apostila de extração
TRANSCRIPT

UNIVERSIDADE DO ESTADO DO RIO DE JANEIRO
DISCIPLINA: OPERAÇÕES UNITÁRIAS III
PROFESSOR: JORGE NAVAES CALDAS
EXTRAÇÃO EM ESTÁGIOS
[Junho de 2011]

SUMÁRIO
1 – Introdução............................................................................................3
1.1 – Coordenadas triangulares equiláteras....................................4
1.2.– Efeito da temperatura.............................................................5
1.3 – Efeito da pressão...................................................................6
1.4 – Sistemas com dois pares parcialmente solúveis....................6
1.5 – Efeito da temperatura.............................................................7
2 – Escolha do solvente...........................................................................8
3 – Métodos de cálculo..........................................................................19
2

1 - IntroduçãoNuma mistura líquida homogênea formada por dois componentes, o
primeiro componente tem maior quantidade e é chamado de solvente; o
segundo componente da mistura (que ocorre em menor quantidade
relativa) é conhecido como soluto.
É possível que, numa determinada condição industrial, se
deseje a separação do soluto dessa mistura. Para isso, é comum a
utilização de um terceiro componente, que é solvente em relação ao
soluto para realizar a separação. Esta operação unitária é denominada
industrialmente de EXTRAÇÃO LÍQUIDO-LÍQUIDO.
Existe uma diferença de solubilidade dos componentes da
mistura em relação ao segundo solvente. Se considerarmos a idealidade,
apenas o soluto que se deseja separar será solúvel no segundo solvente,
enquanto o outro componente apresenta pouca ou nenhuma
solubilidade.
Assim, o único componente a ser transferido da mistura
homogênea inicial para a fase do segundo solvente será o soluto.
Industrialmente, esta operação é realizada em um equipamento
chamado de EXTRATOR ou COLUNA DE EXTRAÇÃO.
Na coluna de extração, a mistura cujo soluto se deseja
remover, chamada de carga da torre, entra em contato com o segundo
solvente através de dispositivos de contato, que podem ser, por exemplo:
pratos, chicanas ou recheios.
Após a carga entrar em contato com o segundo solvente,
ocorre a formação de duas fases líquidas imiscíveis e de diferentes
3

densidades: a primeira, rica no primeiro solvente, chamada de rafinado; a
segunda, rica em soluto e segundo solvente, chamada de extrato.
Numa etapa posterior, ocorre a remoção dos solventes do
rafinado e do extrato, e assim são obtidos os produtos rafinado e
extraído, respectivamente.
Algumas classes de compostos, como os hidrocarbonetos
aromáticos encontram-se em misturas juntamente com hidrocarbonetos
não aromáticos que possuem as mesmas faixas de ebulição. Isto torna a
separação por destilação inviável, de modo que a extração líquido-líquido
é largamente empregada.
Considerando-se três tipos de componente no processo de extração:
Soluto (C) produto que deseja-se extrair da carga.
Co-solvente (A) um ou mais componentes da carga que não devem ser extraído(s).
Solvente (B) produto utilizado para a extração do soluto, o qual nele se solubiliza.
Os dados de equilíbrio para os sistemas ternários, que
representam as concentrações dos constituintes nas fases em equilíbrio,
podem ser lançados em diagramas triangulares dos tipos equilátero ou
retangular, sendo este último o mais apropriado nos cálculos de
engenharia.
O diagrama do triângulo retângulo apresenta a concentração
de soluto (C) na ordenada e a concentração do solvente (B) na abscissa.
A concentração do co-solvente (A) é obtida por diferença.
1.1 - Coordenadas triangulares equiláteras
Estas coordenadas são utilizadas extensamente na bibliografia
química para descrever graficamente as concentrações em sistemas
4

ternários. Uma das propriedades do triângulo retângulo é a soma das
distancias perpendiculares de qualquer ponto dentro do triangulo até
qualquer um dos três lados, é igual à altura do triângulo. Considera-se a
altura como a composição dos 100 % e as distâncias dos três lados, as
porcentagens das frações dos três componentes. Cada vértice do
triângulo representa um dos componentes puros. A distância
perpendicular desde qualquer ponto, como a base AB, representa a
porcentagem de C na mistura k, a distancia da base AC.
Figura 1.1 Coordenadas triangulares equiláteras
1.2 – Efeito da temperatura
Para mostrar o detalhe do efeito da temperatura, requer uma
figura tridimensional, como a figura 1.2 a. Neste diagrama, a temperatura
está representada verticalmente, vê-se que os triângulos isotérmicos são
seccionados através do prisma. Para muitos sistemas deste tipo, a
solubilidade mútua de A e B aumenta com a temperatura, de certa
temperatura t4 a temperatura crítica de solução, A e B se dissolvem
completamente. O aumento da solubilidade a temperaturas mais altas
influi consideravelmente sobre o equilíbrio ternário, isto pode ser
5

mostrado melhor projetando as isotermas até a base do triângulo, como
se mostra na figura 1.2 (b). Não somente decresce a área heterogênea a
temperaturas mais altas sendo que também combinam as linhas
perpendiculares de união. As operações de extração líquida, que
depende da formação de fases líquidas insolúveis, devem levar a
temperaturas inferiores a t4.
Figura 1.2. Efeito da temperatura nos equilíbrios ternários
1.3 – Efeito da pressão
Exceto em pressões muito elevadas, o efeito da pressão sobre
o equilíbrio líquido é tão pequeno que geralmente pode ser ignorado.
Portanto, todos os diagramas aqui mostrados neste documento, a pressão foi
considerada suficientemente elevada para manter o sistema completamente
condensado.
1.4 – Sistemas de três líquidos: dois pares parcialmente solúveis
Um exemplo deste tipo é o sistema cloro benzeno (A) água(B)
metiletilcetona (C), onde A e C são completamente solúveis, onde os
6

pares A-B e B-C apresentam uma solubilidade limitada. Observa-se na
figura 1.3 uma isoterma típica. A temperatura predominante, os pontos
representam as solubilidades de A e B e os pontos H e L as de B e C. As
curvas (rica em A) e JEL (rica em B) são curvas de solubilidade ternárias;
as misturas fora do intervalo destas curvas formam soluções liquidas
homogêneas de uma fase. As misturas como M, dentro da área
heterogênea, formam duas fases líquidas no equilíbrio em E e R, unidas
no diagrama mediante uma linha de união. A curva de distribuição
correspondente está na figura 1.3 (b).
Figura 1.3 Sistema de três líquidos, A-B e B-C parcialmente miscíveis
1.5 – Efeito da temperatura
Geralmente ao aumentar a temperatura, aumenta a
solubilidade, ao mesmo tempo, esse incremento modifica a forma das
linhas de união. A figura 1.4 é típica do efeito que pode-se esperar.
Aumenta a temperatura crítica de solução da mistura binária B-C em t3, o
sistema é similar do tipo que se tinha inicialmente. Também são
possíveis outros efeitos de temperatura.
7

Fig. 1.4 Efeito da temperatura no equilíbrio ternário
2 – Escolha do solventeNa escolha de um solvente para um processo de extração
líquido-líquido, existem vários princípios que podem ser usados como
guia. Eles são geralmente conflitantes, e certamente em nenhuma
substancia simples encontraremos todas as características desejáveis.
Devem ser estabelecidos compromissos, e seguindo critérios quanto a
importância relativa dos vários fatores considerados, estabelecer o
solvente indicado para o caso.
Os fatores normalmente considerados são divididos em três
grupos:
2.1 – Fatores que caracterizam a separação
2.2 – Fatores importantes para o funcionamento do
equipamento de extração
2.3 – Fatores econômicos e outros não classificados
8

2.1 Fatores que caracterizam a separação
2.1.1 Seletividade
É a tradução da facilidade que o solvente tem em dissolver um
componente em detrimento de outro. O melhor solvente neste ponto de
vista é aquele que dissolve o máximo de um componente a um mínimo
do outro.
A seletividade de B por C é definida como a razão de C para A
na fase rica em solvente, dividida pela mesma razão na fase pobre.
βC , A=
(XC )B(X A )B( XC )A( X A )A
ou
ou βC , A=(XC )B . ( X A )A(XC )A . ( X A )B
como
mC=( XC )B(XC ) A
(4.3 ) então βC , A=mC .(X A )A( X A )B
(4.4)
Logicamente β deve sempre ser maior que 1 e quanto maior
melhor, pois mais fácil será a separação. Se β = 1,0 não poderemos
efetuar a separação com o solvente em estudo. (β será igual a 1,0 no
caso de uma tie line passar por B).
As concentrações para as fases ricas em A e B são as
concentrações de equilíbrio, e o valor numérico de beta será o mesmo,
sejam as unidades de concentração usadas, fração mássica ou molar.
9

Do mesmo modo que a volatilidade relativa, beta mostra-se
substancialmente constante para alguns sistemas, e em tais casos pode
ser usado como um modo de correlacionar dados de equilíbrio. Na
maioria dos casos, varia fortemente com a concentração, como mostrado
na figura 2.1.
2.1.2 COEFICIENTE DE DISTRIBUIÇÃO – mc
É importante porque influencia a seletividade equação anterior,
e um coeficiente mais favorável (m >1) conduzirá a uma melhor
seletividade.
2.1.3 CAPACIDADE
Um solvente precisa ter a capacidade de dissolver grandes
quantidades de um soluto, para ser viável. Caso contrário, ele será
provavelmente anti-econômico, mesmo que tenha alta seletividade, pois
uma baixa capacidade de solubilização acarretará uma alta circulação de
solvente no sistema extrator. Por exemplo n-heptano quando usado para
separar cloreto de cobalto da água possui uma forte seletividade em
favor do cloreto de cobalto (β = 9), e a separação é conseqüentemente
excelente. Mas devido a baixa solubilidade de CoCl2 em n-heptano (0,04
%), ele requer um mínimo de 2500 lb de n-heptano para extrair 1 lb de
sal.
10

Fig. 2.1 (fig 4.6 ref. 9 seletividade em sistemas do tipo I e II)
Nas separações da indústria de refino de petróleo, onde A e C
são hidrocarbonetos, é difícil achar um solvente que mostre uma alta
solubilidade para C sem simultaneamente mostrar alta solubilidade para
A, e neste caso alta solubilidade pode significar pobre seletividade. De
fato, isto é incomum nestes processos onde, para aumentar a
seletividade, diminuímos a capacidade de solubilização. Por exemplo, na
preferência de dietileno glicol aquoso ou anidro para separar
hidrocarbonetos aromáticos de parafínicos. Neste caso deve-se
estabelecer um compromisso entre alta seletividade e alta solvência.
2.1.4 Solubilidade do solvente / co-solvente
Em sistemas do tipo I, alto grau de insolubilidade do solvente B
com A é requerida.
11

Isto produz uma alta seletividade através da relação na
equação 2.3 e também como mostrado na figura 2.2, onde notamos que
a faixa de concentração na qual o binário A-C pode ser manipulado,
aumenta consideravelmente. Em ambos os casos mostrados na figura, é
possível usar B para separar soluções de A e C somente dentro dos
limites de A puro a M, pois somente nesta faixa de concentração
resultarão duas fases líquidas pela adição de B.
Fig 2.2 Efeito da solubilidade do solvente na extração
2.2 Fatores importantes para o funcionamento do equipamento
2.2.1 Densidade
A diferença nas densidades das fases contactadas é essencial
e deve ser a maior possível. Não é apenas a taxa de coalescência das
camadas imiscíveis que é aumentada, mas também a capacidade de
promover o contato entre as fases.
É insuficiente analisar meramente as densidades relativas da
solução a ser extraída e do solvente puro, pois a solubilidade mútua dos
dois alterará as densidades para um equipamento de contato contínuo. É
importante estar certo que uma diferença de densidade satisfatória será
obtida para toda a faixa de concentrações existentes no equipamento. As
12

figuras 2.3 e 2.4 indicam situações desejáveis e indesejáveis
respectivamente.
Na figura 2.3 as densidades das camadas saturadas no
sistema do tipo I 1,1,2-acetona-tricloroetano-água (acetona como
substância distribuída) são plotadas, com as camadas em equilíbrio
unidas por uma linha de amarração. A densidade da camada rica em
água é sempre menor que a camada rica em tricloroetano, mas as
alterações nas diferenças de densidade com a concentração de acetona
é razoavelmente grande, já que no PLAIT POINT as densidades das
soluções conjugadas são necessariamente idênticas. A figura 2.4 é do
mesmo tipo de gráfico para o sistema do tipo II, metil etil cetona-água-
tricloroetileno. Note que neste caso há uma reversão de densidades e
enquanto um equipamento em estágios poderá operar com este sistema,
um equipamento de contato contínuo não poderá.
FRANCIS (6) denominou o tipo de sistema onde ocorre
igualdade de densidades nas fases em equilíbrio de “ISOPYNIC”.
Listando aproximadamente 100 sistemas que comportam-se deste modo,
ele enfatizou que estes casos são muito comuns, apesar dos “papers”
originais onde, os sistemas foram igualmente descritos, raramente
salientarem este fato.
“ISOPYNICS” podem ocorrer em todos os tipos de diagrama de
fase e podem algumas vezes desaparecer com uma mudança na
temperatura. O mesmo fenômeno pode também ocorrer em sistemas
multicomponentes importantes industrialmente como aqueles que
envolvem petróleo.
13

Figura 2.3 Figura 2.4
2.2.2 Viscosidade
Deve-se escolher um solvente de viscosidade baixa a fim de se
evitar o arraste das gotículas de fase dispersa e aumentar a capacidade
dos extratores. Menor energia para o bombeamento e agitação, rápida
extração, rápida separação e altas taxas de transferência de calor e
massa são corolários da baixa viscosidade.
2.2.3 Tensão interfacial
A tensão interfacial entre fases imiscíveis que devem ser
separadas deve ser alta para rápida coalescência. Uma tensão interfacial
muito alta exige um maior gasto de energia para dispersão de uma fase
na outra, mas isto normalmente é disponível. Por outro lado, uma tensão
interfacial muito baixa pode levar a formação de uma emulsão estável e,
normalmente muito pouco pode ser feito para modificar isto.
Existem relativamente poucos dados completos sobre tensão
interfacial em sistemas ternários em equilíbrio líquido-líquido, mas é
sabido que tensão interfacial entre dois líquidos parcialmente imiscíveis é
14

usualmente mais alto na ausência do componente distribuído (soluto) e
cai para zero no PLAIT POINT. No caso de pares mutuamente saturados
de dois líquidos, a tensão interfacial tem sido associada às pontes de
hidrogênio característica do líquido e seu volume molar (58). Não é
surpreendente, portanto que DONAHUE e BARTELL (59) foram capazes
de correlacionar empiricamente a tensão interfacial e a solubilidade
mútua, expressa como fração molar xAB e xBA do constituinte menor em
cada líquido saturado. TREYBAL (9) verificou que as coordenadas de
DONAHUE e BARTELL, se modificadas como na figura 4.5 também
podem representar razoavelmente bem sistemas ternários do TIPO I.
Nesta figura, o sistema de coordenadas se reduz àquela de DONAHUE e
BARTEL para binários na ausência do soluto distribuído (XC)A e (XC)B = 0.
Estas tensões interfaciais ternárias são aquelas de MURPHY e
colaboradores (60), excluindo diversos sistemas onde os dados para os
pares não estão de acordo com os valores aceitos. Sabe-se também que
a curva representa bem dados para MEK-ÁGUA-ÁCIDO ACÉTICO e
CICLOHEXANOL-ÁGUA-ÁCIDO ACÉTICO, apesar destes não estarem
plotados para não saturar o gráfico. A bem conhecida regra de
ANTONOFF, que a tensão interfacial entre suas tensões superficiais com
o ar não pode ser aplicada.
Deve ser enfatizado que os dados da figura 11.5, e aqueles
encontrados geralmente na literatura química, são para substâncias
quimicamente puras e devem, portanto ser consideradas como os
valores máximos que podem ser obtidos. A tensão interfacial pode ser
profundamente influenciada (usualmente, mas não sempre, reduzindo)
por quantidades bem pequenas de impurezas, particularmente aquelas
que absorvem na interface líquido-líquido (tenso ativo e assemelhado), e
consequentemente líquidos industriais irão exibir quase sempre valores
menores que os padrões. É, portanto, importante que as medidas sejam
feitas com o sistema líquido-líquido realmente em consideração, usando
15

amostras industriais, quando avaliando solventes para propósitos de
extração. Pode-se saber muito sobre o sistema de um ponto de vista
prático através de um simples teste de agitação dos líquidos em contato
em um funil de decantação. Se líquidos razoavelmente limpos não são
obtidos de uma decantação após a agitação da mistura em 5 ou 10
minutos no máximo, problemas de decantação no equipamento em
escala industrial ocorrendo.
2.3 Fatores econômicos e outros não classificados
2.3.1 Recuperação
Em todos os processos de extração líquida é necessário
remover o solvente extrator dos produtos. Isto é tão importante não só
para evitar a contaminação dos produtos com o solvente, mas também
para permitir a reutilização do solvente. Se destilação é o método a ser
usado, propriedades do solvente, tais como VOLATILIDADE e CALOR
LATENTE DE VAPORIZAÇÃO se tornam importantes. Existem outros
métodos de recuperação de solvente que devem ser considerados, no
entanto. Todo o problema da recuperação de solvente é de importância
capital no sucesso econômico de empreendimento e TREYBAL (9)
apresenta uma exaustiva discussão do assunto.
2.3.2 Reatividade química e estabilidade
Comumente reações químicas entre o solvente e componentes
da solução de alimentação, produzindo substâncias estranhas ao
processo, são indesejáveis desde que reduzem a produção do produto
desejável, problemas de recuperação de solvente são aumentados, e
perdas de solvente podem ocorrer.
O solvente deve ser estável na presença da solução de
alimentação: por exemplo, não devem ser afetados por agentes
16

oxidantes tais como ácido nítrico se este está presente na carga. Não
deve ser suscetível a polimerização ou oxidação pelo ar e não deve ser
afetado por repetidos aquecimentos, mesmo na temperatura máxima
encontrada no equipamento de recuperação por destilação. Solventes
que formam os perigosos e explosivos peróxidos tais como os éteres,
devem ser evitados. Hidrólise, que pode ocorrer, por exemplo, com
solventes clorados na presença de água ou vapor produzindo ácido
clorídrico, deve ser previsto. Em qualquer destes casos, perda de
solvente representa custo de operação.
2.3.3 Corrosividade
De modo a reduzir os custos de equipamento, o solvente não
deve causar severas dificuldades de corrosão com os materiais comuns
de construção ou com aqueles comumente usados para alimentar o
sistema com solvente. A atual viabilidade de materiais não usuais a
custos relativamente baixos, entretanto, tem aberto possibilidades que
estariam fora de questão anos atrás. Plantas comerciais de extração vêm
sendo construídas de plástico ou metal coberto de plástico, e tais
materiais devem ser lembrados se o valor do produto garante tal
investimento.
2.3.4 Pressão de vapor
Comumente se deseja pressão de vapor baixa de modo que
armazenamento e operações de extração sejam possíveis a pressão
atmosférica, ou no máximo moderadamente alta, e deste modo perda de
solvente são mantidas a um mínimo. Exceções a esta condição são
frequentemente feitas com objetivo de facilitar a recuperação de
solvente. Por exemplo, propano líquido e dióxido de enxofre são
comumente usados no refino de produtos de petróleo tirando proveito de
17

suas elevadas seletividades, aproveitando-se também de suas altas
volatilidades na operação de recuperação de solvente.
2.3.5 Ponto de fulgor
Alto ponto de fulgor é, naturalmente, desejável por razões de
segurança.
2.3.6 Toxidez
Materiais altamente venenosos são difíceis de manusear
industrialmente. A não ser que equipamentos complexos de segurança
sejam planejados, com frequente inspeção médica, as substâncias
devem ser evitadas. Solventes que podem deixar resíduos tóxicos em
alimentos e produtos farmacêuticos devem ser evitados nestas
indústrias.
2.3.7 Custos
Custos baixos e grande disponibilidade, usualmente ao mesmo
são, obviamente, atributos desejáveis no solvente. Apesar do solvente
ser recuperado dos produtos, “make-up” de solvente para repor
inevitáveis perdas devem ser providenciadas. Também, grandes
quantidades de um solvente caro retido na unidade representam um
razoável aumento no investimento.
A ampla coleção de dados industriais de MELLAN (52) é
bastante valiosa na estimativa de propriedades outras que não sejam
relativas a seletividade. A aplicação de muitos solventes para separações
extrativas é coberta por patente, e uma cuidadosa pesquisa de patentes
deve ser feita antes de uma escolha final de solvente ser considerada
completa.
18

3 – Métodos de cálculo3.1 Processos de contato em estágios com solvente simples –
Sistema ternário
A separação de componentes de uma solução pode ser feita
por inúmeros caminhos, dependendo da natureza do solvente e do
equipamento empregado. Uma classificação conveniente é a seguinte:
3.1.1 Sistema de solvente simples
Três componentes: dois por separar, e o solvente.
a) Em estágios: nesta categoria estão incluídos todos aqueles
tipos de equipamento onde a mistura e o solvente são
intimamente contatados, até próximo do equilíbrio e então
separados.
b) Contato contínuo: este inclui arranjos onde o solvente e a
mistura são separados e contatados continuamente dentro
do equipamento. Comumente não se atinge o equilíbrio.
3.1.2 Mistura de solventes
Neste tipo estão incluídos aqueles que empregam uma solução
de solventes de dois componentes, onde as relações de solubilidade são
tais que não podemos simplificar para um sistema ternário equivalente.
a) Em estágios
b) Contato contínuo
3.1.3 Solvente duplo (Extração fracionada)
19

Esta categoria inclui aqueles tipos onde a mistura a ser separada é
distribuída entre dois solventes, imiscíveis. O sistema, em última análise,
é quaternário.
a) Em estágios
b) Contato contínuo
Neste trabalho somente teremos contatos em estágios com
solvente simples.
Tabela 3.1 – Analogia entre a extração e a destilação
Operação ou condição na extração Analogia com a destilação
Adição de solvente Adição de calor
Misturador de solvente Refervedor
Remoção do solvente Remoção de calor
Separador de solvente Condensador
Solução rica em solvente saturada
de solvente
Vapor no ponto de orvalho
Solução rica em solvente contendo
mais solvente do que o requerido
para saturação
Vapor superaquecido
Solução pobre em solvente
contendo menos solvente que o
requerido para saturá-la
Líquido abaixo do ponto de bolha
Solução pobre em solvente
saturado com solvente
Líquido no ponto de bolha
Mistura de duas fases líquidas Mistura líquido-vapor
Seletividade Volatilidade relativa
Mudança na temperatura Mudança na pressão
No estudo de vários processos de extração, é útil
frequentemente termos em mente o paralelismo com os processos de
20

destilação, que são geralmente mais familiares. A tabela 3.1 indica esta
analogia.
Nos processos de destilação, uma mistura de duas
substancias, é separada pela criação de duas fases, uma líquida e outra
fase vapor, pela adição de calor, e a separação de ambas em virtude das
concentrações relativas das substâncias serem diferentes nas duas
fases. Subsequente condensação da fase vapor é efetuada pela
remoção do calor. Na extração, duas fases líquidas são formadas pela
adição de um solvente imiscível que se torna análogo ao calor. Desde
que a concentração relativa das substâncias a ser separada é diferente
nas duas fases, a separação física das camadas de líquido produz o
desejado grau de separação. Remoção do solvente da fase rica em
solvente é análoga à condensação do vapor no caso de destilação.
Um estágio teórico (Ideal)
É quando o contato entre duas fases é suficientemente íntimo e
mantido por um período de tempo suficiente, em que a distribuição de
equilíbrio é atingida, de modo que o rafinado e extrato são soluções de
equilíbrio. Neste trabalho o estágio ideal será representado
esquematicamente por um círculo como na figura 3.1
Figura 3.1 – Um estágio ideal
21

3.2 Estágio único
Envolve o uso de um único estágio, onde a solução a ser
separada e o solvente são contatados uma vez e o extrato e rafinado
separados. A operação pode ser em batelada ou contínua. A analogia na
destilação é a vaporização por flash.
1. Coordenadas triangulares: Comumente a solução a ser extraída
consiste somente de uma mistura de A e C, e o solvente extrator é puro
B (S), mas no caso geral os três componentes estão presentes nas
correntes, como o indicado pela posição dos pontos F e S na figura 5.2.
Figura 3.2 – Solução no diagrama ternário
Um balanço material para o sistema fornece F + S = R + E = M
e como E e R estão em equilíbrio, a “TIE LINE” que passa por M resolve
o problema.
É necessário, para que consigamos alguma separação entre os
componentes da mistura M, que esta caia na região de duas fases. Na
figura teremos o rafinado R e o extrato E com composições diferentes
22

possibilitando a separação como demonstramos a regra da alavanca nós
dá:
FS
= MSFM
e RE
=EMRM
A relação S/F é chamada de razão solvente carga.
2. Diagrama de Janecke: Cálculo para o arranjo da figura anterior pode ser seguido no diagrama de Janecke se nós dissermos que as várias correntes que estão em base isenta de solvente são: F, S, E e M
FS
= MSFM
e RE
=EMRM
Figura 3 3 – Diagramas de Janecke e de distribuição
1. Diagrama de distribuição: A figura 3.3 é uma forma do diagrama
de distribuição na qual as concentrações livres de solvente da
solução rica em solvente, são plotadas na ordenada e da solução
pobre em solvente na abscissa. A linha de amarração (“tie line”),
como RE na figura superior é projetada sobre esta coordenada, da
23
No caso de S puro,
teremos XS = ∞ e a linha
será vertical.
JM inclinação – R/E

maneira mostrada, para produzir o ponto de equilíbrio J. O simples
estágio é representado pela “linha de operação” JM, que tem
inclinação – (R/E) de acordo com a equação de balanço.
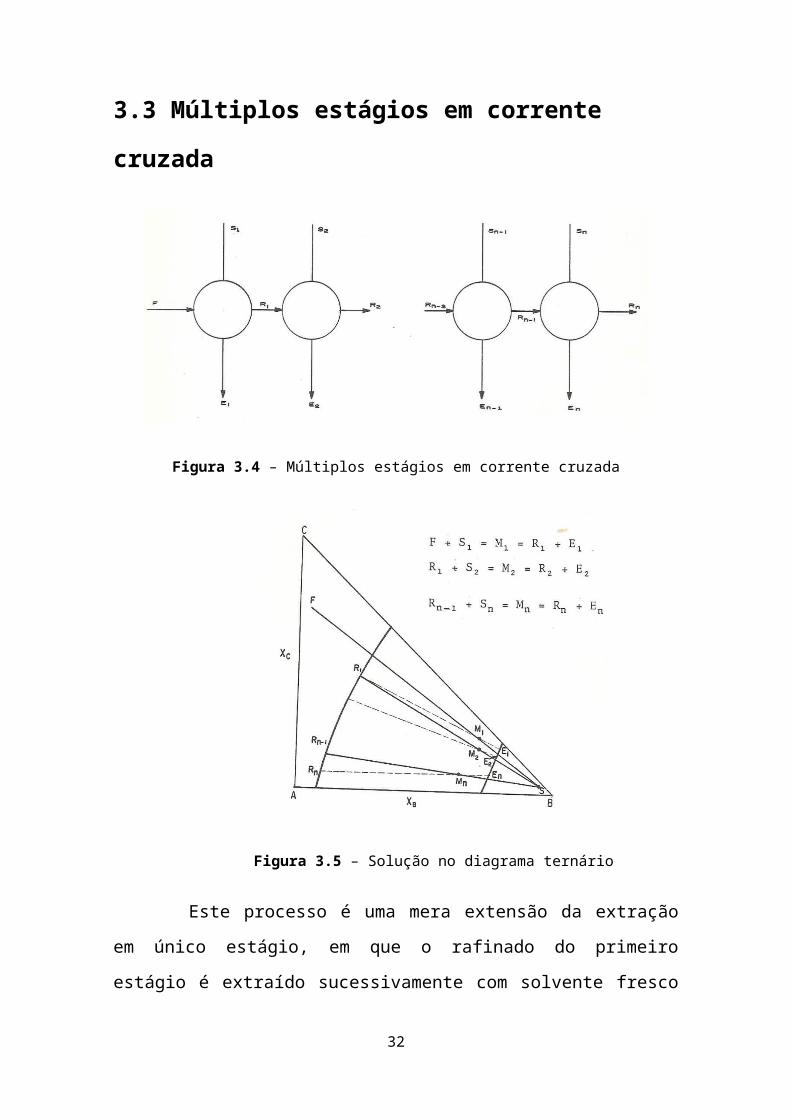
3.3 Múltiplos estágios em corrente cruzada
Figura 3.4 – Múltiplos estágios em corrente cruzada
Figura 3.5 – Solução no diagrama ternário
Este processo é uma mera extensão da extração em único
estágio, em que o rafinado do primeiro estágio é extraído
sucessivamente com solvente fresco de composição fixa. Os métodos de
24

cálculo são mais ou menos óbvios, como podemos ver nos gráficos
seguintes.
No diagrama de Janecke teremos
Figura 3.6 – Solução no diagrama de Janecke
Casos particulares
Variando a temperatura
25

Figura - 3.7
Solventes imiscíveis: Se os líquidos A e B podem ser considerados
completamente imiscíveis, ou se pelo menos suas solubilidades não se
alteram em toda a faixa de concentração da substância distribuída C, nas
condições de processo, os cálculos poderão ser convenientemente
simplificados.
Figura - 3.8
26

Assuma que toda massa do componente A esteja contida na
carga e no rafinado e que o conteúdo de B no extrato de qualquer
estágio é igual ao do solvente que entra no estágio.
Define-se: Y = (xC)B/(xB)B e X = (xC)A / (xB)B
O extrato do m’ésimo estágio terá a concentração ym e o
rafinado Xm. A concentração de C no solvente e na carga será YS e XF. A
massa total das correntes dá:
F = A + CF = A [1 + CF/A] F = A (1 + XF)
Sm = Bm (1+ YS)
Rm = A + Cm = A[1+ (Cm/A)] Rm = A(1+Xm)
Em = Bm (1+ YM)
Para qualquer estágio m, nós podemos fazer o seguinte
balanço para C:
A Xm-1 + Bm Ys = Bm Ym + A Xm
ou Y m−Y S
Xm−Xm−1=−A
Bm
que representa a equação de uma linha reta de inclinação – (A/Bm),
passando pelos pontos (Xm, Ym,) e (Xm-1,YS). Desde que tenhamos
correntes efluindo de estágios de equilíbrio teremos os pontos (Xm,Ym) na
curva de distribuição de equilíbrio.
Lei de Distribuição
No caso especial em que a distribuição obedece a Ym = m X m
Sabemos que para um estágio m+1 o balanço para C dará:
27

B (Ym+1 – YS) = A(Xm – Xm+1)
Resolvendo a equação II simultaneamente com a relação de
equilíbrio para o estagio m+1 para eliminar y m+1 e tendo Ɛ = m B/A,
teremos:
X m+1−X m
Ɛ+1−Y SB /AƐ+1
=0
A qual é uma equação linear de 1ª. ordem, cuja solução é:
que pode ser rearranjado para dar
É a fórmula para a composição do extrato composto será:
Atenção particular é dada à quantidade Ɛ, o fator de extração,
que terá evidentemente, uma importância muito grande em todas as
28

operações de extração. Em virtude da definição de m, Ɛ pode tomar o
significado físico de:
Ɛ=mBA
= quantidadede solutono extratoquantidade desoluto norafinado
Figura 3.9 – Contatos múltiplos em corrente cruzada com solventes imiscíveis, com
quantidades iguais de solvente aplicada a cada estágio. Ɛ é o fator de extração para
cada estágio.
29

3.4 Múltiplos estágios em contracorrente
Na figura 3.10 vemos uma separação com n estágios onde a
corrente de extrato E entra em contato com a corrente de rafinado R em
contracorrente. A carga é denominada F e entra pelo primeiro estágio.
Inicialmente estamos admitindo a inexistência de refluxos e não
considerando os equipamentos de recuperação de solvente.
Figura 3.10 – Conjunto de múltiplos estágios em contracorrente
No caso o número de variáveis a serem especificadas é:
Ni = 2C + 2N + 5
Em geral a influencia da pressão é desprezível, e a torre é
isotérmica ou tem um perfil de temperatura bem definido e controlado.
Pressão em cada estágio..........................................................N
Temperatura em cada estágio...................................................N
F (carga)..............................................................................C + 2
S (solvente)..........................................................................C + 2
Concentração de um componente em E1 ou RN........................1
Total ........................................................................2N + 2C + 5
30

Solução pelo diagrama triangular
Sendo especificadas as variáveis acima podemos calcular, por
exemplo, o número de estágios de uma torre. A sequência para este
cálculo será:
a) Determinamos o ponto M = F + S. isto pode ser feito porque
as composições e as vazões de F e S são conhecidas.
b) O RN (ou E1) pode ser locado no diagrama pois a
concentração de um dos componentes é dada e o ponto
está sobre a curva de equilíbrio.
c) Locado por exemplo E1 podemos locar Rn pois pelo balanço
material global temos:
S + F = M = E1 + Rn
Portanto, M, E1 e Rn estão alinhados e R está sobre a curva de equilíbrio.
d) Determinamos o ponto Δ = F – E1 = Rn – S. Essa expressão
é, consequência direta do balanço material. É interessante
notar que fazendo o balanço material na seção de fundo da
torre até o estagio n, temos: F – E1 = Rn – S = Δ, portanto,
esse ponto que representa a diferença de vazões entre o
rafinado e extrato se mantém ao longo da torre.
O ponto Δ pode ser determinado pela interseção da reta que
passa por F e E1 e da reta que passa por Rn e S.
e) Para a determinação gráfica do número de estágios, o
cálculo tem origem numa das extremidades da torre, por
exemplo, onde fica E1. Conhecido E1 se determina R1
através do traçado da linha de amarração E1R1. Conhecido
R1, como R1 – E2 = Δ, consequentemente, R1, E2 e Δ estão
31
alinhados, portanto traçamos a reta R1Δ e determinamos E2
na interseção com a curva de equilíbrio.
Conhecido E2 determina-se R2 pela linha de amarração e em seguida E3,
ligando R2 com Δ e assim sucessivamente até chegarmos a um Rn com
xC menor que o do Rn. Então contamos o número de linhas de amarração
traçadas para sabermos o número de estágios de equilíbrio.
Figura 3.11 – Solução no diagrama triangular
Observação: Se em vez de conhecermos uma das concentrações, fosse
conhecido o número de estágios teríamos que resolver o problema por
tentativas. Tentaríamos vários E1 até coincidir com o número de estágios
requeridos.
Quando o gráfico fica muito congestionado devido ao grande
número de linhas recorremos ao diagrama de JANECKE e se ainda o
número de estágios é muito grande, recorre-se ao diagrama de
distribuição (que é construído a partir do diagrama triangular). No
diagrama de JANECKE, analogamente ao caso resolvido do diagrama
triangular podemos escrever a expressão do balanço material:
32

M = F + S = RN + E1, porém os pontos só poderão ser locados
se as vazões forem em base livre de solvente.
Naturalmente se o ponto S estiver muito longe da curva de
equilíbrio será mais prático locar o ponto M diretamente determinando
(xB)M pelo balanço material.
M = S + F (vazões sem solvente)
Para o componente B
(xB)M . M = (xB) .S + (xB)F . F
Figura 3.12 – Solução de um sistema contracorrente no diagrama de
Janeckee
33

UTILIZANDO O DIAGRAMA DE DISTRIBUIÇÃO
Para calcular o número de estágios temos que traçar a curva
de operação, que represente o balanço material ao longo da torre. Para
traçar essa curva no diagrama de distribuição lançamos mão do
diagrama triangular.
Um balanço do componente C para o estágio (N) até o (m + 1)
S(Xc)s +Rm(XC)Rm = RN(XC) RN + Em+1 (XC)Em+1
Rearranjando:
Esta é a equação de uma curva, conhecida como CURVA DE OPERAÇÃO, em um sistema de coordenadas (XC)E, VS. (XC)R,
relacionando (X C )E n+1 com (XC)R, desde que S, (XC)S, Rn e (X C )R n
sejam
constantes para toda planta.
Figura 3.13 – Solução pelo diagrama de distribuição
34

Utilizando o diagrama triangular traça-se a curva de equilíbrio
no diagrama de distribuição, conforme visto anteriormente. Do ponto de
operação Δ, linhas tais como ΔEm+1. Estas não necessariamente
coincidentes com as linhas de estágio usadas previamente. As
coordenadas são transferidas para o diagrama de distribuição para
formar a curva de operação. Construção dos estágios entre as curvas de
operação e equilíbrio é mostrada na figura 3.13, supondo que a curva de
equilíbrio já esteja traçada:
Solventes imiscíveis: Como no caso de extração em corrente
cruzada, uma simplificação razoável resulta se for possível considerar os
líquidos A e B substancialmente imiscíveis na faixa de concentração será
usada como no caso anterior, Y = XCB/XBB e X=XCA/XAA, com o A contido
na carga permanecendo constante por todos os rafinados, o conteúdo de
B no solvente e em todos os extratos também ficara constante. Um
balanço para C para o estágio 1 até m, é dado por:
A XF + B Ym+1 = A Xm + B Y1
A qual pode ser rearranjada para fornecer:
Y m+1=ABXM+Y− A
BXF
A qual é a equação de uma linha reta, Ym+1 versus Xm, de
inclinação A/B desde que Y1 e XF constantes para um dado sistema.
Como m representa um estágio qualquer, a linha
corresponderá a uma “reta de operação” e será aplicável ao equipamento
todo e pode ser traçado entre dois pontos cujas coordenadas são (XF,Y1)
e (xn,yS), como na figura 3.14, a linha GK. Cada estágio ideal da
sequência de estágios pode ser considerado operando em corrente
cruzada, com os efluentes em equilíbrio. Para o estágio n, por exemplo,
a linha de operação será dada por:
35

Y n−Y S
Xn−Xn−1=−A
B
A qual é a linha MN da figura 3.14. O retângulo GMPN representa então
o n’ésimo estágio, e retângulos similares representarão os outros, como
mostrado. Na prática, só é preciso traçar a linha GK e os degraus
representando os topos do retângulo para determinar o número de
estágios.
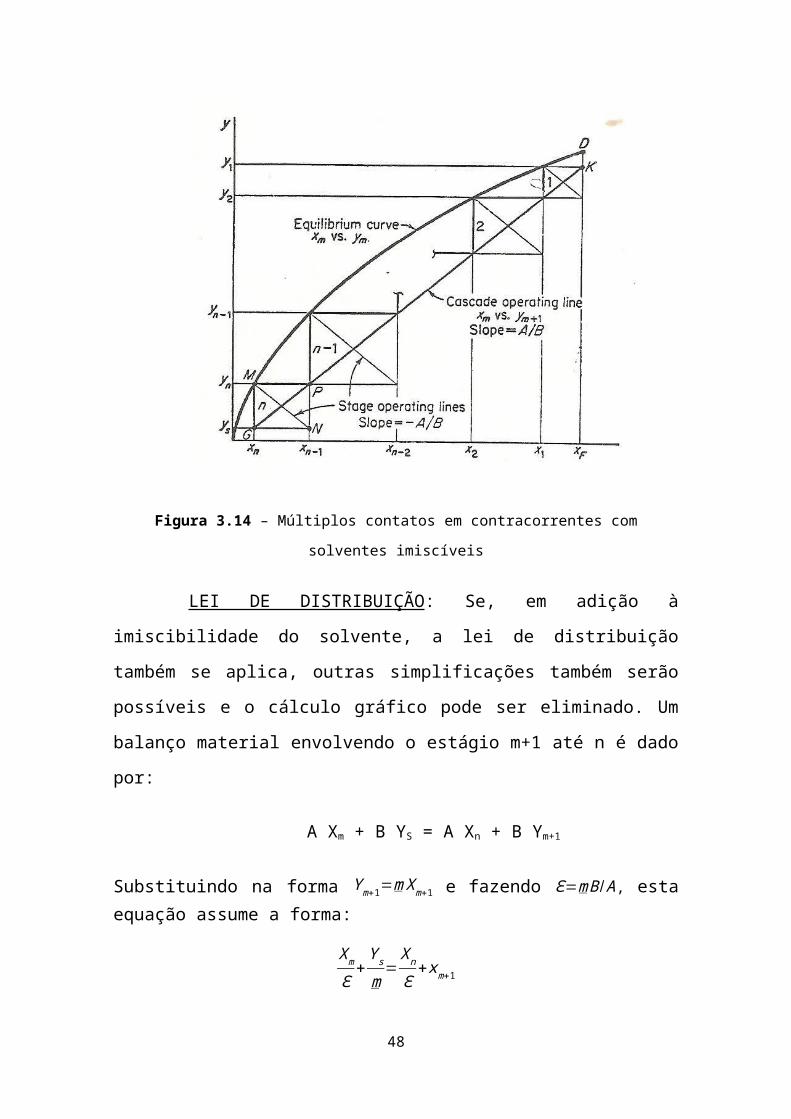
Figura 3.14 – Múltiplos contatos em contracorrentes com solventes imiscíveis
LEI DE DISTRIBUIÇÃO: Se, em adição à imiscibilidade do
solvente, a lei de distribuição também se aplica, outras simplificações
também serão possíveis e o cálculo gráfico pode ser eliminado. Um
balanço material envolvendo o estágio m+1 até n é dado por:
A Xm + B YS = A Xn + B Ym+1
Substituindo na forma Y m+1=m Xm+1 e fazendo Ɛ=mB/A , esta equação assume a forma:
36

Xm
Ɛ+Y s
m=
Xn
Ɛ+xm+1
Esta é uma equação linear de primeira ordem de diferenças
finitas cuja solução é:
Utilizando o diagrama triangular traça-se a curva de equilíbrio
no diagrama de distribuição, conforme visto anteriormente. Do ponto de
operação Δ, linhas tais como ΔEm+1. Estas não necessariamente
coincidentes com as linhas de estágio usadas previamente. As
coordenadas são transferidas para o diagrama de distribuição para
formar a curva de operação. Construção dos estágios entre as curvas de
operação e equilíbrio é mostrada na figura abaixo.
Supondo que a curva de equilíbrio já esteja traçada:
ɛ≠1 Xm=(X F−Y S/m−X n/Ɛ1−1/Ɛ )(1/ Ɛ )m+
Y s /m−Xn/Ɛ1−1/Ɛ
e para Ɛ=1 Xm=X f−m ( Xn−Y S/m )
As quais são muito úteis para o cálculo de concentrações intermediárias
de rafinados sem a necessidade de traçado gráfico. Fazendo-se m = n,
permitindo o rearranjo para as seguintes práticas expressões, quando
Ɛ≠1
XF−X N
X F−Y S/m=ƐN+1−ƐƐ N+1−1
N=
log [ XF−Y S/mX N−Y S/m (1−1Ɛ )+ 1Ɛ ]
log Ɛ
37

E quando Ɛ = 1
XF−X N
X F−Y S/m= NN+1
N=X F−XN
X N−XS /m
Figura 3.15 – Extração em múltiplos estágios em contracorrente com solventes
imiscíveis e coeficiente de distribuição constante.
38

Estas equações são variações das bem conhecidas equações
de KREMSER-BROWN-SOUDERS, expressões originalmente
desenvolvidas para absorção gasosa. Uma solução gráfica para elas é
dada na figura 3.15. Apesar de estritamente aplicável para sistemas
completamente imiscíveis dos solventes A e B e com curva de
distribuição linear, as expressões produzem uma boa aproximação ao
mais correto calculo gráfico para casos de desvio modesto das condições
reais se um fator de extração médio (Ɛ1Ɛn)1/2 for usado.
Razão mínima solvente/carga (para o caso de torre com única carga)
Chamamos razão de solvente em relação à carga, à razão S/F
no caso de torre com uma única carga e S/(F + F’) para uma torre com
duas cargas. A razão solvente carga influi muito na locação do ponto Δ,
porque ela determina a razão de refluxo interna da torre. Em geral,
aumentando-se a razão solvente/carga tornamos as linhas de operação
(linhas que unem S, Rn e Δ) e as linhas de amarração mais afastadas do
paralelismo, ou seja, aumentamos a separação por estágio.
Inversamente, reduzindo a razão S/F fazemos com elas fiquem quase
paralelas e daí reduzimos a separação por estágio. Se essa relação for
reduzida continuamente será atingida uma razão onde em um ponto
qualquer da torre a linha de amarração e a linha de operação coincidem.
Daí não se conseguir passar por esse ponto na construção estágio por
estágio.
Primeiro caso – as linhas de amarração têm inclinação negativa
Conforme diminuímos a razão (S/F) o ponto M caminha no seguimento
SF em direção a (F), como consequências (E1) caminha em direção a P e
Δ caminha para direita, até coincidir com a interseção de uma das “tie-
lines” com a reta determinada por RN e S. Nesse ponto teremos o Δ
correspondente à razão (S/F)min.
39

Figura 3.16 – Primeiro caso
Segundo caso – as linhas de amarração têm inclinação positiva
Neste caso, pode-se reduzir a quantidade de S até o ponto Δ
aparecer no lado esquerdo do diagrama (se as posições de F e P no
diagrama assim o permitirem).
Figura 3.17 – Segundo Caso
Se a posição de (F) não permitir este tipo de análise no
segundo caso, (S/F) mínima corresponde a um (M) tal que E1 = P.
40

A máxima razão solvente/carga será aquela em que a carga
seja totalmente dissolvida.
TORRE COM DUAS CARGAS
Frequentemente duas cargas devem ser injetadas na torre de
extração. Uma relativamente pobre em (C) e outra mais rica. Em vez de
misturarmos as duas em uma única e perder a separação já existente é
preferível mantê-las separadas e injetar a mais rica em (C) no fundo da
torre.
Figura 3.18 – Torre de extração com duas cargas
41

O número de variáveis independentes desse processo é 3C +
2N + 8 que C + 3 variáveis a mais que no caso de uma só carga. C + 2
delas servem para especificar F e a restante para localizar o prato da
carga. Quando se usa computador é mais vantajoso especificar o
número de pratos abaixo da injeção. Quando se calcula o número de
estágios graficamente é mais vantajoso especificar a entrada da carga
como localizada no prato ótimo.
Prato ótimo por definição é aquele com o qual se consegue a
separação desejada com um número mínimo de pratos ou então a
máxima separação com um determinado número de pratos.
Fazendo o balanço material no topo da torre a partir do estágio
n+1 temos:
Rn + S = RN + En+1 ou
RN – En+1 = RN – S = Δ = constante superior
Fazendo agora o balanço no fundo da torre até o estágio M
teremos:
RM + E1 = F’ + EM+1
RM – EM+1 = F’ – E1 = Δ’ = constante inferior
Fazendo o balanço global:
F + F’ + S = RN + E1
(F) + (F’ – E1) = (RN - S)
F + Δ’ = Δ
42

Deste modo percebe-se que os pontos Δ, Δ’ e F ficam sobre
uma mesma reta da expressão 1 temos ainda F + F’ – E1 = RN - S
No cálculo gráfico do número de estágios começa-se por uma
das extremidades da coluna procedendo-se, como foi visto, no caso da
coluna com uma carga. Deve-se nesse procedimento usar o Δ
correspondente a seção. Quando chegamos ao prato de carga mudamos
para o Δ da outra seção da torre e continuamos da mesma maneira.
Quando é especificado que a carga entra no prato ótimo, a mudança de
Δ deve ser feita quando a linha de amarração do estado cruza a linha
ΔΔ’.
Na resolução gráfica temos F e F’ especificados, o que permite
a determinação de FT. Para localizar Δ na seção do rafinado lembramos
que S, RN e Δ estão sobre uma mesma reta e que FT, E1 e Δ também o
estão, Δ está na interseção dessas retas. Para a seção do extrato
determinamos Δ’ pela interseção da reta que passa por Δ e F (Δ, Δ’ e F
alinhados). Pode-se observar fazendo graficamente que se não
mudarmos de Δ quando cruzamos a linha ΔΔ’ o número de estágios para
mesma separação será maior. Graficamente temos:
Figura 3.19 – Solução do caso com duas cargas
Com Δ e Δ’ determinamos o número de estágios
43

Figura 3.20 – Obtenção do número de estágios
No caso de usarmos o Diagrama de Janecke o procedimento
será semelhante, apenas as vazões não incluirão o solvente.
Figura 3.21 – Solução no diagrama de Janecke
44

No diagrama de distribuição haveria duas curvas de operação,
uma para cada seção da torre. Para se obter essas curvas, teríamos que
partir do diagrama triangular. Usar o corresponde à seção e proceder da
mesma forma que para o caso de uma só carga. No traçado para
determinação do número de estágios mudaríamos de curva de operação
ao chegar ao prato de carga.
Figura 3.22 – Solução no diagrama de distribuição
Operações com refluxo
Refluxo de extrato
Nos processos descritos até agora a corrente de extrato que
sai da torre (E1) era removida do mesmo estágio em que F’ entrava.
Embora E1 saísse em equilíbrio com R1 e não com F’, a introdução da
carga neste ponto restringiria a pureza de E1. Isto é notório quando a
carga F’ tem baixa concentração do componente B está extraindo.
45

O remédio para esta situação é substituir a carga F’ por uma
corrente que tenha alta concentração em C, permitindo, assim, que se
consiga uma maior pureza em E1. Isto pode ser conseguido pelo refluxo
de extrato representado abaixo. A carga é deslocada para um estágio
M+1 intermediário.
Figura 3.23 – Esquema com refluxo de extrato
46

Coloquemos no diagrama ternário os pontos que representam
as correntes da figura acima.
Figura 3.24 – Solução no diagrama triangular
Na figura 3.24 retira-se uma quantidade suficiente de solvente de
E1 para deslocar o ponto E’0 para o lado oposto do diagrama. Se todo o
solvente for retirado de E1 obteremos E1 com composição no lado AC do
diagrama.
No exemplo acima, ilustramos com um caso do tipo II por dois
motivos: para mostrar que os princípios da construção gráfica são os
mesmos para os diagramas desse tipo e porque o refluxo de extrato é
normalmente usado para esse tipo de diagrama. Em geral, os refluxos
são necessários quando o solvente é parcialmente imiscível com os dois
componentes da solução e não mostra grande seletividade para nenhum
dos componentes da carga.
47
As equações de balanço material e os pontos Δ são
determinados da mesma maneira que para o caso da torre com duas
cargas. F’ é considerado como uma segunda carga.
Temos então do balanço material apenas da torre
RN + E1 = F + F’ + S
Ou
E1 – F’ = S – RN + F Δ’ = Δ + F (alinhados)
Δ Δ’
Ou E1 – F’ – F = S – RN E1 – ET = Δ (alinhados)
Do balanço material no equipamento de recuperação de solvente.
A locação dos Δs segue então o processo normal.
Δ – está na interseção da reta que passa por RN e S com a reta
que passa por FT e E1.
Δ’ – está na interseção da reta que passa por F’ e E1 com a
reta que passa por Δ e F.
Para se calcular o número de estágios procedemos
analogamente ao da torre com duas cargas. Partimos de qualquer uma
das extremidades e mudamos de Δ quando cruzamos a linha ΔΔ’ (se a
carga foi introduzida no ponto ótimo).
48

RAZÃO DE REFLUXO DE EXTRATO
Normalmente, em torres com duas cargas, é especificada a
razão (F/F’) e com ela podemos locar o ponto Δ’. No caso de refluxo de
extrato, o que é normalmente especificado é a razão (F’/E0). Essa
relação pode ser usada para locar Δ’ na linha por F’ e E1. O balanço
material global determina Δ sobre FΔ’.
Seja dada então a relação F’/ E0; pela regra da alavanca temos
que
E1/SE = F’SE / F’E1 (5.20)
logicamente a determinação dessa relação depende da
especificação dos pontos SE, F’ e E1.
Fazendo um balanço em torno do equipamento de recuperação
de solvente:
E1=SE+F'+E0=SE+(F '
E0+1)E0
( E1SE)SE=SE+( F '
E0+1)E0
SE( E1S E
−1)=( F 'E0 +1)E0
S E
E0=
( F 'E0 +1)( E1S E
−1)Δ'E0
=SE
E0+1=
E0S E
Δ' SE
49

REFLUXO TOTAL DE EXTRATO
Já vimos que Δ’ = E1 – F’ = E0 + SE
Se usarmos refluxo total de extrato teremos E0 = 0 e Δ’ = E1 –
F’ = SE, ou seja, Δ’ coincidirá com SE. Isso implica (PODE SER
COMPROVADO GRAFICAMENTE) em uma maior separação em cada
estágio da seção de extrato (FUNDO) da coluna. Quando Δ’ desce em
direção a SE, Δ move-se em direção a S, pois Δ, Δ’ e F se mantém
alinhados, ou seja, o aumento da razão de refluxo faz as linhas de
operação se afastarem mais do paralelismo e portanto produz o número
de estágios necessários para uma dada separação.
É interessante notar que com refluxo total a carga F sairá
totalmente em RN, já que SE é reciclado e volta a fazer parte de S.
Figura 3.25 – Cálculo da razão de refluxo mínima
50

A vazão mínima de refluxo do extrato é definida como a vazão
de F’ que, se reduzida de um infinitésimo faria o número de estágios
crescer para o infinito. Um número infinito de estágios exige uma zona de
composição constante. Isto ocorre quando uma linha balanço material
coincide com uma (tie line) linha de amarração.
Para se determinar o Δ’min achamos a tie line da seção de
extrato da torre que intercepta a linha E1SE mais próxima de SE. Em
geral, a tie line cujo prolongamento passa por F, é aquela que determina
o Δ’min.
REFLUXO DE RAFINADO
Às vezes, parte do Rn é reciclada e misturada diretamente com
a corrente de solvente para formar S conforme mostrado na figura a
seguir. A razão de refluxo de rafinado é definida como R’N / D. Entretanto,
esse chamado refluxo não é nada mais que o reciclo de uma das fases
do estágio N para o próprio N. Como o equilibrio não é afetado pelas
quantidades relativas dessas fases em equilibrio, o reciclo R’N não altera
a separação do sistema. Em operações práticas não se tem uma
eficiência de 100 % e refluxo de rafinado pode oferecer uma certa
vantagem.
A pré saturação do solvente com o rafinado, fora do estágio N,
de certa forma aumenta o tamanho efetivo da coluna desde que a
mistura não precise ocorrer mais no estágio. Também consegue-se uma
aproximação maior da condição ideal de equilibrio e aumenta-se a
eficiência do último estágio da torre.
Embora o refluxo de rafinado n não afete o cálculo do número
de estágios, ela faz surgir novos pontos no diagrama.
51

FAZENDO O BALANÇO MATERIAL
M = F + S = E1 + RN (I)
M’ = F + S’ = E1 + D (II)
A adição de (R’N) ou (S’) localiza (S) mais próximo da curva de
equilíbrio. O ponto Δ é locado como sempre pela interseção das retas
(RN) e (S) e por (E1) e (F).
Pois
Δ = RN – S = F – E1
Se o refluxo do rafinado tivesse efeito na construção para
determinação do número de estágios, a localização de Δ variaria com a
quantidade de refluxo.
Considere um caso aonde F é completamente especificada e a
posição de E1 foi fixada pela especificação de uma concentração. A
razão solvente/carga S/F foi especificada e entao podemos locar RN. Um
balanço em torno do misturador nos fornece:
RN + S’ = D + S
Δ = RN – S = D – S’ todos os quatro pontos caem em cima da
mesma reta
Portanto, uma variação no refluxo de rafinado não altera a
posição da reta. Apenas a posição de S entre RN e S’ muda com a razão
de refluxo. Portanto, a o ponto de interseção da linha por RN e S e da
linha por F e E1 é independente da razão de refluxo do rafinado.
Deste modo, a redução da razão de refluxo não causa o
aumento do número de estágios e nós não podemos definir uma razão
52

de refluxo de rafinado mínima que faça o número de estágio crescer
indefinidamente.
53