apostila de analise instrumental
TRANSCRIPT

UNIVERSIDADE FEDERAL DA PARAÍBA - UFPB
CENTRO DE CIÊNCIAS EXATAS E DA NATUREZA - CCEN
DEPARTAMENTO DE QUÍMICA - DQ
Análise Química Instrumental
DISCIPLINA: Química Analítica III PROF.: Edvan Cirino da Silva
João Pessoa - 2008

1
INTRODUÇÃO AOS MÉTODOS INSTRUMENTAIS DE ANÁLISE
Classificação: ⇒ Métodos Quantitativos ♦ Métodos Espectroanalíticos ♦ “ Eletroanalíticos ♦ “ Radioanalíticos ♦ “ Termoanalíticos ♦ “ Cromatográficos
⇒ Métodos Qualitativos, de Identificação ou Caracteri zação ♦ Espectrometria no Infravermelho ♦ “ de Ressonância Magnética Nuclear ♦ “ de Massa ♦ “ de Raio X ♦ “ de Ressonância de Spin Eletrônico
⇒ Métodos Espectroanalíticos São aqueles baseados em medidas da absorção e da emissão da radiação UV-Visível por espécies químicas atômicas ou moleculares. ♦ Espectrometria de Absorção Molecular ♦ “ “ “ e Emissão Atômica ♦ “ de Emissão de Fluorescência Atômica e Molecular ♦ Espectrografia de Emissão.
⇒ Métodos Eletroanalíticos São aqueles baseados em medidas de propriedades elétricas (corrente , tensão e resistência ) das espécies químicas. ♦ Potenciometria ♦ Coulometria ♦ Voltametria ♦ Condutometria ♦ Eletrogravimetria
⇒ Métodos Radioanalíticos São os que se baseiam em medidas das radioatividades emitidas por espécies químicas. ♦ Análise por Ativação Neutrônica ♦ Análise por Diluição Isotópica ⇒ Métodos Termoanalíticos Baseiam-se em medidas de calor emitido ou absorvido por espécies químicas. ♦ Termogravimetria ♦ Calorimetria Diferencial Exploratória ⇒ Métodos Cromatográficos

2
São aqueles baseados na combinação de um método instrumental de análise com uma técnica de separação , usando colunas empacotadas ou superfícies porosas . ♦ Cromatografia Gasosa ♦ Cromatografia Líquida OBJETIVOS DO CURSO DE ANÁLISE INSTRUMENTAL ⇒ O objetivo desta disciplina é apresentar e discutir os FUNDAMENTOS
TEÓRICOS, A INSTRUMENTAÇÃO e APLICAÇÕES PRÁTICAS de alguns métodos instrumentais para análise quantitativa de interesse em diversas áreas.
REFERÊNCIAS BIBLIOGRÁFICAS 1. Apostila de Química Analítica Instrumental 2. D. A. Skoog e J. J. Leary - “Princípios de Análise Instrumental ” – 5a Edição – Artmed Editora S.A. Porto Alegre (RS). 3. Otto Alcides Ohlweiler - “Fundamentos de Análise Instrumental ” - Livros Técnicos e Científicos, Rio de Janeiro, Brasil, 1981. 4. M. L. S. S. Gonçalves - “Métodos Instrumentais para Análises de Soluções - Análise Quantitativa ”, Fundação Calouste Gulbenkian, Lisboa, Portugal, 1990. Periódicos de Referência:
♦ Chemical Abstract ♦ Analytical Abstract Revistas Internacionais mais Importantes: Analytica Chimica Acta Analytical Chemistry Critical Reviews in Analytical Chemistry Analytical Procedure Talanta Spectrochimica Acta - Part B The Analyst Analytical Biochemistry Terminologias: ANÁLISE QUÍMICA - consiste na aplicação de um processo ou de uma série de processos para identificar (análise qualitativa) ou quantificar (determinar a quantidade, a concentração, o teor, etc) de uma espécie química (analito) presente em uma amostra.
AMOSTRA ANALÍTICA – pequena porção do material objeto da análise química que representa a composição média qualitativa e quantitativa da população.
AMOSTRAGEM – conjunto de operações que nos permite obter, partindo de uma grande quantidade de material, uma pequena porção (amostra) realmente representativa da composição média do todo.
ANALITO – espécie química presente na amostra cuja concentração se deseja determinar em uma análise. Ex. Cálcio presente no leite, ácido

3
acético no vinagre, colesterol no ovo, cromo do aço inoxidável, etc.
SINAL ANALÍTICO (ou SINAL) -- Resposta instrumental à propriedade do analito (absorbância, intensidade de emissão, etc..))
MATRIZ – compreende todos os constituintes de amostra analítica. Logo, além do analito a matriz da amostra contém os outros componentes chamados “concomitantes ”.
EXATIDÃO – grau de concordância entre o valor (resultado) obtido experimentalmente e o valor esperado (valor mais provável)
PRECISÃO – indica o grau de concordância entre resultados individuais dentro de uma série de medidas. Em outras palavras, a precisão está relacionada com a reprodutibilidade ou repetibilidade das medidas.
SENSIBILIDADE - medida da capacidade de um instrumento (ou método) em distinguir entre pequenas diferenças na concentração do analito.
LIMITE DE DETECÇÃO – é o nível de concentração (ou quantidade) mínima de analito detectável por um instrumento.
SELETIVIDADE - refere-se ao quão um método analítico está livre de interferências de outras espécies presentes na matriz. OBS.: Posteriormente será feita uma descrição quantitativa (matemática) do significado dos termos sensibilidade, limites de detecção e quantificação, bem como de outros termos cujo significado será introduzido oportunamente ETAPAS DE UMA ANÁLISE QUANTITATIVA TÍPICA (1) Amostragem (homogênea ou heterogênea); (2) Escolha do método analítico (instrumental ou clássico); (3) Preparação da amostra (trituração, dissolução, etc); (4) Medida da propriedade do analito (óptica, elétrica, massa, etc); (5) Tratamento de dados (calibração por curva analítica, cálculos, estatístico, etc); (6) Resultados ( interpretação e apresentação) SELEÇÃO DE UM MÉTODO ANALÍTICO
A escolha de um método apropriado para a abordagem do problema analítico requer respostas para as questões: • Que exatidão e precisão são necessárias?• Qual é a quantidade de amostra disponível?• Qual é o intervalo de concentração do analito?• Que componentes da amostra poderão causar interferência?• Quais as propriedades físicas e químicas da matriz? • Quantas amostras serão analisadas?• Recursos disponíveis (instrumentos, pessoal, etc.)
4
É importante ressaltar que, exceto na gravimetria e coulometria, toda análise química quantitativa requer a realização de uma calibração , por meio da qual encontra-se uma relação funcional entre o sinal analítico e a concentração do analito. Este processo encontra-se descrito adiante.
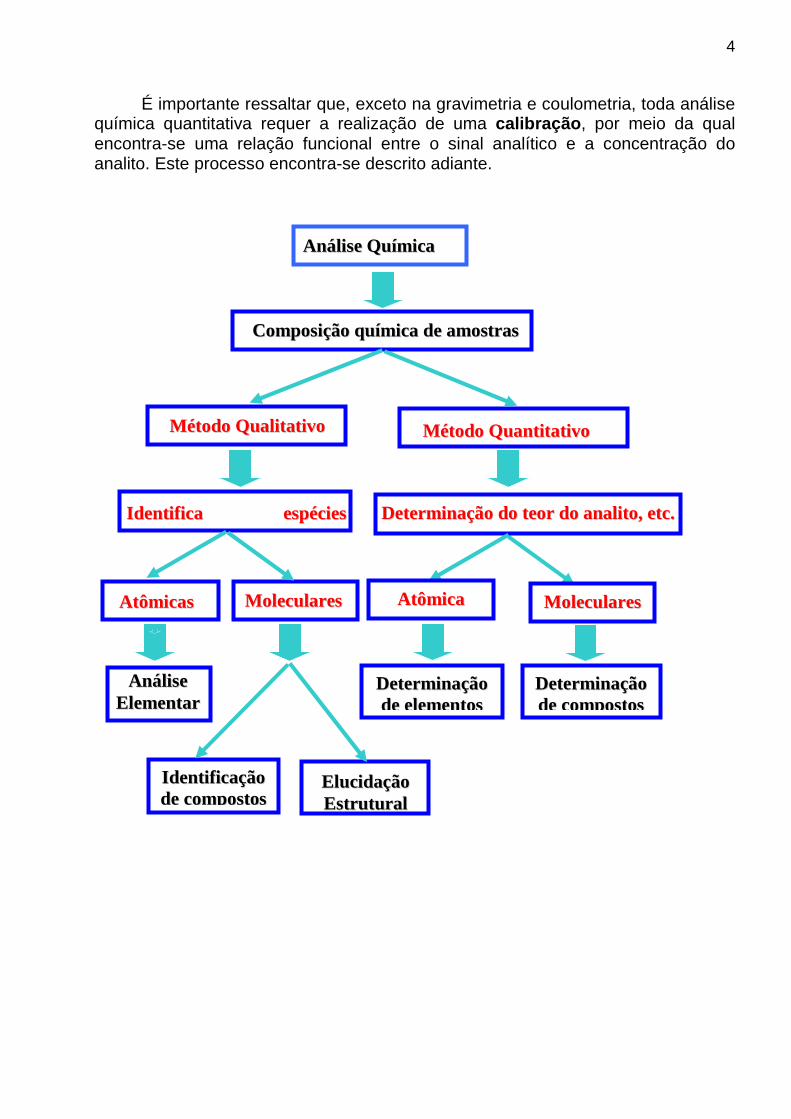
AAnnááll iissee QQuuíímmiiccaa
CCoommppoossiiççããoo qquuíímmiiccaa ddee aammoossttrr aass
MM ééttooddoo QQuuaall ii ttaatt iivvoo MM ééttooddoo QQuuaanntt ii ttaatt iivvoo
II ddeenntt ii ff iiccaa eessppéécciieess químicas
AAttôômmiiccaass MM oolleeccuullaarr eess
AAnnááll iissee EElleemmeennttaarr
EElluucciiddaaççããoo EEssttrr uuttuurr aall
DDeetteerr mmiinnaaççããoo ddoo tteeoorr ddoo aannaall ii ttoo,, eettcc..
AAttôômmiiccaa
II ddeenntt ii ff iiccaaççããoo ddee ccoommppoossttooss
MM oolleeccuullaarr eess
DDeetteerr mmiinnaaççããoo ddee eelleemmeennttooss
DDeetteerr mmiinnaaççããoo ddee ccoommppoossttooss

5
DOMÍNIO DE DADOS Um conceito relevante no contexto dos métodos instrumentais é o de domínio de dados . De fato, para entender como os instrumentos analíticos operam, é fundamental compreender como a informação é codificada . Nesse sentido, pode-se definir domínio de dados como sendo as várias maneiras de codificar a informação eletricamente, ou seja, como voltagem, corrente, carga ou variações dessas grandezas. Os domínios de dados podem ser classificados como:
(i) domínios não-elétricos; (ii) domínios elétricos. Esses tipos de domínios de dados são exemplificados no mapa da figura
abaixo.
Conversões entre domínios de dados durante uma medi da analítica Como ressaltado anteriormente, a medida analítica está associada a um fenômeno (absorção, emissão, potencial elétrico, etc) envolvendo o analito. Todavia, a informação analítica (qualitativa ou quantitativa) reside, em última
MM ééttooddooss AAnnaall íítt iiccooss
GGrr aavviimmééttrr iiccooss TTii ttuulloommééttrr iiccooss
MM ééttooddooss CClláássssiiccooss MM ééttooddooss II nnssttrr uummeennttaaiiss
VVeejj aa aa sseegguuii rr !!

6
análise, em um número que aparece no mostrador do instrumento ou em um gráfico (espectro) que é mostrado, por exemplo, na tela do microcomputador acoplado ao instrumento. Na realidade, qualquer processo de medida analítica pode ser representado por uma série de conversões entre domínios , tal como o ilustrado na figura abaixo. Nesse caso, o exemplo consiste na medida do sinal de fluorescência molecular de uma amostra de água tônica que contém quinino (substância fluorescente). O objetivo é determinar a concentração de quinino a partir da medida de fluorescência quando moléculas de quinino são excitadas com radiação eletromagnética oriunda de um laser.
MEDIDA ANALÍTICA - SINAL E RUÍDO Sabe-se que toda medida analítica é constituída de dois componentes: o sinal e o ruído. O primeiro contém informação sobre o analito e o ruído é a parte indesejada, pois é constituída de informação espúria. Esta pode degradar a exatidão e a precisão de um método, bem como prejudicar o limite inferior da quantidade do analito que pode ser detectada (o limite de detecção). Na figura a seguir (parte a), mostra-se o efeito do ruído sobre um sinal de uma corrente contínua pequena de aproximadamente 10-15 A. Na parte b, mostra-se um gráfico teórico da mesma corrente na ausência de ruído.
Note que a diferença entre os dois gráficos corresponde ao ruído, cuja presença parece ser inevitável nas medidas experimentais. De fato, dados livres de ruídos nunca podem obtidos experimentalmente, pois alguns tipos de ruídos se originam de efeitos quânticos e termodinâmicos cuja manifestação é impossível de ser evitada.

7
Via de regra, a intensidade média do ruído, N, é constante e não depende da magnitude do sinal analítico, S. Conseqüentemente, o efeito do ruído sobre o erro relativo de uma medida diminui com o aumento da magnitude da quantidade medida. Por isso, a relação sinal-ruído, S/N (do inglês: Signal-to-Noise Ratio), é um parâmetro mais útil que o ruído sozinho para descrever qualidade de um método analítico ou a performance de um instrumento. Descrição quantitativa de S/N A intensidade do ruído é apropriadamente descrita pelo desvio-padrão s de várias medidas do sinal analítico S, cuja magnitude é determinada pela média x das medidas. Assim, a relação sinal-ruído S/N é dada por
sx
padrãodesviomédia
NS =
−= .
Note que S/N corresponde ao inverso do desvio-padrão relativo, RSD (do inglês, Relative Standard Desviation). Então,
RSD
1NS =
Para o sinal ruidoso apresentado na figura anterior, o desvio padrão pode ser estimado (com o nível de 99 % de confiança) pela expressão:
5
alsinalsins mínmáx −=
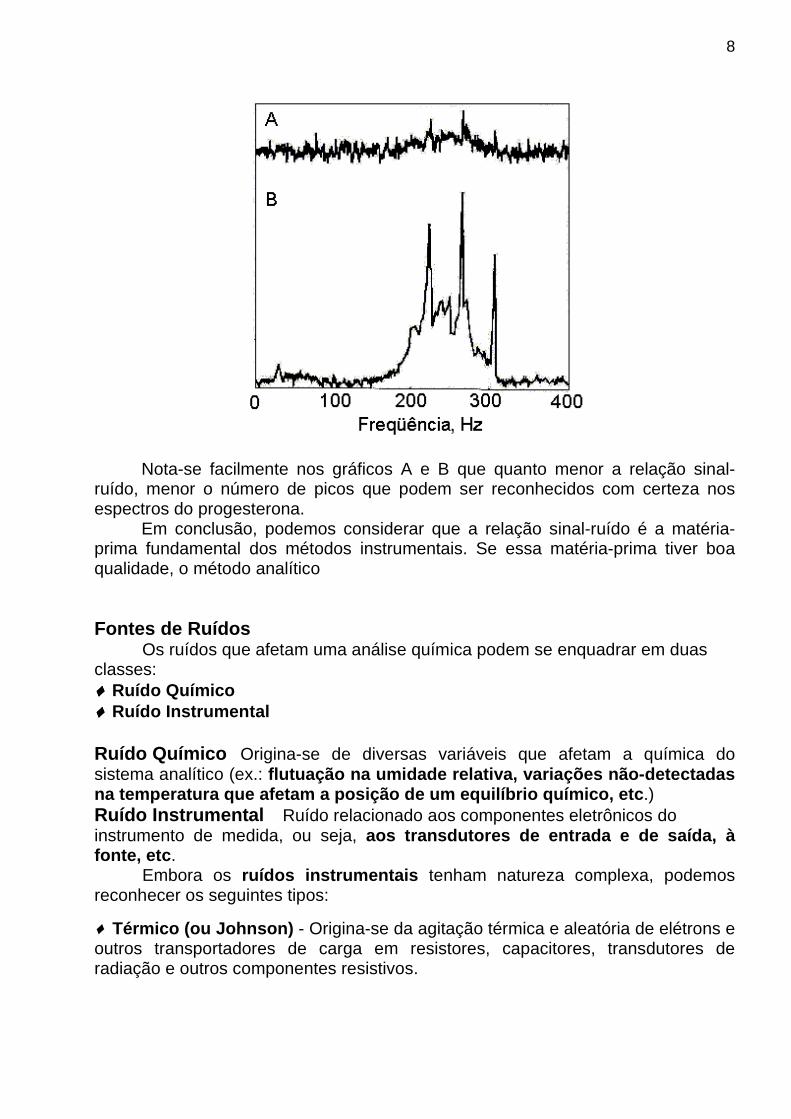
Ao adotar o valor 5 estamos assumindo que as flutuações em torno da média são aleatórias e que seguem uma distribuição normal. A curva normal mostra que 99 % dos dados se encontram entre ± 2,5 σ (desvio-padrão populacional) de sorte que podemos admitir que a diferença entre o valor máximo e o mínimo, com 99 % de certeza, é de 5 σ. Logo, o valor de s dado pela expressão anterior é uma estimativa razoável para o desvio-padão. É importante salientar que, em regra, é impossível detectar um sinal quando S/N é menor que cerca de 2 ou 3. Para ilustrar esse fato, apresentamos, na figura mostrada a seguir, o espectro de RMN para a progesterona com S/N de cerca de 4,3 (gráfico A) e 43 (gráfico B).
8
Nota-se facilmente nos gráficos A e B que quanto menor a relação sinal-ruído, menor o número de picos que podem ser reconhecidos com certeza nos espectros do progesterona. Em conclusão, podemos considerar que a relação sinal-ruído é a matéria-prima fundamental dos métodos instrumentais. Se essa matéria-prima tiver boa qualidade, o método analítico Fontes de Ruídos
Os ruídos que afetam uma análise química podem se enquadrar em duas classes: ♦♦♦♦ Ruído Químico ♦♦♦♦ Ruído Instrumental Ruído Químico Origina-se de diversas variáveis que afetam a química do sistema analítico (ex.: flutuação na umidade relativa, variações não-detect adas na temperatura que afetam a posição de um equilíbri o químico, etc .) Ruído Instrumental Ruído relacionado aos componentes eletrônicos do instrumento de medida, ou seja, aos transdutores de entrada e de saída, à fonte, etc .
Embora os ruídos instrumentais tenham natureza complexa, podemos reconhecer os seguintes tipos:
♦ Térmico (ou Johnson) - Origina-se da agitação térmica e aleatória de elétrons e outros transportadores de carga em resistores, capacitores, transdutores de radiação e outros componentes resistivos.
9
♦ Shot - Ocorre quando elétrons ou outras partículas carregadas atravessam uma junção pn em circuitos eletrônicos (fotodiodo) ou um espaço evacuado entre o anodo e o catodo em fototubos. ♦ Flicker ou 1/f - De origem desconhecida, porém caracteriza-se por apresentar uma magnitude inversamente proporcional à freqüência (f) do sinal observado. Por isso, é também chamado de ruído 1 / f (um sobre f). CALIBRAÇÃO EM ANÁLISE QUÍMICA INSTRUMENTAL Calibração é o processo que busca relacionar o sinal analítico medido com a concentração do analito. A relação funcional (matemática) constitui o modelo de calibração e a representação gráfica do modelo de calibração é denominada curva analítica . Em uma análise química instrumental, quando se deseja construir uma curva analítica necessária para determinar a concentração da amostra, é natural imaginar que a curva deve passar o mais próximo possível dos pontos experimentais. O procedimento mais utilizado a fim de obter esta máxima proximidade é conhecido como método dos mínimos quadrados . Para ilustrar o fundamento do método dos mínimos quadrados, considere a curva de calibração mostrada na figura a seguir:
onde: x1, x2, x3, x4 = concentração das soluções-padrão y1, y2, y3, y4 = leitura instrumental de cada solução padrão yA = leitura da amostra (A) xA = concentração da amostra (A) encontrada através da curva analítica ei = yi - (ye)i = yi – b0 – b1 x i (resíduo) No método dos mínimos quadrados, os valores de b0 e b1 são estimados minimizando-se a soma quadrática dos resíduos (ei) dada por:
Soma quadrática dos resíduos (SQr) = ∑ ⋅−− 2i10i )xbby(

10
Para minimizar a SQr deriva-se (cálculo de 3o grau) a função acima em relação a b1 e b0 e iguala-se as derivadas a zero. Isto leva às seguintes expressões para o cálculo de b1 e b0:
( )∑ ∑∑ ∑ ∑
−⋅
⋅−⋅⋅=
2i
2i
iiii1
x)x(n
yxyxnb e
n
xbyb i1i
0∑ ∑⋅−
=
onde n = no total de medidas OBS: Para avaliar a qualidade do ajuste linear, pode-se tomar como base o valor calculado do “coeficiente de correlação , r(y e,y), entre os valores das leituras instrumentais, y i, e os valores estimados pela equação da reta, (ye)i, dado pela expressão:
( ) 2/12i
2eie
ieie
]yy[]y)y[(
]yy[]y)y[(r
∑ ∑
∑−⋅−
−⋅−=
onde -1 ≤ r ≤ 1, porém em análise química baseada em curva analítica, r só pode apresentar valores compreendidos no intervalo 0 ≤≤≤≤ r ≤≤≤≤ 1.
Para o ajuste linear pode-se também utilizar, de maneira equivalente, a seguinte expressão para o cálculo do coeficiente de correlação, r (x,y) , entre os valores de x (concentração dos padrões) e os valores das leituras instrumentais, y:
2/12i
2i
ii
])yy([])xx([
)]yy()xx[(r
∑ ∑
∑−⋅−
−⋅−=
Quanto mais próximo de 1 estiver o valor de r, calculado usando as expressões apresentas acima, maior é a evidência de que o ajuste linear está sendo eficiente. Por outro lado, um coeficiente de correlação zero (ou próximo de zero) indica que x e y não são linearmente relacionados.
Entretanto, é importante salientar que o valor de r fornece apenas uma idéia da eficiência do ajuste aos dados experimentais, porém não deve ser utilizado para avaliar, com rigor, a qualidade do ajuste. Para isso, deve-se usar o teste F (teste estatístico) da falta de ajuste . Para maiores detalhes sobre esse teste estatístico consultar a referência bibliográfica citada abaixo (∗). Embora o valor de r não possa ser tomado como um critério para avaliação rigorosa da qualidade do ajuste aos dados experimentais, pode-se considerar que o ajuste é aceitável quando r ≥≥≥≥ 0,999. ---------------------------------------------------------------------------------------------------------- (∗∗∗∗) Pimentel, M.F. e Neto, B.B. – “Calibração: Uma Revisão para Químicos Analíticos“, Quím. Nova , 19 (1996) 268. MÉTODO ANALÍTICO - Figuras de Mérito Figuras de mérito são critérios (ou características) numérico(a)s para avaliar a eficiência de um instrumento ou método analítico.A tabela abaixo mostra as figuras de mérito fundamentais que podem ser usadas na escolha de um método analítico.

11
Critério Figura de Mérito
1. Precisão Desvios-padrão absoluto e relativo, coeficiente de variação, variância
2. Tendência Erros sistemáticos absoluto e relativo
3. Sensibilidade Sensibilidades de calibração e analítica
4. Limite de detecção
Branco mais três vezes o desvio-padrão dos sinais do branco
5. Faixa dinâmica Limite de quantificação até o limite de linearidade
6. Seletividade Coeficiente de seletividade
SENSIBILIDADE
Segundo a IUPAC a sensibilidade de calibração é dada pela inclinação (b1) da curva analítica (y = b 0 + b1 x), mas essa definição falha por não considerar a precisão das medidas individuais.
Para resolver esse problema, Mandel e Stiehler propuseram a sensibilidade analítica , g, definida por
γ= b1 / s onde s é o desvio-padrão da medida e b1 representa a inclinação da curva analítica. Sensibilidade Analítica x Sensibilidade de Calibraç ão
Como vantagens da sensibilidade analítica destacam-se:
• menor susceptibilidade aos fatores de amplificaç ão do sinal • seu valor independe das unidades de medida de s. E como desvantagem temos: • dependência da concentração (C), pois s pode var iar com C Limite de Detecção O sinal mínimo distinguível, Sm, do branco é dado por:
Sm = SMbr + k sbr (k = 3 com 95% de confiança*) onde SMbr e sbr são o sinal médio e o desvio-padrão das medidas do branco , respectivamente.
12
Determinação Experimental de S mvRealizam-se 20 a 30 medidas do branco para obter sbr . Por fim, o valor de Cm ou CD, definido quantitativamente como limite de detecção em termos de concentração, é encontrado pela expressão CD = (Sm - SMbr) / b1 = 3 sbr / b1
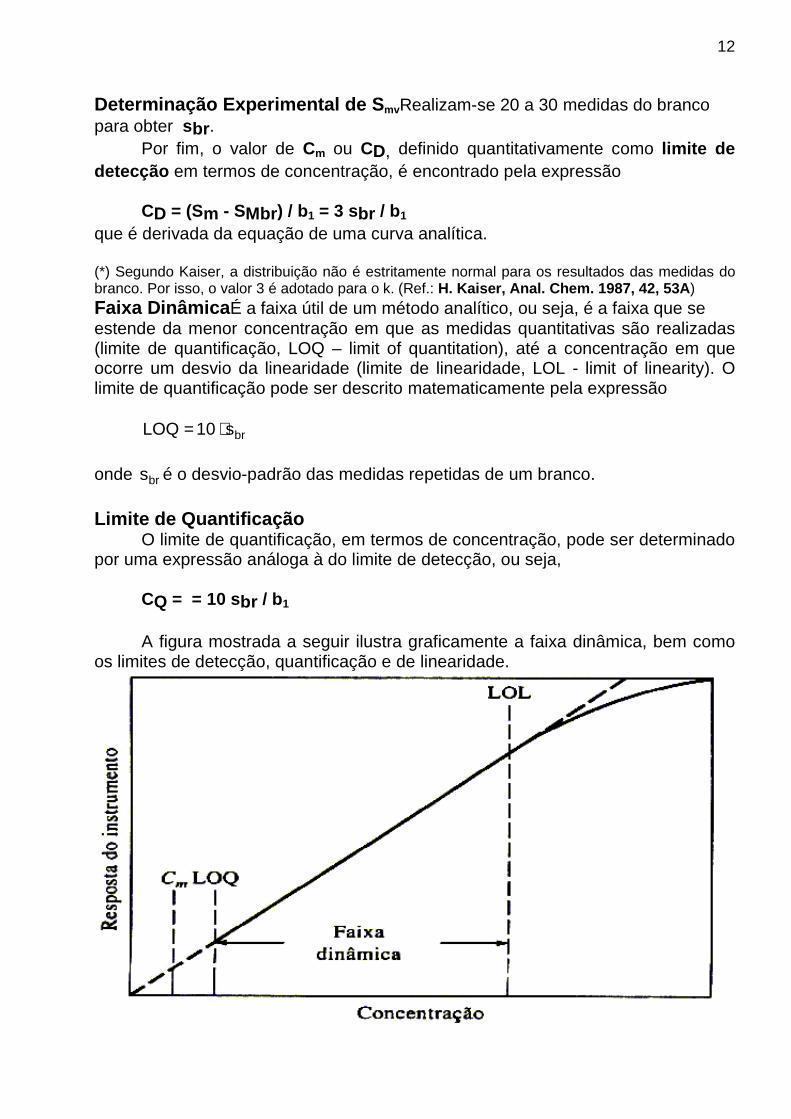
que é derivada da equação de uma curva analítica. (*) Segundo Kaiser, a distribuição não é estritamente normal para os resultados das medidas do branco. Por isso, o valor 3 é adotado para o k. (Ref.: H. Kaiser, Anal. Chem. 1987, 42, 53A ) Faixa Dinâmica É a faixa útil de um método analítico, ou seja, é a faixa que se estende da menor concentração em que as medidas quantitativas são realizadas (limite de quantificação, LOQ – limit of quantitation), até a concentração em que ocorre um desvio da linearidade (limite de linearidade, LOL - limit of linearity). O limite de quantificação pode ser descrito matematicamente pela expressão
brs10LOQ ⋅= onde brs é o desvio-padrão das medidas repetidas de um branco. Limite de Quantificação O limite de quantificação, em termos de concentração, pode ser determinado por uma expressão análoga à do limite de detecção, ou seja, CQ = = 10 sbr / b1
A figura mostrada a seguir ilustra graficamente a faixa dinâmica, bem como os limites de detecção, quantificação e de linearidade.

13
Seletividade Para avaliar quantitativamente a influência dos interferentes
químicos, considere uma amostra que contém um analito A sujeita aos interferentes B e C . Então o sinal instrumental total é dado por
S = mA CA + mB CB + mC CC + Sbronde: - CA, CB e CC são as concentrações das espécies A, B e C - mA, mB e mC são suas sensibilidades de calibração
- Sbr é o sinal do instrumento para o branco Coeficiente de Seletividade O coeficiente de seletividade para A com relação a i (interferente) , ki,A , é dado por:
ki,A = mi / mA
de modo que S = mA (CA + kB,A CB + kC,A CC + ... + ki,A Ci) + Sbr
RADIAÇÃO ELETROMAGNÉTICA - REM
LUZ
Informação química
Qualitativa Quantitativa
MÉTODOS ESPECTROMÉTRICOS - Introdução
• Conceitos, fundamentos e origem da informação • Instrumentação: meio e qualidade da informação • Tratamento de dados: interpretação e extração de informação relevante

14
O que é radiação eletromagnética?
⇒ “É uma forma de energia que se propaga de um ponto a outro em um meio material e pode apresentar características ondulatórias ou corpusculares ”
- Características Ondulatórias - Interferência, reflexão, refração e polarização. - Características Corpusculares - Absorção e emissão da REM por espécies químicas. Propriedades Ondulatórias da REM.
Como onda, a REM compõe-se de um vetor elétrico , E, e um vetor magnético , H que oscilam senoidalmente em planos perpendiculares entre si , e também à direção de propagação da onda. Veja a figura mostrada a seguir:
Propagação da Radiação Eletromagnética
Parâmetros Ondulatórios. O movimento ondulatório é caracterizado pelos seguintes parâmetros: - comprimento de onda ( λλλλ) – distância linear entre dois pontos consecutivos em fase (por exemplo, dois máximos ou dois mínimos da onda); - período (p) – é o intervalo de tempo, em segundos, requerido para dar passagem a dois pontos consecutivos em fase (dois máximos, por exemplo) através de um ponto fixo no espaço; - freqüência ( νννν) – número de ondas que passam por um ponto fixo no espaço por segundo (νννν = 1 / p e tem como unidade o s-1, ciclos por segundo ou hertz (Hz));

15
- velocidade da onda (v i ) – produto da freqüência pelo comprimento de onda: v i = νννν⋅⋅⋅⋅λλλλi (i = meio material qualquer). No vácuo a velocidade de uma onda independe de νννν e alcança o seu valor máximo (c = 3 x 10 8 m/s ); - índice de refração (n i) - é o fator segundo o qual a velocidade da luz é reduzida quando ela se propaga no vácuo e passa a se propagar em um meio material i. Além disso, n i = c / v i de modo que nsólidos > n líquidos > ngases
- amplitude (A) – é a altura máxima da onda; - potência radiante (P) – é a energia que alcança uma dada área do detector por segundo. P pode ser relacionado ao quadrado de A. Propriedades Corpusculares da REM. Para explicar certas interações da REM com o meio material, tais como:
♦ absorção e emissão de radiação por espécies químicas (princípio dos métodos espectroanalíticos); ♦ o efeito fotoelétrico;
passou-se a tratar a REM como constituída de partículas, denominadas de fótons. A energia de um fóton é dado pela equação de Planck:
E = hνννν onde: ♦♦♦♦ h é a constante de Planck (h = 6,6256 x 10-34 J•s)
♦♦♦♦ νννν é freqüência de radiação (em s-1 ou Hz) Se a REM se propaga no vácuo, temos:
E = h c/ λλλλ onde:
♦♦♦♦ c é a velocidade de propagação da REM no vácuo; ♦♦♦♦ λλλλ é o comprimento de onda (1 nm = 10-9 m = 103 pm)
OBS: Para as radiações no visível , ultravioleta e infravermelho , a velocidade de propagação no ar varia de ± 0,1% da velocidade no vácuo. Assim, pode-se usar a equação E = h νννν = h c/λλλλ para interrelacionar νννν, λλλλ e c com a energia de um fóton. INTERFERÊNCIAS ENTRE ONDAS ELETROMAGNÉTICAS As interferências que podem ocorrer entre as ondas eletromagnéticas podem ser: ♦ Construtivas ⇒ quando aumenta amplitude (caso a).
♦ Destrutivas ⇒ quando diminui a amplitude (caso b). OBS: Se ocorrer um cancelamento, a interferência destrutiva é total (caso c).
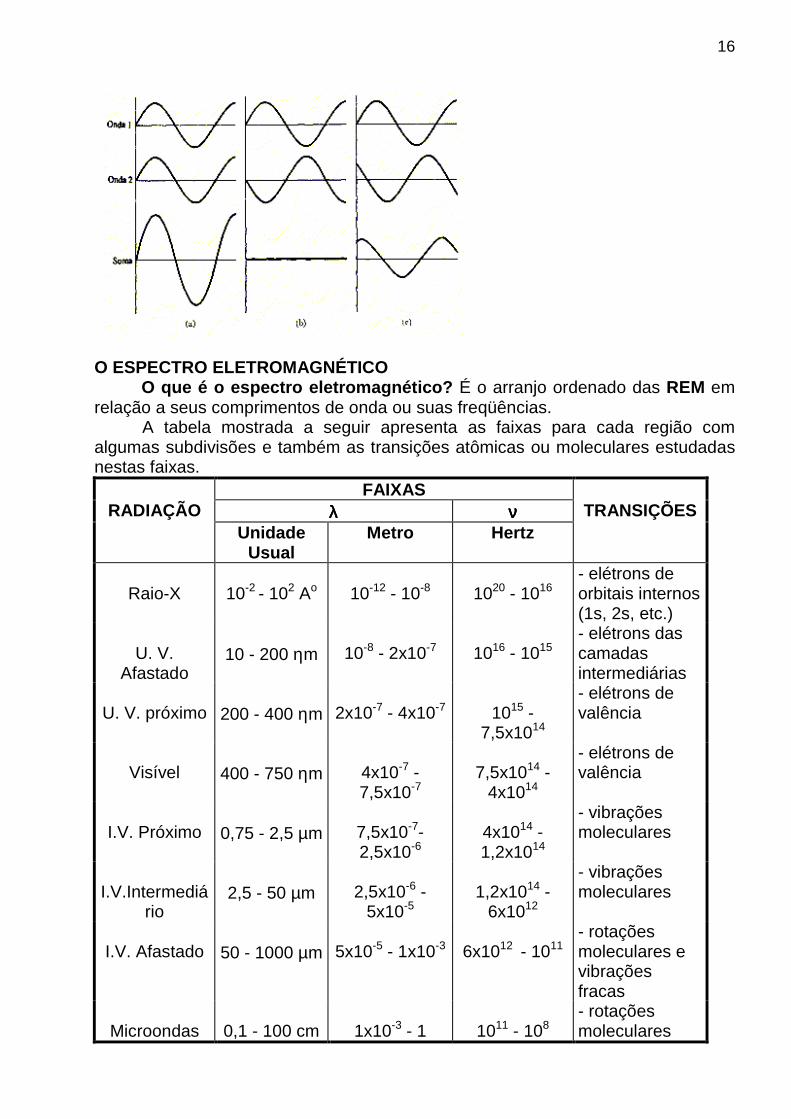
16
O ESPECTRO ELETROMAGNÉTICO O que é o espectro eletromagnético? É o arranjo ordenado das REM em relação a seus comprimentos de onda ou suas freqüências.
A tabela mostrada a seguir apresenta as faixas para cada região com algumas subdivisões e também as transições atômicas ou moleculares estudadas nestas faixas.
FAIXAS RADIAÇÃO λλλλ νννν TRANSIÇÕES
Unidade Usual
Metro Hertz
Raio-X
10-2 - 102 Ao
10-12 - 10-8
1020 - 1016
- elétrons de orbitais internos (1s, 2s, etc.)
U. V.
Afastado
10 - 200 ηm
10-8 - 2x10-7
1016 - 1015
- elétrons das camadas intermediárias
U. V. próximo
200 - 400 ηm
2x10-7 - 4x10-7
1015 -
7,5x1014
- elétrons de valência
Visível
400 - 750 ηm
4x10-7 - 7,5x10-7
7,5x1014 -
4x1014
- elétrons de valência
I.V. Próximo
0,75 - 2,5 µm
7,5x10-7-2,5x10-6
4x1014 - 1,2x1014
- vibrações moleculares
I.V.Intermediá
rio
2,5 - 50 µm
2,5x10-6 -
5x10-5
1,2x1014 -
6x1012
- vibrações moleculares
I.V. Afastado
50 - 1000 µm
5x10-5 - 1x10-3
6x1012 - 1011
- rotações moleculares e vibrações fracas
Microondas
0,1 - 100 cm
1x10-3 - 1
1011 - 108
- rotações moleculares

17
OUTROS CONCEITOS ASSOCIADOS À RADIAÇÃO ELETROMAGNÉT ICA
i) Radiação monocromática - é aquela que contém um único λλλλ. ii) Radiação policromática - contém vários comprimentos de onda λλλλ. iii) Cores primárias da radiação visível - São elas: verde, vermelha e azul. Essas cores originam todas as outras por meio de misturas de acordo com o sistema de adição de cores. iv) Cores secundárias - Resultam da cores primárias combinadas duas a duas em igual intensidade, ou seja, magenta = vermelha + azul amarelo = vermelha + verde ciano = verde + azul v) Cor oposta a uma dada cor secundária - É a cor primária que não entra na composição da secundária.
- a cor verde é oposta ao magenta - a vermelha é oposta ao ciano - a cor azul é oposta ao amarelo
vi) Cor branca - Resulta da combinação balanceada máxima de radiações nas faixas do verde, vermelho e azul, isto é, Cor branca = verde + vermelho + azul com máxima intensidade. Ou ainda a cor branca pode ser dada pela combinação de qualquer cor secundária com sua oposta, ou seja, Cor branca = magenta + verde = amarelo + azul = ciano + vermelho. vii) Cor complementar ⇒ A tabela mostrada a seguir fornece:
♦ as cores da radiação visível em seus intervalos de λλλλ. ♦ e suas cores complementares .
Intervalo aproximado de
λλλλ(nm)
Cor Complemento
400 - 465 violeta verde-amarelo
465 - 482 azul amarelo

18
482 - 487 azul-esverdeado alaranjado
487 - 493 turquesa vermelho-alaranjado
493 - 498 verde-azulado vermelho
498 - 530 verde vermelho-púrpura
530 - 559 verde-amarelado púrpura-
avermelhado
559 - 571 amarelo-verde púrpura
571 - 576 amarelo-esverdeado violeta
576 - 580 amarelo azul
580 - 587 laranja-amarelado azul
587 - 597 alaranjado azul-esverdeado
597 - 617 laranja-avermelhado turquesa
617 – 780 Vermelho turquesa
Como surgem as cores complementares? ⇒ Surgem devido ao fato de que quando um feixe de luz branca (radiações
com todos os λλλλ) incide sobre uma superfície contendo uma substância absorvente, a radiação emergente será um complemento da radiação branca menos a radiação absorvida pela substância.
⇒ Assim, a cor de uma solução colorida que nossos olhos percebem é uma
cor complementar da radiação absorvida. ⇒ Por exemplo, a cor vermelho-púrpura das soluções de KMnO 4 encontra-se
relacionada a uma absorção mais intensa desta substância na região verde (λλλλ = 525 nm ).
⇒ A cor azul-turquesa das soluções de CuSO4•5H2O (AZUL PISCINA ) está
relacionada a uma absorção mais intensa desta substância na região vermelha.
⇒ OBS.: Cor Complementar é um conceito útil em espectrometria absorção
molecular UV-VIS .

19
ESPECTROMETRIA ATÔMICA ÓPTICA Baseia-se na propriedade dos átomos ou íons monoatômicos de absorverem
ou emitirem radiação eletromagnética UV-Vis quando excitados.“O registro gráfico do resultado desse fenômeno é denominado “espectro” ♦ Espectrometria de emissão atômica; ♦ Espectrometria de fluorescência atômica; ♦ Espectrometria de absorção atômica;TIPOS DE ESPECTROS♦ Espectro de raias (ou linhas)*-produzidos por átomos ou íons monoatômicos gasosos♦ Espectro de bandas - gerados por moléculas neutras, íons moléculas e radicais· ♦ Espectro contínuo - produzidos pelos sistemas condensados (ex. sólido incandescente) (* ) Espectro de raias ⇒ de interesse da espectrometria atômica.ORIGEM DOS ESPECTROS ATÔMICOS A figura abaixo mostra uma ilustração do espectro de emissão dos metais alcalinos.
Para uma melhor compreensão de como se originam os espectros acima considere, por exemplo, o caso do sódio cujo diagrama de energias dos orbitais atômicos é mostrado na figura a seguir.
Os átomos gasosos são excitados (térmica ou eletricamente) levando o(s) seu(s) elétron(s) mais externo(s) a níveis energéticos superiores. Quando retornam aos estados de mais baixa energia emitem radiações na região UV-VIS. A Figura a seguir mostra um diagrama dos níveis energéticos para o Na e as possíveis transições.
A emissão de uma raia, por exemplo, é o resultado da transição de um elétron de um nível de energia mais alto para um mais baixo. Além disso, cada raia envolve dois termos espectroscópicos, um do nível energético mais baixo e outro mais alto. Assim, as raias D (dupleto) do sódio são originadas pelas transições:
3 2S1/2 ← 3 2P1/2 (589,6 nm) 3 2S1/2 ← 3 2P3/2 (589,0 nm)
A razão para a formação da raia D do sódio será explicada mais adiante por ocasião da discussão sobre o acoplamento spin-órbita.
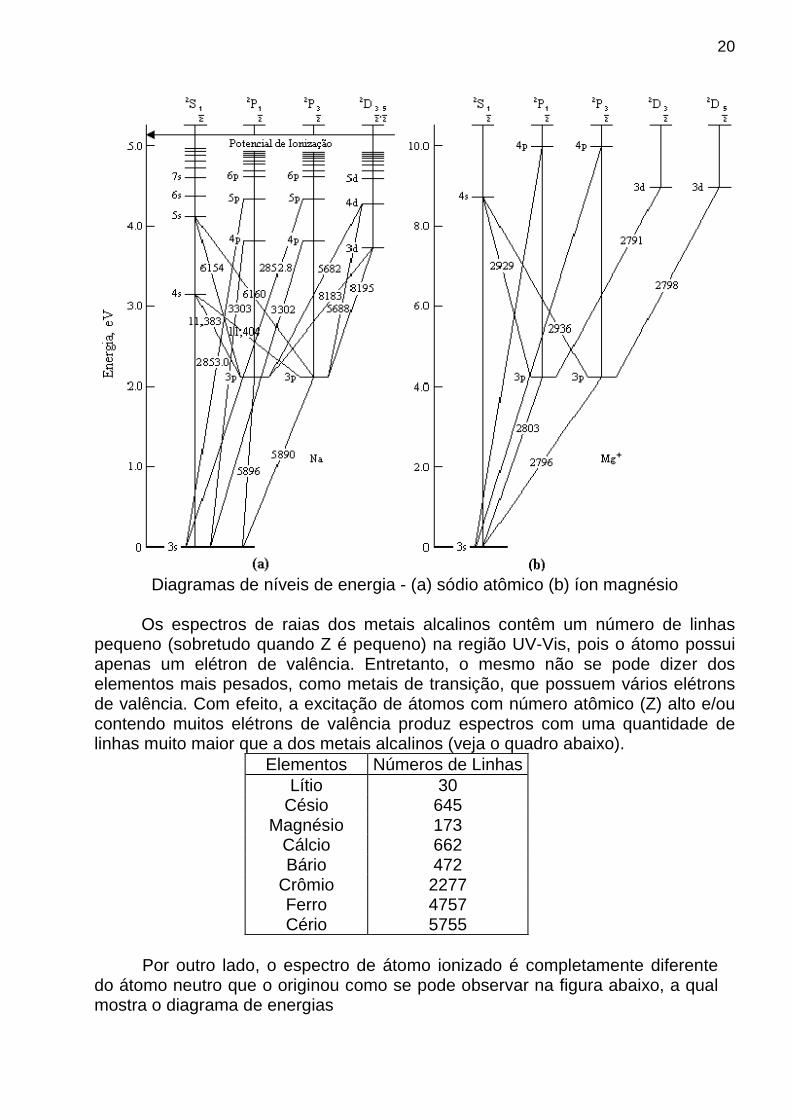
20
Diagramas de níveis de energia - (a) sódio atômico (b) íon magnésio
Os espectros de raias dos metais alcalinos contêm um número de linhas pequeno (sobretudo quando Z é pequeno) na região UV-Vis, pois o átomo possui apenas um elétron de valência. Entretanto, o mesmo não se pode dizer dos elementos mais pesados, como metais de transição, que possuem vários elétrons de valência. Com efeito, a excitação de átomos com número atômico (Z) alto e/ou contendo muitos elétrons de valência produz espectros com uma quantidade de linhas muito maior que a dos metais alcalinos (veja o quadro abaixo).
Elementos Números de Linhas Lítio 30
Césio 645 Magnésio 173
Cálcio 662 Bário 472
Crômio 2277 Ferro 4757 Cério 5755
Por outro lado, o espectro de átomo ionizado é completamente diferente
do átomo neutro que o originou como se pode observar na figura abaixo, a qual mostra o diagrama de energias

21
Se a ionização se deu por perda de um só elétron, o espectro produzido pelo íon assemelha-se muito ao do átomo neutro com Z inferior em uma unidade, porém apresenta as linhas em λ’s menores, a exemplo do Mg+ e Na discutido a seguir.
Diagrama de energias do Mg no estado singlete.
Os espectros dos átomos e íons com mesma configuração eletrônica (isoeletrônicos) são semelhantes, porém as raias aparecem em comprimentos de ondas diferentes. De fato, ao compararmos os diagramas de energias das espécies isoeletrônicas Na (Z=11) e Mg+ (Z=12), verificamos que a energia necessária para promover a transição eletrônica 3s → 3p no Mg+ é cerca de duas vezes a requerida no caso do Na. Embora as espécies tenham a mesma estrutura eletrônica (e assim o mesmo no de elétrons no cerne responsáveis pela blindagem da carga nuclear), o núcleo de Mg+ exerce uma maior atração sobre os elétrons em virtude de sua maior carga nuclear. Conseqüentemente, isso torna mais difícil a transição do elétron do orbital 3s para o 3p, necessitando de uma maior energia (menor λ). RAIA DE RESSONÂNCIA A raia de ressonância corresponde à raia de absorção ou de emissão mais intensa associada à transição de um elétron de valência para a um nível energético imediatamente superior que apresente uma maior probabilidade de transição.

22
Para o Na a raia de ressonância corresponde à chamada raia D (dupleto) que aparece em torno de 590 nm no espectro, cuja emissão é responsável pela cor amarelo-alaranjado das lâmpadas de sódio. A espectrometria atômica utiliza, principalmente, as raias de ressonância embora, às vezes, são usadas outras raias menos intensas ou eventualmente bandas, a exemplo da radiação emitida pelo radical CaOH em chamas frias de ar-gás natural por amostras contendo cálcio. ESTRUTURA FINA DOS ESPECTROS ATÔMICOS – Acoplamento spin-órbita O acoplamento spin-órbita resulta da interação entre o momento magnético do spin (campo magnético do spin eletrônico) e o momento angular orbital (campo magnético devido ao movimento angular do elétron em torno do núcleo). Quando os dois campos têm o mesmo sentido a interação é repulsiva, a qual aumenta a energia eletrônica. Caso contrário, a interação entre os dois campos é atrativa e a energia eletrônica diminui. No caso do Na, por exemplo, quando o de valência é excitado para o orbital 3p experimenta esse acoplamento que desdobra o nível de energia dos orbitais 3p em dois muito próximos (veja o diagrama mostrado anteriormente). Este tipo de interação ocorre tipicamente em átomos contendo elétron desemparelhado em orbitais com momento angular orbital diferente de zero (orbitais p (l=1) principalmente). De um modo geral, observa-se que a intensidade do acoplamento spin-órbita depende: - das orientações relativas de ambos os momentos; - da carga nuclear (Ze). OBS.: (i) No H (Z=1), o acoplamento é muito pequeno em virtude da baixa carga nuclear!!(ii) Os termos espectroscópicos e o acoplamento spin-órbita são discutidos em: P.W.Atkins –“Físico-Química”, Vol. 2, 6ª Edição, LTC, RJ, 1999. ALARGAMENTO DAS RAIAS ESPECTRAIS As raias deveriam ser rigorosamente monocromáticas e dada por:
∆E = EE - EF = hc/λ→ _____________ Eexc ∆E
λ = hc/∆E ____________ Efund
Contudo, a raia se apresenta, na realidade, como uma banda estreita com uma determinada largura, conforme mostra a figura abaixo:

23
O alargamento da raia pode originar-se de três efeitos: - Princípio da Incerteza (Alargamento Natural); - Efeito Doppler; - Efeito de Pressão. Alargamento natural Se os átomos pudessem permanecer um tempo virtualmente infinito nos estados fundamental e excitado, a incerteza das energias dos estados seria desprezível e a transição estaria associada a um único λ. Entretanto, a incerteza da energia de cada estado é complementar ao seu tempo de vida, ou seja, τi .∆Ei = τi.h ∆vi = h/2π ⇒ ∆vi = (1/2π) (1/τi) Assim, a largura natural de uma raia é determinada pelos tempos de vida médios dos estados fundamentais e excitados envolvidos na transição. Conseqüentemente, a uma transição qualquer se associa, efetivamente, a emissão de uma banda cuja meia-largura (∆ν ou ∆λN) (largura natural) é dada por:
∆νN = ∆νq + ∆νp = (1/2π) (1/τi + 1/τj) ou em termos de λ,
∆λN = (λ2/2πc) (1/τi + 1/τj) onde, τi e τj são os tempos de vida médio dos estados i e j envolvidos na transição. As raias mais estreitas são as raias de ressonância. Por exemplo, o ∆λN para a raia de ressonância do mercúrio (253,7 nm) é de 0,00003 nm. Alargamento Devido ao Efeito Doppler A freqüência da radiação absorvida ou emitida por um átomo que se move rapidamente aumenta se o átomo se aproxima do transdutor (detector) e diminui quando ele se afasta do transdutor. Esse fenômeno é conhecido como efeito ou deslocamento Doppler. São típicos alargamentos Doppler na faixa: 0,001 a 0,005 ηm. Alargamento Devido ao Efeito de Pressão Ocorre devido às pequenas variações de energia decorrentes de colisões entre átomos absorvedores ou emissores de radiação e outros átomos, radicais, íons, etc, presentes no meio aquecido.Para concentrações baixas, a meia-largura de uma raia está relacionada, principalmente, ao efeito Dopper; porém no caso de altas concentrações prevalece o efeito de pressão. O efeito Doppler e de pressão estão relacionado à forma de uma raia no ponto de emissão. Contudo, a radiação emitida tem que atravessar a região de excitação até atingir o detector. Assim, a forma da raia pode sofrer modificações

24
relacionadas com os problemas de auto-absorção e auto-reversão discutidos mais adiante. A partir da avaliação dos comprimentos de onda das radiações emitidas (observados nas raias do espectro) é possível descobrir a identidade dos átomos emissores (análise qualitativa elementar). As medidas da intensidade das radiações emitidas (usadas na calibração) fornecem informações para a análise quantitativa elementar.
ESPECTROMETRIA DE EMISSÃO ATÔMICA Fundamentos teóricos
Este método baseia-se na introdução de uma amostra em solução em uma chama ou plasma na forma de um aerossol.A chama ou plasma induz a amostra a emitir radiação eletromagnética na região UV-VIS; a intensidade da luz emitida é proporcional à concentração desta espécie química de interesse, ou seja:
I = k C
onde ♦ C ⇒ concentração do analito nas soluções-padrão (ou amostra) ♦ k ⇒ coeficiente de proporcionalidade que depende da:
- estrutura eletrônica do átomo do analito; - probabilidade de transição associada à raia analítica; - temperatura da fonte de atomização e excitação; - eficiência da atomização; - fatores instrumentais de amplificação.
Na medida da intensidade de uma determinado analito tem-se os seguintes
processos representados diagramaticamente na figura abaixo:
Efeito da Temperatura da Chama na Emissão Atômica
A temperatura da chama ou plasma exerce um papel fundamental na relação entre o número de espécies excitadas e não excitadas. A magnitude deste efeito pode ser derivada a partir da equação de Boltzmann, que é escrita na seguinte forma:

25
∆−=kT
Eexp.
PP
NN ee
00
onde: ♦ Ne e N0 são os números de espécies no estado excitado e no estado
fundamental; ♦ Pe e P0 são os fatores estatísticos que são determinados pelo número
de orbitais em cada nível; ♦ ∆∆∆∆E é a diferença de energia entre os níveis; ♦ k é a constante de Boltzmann (1,38 x 10-23 Joules/Kelvin); ♦ T é a temperatura em Kelvin.
A tabela abaixo apresenta os valores da relação Ne / N0 para as raias de ressonância de alguns elementos a diferentes temperaturas.
Nj/No Raia de Ressonância gj/g0 2000k 3000k 4000k
Cs 852,1 ηm 2 4,44.10-4 7,24.10-3 2,98.10-2 Na 589,0ηm 2 9,86.10-6 5,88.10-4 4,44.10-3 Ca 422,7ηm 3 1,21.10-7 3,69.10-5 6,03.10-4 Zn 213,9ηm 3 7,20.10-15 5,58.10-10 1,48.10-7
Verifica-se que a população de átomos excitados é muito pequena em
relação ao número de átomos no estado fundamental (apenas 0,0001% dos átomos de sódio presentes na amostra são excitados a temperatura de 2000K). Entretanto, esta população aumenta significativamente com um pequeno aumento da temperatura (0,06% à 3000K e 0,4% a 4000K).
Um aumento de 10 Kelvins (2500 para 2510K) na temperatura de emissão relacionada à linha de ressonância do sódio produz um aumento de 4% no número de átomos de sódio excitados. Portanto, os métodos analíticos baseados nas medidas da emissão atômica requerem um controle rigoroso da temperatura de excitação. A CHAMA OU PLASMA A chama ou plasma exerce um papel muito importante na espectrofotometria ou fotometria de emissão atômica. Elas são responsáveis pelas seguintes funções: dessolvatar, vaporizar, atomizar e excitar eletronicamente o átomo em análise . Para cumprir as funções acima a chama ou o plasma deve atingir uma temperatura apropriada, por exemplo, chamas frias (como ar-gás de cozinha, por exemplo) só excitam os alcalinos e alcalinos terrosos. A CHAMA É uma fonte de excitação mais fraca do que o plasma e, normalmente, poucas raias de cada elemento são excitados. A figura abaixo mostra, diagramaticamente, a estrutura de uma chama:
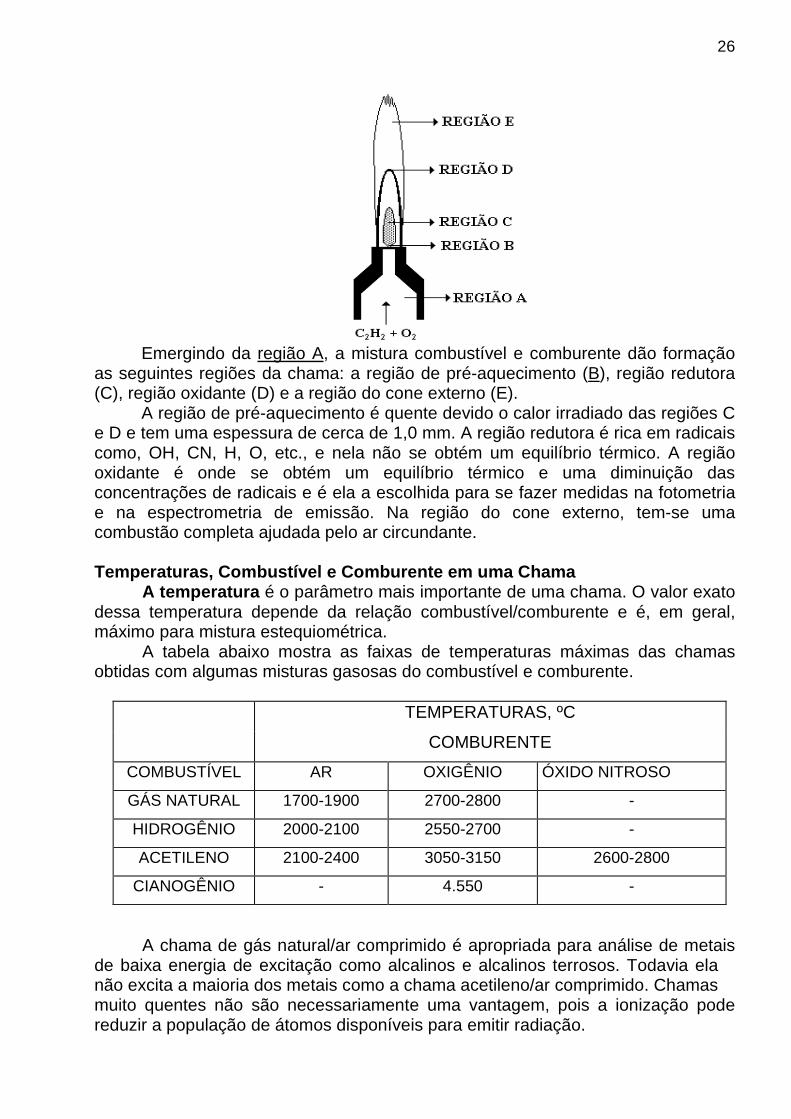
26
Emergindo da região A, a mistura combustível e comburente dão formação as seguintes regiões da chama: a região de pré-aquecimento (B), região redutora (C), região oxidante (D) e a região do cone externo (E). A região de pré-aquecimento é quente devido o calor irradiado das regiões C e D e tem uma espessura de cerca de 1,0 mm. A região redutora é rica em radicais como, OH, CN, H, O, etc., e nela não se obtém um equilíbrio térmico. A região oxidante é onde se obtém um equilíbrio térmico e uma diminuição das concentrações de radicais e é ela a escolhida para se fazer medidas na fotometria e na espectrometria de emissão. Na região do cone externo, tem-se uma combustão completa ajudada pelo ar circundante. Temperaturas, Combustível e Comburente em uma Chama
A temperatura é o parâmetro mais importante de uma chama. O valor exato dessa temperatura depende da relação combustível/comburente e é, em geral, máximo para mistura estequiométrica.
A tabela abaixo mostra as faixas de temperaturas máximas das chamas obtidas com algumas misturas gasosas do combustível e comburente.
TEMPERATURAS, ºC
COMBURENTE
COMBUSTÍVEL AR OXIGÊNIO ÓXIDO NITROSO
GÁS NATURAL 1700-1900 2700-2800 -
HIDROGÊNIO 2000-2100 2550-2700 -
ACETILENO 2100-2400 3050-3150 2600-2800
CIANOGÊNIO - 4.550 -
A chama de gás natural/ar comprimido é apropriada para análise de metais de baixa energia de excitação como alcalinos e alcalinos terrosos. Todavia ela não excita a maioria dos metais como a chama acetileno/ar comprimido. Chamas muito quentes não são necessariamente uma vantagem, pois a ionização pode reduzir a população de átomos disponíveis para emitir radiação.
27
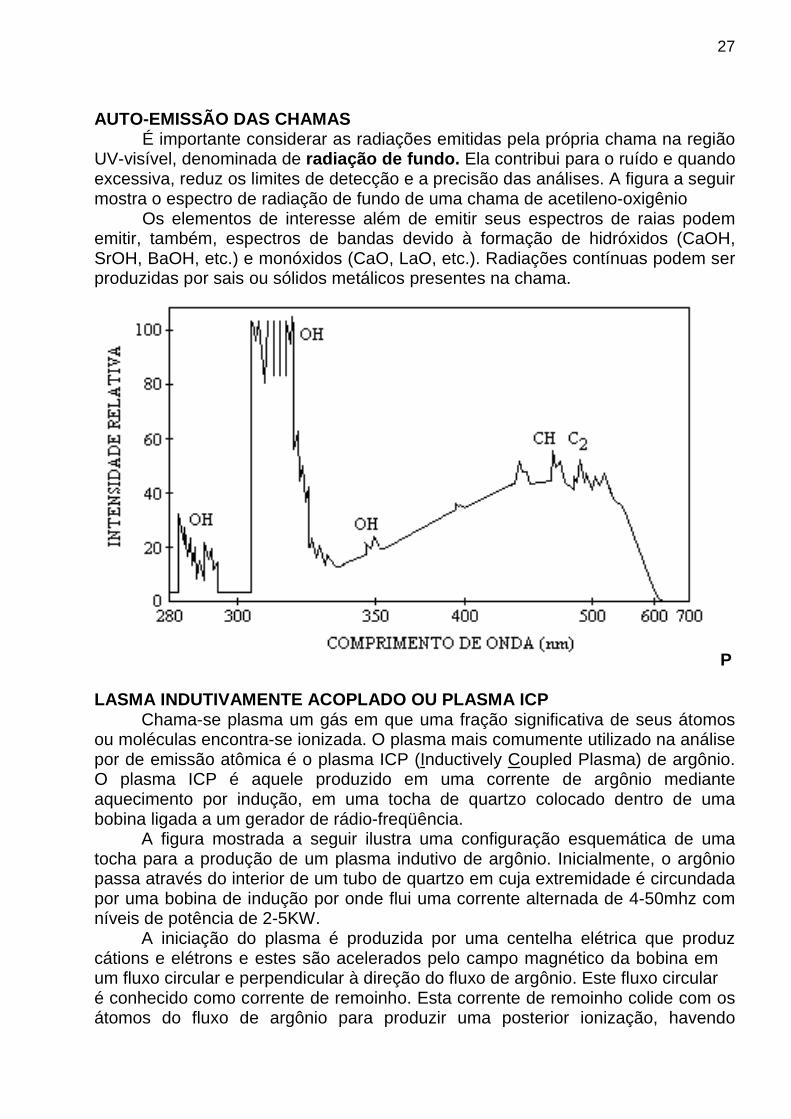
AUTO-EMISSÃO DAS CHAMAS
É importante considerar as radiações emitidas pela própria chama na região UV-visível, denominada de radiação de fundo. Ela contribui para o ruído e quando excessiva, reduz os limites de detecção e a precisão das análises. A figura a seguir mostra o espectro de radiação de fundo de uma chama de acetileno-oxigênio
Os elementos de interesse além de emitir seus espectros de raias podem emitir, também, espectros de bandas devido à formação de hidróxidos (CaOH, SrOH, BaOH, etc.) e monóxidos (CaO, LaO, etc.). Radiações contínuas podem ser produzidas por sais ou sólidos metálicos presentes na chama.
P LASMA INDUTIVAMENTE ACOPLADO OU PLASMA ICP Chama-se plasma um gás em que uma fração significativa de seus átomos ou moléculas encontra-se ionizada. O plasma mais comumente utilizado na análise por de emissão atômica é o plasma ICP (Inductively Coupled Plasma) de argônio. O plasma ICP é aquele produzido em uma corrente de argônio mediante aquecimento por indução, em uma tocha de quartzo colocado dentro de uma bobina ligada a um gerador de rádio-freqüência. A figura mostrada a seguir ilustra uma configuração esquemática de uma tocha para a produção de um plasma indutivo de argônio. Inicialmente, o argônio passa através do interior de um tubo de quartzo em cuja extremidade é circundada por uma bobina de indução por onde flui uma corrente alternada de 4-50mhz com níveis de potência de 2-5KW. A iniciação do plasma é produzida por uma centelha elétrica que produz cátions e elétrons e estes são acelerados pelo campo magnético da bobina em um fluxo circular e perpendicular à direção do fluxo de argônio. Este fluxo circular é conhecido como corrente de remoinho. Esta corrente de remoinho colide com os átomos do fluxo de argônio para produzir uma posterior ionização, havendo

28
aquecimento por efeito Joule e a formação do plasma. As temperaturas no plasma variam 6000 a 10000K.
O isolamento térmico do plasma para evitar superaquecimento do cilindro de quartzo é obtido com uma corrente de argônio introduzida tangencialmente. Este fluxo serve também para centralizar e estabilizar o plasma, dando uma forma toroidal para freqüências em torno de aproximadamente 30MHz. As amostras em solução são aspiradas pneumaticamente e, em forma de aerossol, atinge o plasma. A aspiração pneumática é produzida por um fluxo de argônio, que flui no cone interno da tocha e alimenta o plasma. Ele também é responsável pela formação do aerossol. Um fluxo suporte de argônio é, às vezes, também usado para alimentar o plasma. Vazões típicas de argônio são: 1L/min para aspirar e transportar a amostra, 0-1L/min para o fluxo de suporte e 15L/min para o fluxo de esfriamento. As propriedades físicas e químicas do plasma ICP oferecem algumas vantagens sobre as chamas. um ambiente químico mais limpo. temperaturas mais altas que dissociam completamente os compostos refratários. a faixa linear de concentração é 4 ou mais vezes maior. o espectro é rico em linhas atômicas e iônicas, o que dá uma maior possibilidade de escolha da linha analítica.

29
um baixo sinal de radiação de fundo, o que permite uma maior relação sinal/ruído e um baixo limite de detecção (na faixa de ppb).
O plasma tem também uma radiação de fundo correspondente às raias do argônio, bandas OH e bandas fracas de NO, NH, CN e C2. Todavia, existe uma zona de 1 a 3 cm acima da bobina de indução, onde o plasma é levemente transparente. Esta é a zona de observação analítica. Para muitos elementos a linha iônica é muito mais intensa do que a linha atômica. Para o cálcio a linha de ressonância atômica (422,7ηm) tem no plasma intensidade praticamente desprezível em relação às linhas iônicas 394,4 e 396,2ηm. Este fenômeno é também observado em outros elementos como Ba, Be, Fe, Mg, Mn, Sr, Ti e V, onde as linhas iônicas fornecem um melhor limite de detecção. INSTRUMENTOS PARA MEDIDAS DE EMISSÃO EM CHAMA
Eles apresentam os seguintes componentes essenciais: ♦ Reguladores de pressão e fluxômetros para controle da pressão e
vazão dos gases que alimentam a chama; ♦ Nebulizador-Combustor-Atomizador para introduzir a amostra na
chama em forma de aerossol (nebulizar), dessolvatar, sublimar, atomizar e excitar eletronicamente o átomo ou íon atômico em análise;
♦ Sistema óptico a base de filtro ou monocromador para isolar a radiação desejada;
♦ Detector associado a algum tipo de medidor ou amplificador eletrônico.
A figura abaixo mostra esquematicamente, os componentes básicos de um espectrofotômetro de emissão em chama.
NEBULIZADORES-COMBUSTORES-ATOMIZADORES
Na espectrofotometria de emissão atômica são conhecidos comumente dois tipos de nebulizador-queimador-atomizador:
♦ mistura prévia ♦ consumo total .
Nebulizador-Queimador-Atomizador de Mistura Prévia

30
Eles são caracterizados pela produção do aerossol em uma câmara de condensação para reter as gotículas maiores.
A figura a seguir ilustra um nebulizador-queimador-atomizador de mistura prévia de fluxo concêntrico.
Uma corrente de gás oxidante aspira por ação pneumática (efeito Bernoulli)
a amostra e esta é nebulizada numa câmara, onde, então, se mistura com o gás combustível; as gotículas maiores são recolhidas no fundo da câmara e descartada pelo dreno; somente as partículas menores alcançam a chama. Isto faz com que apenas 5 a 10% da amostra nebulizada atinjam a chama. Nebulizador-Queimador-Atomizador de Consumo Total
É caracterizado pela introdução do aerossol diretamente na chama. No nebulizador-queimador-atomizador de consumo total o aerossol é formado diretamente na chama que é produzida pelos gases combustível e oxidante conduzidos através de canais concêntricos, um em torno do capilar de acesso da solução, para o oxidante e o outro mais externo para o combustível. A corrente do oxidante, ao passar pelo orifício de saída do canal interno, cria uma sucção suficiente para forçar a solução a emergir pelo capilar interno na chama. A figura abaixo mostra um nebulizador-queimador-atomizador de consumo total.

31
Neste dispositivo toda a amostra atinge a chama, porém gotículas maiores atravessam a chama sem serem dessolvatadas. Além do mais ele produz uma chama turbulenta e instável e um sinal analítico muito ruidoso. SISTEMA ÓPTICO Qual a função do sistema óptico?
Sua função é recolher a luz emitida pela chama, isolar a parte interessada (radiação de emissão do analito) e focar esta última sobre o detector. FOTÔMETROS DE EMISSÃO EM CHAMA
Os fotômetros de chama têm suas limitações: usam normalmente chama de baixa temperatura como fonte de excitação. São instrumentos relativamente simples, construídos quase sempre para determinação de Li, Na, K, Ca e Mg . INTERFERÊNCIAS NA ESPECTROFOTOMETRIA DE EMISSÃO EM CHAMA
São problemas que, de alguma maneira, prejudicam as medidas dos sinais de emissão do analito e podem ser classificadas em três categorias:
♦ espectrais; ♦ químicas; ♦ físicas.
INTERFERÊNCIAS ESPECTRAIS
Essas interferências encontram-se relacionadas com as radiações de outros componentes que se inserem na faixa de comprimentos de onda isolada pelo instrumento para o elemento de interesse (analito).
Podem ocorrer principalmente os seguintes tipos de interferência espectral : ♦ sobreposição espectral direta de raias ou bandas;♦ sobreposição por
emissão de radiação contínua;♦ espalhamento de luz;♦ auto-absorção;♦ emissão de radiação de fundo (auto-emissão)Sopreposição Espectral Direta de Raias ou Bandas

32
Ocorre quando a raia analítica (raia do analito) é sobreposta por uma raia de um outro átomo emissor ou por uma banda emitida por uma espécie molecular presente na fonte excitadora.
Como exemplo de sobreposição espectral direta de raia tem-se a sobreposição das linhas 213,858nm do Ni e a 213,851 nm do Cu sobre a linha 213,856 do Zn. Este problema é sério em uma determinação de traços de Zn em amostras contendo Ni e Cu em alta concentração (exemplo: liga metálica Ni-Cu contendo Zn como impureza).
Por outro lado, temos a sobreposição da raia D (589,5 nm) do Na pela banda de CaOH (com centro em 622 nm), como exemplo de interferência direta de banda. Sobreposição por emissão de radiação contínua Promovida por sistemas condensados presentes na fonte de excitação e por alguns elementos devido à recombinação de íons positivos e elétrons livres. Por exemplo: Na+ + e- → Na + hν (contínua) O espectro contínuo do sódio vai de 360 a 602hm e do potássio de 340 a 570hm. Este continuo quando presente vai interferir em todas as raias analíticas presentes nesta região. Espalhamento de luz Causada por partículas presentes na fonte de excitação, reduzindo a intensidade da luz que atinge o detector. Ocorre principalmente em chamas frias onde podem ser produzidas espécies químicas refratárias. Auto-Absorção
Átomos da mesma espécie analítica, presentes na região menos energética da fonte de excitação, encontram-se em estados eletrônicos menos energéticos e são capazes de absorver a radiação emitida na região mais energética. Este fenômeno é chamado de auto-absorção e é responsável pelo enfraquecimento da intensidade da radiação emitida pelo analito. Emissão de Fundo ou Auto-Emissão da Chama
Conforme vimos antes, corresponde às radiações emitidas pela própria chama na região UV-VIS. Ela contribui para o ruído e quando excessiva reduz os limites de detecção e a precisão das medidas. Para eliminar essa interferência utiliza-se o branco para ajustar o zero do aparelho antes de efetuar as medidas dos sinais de emissão dos padrões e amostras. A figura abaixo mostra o espectro de radiação de fundo de uma chama de acetileno-oxigênio INTERFERÊCIAS QUÍMICAS
São aquelas interações químicas entre o analito e outras espécies presentes na solução da amostra que afetam o sinal do analito. Elas normalmente ocorrem através da formação de um composto termicamente estável (refratário) envolvendo o analito. Um exemplo típico de interferência química é a forte depressão do sinal de emissão do cálcio em amostras contendo íons fosfato (PO4
3-), aluminato (AlO2-),
sulfato (SO22-), silicato (SiO4
4-). Esta interferência pode ser eliminada usando agentes mascarantes .
Agentes Mascarantes

33
São espécies químicas adicionadas nas amostras e nas soluções-padrão que tem por objetivo mascarar ou eliminar a interferência química produzida por outras espécies presentes na chama. Os agentes mascarantes podem ser classificados em dois tipos:
♦ agentes mascarantes libertadores; ♦ agentes mascarantes protetores;
Agente Mascarante Libertador
Os agentes mascarantes libertadores reagem preferencialmente com o interferente químico deixando o elemento de interesse livre para ser sublimado e atomizado na chama. Por exemplo, a adição de zircônio, lantânio ou estrôncio elimina a interferência de fosfato na determinação de cálcio, pois estes elementos formam um composto mais estável com o interferente, liberando o cálcio para a excitação. Agente Mascarante Protetor
Os agentes mascarantes protetores previnem a interferência química por formar espécies químicas com o analito mais estáveis e mais facilmente sublimáveis e dissociáveis. Tem sido mostrado que a presença de EDTA elimina a interferência de alumínio, silício, fosfato e sulfato na determinação de cálcio ao formar a espécie Ca – EDTA (complexo). INTERFERÊNCIAS FÍSICAS Essas interferências compreendem: ♦♦♦♦ ionização do analito ♦♦♦♦ interferência ou efeito de matriz Ionização Se durante a excitação ocorrer a ionização, esta reduz a população de átomos neutros na chama e, conseqüentemente, diminui a intensidade de emissão do analito. A ionização pode ser minimizada pela adição de um supressor de ionização . Supressor de Ionização
O supressor de ionização é uma espécie química facilmente ionizável (Cs, Rb, K, Li, Na), que é adicionada em uma grande quantidade (cerca de 1%) nas amostras e nas soluções-padrão com o objetivo de minimizar a ionização do átomo em análise. A diminuição da ionização pode ser entendida partindo das equações:
M ⇔⇔⇔⇔ M+ + e- e Cs ⇔⇔⇔⇔ Cs+ + e-
Como o césio é facilmente ionizado a pressão parcial de elétrons livres na fonte de excitação aumenta deslocando é deslocado, de acordo com o princípio de Le Chatelier, na direção do aumento da pressão parcial do analito M. Efeito da Auto-Absorção e da Ionização sobre a curv a analítica
A auto-absorção e a ionização podem afetar as curvas analíticas produzindo três segmentos distintos em forma de “S”, conforme mostra a figura abaixo. Assim, para concentrações intermediárias de potássio prevalece uma relação linear. Entretanto, para baixas e altas concentrações, observam-se curvaturas com desvios positivos e negativos.

34
Interferência matricial ou efeito de matriz É a influência das propriedades da matriz da amostra (viscosidade, tensão superficial, pressão de vapor, etc) sobre o processo envolvido na medida do sinal analítico. Como ocorre? Para ilustrarmos como ocorre o efeito de matriz em análise quantitativa por fotometria de emissão em chama, considere o exemplo abaixo:
Suponha uma determinação de Na em uma amostra de MEL DE ABELHA,
usando uma curva de calibração construída com soluções-padrão de Na preparadas em água . COMO RESULTADO DA ANÁLISE, TERÍAMOS: ♦ Um menor valor de concentração de Na que o real seria obtido. Isto ocorre porque o Na no mel encontra-se numa matriz muito mais viscosa que o Na das soluções-padrão de calibração, o que diminuirá a taxa de aspiração no aparelho. ♦ A diminuição taxa de aspiração faz como que a leitura do Na da amostra de mel seja menor que a de uma solução padrão de mesma concentração, causando um problema conhecido como EFEITO DE MATRIZ. ANÁLISE QUANTITATIVA
Os seguintes métodos podem ser utilizados na análise quantitativa por emissão atômica:
♦ Método por curva analítica; ♦ Método do padrão interno; ♦ Método por adições de padrão.

35
Uma vez que o método por curva analítica já foi discutido anteriormente, discutiremos aqui o método do padrão interno e o método por adições de padrão . Método do padrão interno
No método do padrão interno uma quantidade conhecida de uma espécie de referência (o padrão interno) é adicionada nas amostras, nas soluções-padrão e no branco. A curva analítica é construída lançando:
♦ nas ordenadas a razão entre sinal do analito e o sinal do padrão interno;
♦ e nas abcissas a concentração das soluções-padrão do analito. OBSERVAÇÃO:
O padrão interno escolhido deve obedecer as seguintes condições: ♦ deve apresentar propriedades físicas, químicas e espectrais
semelhantes ao analito, de modo que ambos sejam igualmente afetados por flutuações da fonte de excitação (chama);
♦ não deve apresentar interferências químicas e espectrais entre si e com os demais componentes da amostra;
♦ não deve estar presente na amostra e no branco;
♦ a sua concentração nas amostras e nas soluções padrão deve ser da mesma ordem de grandeza e deve estar na faixa linear de concentração;
♦ Os sinais do analito e do padrão interno nas amostras e nas soluções padrão devem ser medidos, preferencialmente, em um espectrofotômetro ou fotômetro de emissão em chama multicanal;
♦ Se o padrão interno for escolhido de modo a ter propriedades físicas, químicas e espectroscópicas similares ao analito, ambos os sinais variam proporcionalmente com a variação das condições experimentais e a utilização da relação dos sinais permite corrigir os erros aleatórios. A figura a seguir mostra como isto é possível.
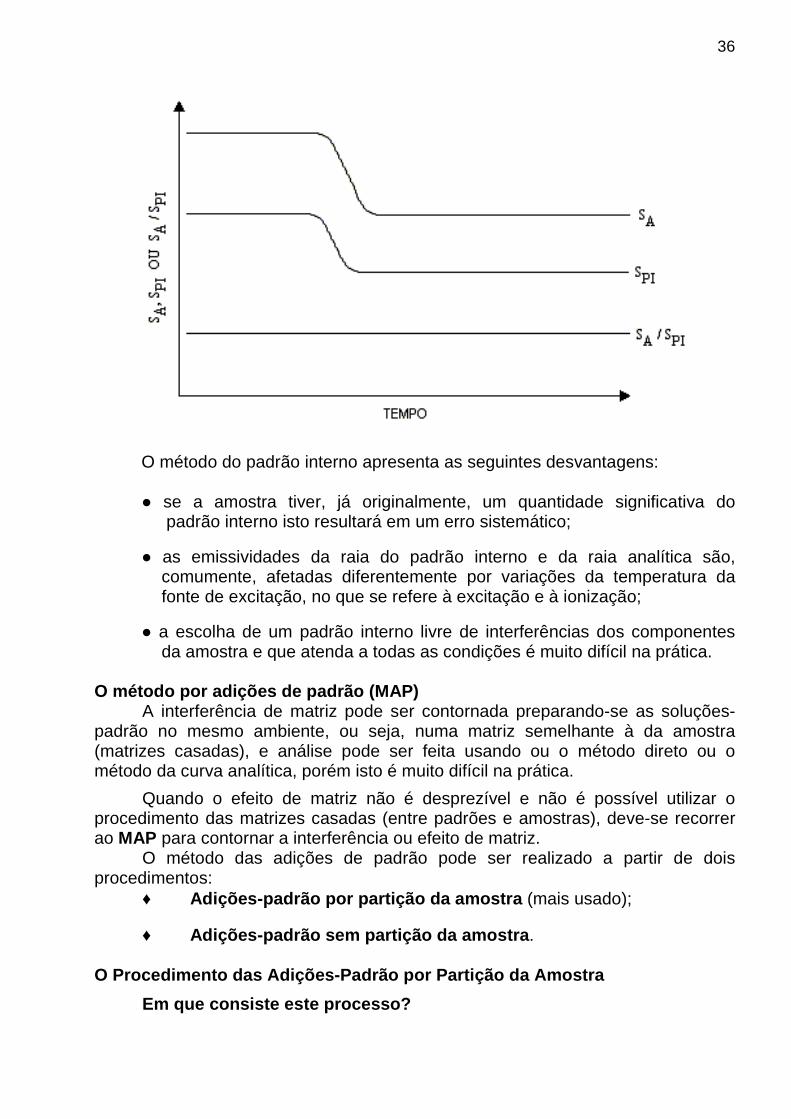
36
O método do padrão interno apresenta as seguintes desvantagens: se a amostra tiver, já originalmente, um quantidade significativa do padrão interno isto resultará em um erro sistemático;
as emissividades da raia do padrão interno e da raia analítica são, comumente, afetadas diferentemente por variações da temperatura da fonte de excitação, no que se refere à excitação e à ionização;
a escolha de um padrão interno livre de interferências dos componentes da amostra e que atenda a todas as condições é muito difícil na prática. O método por adições de padrão (MAP)
A interferência de matriz pode ser contornada preparando-se as soluções-padrão no mesmo ambiente, ou seja, numa matriz semelhante à da amostra (matrizes casadas), e análise pode ser feita usando ou o método direto ou o método da curva analítica, porém isto é muito difícil na prática.
Quando o efeito de matriz não é desprezível e não é possível utilizar o procedimento das matrizes casadas (entre padrões e amostras), deve-se recorrer ao MAP para contornar a interferência ou efeito de matriz.
O método das adições de padrão pode ser realizado a partir de dois procedimentos:
♦ Adições-padrão por partição da amostra (mais usado);
♦ Adições-padrão sem partição da amostra . O Procedimento das Adições-Padrão por Partição da A mostra
Em que consiste este processo?
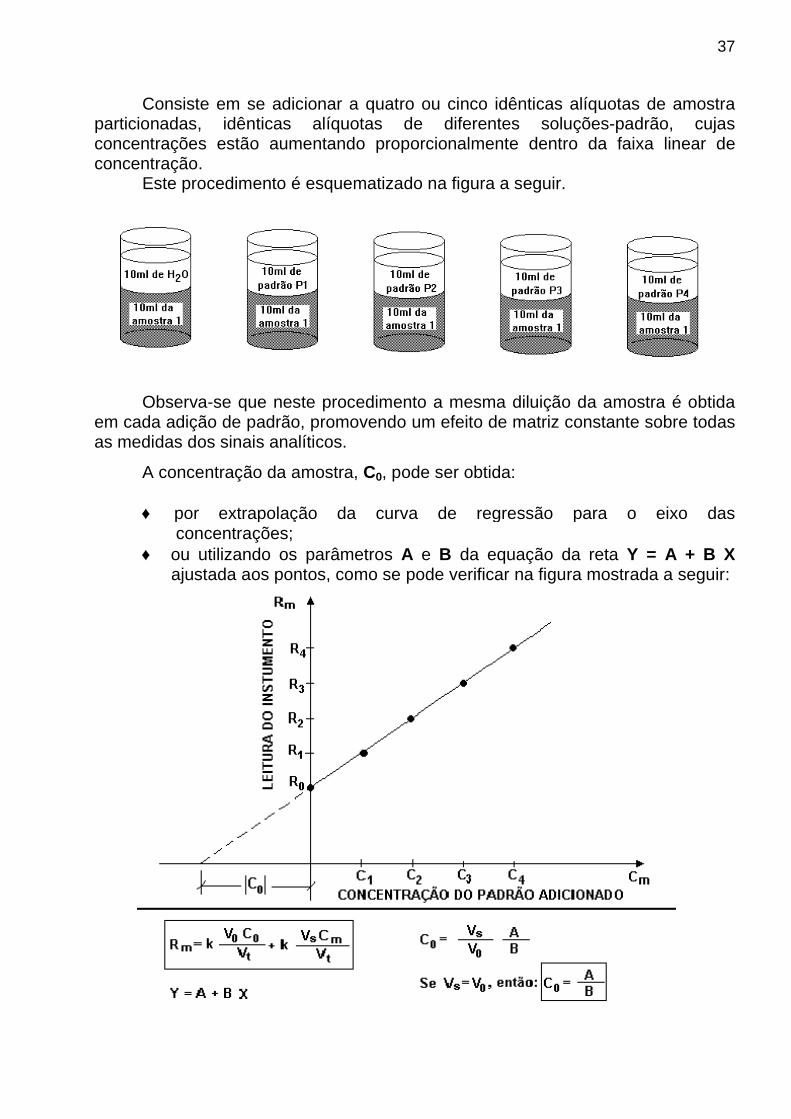
37
Consiste em se adicionar a quatro ou cinco idênticas alíquotas de amostra particionadas, idênticas alíquotas de diferentes soluções-padrão, cujas concentrações estão aumentando proporcionalmente dentro da faixa linear de concentração.
Este procedimento é esquematizado na figura a seguir.
Observa-se que neste procedimento a mesma diluição da amostra é obtida
em cada adição de padrão, promovendo um efeito de matriz constante sobre todas as medidas dos sinais analíticos.
A concentração da amostra, C0, pode ser obtida:
♦ por extrapolação da curva de regressão para o eixo das concentrações; ♦ ou utilizando os parâmetros A e B da equação da reta Y = A + B X ajustada aos pontos, como se pode verificar na figura mostrada a seguir:

38
Curva de Adições-Padrão O Procedimento das Adições-Padrão sem Partição da A mostra
Em que consiste este procedimento?
Consiste em adicionar a uma única alíquota da amostra, alíquotas crescentes de uma mesma solução-padrão, conforme o desenho esquemático da figura mostrada a seguir.
Este método é adequado quando o volume de amostra disponível é limitado
e é freqüentemente utilizado nas técnicas voltamétricas e potenciométricas. Ilustração Gráfica da Aplicação do MAP
A figura, mostrada a seguir, ilustra a aplicação do MAP à análise de quatro amostras que contêm a mesma concentração C0, do analito. O caso ilustrado pela curva A representa uma situação em que o analito se encontra num ambiente sem interferência de matriz. No caso da curva B , o analito encontra-se em um ambiente com efeito de matriz positivo (por exemplo, a determinação de sódio por fotometria de chama em uma amostra contendo etanol). Por outro lado, no caso da curva C , ele encontra-se em um ambiente com efeito de matriz negativo (por exemplo, a determinação de sódio por fotometria de chama em uma amostra contendo glicerol). Em ambiente com efeito de matriz positivo, curva B , observa-se que: R0,s
B e KsB são maiores que
R0,sA e Ks
A, enquanto que em um ambiente com efeito de matriz negativo temos R0,s
C e KsC menores que R0,s
A e KsA.
Não obstante as diferentes matrizes produzam diferentes efeitos de matriz, ou seja, diferentes valores de R0,s e Ks, a aplicação do MAP é capaz de fornecer o mesmo valor esperado da concentração do analito N0 nas amostras, como se pode notar pela extrapolação das curvas A, B e C para o eixo das concentrações das soluções-padrão adicionadas. Por outro lado, se as amostras com efeito de matriz fossem analisadas usando o método da curva
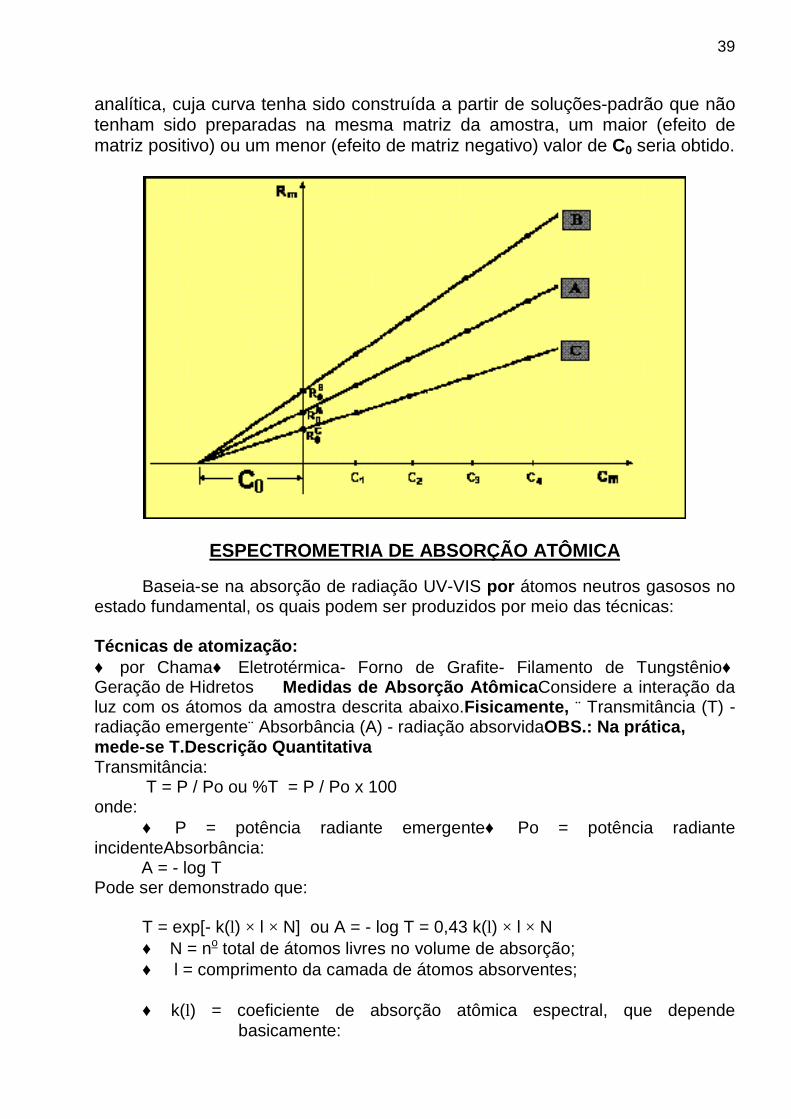
39
analítica, cuja curva tenha sido construída a partir de soluções-padrão que não tenham sido preparadas na mesma matriz da amostra, um maior (efeito de matriz positivo) ou um menor (efeito de matriz negativo) valor de C0 seria obtido.
ESPECTROMETRIA DE ABSORÇÃO ATÔMICA
Baseia-se na absorção de radiação UV-VIS por átomos neutros gasosos no estado fundamental, os quais podem ser produzidos por meio das técnicas: Técnicas de atomização: ♦ por Chama♦ Eletrotérmica- Forno de Grafite- Filamento de Tungstênio♦ Geração de Hidretos Medidas de Absorção Atômica Considere a interação da luz com os átomos da amostra descrita abaixo.Fisicamente, ¨ Transmitância (T) - radiação emergente¨ Absorbância (A) - radiação absorvidaOBS.: Na prática, mede-se T. Descrição Quantitativa Transmitância: T = P / Po ou %T = P / Po x 100 onde:
♦ P = potência radiante emergente♦ Po = potência radiante incidenteAbsorbância: A = - log T Pode ser demonstrado que:
T = exp[- k(l) × l × N] ou A = - log T = 0,43 k(l) × l × N ♦ N = no total de átomos livres no volume de absorção; ♦ l = comprimento da camada de átomos absorventes; ♦ k(l) = coeficiente de absorção atômica espectral, que depende
basicamente:

40
- da estrutura atômica (sobretudo a eletrônica) do analito - da probabilidade de transição; - comprimento de onda da radiação absorvida (l)Contudo, na prática:
A = K C,
onde : ♦ K = definido pela inclinação da curva analítica e depende de: k(l), l, variáveis do processo de atomização, etc. ♦ C = concentração do analito nas soluções-padrão.
Como veremos, os princípios são fundamentalmente os mesmos que os da absorção molecular UV-VIS pela amostra em solução e, portanto, a absorção atômica também é regida pela lei de Beer. Esta técnica apresenta uma boa obediência à lei de Beer, uma vez que as raias da absorção atômica são muito mais estreitas do que as bandas de absorção molecular. Nenhum monocromador consegue separar radiações com largura tão estreita e energia suficientes para excitar átomos e medir a sua absorção. Por este motivo a absorção atômica requer uma fonte de radiação UV-Visível muito mais potente. Além do mais, curvas analíticas não-lineares são inevitáveis quando as medidas de absorbância atômica são feitas com um equipamento de absorção molecular.
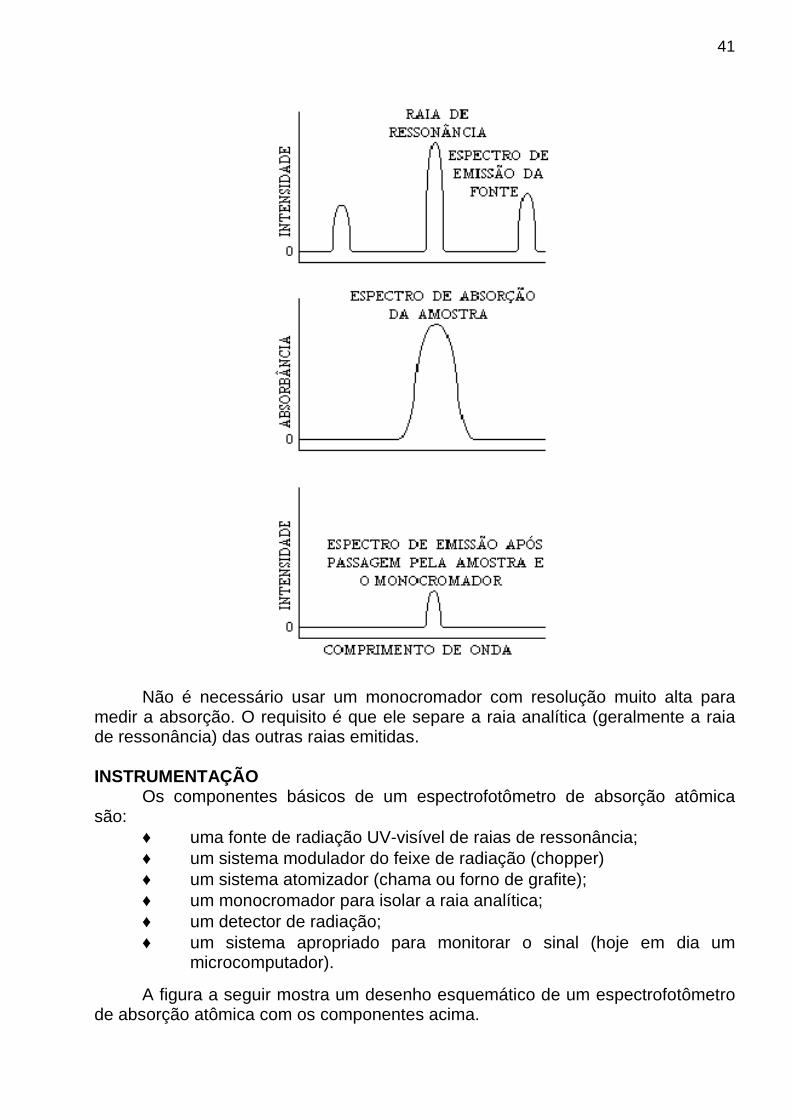
Como resolver este problema? A dificuldade foi resolvida com uma fonte capaz de emitir o espectro de
emissão do elemento de interesse. Por exemplo, uma lâmpada de vapor de sódio para análise de sódio.
Qual é a vantagem da fonte que emite o espectro do elemento de
interesse? Uma fonte de excitação apropriada emite raias com larguras muito menores
do que as das raias de absorção o que permite uma maior linearidade da lei de Beer . A figura a seguir mostra a relação entre o espectro emitido pela fonte, o de absorção e o espectro da emissão após passagem pelo monocromador.
41
Não é necessário usar um monocromador com resolução muito alta para
medir a absorção. O requisito é que ele separe a raia analítica (geralmente a raia de ressonância) das outras raias emitidas. INSTRUMENTAÇÃO
Os componentes básicos de um espectrofotômetro de absorção atômica são:
♦ uma fonte de radiação UV-visível de raias de ressonância; ♦ um sistema modulador do feixe de radiação (chopper) ♦ um sistema atomizador (chama ou forno de grafite); ♦ um monocromador para isolar a raia analítica; ♦ um detector de radiação; ♦ um sistema apropriado para monitorar o sinal (hoje em dia um
microcomputador).
A figura a seguir mostra um desenho esquemático de um espectrofotômetro de absorção atômica com os componentes acima.

42
FONTES DE RAIAS DE RESSONÂNCIA
Elas devem emitir as raias de ressonância do elemento de interesse com largura menor que a raia de absorção e com intensidade e estabilidade suficiente para que as medidas de absorção atômica possam ser realizadas com exatidão satisfatória. A fonte mais usada em espectrofotômetros de absorção atômica é uma lâmpada de catodo oco descrita a seguir. LÂMPADAS DE CATODO OCO
É a mais comum fonte de raia atômica usada na espectrometria de absorção atômica. Por isso, apenas este tipo de lâmpada será descrito aqui.A figura abaixo mostra esquematicamente lâmpadas de catodo oco (LCO) com e sem eletrodos auxiliares.
Ela consiste em um tubo de vidro contendo um gás nobre (argônio ou
neônio) a uma pressão de 1-5mmHg. No seu interior é colocado um catodo cilíndrico oco feito ou recoberto com o elemento de interesse, e um anodo de tungstênio que, em forma circular, envolve a extremidade do catodo. O catodo é envolvido por um tubo de proteção (vidro ou mica) para evitar a formação da descarga elétrica fora da região oca do catodo. A face frontal é de quartzo para raias de ressonância na região UV ou vidro para as raias de ressonância na região visível.
A aplicação de uma alta diferença de potencial, na ordem de 300 V, entre os eletrodos provoca a ionização do gás inerte e uma corrente de 5 a 30 mA é gerada quando os cátions gasosos e os elétrons migram para os eletrodos de carga oposta. Os íons do gás nobre formados são acelerados em direção ao catodo e, colidindo com a superfície da cavidade catódica, produz uma nuvem atômica, por um processo chamado de sputterring (expirrar). Os átomos da nuvem são excitados por colisões com os átomos gasosos energizados e emitem radiações (as raias de ressonância de preferência) quando retornam ao estado fundamental.

43
Um par de eletrodos auxiliares, entre os quais flui uma corrente secundária, produz um maior fluxo de átomos gasosos ionizados para colisão e, conseqüentemente, uma maior intensidade da emissão.
A forma cilíndrica oca do catodo tende a concentrar a radiação em uma região limitada do tubo e aumentar a redeposição dos átomos no catodo em vez das paredes do tubo.
O gás nobre da lâmpada (neônio ou argônio) também produz sua própria emissão e a escolha do gás depende dos elementos do catodo; por exemplo, na lâmpada de arsênio não se pode utilizar neônio em virtude de uma forte raia de emissão deste gás próxima da melhor raia de ressonância do arsênio. MODULADOR DO FEIXE DE RADIAÇÃO
A modulação do feixe da LCO é de fundamental importância para eliminar interferência espectral da radiação de fundo. Ela pode ser feita usando um circuito eletrônico que liga e desliga a lâmpada em ciclos alternados ou através de interruptor rotatatório (chopper) colocado no caminho ótico. O detector recebe dois tipos de sinais: um sinal alternado da fonte (LCO) e um sinal contínuo da chama. Um sistema eletrônico amplifica somente o sinal modulado, ignorando o sinal contínuo. ATOMIZADORES COM CHAMA
Os mesmos tipos de aspiradores-nebulizadores usados na emissão atômica são também utilizados na absorção atômica para a atomização em chama. A principal diferença está na geometria dos queimadores. Veja a figura mostrada a seguir.

44
O queimador mais usual possui apenas uma fenda com um comprimento de 5 a 10 cm., porém o queimador construído com três fendas paralelas muito próximas, conforme mostra a figura acima, apresenta a vantagem de que a chama produzida na fenda central é protegida da difusão perturbadora do ar pelas cortinas periféricas produzidas pelas fendas externas.
Para um caminho ótico menor, o queimador pode normalmente ser girado em um ângulo de 900
. Eles também podem movimentar-se em todas as direções do espaço de forma a se encontrar a melhor posição em que feixe ótico irá atravessar a chama.
Na absorção atômica quase sempre utiliza-se as chamas acetileno-ar e acetileno-óxido nitroso . O queimador usado para chamas de acetileno-ar comprimido é feito de material diferente do material usado no queimador para chamas de acetileno-óxido nitroso. Normalmente, o primeiro queimador utiliza fendas de 10 cm, enquanto o segundo utiliza fendas de 5 cm.
Toda tubulação envolvida no transporte do gás acetileno não deve ser de cobre devido a perigos de explosão. Utiliza-se normalmente aço inoxidável.
Devido a problemas relacionados com a energia de ignição, a chama N20-C2H2 não é acesa diretamente. Primeiro, acende-se uma chama de AR-C2H2, em seguida, simultaneamente, aumenta-se o fluxo de N2O e diminui-se o fluxo de AR até a zero, obtendo-se apenas a chama N20-C2H2. Para apagar a chama o processo inverso deve ser utilizado. ATOMIZADORES ELETROTÉRMICOS Um atomizador eletrotérmico típico, mais conhecido como forno de grafite , foi proposto por L’vov no ínicio da década de 1960 e começou a ser comercializado no início da década de 1970. As figuras, mostradas a seguir, ilustram desenhos esquemáticos de dois tipos de atomizadores eletrotérmicos em diferentes planos no espaço. A amostra é atomizada em um cilindro oco de grafite, denominado de forno de grafite, com 1 a 5 cm de comprimento e 0,3 a 1,0 cm de diâmetro. Este cilindro contém no seu interior uma plataforma, conhecida comumente como plataforma de L’vov , onde é colocada a amostra. A plataforma de L’vov é colocada em uma posição tal que o calor irradia das paredes do forno e a amostra é aquecida por resistividade a uma temperatura uniforme. O forno de grafite possui uma janela de quartzo para a passagem do feixe óptico. O tubo é colocado em uma câmara através do qual flui uma lenta corrente de argônio ou nitrogênio (gás de arraste), para evitar incineração do forno e oxidação dos átomos atomizados, e para arrastar os gases desejados ou não-desejados do centro do atomizador. Para evitar deterioração do forno, as vezes o metano é misturado aos gases N2 e Ar de arraste. O forno é colocado em uma câmara metálica por onde circula água de refrigeração para permitir rápido retorno do forno a temperatura ambiente, após cada amostra ter sido atomizada e o seu sinal analítico requerido ter sido registrado. As amostras líquidas ou em solução são introduzidas na plataforma de L’vov usando uma seringa. As amostras sólidas podem ser pesadas em minúsculas panelas (como as panelas usadas em termogravimetria) e depois
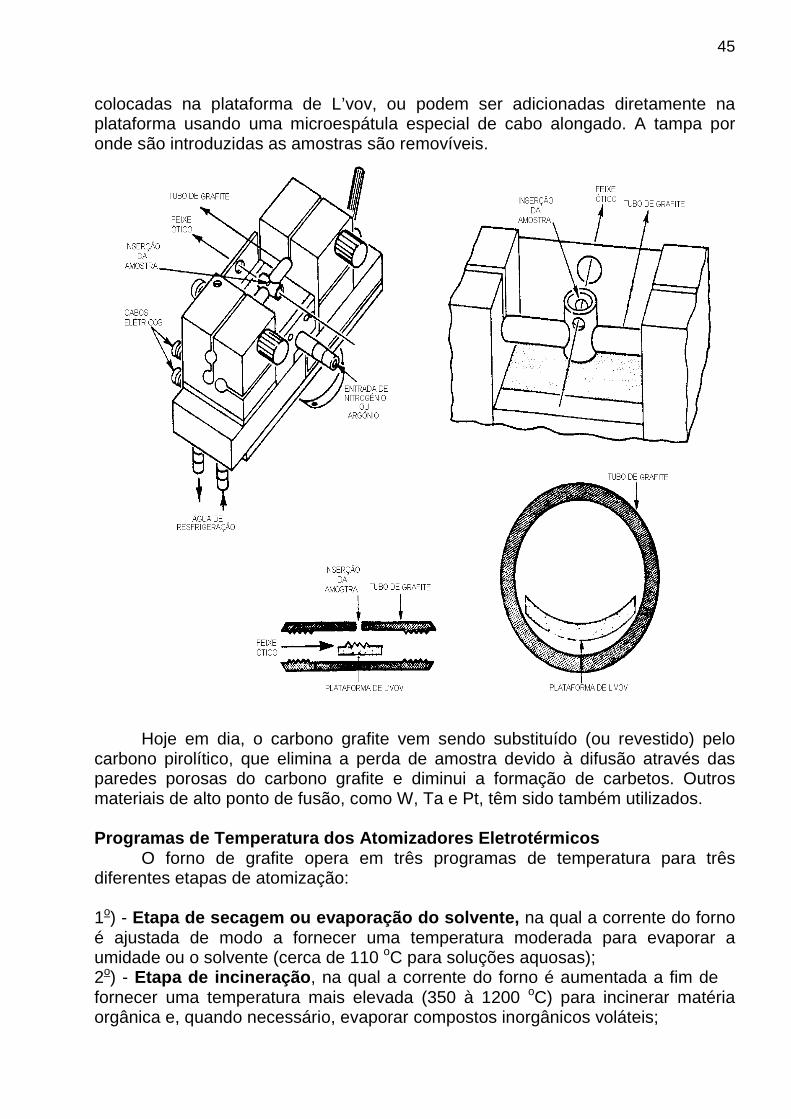
45
colocadas na plataforma de L’vov, ou podem ser adicionadas diretamente na plataforma usando uma microespátula especial de cabo alongado. A tampa por onde são introduzidas as amostras são removíveis.
Hoje em dia, o carbono grafite vem sendo substituído (ou revestido) pelo carbono pirolítico, que elimina a perda de amostra devido à difusão através das paredes porosas do carbono grafite e diminui a formação de carbetos. Outros materiais de alto ponto de fusão, como W, Ta e Pt, têm sido também utilizados. Programas de Temperatura dos Atomizadores Eletrotér micos O forno de grafite opera em três programas de temperatura para três diferentes etapas de atomização: 1o) - Etapa de secagem ou evaporação do solvente, na qual a corrente do forno é ajustada de modo a fornecer uma temperatura moderada para evaporar a umidade ou o solvente (cerca de 110 oC para soluções aquosas); 2o) - Etapa de incineração , na qual a corrente do forno é aumentada a fim de fornecer uma temperatura mais elevada (350 à 1200 oC) para incinerar matéria orgânica e, quando necessário, evaporar compostos inorgânicos voláteis;
46
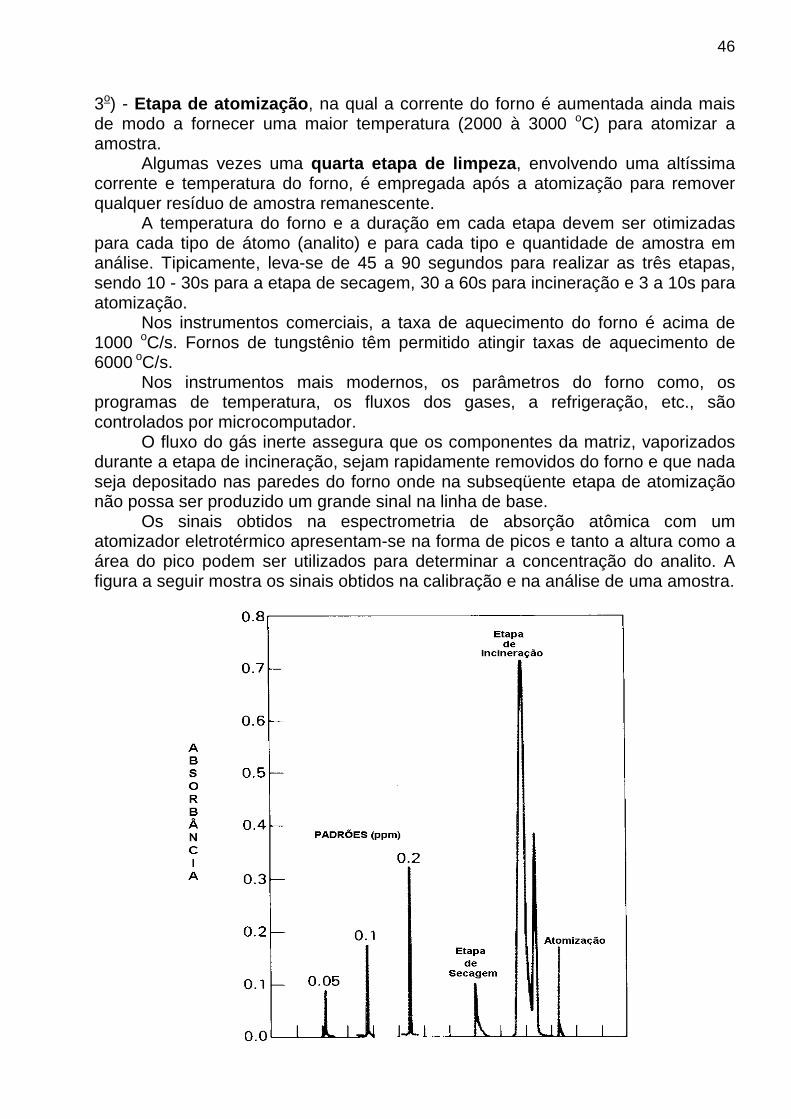
3o) - Etapa de atomização , na qual a corrente do forno é aumentada ainda mais de modo a fornecer uma maior temperatura (2000 à 3000 oC) para atomizar a amostra. Algumas vezes uma quarta etapa de limpeza , envolvendo uma altíssima corrente e temperatura do forno, é empregada após a atomização para remover qualquer resíduo de amostra remanescente. A temperatura do forno e a duração em cada etapa devem ser otimizadas para cada tipo de átomo (analito) e para cada tipo e quantidade de amostra em análise. Tipicamente, leva-se de 45 a 90 segundos para realizar as três etapas, sendo 10 - 30s para a etapa de secagem, 30 a 60s para incineração e 3 a 10s para atomização. Nos instrumentos comerciais, a taxa de aquecimento do forno é acima de 1000 oC/s. Fornos de tungstênio têm permitido atingir taxas de aquecimento de 6000 oC/s. Nos instrumentos mais modernos, os parâmetros do forno como, os programas de temperatura, os fluxos dos gases, a refrigeração, etc., são controlados por microcomputador. O fluxo do gás inerte assegura que os componentes da matriz, vaporizados durante a etapa de incineração, sejam rapidamente removidos do forno e que nada seja depositado nas paredes do forno onde na subseqüente etapa de atomização não possa ser produzido um grande sinal na linha de base. Os sinais obtidos na espectrometria de absorção atômica com um atomizador eletrotérmico apresentam-se na forma de picos e tanto a altura como a área do pico podem ser utilizados para determinar a concentração do analito. A figura a seguir mostra os sinais obtidos na calibração e na análise de uma amostra.

47
Atomização Eletrotérmica Versus Atomização em Chama As medidas de absorção atômica com atomização eletrotérmica apresentam as seguintes vantagens com relação à chama:
- permite analisar pequeníssimos volumes de amostra (de 5 a 100µl de solução); - permite analisar amostras sólidas sem pré-tratamento em muitos casos; - fornece limites de detecção 100 a 1000 vezes maior para a maioria dos elementos (limites de detecção absoluto de 10-8 a 10-13g); - a temperatura pode ser programada para controlar as interferências químicas.
Por outro lado, as medidas são mais demoradas, os efeitos de matriz são superiores e a precisão (5 - 10 %) é menor do que na chama (1 %). Os baixos níveis de detecção da espectrometria absorção atômica com atomizador eletrotérmico são atribuídos aos seguintes fatores:
- a totalidade da amostra é aproveitada; - forma-se uma população de átomos muito maior em um pequeno volume (2 ml); - baixa radiação de fundo. ATOMIZAÇÃO POR GERAÇÃO DE HIDRETOS A atomização em chama não permite determinar elementos como As e Se em níveis de ppb. Todavia, em análise ambiental é de fundamental importância a determinação desses elementos nesses níveis. A atomização por geração de hidretos é uma técnica que permite alcançar estes níveis. A técnica consiste em fazer reagir os elementos que formam hidretos como As, Se, Sb, Bi, Ge, Sn, Te e Pb com o borohidreto de sódio (NaBH4) em meio ácido. Para As(III) a reação é dada por:
O3HsHBO3H→AsO4H3HBH3 233333 +
4 ++++
4A
Uma outra reação pode ter lugar durante a formação do borohidreto dos elementos:
233+
24 4HBOHH+O3HBH +→+−
Os hidretos do analito são produzidos em recipientes em separado e depois são transportados usando um gás inerte em direção ao sistema de detecção. A figura a seguir mostra um sistema em fluxo para atomização por geração de hidretos. O sistema possui uma bomba peristáltica que bombeia continuamente a amostra (ou solução de limpeza), a solução de borohidreto de sódio e ácido clorídrico para um dispositivo misturador. As soluções são bombeadas para um loop onde ocorrem a mistura e reação. O produto reacional vai para um separador gás-líquido onde o hidreto gasoso produzido do elemento em análise é removido do líquido e depois é transportado por um gás inerte, normalmente o nitrogênio, para a cela de absorção por onde passa o feixe óptico.

48
A cela de absorção é adaptada ao queimador e é aquecida por uma pequena chama de ar-acetileno. Durante o aquecimento os hidretos gasosos se dissociam produzindo o elemento no estado gasoso (atomização). A decomposição dos hidretos para formação do átomo envolve a formação de radicais H que são produzidos quando o H2, que também se forma na reação com o borohidreto, se choca com as paredes quentes da cela de absorção e a reação desses radicais com os hidretos formados de acordo com a seguinte reação genérica:
2 xH+M→. xH+xMH
O vapor atômico do analito produzido na cela de absorção é atravessado pelo feixe óptico que vai em direção ao detector. O sinal de absorbância produzido é um pico semelhante ao obtido na atomização eletrotérmica. Entre os elementos que formam hidretos, mercúrio é único que pode ser produzido diretamente em um ambiente frio, devido a sua alta pressão de vapor à temperatura ambiente, na presença do excesso de borohidreto de sódio. As vantagens da técnica de geração de hidretos são:
- os problemas de interferências de matriz são reduzidos, pois os átomos do analito são retirados da matriz da amostra
- podem ser atingidos baixos limites de detecção (ng/ml ≡ ppb e pg/ml ≡ ppt)
MONOCROMADORES
A função primordial do monocromador consiste em isolar de outras raias, a raia de ressonância do elemento de interesse (analito) emitida pela lâmpada. Na absorção atômica, os monocromadores devem cobrir uma faixa espectral que abranja, em um extremo, a raia analítica do arsênio (193,7 nm) e, no outro, a do césio (852,1 nm). TIPOS DE INSTRUMENTOS
Os instrumentos podem ser construídos segundo os modelos de feixe simples ou de feixe duplo.
49
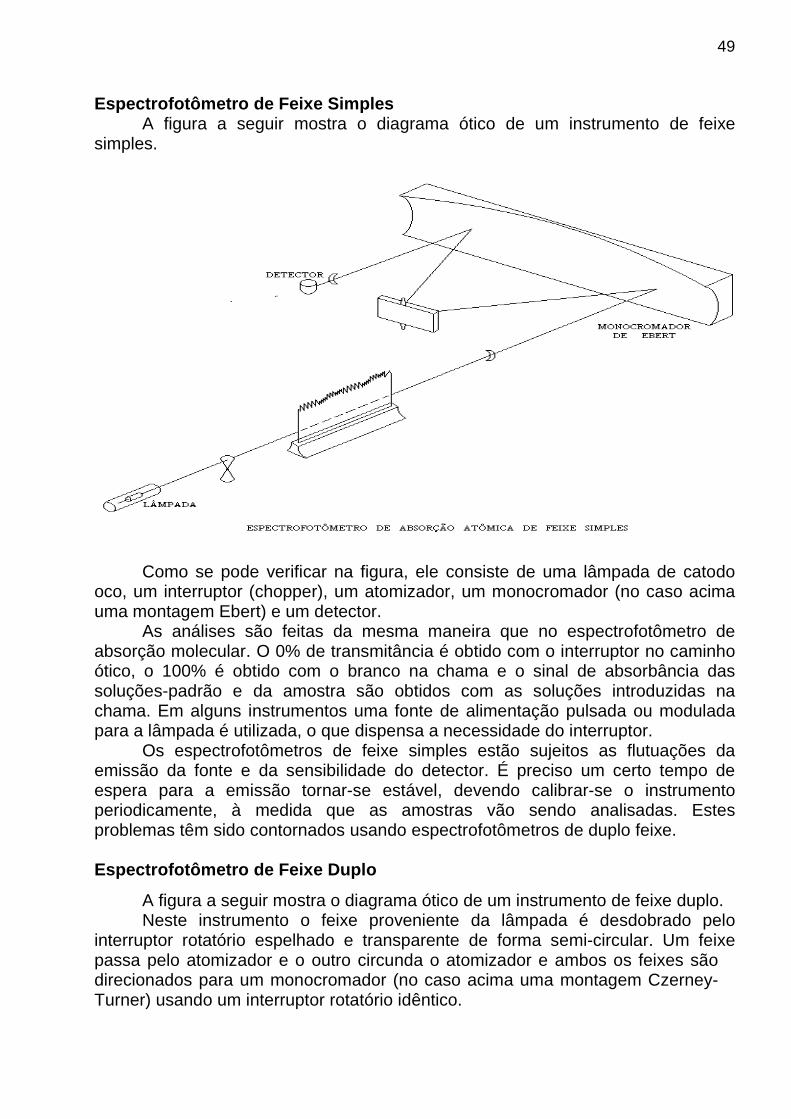
Espectrofotômetro de Feixe Simples A figura a seguir mostra o diagrama ótico de um instrumento de feixe
simples.
Como se pode verificar na figura, ele consiste de uma lâmpada de catodo oco, um interruptor (chopper), um atomizador, um monocromador (no caso acima uma montagem Ebert) e um detector.
As análises são feitas da mesma maneira que no espectrofotômetro de absorção molecular. O 0% de transmitância é obtido com o interruptor no caminho ótico, o 100% é obtido com o branco na chama e o sinal de absorbância das soluções-padrão e da amostra são obtidos com as soluções introduzidas na chama. Em alguns instrumentos uma fonte de alimentação pulsada ou modulada para a lâmpada é utilizada, o que dispensa a necessidade do interruptor.
Os espectrofotômetros de feixe simples estão sujeitos as flutuações da emissão da fonte e da sensibilidade do detector. É preciso um certo tempo de espera para a emissão tornar-se estável, devendo calibrar-se o instrumento periodicamente, à medida que as amostras vão sendo analisadas. Estes problemas têm sido contornados usando espectrofotômetros de duplo feixe. Espectrofotômetro de Feixe Duplo
A figura a seguir mostra o diagrama ótico de um instrumento de feixe duplo. Neste instrumento o feixe proveniente da lâmpada é desdobrado pelo
interruptor rotatório espelhado e transparente de forma semi-circular. Um feixe passa pelo atomizador e o outro circunda o atomizador e ambos os feixes são direcionados para um monocromador (no caso acima uma montagem Czerney-Turner) usando um interruptor rotatório idêntico.

50
O sistema eletrônico mede a relação das intensidades dos dois feixes. As flutuações da emissão da lâmpada e da sensibilidade do detector aparecem tanto no numerador como no denominador e, desta forma, são canceladas. Assim, o instrumento produz uma linha de base constante quase imediatamente com pouca ou nenhuma espera para o aquecimento da lâmpada. Espectrofotômetro de Feixe Duplo com Correção de Ba ckground Usando Fonte Contínua
Para correção de background (absorção pela chama) utiliza-se uma lâmpada de deutério que produz uma radiação contínua que passa, simultaneamente, pelos mesmos dois caminhos do feixe de emissão da fonte, ou seja, um feixe passa pelo atomizador e o outro o circunda.
A figura a seguir mostra o diagrama óptico de um instrumento de feixe duplo com correção de background usando uma fonte contínua.
A absorbância corrigida do analito é dada por: Acorrigida (analito) = A LCO - ALD
onde, ALCO é a absorbância total relacionada ao feixe da lâmpada de cátodo oco e ALD é a absorbância total relacionada ao feixe da lâmpada de deutério. Uma vez que ALCO = ALCO(analito) + A LCO (chama) e ALD = ALD(analito) + A LD (chama), tem-se que: Acorr (analito) = [A LCO(analito) + A LCO (chama)] - [ALD(analito) + A LD (chama)]
Se ALCO (chama) = A LD (chama) e ALD(analito) ≅≅≅≅ 0 (porque o perfil de absorção para o analito é muito estreito comparado com a banda passante da fonte de radiação contínua), obtém-se finalmente que:
Acorr (analito) = A LCO(analito)

51
Apesar do sucesso do sistema de correção de background (ou fundo) com a
lâmpada de deutério, existem algumas limitações: - o ambiente gasoso quente é altamente não-homogêneo de modo que erros negativos ou positivos poderão ocorrer se ambos os feixes não estiverem igualmente alinhados e se outros elementos, presentes na matriz da amostra, absorverem radiações da larga banda passante da fonte contínua. - não se podem fazer correções de fundo para radiações acima de 350nm devido à fraca emissão da lâmpada de deutério acima desse comprimento de onda. Correção de Background Baseada na Auto-Reversão de uma Lâmpada Pulsada Surgiu recentemente na literatura um sistema simples, barato e mais vantajoso do que a correção de background com efeito Zeeman. Chamado de correção de background de Smith-Hieftje , este sistema é baseado no fenômeno da auto-reversão que é produzida quando uma alta corrente é fornecida a lâmpada de catodo oco comum. A lâmpada é alimentada por uma corrente pulsada em períodos de 9,7 ms para corrente normal (6 a 20 mA) e de 0,3ms para uma alta corrente (100 a 500 mA). No período de corrente baixa mede-se a absorbância do analito mais a do background e no período de alta corrente é medida uma pequena absorbância do analito (desprezível) mais a absorbância do background, conforme ilustrado na figura mostrada a seguir. A diferença entre as duas medidas é a absorbância que é registrada pelo instrumento. Se a absorção de background é constante nos dois períodos, tem-se que a diferença de absorbância registrada pelo instrumento é devido apenas ao elemento. Para uma melhor constância, essa diferença pode ser medida por vários ciclos e seu valor médio é a diferença registrada.
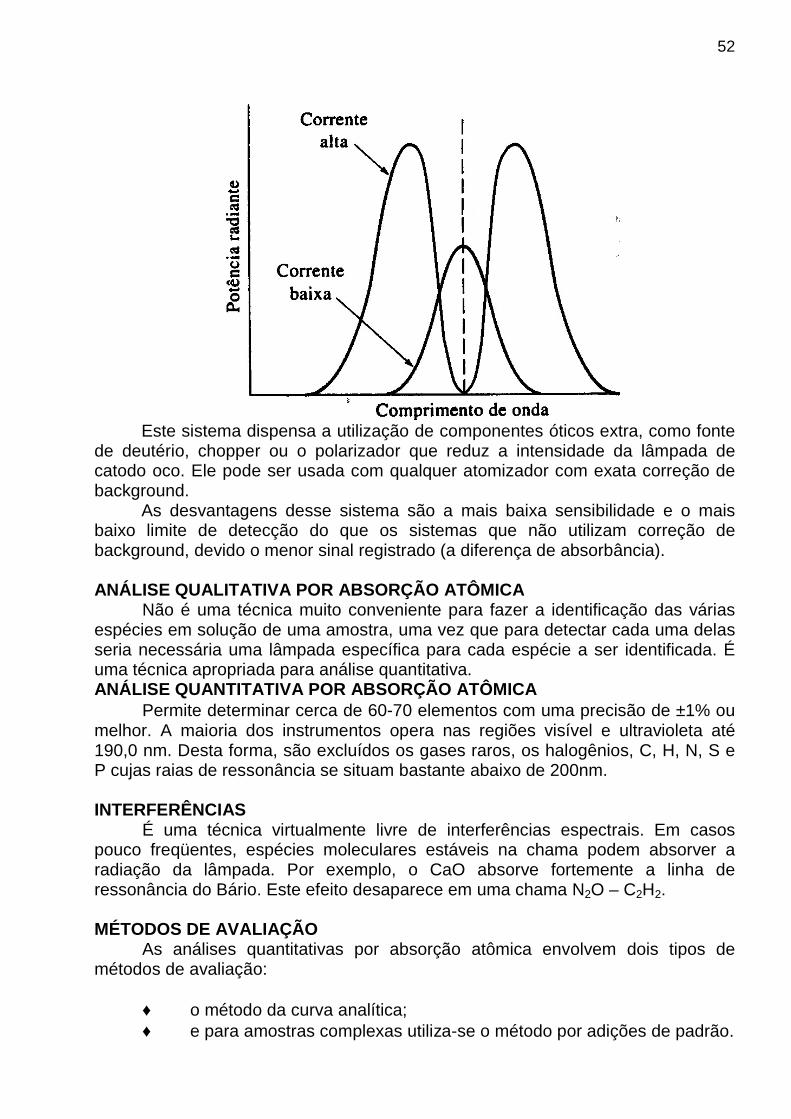
52
Este sistema dispensa a utilização de componentes óticos extra, como fonte de deutério, chopper ou o polarizador que reduz a intensidade da lâmpada de catodo oco. Ele pode ser usada com qualquer atomizador com exata correção de background. As desvantagens desse sistema são a mais baixa sensibilidade e o mais baixo limite de detecção do que os sistemas que não utilizam correção de background, devido o menor sinal registrado (a diferença de absorbância). ANÁLISE QUALITATIVA POR ABSORÇÃO ATÔMICA
Não é uma técnica muito conveniente para fazer a identificação das várias espécies em solução de uma amostra, uma vez que para detectar cada uma delas seria necessária uma lâmpada específica para cada espécie a ser identificada. É uma técnica apropriada para análise quantitativa. ANÁLISE QUANTITATIVA POR ABSORÇÃO ATÔMICA
Permite determinar cerca de 60-70 elementos com uma precisão de ±1% ou melhor. A maioria dos instrumentos opera nas regiões visível e ultravioleta até 190,0 nm. Desta forma, são excluídos os gases raros, os halogênios, C, H, N, S e P cujas raias de ressonância se situam bastante abaixo de 200nm. INTERFERÊNCIAS
É uma técnica virtualmente livre de interferências espectrais. Em casos pouco freqüentes, espécies moleculares estáveis na chama podem absorver a radiação da lâmpada. Por exemplo, o CaO absorve fortemente a linha de ressonância do Bário. Este efeito desaparece em uma chama N2O – C2H2. MÉTODOS DE AVALIAÇÃO
As análises quantitativas por absorção atômica envolvem dois tipos de métodos de avaliação:
♦ o método da curva analítica; ♦ e para amostras complexas utiliza-se o método por adições de padrão.

53
ABSORÇÃO ATÔMICA VERSUS EMISSÃO ATÔMICA De uma maneira geral, os dois métodos se completam em termos de
qualidade, embora a absorção apresente algumas vantagens sobre a emissão atômica:
♦ o número de átomos no estado fundamental é muito maior que o número de átomos excitados, o que dá uma maior sensibilidade;
♦ o efeito da variação da temperatura sobre o número de átomos no estado fundamental não é tão crítico;
♦ o monocromador não necessita apresentar uma alta resolução;
♦ interferência espectral é mínima. As desvantagens da absorção atômica são: ♦ a necessidade de uma lâmpada de catodo oco para análise de um
elemento;
♦ a emissão atômica é melhor para analisar metais alcalinos, alcalinos terrosos, terras raras, Ga, In e Tl, etc;
♦ não permite fazer análise multicomponente simultânea. ESPECTROMETRIA DE FLUORESCÊNCIA ATÔMICA Nesta técnica a solução da amostra é atomizada em uma chama ou um forno de grafite, e os átomos são, então, excitados mediante passagem da radiação de ressonância (ou contínua); mede-se, porém, a potência da fluorescência emitida pelos átomos excitados. Para baixa concentração atômica (C), a potência de fluorescência é diretamente proporcional à concentração.
P = PoKC
onde, Po é potência do feixe incidente e K é um coeficiente de proporcionalidade que depende da eficiência quântica, da absorvidade molar e do percurso ótico. Para uma potência incidente constante tem-se que:
P = K’C INSTRUMENTAÇÃO O espectrofotômetro de fluoresdência atômica nada mais é que um espectrofotômetro de absorção atômica onde se mede a fluorescência atômica em um ângulo de 90o com o feixe de excitação. A figura a seguir mostra um diagrama do sistema básico de um espectrofotômetro de fluorescência atômica.
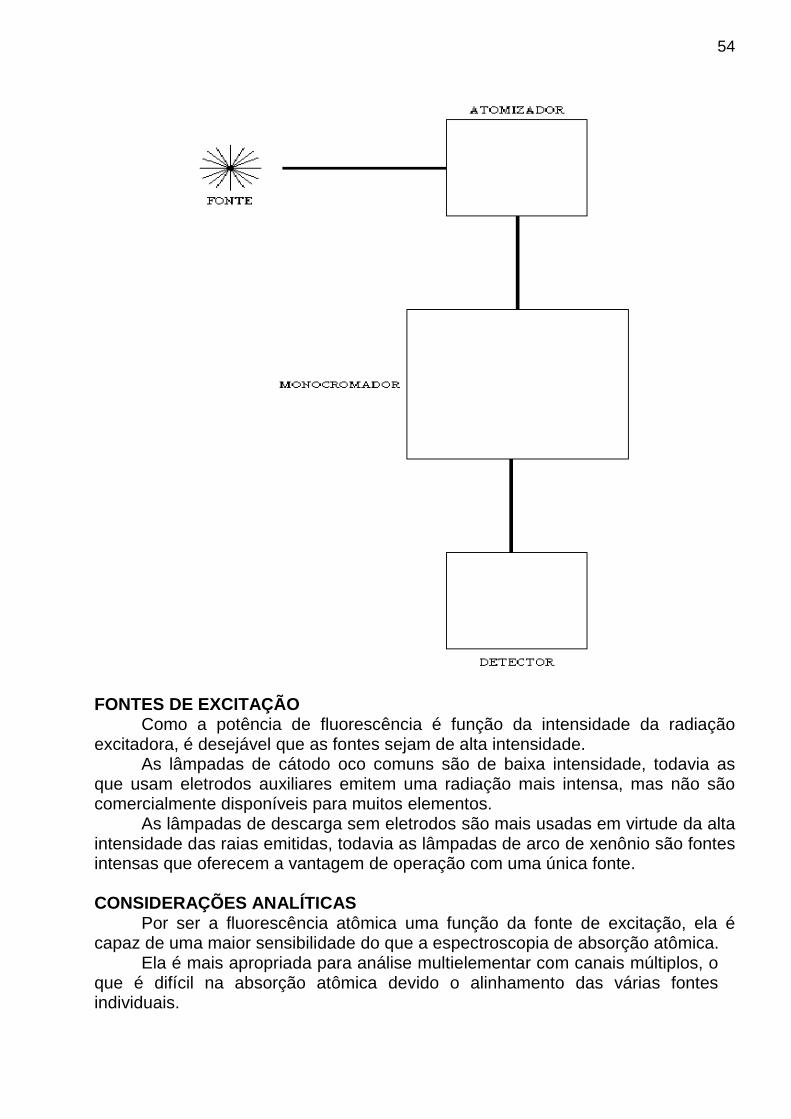
54
FONTES DE EXCITAÇÃO Como a potência de fluorescência é função da intensidade da radiação excitadora, é desejável que as fontes sejam de alta intensidade. As lâmpadas de cátodo oco comuns são de baixa intensidade, todavia as que usam eletrodos auxiliares emitem uma radiação mais intensa, mas não são comercialmente disponíveis para muitos elementos. As lâmpadas de descarga sem eletrodos são mais usadas em virtude da alta intensidade das raias emitidas, todavia as lâmpadas de arco de xenônio são fontes intensas que oferecem a vantagem de operação com uma única fonte. CONSIDERAÇÕES ANALÍTICAS Por ser a fluorescência atômica uma função da fonte de excitação, ela é capaz de uma maior sensibilidade do que a espectroscopia de absorção atômica. Ela é mais apropriada para análise multielementar com canais múltiplos, o que é difícil na absorção atômica devido o alinhamento das várias fontes individuais.

55
ESPECTROMETRIA DE ABSORÇÃO MOLECULAR UV-Vis
Considerações Gerais
A espectroscopia de absorção UV-Vis utiliza radiação eletromagnética cujos comprimentos (λ) se encontram na faixa de 200 a 780 nm. Quando estimulada com esse tipo radiação, a molécula do composto pode sofrer transições eletrônicas por ocasião da absorção de energia quantizada. O registro gráfico da resposta do sistema ao estímulo denomina-se espectro eletrônico de absorção.
A absorção de energia UV-Vis produz modificação na estrutura eletrônica da molécula em conseqüência de transições eletrônicas envolvendo geralmente elétrons π e n (não ligantes) envolvidos em ligações. Isto requer que a molécula contenha pelos menos um grupo funcional insaturado (C=C, C=O, por exemplo) para fornecer os orbitais moleculares π e n. Tal centro de absorção é chamado
cromóforo, sendo responsável principalmente pelas transições *ππ → e
*πn→ . Estas resultam da absorção de radiações eletromagnéticas que se
enquadram em uma região espectral experimentalmente conveniente, ao contrário
das transições *σn→ e *
σσ → que requerem geralmente radiações mais energéticas (λ < 200 nm).
As bandas de absorção podem ser caracterizadas por dois parâmetros fundamentais: a posição e a intensidade. A posição corresponde normalmente ao “λ” da radiação eletromagnética responsável pela transição eletrônica, enquanto a intensidade depende, entre outros fatores, da energia dos orbitais moleculares e probabilidade de transição.
É importante ressaltar que a energia necessária para as transições eletrônicas envolvendo a absorção de radiação UV-Vis depende primariamente do tipo de ligação (ou seja, da presença de cromóforo) e, por último, da estrutura molecular. Todavia, é possível ocorrerem mudanças significativas na posição e intensidade das bandas em decorrência de, por exemplo, uma conjugação entre ligações duplas ou quando o cromóforo C=C encontra-se conjugado com um grupo C=O na estrutura da molécula e vice-versa.
Os espectros de absorção UV-Vis apresentam geralmente bandas largas resultantes da sobreposição dos sinais provenientes de transições vibracionais e rotacionais ao(s) sinal(ais) associado(s) à(s) transição(ões) eletrônica(s). Adicionalmente, quando os espectros são obtidos com a amostra em fase condensada a estrutura espectral fina é removida dando lugar a bandas com perfil liso, tornando os espectros carentes de detalhes e com baixa resolução. Observam-se também alterações tanto na posição como na intensidade das bandas originadas de interações entre suas moléculas e as do solvente. De fato, um aumento na polaridade do solvente promove usualmente deslocamentos da banda para comprimentos de onda maiores (efeito batocrómico) se a transição associada é do tipo ππ → *. Contudo, um deslocamento contrário (efeito hipsocrômico) é observado quando o cromóforo sofre uma transição do tipo
π→n* devido ao aumento da energia entre os dois orbitais moleculares
envolvidos Efeitos envolvendo um aumento (hipercrômico) ou uma diminuição

56
(efeito hipocrômico) na intensidade da banda de absorção também podem ser observados como resultado dessas interações.
A absorção molecular constitui um dos mais amplos caminhos usados pelos químicos analíticos para determinação de espécies moleculares em solução. A grande maioria dos elementos da tabela periódica pode ser determinada usando uma técnica apropriada de absorção molecular . Fundamentos teóricos A energia associada às bandas normalmente observadas nos espectros UV-Vis de uma molécula poliatômica compreende:
Emolécula = Eeletrônica + Evibracional + Erotacional onde a: ♦ energia eletrônica ⇒ associada à distribuição dos elétrons em torno dos núcleos atômicos ♦ energia vibracional ⇒ relaciona-se com a vibração dos átomos ou grupos de átomos em torno das posições de equilíbrio nas ligações ♦ energia rotacional ⇒ encontra-se associada à rotação da molécula em torno do seu centro de gravidade
As três formas de energia são quantizadas como se pode observar no diagrama da figura abaixo:

57
Diagrama esquemático de energias de uma molécula diatômica com
indicação das transições: ∆E = eletônica, ∆J = rotacional pura e ∆V = vibracional-rotacional. Nota-se na figura no diagrama da figura anterior que a diferença entre os
níveis de energia seguem a ordem: ∆∆∆∆Eeletrônica > ∆∆∆∆Evibracional > ∆∆∆∆Erotacional
Por quê? A resposta encontra-se na Mecânica quântic a!!! De fato, a Mecânica Quântica mostra que:
Quanto maior o grau de confinamento da partícula. maior as restrição para o
seu movimento. Assim, maior será o grau de quantização de suas energias. O movimento eletrônico nos orbitais é mais restrito que o vibracional, o qual por sua vez é mais restrito que o rotacional. Logo, os níveis de energia eletrônica são mais espaçados e os níveis rotacionais os menos espaçados entre si.
Origem dos Espectros de Absorção Molecular UV-Vis

58
O processo de absorção é essencialmente o mesmo para espécies orgânicas e inorgâncas, porém existem as algumas peculiaridades em cada classe discutidas a seguir. Os elétrons que contribuem para a absorção UV-VIS das espécies moleculares orgânicas são :
♦ Os elétrons que participam da formação de ligações entre átomos (os elétrons σσσσ e ππππ)
♦ Os elétrons externos dos átomos que não participam da ligação, ou
seja, os elétrons não-ligantes, n ABSORÇÃO POR COMPOSTOS ORGÂNICOS São possíveis quatro tipos de transições eletrônicas nos compostos orgânicos: ♦ transição σσσσ →→→→ σσσσ* ♦ transição n →→→→ σσσσ* ♦ transição n →→→→ ππππ* ♦ transição ππππ →→→→ ππππ*
A figura a segir mostra os níveis energéticos dos orbitais moleculares e os
tipos de transições que ocorrem nos referidos compostos.

59
Transições σσσσ →→→→ σσσσ*. Ocorrem nos hidrocarbonetos que possuem apenas ligações σσσσ e elétrons ligantes . Ex.: Propano ( λmáx 135 ηm).
Transições n →→→→ σσσσ:. Compostos orgânicos saturados contendo átomos com elétrons não-ligantes. Ex. cloreto de metila (λmáx = 173 ηm) e o metanol (λmáx. = 183 ηm).
Transições n →→→→ ππππ* e ππππ →→→→ ππππ*. São as transições mais importantes para espectroscopia UV-VIS dos compostos orgânicos, sendo que: • n →→→→ ππππ* ⇒⇒⇒⇒ compostos contendo orbitais ππππ e heteroátomo com elétrons não-ligados para fornecer os orbitais n (εmáx = 5 a 100 l.cm-1.mol-1);lkl
• ππππ →→→→ ππππ* ⇒⇒⇒⇒ compostos contendo grupo funcional não-saturado (εmáx 100 a 1000 vezes maior)
É importante definir agora alguns termos importantes na discussão dos espectros eletrônicos. Cromóforos São grupos insaturados covalentes responsáveis pela absorção eletrônica (ex. C=C, C=O, NO2, etc)
⇒⇒⇒⇒ Deslocamentos Batocrômico e Hipsocrômico •••• batocrômico ⇒⇒⇒⇒ bandas de absorção π → π* deslocam-se para λmáx.
maior; •••• hipsocrômico ⇒⇒⇒⇒ deslocamento das bandas de absorção
(n →→→→ ππππ*) para λmáx. mais curto;
⇒⇒⇒⇒ Efeitos Hipercrômico e Hipocrômico •••• Efeito hipercrômico ⇒⇒⇒⇒ aumento da intensidade da absorção; •••• Efeito hipocrômico ⇒⇒⇒⇒ diminuição da intensidade da absorção.
⇒⇒⇒⇒ AUXOCRÔMICOS Grupos saturados que, quando ligados a um cromóforo, modificam o comprimento e a intensidade da absorção. Ex.: OH, NH2, Cl, etc).
Este efeito pode ser verificado quando tais grupos substituem átomos de hidrogênio no anel benzênico conforme mostra a tabela abaixo:
CROMÓFORO GRUPO AUXOCROMO SOLVENTE λλλλmáx. (ηm) εεεε
Benzeno -H Ciclohexano 204 7900 Tolueno -CH3 Ciclohexano 207 7000
Clorobenzeno -Cl Etanol 210 7600 Fenol -OH Água 211 6200
Anilina -NH2 Água 230 8600 Tiofenol -SH Hexano 236 103

60
Absorção por multicromóforos não conjugados e conju gados •••• Não conjugados ⇒⇒⇒⇒ cromóforos separados por mais de uma ligação simples. Ex. CH2=CHCH2CH2CH=CH2 (λmáx em 185 ηm e εmax = 20000) •••• Conjugados ⇒⇒⇒⇒ Cromóforos se acham separados por uma ligação simples. Ex. H2C=CH–CH=CH2 (λmáx. em 217 nm e εmax = 21000) Absorção por sistemas aromáticos • Sistemas aromáticos ⇒ espectro com três bandas originadas das transições ππππ →→→→ ππππ*.
Ex. benzeno : uma banda forte em 184 ηm (εmax = 60000) e duas mais fracas, uma em 204 ηm (εmax = 7900) e outra em 256 ηm (εmax = 200). Absorção por espécies inorgânicas Os compostos inorgânicos dos elementos do bloco s e p apresentem bandas de absorção na região UV relacionados a transições n →→→→ ππππ*. Ex.: nitrato (λmáx=313ηm), carbonato (λmáx =217ηm), etc. Absorção por complexos de transferência de carga Muitos complexos devem sua capacidade absorvente a um processo de transferência de carga. Nos complexos de transferência de carga, um dos componentes deve ter a propriedade de doador de elétron e o outro de receptor. A absorção se acha relacionada com a transição de um elétron do doador a um orbital de maior energia do receptor. Assim, o estado excitado é o produto de um espécie de oxi-redução interna. A título de exemplo, consideres os complexos abaixo: [I3]
- Iodo molecular (I 2) com Iodeto (I -) [Fe(SCN)6]
3- Ferro(III) com Tiocianato (SCN -) [Fe(CN)6]
3- Ferro(III) com Cianeto (CN -) →→→→ Azul da Prússia [Fe(fen) 3]
2+ Ferro(II) com 1,10 - Fenantrolina No complexo [Fe(SCN)6]
3-, por exemplo, a absorção se relaciona com a transição de um elétron do íon Tiocianato a um orbital do íon Fe(III). Assim, o complexo resultante é uma espécie excitada com predominância de Fe(II) e o radical SCN. O elétron retorna a seu estado original após um breve período. A maioria dos complexos que apresenta bandas de transferência de carga associadas a um íon metálico que atua com aceptor de elétrons. Uma exceção é o complexo de ferro(II) com o-fenantrolina, onde o ligante é o aceptor e íon metálico é o doador. Alguns complexos orgânicos também podem exibir fortes bandas por transferência de carga como, por exemplo, os complexos de I2 com aminas.

61
A absorção por transferência de carga se caracteriza por absortividades molares muita altas para os máximos de absorção (εmáx. > 10.000).
Absorção por compostos de metais de transição “d” De acordo com as teorias do campo cristalino e do campo ligante , os orbitais d quebram a degenerescência quando formam complexos (veja a figura a seguir).
O valor da diferença de energias dos orbitais “d” , ∆∆∆∆, depende da: ♦♦♦♦ natureza dos ligantes (tamanho, forma, etc) ♦♦♦♦ sua interação com o íon metálico. Com base no estudo do espectro de absorção foi possível colocar alguns
ligantes em uma ordem crescente de valores de ∆∆∆∆, ou seja:
I-<<<<Br -<Cl-<F-<OH-<H2O<SCN-<NH3<etilenodiamina<o-fenantrolina<NO 2-<CN-
Como os valores de ∆ estão aumentando na ordem acima, os valores λmáx de um complexo com CN- (exemplo, [Fe(CN)6]
≡) aparecem em um λmáx menor do que de um complexo com Cl- (exemplo, [Fe(Cl)6]
≡).
Absorção por compostos de metais de transição “f” Os espectros consistem de bandas estreitas e bem definidas, como se pode observar na figura abaixo.
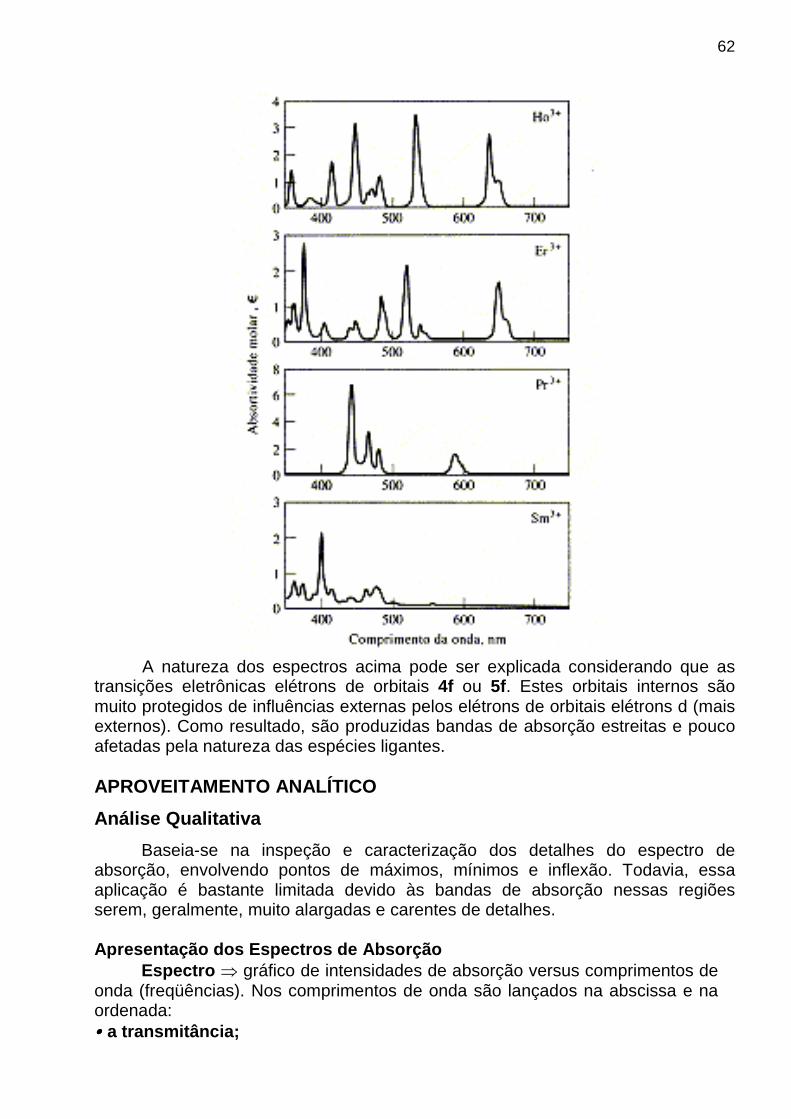
62
A natureza dos espectros acima pode ser explicada considerando que as
transições eletrônicas elétrons de orbitais 4f ou 5f. Estes orbitais internos são muito protegidos de influências externas pelos elétrons de orbitais elétrons d (mais externos). Como resultado, são produzidas bandas de absorção estreitas e pouco afetadas pela natureza das espécies ligantes. APROVEITAMENTO ANALÍTICO
Análise Qualitativa
Baseia-se na inspeção e caracterização dos detalhes do espectro de absorção, envolvendo pontos de máximos, mínimos e inflexão. Todavia, essa aplicação é bastante limitada devido às bandas de absorção nessas regiões serem, geralmente, muito alargadas e carentes de detalhes. Apresentação dos Espectros de Absorção Espectro ⇒ gráfico de intensidades de absorção versus comprimentos de onda (freqüências). Nos comprimentos de onda são lançados na abscissa e na ordenada: •••• a transmitância;
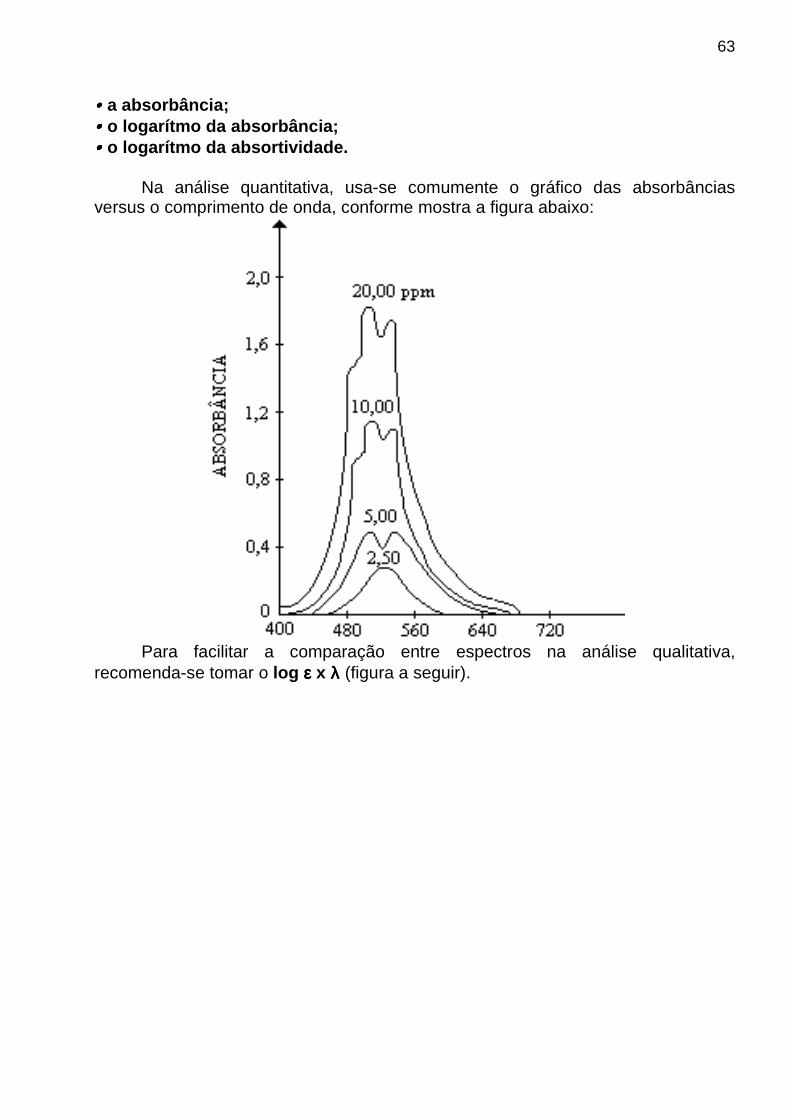
63
•••• a absorbância; •••• o logarítmo da absorbância; •••• o logarítmo da absortividade.
Na análise quantitativa, usa-se comumente o gráfico das absorbâncias
versus o comprimento de onda, conforme mostra a figura abaixo:
Para facilitar a comparação entre espectros na análise qualitativa, recomenda-se tomar o log εεεε x λλλλ (figura a seguir).
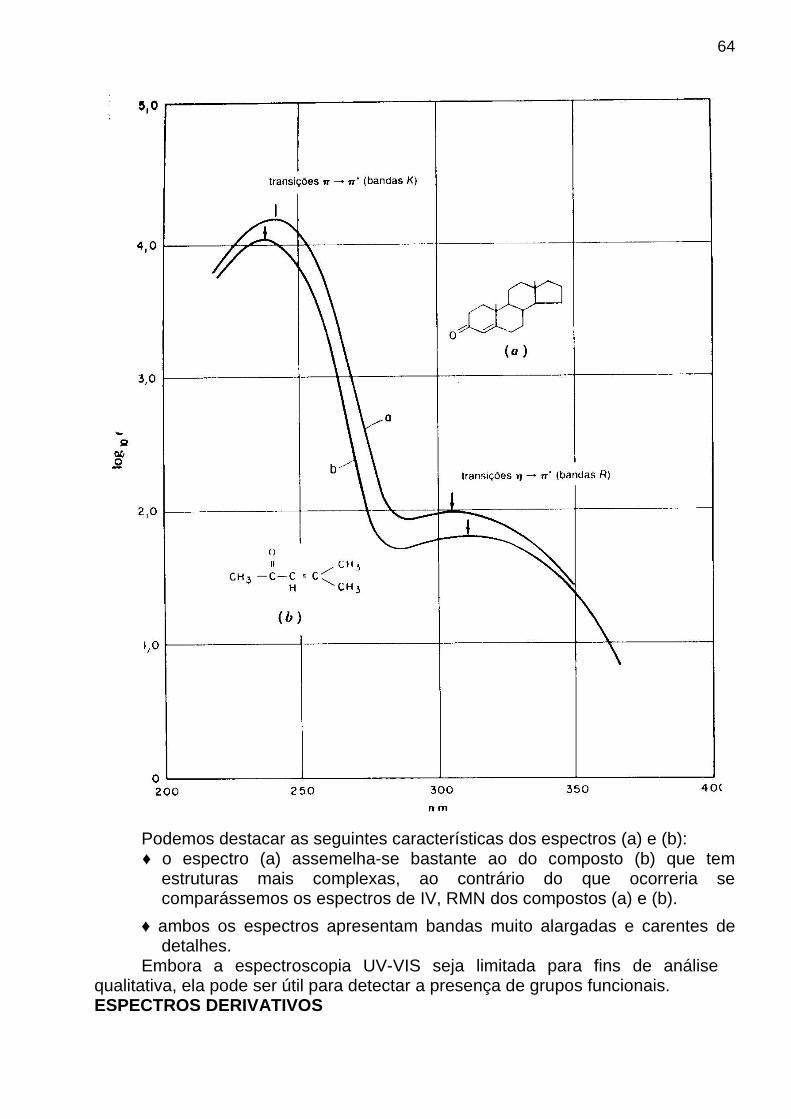
64
Podemos destacar as seguintes características dos espectros (a) e (b): ♦ o espectro (a) assemelha-se bastante ao do composto (b) que tem
estruturas mais complexas, ao contrário do que ocorreria se comparássemos os espectros de IV, RMN dos compostos (a) e (b).
♦ ambos os espectros apresentam bandas muito alargadas e carentes de detalhes.
Embora a espectroscopia UV-VIS seja limitada para fins de análise qualitativa, ela pode ser útil para detectar a presença de grupos funcionais. ESPECTROS DERIVATIVOS
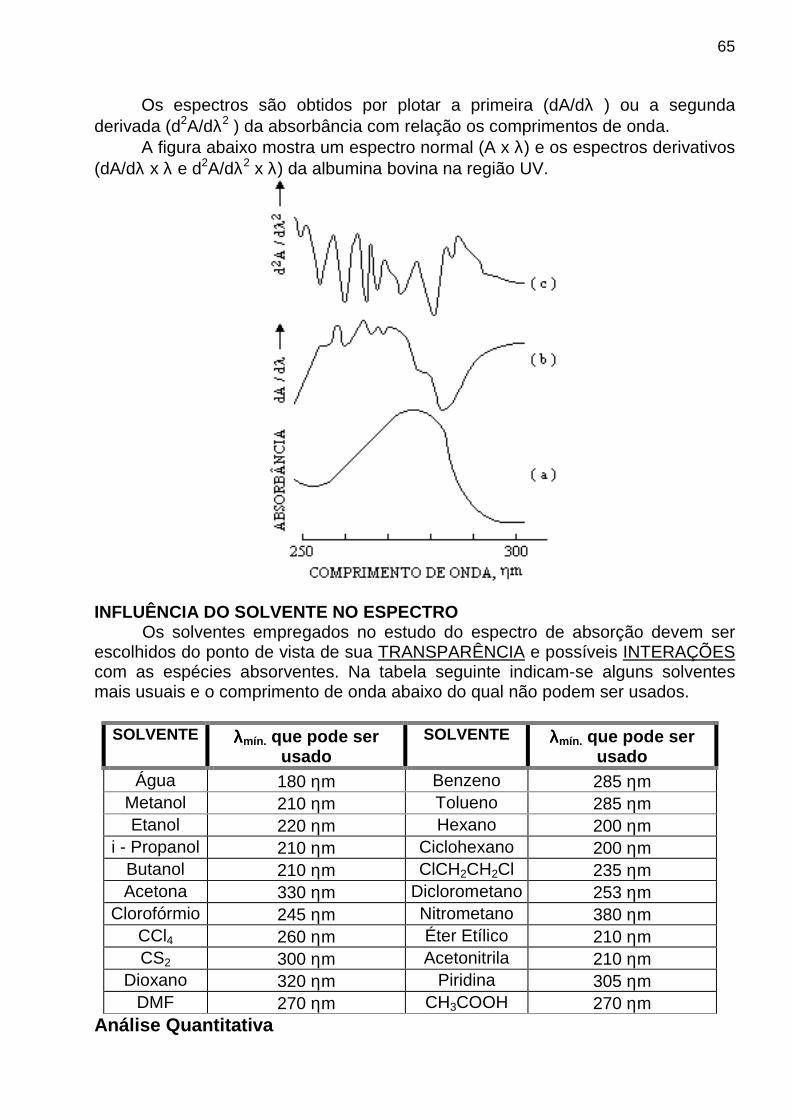
65
Os espectros são obtidos por plotar a primeira (dA/dλ ) ou a segunda derivada (d2A/dλ2 ) da absorbância com relação os comprimentos de onda.
A figura abaixo mostra um espectro normal (A x λ) e os espectros derivativos (dA/dλ x λ e d2A/dλ2 x λ) da albumina bovina na região UV.
INFLUÊNCIA DO SOLVENTE NO ESPECTRO
Os solventes empregados no estudo do espectro de absorção devem ser escolhidos do ponto de vista de sua TRANSPARÊNCIA e possíveis INTERAÇÕES com as espécies absorventes. Na tabela seguinte indicam-se alguns solventes mais usuais e o comprimento de onda abaixo do qual não podem ser usados.
SOLVENTE λλλλmín. que pode ser usado
SOLVENTE λλλλmín. que pode ser usado
Água 180 ηm Benzeno 285 ηm Metanol 210 ηm Tolueno 285 ηm Etanol 220 ηm Hexano 200 ηm
i - Propanol 210 ηm Ciclohexano 200 ηm Butanol 210 ηm ClCH2CH2Cl 235 ηm Acetona 330 ηm Diclorometano 253 ηm
Clorofórmio 245 ηm Nitrometano 380 ηm CCl4 260 ηm Éter Etílico 210 ηm CS2 300 ηm Acetonitrila 210 ηm
Dioxano 320 ηm Piridina 305 ηm DMF 270 ηm CH3COOH 270 ηm
Análise Quantitativa

66
A espectrofotometria de absorção UV-VIS é uma ferramenta muito útil para análise quantitativa. As aplicações são numerosas e cobrem os mais variados tipos de materiais. Estima-se, por exemplo, que 95% das análises quantitativas no campo da saúde (3 milhões por dia) são realizadas usando essa técnica instrumental. Estima-se que quase todos os elementos da tabela periódica podem ser determinados por essa técnica usando uma metodologia adequada. Muitas espécies orgânicas e inorgânicas são absorventes e, portanto, susceptíveis de determinação direta. As espécies que não são absorventes podem ser quantificadas após conversão em espécies absorventes por reagentes apropriados. Para espécies inorgânicas são particularmente importantes os reagentes quelantes que formam complexos corados, estáveis com cátions metálicos. Muitas espécies absorventes apresentam absortividades molares de 10.000 a 40.000 L.cm-1.mol-1, tornando comum a determinação de concentrações na faixa de 10-4 a 10-5 mol L-1, podendo muitas vezes ser estendida, com técnicas apropriadas, até 10-6 ou mesmo 10-7 mol L-1. Os métodos espectrofotométricos quantitativos estão sujeitos a erros relativos de 1 a 3% e possuem seletividade desde moderada a elevada. Todavia, esta seletividade está condicionada a vários fatores experimentais tais como: - escolha do instrumento adequado; - especificidade dos reagentes; - escolha adequada do comprimento de onda para a medida da absorbância. A LEI FUNDAMENTAL DA ESPECTROFOTOMETRIA DE ABSORÇÃO UV-Vis
Os métodos espectrofotométricos são baseados nas medidas da transmitância ou absorbância de uma radiação monocromática que atravessa uma solução contendo uma espécie absorvente e a relação entre estas medidas e a concentração da espécie absorvente.
A relação matemática entre a transmitância ou absorbância e a concentração é conhecida como Lei de Lambert - Beer ou simplesmente Lei de Beer . Transmitância - T
Quando um feixe de radiação monocromática atravessa uma solução que contém uma espécie absorvente, uma parte da energia radiante é absorvida e a outra é transmitida .
A razão da potência radiante do feixe transmitido, P, pela potência radiante do feixe incidente, Po, é conhecida como transmitância, T. Assim: T = P/Po
É também, usual expressar a transmitância percentualmente:
P %T = x 100
Po

67
Absorbância - A. É o logaritmo decimal do inverso da transmitância.
A = log(1/T) = -log(T) = log(P o/P) LEI DE BEER A lei de Beer estabelece uma relação entre a absorbância ou transmitância com a concentração de uma espécie absorvente quando um feixe de radiação monocromática atravessa um recipiente (não absorvente) contendo a espécie absorvente. A expressão matemática é:
A = log(1/T) = -log(T) = abC onde a é uma constante denominada de absortividade (quando a concentração C é dada em gramas por litro) e b é o comprimento do percurso ótico da REM. Quando a concentração for expressa em mols por litro, a absortividade é denominada de absortividade molar εεεε.e a lei de Beer é escrita como:
A = εεεε b C A absortividade molar é uma parâmetro característico de uma espécie absorvente em um meio, a um determinado λλλλ. É expressa em L • mol -1 • cm -1. A sensibilidade de um método espectrofotométrico (ou fotométrico) é governada pela absortividade molar da espécie absorvente. Nos valores tabelados especifica-se o λλλλ e o meio onde se encontra a espécie absorvente. Na realidade a absortividade molar, εεεε, depende do(a): • estrutura eletrônica da molécula absorvente, ou seja, dos tipos de transições possíveis (σσσσ →→→→ σσσσ*, ππππ →→→→ ππππ*, etc) que ela pode sofrer. • probabilidade de transição; • comprimento de onda da radiação incidente (λλλλ); • natureza solvente; • índice de refração do meio (ni). Relação entre Transmitância, Absorbância e Concentr ação. A lei de Beer pode ser expressa simplesmente como:
“ A absorbância de uma espécie absorvente , em um determinado meio, com percurso ótico e comprimento de onda fixos, varia linearmente com a concentração da espécie absorvente (A=εbc), ou a transmitância , nas mesmas condições, varia exponencialmente com a concentração da espécie absorvente (T = 10-εbc)”. As figuras a seguir mostram os gráficos destas relações:

68
A relação linear entre absorbância e concentração faz com que os instrumentos sejam calibrados em valores da absorbância , embora eles meçam na realidade a radiação transmitida. Escalas de Absorbância e Transmitância. ⇒ Por causa da escala ser uniforme (divisões igualmente espaçadas), como
mostra a figura abaixo, os instrumentos com mostradores analógicos apresentam medidas em transmitância.
ESCALA DE TRANSMITÂNCIA
ESCALA DE ABSORBÂNCIA
Os instrumentos mais modernos são digitais com medidas diretas em absorbância . DESVIOS DA LEI DE BEER
As medidas de absorbância em sistemas químicos reais não conduzem a uma completa linearidade sobre todos as faixas de concentração, pois podem ocorrer desvios conforme mostra a figura a seguir:
No desvio positivo o valor da absorbância medida é maior do que o valor esperado pelo aumento da concentração, enquanto no desvio negativo o valor é menor. DESVIOS REAIS, QUÍMICOS E INSTRUMENTAIS
Estes desvios podem ocorrer por problemas decorrentes: ♦♦♦♦ da limitação na derivação da Lei de Beer , desvios reais. ♦♦♦♦ ou por problemas relacionados com a natureza do sistema químico envolvido, desvios químicos, ♦♦♦♦ ou ainda por limitações do instrumento usado, desvios instrumentais.

69
DESVIOS REAIS À medida que se aumenta a concentração da espécie absorvente, dois
fatores que limitam a Lei de Beer podem acarretar desvio da linearidade: ♦ Interações entre Centros Absorventes. ♦ Variação do Índice de Refração.
Interações entre Centros Absorventes
A derivação da Lei de Beer pressupõe que os centros absorventes atuam independentemente uns dos outros, isto é, que eles não manifestam interações recíprocas ou com outros íons ou moléculas presentes. A rigor, a lei se aplica a soluções diluídas (concentração menor que 10-2 mol/L).
Para soluções mais concentradas, a distância média entre os centros absorventes diminui a tal ponto que as interações entre eles afetam a energia necessária para a excitação, alterando a capacidade absorvente e ocasionando um desvio da linearidade. Variação do Índice de Refração. Quando variações da concentração afetam o índice de refração, observa-se desvio da Lei de Beer, pois a absortividade molar depende do índice de refração.
( )
+η
ηε=ε22 2
' , OBS: Para concentrações menores que 10-2 mol/L, η é praticamente constante.
Desvios Instrumentais
São desvios que estão relacionados com os limites dos instrumentos usados na medida da absorbância. Dentre as principais limitações temos:
♦ a largura do feixe de radiação; ♦ a não-linearidade do detector; ♦ a instabilidade da fonte. Nos instrumentos de melhor qualidade, estas deficiências têm sido
amplamente resolvidas. Todavia merece uma atenção particular o problema relacionado a largura do feixe de radiação. LEI DE BEER E LARGURA DO FEIXE DE RADIAÇÃO
A Lei de Beer é derivada supondo um feixe de radiação monocromática. Como nos instrumentos de menor qualidade os feixes apresentam faixas de comprimento de onda, isto pode causar desvios. Uma ilustração deste fato é mostrada nas figuras a seguir:
Na faixa A , εεεε não varia significativamente e o desvio é desprezível (curva A). Na faixa B tem-se uma variação considerável de ε, que determina um desvio apreciável (curva B).
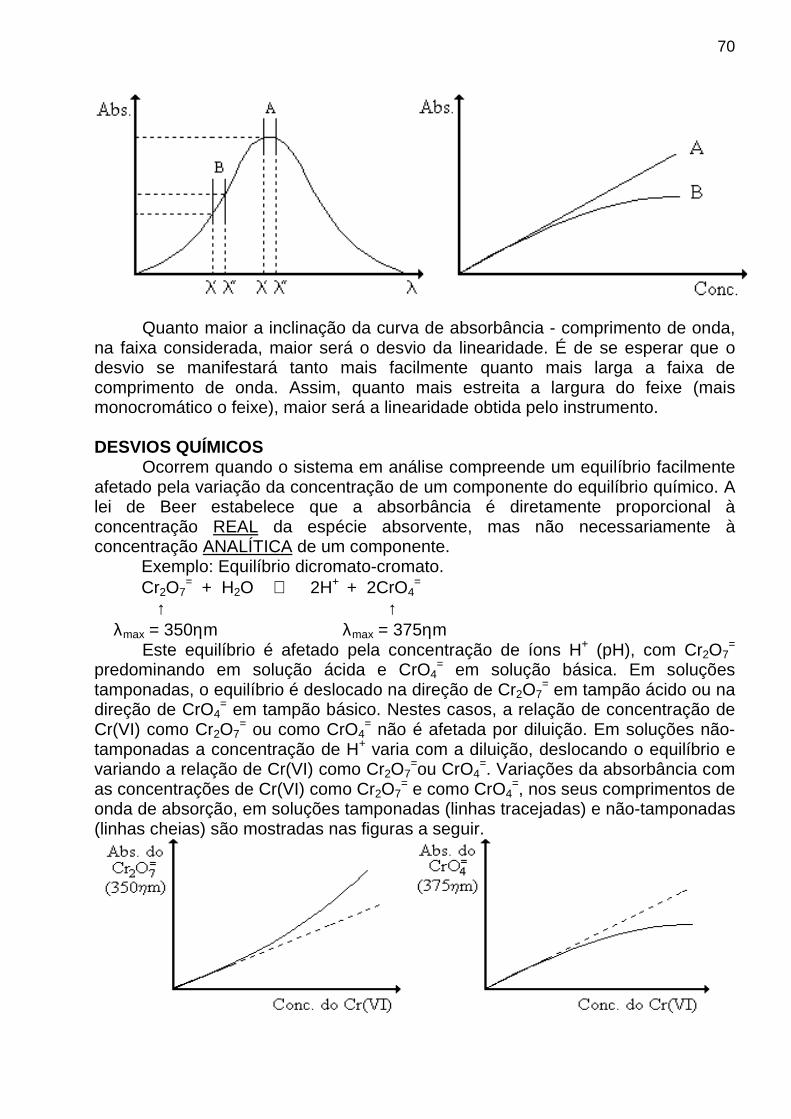
70
Quanto maior a inclinação da curva de absorbância - comprimento de onda,
na faixa considerada, maior será o desvio da linearidade. É de se esperar que o desvio se manifestará tanto mais facilmente quanto mais larga a faixa de comprimento de onda. Assim, quanto mais estreita a largura do feixe (mais monocromático o feixe), maior será a linearidade obtida pelo instrumento. DESVIOS QUÍMICOS
Ocorrem quando o sistema em análise compreende um equilíbrio facilmente afetado pela variação da concentração de um componente do equilíbrio químico. A lei de Beer estabelece que a absorbância é diretamente proporcional à concentração REAL da espécie absorvente, mas não necessariamente à concentração ANALÍTICA de um componente. Exemplo: Equilíbrio dicromato-cromato. Cr2O7
= + H2O ⇔ 2H+ + 2CrO4=
↑ ↑ λmax = 350ηm λmax = 375ηm
Este equilíbrio é afetado pela concentração de íons H+ (pH), com Cr2O7=
predominando em solução ácida e CrO4= em solução básica. Em soluções
tamponadas, o equilíbrio é deslocado na direção de Cr2O7= em tampão ácido ou na
direção de CrO4= em tampão básico. Nestes casos, a relação de concentração de
Cr(VI) como Cr2O7= ou como CrO4
= não é afetada por diluição. Em soluções não-tamponadas a concentração de H+ varia com a diluição, deslocando o equilíbrio e variando a relação de Cr(VI) como Cr2O7
=ou CrO4=. Variações da absorbância com
as concentrações de Cr(VI) como Cr2O7= e como CrO4
=, nos seus comprimentos de onda de absorção, em soluções tamponadas (linhas tracejadas) e não-tamponadas (linhas cheias) são mostradas nas figuras a seguir.
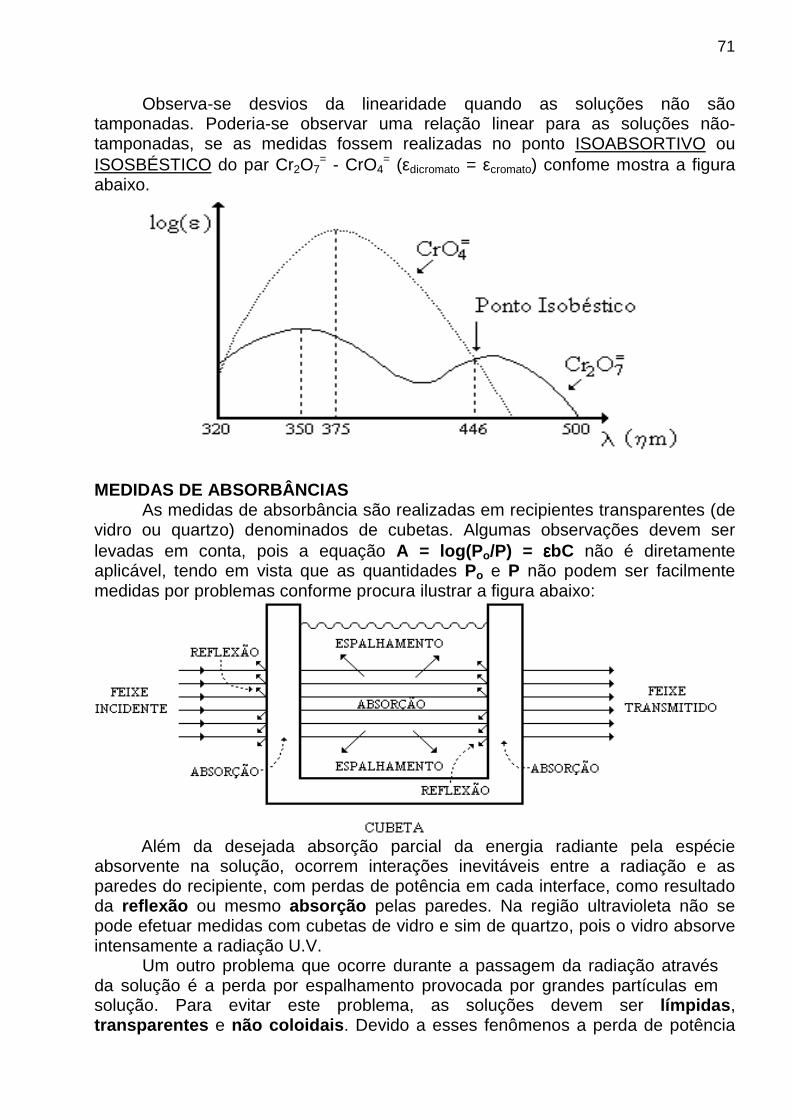
71
Observa-se desvios da linearidade quando as soluções não são tamponadas. Poderia-se observar uma relação linear para as soluções não-tamponadas, se as medidas fossem realizadas no ponto ISOABSORTIVO ou ISOSBÉSTICO do par Cr2O7
= - CrO4= (εdicromato = εcromato) confome mostra a figura
abaixo.
MEDIDAS DE ABSORBÂNCIAS
As medidas de absorbância são realizadas em recipientes transparentes (de vidro ou quartzo) denominados de cubetas. Algumas observações devem ser levadas em conta, pois a equação A = log(P o/P) = εεεεbC não é diretamente aplicável, tendo em vista que as quantidades Po e P não podem ser facilmente medidas por problemas conforme procura ilustrar a figura abaixo:
Além da desejada absorção parcial da energia radiante pela espécie absorvente na solução, ocorrem interações inevitáveis entre a radiação e as paredes do recipiente, com perdas de potência em cada interface, como resultado da reflexão ou mesmo absorção pelas paredes. Na região ultravioleta não se pode efetuar medidas com cubetas de vidro e sim de quartzo, pois o vidro absorve intensamente a radiação U.V.
Um outro problema que ocorre durante a passagem da radiação através da solução é a perda por espalhamento provocada por grandes partículas em solução. Para evitar este problema, as soluções devem ser límpidas , transparentes e não coloidais . Devido a esses fenômenos a perda de potência

72
pode chegar a mais de 10% mesmo em cubetas e solventes considerados transparentes.
Uma maneira de se corrigir os efeitos de interação da radiação com o recipiente consiste em comparar a potência do feixe transmitido através da solução em estudo, com a potência de um feixe que atravessa uma idêntica cubeta, contendo apenas o solvente (correção com o branco). INSTRUMENTAÇÃO PARA MEDIDAS DA ABSORÇÃO UV-VIS Componentes Básicos ⇒ Os instrumentos para medir transmitância ou absorbância envolvem alguns
componentes básicos, quais sejam: ♦ Uma fonte de radiação contínua; ♦ Um dispositivo para separar (“monocromar”) as radiações contínuas; ♦ Um recipiente para amostra; ♦ Um detector para converter a energia radiante em sinal elétrico; ♦ Um mostrador ou registrador para apresentar o sinal elétrico.
Fontes de Radiação
Estes dispositivos devem preencher certos requisitos:
1º) A fonte deve gerar radiação contínua
2º) A fonte deve fornecer um feixe com potência radiante suficiente para permitir uma fácil detecção;
3º) A fonte deve ser convenientemente estável de modo que a potência radiante do feixe permaneça constante durante as medidas de P0 e P.
Nos instrumentos de feixe simples, a estabilidade da fonte é essencial para a
reprodutibilidade das medidas, porém nos instrumentos de feixe duplo, em que P0 e P são medidos simultaneamente, a estabilidade da fonte não é crítica.
Tipos de Fontes de Radiação Visível
A fonte usual é uma lâmpada com filamento de tungstênio . A maioria da energia emitida se concentra no I.V, todavia, a lâmpada é usada para região entre 320 e 2.400 ηηηηm (visível e IV próximo ). A temperatura do filamento varia entre 2000 a 3000K. Utiliza-se normalmente invólucro de vidro. Para a lâmpada produzir radiação estável, é necessário um rigoroso controle da sua fonte de alimentação através de circuitos reguladores. Lâmpada de Tungstênio-Iodo Ela é uma lâmpada de tungstênio comum, contendo, em vez de vácuo, o iodo sublimado. O iodo reage com o tungstênio sublimado formando WI2, que ao se difundir, colide com o filamento quente, decompondo-se com reposição do metal sobre o filamento. Este ciclo químico interno confere à lâmpada uma vida útil duas vezes maior do que a de uma lâmpada comum. Com um invólucro de quartzo esta lâmpada pode operar de 200 a 3000ηm.

73
Fontes de Radiação UV As mais usadas são as lâmpadas de descarga de hidrogênio ou deutério
com janelas de quartzo. Quando se submete o gás hidrogênio ou deutério a uma descarga elétrica, produz-se um espectro contínuo na região UV, cobrindo a faixa de 180ηm, limite de transmissão do quartzo, até 380ηm. Acima de 380ηm a radiação deixa de ser contínua (série de Lyman, Balmer, Paschen, etc).
As lâmpadas de hidrogênio e de deutério cobrem a mesma faixa espectral, mas a potência radiante da lâmpada de deutério é maior. Espectrofotômetros que operam nas regiões UV e VISÍVEL freqüentemente requerem duas fontes:
♦ Uma lâmpada de hidrogênio ou deutério para região UV (180 a 350ηηηηm);
♦ Uma de tungstênio para a região visível (350 a 800ηηηηm). Dispositivos para Separar ou Resolver as Radiações UV-VIS (Seleção dos λλλλ)
As radiações dentro do espectro contínuo podem ser separadas utilizando: ♦ filtros óticos , cujo uso se restringe, praticamente, à região visível; ♦ monocromadores à base de prismas ou redes de difração, que
podem cobrir as regiões U.V. e visível. Filtros Óticos
São dispositivos que selecionam uma faixa espectral relativamente estreita da radiação contínua. As características de desempenho dos filtros, obtidas dos seus espectros de transmitância, são descritas em termos de:
♦ comprimento de onda nominal - aquele correspondente à transmitância máxima do filtro;
♦ largura efetiva da banda – representa a faixa de comprimentos de onda em cujos extremos a transmitância do filtro cai à metade de seu valor máximo (largura à meia altura).
Na figura abaixo, são mostradas as duas características descritas:
Existem dois tipos de filtros óticos:
♦♦♦♦filtro de absorção - são filtros que isolam uma certa banda espectral

74
absorvendo preferencialmente radiações dos demais comprimentos de onda. Eles possuem larguras efetivas de 30 a 50ηm e transmitância máximas que variam de 5 a 30 %;
♦♦♦♦filtro de interferência - o princípio destes filtros está baseado na interferência construtiva e destrutiva que ocorre durante a passagem de um feixe policromático pelo filtro. Os filtros de interferência permitem isolar bandas com larguras efetivas de 10 a 20ηm e transmitâncias máximas de 35 a 75 %.
Portanto, pode-se observar que as características espectrais dos filtros de absorção são inferiores às dos filtros de interferência. De fato, os filtros de interferência possuem maior transmitância máxima e menor largura efetiva de banda. Monocromadores
Os monocromadores servem para separar uma radiação policromática em linhas ou bandas espectrais muito estreitas.
O sistema monocromador consiste nos seguintes compo nentes básicos: ♦ Uma Fenda de Entrada, que recebe a radiação contínua da fonte e
fornece uma estreita imagem ótica; ♦ Uma Lente Colimadora, que torna paralelos os raios propagados
através da fenda de entrada; ♦ Um Elemento de Dispersão (prisma ou rede de difração), que
desdobra a radiação contínua; ♦ Uma Lente de Focagem, para focalizar a radiação desdobrada em
uma fenda de saída; ♦ Uma Fenda de Saída, que isola a linha ou banda espectral de
interesse. Monocromador à Base de Prisma ⇒ A figura a seguir representa, esquematicamente, um monocromador à base
de prisma, mostrando os cinco (5) componentes básicos. A banda espectral desejada é isolada pela fenda de saída mediante conveniente rotação do prisma.
Monocromador de Bunsen
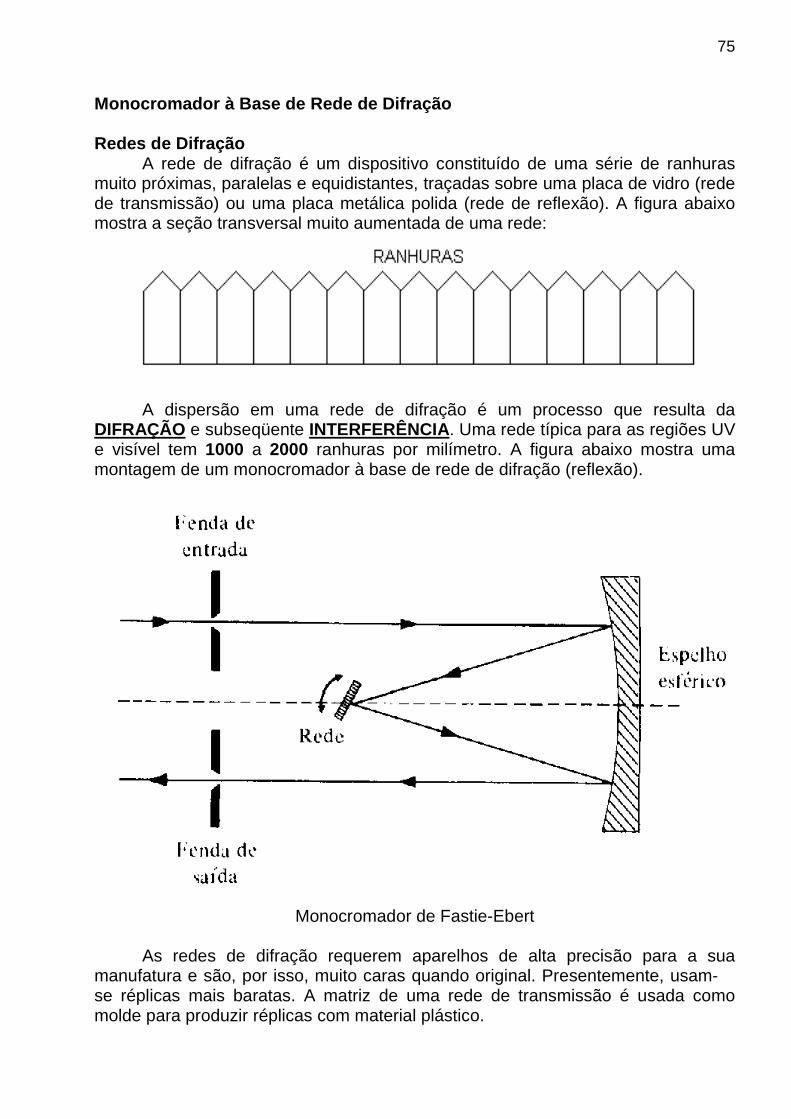
75
Monocromador à Base de Rede de Difração Redes de Difração
A rede de difração é um dispositivo constituído de uma série de ranhuras muito próximas, paralelas e equidistantes, traçadas sobre uma placa de vidro (rede de transmissão) ou uma placa metálica polida (rede de reflexão). A figura abaixo mostra a seção transversal muito aumentada de uma rede:
A dispersão em uma rede de difração é um processo que resulta da DIFRAÇÃO e subseqüente INTERFERÊNCIA. Uma rede típica para as regiões UV e visível tem 1000 a 2000 ranhuras por milímetro. A figura abaixo mostra uma montagem de um monocromador à base de rede de difração (reflexão).
Monocromador de Fastie-Ebert
As redes de difração requerem aparelhos de alta precisão para a sua
manufatura e são, por isso, muito caras quando original. Presentemente, usam-se réplicas mais baratas. A matriz de uma rede de transmissão é usada como molde para produzir réplicas com material plástico.

76
FENDAS As fendas cumprem um papel importante na determinação da qualidade do
monocromador. Cada fenda consiste em duas peças de metal com extremidades aguçadas conforme mostra a figura abaixo:
As extremidades das fendas devem ser rigorosamente paralelas uma à outra e situar-se no mesmo plano. Em alguns instrumentos as aberturas das fendas são fixas, em outros, a abertura é ajustável. A fim de um monocromador separar eficientemente os comprimentos de onda, as fendas devem ser tão estreitas quanto possível. Há, porém, uma abertura ótima para uma fenda, que se fosse tentado estreitá-la ainda mais não se obteria um melhor resultado e, o que é pior, redundaria em redução da potência de radiação, devido a problemas de difração. A largura efetiva da banda de um monocromador depende da dispersão do prisma ou da rede, bem como das aberturas das fendas de entrada e saída. Como a dispersão em um prisma não é uniforme, para se isolar uma dada largura efetiva de banda em um monocromador com prisma, é necessário usar fendas muito mais estreitas no lado dos comprimentos de onda mais longos, do que no lado dos mais curtos. Os monocromadores com rede têm a vantagem de isolar com uma abertura fina, radiação com uma largura efetiva aproximadamente constante para toda a faixa espectral de operação. RECIPIENTES PARA AMOSTRA
Os recipientes usados nas medidas espectrofotométricas são denominados de cubetas. Os instrumentos mais simples (fotômetros ou colorímetros) utilizam cubetas cilíndricas, que são mais baratas. Os espectrofotômetros utilizam normalmente cubetas retangulares com percurso óptico de 1cm. Todavia, encontra-se, comercialmente, cubetas com espessuras de 0,1cm até 10 cm. As cubetas são construídas de material transparente que deixa passar livremente a radiação na região espectral interessada. As cubetas de quartzo são usadas principalmente para medidas na região UV (abaixo de 350ηm), embora elas possam ser usadas também na região visível e no I.V (até 3µm). As cubetas de vidro são usadas apenas na região visível e no I.V. até 2µm.
As cubetas devem ser alojadas em direções perpendiculares à direção do feixe, a fim de reduzir as perdas por reflexão. As cubetas devem se encontrar perfeitamente limpas, pois as impressões digitais, manchas de gordura e qualquer material sobre as paredes da cubeta afetam de maneira acentuada as medidas.

77
Além disso, elas não devem ser secadas mediante aquecimento, pois isso poderia ocasionar a danificação física dela ou uma variação no comprimento do percurso óptico. TRANSDUTORES DE RADIAÇÃO
Os detectores de radiação UV-visível são transdutores de entrada que convertem a energia radiante em sinal elétrico. Os detectores devem apresentar as seguintes características básicas:
♦ Responder à energia radiante dentro da faixa espectral; ♦ Ser sensível para baixos níveis de potência radiante; ♦ Ter resposta muito rápida; ♦ Apresentar uma relação linear entre a potência radiante incidente e o
sinal elétrico produzido RESPOSTA LINEAR DOS TRANSDUTORES
É essencial que o sinal elétrico produzido seja uma função linear da potência do feixe incidente, ou seja,
S = k P + Sr (função de transferência do transdutor) Onde:
- S = sinal elétrico em unidades de corrente, resistência ou tensão.
- k = sensibilidade do detector em termos de resposta elétrica por unidade de potência radiante.
- Sr = sinal residual apresentado na ausência da radiação, o qual pode ser eliminado com circuitos compensadores.
A lei de Beer pode ser expressa como:
A = log (P 0/P) = log (S 0/K/S/K) => A = Log S 0/S
onde:
♦ R é a resposta para a radiação que emerge da amostra ♦ R0 é a do branco.
TIPOS DE TRANSDUTORES
Os tipos de detectores de radiação para operar nas regiões UV-VIS são:
♦ Células Fotoelétricas ou Fototubos; ♦ Tubos Fotomultiplicadores; ♦ Células Fotovoltaicas; ♦ Fotodiodo; ♦ Fototransistor.

78
CÉLULAS FOTOELÉTRICAS OU FOTOTUBOS Neste detector a energia radiante é transformada em corrente elétrica pelo
efeito fotoelétrico . Elas são constituídas de uma superfície catódica, capaz de emitir elétrons quando radiada, e um ânodo, mantido a um potencial positivo em relação ao cátodo, colocados em um tubo de vidro ou quartzo evacuado, conforme mostra a figura abaixo:
O cátodo C é uma peça metálica recoberta com um material fotoemissivo, à base de metais alcalinos (CsSbO; AgOCs; BiAgOCs). Quando a superfície fotossensível do cátodo recebe energia radiante, há emissão de elétrons que são atraídos em direção ao ânodo, resultando uma fotocorrente. A fotocorrente é diretamente proporcional à potência do feixe incidente. TUBOS FOTOMULTIPLICADORES São células fotoelétricas em múltiplos estágios. Eles combinam a emissão fotocatódica com uma enorme amplificação interna da fotocorrente primária, através de um processo multiplicativo de fluxo eletrônico em múltiplos estágios. Conforme mostra a figura abaixo, o tubo consiste em um cátodo (0), uma série de, por exemplo, nove dinodos (1 a 9) e um ânodo (10).
Figura do tubo fotomultiplicador

79
O primeiro dinodo é mantido a um potencial de 90V mais positivo que o cátodo e a mesma diferença de potencial é aplicada entre os dinodos sucessivos até o ânodo. Os elétrons primários emitidos pelo cátodo são acelerados em direção ao primeiro dinodo. Os impactos dos elétrons acelerados contra o dinodo causam a emissão de 2 a 5 elétrons secundários que são acelerados contra o segundo dinodo e assim por diante. Em cada estágio, o número de elétrons é multiplicado por 4, 5 ou mais. A multiplicação final pode ser maior que 108. A fotocorrente resultante pode ainda ser amplificada, conforme mostra a figura abaixo: Os tubos Fotomultiplicadores cobrem a região UV visível (200 - 700 ηm). Eles podem medir potências radiantes 200 vezes mais fracas do que as mensuráveis com fototubos comuns. Eles são usados, somente, com potências radiantes fracas, pois mesmos níveis de radiação moderados podem causar variações irreversíveis nas superfícies dos eletrodos. FOTODIODO O fotodiodo é um diodo semicondutor com junção PN cuja característica é operar em polarização inversa na junção. Na polarização inversa a corrente é praticamente nula, porém, se o cristal for devidamente dopado, o número de portadores aumenta tremendamente sob luz incidente, pois esse fornece energia sob forma de fótons que aumenta o número de portadores minoritários (responsáveis pela condução em polarização reversa). As figuras (1) e (2) abaixo mostram as características e detalhes da construção do fotodiodo. O símbolo está em (3).
A curva mostra que a corrente aumenta com o aumento de intensidade luminosa e é praticamente nula sob ausência de luz. A aplicação do fotodiodo se verifica também em leitura de cartões, circuitos digitais, acopladores ópticos, etc. FOTOTRANSISTOR
O fototransistor é similar ao fotodiodo, mas permite amplificar da corrente. Cada curva na figura encontra-se relacionada a uma intensidade luminosa. IB é a corrente de base gerada pela intensidade luminosa.

80
INSTRUMENTOS PARA ANÁLISE POR ABSORÇÃO UV-VIS
Os instrumentos podem ser classificados em dois grandes grupos: os instrumentos não-fotoelétricos (colorímetros ou comparadores visuais) e os fotoelétricos (espectrofotômetros, fotocolorímetros, fotômetros ou colorímetros). INSTRUMENTOS NÃO-FOTOELÉTRICOS Comparadores visuais Antes de aparecer os instrumentos fotoelétricos, as análises colorimétricas eram feitas por processos de simples comparação visual. Muitos desses sistemas ainda prevalecem devido ao aparelho ser barato e de precisão conveniente para muitas finalidades. A precisão fica em torno de ± 5-10 %, podendo ser melhorada por cuidadosa atenção dos detalhes. O princípio básico da maioria dos comparadores visuais consiste na comparação, sob condições definidas, da cor produzida pela substância em quantidade desconhecida com a mesma cor produzida por soluções de concentração conhecida da mesma substância (soluções padrão). Dentre os comparadores visuais podemos exemplificar: - os Tubos de NESSLER - o Comparador de DUBOSCQ - Os Papéis Indicadores de pH, etc Tubos de Nessler São tubos cilíndricos de vidro com uma marca gravada onde uma série de soluções-padrão é colocada até a altura exata da marca. A amostra preparada nas mesmas condições é colocada em outro tubo e comparada com os padrões olhando-se através das soluções na direção de uma fonte de luz difusa uniforme. Comparador de Duboscq O procedimento visual envolve uma variação da profundidade do líquido através do qual a luz deve passar para alcançar o olho, de modo que a intensidade da cor se igualará à do padrão. Nesse instrumento, mostrado na figura abaixo, as soluções da amostra e do padrão são colocadas em celas de fundo de vidro montadas em um suporte que permite movê-los verticalmente por um dispositivo de coroa e pinhão, munido de uma escala milimétrica. Os Cilindros de vidro fixos com extremidades polidas

81
penetram nas celas por cima. A luz de baixo passa através do fundo das celas atravessa os líquidos e os cilindros. A profundidade dos cilindros é ajustada até que as duas cores se igualem.
Comparador de Duboscq
A partir da posição das escalas de profundidade (igual aqui ao comprimento do percurso ótico) e da concentração da solução do padrão, a concentração da amostra pode ser obtida, se a Lei de Beer é obedecida, por:
AA = AP ⇒ εAbAcA = εBbBcB
como εA = εB ⇒ bAcA = bBcB Deve-se destacar que a relação é obedecida com mais exatidão se o feixe de luz monocromática (obtida com um filtro de cor adequada) for usado, e não a luz branca. OS INSTRUMENTOS FOTOELÉTRICOS
Uma classificação dos instrumentos fotoelétricos considera o tipo de dispositivo usado para selecionar a radiação eletromagnética para as medidas de transmitância ou absorbância. Assim:

82
♦ Quando o dispositivo é um filtro ótico denomina-se o instrumento de fotômetro , fotocolorímetro ou por operarem apenas no visível, colorímetro .
♦ Quando o dispositivo é um monocromador de prisma ou de rede de difração denomina-se de espectrofotômetro .
FOTÔMETRO OU FOTOCOLORÍMETRO
São instrumentos simples, baratos e de fácil manutenção. Eles são usados convenientemente sempre que não se requeiram faixas espectrais muito estreitas. Não costumam operar fora da região visível e não alcançam o grau de precisão dos espectrofotômetros. Os fotocolorímetros são construídos segundo modelos de feixe simples ou duplo. FOTÔMETROS DE FEIXE SIMPLES
A figura abaixo representa um diagrama de um instrumento deste tipo:
Diagrama de um Fotocolorímetro de Feixe Simples
A análise usando um instrumento de feixe simples é feita em três etapas: ♦ Inicialmente, com a célula fotovoltaica bloqueada, o miliamperímetro é
ajustado mecanicamente para 0 % T; ♦ Em seguida, com a cubeta contendo o solvente, regula-se a abertura
variável de modo a obter 100 % T; ♦ Finalmente, substitui-se o solvente pelas soluções-padrão de
calibração e a solução da amostra . O modelo acima requer o funcionamento dos componentes elétricos em
regime razoavelmente estável enquanto se completam as três etapas da medida. A transmitância é medida com uma precisão de cerca de ± 2 % T. FOTÔMETRO DE FEIXE DUPLO Um diagrama de um instrumento deste tipo é mostrado na figura seguir:
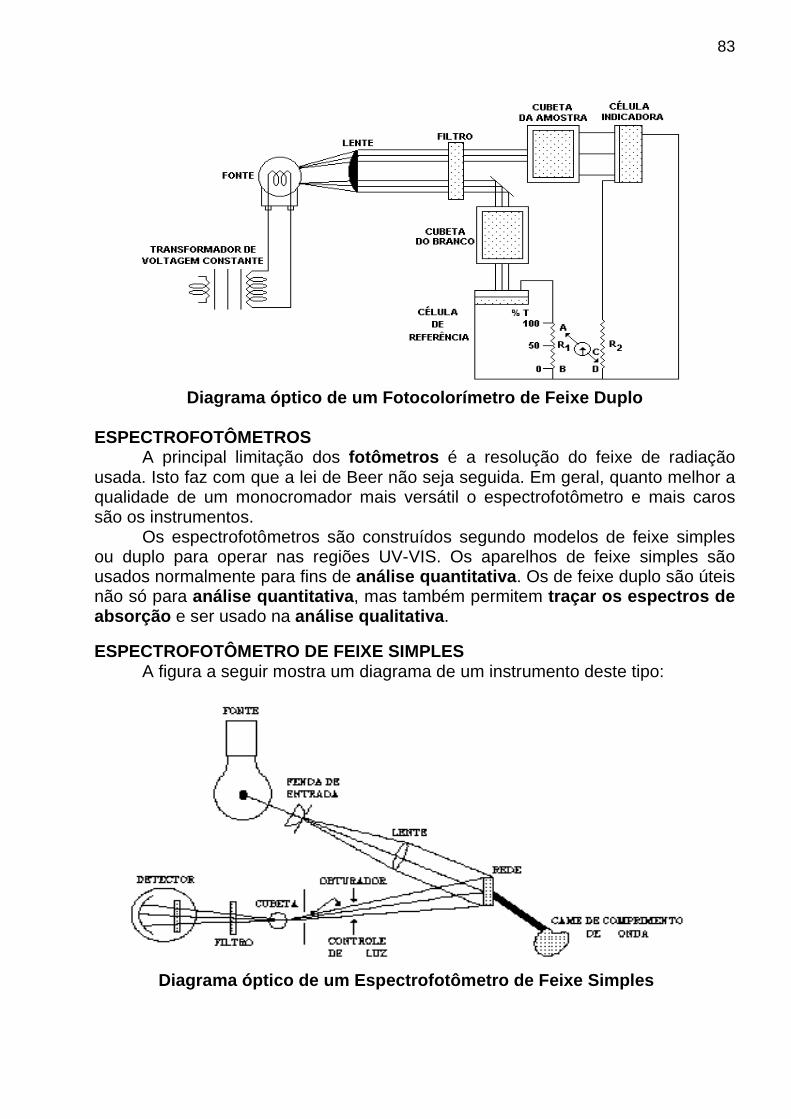
83
Diagrama óptico de um Fotocolorímetro de Feixe Dupl o
ESPECTROFOTÔMETROS
A principal limitação dos fotômetros é a resolução do feixe de radiação usada. Isto faz com que a lei de Beer não seja seguida. Em geral, quanto melhor a qualidade de um monocromador mais versátil o espectrofotômetro e mais caros são os instrumentos.
Os espectrofotômetros são construídos segundo modelos de feixe simples ou duplo para operar nas regiões UV-VIS. Os aparelhos de feixe simples são usados normalmente para fins de análise quantitativa . Os de feixe duplo são úteis não só para análise quantitativa , mas também permitem traçar os espectros de absorção e ser usado na análise qualitativa . ESPECTROFOTÔMETRO DE FEIXE SIMPLES
A figura a seguir mostra um diagrama de um instrumento deste tipo:
Diagrama óptico de um Espectrofotômetro de Feixe Si mples

84
A radiação é selecionada através do giro adequado do came (peça giratória que faz a rede se movimentar). O filtro suplementar é usado para reduzir radiação estranha. ESPECTROFOTÔMETRO DE FEIXE DUPLO
Nestes instrumentos o feixe de radiação proveniente da fonte é dividido em dois: um passa pela cubeta das soluções-padrão ou amostra e o outro pela cubeta que contém o solvente. Nos instrumentos de feixe duplo (espectrofotômetros e fotômetros), as possíveis flutuações da fonte e do detector podem ser corrigidas, pois as medidas de P (radiação que passa pela cubeta da amostra) e P0 (cubeta do branco) são efetuadas simultaneamente. ESPECTROFOTÔMETRO DE VARREDURA
São espectrofotômetros de duplo feixe usados particularmente para traçar os espectros de absorção dos sistemas analíticos. Para isso, eles possuem um mecanismo que faz variar o comprimento de onda do feixe a partir de rotação do prisma ou da rede, como se pode ver na figura mostrada a seguir.
ESPECTROFOTÔMETRO COM ARRANJO DE FOTODIODOS A figura a seguir mostra um diagrama esquemático deste instrumento: O arranjo de fotodiodos é um conjunto de mais de 300 fotodiodos montados em uma pastilha (“CHIP”) semicondutora com 2mm x 18mm de tamanho. Cada fotodiodo é responsável pelo sinal de um comprimento de onda. O feixe colimado passa na amostra, colocada num compartimento aberto e antes de atingir o arranjo de fotodiodos (“photodiode array”), passa por uma rede de difração holográfica onde é dispersado. Isto permite acesso simultâneo a todos os λ (190 a 820 ηm) e uma grande diminuição do tempo de varredura (0,1s) do espectro. Para melhorar a relação sinal/ruído os espectros são varridos durante 1s ou mais e são estocados no microcomputador, calculando depois a média. A utilização de poucos componentes óticos permite que uma única lâmpada de deutério tenha potência suficiente para servir para a região UV e Visível.
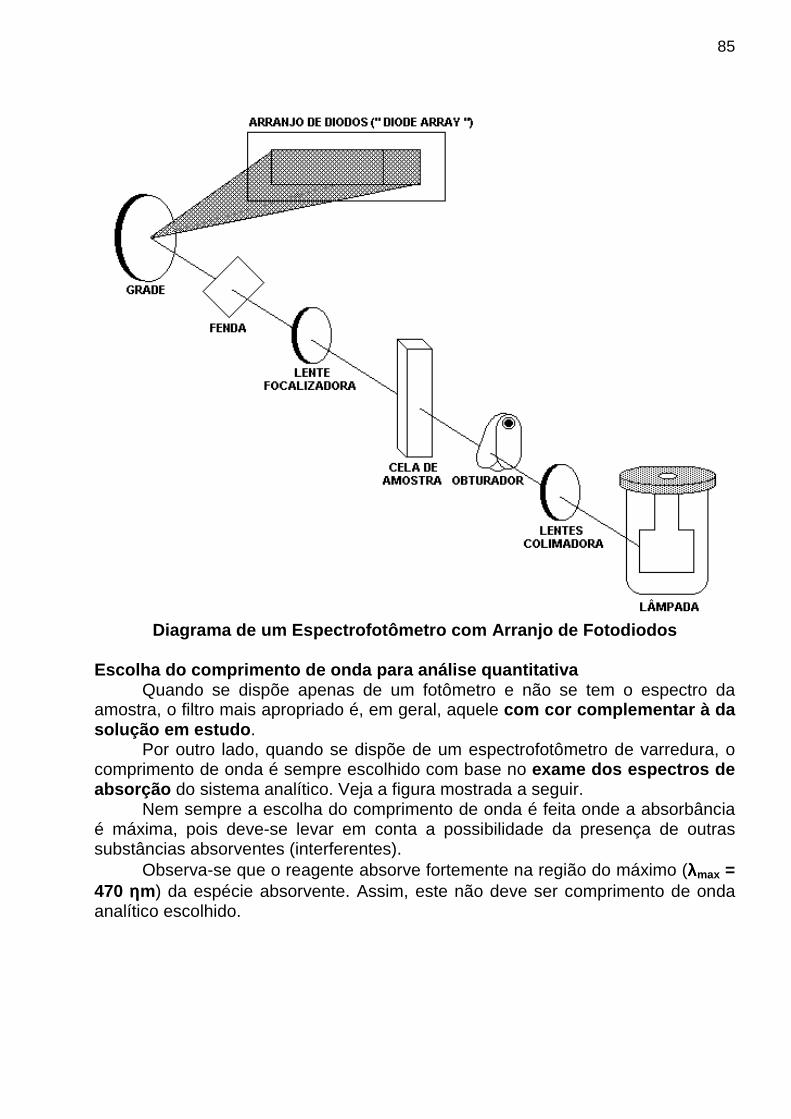
85
Diagrama de um Espectrofotômetro com Arranjo de Fot odiodos
Escolha do comprimento de onda para análise quantit ativa
Quando se dispõe apenas de um fotômetro e não se tem o espectro da amostra, o filtro mais apropriado é, em geral, aquele com cor complementar à da solução em estudo .
Por outro lado, quando se dispõe de um espectrofotômetro de varredura, o comprimento de onda é sempre escolhido com base no exame dos espectros de absorção do sistema analítico. Veja a figura mostrada a seguir.
Nem sempre a escolha do comprimento de onda é feita onde a absorbância é máxima, pois deve-se levar em conta a possibilidade da presença de outras substâncias absorventes (interferentes).
Observa-se que o reagente absorve fortemente na região do máximo (λλλλmax = 470 ηηηηm) da espécie absorvente. Assim, este não deve ser comprimento de onda analítico escolhido.
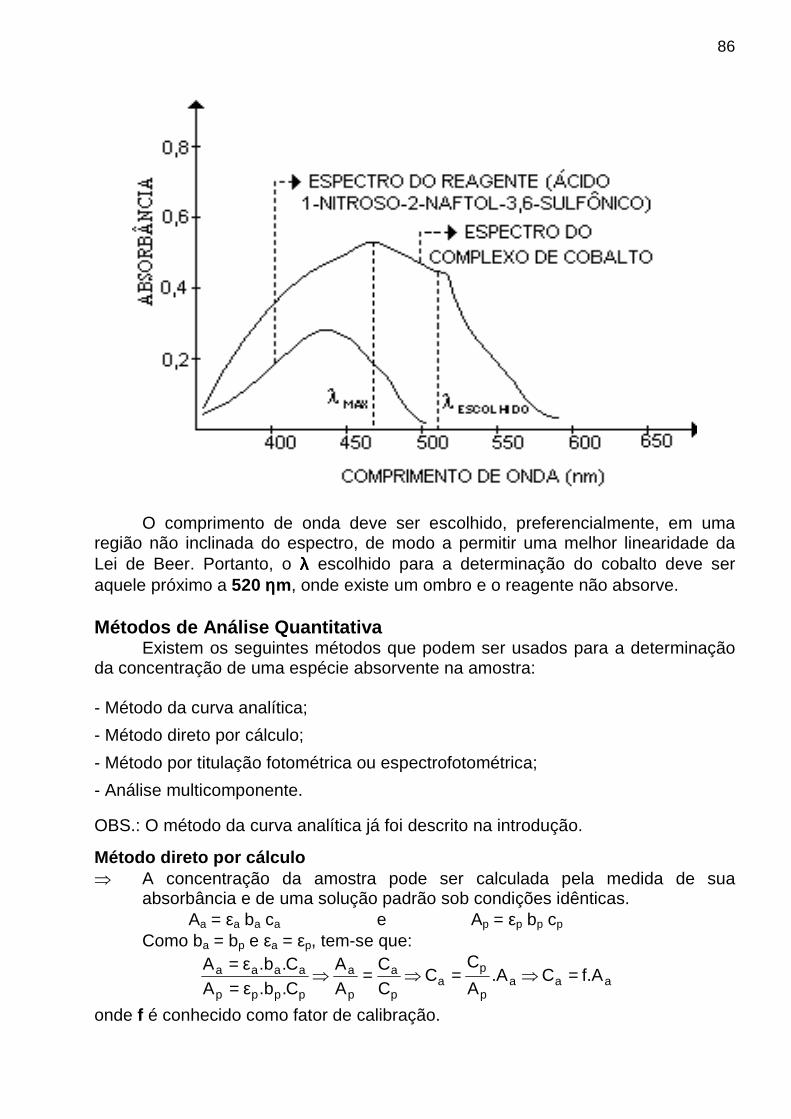
86
O comprimento de onda deve ser escolhido, preferencialmente, em uma região não inclinada do espectro, de modo a permitir uma melhor linearidade da Lei de Beer. Portanto, o λλλλ escolhido para a determinação do cobalto deve ser aquele próximo a 520 ηηηηm, onde existe um ombro e o reagente não absorve. Métodos de Análise Quantitativa
Existem os seguintes métodos que podem ser usados para a determinação da concentração de uma espécie absorvente na amostra: - Método da curva analítica;
- Método direto por cálculo;
- Método por titulação fotométrica ou espectrofotométrica;
- Análise multicomponente.
OBS.: O método da curva analítica já foi descrito na introdução.
Método direto por cálculo ⇒ A concentração da amostra pode ser calculada pela medida de sua
absorbância e de uma solução padrão sob condições idênticas. Aa = εa ba ca e Ap = εp bp cp
Como ba = bp e εa = εp, tem-se que:
aaap
pa
p
a
p
a
pppp
aaaa f.A C A.A
CC
CC
AA
C..bAC..b=A =⇒=⇒=⇒
ε=ε
onde f é conhecido como fator de calibração.
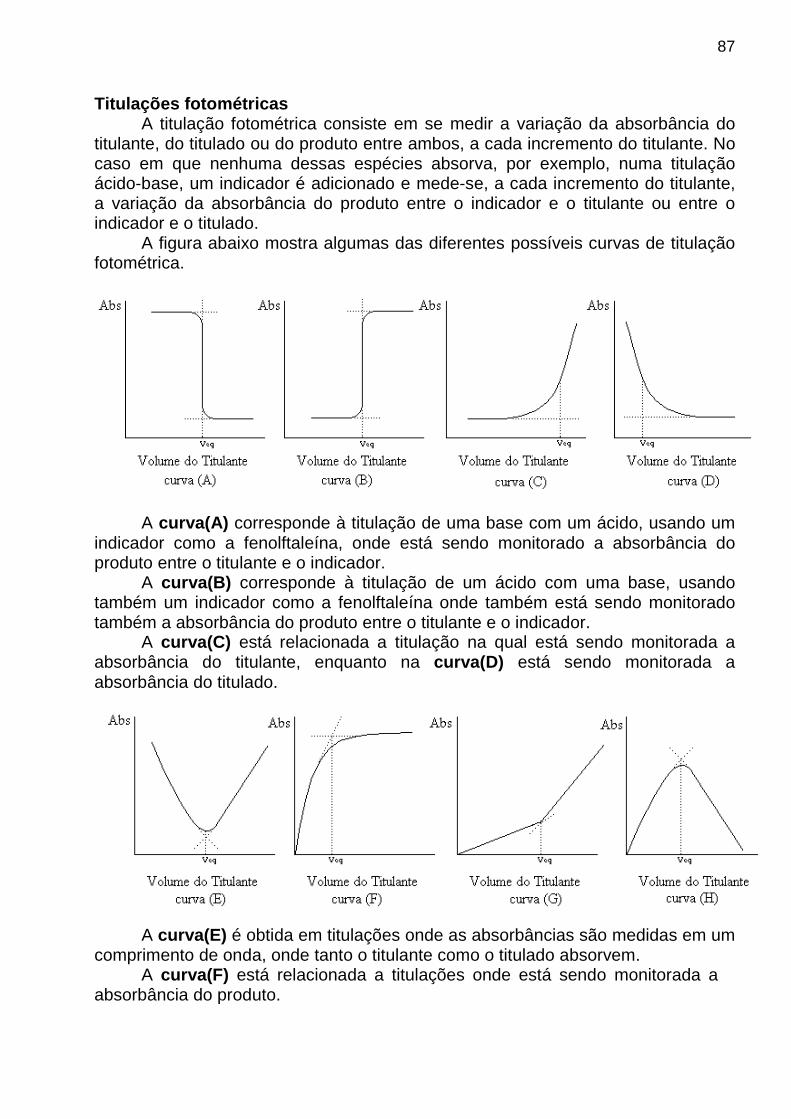
87
Titulações fotométricas A titulação fotométrica consiste em se medir a variação da absorbância do titulante, do titulado ou do produto entre ambos, a cada incremento do titulante. No caso em que nenhuma dessas espécies absorva, por exemplo, numa titulação ácido-base, um indicador é adicionado e mede-se, a cada incremento do titulante, a variação da absorbância do produto entre o indicador e o titulante ou entre o indicador e o titulado. A figura abaixo mostra algumas das diferentes possíveis curvas de titulação fotométrica.
A curva(A) corresponde à titulação de uma base com um ácido, usando um indicador como a fenolftaleína, onde está sendo monitorado a absorbância do produto entre o titulante e o indicador. A curva(B) corresponde à titulação de um ácido com uma base, usando também um indicador como a fenolftaleína onde também está sendo monitorado também a absorbância do produto entre o titulante e o indicador. A curva(C) está relacionada a titulação na qual está sendo monitorada a absorbância do titulante, enquanto na curva(D) está sendo monitorada a absorbância do titulado.
A curva(E) é obtida em titulações onde as absorbâncias são medidas em um comprimento de onda, onde tanto o titulante como o titulado absorvem. A curva(F) está relacionada a titulações onde está sendo monitorada a absorbância do produto.

88
A curva(G) corresponde a titulações nas quais tanto o produto como o titulante absorvem, porém ambos tem diferentes absortividades molares. A curva(H) é produzida em titulações nas quais sucessivos complexos são formados e apenas um complexo absorve. O máximo neste caso representa a completa conversão da forma absorvente. Verifica-se que em todas as curvas de titulação são obtidas duas linhas retas com diferentes inclinações antes e após o ponto de equivalência. Observa-se, também, uma certa curvatura na região em torno do ponto de equivalência devido a problemas relacionados com o equilíbrio químico próximo a este ponto. Por isso, o ponto final de titulação é obtido pela interseção entre os dois segmentos lineares extrapolados. Correção de volume Como a absorbância depende da concentração, é necessário levar em conta o efeito de diluição causado pela adição do titulante. Por isso, as absorbâncias medidas em cada adição de titulante devem ser corrigidas. Isto é feito multiplicando cada absorbância medida pelo fator (V + v i) / V,. de forma que cada absorbância corrigida, CORRA , é calculada pela equação:
+=V
vV iMCORR AA
onde, MA é a absorbância medida a cada volume v i da solução titulante adicionada e V é o volume inicial do titulado. A correção de volume pode ser ignorada quando se utiliza a concentração do titulante muito concentrada em comparação com a concentração do titulado. Por exemplo, no caso em que o volume do titulado V é maior do que 10 ml, o titulante é adicionado usando volumes na faixa de microlitros. Vantagens da titulação fotométrica As titulações fotométricas apresentam uma série de vantagens com relação às análises fotométricas diretas ou por curva de calibração:
• A presença de outras espécies absorventes no mesmo comprimento de onda não é um problema, pois o que importa para a titulação fotométrica, é a variação da absorbância antes e após o ponto final. Em outras palavras, as titulações fotométricas são menos susceptíveis a problemas de interferência espectral quando comparadas aos métodos diretos de calibração. Entretanto, a absorbância das substâncias estranhas não deve ser grande, a fim de que as medidas de absorbância não venham ser feitas na região indesejável da escala (A > 2,0);
• A escolha do comprimento de onda não tem que ser feita necessariamente no comprimento de onda de máxima absorbância, uma vez que o fator importante é a forma da variação da absorbância antes e após o ponto final da titulação. Freqüentemente, comprimentos de onda bem afastados do máximo de absorção são escolhidos, principalmente, para permitir medidas em soluções de alta concentração;

89
• Como é necessário o monitoramente de uma única espécie absorvente, que pode ser a espécie titulada, o titulante ou produto da reação entre ambos ou ainda o produto entre uma dessas espécies e uma substância indicadora, a titulação fotométrica se aplica à determinação de muitas substâncias não-absorventes.
• A localização do ponto final, à base de uma série de medidas requeridas para traçar a curva de titulação implica em uma precisão maior do que a de uma medida isolada. De fato, é possível alcançar uma precisão de 0,5% ou melhor nas titulações fotométricas.
O método é particularmente aplicável quando a variação da cor do indicador, em uma titulação convencional não é nítida em virtude, provavelmente, das reações de titulação não serem completas nas vizinhanças do ponto de equivalência, ou ainda, quando se encontram presentes na amostra substâncias coradas. Dispositivos para titulação fotométrica Além dos componentes dos fotômetros ou dos espectrofotômetros, na realização de uma titulação fotométrica necessita-se de alguns dispositivos extras, tais como: - um recipiente para a titulação com capacidade de 5 a 100 ml; - um agitador magnético ou outro meio apropriado para a agitação da solução - uma bureta ou outro dispositivo automático de adição de volumes. Vale salientar que estes dispositivos não são vendidos juntos com os fotômetros ou espectrofotômetros. Análise multicomponente Este tipo de análise ocorre quando se deseja determinar simultaneamente a concentração individual de dois ou mais componentes absorventes que possuem espectros de absorção sobrepostos. O caso mais simples é o de um sistema contendo duas espécies absorventes, ou seja, dois analitos. Na figura a seguir são mostrados os espectros de absorção de dois componentes, L e M, bem como o espectro da mistura entre ambos.
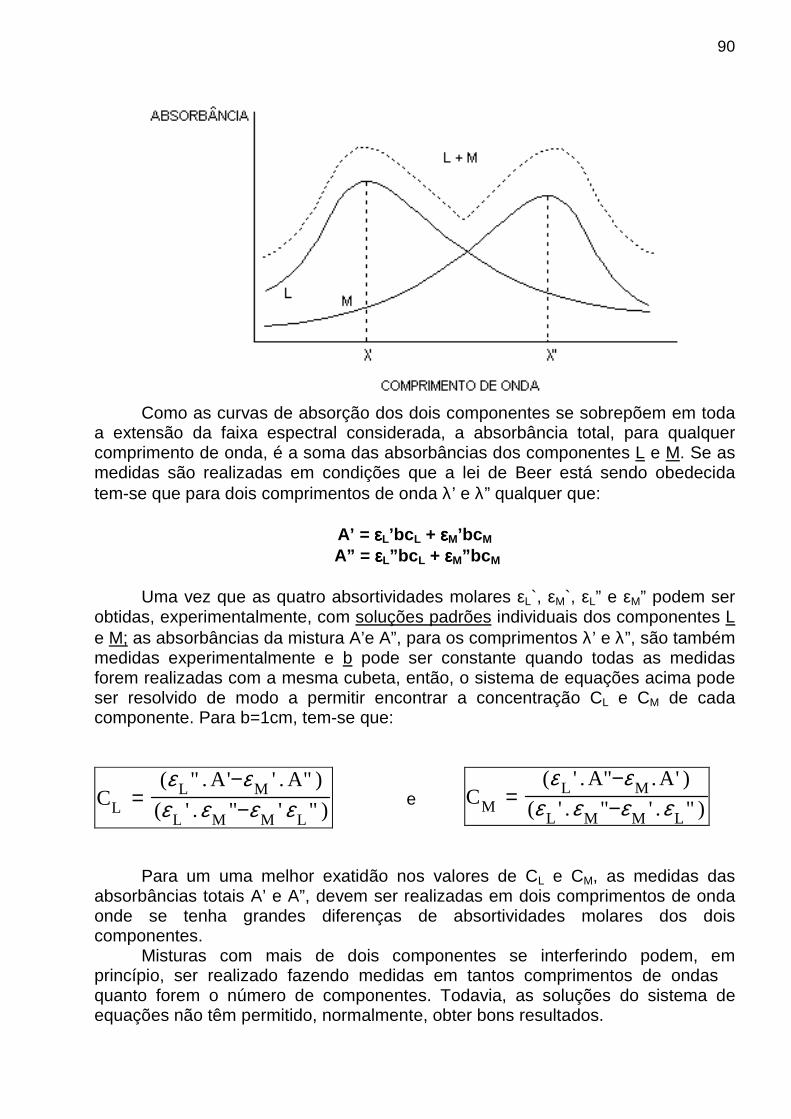
90
Como as curvas de absorção dos dois componentes se sobrepõem em toda a extensão da faixa espectral considerada, a absorbância total, para qualquer comprimento de onda, é a soma das absorbâncias dos componentes L e M. Se as medidas são realizadas em condições que a lei de Beer está sendo obedecida tem-se que para dois comprimentos de onda λ’ e λ” qualquer que:
A’ = εεεεL’bc L + εεεεM’bc M A” = εεεεL”bc L + εεεεM”bc M
Uma vez que as quatro absortividades molares εL`, εM`, εL” e εM” podem ser obtidas, experimentalmente, com soluções padrões individuais dos componentes L e M; as absorbâncias da mistura A’e A”, para os comprimentos λ’ e λ”, são também medidas experimentalmente e b pode ser constante quando todas as medidas forem realizadas com a mesma cubeta, então, o sistema de equações acima pode ser resolvido de modo a permitir encontrar a concentração CL e CM de cada componente. Para b=1cm, tem-se que:
e CA A
ML M
L M M L=
−−
( ' . " . ' )
( ' . " ' . " )
ε εε ε ε ε
Para um uma melhor exatidão nos valores de CL e CM, as medidas das absorbâncias totais A’ e A”, devem ser realizadas em dois comprimentos de onda onde se tenha grandes diferenças de absortividades molares dos dois componentes. Misturas com mais de dois componentes se interferindo podem, em princípio, ser realizado fazendo medidas em tantos comprimentos de ondas quanto forem o número de componentes. Todavia, as soluções do sistema de equações não têm permitido, normalmente, obter bons resultados.
CA A
LL M
L M M L=
−−
( " . ' ' . " )
( ' . " ' " )
ε εε ε ε ε
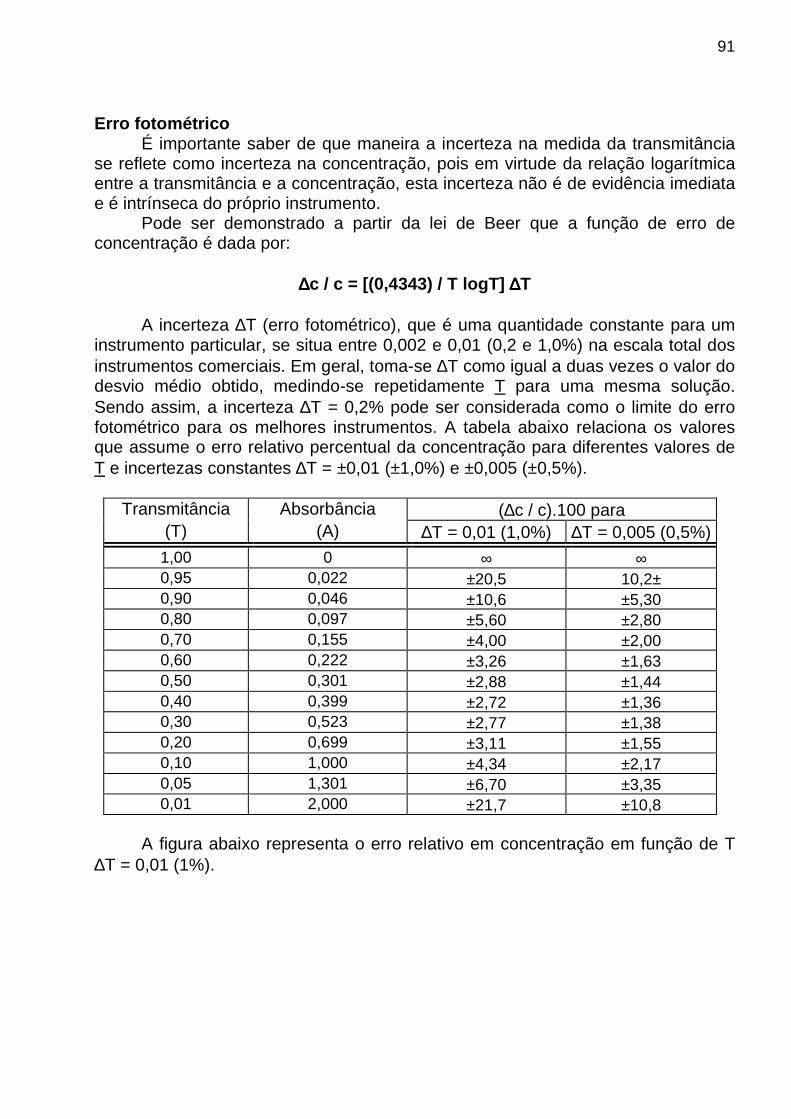
91
Erro fotométrico É importante saber de que maneira a incerteza na medida da transmitância se reflete como incerteza na concentração, pois em virtude da relação logarítmica entre a transmitância e a concentração, esta incerteza não é de evidência imediata e é intrínseca do próprio instrumento. Pode ser demonstrado a partir da lei de Beer que a função de erro de concentração é dada por:
∆∆∆∆c / c = [(0,4343) / T logT] ∆∆∆∆T
A incerteza ∆T (erro fotométrico), que é uma quantidade constante para um instrumento particular, se situa entre 0,002 e 0,01 (0,2 e 1,0%) na escala total dos instrumentos comerciais. Em geral, toma-se ∆T como igual a duas vezes o valor do desvio médio obtido, medindo-se repetidamente T para uma mesma solução. Sendo assim, a incerteza ∆T = 0,2% pode ser considerada como o limite do erro fotométrico para os melhores instrumentos. A tabela abaixo relaciona os valores que assume o erro relativo percentual da concentração para diferentes valores de T e incertezas constantes ∆T = ±0,01 (±1,0%) e ±0,005 (±0,5%).
Transmitância Absorbância (∆c / c).100 para (T) (A) ∆T = 0,01 (1,0%) ∆T = 0,005 (0,5%)
1,00 0 ∞ ∞ 0,95 0,022 ±20,5 10,2± 0,90 0,046 ±10,6 ±5,30 0,80 0,097 ±5,60 ±2,80 0,70 0,155 ±4,00 ±2,00 0,60 0,222 ±3,26 ±1,63 0,50 0,301 ±2,88 ±1,44 0,40 0,399 ±2,72 ±1,36 0,30 0,523 ±2,77 ±1,38 0,20 0,699 ±3,11 ±1,55 0,10 1,000 ±4,34 ±2,17 0,05 1,301 ±6,70 ±3,35 0,01 2,000 ±21,7 ±10,8
A figura abaixo representa o erro relativo em concentração em função de T ∆T = 0,01 (1%).
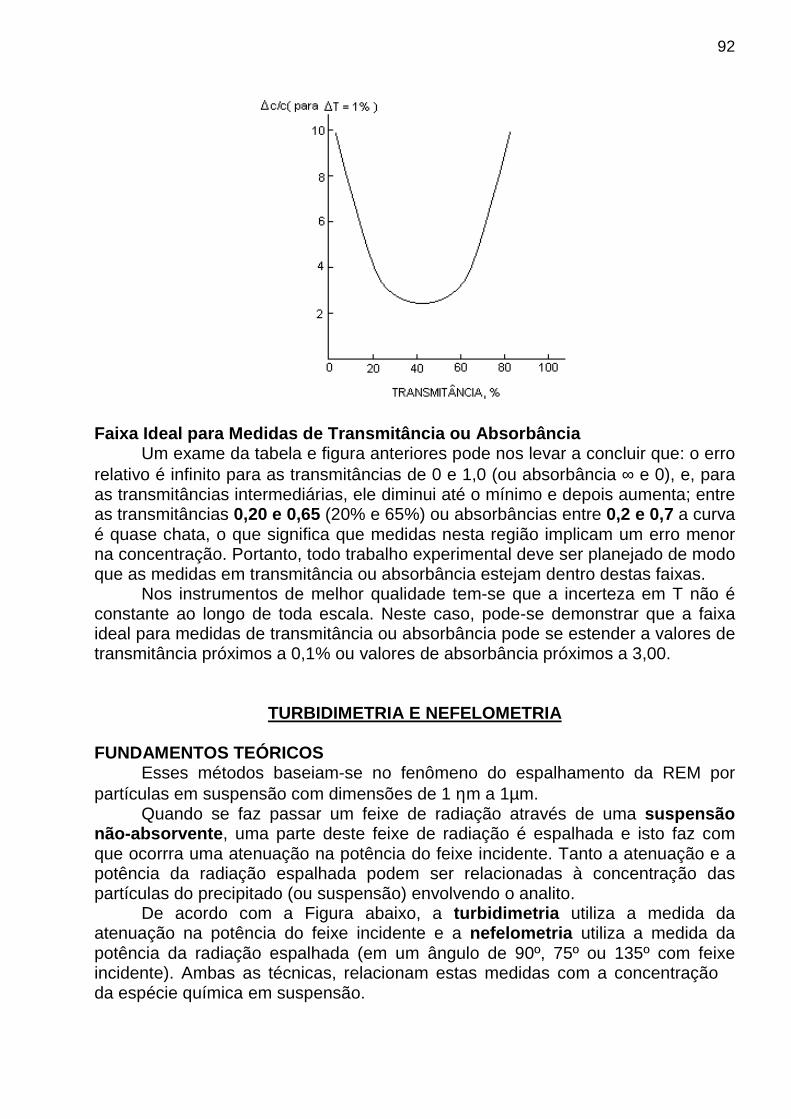
92
Faixa Ideal para Medidas de Transmitância ou Absorb ância Um exame da tabela e figura anteriores pode nos levar a concluir que: o erro relativo é infinito para as transmitâncias de 0 e 1,0 (ou absorbância ∞ e 0), e, para as transmitâncias intermediárias, ele diminui até o mínimo e depois aumenta; entre as transmitâncias 0,20 e 0,65 (20% e 65%) ou absorbâncias entre 0,2 e 0,7 a curva é quase chata, o que significa que medidas nesta região implicam um erro menor na concentração. Portanto, todo trabalho experimental deve ser planejado de modo que as medidas em transmitância ou absorbância estejam dentro destas faixas. Nos instrumentos de melhor qualidade tem-se que a incerteza em T não é constante ao longo de toda escala. Neste caso, pode-se demonstrar que a faixa ideal para medidas de transmitância ou absorbância pode se estender a valores de transmitância próximos a 0,1% ou valores de absorbância próximos a 3,00.
TURBIDIMETRIA E NEFELOMETRIA FUNDAMENTOS TEÓRICOS Esses métodos baseiam-se no fenômeno do espalhamento da REM por partículas em suspensão com dimensões de 1 ηm a 1µm. Quando se faz passar um feixe de radiação através de uma suspensão não-absorvente , uma parte deste feixe de radiação é espalhada e isto faz com que ocorrra uma atenuação na potência do feixe incidente. Tanto a atenuação e a potência da radiação espalhada podem ser relacionadas à concentração das partículas do precipitado (ou suspensão) envolvendo o analito. De acordo com a Figura abaixo, a turbidimetria utiliza a medida da atenuação na potência do feixe incidente e a nefelometria utiliza a medida da potência da radiação espalhada (em um ângulo de 90º, 75º ou 135º com feixe incidente). Ambas as técnicas, relacionam estas medidas com a concentração da espécie química em suspensão.

93
Medidas Turbidimétricas e a Concentração Na turbidimetria, a medida da atenuação da potência do feixe incidente segue uma relação linear com a concentração das partículas responsáveis pelo espalhamento, em uma equação análoga à Lei de Beer, ou seja,
τ = =logP
P kbC0
onde, τ é a turbidância , P0 é a potência do feixe incidente e P é a potência do feixe transmitido. C é a concentração de partículas em suspensão, b é o caminho ótico e k é um coeficiente de proporcionalidade, denominada de turbidez , que depende do tamanho das partículas e do comprimento de onda da radiação incidente. Medidas Nefelométricas e a Concentração Na nefelometria a medida da potência da radiação espalhada Pesp, segue uma relação linear com a concentração das partículas responsáveis pelo espalhamento, em uma equação análoga a fluorometria, ou seja,
Pesp = k P0. C onde, C é a concentração da espécie química em suspensão, P0 é potência da radiação incidente e k é um coeficiente de proporcionalidade que depende do ângulo de observação, do tamanho e forma das partículas, do comprimento de onda da radiação inicidente e dos índices de refração relativo das partículas e do meio. Para uma potência incidente, P0, constante, tem-se que:

94
ζζζζ = k’ C onde, k’é uma constante que depende dos mesmos fatores de k acima, além de depender também da potência do feixe incidente. Faixas de Concentração Versus Turbidimetria e Nefel ometria A medida nefelométrica não é indicada para altas concentrações em virtude da interferência entre partículas. Todavia, como a fluorometria, ela apresenta alta sensibilidade para baixas concentrações e a sensibilidade pode ser aumentada com o aumento da potência do feixe incidente. É difícil detectar pequenas atenuações da radiação incidente, causadas por baixas concentrações, ou seja, a turbimetria não é adequada para medidas de turvações muito fracas (transmitâncias superiores a 90 %). Nestes casos, o método nefelométrico é o mais conveniente. Portanto, nas análises de amostras em suspensão a nefelometria e a turbidimetria se completam com relação as faixas de concentração. A nefelometria é indicada para baixas concentrações, podendo determinar concentrações a nível de ppm, enquanto a turbidimetria é indicada para concentrações mais altas. INSTRUMENTAÇÃO Em princípio, qualquer fotocolorímetro ou espectrofotômetro permite realizar medidas trubidimétricas. Comparadores visuais são também utilizados para medidas de turbidez das águas potáveis. As medidas nefelométricas podem ser realizadas em fluorômetros ou espectrofluorômetros. Turbidímetros e nefelômetros têm sido desenvolvidos usando fonte de luz policromáticas, tal como uma lâmpada de tungstênio. Todavia, para se obter melhores resultados é recomendado usar radiações monocromáticas. Quanto menor o λ da radiação maior é a intensidade do espalhamento. Assim, para se obter uma maior sensibilidade num fotocolorímetro, as medidas devem ser realizadas de preferência com um filtro azul. ANÁLISE QUANTITATIVA Antes de tudo, é importante ressaltar que o precipitado necessita ser pouco solúvel e que ele se forme rapidamente. A adição de colóides protetores , como gelatina, ágar, goma-arábica ou albumina é, às vezes, utilizada para tornar mais estável a suspensão em concentrações muito baixas. Curva Analítica
Na nefelometria e na turbidimetria as amostras são analisadas usando, geralmente, curva analítica. A exata reprodução das condições de precipitação é o aspecto mais crítico nessas análises. Todas as variáveis que podem influir no tamanho das partículas, no momento de sua formação, devem ser cuidadosamente controladas a fim de reproduzir as mesmas condições de fomação da suspensão, tanto para os padrões, como para as amostras.

95
Titulações Turbidimétricas ou Nefelométricas As medidas turbidimétricas e nefelométricas também podem ser empregadas para sinalizar o ponto final em titulações de precipitação. APLICAÇÕES DA TURBIDIMETRIA E NEFELOMETRIA A nefelometria e a turbidimetria se aplicam à determinação de numerosas espécies iônicas mediante a precipitação com reagentes apropriados, em condições que assegurem a formação de uma suspensão estável e reprodutível. A tabela a seguir relaciona algumas das mais importantes aplicações.
ESPÉCIE REAGENTE SUSPENSÃO MÉTODO
Ag
NaCl
AgCl nefelometria turbidimetria
As Te
NaH2PO2
NaH2PO2 As Te
nefelometria turbidimetria
Se Au
SnCl2
SnCl2 Se Au
nefelometria turbidimetria
Cl
AgNO3
AgCl
nefelometria turbidimetria
K Na3Co(NO2)6 K2NaCo(NO2)6 turbidimetria
Na Zn(OAc)2 e UO2(OAc)2
NaZn(OAc2)3(OAc)9
nefelometria turbidimetria
Ca H2C2O4 CaC2O4 turbidimetria
SO4 -2
BaCl2
BaSO4
nefelometria turbidimetria
A aplicação mais largamente empregada da nefelometria e turbidimetria é a determinação do sulfato ou enxofre. Elas são as técnicas preferidas devido, principalmente, às dificuldades de se encontrar um método instrumental simples e barato para análise quantitativa exata desta importante espécie química. A medida da limpidez de águas, bebidas ou medicamentos constitui um exemplo típico de um método simples de determinação nefelométrica ou turbidimétrica.
MÉTODOS ELETROANALÍTICOS Os métodos eletroanalíticos apresentam algumas vantagens sobre os outros métodos de análise quantitativa instrumental. ♦ as medidas são geralmente particular para um determinado estado de oxidação do elemento. É possível determinar através de um dos métodos eletroanalíticos a concentração de por exemplo Fe(II) e Fe(III), enquanto que os métodos espectroscópicos determinam normalmente a concentração do ferro total na amostra. ♦ os instrumentos são relativamente baratos. Custam de $4000 a $5000 com o mais sofisticado custando no máximo $25000, enquanto os espectrofotômetros custam normalmente mais de $25000. ♦ eles fornecem informação sobre atividades das espécies que em estudos fisico-químico e fisiológicos são mais importante do que as suas concentrações.
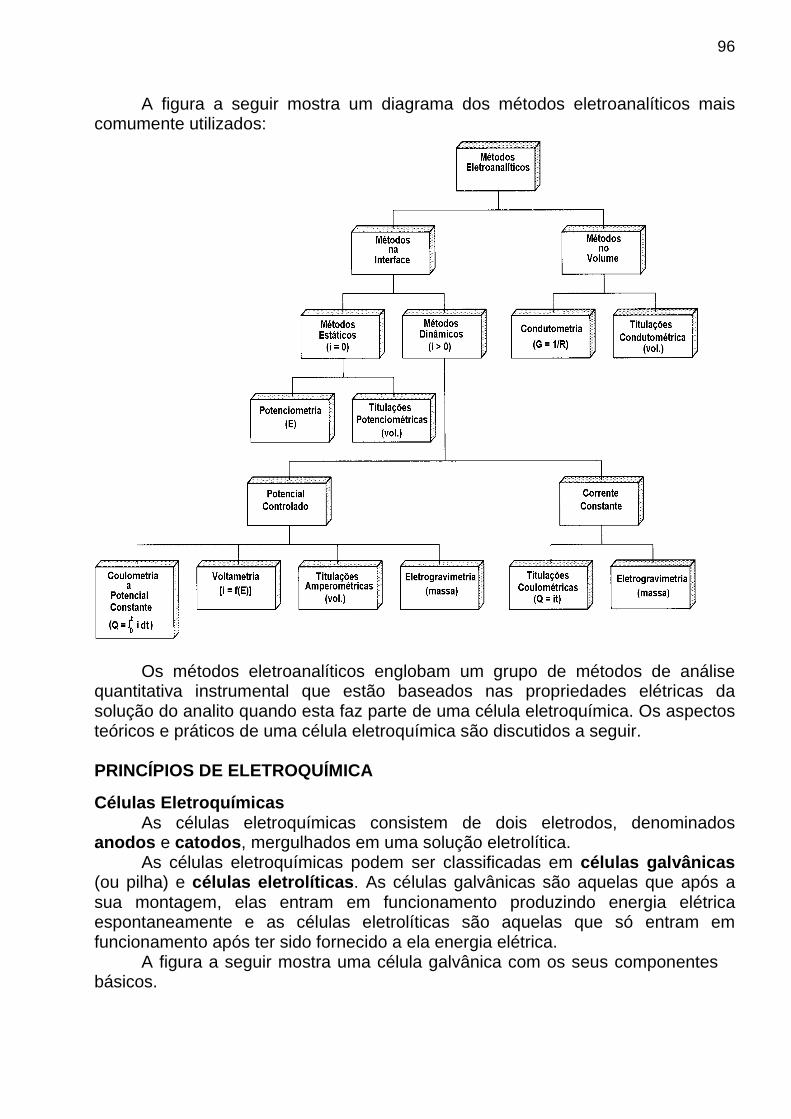
96
A figura a seguir mostra um diagrama dos métodos eletroanalíticos mais comumente utilizados:
Os métodos eletroanalíticos englobam um grupo de métodos de análise quantitativa instrumental que estão baseados nas propriedades elétricas da solução do analito quando esta faz parte de uma célula eletroquímica. Os aspectos teóricos e práticos de uma célula eletroquímica são discutidos a seguir. PRINCÍPIOS DE ELETROQUÍMICA
Células Eletroquímicas As células eletroquímicas consistem de dois eletrodos, denominados anodos e catodos , mergulhados em uma solução eletrolítica. As células eletroquímicas podem ser classificadas em células galvânicas (ou pilha) e células eletrolíticas . As células galvânicas são aquelas que após a sua montagem, elas entram em funcionamento produzindo energia elétrica espontaneamente e as células eletrolíticas são aquelas que só entram em funcionamento após ter sido fornecido a ela energia elétrica. A figura a seguir mostra uma célula galvânica com os seus componentes básicos.

97
Ela é constituída de um eletrodo metálico de zinco mergulhado em uma solução de sulfato de zinco e um eletrodo de cobre mergulhado em uma solução de sulfato de cobre. Estas duas soluções são interligadas por uma ponte salina formada por um tubo em forma de U, contendo no seu interior uma solução saturada de KCl. As extremidades do tubo são tampadas por um material poroso (lã de vidro, asbesto, material polimérico usado em baterias de automóveis) que permite permear os íons K+ e Cl- para restabelecer o equilíbrio de cargas nas duas soluções, durante a reação de oxi-redução. A ponte salina é usada para isolar as duas soluções evitando uma reação direta dos íons cobre com o eletrodo de zinco, mantendo o contato elétrico. Os eletrodos metálicos de zinco e cobre são interligados externamente ao voltímetro por fios elétricos. Ao montar esta cela galvânica, os íons Zn(II) migram do eletrodo de zinco para a solução de sulfato de zinco e os íons Cu(II) migram do seio da solução de sulfato para o eletrodo de cobre, depositando-se sobre este eletrodo. Para restabelecer o equilíbrio de cargas, íons cloreto da ponte salina migram para o seio da solução de sulfato de zinco, enquanto os íons potássio migram para solução de sulfato de cobre. Na semicela do lado esquerdo ocorre a semi-reação de oxidação:
Zn Zn2+ 2e-+ (ÂNODO)
E na semicela do lado direito ocorre a semi-reação de redução:
Cu2+ + 2e- Cu (REDUÇÃO)
A equação da reação total é:

98
Zn Zn2+ +Cu2++ Cu
Para atividades de Zn(II) e Cu(II) igual a 1,00, um potencial de 1,100V é obtido no início da reação. Quando a reação atinge o equilíbrio químico o potencial obtido é igual a 0,000V. A célula galvânica acima torna-se uma célula eletrolítica substituindo o voltímetro por uma bateria com potencial maior do 1,100V e conectando o terminal positivo ao eletrodo de cobre e o negativo ao eletrodo de zinco. A reação invertida é dada por:
+2+2 Cu + Zn Cu Zn ⇔+
Uma célula eletroquímica que invertendo a corrente inverte as reações nos eletrodos é chamada de cela quimicamente reversível. Anodos e Catodos Por definição, o catodo de uma célula eletroquímica é o eletrodo no qual ocorre a semi-reação de redução e o anodo é o eletrodo onde ocorre semi-reação de oxidação. Na célula galvânica mostrada anteriormente, o eletrodo de cobre é o catodo e o eletrodo de zinco é o anodo. Na montagem desta célula como uma célula eletrolítica, o eletrodo de cobre passa a ser o anodo e o eletrodo de zinco passa a ser o catodo. Junções Líquidas Junção líquida é a interface entre duas soluções contendo eletrólitos diferentes ou mesmo eletrólito com concentrações diferentes. Uma célula eletroquímica possui geralmente uma ou mais junções líquidas, embora seja possível montar uma célula sem uma junção líquida. A célula galvânica mostrada anteriormente apresenta duas junções líquidas: uma entre a solução de sulfato de zinco e uma das extremidades da ponte salina e a outra entre a solução de sulfato de cobre e a outra extremidade da ponte salina. A figura a seguir mostra alguns exemplos de junções líquidas.
No caso (a) temos uma junção líquida de eletrólitos iguais com concentrações diferentes. Os íons H+ e Cl- tendem a difundir da esquerda para direita. Porém como a mobilidade do íon H+ é cerca de 4,6 vezes maior do que a do íon Cl-, o lado esquerdo assume uma carga negativa e o lado direito carga positiva.

99
No caso (b) temos uma junção líquida de eletrólitos diferentes com concentrações iguais, tendo uma espécie iônica em comum. Não há praticamente difusão do íon Cl-, mas a mobilidade maior do íon H+ em relação à do íon K+ confere uma carga positiva ao lado esquerdo da junção e negativa ao lado direito. No caso (c) refere-se a eletrólitos diferentes com respeito tanto às concentrações como às espécies iônicas. As diferentes mobilidades das quatro espécies iônicas conferem diferenças de cargas nos dois lados. É justamente este tipo de junção que mais freqüentemente ocorre nos métodos eletroanalíticos. POTENCIAL DE JUNÇÃO LÍQUIDA O potencial de junção líquida é uma diferença de potencial elétrico que se desenvolve em uma junção líquida, resultante de uma separação de cargas devido à difusão das várias espécies iônicas através da interface. O potencial de junção líquida consiste na maior dificuldade dos métodos potenciométricos diretos. Eles não podem ser medidos isoladamente nem tampouco calculados com exatidão. Na prática, reduz-se intensamente o potencial de junção líquida pelo uso de uma ponte salina formada por uma solução concentrada de um eletrólito. Em uma célula eletroquímica, a corrente é carregada indistintamente por todas espécies iônicas e a fração da corrente carregada por uma determinada espécie iônica depende diretamente da concentração relativa e da mobilidade. Assim, quando os íons de um eletrólito particular se acham presentes em concentrações relativamente altas em uma ponte salina, são eles que, de fato, transportam a maior parte da corrente elétrica. Este efeito predomina sobre as diferenças entre as mobilidades e às concentrações dos íons na cela eletroquímica, reduzindo consideravelmente o potencial de junção líquida. A ponte salina mais comumente usada envolve uma solução saturada de KCl (4,2 mol L-1 a 25oC), onde as mobilidades dos íons K+ e Cl- são muito próximas (cerca de 4%). Se o íon cloreto interfere na medida a solução de KCl pode ser substituída por KNO3. A ponte salina simplesmente reduz o potencial de junção líquida, mas nunca consegue eliminá-lo por completo. Célula Eletroquímica sem Junção Líquida Algumas vezes é possível montar uma cela eletroquímica na qual os eletrodos compartilham o mesmo eletrólito e assim eliminam o potencial de junção líquida. A figura a seguir mostra um exemplo de uma cela eletroquímica sem junção líquida.

100
Nota-se que nesta cela a reação de redução e a oxidação ocorre em duas etapas e a reação global de oxi-redução é dada pela equação:
2AgCl(s) + H 2(g) ↔↔↔↔ 2Ag(s) + 2H +(aq) + 2Cl -(aq) Representação Esquemática de uma Cela Eletroquímica Para simplificar a descrição de célula eletroquímica em termos de uma notação, os químicos propuseram algumas regras: (a) São indicados à esquerda o ânodo e as informações relativas a ele. À direita, são colocados na ordem inversa o cátodo e as informações sobre ele; (b) Linhas verticais simples são usadas para representar limites de fases em que são estabelecidos potenciais. Duas linhas verticais indicam uma ponte salina; c) Usa-se vírgula para separar os diversos componentes dispersos em uma solução. A virgula também é usada para separar duas fases nas quais não é estabelecida nenhuma diferença de potencial. Com base nas regras apresentadas, podemos representar as células galvânicas mostradas anteriormente da seguinte forma:
Zn |||| ZnSO4(aZn(2+) = 1,00) |||||||| CuSO4(aCu(2+) = 1,00) |||| Cu Pt, H2(p=1 atm) |||| HCl(0,01M), AgCl(sat) |||| Ag
Estas células galvânicas quando postas para funcionar produzem uma f.e.m, que é dada por:
ECEL = ECÁTODO - EÂNODO + EJ onde EJ é o potencial de junção líquida. Potenciais de Cela – A Equação de Nernst Pode ser demonstrado através da termodinâmica que para a reação descrita pela equação geral a A + c B ↔ c C + d D é válida a equação

101
)(a . )(a
)(a . )(aln
nFRT
biB
aiA
diD
ciC
−= 0CelaCelaE E
onde a = atividade das espécies reagentes (A e B) e produtos (C e D). Além disso, para aA = fA [A], por exemplo, fA é o coeficiente de atividade da espécie A, onde [A] é a sua concentração em mol/L. Para soluções diluídas, f é aproximadamente igual a 1 (um).
A equação anterior é uma forma da equação de Nernst que tem ampla aplicação em eletroanalítica. Medidas de Potenciais de Eletrodo A comunidade química adotou um eletrodo de referência padrão, conhecido como eletrodo padrão ou eletrodo normal de hidrogênio (EPH ou ENH) , cujo potencial, por convenção , foi adotado como sendo exatamente igual a zero em qualquer temperatura. Desta forma, é possível conhecer o potencial isolado de um determinado eletrodo, montando uma cela eletroquímica em que este eletrodo esteja ligado ao eletrodo ENH. A medida do potencial da cela fornece o potencial do eletrodo isolado, já que o potencial do EPH foi adotado como sendo zero. A figura a seguir mostra uma montagem de uma cela eletroquímica usada para medida do potencial de um eletrodo.
Pode-se representar a célula acima da seguinte forma:
Pt, H2(p=1 atm) |||| H+(a=1,00) |||||||| Mn+(a=1,00)|||| M No eletrodo padrão de hidrogênio, a folha de platina deve ser recoberta por uma camada de negro de platina (redução química ou eletroquímica do H2PtCl6 para aumentar a área superficial e assegurar uma semi-reação eletródica rápida e reversível). Quando se associa o ENH a um eletrodo experimental, ele pode atuar como cátodo ou como ânodo espontaneamente (célula galvânica), dependendo dos potenciais relativos entre os dois eletrodos.

102
Potencial de Eletrodo A IUPAC convencionou que: “Potencial de eletrodo E eletrodo é igual à f.e.m de uma célula eletroquímica constituída de um eletrodo padrão de hidrogênio e o eletrodo experimental e que o termo potencial de eletrodo é reservado exclusivamente para as semi-reações escritas como reduções”. Não existe objeção em se usar o termo potencial de oxidação para denotar o potencial do eletrodo para a semi-reação inversa, porém um potencial de oxidação nunca deve ser chamado de potencial de um eletrodo. O sinal do potencial de eletrodo é determinado pelo sinal obtido quando o eletrodo de interesse é acoplado a um eletrodo padrão de hidrogênio em uma cela galvânica . Um sinal positivo para o potencial de eletrodo (Eeletrodo > 0) indica que a semi-reação de redução é espontânea (∆∆∆∆G = - nF∆∆∆∆E) e um valor negativo indica que a semi-reação de oxidação é que é espontânea. Efeito das Atividades nos Potenciais de Eletrodo - Equação de NERNST Considere que o eletrodo experimental da cela eletroquímica anterior foi acoplado a um ENH em uma cela galvânica. Tem-se dois casos: Primeiro caso: Quando o eletrodo experimental atua como ANODO e o ENH atua como CATODO. As semi-reações eletródicas neste caso são dadas por:
M(s) →→→→ Mn+(aq) + ne - (anodo)
2H+(aq) + 2e- →→→→ H2(g) (cátodo)
e a reação global da cela é dada por:
2M(s) + 2nH+(aq) →→→→ 2Mn+(aq) + nH 2(g) A equação de Nernst para esta cela galvânica é dada por:
(a
)(p . )(aln
nFRT
i+H
ni2H
2i+n
M
−=)
E0CelaCelaE
Substituindo os valores das constantes R e F e transformando o logarítmo natural para logarítmo decimal, a equação anterior, a temperatura ambiente (298,15K) torna-se:
(a
)(p . )(alog
n0,059
i+H
ni2H
2i+n
M
−=)
E0CelaCelaE
Segundo Caso: Quando o eletrodo experimental atua como CATODO
As semi-reações eletródicas são dadas por:
H2(g) →→→→ 2H+(aq) + 2e- (anodo)
Mn+(aq) + ne - →→→→ M(s) (catodo)

103
e a reação global da cela é dada por:
2Mn+(aq) + nH 2(g) →→→→ 2M(s) + 2nH+(aq)
A equação de Nernst para esta cela galvânica, a 298,15K, é dada por:
ECela Cela0==== −−−−
ΕΕΕΕ 0,059n log
(a
(a ) . (p ) H
+ i
Mn + i
2H2 i
n
)
ou
ECela Cela0==== ++++
ΕΕΕΕ 0,059n log
(a ) . (p )
(a Mn + i
2H2 i
n
H+ i)
Potencial Padrão de Eletrodo - E 0 Para os dois casos acima, as equações de Nernst revelam que a constante E0 é igual ao potencial do eletrodo quando o termo logaritmo é zero. Esta condição ocorre quando o quociente do termo logaritmo é igual à unidade, ou seja, quando as atividades de todos os reagentes e produtos são iguais à unidade. Assim, o potencial padrão de eletrodo é definido como o potencial de eletrodo experimental acoplado a um ENH, quando todos os reagentes e produtos estão presentes com atividades iguais a 1,000. O potencial padrão de eletrodo é uma constante física importante que fornece uma descrição quantitativa da força diretora de uma semi-reação de um eletrodo que constitui uma cela eletroquímica. Quatro fatores relacionados a esta constante devem ser mantidos em mente: 10) Como o potencial de eletrodo é dependente da temperatura (as propriedades elétricas, como a resistência, variam), a temperatura na qual ela foi medida deve ser especificada.
20) O seu sinal (positivo ou negativo) é o mesmo daquele obtido no condutor elétrico ligado ao eletrodo de interesse, quando este está acoplado a um ENH, formando uma cela galvânica.
30) Como ela é uma medida da força diretora da semi-reação, ela independe de como a equação da semi-reação encontra-se expressa em termos de mols. Por exemplo, o seu valor é o mesmo para as duas semi-reações abaixo. Ag+(aq) + e- ↔ Ag(s) E0 = +0,799V 100Ag+(aq) + 100e- ↔ 100Ag(s) E0 = +0,799V
A tabela a seguir mostra os potenciais padrão de eletrodo para algumas semi-reações. Uma tabela mais extensa pode ser encontrada no HANDBOOK .
Tabela de Potenciais Eletródicos Padrões (25oC) Reação Parcial E0, V
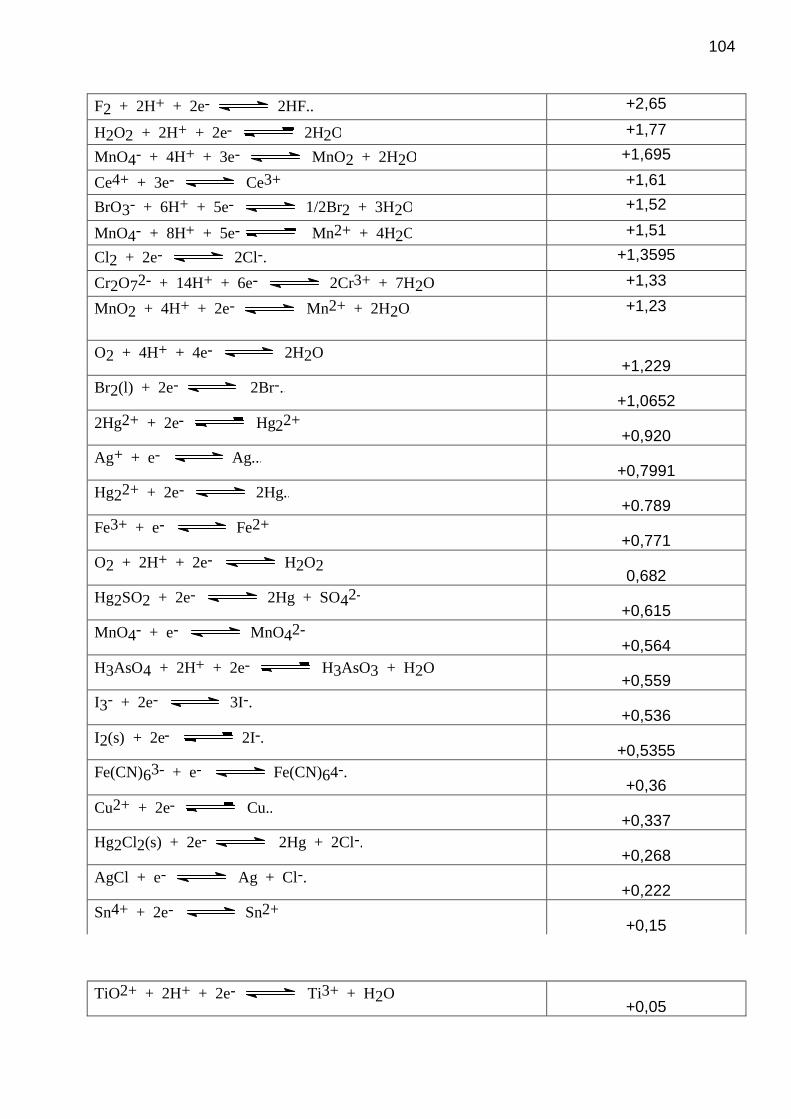
104
F2 + 2H+ + 2e- 2HF.. +2,65
H2O2 + 2H+ + 2e- 2H2O +1,77
MnO4- + 4H+ + 3e- MnO2 + 2H2O +1,695
Ce4+ + 3e- Ce3+
+1,61
BrO3- + 6H+ + 5e- 1/2Br2 + 3H2O +1,52
MnO4- + 8H+ + 5e- Mn2+ + 4H2O +1,51
Cl2 + 2e- 2Cl-. +1,3595
Cr2O72- + 14H+ + 6e- 2Cr3+ + 7H2O +1,33
MnO2 + 4H+ + 2e- Mn2+ + 2H2O.
+1,23
O2 + 4H+ + 4e- 2H2O +1,229 Br2(l) + 2e- 2Br-.. +1,0652 2Hg2+ + 2e- Hg22+. +0,920 Ag+ + e- Ag... +0,7991 Hg22+ + 2e- 2Hg.. +0.789 Fe3+ + e- Fe2+
+0,771 O2 + 2H+ + 2e- H2O2 0,682 Hg2SO2 + 2e- 2Hg + SO42-
+0,615 MnO4- + e- MnO42-
+0,564 H3AsO4 + 2H+ + 2e- H3AsO3 + H2O. +0,559 I3- + 2e- 3I-. +0,536 I2(s) + 2e- 2I-. +0,5355 Fe(CN)63- + e- Fe(CN)64-. +0,36 Cu2+ + 2e- Cu.. +0,337 Hg2Cl2(s) + 2e- 2Hg + 2Cl-.. +0,268 AgCl + e- Ag + Cl-.. +0,222 Sn4+ + 2e- Sn2+
+0,15
TiO2+ + 2H+ + 2e- Ti3+ + H2O. +0,05
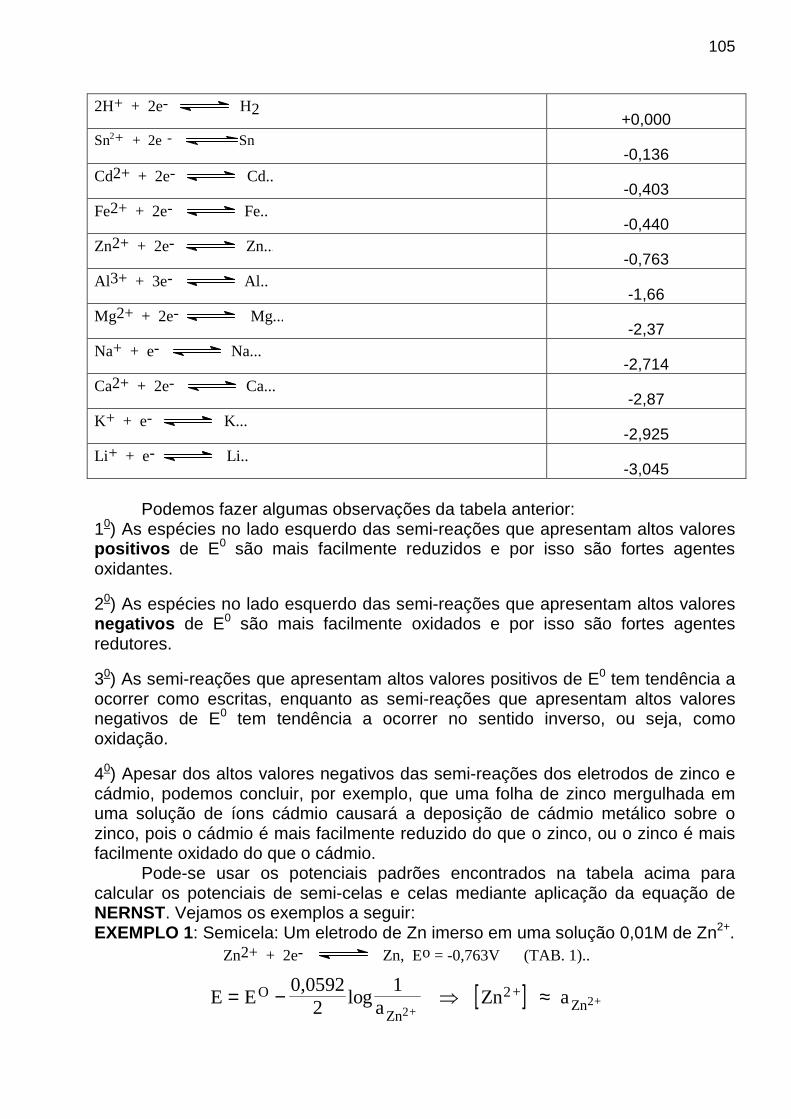
105
2H+ + 2e- H2 +0,000 Sn2+ + 2e- Sn -0,136 Cd2+ + 2e- Cd.. -0,403 Fe2+ + 2e- Fe.. -0,440 Zn2+ + 2e- Zn... -0,763 Al3+ + 3e- Al... -1,66 Mg2+ + 2e- Mg... -2,37 Na+ + e- Na... -2,714 Ca2+ + 2e- Ca... -2,87 K+ + e- K... -2,925 Li+ + e- Li... -3,045 Podemos fazer algumas observações da tabela anterior: 10) As espécies no lado esquerdo das semi-reações que apresentam altos valores positivos de E0 são mais facilmente reduzidos e por isso são fortes agentes oxidantes. 20) As espécies no lado esquerdo das semi-reações que apresentam altos valores negativos de E0 são mais facilmente oxidados e por isso são fortes agentes redutores.
30) As semi-reações que apresentam altos valores positivos de E0 tem tendência a ocorrer como escritas, enquanto as semi-reações que apresentam altos valores negativos de E0 tem tendência a ocorrer no sentido inverso, ou seja, como oxidação.
40) Apesar dos altos valores negativos das semi-reações dos eletrodos de zinco e cádmio, podemos concluir, por exemplo, que uma folha de zinco mergulhada em uma solução de íons cádmio causará a deposição de cádmio metálico sobre o zinco, pois o cádmio é mais facilmente reduzido do que o zinco, ou o zinco é mais facilmente oxidado do que o cádmio. Pode-se usar os potenciais padrões encontrados na tabela acima para calcular os potenciais de semi-celas e celas mediante aplicação da equação de NERNST. Vejamos os exemplos a seguir: EXEMPLO 1: Semicela: Um eletrodo de Zn imerso em uma solução 0,01M de Zn2+.
Zn2+ + 2e- Zn, Eo = -0,763V (TAB. 1)...
[ ]E E0,0592
2 log1
aO
Zn2+= − ⇒ ≈ Zn a2+
Zn2+

106
E0,0592
2log
1
10-2= − − ⇒0 763, E = 0,822V
EXEMPLO 2: Vamos considerar a seguinte célula: Zn | ZnSO4(aZn
2+=0,10) | | CuSO4(aCu
2+=0,10) |Cu.
As semi-reações são dadas por:
Zn2+ + 2e- Zn, Eo = -0,763V (ÂNODO)... Cu2+ + 2e- Cu, Eo = +0,337V (CÁTODO)...
Cálculo de ECÁTODO e EÂNODO:
E E0,0592
2 log1
aCÁTODOO
C 2+= −
u
E +0,3370,0592
2 log1
0,10 E 0,307VCÁTODO CÁTODO= − ⇒ = +
E E0,0592
2 log1
aÂNODOO
2+= −
Zn
E -00,0592
2 log1
0,10 E VÂNODO ÂNODO= − ⇒ = −, ,763 0 793
Se o potencial de junção líquida é considerado desprezível:
ECÉLULA= ECÁTODO – EÂNODO
Então, ECÉLULA = 0,307V – (–0,793V) ⇒ ECÉLULA= +1,100V
Outra maneira de calcular o potencial desta cela é:
Zn2+ + 2e-, Eo = -0,763V...Zn
Cu, Eo = +0,337V...Cu2+ + 2e-
Zn + Cu2+ Zn2+ + Cu, Eot = 0,793 + 0,337 = +1,100V
E0,0592
2 log0,100,10 E VCEL CEL= + − ⇒ = +1100 1100, ,
Efeitos de Reagentes Complexantes e Precipitantes n os Potenciais de Eletrodo Os agentes precipitantes e complexantes que reagem com a(s) espécie(s) participantes da semi-reação de um eletrodo exercem um efeito marcante no potencial do eletrodo como mostram os exemplos a seguir.

107
Exemplo do Efeito de um Reagente Precipitante Um exemplo típico é a variação do potencial de um eletrodo de prata em presença de uma solução saturada de AgI, contendo íons iodetos com atividade 1,00. (Kps para AgI = 8,3 x 10-17).
Ag+ + e- Ag... E0 = +0,7991
+Ag+Aga
1log
10,0592
+0,799a
1log
10,0592
E=E O −=−
Podemos calcular o valor de aag+ a partir do produto de solubilidade. Como,
Kps = ag+ . aI- então, ag+ = Kps/aI
- logo, ps
-IK
a log 0,0592+0,799E −=
que pode ser reescrita como: -Ips a log 0,0592K log 0,0592++0,799E −=
Substituindo os valores de Kps e aI- obtém-se que: 0,151V- =1,00 log 0,059210 x (8,3 log 0,0592++0,799E -17 −= )
Este exemplo mostra que o potencial de redução de íons prata torna-se muito menor quando em presença de íons iodeto. Qualitativamente, esse é um efeito esperado, uma vez que a diminuição na concentração de íons prata diminui também a tendência para a sua redução. Do tratamento acima pode ser verificado que quando a atividade do íon iodeto é igual à unidade, o potencial de eletrodo é igual à soma de duas constantes, ou seja,
V151,0−== psK log 0,0592++0,799E
que representa o potencial padrão de eletrodo para semi-reação:
AgI(s) + e - ↔↔↔↔ Ag(s) + I -(aq) E0 = -0,151V A equação de Nernst para o eletrodo de prata em uma solução saturada com iodeto de prata é dada por:
-I-Iaa log 0,0592E log0592,0151,0E0 ⋅−−=−=
Logo, quando em contato com uma solução saturada de iodeto de prata, o potencial do eletrodo de prata pode ser descrito ou em termos da atividade dos íons prata, com:
Ag+ + e- Ag... E0 = +0,7991

108
e
++++++++
Ag
0
AgAg a
1 log 0,0592-E=E
/
ou em termos da atividade de íons iodeto, com
AgI(s) + e - ↔↔↔↔ Ag(s) + I -(aq) E0 = -0.151V e
-IaE log0592,0151,0 −−=
Exemplo do Efeito de um Reagente Complexante O potencial de um eletrodo de prata em uma solução contendo íons prata e um agente complexante (amônia ou tiossulfato, por exemplo) pode ser tratado de maneira análoga ao exemplo anterior. As reações de formação dos complexos são dadas por:
Ag+ + 2NH3 ↔↔↔↔ [Ag(NH 3)2]+
2]]2)3NH(Ag[
f)a(a
aK
]3NH[]Ag[ +
+=
Ag+ + 2 S 2O3-2 ↔↔↔↔ [Ag(S 2O3)2]
-3 2
Ag
f)a(a
aK
2)3O2S(
3]2)3O2S(Ag[
−
+
+=
onde Kf é a constante de formação do complexo. O potencial do eletrodo para as duas reações é dado por:
2)a(fK
alog0592,0799,0
3NH
]2
)3
NH(Ag[+
+=+Aga log 0,0592+0,799=E
2)a(fK
alog0592,0799,0
2)
3O
2S(
3]
2)
3O
2S(Ag[
−
−
+=+Aga log 0,0592+0,799=E
Considerando as atividades dos ligantes e dos complexos iguais a unidade, o potencial padrão de eletrodo para as semi-reações:
[Ag(NH 3)2]+ + e- ↔↔↔↔ Ag(s) + 2NH 3
[Ag(S 2O3)2]-3 + e- ↔↔↔↔ Ag(s) + 2S 2O3
-2
Pode ser calculado por:

109
f
0
K
1 log 0,0592+0,799=E
Algumas Limitações do Uso de Potenciais de Eletrodo Os potenciais de eletrodo são de grande importância no entendimento dos processos eletroanalíticos. Contudo, há certas limitações para a utilização desses dados que devem ser colocados com clareza. Substituição de Atividades por Concentrações A suposição que as duas quantidades são iguais só é válida para soluções diluídas. Por exemplo, potencial-padrão de eletrodo para a semi-reação Fe3+ + e- ↔ Fe2+ é de +0,771V. O potencial de eletrodo para essa mesma semi-reação medida experimentalmente em uma solução 1M de Fe3+, Fe2+ e ácido perclórico é de +0,732V. A razão para esta discrepância pode ser justificada se escrevermos a equação de Nernst na forma:
3Fe
3
2Fe
2
]Fe[
]Fe[log0592,0
++
++
γ
γ−0E = E
onde, γFe2+ e γFe3+ são os coeficientes de atividades do Fe2+ e Fe3+ na solução. Como γFe2+ > γFe3+, obtém-se um potencial de eletrodo menor do que o
potencial-padrão de eletrodo para semi-reação. Efeito de Reações Laterais O potencial padrão dos eletrodos é afetado pela ocorrência outras reações como, solvatação, associação, dissociação, precipitação, formação de complexos, etc, envolvendo as espécies de interesse que formam o eletrodo. Um exemplo desse caso foi mostrado anteriormente, quando discutido o efeito de agentes precipitantes e complexantes no potencial de eletrodo. Fenômenos como estes podem ser avaliados se os equilíbrios envolvidos e as constantes do processo são conhecidas. Entretanto, nem sempre isso é possível na prática. Um exemplo deste problema é que o potencial de eletrodo para a semi-reação Fe3+ + e- ↔ Fe2+, medida em meio de 1M de ácido perclórico é de +0,732V, em um meio de 1M de ácido clorídrico é de +0,70V e em 1M de ácido fosfórico é de +0,6V. Essas diferenças têm sido atribuídas ao fato do Fe(III) formar complexos mais estáveis com o cloreto e o fosfato do que o Fe(II). Assim, a concentração real de Fe(III) não-complexado em tais solução é menor do que ao do Fe(II) não-complexado. Todavia, estes equilíbrios não são bem estabelecidos e a equação de Nernst para estes sistemas não pode ser proposta com exatidão.
ELETRÓLISE E ELETRODEPOSIÇÃO – Conceitos e Princípi os A eletrodeposição baseia-se na medida da massa do analito eletrodepositado na superfície do eletrodo de trabalho em uma célula eletrolítica. A eletrodeposição envolve usualmente a redução catódica de íons metálicos de acordo com a equação: Mn+ + ne- M
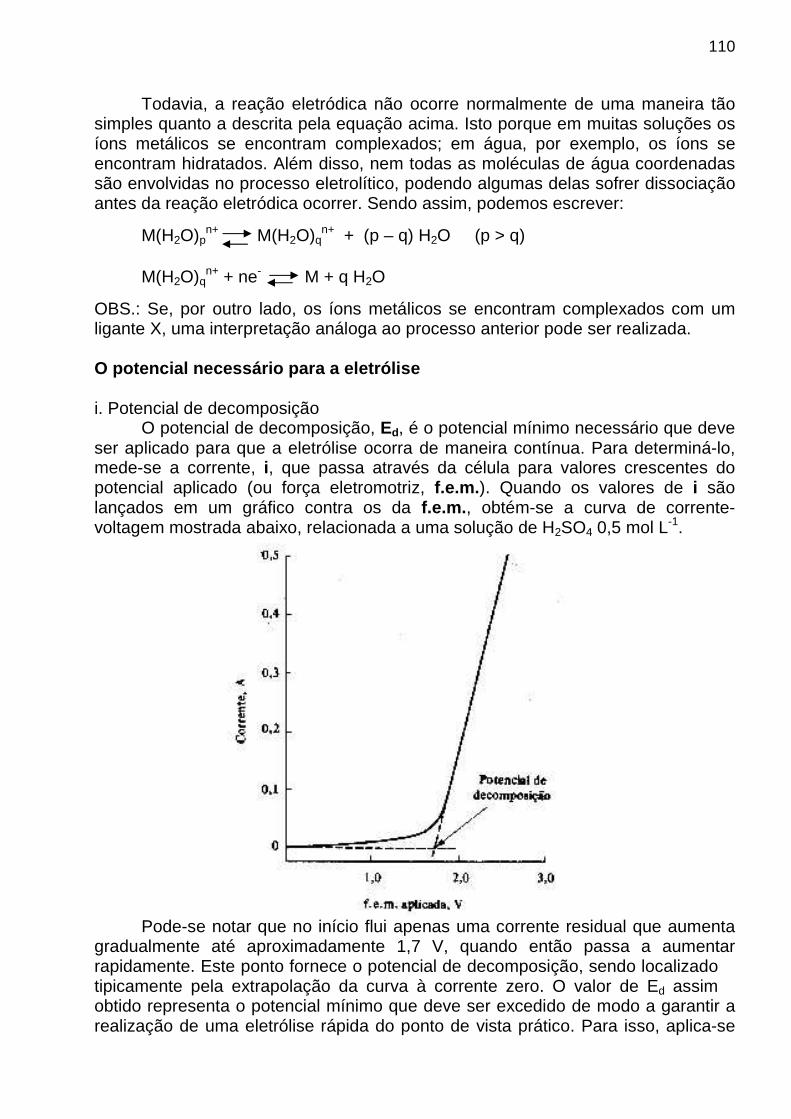
110
Todavia, a reação eletródica não ocorre normalmente de uma maneira tão simples quanto a descrita pela equação acima. Isto porque em muitas soluções os íons metálicos se encontram complexados; em água, por exemplo, os íons se encontram hidratados. Além disso, nem todas as moléculas de água coordenadas são envolvidas no processo eletrolítico, podendo algumas delas sofrer dissociação antes da reação eletródica ocorrer. Sendo assim, podemos escrever:
M(H2O)pn+ M(H2O)q
n+ + (p – q) H2O (p > q) M(H2O)q
n+ + ne- M + q H2O
OBS.: Se, por outro lado, os íons metálicos se encontram complexados com um ligante X, uma interpretação análoga ao processo anterior pode ser realizada. O potencial necessário para a eletrólise i. Potencial de decomposição O potencial de decomposição, Ed, é o potencial mínimo necessário que deve ser aplicado para que a eletrólise ocorra de maneira contínua. Para determiná-lo, mede-se a corrente, i, que passa através da célula para valores crescentes do potencial aplicado (ou força eletromotriz, f.e.m. ). Quando os valores de i são lançados em um gráfico contra os da f.e.m. , obtém-se a curva de corrente-voltagem mostrada abaixo, relacionada a uma solução de H2SO4 0,5 mol L-1.
Pode-se notar que no início flui apenas uma corrente residual que aumenta gradualmente até aproximadamente 1,7 V, quando então passa a aumentar rapidamente. Este ponto fornece o potencial de decomposição, sendo localizado tipicamente pela extrapolação da curva à corrente zero. O valor de Ed assim obtido representa o potencial mínimo que deve ser excedido de modo a garantir a realização de uma eletrólise rápida do ponto de vista prático. Para isso, aplica-se

111
um potencial Ea maior que o Ed de modo que uma corrente, dada pela expressão abaixo (baseada na lei de Ohm), fluirá através da célula.
R
EEi da −=
onde R é a resistência interna da cela. A expressão acima pode ser rearranjada como RiEE da ⋅+= O potencial de decomposição é um termo complexo resultante das interações associadas aos produtos das reações eletródicas. Com efeito, os eletrodos da cela, os quais antes de iniciar a eletrólise eram inertes, transformam-se em eletrodos ativos (polarizados) que se opõem à passagem da corrente através da célula. Na realidade, os eletrodos ativos originam uma célula galvânica cuja f.e.m. se opõe ao potencial aplicado, sendo então caracterizada como f.e.m. de retorno, Er. Quando os eletrodos ativos se comportam reversivelmente, o potencial de decomposição é constituído apenas pela f.e.m. de retorno, podendo esta ser determinada a partir dos potenciais termodinâmicos (E0) dos eletrodos ativos da cela galvânica. Entretanto, quando as condições de reversibilidade não são satisfeitas, o potencial de decomposição observado é significativamente maior que a f.e.m de retorno. Sendo assim, a voltagem adicional que deve ser acrescida a Er de modo a alcançar o valor de Ed é denominada sobrevoltagem ou sobrepotencial, Es, que será melhor descrita adiante. ii. Determinação da f.e.m de retorno Para ilustrar o cálculo de Er, considere a eletrólise de uma solução de um ácido (ou base) efetuada entre eletrodos de platina. Neste caso, ocorrem as seguintes reações durante a eletrólise: 2 H+ + 2 −e H2 (catodo) e H2O ½ O2 + 2 H+ + 2 −e (anodo) A célula galvânica gerada a partir dos produtos da eletrólise pode ser representada como
Pt, H2 (1 atm |||| solução do ácido |||| O2 (1 atm), Pt para a qual a f.e.m de retorno é dada por Er = Ecatodo – Eanodo
Er = 2H2O EE − =
−−− ++ 22 ]H[
1log
20592,0
]H[
1log
20592,0
23,1
Er = 1,23 V Um fato notável no resultado da equação anterior é que na decomposição
eletrolítica de soluções de ácidos ou bases, a eletrólise é efetivamente a decomposição da água. A f.e.m de retorno se mantém em 1,23 V e não depende nem da identidade do ácido (ou base), nem da concentração do eletrólito.
A eletrólise da água se caracteriza por uma estabilização dos potenciais catódico e anódico, não afetando a f.e.m de retorno. Por outro lado, na

112
eletrogavimetria a f.em. de retorno depende da natureza e da concentração do eletrólito como se pode verificar no exemplo ilustrado a seguir.
Considere, por exemplo, a eletrólise de uma solução 0,1 mol L-1 de CuSO4 em meio 0,1 mol L-1 de H2SO4, entre eletrodos de platina.
As reações que ocorrem são: Cu2+ + 2 −e Cu (catodo)
H2O ½ O2 + 2 H+ + 2 −e (anodo) as quais originam a seguinte célula galvânica:
Cu | CuSO4 (0,1 mol L-1), H2SO4 (0,5 mol L-1) | O2 (1 atm), Pt No início da eletrólise, Er tem o valor
Er = Cu2O EE − =
⋅−−− ++ ]Cu[
1log0592,0337,0
]H[
1log
20592,0
23,122
Er = ( )10log0592,0337,01log0592,023,1 ⋅−−⋅− Er = 0,92 V À medida que a eletrólise avança, a f.e.m de retorno aumenta progressivamente devido ao aumento de
2OE e à diminuição de CuE . De fato, a
deposição de cobre no catodo é acompanhada de um decréscimo da concentração de Cu(II) na solução, enquanto a oxidação anódica da água implica um incremento na concentração de íons H+. De acordo com as leis de Faraday, para cada mol de Cu(II) reduzido no catodo, ocorre a oxidação anódica de um mol de moléculas de água produzindo dois mols de íons H+. Assim, quando a concentração de Cu2+ for reduzida para, digamos, 1 x 10-6 mol L-1 (deposição praticamente completa), a concentração de H+ terá atingido cerca de 1,2 mol L-1. Neste caso, o valor de Er será
Er =
⋅−−− −62 10
1log0592,0337,0
)2,1(
1log
20592,0
23,1
Er = 1,1 V Generalizando, em uma eletrodeposição de um metal, M, no catodo e a evolução de oxigênio no anodo a f.em. de retorno, Er, é dada por
−−
−=−= ++ ]M[
1log
n0592,0
E]H[
1log0592,023,1EEE
n0MM2Or
A expressão mostra que Er aumenta gradativamente, enquanto prossegue a eletrodeposição do metal. iii. Sobretensão Como vimos antes, o potencial de decomposição é aproximadamente igual à f.e.m de retorno quando as reações eletródicas são reversíveis. Um exemplo típico é a eletrólise de uma solução de brometo de zinco. Aqui o potencial de decomposição pouco difere de Er determinado a partir dos potenciais da célula galvância formada com os produtos da eletrólise. Todavia, o potencial de decomposição (Ed) é normalmente maior que Er, cujo valor ao se somar com a sobrevoltagem deve alcançar o valor de Ed.
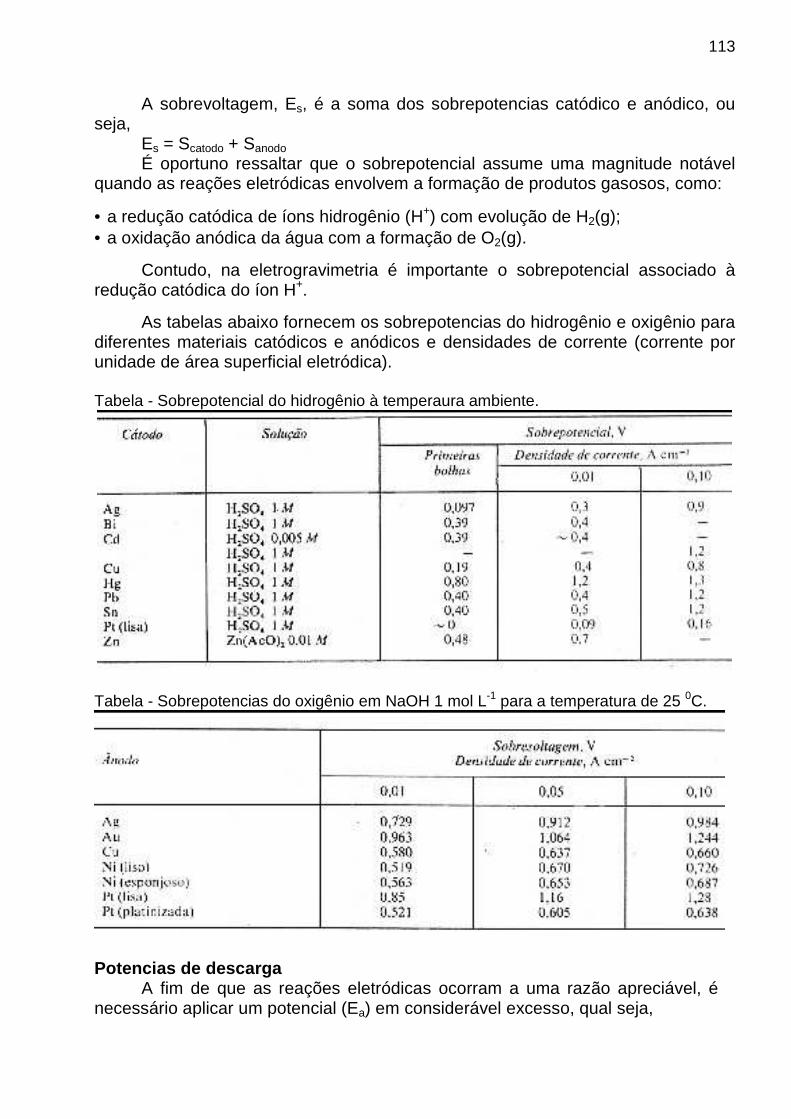
113
A sobrevoltagem, Es, é a soma dos sobrepotencias catódico e anódico, ou seja, Es = Scatodo + Sanodo É oportuno ressaltar que o sobrepotencial assume uma magnitude notável quando as reações eletródicas envolvem a formação de produtos gasosos, como:
• a redução catódica de íons hidrogênio (H+) com evolução de H2(g); • a oxidação anódica da água com a formação de O2(g).
Contudo, na eletrogravimetria é importante o sobrepotencial associado à redução catódica do íon H+.
As tabelas abaixo fornecem os sobrepotencias do hidrogênio e oxigênio para diferentes materiais catódicos e anódicos e densidades de corrente (corrente por unidade de área superficial eletródica). Tabela - Sobrepotencial do hidrogênio à temperaura ambiente.
Tabela - Sobrepotencias do oxigênio em NaOH 1 mol L-1 para a temperatura de 25 0C.
Potencias de descarga A fim de que as reações eletródicas ocorram a uma razão apreciável, é necessário aplicar um potencial (Ea) em considerável excesso, qual seja,

114
Ri)SS()EE(RiEEEcaeletrolíticélula
catodoanodo
galvânicacélula
anodocatodosra ⋅+++−=⋅++=⋅⋅
44 344 2144 344 21
Porém Er = – Ecela eletrolítica = – (Ecatodo – Eanodo) = Eanodo – Ecatodo
Então podemos escrever para a célula eletrolítica
=aE (Eanodo – Ecatodo) + (Sanodo + Scatodo) + i R Ou reagrupando os termos similares como Ea = (Eanodo + Sanodo ) – (Ecatodo – Scatodo ) + i R onde: • (Eanodo + Sanodo ) ⇒ representa o “potencial de descarga anódico”, capaz de forçar a reação anódica; • (Ecatodo – Scatodo ) ⇒ representa o “potencial de descarga catódico”, capaz de promover a reação catódica; • i R ⇒ compensa a queda ôhmica da solução devido à sua resistência. Com o avanço da eletrólise, o potencial de descarga anódico se desloca na direção positiva (ou seja, aumenta), enquanto o potencial de descarga catódico se desloca na direção negativa.
Conseqüentemente, em uma eletrodeposição convencional (sem controle do potencial catódico), a espécie de interesse deve ser a única mais facilmente redutível que o íon hidrogênio. Após a deposição da espécie mais redutível, a estabilização do potencial catódico com a descarga dos íons H+ impede a redução de outras espécies menos redutíveis que o H+.
Por outro lado, se houver várias espécies mais redutíveis que o íon hidrogênio, a técnica convencional não permite evitar a deposição de vários metais. Esse problema pode ser contornado se o potencial catódico for mantido fixo em um nível (valor) em que apenas um dos metais se deposite. Para isso, deve-se empregar a técnica da eletrólise com potencial controlado descrita adiante. Eletrodeposição dos Metais De acordo com os argumentos anteriores, a condição essencial para deposição de um metal é que ele seja mais facilmente redutível que o hidrogênio nas condições em que se processa a eletrólise. Tendo isso em mente, vamos investigar, através dos exemplos a seguir, a viabilidade das seguintes eletrodeposições: a. Eletrólise de uma solução de uma solução de CuSO4 0,1 mol L-1 e 0,5 mol L-1 em H2SO4 entre eletrodos de platina. As reações eletródicas são: Catodo: Cu2+ + 2 −e Cu Anodo: H2O ½ O2 + 2 H+ + 2 −e

115
O objetivo é saber se a descarga do H+ vai atrapalhar a deposição do cobre. O potencial catódico evolui (diminui) de acordo com a equação:
]Cu[
1log
20592,0
337,0E2Cu +⋅−= , sendo 0,3104 V o seu valor no início da
eletrólise. Por outro lado, a evolução anódica de H2 ocorreria em 0 V se não houvesse o sobrepotencial do hidrogênio para o cobre, pois
]H[
1log0592,0EE 0
2H +⋅−= = 0,0 – 0,0592 log 1 = 0,0 V
Entretanto, a descarga do íon H+ ocorrerá no potencial:
Potencial e descarga (H2) =
2H2H SE − = 0,0 – 0,4 = – 0,4 V, onde 0,4 é o
sobrepotencial do hidrogênio para o cobre, admitindo uma densidade de corrente igual a 0,01 A cm-2. Com efeito, a concentração de íons cobre terá baixado a um nível que pode ser determinado como
4,0]Cu[
1log
20592,0
337,0E2Cu −=⋅−= +
⇒ ECu ≈ 1 x 10-25 mol L-1 Conclusão: quando o potencial descarga do H+ for atingido, a deposição do cobre pode ser considerada quase completa. Portanto, a eletrodeposição dos íons Cu2+ é viável nas condições estudadas. b. Considere agora a eletrólise de uma solução 0,1 mol L-1 de ZnSO4 e 0,5 mol L-1 em H2SO4 entre dois eletrodos de platina. Será que essa eletrólise também é viável nas condições estabelecidas? As reações eletródicas desejáveis são: Catodo: Zn2+ + 2 −e Zn Anodo: H2O ½ O2 + 2 H+ + 2 −e O início da deposição do zinco requer o potencial
V792,010
1log
20592,0
763,0E1Zn −=⋅−= −
Por outro lado, a descarga do hidrogênio ocorrerá em: Descarga (H2) =
2H2H SE − = 0,00 – 0,09 = - 0,09 V,

116
onde 2HS = 0,09 V é o sobrepotencial do hidrogênio para a platina, a uma
densidade de corrente igual a 0,01 A cm-2. Uma vez que a descarga do H+ ocorre em um potencial catódico muito menos negativo (maior) que o requerido para iniciar a descarga do íon Zn2+, o zinco não pode ser eletrolisado de uma solução fortemente ácida. Isto porque a evolução de H2 estabiliza o potencial catódico em – 0,09 V. Contudo, a eletrodeposição do zinco torna-se viável em uma solução tamponada a pH = 6,0. Nestas condições, o potencial de descarga do hidrogênio é deslocado bastante na direção negativa por dois motivos:
i) em conseqüência da menor concentração de íons H+; ii) devido ao elevado sobrepotencial do hidrogênio para o zinco, isto é,
V7,0S2H = (p/ Zn).
Sendo assim, o potencial de descarga do íon H+ será:
Pdescarga (H+) =
2H2H SE − = V055,17,010
1log0592,0
6−=−− −
Para este valor, a concentração de íons Zn2+ remanescente na solução é:
V055,1]Zn[
1log
20592,0
763,0E2Zn −=−−= +
⇒ [Zn2+] ≈ 10-10 mol L-1 (que indica uma deposição quase completa!) c. Vejamos agora se é possível inverter a reação espontânea da pilha de Daniel nas condições-padrão.
Pilha de Daniel: Zn + Cu2+ Zn2+ + Cu, Ecela = + 1,1 V (reação espontânea)
Eletrólise: Zn2+ + Cu Zn + Cu2+, Ecela = – 1,1 V (reação não espontânea)
Neste caso, o potencial de descarga do íon H+ é:
Pdescarga = 2H2H SE − = 0,0 – 0,7 = 0,7 V
Por outro lado, a descarga do zinco deverá começar no potencial catódico
V763,011
log2
0592,0763,0EZn −=−−=
Ora, como o potencial de descarga do íon H+ é menos negativo (maior) que o do zinco, logo o H+ se descarregará primeiro e vai atrapalhar a deposição do zinco.
E em relação ao produto anódico da eletrólise, que produtos teremos?
O potencial anódico do cobre é:

117
V34,011
log2
0592,0337,0ECu −=⋅−= . Por outro lado, a descarga de O2 requer o
potencial: Descarga (O2) =
2H2H SE − = 1,23 + 0,58 = 1,81 V.
Conclusão: O cobre se oxidará primeiro no anodo, pois é mais facilmente oxidado que a água nas condições da eletrólise. O que fazer para levar a efeito a deposição do zinc o? Devemos deslocar o potencial de descarga do hidrogênio para um valor mais negativo. Para isso, devemos diminuir a acidez da solução, ou seja, considerando uma solução tamponada ao pH = 6,0 teremos:
Descarga (H+) = V055,17,0)10(
1log
20592,0
26−=−⋅−
−.
Conclusão: nessas novas condições os íons Zn2+ sofrerão redução primeiro, pois o seu potencial de descarga se tornou mais negativo (menor) que o do zinco. Além disso, quando o potencial de descarga do H+ fosse alcançado a concentração de íons zinco restantes na solução seria desprezível, ou seja:
V055,1]Zn[
1log
20592,0
763,0E2Zn −=−−= +
⇒ [Zn2+] = 10-10 mol L-1 (o que indica uma deposição quase completa!)
POTENCIOMETRIA
Introdução A potenciometria baseia-se em medidas do potencial elétrico de células galvânicas. Na realidade, a pontenciometria é uma técnica em que se faz uma aplicação direta da equação de NERNST.
A análise potenciométrica engloba duas técnicas analíticas:
♦ a potenciometria direta; ♦ a titulação potenciométrica.
Na potenciométria direta , é determinada a concentração (ou atividade) de uma dada espécie iônica a partir da medida do potencial de uma cela que contém um eletrodo indicador ou eletrodo de trabalho (normalmente um eletrodo seletivo) e um eletrodo de referência. Os eletrodos seletivos são relativamente livres de interferência e fornece um meio rápido e conveniente para análise quantitativa de grande número de espécies químicas.

118
Na titulação potenciométrica , acompanha-se a variação do potencial do eletrodo indicador (pH ou pX) em relação ao eletrodo de referência em função do volume do titulante adicionado na solução. O equipamento requerido para os métodos potenciométricos é simples e barato e inclui um eletrodo indicador ou de trabalho, um eletrodo de referência e um dispositivo para medir os potenciais, ou seja, um potenciômetro. As características destes dispositivos são discutidas a seguir. Eletrodos de Referência São eletrodos cujo potencial não varia durante as medidas de potencial, ou seja, eles apresentem um potencial constante e completamente insensível à composição da solução em estudo. Em outras palavras, os eletrodos de referência devem basicamente:
♦ ser reversível e obedecer a equação de Nernst; ♦ exibir um potencial constante com o tempo.
Define-se internacionalmente como eletrodo de referência fundamental o eletrodo padrão de hidrogênio (EPH), descrito anteriormente. Entretanto, em virtude das dificuldades práticas no uso do EPH, utilizam-se eletrodos de referência secundários. Estes eletrodos são mais práticos e, portanto, de uso mais corrente.
Dois tipos de eletrodos de referência secundários são mais comumente utilizados: o eletrodo de calomelano saturado e o eletrodo de prata-cloreto de prata . Eletrodo de Calomelano Saturado Ele é formado de dois tubos concêntricos. O tubo interno contém mercúrio em contato com uma pasta de mercúrio/cloreto de mercúrio(I) (calomelano), preparada mediante a trituração destes componentes juntos com uma solução saturada de KCl. O tubo externo contém uma solução saturada de KCl que é responsável pela ponte salina. Os componentes do tubo interno e do tubo externo estão em contato entre si, através de um pequeno orifício existente no tubo interno. A comunicação elétrica entre o mercúrio e o cabo elétrico externo é feita com um fio de platina. O eletrodo de calomelano dispõe de uma abertura lateral que permite repor, sempre que necessário, a solução de cloreto de potássio saturada. Em alguns eletrodos de calomelano, o contato com o meio externo (junção) é feito através de um disco de vidro calcinado, asbesto ou uma fibra porosa. Esta junção apresenta uma alta resistência (2000 a 3000Ω) e uma limitada capacidade de transporte de corrente. Por outro lado, a contaminação da solução do analito por vazamento da solução de cloreto de potássio é mínima. Em outros eletrodos, o contato é feito usando uma luva de vidro esmerilhada, que contém internamente um orifício para permitir o contato com o meio externo. Antes de usar este eletrodo, a luva deve ser deslizada tal que uma ou duas gotas de KCl flua através orifício e umedeça a superfície interna esmerilhada da luva. Esta junção tem baixa resistência, mas tende a vazar pequenas quantidades da solução saturada de cloreto de potássio na amostra.
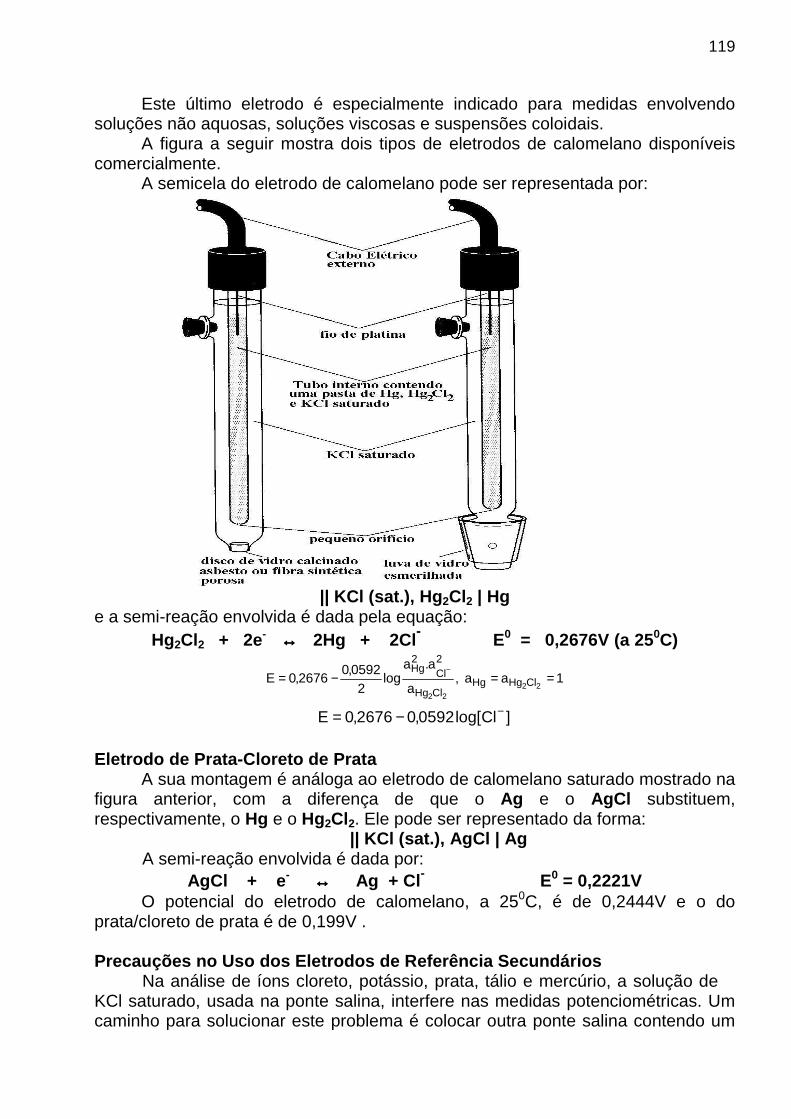
119
Este último eletrodo é especialmente indicado para medidas envolvendo soluções não aquosas, soluções viscosas e suspensões coloidais. A figura a seguir mostra dois tipos de eletrodos de calomelano disponíveis comercialmente. A semicela do eletrodo de calomelano pode ser representada por:
|| KCl (sat.), Hg 2Cl2 | Hg e a semi-reação envolvida é dada pela equação:
Hg2Cl2 + 2e- ↔↔↔↔ 2Hg + 2Cl - E0 = 0,2676V (a 250C)
1aa ,a
a.alog
20592,0
2676,0E22
22
ClHgHgClHg
2Cl
2Hg
==−=−
]Cllog[0592,02676,0E −−= Eletrodo de Prata-Cloreto de Prata A sua montagem é análoga ao eletrodo de calomelano saturado mostrado na figura anterior, com a diferença de que o Ag e o AgCl substituem, respectivamente, o Hg e o Hg2Cl2. Ele pode ser representado da forma:
|| KCl (sat.), AgCl | Ag A semi-reação envolvida é dada por:
AgCl + e - ↔↔↔↔ Ag + Cl - E0 = 0,2221V O potencial do eletrodo de calomelano, a 250C, é de 0,2444V e o do prata/cloreto de prata é de 0,199V . Precauções no Uso dos Eletrodos de Referência Secun dários
Na análise de íons cloreto, potássio, prata, tálio e mercúrio, a solução de KCl saturado, usada na ponte salina, interfere nas medidas potenciométricas. Um caminho para solucionar este problema é colocar outra ponte salina contendo um

120
outro eletrólito não interferente (nitrato de potássio ou sulfato de sódio), entre a amostra e o eletrodo de referência. Eletrodos Indicadores ou Eletrodos de Trabalho Um eletrodo indicador ideal deve responder rapidamente e reprodutivelmente às variações na concentração do analito. Embora não existam eletrodos indicadores absolutamente específicos, alguns são bastante seletivos. Existem dois tipos de eletrodos indicadores ou de trabalho: os eletrodos metálicos ou de oxi-redução e os eletrodos de membrana ou eletrodos seletivos . Eletrodos Indicadores Metálicos ou de Oxi-Redução Eles são baseados estritamente em processos oxi-redução que ocorrem na superfície do eletrodo e permitem determinar a concentração de um número restrito de espécies iônicas. Embora alguns destes eletrodos não sejam de uso satisfatório na potenciometria direta, são utilizados, por outro lado, na titulação potenciométrica. Existem quatro classes de eletrodos indicadores metálicos: os de 1a classe, de 2a classe, de 3a classe e os inertes Eletrodos de 1 a classe São constituídos de um metal imerso numa solução que contém íons do próprio metal. São sistemas do tipo:
M+n + ne- ↔↔↔↔ M e o potencial eletródico é dado por:
]E 0
+−=
n[M
1log
n
0,0592E
que varia com a concentração do metal [Mn+] presente na amostra. Normalmente, esta equação também expressa como:
pME 0
n
0,0592E −=
onde, pM = log (1/[Mn+]). Verifica-se nesta última equação que o potencial do eletrodo fornece uma medida direta do pM, ou seja, ele tem uma relação linear com pM. Praticamente, apenas cobre, prata, mercúrio, cádmio, zinco e chumbo formam eletrodos de primeira classe reversíveis, isto é, capazes de funcionar como eletrodos indicadores de seus próprios íons.
Por outro lado, metais como ferro, tungstênio, níquel, cobalto e crômio não apresentam oxidação/redução reversível e não servem como eletrodos indicadores, pois o potencial medido é influenciado por uma variedade de fatores tais como, formação de óxidos, deformações cristalinas, área da superfície. Eletrodos de 2 a Classe Consistem de um metal revestido por uma substância pouco solúvel do

121
metal e em contato com uma solução contendo o ânion do sal. Exemplos de eletrodos de 2a classe são os eletrodos de prata/haletos de prata sensíveis aos íons haletos.
A reação do eletrodo pode ser re-escrita como: AgX + e - ↔↔↔↔ Ag + X - onde X= Cl -, Br -, ou I -
Como as concentrações das espécies AgX e Ag podem ser consideradas iguais à unidade o potencial deste eletrodo de prata/haleto de prata depende apenas da concentração do íon haleto e equação de Nernst pode ser escrita como:
]Xlog[0592,0EE 0 −−= ou pX0592,0EE 0 +=
Em princípio, um eletrodo de um metal revestido com qualquer sal pouco solúvel do metal, é um eletrodo de 2a classe que responde as variações das atividades ou concentrações do ânion. Eletrodos de 3 a Classe Consistem de um metal em contato com um sal pouco solúvel do metal e um sal menos solúvel de um segundo metal, sendo que os dois sais envolvem um íon comum. Exemplo: Ag|Ag2S, CuS, Cu2+.
+−=
Aga
1log0592,0799,0E
Ag2S 2Ag+ + S2-, KpsAg2S = a2Ag+.aS2-.
CuS Cu2+ + S2-, KpsCuS = aACu+.aS2-
21
+2CuCuS
S2AgAg
a x Kps
Kpsa
=+
)C(25 a
1log
20592,0
Kps
Kpslog
20592,0
799,0E o
2CuCuS
S2Ag
+−+=
De acordo com a Eq. acima, o potencial do eletrodo depende, apenas, de “aCu
2+”. ELETRODOS INERTES Estes eletrodos são constituídos de um metal inatacável (ouro ou platina), em contato com um sistema de oxidação-redução em solução. A função do metal inatacável é de simples condutor. Exemplo: considere um eletrodo de platina mergulhado numa solução do seguinte sistema de oxidação-redução:
Fe+2 ↔ Fe+3 + e-
]Fe[
]Fe[log
10592,0
EE2
30
+
+−=
O eletrodo de platina pode servir como um eletrodo indicador do potencial
produzido pelo sistema redox em relação a um eletrodo de referência, quando

122
ferro(II) estiver sendo titulado por um agente oxidante como, por exemplo, dicromato ou permanganato de potássio. Eletrodos Indicadores de Membrana Seletiva ou Eletr odos Seletivos Os eletrodos seletivos baseiam-se na formação de potenciais elétricos através de uma membrana semipermeável (material que separa duas fases líquidas não igualmente permeável a todos componentes), que devem ser seletivas apenas à espécie química de interesse. O potencial de um eletrodo indicador metálico surge devido a uma reação de oxidação-redução que ocorre na superfície do eletrodo, enquanto no eletrodo de membrana o potencial observado é um tipo de potencial de junção que se desenvolve através da membrana que separa a solução do analito de uma solução de referência. A figura abaixo mostra um diagrama esquemático de um sistema eletroquímico que utiliza um eletrodo de membrana seletivo.
O potencial através da membrana depende das concentrações da espécie química móvel nas soluções externa e interna. Mantendo fixa a concentração da espécie móvel na solução interna, o potencial medido refletirá as variações das concentrações da espécie móvel na solução externa, ou seja, nas amostras. A medida do potencial requer um contato elétrico entre as soluções interna e externa. Isto é feito usando eletrodos de referência (prata/cloreto de prata) nas duas soluções, um interno e outro externo. Um exemplo típico e clássico de um eletrodo seletivo é o eletrodo de vidro (membrana de vidro) usado na determinação potenciométrica de pH. Tipos de Eletrodo de Membrana Seletiva ou Eletrodos Seletivos
1) Eletrodos de Membrana Seletiva Não-Cristalina: - Eletrodo de Membrana de Vidro Sensível a íons H + Exemplo: vidro sensível a H+ - Eletrodo de Membrana de Vidro Sensível a Cátions Exemplos: vidros sensíveis a Na+, Li+, NH4
+

123
- Eletrodos de Membrana Seletiva Líquida Exemplo: líquido trocador iônico para Ca+2 - Eletrodos de Membrana Polimérica Imobilizada com Substância Seletiva Exemplo: matriz de PVC para Ca2+ e NO3
- - Eletrodos de Membrana Polimérica Permeável a Gás Exemplo: matriz de teflon ou polipropileno para NH3 e CO2 - Eletrodos de Membrana Enzimática, Bio-Catalítica ou Bio-Sensora Exemplo: eletrodo enzimático para Uréia,
2) Eletrodos de Membrana Seletiva Cristalina - Eletrodo de Membrana Seletiva Mono-Cristalina Exemplo: LaF3 para F- - Eletrodo de Membrana Seletiva Policristalina Mis ta e Não Mista. Exemplo: Ag2S para S= e Ag+ Eletrodo de Vidro Sensível a Íons H + Este eletrodo é um tipo de eletrodo de membrana cujo fundamento da resposta se deve ao aparecimento de um potencial quando se interpõe uma fina membrana de um vidro especial (SiO2 65% em mols, Li2O 28% em mols, Cs2O 3% em mols e La2O3 4% em mols) entre soluções de pH diferentes . Dois tipos de eletrodos de vidro seletivos a íons H+ são disponíveis comercialmente: ♦ o eletrodo de vidro simples ou não-combinado; ♦ e o eletrodo de vidro combinado. Eletrodo de Vidro Não-Combinado A figura do lado esquerdo a seguir mostra um desenho esquemático de um eletrodo de vidro não-combinado, enquanto a figura do lado direito mostra uma cela eletroquímica para determinação de pH, montada usando este eletrodo:

124
Verifica-se que esta cela eletroquímica necessita de um eletrodo de referência externo para determinação do pH da solução-amostra. Em alguns eletrodos comerciais a solução interna é um tampão contendo íons cloretos. Eletrodo de Vidro Combinado Sensível A cela eletroquímica usada para análise potenciométrica necessita sempre do eletrodo indicador e do eletrodo de referência. É possível incorporar os dois eletrodos em um único dispositivo, de modo que as medidas possam ser realizadas em pequenos volumes. Esses eletrodos são chamados de eletrodos combinados. A figura abaixo mostra um desenho esquemático de um eletrodo de vidro combinado usado para determinação potenciométrica de pH.
Hidratação da Membrana do Eletrodo de Vidro A membrana do eletrodo de vidro deve estar sempre hidratada para que ele funcione adequadamente como um eletrodo sensível a íons H+. Por isso, após a utilização, ele deve ser mantido mergulhado em água destilada ou desionizada. Se ocorrer desidratação da membrana, ela pode ser rehidratada, por exemplo, mergulhando a membrana em uma solução de HCl 1,0 mol/L, durante 24 horas e, depois, lavando-a longamente com água deionizada. Após sua lavagem com água destilada, ela deve ser conservada em água deionizada. Cela Eletroquímica para Medida de pH com um Eletrod o de Vidro A cela eletroquímica usada para medida do pH pode ser representada por: ELETRODO DE VIDRO E
j Ag ||||AgCl(sat),a Cl-||||||||aH+(amostra) ||||membrana ||||aH+(sol. interna),a Cl-, AgCl(sat) ||||Ag

125
Eletrodo de referência externa E
f1 E
f2 Eletrodo de Referência interna
Eref ext. potenciais de fornteira
Eref. int.
(Ef = E
f1 - E
f2)
De acordo com a cela eletroquímica acima, quatro potenciais se desenvolvem durante a medida de pH com o eletrodo de vidro. Dois destes potenciais são os potenciais dos eletrodos de referência interna, Eref. int . e externa, Eref. ext. . O terceiro é o potencial de junção líquida, Ej, produzido na interface entre a ponte salina do eletrodo de referência externa e a amostra. O quarto, e mais importante, potencial é o potencial de fronteira , Ef, que varia com o pH da amostra. Potencial de Fronteira em um Eletrodo de Vidro Sens ível a Íons H + Para servir como um sensor a pH, a membrana de vidro deve conduzir eletricidade. A condução através da membrana está relacionada a dois equilíbrios químico:
H+ + Vidro - ↔↔↔↔ H+vidro - AMOSTRA VIDRO1 VIDRO1
H+vidro - ↔↔↔↔ H+ + Vidro - VIDRO2 Solução interna VIDRO2 onde o subscrito 1 refere-se a interface entre o vidro e a amostra e o subscrito 2 refere-se a interface entre o vidro e solução interna do eletrodo de vidro. A interface onde ocorre uma maior dissociação torna-se negativa com relação a outra e um potencial, denominado de potencial de fronteira, Ef, se desenvolve. A magnitude do potencial de fronteira depende das atividades do íon H+ na amostra e na solução interna do eletrodo. Pode ser demonstrado a partir de considerações termodinâmicas que os potenciais de fronteira Ef1 e Ef2 estão relacionadas as atividades do íon hidrogênio em cada interface por uma equação Nernstiana dada por:
1
'1
11f aa
logn
0592,0kE −= e 1
'2
22f aa
logn
0592,0kE −=
onde k1 e k2 são constantes e a1 e a2 são as atividades do íon H+ na amostra e na solução interna. Os termos a1
’ e a2’ são as atividades do íon H+ na superfície
externa e interna da membrana de vidro. Se as superfícies externa e interna da membrana de vidro possuem, como normalmente acontece, o mesmo número de sítios carregados negativamente, onde o íon H+ pode se dissociar, então, k1 = k2 e a1
’ = a2’. Desta forma, o potencial
de fronteira global, Ef, produzido na membrana de vidro é dada por:

126
E E E naaf f f= − =1 2
1
2
00592, log
Como em um eletrodo de vidro a atividade do íon H+ na solução interna é mantida constante, tem-se que:
121f alog0592,0Kalog0592,0alog0592,0E ++++====−−−−====
ou seja,
1f pHlog0592,0LE −−−−====
Potencial Assimétrico em um Eletrodo de Vidro Sensí vel a Íons H + Se o pH da amostra e da solução interna fosse idêntico, o potencial de fronteira deveria ser teoricamente zero. Entretanto, um potencial diferente de zero, denominado de potencial assimétrico , é normalmente produzido devido à falta de uniformidade nas características físicas e químicas entre as duas interfaces (amostra-vidro e vidro-solução interna) durante a manufatura da membrana. Além disso, devido ao ataque mecânico e químico da superfície externa durante o uso, um potencial assimétrico variável é produzido. Por isso, o eletrodo de vidro deve ser freqüentemente calibrado cada vez que ele for utilizado. Essa calibração é realizada usando soluções tampão de pH conhecido. Potencial de Um Eletrodo de Vidro Sensível a Íons H + O potencial desenvolvido por um eletrodo de vidro tem associado a ele quatro potenciais: os potenciais dos dois eletrodos de referência, o potencial de fronteira, o potencial de junção líquida e o potencial assimétrico. O potencial medido de um eletrodo de vidro é, portanto, dado por:
Evidro = Eref.int. + Ef + Eassim. Uma vez que os potenciais dos eletrodos de referência, o potencial assimétrico e o potencial de junção líquida não devem variar durante a medida de pH, variações no potencial medido com eletrodo de vidro estão relacionadas a variações no potencial de fronteira, que por sua vez estão diretamente relacionadas as variações do pH das amostras, pois o pH da solução interna é mantida sempre constante. Como Ef = L - 0,0592 pH, tem-se que:
Evidro = L’ - 0,0592 pH onde, L’ = L + Eref.int + Eassim. Assim, quando existem diferenças de pH entre a amostra e a solução interna, um potencial na cela eletroquímica de um eletrodo de vidro é produzido e este potencial é utilizado na determinação potenciométrica do pH de uma amostra.

127
Pode ser observado que o potencial de um eletrodo de vidro é dado por uma equação de Nernst similar ao da equação do potencial de um eletrodo indicador metálico, embora os potenciais se desenvolvam por processos completamente diferentes. Um está relacionado com o potencial produzido por processo redox e o outro está relacionado com um potencial de fronteira onde não ocorre uma reação redox. Erro Alcalino nas Medidas de pH com um Eletrodo de Vidro Este erro surge devido ao fato de alguns eletrodos de vidro responderem ao íon hidrogênio e a íons monovalentes como os metais alcalinos. O erro alcalino pode ser explicado considerando o equilíbrio:
H+vidro - + B+ ↔↔↔↔ H+ + B+Vidro VIDRO AMOSTRA AMOSTRA VIDRO onde B+ representa um cátion monovalente, tal como o íon sódio. Observa-se que este erro é sempre negativo, ou seja, o pH medido é menor do que o pH esperado. Para um eletrodo com uma membrana de vidro do tipo CORNING 015, por exemplo, o pH de uma solução tampão de pH 12 contendo 1 mol/L de íons sódio foi medido como sendo 11,3 e para uma solução contendo 0,1 mol/L de íons sódio foi medido como sendo 11,7. A magnitude do erro alcalino depende do cátion e da composição da membrana de vidro. Para a maioria dos eletrodos de vidro este erro passa a ser significativo em medidas de pH acima de 9,0. Todavia, existem eletrodos de vidro especialmente construídos para se trabalhar em meios mais alcalinos (por exemplo, água do mar), onde se podem fazer medidas com erro alcalino desprezível até mesmo para pH maior ou igual a 12. Erro Ácido nas Medidas de pH com um Eletrodo de Vid ro Os eletrodos de vidro acusam valores de pH mais altos do que os verdadeiros em amostras com pH muito baixo, normalmente menor do que 1 ou negativos (soluções de ácidos fortes entre 0,1 e 10 mols/L). A magnitude deste erro positivo, denominado de erro ácido, depende de uma variedade de fatores, não é muito reprodutível e sua causa não é bem entendida. Resistências Elétricas das Membranas de Vidro As membranas de vidro, embora normalmente muito finas (30-100µm), possuem resistências elétricas muito altas 50 a 500 MΩ a temperatura ambiente, as quais aumentam exponencialmente para valores maiores do que GΩs para temperaturas mais baixas. Devido a isso, os eletrodos de vidro devem ser apropriadamente blindados e os circuitos eletrônicos usados para a medida da fem da cela eletroquímica devem ser de altíssima impedância. Eletrodos de Vidro Sensíveis a Cátions Monovalentes O erro alcalino levou alguns pesquisadores a desenvolver membranas de vidro com composições que permitissem que elas não fossem sensíveis a íons H+ e fossem sensíveis a determinados cátions monovalentes.

128
São conhecidos eletrodos de vidro sensíveis a Na+, K+, NH4+, Rb+, Cs+, Li+ e
Ag+, embora são disponíveis comercialmente eletrodos de vidro sensíveis à concentração total de cátions monovalentes, a Na+, Li+ e a NH4
+. A cela eletroquímica é semelhante à usada na determinação de pH e o seu potencial é dado também por uma equação semelhante, ou seja,
Evidro = K - 0,0592 pB onde, B é a atividade ou concentração do cátion monovalente. Efeito da Queda Ôhmica na Medida do potencial de um a Cela Eletroquímica Este efeito pode ser ilustrado pelo exemplo a seguir: EXEMPLO: o potencial de uma cela galvânica, eletrodo de vidro/eletrodo de calomelano , medido em uma solução tampão é teoricamente de 0,800V, considerando que a impedância interna (ou resistência) da cela é de 20MΩ, qual seria o erro relativo na medida do potencial, se ele fosse medido usando um potenciômetro cuja impedância interna (ou resistência) é de 100MΩ.
O circuito eletrônico da medida pode ser considerado como se fosse constituído de uma fonte de potencial da cela, Ecela e de dois resistores em série, o da cela, Rc, e o do potenciômetro, Rp, conforme mostra a figura abaixo:
Pela lei de Ohm, podemos escrever que:
Es = iRc + iRp A corrente do circuito é, para o exemplo, dada por:
A9-x1067,6 610 x 100) + (20
V 0,800 = i =Ω
A queda de potencial no potenciômetro, que é o próprio potencial medido para a cela é:
EP = iRp = 6,67 x 10-9 x 100 x 10 6 = 0,667V e o erro relativo da medida é:

129
Erro Relativo = 0,667 - 0,800
0,800 x 100 = -17%
É fácil mostrar que o erro pode ser reduzido a menos de 0,1% se a impedância interna do potencômetro for mil vezes maior, ou seja, 100GΩ (i=7,998x10-12, Ep=0,7998 e Erro=0.02%). Convenção do Sinal e Equações da Potenciometria Segundo convenção recente da IUPAC, o eletrodo indicador é sempre tratado como o catodo e o eletrodo de referência como anodo, de modo que o potencial de uma cela galvânica é dada por:
Ecela = Eind - Eref + Ej onde Ej é o potencial de junção líquida. Para um cátion Xn+ e para uma ânion Xn-, tem-se que, a 250C, as equações da potenciomentria (equações de NERNST) são dadas por:
E K0,0592
npX E K SpX
cela(cation)
cela (cation) ==== −−−− ⇒⇒⇒⇒ ==== −−−−
E K +0,0592
npX E K +SpX
cela(anion)
cela (anion) ==== ⇒⇒⇒⇒ ====
onde, pX = - log a x; K é uma constante que engloba o potencial do eletrodo indicador, o potencial de referência, o potencial de junção e o potencial de assimetria e S = RT/nF, que a 250C é igual a 59,2mV para um cátion ou ânion monovalente e 29,6mV para um cátion ou ânion divalente, quando a cela galvânica tem um perfeito comportamento Nernstiniano. A equação para o ânion mostra que o potencial medido aumente linearmente com o valor de pX. Entretanto, a equação para o cátion mostra que a medida em que se aumenta pX, o potencial diminui linearmente.
Uma vez que nas curvas de calibração, normalmente, o sinal medido aumenta linearmente com a concentração, para se obter este mesmo comportamento ao se usar um eletrodo seletivo a cátion, alguns instrumentos inverte eletronicamente o sinal medido quando este eletrodo está conectado, usualmente, ao terminal positivo do potenciômetro. Desta forma, o sinal medido aumenta linearmente com o aumento de pX.
Quando o instrumento não faz a inversão eletrônica, uma outra maneira para se obter um aumento do potencial com o aumento de pX é usar na construção da curva de calibração o valor do potencial medido com o sinal invertido. Efeito da Precisão da Medida Potenciométrica na Det erminação de Atividade

130
É importante avaliar como uma incerteza na medida do potencial de uma cela afeta a exatidão da atividade, ax . A maneira como a incerteza nas medidas dos potenciais se reflete em um erro na atividade ax da espécie química em análise pode ser obtida a partir da equação:
∆∆∆∆E E K 0,0592
nlog ax(cation ou anion)
cela==== −−−− ==== ±±±±
que dá:
±±±± ====log aE
0,4343 x 0,0592 / nx
∆∆∆∆
diferenciando chegamos a:
±±±± ====1a
.da 38,9nd( E)x
x∆∆∆∆
substituindo d(∆∆∆∆E) e dax pelos incrementos finitos e multiplicando ambos os lados da equação por 100 encontramos que:
V) (em E)(n33,89x10100x xaxa ∆∆∆∆∆∆∆∆⋅⋅⋅⋅⋅⋅⋅⋅====∆∆∆∆±±±±
ou
mV) (em E)(n3,89100x xaxa ∆∆∆∆∆∆∆∆⋅⋅⋅⋅⋅⋅⋅⋅====∆∆∆∆±±±±
A expressão ±±±±∆∆∆∆ax/ax x 100 é o erro relativo percentual, Er, é portanto:
Er = 3,89 n ∆∆∆∆(∆∆∆∆E) (em mVolt)
Para um potenciômetro cuja a incerteza na medida potenciométrica é de ±1mV ou seja, ∆(∆E)= ±1mV, tem-se que o erro relativo esperado em ax é de cerca de ± 4% para um íon monovalente e de 8% para um íon divalente.
Uma vez que os instrumentos atuais permitem medidas com incertezas de ±0,1mV até ± 0,001mV, tem-se que os erro relativos em aX devido as incertezas nas medidas, devem variar de ±0,4% a 0,004% para íons monovalentes e de ± 0,8 a ± 0,008% para íons divalentes. Atividade versus Concentração

131
A relação entre atividade ax e concentração cx é dada por:
ax= ƒƒƒƒx . cx onde, ƒƒƒƒx é o coeficiente de atividade para um dado íon
O coeficiente de atividade pode ser calculado pela Lei de Debye-Hükel ampliada, ou seja:
− ƒ =+
log.xx
x
AZ IB I
2
1 α
onde Zx e ααααx são a carga e o diâmetro efetivo do íon solvatado; A (=0,511 para água a 25oC) e B (=0,329 para água a 25oC) são constantes que dependem da constante dielétrica do solvente e da temperatura. I é a força iônica da solução que é dada por:
I c Zx x= ∑12
2.
Determinação da Concentração a Partir de Medidas Po tenciométricas. Até agora, temos usado o termo atividade de uma solução, em vez do termo concentração, pois os eletrodos seletivos respondem diretamente a atividade.
O termo atividade embora seja importante na determinação de parâmetros físico-químicos e em estudos biológicos e bioquímicos, pois alguns processos naturais são dependente da atividade, em vez da concentração, os químicos analíticos estão normalmente interessados na determinação da concentração do analito na solução.
A principal razão para isso é que preparação de soluções de atividade exatamente conhecida é difícil devido a problemas de se estabelecer os efeitos de todas outras espécies sobre o íon em investigação. Por outro lado, a preparação de solução de concentração conhecida é relativamente direta.
A suposição de que atividade e concentração são idênticas pode levar a sérios erros, principalmente para espécies polivalente, como mostra as curvas da figura a seguir, obtidas em escalas semi-logarítmicas.
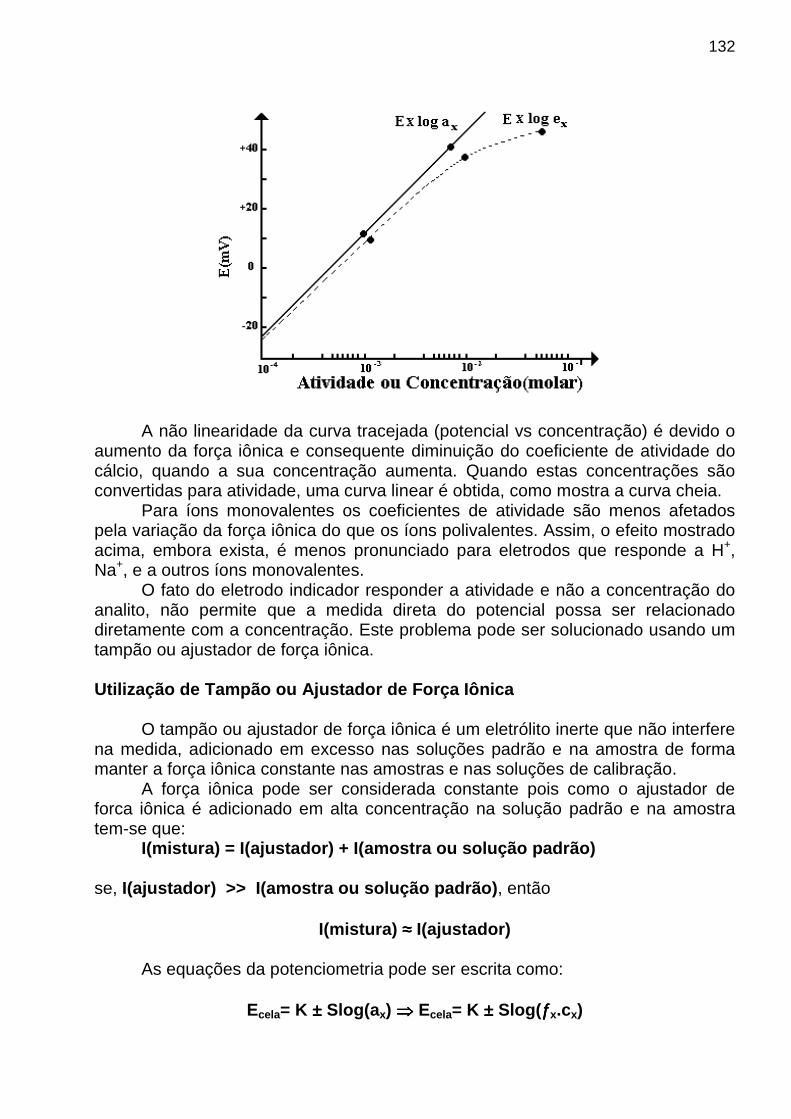
132
A não linearidade da curva tracejada (potencial vs concentração) é devido o aumento da força iônica e consequente diminuição do coeficiente de atividade do cálcio, quando a sua concentração aumenta. Quando estas concentrações são convertidas para atividade, uma curva linear é obtida, como mostra a curva cheia. Para íons monovalentes os coeficientes de atividade são menos afetados pela variação da força iônica do que os íons polivalentes. Assim, o efeito mostrado acima, embora exista, é menos pronunciado para eletrodos que responde a H+, Na+, e a outros íons monovalentes.
O fato do eletrodo indicador responder a atividade e não a concentração do analito, não permite que a medida direta do potencial possa ser relacionado diretamente com a concentração. Este problema pode ser solucionado usando um tampão ou ajustador de força iônica. Utilização de Tampão ou Ajustador de Força Iônica O tampão ou ajustador de força iônica é um eletrólito inerte que não interfere na medida, adicionado em excesso nas soluções padrão e na amostra de forma manter a força iônica constante nas amostras e nas soluções de calibração. A força iônica pode ser considerada constante pois como o ajustador de forca iônica é adicionado em alta concentração na solução padrão e na amostra tem-se que: I(mistura) = I(ajustador) + I(amostra ou solução pa drão) se, I(ajustador) >> I(amostra ou solução padrão) , então
I(mistura) ≈≈≈≈ I(ajustador) As equações da potenciometria pode ser escrita como:
Ecela= K ±±±± Slog(a x) ⇒⇒⇒⇒ Ecela= K ±±±± Slog( ƒƒƒƒx.cx)

133
Se um ajustador de força iônica é usado, tem-se a ƒƒƒƒx, é constante. Assim,
Ecela= K ±±±± Slog ƒƒƒƒx + Slog c x
e o potencial da cela é diretamente proporcional ao logaritmo da concentração, ou seja,
Ecela= K` ±±±± Slog c x onde K’é uma nova constante dada por: K’ = K ±±±± Slog ƒƒƒƒx. Efeito da Temperatura nas Medidas de Potenciais A temperatura além de afetar diretamente as medidas dos potenciais, conforme mostra a equação abaixo,
E KRT
nFacela x= ± 2 303,
log
afeta também os potenciais padrão de eletrodo, de junção líquida, de assimetria, do eletrodo de referência e, também, os coeficientes de atividade, ou seja, afeta o valor de K. Assim, é importante que durante as medidas as amostras e as soluções estejam na mesma temperatura. A tabela a seguir mostra os valores de K a diferentes temperaturas para uma cela de pH.
TABELA T (OC) 0 10 20 30 40 90 K (V) 0,0949 0,1001 0,1053 0,1079 0,1149 0,1337
Compensador de Temperatura Alguns instrumentos são equipados com dispositivos compensadores de temperatura que permite fazer leituras na faixa de 0oC a 100oC. A função principal deste dispositivo é ajustar a escala de medidas para diferentes temperaturas, quando o instrumento além de apresentar a escala em milivolts, apresenta também um escala em px (pH, píon). Com este dispositivo é possível calibrar os instrumentos a uma temperatura e medir a amostra a uma outra temperatura. Compensação Manual e Automática A compensação manual utiliza um resistor variável calibrado de fábrica em relação a temperatura que fornece um potencial ao circuito eletrônico de modo a compensar a variação de temperatura. A compensação automática é conseguida usando um termistor (termômetro de resistência) com apropriado coeficiente de temperatura. O

134
termistor é mergulhado, junto com os eletrodos, na solução em estudo, quando há uma variação de temperatura, as constantes do circuito eletrônico variam de modo a compensar essa alteração. Coeficientes ou Relações de Seletividade Idealmente, um eletrodo indicador deveria ser sensível a apenas uma espécie química e o seu potencial em uma cela deveria seguir a equação:
E= K ±±±± Spx entretanto, alguns eletrodos estão sujeitos a interferências de outros íons, quando eles são mergulhados em uma amostra contendo uma mistura de íons. Neste caso, o potencial da cela contendo este eletrodo é dado pela equação abaixo, chamada de equação de NICOLSKI.
E k 2,303RTnF
log a w ai ij . jiz /zj= ± +
∑
onde ai é a atividade do íon principal de carga zi, aj é a atividade do j-ésimo íon interferente de carga zj e wij é a relação ou o coeficiente de seletividade. A validade dessa equação para mais de um interferente é questionável. Para um eletrodo de Ca2+ interferido por Mg2+ e H+ a equação de NICOLSKI pode ser escrita como:
[ ]E kS w w a
Ca Ca= ± + + +
22log . .
, , a a
Ca+2 +2
Mg+2
Mg+2
H+
H+
Um problema deve ser mencionado sobre a relação de seletividade; os seus valores variam com a variação relativa das concentrações dos íons interferidos e interferentes. Na prática os valores de wij são usados em termos qualitativos para prever se uma interferência é significativa ou não. Métodos Experimentais para Determinação de Coeficie ntes de Seletividade Os seguintes métodos podem ser utilizados: - Método das soluções separadas a atividades ou a potenciais fixados. - Método das soluções misturadas a diferentes atividades do interferido ou do interferente. Embora esses métodos raramente dão valores idênticos, eles são de grandeza comparáveis ou próximos. Método das Soluções Separadas a Atividades ou a Pot enciais Fixados. Nesses métodos, constrói-se uma curva analítica para o interferente e o interferido, e essas curvas são lançadas em um gráfico como mostrado na figura abaixo.
135
Para atividades fixadas tem-se que:
Log Wi, jEj====
−−−− EiS
Para potenciais fixados tem-se que:
Waai j
i
j, ====
Métodos das Soluções Misturadas a Diferentes Ativid ades do Interferente ou do Interferido São construídas curvas mantendo a atividade do interferido constante e variando as atividades do interferente ou vice-versa. As curvas obtidas são mostradas nas figuras a seguir.
Variando-se o interferente ou o interferido tem-se que:

136
Wa
ai j
i
jz zi j
, /( )
====
O mecanismo da interferência que ocorre em eletrodos de membrana sólida tais como os eletrodos seletivos a Br-, I-, Cl-, S=, etc., requer um método diferente para expressar o coeficiente de seletividade. As reações na superfície podem converter um dos componentes da membrana sólida a um segundo componente insolúvel. Como conseqüência, a membrana pode perder sensibilidade ao íon interferido, danificando o eletrodo. Instrumentação para Medidas Potenciométricas Os instrumentos usados na potenciometria são conhecidos como potenciômetros. Uma vez que estes instrumentos são, em sua maioria, calibrados de fábrica para medidas diretas de pH eles são também conhecidos comercialmente com o nome de pHmetro. Existem também instrumentos calibrados de fábrica para a determinação direta da concentração de outros íons ou espécies moleculares e, assim, eles são comercializados como medidores de pX (X=ÍON ou MOLÉCULA) (pCl-, pS=, PO2, etc). Normalmente, esses instrumentos medem também os potenciais de uma cela eletroquímica e podem ser usados, através da construção de curvas de calibração Potencial x pX , para determinar a concentração de uma outra espécie X qualquer. Calibração dos Potenciômetros Usando a Cela Padrão de Weston A medida do potencial de uma cela eletroquímica requer, freqüentemente, o uso de uma cela padrão para calibração dos potenciômetros. Em virtude de sua excelente reprodutibilidade utiliza-se quase universalmente a cela de WESTON. A figura a seguir mostra um diagrama desta cela.
Ela consiste de um recipiente em forma de H, onde o eletrodo negativo, ou seja, o de menor potencial é formado por amálgama de cádmio saturado (12,5% de Cd em massa) e o eletrodo de maior potencial é formado de mercúrio coberto

137
sulfato de mercurio(I). O eletrólito é uma solução saturada de sulfato de cádmio em contato com cristais de excesso de sulfato de cádmio. Essa célula galvânica pode ser representada por:
Cd(Hg)CdSO4.8/3H2O(sat), Hg 2SO4Hg
As reações eletródicas e a reação global é dada por: Cd(Hg) Cd2+ + Hg + 2e-
Hg2SO 4 + 2e- 2Hg + SO42-
Reação Total: Cd(Hg) + Hg2SO4 Cd2+ + 3Hg + SO42-
A uma dada temperatura, as concentrações de todos os componentes do sistema eletródico são constantes; portanto, o potencial da cela de WESTON é praticamente invariante, dependendo muito pouco da temperatura. O potencial. dela, em volts, para temperaturas t variando entre 0o e 40 oC, é dada por:
Et = 1,017300 – 0,0000406(t – 20) – 0,00000095(t – 20)2
Métodos de Análise Qualitativa e Quantitativa por P otenciometria A potenciometria é uma técnica essencialmente de análise quantitativa, embora a resposta de um eletrodo seletivo possa ser usada como indicativo se uma determinada espécie está ou não presente na amostra. Por exemplo, um potenciômetro/medidor de pX pode ser usado para indicar se a espécie X está ou não presente na amostra. Três métodos de análise potenciométrica quantitativa podem ser utilizados para a determinação da concentração de uma espécie química na amostra: ♦ o método da potenciometria direta ou calibração a d ois pontos; ♦ o método da curva de calibração (potencial x pX); ♦ o método da titulação potenciométrica; Método da Potenciometria Direta ou da Calibração a Dois Pontos A determinação da concentração de uma espécie X por potenciometria direta é simples e rápida quando o instrumento já vem calibrado de fábrica para a medida de pX. Procedimento . A escala de pX é, inicialmente, ajustada mergulhando o eletrodo de referência e o eletrodo indicador de X em uma das soluções-padrão e, após ajustar a temperatura desejada com o botão compensador, a escala de pX é ajustada, usando os botões de calibração e de sensibilidade (ou ganho), para ler o valor de pX da solução-padrão. O processo é repetido para outra solução-padrão e novamente para primeira solução-padrão até que seja lido no mostrador os valores esperados para os pX das duas soluções-padrão. Em seguida, os eletrodos são mergulhados na amostra e o valor pX aparece diretamente no mostrador (analógico ou digital). A partir do valor de pXA, pode-se determinar finalmente a concentração do analito na amostra, [XA], a partir da expressão [XA] = 10−−−−pXA. É importante durante a etapa de calibração que as duas soluções-padrão estejam na mesma temperatura e na mesma força iônica (usar um tampão de força

138
iônica). A amostra pode ser medida em outra temperatura, se o potenciômetro possui um compensador de temperatura. Também é importante que as concentrações das amostras estejam dentro da faixa de concentração das duas soluções-padrão. Medir-se pH é a principal aplicação da potenciometria direta. Isto acontece porque é onde ela é mais utilizada e porque o eletrodo de vidro é muito barato. Por isso, a maioria dos potenciômetros é comercializada com escalas de pH. Resumo dos Erros mais comuns na Medida de pH com El etrodos de Vidro Erro Alcalino - não devem ser realizadas medidas para soluções com pH > 11-12. Erro Ácido - não devem ser realizadas medidas para soluções com pH < 0,5 Erros pela Desidratação da Membrana - produz medidas instáveis de pH. Erros no pH dos Tampões - causados pela inadequada preparação das soluções-tampão ou pela deterioração devido à ação de bactérias nas espécies orgânicas dos tampões. Método da Curva de Calibração ou Curva Analítica Usando várias soluções-padrão é construída uma curva analítica de E(cela) x pX. A partir da medida do E(cela) para amostra, o pXA da amostra pode ser obtido ou por interpolação na curva, ou usando a equação da reta obtida pelo método dos mínimos quadrados. Conhecendo pXA, pode-se determinar finalmente a concentração do analito na amostra, [XA], a partir da expressão [XA] = 10−−−−pXA. Titulações Potenciométricas A titulação potenciométrica utiliza a medida do potencial (pH ou pX) de um eletrodo indicador em relação a um eletrodo de referência para acompanhar a variação da concentração do analito durante um processo de titulação objetivando a detecção do ponto final. A figura a seguir mostra o desenho esquemático de um sistema de titulação potenciométrica.

139
Procedimentos para Localização do Ponto Final da Ti tulação São utilizados comumente métodos gráficos para a determinação do ponto final da titulação (P.F.). Os mais comuns são: a) Método da curva de potencial versus volume do titulante; b) Método da curva da primeira derivada versus volume do titulante; c) Método da curva da segunda derivada versus volume do titulante; d) Método da Curva de Gran.
a) Método da Curva de Potencial Versus Volume do Titulante Consiste em traçar uma curva do potencial medido versus o volume adicionado em cada etapa da titulação. Se uma curva sigmóide é obtida, o ponto final é encontrado graficamente no ponto de inflexão conforme mostra a figura a seguir.

140
Esse método tem problemas de exatidão na localização do ponto final de titulação, principalmente quando a curva sigmóide não é bem definida. b) Método da Curva da Primeira Derivada Versus Volume do Titulante Consiste em traçar uma curva da razão entre a variação de potencial (pH ou pX) e a variação do volume adicionado da solução titulante versus o volume médio entre cada adição, ou seja, construir a curva de ∆∆∆∆E/∆∆∆∆V versus V médio , onde 3
iV)1i(ViE)1i(E
−+
−+=∆∆
VE
e 2
VV i)1i( += +
mV
A curva obtida e a localização do ponto final de titulação são mostradas na figura abaixo:
c) Método da Curva da Segunda Derivada Versus Volume do Titulante Consiste em traçar uma curva da razão entre a variação de ∆E/∆V (∆pH/∆V ou ∆pX/∆V) e a variação do volume médio contra a média do volume médio, ou seja, construir a curva de ∆∆∆∆(∆∆∆∆E/∆∆∆∆V)/∆∆∆∆Vm=∆∆∆∆2E/∆∆∆∆V2 (ou (∆∆∆∆(∆∆∆∆pX/∆∆∆∆V)/∆∆∆∆Vm=∆∆∆∆2pX/∆∆∆∆V2) versus MV médio , onde,
( ) ( ) m
2
im1im
i1i
m VE
VV
VE
VE
VVE
∆∆=
−
∆∆−
∆∆
=∆
∆∆∆
+
+ e ( ) ( )
2
VV im1imm
+= +MV
A curva obtida e a localização do ponto final de titulação são mostradas na figura a seguir:
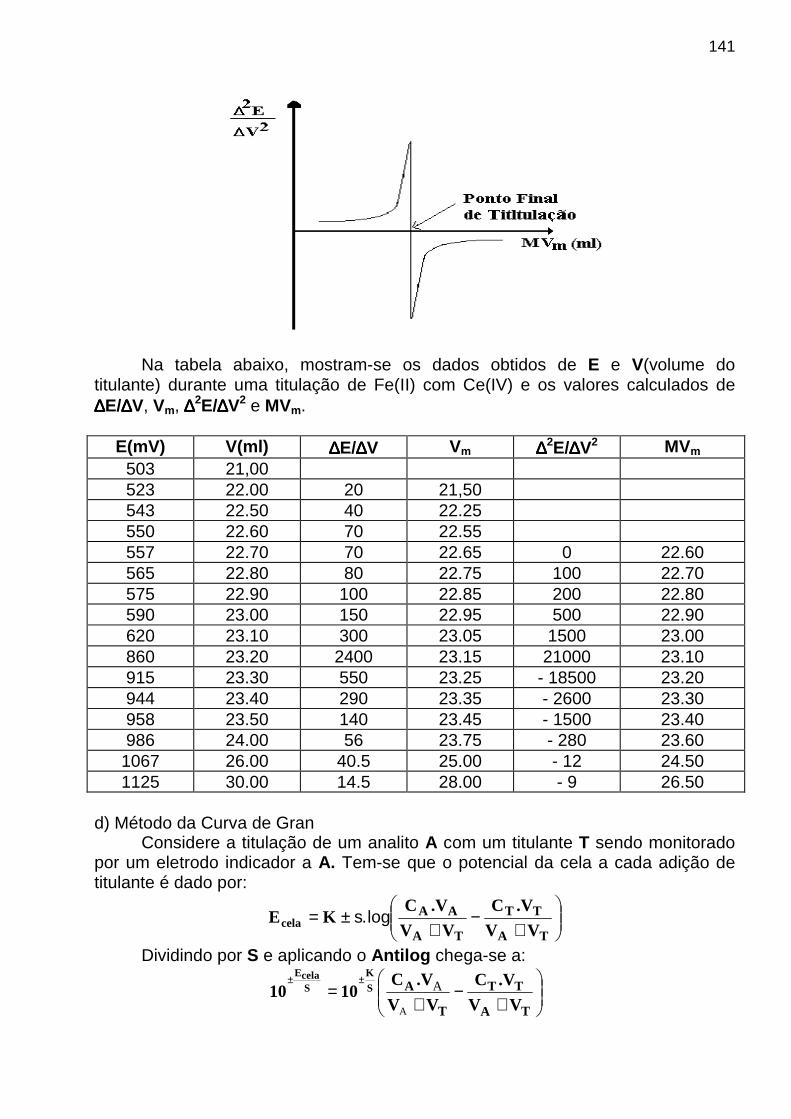
141
Na tabela abaixo, mostram-se os dados obtidos de E e V(volume do titulante) durante uma titulação de Fe(II) com Ce(IV) e os valores calculados de ∆∆∆∆E/∆∆∆∆V, Vm, ∆∆∆∆2E/∆∆∆∆V2 e MVm.
E(mV) V(ml) ∆∆∆∆E/∆∆∆∆V Vm ∆∆∆∆2E/∆∆∆∆V2 MVm 503 21,00 523 22.00 20 21,50 543 22.50 40 22.25 550 22.60 70 22.55 557 22.70 70 22.65 0 22.60 565 22.80 80 22.75 100 22.70 575 22.90 100 22.85 200 22.80 590 23.00 150 22.95 500 22.90 620 23.10 300 23.05 1500 23.00 860 23.20 2400 23.15 21000 23.10 915 23.30 550 23.25 - 18500 23.20 944 23.40 290 23.35 - 2600 23.30 958 23.50 140 23.45 - 1500 23.40 986 24.00 56 23.75 - 280 23.60
1067 26.00 40.5 25.00 - 12 24.50 1125 30.00 14.5 28.00 - 9 26.50
d) Método da Curva de Gran Considere a titulação de um analito A com um titulante T sendo monitorado por um eletrodo indicador a A. Tem-se que o potencial da cela a cada adição de titulante é dado por:
+−
+±=
TA
TT
TA
AA
VV
.VC
VV
.VCKEcela log.s
Dividindo por S e aplicando o Antilog chega-se a:
+−
+=
±±
TA
TT
T
A
VV
.VC
VV
.VC1010 S
KScelaE
A
A
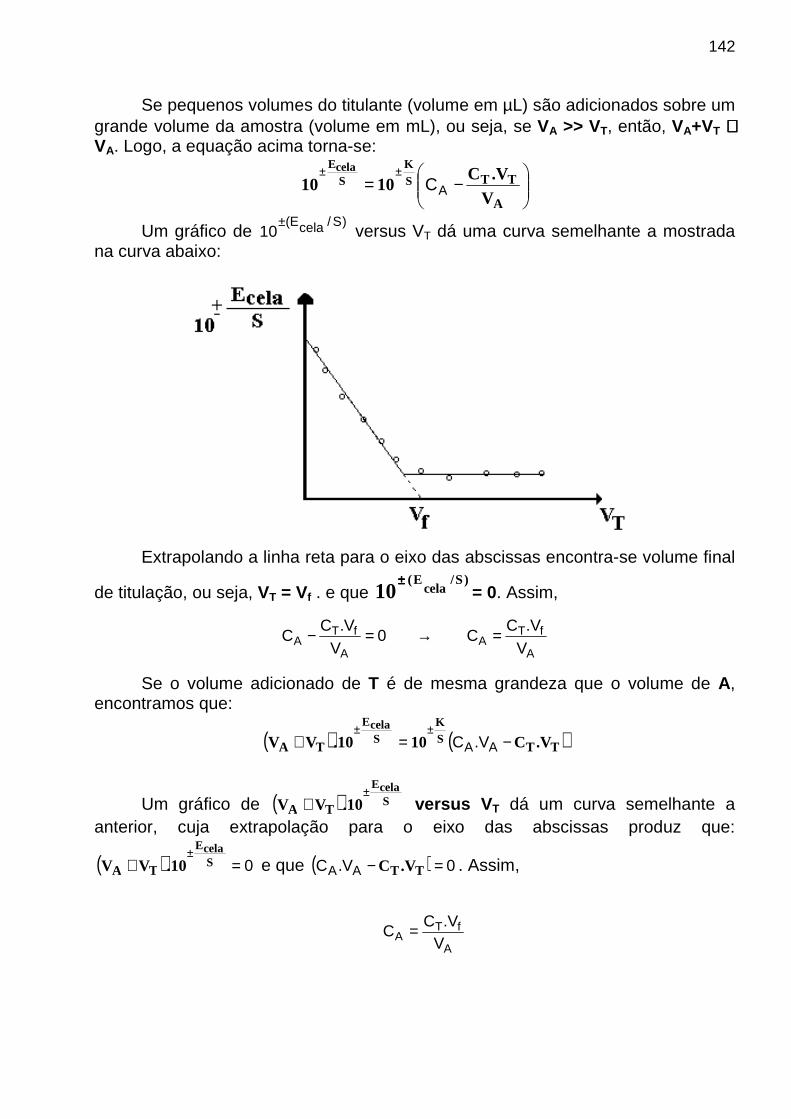
142
Se pequenos volumes do titulante (volume em µL) são adicionados sobre um grande volume da amostra (volume em mL), ou seja, se VA >> VT, então, VA+VT ≅≅≅≅ VA. Logo, a equação acima torna-se:
−=
±±
A
TT
V.VC
1010 SK
ScelaE
AC
Um gráfico de )S/celaE(
10±
versus VT dá uma curva semelhante a mostrada na curva abaixo:
Extrapolando a linha reta para o eixo das abscissas encontra-se volume final
de titulação, ou seja, VT = Vf . e que 10±±±± ( / )E
celaS
= 0. Assim,
A
fTA
A
fTA V
V.CC 0
VV.C
C =→=−
Se o volume adicionado de T é de mesma grandeza que o volume de A, encontramos que:
( ) ( )TTTA .VC10.10 VV SK
ScelaE
−=+±±
AA V.C
Um gráfico de ( ) ScelaE
.10 VV TA±
+ versus V T dá um curva semelhante a anterior, cuja extrapolação para o eixo das abscissas produz que:
( ) 0=+±
ScelaE
.10 VV TA e que ( ) 0V.C AA =− TT .VC . Assim,
A
fTA V
V.CC =

143
CONDUTOMETRIA Introdução A condutometria baseia-se em medidas da condutividade ou condutância elétrica das soluções iônicas. A condução da eletricidade está relacionada com a migração de íons negativos e positivos quando se aplica uma diferença de potencial entre dois eletrodos mergulhados na solução eletrolítica. Os íons negativos migram para o eletrodo positivo e os íons positivos para o eletrodo negativo. A condutância de uma solução iônica depende:
- da natureza dos íons, ou seja, da carga e da mobilidade dos íons; - do número de íons presentes.
Para uma mesma solução de uma espécie iônica, a condutância aumenta com o aumento da concentração desta espécie na solução. A condutometria abrange duas técnicas analíticas: a condutometria direta e a titulação condutométrica .
Na condutometria direta mede-se a condutância da solução quando dois eletrodos inertes, nos quais é aplicada uma diferença de potencial, são mergulhados na amostra.
Na titulação condutométrica mede-se o aumento ou diminuição da condutância durante um processo de titulação. A curva de titulação deve apresentar uma descontinuidade no ponto de equivalência e o volume do ponto final é usado para encontrar a concentração do analito na amostra.
A condutometria direta não pode ser considerada uma técnica específica, pois a condutância elétrica de uma solução é a soma das condutâncias individuais dos íons presentes na amostra. Por outro lado, a titulação condutométrica pode ser considerada uma técnica específica desde que a reação de titulação seja específica. A medida da condutância direta de uma amostra tem sido usada como indicação da existência ou não de uma quantidade elevada de íons. Neste aspecto, por exemplo, esta medida é um parâmetro importante na avaliação da potabilidade de uma água para consumo humano, na qualidade de uma água destilada e/ou deionizada, etc. Para uma amostra de composição conhecida, a medida da condutância tem sido usada para determinação de constantes de ionização, produtos de solubilidade, constantes de formação de complexos, condutâncias equivalentes e efeitos de solventes. Condutância Específica
A resistência, R, de uma solução iônica, como nos condutores metálicos depende da natureza e das dimensões do condutor (veja Figura a seguir). Ela é diretamente proporcional ao seu comprimento l e inversamente proporcional à área da seção transversal A, ou seja,
ρAl
=R

144
onde, a constante de proporcionalidade ρρρρ é a resistência específica do material ou da solução iônica, que é dada em unidades Ω.cm. Uma vez que a condutância, L, de uma solução ou um material é definida como o inverso da resistência, pode-se escrever que:
lA
k=L
onde k (o inverso da resistência específica,1/ρρρρ) é a condutância específica (ou condutividade) com unidades ohm-1.cm-1= S.cm-1, onde S = Siems = ohm-1.
Condutância Equivalente
Para facilitar a comparação das condutâncias de soluções de diferentes
eletrólitos, foi introduzido o conceito de condutância equivalente ΛΛΛΛ. Ela é definida
como a condutância de uma solução contendo um equivalente-grama de um
eletrólito colocada entre eletrodos planos distantes 1cm um do outro e com área
superficial exatamente suficiente para conter todo o volume da solução.
Como o volume da solução e a área do eletrodo não são especificado, pois
depende da solução contendo um equivalente grama do eletrólito, então, por
exemplo, uma solução contendo um 1 e equivalente-grama por litro requereria um
par de eletrodos, cada um com área de superficial de 1000cm2.
Condutância Equivalente versus Condutância Específi ca
A condutância equivalente pode ser derivada da condutância específica e

145
da concentração da solução. O volume, V, de uma solução (em cm3) que contém
um equivalente grama é:
V =1000
C
em que C é a concentração do eletrólito expressa em equivalente-grama por litro.
Em termos da dimensão de uma cela condutimétrica, o volume V é dado por: V =
lA . Como l é fixado como sendo igual a 1cm, tem-se que:
V = A =1000
C
Substituindo a equaçãoL =Al
κκκκ
, onde L=ΛΛΛΛ, na equação acima tem-se que:
ΛΛΛΛ =1000
Cκκκκ
Para a concentração expressa em moles por litro, a condutância equivalente
passa a ser denominada de condutância molar.
Variação da Condutância Equivalente com Concentraçã o
Para um eletrólito forte, a condutância equivalente aumenta medida que a
concentração da solução eletrolítica diminui. O tratamento teórico feito por
Onsager leva, para concentrações < 10-3molar, a expressão:
ΛΛΛΛ = ΛΛΛΛo + (A + B)√√√√C
onde A e B são os fatores que exprimem os efeitos eletroforéticos e o tempo de
relaxação.
As condutâncias equivalentes de um eletrólito tendem para um valor limite
em soluções muito diluídas. A condutância equivalente a diluição infinita é
simbolizada por ΛΛΛΛo. A variação da condutância equivalente com a concentração
depende, em grande extensão, do tipo eletrólito. Na figura abaixo é mostrada as
condutâncias equivalentes de KCl e ácido acético em função da raiz quadrada da
concentração.
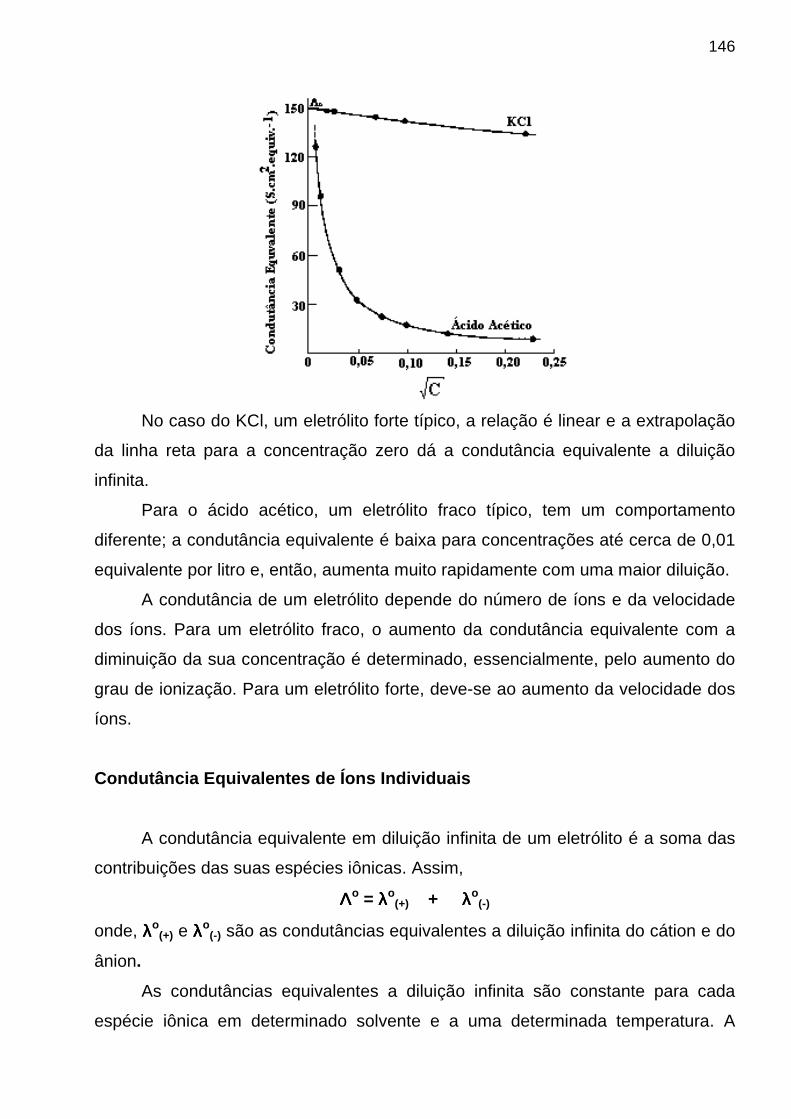
146
No caso do KCl, um eletrólito forte típico, a relação é linear e a extrapolação
da linha reta para a concentração zero dá a condutância equivalente a diluição
infinita.
Para o ácido acético, um eletrólito fraco típico, tem um comportamento
diferente; a condutância equivalente é baixa para concentrações até cerca de 0,01
equivalente por litro e, então, aumenta muito rapidamente com uma maior diluição.
A condutância de um eletrólito depende do número de íons e da velocidade
dos íons. Para um eletrólito fraco, o aumento da condutância equivalente com a
diminuição da sua concentração é determinado, essencialmente, pelo aumento do
grau de ionização. Para um eletrólito forte, deve-se ao aumento da velocidade dos
íons.
Condutância Equivalentes de Íons Individuais
A condutância equivalente em diluição infinita de um eletrólito é a soma das
contribuições das suas espécies iônicas. Assim,
ΛΛΛΛo = λλλλo(+) + λλλλo
(-)
onde, λλλλo(+) e λλλλo
(-) são as condutâncias equivalentes a diluição infinita do cátion e do
ânion.
As condutâncias equivalentes a diluição infinita são constante para cada
espécie iônica em determinado solvente e a uma determinada temperatura. A
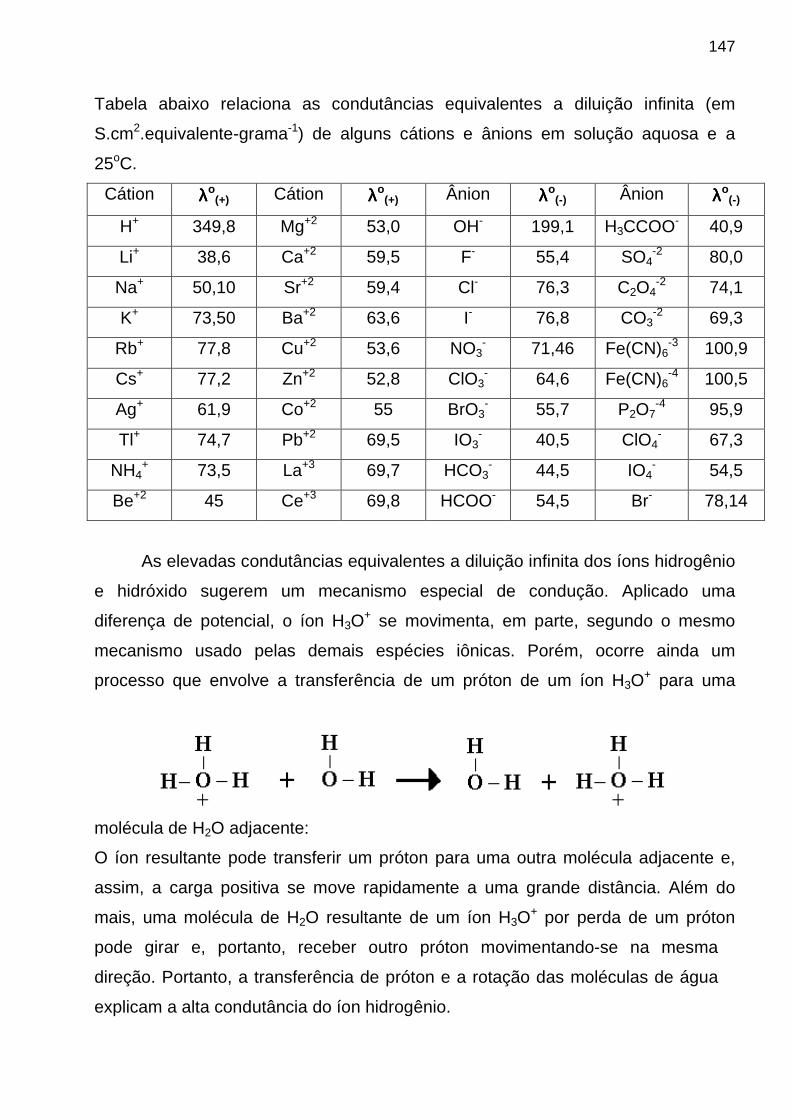
147
Tabela abaixo relaciona as condutâncias equivalentes a diluição infinita (em
S.cm2.equivalente-grama-1) de alguns cátions e ânions em solução aquosa e a
25oC.
Cátion λλλλo(+) Cátion λλλλo
(+) Ânion λλλλo(-) Ânion λλλλo
(-)
H+ 349,8 Mg+2 53,0 OH- 199,1 H3CCOO- 40,9
Li+ 38,6 Ca+2 59,5 F- 55,4 SO4-2 80,0
Na+ 50,10 Sr+2 59,4 Cl- 76,3 C2O4-2 74,1
K+ 73,50 Ba+2 63,6 I- 76,8 CO3-2 69,3
Rb+ 77,8 Cu+2 53,6 NO3- 71,46 Fe(CN)6
-3 100,9
Cs+ 77,2 Zn+2 52,8 ClO3- 64,6 Fe(CN)6
-4 100,5
Ag+ 61,9 Co+2 55 BrO3- 55,7 P2O7
-4 95,9
Tl+ 74,7 Pb+2 69,5 IO3- 40,5 ClO4
- 67,3
NH4+ 73,5 La+3 69,7 HCO3
- 44,5 IO4- 54,5
Be+2 45 Ce+3 69,8 HCOO- 54,5 Br- 78,14
As elevadas condutâncias equivalentes a diluição infinita dos íons hidrogênio
e hidróxido sugerem um mecanismo especial de condução. Aplicado uma
diferença de potencial, o íon H3O+ se movimenta, em parte, segundo o mesmo
mecanismo usado pelas demais espécies iônicas. Porém, ocorre ainda um
processo que envolve a transferência de um próton de um íon H3O+ para uma
molécula de H2O adjacente:
O íon resultante pode transferir um próton para uma outra molécula adjacente e,
assim, a carga positiva se move rapidamente a uma grande distância. Além do
mais, uma molécula de H2O resultante de um íon H3O+ por perda de um próton
pode girar e, portanto, receber outro próton movimentando-se na mesma
direção. Portanto, a transferência de próton e a rotação das moléculas de água
explicam a alta condutância do íon hidrogênio.

148
No caso do íon hidróxido, a transferência de próton obedece ao seguinte
mecanismo:
A ordem das condutâncias equivalentes dos íons dos metais alcalinos,
alcalinos-terrosos e halogênios seguem a ordem inversa do aumento de seus raios
iônicos. Diminuindo o raio iônico aumenta o campo eletrostático (aumenta o
potencial iônico), aumenta camada de hidratação, diminui a mobilidade do íon e,
consequentemente, diminui a condutância do íon.
Exceto para os íons H+ e OH-, os valores das condutâncias equivalentes
para o restante dos íons são normalmente menores do que 80 S.cm2.equivalente-
grama-1. Os valores um pouco mais altos para os íons complexos [Co(NH3)6]+3,
[Fe(CN)6]-3 e [Fe(CN)6]
-4 é que a formação da camada de hidratação é dificultada
pela formação da camada protetora (blindante) dos ligantes NH3 e CN-.
Embora as condutividades equivalentes dos eletrólitos sejam importante
para o químico analítico, as condutividades equivalentes para os íons individuais
são de maior importância, como veremos, por exemplo, na interpretação das
curvas de titulação.
Velocidade e Mobilidades dos Íons
Para soluções diluídas de um eletrólito forte, a corrente e, portanto, sua a
condutividade depende da carga do íon, do número de íons e da velocidade efetiva
dos íons.
A velocidade absoluta com que cada íon se movimenta através de uma
solução depende da natureza do íon, da concentração da solução, da temperatura
e do gradiente de potencial (a diferença de potencial por centímetro na direção do
movimento). Para um gradiente de potencial de 1Volt/cm, a velocidade dos íons é
denominada de mobilidade (em cm/s) e é representada pelo símbolo u.
Em diluição infinita, onde não há interações iônicas, a mobilidade atinge
um valor limite máximo, denominado de mobilidade absoluta . Para sais

149
completamente ionizados, a condutância equivalente limite (a diluição infinita), ΛΛΛΛo,
é proporcional às mobilidades absoluta dos íons, isto é,
ΛΛΛΛo = F(uo+ + uo
-)
onde F é a constante de Faraday.
As condutividades iônicas equivalentes a diluição infinita podem ser
expressa por:
λλλλo(+) = F.uo
+ e λλλλo(-) = F.uo
-
Condução da Corrente
Na medida da condutância de uma solução em uma cela eletroquímica, os
elementos mais comuns no caminho da corrente são: os condutores metálicos, os
contatos metal-metal, as interfaces eletrodo-solução e a própria solução, sendo os
dois últimos os que mais frequentemente limitam a corrente.
Na solução, a corrente pode ser conduzida pela migração dos íons ou pela
polarização do solvente.
Nas interfaces eletrodo-solução, os processos importantes na condução da
corrente são as reações de transferência de elétrons (corrente faradaica) e a carga
da dupla camada elétrica (corrente não-faradaica).
A condução de corrente contínua (cc) através de uma célula eletroquímica
envolve um processo faradaico (reação de oxi-redução) e a condução de corrente
alternada (ca) pode envolver processos faradaico e não-faradaico.
A velocidade de cada reação eletródica e, portanto, a corrente são
controladas, em parte, pela velocidade da reação de transferência de elétrons e ,
em parte, pela velocidade de transporte de uma espécie presente na solução para
as imediações da superfície do eletrodo.
A velocidade da reação de transferência de elétrons depende da natureza e
da concentração da espécie eletroativa e da natureza e do potencial eletrodo. Os
processos responsáveis pelo transporte de massa são: migração, difusão e
convecção.
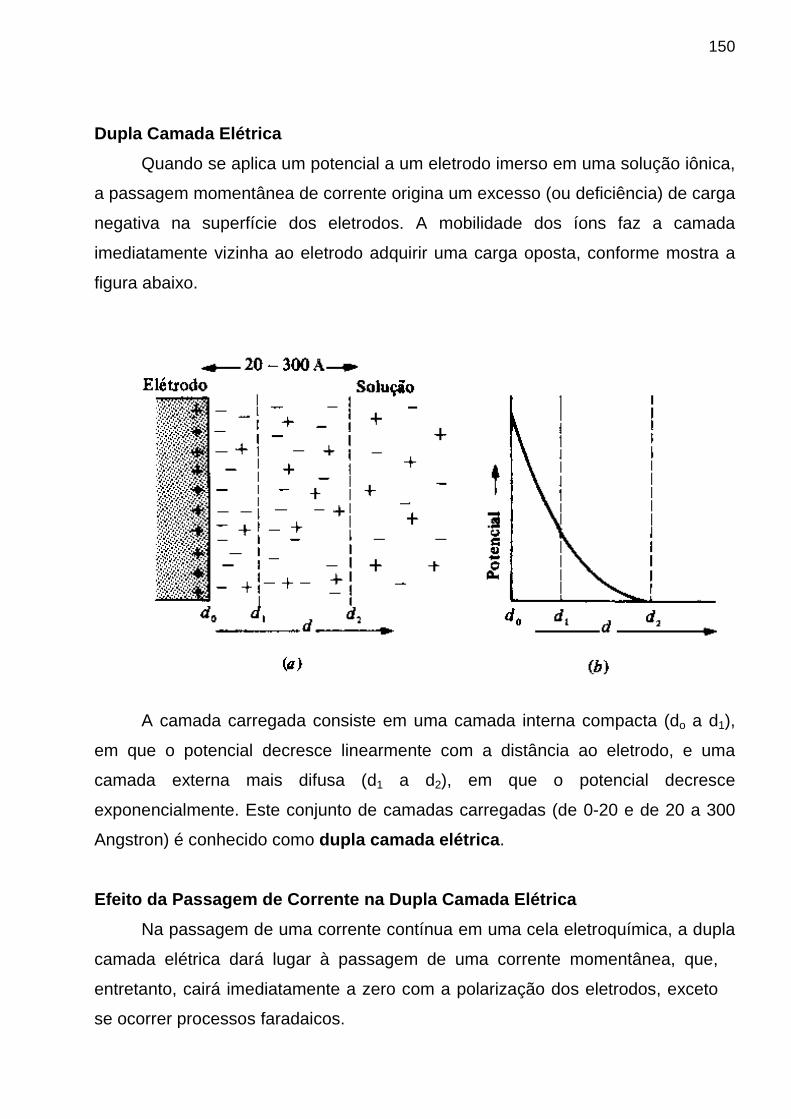
150
Dupla Camada Elétrica
Quando se aplica um potencial a um eletrodo imerso em uma solução iônica,
a passagem momentânea de corrente origina um excesso (ou deficiência) de carga
negativa na superfície dos eletrodos. A mobilidade dos íons faz a camada
imediatamente vizinha ao eletrodo adquirir uma carga oposta, conforme mostra a
figura abaixo.
A camada carregada consiste em uma camada interna compacta (do a d1),
em que o potencial decresce linearmente com a distância ao eletrodo, e uma
camada externa mais difusa (d1 a d2), em que o potencial decresce
exponencialmente. Este conjunto de camadas carregadas (de 0-20 e de 20 a 300
Angstron) é conhecido como dupla camada elétrica .
Efeito da Passagem de Corrente na Dupla Camada Elét rica
Na passagem de uma corrente contínua em uma cela eletroquímica, a dupla
camada elétrica dará lugar à passagem de uma corrente momentânea, que,
entretanto, cairá imediatamente a zero com a polarização dos eletrodos, exceto
se ocorrer processos faradaicos.

151
Na passagem de uma corrente alternada a situação é diferente, a reversão
da relação de cargas a cada meio-ciclo origina uma corrente não-faradaica. A
dupla camada elétrica de um dos eletrodos se carrega em um meio ciclo, enquanto
a do outro eletrodo se descarrega. No seguinte meio-ciclo, a carga e descarga se
invertem em cada um dos eletrodos.
No processo não-faradaico, portanto, ânions e cátions conduzem a corrente
através da solução, alternadamente e cada eletrodo se comporta como um
capacitor. A corrente de capacitância aumenta com a freqüência e com o tamanho
do eletrodo. O controle conveniente destas variáveis permite que, essencialmente,
toda corrente alternada que flui através da célula passe pela interface eletrodo-
solução por processo não-faradaico.
Para frequências na ordem de KHz, a corrente alternada é conduzida
exclusivamente pelo movimento dos íons. Entretanto, para frequências na ordem
de MHz, aparece um mecanismo de condução que resulta da
polarização[induzida(moléculas apolares) e de orientação(moléculas polares)] do
meio dielétrico. A corrente dielétrica, que depende da constante dielétrica do meio,
é diretamente proporcional à frequência.
Medidas da Condutância de Soluções Iônicas
A condutância de uma solução iônica é determinada pela medida da
resistência de uma coluna uniforme da solução que tem a forma definida pela área
dos eletrodos colocados nas extremidades desta coluna. O circuito eletrônico da
cela é ilustrado na figura abaixo.

152
RL1 e RL2 são as resistência dos cabos condutores, que, geralmente, pode ser
ignorada; CP e RS são a capacitância e a resistência entre os eletrodos; Cdc1 e
Cdc2, são as capacitâncias das duplas camadas nos dois eletrodos e Z1 e Z2 são as
impedâncias faradaicas nos dois eletrodos, a eventuais reações eletródicas.
Aplicando um potencial de corrente contínua, nada acontece (salvo por um
breve instante durante a polarização dos eletrodos ou se a tensão for suficiente
para provocar reações eletródicas), uma vez que não pode fluir corrente através de
um capacitor.
Aplicando um potencial de corrente alternada, cada um dos componentes
Cdc passa a ser um caminho tão fácil para a passagem de corrente c-a que não
chega a se estabelecer um potencial ao longo dos componentes Z capaz de fazer
fluir corrente faradaica.
Se Cp puder ser mantido em nível negligenciável e os componentes Cdc em
nível elevado, Rs pode ser avaliado sozinho. Na prática, a capacitância da dupla
camada é aumentada muitas vezes por meio da cobertura dos eletrodos com
negro de platina, que aumenta, bastante, a área superficial e minimiza as correntes
faradaicas por aumentar, bastante a capacitância da dupla camada.
Medidas de Condutância de Soluções Iônicas
A condutância de soluções iônicas é comumente medida com circuitos ponte
de Wheatstone operando com corrente alternada para eliminar a ocorrência de
reações eletródicas, que alterariam a composição da solução e produziria um
medida instável.
Medidas de Condutância de Soluções Iônicas com Pont e de Wheastone
A figura abaixo mostra o circuito eletrônico de um condutivímetro que utiliza
uma ponte de Wheatstone para medida de condutância e que opera com uma
corrente alternada de 1000Hz a um potencial de 6 a 10V pico a pico.
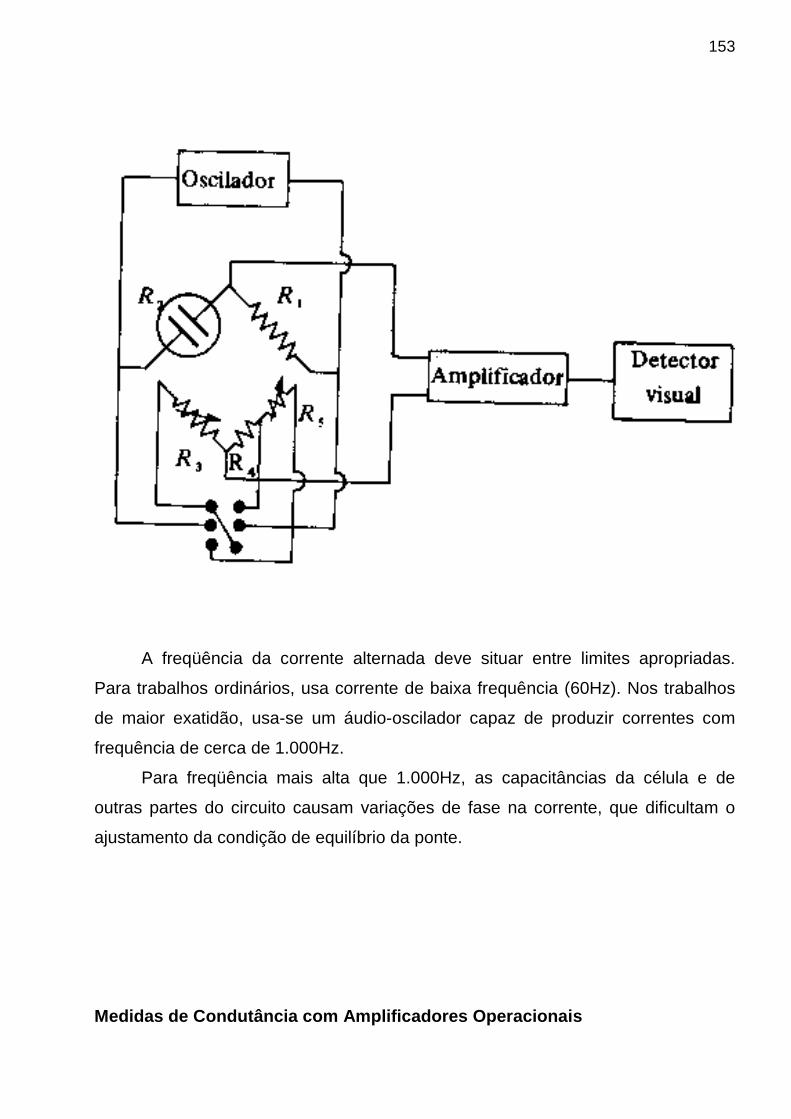
153
A freqüência da corrente alternada deve situar entre limites apropriadas.
Para trabalhos ordinários, usa corrente de baixa frequência (60Hz). Nos trabalhos
de maior exatidão, usa-se um áudio-oscilador capaz de produzir correntes com
frequência de cerca de 1.000Hz.
Para freqüência mais alta que 1.000Hz, as capacitâncias da célula e de
outras partes do circuito causam variações de fase na corrente, que dificultam o
ajustamento da condição de equilíbrio da ponte.
Medidas de Condutância com Amplificadores Operacion ais
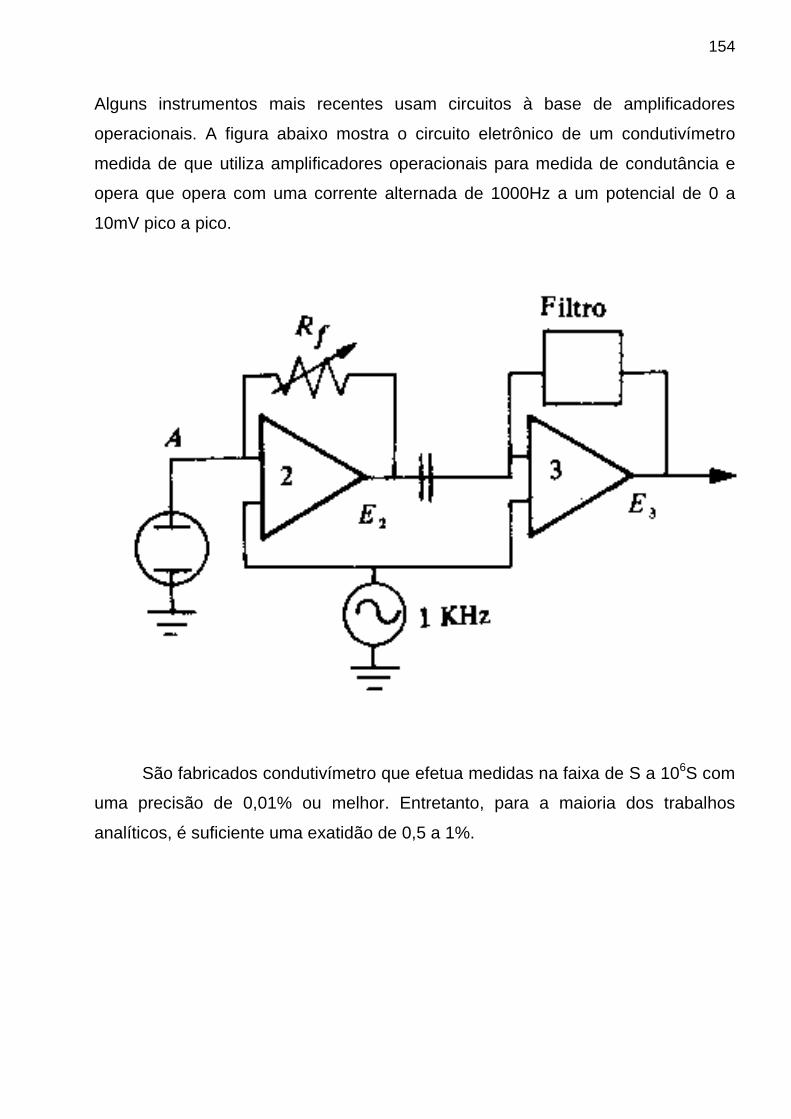
154
Alguns instrumentos mais recentes usam circuitos à base de amplificadores
operacionais. A figura abaixo mostra o circuito eletrônico de um condutivímetro
medida de que utiliza amplificadores operacionais para medida de condutância e
opera que opera com uma corrente alternada de 1000Hz a um potencial de 0 a
10mV pico a pico.
São fabricados condutivímetro que efetua medidas na faixa de S a 106S com
uma precisão de 0,01% ou melhor. Entretanto, para a maioria dos trabalhos
analíticos, é suficiente uma exatidão de 0,5 a 1%.

155
Celas Condutométricas e Medidas de Condutância As celas condutométricas consistem de um recipiente para a solução e dois eletrodos de platina com uma área A em forma de lâminas alinhadas paralelamente e separadas por uma distância l, que definem uma coluna uniforme de solução.
Quando utilizadas para medidas de condutância, as celas necessitam ter uma geometria constante conhecida. Contudo, não é preciso conhecer os valores particulares dos parâmetros l e A, bastando conhecer apenas a relação l/A , conhecida como constante de cela . No caso das celas para titulações condutométricas, não é necessário nem conhecer o valor da constante da cela, l/A , pois o que interessa é acompanhar a variação da condutância que ocorre durante o processo. Neste caso, o essencial é que os eletrodos se mantenham em posições fixas durante a titulação. Calibração das Celas de Condutância Para uma determinada cela usada na medida da condutância, I, A e a razão I/A=θθθθ são constantes, onde θθθθ representa a “constante de cela”. Assim:
θ⋅= Lk
A partir de uma solução-padrão, geralmente de KCl com concentração e condutância específica conhecidas (existem tabelas), pode-se determinar a constante de cela (θθθθ), medindo-se a condutância L da solução-padrão (KCl) em um condutímetro ou condutivímetro. Correção do Solvente nas Medidas de Condutância É importante ressaltar a importância da pureza do solvente (geralmente água) nos trabalhos de condutância de soluções iônicas. Sendo assim, nas medidas de maior exatidão é necessário levar em conta a contribuição do solvente na condutância observada. Considera-se que para a água de alta pureza (água de condutividade), sua condutância específica seja inferior a 1,0 x 10-7 S cm-1 a 25 0C. Por outro lado, água destilada em equilíbrio com dióxido de carbono da atmosfera (água de equilíbrio) possui uma condutância específica de 1,0 x 10-6 S cm-1 a 25 0C.

156
As condutâncias específicas da maioria das soluções encontradas na prática são maiores que 10-3 S cm-1 de modo que, neste caso, a condutância específica do solvente pode ser ignorada. Para as soluções cujas condutividades específicas são menores que 10-3 S cm-1, a contribuição do solvente deve ser descontada especialmente nos trabalhos mais exigentes. Condutância versus Concentração A condutância específica de uma solução diluída de um eletrólito varia quase que linearmente com a concentração do eletrólito conforme é ilustrado nos dados a seguir referentes a soluções de KCl.
Conc.(mols.L -1) 0,0001 0,0010 0.0100 0,1000 1,0000 κκκκ(a 25oC)(S.cm -1) 0,00001489 0,0001469 0,001413 0,01289 0,1119
Para concentrações mais elevadas, a não linearidade no aumento de κκκκ com a concentração ocorre devido a interações iônicas. Para soluções diluídas (<10-2 mol L-1) de um eletrólito forte, a condutância específica para uma faixa de concentração menor do que uma década pode ser dada por:
C.kk ,= Como k = L.θθθθ e k = k’.C , então, L.θθθθ = k ’.C, ou seja, C.kL ,,=
onde k ’’ = k ’ / θθθθ. Este aumento linear está relacionado com aumento do número de íons responsáveis pela condução e este cresce na medida em que se aumenta a concentração do eletrólito. Este comportamento pode não ser observado em eletrólitos fracos, porque o aumento na concentração pode conduzir a uma diminuição do grau de ionização e ao aumento não proporcional da quantidade de íons. Análise Qualitativa por Condutimetria A condutimetria não é uma técnica que permita a identificação dos componentes de uma amostra. Todos os íons presentes numa amostra contribuem indistintamente para a medida da condutância. Portanto, é uma técnica carente de especificidade. Análise Quantitativa por Condutimetria As análises quantitativas podem ser realizadas por condutimetria direta e por titulação condutimétrica . Condutimetria Direta A condutimetria direta baseia-se em medidas da condutância específica. Seu campo de aplicação, na análise quantitativa, é relativamente limitado, em virtude da carência de especificidade da condutância. Aplicações da Condutimetria Direta

157
A determinação da concentração de uma espécie em uma amostra, usando medidas da condutância e uma curva analítica, só pode ser utilizadas em amostras contendo um único eletrólito, o que é difícil na prática.
Curvas analíticas, usando soluções de NaCl (em ppm ou ppb) como padrão, tem sido utilizada em medidores de impurezas de água, salinidade de água do mar, água potável e água salobra.
A medida de condutância pode ser utilizada para determinar a concentração de uma espécie formada em uma reação específica. Por exemplo, o nitrogênio total em matéria orgânica pode ser determinado por conversão em amônia, que é, então, absorvida em solução de ácido bórico para medida de condutância. Pode-se determinar a percentagem de enxofre e carbono em aços ou matéria orgânica, queimando-os e absorvendo o dióxido de enxofre em soluções de peróxido e o dióxido de carbono em solução de hidróxido de bário. Em todos os três casos anteriores, o gás absorvido aumentará ou diminuirá a condutância da solução absorvente e o teor de N, C e O pode ser determinado por curva analítica.
Em geral, a condutividade pode ser utilizada para determinar qualquer elemento existente numa dada amostra, quando esta pode ser convertida numa espécie ionizada e titulada usando uma reação de ácido-base, de precipitação ou complexação. Por ser um método rápido e muito sensível, as medidas de condutância específica têm sido utilizadas para avaliar a pureza de água destilada e deionizada. A condutância específica de uma água de alta pureza deve ser inferior a 0,1µS.cm-1 ou resistência específica de 10MΩ.cm (a 25oC). Água deionizada de um ótimo deionizador Milli-Q-plus apresenta condutância específica de 0,055 µS.cm-1 ou resistência específica de 18,2 MΩ.cm. A água destilada em equilíbrio com o CO2 da atmosfera possui uma condutância específica de 1µS.cm-1 ou resistência específica de 1 MΩ.cm. Correção da Condutividade da Água A pureza da água é importante nos trabalhos sobre condutâncias. As condutâncias específicas da maioria das soluções encontradas, na prática, são maiores do que 10-3S.cm-1 e, então, a condutância específica da água pode ser ignorada. Contudo, no caso de soluções com condutâncias específicas menores do que 10-3S.cm-1, a contribuição da água deve ser descontada. Esta correção só é válida quando os íons do eletrólito presente na solução não reagem com as impurezas iônicas da água. Titulações Condutimétricas A titulação condutimétrica consiste em se acompanhar a variação da condutância no curso de uma titulação. O ponto final é assinalado por uma descontinuidade na curva de condutância versus volume do titulante adicionado. Célula de Titulação Condutimétrica

158
A titulação condutimétrica requer uma célula que possibilite a fácil adição dos incrementos da solução titulante e as sucessivas medidas de condutância. A célula não precisa ser calibrada, ou seja, não é necessária a determinação da constante de célula, basta apenas que os eletrodos permaneçam em posições fixas durante a operação. A figura abaixo mostra uma célula para titulação
condutimétrica. A distância entre os eletrodos pode ser variada de acordo com a condutância
da solução; menor para as soluções de baixa condutância (L = k (A/l)) e maior para as de condutância elevada. Os eletrodos devem ser dispostos em posição vertical, pois, se houver formação de precipitado durante a titulação, não depositará material sólido sobre os eletrodos.
A cada adição de titulante ocasiona um certo aumento de volume e, portanto, um certo efeito de diluição, que se reflete na condutância e deve ser corrigido multiplicando a condutância medida pelo fator (V + v)/V, onde V é o volume da amostra e (V + v) é o volume total da solução após cada adição do titulante.
Na maioria das titulações, é dispensável a termostatização da célula. Porém, nas titulações que envolvem considerável calor de reação, é recomendável termostatizar a célula e colocar um termômetro, controlando a temperatura dentro de uma faixa de ±10C. Para que os erros nas medidas de condutância não sejam superiores a 0,2%, a temperatura deve ser controlada em ±10C.
Para uma maior exatidão na localização do ponto final, é desejável que os dois ramos da curva de condutância vs volume formem um ângulo agudo. Para um ângulo muito obtuso, uma pequena diferença na medida da condutância acarreta um erro considerável na localização do ponto final. Sob condições favoráveis, o ponto final nas titulações condutimétricas é localizado com uma precisão de 0,5%.
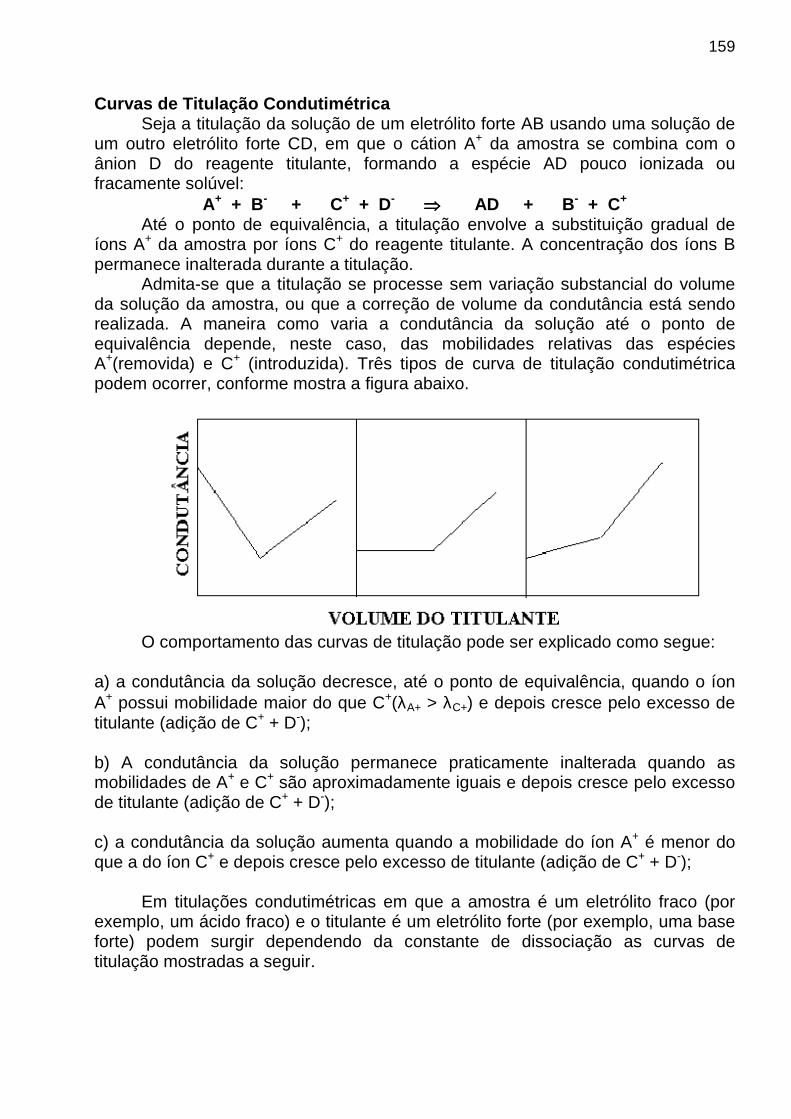
159
Curvas de Titulação Condutimétrica Seja a titulação da solução de um eletrólito forte AB usando uma solução de um outro eletrólito forte CD, em que o cátion A+ da amostra se combina com o ânion D do reagente titulante, formando a espécie AD pouco ionizada ou fracamente solúvel:
A+ + B- + C+ + D- ⇒⇒⇒⇒ AD + B - + C+
Até o ponto de equivalência, a titulação envolve a substituição gradual de íons A+ da amostra por íons C+ do reagente titulante. A concentração dos íons B permanece inalterada durante a titulação.
Admita-se que a titulação se processe sem variação substancial do volume da solução da amostra, ou que a correção de volume da condutância está sendo realizada. A maneira como varia a condutância da solução até o ponto de equivalência depende, neste caso, das mobilidades relativas das espécies A+(removida) e C+ (introduzida). Três tipos de curva de titulação condutimétrica podem ocorrer, conforme mostra a figura abaixo.
O comportamento das curvas de titulação pode ser explicado como segue:
a) a condutância da solução decresce, até o ponto de equivalência, quando o íon A+ possui mobilidade maior do que C+(λA+ > λC+) e depois cresce pelo excesso de titulante (adição de C+ + D-); b) A condutância da solução permanece praticamente inalterada quando as mobilidades de A+ e C+ são aproximadamente iguais e depois cresce pelo excesso de titulante (adição de C+ + D-); c) a condutância da solução aumenta quando a mobilidade do íon A+ é menor do que a do íon C+ e depois cresce pelo excesso de titulante (adição de C+ + D-); Em titulações condutimétricas em que a amostra é um eletrólito fraco (por exemplo, um ácido fraco) e o titulante é um eletrólito forte (por exemplo, uma base forte) podem surgir dependendo da constante de dissociação as curvas de titulação mostradas a seguir.
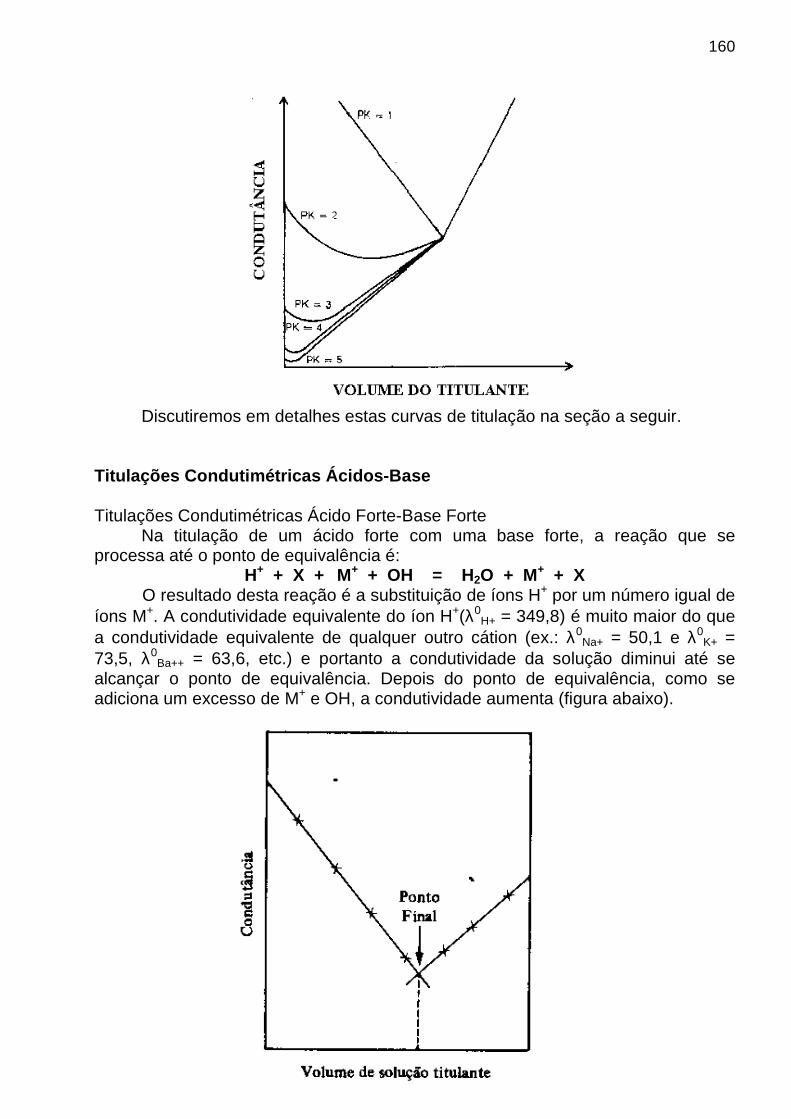
160
Discutiremos em detalhes estas curvas de titulação na seção a seguir.
Titulações Condutimétricas Ácidos-Base Titulações Condutimétricas Ácido Forte-Base Forte Na titulação de um ácido forte com uma base forte, a reação que se processa até o ponto de equivalência é:
H+ + X + M+ + OH = H2O + M+ + X
O resultado desta reação é a substituição de íons H+ por um número igual de íons M+. A condutividade equivalente do íon H+(λ0
H+ = 349,8) é muito maior do que a condutividade equivalente de qualquer outro cátion (ex.: λ0
Na+ = 50,1 e λ0K+ =
73,5, λ0Ba++ = 63,6, etc.) e portanto a condutividade da solução diminui até se
alcançar o ponto de equivalência. Depois do ponto de equivalência, como se adiciona um excesso de M+ e OH, a condutividade aumenta (figura abaixo).

161
Titulações Condutimétricas Ácido Fraco-Base Forte No início da titulação, a reação
HX + M+ + OH = H2O + M+ + X-
produz efetivamente íons X. O aumento resultante na concentração de X, em virtude do equilíbrio
HX = H+ + X
é conseguido à custa da diminuição da concentração de H+. Como a condutividade equivalente do H+ é muito elevada, o decréscimo do termo CH+•λH+ é maior do que o simultâneo crescimento de CM+ •λM+ e CX•λX e, assim, a condutância total da solução diminui. Posteriormente, contudo, a concentração de H+ diminui para um valor tão baixo que a sua contribuição para a condutividade da solução (que agora contém uma concentração relativamente grande de M+ e X) é desprezível.
A condutância total da solução passa por um mínimo e depois começa a aumentar, pois a neutralização de uma maior fração de ácido provoca somente um aumento no número de íons M+ e X e, assim, a condutividade aumenta até o ponto de equivalência.
Além do ponto de equivalência, a curva de titulação reflete o aumento da condutância devido ao aumento do excesso de titulante (eletrólito forte) que tem uma sensibilidade maior do que o aumento de M+ e X.
Como se pode verificar na figura anterior, há uma certa dificuldade em detectar o ponto final de uma titulação ácido fraco-base forte, quando o pK do ácido está entre 1,7 e 2,7, porque a curva antes do ponto final de titulação não é uma reta. Pode resolver essa dificuldade determinando a "linha do sal" obtida efetuando uma titulação do mesmo volume de água com uma solução do sal de sódio (ou potássio) da mesma concentração que a base previamente usada, obtendo-se o ponto final como mostra a figura abaixo.
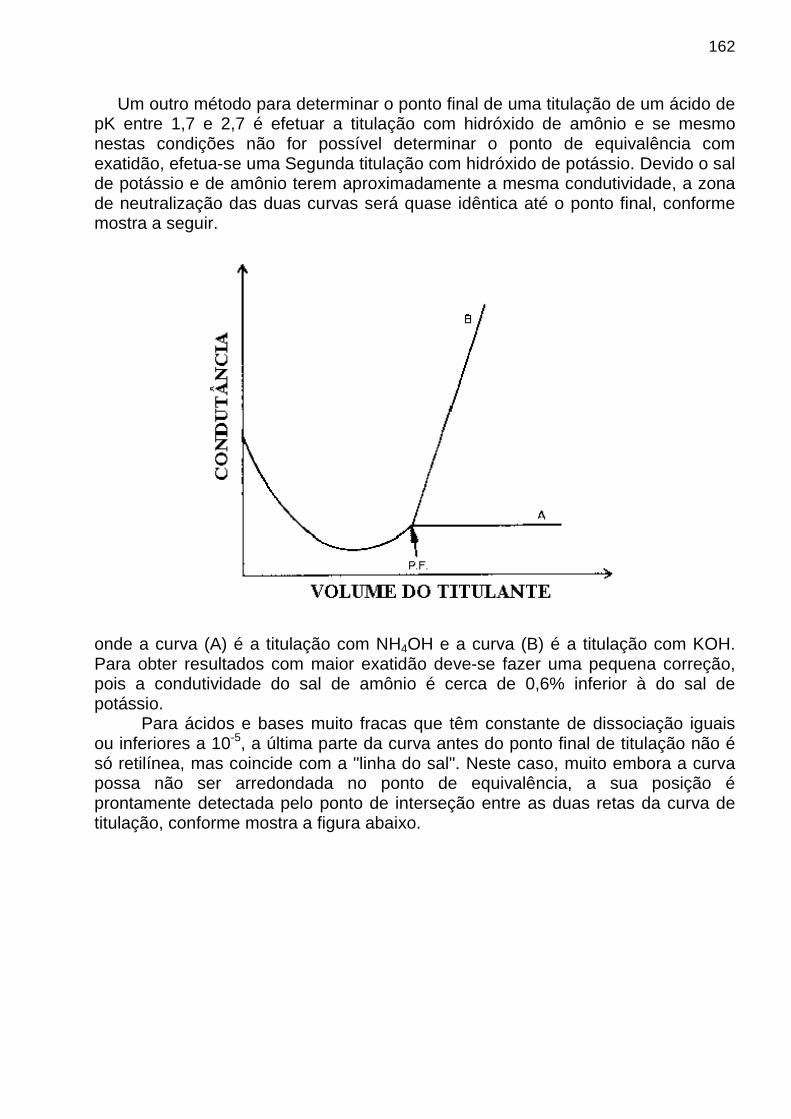
162
Um outro método para determinar o ponto final de uma titulação de um ácido de pK entre 1,7 e 2,7 é efetuar a titulação com hidróxido de amônio e se mesmo nestas condições não for possível determinar o ponto de equivalência com exatidão, efetua-se uma Segunda titulação com hidróxido de potássio. Devido o sal de potássio e de amônio terem aproximadamente a mesma condutividade, a zona de neutralização das duas curvas será quase idêntica até o ponto final, conforme mostra a seguir.
onde a curva (A) é a titulação com NH4OH e a curva (B) é a titulação com KOH. Para obter resultados com maior exatidão deve-se fazer uma pequena correção, pois a condutividade do sal de amônio é cerca de 0,6% inferior à do sal de potássio. Para ácidos e bases muito fracas que têm constante de dissociação iguais ou inferiores a 10-5, a última parte da curva antes do ponto final de titulação não é só retilínea, mas coincide com a "linha do sal". Neste caso, muito embora a curva possa não ser arredondada no ponto de equivalência, a sua posição é prontamente detectada pelo ponto de interseção entre as duas retas da curva de titulação, conforme mostra a figura abaixo.
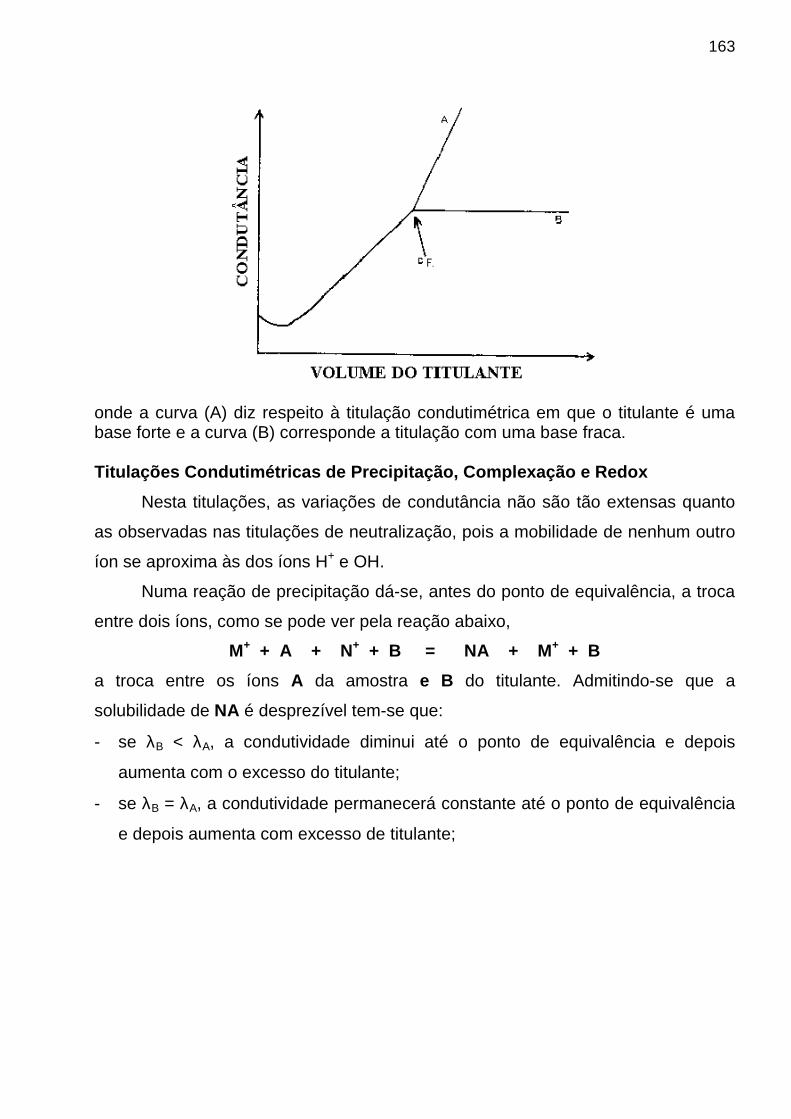
163
onde a curva (A) diz respeito à titulação condutimétrica em que o titulante é uma base forte e a curva (B) corresponde a titulação com uma base fraca. Titulações Condutimétricas de Precipitação, Complex ação e Redox
Nesta titulações, as variações de condutância não são tão extensas quanto
as observadas nas titulações de neutralização, pois a mobilidade de nenhum outro
íon se aproxima às dos íons H+ e OH.
Numa reação de precipitação dá-se, antes do ponto de equivalência, a troca
entre dois íons, como se pode ver pela reação abaixo,
M+ + A + N+ + B = NA + M + + B
a troca entre os íons A da amostra e B do titulante. Admitindo-se que a
solubilidade de NA é desprezível tem-se que:
- se λB < λA, a condutividade diminui até o ponto de equivalência e depois
aumenta com o excesso do titulante;
- se λB = λA, a condutividade permanecerá constante até o ponto de equivalência
e depois aumenta com excesso de titulante;
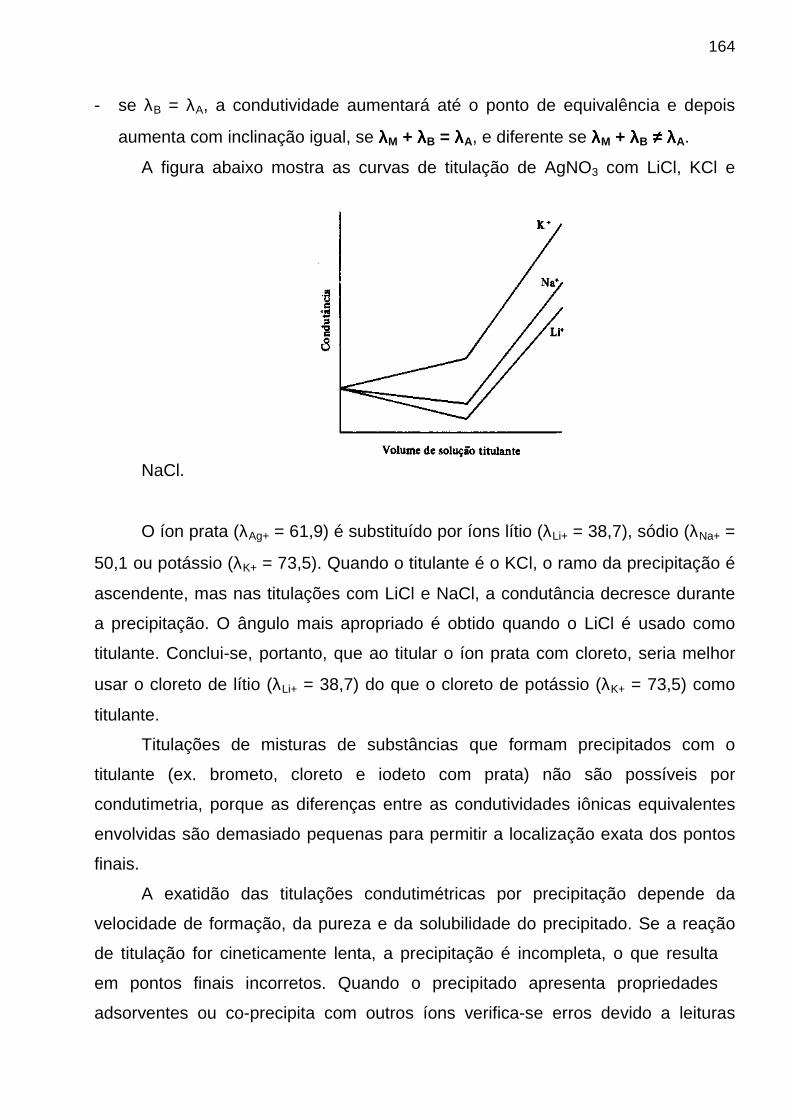
164
- se λB = λA, a condutividade aumentará até o ponto de equivalência e depois
aumenta com inclinação igual, se λλλλM + λλλλB = λλλλA, e diferente se λλλλM + λλλλB ≠≠≠≠ λλλλA.
A figura abaixo mostra as curvas de titulação de AgNO3 com LiCl, KCl e
NaCl.
O íon prata (λAg+ = 61,9) é substituído por íons lítio (λLi+ = 38,7), sódio (λNa+ =
50,1 ou potássio (λK+ = 73,5). Quando o titulante é o KCl, o ramo da precipitação é
ascendente, mas nas titulações com LiCl e NaCl, a condutância decresce durante
a precipitação. O ângulo mais apropriado é obtido quando o LiCl é usado como
titulante. Conclui-se, portanto, que ao titular o íon prata com cloreto, seria melhor
usar o cloreto de lítio (λLi+ = 38,7) do que o cloreto de potássio (λK+ = 73,5) como
titulante.
Titulações de misturas de substâncias que formam precipitados com o
titulante (ex. brometo, cloreto e iodeto com prata) não são possíveis por
condutimetria, porque as diferenças entre as condutividades iônicas equivalentes
envolvidas são demasiado pequenas para permitir a localização exata dos pontos
finais.
A exatidão das titulações condutimétricas por precipitação depende da
velocidade de formação, da pureza e da solubilidade do precipitado. Se a reação
de titulação for cineticamente lenta, a precipitação é incompleta, o que resulta
em pontos finais incorretos. Quando o precipitado apresenta propriedades
adsorventes ou co-precipita com outros íons verifica-se erros devido a leituras

165
inferiores ou encurvamento da curva de titulação. É vantajoso que o precipitado
apresente baixo produto de solubilidade. O produto de solubilidade não deve ser
superior a 5 x 105, na titulação de uma solução 0,1N e maior do 5 x 10-9 na
titulação de uma solução 0,001N
A solubilidade dos precipitados é, frequentemente, reduzida por adição de
etanol (30-40%), que também acelera a precipitação. Todavia, a adição de etanol
provoca uma sensível elevação da temperatura e, por isso, a titulação só deve ser
iniciada depois que a mistura tenha entrado em equilíbrio térmico.
As titulações condutimétricas baseadas em reações de complexação são
menos numerosas e para serem utilizáveis necessitam que o complexo seja
bastante estável. Um titulante muito usado para uma grande variedade de metais é
o EDTA.
Quase todas as titulações redox são efetuadas na presença de um grande
excesso de ácido ou base. Uma titulação típica é a titulação de Fe(II) com
permanganato. Embora a reação resultante
MnO4 + 5Fe2+ + 3H+ = Mn2+ + 5Fe3+ + H2O
Consuma íons H+, diminuindo a condutividade da solução até o ponto de
equivalência, a fração removida é muito pequena, de modo que a variação na
condutividade são normalmente menores do que 1%. Por esta razão, titulações
condutimétricas redox são raramente realizadas.
Aplicações da Condutometria Direta i) avaliação da pureza de água destilada e deionizada:
♦ água de alta pureza ⇒ k < 0,1 µS cm-1 (250C);
♦ água de deionizador Milli-Q-plus ⇒ k ≈ 5,5x10-2 µS cm-1;
♦ água em equilíbrio com CO2 ⇒ 1 µS cm-1 (250C);
ii) Curvas analíticas, usando soluções de NaCl como padrão, tem sido utilizada em medidores de impurezas de água, salinidade de água do mar, água potável e água salobra.

166
Aplicações da Titulação Condutométrica Em geral, a condutividade ou condutância pode ser usada para determinar, a princípio, qualquer espécie existente numa dada amostra se esta for convertida numa espécie ionizada e titulada usando uma reação de: ♦ neutralização (p.e., titulação de ácido acético no vinagre usando NaOH como titulante, etc); ♦ precipitação (p.e., titulação de solução de AgNO3 com cloreto de lítio, sódio ou potássio, etc); ♦ complexação (p.e., alguns metais podem ser titulados com EDTA, etc).