desenvolvimento e validaÇÃo de metodologias …livros01.livrosgratis.com.br/cp100635.pdf ·...
TRANSCRIPT

1
UNIVERSIDADE FEDERAL DA PARAÍBA
LABORATÓRIO DE TECNOLOGIA FARMACÊUTICA – LTF
PROGRAMA DE PÓS-GRADUAÇÃO EM PRODUTOS NATURAIS
E SINTÉTICOS BIOATIVOS
DESENVOLVIMENTO E VALIDAÇÃO DE METODOLOGIAS ANALÍTICAS PARA QUANTIFICAÇÃO DE TIBOLONA
MANIPULADA POR FARMÁCIAS MAGISTRAIS DA PARAÍBA
ALUNO: JOSÉ ALVES CÂNDIDO
ORIENTADOR: Prof. Dr.EDUARDO DE JESUS OLIVEIRA
JOÃO PESSOA – 2008

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
JOSÉ ALVES CÂNDIDO
ORIENTADOR: Prof. Dr.EDUARDO DE JESUS OLIVEIRA
DESENVOLVIMENTO E VALIDAÇÃO DE METODOLOGIAS ANALÍTICAS PARA QUANTIFICAÇÃO DE TIBOLONA
MANIPULADA POR FARMÁCIAS MAGISTRAIS DA PARAÍBA
ÁREA DE CONCENTRAÇÃO: FARMACOQUÍMICA
JOÃO PESSOA - 2008
Dissertação apresentada à coordenação do Programa de Pós-Graduação em Produtos Naturais e Sintéticos Bioativos da Universidade Federal da Paraíba, em cumprimento às exigências para obtenção do Grau de Mestre.

3
JOSÉ ALVES CÂNDIDO
DESENVOLVIMENTO E VALIDAÇÃO DE METODOLOGIAS
ANALÍTICAS PARA QUANTIFICAÇÃO DE TIBOLONA
MANIPULADA POR FARMÁCIAS MAGISTRAIS DA
PARAÍBA
Aprovada em: ______ / ______ / ______
COMISSÃO EXAMINADORA
_______________________________________________________ Prof. Dr.Eduardo de Jesus Oliveira (UFPB - Orientador)
_______________________________________________________ Prof. Dr. Fábio Santos de Souza (UFPB – CCS)
_______________________________________________________ Profª. Drª. Sayonara Maria Lia Fook (UEPB – DF)

4
A minha esposa Miriam.
Ao meu filho Gabriel.
Aos meus pais Cinira e Antônio “in memoriam”.
Aos meus tios Anésia e José “in memoriam”.

5
AGRADECIMENTOS
Ao Prof. Dr. Eduardo de Jesus Oliveira por aceitar a orientação deste trabalho e pela contribuição com as relevantes sugestões. A Agência Estadual de Vigilância Sanitária da Paraíba (AGEVISA), nas pessoas do Prof. Hermano Toscano, Diretor Geral, pela compreensão nas minhas ausências e a Jorge Molina, quando Diretor Geral, pela mesma razão. A Agência Nacional de Vigilância Sanitária (ANVISA) pela cooperação técnica. Aos amigos Sócrates e Roberto, sem a colaboração dos quais este trabalho não teria sido realizado. A minha irmã Maria, minhas sobrinhas e sobrinhos, meu irmão Abelson e em especial ao meu irmão Everaldo, pela força, incentivo, pelas importantes sugestões e a preciosa colaboração na digitação e formatação deste trabalho. Em particular ao sobrinho Dimas, pela estimulante companhia nas corridas, para aliviar o estresse das atividades diárias. Aos professores Givaldo do Departamento de Farmácia, e José Rodrigues do Departamento de Química, pela importante e significativa contribuição na minha formação acadêmica. Aos colegas da AGEVISA, em especial aos da DTMAPT, pela compreensão nos momentos das minhas ausências, especialmente ao colega e amigo João Peixoto, pela força e incentivo para que eu realizasse este projeto. Ao Prof. Dr. Fábio Souza dos Santos pelas valiosas e importantes sugestões. A todos os professores do Programa de Pós-Graduação em Produtos Naturais e Sintéticos Bioativos pelos ensinamentos recebidos. A Coordenação da Pós-Graduação e a secretária Tânia Maria Alves pela execução da parte burocrática deste trabalho. Aos estagiários, Eugênia, Victor, Michelle, Marcela e Sanclayver pela valiosa colaboração neste trabalho. Aos funcionários do LTF, Wellington e Kazuko, pelo apoio técnico. Aos colegas Alexsandro e Joviânia pela companhia e colaboração durante as longas jornadas do trabalho experimental. A todos os colegas da minha turma de Mestrado, e demais colegas do Doutorado pelo companheirismo, pela solidariedade nas horas difíceis, pelos momentos de descontração e pelas amizades que construímos.

6
O melhor soldado não ataca. O lutador superior vence sem violência.
O maior dos conquistadores vence sem esforço. O gerente mais bem-sucedido dirige sem impor.
Isso é chamado não-agressividade inteligente. Isso é chamado superioridade dos homens.
Lao-Tsé, Tao Te King.

7
RESUMO A tibolona (®Livial, Organon) é um fármaco utilizado principalmente na
terapia de reposição hormonal para o tratamento dos sintomas da menopausa,
sendo também comercializada no Brasil em farmácias magistrais na forma de
cápsulas de 2,5 mg. Não há metodologias de análise para formulações a base de
tibolona descritas na literatura, nem métodos oficiais farmacopéicos para
quantificação deste fármaco. Sendo assim, o presente trabalho descreve o
desenvolvimento, a validação e a aplicação de metodologias analíticas à
avaliação da uniformidade de conteúdo de cápsulas de tibolona manipuladas em
farmácias magistrais do Estado da Paraíba. Foram desenvolvidas duas
metodologias analíticas para quantificação da tibolona, sendo a primeira utilizando
CLAE e derivatização pré-coluna com dansilhidrazina e detecção por
fluorescência, e a segunda utilizando CLAE e detecção direta no ultravioleta (207
nm). Todavia, pela simplicidade da técnica, apenas a metodologia por detecção
em UV foi validada e aplicada. Utilizou-se um desenho fatorial fracionário e
análise de regressão linear multivariada a fim de investigar os efeitos das
condições de derivatização da tibolona com dansilhidrazina sobre o rendimento
da reação. Os melhores rendimentos foram obtidos com uma razão molar entre a
dansilhidrazina e a tibolona de 40, um tempo de reação de 20 minutos e uma
temperatura de 48ºC. O método desenvolvido utilizou para a separação do
derivado fluorescente uma coluna monolítica Merck Chromolith C-18
(100mmx4,6mm de DI), e uma fase móvel isocrática que consistiu de um tampão
acetato de amônio 50mM, pH 6.8 e acetonitrila (92,5:7,5 v/v),a um fluxo de 3
mL/min. A detecção do derivado fluorescente (tempo de retenção = 6,69min.) foi
feita utilizando-se um comprimento de onda de excitação de 350nm e de emissão
de 520 nm. O método de quantificação por detecção direta no ultravioleta foi
desenvolvido e validado de acordo com a Resolução RE 899/2003/ANVISA, e
mostrou-se específico, linear na faixa de concentração utilizada (0,001-0,5
mg/mL), preciso (CV% ≤ 5%); exato (85 a 120%) e robusto, dentro das condições
especificadas. A aplicação do método foi realizada através da determinação do
teor da uniformidade de conteúdo das cápsulas de treze farmácias magistrais do
Estado da Paraíba. Os resultados obtidos pela aplicação do método

8
demonstraram que das farmácias analisadas, apenas 38% tiveram suas cápsulas
em conformidade com as especificações de uniformidade de conteúdo (teor entre
85 a 115%). Já com relação ao peso médio, todas as amostras analisadas
estavam dentro dos limites de variação farmacopéicos estabelecidos (± 10%) para
formas de dosagem menores que 300 mg. Conclui-se a partir dos dados obtidos
que as tecnologias de mistura de pós e excipientes utilizados em escala magistral
não estão fornecendo a uniformidade de mistura necessária para a obtenção de
cápsulas de tibolona com dosagem uniforme, e que a falta de uniformidade de
conteúdo nas cápsulas não é reflexo de variações no seu peso médio. Palavras chave: Tibolona; Farmácia Magistral; Cápsulas; Derivatização;
Dansilhidrazina; CLAE; Uniformidade de Conteúdo.

9
ABSTRACT Tibolone (®Livial, Organon) is a drug used chiefly as hormone
replacement therapy for treatment of menopausal symptoms and is also marketed
in Brazil by compounding pharmacies as 2.5 mg capsules. There are no methods
for the analysis of tibolone dosage forms described in the literature or in official
compendia. Thus the present work describes the development, validation and
application of an analytical method for the quantification of this drug to evaluate
the uniformity of dosage forms (2.5 mg hard gelatin capsules) of tibolone sold by
compounding pharmacies in the sate of Paraiba. Two methods were developed for
tibolone quantification, the first one being an HPLC assay after pre-column
derivatization of tibolone with dansylhydrazine and fluorescence detection, and the
second one using HPLC with direct detection in the UV (207 nm). However,
because of its simplicity, only the second method was fully validated and further
used. A fractional factorial design and multiple linear regression were used in
order to evaluate the effects of reaction conditions in the derivatization of tibolone
with dansylhydrazine. The best reaction yield was obtained using a molar ratio of
dansylhydrazine to tibolone of 40, a reaction time of 20 minutes and a temperature
of 48 ºC. The developed method used for the separation of the fluorescent
hydrazone, a monolithic column (Merck Chromolith C-18, 100mmx4,6mm of ID ), a
mobile phase consisting of a 50mM ammonium acetate buffer pH 6.8 and
acetonitrile (92.5:7,5 v/v) delivered isocratically at a flow rate of 3 mL/min. The
detection of the fluorescent derivative (retention time= 6,69 min.) was done using
an excitation wavelength of 350 nm and an emission wavelength of 520 nm. The
method using direct detection in the UV was developed and validated as directed
by the Brazilian regulatory authority (RE 899/2003/ANVISA) and proved to be
selective, linear (from 0,001-0,5 mg/mL), precise (CV% ≤ 5%); exact (85 to 120%)
and rugged. The method was used to determine the uniformity of dosage forms
obtained in 13 compounding pharmacies in the state of Paraiba. The results
demonstrated that only 38% (5 pharmacies) met the specifications of uniformity of
solid dosage forms (assay of individual capsules between 85 to 115%). As for the
weight determination of the dosage forms all pharmacies complied with the
specifications (± 10% for dosage forms with weight below 300 mg). From the

10
results obtained it is possible to conclude that the procedures in use for powder
mixing in compounding pharmacies are not being able to result in uniform powder
for the filling of the capsules, and that the lack of uniformity is not related to weight
variation.
Key words: Tibolone; ketosteroids; hormonal replacement; derivatization;
dansylhydrazine; HPLC

11
LISTA DE FIGURAS
FIGURA 1 – Estrutura química da tibolona ........................................................ 37 FIGURA 2 – Principais metabólitos da tibolona ................................................. 38 FIGURA 3 – Reação de derivatização da tibolona com dansilhidrazina ............ 42 FIGURA 4 – Cromatograma da reação de derivatização mostrando a eluição do pico da tibolona no volume morto. ...................................................................... 46 FIGURA 5 – Cromatograma mostrando picos duplos obtidos para hidrazonas syn e anti quando uma solução de progesterona padrão a 400 μM foi derivatizada com dansilhidrazina .................................................................................................... 50 FIGURA 6 – Formação de hidrazonas syn e anti diastereoisoméricas .............. 51 FIGURA 7 – Cromatograma representativo da separação do derivado fluorescente obtido a partir de uma solução de tibolona a 3 μg/mL, (TR = 6,7 min) ............................................................................................................................. 51 FIGURA 8 – Espectro UV da tibolona na faixa de 190 a 600 nm na concentração de 0,1 mg/mL em solução de metanol/água (70:30) .......................................... 58 . FIGURA 9 – Cromatograma da tibolona com ACN/H2O (90:10) ........................ 58 FIGURA 10 – Cromatograma da tibolona com ACN/H2O (50:50) ..................... 59 FIGURA 11 – Cromatograma tibolona com ACN/H2O (70:30) .......................... 59 FIGURA 12 – Cromatograma representativo da tibolona padrão não degradada ............................................................................................................................ 64 FIGURA 13 – Cromatograma representativo da tibolona padrão submetida à degradação térmica a 50 ºC ............................................................................... 65 FIGURA 14 – Superposição dos cromatograma representativos da tibolona não degradada e degradada a 50 ºC ........................................................................ 65 FIGURA 15 – Superposição dos cromatogramas representativos das curvas de calibração da tibolona padrão obtidas no 1º dia de análise. ............................. 66 FIGURA 16 – Superposição dos cromatogramas representativos das curvas de calibração da tibolona padrão obtidas no 2º dia de análise. .............................. 67 FIGURA 17 – Superposição dos cromatogramas representativos das curvas de calibração da tibolona padrão obtidas no 3º dia de análise. .............................. 68

12
LISTA DE GRÁFICOS
GRÁFICO 1 – Modelamento dos dados da reação de derivatização de tibolona por regressão linear multivariada (RLM) mostrando os valores de rendimento previstos versus observados ............................................................................... 48 GRÁFICO 2 – Efeito das interações dos parâmetros de reação investigados sobre o rendimento da reação de derivatização ........................................................... 48 GRÁFICO 3 a e b – Mapas de resposta de superfície mostrando os efeitos da razão molar dansilhidrazina X tibolona, em relação ao tempo de reação (a) e a temperatura (b), sobre o rendimento da reação de derivatização ....................... 49 GRÁFICO 4 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 1º dia, no intervalo das concentrações estabelecidas..67 GRÁFICO 5 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 2º dia, no intervalo das concentrações estabelecidas.. 68 GRÁFICO 6 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 3º dia, no intervalo das concentrações estabelecidas.. 69 GRÁFICO 7 – Gráfico das médias das três curvas de calibração ....................... 70 GRÁFICO 8 – Gráfico de resíduos dos dados de linearidade ............................. 71 GRÁFICO 9 – Curva de calibração para determinação da precisão e exatidão obtida no 1º dia .................................................................................................... 72 GRAFICO 10 – Curva de calibração para determinação da precisão e exatidão obtida no 2º dia .................................................................................................... 73 GRÁFICO 11 – Curva de calibração para determinação da precisão e exatidão obtida no 3º dia .................................................................................................... 74 GRÁFICO 12 – Resultado da variação individual da uniformidade de conteúdo nas farmácias analisadas .................................................................................... 83

13
LISTA DE TABELAS TABELA 1 – Classificação dos testes de validação segundo a resolução RE nº 899/ANVISA ........................................................................................................ 32 TABELA 2 – Afinidade da tibolona e seus metabólitos por receptores de esteróides sexuais .............................................................................................. 39 TABELA 3 – Métodos de detecção para quantificação da tibolona em estudos farmacocinéticos ................................................................................................. 41 TABELA 4 – Parâmetros experimentais sugeridos pelo desenho fatorial fracionário usando um programa Modde v.4.0 (Umetrix) para derivatização da Tibolona .............................................................................................................. 45 TABELA 5 – Rendimentos previstos para derivatização da tibolona nas condições experimentais sugeridas pelo desenho fatorial fracionário usando um programa Modde v.4.0 (Umetrix) ........................................................................................ 47 TABELA 6 – Condições cromatográficas desenvolvidas para quantificação da tibolona por derivatização com detecção por fluorescência ............................... 52 TABELA 7 – Condições cromatográficas desenvolvidas para quantificação da tibolona por detecção direta com UV .................................................................. 60 TABELA 8 – Resolução dos picos cromatográficos da tibolona não degradada e termodegradada e suas porcentagens de degradação ...................................... 65 TABELA 9 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 1º dia ......................................................... 67 TABELA 10 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 2º dia ......................................................... 68 TABELA 11 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 3º dia ......................................................... 69 TABELA 12 – Média das áreas dos picos cromatográficos obtidos nos três dias para construção da curva de calibração da tibolona padrão .............................. 70 TABELA 13 – Cálculo dos resíduos em relação às três curvas de calibração obtidas na determinação da linearidade ............................................................. 71 TABELA 14 – Valores de precisão e exatidão obtidos para as concentrações B, M e A no 1º dia ........................................................................................................ 72 TABELA 15 – Valores de precisão e exatidão obtidos para as concentrações B, M e A no 2º dia ........................................................................................................ 73

14
TABELA 16 – Valores de precisão e exatidão obtidos para as concentrações, B, M e A no 3º dia .................................................................................................... 74 TABELA 17 – Valores médios de precisão e exatidão obtidos para as concentrações B, M e A nos três dias (inter-dias) .............................................. 75 TABELA 18 – Valores das concentrações B, M e A obtidas na determinação da robustez utilizando 68% de acetonitrila na fase móvel no 1º dia ........ ............... 76 TABELA 19 – Valores das concentrações B, M e A obtidas na determinação da robustez utilizando 72% de acetonitrila na fase móvel no1º dia ......................... 76 TABELA 20 – Valores das concentrações B, M e A obtidas na determinação da robustez utilizando 68% de acetonitrila na fase móvel no 2º dia ........................ 77 TABELA 21 – Valores das concentrações B, M e A obtidas na determinação da robustez utilizando 72% de acetonitrila na fase móvel no 1º dia ........................ 77 TABELA 22 – Valores médios das concentrações, B, M e A obtidas utilizando 68% de acetonitrila na fase móvel nos dois dias, juntamente com a média DP e coeficiente de variação (CV%) ............................................................................ 78 TABELA 23 – Valores médios das concentrações, B, M e A obtidas utilizando 72% de acetonitrila na fase móvel nos dois dias, juntamente com a média DP e coeficiente de variação (CV%) ............................................................................ 78 TABELA 24a – Resultados obtidos na determinação do Peso Médio das cápsulas de tibolona (2,5 mg), manipuladas nas Farmácias A, B, C, D, E, F e G ............. 81 TABELA 24b – Resultados obtidos na determinação do Peso Médio das cápsulas de tibolona (2,5 mg), manipuladas nas Farmácias H, I, J, K, L e M ................... 82 TABELA 25 – Resultados obtidos nas determinações de uniformidade de conteúdo das cápsulas de tibolona (2,5 mg) manipuladas nas Farmácias A, B, C, D, E, F, G, H, I, J, K, L e M ................................................................................. 84

15
LISTA DE ABREVIATURAS E SIGLAS ACN Acetonitrila AGEVISA Agência Estadual de Vigilância Sanitária ANFARMAG Associação Nacional de Farmacêuticos Magistrais ANVISA Agência Nacional de Vigilância Sanitária BP, 2000 British Pharmacopoeia, 2000 C18 Octadecilsilano CG/EM Cromatografia gasosa acoplada a espectrometria de massas CLAE Cromatografia Líquida de alta eficiência CLAE/DAD Cromatografia líquida de alta eficiência acoplada ao detector de arranjo de diodo CLAE/EM Cromatografia líquida de alta eficiência acoplada a espectrometria
de massas CV Coeficiente de Variação DAD Detector com Arranjo de Diodos D.O.U. Diário Oficial da União DP Desvio Padrão DSH Dansilhidrazina EM Espectrometria de massas ENSP Escola Nacional de Saúde Pública FB, 1998 Farmacopéia Brasileira, 1998 FB, 2004 Farmacopéia Brasileira, 2004 FDA Food and Drug Administration INCQS Instituto Nacional de Controle de Qualidade em Saúde IV Infravermelho LOD Limite de detecção LOQ Limite de quantificação NIRS Near Infrared Spectroscopy PAT Processos analíticos tecnológicos PF Ponto de fusão RDC Resolução da diretoria colegiada RE Resolução específica RSD Desvio-padrão relativo SQR Substância química de referência USP United States Pharmacopeia USP, 2000 The United States Pharmacopeia USP 24, 2000 UV Ultravioleta UV-VIS Ultravioleta-visível

16
SUMÁRIO CAPÍTULO I - INTRODUÇÃO 1 INTRODUÇÃO ................................................................................................. 19
1.1 JUSTIFICATIVA ........................................................................................... 22 1.2 OBJETIVOS .................................................................................................. 23
1.2.1 Objetivo Geral ........................................................................................... 23 1.2.2 Objetivos Específicos .............................................................................. 23
CAPÍTULO II - REVISÃO DA LITERATURA 2 REVISÃO DA LITERATURA ........................................................................... 25
2.1 SITUAÇÃO ATUAL DOS MEDICAMENTOS MANIPULADOS NO BRASIL . 25
2.2 PROCESSO DE MISTURA E HOMOGENEIDADE DE CONTEÚDO DE
CÁPSULAS ......................................................................................................... 27
2.3 DESENVOLVIMENTO E VALIDAÇÃO DE METODOLOGIAS ANALÍTICAS 31
2.4 UTILIZAÇÃO DA ANÁLISE FATORIAL COMO FERRAMENTA ESTATÍSTICA
............................................................................................................................. 35
2.5 CARACTERÍSTICAS FISOCOQUÍMICAS E FARMACOLÓGICAS DA
TIBOLONA .......................................................................................................... 36
2.5.1 Características químicas ....................................................................... 37
2.5.2 Ação farmacológica ................................................................................ 37
2.5.3 Indicações ................................................................................................ 39
2.5.4 Apresentações e Dosagens .................................................................... 39
2.5.5 Farmacocinética da Tibolona ................................................................. 39 2.6 TIBOLONA E MÉTODOS PARA SUA DETECÇÃO ..................................... 40
CAPÍTULO III - QUANTIFICAÇÃO DA TIBOLONA POR
DERIVATIZAÇÃO COM DETECÇÃO POR FLUORESCÊNCIA 3 DESENVOLVIMENTO DO MÉTODO .............................................................. 44 3.1 MATERIAIS ................................................................................................... 44
3.2 METODOLOGIA ........................................................................................... 45
3.3 RESULTADOS e DISCUSSÃO ..................................................................... 46

17
CAPÍTULO IV - QUANTIFICAÇÃO DA TIBOLONA POR
DETECÇÃO DIRETA NO UV 4 DESENVOLVIMENTO, VALIDAÇÃO E APLICAÇÃO DO MÉTODO ............. 55
4.1 MATERIAIS ................................................................................................... 54
PARTE I 4.2 DESENVOLVIMENTO DO MÉTODO ........................................................... 56
4.2.1 Metodologia .............................................................................................. 56
4.2.2 Resultados e Discussão .......................................................................... 57
PARTE II 4.3 VALIDAÇÃO DO MÉTODO .......................................................................... 61
4.3.1 Metodologia .............................................................................................. 61
4.3.2 Resultados e Discussão .......................................................................... 63
PARTE III 4.4 APLICAÇÃO DO MÉTODO: ANÁLISE DE CÁPSULAS DE TIBOLONA
MANIPULADA ..................................................................................................... 79
4.4.1 Metodologia .............................................................................................. 79
4.4.2 Resultados e Discussão.......................................................................... 80
CAPÍTULO V - CONSIDERAÇÕES FINAIS
5 CONSIDERAÇÕES FINAIS ................................................................. 88 REFERÊNCIAS ANEXOS

18
CAPÍTULO I
INTRODUÇÃO

19
1 INTRODUÇÃO
A utilização de formulações medicamentosas pelo homem o acompanha
desde os primórdios da humanidade. No mundo primitivo algumas pessoas já
demonstravam um significativo conhecimento terapêutico, utilizando sistemas que
combinavam elementos empíricos, racionais, religiosos e mágicos. Foi na
Mesopotâmia, antes de Cristo que se produziu o primeiro Compêndio
Farmacêutico. A parte traduzida consiste em uma série de fórmulas incluindo seu
modo de preparação. No antigo Egito, foi encontrado o Papiro de Ebers, o mais
importante papiro encontrado, escrito em aproximadamente 1500 a.C. contendo
811 prescrições e 700 drogas mencionadas. Os precursores das farmácias
modernas surgiram no início do Século X, quando ainda eram denominadas de
apotecas ou boticas.
Até a revolução Industrial a farmácia tinha uma importância fundamental na
sociedade, pois os farmacêuticos manipulavam e produziam todos os
medicamentos disponíveis, de acordo com as farmacopéias e as prescrições
médicas. Com o advento da industrialização, as farmácias foram perdendo o
prestígio adquirido do ponto de vista da saúde pública e social, tendo os
farmacêuticos a partir daí se voltado para as atividades industriais (ANTUNES
JÚNIOR, 2002).
Na década de 90 fatos preocupantes aconteceram no mercado
farmacêutico brasileiro; como a descoberta da comercialização de medicamentos
falsificados, fato que se constituiu em um verdadeiro atentado à saúde pública
brasileira, uma vez que levou à morte muitos usuários destes medicamentos.
Como a então Secretaria Nacional de Vigilância Sanitária, órgão responsável pela
regulação sanitária no país, não conseguiu reagir à altura a esta nova realidade
nacional, por não possuir autonomia financeira e administrativa, foi criada a
Agência Nacional de Vigilância Sanitária (ANVISA), apresentando um novo
modelo de gestão, caracterizado por autonomia financeira e administrativa, com
objetivo de promover e proteger a saúde da população. A ANVISA, com o seu
poder regulador, colocou em prática várias atividades e novas regulamentações,

20
apoiada neste período pela então recém-criada Lei dos Genéricos (BRASIL,
ANVISA, 1999e).
Com o padrão de qualidade adquirido com os genéricos, os medicamentos
similares passaram a ser o “nó crítico” do Sistema Nacional de Vigilância
Sanitária, tendo em vista que tais produtos não tinham a mesma eficácia
comprovada obtida pelos genéricos. A ANVISA, em consonância com a política
nacional de medicamentos do Ministério da Saúde redefiniu as regras para o
registro de medicamentos. Estas mudanças foram baseadas em alguns critérios,
como verificação da qualidade quanto à reprodutibilidade, segurança e eficácia
terapêutica; exigência de certificação das Boas Práticas de Fabricação para a
concessão de registro para cada linha de produção de medicamentos; controle da
matéria-prima; ampliação do monitoramento da qualidade dos medicamentos em
comercialização; redução do número de associações irracionais e participação
nas estratégias que facilitam o acesso a medicamentos pela maioria da
população. Todavia, em 2003, a Agência conseguiu estabelecer a transição de
todo mercado de similares para as mesmas exigências de segurança e eficácia
aplicadas aos genéricos, (BRASIL, ANVISA, 2005g).
A manipulação farmacêutica magistral no país passou por grandes
transformações no que se refere a sua regulamentação. Mesmo assim, muitos
dos processos de mudanças ocorridos na indústria farmacêutica não foram
acompanhados pelos medicamentos manipulados, tendo em vista que a análise
dos mecanismos de controle de qualidade, existentes nas farmácias magistrais,
nos leva a supor que são insuficientes para garantir a comprovação da eficácia e
segurança deste tipo de medicamentos, pois os mesmos não passam por testes
de biodisponibilidade, bioequivalência ou mesmo teste de equivalência
farmacêutica.
Os problemas relacionados a desvios de qualidade de medicamentos
manipulados que provocaram diversas internações hospitalares e a morte de
várias pessoas, inclusive crianças (BRASIL, ANVISA, 2008h), fizeram a
autoridade sanitária federal (ANVISA), intervir de forma a aumentar o rigor dos
ensaios de controle da qualidade através de suas resoluções, no sentido de

21
minimizar os riscos provenientes de manipulações mal feitas devido à falta de
controle nos seus processos de manipulação desde a matéria prima até o produto
acabado, para que o segmento magistral possa garantir a eficácia e segurança
desses medicamentos.
Essas dificuldades passam pela característica particular do processo
magistral, que é produzir o medicamento de forma individualizada, aliada à
situação vigente no Brasil, onde ao contrário de outros paises, as farmácias
manipulam todos os grupos de medicamentos – hormônios, antibióticos e
antineoplásicos, inclusive os de alto risco sanitário como os de baixo índice
terapêutico; daí a exigência de se aumentar cada vez mais o rigor nas análises de
controle da qualidade de todo o processo de manipulação.

22
1.1 JUSTIFICATIVA Para a realização do presente trabalho de pesquisa, foi escolhido o fármaco
tibolona por ser considerado um fármaco de baixa dosagem (2,5 mg) e por estar
entre os mais manipulados desta categoria nos estabelecimentos de manipulação
do Estado. Além disso, não existe nenhuma especificação farmacopéica para o
desenvolvimento de metodologias analíticas na determinação de tibolona em
formulações farmacêuticas. Este trabalho pretende assim, dar uma contribuição
importante na determinação do monitoramento da qualidade do medicamento
tibolona manipulado pelas farmácias magistrais da Paraíba, bem como socializar
o trabalho realizado, informando os resultados obtidos e abrindo possibilidades de
colaboração com a AGEVISA, no sentido de que a mesma possa intervir no setor,
de forma educativa, nos casos de resultados de desvios de qualidade, a fim de
que sejam estabelecidas as ações corretivas pelos referidos estabelecimentos e
para que eles possam garantir a qualidade e eficácia dos seus produtos, trazendo
confiança e tranqüilidade para o usuário de medicamentos manipulados.

23
1.2 OBJETIVOS
1.2.1 Objetivo Geral
• Desenvolver e validar metodologias analíticas para quantificar
o medicamento tibolona em cápsulas gelatinosas duras,
manipulado pelas farmácias magistrais da Paraíba.
1.2.2 Objetivos Específicos
• Desenvolver metodologia analítica para a quantificação de tibolona
em formulações farmacêuticas;
• Avaliar diferentes estratégias de detecção para a quantificação da
tibolona por CLAE (fluorescência e ultravioleta);
• Avaliar se a uniformidade de conteúdo das cápsulas de tibolona,
adquiridas em farmácias com manipulação no Estado da Paraíba,
encontra-se dentro dos parâmetros farmacopéicos exigidos;
• Identificar os principais desvios de qualidade encontrados neste
medicamento.

24
CAPÍTULO II
REVISÃO DA LITERATURA

25
2 REVISÃO DA LITERATURA 2.1 SITUAÇÃO ATUAL DOS MEDICAMENTOS MANIPULADOS NO BRASIL
Apesar de Anderson de Oliveira Ferreira afirmar que a suposta ausência de
um controle de qualidade mais rigoroso nas matérias-primas e produtos
acabados, bem como a ausência de controle do processo de produção ser o
maior obstáculo para o crescimento das farmácias magistrais (FERREIRA, 2002)
este segmento no Brasil teve um crescimento bastante significativo nos últimos
oito anos (2000 a 2008). Estima-se que existam na atualidade cerca de cinco mil
e quinhentas farmácias magistrais em todo Brasil (SINAMM, 2008), sendo que na
Paraíba, segundo o cadastro da AGEVISA nesse mesmo período este número
passou de cerca de dezessete para cinqüenta e cinco estabelecimentos, um
crescimento de 323%.
Todo este crescimento deveria ter sido aliado à implantação de sistemas
de garantia da qualidade, à informatização, ao emprego de novas tecnologias e
ao cumprimento das legislações sanitárias, que dentre outros fatores, são alguns
dos caminhos recomendáveis para o crescimento sustentado do setor
(FERREIRA, 2002).
De acordo com Luís Felipe Moreira Lima:
[...] prescrição de formulações magistrais é feita levando-se em conta as características individuais do paciente e a sua patologia, daí os produtos resultantes desta manipulação são específicos para cada caso, estando a sua preparação limitada a poucas unidades, de acordo com as exigências terapêuticas. Por isso, a realização dos testes de biodisponibilidade e bioequivalência, torna-se praticamente inviável, tanto do ponto de vista técnico quanto do ponto de vista da viabilidade econômica [...] (LIMA et al., 1993).
Até o ano 2000 não existiam normas específicas no âmbito nacional que
regulamentassem o setor magistral, e contemplassem procedimentos
operacionais padronizados no sentido de atender às Boas Práticas de
Manipulação. Somente naquele ano, a ANVISA publicou a resolução RDC-

26
33/ANVISA, de 19 de abril de 2000, que aprova o regulamento técnico sobre as
Boas Práticas de Manipulação de Medicamentos em Farmácias (BRASIL,
ANVISA, 2001a). Apesar de esta resolução ter sido considerada um avanço na
regulamentação da manipulação no Brasil, ainda assim deixou algumas lacunas,
dando margem para que as farmácias pudessem manipular qualquer tipo de
medicamento, como aqueles de baixa dosagem incluindo os de baixo índice
terapêutico, sem apresentar um mecanismo de controle adequado que pudesse
garantir a qualidade dos mesmos. Também deu margem para a manipulação de
medicamentos com indicações terapêuticas diferentes daquelas registradas junto
ao órgão sanitário (ANVISA), a exemplo da clonidina, que se encontra registrado
com a única indicação de antihipertensivo e que vinha sendo prescrita e
manipulada na maioria dos casos para uso em crianças e adolescentes, com
baixa estatura, para promover o crescimento (BRASIL, ANVISA, 2003i). A
manipulação da clonidina com este desvio de indicação teve trágicas implicações,
inclusive com o óbito de uma criança de 12 anos, após fazer uso de cápsulas de
clonidina manipuladas em uma farmácia no Distrito Federal no ano de 2003, que
após análise foi constatado um teor de princípio ativo cem vezes maior do que o
declarado (BRASIL, ANVISA, 2003j).
Artigo publicado no site da ANVISA relata que os pesquisadores Gisele Ruf
da Escola Nacional de Saúde Pública (ENSP) e Francisco José Roma
Paumgartten do Instituto Nacional de Controle de Qualidade em Saúde (INCQS),
em palestra proferida como parte da oficina Medicamentos manipulados: Um
desafio para a Vigilância Sanitária, apresentaram dados preocupantes sobre
resultados de laudos laboratoriais de amostras de medicamentos manipulados.
A pesquisadora Gisele Ruf revelou também que entre os anos de 2000 e
2005 o INCQS recebeu amostras de medicamentos manipulados para análises,
relacionados ao relato de 51 casos de gravidade elevada com respeito ao
consumo destes medicamentos, inclusive oito sendo de óbitos, a maioria
crianças, e pelo menos 14 internações hospitalares. Gisele afirma ainda que em
todos os casos, os medicamentos apresentavam ou excesso de quantidade das
substâncias prescritas ou até mesmo algumas que não constavam na prescrição
original.

27
Já o pesquisador Francisco Paumgartten relatou que em 2003, foram
registrados óbitos de quatro crianças por uso inapropriado de medicamentos
manipulados contendo teores de princípios ativos acima do limite estabelecido.
Ainda segundo estes pesquisadores, em 2004 novas mortes ocorreram na Bahia
e em São Paulo. Na sua apresentação, o pesquisador mostrou alguns desvios de
qualidade mais freqüentes, como: excesso ou quantidade insuficiente de princípio
ativo; falta de análise das matérias-primas; heterogeneidade de conteúdo - com
acentuada divergência de quantidade de princípio ativo entre unidades de um
mesmo frasco (BRASIL, ANVISA, 2008d).
A ocorrência destes casos gravíssimos de desvios de qualidade, que
causaram a morte de pessoas pelo uso de medicamentos manipulados, somada a
outros resultados aonde as concentrações de princípio ativo chegavam a mais de
30.000% acima do valor nominal (BRASIL, ANVISA, 2008d), levou a ANVISA a
abrir novas discussões no âmbito nacional, através de consulta pública, sobre a
definição de novas regras, que resultaram na criação da resolução RDC 67,
publicada no Diário Oficial da União (D.O.U.), em 08 de outubro de 2007, que
dispõe sobre Boas Práticas de Manipulação de Preparações Magistrais e
Oficinais Para Uso Humano em Farmácias (BRASIL, ANVISA, 2008b). Mesmo
trazendo um aumento significativo no rigor dos parâmetros de controle de
qualidade, torna-se necessário criar outras alternativas que visem ampliar mais o
rigor das análises desses controles, no sentido de contribuir para tornar os
medicamentos manipulados cada vez mais seguros, eficazes e com qualidade.
2.2 PROCESSO DE MISTURA E HOMOGENEIDADE DE CONTEÚDO DE
CÁPSULAS
Cápsulas são preparações farmacêuticas sólidas, protegidas por um
invólucro normalmente de gelatina ou de outras substâncias adequadas,
apresentando-se em formatos e tamanhos diversos, contendo uma unidade
posológica (dose unitária) do fármaco ativo e destinadas à administração oral.
Podem ser duras ou moles dependendo da natureza do seu invólucro. Nas
farmácias com manipulação, é mais freqüente o emprego de cápsulas de gelatina

28
duras constituídas por dois elementos independentes perfeitamente encaixados.
São muito versáteis podendo ser preparadas extemporaneamente com uma
ampla faixa de dosagem, permitindo ao farmacêutico realizar composições e
individualizar a dose (fórmulas magistrais) de acordo com as necessidades e as
solicitações clínicas.
As vantagens da manipulação na forma de cápsulas incluem a proteção do
fármaco contra agentes externos como pó, ar e luz; elevada resistência física e
mascaramento de características organolépticas desagradáveis dos fármacos.
Por requererem um número de adjuvantes reduzido, o controle de
incompatibilidades é facilitado. Proporcionam estabilidade ao fármaco, devido ao
baixo número de componentes e à ausência de água nas etapas de sua
elaboração, permitindo a incorporação de substâncias incompatíveis. Apresentam
boa biodisponibilidade, visto que o invólucro se dissolve rapidamente no
estômago (10-20 minutos), liberando o material de enchimento (FERREIRA,
2002).
As cápsulas devem atender às exigências de variação de peso e teor de
princípios ativos descritos nas monografias. Para isso devem conter uma
quantidade determinada e uniforme das substâncias ativas, estáveis e
biodisponíveis nesta forma. É, portanto muito importante que a mistura dos
fármacos com os excipientes seja homogênea, para garantir a uniformidade de
dosagem (FARMACOPÉIA BRASILEIRA, 1988).
Um dos fatores que afetam o processo de homogeneização é a
segregação dos pós, considerado um efeito oposto a mistura, onde os
componentes tendem a separação. Esse efeito é muito importante na preparação
de produtos farmacêuticos, sendo responsável por desvios no processo de
manipulação, provocando alteração no resultado do teste de uniformidade de
conteúdo.
A segregação origina-se do fato que as misturas de pós normalmente são
constituídas de partículas que diferem no tamanho, na forma e na densidade;
assim as partículas que apresentam propriedades semelhantes unem-se umas às

29
outras criando na mistura regiões com concentrações maiores do que um
determinado componente da mistura.
Efeito do tamanho da partícula: A diferença de tamanho das partículas
dos componentes de uma formulação é a principal causa de segregação em
mistura de pós. As menores tendem a cair através dos espaços vazios entre as
maiores afetando a homogeneização (segregação por percolação). Por outro lado
as partículas maiores têm energia cinética maior e por isso deslocam-se a
distâncias maiores antes de chegar ao estado de repouso recaindo nas
extremidades da mistura (segregação de trajetória).
Efeito da densidade das partículas: Quando os componentes da mistura
têm densidades diferentes, as partículas mais densas deslocam-se para baixo
mesmo quando de tamanhos iguais. Neste caso, a segregação por percolação
pode ser potencializada se as partículas mais densas também forem as menores
e a segregação de trajetória pode ser aumentada para as partículas de igual
tamanho mas com densidades diferentes.
Efeito da forma das partículas: As partículas esféricas apresentam as
melhores características de fluxo sendo por isso mais fáceis de serem misturadas,
mas podem com facilidade sofrer segregação quando comparadas às não
esféricas. Já as partículas irregulares, ou de forma acicular, permitem
entrelaçamentos, reduzindo a tendência de separação depois de pronta a mistura.
Na mistura de dois pós que se encontram numa formulação em
quantidades desiguais deve-se primeiro triturar o princípio ativo com igual volume
do diluente, reduzindo-se tudo a um pó da mesma tenuidade. A operação é
repetida adicionando-se a mistura de cada vez um volume de diluente igual ao
que ela já ocupa até que todo diluente seja incorporado. Esse processo, chamado
de diluição geométrica, garante que pequenas quantidades de fármacos sejam
distribuídas uniformemente em uma mistura e é utilizado para facilitar e aumentar
a segurança e a precisão da pesagem de fármacos com baixa dosagem e difíceis
de pesar com exatidão (DIVISÃO, 2008).

30
As diluições geométricas normalmente empregadas dependem da faixa de
dosagem do fármaco, podendo ser de 1:10 (acima de 1mg); 1:100 (0,11 a
0,99mg) e 1:1000 (até 0,1mg), (FERREIRA, 2002).
A etapa de mistura é, provavelmente, a unidade operacional mais delicada
no processo de produção de medicamentos. Pois tem como objetivo produzir uma
mistura final com uma distribuição completamente uniforme de princípios ativos e
excipientes, suficientemente homogênea para ser subdividida em doses
individuais garantindo uma dosagem correta, assegurando que cada dose
contenha a proporção correta de cada componente. Assim, o grau de mistura é
um parâmetro importante e a homogeneidade da mistura deve ser rigorosamente
monitorada (SANCHES, 1995).
A espectroscopia no infravermelho próximo, Near Infrared Spectroscopy
(NIRS), é uma ferramenta poderosa na análise não destrutiva, qualitativa e
quantitativa para vários tipos de amostras. Nos últimos cinco anos, o uso da NIRS
aumentou significativamente sua área de aplicação nas análises farmacêuticas,
para identificação de excipientes, matéria-prima e produtos acabados; para
determinação de água em vários ingredientes; para análise de diversas
propriedades em comprimidos e para o controle do processo de fabricação (LI,
2005); (SEKULIC et al., 1998). A NIRS é um sistema de monitoramento on-line
não invasivo medido com base no desvio padrão do espectro obtido. As
informações proporcionadas por imagens químicas de NIRS de diferentes
intervalo de tempo durante todo ciclo de mistura podem ser analisadas
estatisticamente de forma a permitir avaliações quantitativas da uniformidade de
conteúdo resultante do processo de mistura (YIN, 2007).
Tradicionalmente a uniformidade em formulações farmacêuticas é
controlada pela coleta de amostras em diferentes estágios do processo, no
sentido de determinar o princípio ativo usando um método cromatográfico ou
espectroscópico (UV/VIS). Recentemente a espectroscopia de reflectância no
infravermelho próximo (NIRS) vem sendo usada para monitorar o processo de
mistura no sentido de assegurar a uniformidade de misturas constituídas por
alguns excipientes e um princípio ativo. O desempenho desse método em

31
comparação com outros métodos empregados rotineiramente para acompanhar o
processo de mistura e homogeneização, demonstrou ser bastante eficiente
(BLANCO, 2002).
Devido aos métodos convencionais de coleta de amostras, testes e
verificações laboratoriais, serem considerados trabalhosos por muitos fabricantes
e os resultados serem muitas vezes não reprodutíveis, este tipo de processo
analítico tecnológico (PAT), oferece uma solução que proporciona um processo
de controle on-line em tempo real sem ter que coletar amostras; assim, as
desvantagens do equipamento tradicional, tal como o erro de coleta manual pode
ser eliminado e, portanto reduzido o erro de amostragem (YIN, 2007).
Entre os ensaios a que devem submeter-se as cápsulas preparadas,
destacam-se a determinação do peso médio e a uniformidade da dose
(FERREIRA, 2002). A uniformidade das formas de dosagem (doses
unitárias/unidades posológicas) pode ser demonstrada por dois métodos:
Variação de peso e uniformidade de conteúdo.
O método de variação de peso deve ser aplicado para cápsulas somente
no caso de o teor de princípio ativo ser igual ou superior a 50,0 mg por unidade,
ou quando este compreender mais do que 50% em peso da dose unitária
(FARMACOPEIA BRASILEIRA, 1988; THE UNITED STATES
PHARMACOPOEIA, 2004).
2.3 UTILIZAÇÃO DA ANÁLISE FATORIAL COMO FERRAMENTA ESTATÍSTICA
Das etapas do trabalho cientifico, a otimização de parâmetros
experimentais, talvez seja a mais crítica. A otimização desses parâmetros em
química analítica, tem sido feita tradicionalmente através da técnica univariada.
Esse tipo de tratamento estatístico que envolve um grande número de
experimentos, fornece condições que permitem um valor otimizado da resposta.
Porem, as principais desvantagens dessa técnica são o tempo gasto na
otimização individual de cada parâmetro e a falta de avaliação sobre as

32
interações entre as variáveis que afetam o processo em estudo, tendo como
resultado uma otimização ineficiente.
Para isto existe uma explicação muito simples. As variáveis nos sistemas
químicos se correlacionam fortemente através de mecanismos sinérgicos e
antagônicos. Caso este fato seja ignorado o processo de otimização apresenta
pouco valor.
Atualmente as técnicas que envolvem otimização multivariada são as
preferidas para aplicação em química analítica, por permitirem a otimização
simultânea de todos os fatores envolvidos no sistema com um menor número de
experimentos. Mesmo diante dessas vantagens, a utilização de forma efetiva e
crescente das técnicas multivariada na otimização de métodos analíticos, só
ocorreu nas ultimas décadas, demonstrando sua utilidade nos mais variados
campos do conhecimento. Das varias alternativas existentes, as que mais vêm se
destacando, são os sistemas de planejamento fatorial, pois permitem avaliar
simultaneamente o efeito de um grande número de variáveis a partir de certo
número de ensaios experimentais.
Este fato é corroborado pelo grande número de trabalhos encontrados na
literatura consultada, nas mais variadas áreas de estudos, que utilizam a análise
fatorial como ferramenta para o tratamento estatístico dos dados como, por
exemplo, derivatização de esteróides (APPELBLAD et al., 1997), estudos
farmacocinéticos (BEHRE, NIESCHLAG, 1992); (BURGER et al., 1998), (DAVIES,
1991); (GEHRING et al., 2005); (PITSIU et al., 2003); (SASAKI et al., 2005);
(WAJIMA et al., 2003); nível de álcool no sangue (TEPLIN; ABRAN; MICHAELS,
1989), influência dos adjuvantes e da técnica de enchimento sobre as
características de cápsulas (PETRY et al., 1998); e caracterização de misturas de
pós farmacêuticos por infravermelho próximo (MA; ANDERSON, 2008).
Porém, uma grande desvantagem dos planejamentos fatoriais é o aumento
drástico do número de experimentos que acompanha o aumento do número de
variáveis analisadas. Entretanto, o aumento do número de experimentos, sem
haver perda substancial de informações, pode ser reduzido pelo uso dos

33
planejamentos fatoriais fracionários. Nestes casos, com quatro ou mais fatores, os
de integração de ordem mais elevadas podem ser ignorados e somente os efeitos
principais e as interações de dois fatores são analisados (PERALTA-ZAMORA et
al., 2005).
2.4 DESENVOLVIMENTO E VALIDAÇÃO DE METODOLOGIAS ANALÍTICAS
A validação de métodos para quantificação de fármacos é normatizada
pela Resolução RE nº. 899 da ANVISA, publicada no Diário Oficial da União em
29 de maio de 2003, que determina a publicação do “Guia para validação de
métodos analíticos e bioanalíticos” (BRASIL, ANVISA, 2003f).
De acordo com esta resolução, o objetivo de uma validação é demonstrar
que o método é apropriado para a finalidade pretendida, ou seja, a determinação
qualitativa, semi-quantitativa e/ou quantitativa de fármacos e outras substâncias
em produtos farmacêuticos. As informações contidas neste guia se aplicam às
técnicas analíticas que façam uso de métodos cromatográficos (CG e CLAE) e
não cromatográficos.
Devem-se utilizar substâncias de referência oficializadas pela Farmacopéia
Brasileira ou, na ausência destas, por outros códigos autorizados pela legislação
vigente e no caso da inexistência dessas substâncias, será admitido o uso de
padrões de trabalho, desde que a identidade e o teor sejam devidamente
comprovados.
No caso de metodologia analítica não descrita em Farmacopéias ou em
Formulários Oficiais que consistam em ensaios quantitativos, destinados à
determinação de princípios ativos em produtos farmacêuticos ou matérias primas
devidamente reconhecidos pela ANVISA (TABELA 1), a metodologia será
considerada validada, desde que sejam avaliados os seguintes parâmetros:
Especificidade e Seletividade; Linearidade; Intervalo; Precisão; Limite de detecção
(Sensibilidade); Limite de quantificação; Exatidão e Robustez.

34
TABELA 1 – Classificação dos testes de validação segundo a resolução RE nº 899/ANVISA.
CATEGORIA FINALIDADE DO TESTE
I
Testes quantitativos para a determinação do principio ativo em produtos farmacêuticos ou matérias-primas.
II
Testes quantitativos ou ensaio limite para determinação de impurezas e produtos de degradação em produtos farmacêuticos e matérias-primas.
III
Testes de performance (p. ex.: dissolução e liberação de principio ativo).
IV
Teste de identificação.
Fonte: BRASIL,ANVISA, 2003f.
Desta forma, os ensaios analíticos regulados na RE 899, são classificados
em quatro categorias conforme a Tabela 1. Sendo que para cada categoria é
exigido um conjunto de testes de acordo com a sua finalidade (BRASIL, ANVISA,
2003f). A determinação quantitativa de tibolona em produtos farmacêuticos se
enquadra na categoria I da Tabela 1. Assim, para validação do método foram
validados os seguintes parâmetros:
Especificidade – Este parâmetro tem a capacidade de medir exatamente
um composto em presença de outros componentes como impurezas, produtos de
degradação e componentes da matriz. Para análises quantitativas e de
impurezas, a especificidade pode ser determinada pela comparação dos
resultados obtidos de amostras contaminadas com impurezas ou excipientes e
amostras não contaminadas, para demonstrar que o resultado do teste não é
afetado por esses materiais. No caso de não serem disponíveis as impurezas e os
padrões, podem-se comparar os resultados dos testes com os de um segundo
procedimento bem caracterizado (metodologia farmacopéica ou procedimento
validado), incluindo nessas comparações amostras armazenadas sob condições

35
de estresse, como luz, calor, umidade, hidrólise ácido-básica ou oxidação”
(BRASIL, ANVISA, 2003f).
Linearidade – Determina a capacidade de uma metodologia analítica
demonstrar que os resultados obtidos são diretamente proporcionais à
concentração do analito na amostra, dentro de um intervalo estabelecido. É
determinada pelo coeficiente de correlação (r), obtido a partir do gráfico que
relaciona à resposta cromatográfica em função das várias concentrações do
analito ensaiadas através de métodos estatísticos apropriados utilizando o
coeficiente angular, e a soma dos quadrados mínimos da regressão linear. Sendo
adotado como critério mínimo aceitável para (r) um valor maior ou igual a 0,99
(BRASIL, ANVISA, 2003f).
Intervalo – É derivado do estudo da linearidade e depende da aplicação
pretendida do método. É estabelecido através da confirmação de que o método
apresenta exatidão, precisão e linearidade adequadas, quando aplicados a
amostras contendo quantidades de substâncias dentro do intervalo especificado.
É considerada com sendo a faixa entre os limites de quantificação superior e
inferior de um método analítico (BRASIL, ANVISA, 2003f).
Precisão – É a avaliação da proximidade dos resultados obtidos em uma
serie de medidas de uma amostragem múltipla de uma mesma amostra. É
considerada em três níveis: Reprodutibilidade (precisão inter-laboratorial),
precisão intermediaria (precisão inter-corrida) e repetibilidade (precisão intra-
corrida):
A repetibilidade ou precisão intra-corrida é estabelecida pela avaliação da
concordância entre os resultados dentro de um curto período de tempo com o
mesmo analista e a mesma instrumentação. A repetibilidade do método é
verificada com, no mínimo nove determinações, contemplando o intervalo linear
do método, ou seja, três concentrações (baixa, média e alta), com três réplicas
cada ou o mínimo de seis determinações a 100% da concentração do teste.

36
A precisão de um método analítico pode ser expressa como o desvio
padrão ou o desvio padrão relativo - DPR (coeficiente de variação – CV%) de uma
serie de medidas segundo a formula:
x100CMDDPDPR =
Onde: DP é o desvio padrão e CMD a concentração média determinada.
O valor máximo aceitável deve ser definido de acordo com a metodologia
empregada, a concentração do analito na amostra, o tipo de matriz e a finalidade
do método, não se admitindo valores superiores a 5% (BRASIL, ANVISA, 2003f).
Exatidão – Mede a proximidade dos resultados obtidos pelo método em
estudo, em relação ao valor verdadeiro. Deve ser determinada após o
estabelecimento da linearidade, do intervalo e da especificidade do mesmo,
sendo verificada a partir de, no mínimo nove determinações contemplando o
intervalo linear, ou seja; três concentrações, uma baixa, uma média e uma alta
com quatro réplicas cada. (BRASIL, ANVISA, 2003f).
Robustez – É a medida da capacidade do método em resistir a pequenas
e deliberadas variações dos parâmetros analíticos, indicando a confiança do
método no seu uso normal. Constatando-se susceptibilidade à variações nas
condições analíticas; estas devem ser controladas e as precauções necessárias
incluídas no procedimento (BRASIL, ANVISA, 2003f).
Os fatores que devem ser considerados na determinação da robustez são:
Preparo da amostra, espectrofotometria e cromatografia líquida. Com relação a
cromatografia líquida um dos parâmetros que pode ser avaliado entre outros, é
variação da composição da fase móvel.

37
2.5 CARACTERÍSTICAS FISICOQUÍMICAS E FARMACOLÓGICAS DA
TIBOLONA.
Não foi encontrada monografia oficial da tibolona em nenhuma farmacopéia
e nem em outros compêndios oficiais.
2.5.1 Características químicas A Tibolona (Figura 1) é um 3-cetoesteróide sintético de fórmula molecular
C21H28O2, peso molecular 312.45 g/mol registrada no Chemical Abstracts sob o
número CAS 5630-53-5 com a denominação química (7α, 17α)-17-Hidroxi-7-metil-
19-norpreg-5(10)-en-20-in-3-ona (BUDAVARI, S., 1996), também conhecida como
7-metil-noretinodrel (ZUO et al., 2005). É comercialmente disponível como Livial®
(Organon), Libian® (Libbs), Reduclim® (FQM), Tibial® (Neoquímica). Apresenta-se
como um pó cristalino de coloração branca ou esbranquiçada, estável sob as
condições usuais, ponto de fusão 163 ~ 169 ºC, rotação ótica +103° ~ +130°,
praticamente insolúvel em água e solúvel em clorofórmio e metanol (BUDAVARI,
1996).
CH3 OH
C C H
CH3O
FIGURA 1 – Estrutura química da tibolona.
2.5.2 Ação farmacológica Apresenta propriedades androgênicas, estrogênicas e progestagênicas
fracas, que mimetizam a atividade do estrogênio e da progesterona, sendo
utilizado para aliviar os sintomas decorrentes da menopausa.
38
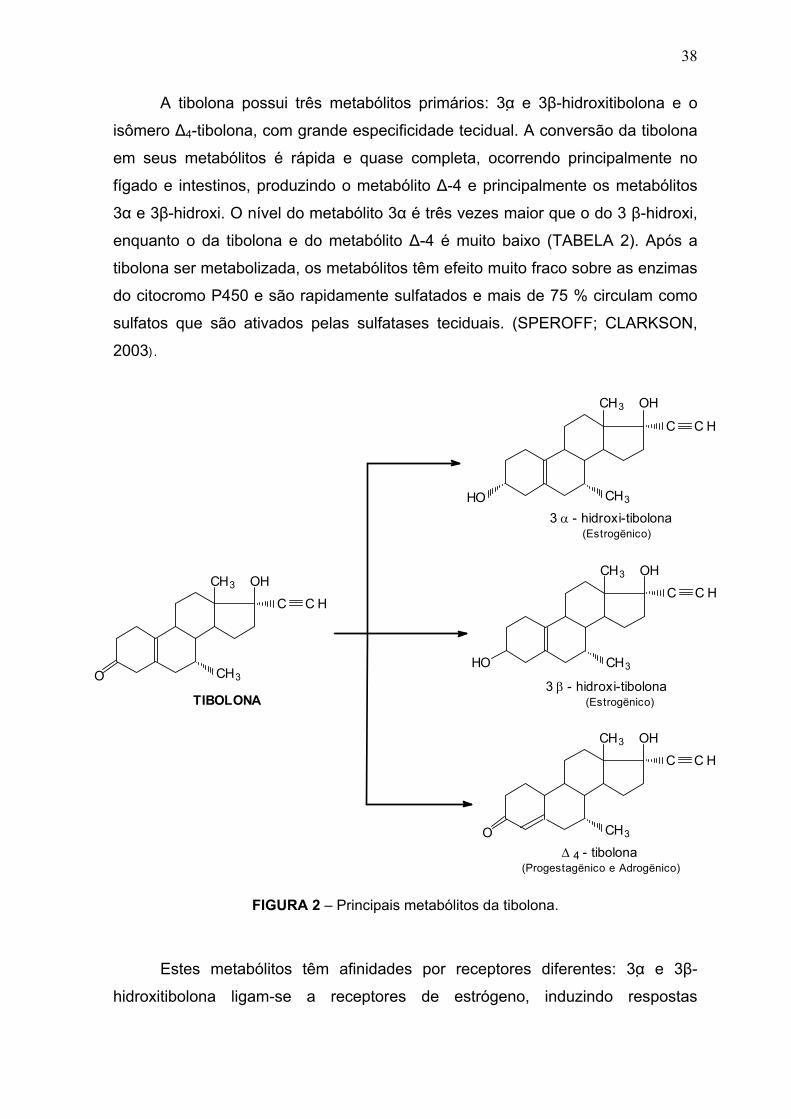
A tibolona possui três metabólitos primários: 3α e 3β-hidroxitibolona e o
isômero Δ4-tibolona, com grande especificidade tecidual. A conversão da tibolona
em seus metabólitos é rápida e quase completa, ocorrendo principalmente no
fígado e intestinos, produzindo o metabólito Δ-4 e principalmente os metabólitos
3α e 3β-hidroxi. O nível do metabólito 3α é três vezes maior que o do 3 β-hidroxi,
enquanto o da tibolona e do metabólito Δ-4 é muito baixo (TABELA 2). Após a
tibolona ser metabolizada, os metabólitos têm efeito muito fraco sobre as enzimas
do citocromo P450 e são rapidamente sulfatados e mais de 75 % circulam como
sulfatos que são ativados pelas sulfatases teciduais. (SPEROFF; CLARKSON,
2003).
CH3 OH
C C H
CH3O
TIBOLONA
HO
O
CH3 OH
C C H
CH3
Δ 4 - tibolona(Progestagënico e Adrogënico)
CH3 OH
C C H
CH3
3 β - hidroxi-tibolona (Estrogënico)
HO
CH3 OH
CH3
3 α - hidroxi-tibolona (Estrogënico)
C C H
FIGURA 2 – Principais metabólitos da tibolona.
Estes metabólitos têm afinidades por receptores diferentes: 3α e 3β-
hidroxitibolona ligam-se a receptores de estrógeno, induzindo respostas

39
semelhantes às do esteróide natural, enquanto que o isômero Δ4 tem elevada
afinidade por receptores de progesterona e andrógenos (BLOOM et al., 2006);
(VERHEUL et al., 2007b).
TABELA 2 – Afinidade da tibolona e seus metabólitos por receptores de esteróides sexuais.
Fonte: SPEROFF; CLARKSON, 2003
2.5.3 Indicações Utilizada na terapia de reposição hormonal para o tratamento dos sintomas
da menopausa e pós-menopausa e como profilaxia contra o desenvolvimento de
osteoporose (ALBERTAZZI; DI MICCO; ZANARDI, 1998).
2.5.4 Apresentações e Dosagens A tibolona 2,5 mg, com o nome de Livial, é comercializada no Brasil pela
Organon, na forma de comprimidos. É manipulada na forma cápsula. As doses
normalmente utilizadas na terapia de reposição hormonal são de 1,25mg a 2,5
mg/dia.
2.5.5 Farmacocinética da Tibolona Após a administração oral, a tibolona é rápida e extensamente absorvida.
Por isso os níveis plasmáticos de tibolona e dos seus metabólitos (isômeros Δ4,
3α e 3β hidróxi) também são baixos, sendo que os níveis dos isômeros 3α e 3β
hidróxi, são mais elevados, porém não ocorre acumulação. O consumo de
Tibolona e metabólitos
Receptores estrogênicos
Receptores progestagênicos
Receptores androgênicos
Tibolona 1.3% 4.9% 3.2%
Metabólito 3 -OH 3.2% — —
Metabólito 3ß-OH 1.7% — —
Isômero - 4 — 12.9% 39.2%

40
alimentos não tem efeitos significativos sobre o grau de absorção. Sua excreção
se dá principalmente na forma de metabólitos conjugados (principalmente
sulfatados); e embora uma parte da quantidade administrada seja excretada na
urina, a maioria é eliminada pelas fezes (VERHEUL et al., 2007a); (VERHEUL et
al., 2007b).
2.6 TIBOLONA E MÉTODOS PARA SUA DETECÇÃO
A estrutura da Tibolona não possui grupos funcionais cromofóricos
suficientemente estendidos, sendo por isso freqüentemente quantificada por
espectrometria de massas. Não há metodologias de análise para a tibolona
descritas na literatura, nem métodos oficiais farmacopéicos e não farmacopéicos
para quantificação deste fármaco em formulações farmacêuticas; embora existam
métodos para estudos de farmacocinética (ZUO et al., 2005).
A Tabela 3 na pagina seguinte, mostra os métodos descritos na literatura
para quantificação da tibolona e seus metabólitos no plasma, na urina, fezes e
tecidos, bem como os meios de detecção utilizados em estudos farmacocinéticos.
É fácil perceber que a detecção para quantificação da tibolona e seus metabólitos,
é feita exclusivamente pelo método de espectrometria de massas. Esse método é
considerado bastante oneroso, por isso não está disponível para uso de rotina na
maioria dos laboratórios analíticos.
Esses fatos sinalizam para a necessidade de se investigar outras
metodologias de quantificação deste fármaco desenvolvendo métodos de
quantificação com detecção por UV ou estratégia de derivatização que permitam
a produção de um derivado detectável por fluorescência, uma técnica
extremamente seletiva e sensível, que encontrará utilidade tanto no controle da
qualidade de medicamentos a base de tibolona, quanto para detecção desta em
fluidos biológicos, como por exemplo, em estudos de
biodisponibilidade/bioequivalência e quando da ocorrência de intoxicações.

41
A reação de derivatização de vários esteróides com função carbonila pela
dansilhidrazina usando ácido trifluorometanosulfônico como catalisador já foi
descrita (APPELBLAD et al., 1997). O método descrito por Appelblad apresentou
algumas limitações, sendo as mais importantes: (a) a necessidade de uma coluna
de concentração para a separação cromatográfica da hidrazona formada e (b) a
formação de hidrazonas diastereoisoméricas syn e anti com alguns dos
esteróides investigados.
TABELA 3 – Métodos de detecção para quantificação da tibolona em estudos farmacocinéticos.
Método de extração
Método de detecção Matriz Limite de detecção/
quantificação
Referência
Líquido-líquido
CG – EM Plasma
0,10-0,94 ng/mL
(tibolona) 0,10-0,73 ng/mL (Isom. Δ4)
0,1 ng/mL (isom. 3α, e 3β hidroxi)
(Quantif.)
TIMMER; DOORSTAM,
2002
Líquido-líquido
Fase-sólida
CG – EM Derivatização Plasma
0,1 ng/mL (tibolona, isom.
Δ4, 3α e 3β hidroxi) (Quantifi.)
TIMMER; VERHEUL;
DOORSTAM, 2002
Fase-sólida CG – EM CL – EM/EM
Tecidos (cérebro, fígado
e coração)
0,1 ng/mL (metab. tibolona)
0,25 ng/mL (tibolona e metab. sulfatados)
(Quantifi.)
VERHEUL; KLOOSTERBOER,
2006
Fase-sólida CG – EM
CL – EM/EM Derivatização
Plasma, urina, bile e fezes
0,1-0,5 ng/mL
(plasma e urina) 0,5-2,0ng/g
(fezes e bile) (Detecção)
VERHEUL, et al., 2007 b
Fase-sólida Líquido-líquido
CG – EM CL – EM
Plasma e tecidos (cérebro, vagina, útero e mama)
0,1-0,5 ng/mL
(plasma) 0,5-2,0 ng/g
(tecidos) (Detecção)
VERHEUL, et al., 2007 a
Líquido-líquido
CG – EM
CG – EMAR Derivatização
Plasma
0,02ng/mL/mL
(Quantifi.)
SON, 2006
Líquido-líquido
CL – EM Derivatização Plasma
100 ng/mL (Quantifi.)
ZUO et al., 2005b
42
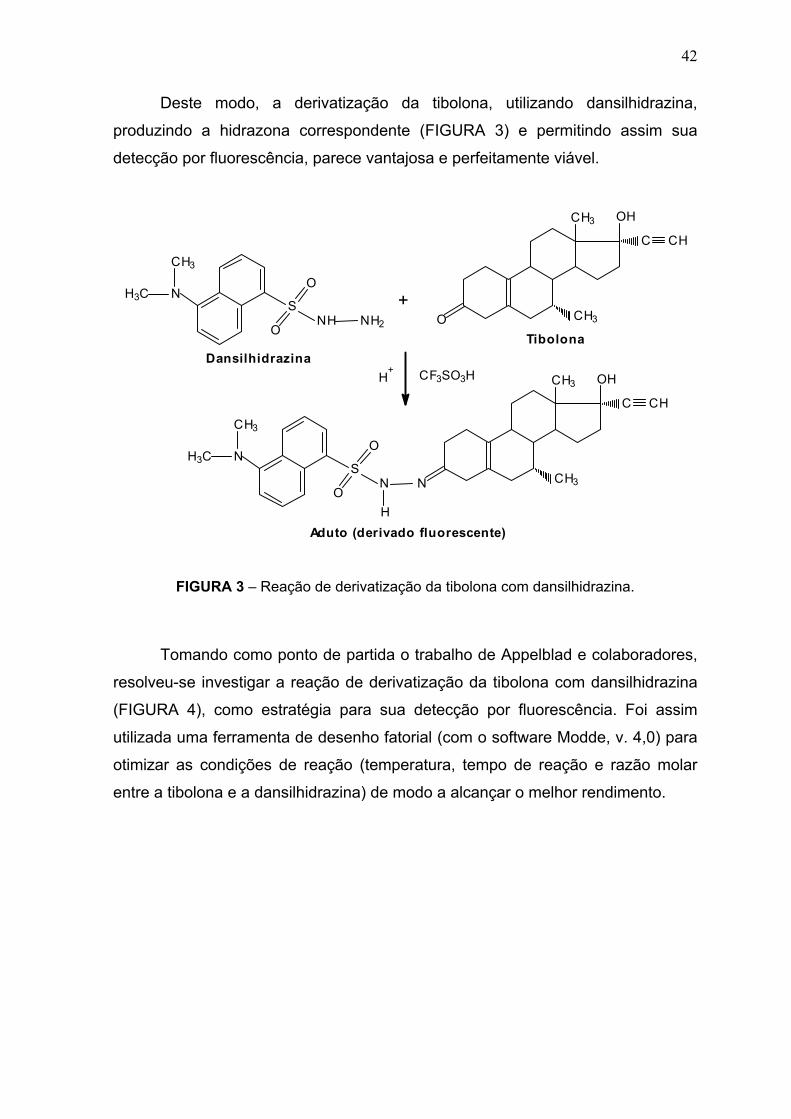
Deste modo, a derivatização da tibolona, utilizando dansilhidrazina,
produzindo a hidrazona correspondente (FIGURA 3) e permitindo assim sua
detecção por fluorescência, parece vantajosa e perfeitamente viável.
S
O
O
NH
N
NH2
CH3
H3C
CH3
OH
C CH
O
CH3
CH3
OH
C CH
CH3
N
H
S
O
O
N
N
CH3
H3C
+
H+ CF3SO3H
DansilhidrazinaTibolona
Aduto (derivado fluorescente)
FIGURA 3 – Reação de derivatização da tibolona com dansilhidrazina.
Tomando como ponto de partida o trabalho de Appelblad e colaboradores,
resolveu-se investigar a reação de derivatização da tibolona com dansilhidrazina
(FIGURA 4), como estratégia para sua detecção por fluorescência. Foi assim
utilizada uma ferramenta de desenho fatorial (com o software Modde, v. 4,0) para
otimizar as condições de reação (temperatura, tempo de reação e razão molar
entre a tibolona e a dansilhidrazina) de modo a alcançar o melhor rendimento.

43
CAPÍTULO III
QUANTIFICAÇÃO DA TIBOLONA POR DERIVATIZAÇÃO COM DETECÇÃO POR
FLUORESCÊNCIA

44
3 DESENVOLVIMENTO DO MÉTODO
3.1 MATERIAIS
Padrões e Reagentes
A tibolona padrão, foi gentilmente cedida pelo laboratório Organon USA,
lote LZ 0655K1. Foram usadas também, água purificada através de um sistema
Milli-Q; Acetonitrila (TEDIA - USA); Metanol (MERCK - Alemanha);
Dansilhidrazina – 5-[dimetilamino]-1-naftalenosulfônil-hidrazina (Sigma-Aldrich -
EUA), Ácido trifuorometanosulfônico (Sigma-Aldrich - EUA), e ainda; Acetato de
amônio. Os solventes eram em grau HPLC.
Equipamentos
Cromatógrafo líquido constituído de um sistema de gradiente de baixa
pressão, equipado com os seguintes módulos: Forno de coluna (CTO - 10AS);
degaseificador (DGU - 14A); detector de fluorescência (RF - 10A); módulo
controlador (SCL-10A); bomba (LC - 10 AD) com válvula solenóide (FCV - 10AL),
todos de marca (SHIMADZU – Tóquio – Japão); e um injetor manual provido
com um loop de amostra de 20 μL (Rheodyne).
3.2 METODOLOGIA
A derivatização da tibolona foi realizada com dansilhidrazina usando ácido
trifluorometanosulfônico como catalisador como sugerido por Appelblad (1977).
Soluções estoque de tibolona e dansilhidrazina (1mg/mL) foram preparadas em
metanol e armazenadas em freezer, mantendo-se estáveis durante todo o período
de armazenamento. As diluições foram realizadas com o mesmo solvente para
obter as concentrações finais necessárias para cada experimento. A
concentração do catalisador variou de 3 a 7 %, uma vez que os dados da
literatura (APPELBLAD et al., 1997) mostram que este parâmetro não influencia
significativamente o rendimento da reação, tendo sido por isso fixada em 3% para

45
os experimentos finais. As reações foram realizadas usando frascos de vidro de
2mL de capacidade e um termobloco para aquecimento.
Para otimização do método, um desenho fatorial fracionário experimental
foi implementado usando o programa Modde v.4.0 (Umetrix) a fim de investigar os
efeitos da Razão molar de dansilhidrazina para Tibolona (20 a 50) a temperatura
de reação (37 a 50 ºC) e o tempo de reação (17 a 30 minutos), no rendimento da
reação de derivatização. Regressão Linear Multivariada foi usada para avaliar os
efeitos de cada parâmetro e suas combinações no rendimento da reação. Depois
de fixar os limites para cada uma das variáveis a serem investigadas, o programa
sugeriu um total de 11 experimentos, variando cada um dos três parâmetros
investigados (TABELA 4). A área do derivado fluorescente nos cromatogramas
obtidos em cada um dos 11 experimentos foi calculada e alimentou o programa
como rendimento a ser otimizado.
TABELA 4 – Parâmetros experimentais sugeridos pelo desenho fatorial fracionário usando um programa Modde v.4.0 (Umetrix) para derivatização da Tibolona.
(*) Razão molar de dansilhidrazina para 1 mol de Tibolona (**) Área do pico da Tibolona (/100.000).
As amostras derivatizadas foram previamente diluídas (1:1) com tampão
fosfato 1M, pH 7.0, para neutralizar o excesso de ácido e diretamente injetadas
em duplicata no cromatógrafo e então cromatografadas usando uma coluna
monolítica (Merck Chromolith comprimento de 100 mm x 4.6 mm DI) com uma
pressão de 117 ± 3 Kgf/cm2, com fluxo de 3 mL/min e um volume de injeção de 20
μ L, utilizando-se como fase móvel, um tampão acetato de amônio 50mM, pH 6.8
Nº de Experimentos
Razão Molar (*)
Temperatura (ºC)
Tempo (min.) Ordem
1 20 37 17,0 7 2 50 37 17,0 8 3 20 50 17,0 1 4 50 50 17,0 11 5 20 37 30,0 6 6 50 37 30,0 4 7 20 50 30,0 9 8 50 50 30,0 5 9 35 35 23,5 3 10 35 35 23,5 10 11 35 35 23,5 2

46
e acetonitrila (92,5:7,5 v/v). O detector de fluorescência foi configurado para usar
um comprimento de onda de excitação de 320 nm e de 520 nm para emissão.
3.3 RESULTADOS e DISCUSSÃO No sentido de confirmar se a estratégia de derivatização para a tibolona
seria uma metodologia eficiente para sua quantificação, foram tomados como
base os resultados obtidos anteriormente por Appleblad (1997), que estudou o
uso de ácido trifluorometanosulfônico como catalisador na reação de
derivatização de vários esteróides com dansilhidrazina, adotando a razão molar
de 50:1, entre o reagente derivatizante e a tibolona, como a melhor proporção.
Appelblad (1997) e seus colaboradores demonstraram que a concentração
do catalisador não é um fator importante na determinação do rendimento desta
reação, porém no presente trabalho a concentração de ácido mostrou ser
fundamental para um tempo de retenção adequado do derivado fluorescente
formado. Por isso, os experimentos foram conduzidos com uma concentração do
ácido fixada em 3%, uma vez que concentrações mais altas causaram uma queda
crucial no tempo de retenção tendo o pico do derivado, nessas concentrações,
eluído no volume morto da coluna (FIGURA 4).
FIGURA 4 – Cromatograma da reação de derivatização mostrando a eluição do pico da tibolona no volume morto quando usada concentrações do ácido, maiores que 3%.
Detector: Ex: 350 nm; Em: 520 nm Tempo: 18,0032 min Amplitude: 0,909 mAU

47
Não foi observada variação no tempo de retenção com a mudança na
proporção do solvente orgânico, indicando provavelmente que o derivado
encontrava-se totalmente ionizado pelo excesso de ácido do meio reacional.
O uso de um desenho fatorial fracionário permitiu uma investigação
sistemática dos parâmetros de reação sobre o rendimento da derivatização como
pode ser observado na Tabela 5 abaixo.
TABELA 5 – Rendimentos previstos para derivatização da tibolona nas condições experimentais sugeridas pelo desenho fatorial fracionário usando um programa Modde v.4.0 (Umetrix).
(*) Razão molar de dansilhidrazina para 1 mol de Tibolona (**) Área do pico da Tibolona (/100.000). O gráfico 1 mostra os resultados do modelamento dos dados da reação de
derivatização da tibolona por Análise de Regressão Linear Multivariada, onde no
eixo das abscissas encontram-se os valores do rendimento observado, e nos
eixos das ordenadas encontram-se os valores do rendimento previsto, oriundos
dos dados da tabela 5. De acordo com os resultados, é possível notar que houve
uma proximidade muito grande entre os valores previstos e os valores
observados, e um coeficiente de correlação linear R2 = 0,9856. Estes resultados
demonstram uma excelente previsibilidade do modelo.
Nº de Experimentos
Razão Molar (*)
Temperatura (ºC)
Tempo (min.) Ordem Rendimento
(**)
1 20 37 17,0 7 8,15 2 50 37 17,0 8 53,5 3 20 50 17,0 1 38,1 4 50 50 17,0 11 57,5 5 20 37 30,0 6 20,1 6 50 37 30,0 4 26,2 7 20 50 30,0 9 56,5 8 50 50 30,0 5 40,2 9 35 35 23,5 3 45,6 10 35 35 23,5 10 45,6 11 35 35 23,5 2 45,6

48
N = 11 R2 = 0.9856 GRÁFICO 1 – Modelamento dos dados da reação de derivatização de tibolona por regressão linear multivariada (RLM) mostrando os valores de rendimento previstos versus observados.
O Gráfico 2 mostra o efeito das interações dos parâmetros investigados
sobre o rendimento da reação de derivatização. De acordo com o gráfico, a razão
molar (RM) e a temperatura (Te) influenciaram de forma relevante quando
MR
Te
TI
MR*
MR
MR
*Te
MR
*TI
-15
-10
-5
0
5
10
Áre
a
N = 11 R2 = 0.9856
GRÁFICO 2 – Efeito das interações dos parâmetros de reação investigados sobre o rendimento da reação de derivatização.
PREV
ISTO
OBSERVADO
49
consideradas de forma isolada, apresentando um efeito positivo. Porém, as
interações entre razão molar/razão molar (RM*RM); razão molar/temperatura
(RM*Te) e razão molar/tempo de reação (RM*TR) mostraram efeitos negativos,
em relação ao rendimento da reação.
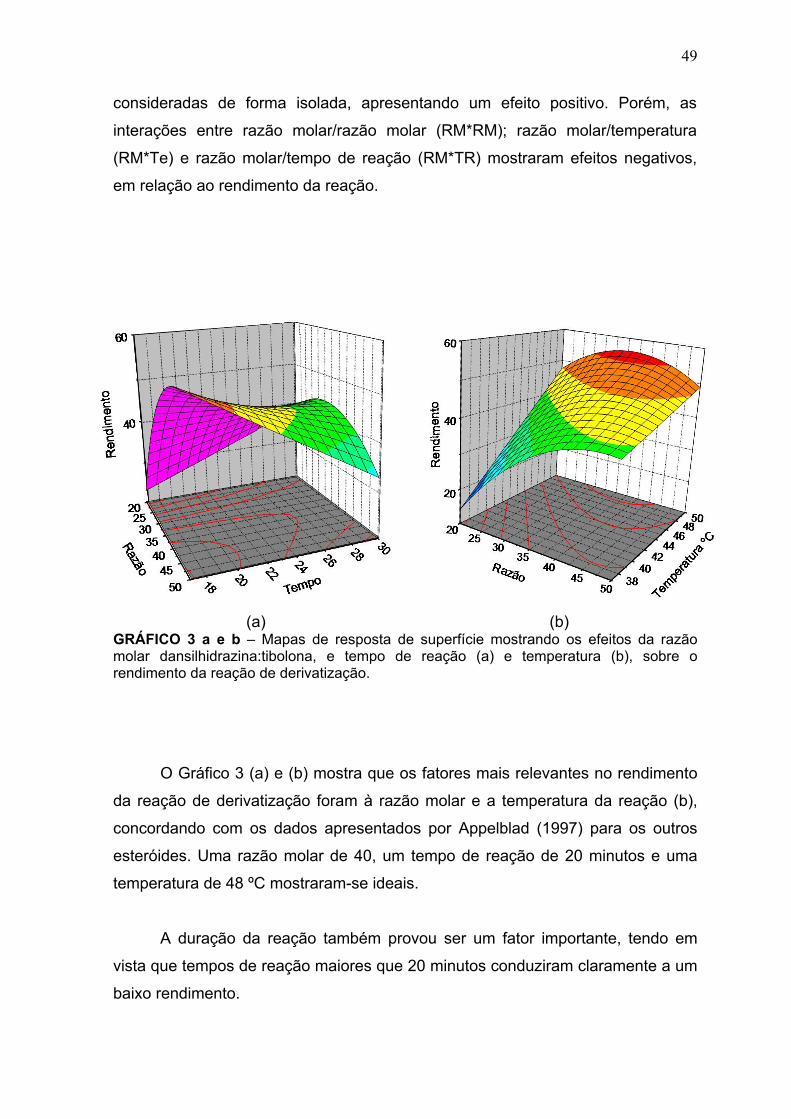
(a) (b) GRÁFICO 3 a e b – Mapas de resposta de superfície mostrando os efeitos da razão molar dansilhidrazina:tibolona, e tempo de reação (a) e temperatura (b), sobre o rendimento da reação de derivatização.
O Gráfico 3 (a) e (b) mostra que os fatores mais relevantes no rendimento
da reação de derivatização foram à razão molar e a temperatura da reação (b),
concordando com os dados apresentados por Appelblad (1997) para os outros
esteróides. Uma razão molar de 40, um tempo de reação de 20 minutos e uma
temperatura de 48 ºC mostraram-se ideais.
A duração da reação também provou ser um fator importante, tendo em
vista que tempos de reação maiores que 20 minutos conduziram claramente a um
baixo rendimento.

50
Progesterona
FIGURA 5 – Cromatograma mostrando picos duplos obtidos para hidrazonas syn e anti quando uma solução de progesterona padrão a 400 μM foi derivatizada com dansilhidrazina. Fonte: APPELBLAD et al., 1997, p. 4910.
Vale a pena notar aqui, como pode ser observado na figura 6 na página
seguinte que a reação da dansilhidrazina com tibolona não parece formar as
hidrazonas sin e anti-diastereoisoméricas que foram relatadas por Appelblad e
colaboradores (1997) para outros esteróides. (FIGURA 5).
Já que a tibolona não apresenta carbonila α-insaturada, provavelmente
seja essa a causa da formação de apenas uma única hidrazona. Isto é muito
vantajoso para os propósitos de quantificação, desde que apenas um único pico é
integrado em vez de ter que usar a soma de dois picos diastereisomericamente
integrados.
Tempo de Retenção (min)

51
syn anti
X Y X
Y
Aduto (derivado fluorescente)
CH3
OH
C CH
CH3
N
H
S
O
O
N
N
CH3
H3C
Syn e anti ?
FIGURA 6 – Formação de hidrazonas syn e anti diastereoisoméricas.
Este aspecto da reação precisa de confirmação através de técnicas de
espectroscópicas e espectrométricas; embora cálculos de computador já tenham
sugerido diferenças na estereoquímica da carbonila que poderiam explicar as
diferenças de regioseletividade observadas entre progesterona e tibolona na
reação com a dansilhidrazina.
FIGURA 7 – Cromatograma representativo da separação do derivado fluorescente obtido a partir de uma solução de tibolona a 3 μg/mL, (TR = 6,69 min.).
Detector: Ex: 320 nm; Em: 520 nm Tempo: 10,9905 min Amplitude: 0,081 mAU
Minutos
mA
U
mA
U

52
Nas condições cromatográficas estabelecidas na metodologia desenvolvida
(TABELA 6), o tempo de corrida pôde ser reduzido de 20 para 11 minutos
permitindo ao derivado fluorescente da tibolona, o pico de interesse, eluir num
tempo de retenção de 6,69 ± 0,2 min, (FIGURA 7) usando uma fase móvel com
7,5% de acetonitrila, sendo sua absorção proporcional ao aumento da
concentração do analito.
TABELA 6 – Condições cromatográficas desenvolvidas para quantificação da tibolona por derivatização com detecção por fluorescência.
Foi possível realizar a reação de derivatização da tibolona usando
condições moderadas, com rendimento quase quantitativo. Este método pode ser
ainda modificado para se obter tempos de retenção ainda mais curtos usando, por
exemplo, uma proporção mais alta do solvente orgânico na fase móvel e assim,
CARACTERÍSTICAS
DESCRIÇÃO
• Cromatógrafo Shimadzu Série 10A vp configurado com os seguintes módulos: Forno (CTO – 10 AS vp), Bomba (LC – 10 AD vp) com válvula solenóide, Degaseificador (DGU 14 A vp), Interface (SCL 10A vp),
• Detector Florescência (RF – 10 A). Regulado para λex=350 nm; λem=520 nm.
• Coluna Monolítica, Merck Chromolith (performance 100 mm x 4.6 mm ID).
• Injetor Manual.
• Volume de injeção 20 μL.
• Fase móvel
Solvente orgânico (acetonitrila) 7,5% com 92,5% de tampão acetato de amônio 50 mM e pH = 6,8.
• Tipo de eluição Isocrática
• Vazão de fluxo 3 mL/min.
• Temperatura 40 ºC

53
diminuir o tempo de análise. Tomando os resultados em conjunto, pode-se
concluir que o método demonstra ser bastante eficiente para quantificação da
tibolona, necessitando para isso ser devidamente validado.
Apesar das vantagens da detecção por fluorescência, em especial a
seletividade e sensibilidade, a necessidade de derivatização pré-coluna e o uso
de um ácido forte como o ácido trifluorometanosulfônico, são inconvenientes
relevantes para aplicação de rotina do método. Por isso resolveu-se avaliar a
possibilidade de desenvolver um método com detecção por ultravioleta para a
quantificação do esteróide.

54
CAPÍTULO IV
QUANTIFICAÇÃO DA TIBOLONA POR DETECÇÃO DIRETA NO UV

55
4 DESENVOLVIMENTO, VALIDAÇÃO E APLICAÇÃO DO MÉTODO 4.1 MATERIAIS
Padrões e Reagentes
A tibolona padrão, foi cedida pelo laboratório Organon USA, lote LZ
0655K1. Foram usados também, água purificada através de um sistema Milli-Q;
Acetonitrila (TEDIA - USA); Metanol (MERCK - Alemanha). Os solventes eram em
grau HPLC.
Equipamentos
• Cromatógrafo líquido (SHIMADZU - Japão), Série 10A vp, com os
módulos: Forno de coluna (CTO - 10AS); degaseificador (DGU - 14h); detector
UV/Vis (SPD - 10AV); módulo controlador (SCL - 10A); bomba (LC - 10AD) com
válvula solenóide (FCV - 10AL); e auto-injetor (SIL - 10AD).
• Cromatógrafo líquido (SHIMADZU - Japão), série 10A vp. Com os
módulos: Forno de coluna (CTO - 10A); degaseificador (DGU - 14A); detector com
arranjo de diodos (SPD - M10A); módulo controlador (SCL - 10A); bomba (LC -
10AD); e Injetor manual provido com um loop de amostra de 20 μL (Rheodyne).
• Espectrofotômetro UV-Vis (VARIAN), modelo Cary 50.
• Balança analítica (SHIMADZU – Japão), modelo AW 220 (Carga
Máxima: 220g, d = 0,1mg).
• Sistema Milli-Q Ultra-Pure Water (MILLIPORE – EUA).

56
PARTE I
4.2 DESENVOLVIMENTO DO MÉTODO
4.2.1 Metodologia Como já descrito anteriormente, o único método descrito na literatura para
quantificação da tibolona (Zuo et al., 2005), utiliza espectrometria de massas, uma
técnica relativamente cara e especializada, fora do alcance de muitos laboratórios
de controle da qualidade. O desenvolvimento de uma metodologia de análise com
detecção no ultravioleta é importante, pois possibilitará o controle deste
medicamento por laboratórios que não dispõem de equipamentos de
espectrometria de massas.
Inicialmente, o padrão de tibolona foi preparado como uma solução
estoque a 1 mg/mL em uma mistura de metanol/água (70:30) que foi armazenada
sob refrigeração. No sentido de otimizar a concentração ideal para obtenção de
uma boa área de pico cromatográfico para a tibolona, confirmou-se o λmax
obtendo-se experimentalmente seu espectro no UV, utilizando um
espectrofotômetro para a faixa do UV-Vis, e ensaiando soluções do analito nas
concentrações de 0.100 mg/mL, 0,050 mg/mL, e 0,025 mg/mL em metanol,
obtidas a partir da solução estoque.
A RE-899/ANVISA recomenda que a linearidade das curvas de calibração
para desenvolvimento de métodos analíticos seja determinada com um intervalo
de, no mínimo, cinco concentrações diferentes do analito a ser testado (BRASIL,
ANVISA, 2003f). Desta forma foi estabelecido um intervalo de oito concentrações
tomando como base a dosagem de 2,5 mg das cápsulas de tibolona manipuladas.
Baseado nesta dosagem (2,5 mg) fez-se uma diluição da solução estoque de
tibolona (1 mg/mL) com acetonitrila:H2O para obter uma solução de concentração
de 0,05 mg/mL a partir da qual estabeleceu-se um intervalo de concentrações de:
0,001; 0,005; 0,010; 0,025; 0,050; 0,100; 0,200 e 0,500 mg/mL diluindo-se esta

57
solução com o mesmo solvente de formas a contemplar o intervalo de 80-120%
da concentração teórica, conforme a RE 899/2003 (BRASIL, ANVISA, 2003f).
Para o desenvolvimento da curva de calibração foi empregada a técnica
cromatográfica de CLAE e como a tibolona dispõe de um cromóforo detectável
(λmax 207 nm), foi possível desenvolver o método baseado na detecção por UV,
tendo em vista que os detectores de UV de comprimento de onda variável são
considerados convenientes e aplicáveis (SNYDER; KIRKLAND; GLAJCH, 1997).
A fim de conseguir uma melhor separação do pico cromatográfico da
tibolona para o desenvolvimento do método foi escolhida a acetonitrila como
solvente orgânico para a fase móvel e a proporção ideal, acetonitrila:água,
Inicialmente foi testada, através de uma corrida cromatográfica em módulo
isocrático, com 90%, 50% e 70% de ACN. Conseguindo-se a melhor separação
com maior pureza do pico da tibolona, com 70% de acetonitrila.
Sendo assim o método foi desenvolvido com a fase móvel acetonitrila:
água (70:30) na forma de eluição isocrática, através de uma coluna Phenomenex
C18 de aço inox com 25 cm de comprimento, 4,6 mm de diâmetro interno e
tamanho de partícula de 5 μm, equipada com uma pré-coluna de 5 cm de
comprimento, com as mesmas especificações, com fluxo de 1 mL/min e utilizando
um detector UV regulado para 207 nm.
O desenvolvimento foi operacionalizado em duplicata e a curva de
calibração foi construída plotando-se os valores médios das áreas dos picos
cromatográficos obtidos em função das concentrações ensaiadas.
4.2.2 Resultados e Discussão Espectro UV da Tibolona
A figura abaixo mostra o espectro da tibolona na concentração de 0,1
mg/mL, considerada ideal por produzir o melhor espectro com os máximos de 207
e 241 nm sendo o λmax 207 nm, com um εmax perfeitamente observável.

58
FIGURA 8 – Espectro UV da Tibolona na faixa de 190 a 600 nm na concentração de 0,1 mg/mL em solução de metanol/água (70:30).
A acetonitrila foi escolhida por ser considerada a melhor alternativa para
escolha inicial de solvente para fase móvel em HPLC de fase reversa (SNYDER;
KIRKLAND; GLAJCH, 1997). Misturas de acetonitrila/água são recomendadas
quando se trabalha com detecção por UV em comprimentos de onda baixo (185 a
210nm), como é o caso da tibolona (207nm). Estas misturas têm viscosidades
muito baixas, resultando em número de pratos um pouco maiores e pressões
menores na coluna, fatores estes bastante vantajosos para separação. (SNYDER;
KIRKLAND; GLAJCH, 1997).
Minutes0 5 10 15 20 25 30 35
mA
U
0
100
200
300
0
100
200
300
FIGURA 9 – Cromatograma da tibolona com ACN/H2O (90:10).
A corrida cromatográfica utilizando 90% de ACN (FIGURA 9) não se
mostrou satisfatória, porque o tempo de retenção foi muito baixo (4,34min) e
próximo do tempo morto da coluna, devido à alta força eluotrópica da mistura.
TR = 4,34 min
Minutos
200 nm 400 nm 600 nm
Comprimento de onda

59
Este efeito pode levar a sobreposição do pico da tibolona com outros
componentes, como produtos de degradação ou impurezas.
Minutes0 2 4 6 8 10 12 14 16 18 20
mA
U
0
100
200
0
100
200
FIGURA 10 – Cromatograma da tibolona com ACN/H2O (50:50).
Já o 2º cromatograma obtido com 50% de ACN (FIGURA 10), exibiu um
tempo de retenção relativamente alto (12,44 min), e a resolução entre o pico da
tibolona e o pico adjacente ainda permite uma alteração no sistema eluente para
uma concentração intermediaria com 70% de acetonitrila.
Minutes0 2 4 6 8 10 12 14 16 18 20
mA
U
0
100
200
0
100
200
FIGURA 11 – Cromatograma tibolona com ACN/H2O (70:30).
O desenvolvimento com 70% de ACN (FIGURA 11), revelou-se ideal,
apresentando uma ótima resolução entre a tibolona e o pico adjacente, alem de
fornecer um tempo de corrida pequeno (6,74 min) que é também outro fator
importante para quantificação de amostras.
Minutos
TR = 12,44 min
Minutos
TR = 6,74 min

60
TABELA 7 – Condições cromatográficas desenvolvidas para quantificação da tibolona por detecção direta com UV.
CARACTERÍSTICAS
DESCRIÇÃO
• Cromatógrafo líquido Shimadzu Série 10A vp,, configurado com os seguintes módulos:
Forno de coluna (CTO - 10AS); degaseificador (DGU – 14A); interface controladora (SCL - 10A); bomba (LC - 10AD) com válvula solenóide (FCV - 10AL). Forno de coluna (CTO - 10AS); degaseificador (DGU - 14A); interface controladora (SCL - 10A); bomba (LC - 10AD);
• Detector UV/Vis (SPD - 10AV) regulado para 207nm. DAD (SPD - M10A).
• Coluna C18 Phenomenex de aço inox (performance 250 mm X
4,6 mm DI, tamanho de partícula de 5 μm) equipada com pré-coluna (5 cm de comprimento), nas mesmas especificações.
• Injetor Automático (SIL - 10AD). Manual, provido com um loop de amostra de 20 μL
(Rheodyne).
• Volume de injeção 20 μL
• Fase móvel Acetonitrila/água (70:30).
• Tipo de eluição Isocrática.
• Vazão de fluxo 1 mL/min.
• Temperatura 40 ºC

61
PARTE II 4.3 VALIDAÇÃO DO MÉTODO
4 .3.1 Metodologia A validação do método proposto foi realizada por meio da avaliação dos
parâmetros de especificidade e seletividade, linearidade, intervalo, precisão e
robustez para o método cromatográfico desenvolvido.
Especificidade e seletividade
A especificidade foi determinada diluindo-se a solução estoque de tibolona
padrão com metanol para a concentração final de 0,025 mg/mL. Esta solução foi
filtrada e submetida à ultrasom por vinte minutos para homogeneização e parte
dela, transferida quantitativamente para um tubo de Eppendorf, sendo submetida
à degradação térmica, em termobloco, a uma temperatura de 120ºC por trinta
minutos. Após resfriamento, foi injetada diretamente em um cromatógrafo líquido
CLAE-DAD/UV-Vis regulado para um λmax de 207 nm nas mesmas condições
cromatográficas estabelecidas anteriormente no desenvolvimento do método
(Tabela 6), em duas repetições para cada amostra analisada. Para avaliar a
especificidade do método foi também cromatografada uma amostra da solução
não degradada (branco), nas mesmas condições.
Linearidade Para a determinação da linearidade, repetiu-se a curva de calibração
previamente construída, em três dias consecutivos, partindo-se de diluições da
solução estoque da Tibolona padrão com acetonitrila/água (70:30) dentro da faixa
de concentrações (0,001; 0,005; 0,010; 0,025; 0,050; 0,100; 0,200 e 0,500
mg/mL), e cromatografadas nas condições já estabelecidas no desenvolvimento
do método.
As curvas obtidas em cada dia tiveram seus coeficientes de correlação
calculados através da equação da reta, pelo método dos mínimos quadrados e
avaliados dentro dos critérios mínimos de aceitação (R2 ≥ 0,99) da RE 899

62
(BRASIL, ANVISA, 2003f). A análise estatística dos dados das curvas obtidas em
dias consecutivos foi realizada utilizando-se análise de variância (ANOVA) e teste
de Tukey a um nível de significância de 95%, utilizando-se o software GraphPad
Prism 4.0.
Precisão (Repetibilidade intra-dia) Após preparar a curva de calibração anteriormente desenvolvida com as
oito concentrações da solução de tibolona padrão estabelecidas (0,001; 0,005;
0,010; 0,025; 0,050; 0,100; 0,200 e 0,500 mg/mL) e constatada sua linearidade, a
repetibilidade intra-dia foi verificada utilizando-se três concentrações baixa,
média e alta, 0,025; 0,100 e 0,500 mg/mL respectivamente. As soluções nessas
concentrações foram obtidas a partir da solução estoque de tibolona por diluição
com acetonitrila/água (70:30) e cromatografadas em CL/UV, em quatro réplicas
para cada concentração, perfazendo um total de 12 determinações. O ensaio foi
repetido em três dias consecutivos.
Com a obtenção da curva de calibração, efetuou-se a regressão linear no
sentido de obter-se a equação da reta. Para calcular os valores reais de tibolona,
substituiu-se o valor de y da equação pelos valores das áreas dos cromatogramas
obtidos para as concentrações, baixa, média e alta. Com esses valores calculou-
se a precisão através do coeficiente de variação aplicando a seguinte fórmula
CV%= DP/média x100.
A precisão inter-dia foi calculada através da média das concentrações
baixa (0,025mg/mL), média (0,1mg/mL) e alta (0,5mg/mL), ensaiadas nos três
dias consecutivos.
Exatidão Para verificar a exatidão utilizou-se a mesma curva de calibração e
regressão linear da precisão; utilizando-se os mesmos dados, as mesmas
condições e os resultados obtidos no experimento anterior, desenvolvido para a
determinação da precisão. A exatidão é expressa pela relação entre a
concentração média experimental e a concentração teórica correspondente, e foi

63
calculada pela diferença porcentual entre as médias das concentrações
experimentais e o valor da concentração teórica de acordo a fórmula abaixo:
x100teórica ãoConcentraç
lexpeimenta média ãoConcentraçExatidão =
A exatidão inter-dia foi calculada através da média dos resultados
encontrados para cada uma das concentrações baixa, média e alta, ensaiadas em
cada dia ao longo dos três dias consecutivos.
Robustez
Construída a curva de calibração anteriormente desenvolvida com as oito
concentrações da solução de tibolona padrão (0,001; 0,005; 0,010; 0,025; 0,050;
0,100; 0,200 e 0,500 mg/mL) estabelecidas e constatada sua linearidade, a
robustez foi verificada utilizando-se soluções com três concentrações: Baixa,
média e alta, 0,025; 0,100 e 0,500 mg/mL respectivamente, obtidas a partir da
solução estoque de tibolona por diluição com acetonitrila/água (70:30) e
cromatografadas em CL UV-Vis, em três réplicas cada perfazendo um total de 9
determinações, variando-se a fase móvel, na proporção de 68 e 72 % de
acetonitrila para 32 e 28 % de água, respectivamente. O ensaio foi repetido em
dois dias consecutivos. A robustez foi avaliada através do cálculo do coeficiente
de variação.
4.3.2 Resultados e Discussão A validação de um método analítico deve garantir por meio de estudos
experimentais, que o método atenda as exigências das aplicações analíticas,
assegurando a confiabilidade dos resultados, devendo para tanto apresentar:
especificidade, linearidade, intervalo, precisão, robustez, sensibilidade, limite de
quantificação e exatidão adequadas à análise (BRASIL, ANVISA, 2003f).
Os parâmetros avaliados no processo de validação foram: especificidade
(seletividade), linearidade, precisão e exatidão, intervalo e robustez.
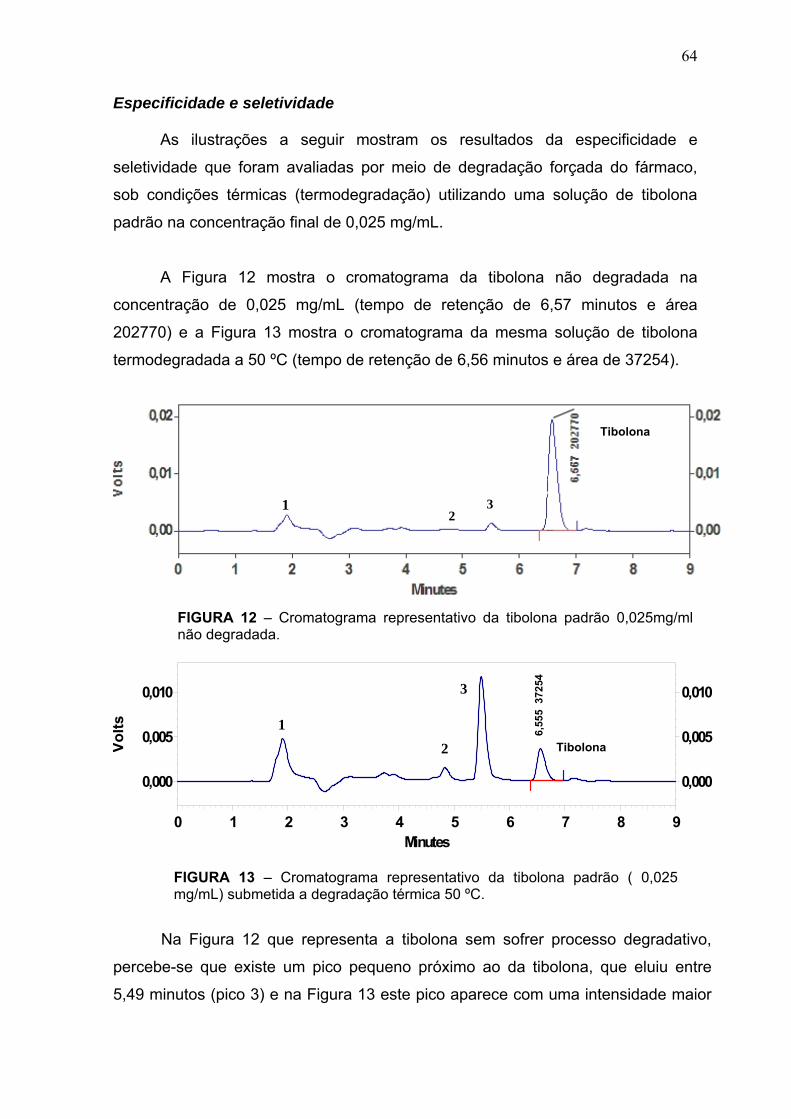
64
Especificidade e seletividade As ilustrações a seguir mostram os resultados da especificidade e
seletividade que foram avaliadas por meio de degradação forçada do fármaco,
sob condições térmicas (termodegradação) utilizando uma solução de tibolona
padrão na concentração final de 0,025 mg/mL.
A Figura 12 mostra o cromatograma da tibolona não degradada na
concentração de 0,025 mg/mL (tempo de retenção de 6,57 minutos e área
202770) e a Figura 13 mostra o cromatograma da mesma solução de tibolona
termodegradada a 50 ºC (tempo de retenção de 6,56 minutos e área de 37254).
FIGURA 12 – Cromatograma representativo da tibolona padrão 0,025mg/ml não degradada.
Minutes0 1 2 3 4 5 6 7 8 9
Volts
0,000
0,005
0,010
0,000
0,005
0,010
6,55
5 3
7254
FIGURA 13 – Cromatograma representativo da tibolona padrão ( 0,025 mg/mL) submetida a degradação térmica 50 ºC.
Na Figura 12 que representa a tibolona sem sofrer processo degradativo,
percebe-se que existe um pico pequeno próximo ao da tibolona, que eluiu entre
5,49 minutos (pico 3) e na Figura 13 este pico aparece com uma intensidade maior
1 2 Tibolona
3
1 2
3 Tibolona
3

65
e no mesmo tempo de retenção da figura 12, e o pico da tibolona degradada
aparece com sua intensidade visivelmente menor do que antes da degradação. Ao
compararmos os cromatogramas antes e depois da degradação pode-se afirmar
que o pico 3, pode ser atribuído ao principal produto de degradação da tibolona.
A Figura 14 abaixo mostra a superposição dos cromatogramas da tibolona
degradada e não degradada, demonstrando que a termodegradação reduziu a área
inicial do pico da tibolona de 82,90% para 16,40% (TABELA 8).
0 1 2 3 4 5 6 7 8 9
0,00
0,01
0,02
0,00
0,01
0,02
M I N U T O S FIGURA 14 – Cromatograma representativo da superposição da tibolona não degradada e degradada a 50 ºC.
A Tabela 8 abaixo mostra os resultados dos cálculos da resolução dos
picos cromatográficos da termodegradação da tibolona demonstrando que a
porcentagem de degradação foi de 66,50 %.
TABELA 8 – Resolução dos picos cromatográficos da tibolona não degradada e termodegradada e suas porcentagens de degradação.
Picos Tempo de retenção
(min) Resolução %
3 5,49 12,71 3,06 (Tib.) 6,55 3,84 82,90
3 5,49 2,69 48,88
Tibolona não
degradada
(Tib.) 6,55 4,03 16,40 % Degradação = % tibolona não degradada – % tibolona degradada
% = 82,90 – 16,40 = 66,50 %
V O L T S
3
2
Tibolona 1

66
O método desenvolvido apresentou-se adequado à análise da tibolona e
seus respectivos produtos de degradação, no qual as resoluções correspondentes
ao pico principal das soluções das amostras degradada e não degradada
apresentam valores superiores a dois, demonstrando que o método é especifico
nas condições pré-estabelecidas, não evidenciando nenhuma interferência entre
os picos. Observou-se também a separação individual dos interferentes
relevantes, neste caso confirmada pela resolução dos analitos que eluem
próximos. O método mostrou-se especifico nas condições analisadas
demonstrando uma resolução adequada entre os picos referentes à tibolona
degradada em relação ao do produto de degradação. Desta forma a
especificidade consiste no parâmetro fundamental nos testes de determinação
quantitativa do teor e seus respectivos produtos de degradação e impurezas.
Linearidade
As ilustrações (figuras, tabelas e gráficos) seguintes apresentam o
resultado da determinação da linearidade da curva de calibração da tibolona nas
concentrações de 0,001; 0,005; 0,010; 0,025; 0,050; 0,100; 0,200 e 0,500 mg/mL
obtida em três dias consecutivos.
As Figuras 15,16 e 17 representam à superposição dos cromatogramas
representativos das curvas de calibração da tibolona padrão, obtidas,
respectivamente nos três dias de análises.
0 1 2 3 4 5 6 7
0
250
500
FIGURA 15 – Superposição dos cromatogramas representativos das curvas de calibração
da tibolona padrão obtidas no 1º dia de análise.
Minutos
mA
U
Dia 26_12_2007
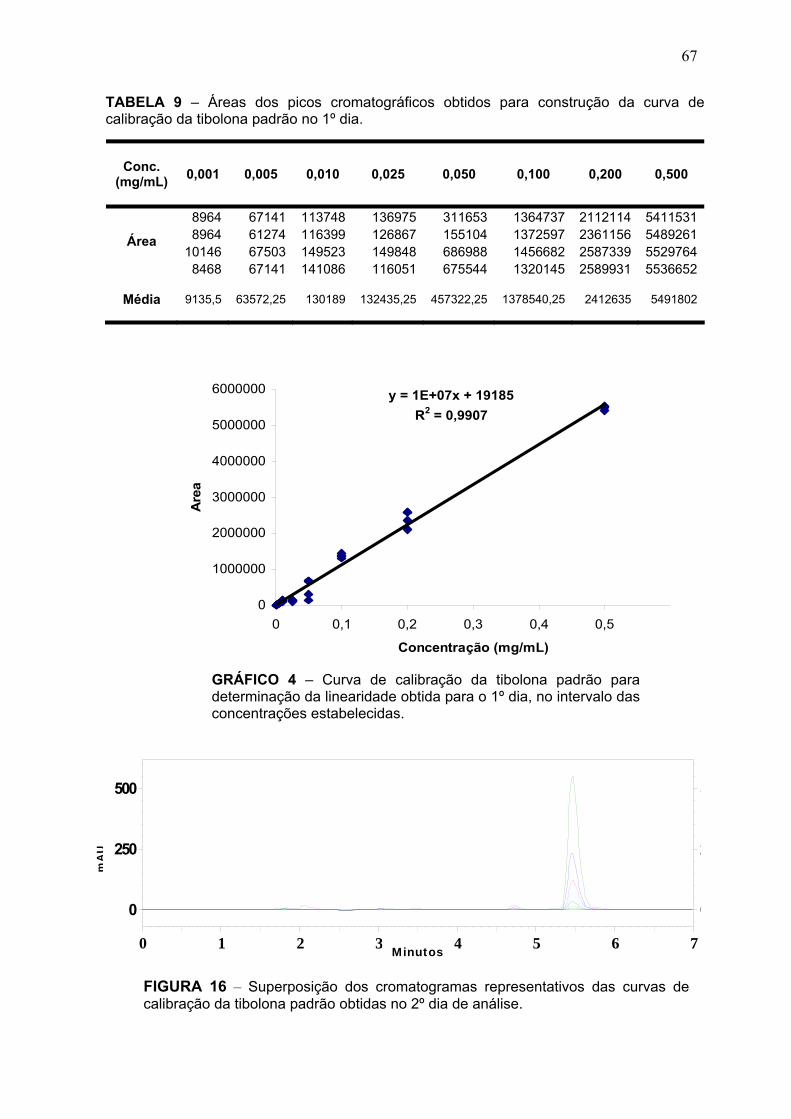
67
TABELA 9 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 1º dia.
Conc.
(mg/mL)
0,001 0,005 0,010 0,025 0,050 0,100 0,200 0,500
8964 67141 113748 136975 311653 1364737 2112114 54115318964 61274 116399 126867 155104 1372597 2361156 5489261
10146 67503 149523 149848 686988 1456682 2587339 5529764Área
8468 67141 141086 116051 675544 1320145 2589931 5536652
Média 9135,5 63572,25 130189 132435,25 457322,25
1378540,25
2412635 5491802
y = 1E+07x + 19185R2 = 0,9907
0
1000000
2000000
3000000
4000000
5000000
6000000
0 0,1 0,2 0,3 0,4 0,5
Concentração (mg/mL)
Are
a
GRÁFICO 4 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 1º dia, no intervalo das concentrações estabelecidas.
0 1 2 3 4 5 6 7
0
250
500
0
2
5
FIGURA 16 – Superposição dos cromatogramas representativos das curvas de calibração da tibolona padrão obtidas no 2º dia de análise.
Minutos
mA
U
Dia 28_12_2007 Dia 28_12_2007

68
TABELA 10 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 2º dia.
Conc.
(mg/mL)
0,001 0,005 0,010 0,025 0,050 0,100 0,200 0,500
15088 74885 155130 326243 547982 1137650 2095643 5597119 5318 76019 160287 330915 545537 1168085 2317151 5612179 8952 80690 159608 320749 569613 1145977 2367180 5581165
Área
9480 82227 167569 326890 576554 1171772 2383121 5638391
Média
9709,5
78455,25
160648,5
326199,3
559921,5
1155871
2290774
5607214
y = 1E+07x + 28517R2 = 0,9993
0
1000000
2000000
3000000
4000000
5000000
6000000
0 0,1 0,2 0,3 0,4 0,5
Concentração (mg/mL)
Are
a
GRÁFICO 5 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 2º dia, no intervalo das concentrações estabelecidas.
Mi t0 1 2 3 4 5 6 7
0
250
500
FIGURA 17 – Superposição dos cromatogramas representativos das curvas de calibração da tibolona padrão obtidas no 3º dia de análise.
Minutos
mA
U
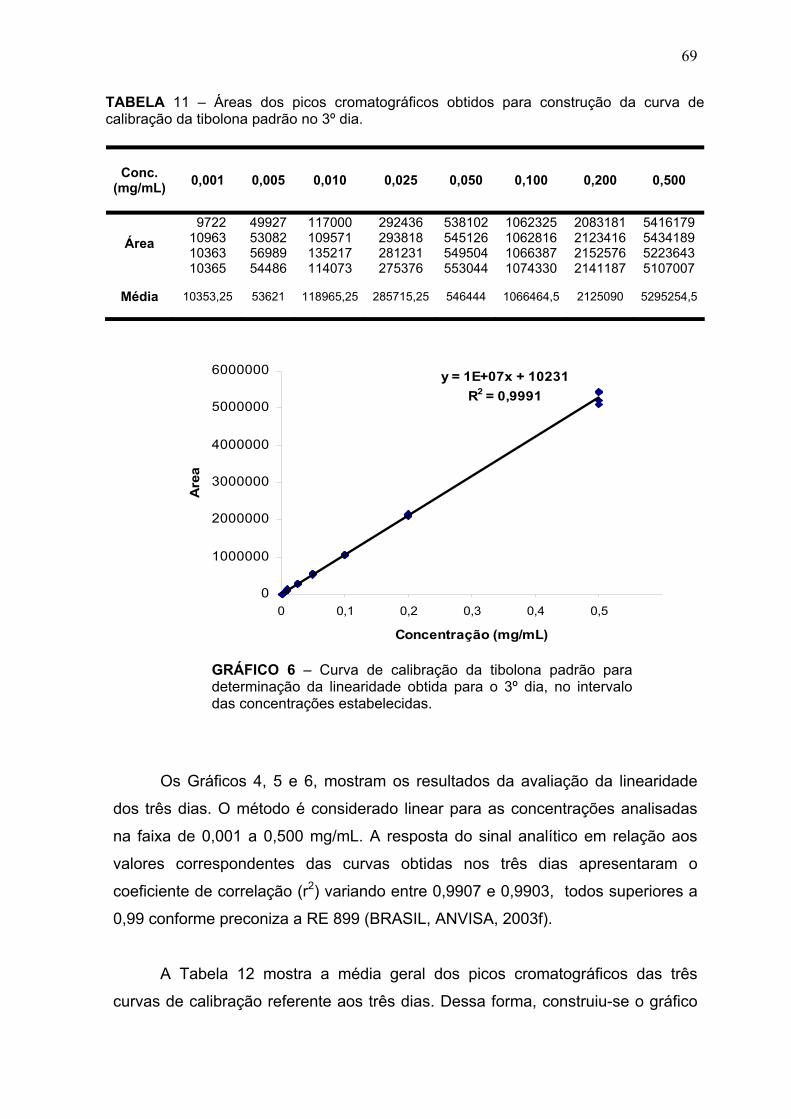
69
TABELA 11 – Áreas dos picos cromatográficos obtidos para construção da curva de calibração da tibolona padrão no 3º dia.
Conc.
(mg/mL)
0,001 0,005 0,010 0,025 0,050 0,100 0,200 0,500
9722 49927 117000 292436 538102 1062325 2083181 5416179 10963 53082 109571 293818 545126 1062816 2123416 5434189 10363 56989 135217 281231 549504 1066387 2152576 5223643
Área
10365 54486 114073 275376 553044 1074330 2141187 5107007
Média 10353,25 53621
118965,25
285715,25 546444
1066464,5
2125090
5295254,5
y = 1E+07x + 10231R2 = 0,9991
0
1000000
2000000
3000000
4000000
5000000
6000000
0 0,1 0,2 0,3 0,4 0,5
Concentração (mg/mL)
Are
a
GRÁFICO 6 – Curva de calibração da tibolona padrão para determinação da linearidade obtida para o 3º dia, no intervalo das concentrações estabelecidas.
Os Gráficos 4, 5 e 6, mostram os resultados da avaliação da linearidade
dos três dias. O método é considerado linear para as concentrações analisadas
na faixa de 0,001 a 0,500 mg/mL. A resposta do sinal analítico em relação aos
valores correspondentes das curvas obtidas nos três dias apresentaram o
coeficiente de correlação (r2) variando entre 0,9907 e 0,9903, todos superiores a
0,99 conforme preconiza a RE 899 (BRASIL, ANVISA, 2003f).
A Tabela 12 mostra a média geral dos picos cromatográficos das três
curvas de calibração referente aos três dias. Dessa forma, construiu-se o gráfico

70
da curva de calibração (Gráfico 7) e fez-se a regressão linear obtendo-se a
equação da reta, sendo o coeficiente de correlação r2 = 0,9994. A análise de
variância (ANOVA) dos dados médios (3 dias consecutivos), com pós teste
(Tukey), mostra que não há diferença significativa na resposta entre os 3 dias
consecutivos, e que as variâncias são iguais (p>0,05).
TABELA 12– Média das áreas dos picos cromatográficos obtidos nos três dias para construção da curva de calibração da tibolona padrão.
Conc. mg/mL)
Média (1º dia)
Média (2º dia)
Média (3º dia)
Media Geral
DP
CV%
0,001 9136 10353 9710 9733,0 608,8 6,25530,005 63572 53621 60575 59256,0 5104,9 8,61510,010 130189 118965 160649 136601,0 21569,1 15,78980,025 317629 285715 326199 309847,7 21334,3 6,88540,050 457322 546444 559922 521229,3 55754,1 10,69670,100 1359625 1066465 1155871 1193987,0 150250,9 12,58400,200 2412635 2125090 2290774 2276166,3 144328,0 6,34080,500 5491802 5295255 5607214 5464757,0 157728,2 2,8863
y = 1E+07x + 28020R2 = 0,9994
0
1000000
2000000
3000000
4000000
5000000
6000000
0,000 0,100 0,200 0,300 0,400 0,500
Concentração (mg/mL)
Áre
a
GRÁFICO 7 – Gráfico das médias das três curvas de calibração.
A análise de variância mostra ainda que, enquanto há diferença
significativa na resposta entre as linhas (níveis de concentração, p<0.0001), não
há diferença significativa entre as colunas (p=0,0927).

71
A Tabela 13 mostra o resultado para o cálculo dos resíduos entre os
valores medidos e os valores calculados a partir da equação de regressão linear.
Os valores de t calculado variaram entre 2,375778 - 87,05821.
TABELA 13 – Cálculo dos resíduos em relação às três curvas de calibração obtidas na determinação da linearidade.
Conc.
(mg/mL)
Xcalculado (teórico)
Média 1 (X1-Xt)
Média 2 (X2-Xt)
Média 3 (X3-Xt)
media (Xm-Xt) Sn*
t cal.**
0,001 28484 19348,5 18130,75 18774,5 18751,25 609,2078 87,058210,005 68484 4911,75 14863 9971,25 9915,333 4975,861 5,636170,010 118484 11705 481,25 42164,5 18116,92 21568,68 2,3757780,025 268484 136048,8 17231,25 57715,25 70331,75 60405,15 3,2932330,050 518484 61161,75 27960 41437,5 43519,75 16698,53 7,3714540,100 1018484 341141,3 47980,5 137387 175502,9 150251,2 3,3037820,200 2018484 394151 106606 272289,8 257682,3 144328 5,0498560,500 5018484 473318 276770,5 588729,5 446272,7 157728,2 8,002688
Limites aceitáveis: * Sn = desvio padrão dos resíduos ** t cal. = (resíduo)/Sn/√n, n = 8.
0.1 0.2 0.3 0.4 0.5 0.6
-50000
0
50000
100000
GRÁFICO 8 – Resíduos dos dados de linearidade.
O Gráfico 8 mostra que não houve uma tendência de distribuição não
aleatória dos resíduos, confirmando a validade do modelo de calibração adotado.

72
Precisão/Exatidão As tabelas e gráficos abaixo representam a precisão/exatidão
determinadas através do parâmetro repetibilidade (intra-dia), avaliada pelo
coeficiente de variação percentual obtido na determinação das áreas dos picos
cromatográficos da Tibolona nas concentrações baixa (B), média (M) e alta (A)
respectivamente (0,025; 0,100 e 0,500 mg/mL) obtidos em três dias consecutivos
(inter-dias).
TABELA 14 – Valores de precisão e exatidão obtidos para as concentrações B, M e A no 1º dia.
Concentração (mg/mL) 0,025 0,100 0,500
0,0237 0,0934 0,4725 0,0256 0,0990 0,4805 0,0252 0,0998 0,4838
Concentrações experimentais
0,0244 0,1005 0,4692
Média 0,0247 0,0982 0,4765 DP 0,0009 0,0032 0,0068
Precisão (%)* 3,4770 3,3074 1,4247 Exatidão (%)** 98,8144 98,1704 95,3008
Limites aceitáveis: * ≤ 5 % (RE 899 – ANVISA/2003) ** 85 a 120 % (RE 899 – ANVISA/2003)
y = 2E+07x - 8014,7R2 = 0,9986
0
1000000
2000000
3000000
4000000
5000000
6000000
7000000
8000000
9000000
0,000 0,100 0,200 0,300 0,400 0,500
Concentração (mg/mL)
Are
a
GRÁFICO 9 – Curva de calibração para determinação da precisão e exatidão obtida no 1º dia.

73
TABELA 15 – Valores de precisão e exatidão obtidos para as concentrações B, M e A no 2º dia.
Concentração (mg/mL) 0,025 0,100 0,500
0,0237 0,1022 0,4809 0,0235 0,1018 0,4736 0,0229 0,1018 0,4842
Concentrações experimentais
0,0218 0,1025 0,4867
Média 0,0230 0,1021 0,4813
DP 0,0009 0,0004 0,0057
Precisão (%)* 3,8331 0,3455 1,1792
Exatidão (%)** 91,8982 102,0648 96,2680 Limites aceitáveis: * ≤ 5 % (RE 899 – ANVISA/2003) ** 85 a 120 % (RE 899 – ANVISA/2003)
y = 2E+07x + 23303R2 = 0,9998
0
2000000
4000000
6000000
8000000
10000000
12000000
0,000 0,100 0,200 0,300 0,400 0,500
Concentração (mg/mL)
Are
a
GRAFICO 10 – Curva de calibração para determinação da precisão e exatidão obtida no 2º dia.

74
TABELA 16 – Valores de precisão e exatidão obtidos para as concentrações B, M e A no 3º dia.
Concentração (mg/mL) 0,025 0,100 0,500
0,0232 0,1026 0,5006 0,0232 0,1017 0,5050 0,0236 0,1020 0,5118
Concentrações experimentais
0,0240 0,1061 0,5083
Média 0,0235 0,1031 0,5064 DP 0,0004 0,0020 0,0048
Precisão (%) 1,5885 1,9856 0,9395 Exatidão (%) 94,0135 103,1215 101,2825
Limites aceitáveis: * ≤ 5 % (RE 899 – ANVISA/2003)
** 85 a 120 % (RE 899 – ANVISA/2003)
y = 2E+07x + 45128R2 = 0,9989
0
2000000
4000000
6000000
8000000
10000000
12000000
0,000 0,100 0,200 0,300 0,400 0,500
Concentração (mg/mL)
Area
GRÁFICO 11 – Curva de calibração para determinação da precisão e exatidão obtida no 3º dia.
A análise das Tabelas 14, 15 e 16 demonstra que o método é preciso e
exato. Foi avaliada a precisão intermediária em três dias e os resultados foram
calculados através do coeficiente de variação. A média dos valores do CV% por
cada dia de análise foi respectivamente, 2.7364%, 1,7859% e 1,5045%,

75
encontrando-se todos abaixo de 5%, conforme as especificações da RE-899. Já a
exatidão, foi avaliada calculando a relação entre a concentração teórica do
fármaco e a concentração média experimental, durante os três dias consecutivos.
A média dos resultados por cada dia de análise foi de 97,4285%, 96,7437% e
99,4725% respectivamente, Estes resultados encontram-se também dentro do
limite permitido pela RE-899, que é de 85% a 115%. (BRASIL, ANVISA, 2003f).
TABELA 17 - Valores médios de precisão e exatidão obtidos para as concentrações B, M e A nos três dias (inter-dias).
Concentração (mg/mL) 0,025 0,100 0,500
1º Dia 0,0247 0,0982 0,4765 2° Dia 0,0230 0,1021 0,4813 3º Dia 0,0235 0,1031 0,5064
Média 0,0237 0,1011 0,4881
DP 0,0009 0,0026 0,0161
PRECISÃO (%)* 3,6813 2,5601 3,2900 EXATIDÃO (%)** 94,9333 101,1333 97,6133
Limites aceitáveis: * ≤ 5 % (RE 899 – ANVISA/2003) ** 85 a 120 % (RE 899 – ANVISA/2003)
A precisão inter-dia foi calculada através do coeficiente de variação
utilizando a média dos três dias das concentrações baixa, média e alta, cujos
valores foram 3,6813, 2,5601 e 3,2900 respectivamente. Todos os valores
encontram-se com coeficiente de variação inferiores a 5%, dentro do limite
preconizado pela RE899. Já a exatidão inter-dia foi calculada através da relação
entre a concentração média experimental e a concentração teórica, utilizando à
média dos três dias das concentrações baixa, média e alta, cujos valores foram
respectivamente, 94,9333%; 101,1333% e 97,6133%, conforme a Tabela 17.
Estes resultados também se encontram de acordo com o que preconiza a RE899,
demonstrando a precisão e exatidão do método. (BRASIL, ANVISA, 2003F).

76
Robustez A robustez foi avaliada através da variação da composição da fase móvel,
variando a fase orgânica em 68% e 72%.
As Tabelas 18, 19, 20 e 21 abaixo, mostram a média das áreas dos picos
cromatográficos das concentrações baixa (B), média (M) e alta (A), 0,25 mg/ml,
0,100 mg/mL e 0,500 mg/mL respectivamente, quando se variou a proporção de
acetonitrila na fase móvel para 68% e 72% ao longo de dois dias consecutivos.
TABELA 18 – Valores das concentrações, B, M e A obtidas na determinação da robustez utilizando 68% de acetonitrila na fase móvel no1º dia, juntamente com a média DP e coeficiente de variação (CV%).
Concentração (mg/mL) 0,025 0,100 0,500
0,0305 0,1218 0,6051
0,0317 0,1255 0,6148 Concentrações experimentais
(mg/mL) 0,0291 0,1251 0,6175
média 0,0304 0,1241 0,6125
DP 0,0013 0,0020 0,0065
CV% 4,3230 1,6134 1,0660
TABELA 19 – Valores das concentrações, B, M e A obtidas na determinação da robustez utilizando 72% de acetonitrila na fase móvel no1º dia, juntamente com a média DP e coeficiente de variação (CV%).
Concentração (mg/mL) 0,025 0,100 0,500
0,0234 0,0912 0,4386
0,0224 0,0924 0,4403 Concentrações experimentais
0,0222 0,0930 0,4443
média 0,0227 0,0922 0,4411
DP 0,0007 0,0009 0,0030
CV% 2,8856 0,9740 0,6700

77
TABELA 20 – Valores das concentrações, B, M e A obtidas na determinação da robustez utilizando 68% de acetonitrila na fase móvel no 2º dia, juntamente com a média DP e coeficiente de variação (CV%).
Concentração (mg/mL)
0,025 0,100 0,500
0,0247 0,0998 0,4917
0,0268 0,1000 0,4930 Concentrações experimentais
0,0244 0,0976 0,4845
média 0,0253 0,0991 0,4898
DP 0,0013 0,0013 0,0046
CV% 5,2191 1,3179 0,9399
TABELA 21 – Valores das concentrações, B, M e A obtidas na determinação da robustez utilizando 72% de acetonitrila na fase móvel no 2º dia, juntamente com a média DP e coeficiente de variação (CV%).
Com os resultados encontrados nas Tabelas 18, 19, 20 e 21, a robustez foi
avaliada utilizando as concentrações da exatidão e calculada através do CV%,
onde os valores de áreas foram adequados nos diferentes níveis de concentração
interdia. Enquanto nos níveis de concentração extremos, os valores de precisão
apresentaram-se maiores que 5%, em relação aos dados intradia. Os dados de
robustez em relação à concentração experimental versus concentração nominal,
apresentaram valores porcentuais acima de 100%, na condição analítica de 68%
no primeiro de análise. Porém, no cálculo da média inter-dia, os valores médios
Concentração (mg/mL) 0,025 0,100 0,500
0,0208 0,1052 0,5537
0,0189 0,1088 0,5220 Concentrações experimentais
0,0208 0,1093 0,6005
média 0,0202 0,1077 0,5587
DP 0,0011 0,0023 0,0394
CV% 5,4114 2,0879 7,0606

78
dos coeficientes de variação quando se variou à concentração para 68% foram de
4,8%, 1,5% e 1%, nas concentrações nominais de 0,025%, 0,1% e 0,5%,
respectivamente (TABELA 22). Para a variação de 72% foram de 4,1%, 1,5% e
3,9%, para as mesmas concentrações nominais anteriores (TABELA. 23). Estes
resultados mostram que os valores médios do CV% ficaram abaixo de 5%,
mesmo nos níveis de concentrações extremos.
TABELA 22 – Valores médios das concentrações, B, M e A obtidas utilizando 68% de acetonitrila na fase móvel nos dois dias, juntamente com a média DP e coeficiente de variação (CV%).
TABELA 23 – Valores médios das concentrações, B, M e A obtidas utilizando 72% de acetonitrila na fase móvel nos dois dias, juntamente com a média DP e coeficiente de variação (CV%).
O método proposto foi considerado robusto no que se refere à precisão e
exatidão quando respeitadas as condições analíticas, como temperatura da
coluna, fluxo da fase móvel, proporções da relação orgânico/aquoso da fase
móvel (acetonitrila/água 70:30) admitindo-se variação de aproximadamente ± 2%.
Sendo assim, estes resultados confirmam que o método pode ser aplicado
garantindo a confiabilidade na quantificação da tibolona.
Concentração (mg/mL) 0,025 0,100 0,500
1º Dia 0,0305 0,1242 0,6125 2º Dia 0,0253 0,0991 0,4898
Média 0,0279 0,1116 0,5511
CV% 4,8 1,5 1,0
Concentração (mg/mL) 0,025 0,100 0,500
1º Dia 0,0227 0,0922 0,4411 2º Dia 0,0202 0,1077 0,5587
Média 0,0214 0,1000 0,4999
CV% 4,1 1,5 3,9

79
PARTE III 4.4 APLICAÇÃO DO MÉTODO: ANÁLISE DE CÁPSULAS DE TIBOLONA MANIPULADA
4.4.1 Metodologia Amostra - Cápsulas de Tibolona 2,5 mg. Como amostra, foi utilizada tibolona manipulada na concentração de 2,5
mg, na forma farmacêutica de cápsulas, procedentes dos treze estabelecimentos
farmacêuticos magistrais que manipulam este medicamento nas cidades de
Campina Grande e João Pessoa no Estado da Paraíba. De cada farmácia
adquiriu-se 20 cápsulas, das quais 10 foram analisadas e as outras 10 foram
mantidas como testemunho. As amostras foram codificadas como FarmA, FarmB,
FarmC, FarmD, FarmE, FarmF, FarmG, FarmH, FarmI, FarmJ, FarmK, FarmL e
FarmM, representando as amostras de cada farmácia analisadas.
Determinação do Peso médio
O peso médio das cápsulas foi determinado segundo a Farmacopéia
Brasileira, pesando-se individualmente 10 cápsulas cheias e em seguida
esvaziando-se quantitativamente cada uma delas em balões volumétricos
individuais de 25mL de capacidade, tendo-se o cuidado de retirar todo conteúdo
restante com auxílio de algodão, que foi imediatamente lavado com uma mistura
acetonitrila/água (70:30) como solvente. Após isso, as cápsulas vazias foram
pesadas e o peso de cada uma foi determinado por diferença seguido da
determinação da variação porcentual do conteúdo das cápsulas em relação à
média. O limite de variação aceitável é de ± 10%, por tratar-se de cápsulas com
até 300mg (FARMACOPEIA BRASILEIRA, 1988).
Uniformidade de conteúdo Os dez balões contendo cada um o conteúdo individual de cada cápsula
tiveram então seus volumes completados com o mesmo solvente até o limite de
sua capacidade, de forma a obter uma concentração final de 0,1 mg/mL. Filtrou-

80
se cada solução obtida e após isso, retirou-se 1mL de cada e diluiu-se para 2mL
com o mesmo solvente, a fim de obter uma solução final de concentração igual a
0,05 mg/mL. Preparou-se a curva de calibração com as oito concentrações da
tibolona padrão conforme já estabelecido e as soluções finais das amostras foram
injetadas em CLAE/UV-VIS em duas réplicas cada, sendo registrados os valores
das áreas dos picos cromatográficos e calculando-se a uniformidade de conteúdo
através da determinação do teor de cada cápsula, bem como a média das áreas,
o desvio padrão e o desvio padrão relativo (coeficiente de variação) das soluções
finais das 13 farmácias amostradas, observando-se que, segundo a Farmacopéia
Brasileira o limite de valor aceitável para uniformidade de conteúdo é de 85% a
120% (FARMACOPEIA BRASILEIRA, 1988).
4.4.2 Resultados e Discussão Determinação do peso médio
A distribuição heterogênea de fármaco em uma cápsula deve ser um dos
fatores de atenção na manipulação de medicamentos, principalmente para
aqueles de baixo índice terapêutico e de baixa dosagem. Por isso a farmácia deve
melhorar cada vez mais o seu processo de mistura e homogeneização, no sentido
de garantir a qualidade dos medicamentos, tornando-os eficazes e seguros.
De acordo com a Farmacopéia Brasileira, o limite de variação do peso
permitido para cápsulas duras e com doses menores que 300 mg é de ±10% do
peso médio dos pós (FARMACOPEIA BRASILEIRA, 1988).
As tabelas seguintes mostram os resultados dos pesos médios, desvios
padrão e coeficiente de variação das cápsulas manipuladas na concentração de
2,5 mg obtidos pela pesagem de 10 unidades de cada uma das 13 farmácias
analisadas.
Os resultados descritos nas Tabelas 24a e 24b nas páginas seguintes
permitem observar que os pesos médios das amostras analisadas encontram-se
dentro dos limites de variação especificados uma vez que das treze amostras

81
TABELA 24a – Resultados obtidos nas determinações do peso médio das cápsulas de tibolona (2,5 mg), manipuladas nas Farmácias A, B, C, D, E, F e G.
Limite aceitável:(*) ± 10% (FB 4. ed. 1988)
FarmA FarmB FarmC FarmD FarmE FarmF
FarmG
Amostras (cápsula)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
1 0,0835 -2,3392 0,1489 -3,0788 0,1472 2,7502 0,1477 -6,4064 0,1210 5,0895 0,1507 41,8219 0,2418 0,6117
2 0,0850 -0,5848 0,1644 7,0103 0,1404 -1,9964 0,1680 6,4571 0,1105 - 4,0299 0,0972 - 8,5263 0,2312 - 3,7989
3 0,0858 0,3509 0,1512 -1,5817 0,1383 -3,4622 0,1504 -4,6955 0,1181 2,5708 0,1019 - 4,1031 0,2364 - 1,6353
4 0,0845 -1,1696 0,1574 2,4539 0,1472 2,7502 0,1670 5,8235 0,1146 - 0,4690 0,0977 - 8,0557 0,2482 3,2747
5 0,0844 -1,2865 0,1539 0,1757 0,1440 0,5165 0,1611 2,0848 0,1089 - 5,4195 0,1018 - 4,1973 0,2411 0,3204
6 0,0791 -7,4854 0,1487 -3,2090 0,1475 2,9597 0,1634 3,5422 0,1133 - 1,5981 0,1071 0,7905 0,2479 3,1498
7 0,0884 3,3918 0,1565 1,8681 0,1443 0,7260 0,1467 -7,0401 0,1222 6,1317 0,1011 - 4,8560 0,2372 - 1,3024
8 0,0881 3,0409 0,1490 -3,0137 0,1363 -4,8583 0,1523 -3,4915 0,1162 0,9206 0,1068 0,5082 0,2395 - 0,3454
9 0,0878 2,6901 0,1529 -0,4752 0,1463 2,1220 0,1609 1,9581 0,1074 - 6,7223 0,1019 - 4,1031 0,2341 - 2,5923
10 0,0884 3,3918 0,1534 -0,1497 0,1411 -1,5077 0,1606 1,7679 0,1192 3,5261 0,0964 - 9,2791 0,2459 2,3176
PM 0,0855 0,1536 0,1433 0,1578 0,1151 0,1063 0,2403 DP 0,0029 0,0049 0,0040 0,0079 0,0051 0,0160 0,0058
CV% 3,4072 3,1675 2,8094 4,9923 4,4343 15,0869 2,4068
Limite de variação * 0,0769 – 0,0940 g 0,1382 – 0,1690 g 0,1289 – 0,1576 g 0,1420 – 0,1736 g 0,1036 – 0,1266 g 0,0957 – 0,1169 g 0,2163 – 0,2643 g
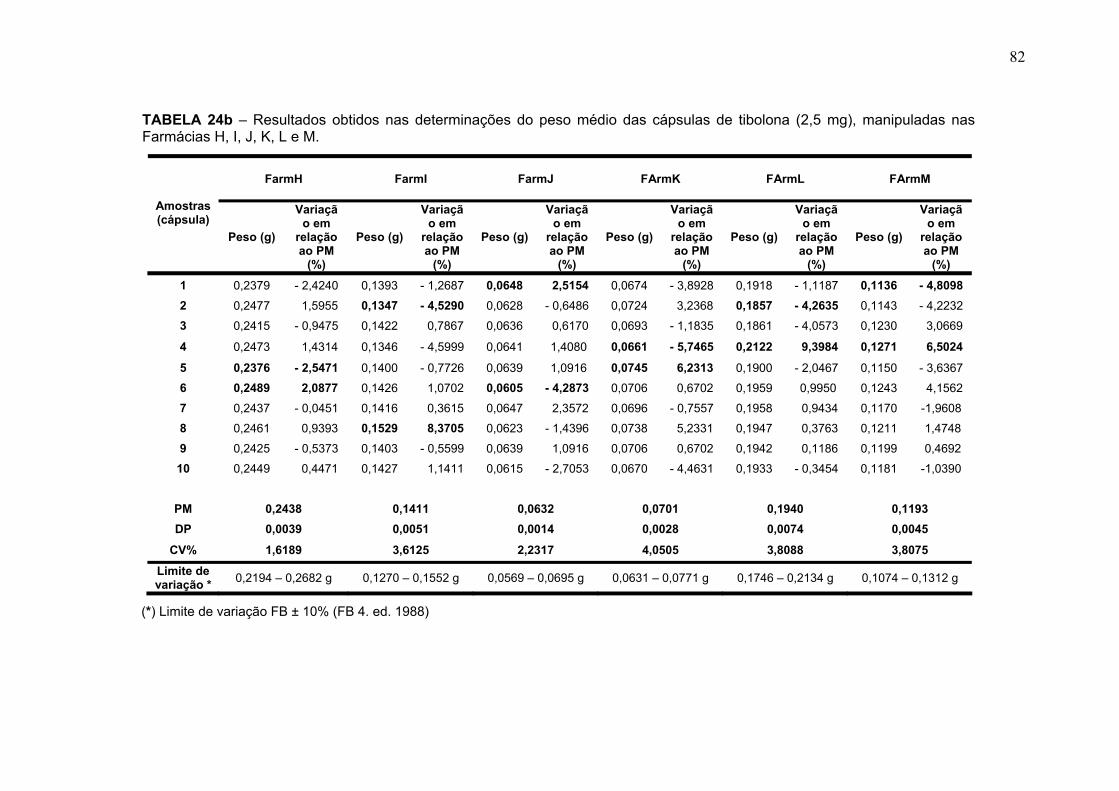
82
TABELA 24b – Resultados obtidos nas determinações do peso médio das cápsulas de tibolona (2,5 mg), manipuladas nas Farmácias H, I, J, K, L e M.
(*) Limite de variação FB ± 10% (FB 4. ed. 1988)
FarmH FarmI FarmJ FArmK FArmL
FArmM
Amostras (cápsula)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
Peso (g)
Variação em
relação ao PM
(%)
1 0,2379 - 2,4240 0,1393 - 1,2687 0,0648 2,5154 0,0674 - 3,8928 0,1918 - 1,1187 0,1136 - 4,8098 2 0,2477 1,5955 0,1347 - 4,5290 0,0628 - 0,6486 0,0724 3,2368 0,1857 - 4,2635 0,1143 - 4,2232
3 0,2415 - 0,9475 0,1422 0,7867 0,0636 0,6170 0,0693 - 1,1835 0,1861 - 4,0573 0,1230 3,0669
4 0,2473 1,4314 0,1346 - 4,5999 0,0641 1,4080 0,0661 - 5,7465 0,2122 9,3984 0,1271 6,5024
5 0,2376 - 2,5471 0,1400 - 0,7726 0,0639 1,0916 0,0745 6,2313 0,1900 - 2,0467 0,1150 - 3,6367
6 0,2489 2,0877 0,1426 1,0702 0,0605 - 4,2873 0,0706 0,6702 0,1959 0,9950 0,1243 4,1562
7 0,2437 - 0,0451 0,1416 0,3615 0,0647 2,3572 0,0696 - 0,7557 0,1958 0,9434 0,1170 -1,9608
8 0,2461 0,9393 0,1529 8,3705 0,0623 - 1,4396 0,0738 5,2331 0,1947 0,3763 0,1211 1,4748
9 0,2425 - 0,5373 0,1403 - 0,5599 0,0639 1,0916 0,0706 0,6702 0,1942 0,1186 0,1199 0,4692
10 0,2449 0,4471 0,1427 1,1411 0,0615 - 2,7053 0,0670 - 4,4631 0,1933 - 0,3454 0,1181 -1,0390
PM 0,2438 0,1411 0,0632 0,0701 0,1940 0,1193 DP 0,0039 0,0051 0,0014 0,0028 0,0074 0,0045
CV% 1,6189 3,6125 2,2317 4,0505 3,8088 3,8075
Limite de variação * 0,2194 – 0,2682 g 0,1270 – 0,1552 g 0,0569 – 0,0695 g 0,0631 – 0,0771 g 0,1746 – 0,2134 g 0,1074 – 0,1312 g
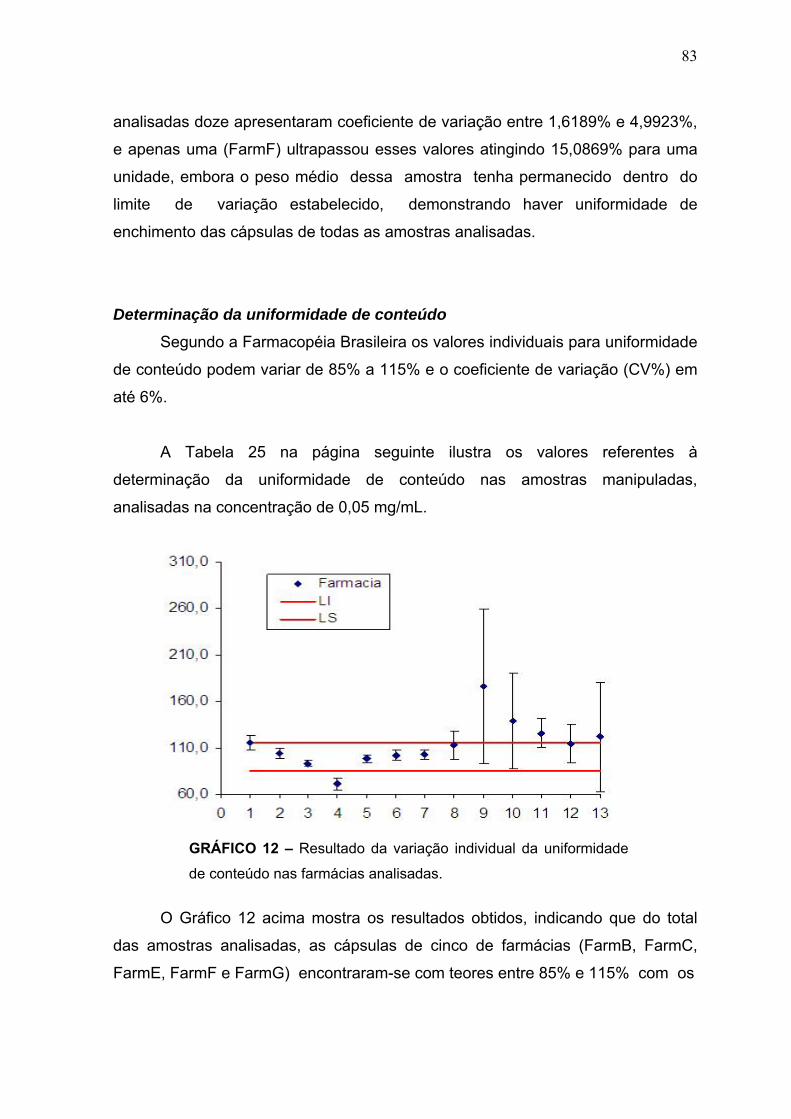
83
analisadas doze apresentaram coeficiente de variação entre 1,6189% e 4,9923%,
e apenas uma (FarmF) ultrapassou esses valores atingindo 15,0869% para uma
unidade, embora o peso médio dessa amostra tenha permanecido dentro do
limite de variação estabelecido, demonstrando haver uniformidade de
enchimento das cápsulas de todas as amostras analisadas.
Determinação da uniformidade de conteúdo
Segundo a Farmacopéia Brasileira os valores individuais para uniformidade
de conteúdo podem variar de 85% a 115% e o coeficiente de variação (CV%) em
até 6%.
A Tabela 25 na página seguinte ilustra os valores referentes à
determinação da uniformidade de conteúdo nas amostras manipuladas,
analisadas na concentração de 0,05 mg/mL.
GRÁFICO 12 – Resultado da variação individual da uniformidade
de conteúdo nas farmácias analisadas.
O Gráfico 12 acima mostra os resultados obtidos, indicando que do total
das amostras analisadas, as cápsulas de cinco de farmácias (FarmB, FarmC,
FarmE, FarmF e FarmG) encontraram-se com teores entre 85% e 115% com os

84
TABELA 25 – Resultados obtidos nas determinações de uniformidade de conteúdo das cápsulas de tibolona (2,5 mg) manipuladas nas Farmácias A, B, C, D, E, F, G, H, I, J, K, L e M.
Nº
Experimentos
FarmA FarmB FarmC FarmD FarmE FarmF FarmG FarmH FarmI FarmJ FarmK FarmL FarmM
1 104,7 100,0 95,8 62,1 99,7 98,4 96,6 115,8 134,0 106,6 115,6 95,1 141,8 2 113,7 113,8 91,2 73,7 90,5 95,9 93,3 101,8 129,5 148,3 123,4 84,3 269,7 3 115,8 98,4 89,8 65,2 97,2 102,3 99,4 95,1 132.0 134,5 151,9 99,2 145,3 4 109,9 109,5 96,0 80,1 98,1 98,4 104,5 97,4 141,5 273,2 108,4 116,6 85,1 5 109,5 106,4 91,5 74,1 101,0 102,4 102,6 115,6 194,5 107,8 118,0 115,2 89,1 6 109,2 99,9 88,6 75,4 100,8 116,8 110,7 133,1 156,5 110,5 115,1 118,5 74,8 7 129,3 96,5 91,7 68,4 103,4 101,9 100,3 141,6 150,5 102,9 115,0 134,5 62,7 8 117,6 108,2 96,4 63,5 98,3 104,5 106,2 115,9 166,4 99,8 133,3 115,1 96,3 9 118,0 108,7 93,9 76,6 91,2 100,3 102,2 111,4 148,0 157,0 150,3 158,1 119,2
10 126,2 100,6 95,9 71,7 98,3 96,7 108,9 100,8 405,9 147,1 127,2 105,7 132,0 Media* 115,4 104,2 93,1 71,1 97,9 101,8 102,5 112,9 175,9 138,8 125,8 114,2 121,6
DP 7,8 5,8 2,9 6,0 4,1 6,0 5,4 15,2 83,1 51,8 15,1 20,9 59,4 CV% 6,7 5,6 3,1 8,5 4,2 5,9 5,2 13,5 47,2 37,4 12,0 18,3 48,8
LI 104,7 96,5 88,6 62,1 90,5 95,9 93,3 95,1 129,5 99,8 108,4 84,3 62,7 LS 129,3 113,8 96,4 80,1 103,4 116,8 110,7 141,6 405,9 273,2 151,9 158,1 269,7
(*) Limite aceitável: 85 – 115% (FB 4. ed. 2004)

85
valores do desvio padrão relativo (CV%) baixos, menores que 6%, resultante de
uma boa homogeneidade da mistura entre as cápsulas individuais, indicando que
os parâmetros de qualidade exigidos foram cumpridos. Em relação às oito
amostras restantes analisadas, apresentaram seus teores e respectivos
coeficientes de variação fora dos limites especificados pela (FARMACOPEIA
BRASILEIRA, 1988).
Analisando estes resultados é possível observar que não foi evidenciada
nenhuma diferença da correlação entre a variação do peso médio e a variação da
uniformidade de conteúdo em função da relação fármaco excipiente, haja vista
que as formulações dos estabelecimentos FarmI, FarmJ, FarmK, FarrmL e
FarmM, com seus valores de pesos médios,141,1mg; 63,2; 70,1 mg; 194.0 mg e
119,3 mg, com seus desvios padrão relativos (CV%), 3,6%; 2,2%; 4,1%; 3,8% e
3,8% em comparação com os dados de uniformidade de conteúdo e seus
correspondentes desvios padrão relativos (CV%) 47,2%; 37,4%; 12,0%; 18,3% e
48,8%, mostrou que as variações na uniformidade de conteúdo, não foram
influenciadas pela variação do processo de enchimento, já que todos os valores
dos pesos médios estão dentro dos limites especificados.
Os dados apresentados na Tabela 25 indicam que os principais fatores que
influenciaram na variação dos teores, estão relacionados com os parâmetros
tecnológicos de mistura e as características físicas e químicas dos componentes
da fórmula. Estes dados nos permitem afirmar que não houve erro de pesagem,
podendo ter ocorrido, falha na homogeneização da mistura, causada por uma
possível segregação dos pós, parâmetro este que depende do tamanho,
densidade e da forma das partículas dos pós envolvidos na mistura. A
segregação fará com que aumente a variação do conteúdo nas amostras
retiradas da mistura, causando a reprovação no teste de uniformidade (AULTON,
2005), além de outros fatores, como nos casos onde a proporção de princípio
ativo é muito pequena em relação aos excipientes, nestes casos a etapa de
mistura é ainda mais crítica e a tendência de ocorrer uma falta de uniformidade
em certas partes da mistura dos pós é muito grande. (MENEGHINI; ADAMS,
2007). Estes resultados se assemelham aos de outros trabalhos publicados que

86
utilizaram à mesma abordagem e os mesmos critérios de aceitação dos limites
especificados pelos compêndios farmacopéicos, em termos de análises de
uniformidade de conteúdo e peso médio de cápsulas manipuladas. (MARCATTO,
et al., 2005); (SILVACOSTA, et al., 2008); (MIOTTO JÚNIOR; ADAMIS, 2004);
(PINHEIRO, et al., 2008)

87
CAPÍTULO V
CONSIDERAÇÕES FINAIS

88
5 CONSIDERAÇÕES FINAIS
Os medicamentos manipulados no Brasil passaram por grandes
modificações nos últimos anos, principalmente no que se refere ao aumento
significativo do número de prescrições a serem manipuladas, pertencentes a
todos os grupos terapêuticos, inclusive os que são considerados de alto risco
sanitário para os usuários, se preparados de maneira a comprometer as Boas
Práticas de Manipulação. Como exemplos podem ser citados aqueles de baixo
índice terapêutico, hormônios, psicotrópicos, antibióticos, onde qualquer desvio ou
erro na manipulação desses produtos poderá causar ineficácia terapêutica, ou
mesmo ser fatal para os usuários.
Casos de intoxicação por medicamentos pertencentes aos grupos citados
foram relatados levando a morte de pessoas inclusive crianças. Estes casos
foram constatados através de resultados de análises laboratoriais no
INCQS/FIOCRUZ, onde mostraram teores de PA variando entre 1.000 a 30.000%
do valor nominal.
Alem disso, estas variações do teor diferem bastante em relação às
cápsulas de uma mesma formulação, como 40 a 180% do valor declarado,
segundo os resultados das análises do INCQS.
Sendo a tibolona um fármaco de baixa dosagem e bastante manipulado e
não apresentando monografia oficial para sua quantificação nas formas
farmacêuticas disponíveis, o desenvolvimento e validação de um método para
esta finalidade foram fundamentais para serem aplicados em farmácias magistrais
e em estudos de bioequivalência.
Os dois métodos desenvolvidos demonstraram ser confiáveis, embora o
método por derivatização com detecção por fluorescência apresente maiores
dificuldades, é fundamental que se aprofunde no sentido de promover sua
validação. Em relação ao método por detecção direta no UV, além de

89
desenvolvido foi também validado e aplicado, apresentando-se confiável,
adequado e sensível, atendendo a todos os parâmetros de validação.
Os resultados da aplicação nas amostras manipuladas nos fazem levar a
algumas reflexões importantes, no tocante ao processo de manipulação magistral
referente aos desvios encontrados na uniformidade de conteúdo. Cerca de 62%
das farmácias analisadas estão com seus teores fora dos limites farmacopéicos
exigidos, onde as possíveis causas podem estar relacionadas aos tipos de
excipientes utilizados e aos processos de mistura de pós.
Independente das causas, esses resultados sinalizam que os usuários
desses medicamentos podem está fazendo uso de doses maiores ou menores do
que as estabelecidas, caracterizando um risco considerável para a saúde,
podendo levar inclusive a processos de intoxicação medicamentosa e até mesmo
a morte.
Apesar de esses resultados serem de apenas um medicamento, eles
coincidem com aqueles analisados pelo INCQS, na detecção de teores fora do
limite permitido pelos compêndios oficiais. . Por outro lado, podemos dizer que o
processo de manipulação é viável, tendo em vista que 38% das farmácias
analisadas estavam dentro dos padrões farmacopéicos que foram estabelecidos
para a realização das análises. Estes fatos nos leva a pensar que as farmácias
precisam reformular os seus processos de manipulação, no que se refere às
técnicas de mistura e homogeneização, sendo mais rigorosa no tocante ao
controle da qualidade, no sentido de obter medicamentos seguros, eficazes e com
qualidade.
Dentro das condições que os métodos foram desenvolvidos e baseados
nos resultados obtidos pode-se afirmar que:

90
Método indireto por derivatização com detecção por fluorescência:
• A redução na concentração do catalisador (ácido
trifluorometanosulfônico) foi importante para obter-se retenção cromatográfica das
hidrazonas formadas.
• Não foi observada a formação de hidrazonas diastereoisoméricas syn
e anti na reação de derivatização da tibolona.
• Os fatores mais relevantes no rendimento da reação de derivatização
foram à razão molar e a temperatura da reação, em acordo com os dados da
Appelblad para os outros esteróides.
• Uma razão molar de 40, um tempo de reação de 20 minutos e uma
temperatura de 48 ºC mostraram-se ideais.
• O método por derivatização desenvolvido mostrou-se adequado, por
isso é fundamental que haja um aprofundamento maior no sentido de que o
mesmo seja validado e possa ser aplicado, apontando para mais uma opção de
quantificação do fármaco tibolona, diante da inexistência, até então, de
metodologias analíticas desenvolvidas para o referido fármaco.
Método direto com detecção por UV
• O método demonstrou ser específico linear dentro das faixas de
concentrações estabelecidas, preciso, exato e robusto, nas condições
especificadas.
• Foi aplicado com êxito na quantificação das amostras de tibolona
manipuladas pelas farmácias magistrais da Paraíba.

91
• Das farmácias analisadas, apenas cerca de 38% estavam com os
teores de uniformidade de conteúdo dentro dos parâmetros farmacopéicos
exigidos, portanto 62% dessas farmácias estão em desacordo com os parâmetros
farmacopéicos e também com a RDC 67/2007/ANVISA, em relação a
uniformidade de conteúdo.
• Os desvios encontrados na uniformidade de conteúdo podem ser
atribuídos aos tipos de excipientes utilizados, bem como aos parâmetros
tecnológicos dos processos de mistura do pós, como falha na homogeneização da
mistura, causada por uma possível segregação dos pós ou por não utilização da
técnica da diluição geométrica no processo, além de outros fatores, como nos
casos onde a proporção de princípio ativo é muito pequena em relação aos
excipientes.
• Esse trabalho é mais uma contribuição na perspectiva de assegurar a
confiabilidade dos medicamentos magistrais, no que se refere a sua eficácia,
segurança e qualidade, desde que sejam manipulados utilizando técnicas
adequadas, em obediência aos compêndios farmacopéicos utilizados e as
legislações sanitárias vigentes.

92
REFERÊNCIAS ALBERTAZZI, P.; DI MICCO, R.; ZANARDI, E. Tibolone: A review. Maturitas The European Menopause Journal, v. 30, p. 299-305, 1998. ANTUNES JUNIOR, D. Farmácia de Manipulação: Noções Básicas. São Paulo: Tecnopress, 2002. 140p. APPELBLAD, P. et al. Derivatization of Steroids with Dansylhydrazine Using Trifluormethanesulfonic Acid as Catalyst. Analytical Chemistry, v. 69. n. 23, p. 4905-4511, Dec. 1, 1977. AQUINO NETO, F. R. de; NUNES, D. da S. e S. Cromatografia: Princípios Básicos e Técnicas Afins. Rio de Janeiro: Interciencia, 2003. 187p. AULTON, M. E. Delineamento de Formas Farmacêuticas. 2. ed. Porto Alegre: Artmed, 2005. 677p. BARROS NETO, B. de; SCARMINIO, I. S.; BRUNS, R. E. Como Fazer Experimentos: Pesquisa e desenvolvimento na ciência e na Tecnologia. 3. ed. Campinas: UNICAMP, 2007. 480p. BARROS, C. B. de. Validação de Métodos Analíticos. Biológico, São Paulo, v. 64, n. 2, p. 175-177, jul./dez. 2002. BEHRE, H. M.; NIESCHLAG, E. Testosterone Buciclate (20 Aet-1) in Hypogonadal Men: Pharmacokinetics and Pharmacodynamics of the New Long-Acting Androgen Ester. Journal of Clinical Endocrinology and Metabolism. v. 75, n. 5, p. 1204-1210, 1992. BLANCO, M.; BAÑO, R. G.; BERTRAN, E. Monitoring Blending in Pharmaceutical Processes by Use of Near Infrared Spectroscopy. Talanta, v. 56, n. 1, p. 203-212, 4 Jan. 2002. BLOOM, M. et al. Metabolism of Tibolone and its Metabolites in Uterine and Vaginal Tissue or Rat and Human Origin. Journal of Steroidal Biochemistry & Molecular Biology, v. 101, p. 42-49, 2006. BRASIL, ANVISA – Agência Nacional de Vigilância Sanitária. RDC nº. 33 de 19 de abril de 2000. Regulamento Técnico sobre Boas Práticas de Manipulação de Medicamentos em Farmácias e seus Anexos. Diário Oficial da União, Brasília, 8 jan. 2001. Disponível em: <http://www.anvisa.gov.br/e-legis/ >. Acesso em: 19 jul. 2008. ______ . RDC nº. 67 de 08 de outubro de 2007. Dispõe sobre Boas Práticas de Manipulação de Preparações Magistrais e Oficinais para Uso Humano em

93
farmácias. Diário Oficial da União, Brasília, 09 out. 2007. Disponível em: <http://www.anvisa.gov.br/e-legis/ >. Acesso em: 19 jul. 2008. ______ . Consulta Pública nº. 31, de 15 de abril de 2005. Proposta de Regulamento Técnico sobre “Boas Práticas de Manipulação de Medicamentos para Uso Humano em Farmácias, e seus Anexos”. Diário Oficial da União, Brasília, 18 abr. 2005. n. 73, Seção I, p. 85. Disponível em: <http://www.anvisa.gov.br/e-legis/ >. Acesso em: 28 ago. 2008. ______ . Fármacos manipulados têm sido consumidos cada vez mais. Texto da Escola Nacional de Saúde Pública (ENSP). Disponível em: <http://www.anvisa.gov.br/divulga/artigos/farmacos.htm>. Acesso em: 15 jun. 2008. ______ . Lei nº. 9787, de 10 de fevereiro de 1999. Altera a Lei nº. 6.360, de 23 de setembro de 1976, que dispõe sobre a vigilância sanitária estabelece o medicamento genérico, dispõe sobre a utilização de nomes genéricos em produtos farmacêuticos e dá outras providências. Diário Oficial da União, Poder Executivo, de 11 de fevereiro de 1999. Disponível em: <http://www.anvisa.gov.br/e-legis/>. Acesso em: 15 abr. 2008. ______ . Resolução RE 899, de 29 de maio de 2003. Determina a publicação do "Guia para Validação de Métodos Analíticos e Bioanalíticos”. Diário Oficial da União, Brasília, 2 jun. 2003. Disponível em: < http://www.anvisa.gov.br/e-legis/>. Acesso em: 19 abr. 2008. ______ . Subsídios à discussão sobre a proposta de regulamentação para farmácias magistrais. Texto apresentado pela Diretoria Colegiada da Anvisa em Audiência Pública realizada na Câmara dos Deputados. Brasília, jun. 2005. Disponível em: <http://www.anvisa.gov.br/divulga/artigos/subsidios.pdf>. Acesso em: 15 jun. 2008. ______ . Uma melhor regulamentação para os medicamentos manipulados no Brasil. Texto da Escola Nacional de Saúde Pública (ENSP). Disponível em: <http://www.anvisa.gov.br/divulga/artigos/farmacos_1.htm>. Acesso em: 28 ago. 2008. ______. CLONIDINA - medicamento manipulado. Alerta SNVS/Anvisa/Ufarm nº 9, de 3 de outubro de 2003. Disponível em: <http://www.anvisa.gov.br/ farmacovigilancia/alerta/federal/2003/federal_9_03.htm>. Acesso em: 09 jun. 2008. ______ . Proibida a manipulação da Clonidina em todo o País. Texto da Escola Nacional de Saúde Notícias da Anvisa. Disponível em:<http://www.anvisa. gov.br/DIVULGA/noticias/2003/031003.htm>. Acesso em: 15 mar. 2008. BRESSOLLE, F.; BROMET-PETIT, M.; AUDRAN, M. Validation of Liquid Chromatographic and Gas Chromatographic Methods: Applications to Pharmacokinetics. Journal of Chromatography B, v. 686, p.3-10, 1996.

94
BRITISH PHARMACOPOEIA. London: Her Magesty’s Stationary Office, v. I, 2003. BUDAVARI, S. (Ed.). The Merck Index. 12. ed. New Jersey: Merck & Co., 1996. 1741p. BURGER, D. M. et al. Low plasma concentration of indinavir are related to virological treatment failure in HIV-1-infected patients on indinavir-containing triple therapy. Antiviral Therapy. v. 3, n. 4, p. 215-220, Nov. 1998. CAZEDEY, E. C. L. et al. Desenvolvimento e Validação de Métodos Analíticos para Determinação de Itraconazol em Produtos Farmacêuticos por CLAE. Química Nova, Rio de Janeiro, v. 30, n. 4, p. 774-776, 2007. COLLINS, H. C.; BRAGA, G. L.; BOMNATO, P. S. (Coord.) Introdução a Métodos Cromatográficos. 4. ed. Campinas: UNICAMP, 1990. 279p. DAVIS, R. L. et al. Amikacin pharmacokinetics in patients receiving high-dose cancer chemotherapy. Antimicrobial Agents and Chemotherapy. v. 25, n. 5, p. 944-947, 1991. DIVISÃO Geométrica e Homogeneização de pós. ANFARMAG Revista do Setor Farmacêutico Magistral, São Paulo, v. 14, n. 70, p. 11-14, jan./fev. 2008. Encarte Técnico n. 22. DUBEY, R. K. et al. Tibolone and Its Metabolites Induce Antimitogenesis in Human Coronary Artery Smooth Muscle Cells: Role of Estrogen, Progesterone and Androgen Receptors. Journal of Clinical Endocrinology & Metabolism, v. 89, n. 2, p. 852-859, 2004. EL-HAGRASY, A. S. et al. Near-infrared Spectroscopy and Imaging for the Monitoring of Powder Blend Homogeneity. Journal of Pharmaceutical Sciences. v. 90, n. 9, p. 1298-1307, 2001. ERMER, J.; MILLER, J. H. McB. (Eds.) Method Validation in Pharmaceutical Analysis: A Guide to Best Practice. Weinheim: Willey-VCH, 2005. 527p. FARMACOPÉIA BRASILEIRA. 4. ed. Parte I. São Paulo: Atheneu, 1988. FARMACOPÉIA BRASILEIRA. 4. ed. Parte II. São Paulo: Atheneu, 1996. FERREIRA, A. de O. Guia Prático da Farmácia Magistral. 2.ed. Juiz de Fora: OESP Gráfica. 2002. 845p. GEHRING, R. et al. Multivariate meta-analysis of pharmacokinetics studies of ampicillin trihydrate in cattle. American Journal of Veterinary Research. v. 66, n. 1, p. 108-112, Jan. 2005. GIL, E. de S. (Org.). Controle físico-químico de Qualidade de Medicamentos. 2. ed. São Paulo: Pharmabooks, 2007.485p.

95
HARRIS, D. C. Cromatografia Liquida/Espectrometria de Massa. In:_Análise Química Quantitativa. 5. ed. Rio de Janeiro: LTC, 2001. 862p. Cap. 25, p. 640-649. HYYTIAINEN, M. et al. Trifluoromethanesulfonic Acid as a Catalist for the Formation of Dansylhydrazone Derivatives. Journal of Chromatography A, v. 740, p.279-283, 1996. ITO, M. et al. Development of a Method for the Determination of Caffeine Anhydrate in Various Designed Intact Tablets by Near-infrared Spectroscopy: A Comparison Between Reflectance an Transmittance Technique. Journal of Pharmaceutical and Biomedical Analysis. v. 47, p. 819-827, 2008. KACHIGAN, S. K. Multivariaate Statistical Analisys: A conceptual Introduction. 2. ed. Neuw York: Radius Press, 1991. 303p. LACHMAN, L.; LIEBERMAN, H. A.; KANIG, J.L. Teoria e pratica na Industria Farmacêutica. Lisboa: Calouste Gulbenkian, 2001. v. 1 e 2, 1517p. LI, W.; WOROSILA, G. D. Quantification of Active Pharmaceutical Ingredientes and Calibration Models by Near-Infrared Spectroscopy. International Journal of Pharmaceutics, v. 295, p.213-219, 2005. LIMA, L. F. M. et al. Vigilância Sanitária de Medicamentos e Correlatos. Rio de Janeiro: Qualitymark, 1993. 329p. MA, H.; ANDERSON, C. A. Caracterization of Pharmaceutical Powder Blends by NIR Chemical Imaging. Journal of Pharmaceutical Sciences, v. 97, n. 8, p. 3305-3320, Aug. 2008. MATHEWS, L. et al. Monitoring Blend Uniformity with Effusivity. Pharmaceutical Technology, p. 80-84, Abr. 2002. MENDHAM, J. et al. (Rev.). Vogel Análise Química Quantitativa. 6. ed. Rio de janeiro: LTC, 2002. 462p. MILLER, J. N.; MILLER, J. C. Estadistica y Químiometria para Química Analitica. 4. ed. Madrid: Prentice Hall, 2002. 196p. O X DA MANIPULAÇÃO. ANVISA Boletim Informativo, Brasília, n. 56, p.6-8, jun. 2005. PAUMGARTTEN, F. J. Papel das farmácias magistrais deve ser complementar. ANVISA Boletim Informativo, Brasília, n. 56, p.4-5, jun. 2005. PERALTA-ZAMORA, P.; MORAIS, J. L.; NAGATA, N. Por Que Otimização Multivariada? Engenharia Sanitária e Ambiental, Rio de Janeiro, v. 10, n. 2 p. 106-110, abr./jun. 2005.

96
PETRY, R. D. et al. Influencia de Adjuvantes e Técnica de Enchimento Sobre as Características Farmacêuticas de Cápsulas de Gelatina Duras contendo Teofilina. Caderno de Farmácia, v. 14, n. 1/2, p. 13-19, 1998. PITSIU, M. et al. Retrospective population pharmacokinetic analysis of cetrizine in children aged months to 12 years. British Journal of Clinical Pharmacology. v. 57, n. 4, p. 402-411, Apr. 2004. PORTA, V. et al. Método Analítico para a Determinação de Meloxican em Plasma humano por Cromatografia Líquida de Alta Eficiência (CLAE). Revista Brasileira de Ciências Farmacêuticas, São Paulo, v. 41, n. 2, p. 215-222, abr./jun., 2005. RILEY, C. M.; ROSANSKE, T. W. (Eds.). Development and Validation of Analitycal Methods. Oxford: Pergamon, 1996. 442p. SAKAMOTO, T.; FUJIMAKI, Y.; HIYAMA, Y. Studies on the Influence of Uniformity of Particle Size of Powder, Trapping and Sample Replacement for Diffusion Reflectance Quantitative NIR Spectrometric Analysis. Pharmazie, v. 62, n. 11, p. 841-846, Nov. 2007. SÁNCHES, F. C. et al. Monitoring Powder Blending by NIR Spectroscopy. Fresenius Journal of Analytical Chemistry, v. 352, p. 771-778, 1995. SASAKI, Y. et al. A Pharmacokinetic and Pharmacodynamic Analysis of CTP-11 and its Active Metabolite SN-38. Cancer Science. v. 86, n. 1, p. 101-110, Aug. 2005. SEKULIC, S. S. et al. Automated System for the on-Line Monitoring of Powder Blending Processes Using Near-Infrared Spectroscopy. Part II: Quantitative Approaches to Blend Evaluation. Journal of Pharmaceutical and Biomedical Analysis, v. 17, p. 1285-1309, 1998. SILVA, A. V. da; GOMES, S. F. de O. Desenvolvimento e Validação de Metodologia Analítica. ANFARMAG Revista do Setor Farmacêutico Magistral, São Paulo, v. 14, n. 70, p. 2-10, jan./fev. 2008. Encarte Técnico n. 22. SINAMM: Novos Paradigmas para a Farmácia Magistral. ANFARMAG Revista do Setor Farmacêutico Magistral, São Paulo, v. 14, n. 72, p. 8-21, mai./jun. 2008. SKOOG, D. A. et al. Fundamentos de Química Analítica. 8.ed. São Paulo: Tomson, 2006. 999p. SNYDER, L. R.; KIRKLAND, J. J. GLAJCH, J. L. Practical HPLC Method Development. 2. ed. New York: Willwy & Sons, 7997.745p. SON, J. et al. Improved Detectability in Pharmacokinetic Study of Tibolone by Gás Chromatography-High Resolution Mass Spectrometry with selected Íon Monitoring. Talanta, v. 70, p. 37-42, 2006.

97
SPEROFF, L.; CLARKSON, T. B. Cover Story: Is tibolone a viable alternative to HT? Contemporary Ob/Gyn. V. 48, n.1, p. 54-68, Aug. 2003. SWEGLE, J. M. Tibolone: A Unique Version of Hormone Replacement Therapy. The Annals of Pharmacotherapy. v. 38, n.. 5, p. 874-881, 2004. TAPLIN, L. A.; ABRAN, K. M.; MICHAELS, S. K. Blood alcohol level among emergency room patients: a multivariate analysis. Journal of Studies on Alcohol. v. 50, n. 5, p. 441-447, Sep. 1989. THE UNITED STATES PHARMACOPOEIA. 28. ed. Rockville: United States Convention, 2004. TIMMER. C. J.; DOORSTAM, D. P. Effect of Renal Impairment on the Pharmacokinetics of a Single oral Dose of Tibolone 2,5 mg in Early Postmenopausal Women. Pharmacotherapy, v. 22, n.2, p. 184-153, 2002. TIMMER. C. J.; VERHEUL, H. A. M.; DOORSTAM, D. P. Pharmacokinetics of Tibolone in Early and Late Postmenopausal Women. British Journal of Clinical Pharmacology, v. 54, n. 2, p. 101-106, Aug. 2002. TU, WEI-DA et al. The Effect of Powder S on Induction Behavior and Binder Distribution During High Shear Melt Agglomeration of Calcium Carbonate. Powder Technology, v. 184, p. 289-312, 2008. VERHEUL, H. A. M. et al. Pharmacokinetic Parameters of Tibolone and Metabolites in Plasma, Urine, Feces and Bile from Ovariectomized Cynomolgus Monkeys after a Single Dose or Multiple Doses of Tibolone. Drug Metabolism and Disposition, v. 35, n. 7, p. 1112-1118, 2007. ______ . Selective Tissue Distribution of Tibolone Metabolites in Mature Ovariectomized Female Cynomolgus Monkeys after Multiple Doses of Tibolone. Drug Metabolism and Disposition, v. 35, n. 7, p. 1105-1111, 2007. VERHEUL, H. A. M.; KLOOSTERBOER, H. J. Metabolism of Exogenous Sex Steroids and Effect on Brain Functions with Focus on Tibolone. Journal of Steroidal Biochemistry & Molecular Biology, v. 102, p. 195-204, Dec. 2006. VOS, R. M. E. et al. The in Vivo Human Metabolism of Tibolone. Drug Metabolism and Disposition, v. 30, p. 106-112, 2002. WAJIMA, T. et al. Prediction of human pharmacokinetics from animal data and molecular structural parameters using multivariate regression analysis: Oral clearance. Journal of Pharmaceutical Sciences. v. 22, n. 12, p. 2427-2440, Sep. 2003. WARGO, D. J.; DRENNEN, J. K. Near-Infrared Spectroscopic Characterization of Pharmaceutical Powder Blends. Journal of Pharmaceutical and Biomedical Analysis, v. 14, p. 1415-1423, 1996.

98
YIN, T. A Guide to Blend Uniformity. Journal of GXT Compliance, 10 Out. 2007. YU, W.; HANCOCK, B. C. Evaluation of Dynamic Image Analysis for Characterizating Pharmaceutical Excipient Particles. International Journal of Pharmaceutics. v. 361, p. 150-157, 2008. ZARBIELLI, M. G.; MACEDO, S.; MENDEZ, A. L. Controle de Qualidade de Cápsulas de Piroxicam Manipuladas em Farmácias do Município de Erechim (RS). Infarma, v. 10, n. 1/2, 2007. ZUO, M. et al. p-Toluenesulfonyl Isocianate as a Novel Reagent to Enhance the Eletrospray Ionization and its Application in the Determination of Two Stereo isomers of 3-Hydroxyl-7-methyl-norethylnodrel in Plasma. Journal of Chromatography B, v. 814, p.331-337, 2005. ______ . Stereoseletivity in Metabolic 3-reduction of Tibolone in Healthy Chinese Female Volunteers. Acta Pharmacologica Sinica, v. 26, n. 12, p. 1527-1530, Dec. 2005.

99
A N E X O S

100
ANEXO a
Derivatization of tibolone with dansylhydrazine using trifluoromethanesulfonic acid as a catalyst: Quantification by HPLC with fluorescence detection
Cândido J.A., Figueiredo E.A. and Oliveira, E.J.
Programa de Pós Graduação em Produtos Naturais e Sintéticos Bioativos: Laboratório de Tecnologia Farmacêutica,Universidade Federal da Paraíba, Caixa Postal 5080, CEP:
58051-970, João Pessoa, Paraíba, Brazil. Introduction Tibolone (®Livial, Organon) is a synthetic steroid (1) used in hormone replacement therapy for the relief of menopausal symptoms (Swegle, 2004). Tibolone has a poor chromophore and thus it is often quantified using mass spectrometric detection. The derivatization of several steroids with dansyl hydrazine using trifluoromethanesulfonic acid as a catalyst has been described (Appleblad et. al., 1997). While the derivatization reaction can be accomplished using mild reaction conditions, with almost quantitative yield, the method described by Applebad had several limitations, the most important ones being: (a) the need for a concentration column for the chromatographic separation of the hydrazone and (b) the formation of diastereomeric syn- and anti-hydrazones with some of the steroids investigated. Our aim was to develop a sensitive and simple method for quantification of tibolone using fluorescence detection by investigating the derivatization of tibolone with dansyl hydrazine (2) in order to form a fluorescent hydrazone adduct (3), without the limitations previously described for other steroids. Methods Derivatization of the steroid Tibolone (kindly donated by Organon) was derivatized with dansyl hydrazine (Sigma-Aldrich) using trifluoromethanesulfonic acid (Sigma-Aldrich) as a catalyst. Tibolone and dansylhydrazine stock solutions (1mg/mL) were prepared in methanol and then diluted to give the final concentrations needed for each experiment. The concentration of the catalyst was fixed at 5%. The reactions were done using 2mL screw-capped vials and a termoblock for heating. A fractional factorial experimental design was implemented using Modde v.4.0 (Umetrix) to investigate the effects of the following parameters on the yield of the derivatization reaction: molar ratio of dansyl hydrazine to tibolone (range: 20 to 50), reaction temperature (range: 37 to 50 ºC) and reaction time (range: 17 to 30 minutes). Multivariate linear regression was used to evaluate the effects of each parameter and their combinations on the reaction yield. After setting the limits for each parameter to be investigated, the software suggested a total of 11 experiments (Table 1) varying each of the 3 parameters investigated. The area of the fluorescent derivative in the chromatogram obtained by injecting samples from each of the 11 experiments was calculated and fed to the software as yield to be optimized. Chromatographic separation The HPLC system consisted of a low gradient chromatograph (from Shimadzu, Japan) with a column oven (CTO-10AS), a degasser (DGU-14A), a fluorescence detector (RF-10A), a controller module (SCL-10A) and a

101
manual injector fitted with a 20 μL sample loop. The derivatization samples were chromatographed using a monolithic column (Merck Chromolith performance 100 mm x 4.6 mm ID) with a flow rate of 3 mL/min and an injection volume of 20 μL. The mobile phase consisted of 50mM ammonium acetate buffer pH 6.8 and acetonitrile (92,5:7,5,v/v). Samples of the reaction medium were diluted 1:1 with 1M phosphate buffer pH 7.0 and directly injected into the chromatograph. The fluorescence detector was configured to use an excitation wavelength of 320 nm and an emission wavelength of 520 nm. Results Table 1. Experimental conditions suggested for the derivatization of tibolone using a fractional factorial design with Modde v.4.0 (Umetrix) Exper. Number Molar ratio* Temperature (ºC) Time(min.) Order 1 20 37 17.0 7 2 50 37 17.0 8 3 20 50 17.0 1 4 50 50 17.0 11 5 20 37 30.0 6 6 50 37 30.0 4 7 20 50 30.0 9 8 50 50 30.0 5 9 35 35 23.5 3 10 35 35 23.5 10 11 35 35 23.5 2 * Molar ratio of dansyl hydrazine to tibolone.

102
Figure 1. Surface response maps showing the effects of molar ratio of dansylhydrazine to tibolone, temperature and reaction time, on the yield of the derivatization reaction.
S
O
O
NH
N
NH2
CH3
H3C
CH3
OH
C
O
CH3
CH3
OH
C
CH3
N
H
S
O
O
N
N
CH3
H3C
+
H+ CF3SO3H
DansilhidrazinaTibolona
Aduto (derivado fluorescente)(3) Fluorescent adduct
(2) Dansyl hydrazine (1) Tibolone

103
Figure 2. Chromatogram showing the detection of the fluorescent derivative of tibolone (3 μg/mL, rt = 6.7 minutes). Figure 3. Multivariate linear regression (MLR) modeling of data showing predicted versus observed values for yield The use of a fractional factorial design allowed a systematic investigation of reaction parameters on the yield of the derivatization reaction. Figure 1 shows the relationship between these parameters and yield using surface response maps. The highest reaction yield was achieved using temperatures at around 40-47 ºC, and molar ratio of dansyl hydrazine to tibolone of 30-40. The duration of the reaction proved to be an important factor, with reaction times greater than 20 minutes clearly leading to poor yield. It is worth noting that the reaction of dansyl hydrazine with tibolone does not appear to

104
form the diastereomeric syn- and anti-hydrazones that were detected with other steroids, for example progesterone (Appelblad et. al., 1997). Indeed, the single peak in the chromatogram of the reaction medium (Figure 2) suggests that a single isomer is formed. This is advantageous for quantification purposes, since a single peak is integrated instead of having to use the sum of two integrated diastereomeric peaks. This aspect of the reaction needs confirmation by spectroscopic techniques, but computer calculations suggests differences in the stereochemistry of the carbonyl that could explain the differences in regioselectivity observed. In our work the concentration of trifluoromethanesulfonic acid proved to be crucial for a proper retention of the fluorescent derivative formed. Since Appelbad (Appelblad et al., 1997) showed that the catalyst concentration is not an important factor in determining the yield of this reaction, our experiments were conducted with a fixed concentration of the acid (5%). Higher concentrations caused a marked drop in retention, with the peak of the derivative eluting at the void volume of the column. The modelling of data by multivariate linear analysis showed an excellent predictability of the model (r2=0,99). Finally, the use of a monolithic column with a flow rate of 3mL/min. allowed tibolone to elute at 6.9 min. using a mobile phase with 7.5% of acetonitrile. The method can be optimized to have shorter retention times using a higher proportion of organic modifier, decreasing even further analysis time. Conclusions 1. The reaction of tibolone with dansylhydrazine using trifluoromethanesulfonic acid as a catalyst does not appear to form diastereomeric syn- and anti-hydrazones . 2. The concentration of trifluoromethanesulfonic acid (catalyst) at 5% is crucial for the proper retention of the derivative formed. 3. The optimum yield was achieved using a molar ratio of dansyl hydrazine to tibolone of 40, a reaction time of 20 minutes and a temperature of 48 ºC. Acknowledgements The authors wish to thank Organon for donating tibolone reference standard and CNPq for financial support References Swegle, J.M. (2004). Tibolone: A Unique Version of Hormone Replacement Therapy. The Annals of Pharmacotherapy. 38 (5), 874-881. Appelblad, P. et al. (1997). Derivatization of Steroids with Dansylhydrazine Using Trifluoromethanesulfonic Acid as Catalyst. Analytical Chemistry. 69, 4905-4911.

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo