danielle juais - biblioteca digital de teses e ... · de química, universidade de são paulo, são...
TRANSCRIPT

UNIVERSIDADE DE SÃO PAULO
INSTITUTO DE QUÍMICA
Programa de Pós-Graduação em Química
DANIELLE JUAIS
Produção de Polímeros
Derivados de Fontes
Renováveis via Catálise
Enzimática
São Paulo
Data do Depósito na SPG:
06/03/2009

DANIELLE JUAIS
Produção de Polímeros
Derivados de Fontes Renováveis
via Catálise Enzimática
Dissertação apresentada ao Instituto de
Química da Universidade de São Paulo para
obtenção do Título de Mestre em Química
(Química Orgânica)
Orientador: Prof. Dr. Luiz Henrique Catalani
São Paulo
2009

Danielle Juais
Produção de Polímeros Derivados de Fontes Renováveis via Catálise Enzimática.
Dissertação apresentada ao Instituto de Química
da Universidade de São Paulo para obtenção do
Título de Mestre em
Química (Química Orgânica)
Aprovado em: _____________
Banca Examinadora
Prof. Dr. _________________________________________________
Instituição: ________________________________________________
Assinatura: ________________________________________________
Prof. Dr. _________________________________________________
Instituição: ________________________________________________
Assinatura: ________________________________________________

Dedico este trabalho aos meus pais, Abrahão C. Juais e Maria de Fátima
Juais, pelo amor, apoio e incentivo, e ao meu namorado, Renan Ferrarini,
por ser um verdadeiro porto seguro no meio da tempestade, e por
proporcionar os melhores momentos da minha vida.

AGRADECIMENTOS
Agradeço a Deus, por sempre ser um socorro presente no meio de tantas
dificuldades.
Às agências de fomento à pesquisa, CNPq, pela bolsa de estudos, e Fapesp
e Capes, pelo suporte financeiro indireto.
Ao Prof. Dr. Luiz Henrique Catalani, pela oportunidade de fazer parte do seu
grupo de pesquisa.
Aos colegas e ex-colegas do Laboratório de Biomateriais Poliméricos, Alliny,
Antônio, Fábio, Guilhermino, Janaína, Mariana, Patrícia, Renata, Ricardo, Romeu,
Vânia, ao técnico Luiz e à secretária Sílvia pelo companheirismo, amizade,
conselhos, discussões e inúmeras risadas. Meu especial agradecimento ao Romeu,
por toda ajuda prestada, e pela paciência ao me ensinar.
Ao Prof. Dr. Richard A. Gross, do Laboratório do Centro NSF de Biocatálise e
Bioprocessamento de Macromoléculas, da Polytechnic University, em Nova York, e
seus alunos, pela realização das análises de DSC e TG.
Ao Prof. Dr. João V. Comasseto, bem como aos seus alunos, pela realização
das análises de CG-MS, empréstimo de materiais, pela amizade e paciência na
resolução de todas as minhas dúvidas com relação à síntese orgânica.
Aos funcionários do IQUSP, sobretudo da Central Analítica e da Secretaria de
Pós-Graduação, principalmente ao Milton e à Cibele, pela amizade e eficiência.

À minha família, sobretudo aos meus pais e irmãos, Júlio, Carlos, Francisco,
Débora e Helen e à minha sobrinha Letícia, que são a base que sustenta minha
vida.
À dona Neide Dacunti Favorito, por ser uma verdadeira mãe para mim em
São Paulo.
Aos amigos do IQ-USP, Aline, Lollo, Vítor Z. e Bruno, pela amizade e bons
momentos compartilhados.
Aos meus amigos do Departamento de Química da UFPR, principalmente à
Karen, Ju, Kimura, Carol, Samanta, Tico, Zé, Marcela, Zezé, Alcindo, Cida, Janjão,
Soraia, Emir, Joel, Brazuka, Beiço, Flávio, Gustavo, Vítor, Alfredo e Sílvia, entre
tantos outros, que com certeza contribuíram para mais essa vitória.
A todos que de alguma maneira colaboraram para a execução desse trabalho.

"Aprendi através da experiência amarga a suprema lição: controlar minha ira é
torná-la como o calor que é convertido em energia. Nossa ira controlada
pode ser convertida numa força capaz de mover o mundo." (Ghandi)

RESUMO
Juais, D. Produção de Polímeros Derivados de Fontes Renováveis via Catálise
Enzimática. 2009. 116p. Dissertação – Programa de Pós-Graduação em Química.
Instituto de Química, Universidade de São Paulo, São Paulo.
A busca por materiais derivados de fontes renováveis e com características
como biocompatibilidade e biodegradabilidade tem crescido significativamente nos
últimos anos. A utilização de enzimas na polimerização representa um grande passo
para a obtenção destes, visto que possibilitam a produção de polímeros evitando a
utilização de catalisadores tóxicos e, assim, melhorando sua biocompatibilidade.
O presente trabalho descreve a utilização de monômeros funcionais derivados
de fontes renováveis na produção de poliésteres hidrolisáveis via catálise
enzimática. As sínteses de polímeros produzidos a partir de isosorbídeo e ácidos
dicarboxílicos ou derivados - como seus ésteres alquílicos e vinílicos - foram feitas
utilizando a lipase de Candida antarctica – Fração B como catalisador. As
polimerizações foram realizadas por policondensações em massa e em solução,
utilizando-se diferentes solventes e diferentes técnicas para remoção de
subprodutos de reação. A principal abordagem foi o estudo das diferentes condições
reacionais realizadas, variando-se o tempo de reação, tipo do monômero, solvente
utilizado (se for o caso) e tipo de técnica para remoção de subprodutos visando o
aumento da massa molar dos polímeros.
A condição que forneceu os materiais com maiores massas molares foi a
policondensação em solução, utilizando a mistura cicloexano:benzeno como

solvente. Tendo por objetivo investigar profundamente a condição ótima obtida, e
estabelecer padrões de comparação com outros sistemas, foram estudados, nessa
condição, parâmetros como tempo de reação, efeito do tamanho da cadeia
carbônica do monômero, grupo de saída, solubilidade dos polímeros e diluição do
sistema.
Os materiais obtidos foram caracterizados por cromatografia por exclusão de
tamanho (SEC), termogravimetria (TG), calorimetria exploratória diferencial (DSC),
espectroscopia no infravermelho, difração de raios-X, e Ressonância Magnética
Nuclear (RMN) de 1H e 13C.
Através deste trabalho foi provado que, embora apresente uma cinética de
reação lenta, a polimerização enzimática deste diol secundário estericamente
impedido é possível, fornecendo poliésteres com massas molares similares às
obtidas via catálise química. Todos os resultados obtidos neste trabalho são inéditos
no que diz respeito à polimerização enzimática de dióis secundários impedidos, mais
especificamente de isosorbídeo.
Palavras-chave: isosorbídeo, catálise enzimática, dióis secundários,
policondensação em solução.

ABSTRACT
Juais, D. Production of polymers derived from renewable sources by enzyme
catalysis. 2009. 116p. Masters Thesis – Graduate Program in Chemistry. Instituto
de Química, Universidade de São Paulo, São Paulo.
The search for materials derived from renewable sources, with characteristics
such as biocompatibility and biodegradability has grown significantly in recent years.
The use of enzymes in the polymerization is a major step for the attainment of these
materials, since it allows the production of polymers while avoiding the use of toxic
catalysts and thus improving its biocompatibility.
This paper describes the use of functional monomers derived from renewable
sources in the production of hydrolysable polyesters by enzyme catalysis The
synthesis and characterization of polymers derived from isosorbide and dicarboxilic
acids or derivatives - such as alkyl and vinyl esters - were carried out using the lipase
from Candida antarctica - Fraction B as catalyst. The polymerizations were
accomplished by polycondensations in bulk and in solution, using different solvents
and different techniques for removal of reaction byproducts. The main approach was
to study the different reaction conditions, by varying the reaction time, monomer type,
solvent used (if applicable) and the type of technique for removal of byproducts,
aiming at maximizing polymer molar mass.
The condition that provided the material with higher molecular weight was the
solution step-growth polymerization, using a mixture cyclohexane:benzene as
solvents. Aiming to thoroughly investigate the optimum condition obtained, and to

establish standards for comparison with other systems, it was studied, in this
condition, parameters such as reaction time, effect of monomer carbon chain length ,
leaving group, polymers solubility of and dilution of the reaction system.
The materials were characterized by gel permeation chromatography (SEC),
thermogravimetry (TG), differential scanning calorimetry (DSC), infrared
spectroscopy, X-ray diffraction and 1H and 13C Nuclear Magnetic Resonance (NMR).
Through this work it was proved that, in spite of a slow reaction kinetics, the
enzymatic polymerization of this hindered secondary diol is possible, providing
polyester with molecular weight similar to those obtained by chemical catalysis. All
results obtained in this work are unprecedented with respect to the enzymatic
polymerization of hindered secondary diols, more specifically of Isosorbide.
Key words: Enzyme catalysis, isosorbide, secondary diols, solvent-based step-
growth polymerization

LISTA DE ABREVIATURAS E SIGLAS
CAL-B: Lipase de Candida antarctica – Fração B.
CDCl3: Clorofórmio deuterado
DSC: Calorimetria Exploratória Diferencial
FDA: Food and Drugs Administration
FTIR: Infravermelho com Transformada de Fourier
IBGE: Instituto Brasileiro de Geografia e Estatística
ITB: Iterative Tandem Catalysis – Catálise Acoplada Iterativa
MEK: 2-Butanona
MM: Lipase de Mucor miehei
𝐌 𝐧: Massa molar numérica média
𝐌 𝐰: Massa molar ponderal média
N-435: Lipase de Candida antarctica – Fração B – Novozym 435
P.E.: Ponto de ebulição
PD: Índice de polidispersividade (PD=𝑀 𝑤 /𝑀 𝑛 )
PET: Poli(tereftalato de etileno)
PF: Lipase de Pseudomonas fluorescens
PPL: Lipase de pâncreas de porco
PS: Lipase de Pseudomonas sp.
PTFE: Poli(tetrafluoretileno)
RMN 1H e 13C: Ressonância Magnética Nuclear de hidrogênio e carbono
SEC: Cromatografia por exclusão de tamanho
Tg: Temperatura de Transição Vítrea
TG: Termogravimetria
THF: Tetraidrofurano
Tm: Temperatura de Fusão Cristalina
TMS: Tetrametilsilano
WAXD: Difração de raios-X de alto ângulo.

ÍNDICE DE FIGURAS
FIGURA 1 - ESTRUTURAS DOS 1,4: 3,6 - DIANIDROEXITÓIS. ____________________________________ 20
FIGURA 2 - ESTRUTURA TRIDIMENSIONAL DA CAL-B (WWW.NOVOZYMES.COM) _____________________ 31
FIGURA 3 - RESULTADOS OBTIDOS ATRAVÉS DA POLIMERIZAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA.
___________________________________________________________________________ 64
FIGURA 4 - ESTUDO COMPARATIVO ENTRE AS TÉCNICAS EMPREGADAS. ___________________________ 65
FIGURA 5 - ESTUDO COMPARATIVO DOS GRAUS DE POLIMERIZAÇÃO OBTIDOS EM CICLOEXANO E NA MISTURA
CICLOEXANO:BENZENO. _________________________________________________________ 69
FIGURA 6 - VANTAGEM DO USO DA MISTURA DE CICLOEXANO E BENZENO _________________________55
FIGURA 7 - ESTUDO DO CRESCIMENTO DA MASSA MOLAR DO POLI(ADIPATO DE ISOSORBILA) EM FUNÇÃO DO
TEMPO DE REAÇÃO. ____________________________________________________________57
FIGURA 8 - TROCA DE BENZENO POR TOLUENO NA MISTURA DE SOLVENTES. _______________________ 75
FIGURA 9 - AVALIAÇÃO DAS MASSAS MOLARES DO POLI(ADIPATO DE ISOSORBILA) QUANTO AO ÁLCOOL DE
SAÍDA. ______________________________________________________________________ 77
FIGURA 10 - EFEITO DA PROPORÇÃO SOLVENTE: ISOSORBÍDEO. ________________________________ 82
FIGURA 11- EFEITO DA CONCENTRAÇÃO DE BENZENO NA MISTURA DE SOLVENTES. __________________ 84
FIGURA 12 - CURVAS DE DSC DOS MATERIAIS OBTIDOS ______________________________________ 87
FIGURA 13 - CURVAS DE ANÁLISE TERMOGRAVIMÉTRICA DOS MATERIAIS OBTIDOS. __________________ 88
FIGURA 14 - ESPECTRO DE 1H-RMN DO POLI(SUCCINATO DE ISOSORBILA)_________________________74
FIGURA 15 - ESPECTRO DE 13
C-RMN DO POLI(SUCCINATO DE ISOSORBILA). _______________________ 92
FIGURA 16 - ESPECTRO DE 1H-RMN DO POLI(GLUTARATO DE ISOSORBILA) ._______________________ 76
FIGURA 17- ESPECTRO DE 13
C-RMN DO POLI(GLUTARATO DE ISOSORBILA). _______________________ 94
FIGURA 18 - ESPECTRO DE 1H-RMN DO POLI(ADIPATO DE ISOSORBILA).__________________________ 78
FIGURA 19 - ESPECTRO DE 13
C-RMN DO POLI(ADIPATO DE ISOSORBILA). _________________________ 96
FIGURA 20 - ESPECTRO DE 1H-RMN DO POLI(SUBERATO DE ISOSORBILA). ________________________ 97
FIGURA 21 - ESPECTRO DE 13
C-RMN DO POLI(SUBERATO DE ISOSORBILA). ________________________ 98
FIGURA 22 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SEBACATO DE ISOSORBILA). ___________ 99
FIGURA 23 - ESPECTRO DE 13
C-RMN DO POLI(SEBACATO DE ISOSORBILA). _______________________ 100
FIGURA 24 - ESPECTRO DE 1H-RMN DO POLI(DODECANODIOATO DE ISOSORBILA). _________________ 101
FIGURA 25 - ESPECTRO DE 13
C-RMN DO POLI(DODECANODIOATO DE ISOSORBILA). _________________ 102
FIGURA 26 - DIFRATOGRAMAS DE RAIOS-X DOS POLIÉSTERES OBTIDOS. _________________________ 103
FIGURA 27 - ESPECTRO NO INFRAVERMELHO DO POLI(SUCCINATO DE ISOSORBILA) _________________ 106
FIGURA 28 - ESPECTRO NO INFRAVERMELHO DO POLI(GLUTARATO DE ISOSORBILA) _________________ 106
FIGURA 29 - ESPECTRO NO INFRAVERMELHO DO POLI(ADIPATO DE ISOSORBILA) ___________________ 107
FIGURA 30 - ESPECTRO NO INFRAVERMELHO DO POLI(SUBERATO DE ISOSORBILA) __________________ 107
FIGURA 31 - ESPECTRO NO INFRAVERMELHO DO POLI(SEBACATO DE ISOSORBILA) __________________ 108
FIGURA 32 - ESPECTRO NO INFRAVERMELHO DO POLI(DODECANODIOATO DE ISOSORBILA) ____________ 108

ÍNDICE DE ESQUEMAS
ESQUEMA 1 - SÍNTESE DO ISOSOSORBÍDEO A PARTIR DA D-GLICOSE. ____________________________ 19
ESQUEMA 2 - REAÇÕES DE POLIMERIZAÇÃO CATALISADAS POR LIPASES. _________________________ 29
ESQUEMA 3 - MECANISMO DO SÍTIO CATALÍTICO DA CAL-B. ___________________________________ 30
ESQUEMA 4 - SÍNTESE DO POLI(ADIPATO DE ISOSORBILA), ATRAVÉS DE POLICONDENSAÇÃO EM MASSA. AS
REAÇÕES FORAM REALIZADAS SOB VÁCUO E SOB PRESSÃO AMBIENTE. _______________________ 52
ESQUEMA 5 - SÍNTESE DE POLÍMEROS UTILIZANDO DERIVADOS DIVINÍLICOS. _______________________ 56
ESQUEMA 6 – SÍNTESE DE POLÍMEROS EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA. ________________ 59

ÍNDICE DE TABELAS
TABELA 1 - ESPECIFICAÇÕES TÉCNICAS DA CAL-B – NOVOZYM-435 – WWW.NOVOZYMES.COM _______ 31
TABELA 2 - ÁCIDOS E ÁLCOOIS UTILIZADOS NA SÍNTESE DOS DIÉSTERES.__________________________ 22
TABELA 3 - TEMPOS UTILIZADOS NAS POLIMERIZAÇÕES EM MASSA SOB PRESSÃO REDUZIDA. ___________ 40
TABELA 4 - POLIMERIZAÇÃO EM SOLUÇÃO SOB PRESSÃO REDUZIDA. _____________________________ 25
TABELA 5 – POLÍMEROS PRODUZIDOS A PARTIR DE POLIMERIZAÇÃO COM MISTURAS AZEOTRÓPICAS DE
SOLVENTES. __________________________________________________________________ 43
TABELA 6 – MISTURAS AZEOTRÓPICAS BINÁRIAS. ___________________________________________ 43
TABELA 7 - MISTURAS AZEOTRÓPICAS TERNÁRIAS _ _________________________________________28 TABELA 8 - RESULTADOS OBTIDOS NAS REAÇÕES DE POLICONDENSAÇÃO EM MASSA SOB PRESSÃO REDUZIDA. _________________________________________________________________________37
TABELA 9 – RESULTADOS OBTIDOS PARA POLICONDENSAÇÕES REALIZADAS SOB PRESSÃO AMBIENTE. ____ 54
TABELA 10 - RESULTADOS OBTIDOS PARA POLICONDENSAÇÕES UTILIZANDO MONÔMEROS DIVINÍLICOS. ___ 57
TABELA 11 - VALORES DE LOG P PARA DIFERENTES SOLVENTES ________________________________46
TABELA 12 - RESULTADOS OBTIDOS PARA POLIMERIZAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA. _ 63
TABELA 13 - POLIMERIZAÇÃO EM SOLUÇÃO SOB PRESSÃO REDUZIDA. ____________________________ 66
TABELA 14 - POLÍMEROS DA SÉRIE DE ÉSTERES DOS DIÁCIDOS CARBOXÍLICOS EM CICLOEXANO__________51
TABELA 15 - POLÍMEROS DA SÉRIE DE ÉSTERES DOS DIÁCIDOS CARBOXÍLICOS EM CICLOEXANO: BENZENO 6:1.
___________________________________________________________________________ 68
TABELA 16 - ESTUDO DO CRESCIMENTO DA MASSA MOLAR DO POLI(ADIPATO DE ISOSORBILA) EM FUNÇÃO DO
TEMPO DE REAÇÃO. ____________________________________________________________ 71
TABELA 17 - RESULTADOS OBTIDOS COM A TROCA DE TOLUENO POR BENZENO NA MISTURA DE SOLVENTES. 74
TABELA 18 - MASSAS MOLARES DO POLI(ADIPATO DE ISOSORBILA) OBTIDAS VARIANDO-SE O ÁLCOOL DE SAÍDA.
___________________________________________________________________________ 76
TABELA 19 - COMPOSIÇÃO DOS AZEÓTROPOS VARIANDO-SE A CADEIA CARBÔNICA DO ÁLCOOL. _________ 79
TABELA 20 - SOLUBILIDADE DO POLI(ADIPATO DE ISOSORBILA) COM MW~30KDA. ____________________ 80
TABELA 21 - RESULTADOS OBTIDOS VARIANDO-SE A PROPORÇÃO ISOSORBÍDEO: SOLVENTE.____________65
TABELA 22 - RESULTADOS OBTIDOS VARIANDO-SE A CONCENTRAÇÃO DE BENZENO NA MISTURA DE
SOLVENTES. __________________________________________________________________ 83
TABELA 23 - TEMPERATURA DE TRANSIÇÃO VÍTREA DOS POLIÉSTERES OBTIDOS VIA CATÁLISE ENZIMÁTICA E VIA
CATÁLISE QUÍMICA. _____________________________________________________________ 87
TABELA 24 - PERDA DE MASSA DOS MATERIAIS OBTIDOS______________________________________71 TABELA 25 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SUCCINATO DE ISOSORBILA).___________74
TABELA 26 - DESLOCAMENTOS QUÍMICOS DE 13C-RMN DO POLI(SUCCINATO DE ISOSORBILA). _________ 92
TABELA 27 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(GLUTARATO DE ISOSORBILA) ___________76
TABELA 28- DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(GLUTARATO DE ISOSORBILA). __________ 94
TABELA 29 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(ADIPATO DE ISOSORBILA). _____________ 95
TABELA 30 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(ADIPATO DE ISOSORBILA). ____________ 96
TABELA 31 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SUBERATO DE ISOSORBILA). ___________ 97
TABELA 32 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(SUBERATO DE ISOSORBILA). ___________ 98
TABELA 33 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SEBACATO DE ISOSORBILA). ___________ 99
TABELA 34 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(SEBACATO DE ISOSORBILA). __________ 100
TABELA 35 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(DODECANODIOATO DE ISOSORBILA). ____ 101
TABELA 36 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(DODECANODIOATO DE ISOSORBILA). ____ 102
TABELA 37 - PICOS DE ABSORÇÃO NO INFRAVERMELHO. _____________________________________ 105

SUMÁRIO
1 - INTRODUÇÃO. ________________________________________________________________ 17
1.1 - POLÍMEROS DERIVADOS DE FONTES RENOVÁVEIS: ___________________________________ 17
1.2 - DIANIDROEXITÓIS: __________________________________________________________ 18
1.2.1 – Isosorbídeo: _________________________________________________________ 21
1.3 - POLIMERIZAÇÃO POR CATÁLISE ENZIMÁTICA. _______________________________________ 24
1.3.1 - Lipases: ____________________________________________________________ 28
1.3.1.1 – Lipase de Candida antarctica – Fração B - CAL-B: ________________________________ 29
2 – OBJETIVOS. _________________________________________________________________ 34
3 – MATERIAIS E MÉTODOS. ______________________________________________________ 35
3.1 – MATERIAIS: ________________________________________________________________ 35
3.2 – MÉTODOS: ________________________________________________________________ 36
3.2.1 - Preparação e purificação de reagentes, solventes e monômeros. _________________ 36
3.2.1.1 - Purificação do acetato de etila. ____________________________________________ 36
3.2.1.2- Purificação do clorofórmio. ________________________________________________ 36
3.2.1.3- Purificação do isosorbídeo (IS). ____________________________________________ 37
3.2.1.5- Purificação do ácido adípico. ______________________________________________ 37
3.2.1.6- Síntese dos Ésteres dos Ácidos Dicarboxílicos _______________________________ 37
3.2.1.7- Síntese do Adipato de Divinila. _____________________________________________ 38
3.2.2 - Polimerizações _________________________________________________________ 39
3.2.2.1- Polimerização em massa sob pressão ambiente. _____________________________ 39
3.2.2.2 - Polimerização em massa sob pressão reduzida. _____________________________ 39
3.2.2.3 - Polimerização em solução sob pressão reduzida. ____________________________ 40
3.2.2.4 - Polimerização em massa sob pressão ambiente utilizando monômeros divinílicos. 41
3.2.2.5 - Polimerização com solvente sob refluxo em sistema aberto utilizando monômeros
divinílicos. ______________________________________________________________________ 41
3.2.2.6 - Polimerização com misturas azeotrópicas de solvente. _______________________ 42
3.2.3 - Purificação dos polímeros obtidos. _________________________________________ 44
3.2.4 – Teste de solubilidade dos polímeros obtidos: _________________________________ 45
3.2.5 - Caracterização dos polímeros obtidos. ______________________________________ 45
3.2.5.1 - Cromatografia por exclusão de tamanho (SEC): ___________________________________ 45
3.2.5.2 – Calorimetria exploratória diferencial (DSC): ______________________________________ 46
3.2.5.3 – Análise termogravimétrica (TG): _______________________________________________ 46
3.2.5.4 – Espectroscopia no Infravermelho: _____________________________________________ 47
3.2.5.5 – Difração de Raios-X: ________________________________________________________ 47
3.2.5.6 – Ressonância Magnética Nuclear de 1H e
13C: ____________________________________ 47

4 – RESULTADOS E DISCUSSÃO: __________________________________________________ 49
4.1 - POLICONDENSAÇÃO EM MASSA, SOB PRESSÃO AMBIENTE E SOB PRESSÃO REDUZIDA: __________ 51
4.1.1 – Policondensação em massa sob pressão reduzida: ____________________________ 53
4.1.2 - Policondensação em massa sob pressão ambiente: ____________________________ 54
4.1.3 - Considerações gerais: ___________________________________________________ 55
4.2 - POLICONDENSAÇÃO UTILIZANDO MONÔMEROS DIVINÍLICOS: _____________________________ 56
4.3 - POLICONDENSAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA: _______________________ 59
4.3.1 - Polimerização em solução sob pressão reduzida. ______________________________ 66
4.4 - INVESTIGAÇÃO DO SISTEMA CICLOEXANO:BENZENO. ___________________________________ 67
4.4.1 – Estudo comparativo entre sistemas utilizando cicloexano e misturas cicloexano:benzeno
6:1. ________________________________________________________________________ 67
4.4.1.1 - Vantagem do uso da mistura de cicloexano e benzeno: _____________________________ 69
4.4.2 – Estudo do crescimento da massa molar em função do tempo de reação: ___________ 71
4.4.3 – Troca de benzeno por tolueno na mistura: ___________________________________ 73
4.4.4 - Avaliação das massas molares quanto ao álcool de saída. ______________________ 75
4.4.5 - Teste de solubilidade dos materiais obtidos: __________________________________ 79
4.4.6 - Efeito da proporção monômero: solvente ____________________________________ 81
4.4.7 - Efeito da concentração de benzeno na mistura: _______________________________ 82
4.5 - CARACTERIZAÇÃO DOS POLIÉSTERES OBTIDOS. ______________________________________ 85
4.5.1 – Calorimetria Exploratória Diferencial (DSC): __________________________________ 85
4.5.2 – Análise Termogravimétrica: _______________________________________________ 87
4.5.3 – Ressonância Magnética Nuclear: __________________________________________ 89
Espectro de 1H-RMN do Poli(succinato de isosorbila). _____________________________________ 91
Espectro de 13
C-RMN do Poli(succinato de isosorbila). ____________________________________ 92
Espectro de 1H-RMN do Poli(glutarato de isosorbila). ______________________________________ 93
Espectro de 13
C-RMN do Poli(glutarato de isosorbila). _____________________________________ 94
Espectro de 1H-RMN do Poli(adipato de isosorbila). _______________________________________ 95
Espectro de 13
C-RMN do Poli(adipato de isosorbila). ______________________________________ 96
Espectro de 1H-RMN do Poli(suberato de isosorbila). ______________________________________ 97
Espectro de 13
C-RMN do Poli(suberato de isosorbila). _____________________________________ 98
Espectro de 1H-RMN do Poli(sebacato de isosorbila). _____________________________________ 99
Espectro de 13
C-RMN do Poli(sebacato de isosorbila). ____________________________________ 100
Espectro de 1H-RMN do Poli(dodecanodioato de isosorbila). _______________________________ 101
Espectro de 13
C-RMN do Poli(dodecanodioato de isosorbila). ______________________________ 102
4.5.4 – Difração de Raios-X: ___________________________________________________ 103
4.5.5 – Espectroscopia no Infravermelho: _________________________________________ 104
5 - CONCLUSÕES: ______________________________________________________________ 109
6 - REFERÊNCIAS: ______________________________________________________________ 111

17
1 - INTRODUÇÃO
1.1 - POLÍMEROS DERIVADOS DE FONTES RENOVÁVEIS
A biomassa constitui em uma fonte renovável de matérias-primas que dão
origem a novos compostos menos poluentes, capazes de substituir os derivados do
petróleo. A utilização efetiva da biomassa é um alvo de investigação atraente e
desafiador.
O uso de fontes renováveis como uma alternativa para produtos derivados de
combustíveis fósseis vem ganhando preferência, principalmente devido à sua
disponibilidade inesgotável e às considerações ambientais, como por exemplo, o
potencial “sequestro de carbono” (van Dam, et al., 2005). A utilização de derivados
de fontes renováveis em detrimento do uso de produtos de fontes não-renováveis é
um dos 12 princípios da química verde (Lenardão, et al., 2003). Muitos governos
estão desenvolvendo políticas que visam o desenvolvimento de uma sociedade
sustentável através da exploração de produtos renováveis (Clinton, 2000). Em 1998,
o Departamento de Energia dos Estados Unidos estabeleceu uma meta para que,
até 2020, haja um aumento de 10% na produção e no uso de produtos químicos
básicos derivados de plantas, e que até 2050, o aumento seja de 50%. A
substituição do uso do petróleo pelo uso de fontes renováveis para a produção
desses produtos pode gerar uma redução de até 17% da utilização do petróleo e,
consequentemente, diminuir a dependência mundial desse combustível (Executive
Steering Comittee, 1999).

18
O desenvolvimento de polímeros derivados de biomassa é um dos mais
importantes e urgentes assuntos do ponto de vista da preservação ambiental,
considerando-se o fato de que a grande maioria dos polímeros utilizados para fins
comerciais origina-se de monômeros derivados do petróleo. A poluição ambiental
causada pela produção e o descarte de polímeros derivados do petróleo tem
levantado um crescente interesse nas indústrias químicas. Assim, polímeros
derivados do amido ou outros carboidratos são de grande interesse, por serem
provenientes de fontes inteiramente renováveis. Consequentemente, não é
surpreendente o grande aumento das atividades de pesquisa nesse campo nos
últimos anos (Kricheldorf, 1997; Chatti, et al., 2006).
1.2 - DIANIDROEXITÓIS:
O milho é uma fonte de amido, matéria-prima natural, renovável, versátil e
biodegradável, e vem substituindo o petróleo em muitas aplicações industriais,
desde plásticos até para produção de etanol. No Brasil, a safra de milho em 2007 foi
de 38,3 milhões de toneladas (IBGE, 2007). Os Estados Unidos são responsáveis
por 44% da produção mundial de milho, com lavouras ocupando 77 milhões de
acres. Entre todas as lavouras, o milho é a principal fonte de amido, devido ao seu
amplo suprimento e baixo custo relativo a outras fontes de amido ou açúcar, e
também porque existem sistemas bem estabelecidos de transporte e
armazenamento (Busche, et al., 1983).
Os carboidratos utilizados na obtenção de polímeros biodegradáveis são em
grande parte provenientes do amido. No entanto, o uso direto de derivados

19
monoméricos de carboidratos é extremamente difícil devido ao número de
funcionalidades hidroxila nestes compostos. Esse problema pode ser contornado
utilizando-se 1,4:3,6-dianidroexitóis.
Dianidroexitóis são conhecidos produtos da indústria do milho, obtidos através
da hidrogenação seguida de dupla desidratação de açúcares como a D-glicose
(Esquema 1) (Stross, et al., 1992). São produzidos mundialmente 650 mil toneladas
por ano desses compostos (Sheldon, 1993), e seu potencial tem sido explorado para
a síntese de diversos polímeros como poliéteres (Kricheldorf, et al., 1995) (Chatti, et
al., 2002), poliuretanos (Bachmann, et al., 1998), policarbonatos (Kricheldorf, et al.,
1997) e poliésteres (Kricheldorf, et al., 2003).
CHO
OH
H
CH2OH
OH
OH
OH
H
H
H
H
CH2OH
CH2OH
H
H
H
OH
OH
HO HOcat.
hidrogenação
dupladesidratação
H+ O
OHO
OH
1
2
3
4
5
6
1
2
3
4
5
6
123
456
D-Glicose Sorbitol Isosorbídeo
H
H
ESQUEMA 1 - SÍNTESE DO ISOSOSORBÍDEO A PARTIR DA D-GLICOSE.
Estes compostos quirais são obtidos a partir do amido como três isômeros de
acordo com as configurações relativas das duas funções hidroxila (Flèche, et al.,
1986) (Hopton, et al., 1969): isosorbídeo, isomanídeo, e isoidídeo, respectivamente
derivados do D-glucitol, D-manitol e L-iditol (Figura 1). Analisando-se as estruturas
tridimensionais destes compostos, pode-se observar que os dianidroexitóis têm dois
anéis de cinco membros aproximadamente planares, fundidos em cis, em forma de
“V”, com os grupos hidroxila nas posições C-2 e C-5, que podem ser “endo” ou “exo”,

20
dependendo de sua posição, dentro ou fora da “cunha”. No isosorbídeo, uma das
hidroxilas está na configuração “endo” (C-5) e a outra “exo” (C-2), no isomanídeo,
ambas estão na configuração “endo” e no isoidídeo, ambas estão na configuração
“exo”. A reatividade relativa destes compostos pode ser explicada através do
impedimento estérico que ocorre devido às posições dos grupos hidroxilas nas
moléculas.
FIGURA 1 - ESTRUTURAS DOS 1,4: 3,6 - DIANIDROEXITÓIS.
O interesse nesse tipo de compostos como monômeros para a síntese de
vários policondensados tem aumentado muito nos últimos tempos. Essa tendência
se deve a vários motivos (Kricheldorf, 1997):
Podem ser preparados a partir de sacarídeos e são, portanto, monômeros
derivados de fontes renováveis;

21
São substâncias atóxicas e seus poliésteres derivados de ácidos
dicarboxílicos alifáticos são sensíveis à hidrólise, degradam-se em
questão de meses em contato com a água, e por isso são considerados
materiais biodegradáveis muito úteis;
São compostos quirais e pertencem ao grupo de monômeros quirais de
menor custo (particularmente o isosorbídeo). As aplicações potenciais de
polímeros quirais são rentáveis e incluem membranas, fases estacionárias
para cromatografia, entre outras.
1.2.1 – ISOSORBÍDEO:
O isosorbídeo é o dianidroexitol mais abundante, visto que é derivado da
glicose via sorbitol. É biodegradável e recomendado pelo FDA1 como um material
seguro. É quimicamente e termicamente estável. Poliésteres derivados de
isosorbídeo não racemizam ou decompõem quando aquecidos por poucos minutos
acima de 350°C. Em princípio, o isosorbídeo pode ser diretamente incorporado em
poliésteres comerciais como o poli(tereftalato de etileno) (PET), que é um polímero
amplamente usado na fabricação de embalagens para alimentos e bebidas. Devido
à sua rígida estrutura molecular, o isosorbídeo enrijece as cadeias de PET e
aumenta a temperatura de transição vítrea do PET para 85 a 90ºC, dependendo do
grau de incorporação, o que permite que frascos feitos com esses copolímeros
possam ser envasados à quente, com produtos que necessitam ser pasteurizados,
ou que são tão viscosos que necessitam calor para reduzir o tempo de envase.
1 FDA: Food and Drug Administration - agência americana reguladora de alimentos, medicamentos,
vacinas e cosméticos para usos em seres humanos.

22
Frascos normais de PET simplesmente se distorcem quando são envasados à
quente. (Adelman, et al., 2003).
Os custos preliminares estimados mostram que o isosorbídeo tem um preço
competitivo com monômeros derivados de petróleo, usados para síntese de
plásticos (Kricheldorf, et al., 1997; Adelman, et al., 2003). Assim como no PET
modificado, há muitas aplicações potenciais para o isosorbídeo na indústria de
polímeros. O uso do isosorbídeo derivado do milho irá reduzir potencialmente a
quantidade de petróleo necessária para a produção de plásticos. Algumas
aplicações potenciais para os materiais poliméricos derivados do isosorbídeo são
(Malhotra, et al., 2007):
Resinas de isosorbídeo biglicidil éter para substituir resinas epóxis de
bisfenol-A, usadas como rótulos em latas de alimentos e bebidas. O bisfenol-
A é suspeito de ser xenoestrógeno e sua substituição por um diol derivado de
fontes naturais tem grandes vantagens;
Resinas de policarbonatos de isosorbídeo como plásticos moldáveis, com
altos valores de temperatura de transição vítrea, transparência e boa
resistência aos raios UV, podem ser potenciais candidatos para substituição
de policarbonatos nas embalagens de CD;
Poliuretanos amorfos insolúveis baseados em isosorbídeo como resinas de
separação quiral, de grande valor potencial no mercado de fármacos e
química fina, para resolução de moléculas enantioméricas, explorando a
quiralidade desse composto;
Copoliésteres de isosorbídeo de estereoquímica controlada como potenciais
fibras de alto desempenho e resinas de engenharia, podendo-se criar novas

23
classes de poliésteres de performance controlada, através do controle da
quiralidade do polímero ao longo da cadeia;
Resinas de poliésteres de isosorbídeo com ácidos tereftálico e isoftálico, e
copolímeros com monômeros como L-lactídeo, caprolactona, etc. para usos
em aplicações biomédicas. Essas aplicações variam de resinas
ambientalmente corretas para liberação de fármacos até novos materiais de
sutura degradáveis.
O primeiro registro da síntese do isosorbídeo data do ano 1946 (Montgomery, et
al., 1946), e o uso do isosorbídeo como monômero para síntese de poliésteres
insaturados foi publicado primeiramente por Jenkins e colaboradores (Jenkins, et al.,
1963). Um dos primeiros trabalhos relevantes utilizando isosorbídeo como
monômero foi feito por Thiem e Lüders (Thiem, 1984), que utilizaram um cloreto de
ácido dicarboxílico aromático para a síntese de politereftalatos. Storbeck e
colaboradores (Storbeck, et al., 1993) aperfeiçoaram as condições de reação usadas
por Thiem e Lüders, utilizando piridina como aceptor de HCl (subproduto da
policondensação). Braun and Bergmann (Braun, et al., 1992) sintetizaram
poliésteres a partir de isosorbídeo e de cloretos de ácidos dicarboxílicos aromáticos
e alifáticos. Okada e colaboradores (Okada, et al., 1995; Okada, 1996) também
sintetizaram poliésteres alifáticos utilizando isosorbídeo e cloretos de ácidos
dicarboxílicos alifáticos. Brandenburg e colaboradores (Brandenburg, et al., 2004)
sintetizaram uma série grande de poliésteres alifáticos e aromáticos a partir de
ácidos dicarboxílicos com uma série de dióis primários e isosorbídeo. Por
transesterificação, sintetizaram copolímeros de um poliéster com isosorbídeo
incorporado à sua estrutura química, com outro, sem conter isosorbídeo.

24
A variedade de trabalhos que vem sendo publicados comprova que a
incorporação de isosorbídeo na cadeia polimérica de poliésteres tem provado ser
uma abordagem interessante para obter produtos biodegradáveis com diferentes
propriedades.
1.3 - POLIMERIZAÇÃO POR CATÁLISE ENZIMÁTICA
Organismos vivos produzem diferentes macromoléculas (polissacarídeos,
polinucleotídeos, proteínas, poliésteres) para suas necessidades metabólicas. A
síntese dessas substâncias geralmente envolve reações de polimerização via
catálise enzimática in vivo. Uma atividade que tem crescido nos últimos anos é o
uso de enzimas isoladas. Essas enzimas, originadas de organismos vivos, têm
encontrado diversas aplicações como catalisadores para a síntese de polímeros in
vitro.
Devido às suas características como diversidade, especificidade e
biodegradabilidade, polímeros de origem enzimática têm recebido cada vez mais
atenção como candidatos a aplicações industriais (Dalla Vecchia, et al., 2004). Em
muitos casos, enzimas levam à síntese de polímeros que eram impraticáveis ou
impossíveis de serem obtidos através da química convencional (Gross, et al., 2001).
Além disso, os polímeros são sintetizados a partir de fontes renováveis e de
matérias-primas de baixo custo, e as polimerizações ocorrem sob condições brandas
com mínimo impacto ambiental.
Outras vantagens do uso de enzimas na polimerização:

25
Alta eficiência na conversão de substratos devido à sua alta seletividade
para uma dada transformação orgânica;
Alta enantio e regiosseletividade;
Possibilidade de reciclagem do catalisador;
Enzimas têm a habilidade de serem usadas na maioria dos meios
reacionais;
Evitam o uso de catalisadores potencialmente tóxicos.
Avanços recentes na enzimologia não-aquosa têm expandido
significativamente as condições potenciais nas quais as reações de polimerização
via catálise enzimática podem ser executadas. Exemplos de meios reacionais
alternativos incluem vários solventes orgânicos, misturas bifásicas de solventes
orgânicos com misturas aquosas, misturas azeotrópicas, sistemas de micelas
reversas, e fluidos supercríticos. Meios de reação não tradicionais têm oferecido
novas opções para o controle do peso molecular, assim como da morfologia e
arquitetura dos produtos poliméricos. Devido ao fato de que esterificações e
transesterificações enzimáticas têm se mostrado efetivas em solventes orgânicos,
muitas reações enzimáticas em meios não-aquosos têm sido reportadas (Klibanov,
1990). Algumas enzimas hidrolíticas são estáveis mesmo em solventes orgânicos, e
podem ser empregadas em certas condensações difíceis de serem realizadas em
meio aquoso. Por isso, a indústria química e os químicos orgânicos vêm mudando
sua percepção quanto ao uso de enzimas em síntese, utilizando solventes
orgânicos. A visão tradicional era baseada no conhecimento convencional de que o
uso de enzimas era restrito a meios aquosos e temperaturas próximas à ambiente,
que as enzimas eram muito caras e frágeis como catalisadores, e que eram

26
extremamente específicas (baseados no paradigma uma enzima/ uma reação/ um
substrato).
Os estudos pioneiros de Klibanov e colaboradores, desde 1984 (Zaks, 1984;
Klibanov, 1986), utilizando enzimas em solventes orgânicos contendo pouca água
ou em meios completamente anidros constituem numa mudança no ponto de vista
anterior dos químicos orgânicos. Hoje, já é muito bem aceito que as enzimas não só
são capazes de funcionar perfeitamente em solventes orgânicos, como já se provou
que as enzimas são mais estáveis termicamente em meio orgânico do que em água.
Como a maioria dos compostos orgânicos é solúvel em solventes orgânicos e muito
pouco solúveis em água, os químicos orgânicos rapidamente reconheceram o
potencial das enzimas como catalisadores. O uso de enzimas em meios não
aquosos oferece diversas vantagens (Dordick, 1991):
Aumento da solubilidade de substratos apolares;
Aumento da termoestabilidade;
Fácil recuperação dos produtos através de solventes de baixos pontos
de ebulição;
A imobilização é frequentemente desnecessária, pois a enzima é
insolúvel;
Fácil recuperação da enzima por filtração ou centrifugação;
Se a imobilização for necessária para melhor desempenho, a simples
adsorção em superfícies não porosas é suficiente;
Supressão de reações laterais “água-dependentes”;

27
Deslocamento do equilíbrio termodinâmico favorecendo a síntese em
detrimento da hidrólise em reações como a esterificação;
A possibilidade de realizar reações com outros nucleófilos que não
seriam praticáveis em água, como por exemplo, transesterificação;
Fácil integração de passos enzimáticos com passos químicos
convencionais em sínteses multietapas.
A polimerização por catálise enzimática vem se desenvolvendo rapidamente,
principalmente nas duas últimas décadas, através do trabalho de autores como
Gross e colaboradores (Gross, et al., 2001) e Kobayashi e colaboradores
(Kobayashi, et al., 2001), entre outros. A síntese de poliésteres alifáticos por
processos enzimáticos tem sido amplamente estudada do ponto de vista da ciência
de materiais biodegradáveis (Albertsson, et al., 1995).
Certamente, ainda existem muitos desafios técnicos para o desenvolvimento
de processos comerciais que envolvem polimerizações por catálise enzimática. A
identificação de uma enzima com as capacidades sintéticas específicas requer uma
intensa pesquisa. Comparando com catalisadores químicos, reações com enzimas
geralmente ocorrem com baixa cinética de reação. Além disso, o isolamento e a
purificação dos produtos poliméricos por processos de baixo custo também são um
desafio. Um melhor entendimento dos mecanismos de reação pode identificar
barreiras e limitações dos processos. Contudo, existem muitas razões para o
otimismo que existe no que diz respeito a uma revolução que está começando, onde
a catálise enzimática in vitro será incorporada em numerosos processos
relacionados a polímeros.

28
1.3.1 - LIPASES:
As lipases são enzimas pertencentes ao grupo das hidrolases, que catalisam
a hidrólise de ésteres de ácidos graxos, normalmente em ambientes aquosos de
organismos vivos. São encontradas em tecidos de vários animais e plantas e podem
ser produzidas por fermentação de diversas espécies de microrganismos, tais como
fungos, leveduras e bactérias. Têm sido amplamente utilizadas em síntese orgânica
devido à sua alta disponibilidade e baixo custo, e têm sido extensivamente
investigadas com relação às suas propriedades bioquímicas, fisiológicas e, mais
recentemente, para aplicações industriais (Pandey, et al., 1999).
Algumas lipases são estáveis em solventes orgânicos e podem ser usadas
como catalisadores para esterificações e transesterificações. Essa catálise
possibilita a produção de poliésteres e policarbonatos através de vários métodos de
polimerização. Reações típicas de polimerizações catalisadas por lipases estão
resumidas no Esquema 2 (Kobayashi, et al., 2001). Biotransformações de várias
combinações de derivados de ácidos dicarboxílicos e glicóis para obtenção de
poliésteres foram publicadas nos últimos 20 anos (Bornschauer, et al., 2002). Ácidos
dicarboxílicos e seus derivados, ésteres ativados e desativados, anidridos cíclicos e
polianidridos foram reportados como monômeros para a síntese de poliésteres sob
condições reacionais brandas.

29
CO
O
R
LipaseORC
O
**
n
Polimerização de Lactonas por Abertura de Anel
Policondensação de Ácidos Carboxílicos ou Derivados com Glicóis
XO2CRCO2X + HOR'OH LipaseCRC OR'
O O
* *
n
Policondensação de Oxiácidos e seus Ésteres
X= H, Alquil, Alquil halogenado, Vinil, etc.
HORCO2X
-XOH
Lipase
-XOH ORC **n
X= H, Alquil, Alquil halogenado, Vinil, etc.
O
ESQUEMA 2 - REAÇÕES DE POLIMERIZAÇÃO CATALISADAS POR L IPASES.
1.3.1.1 – Lipase de Candida antarctica – Fração B - CAL-B:
Muito tem sido publicado sobre o uso de lipases para catalisar
biotransformações, muitas dessas reações são realizadas em meios não aquosos.
Certamente, a CAL-B, que é a lipase de Candida antarctica - fração B, tem sido a
lipase mais amplamente utilizada (Anderson, et al., 1998). Com sua estrutura
completamente elucidada (Uppenberg, et al., 1994) e mecanismo do sítio catalítico
bem conhecido (Faber, 1997; Kwon, et al., 2007) - Esquema 3, essa enzima tem
sido amplamente usada nas pesquisas e na indústria, principalmente por possuir
uma característica chamada promiscuidade catalítica (Copley, 2003), que é a
habilidade do sítio ativo da enzima em catalisar diferentes transformações químicas.

30
O O
Asp
N NH
His
OH
Ser
R O
O
OH
Passo 1
Passo 2O O
Asp
HN N
His
HO
Ser
R
O
R2 R3
OH
Intermediário acil-enzima
Nucleófilo
R2 R3
O
O
R
ESQUEMA 3 - MECANISMO DO SÍTIO CATALÍTICO DA CAL-B.
Nos trabalhos de Uppenberg (Uppenberg, et al., 1994), e Cygler e Shrag
(Cygler, et al., 1997), foi mostrado que o sítio ativo de uma lipase é dividido em um
lado ligante-acila e outro lado ligante-álcool, o lado álcool sendo substrato-
específico, e o lado acila é descrito como sendo relativamente não-específico. Um
número considerável de trabalhos vem sendo publicado sobre a
estereoespecificidade do lado álcool do sítio ativo das lipases. Como um exemplo, a
CAL-B é altamente específica para alcoóis secundários, devido à cavidade oxiânion,
que estabiliza o intermediário tetraédrico álcool-acil-enzima (Uppenberg, et al.,
1995). As lipases, no geral, são capazes de hidrolisar triglicerídeos em uma interface
óleo-água, onde sua atividade é drasticamente aumentada. A CAL-B, apesar de não
ser tão eficiente como as outras lipases na hidrólise de triglicerídeos, é de particular
interesse porque demonstra alta estereoespecificidade em substratos quirais durante
a hidrólise ou a síntese orgânica.

31
FIGURA 2 - ESTRUTURA TRIDIMENSIONAL DA CAL-B (WWW.NOVOZYMES.COM)
Neste trabalho, a CAL-B utilizada foi a NOVOZYM-435, que é a lipase de
Candida antarctica imobilizada em matriz de poliacrilato, que confere uma maior
termoestabilidade à enzima. As propriedades da NOVOZYM-435 estão resumidas
na Tabela 1.
TABELA 1 - ESPECIFICAÇÕES TÉCNICAS DA CAL-B – NOVOZYM-435 – WWW.NOVOZYMES.COM
Densidade 430 kg/m3
Diâmetro das partículas 0,3-0,9 mm
Quantidade de água 1-2% m/m
Peso Molecular 33 kDa
pH ótimo 7,0
Estabilidade ao pH 7,0 – 10,0
Temperatura ótima ~70ºC
Atividade declaradaa 10000 PLU/g
a – A atividade é dada pela síntese de laureato de propila (PLU=Propyl Laurate Units). 1 PLU
corresponde à formação de 1 µmol de éster por minuto.

32
Muitos trabalhos vêm sendo publicados abordando a síntese de poliésteres
de glicóis e diácidos e seus derivados via catálise enzimática utilizando lipases. No
entanto, até o presente momento não há registros de polimerizações de dióis
secundários via catálise enzimática propriamente dita. Inclusive, quando se realiza a
polimerização enzimática de polióis como os alditóis (Hu, et al., 2006), que possuem
em sua estrutura tanto hidroxilas primárias quanto secundárias, a enzima realiza a
polimerização através das hidroxilas primárias e, como em se tratando de hidroxilas
secundárias, até então só era conhecida a seletividade da CAL-B por configurações
(R) , somente as hidroxilas (R) secundárias reagiram, aumentando a massa molar do
polímero através de ramificações na cadeia, em concordância com os estudos de
Hult e colaboradores (Rotticci, et al., 1998).
Estudos recentes realizados por Palmans e colaboradores (Kanca, et al.,
2008; van As, et al., 2007), mostram a polimerização de misturas racêmicas de
monômeros contendo hidroxilas secundárias através de Catálise Acoplada Iterativa
(Iterative Tandem Catalysis – ITC), mais especificamente resolução cinética
dinâmica, que é a conversão química na qual dois ou mais catalisadores trabalham
juntos para obter a reação química desejada. A resolução cinética dinâmica tem sido
amplamente utilizada para obtenção de compostos enantiomericamente puros.
Nesses trabalhos de polimerização, os autores utilizaram simultaneamente, CAL-B e
um catalisador de rutênio. Assim, a enzima desempenhava o papel de esterificar os
alcoóis com configuração (R), enquanto o catalisador basicamente convertia as
hidroxilas (S) em (R), originando uma reação em cascata, obtendo polímeros com
massas molares de 15 kDa.
Nesse trabalho, reportamos a utilização de isosorbídeo [(1R,4R,5R, 8S)-2,6-
dioxabiciclo[3.3.0]octan-4,8-diol], um diol com hidroxilas nas configurações (R) e (S)

33
na mesma molécula, com ácidos dicarboxílicos e seus derivados, via catálise
enzimática propriamente dita, utilizando como catalisador apenas a lipase de
Candida Antarctica, fração B (CAL-B), para a obtenção de poliésteres.

34
2 – OBJETIVOS
Tendo em vista as recentes políticas de incentivo à exploração da biomassa
para obtenção de materiais “ambientalmente corretos”, visando à redução do uso de
compostos derivados do petróleo, e considerando ampla disponibilidade e as
potencialidades químicas e econômicas do isosorbídeo descritas anteriormente,
aliadas às múltiplas vantagens que a catálise enzimática pode proporcionar na
obtenção de polímeros biodegradáveis e biocompatíveis, o objetivo principal desse
trabalho foi desenvolver metodologias de preparo de poliésteres de isosorbídeo via
catálise enzimática.
Dentro deste escopo, tendo por finalidade a obtenção de materiais com altos
valores de massas molares, exploramos diferentes parâmetros como: técnicas de
policondensação, solventes, tempo de reação, grupo de saída, solubilidade e
diluição dos sistemas e tipos de monômeros, para a produção de polímeros com
diferentes características.
A grande importância do trabalho, e o grande desafio proposto, nesse caso,
consistem no fato de que, até o presente momento, não existem registros de êxito
em polimerizações de dióis secundários impedidos estericamente, como é o caso do
isosorbídeo, através de catálise enzimática, o que possibilitaria uma infinidade de
aplicações dos polímeros, principalmente porque os materiais obtidos são livres de
quaisquer resíduos de metais pesados, provenientes dos catalisadores
convencionais.

35
3 – MATERIAIS E MÉTODOS
3.1 – MATERIAIS:
Cicloexano, benzeno, tolueno, n-heptano, MEK (Merck), acetona, n-hexano,
clorofórmio, metanol (Synth e Vetec), acetato de etila (Synth), foram secos e
destilados previamente. O éter difenílico (Aldrich) foi previamente destilado.
Pentóxido de fósforo anidro (Sigma-Aldrich e Vetec), sulfato de magnésio
anidro (J. T. Baker), cloreto de cálcio anidro (Synth e Casa Americana) e bicarbonato
de sódio (Synth), foram utilizados para secar e purificar os solventes e monômeros.
O acetato de vinila (Aldrich), acetato cúprico (Malinckrodt), acetato mercúrico
(Merck) e acetato de sódio (Synth) foram utilizados na síntese dos monômeros
divinílicos sem purificação prévia.
Os ácidos succínico, adípico, glutárico, subérico, sebácico (Aldrich) e
dodecanodióico (Merck), foram utilizados conforme recebido, nas sínteses dos seus
respectivos diésteres. Nas polimerizações onde se utilizou o ácido propriamente dito,
este foi purificado por recristalização conforme descrito abaixo.
O isosorbídeo (Cerestar e Aldrich) foi previamente recristalizado em acetato
de etila e seco antes de ser utilizado.
A enzima CAL-B (Novozym-435) foi gentilmente cedida pela Novozymes do
Brasil, e utilizada conforme recebido.
As peneiras moleculares de 3Å e 4Å (Aldrich) foram previamente ativadas em
mufla por 6 horas a 300ºC.
Na síntese e purificação dos monômeros, a pureza dos compostos foi
determinada por CG e CG-MS, no Laboratório de Compostos de Selênio e Telúrio,

36
no IQ-USP. As análises por cromatografia a gás (CG) foram efetuadas nos
cromatógrafos SHIMADZU GC-2010, equipado com um detector de ionização de
chama, e os espectros de massa de baixa resolução foram obtidos em um
instrumento Shimadzu CG-MS-17A/QP5050A. Os gases de arraste foram nitrogênio
para CG, e hélio para CG-MS e a coluna empregada foi de sílica fundida revestida
com goma de poli SPB-35 (35% difenil / 65% dimetilsilicone) . A rampa de
aquecimento foi: temperatura inicial 100ºC, gradiente de 10ºC/min, temperatura final:
250ºC.
3.2 – MÉTODOS:
3.2.1 - PREPARAÇÃO E PURIFICAÇÃO DE REAGENTES, SOLVENTES E
MONÔMEROS.
3.2.1.1 - Purificação do acetato de etila.
O acetato de etila foi seco em CaCl2, refluxado, duplamente destilado de
P2O5 e guardado sob peneira molecular de 3Å ativada.
3.2.1.2- Purificação do clorofórmio.
O Clorofórmio (Synth) utilizado para as análises de SEC foi tratado com
NaHCO3, seco com CaCl2 anidro e filtrado em membrana de PTFE modificado de
0,45 µm de poro.

37
3.2.1.3- Purificação do isosorbídeo (IS).
O isosorbídeo foi purificado por recristalização em acetato de etila
previamente tratado, seco sob vácuo em dessecador com pentóxido de fósforo por
72h (até massa constante). O rendimento da recristalização foi, em média, 85%. A
verificação da pureza do material obtido foi feita através de cromatografia gasosa
(pureza >99,8%).
Antes de ser utilizado nas polimerizações, o isosorbídeo recristalizado foi,
ainda, seco sob vácuo por 48h, em dessecador com pentóxido de fósforo até peso
constante.
3.2.1.5- Purificação do ácido adípico.
O ácido adípico foi recristalizado em solução de etanol: água 2:1 e seco em
dessecador sob vácuo por 24 h.
3.2.1.6- Síntese dos ésteres dos ácidos dicarboxílicos.
Os ésteres dos ácidos dicarboxílicos foram sintetizados de acordo com
Klostergaard (Klostergaard, 1958) (dados na Tabela 2).
A uma mistura de 70 mmol do diácido de interesse em 700 mmol do álcool
anidro, adicionou-se ácido sulfúrico até solubilização do ácido, e 50 mL de hexano,
refuxou-se por 30 minutos. Ao fim da reação, a mistura foi lavada com solução
saturada NaHCO3. Após a separação de fases, lavou-se a fase aquosa com hexano
(3X30 mL). As fases orgânicas combinadas foram secas com Na2SO4 e a mistura foi
concentrada em rotoevaporador. Os produtos foram purificados por destilação a
vácuo, e a pureza foi determinada por cromatografia gasosa (em todos os casos,
maior que 99,5%).

38
TABELA 2 - ÁCIDOS E ÁLCOOIS UTILIZADOS NA SÍNTESE DOS DIÉSTERES.
Ácido Álcool
Rendimento após
purificação
Succínico Etanol 62%
Succínico Metanol 55%
Glutárico Etanol 63%
Adípico Metanol 65%
Adípico Etanol 78%
Adípico n-Propanol 56%
Adípico n-Butanol 51%
Subérico Etanol 72%
Sebácico Etanol 70%
Dodecanodióico Etanol 67%
3.2.1.7- Síntese do adipato de divinila e succinato de divinila.
Os ésteres divinílicos foram preparados através de uma adaptação do
procedimento descrito por Kikukawa e colaboradores. (Kikukawa, et al., 1972).
Em um balão de 125 mL, foram adicionados 68 mmol de ácido adípico (ou
ácido succínico), 330 mg de acetato mercúrico, 20 mg de acetato cúprico e 70 mL de
acetato de vinila. A mistura foi aquecida sob refluxo por 15 minutos. Adicionou-se
então, gota a gota, 70µL de ácido sulfúrico concentrado. A mistura foi deixada sob
refluxo por 16h. Adicionou-se cerca de 5 g de acetato de sódio e a solução foi
filtrada. Lavou-se com solução saturada de NaHCO3 até cessar a efervescência.
Extraiu-se com 3 X 40 mL de hexano, as fases orgânicas foram combinadas,
lavadas com água, secas com Na2SO4 e concentradas. O rendimento foi de
aproximadamente 40%. O produto foi purificado por destilação a vácuo (destilado
duas vezes).

39
3.2.2 - POLIMERIZAÇÕES
3.2.2.1- Polimerização em massa sob pressão ambiente.
Em um balão de fundo redondo de 50 mL, adicionou-se 10 mmol de
isosorbídeo previamente seco, pesado analiticamente, e 10 mmol de adipato de
dietila, adicionou-se a enzima (CAL-B N-435) numa proporção de 10% relativa à
massa total dos monômeros. O balão foi fechado com septo de borracha, e duas
agulhas foram inseridas nesse, uma para entrada e outra para saída de N2. A
mistura foi submetida a aquecimento em banho de óleo, a uma temperatura de 85ºC,
sob fluxo de nitrogênio e agitação magnética durante o tempo indicado. Decorrido
esse tempo, a mistura foi suspensa em clorofórmio, filtrada para remoção da enzima
e o solvente foi evaporado no rotoevaporador.
3.2.2.2 - Polimerização em massa sob pressão reduzida.
Em um balão de fundo redondo de 50 mL, adicionou-se 10 mmol de
isosorbídeo previamente seco, pesado analiticamente, e 10 mmol de adipato de
dietila, adicionou-se a enzima (CAL-B N-435) numa proporção de 10% relativa à
massa total dos monômeros. A mistura foi então submetida a aquecimento em
banho de óleo a 70ºC, sob agitação magnética, primeiramente sob pressão
ambiente, para formação de oligômeros, depois sob vácuo, para deslocamento do
equilíbrio através da remoção de subprodutos de reação (etanol, no caso), pelo
tempo especificado abaixo (Tabela 3). Após esse tempo, a mistura foi suspensa em

40
clorofórmio, filtrada para remoção da enzima e o solvente foi evaporado no
rotoevaporador.
TABELA 3 - TEMPOS UTILIZADOS NAS POLIMERIZAÇÕES EM MASSA SOB PRESSÃO REDUZIDA.
Tempo sob pressão ambiente (h) Tempo sob pressão reduzida (~2mmHg) (h)
4,5 48
24 48
86 36
Monômeros: isosorbídeo e adipato de dietila, razão molar 1:1. Temperatura: 70ºC. Enzima: CAL-B N-
435 10% em massa de monômeros.
3.2.2.3 - Polimerização em solução sob pressão reduzida.
Em um balão de fundo redondo de 50 mL, adicionou-se 10 mmol de
isosorbídeo previamente seco, pesado analiticamente, e 10 mmol do monômero
descrito na Tabela 4, e 10 mL de difenil éter, adicionou-se a enzima (CAL-B N-435)
numa proporção de 10% relativa à massa total dos monômeros. A mistura foi então
submetida a aquecimento em banho de óleo, a 70ºC, sob agitação magnética,
primeiramente sob pressão ambiente, depois sob vácuo (~2mmHg), para
deslocamento do equilíbrio através da remoção de subprodutos de reação (etanol,
no caso), pelo tempo especificado abaixo. Após esse tempo, a temperatura foi
elevada para 110ºC para a evaporação do difenil éter, ainda sob vácuo, e a mistura
foi suspensa em clorofórmio ou THF, filtrada para remoção da enzima e o solvente
foi evaporado no rotoevaporador.

41
TABELA 4 - POLIMERIZAÇÃO EM SOLUÇÃO SOB PRESSÃO REDUZIDA.
Monômero A
Tempo sob pressão
ambiente a 70ºC
(h)
Tempo sob vácuo a
70ºC
(h)
Tempo sob vácuo a
110ºC
(h)
Adipato de dietila 24 72 6
Succinato de dietila 24 72 6
Monômero B: Isosorbídeo, razão molar A:B=1:1, enzima: CAL-B N-435 10% em massa de
monômeros.
3.2.2.4 - Polimerização em massa sob pressão ambiente utilizando
monômeros divinílicos.
Em um balão de fundo redondo de 50 mL, adicionou-se 10 mmol de
isosorbídeo previamente seco, pesado analiticamente, e 10 mmol de adipato de
divinila, adicionou-se a enzima (CAL-B N-435) numa proporção de 10% relativa à
massa total dos monômeros. O balão foi fechado com septo de borracha, e duas
agulhas foram inseridas neste, uma para entrada e outra para saída de N2. A mistura
foi submetida a aquecimento em banho de óleo, a uma temperatura de 70ºC, sob
fluxo de nitrogênio e agitação magnética durante 110 h. Decorrido esse tempo, a
mistura foi suspensa em clorofórmio, filtrada para remoção da enzima e o solvente
foi evaporado no rotoevaporador.
3.2.2.5 - Polimerização com solvente sob refluxo em sistema aberto utilizando
monômeros divinílicos.
Em um balão de fundo redondo de 50 mL, adicionou-se 10 mmol de
isosorbídeo previamente seco, pesado analiticamente, e 10 mmol do monômero

42
(adipato de divinila ou succinato de divinila), e 40 mL de hexano, adicionou-se a
enzima (CAL-B N-435) numa proporção de 10% relativa à massa total dos
monômeros. A mistura foi, então, submetida a aquecimento sob refluxo, sob fluxo de
nitrogênio, em banho de óleo, durante 7 dias. Decorrido esse tempo, adicionou-se
clorofórmio, a mistura foi então filtrada para remoção da enzima e o solvente foi
evaporado por rotoevaporação.
3.2.2.6 - Polimerização com misturas azeotrópicas de solvente.
Em um tubo reacional de 80 mL, adicionou-se 10 mmol de isosorbídeo
previamente seco, pesado analiticamente, e 10 mmol do monômero descrito na
Tabela 5, e 65 mL de solvente (ou mistura de solventes) especificado abaixo,
adicionou-se a enzima (CAL-B N-435) numa proporção de 10% relativa à massa
total dos monômeros. Um sistema Dean-Stark foi adaptado ao tubo. O Dean-Stark
foi preenchido de duas maneiras diferentes: nas reações onde o subproduto é água
ou metanol, com peneira molecular de 3Å; nas reações onde o subproduto é o
etanol, n-propanol ou n-butanol, com peneira molecular de 4Å. A mistura foi, então,
submetida a aquecimento sob as temperaturas indicadas, durante o tempo
especificado abaixo. As peneiras moleculares foram trocadas de 24 em 24 horas.
Decorrido esse tempo, adicionou-se clorofórmio, a mistura foi então filtrada para
remoção da enzima e o solvente foi evaporado por rotoevaporação.

43
TABELA 5 – POLÍMEROS PRODUZIDOS A PARTIR DE POLIMERIZAÇÃO COM MISTURAS AZEOTRÓPICAS DE
SOLVENTES.
Monômero Solvente Temp. (ºC) Tempo (h)
Ácido adípico
Benzeno 80 168
Clorofórmio 75 168
MEK: hexano 4:1 75 168
Adipato de dietila
Acetona: Clorofórmio 4:1 75 168
Benzeno 75 168
Clorofórmio 75 168
Clorofórmio: hexano 3:2 75 168
Diclorometano 75 168
n-Hexano 75 168
Tetracloreto de Carbono 75 168
Cicloexano 85 168
Cicloexano: Benzeno 4:1 85 168
Cicloexano: Benzeno 6:1 85 168
n-Heptano 90 168
Succinato de dietila Acetona: Clorofórmio 4:1 75 168
n-Hexano 70 168
As Tabelas 6 e 7 mostram os pontos de ebulição e a composição das
misturas azeotrópicas utilizadas nos experimentos (Lide, 2005):
TABELA 6 – MISTURAS AZEOTRÓPICAS BINÁRIAS.
Subproduto Solvente P.E.
(⁰C)
% de subproduto
na mistura
P.E. da
mistura
Etanol
P.E.: 78.3⁰C
Benzeno 80.1 31,7 67.9
n-Hexano 68.95 21 58.68
n-Heptano 98.5 51 70.9
Cicloexano 80.8 29.2 64.8
Tetracloreto de Carbono 76.8 15.8 65.1
Diclorometano 83.7 37 70.5
Água
P.E.: 100⁰C
Benzeno 80.1 8.83 69.25
Clorofórmio 61 2.8 56.1

44
TABELA 7 – MISTURAS AZEOTRÓPICAS TERNÁRIAS
Subproduto Solvente PE (⁰C)
% de
solvente na
mistura
% de
subproduto
PE da
mistura
Água
PE:100⁰C
n-Hexano 79.6 26 19 59
2-Butanona (MEK) 68.7 55
Etanol
PE:78.3ºC
Clorofórmio 61 65.3 10,4 63.2
Acetona 56.1 24.3
Clorofórmio 61 56.1 9,5 57.3
n-Hexano 79.6 34.4
Cicloexano 80.8 57,6 29,6 64,7
Benzeno 80.1 12,8
3.2.3 - PURIFICAÇÃO DOS POLÍMEROS OBTIDOS.
Os materiais obtidos foram purificados por dissolução e precipitação,
conforme descrito abaixo: (Uyama, et al., 1999)
Em um béquer, o material impuro foi dissolvido na menor quantidade possível
de clorofórmio, e, em seguida, adicionado gota a gota em uma grande quantidade de
metanol (dez vezes o volume de clorofórmio utilizado), sob agitação vigorosa. O
polímero permanece então aderido às paredes do béquer. O sobrenadante foi então
eliminado e os materiais foram secos, primeiramente em dessecador sob vácuo por
1 h, e posteriormente em estufa sob vácuo a 45ºC por 48 h.

45
3.2.4 – TESTE DE SOLUBILIDADE DOS POLÍMEROS OBTIDOS:
Com o objetivo de se determinar a solubilidade de uma amostra padrão de
poli(adipato de isosorbila) com 𝑀 𝑤~30000 Da, uma grande quantidade de polímero
foi adicionada a 2 mL de diferentes solventes de forma que o polímero formasse
uma solução supersaturada com o solvente em questão, e a solução foi mantida sob
agitação magnética por 48h. Decorrido este tempo, uma alíquota de 1,00 mL foi
retirada e colocada em um béquer previamente pesado em balança analítica. O
solvente foi então evaporado sob vácuo por 12 h, e o béquer foi novamente pesado.
A diferença entre as massas obtidas é a solubilidade do material em 1 mL de
solvente.
3.2.5 - CARACTERIZAÇÃO DOS POLÍMEROS OBTIDOS.
3.2.5.1 - Cromatografia por exclusão de tamanho (SEC):
As massas molares médias 𝑀 𝑤 e 𝑀 𝑛 , bem como a razão 𝑀 𝑤 / 𝑀 𝑛 que é o
índice de polidispersividade (PD), foram determinados por cromatografia por
exclusão de tamanho, realizadas em um cromatógrafo Shimadzu modelo Class-VP
10A, equipado com um detector de índice de refração RID-10A com a cela a 35ºC,
forno CTO-10A a 35ºC, loop de 20 µL, bomba LC-10AD, com fluxo de 1 mL por
minuto. Foram utilizadas quatro colunas Waters tipo Styragel® HR6 (WAT054468),
HR4 (WAT044225), HR3 (WAT044222), e HR1 (WAT04234). A fase móvel utilizada
foi clorofórmio. Os valores de massas molares foram reportados com relação a
padrões de poliestireno (com 𝑀 𝑤= 820, 2460, 5120, 13200, 29300, 47500 e 216000),

46
que foram utilizados na construção de curvas de calibração. A concentração das
amostras (injetadas) foi de aproximadamente 20 mg mL-1.
3.2.5.2 – Calorimetria exploratória diferencial (DSC):
Os ensaios de DSC foram realizados no Laboratório do Centro NSF de
Biocatálise e Bioprocessamento de Macromoléculas, na Polytechnic University, em
Nova York, em um calorímetro TA Instruments DSC2920, calibrado com padrão de
Índio. A taxa de aquecimento foi de 10ºC/min, sob proteção de atmosfera de
nitrogênio sob fluxo de 50mL/min, e a varredura foi feita de -70 a 200ºC.
Primeiramente, a amostra foi resfriada a -70ºC e então aquecida a 10ºC/min até
200ºC, mantida a 200ºC por 3 minutos e, em seguida, resfriada a -70ºC a 10ºC/min.
Assim que a amostra foi resfriada, esta foi aquecida novamente até 200ºC. O
software “TA Advantage Control” foi usado para controlar as condições
experimentais e o software “TA Universal Analysis” para analisar os resultados.
3.2.5.3 – Análise termogravimétrica (TG):
As análises termogravimétricas foram realizadas no Laboratório do Centro
NSF de Biocatálise e Bioprocessamento de Macromoléculas, na Polytechnic
University, em Nova York, no equipamento TA Instruments TGA2950. Cada amostra
(10 mg) foi aquecida de 40 a 750ºC, a uma taxa de aquecimento de 10ºC por minuto
sob proteção de atmosfera de nitrogênio com fluxo de 50 mL/min. Os dados foram
analisados utilizando o software “TA Universal Analysis”.

47
3.2.5.4 – Espectroscopia no Infravermelho:
Os ensaios de espectroscopia no infravermelho foram realizados no
equipamento FTIR Bomem MB 100 (faixa de operação de 350 – 4000 cm-1,
resolução de 2 cm-1). As amostras foram analisadas em filmes preparados em
clorofórmio e secos em estufa a vácuo.
3.2.5.5 – Difração de Raios-X:
Os ensaios de difração de raios-X de alto ângulo (WAXD) foram obtidos no
equipamento X-Ray Difratometer Miniflex da Rigaku. Os ensaios foram realizados
usando um feixe monocromático () de raios X de 0,194 nm (FeK) ou 0,154 nm
(CuK), voltagem de 30 kV, corrente de 15 mA, intervalo de ângulo 2 tipicamente de
3 a 60 e leitura de 2 de 0,02 em 0,02. As amostras na forma de pó sofreram
tratamento térmico prévio (recozimento; annealing) em estufa a vácuo por 48 h a
90ºC.
3.2.5.6 – Ressonância Magnética Nuclear de 1H e 13C:
Para fins de determinação estrutural, as análises de ressonância magnética
nuclear (RMN) de 1H e 13C foram realizadas em um equipamento Bruker DPX 300
(300 e 75 MHz, respectivamente). As amostras (20-60 mg e 20-150 mg para 1H e

48
13C NMR, respectivamente) foram dissolvidas em CDCl3 (0,5-0,7 mL), que continha
tetrametilsilano (TMS) como referência interna, e em alguns casos, os espectros
foram calibrados pelo deslocamento químico do CDCl3, no caso do13C, ou CHCl3 no
caso do hidrogênio. Os deslocamentos químicos () são dados em ppm.

49
4 – RESULTADOS E DISCUSSÃO:
Geralmente, a policondensação é uma tarefa mais difícil do que polimerização
por adição, já que massas molares altas só são alcançadas quando há alto grau de
conversão (>98%) (Edlund, et al., 2003). Um balanço estequiométrico exato é de
fundamental importância no caso e, enquanto esse balanço é prontamente obtido
quando são utilizados monômeros do tipo AB, como hidróxi-ácidos, por exemplo,
torna-se um grande problema prático quando se utilizam monômeros do tipo AA e
BB, como diácidos e dióis. Em um sistema contendo apenas monômeros
bifuncionais, com dois tipos de grupos A e B, presentes em números NA e NB, se
NA<NB de forma que a razão seja r = NA/NB, o número total de monômeros é ½
(NA+NB ) = ½ NA(1+1/r). Na amplitude da reação p (definida por A grupos; para B
grupos, amplitude de reação = rp), o número total de terminações de cadeias é NA
(1-p) + NB (1-rp) = NA [1-p + (1 - rp) /r]. Como esse é o dobro do número de
moléculas presentes, então o grau de polimerização aproximado será (Billmeyer,
1984):
𝐺𝑃 𝑛 =
𝑁𝐴(1 +1𝑟)/2
𝑁𝑎 1 − 𝑝 +1 − 𝑟𝑝
𝑟 /2=
1 + 𝑟
1 + 𝑟 − 2𝑟𝑝
Como 𝑝 → 1
𝐺𝑃 𝑛 =
1 + 𝑟
1 − 𝑟
Essa equação mostra claramente a necessidade de um controle
estequiométrico fino na reação, para obtenção de materiais com altos graus de
polimerização. Erros na pesagem, a presença de impurezas monofuncionais,
Equação 1.

50
reações laterais ou o excesso de um dos reagentes podem ser sérios fatores
limitantes do grau de polimerização a ser obtido.
As reações de polimerização consideradas neste trabalho são condensações
do tipo esterificação ou transesterificação de monômeros do tipo AA e BB. Em
ambos os casos, trata-se de um processo complexo com várias sub-reações
seqüenciais em equilíbrio. Desta forma, uma das maneiras mais aplicadas de
obtenção de altos rendimentos e, nestes casos, altos valores de massa molar é o
deslocamento do equilíbrio em direção aos produtos através da remoção de um dos
mesmos. A remoção de água ou de álcool volátil é a maneira mais simples nestes
casos.
Os materiais produzidos durante este trabalho são inéditos no que diz
respeito à síntese de poliésteres de isosorbídeo via catálise enzimática, tanto por
policondensação em massa como em solução.
Neste trabalho, realizou-se uma varredura de condições experimentais e de
métodos para deslocamento do equilíbrio, partindo-se, paralelamente, de três frentes
de trabalho diferentes, todas a partir de catálise enzimática: (i) policondensação em
massa, sob pressão ambiente e sob pressão reduzida, (ii) policondensação
utilizando monômeros divinílicos, e (iii) policondensação em solução com destilação
azeotrópica, com o objetivo de se encontrar a condição experimental ideal, que
proporcionasse a obtenção dos maiores valores de massas molares e os melhores
rendimentos.

51
4.1 - POLICONDENSAÇÃO EM MASSA, SOB PRESSÃO AMBIENTE E SOB PRESSÃO
REDUZIDA:
Sínteses enzimáticas de poliésteres por reações entre diácidos ou derivados
e dióis primários através de polimerizações em massa, catalisadas por Lipase de
Candida antarctica foram publicadas por diversos autores (Uyama, et al., 1998;
Binns, et al., 1998; Uyama, et al., 2000), obtendo polímeros ou oligômeros em
condições brandas, a despeito da mistura heterogênea formada entre o catalisador e
os monômeros. O comportamento da polimerização, no caso, depende fortemente
do tamanho da cadeia de ambos os monômeros. Normalmente, massas molares
maiores são obtidas quando se utiliza vácuo para remoção de água ou álcool do
meio, deslocando-se o equilíbrio na direção dos produtos. Esse tipo de processo tem
chamado atenção por ser um método de síntese ambientalmente correto, pois evita
o uso de solventes orgânicos.
Binns e colaboradores (Binns, et al., 1999) polimerizaram, via catálise
enzimática em larga escala, dióis primários e diácidos, em massa, utilizando vácuo
controlado para deslocamento de equilíbrio, e obtiveram massas molares médias
(𝑀 𝑤 ) de 1500 Da. Outro trabalho dos mesmos autores (Binns, et al., 1998) mostra
que, tanto utilizando monômeros AA e BB como monômeros AB, a polimerização em
massa rende apenas oligômeros.
Via catálise química, Okada e colaboradores (Okada, 1996) sintetizaram
poliésteres de isosorbídeo em massa, utilizando cloretos de ácidos dicarboxílicos,
sob vácuo a 160ºC, obtendo, para o poli(adipato de isosorbila), massa molar de 26
kDa. Braun e Bergmann (Braun, et al., 1992) realizaram policondensações de uma

52
série de cloretos de ácidos dicarboxílicos com isosorbídeo e isomanídeo, obtendo
resultados similares. Devido ao fato de que, no caso ambos os monômeros são
líquidos e que, sob altas temperaturas, a viscosidade do sistema cai drasticamente,
facilitando a agitação, massas molares maiores são observadas.
Até o presente momento, não encontramos na literatura nenhum estudo
tratando de polimerização de isosorbídeo com diésteres alquílicos, tanto por catálise
química, quanto por catálise enzimática, para que haja uma comparação efetiva
entre sistemas similares.
Neste capítulo do trabalho, em uma primeira abordagem, foram realizadas
policondensações em massa (sem a utilização de solventes) para a síntese do
poli(adipato de isosorbila), utilizando adipato de dietila como monômero. Essas
reações foram realizadas de duas formas distintas: sob pressão reduzida e sob
pressão ambiente.
O
O
O
OO
OHO
OH
+O
O
O
OO
OO
CAL-BH
H
*
H
H-EtOH
ESQUEMA 4 - SÍNTESE DO POLI(ADIPATO DE ISOSORBILA), ATRAVÉS DE POLICONDENSAÇÃO EM MASSA. AS
REAÇÕES FORAM REALIZADAS SOB VÁCUO E SOB PRESSÃO AMBIENTE.

53
4.1.1 – POLICONDENSAÇÃO EM MASSA SOB PRESSÃO REDUZIDA:
As polimerizações foram realizadas em massa utilizando vácuo para remoção
dos subprodutos de reação. Num primeiro momento, a reação ocorreu à pressão
ambiente, para que ocorresse a conversão prévia dos monômeros em oligômeros.
Após esse tempo, a reação foi submetida ao vácuo por mais um período de tempo, à
mesma temperatura. Os resultados estão apresentados na Tabela 8.
TABELA 8 - RESULTADOS OBTIDOS NAS REAÇÕES DE POLICONDENSAÇÃO EM MASSA SOB PRESSÃO REDUZIDA.
Tempo
fase A (h)
Tempo
fase B (h)
𝑴 𝒏
(Da)
𝑴 𝒘
(Da) PD
4,5 48 400 630 1,6
24 48 870 1700 1,9
86 36 1100 2100 1,9
Temperatura: 70ºC. Fase A: sob pressão ambiente, fase B: pressão: 2 mmHg. Monômeros: Adipato
de dietila e isosorbídeo, razão molar 1:1, quantidade de enzima: 10% em massa de monômeros.
Fazendo-se uma breve análise dos valores de massas molares obtidos,
percebe-se que massas molares maiores são obtidas quanto maior é o tempo de
oligomerização dos materiais. Uma justificativa razoável para esses dados é que
uma prévia oligomerização transforma os monômeros em moléculas maiores, que
não sofrem evaporação sob vácuo, mesmo com temperaturas maiores. Quando o
tempo da fase A (temperatura ambiente) é menor, quantidades variadas de
isosorbídeo ou adipato de dietila não consumidos podem ser removidos na fase B
(sob vácuo), desequilibrando a equimolaridade dos monômeros no meio,
comprometendo o crescimento da cadeia.

54
4.1.2 - POLICONDENSAÇÃO EM MASSA SOB PRESSÃO AMBIENTE:
Paralelamente a esses estudos, realizou-se, também, a síntese do
poli(adipato de isosorbila), via polimerização em massa, utilizando adipato de dietila
como monômero, mas sem a utilização do vácuo para remoção do etanol. Para que
ocorresse o deslocamento do equilíbrio, submetemos a reação a uma maior
temperatura (85ºC), já que o etanol tem ponto de ebulição igual a 78,2ºC. Utilizou-se
sistema aberto sob fluxo de nitrogênio para facilitar a remoção dos vapores de
etanol. Os resultados obtidos encontram-se na Tabela 9.
TABELA 9 – RESULTADOS OBTIDOS PARA POLICONDENSAÇÕES REALIZADAS SOB PRESSÃO AMBIENTE.
Tempo (h) 𝑴 𝒏 (Da) 𝑴 𝒘 (Da) PD
72 880 2000 2,3
168 980 3800 3,9
Temperatura: 85ºC, sob N2. Monômeros: Adipato de dietila e isosorbídeo, razão molar 1:1,
quantidade de enzima: 10% em massa de monômeros.
Os resultados mostram que, através desse método, é possível se obter
maiores valores de massas molares (𝑀 𝑤 ), apesar do fato de que os valores de
polidispersividade serem altos, o que não é desejável, visto que estudos
comparativos indicam que polimerizações em massa tendem a apresentar valores
de polidispersividade menores que 2, e substancialmente menores do que aquelas
realizadas em solução (Binns, et al., 1998). Uma possível explicação para as
massas molares mais altas em comparação com o caso anterior, é a diminuição da
viscosidade do sistema, devido ao aumento da temperatura.

55
4.1.3 - CONSIDERAÇÕES GERAIS:
Comparando-se os resultados obtidos através das duas técnicas utilizadas,
observa-se que, ao utilizar o sistema aberto e uma maior temperatura, foram obtidas
massas molares maiores. No entanto, aumentam também os índices de
polidispersividade dos materiais.
As duas técnicas resultaram apenas em oligômeros. O principal fator limitante
do crescimento das massas molares, nesse caso, é o fato de que, com o aumento
das massas molares, a viscosidade do sistema aumenta, dificultando a agitação do
meio reacional, e como não é possível um aumento muito grande na temperatura do
sistema, que causaria a desnaturação da enzima, não é possível diminuir a
viscosidade do meio. Melhores resultados poderiam ser obtidos com o uso de
agitação mecânica forçada (alto torque), que facilitaria a continuidade das reações,
porém com grande risco de pulverização do catalisador, dificultando sua remoção.

56
4.2 - POLICONDENSAÇÃO UTILIZANDO MONÔMEROS DIVINÍLICOS:
O
O
O
O
O
OHO
OH
+
CAL-BH
H
O
O
O
OO
OO
**
H
HH
O_n
n= 1 ou 3
ESQUEMA 5 - SÍNTESE DE POLÍMEROS UTILIZANDO DERIVADOS DIVINÍLICOS.
Em outro estudo que foi realizado, utilizamos como monômeros os derivados
divinílicos dos diácidos. O uso de ésteres vinílicos como agentes acilantes em
reações catalisadas por lipases tem se mostrado muito eficiente, já que reação de
transesterificação com estes substratos gera o álcool vinílico, que gera rapidamente
o acetaldeído, no equilíbrio ceto-enólico a que está submetido, deslocando o
equilíbrio para os produtos.
A polimerização enzimática de ésteres divinílicos e glicóis foi demonstrada
pela primeira vez em 1994 por Kobayashi e Uyama (Uyama, et al., 1994), utilizando
Lipase PF, a 45ºC, em éter diisopropílico, obtendo polímeros com massas molares
de 27 kDa, e comparando, sob condições reacionais similares com o uso de ésteres
dietílicos, que não renderam materiais poliméricos, indicando a alta
polimerizabilidade de derivados divinílicos perante um catalisador enzimático. Em
outro trabalho, esses mesmos autores realizaram a polimerização enzimática de
α,ω-glicóis com diferentes tamanhos de cadeias com adipato de divinila e sebacato
de divinila, utilizando diferentes lipases, mostrando a alta atividade catalítica das
enzimas frente a derivados divinílicos. Russel e colaboradores (Chaudhary, et al.,
1997), mostraram que, utilizando CAL-B como catalisador, a polimerização em
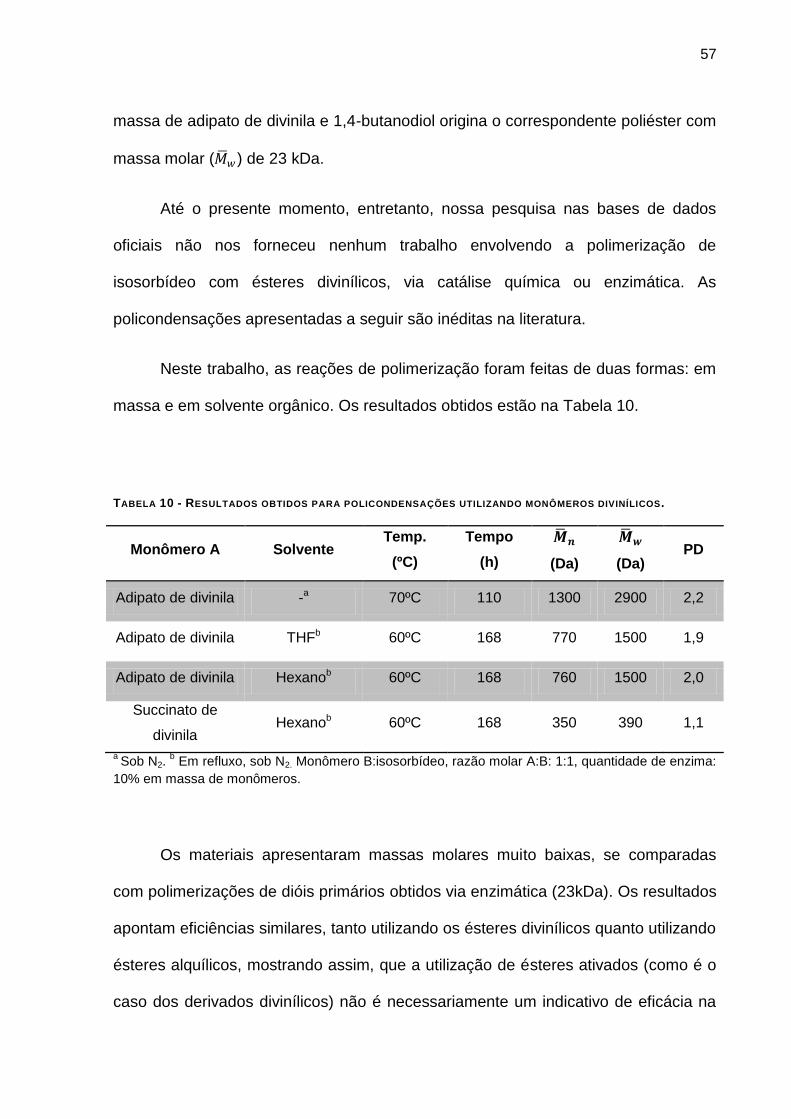
57
massa de adipato de divinila e 1,4-butanodiol origina o correspondente poliéster com
massa molar (𝑀 𝑤 ) de 23 kDa.
Até o presente momento, entretanto, nossa pesquisa nas bases de dados
oficiais não nos forneceu nenhum trabalho envolvendo a polimerização de
isosorbídeo com ésteres divinílicos, via catálise química ou enzimática. As
policondensações apresentadas a seguir são inéditas na literatura.
Neste trabalho, as reações de polimerização foram feitas de duas formas: em
massa e em solvente orgânico. Os resultados obtidos estão na Tabela 10.
TABELA 10 - RESULTADOS OBTIDOS PARA POLICONDENSAÇÕES UTILIZANDO MONÔMEROS DIVINÍLICOS.
Monômero A Solvente Temp.
(ºC)
Tempo
(h)
𝑴 𝒏
(Da)
𝑴 𝒘
(Da) PD
Adipato de divinila -a 70ºC 110 1300 2900 2,2
Adipato de divinila THFb 60ºC 168 770 1500 1,9
Adipato de divinila Hexanob 60ºC 168 760 1500 2,0
Succinato de
divinila Hexanob 60ºC 168 350 390 1,1
a Sob N2.
b Em refluxo, sob N2. Monômero B:isosorbídeo, razão molar A:B: 1:1, quantidade de enzima:
10% em massa de monômeros.
Os materiais apresentaram massas molares muito baixas, se comparadas
com polimerizações de dióis primários obtidos via enzimática (23kDa). Os resultados
apontam eficiências similares, tanto utilizando os ésteres divinílicos quanto utilizando
ésteres alquílicos, mostrando assim, que a utilização de ésteres ativados (como é o
caso dos derivados divinílicos) não é necessariamente um indicativo de eficácia na

58
polimerização, quando se trata de dióis secundários. Assim como no caso das
outras polimerizações em massa realizadas, o fator viscosidade acaba dificultando a
agitação do sistema reacional, impedindo que as cadeias continuem a crescer.
No que diz respeito às policondensações em solução, observa-se que,
mesmo com tempos maiores de reação, as massas molares obtidas são muito
baixas.
Através dessas técnicas, assim como nas polimerizações em massa
utilizando derivados dietílicos, obtivemos apenas oligômeros. Além do relativo
insucesso nos resultados obtidos, alguns outros fatores contribuíram para o
abandono dessa linha de experimentos, como: (i) a dificuldade na preparação dos
monômeros divinílicos, com baixos rendimentos e difícil purificação, (ii) a
instabilidade destes frente às temperaturas mais altas, e (iii) o maior custo relativo
que o uso desses monômeros agrega à síntese dos polímeros, o que limitaria o
impacto tecnológico potencial desses materiais (Mahapatro, et al., 2003).

59
4.3 - POLICONDENSAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA:
RO
O
O
OR
O
OHO
OH
+O
O
O
OO
OCAL-B
H
H
R = CH2CH3 ou HO**
H
H
- ROHn n= 1 ou 3
Dean -Stark
Solvente
ESQUEMA 6 – S ÍNTESE DE POLÍMEROS EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA.
A destilação azeotrópica é um método amplamente utilizado em reações de
esterificação e transesterificação para deslocamento do equilíbrio reacional na
direção dos produtos. Muitos ácidos dicarboxílicos e seus ésteres alquílicos estão
disponíveis comercialmente. Entretanto, eles frequentemente mostram baixa
reatividade perante as lipases. Por isso, o desenvolvimento de equipamentos para
criar condições favoráveis para que a polimerização ocorra é muito importante para
a obtenção de polímeros com altos valores de massas molares. Por exemplo, Binns
e colaboradores (Binns, et al., 1993) (Binns, et al., 1993) desenvolveram um
equipamento que possibilitou o uso de peneiras moleculares para a remoção de
água na síntese de poliésteres de 1,4-butanodiol e ácido adípico, utilizando lipase de
Mucor miehei (MM), obtendo poliésteres com grau de polimerização de
aproximadamente 20.
Para a utilização deste processo, a primeira condição que um solvente a ser
utilizado deve atender é que este precisa formar um azeótropo conhecido com o
subproduto a ser removido do sistema (água ou etanol, que são os subprodutos de
reação quando usamos o ácido ou o derivado dietílico, respectivamente). Neste
trabalho, as reações foram realizadas utilizando sistemas Dean-Stark adaptados,

60
preenchidos com peneiras moleculares (Binns, et al., 1998) para remoção de
subprodutos de reação pela formação de azeótropos conhecidos (Tabelas 6 e 7)
Alguns autores vêm desenvolvendo estudos de polimerização por catálise
enzimática em solução, estabelecendo quadros comparativos entre diferentes tipos
de solventes. Alguns exemplos dos efeitos do solvente na síntese de poliésteres:
Shuai e colaboradores (Shuai, et al., 1999) publicaram a policondensação catalisada
por PPL de ácido 3-hidróxibutírico e ácido 12-hidróxidodecanóico em éter dietílico à
temperatura ambiente e em tolueno a temperaturas de 55 e 75ºC obtendo massas
molares (𝑀 𝑤 ) de 230 e 2900, quando se utilizou éter dietílico e tolueno,
respectivamente. Gross e colaboradores (Mahapatro, et al., 2003) realizaram
policondensações catalisadas por CAL-B de dióis primários e diácidos em solventes
com alto ponto e ebulição, obtendo massas molares mais altas quando se utilizou
éter difenílico como solvente (𝑀 𝑤= 27700).
É conhecido o fato de que o meio reacional é um dos fatores mais
importantes da catálise enzimática (Dordick, 1989). Estudos indicam que, com uma
escolha prudente de solventes, pode-se manipular a reação catalisada por enzimas
em um meio não-aquoso (Zaks, 1988). A propriedade do solvente que pode ser
analisada aqui é a sua hidrofobicidade, expressa como log P, onde P é o coeficiente
de partição entre o 1-octanol e a água. Geralmente é aceito que a atividade da
lipase é maior quanto maior o valor de log P do solvente (Kumar, 2000).
Laane e colaboradores (Laane, et al., 1987) classificaram os solventes em
três grupos, de acordo com os valores de log P (Tabela 11): no primeiro grupo estão
os solventes com log P menor que 2. Gutman e colaboradores (Gutman, 1993)
afirmam que nesse grupo, os solventes não produziriam polímeros de maiores

61
massas molares do que se as reações fossem realizadas por polimerização em
massa. Uma explicação para esse fato seria a aparente inativação da enzima que
aconteceria devido às mudanças conformacionais induzidas pelas interações entre o
solvente polar e a enzima. Esses solventes, mais hidrofílicos, retiram a camada
d’água necessária para que a enzima mantenha sua configuração, fazendo com que
a enzima perca sua atividade. O segundo grupo de solventes, com log P entre 2 e 4,
possuiriam propriedades intermediárias e levariam a melhores resultados. O terceiro
grupo de solventes, que possuem log P maior que 4, cuja utilização resultaria nas
maiores massas molares obtidas e nas maiores taxas de conversão dos monômeros
em polímeros, devido a um menor efeito sobre a conformação da enzima, deixando-
a em um estado ativo.
Dong e colaboradores (Dong, et al., 1999) estudaram a polimerização por
abertura de anel da ε-caprolactona a 37ºC por 240 h em uma série de solventes,
utilizando lipase PS como catalisador. Polimerizações enzimáticas em solventes com
log P < 2 (THF, clorofórmio, 1,2-dicloroetano) resultaram em baixos graus de
conversão e massas molares baixas. Por outro lado, para solventes com log P entre
2 e 4 (benzeno, tolueno, éter diisopropílico e cicloexano), os graus de conversão e
as massas molares foram significativamente maiores.

62
TABELA 11 - VALORES DE LOG P PARA DIFERENTES SOLVENTES.
Grupo Solvente log P
1
Acetonitrila -0,33
Acetona -0,23
THF 0,49
MEK 0,86
Diclorometano 1,01
2
Clorofórmio 2,0
Benzeno 2,0
Tolueno 2,5
Tetracloreto de Carbono 2,86
Cicloexano 3,2
n-Hexano 3,5
3 n-Heptano 4,0
Difenil éter 4,3
Nossos dados parecem confirmar em parte esta observação onde, apesar de
não obtermos os valores de log P para as misturas, a presença de cicloexano
parece conduzir a maiores valores de 𝑀 𝑤 e 𝑀 𝑛 . Comparando os valores de massas
molares obtidos com diversos solventes (Tabela 12), observou-se que os melhores
resultados foram obtidos quando se utilizou misturas de cicloexano e benzeno como
solventes, onde o cicloexano, que tem um alto valor de log P, está em maiores
proporções.

63
TABELA 12 - RESULTADOS OBTIDOS PARA POLIMERIZAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA.
Monômero A Solvente Temp. (ºC) 𝑴 𝒏 (Da) 𝑴 𝒘 (Da) PD
Ácido adípico
Benzeno 80 210 230 1,1
Clorofórmio 75 250 420 1,7
MEK: hexano 4:1 75 1100 2700 2,5
Adipato de
dietila
Acetona:
Clorofórmio 4:1 75 390 480 1,2
Benzeno 75 710 760 1,1
Clorofórmio 75 560 820 1,5
Clorofórmio:
hexano 3:2 75 920 2600 2,8
Diclorometano 75 860 900 1,0
n-Hexano 75 3200 6400 2,0
n-Heptano 90 240 420 1,7
Tetracloreto de
Carbono 75 380 430 1,7
Cicloexano 85 8100 21000 2,6
Cicloexano:
Benzeno 4:1 85 13000 31000 2,4
Cicloexano:
Benzeno 6:1 85 17000 37000 2,2
Succinato de
dietila
Acetona:
Clorofórmio 4:1 75 300 330 1.1
n-Hexano 75 600 1800 3,0
Tempo: 168h. Sistemas Dean-Stark com peneiras moleculares. Monômero B: isosorbídeo. Razão
molar A:B: 1:1. Quantidade de enzima: 10% em massa de monômeros.
Muitos esforços têm sido feitos para generalizar como substâncias orgânicas
interagem com as moléculas da enzima e como elas influenciam na estabilidade
enzimática. A hipótese corrente (Zaks, 1988) é que, quando uma enzima é colocada
em um solvente orgânico hidrofóbico, é cineticamente (mas não
termodinamicamente) “congelada” em sua conformação nativa, preservando a
camada d’água essencial para manter sua atividade. Obviamente, a atividade ainda

64
é menor do que quando se utiliza a enzima em meio aquoso, devido à perda da
flexibilidade que ocorre no “congelamento” da enzima, mas esse enrijecimento tem a
vantagem de proporcionar uma maior termoestabilidade à enzima (Zaks, 1984).
A Figura 3 mostra, comparativamente, os resultados obtidos utilizando essa
técnica. Podemos observar uma grande vantagem quando se utiliza cicloexano e em
sistemas de solvente contendo cicloexano em maiores proporções.
FIGURA 3 - RESULTADOS OBTIDOS ATRAVÉS DA POLIMERIZAÇÃO EM SOLUÇÃO COM DESTILAÇÃO AZEOTRÓPICA.
0 10000 20000 30000 40000
Benzeno
Clorofórmio
MEK: hexano 4:1
Acetona: Clorofórmio 4:1
Benzeno
Clorofórmio
Clorofórmio: hexano 3:2
Diclorometano
n-Hexano
n-Heptano
Tetracloreto de Carbono
Cicloexano
Cicloexano: Benzeno 4:1
Cicloexano: Benzeno 6:1
Acetona: Clorofórmio 4:1
n-Hexano
Ácid
o a
díp
ico
Ad
ipa
to d
e d
ietila
Succin
ato
de d
ietila
230
420
2700
480
760
820
2600
900
6400
420
430
21000
31000
37000
330
1800
Mo
nô
me
ro A
Tempo de reação:168hTemperatura: 75-85ºCEnzima: CAL-B N-435 -10% emmassa de monômerosMonômero B: Isosorbídeorazão A:B: 1:1
Mw
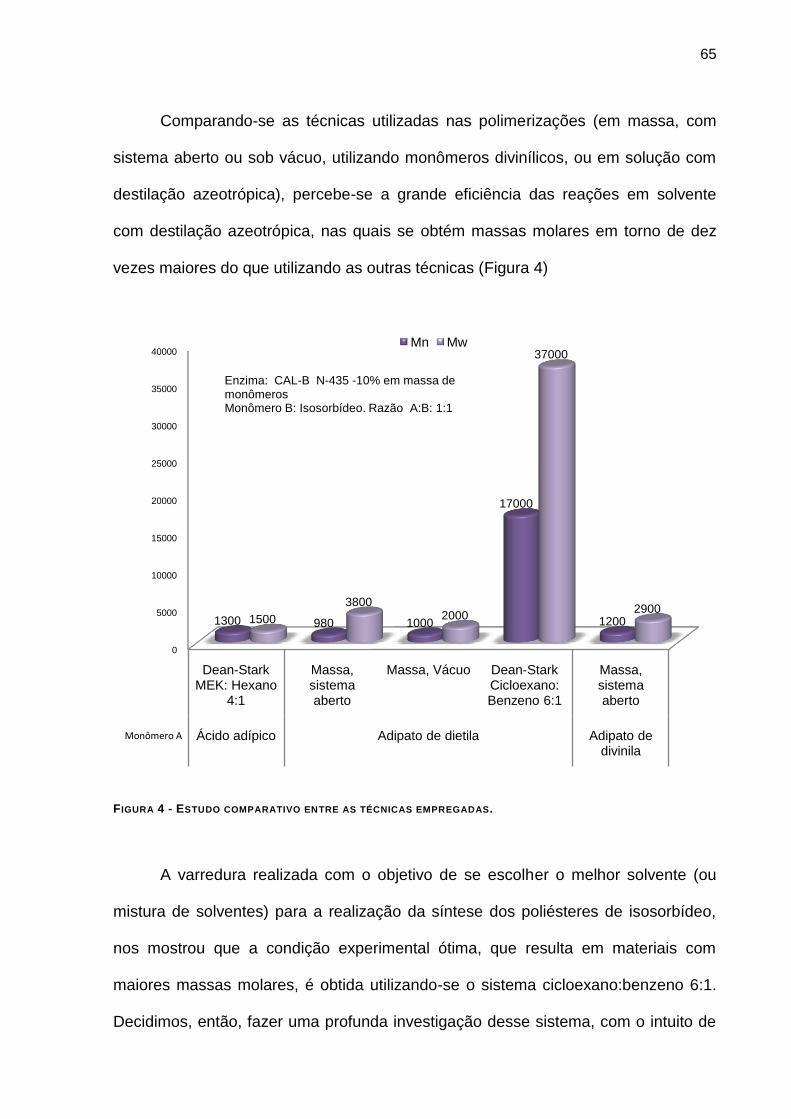
65
Comparando-se as técnicas utilizadas nas polimerizações (em massa, com
sistema aberto ou sob vácuo, utilizando monômeros divinílicos, ou em solução com
destilação azeotrópica), percebe-se a grande eficiência das reações em solvente
com destilação azeotrópica, nas quais se obtém massas molares em torno de dez
vezes maiores do que utilizando as outras técnicas (Figura 4)
FIGURA 4 - ESTUDO COMPARATIVO ENTRE AS TÉCNICAS EMPREGADAS.
A varredura realizada com o objetivo de se escolher o melhor solvente (ou
mistura de solventes) para a realização da síntese dos poliésteres de isosorbídeo,
nos mostrou que a condição experimental ótima, que resulta em materiais com
maiores massas molares, é obtida utilizando-se o sistema cicloexano:benzeno 6:1.
Decidimos, então, fazer uma profunda investigação desse sistema, com o intuito de
0
5000
10000
15000
20000
25000
30000
35000
40000
Dean-Stark MEK: Hexano
4:1
Massa, sistema aberto
Massa, Vácuo Dean-Stark Cicloexano: Benzeno 6:1
Massa, sistema aberto
Ácido adípico Adipato de dietila Adipato de divinila
1300 980 1000
17000
12001500
38002000
37000
2900
Mn Mw
Monômero A
Enzima: CAL-B N-435 -10% em massa de monômerosMonômero B: Isosorbídeo. Razão A:B: 1:1

66
aperfeiçoar o método, bem como estabelecer uma comparação entre sistemas que
utilizam cicloexano puro e em mistura com benzeno e entender qual o papel do
benzeno nas misturas. O estudo completo está descrito na seção 4.4.
4.3.1 - POLIMERIZAÇÃO EM SOLUÇÃO SOB PRESSÃO REDUZIDA.
Ainda na tentativa de realizar polimerizações utilizando um solvente com alto
valor de hidrofobicidade (grupo 3), foram feitas policondensações em solução
utilizando éter difenílico como solvente. Primeiramente, as reações foram
conduzidas sob pressão ambiente e, em um segundo momento, sob vácuo, para
remoção do etanol do sistema. Os resultados (Tabela 13) mostram que a
polimerização não foi possível para polímeros derivados de isosorbídeo utilizando
esse solvente, apesar de diversos trabalhos terem sido publicados com excelentes
resultados na polimerização de dióis primários em éter difenílico, via catálise
enzimática (Linko, 1995; Mahapatro, et al., 2003; Azim, 2006).
TABELA 13 - POLIMERIZAÇÃO EM SOLUÇÃO SOB PRESSÃO REDUZIDA.
Monômero
A
Tempo
Fase A Tempo
Fase B
Tempo
Fase C 𝑴 𝒏 (Da) 𝑴 𝒘 (Da) PD
Adipato de
dietila 24 72 6 430 480 1,1
Succinato de
dietila 24 72 6 260 270 1,0
Solvente: Difenil éter. Fase A: pressão ambiente, temp: 70ºC Fase B: pressão: 3 mmHg, temp. 70ºC
Monômero B: isosorbídeo. Razão molar A:B: 1:1. Quantidade de enzima: 10% em massa de
monômeros.

67
4.4 - INVESTIGAÇÃO DO SISTEMA CICLOEXANO:BENZENO.
4.4.1 – ESTUDO COMPARATIVO ENTRE SISTEMAS UTILIZANDO CICLOEXANO E
MISTURAS CICLOEXANO:BENZENO 6:1.
Com o objetivo de compararmos as possíveis vantagens do uso das misturas
cicloexano e benzeno com relação ao cicloexano puro, uma série de seis ésteres
etílicos de ácidos dicarboxílicos foi testada tanto com o solvente puro quanto com a
mistura de solventes. As reações foram realizadas a uma temperatura de 85ºC,
durante 7 dias. Os ésteres utilizados foram: succinato de dietila, glutarato de dietila,
adipato de dietila, suberato de dietila, sebacato de dietila e dodecanodioato de
dietila, que são ésteres dos diácidos de 4, 5, 6, 8, 10 e 12 carbonos
respectivamente. Os resultados são apresentados a seguir:
A Tabela 14 e a Tabela 15 mostram os resultados obtidos quando se utilizou
cicloexano e cicloexano:benzeno 6:1, respectivamente, como solventes:
TABELA 14 - POLÍMEROS DA SÉRIE DE ÉSTERES DOS DIÁCIDOS CARBOXÍLICOS EM CICLOEXANO.
Monômero A 𝑴 𝒏 𝑴 𝒘 PD GPw Rendimentoa
Succinato de dietila 1600 3000 1,9 13 71%
Glutarato de dietila 9300 16000 1,7 67 56%
Adipato de dietila 8100 21000 2,5 81 74%
Suberato de dietila 3900 10000 2,5 35 61%
Sebacato de dietila 3900 9000 2,3 29 51%
Dodecanodioato de dietila 11000 21000 2,0 63 75%
a – Rendimento após purificação. Temperatura do banho: 85ºC. Tempo:168h. Solvente: cicloexano. Monômero
B: isosorbídeo. Razão molar A:B: 1:1. Quantidade de enzima: 10% em massa de monômeros.
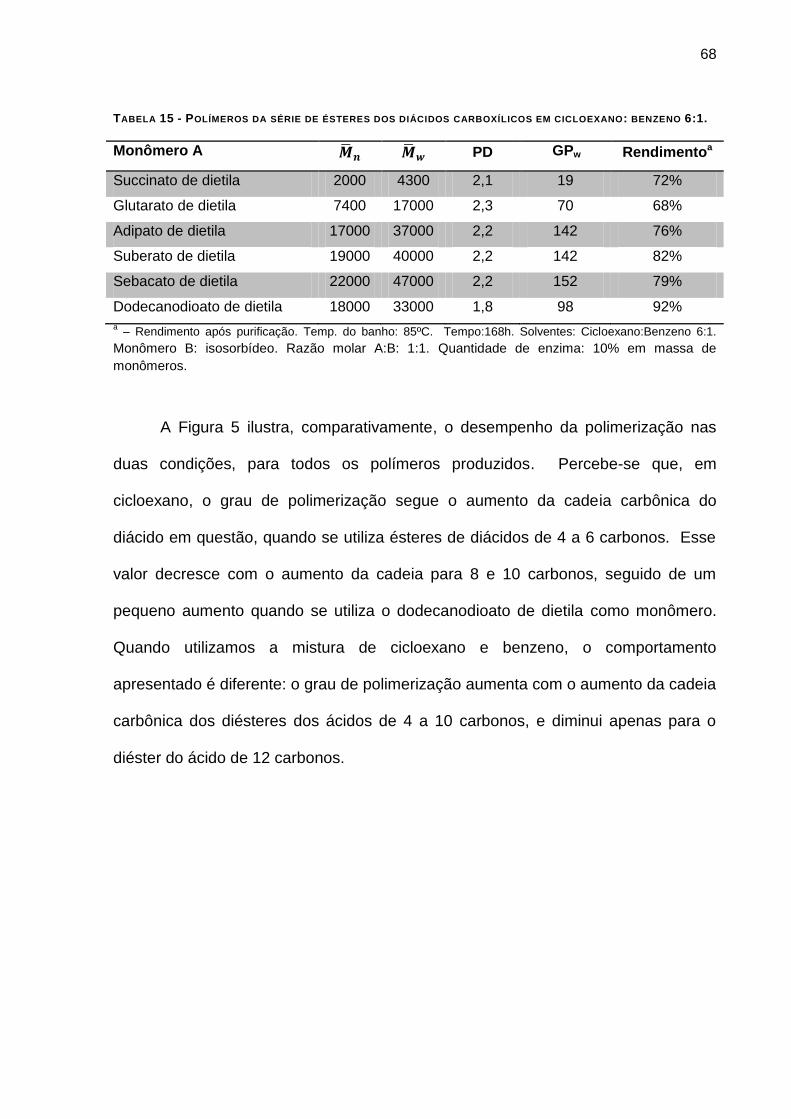
68
TABELA 15 - POLÍMEROS DA SÉRIE DE ÉSTERES DOS DIÁCIDOS CARBOXÍLICOS EM CICLOEXANO: BENZENO 6:1.
Monômero A 𝑴 𝒏 𝑴 𝒘 PD GPw Rendimentoa
Succinato de dietila 2000 4300 2,1 19 72%
Glutarato de dietila 7400 17000 2,3 70 68%
Adipato de dietila 17000 37000 2,2 142 76%
Suberato de dietila 19000 40000 2,2 142 82%
Sebacato de dietila 22000 47000 2,2 152 79%
Dodecanodioato de dietila 18000 33000 1,8 98 92%
a – Rendimento após purificação. Temp. do banho: 85ºC. Tempo:168h. Solventes: Cicloexano:Benzeno 6:1.
Monômero B: isosorbídeo. Razão molar A:B: 1:1. Quantidade de enzima: 10% em massa de
monômeros.
A Figura 5 ilustra, comparativamente, o desempenho da polimerização nas
duas condições, para todos os polímeros produzidos. Percebe-se que, em
cicloexano, o grau de polimerização segue o aumento da cadeia carbônica do
diácido em questão, quando se utiliza ésteres de diácidos de 4 a 6 carbonos. Esse
valor decresce com o aumento da cadeia para 8 e 10 carbonos, seguido de um
pequeno aumento quando se utiliza o dodecanodioato de dietila como monômero.
Quando utilizamos a mistura de cicloexano e benzeno, o comportamento
apresentado é diferente: o grau de polimerização aumenta com o aumento da cadeia
carbônica dos diésteres dos ácidos de 4 a 10 carbonos, e diminui apenas para o
diéster do ácido de 12 carbonos.
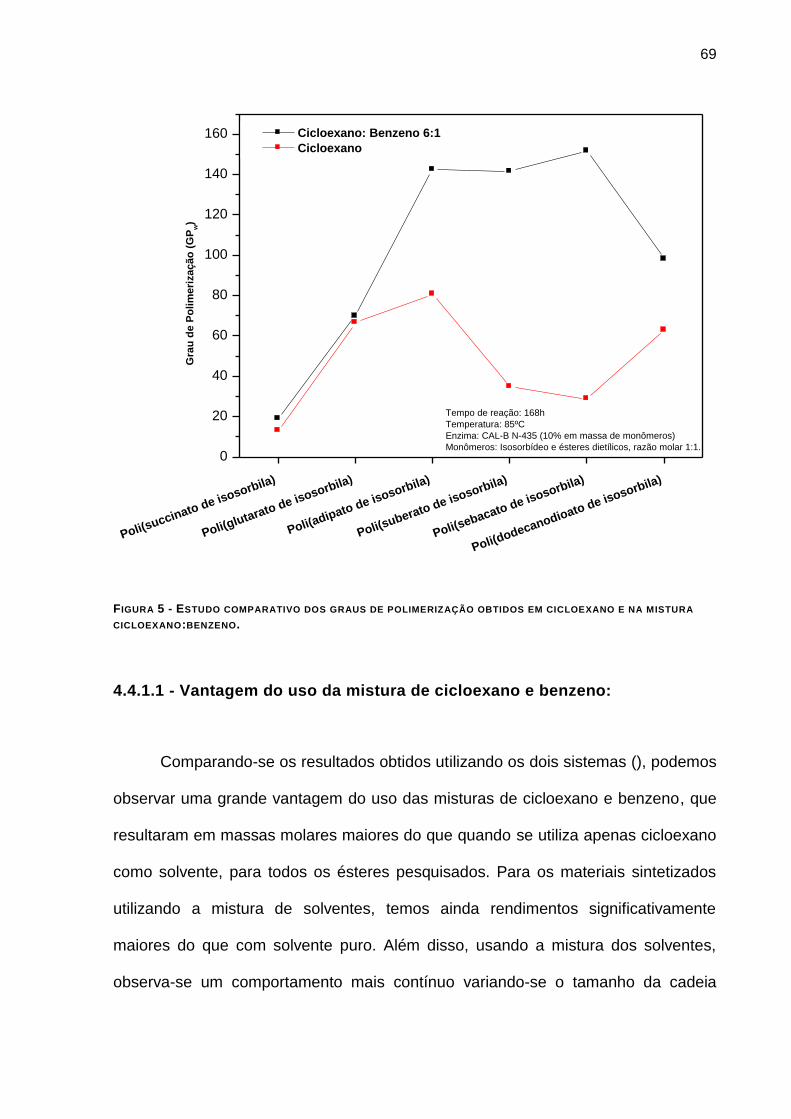
69
Poli(succinato de isosorbila)
Poli(glutarato de isosorbila)
Poli(adipato de isosorbila)
Poli(suberato de isosorbila)
Poli(sebacato de isosorbila)
Poli(dodecanodioato de isosorbila)
0
20
40
60
80
100
120
140
160
Tempo de reação: 168h
Temperatura: 85ºC
Enzima: CAL-B N-435 (10% em massa de monômeros)
Monômeros: Isosorbídeo e ésteres dietílicos, razão molar 1:1.
Gra
u d
e P
olim
eri
zação
(G
Pw)
Cicloexano: Benzeno 6:1
Cicloexano
FIGURA 5 - ESTUDO COMPARATIVO DOS GRAUS DE POLIMERIZAÇÃO OBTIDOS EM CICLOEXANO E NA MISTURA
CICLOEXANO:BENZENO.
4.4.1.1 - Vantagem do uso da mistura de cicloexano e benzeno:
Comparando-se os resultados obtidos utilizando os dois sistemas (), podemos
observar uma grande vantagem do uso das misturas de cicloexano e benzeno, que
resultaram em massas molares maiores do que quando se utiliza apenas cicloexano
como solvente, para todos os ésteres pesquisados. Para os materiais sintetizados
utilizando a mistura de solventes, temos ainda rendimentos significativamente
maiores do que com solvente puro. Além disso, usando a mistura dos solventes,
observa-se um comportamento mais contínuo variando-se o tamanho da cadeia
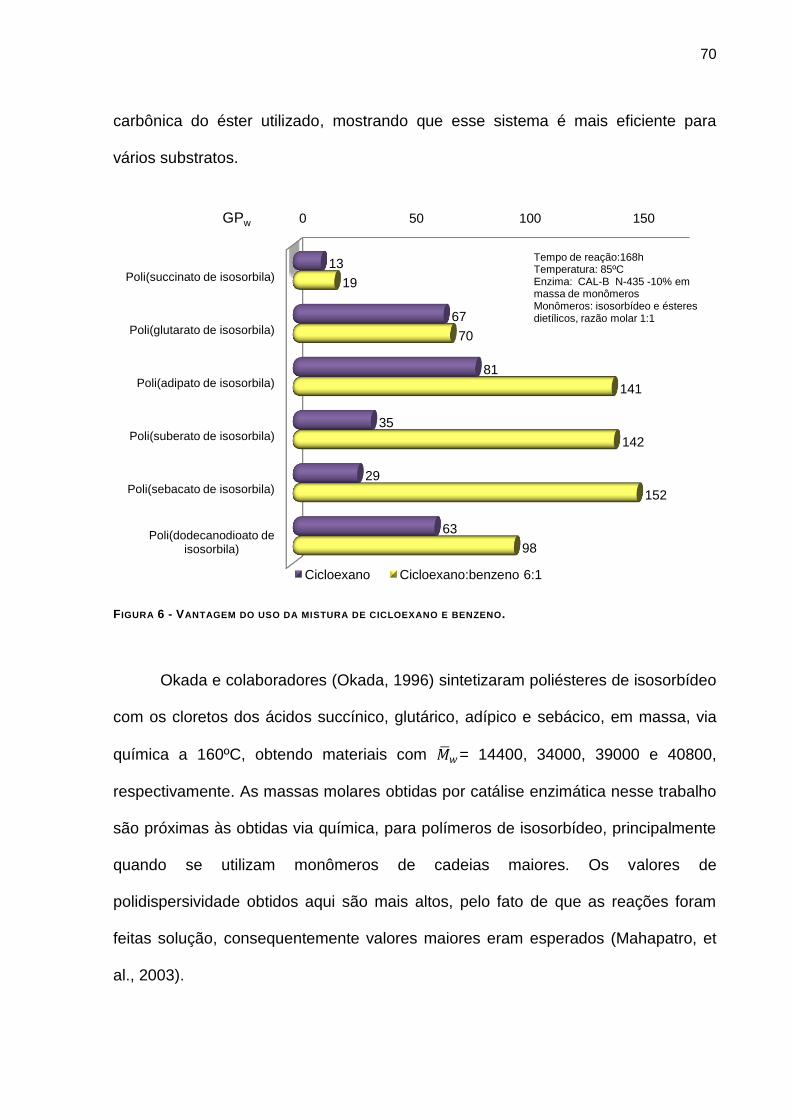
70
carbônica do éster utilizado, mostrando que esse sistema é mais eficiente para
vários substratos.
FIGURA 6 - VANTAGEM DO USO DA MISTURA DE CICLOEXANO E BENZENO.
Okada e colaboradores (Okada, 1996) sintetizaram poliésteres de isosorbídeo
com os cloretos dos ácidos succínico, glutárico, adípico e sebácico, em massa, via
química a 160ºC, obtendo materiais com 𝑀 𝑤= 14400, 34000, 39000 e 40800,
respectivamente. As massas molares obtidas por catálise enzimática nesse trabalho
são próximas às obtidas via química, para polímeros de isosorbídeo, principalmente
quando se utilizam monômeros de cadeias maiores. Os valores de
polidispersividade obtidos aqui são mais altos, pelo fato de que as reações foram
feitas solução, consequentemente valores maiores eram esperados (Mahapatro, et
al., 2003).
0 50 100 150
Poli(succinato de isosorbila)
Poli(glutarato de isosorbila)
Poli(adipato de isosorbila)
Poli(suberato de isosorbila)
Poli(sebacato de isosorbila)
Poli(dodecanodioato de isosorbila)
13
67
81
35
29
63
19
70
141
142
152
98
Cicloexano Cicloexano:benzeno 6:1
GPw
Tempo de reação:168hTemperatura: 85ºCEnzima: CAL-B N-435 -10% em massa de monômerosMonômeros: isosorbídeo e ésteres dietílicos, razão molar 1:1

71
Através da catálise enzimática, já foi demonstrado por Gross e colaboradores
(Mahapatro, et al., 2003), que diácidos de cadeias carbônicas maiores (C6 e C8)
apresentam reatividades mais altas do que cadeias menores (C4 e C5). Nossos
resultados concordam com esses estudos, pois utilizando diácidos de C4 a C6, em
cicloexano, o mesmo comportamento é observado, onde o grau de polimerização
aumenta com o aumento da cadeia carbônica. No caso da mistura
cicloexano:benzeno, a tendência é observada de C4 a C10.
4.4.2 – ESTUDO DO CRESCIMENTO DA MASSA MOLAR EM FUNÇÃO DO TEMPO DE
REAÇÃO:
Tendo como principal objetivo estabelecer o tempo ótimo de reação, realizou-
se um estudo do aumento da massa molar do poli(adipato de isosorbila) em função
do tempo de reação. As reações foram realizadas em cicloexano:benzeno 6:1, em
banho de óleo a 85ºC.
TABELA 16 - ESTUDO DO CRESCIMENTO DA MASSA MOLAR DO POLI(ADIPATO DE ISOSORBILA) EM FUNÇÃO DO
TEMPO DE REAÇÃO.
Tempo (h) 𝑴 𝒏 𝑴 𝒘 PD Rendimentoa
12 1700 5000 2,9 60%
24 1800 5000 2,8 68%
48 1800 6000 3,3 74%
72 2800 11000 3,7 78%
96 5500 18000 3,3 78%
120 8100 19000 2,4 81%
144 10000 25000 2,4 84%
168 14000 32000 2,3 87%
192 14000 33000 2,3 87% a – Rendimento após purificação. Temperatura do banho: 85ºC. Solventes: Cicloexano:benzeno 6:1.
Monômeros: adipato de dietila e isosorbídeo. Razão molar 1:1. Quantidade de enzima: 10% em
massa de monômeros.
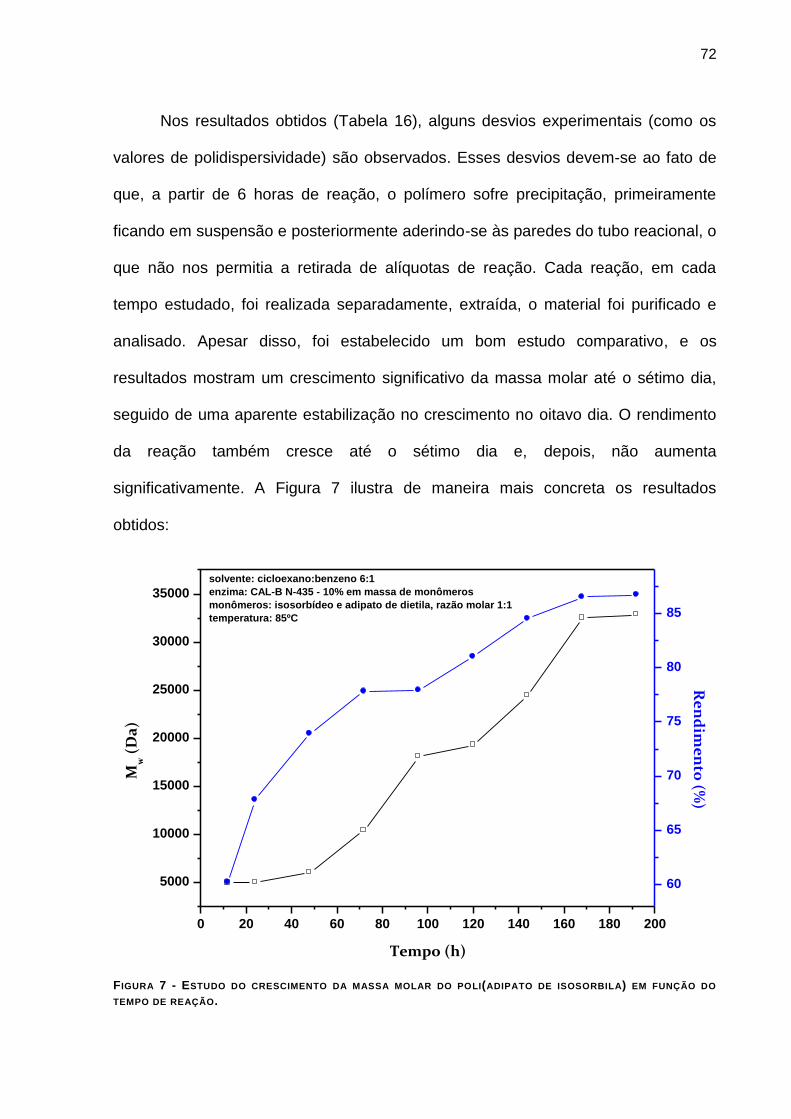
72
Nos resultados obtidos (Tabela 16), alguns desvios experimentais (como os
valores de polidispersividade) são observados. Esses desvios devem-se ao fato de
que, a partir de 6 horas de reação, o polímero sofre precipitação, primeiramente
ficando em suspensão e posteriormente aderindo-se às paredes do tubo reacional, o
que não nos permitia a retirada de alíquotas de reação. Cada reação, em cada
tempo estudado, foi realizada separadamente, extraída, o material foi purificado e
analisado. Apesar disso, foi estabelecido um bom estudo comparativo, e os
resultados mostram um crescimento significativo da massa molar até o sétimo dia,
seguido de uma aparente estabilização no crescimento no oitavo dia. O rendimento
da reação também cresce até o sétimo dia e, depois, não aumenta
significativamente. A Figura 7 ilustra de maneira mais concreta os resultados
obtidos:
0 20 40 60 80 100 120 140 160 180 200
5000
10000
15000
20000
25000
30000
35000
Tempo (h)
Mw (
Da
)
60
65
70
75
80
85
solvente: cicloexano:benzeno 6:1
enzima: CAL-B N-435 - 10% em massa de monômeros
monômeros: isosorbídeo e adipato de dietila, razão molar 1:1
temperatura: 85ºC
Re
nd
ime
nto
(%)
FIGURA 7 - ESTUDO DO CRESCIMENTO DA MASSA MOLAR DO POLI(ADIPATO DE ISOSORBILA) EM FUNÇÃO DO
TEMPO DE REAÇÃO.

73
Esses resultados nos permitiram concluir que sete dias (168 h) podem ser
considerados como tempo ótimo para as reações em cicloexano:benzeno.
De acordo com Sheldon (Sheldon, 1996), uma justificativa para o
crescimento limitado de polímeros produzidos por policondensação via catálise
enzimática, que representa uma séria limitação para o método, é o aparecimento de
limitações difusionais com o aumento da massa molar do polímero. A enzima deve
gerar continuamente o intermediário acil-enzima, através da reação com a função
éster terminal do polímero em crescimento. A eficiência desse processo cai
rapidamente conforme aumenta a massa molar do polímero, porque a difusibilidade
do material no sítio da enzima decresce, limitando o crescimento do polímero.
4.4.3 – TROCA DE BENZENO POR TOLUENO NA MISTURA:
Devido à sua alta toxicidade (Ferreira da Costa, 2002), o uso do benzeno é
evitado em todo o mundo. Embora tenhamos obtido excelentes resultados utilizando
pequenas quantidades desse solvente, ainda assim um estudo foi realizado trocando
benzeno por tolueno, pois este apresenta uma toxicidade muito menor, na mesma
porporção (6:1) na mistura, na tentativa de obtermos massas molares análogas
àquelas obtidas utilizando benzeno. A hipótese levantada era que, apesar de não
formar um azeótropo conhecido com o cicloexano e o etanol (o azeótropo destilado,
no caso, seria o binário de cicloexano e etanol), o tolueno, que possui um valor de
log P ainda maior que o benzeno (Tabela 11), e estrutura muito parecida com este,
deveria apresentar um comportamento, frente à enzima, similar ou superior ao do
benzeno. De fato, diversos trabalhos, como os de Mahapatro e colaboradores

74
(Mahapatro, et al., 2003), Kumar e Gross (Kumar, 2000), e de Binns e colaboradores
(Binns, et al., 1998), mostram a eficiência da utilização de tolueno como solvente em
reações de polimerização via catálise enzimática, o que motivou ainda mais este
estudo.
As reações foram realizadas utilizando dois diésteres diferentes: adipato de
dietila e sebacato de dietila. Os resultados estão apresentados na Tabela 17.
TABELA 17 - RESULTADOS OBTIDOS COM A TROCA DE TOLUENO POR BENZENO NA MISTURA DE SOLVENTES.
Polímero 𝑴 𝒏 𝑴 𝒘 PD Rendimentoa
Poli(adipato de isosorbila) 9000 20000 2,3 78%
Poli(sebacato de isosorbila) 8400 18000 2,1 71%
a – Rendimento após purificação. Temp. do banho: 90ºC. Tempo:168h. Solventes: Cicloexano:Tolueno 6:1.
Monômeros: adipato de dietila ou sebacato de dietila e isosorbídeo. Razão molar 1:1. Quantidade de
enzima: 10% em massa de monômeros.
FIGURA 8 - TROCA DE BENZENO POR TOLUENO NA MISTURA DE SOLVENTES.
0
10000
20000
30000
40000
50000
Poli(adipato de isosorbila) Poli(sebacato de isosorbila)
37000
47000
20000 1800021000
9000Mw(D
a)
Cicloexano: benzeno 6:1 Cicloexano: tolueno 6:1 Cicloexano
Tempo de reação:168hEnzima: CAL-B N-435 - 10% em massa de monômerosMonômeros: isosorbídeo e adipato ou sebacato de dietila, razão molar:1:1.
(85ºC) (90ºC) (85ºC)

75
Comparando os resultados obtidos com as massas molares obtidas quando
se utiliza cicloexano puro e a mistura de cicloexano e benzeno percebe-se que, no
caso, em se tratando de aumento de massa molar, não há vantagem na troca dos
solventes nas misturas, pois os valores obtidos são muito menores do que quando a
mistura com benzeno é utilizada. Apesar disso, a mistura cicloexano:tolueno rendeu
resultados similares ou melhores (no caso do poli(sebacato de isosorbila)) do que
quando se utilizou cicloexano puro. Isso nos permite concluir que, mesmo que a
polimerização utilizando tolueno na mistura seja viável, os resultados ainda são
muito inferiores aos obtidos com a utilização do sistema cicloexano:benzeno.
4.4.4 - AVALIAÇÃO DAS MASSAS MOLARES QUANTO AO ÁLCOOL DE SAÍDA.
Ainda no intuito entender como funciona o sistema cicloexano:benzeno,
realizou-se um estudo para se avaliar as massas molares em função do álcool de
saída, ou seja, variando o tamanho da cadeia carbônica da porção éster terminal
dos monômeros (ésteres do ácido adípico).
Para esse estudo, foram realizadas policondensações utilizando os adipatos
de metila, etila, n-propila e n-butila. Os resultados encontram-se na Tabela 18.

76
TABELA 18 - MASSAS MOLARES DO POLI(ADIPATO DE ISOSORBILA) OBTIDAS VARIANDO-SE O ÁLCOOL DE SAÍDA.
Álcool 𝑴 𝒏 𝑴 𝒘 PD Rendimentoa
Metanolb 11000 28000 2,6 79%
Etanolc 17000 37000 2,2 75%
n-Propanold 2600 4800 1,8 59%
n-Butanole 330 460 1,4 30%
a – Rendimento após purificação. Temp. do banho: 85-90ºC. Tempo:168h. Solventes: Cicloexano:Benzeno 6:1.
Alcoóis provenientes dos adipatos de bdimetila ,
cdietila,
ddi-n-propila e
edi-n-butila, nas transesterificações com
isosorbídeo, razão molar 1:1. Enzima: CAL-B N-435, 10% em massa de monômeros.
A Figura 9 ilustra comparativamente os resultados obtidos. Fazendo uma
breve análise dos resultados, observa-se que, utilizando os ésteres de n-propila e n-
butila, os valores de massas molares são muito baixos e, no caso do éster de butila
não houve polimerização. Já utilizando os ésteres de metila e etila, obtivemos
polímeros de massas molares consideráveis.
Alguns fatores devem ser considerados aqui. Primeiramente, levemos em
consideração o azeótropo formado (Tabela 19): quando o azeótropo ternário n-
butanol/cicloexano/benzeno é formado, temos uma temperatura de ebulição de
aproximadamente 77ºC, e apenas 4% do álcool na mistura. Quando se trata do
azeótropo n-propanol/cicloexano/benzeno, a composição do azeótropo é muito
parecida, com baixas quantidades do álcool de saída, porém, com uma temperatura
de ebulição cerca de 4ºC menor do que o azeótropo com o n-butanol. Esse fato
pode explicar a maior eficiência na remoção do n-propanol do meio reacional, pois
este azeótropo necessita de menos energia para ser removido do sistema. Assim,
pode-se concluir que a transesterificação utilizando ésteres de n-propila e n-butila
não resulta em valores significativos de massas molares, porque a remoção do
álcool de saída é muito baixa nestes casos. Ainda, outro fator a ser levado em conta,
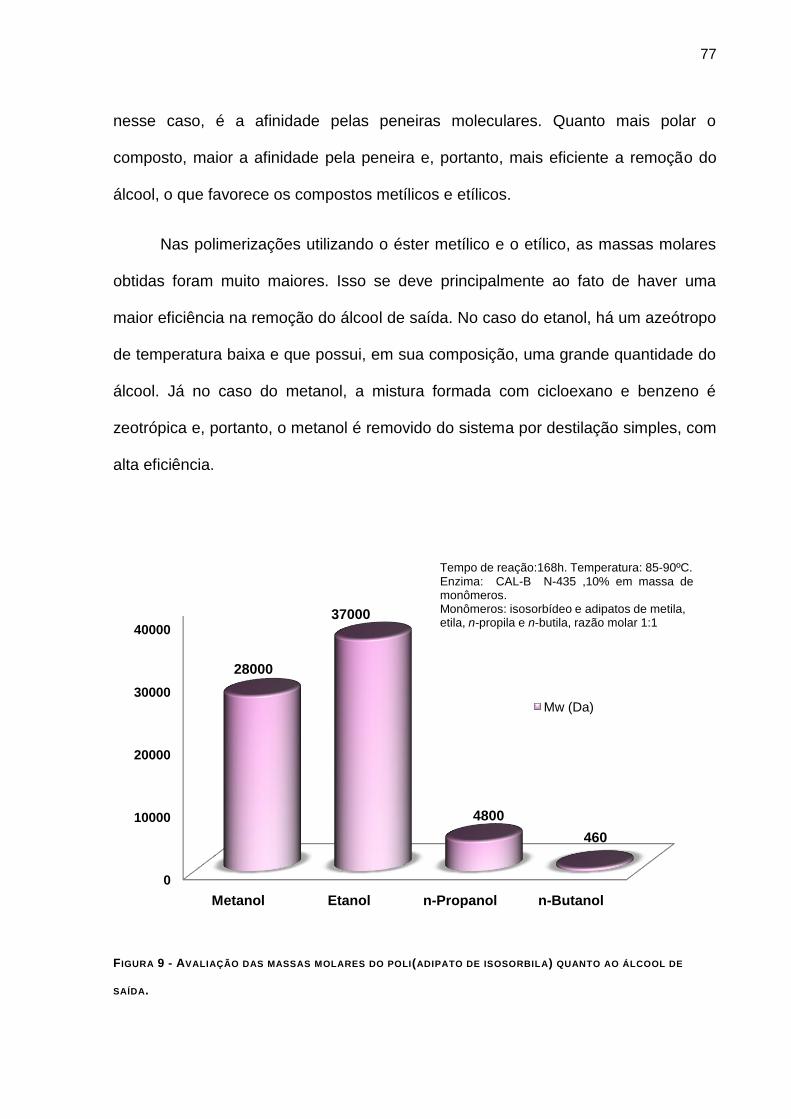
77
nesse caso, é a afinidade pelas peneiras moleculares. Quanto mais polar o
composto, maior a afinidade pela peneira e, portanto, mais eficiente a remoção do
álcool, o que favorece os compostos metílicos e etílicos.
Nas polimerizações utilizando o éster metílico e o etílico, as massas molares
obtidas foram muito maiores. Isso se deve principalmente ao fato de haver uma
maior eficiência na remoção do álcool de saída. No caso do etanol, há um azeótropo
de temperatura baixa e que possui, em sua composição, uma grande quantidade do
álcool. Já no caso do metanol, a mistura formada com cicloexano e benzeno é
zeotrópica e, portanto, o metanol é removido do sistema por destilação simples, com
alta eficiência.
FIGURA 9 - AVALIAÇÃO DAS MASSAS MOLARES DO POLI(ADIPATO DE ISOSORBILA) QUANTO AO ÁLCOOL DE
SAÍDA.
0
10000
20000
30000
40000
Metanol Etanol n-Propanol n-Butanol
28000
37000
4800
460
Mw (Da)
Tempo de reação:168h. Temperatura: 85-90ºC.Enzima: CAL-B N-435 ,10% em massa demonômeros.Monômeros: isosorbídeo e adipatos de metila,etila, n-propila e n-butila, razão molar 1:1

78
Outro importante fator que deve ser levado em consideração, é a
difusibilidade do álcool de saída no sítio ativo, e sua afinidade com o meio apolar
externo à enzima. Yang e Russel (Yang, 1996) afirmam que, para que haja uma
eficiente conversão dos substratos, deve haver a entrada do substrato no sítio ativo
da enzima, através da micro-fase aquosa e certa afinidade do produto (no caso o
álcool de saída) em sair do sítio ativo da enzima para que a reação prossiga.
Quando o álcool de saída é o metanol, este por ser mais polar, tem uma menor
afinidade pelo meio reacional apolar que circunda a enzima, e tende a ficar mais
retido por afinidade tanto com os resíduos hidrofílicos da enzima, quanto com as
moléculas de água presentes no microambiente enzimático. O etanol é um
composto um pouco menos polar, então se pode dizer que suas moléculas possuem
uma maior facilidade de saída do ambiente enzimático, e maior afinidade com o
meio apolar externo do que o metanol. Obviamente, se considerássemos apenas o
fator polaridade, essa explicação favoreceria os polímeros formados a partir dos
ésteres propílicos e butílicos mas, conforme explicado anteriormente, o
favorecimento que a polaridade dos compostos ofereceria no aumento da massa
molar é suprimido pelo fator azeotrópico.
Por isso, invariavelmente, as policondensações utilizando diésteres etílicos
rendem materiais de massas molares mais altas do que quando são utilizados
diésteres metílicos, apesar do metóxido ser conhecidamente um melhor grupo
abandonador do que o etóxido (pKa metanol= 15,9 pKa etanol: 15,5) (Vollhardt, et al.,
2002).

79
TABELA 19 - COMPOSIÇÃO DOS AZEÓTROPOS VARIANDO-SE A CADEIA CARBÔNICA DO ÁLCOOL.
Solventes: Proporção do álcool na
mistura (%)
Temperatura do
Azeótropo (°C)
Metanol /Cicloexano /Benzeno Mistura Zeotrópica
Etanol /Cicloexano /Benzeno 29,6 64,7
n-Propanol /Cicloexano /Benzeno 5,5 73,8
n-Butanol /Cicloexano /Benzeno 4 77,4
Esse estudo mostra que uma eficiente remoção do álcool do sistema (e o
conseqüente deslocamento do equilíbrio na direção dos produtos), como era de se
esperar, é de fundamental importância para o desempenho das reações de
policondensação.
4.4.5 - TESTE DE SOLUBILIDADE DOS MATERIAIS OBTIDOS:
Os estudos 4.4.5, 4.4.6 e 4.4.7, têm por objetivo investigar o papel que o
benzeno desempenha na mistura de solventes utilizada. O principal fato que motivou
esses estudos foi a ocorrência de precipitação do material com poucas horas de
reação, inicialmente formando uma suspensão e, em seguida, acontece a aderência
dos polímeros nas paredes do tubo reacional, tornando o meio heterogêneo.
As grandes questões originadas aqui são: como a massa molar do polímero
continua a crescer se temos, com o passar do tempo, a precipitação do poliéster
sintetizado, e a conseqüente formação de um meio reacional heterogêneo? Como
explicar o fato de que a enzima consegue desempenhar seu papel catalítico,
fazendo com que as cadeias insolúveis continuem tendo sua massa molar
aumentada, se esta se encontra imobilizada em uma matriz polimérica que por si só

80
já está heterogênea no meio, e o fato de o polímero não ser solúvel no meio
reacional e precipite quando há o crescimento da massa molar, tornam o meio
duplamente heterogêneo?
Para tentarmos responder a essas questões, primeiramente, foi realizado um
estudo da solubilidade dos materiais obtidos, tanto em solventes puros, como nas
misturas de solventes utilizadas. Os resultados são apresentados na Tabela 20.
TABELA 20 - SOLUBILIDADE DO POLI(ADIPATO DE ISOSORBILA) COM MW~30KDA.
Solventes Solubilidade (mg/mL)
Clorofórmio 298
Cicloexano menor que 0,1
Tolueno 4
Benzeno 18
Hexano menor que 0,1
Hexano:clorofórmio 3:2 1,2
Cicloexano:tolueno 6:1 menor que 0,1
Cicloexano:benzeno 6:1 0,6
Os resultados mostram a baixa solubilidade do material em hidrocarbonetos e
suas misturas. Porém, apesar de pequena, observa-se que, em benzeno ou na
mistura contendo benzeno, a solubilidade do material é maior do que em solventes
puros ou nas outras misturas utilizadas (exceto quando se usa clorofórmio, mas
esse sistema já mostrou baixa eficiência na policondensação). No entanto, apenas
com esses resultados não se pode dizer se o papel que a presença do benzeno
desempenha no sistema é de solubilidade.

81
4.4.6 - EFEITO DA PROPORÇÃO MONÔMERO: SOLVENTE
Para tentar responder à questão apresentada no item anterior, se o papel do
benzeno é a influência na solubilidade, foi realizado um estudo em cicloexano e
benzeno, para avaliar o efeito da diluição do sistema, variando-se a proporção
isosorbídeo: solvente. Os resultados estão na Tabela 21.
TABELA 21 - RESULTADOS OBTIDOS VARIANDO-SE A PROPORÇÃO ISOSORBÍDEO: SOLVENTE.
Proporção IS:Solvente 𝑴 𝒏 𝑴 𝒘 PD Rendimentoa
1:20 4800 17000 3,5 82%
1:30 8400 18000 2,1 85%
1:40 8800 20000 2,3 82%
1:50 9100 21000 2,3 83%
1:60 19000 40000 2,1 92,%
1:80 18000 45000 2,5 92%
a – Rendimento após purificação. Temp. do banho: 85ºC. Tempo:168h. Solventes: Cicloexano:Benzeno 6:1. A
proporção é calculada em g de isosorbídeo/ mL de solvente. Monômeros: isosorbídeo e adipato de dietila, razão
molar 1:1. Enzima: CAL-B N-435, 10% em massa de monômeros.
Os resultados mostram uma dependência direta da massa molar com a
diluição do sistema. Massas molares maiores foram obtidas utilizando sistemas mais
diluídos. A Figura 10 ilustra comparativamente o efeito que a diluição exerce no
sistema:

82
FIGURA 10 - EFEITO DA PROPORÇÃO SOLVENTE: ISOSORBÍDEO.
Esse comportamento nos permite concluir que o a solubilidade do polímero no
meio reacional desempenha importante papel no crescimento da cadeia, como era
de se esperar. Quanto maior a quantidade de solvente, mais eficiente é o sistema. A
hipótese proposta nesse caso é que o aumento da solubilidade do sistema
possibilita uma interface que, em equilíbrio com a fase sólida presente no meio
reacional, permite que as porções ésteres terminais dos polímeros entrem no sítio
ativo da enzima e sofram a transesterificação, possibilitando a fração solúvel a
crescer.
4.4.7 - EFEITO DA CONCENTRAÇÃO DE BENZENO NA MISTURA
Realizou-se um estudo para determinar a concentração de benzeno ideal na
mistura, também na tentativa de diminuir a quantidade de benzeno no sistema,
0
10000
20000
30000
40000
50000
20 X 30 X 40 X 50 X 60 X 80 X
17000 1800020000
31000
40000
45000
Mw(D
a)
A proporção é calculada em g de isosorbídeo/ mL de solvente.
Monômeros: adipato de dietila e isosorbídeo, razão molar 1:1.
Solvente: cicloexano:benzeno 6:1. Tempo de reação:168h.Temperatura: 85ºC. Enzima: CAL-B N-435 -10% em massa de monômeros.

83
devido ao potencial tóxico desse solvente. Para isso, foram feitas reações em
misturas de solventes com 20%, 14%, 11% e 9% de benzeno. Os resultados estão
na Tabela 22.
TABELA 22 - RESULTADOS OBTIDOS VARIANDO-SE A CONCENTRAÇÃO DE BENZENO NA MISTURA DE SOLVENTES.
Concentração de Benzeno
(Proporção benzeno:cicloexano) 𝑴 𝒏 𝑴 𝒘 PD Rendimentoa
20% (1:4) 13000 31000 2,4 61%
14% (1:6) 18000 40000 2,2 76%
11% (1:8) 16000 36000 2,2 76%
9% (1:10) 14000 32000 2,3 86% a – Rendimento após purificação. Temp. do banho: 85ºC. Tempo:168h. Monômeros: isosorbídeo e adipato de
dietila, razão molar 1:1. Enzima: CAL-B N-435, 10% em massa de monômeros.
As massas molares obtidas mostram que melhores resultados são obtidos
quando são utilizadas misturas com 14% de benzeno (proporção 1:6
cicloexano:benzeno). Utilizando quantidades maiores e menores que essa, as
massas obtidas são inferiores. A Figura 11 mostra comparativamente como a
concentração de benzeno influencia na obtenção dos materiais com diferentes
massas molares.
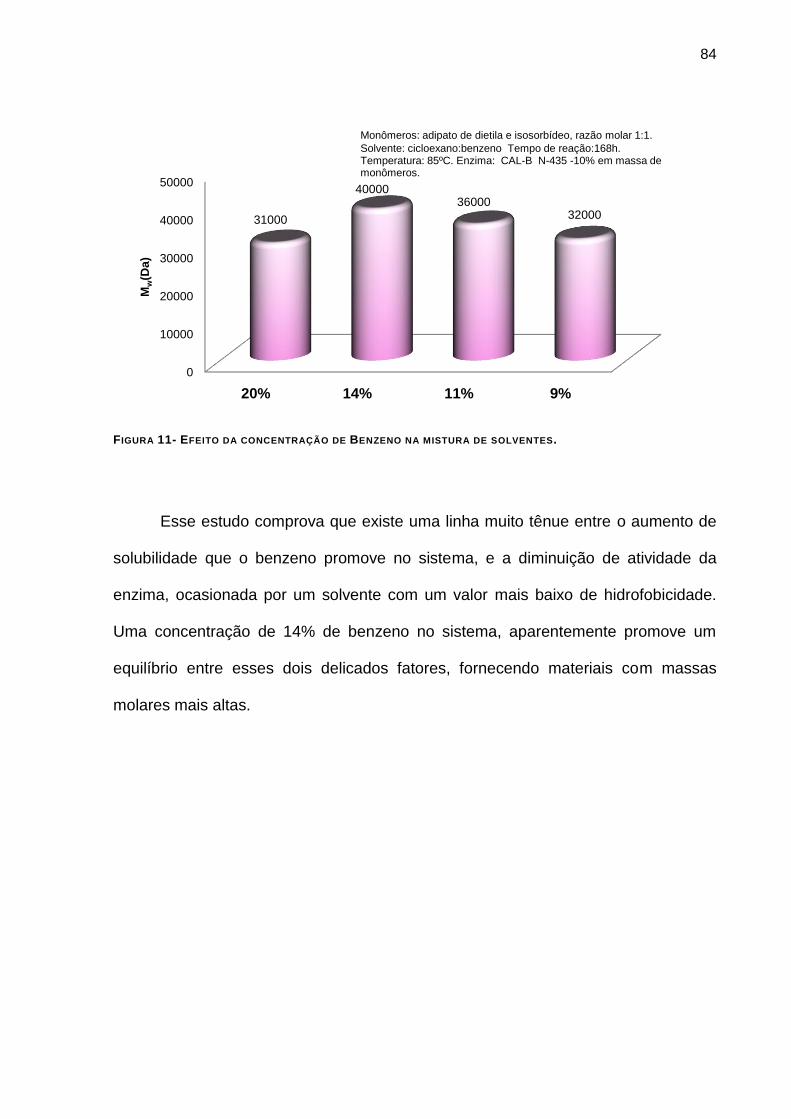
84
FIGURA 11- EFEITO DA CONCENTRAÇÃO DE BENZENO NA MISTURA DE SOLVENTES.
Esse estudo comprova que existe uma linha muito tênue entre o aumento de
solubilidade que o benzeno promove no sistema, e a diminuição de atividade da
enzima, ocasionada por um solvente com um valor mais baixo de hidrofobicidade.
Uma concentração de 14% de benzeno no sistema, aparentemente promove um
equilíbrio entre esses dois delicados fatores, fornecendo materiais com massas
molares mais altas.
0
10000
20000
30000
40000
50000
20% 14% 11% 9%
31000
4000036000
32000
Mw(D
a)
Monômeros: adipato de dietila e isosorbídeo, razão molar 1:1.
Solvente: cicloexano:benzeno Tempo de reação:168h.Temperatura: 85ºC. Enzima: CAL-B N-435 -10% em massa de monômeros.

85
4.5 - CARACTERIZAÇÃO DOS POLIÉSTERES OBTIDOS.
Todos os materiais obtidos foram analisados por cromatografia por exclusão
de tamanho. Além dessa técnica, polímeros foram caracterizados através das
seguintes técnicas analíticas: calorimetria exploratória diferencial, análise
termogravimétrica, ressonância magnética nuclear, difração de raios-X e
espectroscopia no infravermelho. Foram analisados os polímeros de maiores
massas molares obtidas, um de cada estrutura (unidade repetitiva) utilizada.
4.5.1 – CALORIMETRIA EXPLORATÓRIA DIFERENCIAL (DSC):
-25 0 25 50 75
En
do
Temperatura (0C)
Poli(Succinato de isosorbila)
Poli(Glutarato de isosorbila)
Poli(Adipato de isosorbila)
Poli(Suberato de isosorbila)
Poli(Sebacato de isosorbila)
Poli(Dodecanodioato de isosorbila)
FIGURA 12 - CURVAS DE DSC DOS MATERIAIS OBTIDOS

86
As análises feitas por DSC (Figura 12) mostram que, de modo geral, a
temperatura de transição vítrea dos materiais diminui conforme aumenta a cadeia
carbônica apolar da unidade repetitiva. Apenas o poli(adipato de isosorbila) mostrou
um aumento inesperado na Tg em comparação com o poli(glutarato de isosorbila).
Esse desvio também foi observado por Okada e colaboradores. (Okada, 1996).
Em comparação com os polímeros obtidos via catálise química, os
materiais sintetizados via catálise enzimática apresentaram valores de Tg maiores
para os polímeros com as porções éster de 4 e 5 carbonos, (Tabela 23) (Okada,
1996), o que não era esperado, por possuírem massas molares menores do que as
obtidas via química. Esse comportamento pode sugerir que as cadeias de polímeros
sintetizados via catálise enzimática encontram-se mais organizadas e, por isso, têm
seu grau de liberdade diminuído, aumentando a Tg desses materiais. Os outros
polímeros apresentaram comportamentos esperados com relação às temperaturas
de transição vítrea obtidas: o poli (adipato de isosorbila) obtido via enzimática
apresentou massa molar e Tg menores do que o mesmo material obtido via catálise
química; o poli(sebacato de isosorbila) apresentou Tg maior via enzimática, mas a
massa molar deste também foi maior.
Nenhum dos materiais analisados apresentou temperatura de fusão cristalina
(Tm), comprovando que estes poliésteres apresentam estrutura amorfa.
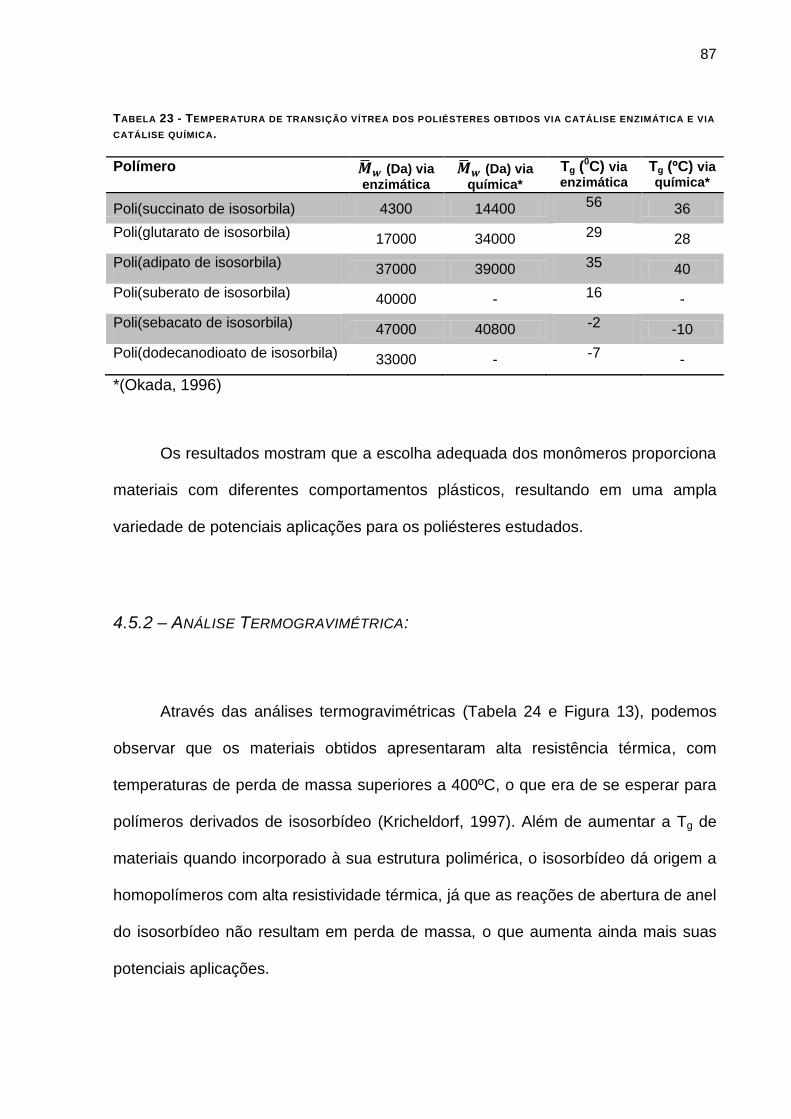
87
TABELA 23 - TEMPERATURA DE TRANSIÇÃO VÍTREA DOS POLIÉSTERES OBTIDOS VIA CATÁLISE ENZIMÁTICA E VIA
CATÁLISE QUÍMICA.
Polímero 𝑴 𝒘 (Da) via
enzimática 𝑴 𝒘 (Da) via
química*
Tg (0C) via
enzimática Tg (ºC) via química*
Poli(succinato de isosorbila) 4300 14400 56 36
Poli(glutarato de isosorbila) 17000 34000 29 28
Poli(adipato de isosorbila) 37000 39000 35 40
Poli(suberato de isosorbila) 40000 - 16 -
Poli(sebacato de isosorbila) 47000 40800 -2 -10
Poli(dodecanodioato de isosorbila) 33000 - -7 -
*(Okada, 1996)
Os resultados mostram que a escolha adequada dos monômeros proporciona
materiais com diferentes comportamentos plásticos, resultando em uma ampla
variedade de potenciais aplicações para os poliésteres estudados.
4.5.2 – ANÁLISE TERMOGRAVIMÉTRICA:
Através das análises termogravimétricas (Tabela 24 e Figura 13), podemos
observar que os materiais obtidos apresentaram alta resistência térmica, com
temperaturas de perda de massa superiores a 400ºC, o que era de se esperar para
polímeros derivados de isosorbídeo (Kricheldorf, 1997). Além de aumentar a Tg de
materiais quando incorporado à sua estrutura polimérica, o isosorbídeo dá origem a
homopolímeros com alta resistividade térmica, já que as reações de abertura de anel
do isosorbídeo não resultam em perda de massa, o que aumenta ainda mais suas
potenciais aplicações.
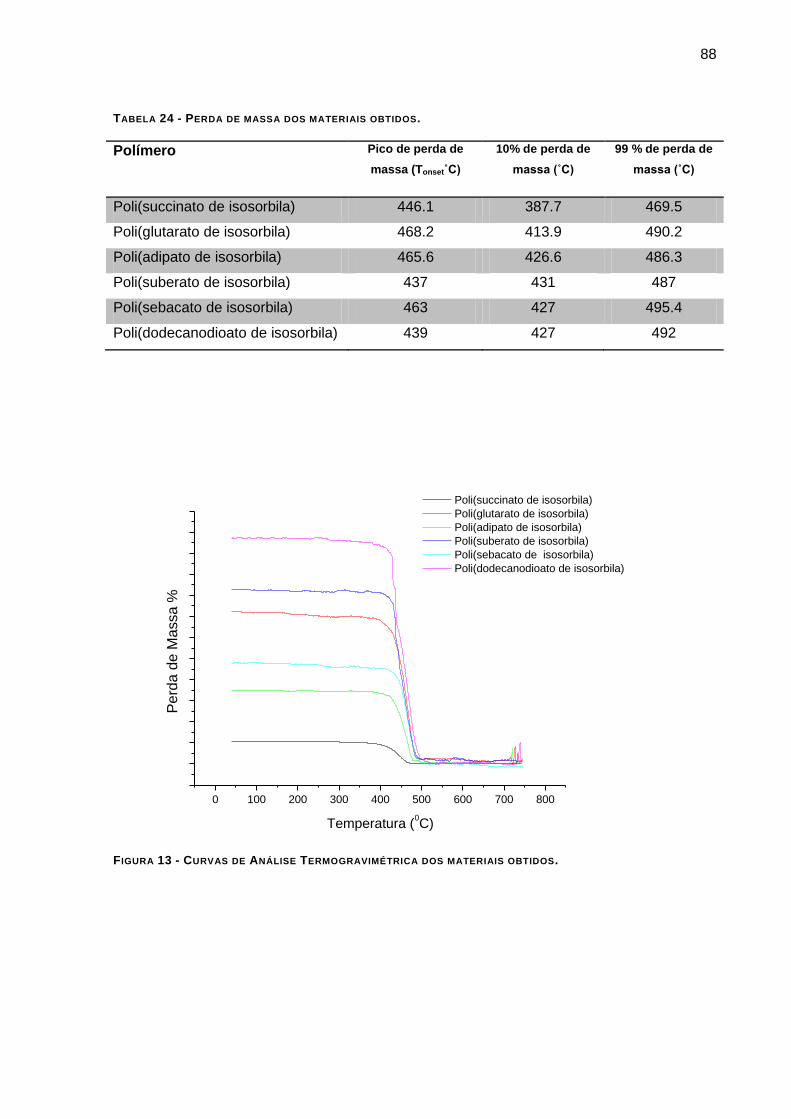
88
TABELA 24 - PERDA DE MASSA DOS MATERIAIS OBTIDOS.
Polímero Pico de perda de
massa (Tonset˚C)
10% de perda de
massa (˚C)
99 % de perda de
massa (˚C)
Poli(succinato de isosorbila) 446.1 387.7 469.5
Poli(glutarato de isosorbila) 468.2 413.9 490.2
Poli(adipato de isosorbila) 465.6 426.6 486.3
Poli(suberato de isosorbila) 437 431 487
Poli(sebacato de isosorbila) 463 427 495.4
Poli(dodecanodioato de isosorbila) 439 427 492
0 100 200 300 400 500 600 700 800
Temperatura (0C)
Poli(succinato de isosorbila)
Poli(glutarato de isosorbila)
Poli(adipato de isosorbila)
Poli(suberato de isosorbila)
Poli(sebacato de isosorbila)
Poli(dodecanodioato de isosorbila)
FIGURA 13 - CURVAS DE ANÁLISE TERMOGRAVIMÉTRICA DOS MATERIAIS OBTIDOS.
Pe
rda
de M
assa %

89
4.5.3 – RESSONÂNCIA MAGNÉTICA NUCLEAR:
Com o objetivo de se confirmar as estruturas das unidades repetitivas dos
poliésteres produzidos nesse trabalho, foram realizadas análises de ressonância
magnética nuclear de carbono e de hidrogênio, de cada uma das seis estruturas
poliméricas sintetizadas. Em alguns espectros, pode-se observar a presença dos
hidrogênios dos grupos terminais etila e hidroxila (marcados como * e **). Em outros,
essa observação não é possível devido à sobreposição dos picos por outros de
maior intensidade.
Para fins de comparação, abaixo mostramos as atribuições de RMN 1H e/ou
13C do resíduo de isosorbídeo dos produtos encontrados na literatura.
(Okada, et al., 1995)
O
O
OO
H
H
*
O
x
1
35
4
6
2*
O
13C-RMN (67,8 MHz, CDCl3) (ppm): 70 (C-6); 72 (C-1); 73 (C-5); 77 (C-2); 81
(C-4); 86 (C-3).
(Chatti, et al., 2006)
O
O O
O
CO *
*
y
2
5
43
1+1'
6+6'
1H-RMN (100 MHz, CDCl3) (ppm): 4,1-3,9 (H-1,H-1’,H-6,H-6’); 4,5 (H-4); 4,8 (H-
3); 5,1 (H-2, H-5).

90
(Sablong, et al., 2008)
O
O
H
HO
O 21
34
65
O(CH2)4OH
O
O
O
HO(CH2)4O
O
1H-RMN (400 MHz, CDCl3) (ppm): 4,05 (d 2H; H-6a,b); 4,08 (s largo; 2H; H-1a,b);
4,68 (d) = 4,8 Hz; 1H; H-3); 5,06 (dd 1H; H-4); 5,40 (dt 1H; H-5); e 5,42 (s largo; 1H;
H-2).
13C-RMN (100 MHz, CDCl3) (ppm): 70,8 (C-6); 73,3 (C-1); 74,9 (C-5); 78,8 (C-2);
81,1 (C-4); e 86,1 (C-3).
Os espectros de RMN de hidrogênio e carbono-13 de cada estrutura, com
seus respectivos deslocamentos químicos e atribuições, estão apresentados a
seguir:
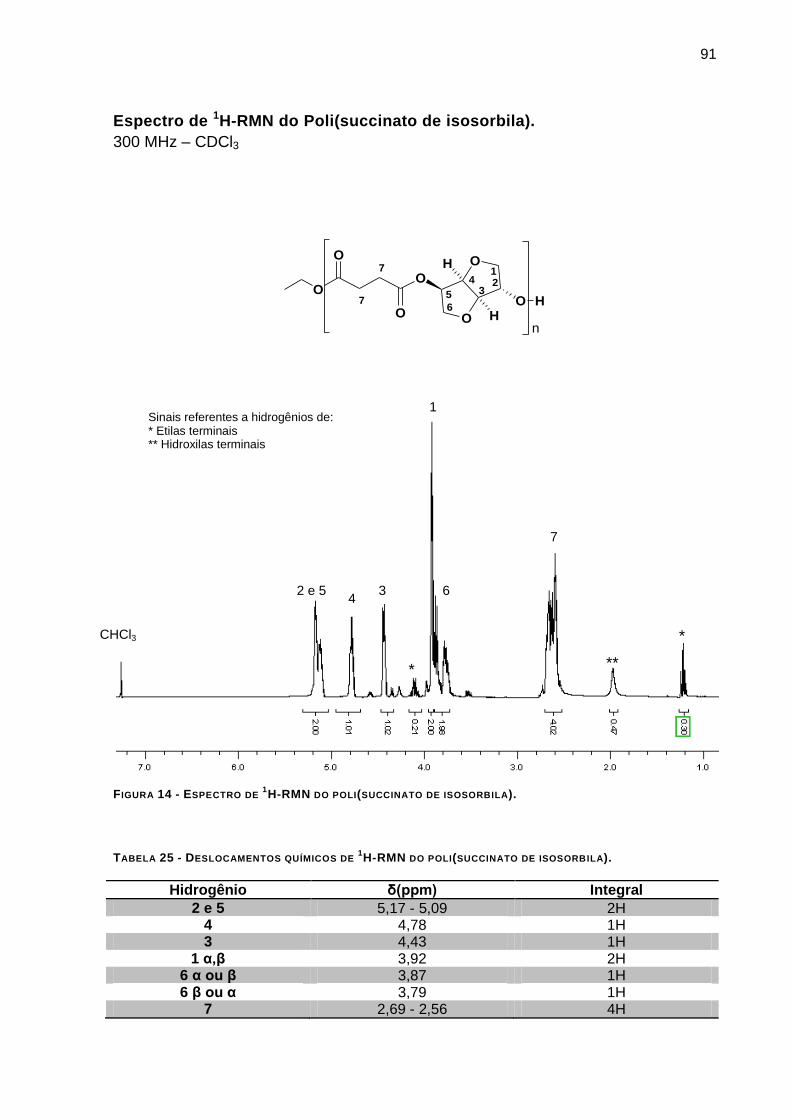
91
Espectro de 1H-RMN do Poli(succinato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
O
O
H
H
12
34
5
67
7
H
n
FIGURA 14 - ESPECTRO DE 1H-RMN DO POLI(SUCCINATO DE ISOSORBILA).
TABELA 25 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SUCCINATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,17 - 5,09 2H 4 4,78 1H 3 4,43 1H
1 α,β 3,92 2H 6 α ou β 3,87 1H 6 β ou α 3,79 1H
7 2,69 - 2,56 4H
CHCl3
Sinais referentes a hidrogênios de: * Etilas terminais ** Hidroxilas terminais
*
* **
1
2 e 5 4
3
7
6
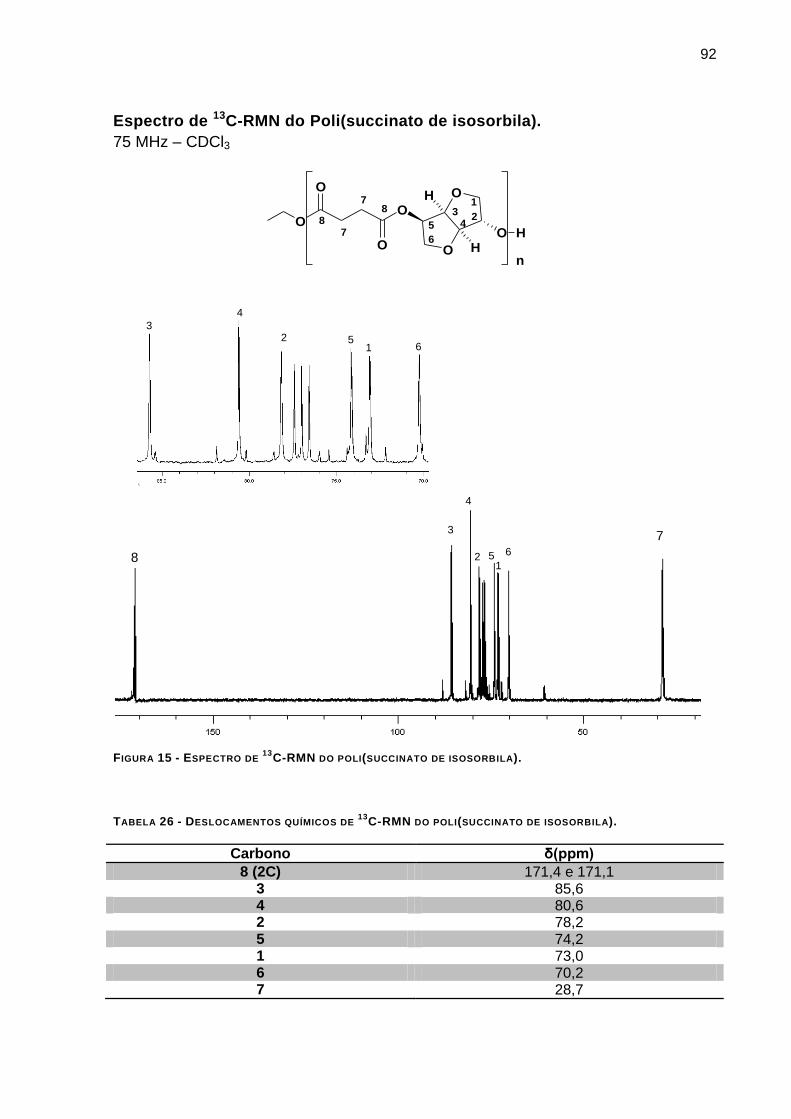
92
Espectro de 13C-RMN do Poli(succinato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
O
O
H
H
1
2345
67
7
88
H
n
FIGURA 15 - ESPECTRO DE 13
C-RMN DO POLI(SUCCINATO DE ISOSORBILA).
TABELA 26 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(SUCCINATO DE ISOSORBILA).
Carbono δ(ppm)
8 (2C) 171,4 e 171,1 3 85,6 4 80,6 2 78,2 5 74,2 1 73,0 6 70,2 7 28,7
2
4
3 7
6 8 5
1
2
4
3
6 5
1
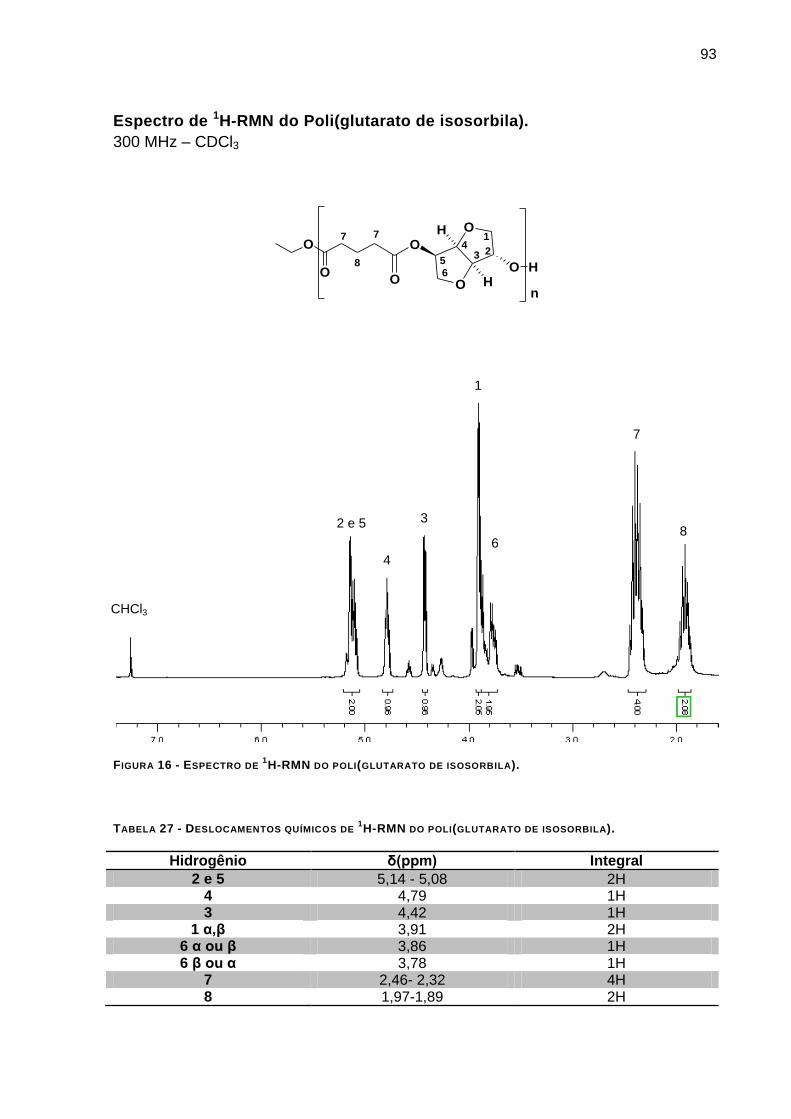
93
Espectro de 1H-RMN do Poli(glutarato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
H
H
1
234
5
6
7 7
8
O
O H
n
FIGURA 16 - ESPECTRO DE 1H-RMN DO POLI(GLUTARATO DE ISOSORBILA).
TABELA 27 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(GLUTARATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,14 - 5,08 2H 4 4,79 1H 3 4,42 1H
1 α,β 3,91 2H 6 α ou β 3,86 1H 6 β ou α 3,78 1H
7 2,46- 2,32 4H 8 1,97-1,89 2H
CHCl3
1
2 e 5
4
3
7
6
8
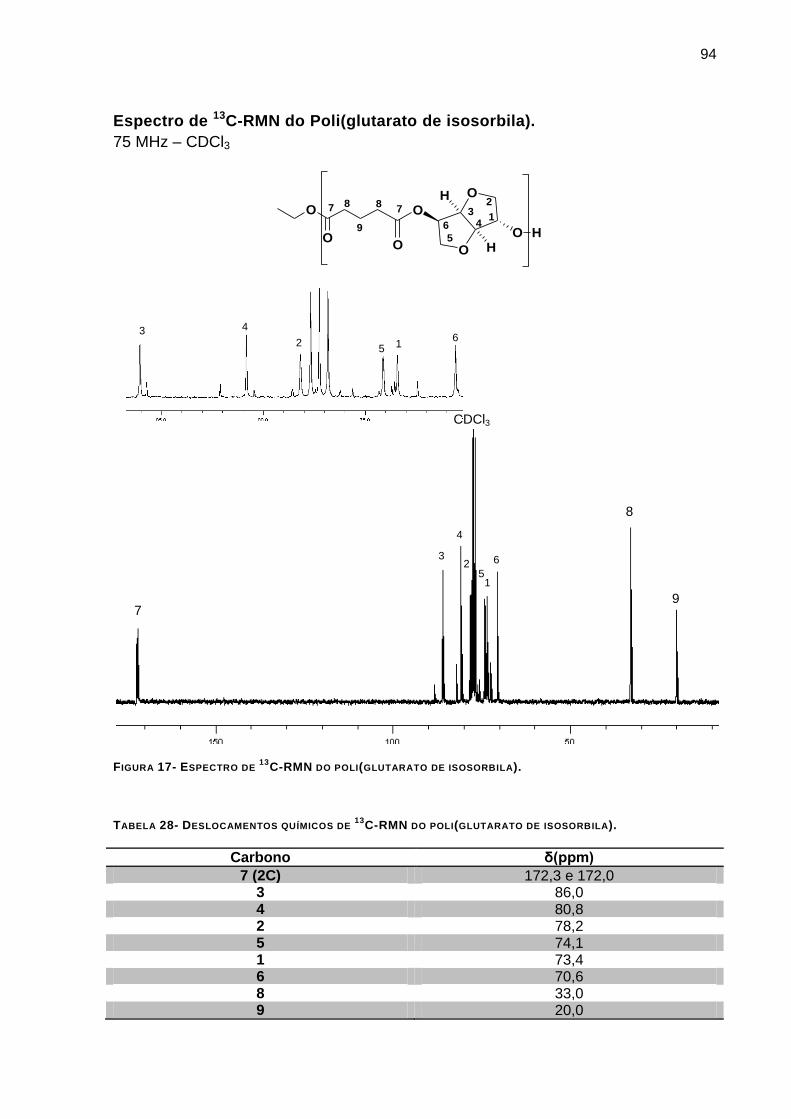
94
Espectro de 13C-RMN do Poli(glutarato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
H
H
1
23
4
5
6
7 78
O
O H
8
9
FIGURA 17- ESPECTRO DE 13
C-RMN DO POLI(GLUTARATO DE ISOSORBILA).
TABELA 28- DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(GLUTARATO DE ISOSORBILA).
Carbono δ(ppm)
7 (2C) 172,3 e 172,0 3 86,0 4 80,8 2 78,2 5 74,1 1 73,4 6 70,6 8 33,0 9 20,0
2
4
3
7
6
8
5 1
9
CDCl3
4 3 6
5 1 2
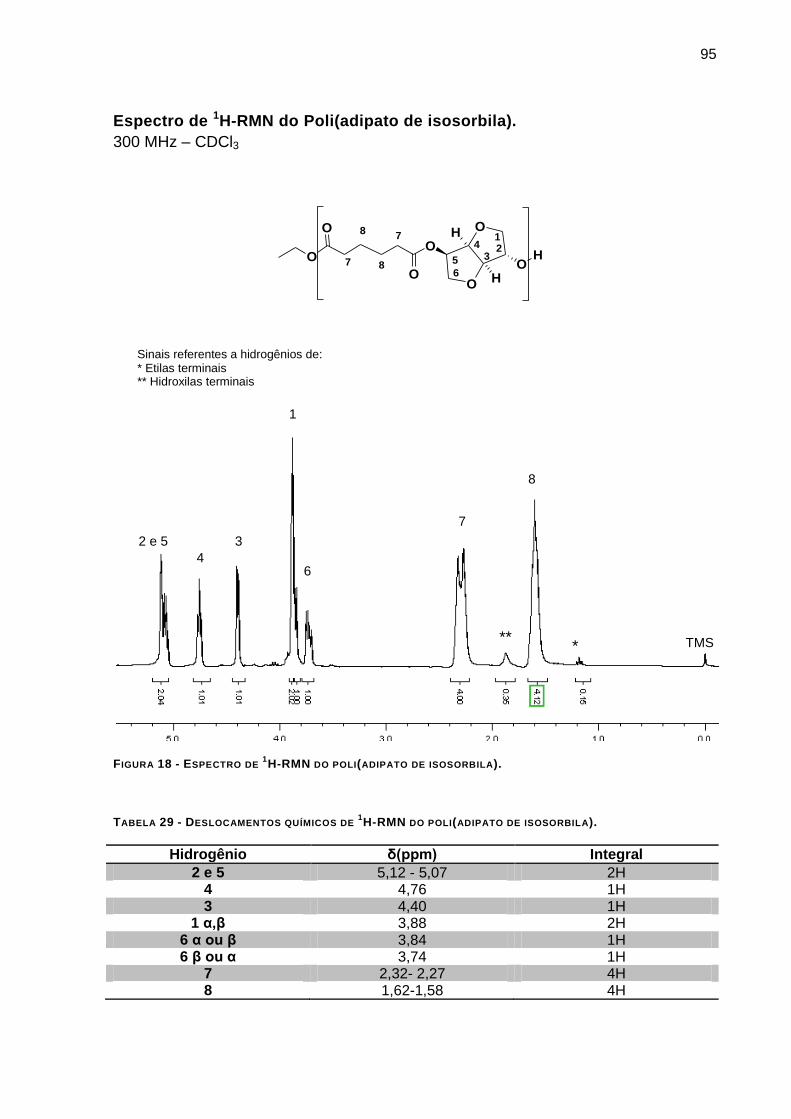
95
Espectro de 1H-RMN do Poli(adipato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
7
8HO
O
7
8
FIGURA 18 - ESPECTRO DE 1H-RMN DO POLI(ADIPATO DE ISOSORBILA).
TABELA 29 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(ADIPATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,12 - 5,07 2H 4 4,76 1H 3 4,40 1H
1 α,β 3,88 2H 6 α ou β 3,84 1H 6 β ou α 3,74 1H
7 2,32- 2,27 4H 8 1,62-1,58 4H
Sinais referentes a hidrogênios de: * Etilas terminais ** Hidroxilas terminais
* ** TMS
1
2 e 5
4
3
7
6
8
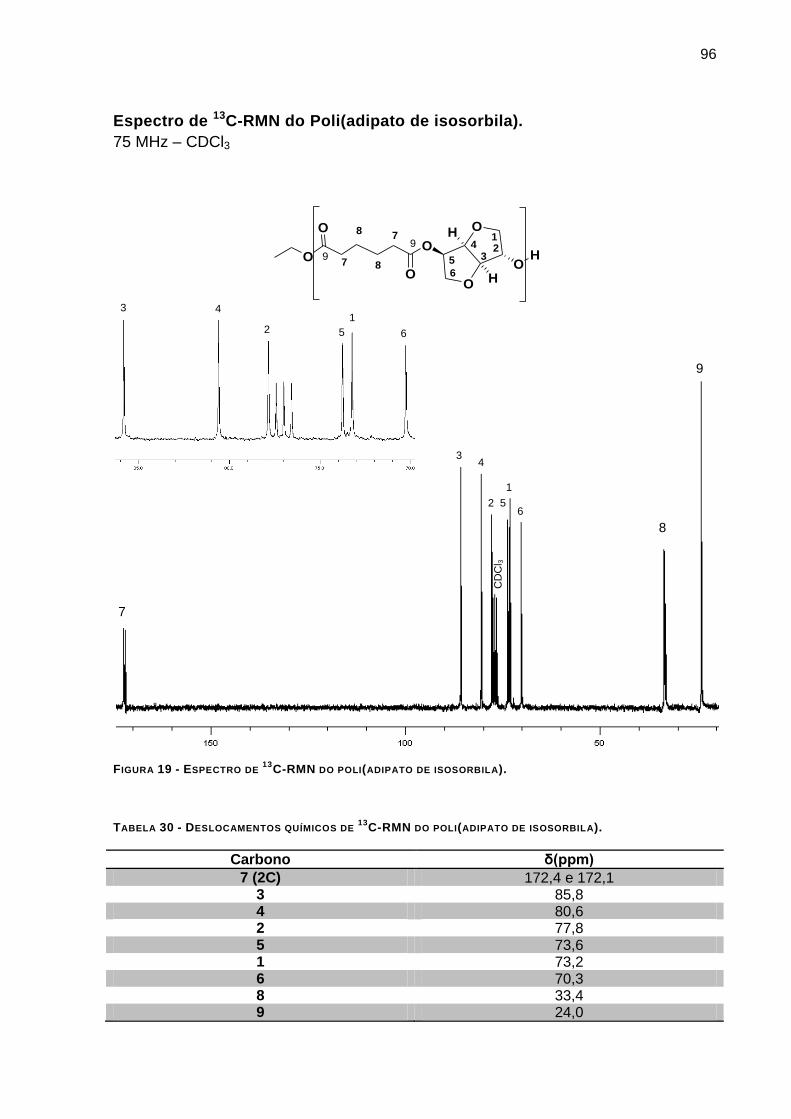
96
Espectro de 13C-RMN do Poli(adipato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
7
8H9O
O
7
8
9
FIGURA 19 - ESPECTRO DE 13
C-RMN DO POLI(ADIPATO DE ISOSORBILA).
TABELA 30 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(ADIPATO DE ISOSORBILA).
Carbono δ(ppm)
7 (2C) 172,4 e 172,1 3 85,8 4 80,6 2 77,8 5 73,6 1 73,2 6 70,3 8 33,4 9 24,0
2
4 3
7
6
8
5
1
9
CD
Cl 3
2
4 3
6 5
1

97
Espectro de 1H-RMN do Poli(suberato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
7
8H
9O
O
7
8 9
FIGURA 20 - ESPECTRO DE 1H-RMN DO POLI(SUBERATO DE ISOSORBILA).
TABELA 31 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SUBERATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,15 - 5,08 2H 4 4,79 1H 3 4,44 1H
1 α,β 3,91 2H 6 α ou β 3,88 1H 6 β ou α 3,75 1H
7 2,35- 2,25 4H 8 1,62-1,57 4H 9 1,31-1,30 4H
1
2 e 5 4
3
7
6
8
9
Sinais referentes a hidrogênios de: * Hidroxilas terminais
*
CHCl3

98
Espectro de 13C-RMN do Poli(suberato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
78
H9
O
O
78 9
10
10
FIGURA 21 - ESPECTRO DE 13
C-RMN DO POLI(SUBERATO DE ISOSORBILA).
TABELA 32 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(SUBERATO DE ISOSORBILA).
Carbono δ(ppm)
7 (2C) 172,8 e 172,6 3 85,9 4 80,6 2 77,8 5 73,7 1 73,3 6 70,2 8 33,8 9 28,5
10 24,5
2
4
7
6
8 5 1
9
CD
Cl 3
10
3
2
4 6 5
3 1
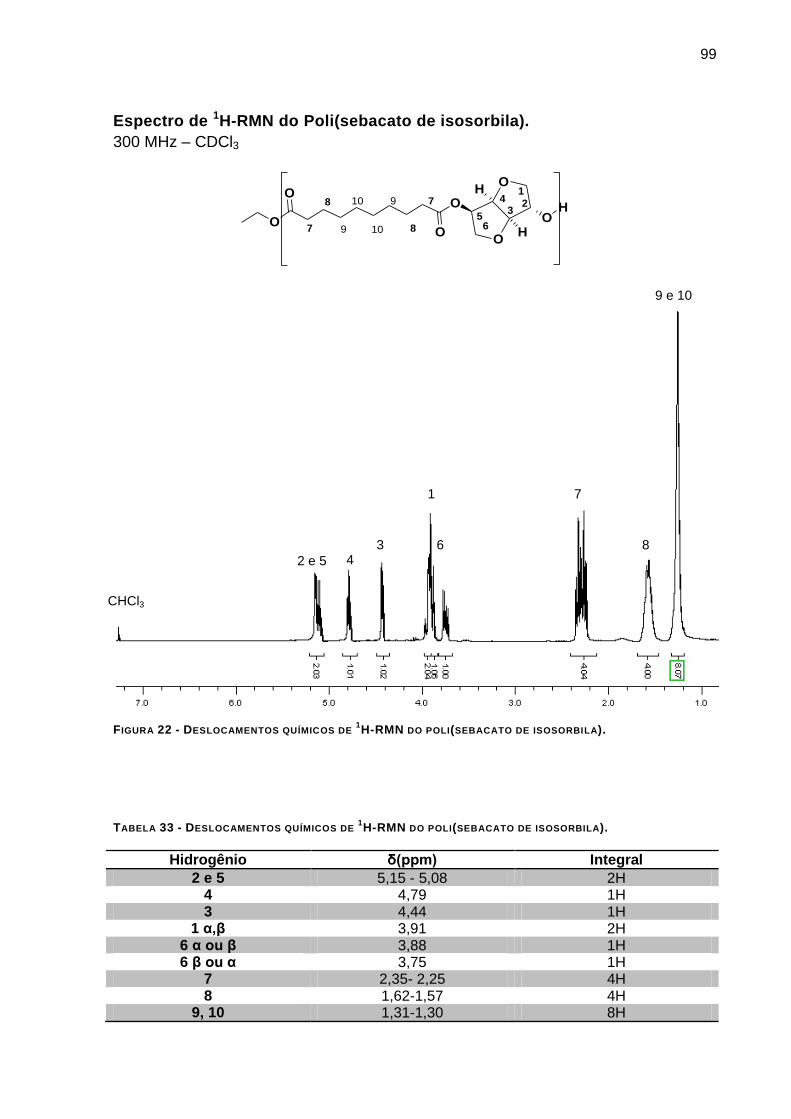
99
Espectro de 1H-RMN do Poli(sebacato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
H
H
1
23
4
56
7
8
H
9O
O
7
8 9
10
10
FIGURA 22 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SEBACATO DE ISOSORBILA).
TABELA 33 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(SEBACATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,15 - 5,08 2H 4 4,79 1H 3 4,44 1H
1 α,β 3,91 2H 6 α ou β 3,88 1H 6 β ou α 3,75 1H
7 2,35- 2,25 4H 8 1,62-1,57 4H
9, 10 1,31-1,30 8H
1
2 e 5 4 3
7
6
8
9 e 10
CHCl3
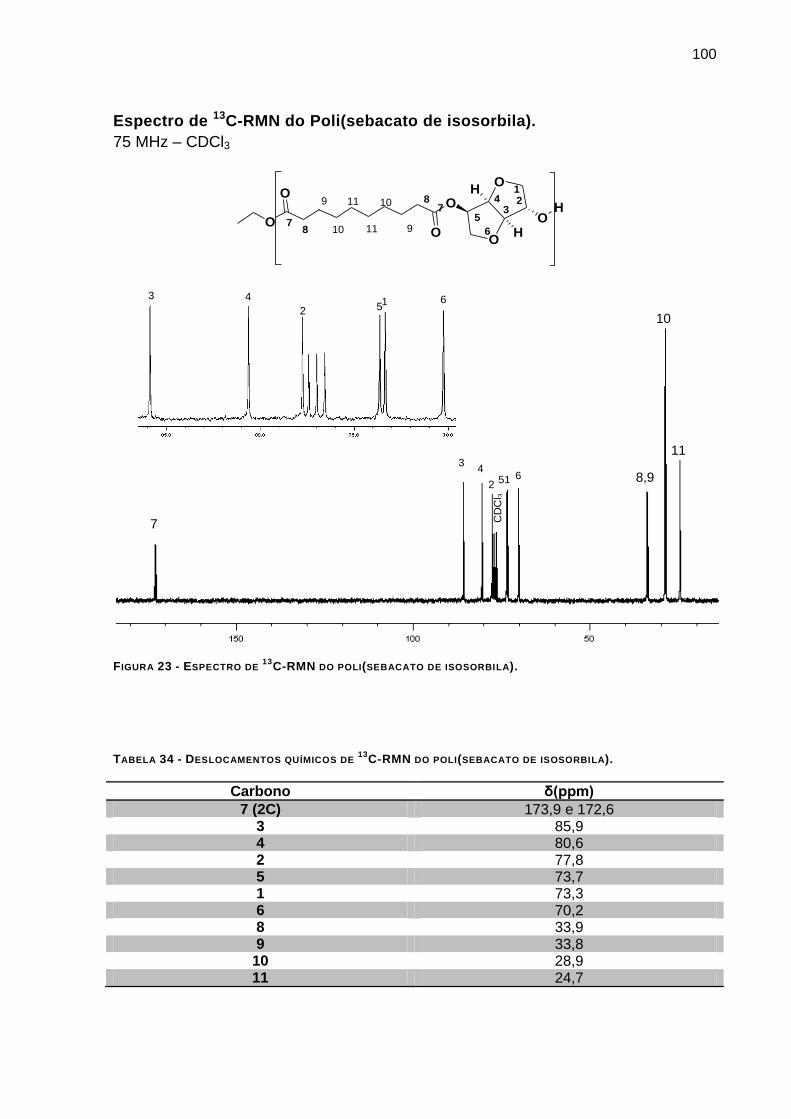
100
Espectro de 13C-RMN do Poli(sebacato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
78
H9
O
O
78 9
10
10
11
11
FIGURA 23 - ESPECTRO DE 13
C-RMN DO POLI(SEBACATO DE ISOSORBILA).
TABELA 34 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(SEBACATO DE ISOSORBILA).
Carbono δ(ppm)
7 (2C) 173,9 e 172,6 3 85,9 4 80,6 2 77,8 5 73,7 1 73,3 6 70,2 8 33,9 9 33,8
10 28,9 11 24,7
2
4 3
7
6 8,9 51
11
CD
Cl 3
10
4 3 6 5 2 1
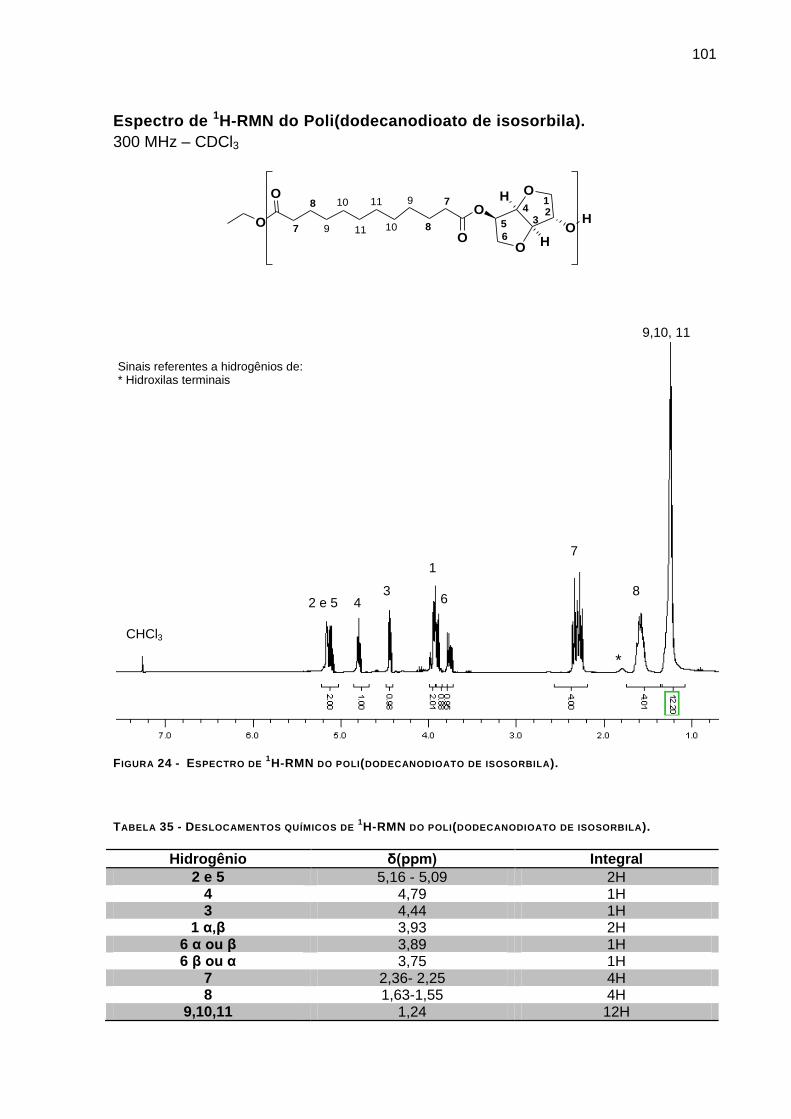
101
Espectro de 1H-RMN do Poli(dodecanodioato de isosorbila).
300 MHz – CDCl3
O
O
O
O
O
H
H
12
34
5
6
7
8H
9O
O
7
8 9
10
10
11
11
FIGURA 24 - ESPECTRO DE 1H-RMN DO POLI(DODECANODIOATO DE ISOSORBILA).
TABELA 35 - DESLOCAMENTOS QUÍMICOS DE 1H-RMN DO POLI(DODECANODIOATO DE ISOSORBILA).
Hidrogênio δ(ppm) Integral
2 e 5 5,16 - 5,09 2H 4 4,79 1H 3 4,44 1H
1 α,β 3,93 2H 6 α ou β 3,89 1H 6 β ou α 3,75 1H
7 2,36- 2,25 4H 8 1,63-1,55 4H
9,10,11 1,24 12H
1
2 e 5 4 3
7
6
8
9,10, 11
Sinais referentes a hidrogênios de: * Hidroxilas terminais
*
CHCl3
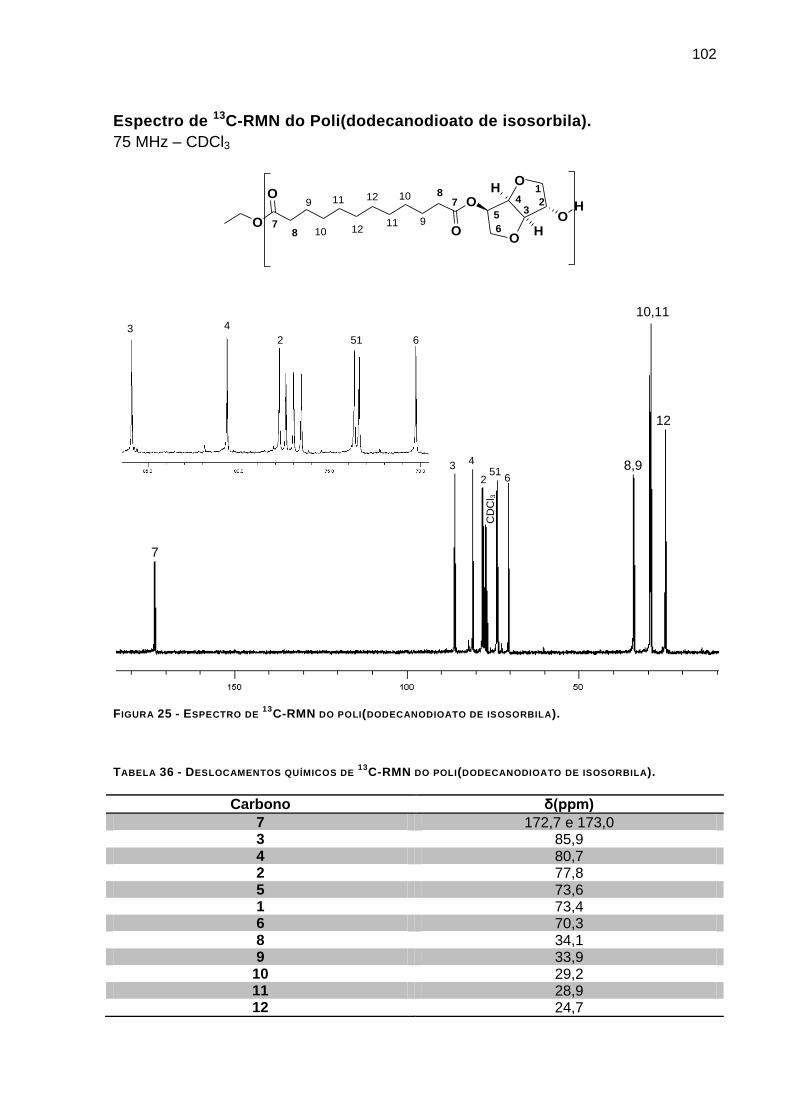
102
Espectro de 13C-RMN do Poli(dodecanodioato de isosorbila).
75 MHz – CDCl3
O
O
O
O
O
H
H
1
23
4
5
6
78
H9
O
O
78
9
10
10
11
1112
12
FIGURA 25 - ESPECTRO DE 13
C-RMN DO POLI(DODECANODIOATO DE ISOSORBILA).
TABELA 36 - DESLOCAMENTOS QUÍMICOS DE 13
C-RMN DO POLI(DODECANODIOATO DE ISOSORBILA).
Carbono δ(ppm)
7 172,7 e 173,0 3 85,9 4 80,7 2 77,8 5 73,6 1 73,4 6 70,3 8 34,1 9 33,9
10 29,2 11 28,9 12 24,7
4 3
7
6 8,9
12
CD
Cl 3
10,11
2 51
4 3 51 6 2
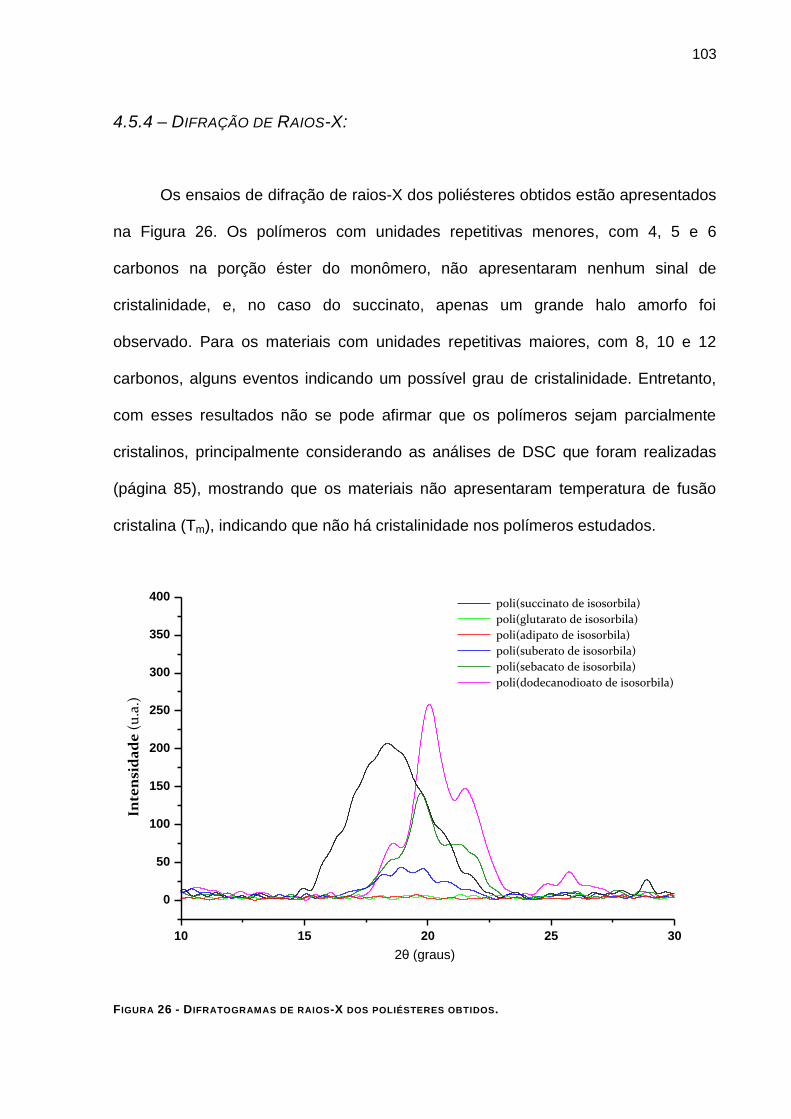
103
4.5.4 – DIFRAÇÃO DE RAIOS-X:
Os ensaios de difração de raios-X dos poliésteres obtidos estão apresentados
na Figura 26. Os polímeros com unidades repetitivas menores, com 4, 5 e 6
carbonos na porção éster do monômero, não apresentaram nenhum sinal de
cristalinidade, e, no caso do succinato, apenas um grande halo amorfo foi
observado. Para os materiais com unidades repetitivas maiores, com 8, 10 e 12
carbonos, alguns eventos indicando um possível grau de cristalinidade. Entretanto,
com esses resultados não se pode afirmar que os polímeros sejam parcialmente
cristalinos, principalmente considerando as análises de DSC que foram realizadas
(página 85), mostrando que os materiais não apresentaram temperatura de fusão
cristalina (Tm), indicando que não há cristalinidade nos polímeros estudados.
10 15 20 25 30
0
50
100
150
200
250
300
350
400
poli(succinato de isosorbila)
poli(glutarato de isosorbila)
poli(adipato de isosorbila)
poli(suberato de isosorbila)
poli(sebacato de isosorbila)
poli(dodecanodioato de isosorbila)
Inte
nsi
da
de
(u
.a.)
FIGURA 26 - D IFRATOGRAMAS DE RAIOS-X DOS POLIÉSTERES OBTIDOS.
2θ (graus)

104
Contudo, os resultados das análises de DSC e raios-X sugerem que pode
haver certa organização dos sistemas poliméricos, resultado de uma regularidade
estrutural, mesmo não havendo dados concretos sobre a cristalinidade dos
materiais. A hipótese levantada, nesse caso, é que, como as duas hidroxilas do
isosorbídeo possuem configurações diferentes (como se, ao invés de um monômero
AA, tivéssemos um monômero AA’), a enzima poderia ter preferência por uma das
hidroxilas e, por isso, a reação ocorreria primeiramente pela formação de dímeros
através da “hidroxila preferencial”, do tipo AA’-BB. A subsequente polimerização
através destes dímeros primários geraria cadeias poliméricas do tipo -[AA’-BB]n-,
justificando a presença dessa regularidade estrutural.
4.5.5 – ESPECTROSCOPIA NO INFRAVERMELHO:
As análises feitas por espectroscopia no infravermelho nos forneceram
espectros muito característicos e semelhantes entre si, devido à similaridade
estrutural das unidades repetitivas dos poliésteres em questão. Os principais picos
de absorção dos polímeros encontram-se na Tabela 37.
Algumas considerações importantes podem ser feitas com base nos
espectros obtidos (Figura 27 a Figura 32): o pico em aproximadamente 3600 cm-1 é
atribuído à hidroxila terminal do álcool. De uma forma geral, a intensidade dos picos
de absorção de hidroxila diminui em proporção com o pico de absorção de C-H
conforme aumenta o grau de polimerização do poliéster em questão, chegando a
desaparecer no espectro do poli(sebacato de isosorbila), devido à diminuição
significativa dos grupos hidroxila terminais por meio da formação do poliéster com

105
maior grau de polimerização. Os picos em aproximadamente 2900 cm-1 são
atribuídos ao estiramento da ligação carbono-hidrogênio, presente em todas as
estruturas poliméricas em questão. Por volta de 1730 cm-1, temos o pico atribuído à
carbonila de éster, também em todos os polímeros estudados, com intensidade
muito alta. Em aproximadamente 1400 cm-1, observamos um pico também
importante, atribuído à deformação das ligações C-H em -CH3, mostrando então a
porção éster terminal das cadeias poliméricas, já que o monômero envolvido foi o
derivado dietílico dos ácidos dicarboxílicos.
TABELA 37 - PICOS DE ABSORÇÃO NO INFRAVERMELHO
Grupo SUC (19) GLU (69) ADI (142) SUB (142) SEB (151) DOD (98)
O-H 3641 3645 3533 3622 - 3620
C-H
(estiramento) 2983 2939 2937 2935 2929 2854
C=O 1737 1739 1731 1730 1737 1739
CH3
(deformação) 1367 1460 1461 1463 1463 1463
C-C 1155-877 1174-617 1093-617 1164-734 1168-975 1095-885
Valores em cm-1. O grau de polimerização dos materiais analisados está entre parênteses. SUC= Poli(succinato de isosorbila), GLU= Poli(glutarato de isosorbila), ADI= Poli(adipato de isosorbila), SUB= Poli(suberato de isosorbila), SEB= Poli(sebacato de isosorbila), DOD= Poli(dodecanodioato de isosorbila).
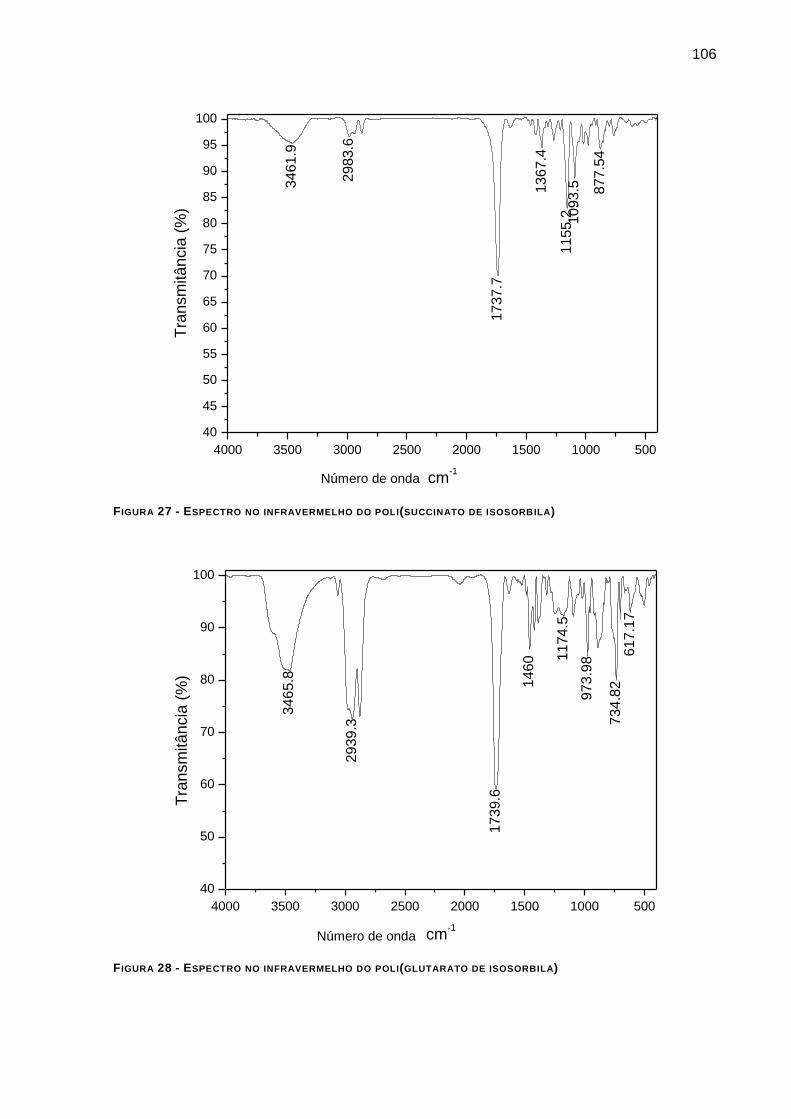
106
4000 3500 3000 2500 2000 1500 1000 500
40
45
50
55
60
65
70
75
80
85
90
95
100
877.5
4
1093.5
1155.2
1367.4
1737.7
2983.6
3461.9
T
ransm
itância
(%
)
cm-1
FIGURA 27 - ESPECTRO NO INFRAVERMELHO DO POLI(SUCCINATO DE ISOSORBILA)
4000 3500 3000 2500 2000 1500 1000 500
40
50
60
70
80
90
100
617.1
7734.8
2
973.9
81174.5
1460
1739.6
2939.3
3465.8
Tra
nsm
itân
cia
(%
)
cm-1
FIGURA 28 - ESPECTRO NO INFRAVERMELHO DO POLI(GLUTARATO DE ISOSORBILA)
Número de onda
Número de onda
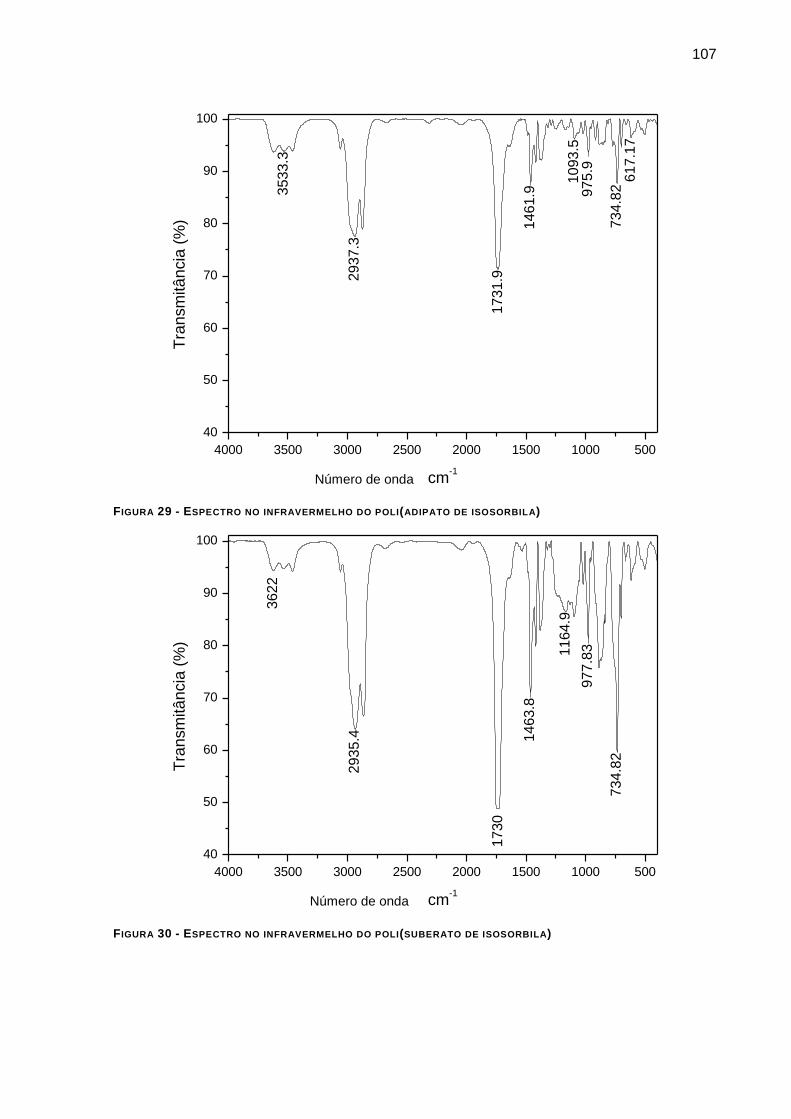
107
4000 3500 3000 2500 2000 1500 1000 500
40
50
60
70
80
90
100
617.1
7734.8
2975.9
1093.5
1461.9
1731.92
937.3
3533.3
Tra
nsm
itância
(%
)
cm-1
FIGURA 29 - ESPECTRO NO INFRAVERMELHO DO POLI(ADIPATO DE ISOSORBILA)
4000 3500 3000 2500 2000 1500 1000 500
40
50
60
70
80
90
100
734.8
2
977.8
31164.9
1463.8
1730
2935.4
3622
Tra
nsm
itância
(%
)
cm-1
FIGURA 30 - ESPECTRO NO INFRAVERMELHO DO POLI(SUBERATO DE ISOSORBILA)
Número de onda
Número de onda
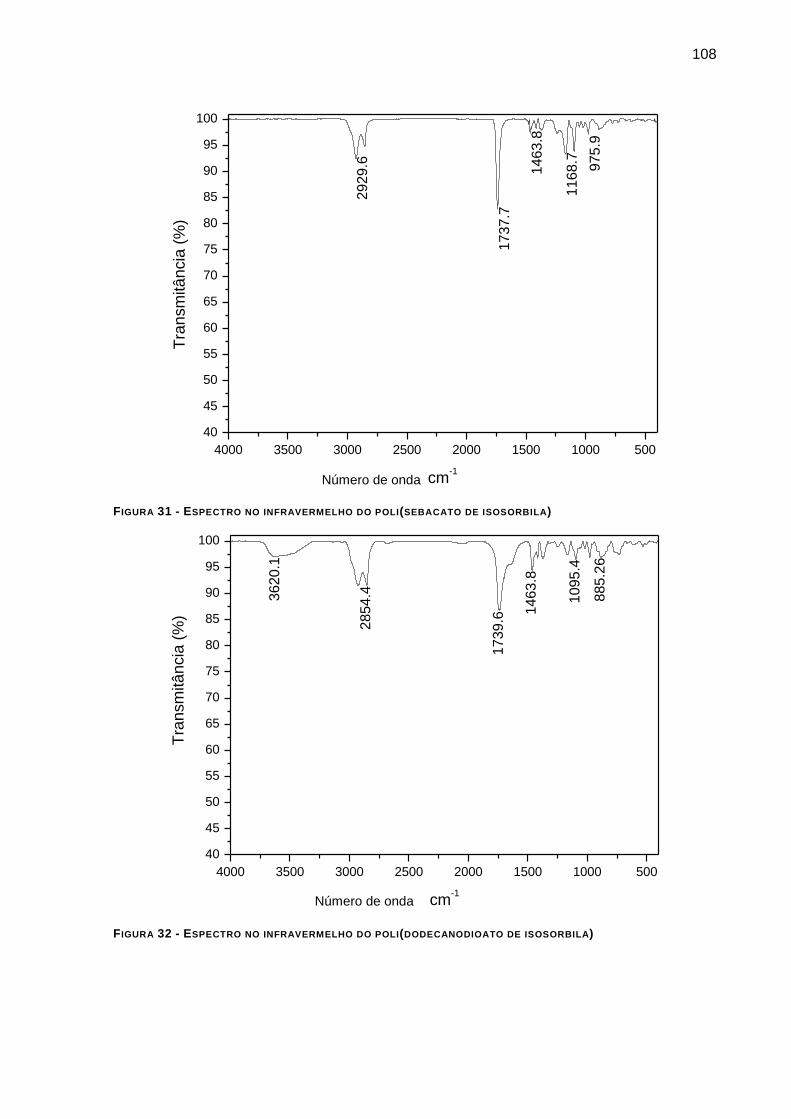
108
4000 3500 3000 2500 2000 1500 1000 500
40
45
50
55
60
65
70
75
80
85
90
95
100
975.9
1168.7
1463.8
1737.7
2929.6
Tra
nsm
itância
(%
)
cm-1
FIGURA 31 - ESPECTRO NO INFRAVERMELHO DO POLI(SEBACATO DE ISOSORBILA)
4000 3500 3000 2500 2000 1500 1000 500
40
45
50
55
60
65
70
75
80
85
90
95
100
885.2
6
1095.4
1463.8
1739.62854.43620.1
Tra
nsm
itância
(%
)
cm-1
FIGURA 32 - ESPECTRO NO INFRAVERMELHO DO POLI(DODECANODIOATO DE ISOSORBILA)
Número de onda
Número de onda

109
5 - CONCLUSÕES:
Atualmente, existe uma enorme demanda por polímeros derivados de fontes
renováveis, devido aos benefícios oferecidos pelo uso desses materiais, que
substituem com vantagens os polímeros derivados do petróleo, principalmente por
terem custos de produção semelhantes aos materiais provenientes de combustíveis
fósseis. Esse trabalho representa um grande avanço na área, por possibilitar a
síntese via enzimática de polímeros de isosorbídeo, um diol secundário e
estericamente impedido, obtendo materiais com características muito interessantes,
de massas molares similares às obtidas via catálise química, sendo a catálise
enzimática uma alternativa “limpa” de síntese de polímeros.
Esses materiais foram produzidos com sucesso em altos rendimentos,
utilizando lipase de Candida antarctica – Fração B como catalisador, através de
diversas metodologias: policondensação em massa sob pressão ambiente e sob
pressão reduzida; policondensação em massa e em solução utilizando monômeros
divinílicos e policondensação em solução com destilação azeotrópica.
Ao contrário do que previam todas as expectativas iniciais, a técnica que se
mostrou mais eficiente de polimerização enzimática do isosorbídeo foi a
polimerização em solução com destilação azeotrópica fornecendo materiais com
massas molares maiores. Diz-se contrário às expectativas, pelo fato de que, dentro
da varredura de condições realizadas, misturas de cicloexano e benzeno, onde os
polímeros são praticamente insolúveis, mostraram-se mais eficientes do que
solventes onde a solubilidade dos polímeros é maior. Ainda serão necessários
outros estudos para elucidar o funcionamento desse sistema, onde os polímeros

110
continuam tendo suas massas molares aumentadas, mesmo quando o sistema
polímero:solvente passa a ser heterogêneo. Dentro dessa investigação, alguns
avanços já foram feitos, como a determinação da importância da diluição do sistema,
a solubilidade dos polímeros nos solventes em questão, e a concentração de
benzeno ideal para a obtenção de massas molares maiores. Com isso, alguns
pontos foram esclarecidos, mas ainda devemos aprofundar os conhecimentos nesse
assunto.
Embora tenham apresentado uma cinética de reação lenta, provou-se que
polimerizações de dióis secundários, como o isosorbídeo, são possíveis de serem
realizadas via catálise enzimática, com resultados muito promissores. Todos os
resultados obtidos são inéditos para polímeros derivados de isosorbídeo, abrindo,
assim, uma nova linha de pesquisas nessa área, ampliando ainda mais a variedade
de potenciais aplicações desse diol.

111
6 - REFERÊNCIAS:
Adelman, D.J., Charbonneau, L.F. e Ung, S. 2003. Process for Making
Poly(ethylene-co-isosorbide) Terephtalate Polymer. US6,656,577 USA, 2003.
Albertsson, A. C. e Karlsson, S. 1995. Degradable polymers for the future. Acta
Polym. 1995, Vol. 46, 114.
Anderson, E.M., Larsson, K.M. e Kirk, O. 1998. The use of Candida antarctica
Lipase B in Organic Synthesis. Biocatal. Biotransform. 1998, Vol. 16, 181.
Azim, H., Dekhterman, Z.J., Gross, R.A. 2006. Candida Antarctica Lipase B-
Catalyzed Synthesis of Poly(butylene succinate): Shorter Chain Building Blocks Also
Work. Biomacromolecules. 2006, Vol. 7, 3093.
Bachmann, F., Ruppenstein, M. e Thiem, J. 1998. Synthesis of a novel starch-
derived AB-type polyurethane. Macromol. Rapid Commun. 1998, Vol. 19, 21.
Billmeyer, F.W. 1984. Textbook of Polymer Science. New York : John Wiley & Sons,
1984.
Binns, F. Harffey, P., Roberts, S. e Taylor, A. 1999. Studies Leading to the Large
Scale Synthesis of Polyesters Using Enzymes. J. Chem. Soc., Perkin Trans.1. 1999,
2671.
Binns, F., et al. 1993. Enzymic polymerization of an unactivated diol/diacid system.
J. Chem. Soc. Perkin Trans. 1: Org. Bio-Org. Chem. 1993, Vol. 8, 899.
Binns, F., et al. 1998. Studies of Lipase-Catalyzed Polyesterification of an
Unactivated Diacid-Diol System. J. Polym. Sci. Part A: Polym. Chem. 1998, Vol. 36,
2069.
Bornschauer, U.T. e Kazlauskas, R.J. 2002. Hydrolases in Organic Synthesis. New
York : Wiley -VCH, 2002.
Brandenburg, C.J. e Hayes, R.A. 2004. Process to Produce Polyesters which
Incorporate Isosorbide. (Du Pont). US 6,818, 730 USA, 2004.
Braun, D. e Bergmann, M. 1992. Polymers from 1,4:3,6-dianhydrosorbitol. . J. Prakt.
Chem. 1992, Vol. 334(4), 298.
Busche, R.M., Donald, C.C. e Hardy, R.W.F. 1983. Production of Feedstock
Chemicals. Science. 1983, Vol. 219, 733.
Chatti, S., et al. 2002. Efficient synthesis of polyethers from isosorbide by
microwave-assisted phase transfer catalysis. Eur. Polym. J. . 2002, Vol. 38, 1851.

112
Chatti, S., et al. 2006. Synthesis and Properties of new Poly(ether-ester)s
Containing Aliphatic Diol Based on Isosorbide: Effects of the Microwave-assisted
Polycondensation. Eur. Poyml. J. 2006, Vol. 42, 410.
Chaudhary, A.K., Lopez, J. e Russel, A.J. 1997. Biocatalytic Solvent-Free
Polymerization To Produce High Molecular Weight Polyesters. . Biotechnol. Prog.
1997, Vol. 13, 318.
Clinton, W.J. 2000. Developing and Promoting Biobased Products and Bioenergy.
White House: The Executive Order. 2000, nº13134.
Copley, S.D. 2003. Enzymes with Extra Talents: Moonlighting Functions and
Catalytic Promiscuity. Curr. Op. Chem. Biol. 2003, Vol. 7, 265.
Cygler, M. e Schrag, J. 1997. Structure as a Basis for Understanding Interfacial
Properties of Lipases. Methods Enzymol. 1997, Vol. 284, 3.
Dalla Vecchia, R., Nascimento, M.G. e Soldi, V. 2004. Aplicações Sintéticas de
Lipases Imobilizadas em Polímeros. Química Nova. 2004, Vol. 27(4), 623.
Dong, H., Gui, C.S. e Shen, J.C. 1999. Study on the enzymic polymerization
mechanism of lactone and the strategy for improving the degree of polymerization. J.
Polym. Sci. Part A: Polym. Chem. 1999, Vol. 37(9), 1265.
Dordick, J.S. 1989. Enzymatic Catalysis in Monophasic Solvents. Enzyme Microb.
Technol. 1989, Vol. 11, 194.
—. 1991. Principles and Applications of Nonaqueous Enzymology. [A. do livro] H.W.
Blanch e D.S. Clark. Applied Biocatalysis. New York : Marcel Dekker, 1991.
Edlund, U. e Albertsson, A.C. 2003. Polyesters Based on Diacid Monomers. Adv.
Drug. Del. Rev. 2003, Vol. 55, 585.
Executive Steering Comittee, Renewables Vision 2020. 1999. The Technology
Roadmap for Plant/Crop-Based Renewable Resources 2020. Washington, DC : U.S.
Department of Energy, 1999. DOE/GO - 10099-706.
Faber, K. 1997. Biotransformations in Organic Chemistry. Berlin : Springer-Verlag,
1997.
Ferreira da Costa, M. A., Barrozo da Costa, M. F. 2002. Benzeno: uma questão de
saúde pública. Interciencia. 2002, Vol. 27, 201.
Flèche, G. e Huchette, M. 1986. Isosorbide. Preparation, properties and chemistry.
Starch/Staerke. 1986, Vol. 38 (1), 26.
Gross, R.A., Kumar, A. e Kalra, B. 2001. Polymer Synthesis by In Vitro Enzyme
Catalysis. Chem. Rev. 2001, Vol. 101, 2097.

113
Gutman, A. L., Knani, D., Kohn, D. 1993. Enzyme Catalyzed Ring-Opening
Polymerization of Ethylene Isopropyl Phosphate. J. Polym. Sci. Part A: Polym. Chem.
1993, Vol. 31, 1221.
Hopton, F.J. e Thomas, G.H.S. 1969. Conformations of some dianhydrohexitols.
Can. J. Chem. 1969, Vol. 47, 2395.
Hu, J., Gao, W. e Gross, R.A. 2006. "Sweet Polyesters": Lipase-Catalyzed
Condensation-Polymerization of Alditols. Macromolecules. 2006, Vol. 39, 6792.
IBGE. 2007. Levantamento Sistemático da Produção Agrícola. www.ibge.gov.br.
2007.
Jenkins, Victor F., Mott, Anthony e Wicker, Ronald J. 1963. Unsaturated
polyesters and coatings therefrom. GB 927786 Brit. Pat., 1963.
Kanca, U., et al. 2008. Iterative Tandem Catalysis of Racemic AB Monomers. J.
Pol.Sci. Part A: Pol. Chem. 2008, Vol. 46, 2721.
Kikukawa, K., Nozakura, S. e Murahashi, S. 1972. Cyclopolymerization of Divinyl
Esters of Dibasic Acids and Structure of Poly(Vinyl Alcohol) Derived from the
Polymers. J. Polym. Sci. Part A - Polym. Chem. 1972, Vol. 10, 139.
Klibanov, A.M. 1990. Asymmetric Transformations Catalyzed by Enzymes in
Organic Solvents. Acc. Chem. Res. 1990, Vol. 23, 114.
—. 1986. Enzymes that Work in Organic Solvents. Chemtech. 1986, 354.
Klostergaard, H. 1958. Esterification with Trapping Phase. J. Org. Chem. 1958, Vol.
23, 108.
Kobayashi, S., Uyama e Kimura, H. 2001. Enzymatic Polymerization. Chem. Rev.
2001, Vol. 101, 3793.
Kricheldorf, H. R. e Al Masri, M. 1995. New polymer syntheses. LXXXII. Syntheses
of poly(ether-sulfone)s from silylated aliphatic diols including chiral monomers. J.
Polym. Sci. Part A: Polym. Chem. 1995, Vol. 33(15), 2667.
Kricheldorf, H. R. e Gomourachvili, Z. 1997. Polyanhydrides. Part 10. Aliphatic
polyesters and poly(ester-anhydride)s by polycondensation of silylated aliphatic diols.
Macromol. Chem. Phys. 1997, Vol. 198(10), 3149.
Kricheldorf, H.R. 1997. "Sugar Diols" as Building Blocks of Polycondensates. Rev.
Macromol. Chem. Phys. 1997, Vol. C37(4), 599.
Kricheldorf, H.R., et al. 2003. Macrocycles 27: Cyclic aliphatic polyesters of
isosorbide. J. Polym. Sci. Part A: Polym. Chem. 2003, Vol. 41, 3414.

114
Kumar, A., Gross, R.A. 2000. Candida antarctica Lipase B Catalyzed
Polycaprolactone Synthesis: Effects of Organic Media and Temperature.
Biomacromol. 2000, Vol. 1, 133.
Kwon, C.H., Shin, D.Y. e Kang, J.W. 2007. Molecular Modeling and its Experimental
Verification for the Catalytic Mechanism of Candida antarctica Lipase B. J. Microbiol.
Biotechnol. 2007, Vol. 17(7), 1098.
Laane, C., et al. 1987. Rules for Optimization of Biocatalysis in Organic Solvents.
Biotechnol. Bioeng. 1987, Vol. 30, 81.
Lenardão, E.J., et al. 2003. "Green Chemistry" - Os 12 Princípios da Química Verde
e sua Inserção nas Atividades de Ensino e Pesquisa. Química Nova. 2003, Vol.
26(1), 123.
Lide, David R. 2005. CRC Handbook of Chemistry, Internet Version.
<http://www.hbcpnetbase.com>. [Online] 2005.
Linko, Y.Y., Wang, Z.L., Seppala, J. 1995. Lipase-catalyzed synthesis of poly(1,4-
butyl sebacate) from sebacic acid or its derivatives with 1,4-butanediol. J. Biotechnol.
1995, Vol. 40, 133.
Mahapatro, A., Kalra, B. e Gross, R.A. 2003. Lipase-Catalyzed Polycondensations:
Effect of Substrates and Solvent on Chain Formation, Dispersity and End-Group
Structure. Biomacromolecules. 2003, Vol. 4, 544.
Malhotra, S.V., Kumar, V. e Jaffe, M. 2007. Corn-Based Chemistry and its
Applications. The Bridge. 2007, Vol. 37(4), 17.
Montgomery, R. e Wiggins, L. F. 1946. Anhydrides of polyhydric alcohols. IV.
Constitution of dianhydrosorbitol. J. Chem. Soc. 1946, 390.
Okada, M., Okada, Y. e Aoi, K. 1995. Synthesis and Degradabilities of Polyesters
from 1,4:3,6-Dianhydrohexitols and Aliphatic Dicarboxylic Acids. . J. Polym. Sci. Part
A. Polym. Chem. 1995, Vol. 33, 2813.
Okada, M., Okada, Y., Aoi, K. 1996. Biodegradable Polymers Based on Renewable
Resources: Polyesters Composed of 1,4:3,6-Dianhydrohexitol and Aliphatic
Dicarboxylic Units. J. Appl. Polym. Sci. 1996, Vol. 62, 2257.
Pandey, A., et al. 1999. The Realm of Microbial Lipases in Biotechnology.
Biotechnol. Appl. Biochem. 1999, Vol. 29, 119.
Rotticci, D., Orrenius, C. e Hult, K. 1998. Molecular Recognition of sec-Alcohol
Enantiomers by Candida antarctica Lipase B. J. Mol. Catal. B: Enzymatic. 1998, Vol.
5, 267.

115
Sablong, R.l, Duchateau, R, e van Haveren, J. 2008. Incorporation of Isosorbide
into Poly(butylene terephthalate) via Solid-State Polymerization. Biomacromolecules.
2008, Vol. 9(11), 3090.
Sheldon, R.A. 1993. Chirotechnology: Industrial Synthesis of Optically Active
Compounds. New York : Marcel Dekker, 1993. p. 145.
—. 1996. Large-scale enzymatic conversions in non-aqueous media. [A. do livro]
A.M.P Koskinen e A.M. Klibanov. Enzymatic Reactions in Organic Media. Glasgow :
Blackie Academic & Professional, 1996.
Shuai, X., Jedlinski, Z. e Kowalckuk, M. 1999. Enzymic synthesis of polyesters
from hydroxyl acids. Eur. Polym. J. 1999, Vol. 35(4), 721.
Storbeck, R., Rehahn, M. e Ballauff, M. 1993. Synthesis and Properties of High-
Molecular-Weight Polyesters Based on 1,4:3,6-Dianhydrohexitols and Terephthalic
Acid. . Makromol. Chem. 1993, Vol. 194, 53.
Stross, P. e Hemmer, R. 1992. 1,4:3,6-Dianhydrohexitol. Adv. Carbohyd. Chem.
Biochem. 1992, Vol. 49, 93.
Thiem, J. Lüders, H. 1984. Condensation Reactions. Synthesis of
Polyterephthalates Derived from Dianhydrohexitols. . Polym. Bull. 1984, Vol. 11, 365.
Uppenberg, J., Hansen, M.T. e Jones, T.A. 1995. Crystallographic and Moleculas
Modelling Studies of Lipase B from Candida antarctica Reveal a Estereoespecific
Pocket for Secondary Alcohols. Biochem. 1995, Vol. 34, 16838.
—. 1994. The Sequence, Crystal Structure Determination and Refinement of Two
Crystal Forms of Lipase B from Candida antarctica. Structure. 1994, Vol. 2, 293.
Uyama, H. e Kobayashi, S. 1994. Lipase-catalyzed polymerization of divinyl adipate
with glycols to polyesters. Chem. Lett. 1994, 1687.
Uyama, H., Yaguchi, S. e Kobayashi, S. 1999. Lipase-Catalyzed Polycondensation
of Dicarboxilic Acid-Divinyl Esters and Glycols to Aliphatic Polyesters. J. Polym. Sci -
Part A: Polym. Chem. 1999, Vol. 27, 2737.
Uyama, H.i, Inada, K. e Kobayashi, S. 1998. Enzymic polymerization of dicarboxylic
acid and glycol to polyester in solvent-free system. Chemistry Letters. 1998, Vol. 12,
1285.
—. 2000. Lipase-catalyzed synthesis of aliphatic polyesters by polycondensation of
dicarboxylic acids and glycols in solvent-free system. Polym. J. 2000, Vol. 32(5), 440.
van As, B.A.C., Palmans, A.R.A. e Meijer, E.W. 2007. Iterative Tandem Catalysis of
Secondary Diols nd Diesters to Chiral Polyesters. Chem. Eur. J. 2007, Vol. 13, 8325.

116
van Dam, J.E.G., Klerk-Engels, B. e Rabbinge, R. 2005. Securing Renewable
Resource Supplies for Changing Market Demands in a Bio-based Economy.
Industrial Crops and Products. 2005, Vol. 21, 129.
Vollhardt, P.C. e Schore, N.E. 2002. Organic Chemistry. New York : W.H. Freeman
and Company, 2002.
Yang, Z., Russel, A.J. 1996. Fundamentals of Non-Aqueous Enzymology. [A. do
livro] A.M.P. Koskinen e A.M. Klibanov. Enzymatic Reactions in Organic Media.
Glasgow : Blackie Academic & Professional, 1996.
Zaks, A., Klibanov, A.M. 1988. Enzymatic Catalysis in Nonaqueous Solvents. J.
Biol. Chem. 1988, Vol. 263, 3194.
—. 1984. Enzymatic Catalysis in Organic Media at 100ºC. Science. 1984, Vol. 224,
1249.
—. 1988. The Effect of Water on Enzyme Action in Organic Media. J. Biol. Chem.
1988, Vol. 263, 8017.