apostila de qualidade cap ii
TRANSCRIPT

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 1/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 1 de 130
3- VALIDAÇÃO DE PROCESSOS:A VALIDAÇÃO NA INDÚSTRIA FARMACÊUTICA
O profissional que trabalha na área de validação dentro de uma indústria farmacêutica,
pode ser considerado como aquele de maior importância dentro da indústria, devendo estar capacitado acerca de todas as atividades realizadas pela empresa. Despertar a atenção da grandeimportância deste profissional faz com que se torne necessário entender o que é validação e qual ésua importância para a condução de qualquer tipo de atividade realizada dentro de uma plantafarmacêutica podendo influir de forma definitiva na qualidade do produto desejado.Daí se pergunta; Porque validar um processo de fabricação, limpeza, método de análise ousistema computacional que controle uma operação qualquer? O que determina esta necessidade?No caso da preparação de uma forma farmacêutica estéril, onde se detectou um fator decontaminação de 1,0 % do total das amostras analisadas (com base em 20 unidades conformepreconiza a USP), podemos afirmar que posteriormente, 8 vezes em 10 será comercializada umaamostra contaminada. Desta forma a garantia de que um produto farmacêutico qualquer sejaproduzido de forma segura, eficaz e reprodutível, faz com se torne extremamente importante,validar não só seu processo de fabricação, mas também, todas as demais atividades que possaminfluenciar na qualidade final do mesmo.
Assim sendo, como se pode definir o termo validação?
Segundo o FDA, Food and Drug Administration - USA, a validação seria a evidencia documentadade que um sistema se encontra em grau de fazer aquilo que se propõem de forma consistente edentro das especificações e atributos de qualidade preestabelecidos.
* Ou ainda; Somente uma documentação clara e evidente pode assegurar um alto nível desegurança a um processo específico, afim de que este esteja em grau de produzir de maneiraconstante e uniforme um produto com correspondência a suas especificações e características dequalidade.
Que vantagem se obtém com a implantação de um programa de validação?
Redução de perdas no processo; Menor incidência de desvios; Maior racionalização das atividades desenvolvidas; Redução dos níveis dos estoques de segurança; Criação de bases sólidas para o desenvolvimento de programas de treinamento.
Atualmente, podemos conduzir a validação de acordo com três diferentes abordagens;
- Validação Prospectiva: é conduzida antes do início da inserção de um produto na linha deprodução e comercialização, seja ele novo ou produto já em linha que tenha sofrido modificaçõessignificativas no seu processo de fabricação, tais como; modificação de equipamentos, processode fabricação, matérias-primas críticas ou dimensões de lote. Pode ser considerada como aabordagem utilizada antes do sistema entrar em funcionamento.
- Validação retrospectiva: abordagem que toma como base de dados, o histórico de produção de
lotes pregressos. Somente produtos fabricados por muito tempo na empresa podem ser validadospor esta metodologia. Deve se basear no mínimo, nas informações de 20 a 40 lotes consecutivos.Considera-se serem as informações existentes na empresa, suficientes para se atender àsexigências legais e de registro. Deve se observar a total qualificação de equipamentos einstalações da empresa quando da fabricação do primeiro lote considerado neste intervalo.
- Validação Concorrente: conduzida contemporaneamente ao processo produtivo e distribuição doproduto. Se aplica a produtos já a venda no mercado, mas que não possuam dados suficientespara suportar uma abordagem de validação retrospectiva.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 2/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 2 de 130
A implementação do programa de validação dentro da indústria farmacêutica transcorrerásegundo as seguintes etapas:
1- Desenvolvimento do Plano mestre de validação e validação de projeto.
2- Etapa de pré-qualificação:
- Definição das necessidades da empresa;- Seleção de fornecedores;- Inspeção de fornecedores;- Inspeção prévia da planta;- Comissionamento de equipamentos;- Definição das características do sistema.
3- Etapa de qualificação/ validação:- Qualificação de instalações;- Qualificação operacional (instalação, operação e performance);- Validação de processos. Sistema validado
4- Re-qualificação periódica e eventual revalidação.
1- PREPARAÇÃO DO PLANO MESTRE DE VALIDAÇÃO: A proposta deste documento seriaservir de base para o desenvolvimento de todo o programa de validação da empresa. Buscaexplicitar o entendimento de todas as atividades a serem desenvolvidas, assim como, determinar as responsabilidades não só sobre estas atividades, como também, por todo o processo devalidação propriamente dito. Este documento deverá incluir, no mínimo, os seguintes itens:
Aprovações e responsabilidades Abrangências Glossário de termos Esboço preliminar do design da planta Qualificação e especificação das matérias-primas Descrição do processo Divisão de áreas e suas classificações Descrições dos serviços necessários Descrição dos equipamentos Sistemas automatizados Arquivo do histórico dos equipamentos Documentos de construção (system master file) Protocolos necessários POPs Agenda do processo de validação Monitoramento ambiental e tipo de tratamento de efluente
Procedimentos analíticos e sua validação Programa de calibração de equipamentos .......... de treinamento .......... de manutenção preventiva .......... de controle de modificação .......... de controle de documentos Determinação do pessoal chave Matrizes de documentos e exemplos de protocolos, relatórios, NOPs e POPs

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 3/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 3 de 130
É necessário considerar diversos manuais de BPF como os do FDA, ANVISA e da CEE,quando se inicia a confecção do plano mestre de validação. Este deve conter ainda um índice queenumera todos as etapas a serem seguidas e um glossário de termos para que toda a equipe devalidação tenha a perfeita compreensão da terminologia utilizada. Pode se utilizar inclusive,glossários de termos de legislações vigentes relacionadas às BPFc.
Na página de aprovação do plano mestre de validação (PMV.), será necessária aparticipação das seguintes áreas:
Área Nome Assinatura DataProdução __/__/__
Engenharia __/__/__Segurança __/__/__Assuntos
regulatórios __/__/__
Gerencia devalidação
__/__/__
Como exemplos de termos para um glossário do PMV temos:
- Critério de aceitação: Nível de qualidade aceitável para um dado produto, lote ou unidadefabricada, assim como, seu critério ou nível de aprovação e rejeição.
- Change control: Sistema de controle formal pelo qual pessoas qualificadas de determinadasáreas, revêem mudanças atuais ou propostas das mesmas, desde que estas afetem diretamentena qualidade do produto e no seu status validado.
- D value: O tempo necessário para a uma dada temperatura se reduzir o número demicrorganismos a 90%.
- Áreas críticas: Locais onde produtos são mais expostos ao ambiente, em especial produtos
estéreis.
- Variáveis críticas de processo: Etapas do processo de fabricação de maior importância para oproduto em termosde sua qualidade. Áreas críticas podem possuir no máximo 100 000partículas/m3 de ar que sejam maiores que 0.5 m. Dependendo do tipo de produto fabricado,este valor pode ser muito menor.
- Worst case (pior caso): Conjunto de condições localizadas em torno de limites máximos emínimos nos quais existe a maior chance do processo não funcionar, quando comparado àscondições ideias.
Seguindo-se os itens necessários para a confecção do PMV, consideraremos a confecçãodos diagramas preliminares e o desing da planta. Esta etapa pode ser também denominada como
design validation, onde se deve descrever o fluxo de materiais da empresa, pessoal e produtos aserem fabricados, descrevendo como estes se enquadram às boas normas de fabricação vigentes.Cada área produtiva deve ser classificada de acordo com a atividade a ser desenvolvida, tendo aperfeita descrição e previsão dos serviços necessários para o bom desenvolvimento das atividadesrealizadas.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 4/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 4 de 130
Um exemplo disto pode ser visto no controle do ar exaurido e insuflado em cada setor;-Filtros HEPA para áreas estéreis - classes 100 e 10 000-Controle de umidade relativa e temperatura 20 oC/ 25-30% UR efervescentes e 30oC/ 60%UR paracápsulas gelatinosas duras.
Outro aspecto a se abordar seria a qualificação dos fornecedores de matérias-primas.Neste ponto serão definidos os critérios de aceitação para cada material a ser utilizado naempresa, estipulando-se suas características e testes a serem realizados. Com materiaisdestinados à fabricação de sólidos orais, como, por exemplo, deve ser avaliado o tamanho departícula, densidade aparente, umidade e teor entre outras propriedades. Certificados de análisedevem ser obtidos de todos os fornecedores, especificando-se as condições ideais dearmazenagem ressaltando-se que produtos com monografias descritas em farmacopéias devemter as mesmas como referências mínimas. Os certificados de análise devem ser de três diferenteslotes e o processo de fabricação e os principais subprodutos de síntese devem ser deconhecimento do comprador. Os fornecedores são obrigados a manter as embalagens dosmateriais fornecidos de acordo com as especificações da empresa. A possibilidade de programasde re-engenharia de fornecedores de matérias-primas e fornecimento tipo “just in time“ devem ser considerados.
Na descrição do processo, devem ser mostradas todas as suas etapas, determinação deetapas críticas, devendo isto ser feito para todos os produtos a serem fabricados. Deve se incluir da recepção de matéria-prima até a embalagem final. Diagramas de blocos deverão ser utilizadospara tal. Deve ser prevista, inclusive a forma de empilhamento dos produtos acabados.Na classificação de salas e áreas se considera as atividades a serem realizadas, número depessoas envolvidas, grau de contaminação máximo permitido, determinando-se ainda, quaissistemas serão utilizados para este controle. Estes sistemas são classificados como serviçosdevendo englobar a produção de água (purificada ou para injetáveis), ar condicionado, exaustão,sistemas elétricos, vácuo, ar comprimido e sistemas de segurança.
Todos os equipamentos da planta produtiva devem ser enumerados. Seus programas decalibração e manutenção preventiva devem ser previamente estabelecidos. Especial cuidado deveser dispensado a sistemas eletrônicos e informatizados presentes.
A qualificação de projeto refere-se à avaliação prévia realizada antes da compra de umequipamento ou reforma / construção de uma sala ou setor para checar se tais sistemas seenquadram dentro das BPFc.
2- ETAPA DE PRÉ-QUALIFICAÇÃO nesta etapa se realiza a revisão de todos os serviços, áreasde produção, fluxo de produção, equipamentos e sistemas computacionais. Os fornecedoresdevem ser inspecionados quanto às condições de produção, reprodutibilidade e assepsiabuscando uma confirmação do programa de fornecimento previsto. Posteriormente será iniciada aconfecção dos protocolos de validação. Nestes serão estabelecidos os sistemas a seremvalidados, contemplando, métodos de análise, fabricação, sistemas computadorizados e etc....Todos os documentos associados aos protocolos deverão ser igualmente confeccionados. Natabela abaixo são exemplificados os protocolos mínimos necessários para a implantação de umprograma de validação (Tabela 1).

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 5/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 5 de 130
Tabela 1: Protocolos de validação a serem implementados em um programa de validação:
Objetivo QI QO VP
Fluxo S N NGerador de luz S S NAr condicionado S S NÁgua S S SExaustão S S NNitrogênio S S NÁgua fria S S NVapor purificado S S NVácuo de processo S N NAr comprimidopurificado
S S N
Ar comprimidode processo
S N N
Vácuo purificado S S NVácuo de secagem S N NSistemascomputacionais
S S S
Processos defabricação
S S S
Métodos analíticos N S S
Todos os POPs devem ter sido revisados e confeccionados e posteriormente, reavaliadasde acordo com os resultados obtidos da implantação do protocolo. A empresa deve possuir umsuporte analítico bastante versátil para a implantação do programa de validação. Todos osequipamentos devem ser previamente calibrados e qualificados antes de se iniciar a validação deum processo. Empresas devidamente cadastradas pelo INMETRO-RBC podem ser usadas para a
calibração destes equipamentos. O treinamento dos funcionários deve ter sido iniciado e osmesmos devem ter conhecimentos suficientes sobre as BPFc. Um completo histórico dosequipamentos existentes na empresa, contendo especificações, certificados, manuais, curvas deperformance, ordem de compra, informações sobre o fornecedor devem ser organizados,economizando tempo fundamental para o processo de validação.
Em um protocolo de validação deve constar: Objetivos Responsabilidades Etapas críticas do processo com os equipamentos utilizados Parâmetros a serem medidos e variações aceitas Metodologia analítica para teste do sistema e critérios de aceitação Descrição detalhada do sistema e equipamentos Aprovação / responsabilidades das áreas envolvidas

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 6/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 6 de 130
3- ETAPA DE QUALIFICAÇÃO: o primeiro passo a ser executado nesta etapa será a qualificaçãodas instalações. A planta produtiva e áreas de serviços é avaliada segundo sua adequação àsBPFc, comparando-se a correspondência prática entre o projeto e sua execução, considerando-se
fluxo de materiais, pessoal, produção e material balance, classificação ambiental, pressurização deárea, e adequação de serviços e materiais de construção. Deverão ser realizados testes deestanquedade dos filtros de ar, avaliada a pressão diferencial das salas, a classificação das salasem repouso, a presença de todos os serviços necessários nas suas quantidades reais (vácuo, ar comprimido, eletricidade entre outros), a checagem, da possibilidade de limpeza e manutenção dosequipamentos, assim como a avaliação dos programas de manutenção em geral.
Em resumo, nesta etapa se observa:
- Limpeza e manutenção;- Adequação do espaço aos equipamentos e materiais a serem utilizados;- Adequação da área (classificação);- Controle de contaminação;- Sistemas computacionais e sua instalação;- Exame de toda a documentação;- Descartes de resíduos e possibilidade de manutenção;- Conferência da correspondência dos materiais e aparelhos às especificações do projeto;- Checagem dos aspectos de segurança EPC e EPI (equipamentos de proteção individuais ecoletivos.
Estando as instalações adequadas, se inicia a segunda etapa de qualificação, a chamadaqualificação operacional ou qualificação de equipamentos. Nesta etapa são verificadas ascondições operacionais dos equipamentos utilizados na produção e seus sistemas de alarme esegurança. Para a qualificação de um equipamento temos:
Design Qualification: Confirma se os requisitos de BPFc e produção do equipamento ousistema foram atendidos quando de sua compra. Deve se observar:
Definir as exigências básicas do projeto em termos simples; Especificar os parâmetros de processo a serem monitorados em detalhes e rever estas
especificações com o usuário final;
Checar com o fabricante o controle de fabricação do equipamento e o registro / teste demodificações críticas;
Inspecionar e testar o equipamento antes de seu embarque; FAT factory accetability teste,primeira etapa do comissionamento;
Preparar relatório final de qualificação de design.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 7/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 7 de 130
Tendo sido finalizada a etapa de QD, se espera que o plano mestre referente a esta
atividade tenha sido concluído. Este documento deve ser considerado como um documentoque se atualiza constantemente, a cada nova informação obtida durante a validação. Suavalidade média é de um ano. Nele deve constar além dos itens já citados:
Lista preliminar de equipamentos a serem validados;
Diagramas dos equipamentos;
Dados dos locais de instalação;
Agenda preliminar das adaptações dos serviços mecânicos e elétricos a serem realizados;
Resumo das especificações dos equipamentos;
Determinação das inter-relações entre diversas peças de um equipamento e diferentesequipamentos de uma linha (family tree);
Qualificação do fornecedor do equipamento (experiência, familiarização com a empresa e
confidencialidade).Nesta etapa se observa os conceitos básicos de BPEc e deve ser conduzida com aparticipação da equipe de engenharia da empresa e o corpo técnico do fornecedor.
Qualificação de instalação: Realiza-se a segunda etapa de comissionamento SAT (siteaccetability test) – testar o funcionamento do equipamento no seu destino. Confirma se oscomponentes específicos do equipamento foram instalados corretamente; voltagem; sentido derotação, vazão de água purificada etc... segundo suas especificações, assim como, se os mesmosse encontram calibrados e estas atividades documentados. Checa-se aspectos de manutençãolimpeza do equipamento no local instalado. Correlaciona-se se os componentes dos equipamentoscorrespondem ao projeto do mesmo e seu manual e ainda, se certifica os materiais utilizados nasua construção, garantindo a identidade dos mesmos. Como exemplo, se cobra a certificação doaço inox e soldas orbitais utilizadas em um duto para condução de WFI.
Esta etapa garante que o equipamento foi construído de acordo com a solicitação daempresa e instalado conforme a especificação do fabricante. Checa-se inclusive se os circuitoseletrônicos do equipamento e demais itens. È fundamental que haja a identificação doequipamento pelo uso de numeração seqüencial (TAG).
Procedimento para a qualificação de instalação: Cheque o número de série do equipamento antes de sua instalação; Garanta que o mesmo foi embalado segundo o especificado; Cheque que se todos os acessórios e manuais foram enviados junto com o equipamento; Verificar se toda a instalação foi realizada conforme projetado, inclusive a conexão com os
serviços necessários; Instituir e iniciar a rotina de calibração; Testes de IQ:
Confeccionar o módulo de qualificação das instalações do protocolo de validação. Nele deveconter :
Descrição do sistema; Esquemas eletrônicos e mecânicos;
Manual do equipamento; Componentes; Lista dos instrumentos de medida; Relatórios técnicos dos fabricantes;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 8/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 8 de 130
Testes e checagens realizadas.
Qualificação operacional: teste de funcionamento do sistema ou equipamento,verificando se ele funciona conforme previsto antes do início das operações; avalia-se ofuncionamento das várias partes do sistema de produção no intervalo de calibração. Comoexemplo, tomamos a pesagem de uma massa a ser homogeneizada em turbo-emulsificador, ondetemos limites máximos e mínimos de peso; verificação do real volume de um tanque.- Verificação de sistemas de alarmes e dispositivos de segurança: Temperaturas máximas detrabalho para um tanque de xarope ou homogeneizador de supositórios; variação da espessura decomprimidos e não enchimento de cápsulas; interrupção do funcionamento das hélices demisturador em sigma quando se levanta sua tampa.- Verificação dos sistemas automáticos e computadorizados dos equipamentos ou sistemas;- Verificação de umidade relativa, temperatura, partículas por metro cúbico, pressurização e outros
aspectos que influenciam no processo em funcionamento simulado;- Checagem se os valores medidos pelo equipamento (RPM, temperatura e etc … correspondem arealidade).
Qualificação de performance:
-Serão utilizado nesta etapa, placebos nas Quantidades idênticas às reais. Funcionamento da linha de embalagem com placebos de tamanho igual ao original; Checagem de velocidade de fluxo, temperatura e pressão em sistemas de envase com líquidos
com água; Abertura e fechamento de válvulas de um tanque e sua movimentação, quando carregado com
água; Em alguns casos, quando o equipamento é muito simples, nesta etapa se observará apenas
requisitos de BPFc em relação aos mesmos , principalmente se estes são muito antigos.
Algumas observações devem ser feitas;
O nível de atenção dedicado ao equipamento ou acessório é proporcional a necessidade daempresa e, sobretudo, ao impacto do mesmos a qualidade final do produto. Para o seu bomfuncionamento, o equipamento deve ter um elevado grau de controle direcionado ao mesmo,sendo fundamental a implementação de um eficiente programa de calibração, manutençãopreventiva. Em alguns casos estes serão os únicos pontos de controle de BPFc. Maior dificuldadede validação será observada em equipamentos com sistemas eletrônicos ou computacionais.
Cada aparelho ou linha de produção deve possuir um logbook para o registro de seu uso,produtividade, limpeza, calibração e manutenção. Uma linha de produção pode ter sua validaçãoresumida da seguinte forma;
1- Definição do uso;2- Definição das características técnicas;3- Delineamento do desing do sistema;4- Compra;5- QI;6-QO;7-Qualificação performance.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 9/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 9 de 130
Os seguintes documentos serão gerados nesta etapa:
Protocolo de validação Desing dos equipamentos Módulo de execução de teste Relatório de validação Procedimentos de utilização dos equipamentos Documentação de treinamento sobre utilização dos equipamentos

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 10/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 10 de 130
PROTOCOLO DE QUALIFICAÇÃO DE TANQUE DE PREPARO DE SOLUÇÃO
1 – Objetivo
Determinar as normativas utilizadas para qualificar os equipamentos do laboratório Tabajara,objetivando o atendimento das exigências da Agência Nacional de Vigilância Sanitária - ANVISA,em especial o disposto na RDC 210 de 2003. Estabelecer os requisitos necessários para que aqualificação de equipamentos realizada na empresa tenha um suporte técnico confiável, sendoacompanhada de evidências documentadas dos trabalhos desenvolvidos.
2 - AbrangênciasQualificação dos equipamentos utilizados nos processos produtivos, incluindo máquinas deenvase, dornas, etc., do Laboratório Tabajara. A qualificação de equipamentos será dividida emtrês etapas: instalação, operação e performance. As qualificações podem ser realizadas por equipamento, por áreas, contemplando os equipamentos em determinada área, ou por grupo deequipamentos, de acordo com a similaridade. Todas as etapas de qualificação devem ser documentadas em cada estudo.
3 - Responsabilidades
10 Garantia da Qualidade
Definir a abordagem para qualificação de equipamentos e sistemas conforme prioridadesdo programa de validação da empresa, de acordo com a criticidade do equipamento /sistema (ou grupo de equipamentos);
Estabelecer em conjunto com o Controle de Qualidade, os experimentos envolvidos naqualificação de operação e / ou performance;
Preparar junto ao setor responsável pelo equipamento em qualificação, os protocolos de
validação e matrizes de documentos a serem utilizados na qualificação (POP’s, Relatóriosde validação, livros de registro, especificações técnicas, manuais, etc.); Responder por resultados inesperados; Responsabilizar-se pela aprovação de toda a documentação produzida durante a
implantação do programa de validação; Determinar a participação de pessoas de outros setores como colaboradores do programa
de validação.
Controle de Qualidade
Oferecer suporte técnico para análises e coordenação para as análises relacionadas àsetapas de qualificação de operação e / ou performance;
Realizar as devidas amostragens; Realizar os testes f ísico-químicos necessários;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 11/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 11 de 130
ELABORAÇÃO: VERIFICAÇÃO: APROVAÇÃO:
Produção
Garantir a coerência nos protocolos e relatórios de validação; Auxiliar na execução de amostragens, quando necessário; Manter atualizada a lista de assinaturas dos envolvidos no processo; Checar a qualificação, manutenção e calibração prévia, dos instrumentos e equipamentos.
Manutenção
Dar suporte técnico para realização, verificação e aprovação das especificações, critérios
de aceitação e resultados obtidos na qualificação; Manter os equipamentos em perfeito estado de conservação seguindo todas as
determinações do programa de manutenção preventiva dos equipamentos. Co-executar com o setor produtivo a qualificação de instalação.
4- Guarda da Documentação Todos os documentos originais durante a validação, ficam sob a responsabilidade do
executante da validação; Os documentos concluídos serão arquivados na Garantia da Qualidade, após terem sido
devidamente aprovados.5- Memorial descritivo de equipamento:
5.1- Especificações Funcional: tanque de aço inox destinado a preparação de anestésico local
Tabajara antes de sua filtração esterilizante.5.2 – Especificações técnicas:1- No de TAG: 1642- Material de construção: aço inoxidável AISI 3043- Modelo: NC4- Fabricante: Quiminox5- Numero de serie: NC6- Data de fabricação: NC7- Localização: 4o Andar 8- Ativo fixo: 17949- Tensão de rede: 220 V10- Tensão de comando: NC11- Freqüência de rede: 60 Hz12- Pressão de ar comprimido: 200 m3/h13- Capacidade: 650 L
5.3 – Descrição de funcionamento: verificar se o painel de comando se encontra devidamenteenergizado e se o ar comprimido se encontra ligado. O equipamento é ligado, acionando ofuncionamento da agitação; o tanque é pressurizado com nitrogênio filtrado e a descarga deproduto é feita em válvula de alavanca no fundo do tanque. O mesmo possui sistema de clean inplace por vapor puro. Após a remoção do produto, o tanque é limpo, desmontado e esterilizado.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 12/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 12 de 130
5.4- Descrição do sistema / equipamento: tanque em aço inox 304, de capacidade de 650 L,provido de agitação mecânica impulsionada por motor elétrico. Contém 02 válvulas de diafragma(numeração 001 e 002), válvula de esfera 001, válvual de pressão 001 e manômetro para controle depressão. Provido de tubulação para fornecimento de serviços, vapor, nitrogênio e ar comprimido.
5.5- Controle de modificações: qualquer alteração no equipamento somente poderá ser realizadacom a aprovação da Coordenação de Garantia da Qualidade, avaliando se a forma de como estaalteração impactará no status qualificado deste equipamento e a necessidade de uma novaqualificação.
5.6 – Qualificação de instalação. Data de realização ___/___/___ :
Ponto observado Especificação Observado Conforme( S ) ( N )
TANQUE DE MISTURAExiste manual técnico doequipamento?
Presente Sim ( )
Existe certificado do material deconstrução do tanque ?
Aço Inox 316 Teste realizadopositivo
( )
O Tanque consegue manter estanque o nitrogênio insuflado ?
Estanquedade Mantém a pressão nomanômetro
( )
Havia certificado de calibração domanômetro
Calibrado Calibração vigente ( )
Foi realizado teste hidrostático? Não apresentavazamentos no tanque
Tanque semvazamentos
( )
O equipamento está identificado ? Número de TAGpresente
TAG presente ( )
As superfícies externas do tanquesão adequadas
Soldas decapadas epassivadas.Acabamento
Conforme ( )
O tanque possui capacidade 650 L 650 L Capacidade de 650L na marcaespecificada
( )
Tampa do tanque Não deve apresentar sinais de corrosão nemdanos que possamcausar malfuncionamento
Conforme asespecificações
( )
Vedação da tampa O silicone da tampautilizado como vedaçãose encontra em bomestado
Conformeespecificado
( )
As superfícies internas do tanque seencontram em bom estado
Soldas decapadas epassivadas.Acabamento polido
Conformeespecificado
( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 13/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 13 de 130
Ponto observado Especificação Observado Conforme( S ) ( N )
AGITADOR11 O agitador está identificado? TAG presente OK ( )
Fabricante identificado? WEG OK ( )
Qual a potência do motor? 0,37 Kw OK ( )Qual a rotação do motor? 1730 rpm OK ( )Qual a tensão de alimentaçãoelétrica?
220 V OK ( )
Qual a freqüência de funcionamento? 60 Hz OK ( )Qual o material das hastes e hélices? Aço inox 316 OK ( )Estado geral das hastes: Sem falhas e com fixação
perfeita. Não deveapresentar desgastes oudanos que possam levar acontaminação
OK ( )
Estado geral das hélices: Sem falhas e com fixaçãoperfeita. Não deveapresentar desgastes oudanos que possam levar acontaminação
OK ( )
Ponto observado Especificação Observado Conforme( S ) ( N )
VÁLVULA DE DIAFRAGMA12 A válvula está identificado? TAG presente OK ( )
Funcionalidade: Entrada de vapor notanque
NA NA
Fabricante da válvula: Sisto OK ( )Qual o material da válvula? Aço inox 316 OK ( )Qual o material da junta? Silicone OK ( )
Conexão 1 pol TC OK ( )Estado geral da válvula: Não deve apresentar falhas de conexão ouvazamentos
OK ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 14/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 14 de 130
Ponto observado Especificação Observado Conforme( S ) ( N )
VÁLVULA REGULADORA DE PRESSÃO13 A válvula está identificado? TAG presente OK ( )
Funcionalidade: Regular a pressão denitrogênio no tanque
NA NA
Fabricante da válvula: CKD OK ( )Modelo da válvula R 1000-BG-NT série 1622Qual o material da válvula? Aço inox 316 OK ( )Ponto observado Especificação Observado Conforme
( S ) ( N )MANÔMETRO
14 Componente está identificado? TAG presente OK ( )
Funcionalidade: Controlar a pressão denitrogênio no tanque
NA NA
Fabricante do manômetro: Fambras OK ( )Escala 0-11 Bar Há certificado de calibração? Presente e válido OK ( )
Estado geral da válvula: Não deve apresentar falhas de fixação e nãoapresenta danos quepossam causar malfuncionamento
OK ( )
Ponto observado Especificação Observado Conforme( S ) ( N )
SEGURANÇA15 O local possui dimensões
adequadas?
Sim OK ( )
A instalação do equipamento atendeas condições de uso
Sim OK ( )
Todos os EPIs e EPCs necessáriosestavam presente no setor: PPRA OK ( )
Existia POP de operação no setor Sim OK ( )Existia registro de treinamento depessoal no setor
Sim OK ( )
Ponto observado Especificação Observado Conforme( S ) ( N )
UTILIDADES16 As utilidades estão conectadas de
forma a facilitar a limpeza?
Sim OK ( )
O fornecimento de WFI estáfuncionando conforme previsto?
Sim OK ( )
O fornecimento de N2 estáfuncionando conforme previsto?
PPRA OK ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 15/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 15 de 130
Ponto observado Especificação Observado Conforme( S ) ( N )
UTILIDADESO fornecimento de vapor puro estáfuncionando conforme previsto?
Sim OK ( )
Estado da pintura das tubulações Não deve apresentar falhas e pontos decorrosão; deve seguir padrão de cor
OK ( )
Limpeza externa Não deve apresentar sujidades na parte externado componente
OK ( )
5.7 – Qualificação operacional/performance. Data de realização ___/___/___ :
Ponto observado Especificação Observado ( S ) ( N )Conforme
Cor
TANQUE DE MISTURAO Tanque mantém a pressão deoperação quando carregado ?
1,5 Bar por 5minutos Mantém a pressão nomanômetro
( )
Foi observado algum vazamento desolução anestésica com o tanquepressurizado?
Nãoapresentarvazamentosno tanque
Tanque semvazamentos
( )
Qual a rotação do motor durante afabricação do produto?
1730 -1300rpm OK ( )
Qual a freqüência de funcionamentodurante a fabricação do produto?
60 Hz OK ( )
VÁLVULA DE DIAFRAGMA17 A válvula de vapor apresentava
vazamento de vapor durante sua
utilização?
Sem vazamentos OK ( )
A válvula abre e fecha comfacilidade?
Conforme OK ( )
A vazão de vapor atende asnecessidades para limpeza dotanque, cobrindo toda sua superfícieinterna?
Conforme OK ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 16/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 16 de 130
Ponto observado Especificação Observado ( S ) ( N )Conforme
Corr
VÁLVULA REGULADORA DE PRESSÃO18 A válvula de controle de pressão
de nitrogênio apresentava
vazamento durante sua
utilização?
Manutenção de pressãono manômetro com a
entrada de nitrogênio notanque fechada
OK ( )
A vazão de nitrogênio atende asnecessidades para o funcionamentodo tanque?
5,0 bar de pressão nomanômetro
OK ( )
A válvula abre e fecha comfacilidade?
Conforme OK ( )
A vazão da linha de WFI se encontraconforme especificado ?
OK ( )
5.8 – Programa de manutenção preventiva: O equipamento em qualificação se encontra dentrodo programa de manutenção preventiva/corretiva da empresa, estando disponível todas as peçasde manutenção necessárias para a boa condução do mesmo.
5.9 - Não conformidades observadas: _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _________________________________________
5.10 - Observações: _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________
_______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _________________________________________

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 17/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 17 de 130
5.11- Conclusões:
_______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________
_______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _________________________________________
6 – Re-qualificação
Deve se revalidar os equipamentos a cada 03 anos ou realizar a revalidação completa em casosde:
Alterações nos equipamentos; Alterações de especificações técnicas, funcionais ou de desempenho; Alterações de especificações do processo de fabricação; Alterações de sistemas auxiliares. Remanejamento dos Equipamentos. Alterações de Lay Out.
8 - Bibliografia
Berry, Ira R. ; Nash, Robert A . Pharmaceutical Process Validation 2th edition, Revised andExpanded, 1996;
Plano Mestre de Validação Tabajara

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 18/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 18 de 130
A última etapa da validação da planta produtiva seria a validação de processo defabricação. A validação do processo de fabricação pode ser conduzida por 03 diferentesabordagens. Uma delas se baseia em dados históricos, enquanto as outras duas se baseiam emdados experimentais. A primeira é conhecida como validação retrospectiva, enquanto as outrasduas, como validação prospectiva (realizada antes do sistema entrar em funcionamento ou do
produto entrar no mercado) e a outra validação concorrente, realizada concomitantemente aofuncionamento do sistema.
Validação retrospectiva - Escolha do produto. O produto deve ser escolhido, considerando queseu primeiro lote foi fabricado com instalações e equipamentos qualificados, pelos seguintesaspectos:
Processo estável e robusto; Sem alteração por um período de tempo longo; 20 lotes consecutivos (arbitrário); Sem alteração de excipientes ou ativos; Sem alteração de equipamentos; Sem alteração de processo de fabricação.
A Garantia da Qualidade, em colaboração com a produção é a principal responsável pelotrabalho (veracidade dos fatos). A preparação do procedimento escrito contemplará asresponsabilidades do grupo de validação, os produtos a serem validados por ordem de prioridade(vendas, fora de linha, teor de ativo e tipo de formulação), a seleção de etapas críticas eparâmetros a serem medidos, a periodicidade de reuniões do grupo de trabalho e seu líder o“Follow up “ para achados inesperados e aprovações e a localização dos arquivos.O protocolo de validação deve conter:
Dados coletados; No de lotes estudados; Tratamento estatístico; Agenda de validação e data de aprovação.
Considerações gerais:
Considerar informações do SAC; Não observar rendimento como medida (soma de influências); Qualificação de fornecedores (ajuste de especificações) e variabilidade de
características das matérias-primas deve ser observado. Quando seiniciaram?
Loog Books e alterações da planta = Pode desqualificar um processo; Avaliar a veracidade dos batch records. Rejeitados ou reprocessos devem
ser excluídos.
Finaliza-se a validação, com a confecção do respectivo relatório de validação. Neste deve secalcular médias e desvios padrão de cada ponto de checagem de BPF e verificar se estesenquadram nas especificações do produto. Buscar estabelecer planilhas para as diferentesvariáveis monitoradas no desafio de processo estabelecido (Média, desvio padrão e teste T).

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 19/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 19 de 130
Validação concorrente
Preparar o diagrama de processo com as variáveis possíveis para cada operação unitária; Preparar fluxograma de processo; Determinar os pontos críticos e limites de especificação;
Acompanhar cada passo do processo – checagem de BPF; Procedimento de teste e amostragem (desafio) ; Correr 03 lotes consecutivos e dentro das especificações; O processo se inicia na pesagem e finaliza na embalagem secundária.
Validação prospectiva
Etapas iniciais: Desenvolvimento da formulação; Desenvolvimento do processo. Desing do processo: Preparar diagrama de processo; Matriz de influências; Procedimentos experimentais; Protocolos. Caracterização: Identificar as variáveis críticas para cada etapa; Estabelecer as tolerâncias máximas e mínimas;
Verificação: Ajustar o protocolo de validação; Determinar as variações do processo em condições de operação; Prepara documentos de transferência de processo; Finalizar as especificações de processo; Correr 3 lotes pilotos; Proceder a validação formal.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 20/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 20 de 130
EXEMPLO DE PROTOCOLO DE VALIDAÇÃO DE PROCESSO
1.Objetivo: descrever o procedimento experimental a ser utilizado na validação do processo defabricação do produto aplicando-se para tal a abordagem de validação concorrente, conforme
preconiza a rdc 210. o objetivo principal deste estudo de validação é fornecer evidênciasdocumentadas de que o processo de manipulação e embalagem está sendo realizado de acordocom as BPF atendendo ainda especificações e atributos de qualidade pré determinadas em seuregistro.
2. Abrangência: Este procedimento é aplicável ao processo de fabricação dos produtosChophytol e Passiflorine na unidade de produção da Millet Roux.
3. Responsabilidades:Setor de Garantia de Qualidade:
Revisão do procedimento fabricação e seu fiel cumprimento (Ordem de fabricação);
Revisão dos procedimentos operacionais de cada equipamento utilizado no processo;
Revisão de toda a documentação de qualidade relacionada ao processo de fabricação do
produto; Estabelecer as etapas críticas, testes a serem realizados, limites de aceitação e estratégia de
amostragem utilizada na etapa de desafio do processo de fabricação do produto; Checagem da prévia qualificação das matérias-primas, instalações e equipamentos utilizados
na fabricação do produto fabricado;
Checagem da prévia validação das metodologias analíticas utilizadas nos testes de desafio econtrole de qualidade do produto Fabricado;
Preparar o protocolo e o relatório de validação e os protocolos de change control referentes àfabricação do produto Fabricado;
Estabelecer o programa de treinamento referente à certificação de operadores para afabricação produto Fabricado.
Setor de Produção Farmacêutica:
Disponibilizar materiais, pessoal e utensílios necessários para a validação; Disponibilizar logbooks e demais documentações de qualidade sob a guarda deste setor;
Coordenar a execução dos três lotes consecutivos, alvos do estudo de validação;
Auxiliar no estabelecimento dos critérios de aceitação;
Auxiliar na amostragem durante a fabricação do produto
Revisar o protocolo e o relatório de validação preparados;
Aprovar o protocolo e o relatório de validação.
Controle de Qualidade;
Realização das amostragens; Realização dos testes físico-químicos e microbiológicos necessários;
Aprovação dos ensaios realizados durante a validação; Revisar o protocolo e o relatório de validação preparados;
Aprovar o protocolo e o relatório de validação.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 21/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 21 de 130
Engenharia:
Supervisão do serviço de montagem e manutenção de equipamentos e utilidades utilizados nafabricação do produto Fabricado;
Auxilio na checagem dos protocolos de qualificação de equipamentos e instalações;
Supervisão do programa de calibração de equipamentos de medição utilizados no processo defabricação produto Fabricado.
4. Considerações Preliminares: Todo esforço de validação aqui desenvolvido segue aabordagem concorrente, conforme descrito na RDC 210. As diretrizes gerais a serem seguidas seencontram relacionadas no protocolo geral de validação concorrente PPO XX-23. É pré-requisitopara a validação do processo de fabricação, a qualificação de todas matérias-primas, dosequipamentos utilizados na produção, das instalações onde este produto é fabricado e ainda,devem ser validados todos os procedimentos de limpeza de equipamentos, utensílios e área e asmetodologias analíticas empregadas na análise de produto final, intermediário e nos testes dedesafio de processo.Os seguintes documentos são utilizados como base para o desenvolvimento desteprotocolo:
Os equipamentos devem ser qualificados segundo o protocolo geral PPO XP-002; As instalações devem ser qualificadas segundo o protocolo geral PPO XP-003; As matérias-primas devem ser qualificadas segundo o protocolo geral PPO XP-002; Os métodos analíticos devem ser validados segundo o protocolo geral PPO XP-003; Os procedimentos de limpeza devem ser validados segundo o protocolo geral PPO XP-004; A validação do processo de fabricação segundo a abordagem concorrente segue o protocolo
geral PPO XP-005; Deve Ter sido observada a manutenção de todos os equipamentos de produção segundo o
PPO XP- 006; Todos os instrumentos de medição utilizados para monitoração e teste do processo devem
estar calibrados por órgão credenciado na RBC INMETRO segundo o PPO XP-007.
Para a validação do processo de fabricação, serão utilizados 3 lotes consecutivos. O produto final
produzido deve atender às especificações farmacopéicas (quando o caso) ou aquelasdeterminadas pela empresa como adequadas. Caso um lote se encontre fora destasespecificações ou não atenda os valores especificados como limites de aceitação no teste desafiode processo, deve-se fazer a alteração necessária no mesmo (emitir protocolo de controle demodificação) e outros três novos lotes devem ser testados para que o processo de validação sejaconsiderado como concluído.
4.2 Revalidação:O processo deverá ser revalidado a cada 20 lotes, podendo-se optar para esta revalidação pelaabordagem retrospectiva ou, no caso do processo estar apresentado desvios acima dos esperadospelo estado de controle estatístico, pela abordagem concorrente. Durante este período (decorrer de 20 lotes), o processo deve ter mantido seu status validado sem alterações. Devem ser confrontados os dados obtidos no programa de estabilidade (conduzido segundo PPO XP-024)
referente a estes 20 lotes com os resultados de revalidação de forma a se confirmar aadequabilidade do processo de fabricação.
Determina a necessidade de revalidação e conseqüentemente a emissão de um protocolo decontrole de modificação: Substituição de matéria-prima ou fornecedor qualificado; Troca de equipamento utilizado no processo; Alterações no procedimento de fabricação; Alterações nas dimensões de lote; Alterações do local onde o produto era fabricado;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 22/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 22 de 130
4.3 Amostragem: O processo de amostragem segue a normatização proposta no Militar Standart e na NBR 5426, nos planos de amostragem e procedimentos na inspeção por atributos. ABNT-Brasil, 1989.
5. Procedimento
5.1 Descrição do produto:
5.2 Avaliação de especificação e qualificação das matérias-primas utilizadas no processo.Todas as matérias-primas utilizadas no processo de fabricação do produto FABRICADO CREMECAPILAR tem suas especificações obtidas da Farmacopéia Européia 2000, Americana (USP 25)ou, em sua ausência, do Manual da CTFA.
Matéria PrimaEspecificação de Matéria Prima
Parâmetros críticos Especificaçãocompleta
Matéria- prima USP
Código 1XXXX-XFORNECEDOR QUALIFICADO S ( ) N ( )
Teor: XX% - PPO XX-01

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 23/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 23 de 130
5.3 Qualificação das instalações destinadas à fabricação do Fabricado 60 mL: Checar aadequação da área destinada à fabricação do produto em termos das BPFs (Boas Práticas deFabricação Vigentes), assim como de todos os serviços utilizados durante o processo.
Descrição Serviços utilizados Classificação
ambiental
Área Qualificada?
S ( ) N ( )PPO/Relatório No :
Verificado por:
Data:Sala de Pesagem HVAC
Qualificado?S ( ) N ( )
PPO ____________
Classe DP= 15 Pa S ( ) N ( )
PPO ____________
___/___/__
Boxe de FluxoLaminar parapesagem dematérias-primas
HVAC
Qualificado?S ( ) N ( )
PPO ____________
Classe AP= NA S ( ) N ( )
PPO ____________
___/___/__
Sala de Lavagem HVAC e PW
HVAC Qualificado?S ( ) N ( )
PPO ____________
PW Qualificada?S ( ) N ( )
PPO ____________
NCP=NA S ( ) N ( )
PPO ____________
___/___/__
Área de produção HVAC, PW e ar comprimido
HVAC Qualificado?S ( ) N ( )
PPO ____________
PW Qualificada?S ( ) N ( )
PPO ____________
Ar comp.Qualificado?S ( ) N ( )
PPO ____________
Classe DP=15PA S ( ) N ( )
PPO ____________
___/___/__

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 24/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 24 de 130
Descrição Serviços utilizados Classificaçãoambiental Área Qualificada?S ( ) N ( )PPO/Relatório No :
Verificado por:Data:
Sala de envase HVAC e Ar comprimido
HVAC Qualificado?S ( ) N ( )
PPO ____________
Ar comp.Qualificado?S ( ) N ( )
PPO ____________
Classe DP=NA S ( ) N ( )
PPO ____________
___/___/__
5.4 Equipamentos acessórios e utensílios: Checar a identificação, o status de limpeza doequipamento ou utensílio (verificar se o processo de limpeza é validado) e se o equipamento foiqualificado. No caso de alguma não conformidade, registrar no campo de observações e comunicar a Garantia da Qualidade para que as providências necessárias sejam tomadas
IDENTIFICAÇÃO AVALIAÇÃOItem
Descrição TAG Calibradoem:
Próximacalibração:
Qualificação:S ( ) N ( )PPO/Relatório No :
Verificado porData:
01 Tanque cilíndrico Sans Souci750 L com misturador emhélice Weg 10 Hp 3420 rpm3F em aço inox 304
T2 ___/___/__ ___/___/__ S ( ) N ( )
PPO ____________
___/___/__
02030405

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 25/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 25 de 130
5.5 Descrição do processo
A manipulação dos produtos fabricados deve ser descrita detalhadamenteapontando-se inclusive pontos críticos de controle e limites de aceitação:
5.6 Fluxograma de processo:A fabricação do produto Fabricado deve seguir o diagrama de fluxo do processo abaixo:Legenda: As setas representam os testes que serão realizados para a validação do processo. Osquadros amarelos representam os processos e equipamentos envolvidos, enquanto os verdes asmatérias-primas e ativos envolvidos:
5.7 Avaliação da reprodutibilidade do processo: Realizar a fabricação de três lotesconsecutivos de forma a se garantir a reprodutibilidade do processo. Nas etapas críticas doprocesso, deve se desafiar o mesmo em 03 valores de suas variáveis críticas, um mínimo, ummáximo e um valor intermediário, sendo o primeiro lote realizado com todos os parâmetros dodesafio em seus valores mínimos, no segundo o valor intermediário e o terceiro no valor máximo.Todas as atividades de fabricação devem ser acompanhadas, monitoradas e controladas e os
registros realizados nas tabelas abaixo. Caso se obtenha um produto fora das especificações devese iniciar novamente o estudo, realizando-se, no entanto, as modificações necessárias para o seuajuste.5.7.1 Checagem do processo de pesagem: Realizar a checagem da pesagem das matérias-primas a serem utilizadas no processo, observando sua procedência, procedimento de pesagem(limpeza da sala e dos boxes de pesagem, seus registros e procedimentos), calibração dasbalanças e liberação pelo Controle de Qualidade. Este procedimento deve ser realizado em cadaum dos três lotes consecutivos fabricados durante o esforço de validação.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 26/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 26 de 130
OF Nº: ______ Peso do Lote 1000 Kg
Produto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___BALANÇA UTILIZADA: TAG__________ BALANÇA UTILIZADA: TAG__________CALIBRADA ? S ( ) N ( ) CALIBRADA ? S ( ) N ( )PRÓXIMA CALIBRAÇÃO EM: ___/___/___ PRÓXIMA CALIBRAÇÃO EM: ___/___/___SALA LIMPA EM: ___/___/___ PPO No : __________ SALA LIMPA EM: ___/___/___ PPO No : __________BOXE LIMPO EM ___/___/___ PPO No : __________ BOXE LIMPO EM ___/___/___ PPO No : __________
ESPECIFICADO ENCONTRADOProduto Código Quantidade a
pesar Quantidade
PesadaConferido por: Laudo de análise
No
Verificado por: Data:
Observações:

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 27/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 27 de 130
5.7.2 Amostragem durante o processo:
Descrever cada etapa de amostragem e como a mesma deve ser realizada.
Rotulagem das amostras: todas as amostras colhidas durante o esforço de validação devem ser
rotuladas segundo o modelo descrito abaixo, e encaminhadas imediatamente para o laboratório decontrole de qualidade para análise.
AMOSTRA PARA ANÁLISE
LOTE DESTINADO À VALIDAÇÃO DE PROCESSOPRODUTO:____________________________________
LOTE:___________EQUIPAMENTO:________________
FASE:______________PESO:___________________
VOLUME:________________________________

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 28/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 28 de 130
5.7.3 Checagem da adequação das instalações: Durante a fabricação de cada um dos lotesem teste durante o esforço de validação, deve se realizar uma checagem em relação à adequaçãodas instalações utilizados na fabricação do Fabricado segundo as informações abaixo:
OF Nº: ______ Peso do Lote: 1000 KgProduto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
Local Especificação Encontrado Laudo ouregistro? (S) (N)
Verificado por:Data:
1- Almoxarifadode matérias -primas
Limpeza do local,ausência de sobras deMP e etiquetas delimpeza.
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
( ) ( )
2- Sala depesagem Limpeza do local,ausência de sobras deMP e etiquetas delimpeza e pesagem.Qualificação daInstalação. Ambienteclasse D.
Conforme ( )Não Conforme ( )
Registro delimpeza RL XP-01e PPO XP-002
( ) ( )
3- Sala deLavagem
Limpeza da sala eorganização do local.Ausência de utensílios emateriais estranhos aoprocesso.
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
( ) ( )
4- Sala deFabricação
Limpeza da sala eorganização do local.Qualificação dainstalação. Ambienteclasse D.
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
e PPO XP-002
( ) ( )
5- Sala deenvase
Limpeza da sala eorganização do local.Qualificação dainstalação. Ambienteclasse D.
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
e PPO XP-002
( ) ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 29/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 29 de 130
OF Nº: ______ Peso do Lote 1000 Kg
Produto: Fabricado 60 mL Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
Local Especificação Encontrado Laudo ouregistro? (S) (N)
Verificado por:Data:
1- Boxe depesagem defluxo laminar
Limpeza do local,ambiente classe A,qualificação dainstalação e ausência deutensílios no local.
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
e PPO XP-002
( ) ( )
5.7.4 Checagem da adequação dos equipamentos: Durante a fabricação de cada um doslotes em teste durante o esforço de validação, deve se realizar uma checagem em relação àadequação dos equipamentos utilizados na fabricação do Fabricado segundo as informações
abaixo:
OF Nº: ______ Peso do Lote 1000 Kg
Produto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
Equipamento Especificação Encontrado Laudo ouregistro? (S) (N)
Verificado por:Data:
Conforme ( )
Não Conforme ( )
Registro delimpeza RL XP-01
e PPO XP-022
( ) ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 30/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 30 de 130
5.7.5 Checagem do processo: neste etapa são avaliadas a reprodutibilidade do processo e aadequação do mesmo às Boas Práticas de Fabricação dos três lotes fabricados no esforço devalidação.OF Nº: ______ Peso do Lote 1000 Kg
Produto: Código: 10001Lote :_____________ Data de Fabricação: ___/___/___
Etapa Parâmetroobservado
Especificado Obtido Pontos críticos Realizado por:Data:
Controle de processo;Máquina: Lote: Limite ideal: 0.2% Início do envase:Final do envase:
Hora decoleta
P1 P2 P3 P4 P5 P6 P7 P8 P9 P10
Peso Médio:DPR:
Maior peso:Menor peso:Densidade:Observações e ajustes de máquina realizados:

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 31/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 31 de 130
5.7.6 Embalagem secundária:Especificações: O quadro abaixo lista as especificações das matérias-primas, material deembalagem e produto acabado relacionados à produção do produto.
OF Nº: ______ Peso do Lote 1000 KgProduto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
Material de Embalagem Especificação Material deEmbalagem
Conforme
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )
Cartucho S ( ) N ( )

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 32/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 32 de 130
5.7.7 Checagem do processo de embalagem secundária:OF Nº: ______ Peso do Lote 1000 Kg
Produto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
O rendimento do processo de embalagem obteve um rendimento de no mínimo igual a98% ?
Sim ___Não___
Se não, explique na seção de Comentários.
Todos os critérios de aceitação foram atingidos? Sim ___Não___
Se não, explique na seção de Comentários.Comentários:
Etapa deFabricação
Parâmetro Parâmetro de processo Valor Obtido Passou/Falhou
Fechamento dosfrascos
Pressão da rosqueadora4,0 a 6,5 [kgf/cm2]
Colocação de Bula
Frasco sem bula
NA NA
Encartuchamento Cartuchos íntegros efechados
NA NA
Rotulagem Velocidade da Linha NA

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 33/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 33 de 130
5.7.8 Pessoal: Deve ser avaliado como parte da validação do processo, se o pessoal envolvido seencontra treinado/certificado ou não para a atividade a ser executada.
OF Nº: ______ Peso do Lote 1000 Kg
Produto: Código: 10001
Lote :_____________ Data de Fabricação: ___/___/___
Colaborador Treinado para oprocesso
Existe registro? Verificado por:Data:
19 Roberto
Carlos
Montanha
Sadam
Osama BinLaden
7.7.9 Monitoramento ambiental: Neste ponto, se avalia se o local de fabricação do produto emquestão se encontra apto em termos de carga biológica para a sua execução.
Sala/Área Especificação Encontrado Verificado por:Data:
20 Sala de
manipulação 1000 ufc/mL
21 Sala de
envase10 000 ufc/mL
Sala embalagemsecundária
NA
NA = Não se aplica; contagem feita por plaqueamento.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 34/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 34 de 130
8. Resultados: Os resultados obtidos devem ser listados na forma de relatório de validação, paracada processo específico, onde os dados acima tabelados são compilados e conclusões sobre aadequabilidade do processo, sua reprodutibilidade, possíveis ajustes e a periodicidade e motivos
que levem a necessidade de sua re-validação são enumerados. 9. Relatório de validação:Deve constar no relatório de validação:
Produto;
Formulação;
Lotes testados;
Identificação dos equipamentos e áreas;
Plano de amostragem;
Método analítico utilizado;
Procedimento de limpeza;
Número de testes;
Descrição completa da amostragem; Critério de aceitação utilizado e justificativa;
Resultados obtidos;
Tratamento estatístico e análise de resultados;
Conclusão; conforme ou não;
Recomendações;
Assinaturas.
10. Aprovações:
Protocolo de Validação XXX Aprovado CQ:
____________________________________ Data ___/____/____
Protocolo de Validação XXX Aprovado GQ:
____________________________________ Data ____/____/____
Protocolo de Validação XXX Aprovado Produção:
____________________________________ Data ____/____/____
Protocolo de Validação XXX Aprovado Engenharia:
____________________________________ Data ____/____/____
11. Glossário de Termos: Listar todos os termos técnicos utilizados relacionados ao processo defabricação a ser validado para se obter homogeneidade na troca de informações eentendimento de todos os envolvidos.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 35/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 35 de 130
12. Anexos: São inseridos nesta seção PPOs de todas as atividades do processo que esta sendovalidado, os métodos de análise utilizados, protocolos de qualificação de instalações eequipamentos e matrizes de relatório de validação.
13. Referências Bibliográficas: PVG 01/01- Confecção, distribuição e normas gerais para a confecção de documentos de
validação;
Berry, Ira R. ; Nash, Robert A . Pharmaceutical Process Validation 2th edition, Revised and
Expanded, 1996;
Guide to inspections Validation of cleaning process. FDA, 2000;
Guide to inspections of Process Validation. FDA, 2000;
Hwang, R. Process Desing and Data Analysis for Cleaning Validation, Pharmaceutical
Tecnology, 112-115, jan, 1997.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 36/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 36 de 130
VALIDAÇÃO DE PROCESSO DE LIMPEZA:
Busca contemplar a validação de todos os processo de limpeza utilizados dentro da plantaprodutiva não só para as áreas de produção como também para todos os equipamentos eutensílios utilizados. Serão sujeitos a validação os processo de limpeza empregado, não só paraos equipamentos de produção (granuladores, compressoras, reatores e etc...), como para aslinhas de produção (blistadeiras, linhas de embalagem e similares), containers de matérias-primase equipamentos de laboratório. Normalmente, o grau de atenção dispensado, assim como, o rigor nos critérios de aceitação serão proporcionais ao impacto do equipamento ou sistema em questãona qualidade final do produto.
A primeira consideração a ser feita seria:
O QUE É LIMPO ?? Mais ainda; QUANTO LIMPO É O LIMPO????
A resposta a esta questão, aparentemente óbvia, mostra a complexidade do que seria avalidação de um processo de limpeza e sua importância. A eficiente limpeza de um equipamentoou área poderá evitar problemas gravíssimos, como;
DIAZEPAM
x
CITRATO DE SILDENAFIL
O FDA requer que todos os dados referentes a a validação dos processos de limpeza utilizadossejam capazes de demonstrar que houve a remoção total de todos os resíduos ou impurezaspresentes até um nível “aceitável”. Este critério de aceitação deve ser determinado pela empresa
em função das características de cada produto e de cada linha de produção. Estes procedimentosdevem ser conduzidos de forma tal que se considere o equipamento em questão, o tipo de produtonele processado e a possíveis interações deste com os agentes de limpeza.Logicamente, os critérios de limpeza serão notavelmente diferentes quando se trata da preparaçãode um shampoo em um reator de aço inox comparando-se a uma enchedeira de ampolas parainjetáveis. O Federal register em seu CFR 211.67 preconiza:Equpment cleaning and maitenance: A planta produtiva e os equipamentos de produção deverãoser limpos, mantidos e sanitizados com periodicidade adequada com a finalidade de prevenir avarias ou contaminações que possam alterar a segurança, identidade, título, qualidade ou purezade um produto farmacêutico, fazendo com que este não atenda requisitos oficiais e deestabilidade.Para se exemplificar a importância da validação de um processo de limpeza, em 1996 das 55warning letters emitidas pelo FDA, 21 destas se relacionavam com problemas associados a
cleaning validation. Nesta, subentende-se a limpeza química e microbiólogica.
Ao se iniciar o programa de validação de limpeza, deve ser confeccionado um documentoescrito que enumere todos os aspectos a serem abordados. Este é conhecido como Writtencleaning program. Neste deve constar:
Protocolo de validação Plano de validação Responsabilidades Documentação correlata

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 37/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 37 de 130
Detalhamento sobre os processos, equipamentos e materiais utilizados Métodos analíticos e sua validação Desvios em relação aos outros lotes precedentes Programas de treinamento Proteção dos equipamentos limpos Explicação racional dos limites de aceitação
Inspeção dos equipamentos limpos antes da validação Prazo máximo de limpeza e etiquetagem Registro e conservação dos documentos produzidos
O programa de validação deve ser adaptado ao tipo de equipamento e de produto emquestão, existindo diferentes cenários a serem confrontados;-Produtos biotecnológicos-Fármacos-Formas farmacêuticas finais
Deve ser validado ainda, o tempo limite entre a conclusão do processo e o início da
limpeza. Devem haver métodos específicos de análise para se adequar ao processo de validação,devendo ser comprovada a sensibilidade dos mesmos. Técnicas de amostragem específicasdevem ser desenvolvidos para cada caso específico.
A nível químico devemos analisar:
-Resíduos de princípio ativo-Resíduos de excipientes-Produtos de degradação-Resíduos de detergente e ou sanitizantes
A nível microbiológico devemos analisar:
-Água-D value-Curva de descontaminação
De forma similar ao que se observou para a validação de processos produtivos, a validação de umprocesso de limpeza se iniciará com a confecção do validation master plan. Neste documento,deverá constar:
Diagrama do fluxo de produção Equipamentos a serem limpos Análise crítica do processo de limpeza Procedimento analítico utilizado e técnica de amostragem Formato do protocolo de validação de limpeza
Recursos envolvidos e qualificação do pessoal Definição de critérios a serem aplicados para novos produtos Protocolos de suporte ( recovery factor; níveis microbiológicos e resíduos de detergente)
Recovery factor: Parâmetro utilizado para se avaliar a eficiência do processo de amostragem; ocálculo do fator de recuperação deve ser realizado nos seguintes aspectos;
-Parte do equipamento a testar -Tipo de material-Técnica de recuperação do resíduo

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 38/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 38 de 130
Ainda deve ser ressaltado: O produto deverá ser recuperado com as mesmas técnicas deamostragem para o caso real, sendo o equipamento sujo com o mesmo produto nas condiçõesmais próximas o possível daquelas tidas como reais.Esta determinação deverá ser feita em triplicata, devendo o mesmo ser superior a 60% daquantidade teórica para que se considere a técnica como adequada. Todos os procedimentos
operacionais devem ser seguidos conforme a realidade de produção.Critérios de aceitação: Os critérios de aceitação para a validação de limpeza são os padrões eespecificações com os quais o procedimento de limpeza deve ser confrontado para demonstrar aeficácia de remoção de princípios ativos, excipientes ou detergentes do equipamento ou área,garantindo ainda que a presença de microrganismos se encontre abaixo dos limites pré fixados.Estes critérios/limites de aceitação serão determinados com base nos seguintes pontos:
-Tipo de produção: Estéril, sólido, cosmético etc... .-Tipo de limpeza: Manual ou automática-Natureza do produto: Potência, toxicidade, solubilidade, alergenicidadeAntes de se buscar verificar o atendimento dos limites de aceitação, o procedimento de limpezadeve observar;
-Nenhum resíduo deve ser detectado visualmente após a limpeza-A quantidade limite da substância deve ser detectável pelo método analítico proposto-Após sua finalização o método de limpeza deve garantir a completa remoção do agente delimpeza-não mais de 0,1% da dose normal terapêutica do produto fabricado poderá estar presente na dosemáxima terapêutica do produto processado posteriormente após a limpeza
Caso este critério não seja aplicável por alguma razão, temos:
Não mais de 10 ppm de cada produto pode ser detectado nos produtos posteriormente fabricadosapós limpeza, 10 mg de produto por quilo de produto posteriormente fabricado.
Caso se considere o uso do produto e sua via de administração teremos:
Produtos de uso tópico: 0,1 - 0,01%
Produtos de uso oral: 0,01-0,001%
Produtos estéreis: 0,001- 0,0001%
Nestes limites devemos considerar ainda, o volume do lote fabricado, anterior e posteriormenteassim como a área do equipamento utilizado.Quando se considera a toxidade do produto, podemos ter como limite de aceitação a chamadaNOEL ( non observable effective level) que é calculada dividindo-se a dose ativa mais baixa dofármaco em questão por um fator de segurança, em geral 40, de acordo com o tamanho do lote.
Como são escolhidos os produtos para se desenvolver um procedimento de limpeza? Como sedetermina a ordem de prioridades da empresa?Nesta escolha, serão escolhidos os produtos para os quais não serão utilizados precedentes parasua limpeza, sendo observado:
-Política da empresa-Uso do método desenvolvido em produtos futuros-Solubilidade em água do produto-Afinidade com os excipientes-Solubilidade dos excipientes

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 39/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 39 de 130
-Toxicidade-Restrições ao emprego do produto
Com base no exposto, podemos afirmar que produtos muito tóxicos, como citostáticos edigitálicos, -lactâmicos ou psicotrópicos, não deverão utilizar precedentes, devendo ser seusprocessos de limpeza previamente validados.
Todos os produtos tidos como insolúveis (1,0 mg/L) e muito pouco solúveis ( 1mg/100mL) serãoalvos de validação. Deve ainda ser considerada a hidrofilia dos excipientes utilizados e o uso desubstâncias solubilizadoras que fazem com que haja a tendência a formar incrustações quando douso de água para limpeza (precipitação do ativo).A ED 50 do produto deve ser outro parâmetro a se considerar, devendo se organizar as prioridadesprimeiro com este parâmetro, seguido da solubilidade em água.
PREPARAÇÃO DO PROTOCOLO DE VALIDAÇÃO:
Neste documento, será descrito o processo a ser seguido quando da execução doprocedimento de limpeza e todas as demais informações a ele correlacionadas;
Descrição do objetivo do trabalho responsabilidades Descrição do produto:MatrizSolubilidade em águaDados toxicolígicos e DL 50
Dose terapêutica mínimaPosologia diária
Desenvolvimento do critério de aceitação
Descrição do processo de fabricação evidenciando-se os pontos críticos
Descrição dos equipamentos contendo suas geometrias e superfície total
Equipamento superfície decontato total(cm2)
Material Local de limpeza NOP
Misturador 20.000 aço inox 316 sala de lavagem 04-07
Peneirador mecânico
7.800 rede de tefloncom suporte em
aço inox
sala de lavagem 04-12
Granulador Diosna
130.000 aço inox in place 04-04
Bomba dosador 650 aço inox setup room 04-20

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 40/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 40 de 130
Determinar critérios de aceitaçãoBest caseWorst case – qual ativo deve ser monitorado; em geral o mais tóxico, menos solúvel emágua e tivo na menor dosagem
redução a 10 ppm Tipo de amostragemPlaceboSwabImersão
Método de análise utilizado e sua validação Avaliação dos resultados e indicação das ações eventualmente necessárias Conclusões e aprovação final do protocolo
Observações gerais:
produtos que se degradam facilmente tendo seus produtos de degradação a capacidade decatalisar o processo, devem receber atenção especial. Exemplo: Nifedipina - degrada com a luzUV (nitro derivados) e com a luz solar (nitroso derivados). Se a limpeza não for eficiente depois doterceiro lote haverão problemas.
Deve ser previsto no processo validado a periodicidade para um processo de sanitizacão total.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 41/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 41 de 130
VALIDAÇÃO DE MÉTODOS DE ANÁLISE
Considerando-se que atualmente, segundo os atuais padrões de qualidade exigidos na
maioria dos compêndios de boas práticas de fabricação vigentes em todo o mundo, para que umproduto farmacêutico tenha garantida sua eficácia e segurança, os métodos de análise utilizadospara assegurar a conformidade do fármaco em questão, em relação às suas especificaçõespreviamente estabelecidas, devem atender a padrões de exatidão e reprodutibilidade, garantidoscom a validação dos mesmos.Há vários conjuntos de normativas publicadas com o objetivo de fornecer diretrizes a seremseguidas quando se deseja validar um método analítico. A ICH (International Conference onHarmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use)publicou 02 (dois) guidelines; a USP (United States Pharmacopeia) tem um capítulo exclusivosobre o assunto; O FDA ( Food and Drug Administration), publicou em 1998 o guia de diretrizessobre a validação de procedimentos analíticos; segundo o Federal Food, Drug and Cosmetic Act,as monografias existentes na farmacopéias dos Estados Unidos da América constituem padrõeslegais, tendo todos os métodos analíticos a serem validados que seguir estes padrõesfamacopéicos vigentes.A validação de um método analítico é um processo que consiste de quatro etapas distintas: Validação do Software; Qualificação do Hardware (Instrumentação); Validação do Método; System Suitability Testing ;Sendo que o ICH já incorpora o System Suitability Testing ao item Validação do Método.
O processo começa com um software validado e o equipamento qualificado, daí entãopassa-se à etapa de desenvolver e validar o método analítico, fechando-se todo o processo numavalidação total. Cada passo é crítico no sucesso do processo como um todo. Antes de iniciar atarefa de validação de um método analítico é necessário qualificar o equipamento, pois docontrário se durante a execução da validação um problema for encontrado, será difícil identificar afonte do mesmo.Existem vários parâmetros a serem avaliados dentro da validação de um determinado método
analítico. Este, após Ter sido testado e atendendo aos critérios de aceitação, obteremos oendosso de que o método está validado e é confiável. Este conjunto de parâmetros é aplicável aqualquer tipo de metodologia analítica, seja ela cromatográfica, espectroscópica ou titulométrica.Na execução de um projeto de validação pode ser utilizado a terminologia e o conjunto deparâmetros propostos pela ICH por ser mais abrangente e segundo informações, o FDA já estábuscando harmonizar os guidelines da ICH e a USP eventualmente publicá-lo.
Na avaliação do ICH ( International Conference on Harmonization ) estes são os tipos deprocedimentos analíticos a serem validados:
1) Testes de IdentificaçãoServem para garantir a identidade do fármaco ou outra substância de interesse em uma amostra.
São normalmente efetuados por comparação de uma propriedade da amostra (i.e., espectro,comportamento cromatográfico, reatividade química, etc) com a de um padrão de referência.
2) Testes para quantificação de impurezas/ Testes de limites para controle de impurezasPodem ser tanto um teste quantitativo quanto um teste limite para a impureza na amostra. Os doistestes são feitos para refletir com exatidão, as características de pureza da amostra. Ascaracterísticas da validação são diferentes de um tipo de teste para outro.
3) Testes QuantitativosSão procedimentos utilizados para se medir a quantidade de fármaco ou outra substância de
interesse em uma formulação farmacêutica, ou a dosagem da matéria-prima pura (determinação

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 42/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 42 de 130
de teor ). Os mesmos parâmetros de validação também podem ser aplicados a determinações
associadas a outros procedimentos analíticos ( i.e., teste de dissolução).
O objetivo para o qual o método analítico se destina também influenciará na definição de quais os
parâmetros de validação precisam ser avaliados.
Os parâmetros típicos a serem observados na validação de um método analítico qualquer são osseguintes:
Exatidão;
Precisão ( Repetibilidade, Precisão Intermediária e Reprodutibilidade);
Especificidade/Seletividade;
Limite de Detecção;
Limite de Quantificação;
Linearidade;
Range; Robustez;
System Suitability.
A abrangência do método e seus parâmetros de validação a serem avaliados devem ser definidos
no início do processo e as seguintes perguntas devem ser respondidas mais breve o possível :
Que substâncias são de interesse na utilização do método?
Quais são os níveis de concentração esperados?
Quais são as matrizes das amostras?
Existem substâncias interferentes de interesse? Se sim, elas devem ser detectadas equantificadas?
Existem exigências regulatórias ou legislações específicas?
A informação gerada será quantitativa ou qualitativa?
Quais são os limites de detecção e quantificação requeridos?
Qual é a faixa de concentração esperada?
Quão robusto o método deve ser?
Que tipo de equipamento deve ser utilizado? O método é específico para um instrumento ou
ele deve ser utilizado pôr todos os instrumentos do mesmo tipo?
O método será utilizado pôr um específico laboratório ou deverá ser aplicável em qualquer
laboratório?
Que habilidades (capacidade técnica) terão os futuros usuários do método?
A tabela abaixo lista os parâmetros mais importantes utilizados na validação dos diferentes tiposde métodos analíticos.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 43/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 43 de 130
Tabela: Parâmetros avaliados em diferentes métodos analíticos
Tipo de Procedimento Identificaçãoo Teste p/ Impurezas Teor
Parâmetros Quantitativo limite
Exatidão - + - +
Precisão
Repetibilidade - + - +
Precisão Intermediária - + (1) - + (1)
Especificidade (2) + + + +
Limite de detecção - - (3) + -
Limite de Quantificação - + - -
Linearidade - + - +
Range (faixa) - + - +
- significa que este parâmetro não é normalmente avaliado;+ significa que este parâmetro é normalmente avaliado.
(1) Em casos onde a reprodutibilidade (vide item “Definições e Termos”) tiver sido feita a precisãointermediária não é necessária;
(2) A falta de especificidade de um método analítico pode ser compensada por outro
procedimento analítico que sirva de apoio;(3) Pode ser necessário a determinação do limite de detecção em alguns casos.
Os parâmetros Robustez e ”System Suitability Testing “ não estão na tabela mas devem ser
avaliados na execução da validação do método analítico.
Ainda segundo o ICH uma posterior revalidação pode ser necessária nas seguintes circunstâncias: mudanças na síntese da matéria-prima(fármaco); mudanças na composição da formulação farmacêutica; mudanças no método analítico.O grau de revalidação necessária vai depender da natureza das mudanças.
Também se considera como primordial o uso de padrões de referência para a obtenção de dadosexatos. Dois tipos de padrões podem ser encontrados:
Padrões compendiais; obtidos de fontes confiáveis como a USP, não necessitando decaracterização posterior, apenas cuidados específicos quando de sua manipulação; Padrões não compendiais: obtidos com certo esforço, devendo ser cuidadosamente
caracterizados para se garantir sua identidade, potência e pureza. Devem ser utilizados paratal, métodos absolutos para seu doseamento.
No caso de análises cromatográficas, pode se admitir o uso de padrões internos e externos paraquantificação da substância em estudo. O padrão externo de ser utilizado quando a amostra éanalisada em um cromatograma separado do padrão. Este método se baseia na comparação entrea resposta da amostra e do padrão pôr suas áreas na cromatografia líquida ou pôr intensidade ouabsorção da mancha em cromatografia da camada fina.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 44/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 44 de 130
Com o padrão interno, um composto de pureza conhecida é diretamente adicionado a amostra,não devendo interferir nas características da mesma, devendo se relacionar às áreas do padrãointerno e à da amostra, nisto sua concentração ser conhecida. É muito utilizado esta técnica paramatrizes biológicas e pequenas faixas de concentração.TERMOS E DEFINIÇÕES1. Método Analítico
O método analítico se refere ao modo de executar a análise. Ele deve descrever em detalhes ospassos necessários para se executar cada teste analítico. Ele deve incluir mas não se limita a :a amostra, o padrão de referência, as preparações dos reagentes, o uso dos equipamentos evidraria, geração da curva de calibração, o uso das fórmulas para cálculos, etc.
2. Validação de Método AnalíticoEvidência documentada, obtida por meio de experimentação que demonstra ser o métodoanalítico adequado e confiável para o objetivo ao qual se propõe.
3. EspecificidadeEspecificidade é a habilidade de medir/determinar com exatidão e especificamente a substânciade interesse na presença de outros componentes que possam estar presentes. Normalmenteestes podem incluir: impurezas, produtos de decomposição, ingredientes ativos.Deve-se garantir que a resposta encontrada seja devido à substância de interesse somente.
4. LinearidadeA linearidade de um método analítico é a sua habilidade (dentro de uma faixa definida) de obter resultados que são diretamente proporcionais à concentração (quantidade) da substância deinteresse na amostra analisada.Para o estabelecimento de linearidade, o uso mínimo de 05 (cinco) concentrações érecomendado.
5. Limite de DetecçãoO limite de detecção de um método analítico é a menor quantidade da substância de interessena amostra que pode ser detectada mas não necessariamente quantificada como um valor
exato.
6 Limite de QuantificaçãoO limite de quantificação de um método analítico é a menor quantidade da substância deinteresse na amostra que pode ser determinada quantitativamente com adequada exatidão eprecisão.O limite de quantificação é um parâmetro para ensaios quantitativos para baixos níveis decompostos nas amostras, e particularmente utilizado para determinação de impurezas eprodutos de degradação.
7 Exatidão (Acurácia)A exatidão é a medida de proximidade de concordância entre o valor que é aceito como valor verdadeiro convencional ou valor de referência aceito e o valor encontrado (valor esperado x
valor encontrado). É medido como a percentagem de substância recuperada na análise, ao secontaminar placebos (simulação da matriz na qual não se encontra a substância em análise)com várias quantidades diferentes da substância em estudo.Para documentação da exatidão o guideline do ICH sobre metodologia recomenda efetuar nomínimo nove determinações , utilizando no mínimo 3 níveis de concentração cobrindo a faixaespecificada em triplicata.
8. Precisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 45/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 45 de 130
A precisão de um procedimento analítico expressa a proximidade de concordância (grau deespalhamento) entre uma série de determinações obtidas de múltiplas análises de uma mesmaamostra homogênea sob as condições prescritas no método analítico.A precisão pode ser considerada em 03 (três) níveis: Repetibilidade; Reprodutibilidade;
Interlaboratorial.A precisão deve ser investigada utilizando-se amostras autênticas e homogêneas. Contudo senão é possível obter uma amostra homogênea, a precisão pode ser investigada usando-seamostras artificialmente preparadas ou uma solução amostra.A precisão de um método analítico é normalmente expressa como variança, desvio padrão oucoeficiente da variação de uma série de medidas.
8.1RepetibilidadeA repetibilidade expressa a precisão sob as mesmas condições operacionais durante um curtointervalo de tempo.
8.2 ReprodutibilidadeA reprodutibilidade expressa variações dentro do mesmo laboratório: diferentes dias, analistas eequipamentos diferentes.
8.3 InterlaboratorialO estudo interlaboratorial expressa a precisão entre diferentes laboratórios (estudoscolaborativos, normalmente aplicados para padronização de metodologia).
9. Faixa (Range)A faixa de um método analítico é o intervalo entre as concentrações (quantidades) inferior esuperior da substância de interesse na amostra (incluindo estas concentrações) para o qual ométodo tem um adequado nível de precisão, exatidão e linearidade.
22 RobustezRobustez é a capacidade de um método de permanecer inalterado ao sofrer pequenas mas
deliberadas variações em alguns de seus parâmetros. A robustez de um método é avaliadovariando-se parâmetros tais como: percentual de solventes orgânicos, pH ou força iônica emuma fase móvel (CLAE), ou ainda a temperatura, tempo de extração da amostra, etc. edeterminando-se o efeito (se houver) nos resultados obtidos ao se utilizar o método.De acordo com os guidelines do ICH, a robustez de um método analítico deve ser avaliada noinício do processo de desenvolvimento do mesmo. E ainda, se os resultados de um métodosão suscetíveis a variações em seus parâmetros, estes devem ser adequadamentecontrolados e deve-se documentar as precauções cabíveis na utilização do método.
23 System Suitability Testing
System suitability testing é uma parte integral de muitos métodos analíticos. Os testes sãoelaborados no conceito de que o equipamento, as partes eletrônicas, operações analíticas eamostras a serem analisadas constituem um sistema único (um todo) e podem ser avaliadoscomo tal. Os parâmetros de teste para avaliação de System Suitability são estabelecidosindividualmente para cada método analítico a ser validado, dependendo das suascaracterísticas. Se presta como forma de avaliar o “status” validado do método analítico.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 46/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 46 de 130
Quando se deve re-validar um método analítico???
Quando se altera a matriz; excipiente, detergente ou sanitizante; Quando se altera o equipamento ou sistema que esta sendo limpo; Quando se faz alterações no método; substituição de um detetor de UV por um de índice
de refração, por exemplo; Aparecimento de desvios não aceitáveis no mesmo.
SEMPRE É IMPORTANTE LEMBRAR QUE A AUTOMAÇÃO DE AMOSTRADORES,SISTEMAS DE INJEÇÃO OU QUALQUER OUTRO ASSESSÓRIO DO EQUIPAMENTO DE
ANÁLISE PODE, PELA PRESENÇA DE COMPONENTES ELETRÔNICOS E OU SOFTWEAR,TORNAR A VALIDAÇÃO DO MÉTODO MAIS DIFÍCIL.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 47/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 47 de 130
Boas Práticas de Fabricação Vigentes BPFv:
Pode se classificar as BPF como sendo um conjunto de normativas, tradicionalmenteligadas aos órgãos reguladores sanitários que visam garantir a qualidade, eficácia terapêutica,
segurança e reprodutibilidade de resultados de um produto farmacêutico ou qualquer outro sujeitoa controle sanitário. O primeiro guia de fabricação de medicamentos data de 1963, tendo sidoemitido pelo FDA sob o nome de GMP (good manufacturing pratices). A necessidade de se criar este guia, surgiu com o reconhecimento em 1938 de acidentes causados pôr elixires desulfanilamida com etileno glicol, onde o aumento acidental da concentração deste excipiente levouao óbito de alguns usuários. Daí a necessidade de se regulamentar a fabricação de ummedicamento e garantir conseqüentemente sua segurança e eficácia. A confirmação final destanecessidade veio com a descoberta dos efeitos secundários teratogênicos da talidomida e dasfreqüentes contaminações cruzadas durante a fabricação de pinicilinâmicos e dietilbestrol. Assim,na sua X assembléia mundial em 1967, a OMS (Organização Mundial de Saúde), solicitou oestabelecimento de Normas Corretas de Fabricação e controle de medicamentos, sendorecomendado ao seus membros a aplicação das mesmas, tornando-as obrigatórias em todomundo em 1971.
Resumo sucinto da evolução das BPF; Em 1978 surge o conceito de validação 1982; Novo manual OMS Em 1983 surgem as GMP britânicas 1988 GMP da Comunidade européia 1992; Edição de novo manual da OMS 1993-5 GAMP; Boas Práticas de Fabricação automatizadas.
E o que vem a ser as BPF ??Pode se afirmar que este conjunto de normativas é um dos componentes da garantia de
qualidade que visa assegurar a fabricação de maneira homogênea e controlada de qualquer tipode medicamento, com padrões de qualidade adequados e de acordo com os requisitos legais paraa sua comercialização. Esta qualidade deve ser obtida em todos as etapas de fabricação doproduto, não apenas, averiguada no seu ponto final. Além deste parâmetro, as BPF, buscamgarantir a eficácia e segurança destes medicamentos. Eficácia : Capacidade de um medicamento qualquer de conter a quantidade e tipo de princípio
ativo idêntica a rotulada e a capacidade de liberar este princípio ativo quando administrado, deacordo com a biodisponibilidade prevista.
Segurança: Garantia da perfeita atuação do medicamento quando administrado, em termos desua atividade farmacológica e efeitos colaterais.
Se este conjunto de normativas pertence a Garantia de qualidade, o que vem a ser esta? Gerenciamento do conjunto de questões e atividades que envolvem individual ou
coletivamente a qualidade do produto fabricado, viabilizando sua utilização final. A política de qualidade da empresa seria a gestão da qualidade.
Pode se tomar como objetivos básicos de qualquer programa de BPFv:
Evitar contaminações, externas e cruzada;
Evitar misturas acidentais;
Garantir a rastreabilidade da fabricação e distribuição de um produto qualquer;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 48/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 48 de 130
Garantir a identidade e teor do constituinte ativo ou do produto como um todo.
As Boas Práticas de Fabricação, seriam nada mais que o conjunto de regrasdocumentadas que seguidas garantiriam o funcionamento da empresa segundo a ótica daqualidade e segurança total relacionada ao produto. Estas regras serão traduzidas na empresa soba forma de Procedimentos, Normas operacionais padrão ( POP e NOP), instruções de trabalho,
assim como , através da definição clara e registro de todos os processos que reflitam na qualidadefinal do produto com a sua sistemática revisão. Um outro fator fundamental para o bomcumprimento de um programa de BNF será a formação e o treinamento constantes e eficientespara todos os funcionários da empresa diretamente envolvidos com a qualidade do produto,componente fundamental base para melhoria contínua na empresa. A aplicação de conceitoscomo “rotation job” ajudarão ao funcionário a conhecer o processo produtivo como um todo(mudança periódica da atividade exercida pôr este funcionário) tornando-o mais eficiente nocumprimento das metas de qualidade de empresa e no cumprimento das NOP e dos POPexistentes. Ao fim de cada processo, se realiza a chamada reconciliação de lote, onde sãochecados e conferidos todos os documentos produzidos, observando o fiel cumprimento de todasas atividades realizadas na fabricação do produto, garantindo sua qualidade e rastreabilidade doprocesso como um todo. Ao fim o chamado batch release (documento que autoriza acomercialização do lote) e assinado pelo diretor técnico ou a quem este delegou a tarefa.
A aplicação da filosofia das BPF em cada empresa no seu dia a dia, de forma dinâmica seadequando às necessidades da mesma, produz uma diferenciação entre as BPF propriamenteditas e as BPFv (Boas Práticas de Fabricação vigentes) que seriam análogo à grande e a pequenaBPF, esta que muda a cada dia e não pode ser resumida em um manual, pois este se tornariaantiquado em pouquíssimo tempo.
Desta forma, podemos considerar como requisitos básicos para a implantação de um programa deBPF os seguintes aspectos:
Treinamento de pessoal em termos técnicos, de higiene pessoal e comportamento no interior da fábrica;
Espaço e instalações;
Materiais, recipientes e rotulagem;
Documentação de qualidade eficiente e válida e a documentação auxiliar; Armazenamento adequado;
Controle do processo de fabricação e da qualidade do produto final;
Análise e gerenciamento de risco.
1- Pessoal: Todo o pessoal envolvido na produção e controle de medicamentos, deve ter capacitação adequada para tal. Estes devem conhecer e compreender a responsabilidade que lhesé atribuída. Estes devem se comportar adequadamente na planta produtiva, estar devidamenteuniformizados e assumir adequadas normas de higiene pessoal.
2- As instalações devem possuir arranjo de tal forma idealizado que possibilite; fácil limpeza emanutenção; Evite contaminações cruzadas e externas; Evite efeitos perniciosos da contaminação
externa na qualidade do medicamento. Devem ser criados circuitos capazes de evitar misturasacidentais e contaminações. As instalações devem promover um grau adequado de isolamento daárea externa à fábrica, evitando inclusive a entrada de vetores na mesma.
3- Deve se evitar o aproveitamento de frascos ou recipientes de matérias-primas. Caso se utilizeeste procedimento, a limpeza dos mesmos deve ser validada, e seus rótulos totalmente retirados.Deve se anexar a respectiva etiqueta de limpeza nestes frascos. A rotulagem de todos os produtosiniciais, finais e intermediários no interior da fábrica, deve buscar identificar seu status; quarentena,liberado, rejeitado ou em processo. Ainda cabe identificar o produto e todas as demais informaçõesimprescindíveis para assegurar a rastreabilidade do processo de fabricação.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 49/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 49 de 130
4 - Todas as operações diretamente relacionadas à produção devem possuir normas operacionaispadrão, sendo as mesmas escritas em linguagem clara e imperativa; pe, Pese 50 Kg de amido em
balança digital com registrador......Operações correlatas podem ter suas NOP substituídas pôr programas de treinamento, como a lavagem de mãos pôr exemplo. Documentos auxiliares comocartas de controle de processo, folhas de pesagem e etc... devem ser confeccionadas de forma aauxiliar no controle da execução das atividades produtivas da empresa.
5- Os almoxarifados devem ser organizados de forma lógica e ordenada, divididos em ruas eapartamentos ou outro sistema de identificação que permita a fácil localização do materialarmazenado; deve ser previsto espaços individualizados para quarentena, expedição, produtosrejeitados, recolhidos e devolvidos e recepção de matérias-primas; deve haver o controle de suatemperatura e umidade relativa e uma política explícita de que os primeiros materiais a entrar serãoos primeiros materiais a sair (FIFO ou PEPS); Ainda deve se prever um local em separado para osmateriais de embalagem, sendo os impressos armazenados em local fechado e de acessocontrolado, e um local fechado para a armazenagem dos produtos psicotrópicos. Produtos termosensíveis devem ser armazenados em câmaras frigoríficas. O almoxarifado deve possuir umsistema de monitoramento e controle de temperatura e umidade relativa em seu interior, salvo odestinado à materiais de embalagem.
6- É obrigatória a presença de controle de processo no interior da planta produtiva, podendo seadmitir a terceirização de parte do controle de qualidade apesar de não adequado (Dec. Lei 3961de out. 2001). É necessária a existência de um setor de documentação técnica. O controleestatístico de processo deve dar suporte ao programa de qualidade da empresa seja por meio docálculo de capabilidade de processo, seja por meio do suporte do programa de gerenciamento eanálise de risco, fundamental para a consolidação da implantação do programa de BPF.
Entre os vários documentos utilizados para o eficiente para o cumprimento das BPF, temos
Especificações de matérias-primas e de materiais de embalagem; Fórmula padrão para cada um dos produtos fabricados (ver modelo a seguir); Normas operacionais de fabricação e embalagem de cada produto; Normas de pesagem; Amostragem; Distribuição de produtos acabados; Manual de análise; Manual da qualidade; Manual de BNF; Plano Mestre de Validação; Procedimentos de limpeza de equipamentos e áreas;
Procedimentos de montagem e calibração de equipamentos; Procedimentos de retirada de produto do mercado e devolução; Programa de manutenção preventiva; Protocolos e Relatórios de validação e qualificação; Fichas e registros de dados/atividades.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 50/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 50 de 130
A identificação de salas, atividades realizadas e outras informações de segurança, como o uso deEPIs, EPCs e similares, podem ser feitas com placas indicativas, que auxiliarão na manutenção do
correto fluxo de produção, auxiliando ainda na educação dos colaboradores da empresa emtermos das BPFs e medidas similares. Abaixo se verifica alguns exemplos destas identificações.
EXEMPLOS DE PLACAS INDICATIVAS DE STATUS DE PRODUÇÃO
EXEMPLO DE PLACAS INDICATIVA DE ATIVIDADES
AVISO
Uso Obrigatório de Toucae Máscara nesta área
SETOR DE SÓLIDOS ORAIS
PRODUTO _________________________________
LOTE ______________________________________
FASE _______________________________________
OPERADOR ____________________ DATA __/__/__

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 51/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 51 de 130
Boas Práticas de Fabricação Vigentes (BPFv) e a organização da unidade fabril.
Uma planta industrial destinada a fabricação de produtos sujeitos a controle sanitário deveser projetada e construída de forma a se atender as boas práticas de fabricação vigentesespecíficas para cada setor. Neste projeto, deve se tomar como objetivo evitar a todo custocontaminações cruzadas, contaminação do produto fabricado por agentes estranhos de qualquer natureza, retro cruzamentos e misturas acidentais, mantendo-se as condições de higiene e limpezada fábrica as mais elevadas o possível. Ainda se busca com o emprego das BPFv garantir atravésde documentação lógica e funcional, que todo o processo fabril (desde a recepção de matérias-primas até a distribuição do produto) possam ser facilmente rastreados (rastreabilidade). Estasexigências transformam a construção de uma indústria farmacêutica, assim como os equipamentosnela utilizados, de custo extremamente elevado. Uma estimativa prévia (ano de 2002), mostra que,contando com um grau mínimo de automação, uma planta produtiva farmacêutica pode chegar avalores próximos a U$ 800,00 por metro quadrado. Ao mesmo tempo, se busca atender àsexigências pertinentes ao código sanitário em vigor.
O desing de uma indústria farmacêutica, deve observar os aspectos inerentes à engenharia deprocesso, construção civil, fluxo da planta produtiva (arquitetura e Tecnologia Farmacêutica),serviços ambientais e validação de todos os processos que influenciarão diretamente na qualidadefinal do produto fabricado. Esta construção deve ser conduzida sempre com a intensa cooperaçãodo serviço de engenharia da empresa e a área técnico produtiva, associando as necessidades doprocesso de fabricação com a possibilidade de se viabilizar os mesmos em termos da engenhariae arquitetura. Seqüencialmente, os estágios relacionados a condução de um projeto destenatureza seriam:
1- Estudo da viabilidade do projeto;2- Engenharia de processo;a- fluxo de pessoal b- equipamentos e serviços necessários3- Material flow e material balance;
4- Construção e validação.
VIABILIDADE:Deve ser realizado um estudo para determinar o range de produtos a serem fabricados pela
empresa, suas quantidades especificações e a demanda pelos mesmos. Esta demanda deve ser previamente estudada e um profundo estudo de mercado deve ser realizado. Os limites financeirosdo projeto devem ser estabelecidos, tomando-se por base o retorno do investimento realizado(lembrar sempre que o custo de validação da planta produtiva se encontra em torno de 10% dovalor do investimento total).
Deve se observar ao mesmo tempo, a disponibilidade de pessoal qualificado no local ondese pretende instalar a planta produtiva, como também, o fácil recebimento de matérias-primas eescoamento da produção. A legislação sanitária vigente deve ser igualmente observada, comotambém a necessidade de se atender a uma legislação específica como as normativas do FDA ou
de Pais fora do âmbito do Mercosul. Estas normativas determinarão o grau de exigência que sedeve aplicar a planta produtiva e seus processos, e conseqüentemente o custo a ela associado.
A avaliação inicial do Project team considerará a posição de mercado do produto e osseus requisitos para fabricação, técnicos e legais. Uma visão geral do projeto deve ser traçadapara permitir sua inicialização (braimstorm). Diversos projetos podem ser aprovados com base eminformações gerais, a partir das quais, seus responsáveis discutirão os detalhes para realização.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 52/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 52 de 130
Serão observados os seguintes pontos:
Aspectos mercadológicos:
Produtos a serem fabricados;
Quantidades;
Projeção da produção e ampliações; Escoamento da produção / público alvo - mercado para o produto.
Aspectos regulatórios:
Qual agência deve aprovar o projeto? Qual normativa de BPF a seguir? Quais os padrões de qualidade e validação necessários para comercializar o produto
fabricado? Destinam-se os mesmos a exportação?
LOCALIZAÇÃO:
A área escolhida para a instalação da planta produtiva deve apresentar uma vizinhança
verde, de forma a evitar altos níveis de contaminação do ar. Uma área de grande atividadeindustrial (poluentes) pode apresentar um elevado grau de contaminação no ar, dificultando seubeneficiamento. Deve se adequar em termos de posturas municipais a atividade desenvolvida e olocal escolhido deve possuir todos os serviços públicos necessários, como água, luz, esgoto eestradas, possibilitando um fácil escoamento da produção. O local deve ser preferencialmentepouco habitado, de forma a evitar complicações para a implantação do projeto. Este pode ser restrito no que diz respeito ao uso de produtos inflamáveis ou de toxicidade elevada. Um cuidadoespecial deve ser dado à questão da legalização da empresa frente aos órgãos reguladores dequestões ambientais.
DISTRIBUIÇÃO DE ESPAÇO:
A boa distribuição de espaço de uma indústria farmacêutica se mostra como o ponto chave
para sua operacionalidade. Devem ser destinadas áreas de tamanho e arranjo adequados paracada uma das operações a serem realizadas em seu interior. Entre estas, temos as áreas deserviços (tratamento de água, produção de vapor, ar comprimido, eletricidade e etc..),administração, controle de qualidade, produção, almoxarifados e etc... Este espaço e osequipamentos nele alocados, deve ser proporcional a produção esperada, comportando inclusive,futuras expansões (previstas no material balance). A característica de cada área, em termos demateriais de construção e de qualidade ambiental, dependem do produto a ser fabricado em cadauma delas e a qual etapa de fabricação elas se referem. Obviamente, os almoxarifados e a áreaprodutiva ocuparão a maior parte da planta fabril. Cerca de 40% da área útil total da empresacorresponde a área produtiva, cabendo obviamente aos almoxarifados os 60% restantes, ou seja,uma média de 2/3 da área total. Estes almoxarifados devem ser arranjados em posições opostasna planta produtiva, a matéria-prima entra por uma das extremidades enquanto o produto acabadosai pela outra, possuindo o primeiro, recepção de matérias-primas, quarentena e no segundo,
expedição individualizada. Resumidamente, podemos distribuir o espaço dentro da fábrica entreas seguintes áreas abaixo:

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 53/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 53 de 130
1- Áreas de serviço e apoio: Estas áreas correspondem aos refeitórios, enfermarias, produção devapor, água, vácuo, ar comprimido, eletricidade, manutenção e engenharia, segurança,documentação técnica e sanitários. Os sanitários devem possuir condições de higiene econstrução compatíveis com a área no qual estes se encontram.
2- Administração e marketing;3- Controle de Qualidade (ideal se localizar fora da planta produtiva, porém no mesmo edifício);
4- Produção e controle de processo (no mesmo local; áreas especiais para penicilinâmicos,cefalosporínicos, hormônios e citostáticos);
5- Almoxarifados
Cada uma destas áreas deve ser ordenada e construída de forma a se evitar o contato doproduto em fabricação com o ambiente externo ou superfícies contaminadas, buscando não sealterar sua qualidade, segurança e eficácia.
Devem ser inseridas barreiras capazes de controlar este contato, assim como, evitar que oproduto fabricado ou subprodutos de seu processo de fabricação se transfiram a outros pontos doambiente íntimo da fábrica ou ao meio ambiente. Desta maneira, podemos afirmar que o localdestinado à fabricação de um produto farmacêutico deve ser o mais isolado possível. As paredesda fábrica, o interior de um equipamento ou ainda, um isolador específico, poderão funcionar comobarreiras. A classificação do ar ambiental da fábrica, visto sua direta influencia na qualidade finaldo produto, deve ser determinada de forma a adequar as suas aplicações a cada tipo de produto.
O ambiente que circunda o produto pode ser classificado como se segue:
Ambiente exterior ou área preta: tudo aquilo que rodeia a edificação da fábrica, sendo o mesmogovernado de forma direta pelas leis naturais e outros processos artificiais como a poluição. Não seexerce nenhum tipo de controle sobre o mesmo, salvo o controle de insetos e roedores. Devem ser plantadas na área que circunda a planta fabril, arbustos ou árvores que espantem insetos, como aarruda, por exemplo. Sempre evitar árvores frutíferas ou flores, que os atraem. A expedição da
fábrica pode ser considerada como área preta. Em geral são ambientes não classificados emtermos da qualidade de seu ar.
Ambiente próximo ou área cinza: formado no interior da planta produtiva de forma artificial, compontos de controle capazes de minimizar efeitos externos. Ainda é influenciado por eventosalheios e ou correlatos ao processo produtivo. Exemplos de áreas cinzas são observados na áreade embalagem secundária do produto ou corredores de circulação em plantas produtivas dequalidade muito reduzida; ou seja, onde o produto não se encontra diretamente exposto. Esteambiente já pode ser classificado, sendo sua classificação em geral, no mínimo classe 300.000.
Ambiente contíguo ou área branca: representa a área onde o produto entra em contato diretocom o ambiente, como áreas de fabricação e a parte interna de equipamentos. Possuem controle
total, não sofrendo nenhum tipo de influência de qualquer natureza, salvo aquela relacionada apessoas inadequadamente preparadas para a execução do trabalho de produção ou vestindouniformes inadequados. Podem chegar a níveis extremamente elevados de controle, como no casode áreas estéreis. Possuem pressurização do ar, sendo sempre a pressão maior no ambiente demaior limpeza ( um valor de P aceito seria de 8,0 pa). Em geral, estes ambientes são de classe1000 ou 10.000. Áreas estéreis, onde se observa o envase do produto obtido por filtraçãoesterilizante, devemos ter uma classificação tipo 100 (ver a classificação de ambientes defabricação na legislação atual de BPF). A tendência atual é permitir como classificação ambientalmínima um valor igual a 100 000, o que requer uma maior sofisticação nos sistemas de tratamentode ar a serem utilizados na unidade fabril.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 54/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 54 de 130
A intercomunicação destes ambientes deve ser realizada por interlocks ou passthroug, osquais devem sempre possuir pressão positiva da área mais controlada para a menos controlada,evitando qualquer tipo de troca entre elas. Deve haver normas de como se comportar ao se passar de um compartimento para outro. Suas portas nunca podem abrir ambas ao mesmo tempo (porta
da parte menos controlada abre primeiro, seguido da porta da área mais controlada). Esta pressãoé maior na área mais controlada do que no interior do interlock, e neste local, a pressão é maior que a da área não controlada. A forma de se controlar efetivamente cada um destes ambientesserá obtida através da correta construção da planta produtiva, inclusive, através da correta escolhados materiais utilizados para este fim e da forma como estes são empregados. A construção daplanta produtiva deve sempre evitar o cruzamento de matérias-primas com produtos acabados e amistura ou contaminação de um produto por outro que esteja sendo fabricado ao mesmo tempo naempresa. Isto se determina, quando se idealiza o fluxo de produção da fábrica.Para alguns tipos de fármacos, visto o grande risco ao operador e ao paciente que pode vir autilizar um produto contaminado, deverão ser fabricados em recintos totalmente isolados ededicados exclusivamente a esta categoria de produtos. Estes seriam fármacos muito tóxicos e /ou ativos em baixas concentrações, a saber:
Antibióticos -lactâmicos (penicilinâmicos e cefalosporínicos em separados):
Sensibilização;
Alergia;
Asma;
Choque anafilático;
Toxicidade elevada.
Hormônios:
Toxicidade;
Dose farmacológica muito baixa;
Citostáticos e quimioterápicos em geral:
Atividade teratogênica; Mutagênico; Uso de pressão negativa no local
Extremamente tóxicos.
Tanto a área de fabricação, quanto o sistema de HVAC devem ser exclusivos para afabricação deste tipo de produto (WHO e FDA). Ainda se incluiriam nesta classe de produtos,aqueles de origem biológica como vacinas, preparações bacterianas e qualquer tipo de preparaçãocom microrganismos vivos.
Na determinação do fluxo de produção ideal deve se considerar o fluxo de materiais e o fluxo depessoal, a saber;
Fluxo de pessoal: deverá ser observado o número de funcionários que trabalhará na empresa,suas qualificações e necessidades reais para fabricação de cada produto em suas áreas
previamente determinadas; para produzir um comprimido deve circular n pessoas na sala degranulação passando pelo vestiário com espaço para lavagem de equipamentos, etc... . A reduçãodo número de funcionários ao mínimo necessário receberá uma importante atenção; esta poderáser obtida através do desenho eficiente do layout de equipamentos e de utilidades. Serádeterminado o número de homens / hora necessários para cada atividade, sua movimentação ecada equipamento a ser instalado, sendo a área para tal destinada, otimizada com estes dados. Nodesenho do fluxo de produção ideal, além da movimentação de pessoal já prevista anteriormente,deve se prever a circulação de ar, presença de eventuais visitantes, armazenamento de semi-elaborados e locais destinados a itens contaminados, como lixo, ferramentas, manutenção de

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 55/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 55 de 130
equipamentos, limpeza e produtos inutilizados. Sempre se busca otimizar o fluxo de forma a seevitar retrocruzamentos ou misturas acidentais.Em cada um dos compartimentos descritos acima, torna-se ainda necessário se prever áreas demanutenção, circulação, lavagem, controle em processo e armazenagem de semi-elaborados.
Fluxo de materiais: deve ser identificado o fluxo de materiais dentro da empresa durante a
confecção de sua planta baixa. Isto determinará a viabilidade operacional da mesma. Nesta análisepoderá ser contemplado;
Para os almoxarifados de matérias-primas: Recepção de matérias-primas e limpeza; Quarentena; Armazenamento; Armazenamento de impressos; Armazenamento de produtos controlados; Pesagem (área classificada de 100 a 10 000); Área com refrigeração; Produtos rejeitados; Liberação; Unidade de coleta de dados – PCP.
Para a produção: Unidades de fabricação; Áreas de apoio (limpeza, controle em processo etc..); Armazenagem de materiais em processo e semi - elaborados; Embalagem primária; Embalagem secundária.
Almoxarifado de produto acabado: Quarentena; Armazenagem; Produtos especiais (port. 344);
Expedição individualizada; Produtos recolhidos; Arquivo retenção (temp 30o C e UR 65%).
Áreas de serviços: HVAC ( calor, ventilação e ar condicionado); Produção de água; Ar comprimido e breathing air; Vácuo; Vapor e vapor limpo (WFI) x (caldeiras e dutos); Eletricidade; Efluentes;
Gases especiais.
Área de suporte: Engenharia; Manutenção; Enfermaria; Escritórios; Recepção; Refeitórios; Administração;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 56/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 56 de 130
SAC.
O material balance da empresa é considerado como uma das bases para o seu bomfuncionamento e desenho de seu layout, seguido do posterior estabelecimento do fluxo demateriais. Deve ser feita uma estimativa do volume de produção, de possíveis expansões e
aumento de produtividade, avaliando-se ao mesmo tempo, quanto deve ser a capacidade dearmazenagem total da empresa, tanto de materiais quanto de produtos acabados / intermediários.Neste design, se projeta a área para cada setor da empresa e suas localizações, arranjando asmesmas de forma lógica, seqüencial, unidirecional de forma que evite retrocruzamentos oumisturas acidentais. Um produto fabricado em um ambiente jamais pode contaminar outro produtofabricado em um ambiente distinto da fábrica. Deve se restringir o retorno de material jáprocessado para uma área anterior e restringir o acesso de pessoal a cada setor (somente opessoal necessário para a execução do trabalho). Os fluxos de produção tidos como ideais são oschamados fluxos unidirecionais, arranjando-se seqüencialmente cada área produtiva, com osalmoxarifados de matéria-prima e produto acabado dispostos em lados opostos da plantaprodutiva. Estes fluxos unidirecionais podem se apresentar como sendo lineares ou em forma deU. Neste último caso, os almoxarifados se arranjarão em cada uma das extremidades do U. Umfluxo produtivo poderá se arranjar ainda, vertical ou horizontalmente. Nos fluxos de produçãoverticais, se utiliza a gravidade como força motriz do sistema. No caso do fluxo horizontal, seutilizará o vácuo ou outra fonte de energia como força motriz. Em ambos os casos estas formas dealimentação serão responsáveis por uma maior automação do processo fabril.
Almox. MP pesagem granulação compressão revestimento embalagem Almox.Prod. acabado
N/C 10000 100000100001000010000 N/C
Figura: representação de um fluxo de produção horizontal; granulação

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 57/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 57 de 130
10000
N/C
10000Almox. MP revestimento embalagem Almox. Prod.
Acabado
N/C
Pesagem
Mistura
Compressão
10000
10000
Figura: representação de um fluxo de produção vertical; compressão direta e granulação.
Abaixo se visualiza um exemplo de parte de um fluxo de produção vertical para produção deinjetáveis.
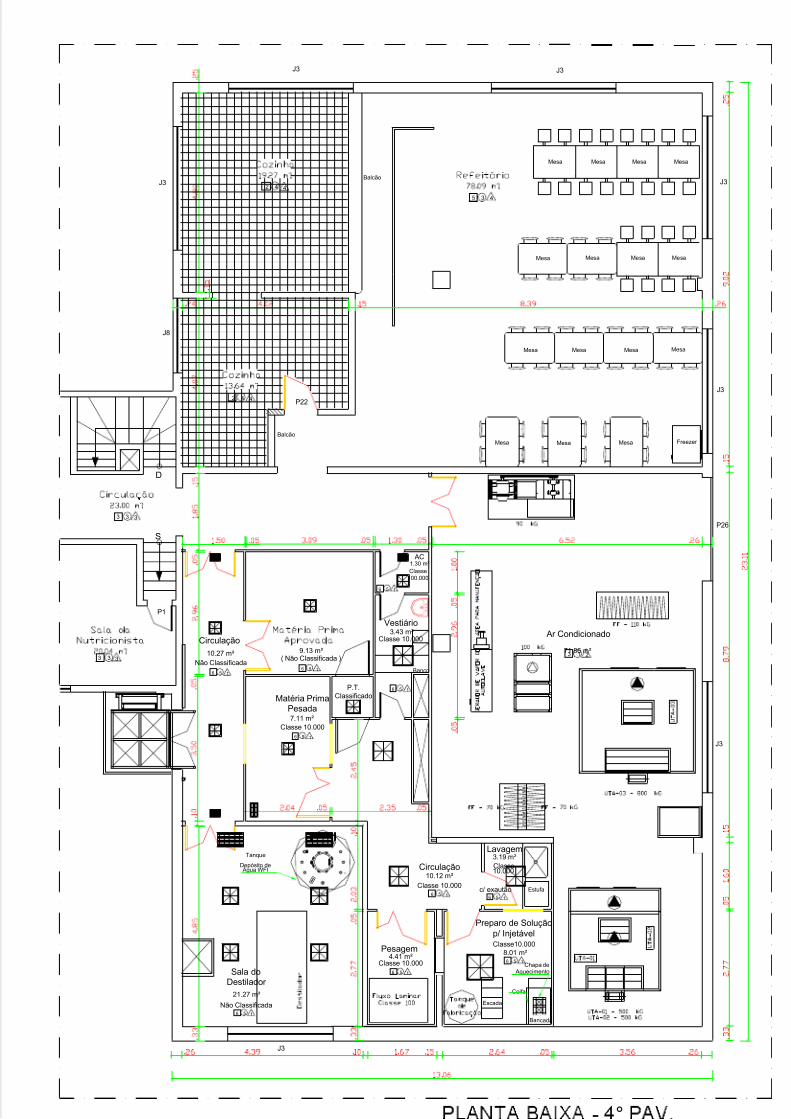
5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 58/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 58 de 130
J3
J3
P26
J3
J3
J3
J3
J8
P22
S
D
P1
Ar Condicionado
71.86 m²3 3
3 3
Mesa Mesa Mesa
MesaMesaMesaMesa
Mesa Mesa Mesa Mesa
MesaMesaMesaMesa
Balcão
BalcãoFreezer
J3
2 4
2 4
5 3
Preparo de Soluçãop/ Injetável Classe10.000
8.01 m²
Chapa deAquecimento
Bancada
Estufa
Vestiário
Banco
Circulação
10.27 m²
Sala doDestilador
21.27 m²
Matéria PrimaPesada7.11 m²
9.13 m²
3.43 m²
AC1.30 m²
Pesagem4.41 m²
Lavagem3.19 m²
P.T.Classificado
Circulação10.12 m²
Classe 10.000
Classe 10.000
Classe 10.000
Classe 10.000
Classe100.000
( Não Classificada )Não Classificada
Não Classificada
Classe10.000
c/ exautão
3
3
4
4
4
3 1 2
6 5 1
- °
Coifa
Escada
Tanque
Depósito deÁgua WFI
6 5 1
6 5 1
6 5 1
6 5 1
6 5 1
6 5 1
6 5 16 5 1
6 5 1

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 59/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 59 de 130
No desenho do fluxo de produção ainda deve ser prevista a separação da planta produtiva por seu grau de limpeza. A entrada da planta sempre se encontra próxima a áreas mais sujas e seuinterior próximo a áreas mais limpas. Áreas críticas de processo, como setores de produção de
líquidos estéreis, jamais devem ser colocadas junto a paredes externas, evitando-se problemas emfuturas expansões. Jamais se instalam ralos nestas áreas. Nas demais, apenas ralos sifonados epreferencialmente em aço inox.
O documento produzido neste processo, a planta de fluxo, em adição ao relato dos critérios deengenharia utilizados no projeto e as operações de segurança, irão garantir o perfeitofuncionamento da fábrica. Para os sistemas de serviços devem ser confeccionados diagramasindividuais constando níveis de consumo, rotas de eliminação de efluentes e classificação de cadaum destes. Finalmente, quando do desenvolvimento do design do fluxo de produção, deve seprever uma possível redução de espaço quando do uso de sistemas automatizados, assim como,redução do transito de pessoal e número de equipamentos, aumentando ao mesmo tempo aprodutividade destes sistemas fechados.
MATERIAIS DE CONSTRUÇÃO E CORRELATOS:
A forma de se controlar efetivamente cada um dos ambientes no interior da fábrica,decorre da correta escolha dos materiais de construção utilizados para este fim e da forma comoestes são empregados. Diferentes coeficientes de dilatação dos materiais utilizados na construçãoda planta produtiva, por exemplo, podem originar o aparecimento de rachaduras e de pequenasfissuras, estimulando o crescimento microbiano (aumento indireto da porosidade da superfície).Sendo assim, podemos resumir as seguintes características como fundamentais para estesmateriais:
Paredes, pisos, portas e janelas devem ser herméticas, permitindo que a troca de energia emassa ocorra apenas pelos locais e formas previstas pelo processo e nas condições jáestabelecidas. Estas não devem favorecer o crescimento de MOs (madeira, papéis e outrasfontes de C. N e H). Deve ser prevista a troca de gases (evaporação de água) pelo piso, emespecial o piso epoxi. Terrenos muito úmidos podem gerar problemas de bolhas no caso de
pisos com revestimento epoxi. Estes pisos devem ser monolíticos e constituídos em resinasepoxi, ou ainda, de korodur, granitina, placas de PVC soldadas ou moldadas.
Resinas epoxi: Ideal para áreas de suporte; apresenta custo não muito elevado. Boaresistência química e frágil em termos mecânicos.
Placas de PVC soldada: Resistência química elevada. Possuem tamanho variável sendo muitoversáteis. Ideal para áreas assépticas. Compatíveis com paredes monolíticas. É custoso e debaixa resistência mecânica. Mancha facilmente.
Stom Hard ou terrazzo epoxi: Pavimentos inteiramente revestidos de massa epóxi contendopedaços de granito ou quartzo para evitar a formação de bolhas. Possui grande resistênciamecânica e química, inclusive frente a agentes básicos ou ácidos. É durável, resistente erecomendado para áreas de eleva da qualidade. Sua porosidade é reduzida; por sua grandedurabilidade, se justifica sua larga utilização.
Granitina: Piso de cimento mesclado de pedaços de granito. Possui elevada porosidade sendo
necessário para tal, seu posterior revestimento com resinas acrílicas ou cera. Resistênciaquímica moderada. Pode manchar facilmente, porém possui elevada resistência mecânica.Recomendável para áreas de embalagem secundária.
Paredes: Uso de massa e tinta epóxi; Durável, de custo reduzido e resistente ao impacto. Idealpara áreas de tráfico intenso. Como desvantagem, este revestimento tem a tendência aapresentar rachaduras com o tempo, não sendo ideal para áreas estéreis.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 60/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 60 de 130
Revestimentos de PVC sem juntas; Aplicado por spray sobre blocos de concreto da parede;possuem elevadas resistências químicas, mecânicas, tendo ainda uma grande flexibilidade,evitando rachaduras. Seu custo é elevado, sendo apenas recomendado para áreas críticas.
Placas de PVC soldadas; Simulam o revestimento anterior, porém, mais fino. Desta forma, se
torna mais susceptível ao tráfego mais intenso. Indicada para áreas assépticas. Esterevestimento e o uso de PVC moldado é um dos tipos mais caros disponíveis até o presentemomento.
Painéis pré-fabricados: Pode se utilizar para este fim, divisórias de resinas fenólicas revestidasde melamina, que possuem recheio de poliuretana anti-inflamável, lâ de rocha ou isopor.Estas quais apresentam baixa porosidade e elevada resistência química. Painéis de aço inox316 ou vidro blindex são outras alternativas nesta área. Sua escolha é baseada no custo finaldo produto, sendo os painéis de aço inox os de custo mais elevado. Possuem versatilidade emobilidade, sendo mais facilmente adequável a futuras expansões. O alumínio anodizado égeralmente utilizado para arremates de junções entre piso, paredes e tetos.
Para os tetos temos: placas de PVC; de resinas fenólicas; revestimentos epóxi; de poliéster; eplacas de PVC soldadas. Lembrar sempre da necessidade da presença de um teto falso auto-sustentável para que por este circule todos os dutos de vapor, eletricidade, água, ar
comprimido e similares, para que se faça a manutenção sem a necessidade de ingresso nointerior da planta produtiva. Portas: atualmente se utilizam portas com a mesma constituição das divisórias sanitárias
acima citadas ou o revestimento de aço inox como tipos ideais. Todos os efluentes da empresa devem ser beneficiados antes de serem lançados ao meio
ambiente. Lavadores de pós, ciclones ou precipitadores elétricos, são utilizados para tratar o ar exaurido da fábrica; incineradores de alta temperatura são utilizados para áreas que trabalhamcom produtos biológicos, antibióticos ou citostáticos. Ainda deve se prever piscinas detratamento de efluentes com coagulantes (químico; policloreto de alumínio e aluminato desódio) e lodo ativado (microbiolóico).
Todos os materiais de construção utilizados devem resistir ao atrito, abrasão e as maisexigentes condições de trabalho. Devem ainda ser de fácil limpeza e de elevada resistênciatérmica, química e mecânica.
Janelas; Evitar sempre que possível. Usar panos de vidro ou clarabóias para aproveitar ailuminação natural. Não devem estar presentes em áreas brancas. É recomendável se evitar janelas em áreas cinzas e pretas visto que esta providência pode facilitar o controle de entradade aves, roedores e insetos.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 61/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 61 de 130
Foto de piso autonivelante em epóxi com microesferas de vidro
Todo tipo de tubulação, elétrica, hidráulica ou de qualquer outra natureza, deve ser embutidano teto ou nas paredes para se evitar acúmulo de sujeira. As tomadas devem ser blindadas e a 30cm do chão ou devem provir do teto falso. As placas indicativas para a restrição de acesso a certasáreas, para a indicação do uso de EPI (equipamento de proteção individual) e EPC (equipamentode proteção coletiva), sobre o risco de trabalho no local e controle de produção, entre outros.Aspectos referentes á segurança devem ser descritos no manual de biosegurança do laboratório. Estas placas seguem padronização internacional; AVISO-AZUL; EMERGÊNCIA/PERIGO-
VERMELHO; SEGURANÇA-VERDE; ATENÇÃO-AMARELO.
Toda a tubulação da fábrica deve possuir cor padronizada para cada utilização: Branco- Vapor Preto - InflamáveisAmarelo- Gases não liquefeitosVermelho- Equipamentos de incêndioAzul- Ar comprimidoVerde- ÀguaPlatina- VácuoAlumínio- Gases liquefeitos e inflamáveisCinza escuro- eletrodutosMarrom- Materiais fragmentados

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 62/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 62 de 130
Resumo das características dos materiais de construção utilizados na construção de uma plantafarmacêutica:
Durabilidade elevada;
Resistência mecânica;
Resistência química;
Baixo custo;
Acabamento sofisticado;
Baixa porosidade;
Manter seu aspecto inalterado frente as mais adversas condições.
Apesar de se considerar a construção de paredes como a melhor forma de se isolar áreasespecíficas de uma indústria farmacêutica, em almoxarifados ou no setor de embalagemsecundária, podemos admitir que a separação de certas áreas seja realizada por divisórias oucorrentes para demarcação. Ainda se admite o uso da tecnologia de isoladores de equipamentos,até mesmo para áreas estéreis.
Produtos de elevada toxicidade e atividade como -lactâmicos (penicilinâicos
cefalosporínicos), hormônios, citostáticos devem ser fabricados em áreas da planta produtiva individualizada e isolada das demais, possuindo ainda exaustão e refrigeraçãoindividualizadas, sendo o ar condicionado filtrado por filtros hepa para sua reutilização e oar exaurido incinerado antes de ser eliminado. Produtos cosméticos devem ser fabricadosem uma planta produtiva separada segundo a legislação vigente, apesar da permissividadedos órgãos fiscalizadores.
Áreas comuns como banheiros, refeitórios, escritórios e similares, por serem muito susceptíveisa contaminação devem se localizar fora da área produtiva. Mesmo assim, devem receber oscuidados de higienização e sanitização idênticos a área para a qual se destinam; áreas brancassão atendidas por banheiros com limpeza semelhante estas áreas, mesmo sendo segregados.
O controle de qualidade deve se localizar em local fora da planta produtiva, devendo haver, noentanto, no interior da mesma, ilhas de controle em processo.
Equipamentos: aparelhos de ação mecânica destinado a atuar sobre diversos materiais queformam o produto entrarão diretamente em contato com o mesmo, podendo representar umagrande fonte de contaminação. Assim, ele não poderá alterar química ou fisicamente estesmateriais por ele processados. Os equipamentos devem ser de fácil limpeza, desmontadofacilmente, fácil sanitização e esterilização. Todas as suas partes que entram em contato diretocom o produto devem ser confeccionadas em aço inox 316 AISI ou teflon. Devem sempre quepossível, isolar o ambiente onde o produto é exposto no seu interior, como por exemplo, máquinasde compressão fechadas como a FETTE P 3000. Suas partes mecânicas devem sempre seencontrar em excelente estado de manutenção, não gerando partículas, fumaça ou jogando óleopara o ambiente.
Sempre que possível, localizar a parte mecânica do equipamento em local fora da áreaprodutiva, evitando contato do produto com seus rejeitos. Um exemplo clássico seria a exposiçãode apenas as portas de uma estufa ou autoclave para a área produtiva e sua parte mecânica parauma área de serviço.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 63/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 63 de 130
Compressora Fette 4090i.
Áreas para serviços gerais e apoio:São destinadas a fabricação de produtos utilizados em grande quantidade na fábrica,
porém não diretamente envolvidos na confecção do medicamento (a exceção da água). Entre
estes produtos, podemos citar a água, o vapor (limpo ou não), o ar comprimido (breathing air), o ar condicionado, o vácuo e etc... . Estes setores se localizaram fora da planta produtiva, sendo osprodutos gerados levados ao interior da fábrica por dutos padronizados que passarão por um tetofalso presente na planta. Devem existir procedimentos escritos para a limpeza destes dutos emespecial para os dutos de ar condicionado, grande fonte de contaminação. Todos estes setoresdevem constar na planta baixa da empresa encaminhada para ANVISA e VISA Estadual.
Um especial cuidado deve ser direcionado ao setor de tratamento de água, queapesar de se localizar em uma área de serviços produz uma matéria-prima farmacêutica,devendo o mesmo apresentar condições de higiene e manutenção adequados.
O ar que ingressará no interior da planta produtiva deve sempre ser beneficiado de acordocom o tipo de área para o qual este se destina, área preta, branca ou cinza. Deve se proceder aretirada de contaminantes físicos, químicos e biológicos do mesmo. Este ainda deve ser ajustadoem termos de sua temperatura (ideal 22-25o C) e sua umidade cerca de 45-60%, (abaixo destevalor, apenas em áreas críticas, como a destinada ao manuseio de efervescentes ou similares).Não é indicado o uso de sistemas de refrigeração que capturem o ar diretamente do ambiente (ar condicionado de parede ou similar) onde não temos controle de sua qualidade. O ideal seria o uso
de centrais de refrigeração onde este ar é filtrado e controlado devidamente. Devem existir procedimentos escritos para o controle da limpeza do sistema de dutos e grelhas (fonte potencialde contaminação microbiológica), assim como para o beneficiamento do ar propriamente dito.Estes devem ser sempre atualizados e validados. Para a preparação de formulações estéreis ou
5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 64/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 64 de 130
em ambientes classificados, o ar deve ser filtrado também por filtros absolutos do tipo HEPA, ondeirá se requerer um controle ainda mais rigoroso de sua qualidade e da manutenção e limpeza dosistema. O controle da qualidade final deste ar pode ser realizado pelo uso de cartas psicométricas(UR e temperatura), pelo monitoramento microbiológico do ambiente (plaqueamento, swab, slit toagar e biotest) e ainda pelo uso de contadores de partículas (fotômetros a laser).
Relação ideal para refrigeração 01 TR (12000 btu) = 15 m
2
O controle da umidade relativa e da temperatura deverá ser mais rigoroso em áreasdestinadas a preparação de produtos estéreis, onde o calor e a água favorecem o crescimentomicrobiano. Lembrar que para a preparação de efervescentes, a temperatura e a umidade relativadevem ser mantidos sempre a 20o C e 38% de UR. Para a preparação de cápsulas gelatinosasduras, os valores ideais estariam entre 25o C e 40-60% de UR. A exaustão ou renovação do ar nointerior da planta produtiva deve ser realizada de acordo com as exigências de cada setor. Salasde granulação ou compressão devem ter uma exaustão extremamente eficiente para se remover grande número de particulados gerados. Já um processo que trabalhe com substâncias ativas embaixas concentrações, o ar exaurido deve ser incinerado a 200 o C antes de lançado ao ambiente.O ar exaurido pode ser reutilizado, o que representará uma economia de 85 % da temperatura doar condicionado. Entretanto, para esta re-utilização, devem ser empregados filtros HEPA ousimilares para filtrar o ar devendo este processo ser cuidadosamente monitorado e validado. Emgeral, especialmente no Brasil em pequenas empresas, o ar exaurido é eliminado. Em uma áreaonde o processo não gera muitas partículas, a exaustão ideal se encontra em torno de 20 vezes ovolume de ar da sala por jornada de trabalho. O próprio aparelho de ar condicionado podeproporcionar exaustão. Esta pode ser conectada ainda na caixa de retorno do aparelho, devendoneste caso se tomar a precaução de utilizar filtros HEPA para o reaproveitamento do ar. Para umaboa exaustão, deve se pensar em 5 a 8 m3 /min, contrastando com os 3 a 4 m3 /min normalmenteobservado como retorno do sistema de ar condicionado. A exaustão para uma boa renovação doar e para fins de classificação ambiental deve ser feita pela parte inferior a sala preferencialmente.O sistema de insuflamento de ar deve ser controlado em balanço com a exaustão para se garantir a pressurização da área. Em média, pela NBR 6401, pode se considerar como condição deconforto a temperatura de 24 a 26º C e a UR de 40 a 65 %.Segundo o Federal Standard 209 E e ISO 14644 o seguinte grau de renovação é recomendado:
Número de renovações por hora Classificação do ambiente> 120 1000>40 10.000>20 100.000
A seguinte classificação de ambiente é descrita por este mesmo código;
Classe No de partículas por m3 de ar;100 0,5 m – 3530 e 0 Mos viáveis
1000 0,5 m – 35300 e 5 Mos viáveis10.000 0,5 m – 353000 e 100 Mos viáveis
100.000 0,5 m – 3530000 e 500 Mos viáveis

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 65/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 65 de 130
LABORATÓRIO TABAJARA LTDA.ENVELOPE DE RECONCILIAÇÃO DE LOTES
PRODUTO:_____________________________________
LOTE:__________________________________________
DATA DE FABRICAÇÃO:__/__/__
CONTEÚDO:REQUISIÇÃO DE MAT. PRIMASREQUISIÇÃO DE MAT. EMBAL.
CHEK LIST DE PESAGEM
CARTA DE PROCESSO
CARTAS DE CONTROLEDE PROCESSO
BOLETIM DE ANÁLISE DEPRODUTO FINAL E SEMIELABORADOGUIA DE REMESSA DEPRODUTO ACABADO
ETIQUETAS DE LIMPEZA
ETIQUETAS DE PESAGEM
ORDEM DE FABRICAÇÃO
AMOSTRAS DE MATERIAL DEEMBALAGEM
CHEK LIST DE EMBALAGEM
CONFERIDO POR: ____________________________/__/__
GARANTIA DA QUALIDADE: __________________/__/__

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 66/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 66 de 130
tabajara fórmula padrãoDepartamento de Produção / Fabricação Pág.: 1 de 5Produto: Oralix Spray Granel Código: 505570Lote:____________ Lote Padrão: 300 litros x unid. devendaRendimento Padrão: 99% Quantidade do Lote: ______________
Hora de Início: _________ Data: ____/____/____
Hora do Final: __________ Validade xx/xx/xx
Processo de Fabricação
1 - Considerações GeraisO manipulador ao receber o pallet contendo os insumos para o lote em fabricação deveinspecionar.- Se as gaiolas contendo as matérias-primas estão lacradas.- Se a ordem de produção está acompanhando as matérias - primas.
- Se o check-list de Pesagem de Matérias-Primas para a Produção de Medicamentos naCentral de Pesagem foi devidamente preenchido pelo pesador.- Se as etiquetas de pesagem das matérias-primas estão preenchidas e de acordo com aidentificação da Ordem de Produção.- Se o número de volumes é o mesmo contido na Ordem de Produção.- A aparência física dos volumes (se estão limpos, lacrados e aprovados).- O manipulador deve seguir rigorosamente todas as etapas descritas e vistá - las.- Caso ocorra qualquer anormalidade, das acima ou outras, comunicar imediatamente aoresponsável pelo setor.- Anexe todas as etiquetas de pesagem na folha anexa.- Verifique se o reator foi limpo de acordo com o procedimento de limpeza adequado.- Anexe a etiqueta de limpeza no reator e solicite a liberação do Controle de Qualidade.
2 - Considerações Especiais- Evite contato direto da pele e dos olhos com matérias-primas.- Evite inalar as matérias-primas.- Evite contato direto com Ferro.- Utilize ao manipular as substâncias, materiais de proteção individual.
3 – Equipamentos utilizados no processo- Tanque 500L, provido de reator - Máquina de envase Erly 2 bicos- Máquina recravadeira- Fechadora automática de Caixas de Embarque – 3MElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx.Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 67/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 67 de 130
tabajara fórmula padrãoDepartamento de Produção / Fabricação Pág.: 2 de 5Produto: Oralix Spray Granel Código: 505570Lote:____________ Lote Padrão: 300 litros x unid. devendaRendimento Padrão: 99% Quantidade do Lote: ______________
Hora de Início: _________ Data: ____/____/____
Hora do Final: __________ Validade xx/xx/xx
4 - Procedimento
1ª - Selecione previamente os materiais que serão utilizados nesta fabricação, deixando-os emlocal separado dos demais.
_______________________ ______________Manipulador Data
2ª - Solicitar a aprovação da água ao Controle de Qualidade.
_______________ _________________ ________________Manipulador Aprovação C.Q. Data
3ª - Solicitar a aprovação da água de lavagem do tanque de fabricação.
_______________ _________________ ________________Manipulador Aprovação C.Q. Data
4ª- Verifique se o equipamento foi liberado pelo Controle de Qualidade (visto no documento delimpeza).
_______________________ ______________Manipulador Data
5ª - Em um tanque reator de aço inoxidável com capacidade para 500L, provido deagitador móvel ou fixo, transfira 60 litros de água desmineralizada e dissolva o FosfatoBissódico, agitando por 20 minutos.
Início da Agitação:_______:_______ Fim da agitação: ________:_______
Quantidade água transferida:_______ L
_______________________ ______________Manipulador Data
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx.Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 68/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 68 de 130
tabajara fórmula padrãoDepartamento de Produção / Fabricação Pág.: 3 de 5Produto: Oralix Spray Granel Código: 505570Lote:____________ Lote Padrão: 300 litros x unid. devendaRendimento Padrão: 99% Quantidade do Lote: ______________
Hora de Início: _________ Data: ____/____/____
Hora do Final: __________ Validade xx/xx/xx
6ª - Adicione o Ácido Lático. _______________________ ______________
Manipulador Data
7ª - Adicione à etapa 6 o Quinosol e o EDTA. Agite por 20 minutos.
Início da Agitação:_______:_______ Fim da agitação: ________:_______
_______________________ ______________Manipulador Data
8a. – Adicione à etapa 7 o Maltitol, a Sacarina, o Hidrolato de Malva, o Cloreto de Cetilpiridina e oscorantes. Agite por 10 minutos.
Início da Agitação:_______:_______ Fim da agitação: ________:_______
_______________________ ______________Manipulador Data
9a. – Em outro recipiente, dissolva no Álcool a Tirotricina, o Mentol, o Methocel, a Lidocaína,Cânfora e o Óleo de Menta dissolvido no Surfon. Agite por 30 minutos.
Início da Agitação:_______:_______ Fim da agitação: ________:_______
_______________________ ______________Manipulador Data
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx.Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 69/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 69 de 130
tabajara fórmula padrãoDepartamento de Produção / Fabricação Pág.: 4 de 5Produto: Oralix Spray Granel Código: 505570Lote:____________ Lote Padrão: 300 litros x unid. devendaRendimento Padrão: 99% Quantidade do Lote: ______________
Hora de Início: _________ Data: ____/____/____
Hora do Final: __________ Validade xx/xx/xx
10a. – Adicione a solução 9 no reator.
_______________________ ______________Manipulador Data
11a. – Verifique o pH, ajuste se necessário, com solução de Ácido Lático 20% ou Hidróxido deSódio 10%. Faixa de pH 3,8 – 4,5
pH : _______________ __________________________ Analista Controle de Qualidade
Correção:
SIM NÀO Quantidade (Kg/L) Ácido Lático 20% ( ) ( ) _____________________
Hidróxido de Sódio 10% ( ) ( ) _____________________
_______________________ ___________Manipulador Data
12ª - Caso seja efetuada a correção, solicitar nova verificação de pH ao Controle de Qualidade.Faixa de pH 3,8 – 4,5.
pH:___________________ _____________________________ Analista de Controle de QualidadeElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx.Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 70/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 70 de 130
tabajara fórmula padrãoDepartamento de Produção / Fabricação Pág.: 5 de 5Produto: Oralix Spray Granel Código: 505570Lote:____________ Lote Padrão: 300 litros x unid. devendaRendimento Padrão: 99% Quantidade do Lote: ___________
Hora de Início: _________ Data: ____/____/____
Hora do Final: __________ Validade xx/xx/xx13ª - Complete com água desmineralizada até o volume de 300 litros.
_______________________ ______________Manipulador Data
14ª - Faça a filtração em filtro de 1 micra nominal.
_______________________ ______________Manipulador Data
15ª - Solicite por requisição a análise química ao Controle de Qualidade.
_______________________ ______________Manipulador Data
16ª - Liberação para o envase.
_______________________ ______________Controle de Qualidade Data
17ª - Retire as etiquetas de limpeza e anexe à documentação.
_______________________ ______________Manipulador Data
Entregue por: ___________________________________ em ___/___/___
Recebido por: __________________________________ em ___/___/___
___________________________ _________________________ Responsável Fabricação Responsável Embalagem
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx.Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 71/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 71 de 130
LABORATÓRIO TABAJARA.
Departamento de Garantia da Qualidade - Setor Fabricação Pág.: 8/13
FÓRMULA PADRÃO FP 001 OF Nº: ______
Produto: Bacterian SP – 50 mL Código: 10001
Elaborado Digitado Revisado
10.06.00
Aprovado
10.06.00
Próx. Revisão
06.01
ETIQUETAS DE PESAGEM E DE LIMPEZA Cole nesta e na próxima página, as etiquetas depesagem e limpeza;
VER ANEXO
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx. Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 72/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 72 de 130
LABORATÓRIO TABAJARA.
Departamento de Garantia da Qualidade - Setor Fabricação Pág.: 9/13
FÓRMULA PADRÃO FP 001 OF Nº: ______
Produto: Bacterian SP – 50 mL Código: 10001
Elaborado Digitado Revisado
10.06.00
Aprovado
10.06.00
Próx. Revisão
06.01
ETIQUETAS DE PESAGEM E DE LIMPEZA Cole nesta página, as etiquetas de pesagem elimpeza;
VER ANEXO
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx. Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 73/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 73 de 130
TABAJARADepartamento de Garantia da Qualidade - Setor Fabricação Pág.: 10/13FÓRMULA PADRÃO FP 001 OF Nº: ______
Produto: Bacterian SP – 50 mL Código: 10001Lote : _ . _ . _ . _ . _ . _ Data de Fabricação: ___/___/___
Rendimento teórico: 100 L prazo de validade: 36 meses
Forma farmacêutica: solução oral Densidade a 28o C: _____
Volume médio teórico: 51 mL Concentração de princípo ativo (thimerosal): 0,1g por mL.Fórmula padrão: 100 L = 1960 und/ 51 mlFórmula centesimal: 100 L = 1960 und / 51 ml
Código Descrição Un Teor Requis. %P/P
Requisitadopara 100 l
Entregue Registro
2.00309-0 Água purif. L 100% 29.000 29.0002.00326-0 Álcool etílico
(d=0,808)
L 96% 60.000 60.000
2.00319-3 Acetona L 99% 11.000 11.0002.01674-6 Timerosal Kg 98% 0.030 0.1
5.00819-0 FrascoBacterian
Un _ 1960 1960
5.00900-5 Caixa depapelão
Un _ 163 163
5.01354-9 Tampa comaplicador
Un _ 1960 1960
5.00962-5 CatuchoBacterian
Un _ 1960 1960
5.00000-0 Fluoresceina Kg 100% 0,001 0,001
5.00000-0 Vermelhoamarante Kg 100% 0,001 0,001
5.00000-0 Lacre docartucho
Un _ 1960 1960
ElaboradoJAV
DigitadoTAG
RevisadoBAS
AprovadoAMOR
Próx. Revisão

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 74/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 74 de 130
LABORATÓRIO TABAJARA.Departamento de Garantia da Qualidade - Setor Fabricação Pág.: 11/13FÓRMULA PADRÃO FP 001 OF Nº: ______ Produto: Bacterian SP – 50 mL Código: 10001
Lote : _ . _ . _ . _ . _ . _ CARTA DE CONTROLE EM PROCESSOLÍQUIDOS:
Teste a realizar especificaçãoaspecto Líquido Límpido isento de precipitadocor Laranja clarodensidade 0.880-0.900pH 6.0-7.0volume médio 51 mL
Controle do enchimento:Máquina No : operador:
data hora volumemedido
fechamento vácuo rejeição
1- __/__/__ 2- __/__/_ _ 3- __/__/__ 4- __/__/__ 5- __/__/__ 6- __/__/__ 7- __/__/__ 8- __/__/__ 9- __/__/__ 10-__/__/__
Testes específicos:data hora densidade pH cor aspecto1- __/__/__ 2- __/__/_ _ 3- __/__/__ 4- __/__/__ 5- __/__/__ 6- __/__/__ 7- __/__/__ 8- __/__/__ 9- __/__/__ 10-__/__/__ Realizar teste de vazamento em dessecador sob vácuo com 10 amostras.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 75/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 75 de 130
LABORATÓRIO TABAJARA.Departamento de Garantia da Qualidade - Setor Fabricação Pág.: 12/13FÓRMULA PADRÃO FP 001 OF Nº: ______ Produto: Bacterian SP – 50 mL Código: 10001Lote : _ . _ . _ . _ . _ . _
Observações:____________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________
__________________/__/__ Supervisor de Produção
_________________/__/__ Garantia da Qualidade

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 76/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 76 de 130
BOLETIM DE ANÁLISE MICROBIOLÓGICA
ITEM ESPECIFICAÇÃO RESULTADO ANALISTAÁGUA PURIFICADA 100 UFC/mL Conforme Carlos __/__/__AR AMBIENTAL 40 000 ufc/mL Conforme Carlos __/__/__
Resultados obtidos conforme POP MI010202
Data __/__/___
Coordenador de Microbiologia: ____________________
( ) Aprovado
( ) Reprovado

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 77/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 77 de 130
CHECK LIST EMBALAGEM - LÍQUIDOS
PRODUTO: ____________________________________ LOTE: ___________________
VD ( ) AG ( ) MAQUINÁRIO UTILIZADO _______________________________
CONTROLE DE OPERAÇÃO:PRODUTOS ANTERIORES LOTE LINHA
INÍCIO FINALDATA HORA DATA HORA
OBSERVAÇÕES RENDIMENTO (UNIDADES)Amostra/Arquivo: ________und Rend. Previsto:
DIAS I M F SETOR ASS Quantidade real:
DIFERENÇA:Obs:
Encarregado de Linha:_____________________________________ Data: ___/___/___
Analista de Controle:______________________________________ Data: ___/___/___
Perda de Granel na embalagem (Kg):____________________________
Processo de embalagem de acordo ao estabelecido ?
Chefe de embalagem: ______________________________________ Data ___/___/___
Controle de Qualidade _____________________________________ Data ___/___/___

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 78/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 78 de 130
TABAJARA.Boletim de análise No: Produto:Código: Data de fabricação:Lote: Quantidade:Solicitante: Fase: (Semi elaborado, enchimento, produto final)Comentários:
RESULTADOS ANALÍTICOSÍtens solicitados Resultados esperados Resultados encontrados
Observações:____________________________________________________________________ _______________________________________________________________________________ _______________________________________________________________________________ _______________________________________________________
___________________________/__/__ Analista:
___________________________/__/__ Supervisor:

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 79/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 79 de 130
Check-List de Controle em Processo
SÓLIDOS ORAIS – EMBALAGEM
Produto: ________________________ Lote:_________________ ( )AG ( )OR
TABAJARA ( ) Fabricação: _____/_____ Validade: _____/_____ Cliente:___________________ OP Nº:________________ Início do processo: _____/_____/_____ Fim do processo: _____/_____/_____ Responsável da linha:_______________ Garantia da Qualidade:________________
ÍTENS A SEREM INSPECIONADOS OK ÑOK01- O local deve estar limpo e devidamente ordenado02- Presença de etiqueta de limpeza assinada pelo responsável da limpeza03- A cabina ou linha deve estar devidamente identificada04- Ausência de materiais do lote anterior 05- O peso deve ser controlado a cada 30 minutos e registrado em folha própria06- Executar o teste de vácuo a cada 30 minutos .
07- Conferir a codificação do material da embalagem primária e secundária de acordocom a OP08- A impressão deverá ser legível09- Anexar blister ou strip (vazio), ao documento após liberação de linha diária10- Anexar cartucho codificado e bula ao documento, após liberação de linha diária.11- Verificar a codificação da caixa de embarque
Data Hora Responsável Data Hora Responsável Data Hora Responsáve
NA = não aplicável Quantidade de amostra retirada para análise: _______________
Observações:_____________________________________________________________________

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 80/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 80 de 130
AMOSTRA DE MATERIAL DE EMBALAGEMCARIMBO- CARTUCHO / BULA DOBRA
Produto:________________________ Lote: __________ Data ___/___/___

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 81/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 81 de 130
Departamento de Controle de QualidadeCheck-List de Reconciliação
Nome do Produto:__________________ Lote:__________________ OP Nº: ________________
Data saída: _____/_____/____ Data de entrada:_____/_____/____ DOCUMENTAÇÃO QUE COMPÕE UM LOTE SITUAÇÃO
OP DE FABRICAÇÃORótulo de Aprovação do ProdutoLaudo de Análise MicrobiológicaLaudo de Análise Físico-QuímicaRótulo de limpeza do tanqueDossiê de Fabricação com fórmula centesimalPick-List de Matéria-prima (conferir com a folha de peso de matéria-prima)Folha de peso correspondente a matéria-primaGuia do Produto
OP DE EMBALAGEM
Folha de Ordem de ProduçãoFolha de acompanhamento de produçãoPick-List de requisição de materiais (conferir com a OP)Pick-List de devolução de materiais (conferir com OP)Relatório de Desvio se necessárioGuia de embalagemCheck_list de inspeçãoFolha de Controle de líquidosFolha de Controle de blister Folha de pesoGráficoRótulo de limpeza da máquinaLiberação de linha
Amostra do produtoAmostra de embalagemLiberação de caixas (conferir com a OP)Protocolo de liberação de caixas.
A OP DE FABRICAÇÃO E A OP DE EMBALAGEM DEVEM SER ARQUIVADAS JUNTAS
OBS.:

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 82/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 82 de 130
OBSERVAÇÕES GERAIS 1: CONTROLE DE INSTRUMENTOS DE MEDIÇÃO
ISO 17 025.
Exigência de certificado de calibração de vidrarias para medição em laboratórios de análise. A
validade de uma calibração é de 10 anos. Isto é um requisito fundamental em termos de BPFv.Além da calibração, deve se exigir os seguintes pontos:
Deve haver registro de controle de uso (desgaste) – logbook de controle;
Registro de manutenção;
Registro de calibração;
NOPs/SOPs para o uso correto.
Características do material de vidro utilizado: O vidro deve ser de alta precisão e de elevadaprecisão na medição. Ex: 25 mL 0.2500mL; para uma proveta, em termos de comparação, seaceita até 10% de variação.VIDRO CLASSE A.
VIDRO CLASSE B: A tolerância de variação de volume deste tipo de vidro é o dobro daquelaverificada para o vidro classe A .Alguns Fornecedores recomendam que a calibração da vidraria se faça a cada 3 anos. Porém, a
ASTM recomenda que esta se faça a cada 10 anos. DIN ISO 10012; Depende das condições deuso.Não se admite o uso de vidraria ou qualquer outro tipo de vidraria não calibrada em um laboratóriode análise.
OBSERVAÇÕES GERAIS 2: CERTIFICAÇÃO DE PESSOAL.
A certificação de pessoal pode ser resumida da seguinte forma:
Pensar na empresa de forma racional;
Revisar todos os procedimentos: Elaborar precisamente o organograma da empresa com a definição de todos os cargos;
Valorizar cada cargo ou função e criar mecanismos para tal;
Promover a prática de roation job.
Deve se pensar em fornecer treinamento básico em BPFv constantemente para todos osfuncionários, não só para os recém contratados.
Vantagens da certificação:
Identifica-se a qualificação necessária para conduzir os processos em curso; Assegura o domínio das tecnologias empregadas na fábrica;
Garantir a terceiros (FDA ou clientes) que o pessoal da fábrica se encontra apto para arealização do trabalho proposto.
Antes de se iniciar a qualificação deve se avaliar:
OBJETIVOS DA EMPRESA X ESTADO ATUAL
Da diferença obtida entre estes dois pontos se pode traçar um plano de ação. Este plano de açãodeverá focar:
Quais são as funções existentes na empresa?

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 83/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 83 de 130
Estas são coerentes com as atividades a serem desenvolvidas?
Qual o nível de conhecimento, comportamento e performance dos funcionários?
Qual a política de recursos humanos da empresa?
PLANO DE AÇÃO:
Atividades que no futuro serão necessárias para atendimento das BPFv- Governarão asmudanças;
Plano de otimização das atividades da empresa;
Competência pessoal x cargo exercido;
Grau de competência;
Forma de acabar com a diferença entre o ideal e os objetivos.
Deve se focar na importância das interfaces de cada uma das tarefas realizadas em umamesma linha de produção. Deve se descrever as atividades, obrigações e conhecimentosreferentes a cada posto de trabalho. Evita-se assim:
Excesso de responsabilidades e comprometimento do produto final; Organograma irreal e sem correspondência com o conhecimento dos funcionários.
Pode se correlacionar a um funcionário, ao invés de uma função, um procedimento operacionalespecífico. Deve existir uma ficha de funções escrita para o pessoal que ocupa uma cargo deresponsabilidade.A certificação deve ser conduzida por: Processo; Parte do processo; Produto.
A certificação da atividade não deve se restringir apenas a execução da mesma, mas sim aoconhecimento teórico a ela relacionada e ao cumprimento das BPFv.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 84/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 84 de 130
Salas Limpas e tecnologia de produção de formas farmacêuticas estéreis:
Como principal característica de uma forma farmacêutica estéril, obviamente, podemosressaltar a necessidade de uma total ausência de microorganismos viáveis e pirogênio nasmesmas, isto é , sua esterilidade apirogenia. Dentre as diversas técnicas capazes de conduzir apreparação de um produto estéril, deve sempre ser observado que, em nenhuma hipótese ométodo de esterilização seja capaz de alterar a constituição do produto em questão, nem aestrutura molecular do fármaco veiculado, assim como, não deve deixar resíduos tóxicos nomesmo. Neste caso, considera-se como técnicas mais comuns para se obter a esterilidade de umproduto farmacêutico, o uso do calor úmido ou autoclavagem, o uso de radiação gama de Co60 , acondução asséptica do processo através da sua fabricação em salas limpas,assim como, o uso dafiltração esterilizante. Uma alternativa para a preparação de pós estéreis, na qual também seobserva a condução asséptica de sua fabricação associada à filtração esterilizante seria aliofilização.
As maiores restrições ao uso da esterilização terminal (autoclaves), se relacionam
principalmente à instabilidade de alguns ativos ao calor; ao mesmo tempo, deve se ter em mente obaixo poder de penetração da radiação Co60 restringindo severamente o tamanho do frasco quese pode esterilizar por esta técnica, assim como, a quantidade de produto nele inserido. Destaforma, para produtos que não podem sofrer esterilização terminal, todo seu manuseio deve seprocessar em local onde se possa evitar qualquer tipo de contaminação, em especial durante seuenchimento, sendo esta etapa tida como etapa crítica do processo. Este ambiente especial échamado de sala limpa (clean room do inglês), devendo possuir características específicas deconstrução, rigoroso controle do tipo de ar nela insuflado, do comportamento dos operadores emseu interior e dos utensílios nela contidos, tendo sido todos estes itens idealizados de forma a seevitar que os mesmos passem a representar uma potencial fonte de contaminação para o produtoaqui exposto.
MAIS SOBRE ESTERILIZAÇÃO: Todo tipo de produto farmacêutico pode sofrer alguma
contaminação. Esta pode se originar das matérias-primas utilizadas ou pelo seu manuseio durantea fabricação em qualquer uma de suas etapas. Uma das principais fontes de contaminação poderiaadvir de uma das matérias-primas mais utilizadas para este tipo de produto, a água. A esterilidadeno sentido da palavra, seria a completa ausência de microrganismos viáveis. Esta pode ser obtidapela exposição da preparação a agentes de esterilização variados, sejam estes químicos oufísicos, tais como o calor úmido ou seco e a radiação ionizante, entre outros. Em verdade, não seobtém a morte total e imediata dos microrganismos viáveis em um processo de esterilização. Oque se obtém, é a diminuição exponencial deste número com o tempo, sendo a esterilidadecompleta somente obtida com um tempo de exposição infinito. Ao mesmo tempo, os teste deesterilidade são realizados em uma amostra representativa do lote de um produto farmacêutico,sendo somente possível se garantir esta esterilidade com o consumo de todo lote. Assim, é deextrema importância que se observe rigorosamente as normas de BPFc durante todo processofabril, de forma a se garantir a esterilidade do produto. A USP afirma que um produto injetávelesterilizado por autoclave deve apresentar 1 MO viável de 1 000 000 inicialmente presentes(redução à 10 -6). Com produtos termoestáveis, pode se expor o mesmo ao calor e o temponecessário para se alcançar a remoção total dos MOS viáveis (over kill). Caso contrário, se ajustao tempo de processo de forma a se obter uma carga bacteriana aceitável. Considera-se comoaceitáveis, valores mínimos de redução de 10 -6 sendo este considerado como o sterilityassunrance level SAL. Como tal, este parâmetro é tido como fundamental na validação doprocesso de esterilização, não sendo obtido até o momento um método validado com base no usode radiação de Co60 .

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 85/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 85 de 130
Em geral, deve se buscar nos processos de esterilização os seguintes objetivos: Adequabilidade de processo; Uniformidade de resultados; Reprodutibilidae.
Suas principais características seriam: Elevada atividade antimicrobiana; Controle de seus parâmetros para regular acuradamente o processo; Compatibilidade com os materiais esterilizados; Ausência de risco para o operador; Tempo de processo e custo reduzidos.
Quanto aos métodos de esterilização de forma geral, podemos dividi-los em dois grandesgrupos;Processos de controle paramétrico: processo no qual os seus parâmetros físicos durante o ciclo deesterilização podem ser acuradamente medidos, sendo o mesmo validado, controlado e verificadoem sua totalidade.
Dois fatores podem ser considerados como fundamentais para se obter uma esterilidadeadequada; A carga bacteriana inicial e a cinética de inativação dos microorganismos presentes.Como outros aspectos relevantes podemos citar: A redução da carga bacteriana é diretamente proporcional ao tempo de exposição do agente
de esterilização; O processo de esterilização segue uma cinética de primeira ordem; K = 1/t . ( logNo – log Nt)
;principal objetivo seria determinar K (No = carga bacteriana inicial e Nt = final); Se utiliza normalmente o D value; tempo necessário para reduzir a população microbiana a
90% ou 1 ciclo logarítimico, sobrevivência de 1 em 10 como parâmetro de checagem daeficácia do processo;
Como processos paramétricos, temos a esterilização por vapor saturado, calor seco e radiaçãoinonizante. Como segundo grupo de processos de esterilização, temos os processos validadoscom bio indicadores. Neste caso, se utiliza um indicador biológico para se garantir a esterilidade aser obtida no processo. Em geral, este bio indicador é uma suspensão de bactérias em um suporte
inerte ou um meio de cultura estéril empregado na validação por media fill. Como exemplo destetipo de processo, temos a esterilização com agentes químicos e a filtração esterilizante. Em ambosos casos, a cinética de esterilização verificada é de ordem um. Logo podemos propor que umapopulação bacteriana se reduziria segundo a equação abaixo;K = 1/t . (log No – log N); se desejamos o tempo para reduzir em 90% esta população, ( Dvalue) podemos afirmar que N = No . 0,1 donde;K = 1/t . ( log No – log No . 0,1); K = 1/t . (1) ; t = 1/K e D = 1/K, por isto se deve calcular K,calculando a partir deste valor o tempo para se alcançar o SAL de 10 –6.
RESUMO DAS CONDIÇÕES PARA ESTERILIZAÇÃO DEUM PRODUTO FARMACÊUTICO
TÉCNICA CONDIÇÕES DE PROCESSOCalor seco 140-170 o C e 60-180 min
Calor úmido sob pressão 121-132o C 5-45 min e 1,336 Kg/cm2 de pressãoÓxido de etileno 25-75o C , 450-1000 mg/L de gás, 50-80% de UR e 1-2 h
Radiação 60Co ou 137 CS 1,5-3,5 Mrad por 15 Seg.Formol vapor 70-80o C, 8-15 mg/L de gás e 2-6 h.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 86/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 86 de 130
Um ciclo de esterilização deve de uma forma geral, reunir ao menos, os seguintes objetivos: Adequabilidade de processo; Uniformidade de processo; Reprodutibilidade.
As seguintes características são desejadas:
Elevada atividade antibacteriana; Possibilidade de controlar e medir de modo preciso os parâmetros que regulam o processo; Compatibilidade com os materiais que estão sendo esterilizados; Ausência de riscos para o operador do processo; Custo e tempo reduzidos.
Segundo as normativas do FDA e ANVISA, sempre que possível deve se optar pelo uso de esterilização terminal por autoclave, devendo este processo ser validado anualmente.Caso contrário, deve se optar pela utilização da chamada f iltração esterilizante.
Filtração esterilizante: Esta seria o uso de filtração de alta eficiência para remover de umapreparação qualquer tipo de bactérias (viáveis ou não), pirogênios e qualquer outro tipo demicroorganismo nocivo, buscando se obter como conseqüência sua esterilização. É indicada comoalternativa para fármacos que não possam sofrer nenhum outro tipo de processo de esterilizaçãoterminal, sendo aprovado pelo FDA apenas nesta hipótese. É necessário neste tipo de processo
que todas as suas etapas sejam conduzidas em um ambiente livre de fontes de contaminaçãoconhecidos como salas limpas. Deve ser utilizado um filtro de classificação absoluta de 0,1 micra, oqual é capaz de remover todos os Mos viáveis ou não, assim como pirogênio, produto de seumetabolismo sintetizado em condições extremas, especificamente quando da morte demicrorganismo gram negativos. Este filtro pode ser constituído de materiais diversos, podendo ser na maioria dos casos, reutilizados pós esterilização em autoclave. Entretanto, deve se validar onúmero máximo de autoclavações que este filtro é submetido, para que o mesmo não liberepartículas no material filtrado já em sua embalagem final. O produto da filtração é conduzido emtubulação de teflon ou aço inox 316 eletroolido (sanitário) para reator de armazenagempressurizado de aço inox 316 sob atmosfera de nitrogênio ou ar estéril, sendo este encaminhadoposteriormente para enchimento. O processo é conduzido em uma sala limpa até que o produtoesteja embalado (embalagem primária). Uma atenção especial deve ser dada para a remoção depirogênio .
Para se assegurar a eficiência do processo, um dos testes realizados, seria o teste deintegridade de membranas de filtração esterilizante; Este teste se destina a garantir o bomfuncionamento das membranas utilizadas, avaliando se existe na matriz filtrante porosa algumcapilar ou orifício de diâmetro maior do que aquele rotulado em sua classificação absoluta, ou seexiste algum tipo de vazamento no sistema. Este teste se baseia na existência de uma elevadatensão interfacial entre o líquido presente nos capilares do filtro e a parede destes, fazendo comque o líquido fique ali retido. Para que se possa deslocar este líquido, deve ser aplicada uma forçacapaz de sobrepor a tensão interfacial, sendo esta força inversamente proporcional ao tamanho doporo. Assim, para que se possa determinar a integridade do filtro, deve ser aplicada uma pressãoao sistema, não devendo esta ser menor do que o valor esperado para o diâmetro previamenterotulado. Este teste pode ser quantificado pela seguinte relação:P = K . ( 4 . cos / D)P = pressão do ponto de bolha;K = Fator de correção de forma (experimental);D = diâmetro do poro; = Tensão superficial do líquido; = ângulo de contato líquido/filtro.
O teste deve ser conduzido com a membrana cheia do líquido e com o sistema totalmentevedado. Se bombeia o líquido até o enchimento do frasco inferior, fechando-se a válvula deentrada; Inicia-se o insuflamento de nitrogênio estéril abrindo as válvulas A e B até igualar apressão. Fecha-se a válvula B. Não pode haver variação no manômetro 1 ou borbulhamento no

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 87/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 87 de 130
frasco de coleta. Apesar de cada sistema ter seu valor ideal de pressão de trabalho, o valor de 20psi (pounds/ pol2) é tida como ideal (ver figura abaixo).
N2A B
12
3
C
Esquema de funcionamento do teste de bolha
= ventilação
Na rotina de produção, o teste de bolha pode se tornar praticamente inaplicável, já
que em grandes volumes de líquido sendo envasados, a avaliação apenas no final ou antes de seiniciar o processo ao garante a efetividade do processo de esterilização. Desta forma, a alternativamais racional para se avaliar a integridade do sistema de filtração seria o uso do teste de pressãodifusiva. Este teste se baseia no fato de que, uma vez cheios de líquido, o ar sob pressão passarápelos poros a uma pressão diferencial menor do que aquela verificada para o ponto de bolha.Quando o filtro é muito pequeno, a pressão a ser utilizada será extremamente pequena. Porém,nos filtros usualmente utilizados em fabricação asséptica, esta pressão é grande o bastante paraser medida, sendo o teste sensível o bastante para avaliar a integridade do filtro. Neste teste, se
aplica uma pressão igual a 80% do valor equivalente ao ponto de bolha, onde se o fluxo de ar aumentar levar a uma pressão mais baixa que a esperada, teremos um poro de valor maior que orotulado, ou seja, perfurações ou vazamentos no sistema.
Não só bactérias ou fungos devem ser removidos da preparação neste tipo de processo.Também deve se obter a remoção total de pirogênios do meio, de forma a se garantir a segurançado produto. O pirogênio se origina do metabolismo usual do microorganismo, sendo geradosespecialmente quando de sua morte. Possui estrutura basicamente lipo-polissacaídica, sendo seurepresentante mais importante a endotoxina. Sua estrutura é extremamente variada, apresentandoainda elevado peso molecular cerca de (106 g/mol) e uma cadeia lateral polissacarídica,responsável pela sua antigenicidade. Todavia, alguns autores atribuem sua maior toxicidade a suacadeia lipídica. Estas substâncias podem formar vesículas ou micelas em meio aquoso emespecial na presença de cálcio e magnésio, sendo seu tamanho final influenciado pela presença ounão de tensoativos.
A presença de pirogênio em um preparação injetável pode levar a febre, choque e mortedo paciente. Produz resposta fisiológica em concentrações acima de 0,1 mg/Kg. Para se obter esta concentração, é necessário uma carga bacteriana de 106 bactérias / mL . Classicamente, seavalia a presença de pirogênio em uma preparação injetável pelo teste de injeção do produto emcoelhos, onde se monitora posteriormente o aumento de temperatura destes animais (mínimo de10). Entretanto, a baixa reprodutibilidade do teste e os problemas referentes à manutenção de umbiotério dentro de uma indústria farmacêutica, fizeram com que este procedimento entrasse emdesuso. Atualmente se utiliza o teste de pirogênio “in vitro” baseado no uso de um reativoconstituído de um lisado de Aemebócitos de um caranguejo conhecido como Limulus polipheno, oqual possui a proteína coagulina a qual é capaz de formar um gel róseo quando em contato com

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 88/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 88 de 130
materiais contaminados por pirogênio (teste do coágulo qualitativo com sensibilidade de 0,12UE/mL). Este teste, ainda pode ser conduzido das seguintes formas: Teste do substrato cromogênico; Ensaio turbidimétrico; Quantificação cromatgráfica; Determinação da proteína de Lowry.
No teste do substrato cromogênico, se utiliza um substrato sintético contendo a seqüênciade amino ácidos similar a da coagulina. Este produto em contato com o pirogênio produz umareação colorimétrica quantitativa, podendo a concentração de pirogênio ser determinadaespctroscopicamente.Já no ensaio turbidimétrico, se utiliza o mesmo reativo que no teste do coágulo, se verificando oaumento da turbidez do meio com o decorrer do tempo. No teste da proteína de Lowry se mede aquantidade de coágulo formado a 660 nm, comparando-se a uma reta de calibração com o padrãode positivo lambda 2 da USP. Pode se propor ainda a utilização da cromatografia líquida depermeação em gel como forma de quantificar este metabólito.
Considerações gerais:
O teste pode ter variações por ser o reagente de origem natural;
Ao modificar o lote ou fabricante do kit, é recomendável validar o teste com o positivo lâmbda 2da USP e repetir o teste com um produto já analisado pelo kit anteriormente existente;
A presença de cálcio ou magnésio no produto pode levar a falsos positivos;
Ácidos nucleicos, polissacarídeos e cianocobalamina, também podem levar a falsos positivos;
Sensibilidade do teste é de 0,12 UE/mL .
Como consenso, temos que na filtração esterilizante, para que esta possa reter eficientemente pirogênios, deve ser conduzida utilizando-se filtros positivamente carregados ou dematerial lipofílico. Em ambos os casos, se obtém um aumento da afinidade do pirogênio pelasuperfície do filtro, aumentando a capacidade de retenção, independente do tamanho da estruturaformada pelo pirogênio em solução (em geral de 0,1 a 0,3 micra). Como exemplo de filtros paraesta finalidade, temos os filtros de Nylon, teflon ou com aminas quaternárias quimicamente ligadasa estrutura do polímero. No caso da filtração esterilizante usual, se utilizam filtros de acetato decelulose, polissulfonas, policarbonato, teflon entre outros. O filtro ideal deve resistir a autoclavageme possuir uma boa performance de filtração ( ver tabela abaixo).
Controle em processo:
1- Integridade de ampolas: Teste com solução de azul de metileno em autoclave com vácuoem dois cilcos de 5,0 minutos;alternativa: uso de corrente elétrica de alta voltagem “ in line”para detectar furos ou fissuras nas ampolas;
2- Revisar em fundo branco/preto para detectar partículas em suspensão; uso de laser paradetectar partículas em suspensão;
3- Volume- checagem do volume de enchimento;4- pH e densidade.
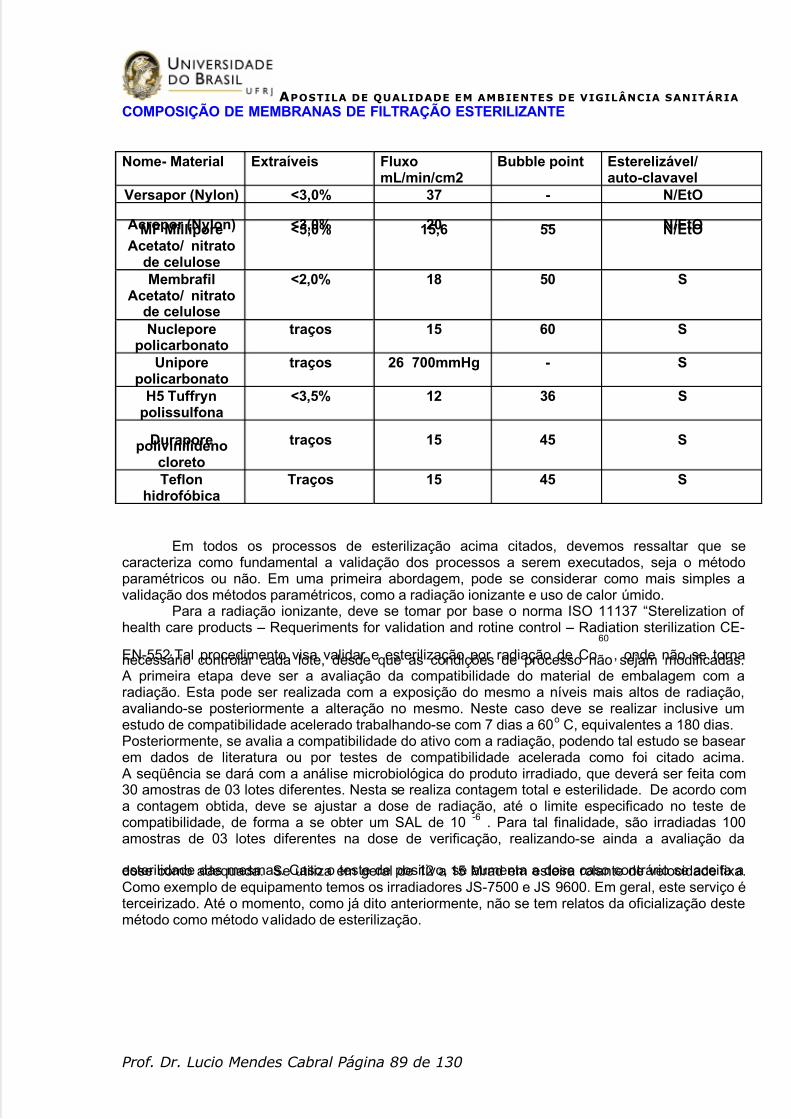
5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 89/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 89 de 130
COMPOSIÇÃO DE MEMBRANAS DE FILTRAÇÃO ESTERILIZANTE
Nome- Material Extraíveis FluxomL/min/cm2
Bubble point Esterelizável/auto-clavavel
Versapor (Nylon) <3,0% 37 - N/EtO
Acropor (Nylon) <3,0% 20 - N/EtOMF MilliporeAcetato/ nitrato
de celulose
<5,0% 15,6 55 N/EtO
MembrafilAcetato/ nitrato
de celulose
<2,0% 18 50 S
Nucleporepolicarbonato
traços 15 60 S
Uniporepolicarbonato
traços 26 700mmHg - S
H5 Tuffrynpolissulfona
<3,5% 12 36 S
Duraporepolivinilidenocloreto
traços 15 45 S
Teflonhidrofóbica
Traços 15 45 S
Em todos os processos de esterilização acima citados, devemos ressaltar que secaracteriza como fundamental a validação dos processos a serem executados, seja o métodoparamétricos ou não. Em uma primeira abordagem, pode se considerar como mais simples avalidação dos métodos paramétricos, como a radiação ionizante e uso de calor úmido.
Para a radiação ionizante, deve se tomar por base o norma ISO 11137 “Sterelization of health care products – Requeriments for validation and rotine control – Radiation sterilization CE-
EN-552.Tal procedimento visa validar e esterilização por radiação de Co60
, onde não se tornanecessário controlar cada lote, desde que as condições de processo não sejam modificadas.A primeira etapa deve ser a avaliação da compatibilidade do material de embalagem com aradiação. Esta pode ser realizada com a exposição do mesmo a níveis mais altos de radiação,avaliando-se posteriormente a alteração no mesmo. Neste caso deve se realizar inclusive umestudo de compatibilidade acelerado trabalhando-se com 7 dias a 60o C, equivalentes a 180 dias.Posteriormente, se avalia a compatibilidade do ativo com a radiação, podendo tal estudo se basear em dados de literatura ou por testes de compatibilidade acelerada como foi citado acima.A seqüência se dará com a análise microbiológica do produto irradiado, que deverá ser feita com30 amostras de 03 lotes diferentes. Nesta se realiza contagem total e esterilidade. De acordo coma contagem obtida, deve se ajustar a dose de radiação, até o limite especificado no teste decompatibilidade, de forma a se obter um SAL de 10 -6 . Para tal finalidade, são irradiadas 100amostras de 03 lotes diferentes na dose de verificação, realizando-se ainda a avaliação da
esterilidade das mesmas. Caso o teste de positivo, se aumenta a dose caso contrário se aceita adose como adequada. Se utiliza em geral de 12 a 15 Mrad em esteira rolante de velocidade fixa.Como exemplo de equipamento temos os irradiadores JS-7500 e JS 9600. Em geral, este serviço éterceirizado. Até o momento, como já dito anteriormente, não se tem relatos da oficialização destemétodo como método validado de esterilização.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 90/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 90 de 130
No caso da qualificação de estufas e autoclaves, o seguinte procedimento deve ser utilizado:Autoclaves: Utilização de sensores de temperatura para se verificar a temperatura no interior daautoclave e sua correlação com os registros externos de pressão e temperatura.Equipamentos utilizados:
Registrador gráfico microprocessado de temperatura;
Termopares tipo T calibrados com termômetros padrão; Bomba hidráulica manual;
Manômetro de teste Wika de 0-7 Kgf/cm2 previamente calibrado.Procedimento:
Distribuir os sensores de temperatura no interior do autoclave geometricamente, estando amesma vazia;
Colocar um sensor perto da manovacuômetro;
Executar 3 corridas em condições normais de esterilização;
Correlacionar os valores obtidos com as temperaturas aceitas.Valores de aceitação: A diferença entre a temperatura máxima e média e a diferença entre a
temperatura mínima durante a avaliação da esterilização não deverá ultrapassar 1o C.A temperatura mínima não deverá ser menor que 121 o C +/- 1.
Avaliação de penetração de calor: Deve se executar o teste conforme descrito acima, poréminserindo os sensores de temperatura no interior dos frascos que serão utilizados na fabricaçãousual do produto ou na impossibilidade, utilizar frascos erlenmayer de 150 mL.
Quando se considera a validação de estufas de esterilização e depirogenização, o mesmoprocedimento acima deve ser seguido. Deve se ressaltar que existem sensores de temperatura outermopares que não possuem fios, sendo as informações armazenadas no mesmo eposteriormente transferidas a um computador. Todos os equipamentos descritos se encontrailustrados no anexo. No caso de despirogenização de material de vidro, o método mais aceito paraeste fim seria o uso de calor seco a 250-300º C por 8 horas (em estufa ou esteira), ou ainda, o usode ultrassom. Entretanto, a autoclavagem, desde que validada em termos da remoção de pirogênioaplicando-se para tal o padrão de endotoxina da USP, pode ser uma alternativa viável,principalmente para tubetes ou carpules utilizados em injetáveis odontológicos. A preparação daembalagem plástica em ambiente estéril ou técnica de Blow-fill também é uma alternativa
tecnológica viável, apesar de mais cara.
Validação do sistema de filtração esterilizante:Para a validação de um processo conduzido em uma área limpa, deve ser considerada a
necessidade de se fabricar 06 lotes nas exatas condições de processo, porém substituindo oproduto por um meio de cultura esterilizado, como o agar com tripticaseina e o tioglicolato. Desteslotes, 03 devem ser conduzidos com operadores ou sistemas com não conformidades propositais,de forma a correlacionar o número de partículas dispersas com estas atividades. Nos demais 03lotes, deve se seguir rigorosamente à rotina de fabricação do produto, buscando a maior similaridade possível com a realidade. As seguintes considerações devem ser feitas: Usar igual volume de meio em relação ao produto real; As tampas dos frascos devem entrar em contato com o meio para avaliar seu manuseio e
esterilidade (virar frasco);
Determinar limites de alerta/ ação a partir das contagens de partículas; Determinar processo ideal de limpeza (figura 8 ou movimentos verticais descendentes entreoutros); validar o detergente e agente de sanitização ideais; rodízio de sanitizante quinzenal.
Não se deve observar nenhum crescimento nos frascos utilizados ( em geral 10% da dimensãodo lote real) sendo o limite de alerta uma contaminação em 0,1 % dos frascos.
Sistemas de filtração de ar de alta performance (hepa):
Por definição, o ar contido em uma sala limpa é classificado de acordo com o número departículas presentes por metro cúbico, sendo estas partículas orgânicas ou inorgânicas, viáveis ou

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 91/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 91 de 130
não. Segundo o Federal Standard 209 E (Padrão americano federal), se define este nível departículas através de uma classificação crescente iniciada com a classe 10 seguindo-se a classe100, 1000, 10 000 e 100 000, classificação máxima aceita para um ambiente fabril farmacêutico.O número a que se refere esta classificação equivale ao número de partículas menores ou iguais a0,5 m presentes por ft3 de ar. Outros tamanhos de partículas também são regulamentados emcada classe conforme ilustra a tabela abaixo. Outras classificações, inclusive a citada na RDC
210/03 ANVISA também regulamentam o número máximo de partículas em suspensão. Aindaregulamenta este tipo de classificação a ISO 14 644-1. A seguir, é descrita a constituição do Ar segundo o FR 209 E e sua correspondência na RDC 210 da ANVISA.
Classe No de partículas por ft3 de ar;100 (A) e (B) 0,5 m – 3530 e 0 Mos viáveis
1000 0,5 m – 35300 e 5 Mos viáveis10.000 (C ) 0,5 m – 353000 e 100 Mos viáveis100.000 (D) 0,5 m – 3530000 e 500 Mos viáveis
A obtenção deste ar com a classificação desejada decorre da utilização de filtros especiaisconhecidos como filtros HEPA. Estes são filtros destinados à remoção de todas as partículas do ar
que possuam tamanho menor ou igual a sua classificação de poro absoluta. Possuem capacidadede retenção de 99,999% tendo diversos parâmetros governando seu funcionamento. Entre estestemos:
Velocidade do fluxo de ar: Esta medida deve ser realizada por meio deanemômetro, devendo ser o valor ideal de velocidade em torno de 0,5 f 3/ min, segundoo Federal Standard 209 D. Porém, o funcionamento normal deste filtro em uma salalimpa pode levar a um aumento desta velocidade, conforme visto na tabela abaixo:
Velocidade ideal de funcionamento de filtros HEPA
Atividade realizada Velocidade do ar em f 3 /minValor ideal 0,5
Abertura de porta 0,72Fluxo laminar 30
Pessoal caminhando 105HVAC 145
Ar condicionado 250
Isto demonstra que é muito mais vantajoso, deixar o filtro HEPA funcionando a maior velocidade possível para se obter o ambiente com a classificação desejada. Acreditava-se que ouso de velocidades de ar maiores que 30 f 3/min levariam a turbulência em um fluxo laminar de ar.Porém, não existe nenhuma prova científica para esta afirmação, reforçando a proposta inicial.
Em teoria, quanto maior for o fluxo de ar sobre o filtro, menor seria sua capacidade defiltração (partículas livres, aerossóis e bactérias). Porém, a prática mostra que um maior fluxo de ar não compromete seu funcionamento, em especial porque as partículas de 0,1 a 0,5 m,consideradas como críticas no processo possuem um baixo poder de penetração. Consdera-secomo fundamental para o bom funcionamento deste filtros a avaliação de sua integridade. Esteteste era feito empregando-se um equipamento que gera um aerossol de dioctil ftalato (DOP), oqual deve ser colocado antes do filtro em teste, ajustando-se a velocidade do fluxo de aerossolpara se ajustar o tamanho de partícula a ser obtida. Este aerossol deve ser totalmente retido pelofiltro, sendo a passagem de algum tipo de resíduo detectada por fotômetro contador de partículas,manual ou fixado na saída do filtro. Como este equipamento é capaz de gerar até 5.0 x 10 6 partículas por cm 3 de ar, podem ser testados filtros de eficiência igual a 0,001 %.. Porém, estegerador de DOP é muito pesado e grande, sendo difícil se colocar o mesmo na saída de um fluxolaminar ou em alguns pontos específicos de uma sala limpa. Desta forma, se utilizam os fotômetros

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 92/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 92 de 130
digitais manuais para se avaliar o número de partículas que saem do filtro (modelo ROYCOCOUNTER por exemplo) para garantir seu perfeito funcionamento. É importante que emconcomitância com o uso do fotômetro, se empregue algum método microbiológico que possacorrelacionar esta contagem de partículas com o grau de microorganismos viáveis. Pode se utilizar para este fim placas de agar nos locais críticos (saída do filtro, mais perto dos equipamentos deprocesso, mais perto do produto e onde há maior tráfego de operadores). Outra alternativa seria o
amostrador centrífugo de ar que recolhe constantemente o ar do ambiente inoculando o mesmoem fitas de agar indicando qualquer possível contaminação. Este é utilizado por dois períodos de30 minutos durante a jornada de 08 horas de trabalho.
Uma outra aplicação deste tipo de filtro seria a obtenção de um fluxo laminar de ar paraque se possa garantir a assepsia de um processo de enchimento de produtos estéreis. Este fluxolaminar de ar seria um “êmbolo “ de ar purificado que faria um efeito de varredura de todas aspartículas viáveis ou não por onde passar. Desta forma, se efetuaria uma limpeza desta área,garantindo a esterilidade do produto.
Calor, anteparos de geometria irregular e movimentos bruscos no interior da região defluxo laminar podem levar a formação de turbulência e prejudicar seu efeito de limpeza. Empequenos ambientes como em uma capela de fluxo laminar, torna-se mais fácil se garantir um fluxolaminar de ar. Em equipamentos de enchimento se utilizam cortinas de materiais poliméricos parase obter um fluxo da ar laminar naquela região, devendo o filtro HEPA estar instalado a 1,5 m doequipamento.
Exemplo de fluxo laminar
Dificilmente se consegue obter um fluxo laminar de ar perfeito. O operador neste sistema éum ponto crítico, devendo se manter o mais afastado o possível do local. Na parte inferior (piso) dacabine de fluxo laminar deve haver uma placa perfurada de aço inox com sucção de ar para seassegurar a qualidade do fluxo. A velocidade de ar insuflado deve ser igual a do ar removido,sendo a diferença máxima de temperatura entre o ar que entra e o ar que sai de 5o C no máximo,
de forma a se evitar turbulências. Segundo o Federal Standard 209 E (b 55295) e ISO 14644-1 oseguinte grau de renovação de ar é recomendado para uma indústria farmacêutica:
Número de renovações por hora Classificação do ambiente120-600 10040-50 10.00020-30 100.000

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 93/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 93 de 130
De uma forma geral, este valor pode estar 30% acima do valor ideal. Porém, isto não éconsiderado como super dimensionamento, e sim, medida de segurança. Quando existe algumafonte de calor no local, como em um túnel de esterilização por exemplo, 35 renovações por hora
são suficientes para manter a classificação.Em termos da pressurização de um ambiente classificado, em uma eclusa de 5,0 m3 (1,42 x 1,42 x2,50), deve receber 120 trocas por hora para se obter uma pressurização de 1,3 mm de coluna deágua ou 13 pa (pascals), ideal para garantir pressurização diferencial segundo o FDA. Porém umvalor de 8,0 pa pode ser aceitável se o sistema não for crítico. Já a CEE, fala em termos de BPF,em 29 pa como valor mínimo aceito. O consenso indica que é a validação da instalação a capaz dedeterminar o valor ideal de pressão diferencial.
Salas limpas: comportamento do pessoal de trabalho:
O comportamento do pessoal dentro de uma sala limpa é ponto de fundamentalimportância para o sucesso do processo fabril, garantindo conseqüentemente, a esterilidade doproduto fabricado. Dentre os diversos aspectos a serem considerados, o uniforme dos operadorespoderia ser considerado como um dos pontos de maior relevância. O mesmo deve atuar como umabarreira, evitando que o operador contamine o produto ou ambiente com os milhões de partículas,perdigotos ou microorganismos que o mesmo libera. Estes contaminantes podem se encontrar emseus cabelos , pele, cosméticos e vestimentas internas. O uniforme além de confortável para ooperador,deve representar uma barreira para 100% da superfície da pele do operador e seuscabelos, evitando também que se formem bolsas de ar quente no interior do mesmo, o que poderiaacelerar o processo de troca de ar do interior para o exterior, levando a contaminações. Estesuniformes devem ser confeccionados em materiais que funcionem como boas barreiras, nãoliberem partículas ao ambiente e possuam pontos de troca de ar (interior/ área externa), filtrando oar que estava em contato com o operador. A parte mais crítica deste uniforme seria as luvas, vistoserem estas que mais se aproximam do produto. Se recomenda o uso de luvas de latexresistente, e mais, sempre o uso de dois pares.
O operador deve ser uma pessoa calma, de personalidade fácil, não deve usar barba,
cabelos longos, deve ter hábitos de higiene rigorosos, ser meticulosa, não deve usar jóias, relógiosou cosméticos na área fabril, e uma vez dentro da mesma, deve falar o mínimo possível e evitar movimentos bruscos, movimentando-se também o mínimo possível. Estes movimentos podemlevantar partículas presas ao chão ou em outras superfícies, levando a um aumento decontaminação. É previsto inclusive, um intervalo de operação de 15 min a cada duas horas para seremover este excesso de partículas do ambiente (cleansing time).
Estando o operador doente (inclusive resfriado ou mulheres menstruadas) estedeve ser deslocado para outra função. Se dá preferencia ao emprego de mulheres neste setor por serem mais calmas e meticulosas.
resumo do perfil do operador de sala limpa: Calmo e ameno; Não ceder a pressão;
Não usar barba e cabelos longos; Unhas curtas; Boas práticas de higiene; Não fumante; Não usar jóias ou cosméticos Não trabalhar doente.
aspectos de construção e processo:O primeiro aspecto a ser considerado quando da construção de uma sala limpa seria seu
custo extremamente elevado, o que faz com que se pense em idealizar o local com a menor área

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 94/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 94 de 130
possível. Um outro ponto fundamental seria seu sistema de HVAC (do inglês, ar condicionado,ventilação e exaustão) do qual decorre a qualidade do ar ambiental. Dentre os cuidados a seremobservados neste setor temos: Manutenção, limpeza e sanitização periódica dos filtros dutos e pré-filtros; Ordenar as salas por classificação de ambiente; Validação rigorosa e completa do sistema de produção e transporte de WFI e vapor puro
(cuidado com a formação de rouge); Uso obrigatório de interlocks ou air locks e passthrought; Pisos e paredes anti-estáticos, de fácil limpeza e não porosos (PVC soldado ou em placas e
resinas fenólicas revestidas de melamina e recheio de lã de rocha ou poliuretano anti-chamaem forma de divisórias);
Paredes de aço inox para exposição a água ou vapor; Em alguns casos, pisos de resina epóxi tipo stom hard; Painéis de controle de equipamentos e serviços em aço inox 316; Uso de sistemas de controle e monitoração a distância; Usar ciclones ou outro tipo de precipitador para o ar de alimentação; Recuperar o ar exaurido filtrando o mesmo em HEPA (85% de economia do sistema de HVAC)
validação rigorosa; UR entre 40-45% e temp. 20o C (abaixo deste valor gera desconforto e leva ao risco de
explosões); Avaliar periodicamente a pressurização das áreas. Maior pressão na área mais limpa e
decresce para área mais suja; Em geral a classificação ambiental requerida seria áreas classe 100 (enchimento) e 10000 na
preparação de produtos estéreis m sistemas abertos e 100 000 em sistemas fechados.
Tipos mais comuns de sanitizantes:Quaternários de amônio: Não corrosivos e ideais para a superfície por evitar deposição de MOs;não é indicado para locais onde o produto esteja exposto. São potencializados pelo EDTA e nãosão ativos contra vírus, fungos , leveduras e esporos.Biguadinas: Catiônico de baixa toxicidade e não agressivo. É mais efetivo contra fungos eleveduras sendo comparável aos quaternários de amônio;Álcool a 70% : Assepsia das mãos somente; baixa eficácia;Iodo: Mancha superfícies e é muito reativo;
Fenol: Agressivo, tóxico e corrosivo;Hipoclorito: Corrosivo, porém de amplo espectro de atuação; irritante em determinadasconcentrações; usado principalmente na forma inorgânica (hipoclorito 170 ppm), sendo aindadisponível na forma orgânica (monocloro amina por exemplo).Ácido peracético e peróxido de hidrogênio: Oxidante forte a baixas concentrações. Até hoje não seconhece nenhum mecanismo de resistência de diferentes microroganismos a estes agentes. Oácido é utilizado entre 0,5 a 2,0 % e o peróxiodo de 1,0 a 10% , inclusive na forma de vapor.Somente o ácido peracético deixa resíduo (ácido acético) o qual é pouco tóxico e facilmenteremovido.
Glutaraldeido e formaldeído 0,40% : Esterilização de instrumentos em solução; em francodesuso.
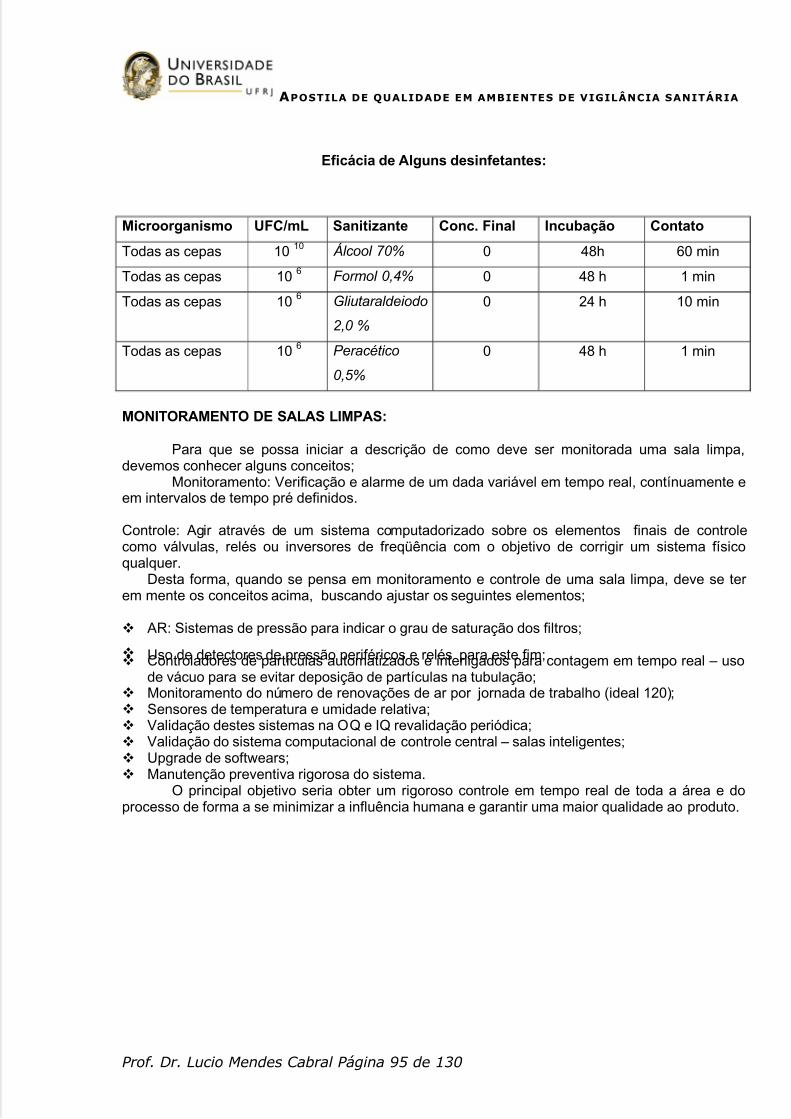
5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 95/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 95 de 130
Eficácia de Alguns desinfetantes:
Microorganismo UFC/mL Sanitizante Conc. Final Incubação Contato
Todas as cepas 10 10 Álcool 70% 0 48h 60 min
Todas as cepas 10 6 Formol 0,4% 0 48 h 1 min
Todas as cepas 10 6 Gliutaraldeiodo
2,0 %
0 24 h 10 min
Todas as cepas 10 6 Peracético
0,5%
0 48 h 1 min
MONITORAMENTO DE SALAS LIMPAS:
Para que se possa iniciar a descrição de como deve ser monitorada uma sala limpa,devemos conhecer alguns conceitos;
Monitoramento: Verificação e alarme de um dada variável em tempo real, contínuamente eem intervalos de tempo pré definidos.
Controle: Agir através de um sistema computadorizado sobre os elementos finais de controlecomo válvulas, relés ou inversores de freqüência com o objetivo de corrigir um sistema físicoqualquer.
Desta forma, quando se pensa em monitoramento e controle de uma sala limpa, deve se ter em mente os conceitos acima, buscando ajustar os seguintes elementos;
AR: Sistemas de pressão para indicar o grau de saturação dos filtros;
Uso de detectores de pressão periféricos e relés para este fim; Controladores de partículas automatizados e interligados para contagem em tempo real – uso
de vácuo para se evitar deposição de partículas na tubulação; Monitoramento do número de renovações de ar por jornada de trabalho (ideal 120); Sensores de temperatura e umidade relativa; Validação destes sistemas na OQ e IQ revalidação periódica; Validação do sistema computacional de controle central – salas inteligentes; Upgrade de softwears; Manutenção preventiva rigorosa do sistema.
O principal objetivo seria obter um rigoroso controle em tempo real de toda a área e doprocesso de forma a se minimizar a influência humana e garantir uma maior qualidade ao produto.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 96/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 96 de 130
Liofilização:
É um processo que visa remover a água presente em uma solução ou suspensão fazendo
com que esta passe diretamente da fase sólida para a fase de vapor, diminuindo desta maneira, aquantidade de calor que deve ser cedida ao sistema, preservando fármacos termossensíveis. Comesta técnica, pode se obter produtos com baixos teores de umidade (ca. 2,0% ou menos) e deelevada porosidade, o que os torna de fácil dissolução.Para que se possa obter a passagem da água de sua fase sólida para a fase de vapor, o materialem questão deve ser processado em condições de pressão e temperatura, abaixo daquelasobservadas no ponto triplo da água (Ge. 4,8 mm Hg e 0,0098o C). Ao se analisar curavas depotencial químico x pressão, podemos observar que a fase a qual apresenta menor potencialquímico, é sempre a fase mais estável. Assim, abaixo da temperatura de fusão, o líquido apresentamenor potencial químico que o sólido. Acima desta temperatura, o líquido se congelariaespontaneamente, pois isto levaria a diminuição da energia de Gibbs, estabilizando o sistema.Com a variação da pressão, tendo sido esta reduzida, se verifica um deslocamento natemperatura de fusão e uma redução grande em seu ponto de ebulição. Assim, reduzindo apressão a um valor extremamente baixo, o ponto de ebulição do líquido pode ficar abaixo do pontode fusão do sólido. Neste caso, o líquido não possui estabilidade em temperatura alguma,verificando-se a sublimação do sólido. Existe um valor de pressão na qual as três curvas seinterceptam a uma mesma temperatura. Esta pressão define o ponto triplo da substância, onde astrês fases coexistem. Abaixo deste valor, podemos verificar o fenômeno anteriormente citado, asublimação. Quanto maior for o ponto de fusão e menor a diferença entre os pontos de fusão eebulição, maior será a pressão abaixo da qual se observará a sublimação (eq 1);
Eq 1 : Ln p = -10,8 ( Tb – Tm / Tm) ; Tb = ponto de ebulição e Tm = ponto de fusão.
As principais vantagens do processo de liofilização podem ser resumidas como;
Pequenos danos ao fármaco veiculado; possibilidade de trabalho com substânciastermolábeis;
Formação de estrutura porosa de fácil ressolubilização; Dosagem mais precisa e acurada; Produto pode ser manuseado em condições 100% assépticas.
Apesar destas vantagens, podemos afirmar que as desvantagens deste processo estão ligadasapenas a aspectos de custo e tempo de processo, sendo estes;
Elevado custo do equipamento; Elevado custo energético ( 3 x maior que o uso de uma estufa); Tempo de processo entre 24 a 72 h.
Logo a aplicação deste processo se restringirá;
Materiais instáveis em solução; Materiais termolábeis; Produtos estéreis; Dosagem precisa; Injetáveis extemporâneos; Ou seja produtos estéreis de alto valor agrageado; Citostáticos, antibióticos , vacinas e produtos de biotecnologia.
O processo de liofilização será conduzido em três etapas distintas; o congelamento prévio domaterial abaixo de sua temperatura eutética (onde este se mostra totalmente cristalizado; no

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 97/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 97 de 130
caso de produtos amorfos se fala em temperatura de colapso), a sublimação de água parareduzir o valor de umidade residual a 4,0% p/p (secagem primária) e a secagem secundária,onde se leva o material a valores de umidade residual até o desejado, isto é , valores menoresque 1,0% p/p.
Estas fases podem se sobrepor em alguns momentos e em alguns casos, até mesmo emtodo o ciclo. A presença de solventes orgânicos, em geral o etanol, faz com que o material não
congele na temperatura usual do processo, tornando o mesmo inviável. Na fase inicial desecagem, os vapores de água passarão pelos interstícios deixados pelos cristais de gelo que já sublimaram. Desta maneira, o formato destes cristais é fundamental para o controle doprocesso; cristais pequenos e descontínuos, tornam a passagem do vapor mais difícil,aumentando sobremaneira o tempo total do processo. Logo, estes devem, ser longos edistribuídos por toda a massa, da parte mais inferior até a superfície, o que pode ser obtidocom o controle da fase inicial de congelamento.
Este congelamento poderia ser obtido pelo uso de vácuo, visto que este deve estar presente no equipamento para seu uso posterior óbvio. Entretanto, ao se remover moléculasde maior energia por vácuo, o que levaria a uma abaixamento da temperatura, podemos Ter aformação de espuma e de pequenos cristais de gelo, o que é extremamente prejudicial aliofilização industrial (tempo). A formação de pequenos cristais de gelo também pode ser observada quando se utiliza refrigerantes consumíveis como o N2 líquido para umcongelamento rápido. Desta maneira, se recomenda o uso de um líquido refrigerante quecircule em torno dos frascos contendo o material a ser congelado, o qual pode levar aocongelamento progressivo do material (manter por cerca de 2 h abaixo da temp. eutética),sendo este mesmo líquido utilizado posteriormente na secagem primária, visto que ao seaplicar vácuo ao sistema, a temperatura tenderia a abaixar ainda mais, congelando emdemasia o material interrompendo consequentemente a sublimação do mesmo. Assim, omesmo fluido que congela o material por convecção, difusão e irradiação, deve mante-loaquecido a cerca de 30-40 oC para que se observe uma taxa de remoção de água 1 mL degelo removido por 1 m3 de vapor.Para garantir o bom funcionamento da bomba de vácuo, o coração do sistema, deve haver uma câmara de congelamento para capturar todo vapor de água formado não permitindo queestes a alcancem evitando futuros danos. O processo segue então até se obter o teor final deágua desejado, sendo o tempo total, determinado em estudos piloto o por sensores de águapresentes na câmara de secagem ou no condensador de gelo.
Os liofilizadores modernos podem ser então constituídos pelos seguintes componentes:
Câmara de secagem separada de um condensador de gelo para se evitar contaminaçãocruzada e permitir a determinação do ponto final do processo;
Ambos confeccionados em aço inox 316 que resistam a pressão e sanitização por vapor; Sistema de circulação de fluido refrigerante; Instrumentação de controle e monitoração de temp. , vácuo e demais variáveis do processo; Sistema de fechamento de frascos pneumático ou hidráulico; Sistema automático de carga e descarga.
Para produtos de baixo valor agregado, pode se colocar a câmara de secagem junto aocondensador de gelo para se minimizar custos.
Detalhes técnicos :
60 a 90 m2 da áreas de bandejas para secagem (alguns com cerca de 200 m2 para alimentos); Parede dupla e bomba elétrica de circulação do fluido refrigerante; Utiliza freon 22 para congelamento; Compressor tipo Worthington com capacidade mínima de 300 Kg de condensado; Bombas de vácuo (02) Edwards de 600 mc/h; Válvula para controle de entrada de vapor de água no condensador de gelo;

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 98/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 98 de 130
Comando computadorizado a distância (em geral em área não asséptica, diferente da área deprocesso, classe 1000);
Sistemas de CIP e SIP; Drenos para água condensada na sanitização por vapor.
Esquema resumido de um liofilizador industrial
Boas normas de fabricação relacionadas ao processo de liofilização:
Cada equipamento destinado a liofilização, deve ser construído e desenhado de forma afacilitar a sua operação, limpeza, manutenção e demais operações correlatas, visando ao mesmotempo, atender às Boas Normas de Fabricação Vigentes. Desta maneira, este equipamento deveobservar;
Que em sua construção todas as superfícies que entrem em contato com o produto não sejamreativas, adsorventes e favoreça o crescimento microbiano, garantindo a integridade deproduto;
Ser construído de forma que nenhuma das substâncias ou unidades funcionais como óleo,vapor ou similares entrem em contato com o produto; Construído de forma a facilitar carga, descarga, controle, limpeza manutenção e qualquer outro
tipo de atividade similar; Ser seguro ao ambiente e ao operador; Ser idealizado de forma a reduzir ao máximo seu consumo de energia.
No que se relaciona a integridade do produto fabricado, podem ser ressaltados osseguintes aspectos em relação aos liofilizadores industriais:
Camara de pré-congelamento
Camara de liofilização
óleo quente
óleo frio
Válvula decontrole de vácuo
condesador degelo
bomba de vácuo
bomba de vácuo
respiro 0,22 micra
unidade de aquecimentoresfriamento e circulação

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 99/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 99 de 130
Sistema de carga e descarga veloz e que não gere particulados por atrito (uso da carregadoresde bandejas com rodas de teflon);
Sistema de alarme e monitoração eficiente e informativo; Sistema inteligente de controle e monitoramento de temperatura (auto ajuste); Duplicação de todos os componentes fundamentais do sistema; condensador de gelo, bombas
de vácuo, teclados e sistemas de controle, sistemas de controle manuais e em alguns casos a
câmara de secagem; Uso de portas opostas para a carga em área classe 1000 e descarga em área classe 10 000
ou 100 000.
Em relação a segurança do operador; Sistema de carga que previna a liberação de substâncias tóxicas (carregador de bandejas); Controle do processo computadorizado e a distância; Interlocks de segurança; Todos os equipamentos auxiliares (não fazem parte do liofilizador) devem funcionar a baixa
tensão 24 V; Sistema de interrupção de potência individual para cada unidade do sistema; Portas com amplo grau de abertura (125o ) para se facilitar o acesso.Consumo de energia:
Aquecimento com controle preciso de temperatura; Condensador de gelo com temperatura programável; evita o desligamento total, que aumenta o
gasto de energia para se alcançar novamente temperaturas de - 75 o C; Usar dois condensadores (alternados e sem desligamento total); Isolamento térmico e de vácuo eficiente; Usar pequenos volumes por vial para se obter maior economia no processo (1/3 do volume).
Características gerais:
Superfície de secagem com cerca de 63 m2 em 3 módulos; 30 bandejas de carga; Distância entre as bandejas regulável 11 a 13 cm; Temperatura mínima das bandejas -55 oC; Condensador a - 75 o C , secundário a - 85 oC; Esterilização por vapor a 126 oC Uso de hipoclorito a 170 ppm; Potência instalada de 750 KW.
Cada uma destas unidades podem ser consideradas como liofilizadores, esterilizadores,um túnel de separação e um sistema integrado de carga e descarga.
Detalhes construtivos: Câmara de secagem, bandejas partes internas da câmara e do condensador em aço inox 316
eletropolido; Espessura média de 320 mesh para a câmara e 240 mesh para o condensador; Drenagem total de água de seu interior; permite CIP (clean in place) e SIP (sterilization in
place); Soldaduras orbitais em toda a parte interna; Pistolas e aplicadores de vapor e água internos para CIP/SIP; 40 sensores internos de temperatura.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 100/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 100 de 130
Esquema de um sistema de carga e descarga:
Fechamento hidráulico
Suportes internos
Bandejas

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 101/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 101 de 130
Fotômetro para contagem de partículas

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 102/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 102 de 130
Cartuchos para filtração esterilizante
Exemplo de Máquina de Envase de ilíquidos injetáveis

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 103/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 103 de 130

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 104/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 104 de 130
Visão Geral de uma Sala Limpa

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 105/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 105 de 130
Envase de pós injetáveis na FURP
Visão geral de salas limpas e uniformes utilizados neste setor.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 106/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 106 de 130
Tratamento de Água - princípios de filtração:
Filtração, por definição, é o processo pelo qual se separam as partículas em suspensão deum fluido (gás ou liquido), pela passagem do fluido através de um material permeável. Oprocesso de filtração tem, hoje em dia, uma importância fundamental na indústria moderna, sendo
que é difícil imaginar uma indústria que não tenha algum tipo de dependência em relação aosprocessos de filtração.O processo de filtração tem suas raízes na antigüidade: documentos chineses descrevem formasprimitivas de filtrar; os escritos hebreus mencionam também processos de filtração. Os egípciosdesenvolveram técnicas de filtração para tinturas e cerveja. Durante a Idade Média, os processosde filtração por gravidade foram de aplicação corrente. As primeiras patentes sobre equipamentosde filtração datam do final do século XVIII, na Europa, reinvindicando a utilização de materiaisporosos, filtros de leito de areia com camadas de diferente granulometria e, inclusive, comprocesso de regeneração por fluxo reverso.Após a metade do século XIX, têm sido registradas numerosas patentes para filtros contínuos,como os de tambor, assim como diferentes mecanismos de limpeza dos meios filtrantes. Por voltade 1950 apareceram os primeiros filtros para gases com limpeza do meio filtrante pelo efeitos de"jet-pulse". Os primeiros filtros empregados para injetáveis e vacinas foram idealizados por Chamberlain (1884), misturas de asbestos e celulose foram inventadas por Schrnitthenner (1915) ea primeira membrana porosa de celulose por Zsigmondy entre 1914 e 1918, sendo que a fábricaSartorius a comercializa desde 1929 (Sartorius -Werke Gottingen). Hoje em dia, os campos deutilização de filtros são tão diversos que é comum o seu emprego para filtrar tanto líquidoscriogênicos como em altas temperaturas, na separação de partículas em processos industriais,como na separação de componentes de alto peso molecular de outros de peso molecular menor,resistindo à ação de poderosos solventes ou fluidos altamente corrosivos. Devido ao continuoaumento das exigências, as técnicas de filtração tiveram um extenso desenvolvimento, forçandouma especialização, em função das inúmeras aplicações.
Cada filtro em especial, de uma finalidade concepção, incorporando umatecnologia especifica, responderá de forma diferente à mudanças nos parâmetrosoperacionais. Alguns filtros podem ser mais sensíveis que outros a pulsações de pressão eperder a eficiência pela descarga do contaminante retida na sua estrutura, podendo fazê-lo,inclusive a uma determinada freqüência de pulsações de pressão. Nenhum sistema de teste
de filtros, atualmente em uso, tem a possibilidade de gerar impulsos de pressão oscilantesde amplitude e freqüência controláveis.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 107/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 107 de 130
CLASSIFICAÇÃO DAFILTRAÇÃO
VELOCIDADE DECRESCIMENTO DO
BOLO
CONCENTRAÇÃO DESÓLIDOS EM PESO
TIPO DE FILTRAÇÃO
1- Filtração rápida2- Filtração média
3- Filtração lenta4- Clarificação comleito
5- Clarificação
Rápido: cm/seg.Médio: cm/min
Lento: cm /hBolo desprezível
Bolo desprezível
40%2-10%
0.1-2,0%0,01-0,5%
0.01-0.1%
Tambores e esteiraBandejas e tambores
Filtros prensaLeitos granulares
Cartuchos
Outro parâmetro é o tamanho da partícula a ser separada do fluído. Foi comprovada, através detestes, urna interação das características das partículas e material do particulado, o fluido e omaterial do meio filtrante, por exemplo, um filtro quando submetido ao mesmo diâmetro departículas de óxido de ferro ou silício, comportou-se de forma diferente para ambos os materiaiscom diferentes características de retenção.
Processos de filtracão
Os sólidos em suspensão são separados dos fluidos por três mecanismos principais: impactaçãoinercial, interceptação por difusão e interceptação direta. Mesmo que todos estes mecanismos defiltração estejam operacionalmente presentes, a sua importância relativa e a importância de cadaum muda. Tanto a impactação inercial como a interceptação por difusão são muito menos efetivoscom líquidos do que com gases. Visto que a densidade das partículas é muito mais próxima doliquido do que a do gás, o" desvio de uma partícula em suspensão de uma linha de fluxo liquido émuito menor e, em conseqüência, a impactação da partícula com a estrutura do meio filtrante émais provável. No entanto, a impactação, em muitos sistemas, não é seguida pela adesão daspartículas na superfície do meio filtrante. A interceptação por difusão em líquidos é muito limitadadevido a que o movimento Browniano, praticamente, não se produz em suspensões liquidas deforma tão pronunciada quanto nas suspensões gasosas.
Impactação Inercial
As partículas numa corrente fluida têm massa e velocidade, vez que têm um momento associadocom o movimento do fluído. Urna Assim como o liquido e as partículas em. suspensão passampelo meio filtrante, o fluxo do liquido fará o caminho de menor resistência e fluir. em torno dasfibras. As partículas, devido ao seu momento maior, tenderão a mover-se em linha reta e, comoresultado, as partículas localizadas no, ou perto, do centro da linha de fluxo se chocarão ouimpactarão nas fibras do meio filtrante, sendo removidas do fluxo. A Figura 1 ilustra este processo.As linhas de fluxo, mostradas como linhas continuas, fluem em volta das fibras, enquanto aspartículas continuam o seu caminho, mostrado com linhas tracejadas, e batem contra as fibras.Geralmente, as partículas maiores desviam-se mais facilmente das linhas de fluxo do que asmenores. Na prática, no entanto, devido à diferença de densidade entre partículas e o fluído ser pequena, o desvio da linha de fluxo líquido é muito menor, e a impactação inercial em filtração delíquidos tem um papel relativamente pequeno.
Interceptação por DifusãoPara partículas extremamente pequenas (e aquelas de pequena massa), a separação pode ser resultado da interceptação por difusão. Neste processo, as partículas estão em estado de colisãocontinua com as moléculas do fluído. Estas colisões freqüentes fazem com que as partículas emsuspensão se movimentem de forma aleatória em torno das linhas de fluxo do fluido. Estemovimento, que pode ser observado microscopicamente, é chamado de movimento Brownianow.O movimento Browniano faz com que estas partículas pequenas sejam desviadas das linhas defluxo de fluído, aumentando aparentemente o seu diâmetro e aumentando, conseqüentemente asua probabilidade de bater com a superfície do filtro e serem removidas. Assim, como naimpactação inercial, a interceptacão por difusão.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 108/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 108 de 130
tem um papel mínimo na filtração de líquidos. Isto deve-se ao fato de que a característica naturalinerente do fluxo do liquido tende a reduzir o movimento lateral ou os desvios das partículas alémdas linhas de fluxo do fluido.Interceptação DiretaEnquanto os mecanismos de impactação inércia e intercepta cão por difusão não são tão efetivosem filtros para serviço com líquidos como em filtros para gases, a interceptacão direta é igualmente
efetiva para ambos os fluídos e é, portanto, o mecanismo desejado para separar partículas delíquidos. Num meio filtrante, é observada a presença, não de uma fibra, mas de um conjunto comgrande número destas fibras. Estas fibras definem as aberturas pelas quais passa o fluido. Separtículas em suspensão no fluido são maiores que os poros ou aberturas no meio filtrante, elasserão removidas como resultado da intercepta cão direta nos furos. A interceptação direta éfacilmente visualizada num filtro de tela ou malha de arame, com poros uniformes e semprofundidade ou espessura, uma vez que a partícula passa pelas aberturas ela continua nacorrente do fluido. Mesmo assim, o filtro coletará uma significativa porção de partículas de diâmetromenor que o diâmetro dos poros do meio filtrante.Na prática, a maioria das partículas em suspensão, mesmo as muito pequenas, quando olhadas dediversas direções, são de forma irregular e podem afazer pontes nas aberturas.O efeito ponte pode também ocorrer se duas ou mais partículas atingem o furo simultaneamente.Uma vez que a partícula é detida num poro, este poro é, pelo menos parcialmente, ocluso econsequentemente capaz de separar as partículas ainda menores da corrente do fluido, menoresque os Poros do Meio.Interações especificas da superfície podem causar a aderência de partículas pequenas nasuperfície interna dos poros do meio filtrante. Por exemplo, uma partícula consideravelmentemenor que o poro, tem a possibilidade de aderir-se ao poro, visto que asduas superfícies estão opostamente carregadas. Podem existir outros tipos de interação, tais comoligação de hidrogênio e forças de Van der Waals.
Auxiliares Filtrantes para LíquidosÉ possível aumentar a efetividade de remoção de partículas de um líquido por um filtro, comdiversos métodos que serão apresentados resumidamente, a seguir.
Deposição eletrostáticaA maioria das partículas tem carga negativa. Os meios filtrantes têm, normalmente, cargas quepodem afetar tanto a eficiência de remoção das partículas, como a eficiência de retenção daspartículas no filtro. É possível aumentar os mecanismos de captura de partículas de um filtro pelaindução da carga desejada (usualmente positiva) nas fibras. A entre partículas e fibras emcondições de Zeta potencial diferente. A partícula tem uma carga superficial negativa comoresultado da dupla camada de íons, formada na sua superfície. O meio filtrante tem um Zetapotencial positivo induzido que promove a remoção das partículas. A intensidade da carga, tantoda partícula, como das fibras, é importante. Em geral, assim como a intensidade da carga aumentae o tamanho das partículas diminui a eficiência de captura aumenta. O beneficio óbvio de um filtrode superfície carregada é a habilidade de remoção de partículas muito finas por um meio filtranteque tem poros relativamente grandes, baixa perda de pressão e uma grande capacidade de
retenção de contaminantes. Este procedimento não deve ser confundido com a filtração de pré-cobertura, onde o auxiliar filtrante é depositado sobre o filtro e a suspensão é forçada a fluir atravésdele. Assim como no caso da floculação, o objetivo do auxiliar filtrante é atingir umapermeabilidade desejada no bolo.Um dos tipos mais comuns de auxiliar de filtração, é visto nas terras diatomáceas, que consisteem depósitos sedimentos fossilizados. Os esqueletos de diatomáceas têm uma ampla variedadede formas e é esta propriedade que permite que ela produza bolos filtrantes de altapermeabilidade. Outros auxiliares filtração são a perlita (rocha vulcânica fundida resfriada emágua), carvão e celulose. A filtração com auxiliar filtrante não é comum na clarificação de sólidosmas , quando empregada, pode ser encontrando associada à filtração por cartuchos. Os filtros de

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 109/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 109 de 130
cartucho são empregados como filtros armadilha" a jusante dos filtros de pré-camada paracapturar qualquer auxiliar filtrante que possa vazar através do meio filtrante.
Filtração por cartuchosNo Sistema de filtração existem parâmetros que influenciam o processo de seleção doequipamento de separação para a remoção de sólidos de fluídos por meio de filtração, seja o
fluído líquido ou gás. A concentração de sólidos é outro parâmetro a se considerar neste processo,sendo a concentração máxima em um processo de clarificação igual a 0,5% em peso. Deve aindaser considerado na escolha do tipo de cartucho a ser utilizado, o material que o constitui, aadequabilidade a finalidade do sistema sua classificação ( se absoluta ou nominal) e a área útil defiltração.
Cartuchos CoalescentesOs cartuchos coalescentes são elementos filtrantes projetados para a remoção de aerossóislíquidos de fluxos de ar ou gases comprimidos. O funcionamento do elemento coalescentebaseado na capacidade de interceptar as gotículas de aerossóis líquidos, fazendo com que elas seaglomerem ou coalescam, formando gotículas maiores, de tal tamanho que possam ser separadasda corrente do gás por gravidade. As características desejáveis para um elemento coalescenteótimo são:- Alta eficiência de separação para aerossóis de 0.1 até 0.6J1m, numa ampla faixa deconcentração variável do aerossol no fluido afluente. A concentração de aerossóis na saída nãodeverá exceder 0.05 partes por milhão em peso, para proteção dos componentes a jusantes(instrumentos, motores, compressores, leitos absorventes, etc.). Baixa perda de pressão apóssaturação com aerossol liquido. A resistência ao fluxo do sistema deverá ser mínima para manter baixas as perdas de pressão do sistema e reduzir o consumo de energia.Grande capacidade de acumulação de contaminantes para permitir a acumulação decontaminantes sólidos mantendo baixas perdas de pressão. Os contaminantes sólidos sãoprovenientes do gás aspirado, corrosão, oxidação e desgaste do compressor. O fluxo do gás noselementos coalescentes é inverso ao fluxo dos cartuchos filtrantes convencionais, isto é, o gásentra pelo núcleo central do elemento e sai pela parte externa, desta forma a velocidade de saídado gás é menor, não permitindo o arraste de gotículas. Devido ao fluxo inverso, o meiocoalescente deverá ser rigidamente suportado para resistir aos elevados diferenciais de pressão,normalmente em volta de 3kg/cm2 (30 psid) .
O meio filtrante coalescente tem normalmente poros menores no lado de entrada do fluxo do gás,aumentando para o lado de saída, para permitir a passagem de gotículas cada vez maiores,resultado da aglomeração. Após um suporte metálico de chapa perfurada, pode haver ainda umacamada de espuma plástica que desempenha o papel de filtro secundário e permite a drenagemdo "liquido, não permitindo a sua reentrada na corrente de gás.
FILTRAÇÃO POR MEMBRANA
As membranas são definidas como uma lâmina fina de material permeável, flexível e possível deser p1isada. Este material é relativamente novo se comparado com asbestos ou cerâmica porosa,empregados antigamente para esterilização de líquidos Estas membranas foram desenvolvidas a
partir de diferentes materiais poliméricos como certos ésteres de celulose com este principalobjetivo. O procedimento de fabricação altamente sofisticado, resulta na obtenção de uma folha dematerial de espessura em torno de 120micra, com uma estrutura porosa bem definida, e umvolume poroso de 80% aproximadamente. Uma análise microscópica deste material mostra umatortuosidade elevada para seus poros, longe de uma estrutura perfeitamente disposta na superfíciedo mesmo. Apesar da irregularidade do meio, este funciona como filtro absoluto para osdiâmetros de partículas que foi regulamentado, mantendo sua integridade mesmo sob pressão.Após a Segunda Guerra, tiveram lugar uma série de desenvolvimentos para fabricar membranasde outros materiais poliméricos, inclusive porque as membranas de éster de celulose, que sãoquebradiças, impediam o plissado para formação de cartuchos. Ao mesmo tempo, e em paralelo,

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 110/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 110 de 130
foram desenvolvidas membranas fibrosas". Todos os esforços de desenvolvimento foram sempredirigidos para obter membranas com o mesmo objetivo final: filtros com qualificação de retençãoabsoluta de 100% para determinado tamanho partícula. Para filtração de fluídos, isto significaprincipalmente, 0.2 micra. Podemos classificar as membranas em dois tipos: a- membranas
plásticas e membranas fibrosas.a- Fabricadas por processos sofisticados, envolvem a remoção de uma solução dê polímero deuma certa quantidade de material volátil, transportando este filme por uma câmara com ambientecontrolado. Os materiais podem ser: acetatos de celulose, acetatos múltiplos de celulose, nitratosde celulose, ésteres de celulose, misturas de polímeros de celulose, Nylon 6, Nylon 66, PVC,PVDF, Teflon, etc. Os materiais mais quebradiços são empregados, ainda hoje, em forma dediscos e materiais flexíveis são plissados e formados como cartuchos filtrantes.O diâmetro dos poros pode ser controla, e são fabricados, desde 100 micra até para aplicações deultrafiltração e osmose reversa, com poros de 0,000l micra.b- membranas fibrosas: A fabricação deste tipo de membrana envolve a dispersão controlada,estruturação e ligação entre microfilamentos sintéticos inorgânicos, geralmente um materialcristalino. Estes microfilamentos possuem diâmetro extremamente uniformes. As microfibras sãointerligadas por meio de resinas sintéticas. As membranas de micro fibras são de maior espessuraque as membranas plásticas, mas isto não significa que possam ser classificadas como filtros deprofundidade, já que os mecanismos de retenção de partículas são os mesmos que os demembrana plástica. Com este procedimento é possível obter filtros de membrana que possuamqualificação absoluta para as partículas desejadas e microorganismos com suficiente rigidez paraevitar a descarga das partículas retidas, assim como a migração do próprio meio filtrante, sendoestas as características requeridas para filtros com grau de retenção absoluto. A membrana éplissada e formada como cartucho filtrante, com características Únicas como insensibilidade àtemperatura, capacidade de esterilizacão por fluxo direto de vapor, ampla compatibilidade química,podendo ser re-esterilizada, e de baixo Custo.
Classificação de filtros:
Meios Filtrantes de Matriz Porosa não Rígida: Os meios filtrantes com poros que não são fixos,
dependem para sua função dos mecanismos de filtração de impactação inercial e/ou interceptaçãopor difusão para retirar as partículas dentro de seus interstícios e espaços da sua estrutura interna.Exemplos deste tipo de materiais são os feltros, tecidos, placas filtrantes e mantas de fibra devidro. Estes filtros são construídos com meios filtrantes com matriz não rígida, e de espessurasuficiente para reter partículas de determinado tamanho, com base num processo estatístico finito.Pode-se deduzir que as partículas retidas podem ser soltas, se a estrutura do meio filtrante é talque as dimensões do poro aumentem assim como aumenta a pressão diferencial. Tambémqualquer tipo de feltro pode reter partículas menores que a dimensão de seus poros, porém sob ascondições de fluxo de impulso ou variável, estas partículas mais finas tem maior probabilidade deserem soltas por um filtro em que os seus poros aumentem de tamanho. Isto é sempre umproblema com meios filtrantes de porosidade não rígida. Este tipo de meio tem sempre muitoscaminhos tortuosos para o fluído e, naturalmente, as passagens mais estreitas são as queentopem antes com partículas e, como resultado, o fluído é obrigado a tomar o caminho das
passagens mais abertas. Visto que a estrutura do meio não é integral, o aumento de pressão naspassagens maiores pode originar a separação do meio e, como conseqüência, tornar estaspassagens maiores ainda, criando canais de fluxo. O resultado óbvio é que o efeito decanalização afeta adversamente o desempenho do filtro. Os meios porosos não rígidos nãodependem somente da captura das partículas, mas também do mecanismo de adsorção para reter as partículas capturadas. Estas forças são moleculares e de superfície, e a partícula permaneceráretida até que a força de arraste do fluído seja menor que a força de retenção. Porém, depois queo filtro seja submetido ao fluxo por algum tempo, e tenha coletado certa quantidade de materialparticulado, um brusco aumento de velocidade do fluído ou de pressão, poderá vencer as forças deretenção e originar a soltura de algumas partículas. Isto é chamado de descarga e ocorre

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 111/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 111 de 130
freqüentemente quando o filtro está em operação por algum tempo. Este efeito pode dar a falsaimpressão de que o filtro tem urna longa vida de serviço. Além disso, a maioria dos meios filtrantesde matriz não rígida, estão sujeitos à migração do meio. Isto significa que partes componentes domeio filtrante são separadas e arrastadas pelo fluxo do fluído, contaminando o fluído que passoupelo filtro. A migração do meio filtrante é, às vezes, incorretamente considerada como a liberaçãoda contaminação adquirida pelo meio durante a sua fabricação.
Meios Filtrantes de Matriz Porosa Rígida: Os meios filtrantes de poros rígidos ou fixos, podemser de uma camada simples de meio filtrante, ou compostos de múltiplas camadas. Estes meiosdependem, fundamentalmente, do mecanismo de interceptação direta para a retenção departículas, e são fabricados de forma que a sua estrutura não se deforme pela passagem tortuosado fluido. Estes filtros podem reter partículas por adsorção como resultado da impactação inercial eda interceptação por difusão. Também podem ter poros maiores que a qualificação do seu grau deretenção. O tamanho do poro é controlado na sua fabricação, de forma que possa ser garantida aretenção quantitativa de partículas de tamanho determinado. Por outro lado, fazendo o meiofiltrante com suficiente espessura, pode-se conseguir que a soltura das partículas retidas, que sãomenores que o grau de retenção especificado, ou seja minimizada, inclusive em condições de fluxode impulso. Os chamados filtros de superfície ou peneira podem ser qualificação neste grupo,sempre que tenham estrutura rígida, no entanto, os filtros de malha podem ter estruturas tantofixas como não fixas, e serão melhor classificados num dos dois grupos.
Conclusão sobre Tipos de Meio Filtrante: Depois de ter analisado a informação anterior,podemos chegar à conclusão de que classificar os filtros como de profundidade ou de superfícienão tem nenhum significado. Quase todos os filtros, aparentemente, são de profundidade quandoanalisados sob o microscópio, incluindo as membranas de filtração estéril.
Uma classificação mais significativa dos filtros é a seguinte:a) Filtros com estrutura porosa não rígida, poros não fixos, nos quais os poros aumentam dedimensão com aumento de pressão (bobinados, os feltros de baixa densidade, os prensados).b) Filtros de estrutura porosa rígida com poros fixos que não aumentam de tamanho com a pressão(a maioria dos filtros de membrana).Os filtros de estrutura porosa rígida são superiores em muitos sentidos. Eles combinam uma altacapacidade de retenção por unidade de área, tanto para partículas maiores quanto para as
menores que o tamanho determinado, com uma mínima soltura das partículas menores retidas sobcondições de fluxo de impulso. Estes filtros podem ser qualificados como absolutos para otamanho de partículas determinado. Os filtros de estrutura porosa não rígida não podem ter qualificação absoluta para tamanhos de partículas determinado, estão sujeitos à migração do meiofiltrante e certas descargas de partículas sob condições de fluxo de impulso. Uma comparação dacapacidade de retenção de contaminantes, entre filtros de estrutura porosa rígida e filtros deestrutura porosa não rígida, não tem muito significado, devido a que, normalmente, a qualificaçãode retenção nominal dos filtros de estrutura porosa não rígida não têm relação com o seudesempenho em serviço. Com o fato de não ter significado se o filtro é de superfície ou deprofundidade, surge então a questão de como julgar um filtro. Somente duas perguntas têmimportância para o usuário de filtros:1- O filtro em questão está sujeito à descarga do particulado retido, canalizado ou migração do
meio? Se assim for, então para a maioria das aplicações é imprudente o uso deste filtro. 2- O
fabricante garante que o filtro removerá, confiavelmente e com segurança, todas as partículaspara as quais foi selecionado, do fluído em questão?
Classificação de filtros pela sua retenção: vários sistemas de classificação foram desenvolvidospara descrever a aptidão de retenção dos elementos filtrantes. Desafortunadamente, até omomento não existe um sistema único aceito em geral, e isto tem como conseqüência a confusãodo usuário de filtros. Os diversos sistemas em uso são:Classificação nominal: Muitos fabricantes de filtros baseiam-se na classificação nominal de seusfiltros feitos assim pela NFPA (National Fluid Power Association): "Um valor arbitrário, emmicrômetros, fixado pelo fabricante do filtro, com base na remoção de uma determinada

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 112/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 112 de 130
percentagem de todas as partículas de tamanho determinado ou maiores, raramente é bemdefinido e não é possível de reproduzir." Na prática, o contaminante" é introduzido antes doelemento filtrante e o fluido efluente do filtro é analisado miroscopicamente. Um determinado valor "nominal" de retenção de um filtro, significa que 98% em peso do contaminante mencionadoanteriormente foi removido, e que 2% em peso do contaminante passou pelo filtro.
Nota-se que este teste é gravimétrico e não de contagem de partículas. Uma contagem departículas antes e após o fi ltro tem maior significado, para avaliar o desempenho do filtro. Os váriostestes utilizados para classificar nominalmente" filtros de porosidade não fixa, tem dado resultadosnebulosos e confusos. Os problemas típicos são:(a) A remoção de 98% do contaminante em peso é determinada pelo emprego de umdeterminado contaminante a uma concentração e fluxo definidos. Se alguma das condições deteste é alterada, os resultados do teste são alterados significativamente. Não se tem umadefinição, pelo teste, dos 2% de contaminante que passaram pelo filtro. Não é incomum que umfiltro com retenção nominal de l,0 micra deixe passar partículas de 30 até 80 micra. Os dados dostestes não são possíveis de serem reproduzidos pelos diferentes laboratórios de testes. Algunsfabricantes não têm como base, para sua classificação nominal, o valor de 98% de remoção decontaminantes em peso, porém empregam valores de 95%, 90% e ainda menores. Asconcentrações muito altas e contaminantes empregados neste tipo de teste não são típicas parasistemas de condições normais, e como resultado produzem valores ilusoriamente elevados deeficiência. Poderia se pensar que filtros com classificação nominal de 1,0 micra deveriam reter todas ou quase todas as partículas de 1,0 micra ou maiores. Alguns fabricantes continuam autilizar a classificação nominal por duas razões: a primeira é que faz parecer que seus filtros sãomais finos do que realmente parecem, a segunda é, que é impossível de se classificar absolutamente filtros com matriz porosa que não é rígida.Classificação absoluta: A NFPA define a classificação absoluta assim: "O diâmetro da partículadura, esférica, que pode passar pelo meio filtrante sob condições de teste, é uma indicação doporo de maior abertura do elemento filtrante. Este tipo de classificação pode ser feita só com meiosfiltrantes de estrutura rígida e integral. O teste original e o termo de classificação foram propostospelo Dr. D. B. Pall, nos anos 50, sendo reconhecidos pela Comissão de Filtros A-6 da SAE eadotados com pequenas modificações. Um ponto que confunde o usuário de filtros comclassificação absoluta é que, quando se mede a contaminação após o filtro, são invariavelmenteencontrados contaminantes maiores que a porosidade de classificação absoluta. A primeira vista,
isto pode trazer dúvidas sobre o conceito de classificação absoluta. Porém, devemos concordar que é praticamente impossível tomar amostras, transferi-las e fazer o teste sem adicionar umacerta quantidadede contaminante. Mesmo um filtro novo é contaminado quando retirado da sua embalagem. Tudoisto é chamado de contaminante básico ou inicial", e antes de se realizar um teste, um laboratórioexperiente determinará o valor desta contaminação inicial. Os testes podem ser invalidados se a contaminação básica ou inicial estiver acima do valor pré-determinado. Existemdiversos tipos de testes, devidamente reconhecidos, para estabelecer a classificação absoluta defiltros. O tipo de teste empregado depende do fabricante, do tipo de meio filtrante a ser testado e,algumas vezes, da indústria usuária. Em todos os casos, o filtro é desafiado pelo bombeamento,através dele, de uma suspensão de um contaminante reconhecido (por exemplo: esferas de vidroou suspensão de bactérias) , e tanto o af luente como o efluente são examinados para determinar apresença do contaminante de teste. Este tipo de teste é destrutivo, o filtro testado não poderá ser
usado depois em serviço. Consequentemente, foram estabelecidos testes de integridade parafiltros que não são destrutivos mas correlatos com o teste destrutivo de classificação. Em outraspalavras, se o filtro tem seu teste de integridade aprovado pelo teste não destrutivo, isto significaráque ele poderá ser aprovado no teste destrutivo. Porém, após a aprovação pelo teste deintegridade, o elemento filtrante poderá ser posto em serviço e ele fornecerá ao usuário osresultados pretendidos pelo fabricante do f iltro.Testes de esferas de vidro: Uma suspensão de esferas de vidro (todas calibradas dentro defaixas de dimensão pré determinada) é passada pelo filtro e são coletadas amostras do f luído apóso filtro numa membrana analítica. As esferas de vidro são empregadas pelo fato de seremclaramente distinguíveis da contaminação básica gerada durante o próprio teste. As esferas que

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 113/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 113 de 130
passaram pela filtro são examinadas ao microscópio para determinar a esfera de vidro de maior diâmetro que passou pelo filtro. Este diâmetro estabelece a classificação absoluta do filtro.Sistema de índice beta: Enquanto a classificação absoluta é claramente muito mais útil que aclassificação nominal, um método de teste foi recentemente desenvolvido que expressa aclassificação do filtro por designação de valor d.e índice Beta () . Os índices Beta sãodeterminados empregando um teste desenvolvido pela Oklahoma State University chamado OSU
F-2 Filter Performance Test, ou simplesmente, Teste F-2-. O teste foi originalmente desenvolvidopara ser empregado com filtros de sistemas hidráulicos e de lubrificação, este método foi adaptadopela PALL CORPORATION para fazer testes rápidos, semi automaticamente com filtros que sãoutilizados com sistemas de líquidos aquosos, óleos e outros fluidos. O sistema de teste original éutilizado para classificar filtrosque operam em sistemas de re-circulação do fluido, a PALL CORPORATION adaptou também ométodo de teste para filtros que operam em sistemas de passagem simples. O sistema de índiceBeta é de conceito simples e pode ser empregado para medir e predizer o desempenho de umaampla variedade de cartuchos filtrantes, operando nas condições de teste especificadas. Se acontagem total de partículas é medida para diferentes diâmetros de partículas, tanto no fluido quechega ao filtro, como no que sai dele, poderá ser traçado um perfil da eficiência de remoção decada filtro.
Sistema de teste para filtros microbiológicos: O teste de filtros esterilizantes microbiológicoscom desafio de microorganismos é um teste destrutivo, o elemento filtrante não podendo ser utilizado após o teste. Este teste é realizado para validar o filtro e fazer a correlação dosparâmetros para testes de integridade não destrutivos a serem realizados para controle dequalidade do fabricante e pelo usuário. Está bem estabelecido que a eficiência de retenção de umdeterminado filtro é maior quando o fluído é gás ou ar do que quando o fluído é um líquido.consequentemente, o teste de desafio com líquido como fluído é o mais severo teste parademonstrar a capacidade de retenção de um filtro. Conforme a última definição do FDA, um filtrode, grau esterilizante é um filtro que produz um efluente estéril quando desafiado comPseudomonas diminuta (ATCC 19146) com concentração mínima de 107 organismos por cm2 deárea efetiva do filtro. Este é o microrganisrno empregado para testar filtros cuja porosidade for definida como de 0,22 micra. Para o teste de filtros de 0,45 micra, se utiliza como teste a Serratia marcescens (ATCC 14756). Para filtros de 0,6 micra, é utilizada cepas de Saccharomycescerevisiae. Para filtros de 0,1 micra, é empregado Mycoplasma-Acholeplasma.
Para filtros de 0,05 micra é utilizado Bacteriphage Tl de E.Coli .Testes para Desafio Bacteriológico com Líquido o sistema está composto de filtros da água deentrada, bomba de circulação de vazão variável (deslocamento positivo) , controlador de vazãoautomático (normalmente ajustado para 2l rpm por cada cm2 de superfície de filtro), dosador debactérias de desafio, carcaça e filtro de teste, manômetro e vários suportes e membranas deanálise e controle. A verificação da esterilidade é feita pelo plaqueamento das membranas de testeem agar Mueller-Jlinton, posteriormente incubado por 72 horas para observar a presença ouausência de colônias de microrganismos.Teste para Desafio Microbioló9ico com Aerossol: O sistema de teste com aerossol inclui ageracão de aerossol mono-disperso de uma solução de Bacteriphage TI que é direcionado para ofiltro em teste misturado com ar seco ou úmido. Todo o aerossol que sai do filtro é passado por recipientes impactadores de liquido que contém um liquido recuperador estéril. Ao finalizar o teste,o conteúdo completo dos impactadores de líquidos é testado para detectar a presença do
bacteriófago pela técnica de cultura.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 114/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 114 de 130
Nos dias atuais, a água destinada ao uso farmacêutico é tida sem, sombra dedúvidas, como uma das matérias-primas mais utilizadas na fabricação de diversas formasfarmacêuticas, influindo diretamente na qualidade, segurança e eficácia das mesmas. Isto éverdadeiro mesmo quando a água é usada para:
-Formulação de produtos (líquidos, semi-sólidos, emulsões,parenterais e suspensões)-Processamento (granulação por via úmida e revestimento)-Usada como enxágüe final de uma superfície de contato do produto-Limpeza de área estéril
Apesar de ser possível se utilizar água de fonte natural, as indústrias farmacêuticas deuma forma geral, recebem seu abastecimento de água da rede pública, não havendo uma grandepreocupação com sua classificação. Todavia uma análise de custos deve ser realizada para seavaliar as vantagens de utilizar água da rede pública ou de fontes naturais. Talvez o custoadicional de tratamento necessário para seu uso farmacêutico, não compense a economia obtida.De qualquer forma, é fundamental para se iniciar o processo de tratamento de águas, que seconheça o seu histórico onde se avalia a sua qualidade em termos microbiológicos e físico-químicos durante todo o ano para se idealizar seu processo de beneficiamento.Abaixo se esquematiza o processo de tratamento de água de uma fonte natural até a distribuiçãoda mesma.
Esquema de tratamento e distribuição de água da rede pública.
De forma a tornar a empresa parcialmente independente do fornecimento da rede pública,esta obrigatoriamente deve possuir uma cisterna. Esta cisterna deve ser limpa periodicamente(semestral) e deve ter sua qualidade microbiológica controlada (semanal). Para se evitar variações
grandes na qualidade desta água e para diminuir a periodicidade de limpeza destas cisternas, devehaver uma pré-filtração da água no ponto de alimentação. Como o cloro que chega neste pontofinal pode variar de acordo com o estado da tubulação de alimentação deve ainda ser previsto apossibilidade de se adicionar cloro a água da cisterna para o controle da contaminação bacteriana.Para isto se utiliza uma bomba dosadora de cloro e como pré-filtro Filtros, os de carcaça de açoinox e com recheio de areia ou quartzo. Estes devem ser retrolaváveis e deve se prever suahigienização periódica. A água então armazenada deve ser semanalmente analisada em termosmicrobiológicos, fisico-químicos e quanto ao seu teor de cloro.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 115/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 115 de 130
Deve ser observado:Histórico da qualidade da água com o decorrer do anoProcedimento escrito de retrolavagem dos filtros (operação e periodicidade)Procedimento escrito de operação da bomba dosadora de cloroProcedimento escrito de limpeza das cisternas e ficha de controle de limpeza
Análise semanal da água da cisterna (microbiologia e físico-química) O segundo passo no processo de tratamento de águas para fins farmacêuticos
seria a remoção do cloro presente na mesma, o qual se adicionou para o controle da qualidademicrobiológica. Tanto o processo de pré-purificação como o de remoção de cloro são processos defiltração. Caso as tubulações estejam contaminando a água fornecida, o programa de lavagem a
jato das mesmas deve ser iniciado, conduzindo a um fornecimento de água que possua um níveladequado de cloro no interior das tubulações, principalmente após um baixo período de uso, poisisto pode ter contribuído p/ o desenvolvimento de MOs (baixo período de uso pode levar umadiminuição do nível de cloro). Uma alternativa a filtração como forma de se remover cloro seria suareação com metabissulfito de sódio. Como a filtração estará presente em diversos métodos depurificação de águas a serem utilizados, devemos conhecer um pouco mais deste processo.
A filtração pode ser dividida em três classes:
-pré-filtração para remover grandes partículas, objetivando prevenir a contaminação com partículasdo equipamento de tratamento de água, como também, para prolongar a vida útil do equipamentode filtração final. Inclui-se nesta classe os filtros de areia (ou quartzo). Estes filtros removempartículas de 40 m e maiores e podem ser laváveis (retrolavagem).
-microfiltros (0,22 m) usado p/ remover muitas bactérias.
- Ultra filtração- a faixa de porosidade (0,001-0,01 m) fica entre microfiltração e osmose reversa(outros sistemas de filtração de alta eficiência). Este equipamento removerá partículas e bactérias,mas não remove pirogênios dependendo do tamanho do poro da membrana UF e da natureza dopirogênio contaminante. Não removem sais dissolvidos. Filtros constituem uma sessão adicional nosistema de tubulação e são sujeitos a condições de fluxo baixo ou intermitentes. Se não existeagente bacteriocida residual na água, qualquer MO corrente abaixo de um microfiltro ou UF podemultiplicar-se. É importante que em qualquer sistema que empregue filtros, o controlemicrobiológico seja efetuado e toda tubulação corrente abaixo com os dutos e filtros esterilizadosefetivamente.
Nos sistemas onde usam-se filtros devem ser observados determinados aspectos:-microfiltros devem ser instalados apenas onde existe uma necessidade de reduzir o nívelbacteriano. Eles não devem ser utilizados indiscriminadamente como uma política de segurança.- uso de um filtro não deveria corrigir um defeito microbiológico na operação de um outrocomponente no sistema. Os filtros devem ser selecionados p/ adequar a uma aplicação particular -a instalação deve ser adequada p/ permitir a melhor utilização da capacidade do filtro-após seleção do filtro, o mesmo deve ser validado p/ garantir que não existirá migração, liberaçãode partículas sob o impacto das condições variáveis de fluxo, pressão, temperatura e choque
hidráulico.-deverá existir um procedimento para a inspenção do filtro, substituição, teste de integridade emonitoramento.-filtros usados p/ controle microbiolgico e todos os sistemas de distribuição corrente abaixo devemser esterilizados periodicamente p/ proteger o filtro de se tornar um meio de crescimentobacteriano.-se o filtro é tido como ineficaz, mesmo quando usado adequadamente, o mesmo deve ser retirado.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 116/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 116 de 130
Algumas considerações a serem feitas em relação ao equipamento de filtração:-porosidade da membrana-teste de integridade da membrana-queda de pressão que cruza a membrana
-método de sanitização na montagem do filtro-remoção de resíduo químico da sanitização-liberação de partículas da membrana filtranteComo já dito anteriormente, para se tornar parcialmente independente do sistema público deabastecimento, toda empresa deve possuir uma cisterna. A qualidade microbiológica da água nelapresente é controlada pela adição de cloro. Assim, após prévia filtração com filtros de areia pararemoção de particulados, devemos proceder à remoção do cloro na água. Duas possibilidades seencontram disponíveis; uso de bissulfito de sódio 1,0 % p/v e o uso de carvão ativado, maisdifundido que a técnica anterior.
Filtração com Carbono ativado:
Técnica freqüentemente usada para retirar cloro e constituintes orgânicos da água(mecanismo de adsorsão) para proteger as resinas trocadoras de íons e certos tipos deequipamento de osmose reversa da degradação prematura, assim como diminuir a corrosão desuperfícies de aço inox. Uma vez que o cloro tenha sido retirado, não existe agente residualbacteriocida na água e, desta forma, a população microbiológica pode se multiplicar rapidamente.
Filtros multimeios de areia ou carvão ativado

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 117/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 117 de 130
C + 2Cl2 + 2H2O 4HCl + CO2
Figura- Mecanismo de adsorsão do cloro pelo carvão.
Os leitos de carbono podem torna-se áreas de geração de MOs devido a alta áreasuperficial e baixo fluxo. A camada inicial ainda tem cloro ativo, mas nas camadas mais inferiores oteor de cloro diminui, permitindo a geração de MOs. Os filtros de carbono devem ser monitoradosquanto ao nível microbiológico de forma similar àquela dos equipamentos de abrandamento ondeverificamos uma grande tendência ao crescimento de microrganismos. Os filtros de carbono e deareia devem ser monitorados quanto:
-freqüência de lavagem e tempo de clarificação-recirculação contínua-exigências para sanitizar o leito-utiliza-se NaOH, Formol, ácido acético e vapor a 100o C-substituição do carbono
-monitorar a queda de pressão
Devem existir procedimentos de sanitização dos filtros escritos com a periodicidada dos mesmos(devidamente validados). A capacidade adsortiva do carvão é considerada como praticamenteirrelevante, em vistas a sua rápida saturação e difícil regeneração (acima de 200 o C).
A partir da retirada do cloro da água, o tratamento subseqüente será determinado pelo tipode água que se deseja obter, sendo esta classificada de formas diversas.
Filtro de carvão ativado

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 118/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 118 de 130
Consolidação da classificação de águaNa verdade existe similaridade entre as diversas classificações para água. Na
indústria, teremos em geral os seguintes tipos:1- água purificada:
a) sem adições de substânciasb) difícil controlar nível MOs (a menos que seja manipulada de forma similar WFI).Muitas operações podem usar água p/ beber (mais fácil controlar) ao invés de usar água purificadasegundo USP.
2- água p/ limpeza e enxague não tem exigências para a pureza química alémdaquelas descritas para água de beber. Por isso pode se usar esta água p/ enxágüe e limpezainiciais, pois:
-é mais barato e fácil de controlar -controle microbiológico pode ser feito usando cloração ou água quente (fazer
validação p/ verificar se traços de impurezas químicas presentes na água foram removidas). Águapotável p/ beber pode ser usada p/ lavar:
-portas-paredes-e outras superfícies não usadas na produção ao qual também pode usar sabão ou
outros agentes de limpeza (superfície usadas na produção- misturadores ).
Em geral, a classificação mais utilizada para a água com fins farmacêuticos pode ser considerada como sendo a utilizada pela USP. Nesta, a água é classificada como água purificada(PW), água para injetáveis (WFI) e , apesar de pouco empregada, água estéril para injetáveis.Estes dois tipos principais de água PW e WFI possuem as características mostradas na tabelaabaixo:
Tabela: classificação comparativa da água segundo a USP, JP, Int Ph e Eur Pharm.
Eur. Pharm. JP USP Int. Ph.
pH 5,0-7,0 5,0-7,0 5,0-7,0 Passa no testeCloretos <0,5 Passa no teste NA Passa no testeSulfato Passa no teste Passa no teste NA Passa no testeAmônio <0,2 <0,05 NA Passa no testeCa/Mg Passa no teste NA NA Passa no testeNitratos <0,2 Passa no teste NA Passa no testeNitritos NA Passa no teste NA NACondutividadeS/cm
NA NA 1.3 NA
Sólidos <10 <10 NA <10TOC 0,5 mg/L Passa no teste 0,5 mg/L NAMetais pesados NA NA NA Passa no teste
WFI: USP e Eur Pharm. 1,3 S/cm e 0,5 mg/L de TOC. Teste de pirogênio negativo.LAL: Teste “in vitro “ para a detecção de pirogênios. É constituído de um lisado de células deamebócitos de um caranguejo chamado Limilus polipheno , o qual após incubação de 60 min. a37 graus com inoculação da substância em teste produz um gel firme róseo. É tido como 10 a 5vezes mais sensível que o teste de coelho.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 119/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 119 de 130
A próxima etapa em um sistema de tratamento de águas seria a retirada dos íonspresente na mesma. Estes íons são prejudiciais, pois a precipitação destes em especial, cálcio emagnésio pode:
-danificar caldeiras (deposição sobre as superfícies) - diminui a eficiência de trocas
caloríficas (dureza permanente e temporária).-promover a corrosão de superfícies-precipitação de princípios ativos - diminui a dose farmacêutica
Esta remoção de íons pode ser realizada por:
1- trocadores de íons2- destilação
Trocadores de íonsA troca iônica é uma técnica muito utilizada na purificação de águas. Os principais
equipamentos são:-Abrandadores-Trocadores de Cátions-Trocadores de Ânions-Trocadores de Leito MistoÁgua “softening” (abrandada)A dureza da água se dá pela presença de íons Ca++ e Mg++ (como também por Fe++ ,
Ba++ e outros íons). A dureza pode ser dividida em:a) dureza temporária- os íons se precipitam por aquecimento e é formada
principalmente por carbonatos e bicarbonatos de cálcio e magnésio.b) dureza permanente- não precipta com o aquecimento - é formada principalmente
por cloretos, nitratos e sulfatos de cálcio e magnésio.Softening é um mecanismo onde os íons duros de Mg++ e Ca++ são trocados por íons
de sódio em uma coluna de resinas (Figura 3).
Esquema de uma resina trocadora de íons Mg++ e Ca++.
O abrandamento é um processo relativamente barato, pois as resinas sãoregeneradas por uma solução de salmoura (vida média das resinas 3-5 anos) (Figura 4).
Mecanismo de regeneração de resinas trocadoras de íons Mg++ e Ca++.
Contudo, se o nível de cloro residual está baixo na água, pode ocorrer umcrescimento microbiológico neste equipamento. A grande área de superfície e o baixo fluxo quesão freqüentemente encontrados são favoráveis ao crescimento destes microorganismos. O uso deNaCl como agente regenerador das resinas têm pouco efeito bacteriocida.
C a+ +
Mg+ +
+ 2NaC l + 2NaR R 2
(excesso)
C a+ +
Mg
+ + C l2
C a
+ +
Mg+ +
(HC O3
)2
SO4
C l2
+ 2Na C a
+ +
Mg+ +
R 2 + N a2
(HC O3 )2
SO4
C l2
R

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 120/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 120 de 130
A qualidade da água deveria ser monitorada periodicamente quanto ao nível de cloroe conteúdo microbiológico. Os equipamentos de softening devem ser observados quanto:
-freqüência de regeneração de resinas-exigências para regenerar o leito-substituição das resinas
-monitoramento quanto a queda de pressão e durezaTrocadores de CátionsDesmineralização, por trocadores de íons (DI), removerá impurezas iônicas
dissolvidas. Dois ciclos são importantes na purificação da água por trocadores de cátions:a) ciclo do sódio - abrandamentob) ciclo do hidrogênio - desmineralizaçãoAs resinas tem maior afinidade por íons de maior valência.A reação que se processa durante a operação está descrita na figura abaixo (Figura
5):
Res-H + X Res-X + H+++ +
(SO3H ou SO3Na)
Esquema de operação das resinas trocadoras de cátions.
Algumas observações devem ser feitas, tais como:- O pH final da operação é ácido- Este aumento na acidez pode acarretar uma diminuição da alcalinidade (ver Figura 6
abaixo):
HCO3 + H H2CO3 H2O + CO2- +
Mecanismo proposto para a diminuição da alcalinidade da água.
- Íons H+ podem deslocar íons sódio da resina , levando a formação de NaOH quandoencontrar com o efluente do leito aniônico, aumentando a condutividade da água.
Trocadores de ânionsA resina nesse caso se encontra na forma básica (figura 7).
(N(CH3)3 OH)
Esquema de operação das resinas trocadoras de ânions.
Trocadores de leito mistoO desenvolvimento dos trocadores de leito misto propiciou a geração de água com
baixíssimo níveis de eletrólitos, alem de tornar o processo mais barato e os equipamentos menosvolumosos. No leito misto não há nenhuma preocupação com formação de água com teores ácidosou básicos, pois o produto da troca é a água.
Res-OH + A Res-A + OH- - - -

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 121/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 121 de 130
ResH+ ResOH-
Na+
Cl-
H+
H+
H+
H+
OH-
OH-
OH- OH
-
"troca" "troca"
ResH+ ResOH-
H+
H+
H+
Na+
OH-
OH-
OH-
Cl-
OH-
H+
H2O
Figura 8- Esquema de operação de trocadores de leito misto.
Esquema de unidade de troca iónica
Na +
K+
H
HH
HHH
H
HH
Na+
Na +
Na +
Na +
Na+
Na +
K+
K+
K+
K +
K+
K+
K+
H+
H+
H
SO 42-
OH -
SO 42-
SO 42-
CI -
SO 42-
CI -
C I -CI -
C I -CI -
20H -
SO 42-
OH
OHOH
OH
CI -

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 122/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 122 de 130
As unidades de DI (trocadores de íons ou desionizadores) são excelentes áreas paraa geração de bactérias. O processo de regeneração permite a remoção de algumas bactérias dasresinas por causa da alta velocidade de fluxo no processo de lavagem e a ação de sanitização dotratamento químico usado para regenerar as resinas (uso de soda e ácidos para regeneração). Apopulação bacteriana tende a aumentar quando o período de serviço aumenta, mas a temperatura
e a velocidade de fluxo também afetará a velocidade de crescimento.DI não faz nada p/ aumentar a qualidade microbiológica da água e usualmentecontribui p/ a degradação bacteriológica da mesma. A alta carga de bactérias também produzproblemas de pirogenia.
Podem existir impurezas na água fornecida que não podem ser efetivamenteremovidas a menos que se utilize DI. Enquanto o uso de trocadores de íons é menos desejável doque outros métodos de tratamento de água do ponto de vista do controle microbiológico, existevárias considerações que podem ajudar a restringir o crescimento microbiológico na unidade de DI,tais como:
-colocar lâmpada UV na entrada dos leitos-tamanho do equipamento deve garantir regeneração frequente, embora não seja
ideal do ponto de vista econômico.-a qualidade química da água deve ser usada p/ determinar quando a unidade deve
ser regenerada-Trabalhar a altas temperaturas (60-70 graus).Os trocadores de íons podem ser pequenos (portáteis) e fixos.
Uso de reservatórios: os tanques são coletados e transportados para uma área deserviço central onde ocorre a regeneração. As resinas dos tanques são misturados e regeneradosem massa. As resinas são retornados aos pequenos tanques e desta forma, estão prontos p/iniciar todo o processo novamente. Os tanques de resinas devem ser sanitizados enquanto vazios.
Equipamentos de deionização (DI) portáteis incluem algumas considerações:-pressanitização antes do uso -pelo fornecedor -contaminantes a partir das resinas devem ser avaliados-contaminantes a partir da água de enxágüe-controle microbiológico em uso intermitente
Normalmente, os deionizadores fixos são planejados p/ serem regenerados no localde uso.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 123/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 123 de 130
Controle do ar
Controle elétrico
Fornecimentode H2O
Deionizador
H2O deionizada
RejeitoRegeneradores químicos(HCl, H2SO4,NaOH)
Figura 9- Deionizador
Algumas considerações devem ser feitas em relação aos deionizadores fixos:-medir a qualidade e condições em vários estágios (após fixar o leito por exemplo;
microbiologia e fisico-química).-variação das condições durante os ciclos de serviço-condições do leito (nível microbiológico)-possíveis reciclagems continuas da água através do leito de resina.-qualidade dos regeneradores químicos-condições e qualidade do ar fornecido-sílica coloidal e dissolvida -não detectado por condutividade-aminas provenientes das resinas
-sanitização e regeneração -freqüência de regeneração
DestilaçãoA destilação se baseia no princípio da mudança de fase. A água ao passar para o
estado vapor fica livre da maior parte dos seus contaminantes. Desta forma, ao condensar tem-seuma água mais pura. Contudo, muitos contaminantes (CO2 e outros gases, além de substânciasorgânicas com baixo ponto de ebulição) destilam com a água e, portanto, contamina a mesmaapós a condensação. Desta forma, ao final do processo é difícil obter uma água totalmente estéril,apirogênica e livres de metais.
A destilação é o método mais largamente aceito para a produção de água com finsfarmacêutico. Se a unidade de destilação está operando satisfatoriamente, devidamente limpa,sanitizada com estes processos validados, a mesma deveria fornecer água livre de pirogênio. Ostrocadores de calor e a instalação dos filtros dos suspiros (área de ventilação; uso de filtros 0,22micra) são alguns componentes dos destiladores que devem ser observados. A presença debuflles torna mais difícil a passagem de íons ou gotículas para o condensador. Incrustações, asquais reduzem a eficiência do sistema, devem ser evitadas. A destilação é o método mais seguro,mas também é o mais caro p/ o tratamento de água. Uma grande quantidade de energia é gastapara a mudança de estado da água para a forma de vapor. É um dos métodos mais difundidospara a preparação de água para injetáveis.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 124/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 124 de 130
Osmose ReversaA osmose reversa é um fenômeno natural fisíco-químico. Quando duas soluções, com
diferentes concentrações, são colocadas num mesmo recipiente separados por uma membranasemi-permeável, ocorrendo naturalmente a passagem do solvente da solução mais diluída para a
solução mais concentrada até que se encontre um equilíbrio. Neste ponto a coluna de solução dolado da solução mais concentrada estará acima da coluna mais diluída. A esta diferença entrecolunas de solução se denominou pressão osmótica (Figura 10b). A osmose reversa é obtidaatravés da aplicação mecânica de pressão superior a pressão osmótica do lado da solução maisconcentrada. Em comparação à outros métodos, a osmose reversa é capaz de remover contaminantes em um intervalo de 0,1 a 10 000 nanômetros, sendo um dos mais abrangentesmétodos de filtração.
Esquema do princípio da Osmose Reversa.
O tratamento com osmose reversa permitirá a remoção:-sais dissolvidos-partículas-bactérias-materiais pirogênicosEnquanto as propostas de BPF permitem o uso de membranas de OR para a
produção de água purificada, para aplicações parenterais existem controvérsias que não permitemsuportar esta alegação. No sentido teórico, OR deveria prevenir a passagem de todas as bactériase pirogênios. A sobrecarga biológica da água de alimentação e sua qualidade, irão influenciar aeficiência do sistema, levando a entupimentos e perda de eficiência. O cloro, quando presente,levará a degradação da membrana.
Esquematicamente, podemos idealizar u sistema de osmose reversa como se seguena figura abaixo.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 125/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 125 de 130
Equipamento de OR.
Como o processo de Osmose Reversa é uma filtração muito fina, o fluxo de água dealimentação deve ser tangencial para minimizar o entupimento da membrana (Figura 13).
Alimentação tangencial
Água dealimentação R e jeito
F iltr ado
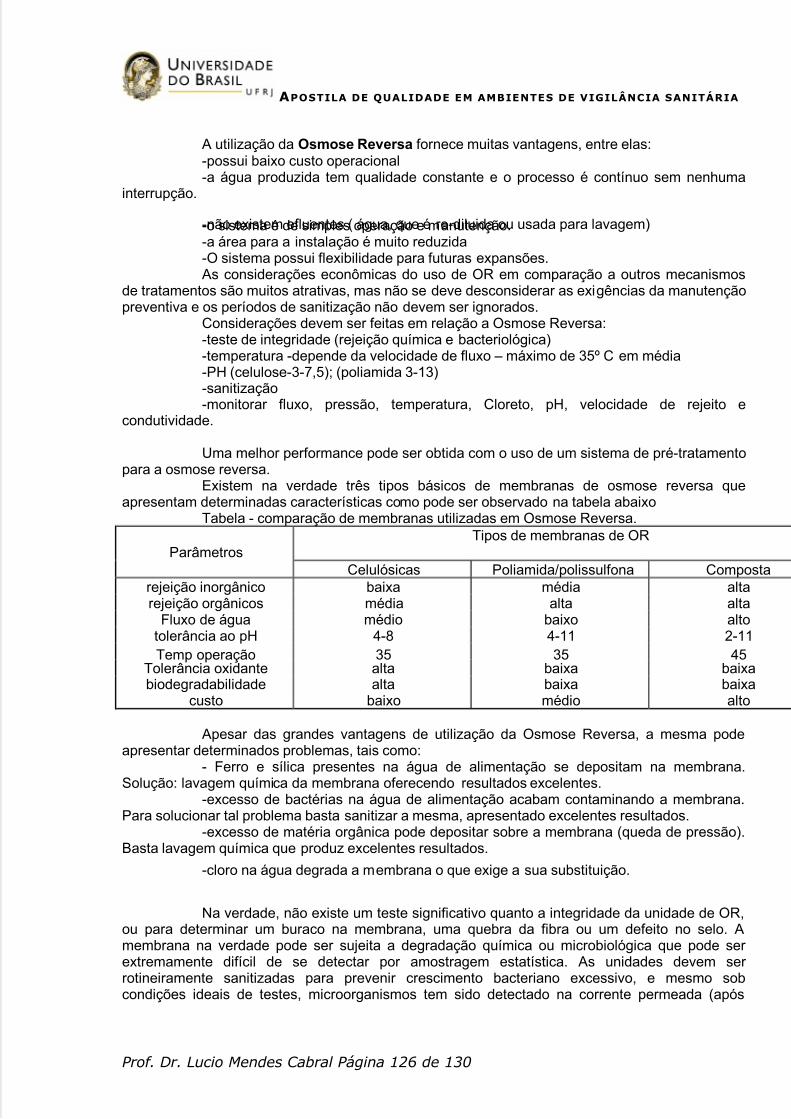
5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 126/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 126 de 130
A utilização da Osmose Reversa fornece muitas vantagens, entre elas:-possui baixo custo operacional-a água produzida tem qualidade constante e o processo é contínuo sem nenhuma
interrupção.
-não existem efluentes ( água, que é re-diluida ou usada para lavagem)-o sistema é de simples operação e manutenção.-a área para a instalação é muito reduzida-O sistema possui flexibilidade para futuras expansões.As considerações econômicas do uso de OR em comparação a outros mecanismos
de tratamentos são muitos atrativas, mas não se deve desconsiderar as exigências da manutençãopreventiva e os períodos de sanitização não devem ser ignorados.
Considerações devem ser feitas em relação a Osmose Reversa:-teste de integridade (rejeição química e bacteriológica)-temperatura -depende da velocidade de fluxo – máximo de 35º C em média-PH (celulose-3-7,5); (poliamida 3-13)-sanitização-monitorar fluxo, pressão, temperatura, Cloreto, pH, velocidade de rejeito e
condutividade.
Uma melhor performance pode ser obtida com o uso de um sistema de pré-tratamentopara a osmose reversa.
Existem na verdade três tipos básicos de membranas de osmose reversa queapresentam determinadas características como pode ser observado na tabela abaixo
Tabela - comparação de membranas utilizadas em Osmose Reversa.
ParâmetrosTipos de membranas de OR
Celulósicas Poliamida/polissulfona Compostarejeição inorgânico baixa média altarejeição orgânicos média alta alta
Fluxo de água médio baixo altotolerância ao pH 4-8 4-11 2-11Temp operação 35 35 45
Tolerância oxidante alta baixa baixabiodegradabilidade alta baixa baixa
custo baixo médio alto
Apesar das grandes vantagens de utilização da Osmose Reversa, a mesma podeapresentar determinados problemas, tais como:
- Ferro e sílica presentes na água de alimentação se depositam na membrana.Solução: lavagem química da membrana oferecendo resultados excelentes.
-excesso de bactérias na água de alimentação acabam contaminando a membrana.Para solucionar tal problema basta sanitizar a mesma, apresentado excelentes resultados.
-excesso de matéria orgânica pode depositar sobre a membrana (queda de pressão).Basta lavagem química que produz excelentes resultados.
-cloro na água degrada a membrana o que exige a sua substituição.
Na verdade, não existe um teste significativo quanto a integridade da unidade de OR,ou para determinar um buraco na membrana, uma quebra da fibra ou um defeito no selo. Amembrana na verdade pode ser sujeita a degradação química ou microbiológica que pode ser extremamente difícil de se detectar por amostragem estatística. As unidades devem ser rotineiramente sanitizadas para prevenir crescimento bacteriano excessivo, e mesmo sobcondições ideais de testes, microorganismos tem sido detectado na corrente permeada (após

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 127/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 127 de 130
passar pela membrana). Procedimentos devem ser escritos cuidadosamente p/ garantir que oequipamento está operado, monitorado, mantido e sanitizado de forma eficaz.
Para a preparação de água em grandes volumes, a unidade de OR deve possuir umaelevada área de filtração. Assim, duas configurações de membranas atendem a esta exigência:
- Fibra oca: É composta de uma série de capilares no interior dos quais passa a águaa ser filtrada, sendo a mesma recolhida em seu exterior.
- Spiral Wound: Nesta configuração, temos uma membrana em forma de envelopeenrolada em um tubo de aço coletor; a água passa por sobre a membrana enrolada, sendopermeada para o interior do tubo coletor.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 128/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 128 de 130
UltrafiltraçãoAlguns equipamentos de ultrafiltração (UF) são muito similares ao equipamento deosmose reversa, exceto que a porosidade da membrana é grandemente diferente. Então, muitasconsiderações fixadas acima também podem servir para o equipamento de UF.
Algumas membranas ultrafiltrantes são confeccionadas a partir de polisulfonas aosquais exibem melhor estabilidade de pH e toleram certos agentes químicos e de limpeza.
A maioria dos equipamentos de tratamento de água foram planejados e desenvolvidospara as indústrias onde a maior preocupação era a pureza química enquanto a qualidademicrobiológica era relegada à segundo plano. Estes equipamentos, desenvolvidos para fornecer água p/ caldeira e aplicações relacionadas, deve ser cuidadosamente analisados quando foremutilizados em aplicações farmacêuticas. Outros equipamentos foram planejados para o tratamentode água salobra ou água do mar. Utilizar equipamentos construídos com aço inoxidável e colocar alguns componentes que permitem a limpeza não torna estes equipamentos adequados para a
produção de água para fins farmacêuticos. Na verdade, o equipamento ao ser planejado deverespeitar as legislações vigentes para que a Água produzida possa ter as características desejadase, portanto, o mesmo deve estar sempre sujeito a análises e, caso seja comprovado que o mesmonão desempenha as suas funções de forma eficiente, este equipamento deve ser substituído.
A escolha do equipamento deveria ser conduzida, levando-se em consideração:-tamanho e capacidade (a partir do ponto de vista microbiológico e arquitetura)-capital de investimento-custos operacionais-exigência de mão-de-obra (depende do tamanho do equipamento que influencia no
crescimento de MOs).Alguns procedimentos que aumentam a qualidade química da água (redução do nível
de cloração) podem diminuir a qualidade microbiológica.O objetivo do design deveria ser a construção de um equipamento que satisfaça as
exigências da qualidade da água enquanto controla a qualidade através do processo detratamento desde o local de fornecimento da água até ao uso final. Não se deve permitir que umsistema tenha um crescimento microbiológico descontrolado, enquanto espera-se somente corrigir o problema no processo de tratamento final é inaceitável.

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 129/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Prof. Dr. Lucio Mendes Cabral Página 129 de 130
Onde o problema de contaminação microbiológica for muito grave, o aquecimento e aestocagem da água a 80o C se mostra efetivo para controlar este problema (eleva a qualidade
microbiológica). Existe a vantagem de não adicionar substâncias químicas, não existirá problemasde resíduos químicos. Este método é caro, gasto de energia e exige que os tanques de estocageme a tubulação do sistema de distribuição sejam isolados para conservar o calor. A água nestatemperatura é de difícil manipulação e certos componentes que entram na formulação dedeterminados medicamentos não podem ser formulados com a água nesta temperatura. Se a águadeve ser resfriada antes de ser utilizada, existe um grande potencial para o desenvolvimento deMOs e pirogênios na porção do sistema que está operando a uma temperatura mais baixa. A figuraabaixo (Figura 16) mostra um sistema simples de recirculação de água aquecida. Constantecontrole e um esforço de validação devem ser destinados aos pontos de refrigeração de água dosistema.
Sistema básico de recirculação de água aquecida
Pontos de uso Bomba
Tanque deestocagem Jaqueta de
aquecimento
T
Válvula de controle de fluxoOR
T P

5/10/2018 Apostila de Qualidade Cap II - slidepdf.com
http://slidepdf.com/reader/full/apostila-de-qualidade-cap-ii 130/130
APOSTILA DE QUALIDADE EM AMBIENTES DE VIGILÂNCIA SANITÁRIA
Exemplo de destilador sob pressão