fenotipo tipico (early onset<2 anni/late onset)
• Crisi epilettiche precoci refrattarie al la terapia AED
(epi lessia mioclonoastatica; assenze atipiche; apnee)
• Microcefal ia evolutiva con ipotonia
• Ritardo mentale e cognit ivo evolutivo
• Spasticità, atassia, disartria, opsoclono, disturbi del
movimento (mioclono /
• Coreoatetosi), discinesia parossistica indotta
dal l ’esercizio .

GLUT-1 deficiency
syndrome
Fenotipo atipico
Fenotipo atipico:
• disturbo del movimento /ritardo mentale
senza epilessia
• Assenze (precoci<4 anni) con sviluppo
regolare.

GLUT-1 deficiency
syndrome
Diagnosi e terapia
Clinica
Ipoglicorrachia- rapporto glucosio
liquor/glicemia (0,33,vn:0.65)
Esame molecolare: mutazioni in eterozigosi
in gene SLC2A1 (Location: 1p34.2)
Terapia: dieta chetogena
METEMOGLOBINEMIA
AUTOSOMICA
RECESSIVA
EREDITARIA TIPO II

Metaemoglobinemia autosomica
recessiva ereditaria tipo II
• Malattia autosomica recessiva ereditaria causata da
def icit di NADH citocromo b5 reduttasi (diaforasi 1).
• Prima malattia ereditaria enzimatica identif icata
• Non è categorizzata come malattia congenita del
metabolismo.
• NADH-Citocromo b5 reduttasi: localizzato sul
cromosoma 22q13-qter e contiene 9 esoni.
• Descritte 43 mutazioni in circa 60 pazienti

Metaemoglobinemia autosomica
recessiva ereditaria tipo II
Forma legata a membrane:
• local izzazione microsomiale, reticolo endoplasmatico e
mitocondriale: metabolismo
• acidi grassi, biosintesi colesterolo, farmaci, previene
la perossidazione l ipidica del le
• membrane con Coenzima Q e ascorbato.
Forma idrosolubile:
• eritrociti e batteri
• r iduzione del la metaemoglobina

Metaemoglobinemia autosomica
recessiva ereditaria tipo II
Es is tono 2 forme c l in iche:
Tipo I:
• c ianos i come un ico s intomo
• def ic i t l im i tato a l la forma er i t roc i tar ia
• missense mutat ions- r iduz ione de l la s tab i l i tà ce l lu lare
Tipo II:
• c ianos i , grave r i tardo g lobale , coreatetos i -ba l l ismo, microcefa l ia
progress iva, decesso ne l I decennio d i v i ta .
• def ic i t d i ambedue le forme
• sp l ic ing, stop-codon o f rameshi f t , mutaz ioni d ist rutt ive con
perd ita de l la funz iona l i tà enz imat ica

Metaemoglobinemia autosomica
recessiva ereditaria tipo II
Danno cellulare per perossidazione l ipidi di membrana
causa ipofunzionalità del sistemaNADH-citocromo b5
reduttasi in sinergia con coenzima Q10 ed ascorbato e
con conseguente carenza di cerebroside, riduzione della
miel ina in pochi pazienti di cui sono disponibi l i r isultati
autoptici .
Stress ossidativo mitocondriale, morte cel lulare-
apoptosi:atrof ia cerebrale-cerebellare.

Metaemoglobinemia autosomica
recessiva ereditaria tipo II - Terapia
• Vitamina C: 100mg/Kg /die per os antiossidante con
sistema NADH cit b5-riduce la metaemoglobina
(faci l ita trasporto di gruppi aci l i intramembrana
mitocondriale).
• Blu di metilene: 3mg /Kg /die per os; 1-3 mg /Kg e.v.
NB: valutare se def icit di G6PD, perchè è necessario
per formazione di leucometi lene (antidoto reale di
metaemoglobina)
• Ossigeno iperbarico-trasfusioni

Metaemoglobinemia autosomica
recessiva ereditaria tipo II - Conclusioni
• Cianosi al la nascita: dosaggio metaemoglobina
• Se elevata: prove di funzionalità biochimica ed esame
molecolare
• Avviare terapia specif ica
• Consigl io genetico ed esame molecolare in gravidanza

Pantothenate kinase-
associated neurodegeneration
(PKAN) with brain
iron accumulation: PANK2
gene;20p13-p12.3
• Pr ima de i 10 ann i : corea-d is ton ia
park inson ismo-s indrome
p i ramida le - ret in i te p igmentosa
• RMN ence fa lo : r idot to segna le inT2
in g .pa l l i do e s .n igra; occh io d i
t ig re: area iper in tensa a l l ’ in terno
d i a rea ipo intensa
• Raramente Acantoc i tos i

FOXG1 GENE
SYNDROME

FOXG1 GENE
SYNDROME

COREE
EZIOLOGIA
Primarie: genetiche e neurodegenerative
Neurometaboliche
Disimmuni
Infettive
Metaboliche - Tossiche

Coree neurometaboliche
• Mala t t i e m i tocondr ia l i : MELAS
• Ac idur i e o rgan i che: met i l -ma l on i co ac idur i a , ac idur i a prop i on i ca , g lu ta r i c o -
ac idur i a .
• Ipe rg l i c i nemia non che to t i c a
• Lesh-Nyan
• Mala t t i a d i W i l son -ace ru l op la sm inemia
• Dis turb i neuro t ra sme t t i t o r i a l i
• Cero ido l i po f usc i no s i
• Def i c i t d i c rea t ina
• Ga la t to sem ia
• Fen i l che tonur ia

COREE
EZIOLOGIA
Primarie: genetiche e neurodegenerative
Neurometaboliche
Disimmuni
Infettive
Metaboliche - Tossiche

Coree disimmuni
• Corea di Sydenham
• Sindrome da anticorpi anti fosfol ipidi
• LES
• Coree paraneoplastiche
• Corea post-vaccinale
• Corea gravidarum
• Da anticorpi anti NMDAR (N-Meti l-D-aspartate
receptor) in siero /CSF

COREA DI
SYDENHAM

Corea di
Sydenham
Definizione
• Diso rd ine neuro log ico magg iore de l la f ebbre reumat ica acu ta.
• A d i s tanza d i mes i da in fez ione da s t reptococco beta-emol i t i co de l
g ruppo A ( range: da 1 se t t imana a 3 ann i ! )
• La fo rma d i co rea p reva lente in e tà evo lut iva
• Mani fes taz ion i c l in i che: corea , emicorea, esord io con “ t i c ”=corea
f rammentata , c r is i ocu log i re , d i s turb i comportamenta l i , d isar t r ia ,
i r requ ietezza ( f idget ing) .
• Corea f l acc ida o corea para l i t i ca/ ipoton ia severa con segn i spec i f i c i
come i l “segno de l mungitore” e l a t r ip l i ce f l ess ione .
• Può essere l ’ un i ca man i fes taz ione de l la f ebbre reumat ica .

Corea di
Sydenham
Terapia
• Trattamento f isiopatologico della corea di
Sydenham: corticosteroideo /immunoglobuline
• Trattamento del disturbo del movimento:
incremento della trasmissione gabaergica e
col inergica: acido valproico, carbamazepina
• Modulazione della trasmissione dopaminergica:
neurolettici .
• Prof i lassi antibiotica: benzatin-penici l l ina i.m.
• Terapia disturbo dell ’umore, tratti OC: SSRI, presa
in carico psicologica.

COREE
EZIOLOGIA
Primarie: genetiche e neurodegenerative
Neurometaboliche
Disimmuni
Infettive
Metaboliche - Tossiche

Coree infettive
• Herpes symplex
• Mycoplasma p.
• Encefalopatia HIV
• Endocardite batterica
• Toxoplasmosi
• Cisticercosi
• Sifilide

Choreatetosis in relapsing herpes symplex
encephalitis
• Coreoatetosi a distanza di 2-4 settimane dalla f ine del trattamento
antivirale.
• Semeiologia: disturbo della coscienza, coreoatetosi crisi epilettiche.
• Choreatetosis in relapsing herpes symplex encephalitis
• Durata, dosi di antivirali non idonee
• Riattivazione Herpes virus?
• Meccanismo immunomediato?

COREE
EZIOLOGIA
Primarie: genetiche e neurodegenerative
Neurometaboliche
Disimmuni
Infettive
Metaboliche - Tossiche

Coree metaboliche-tossiche
• Ipo- ipernatr iemia
• Pos tpump chorea .
• Ipoca lcemia
• Iper t i ro id i smo-Ipoparat i ro id ismo
• Insuf f i c ienza rena le -epat ica
• Por f i r i a acu ta - in termi t tente (pos ter ior revers ib le encepha lopathy
syndrome)
• In toss icaz ione da monoss ido d i carbon io/mercur io
• Farmac i : an tagonis t i D2; AED (PHT;CBZ;VPA); an fe tamine; bac lo fen;
l i t i o ; an t idepress iv i t r i c ic l i c i ; s te ro id i ; contraccet t i v i o ra l i ;
c i c lospor ina

Postpump chorea: movimenti coreatetoidi entro
due settimane da un bypass cardiopolmonare
• assoc iaz ione con ipotermia profonda, arresto
cardiocirco lator io
• assenza d i anomal ie macroscopiche a RMN/TAC (atrof ia
nuc lei del la base e non les ioni focal i ) suggerendo un’ ipotesi
tromboembol ica o biochimica
• r itardo di svi luppo /cognit ivo possib i le
• prognosi r iservata.

APPROCCIO
CLINICO

WORK UP
DIAGNOSTICO
NELLE COREE
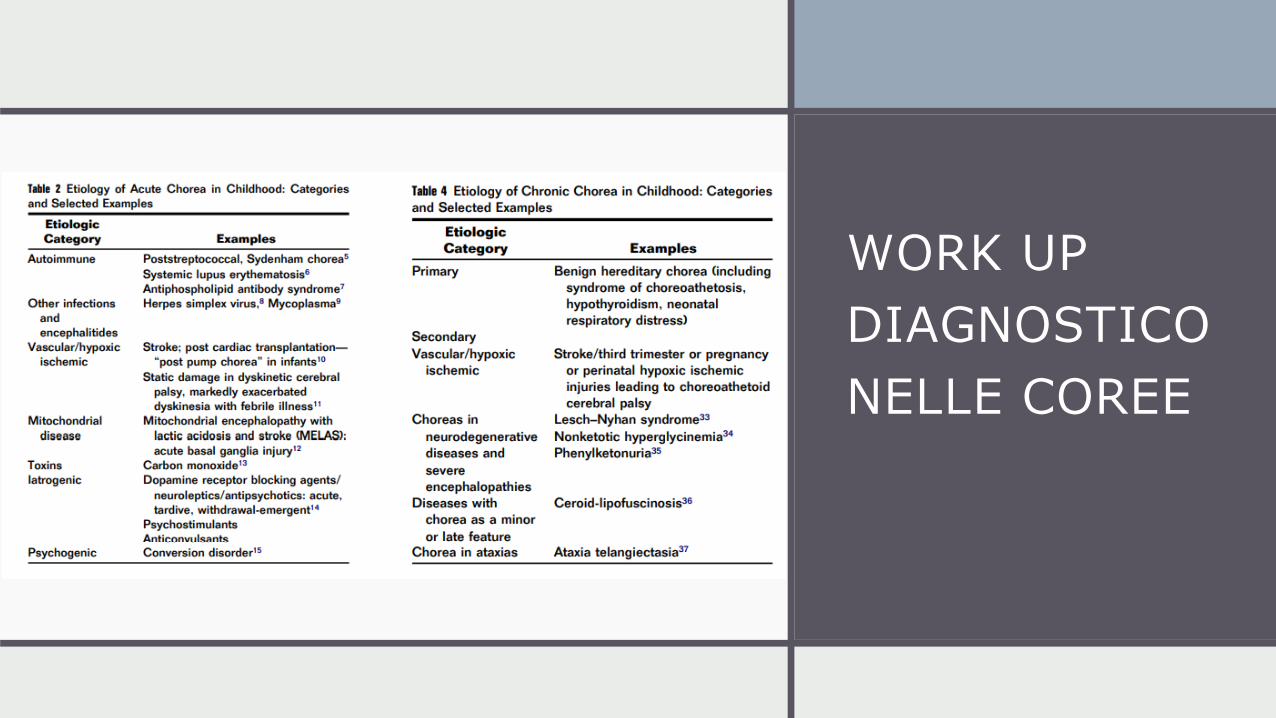
WORK UP
DIAGNOSTICO
NELLE COREE


BIBLIOGRAFIA

GRAZIE DELL’ATTENZIONE