biologia estrutural - laboratory of biomolecular systems · estruturais a serem na montagem de...
TRANSCRIPT

wfdaj.sites.uol.com.br
© 2006 Dr. Walter F. de Azevedo Jr.
Biologia EstruturalQualidade de modelos estruturaisProf. Dr. Walter F. de Azevedo Jr.
wfdaj.sites.uol.com.br
© 2006 Dr. Walter F. de Azevedo Jr.
Biologia EstruturalResumo SCOP Modelagem molecular
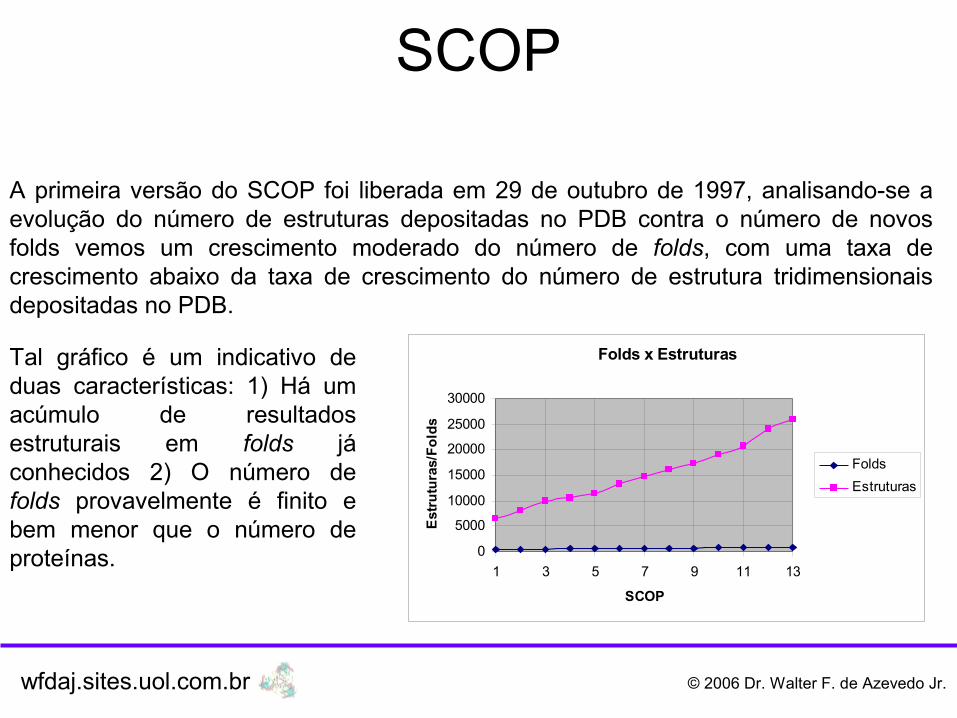
A primeira versão do SCOP foi liberada em 29 de outubro de 1997, analisando-se a evolução do número de estruturas depositadas no PDB contra o número de novos folds vemos um crescimento moderado do número de folds, com uma taxa de crescimento abaixo da taxa de crescimento do número de estrutura tridimensionais depositadas no PDB.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
SCOP
Tal gráfico é um indicativo de duas características: 1) Há um acúmulo de resultados estruturais em folds já conhecidos 2) O número de folds provavelmente é finito e bem menor que o número de proteínas.
Folds x Estruturas
0
5000
10000
15000
20000
25000
30000
1 3 5 7 9 11 13
SCOP
Est
rutu
ras/
Fold
s
FoldsEstruturas

Comparando-se a evolução do crescimento do número de famílias e superfamílias vemos uma taxa de crescimento mais próxima da taxa de crescimento das estruturas, mas mesmo assim, ainda mais baixo.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
SCOP
Estruturas X Superfamílias e Famílias
0
5000
10000
15000
20000
25000
30000
1 3 5 7 9 11 13
SCOP
Estr
utur
as/S
uper
amíli
as/F
amíl
ias
SuperfamíliasFamíliasEstruturas

Modelagem Molecular
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Algoritmo
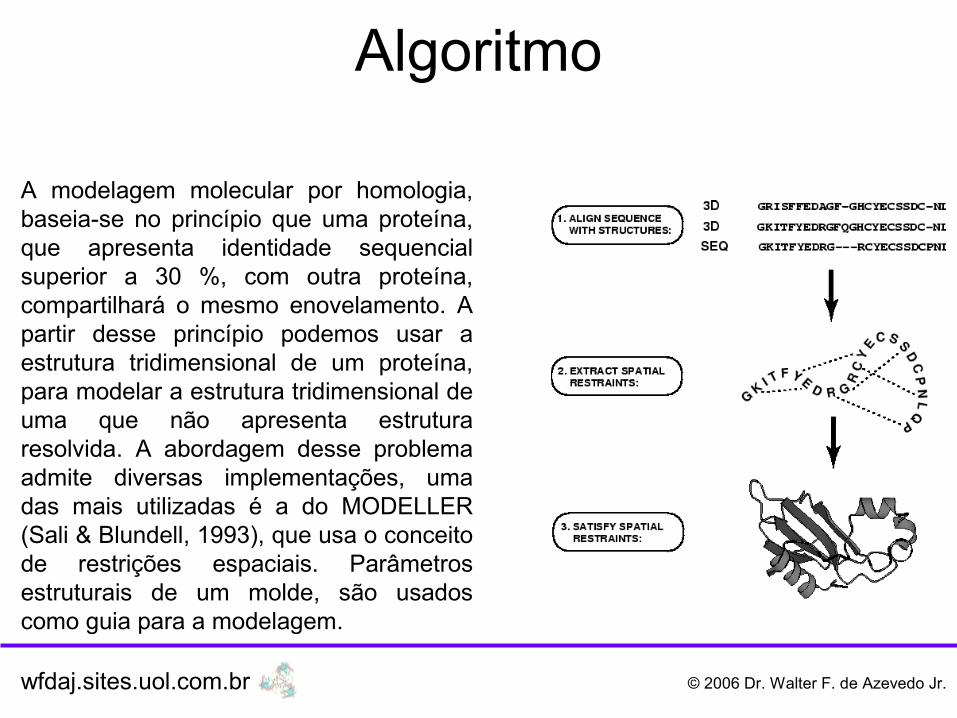
A modelagem molecular por homologia, baseia-se no princípio que uma proteína, que apresenta identidade sequencial superior a 30 %, com outra proteína, compartilhará o mesmo enovelamento. A partir desse princípio podemos usar a estrutura tridimensional de um proteína, para modelar a estrutura tridimensional de uma que não apresenta estrutura resolvida. A abordagem desse problema admite diversas implementações, uma das mais utilizadas é a do MODELLER (Sali & Blundell, 1993), que usa o conceito de restrições espaciais. Parâmetros estruturais de um molde, são usados como guia para a modelagem.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Algoritmo
A modelagem por homologia pode seguir o algoritmo mostrado à direita. Nesta abordagem o usuário seleciona um molde para a modelagem, é iniciado o alinhamento do molde com a sequência a ser modelada, para posteriormente ser construído o modelo. O modelo construído é avaliado, caso satisfaça a uma condição padrão é considerado adequado, caso contrário é selecionado outro molde, se possível. O processo de modelagem permite que sejam selecionados diferentes conjuntos de parâmetros estruturais a serem na montagem de modelos distintos.
Início
Identificação de moldes
Seleção de moldes
Alinhamento
Construção domodelo
Avaliação do modelo
Modelo OK?
Fim
Sim
Não

Início
Identificação de moldes
Seleção de moldes
Alinhamento
Construção domodelo
Avaliação do modelo
Modelo OK?
Fim
Sim
Não
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Algoritmo
O algoritmo do PARMODEL usa uma implementação paralela do MODELLER, o que permite automatizar o processo de modelagem usando programação de grão grosso, onde as modelagem são divididas entre os nós de um cluster de computadores do tipo Beowulf. Cada modelagem usa um conjunto distinto de restrições espaciais, o que permite varrer diversas condições iniciais, gerando modelos distintos para a mesma sequência, sem contudo ocorrer uma mudança drástica na estrutura. Podemos obter modelos com melhor qualidade estereoquímica, variando-se as restrições espaciais.
Último modelo ?
Não Selecionao melhormodelo
Fim
Sim
Uchoa et al., BBRC (2004)Silveira et al., J. Mol. Mod. (2005)
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Algoritmo do Parmodel
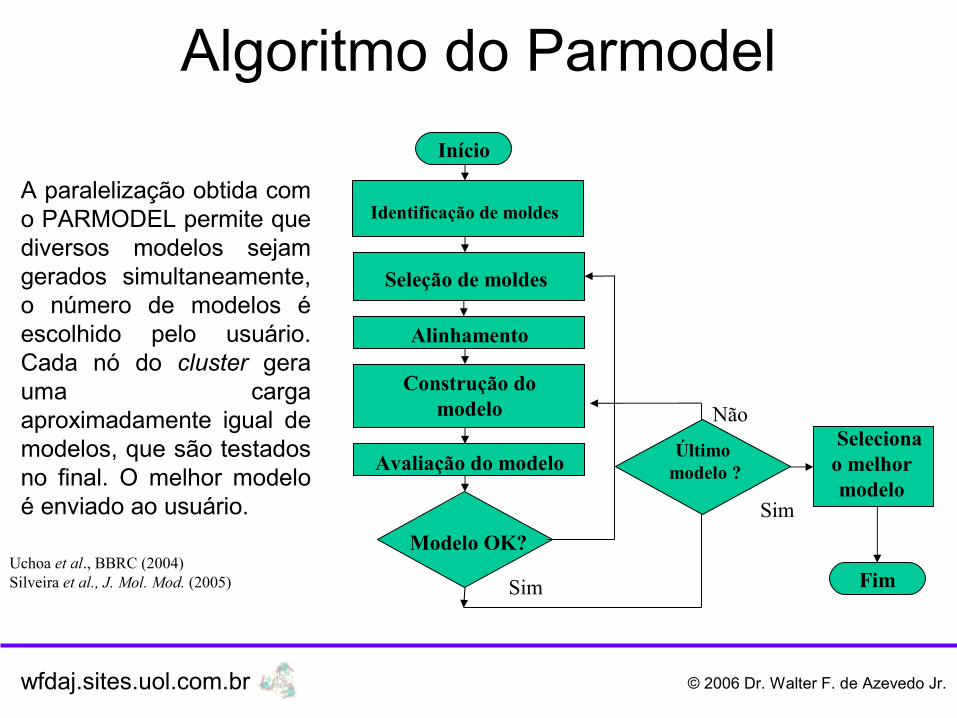
A paralelização obtida com o PARMODEL permite que diversos modelos sejam gerados simultaneamente, o número de modelos é escolhido pelo usuário. Cada nó do cluster gera uma carga aproximadamente igual de modelos, que são testados no final. O melhor modelo é enviado ao usuário.
Início
Identificação de moldes
Seleção de moldes
Alinhamento
Construção domodelo
Avaliação do modelo
Modelo OK?
Sim
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Computação Paralela
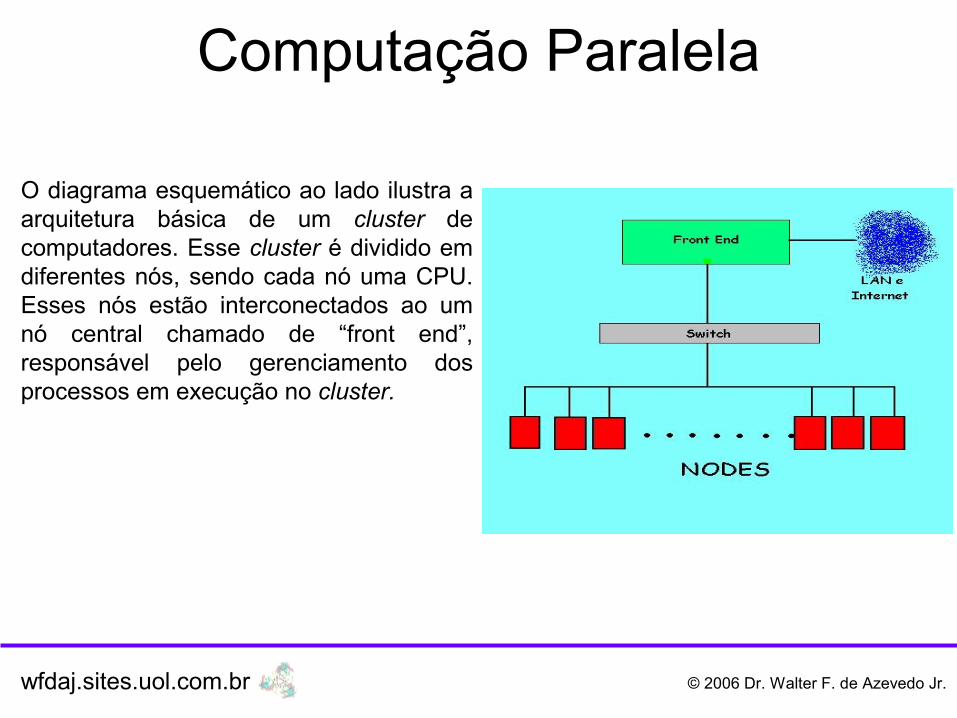
O diagrama esquemático ao lado ilustra a arquitetura básica de um cluster de computadores. Esse cluster é dividido em diferentes nós, sendo cada nó uma CPU. Esses nós estão interconectados ao um nó central chamado de “front end”, responsável pelo gerenciamento dos processos em execução no cluster.

Computação Paralela
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
A figura ao lado mostra 2 clusters do tipo Beowulf usados para modelagem molecular por homologia. O programa PARMODEL, para modelagem molecular por homologia, está instalado no clusterda direita.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Página de entrada do PARMODEL. O PARMODEL é um servidor web dedicado à modelagem molecular por homologia e análise da qualidade estrutural de modelos de proteínas. Clicando-se na opção “parmodel modeling” temos acesso à página de entrada para modelagem molecular por homologia on-line. A interface do PARMODEL é simples e intuitiva, permitindo acesso a uma ferramenta poderosa de modelagem molecular de forma rápida.
PARMODEL
Referência:UCHOA et al. .Biochem. Biophys. Res. Commun. 2004;325(4):1481-1486.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Página de entrada do PARMODEL. A página de entrada do PARMODEL descreve brevemente as principais ferramentas presentes no servidor web. Além do “parmodel modeling”, temos interfaces para otimização do modelo estrutural (parmodel optimization) e análise da qualidade de modelos de proteínas (parmodel assesment).

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Para modelagem molecular por homologia o PARMODEL solicita ao usuário o preenchimento de um formulário breve, com as informações necessárias para modelagem.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. O PARMODEL retornará os resultados da modelagem molecular por homologia por e-mail, assim como primeira etapa é solicitado ao usuário seu e-mail e uma identificação do usuário e da instituição.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Variando-se as restrições espaciais é possível a gerar até 1000 modelos para cada proteína a ser modelada. Caso o usuário deseje gerar 1000 modelos é necessário indicar que o número de modelos será de 1 até 1000, como indicado ao lado. Duas informações fundamentais para a modelagem molecular por homologia. 1) A sequência de aminoácidos da proteína a ser modelada (no formato fasta). 2) Arquivo PDB com as coordenadas atômicas do molde (template). Nesta versão é possível o uso de até 8 moldes. Os dois slides a seguir mostram os arquivos exemplos usados neste modelagem.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. A sequência da proteína a ser modelada é carregada no PARMODEL no formato fasta. Neste formato a primeira linha começa com o símbolo “>” maior que, normalmente seguido com comentário sobre a proteína, identificação, código de acesso, organismo etc. Da segunda linha em diante fica a sequência de aminoácidos usando o código de uma letra. Na figura ao lado temos o arquivo fasta para a sequência de aminoácidos da CDK3 humana, a proteína a ser modelada neste exemplo.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. O arquivo com as coordenadas atômicas do molde (template), neste caso o arquivo PDB (1HCL) com a estrutura da CDK2 humana na forma apo, a ser usado para a modelagem da CDK3. A CDK2 apresenta identidade sequêncial de 74,8 % com a CDK3, o que indica que a CDK2 é um bom molde para a modelagem da CDK3.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Na fase seguinte o usuário indica o método usado para obtenção do molde (template). Há 3 opções: Difração de raios X, NMR e modelagem. No exemplo aqui analisado a opção é difração de raios X. Outro campo a ser preenchido é sobre a presença de ligantes, neste caso coloca-se “0” visto que estamos modelando a enzima na forma apo.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. A etapa seguinte permite ao usuário indicar a forma como será avaliado os modelos gerados. Lembramos que podemos gerar diversos modelos (até 1000), para a mesma proteína. O PARMODEL retornará ao usuário o melhor modelo, segundo um critério estabelecido pelo usuário, como o PROCHECK ou o VERIFY3D, ou usarmos a função objetiva do programa MODELLER, como mostrado na figura ao lado. Depois de selecionar a forma de avaliação do modelo clicamos em “submit”, e termos a mensagem mostrada no slide a seguir.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. A modelagem foi aceita e os resultados serão enviados via e-mail para o usuário.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. É possível avaliar a evolução do processo de modelagem na página de entrada (www.biocristalografia.df.ibilce.unesp.br/bioxtal1.php ) escolhendo-se a opção “cluster”.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. A partir do uso da ferramenta “Ganglia Cluster Toolkit”, usada para avaliar o desempenho de clusters de computadores podemos verificar a entrada da modelagem solicitada ao PARMODEL, caso o clusteresteja sem modelagens no momento da solicitação, como mostrado na figura ao lado. O gráfico da direita acima indica o número de processos atualmente sendo executados no cluster. O segundo gráfico abaixo indica a porcentagem da CPU sendo usado. À esquerda é indicado o número de CPUs sendo usados no cluster, 15 no total.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Na página seguinte é mostrado o tráfego de rede e a velocidade do tráfego de rede, bem como um gráfico de pizza indicando o uso do cluster.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. O final da página indica o uso de cada nó (CPU) do cluster. Cinza indica uso, branco indica sem uso. A situação indicada mostra sem uso, esta é a situação onde não nenhuma proteína está em processo de modelagem.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Vamos analisar o uso do cluster logo após a entrada da modelagem da CDK3. Poucos minutos após a submissão da modelagem vemos claramente um aumento nos gráficos do número de processos (gráfico de cima) e da porcentagem de CPU usada (segundo gráfico de cima para baixo)

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. No terceiro gráfico vemos um pico do tráfego de rede, devido ao início da modelagem, e depois uma velocidade de transferência constante (quarto gráfico) devido à comunicação dos diferentes nós durante a modelagem. O PARMODEL divide a modelagem igualmente entre os nós, armazena os resultados e retorna ao usuário o arquivo PDB com o melhor modelo. Vemos também uma mudança no gráfico de pizza da esquerda, indicando um aumento do uso do cluster.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Os dezesseis gráficos do final da página indicam o uso de cada CPU, há uma CPU não sendo usada, e as outras quinze sendo usadas intensamente.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Ao final da modelagem vemos que o uso do clustervolta ao mínino.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
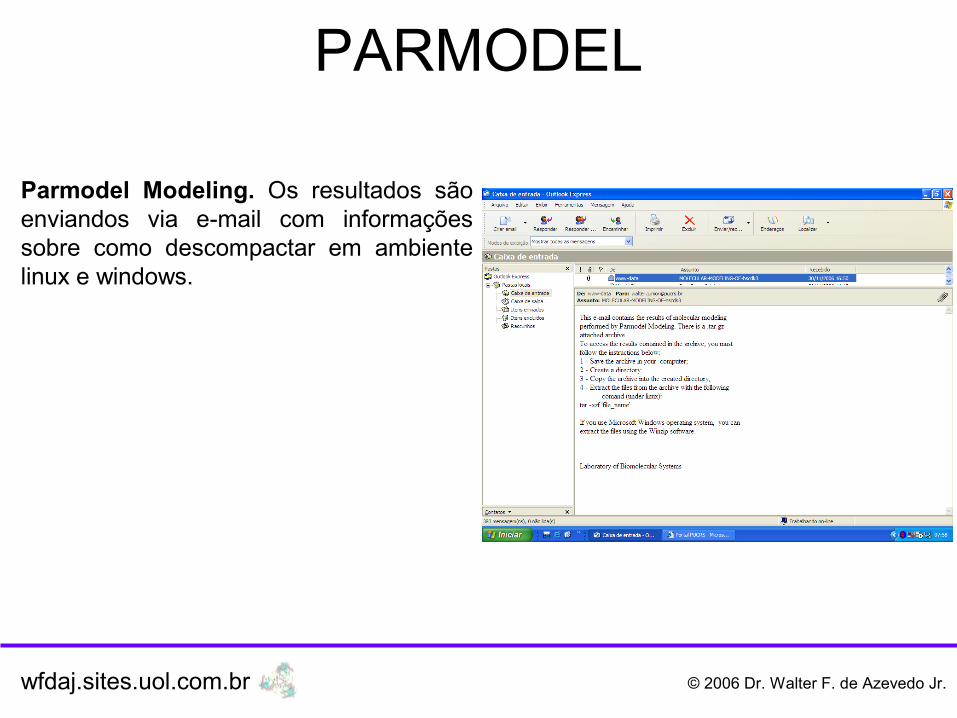
PARMODEL
Parmodel Modeling. Os resultados são enviandos via e-mail com informações sobre como descompactar em ambiente linux e windows.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. Em linux basta copiar o arquivo para um diretório e usar o comando tar –xvf “nome-do-arquivo”. Em windows pode usar o winrar ou winzip ou qualquer outra programa para descompactar. O arquivo compactado apresenta três arquivos, mostrados no próximo slide.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. O arquivo alignment.pap com o alinhamento da sequência de aminoácidos do modelo e do template, usado na modelagem. O arquivo pdb com o melhor modelo e o arquivo rank com o valor da função objetiva do MODELLER.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
PARMODEL
Parmodel Modeling. A função objetiva é listada da menor para a maior, com o respectivo modelo. Quanto menor a função objetiva do MODELLER melhor é o modelo. O arquivo PDB enviado ao usuário é aquele que apresenta menor valor para a função objetiva.

DBMODELING
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
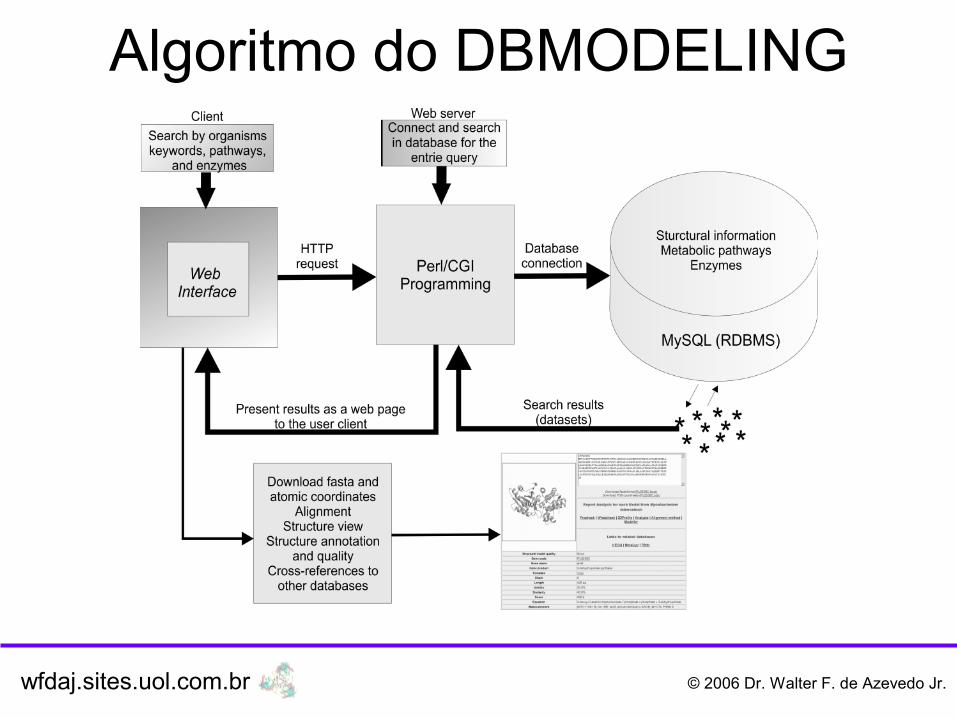
Algoritmo do DBMODELING

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
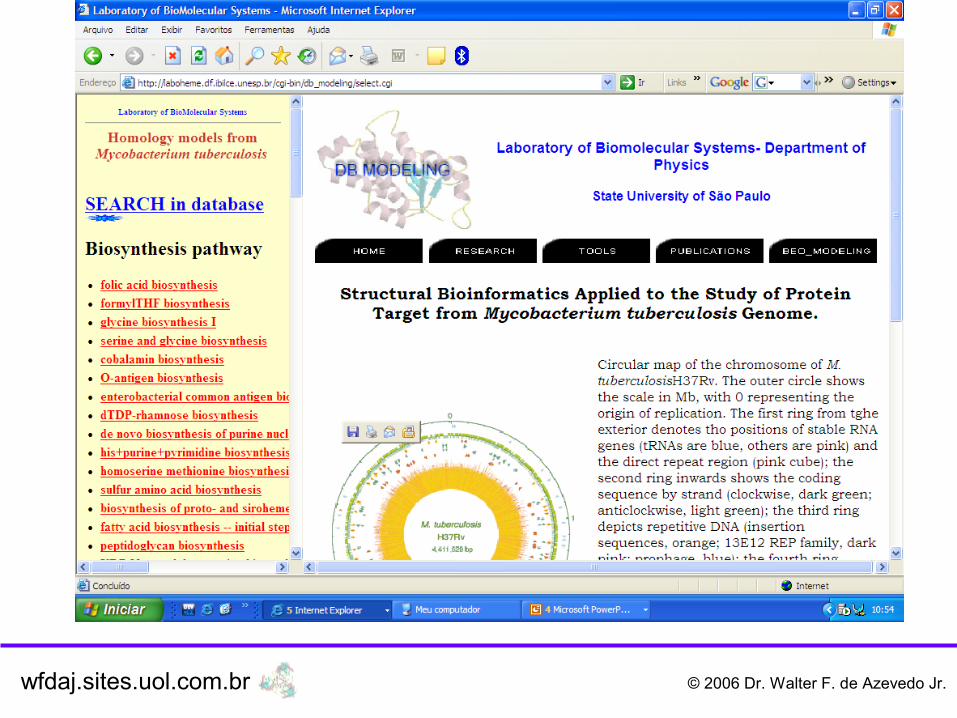
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
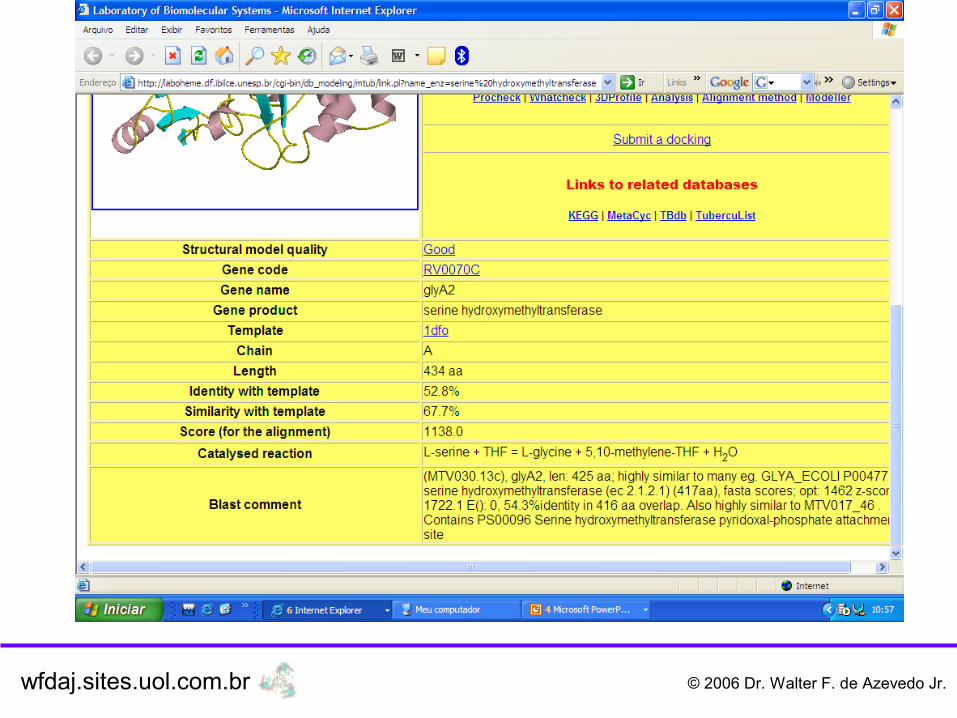
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

BOURNE, P. E. & WEISSIG, H. Structural Bioinformatics. John Wiley & Sons, Inc. New Jersey, 2003.
LASKOWSKI, R. A., MACARTHUR, M. W., MOSS, D. S., THORNTON, J. M PROCHECK: a program to check the stereochemical quality of protein structures. J. of Appl. Cryst. 26(2), 283-291, (1993).
LESK, A. M. Introduction to Protein Architecture. Oxford University Press, New York, 2001.
DA SILVEIRA, N. J. F., UCHOA, H. B., PEREIRA, J. H., CANDURI, F., BASSO, L. A., PALMA, M. S., SANTOS, D. S., & DE AZEVEDO, W. F. Jr. Molecular models of protein targets from Mycobacterium tuberculosis. Journal of Molecular Modeling 11, 160-166, 2005.
UCHOA, H. B., JORGE, G. E., DA SILVEIRA, N. J. F, CAMERA, J. C. Jr., CANDURI, F., DE AZEVEDO, W. F.Biochem. Biophys. Res. Commun. 2004;325(4):1481-1486.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Referências